
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 29 September 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.1020948
This article is part of the Research TopicCytotoxic Impact of Tau Oligomers and RNA-Binding Proteins (RBPs) in Neurodegenerative Diseases.View all 4 articles
 Cinzia Tiloca1
Cinzia Tiloca1 Stefano Goldwurm2
Stefano Goldwurm2 Narghes Calcagno1,3
Narghes Calcagno1,3 Federico Verde1,4
Federico Verde1,4 Silvia Peverelli1Daniela Calini1Anna Lena Zecchinelli2Davide Sangalli1,5
Silvia Peverelli1Daniela Calini1Anna Lena Zecchinelli2Davide Sangalli1,5 Antonia Ratti1,6
Antonia Ratti1,6 Gianni Pezzoli2
Gianni Pezzoli2 Vincenzo Silani1,4
Vincenzo Silani1,4 Nicola Ticozzi1,4*
Nicola Ticozzi1,4*Background: Aggregates of TAR DNA-binding protein of 43 kDa (TDP-43) represent the pathological hallmark of most amyotrophic lateral sclerosis (ALS) and of nearly 50% of frontotemporal dementia (FTD) cases but were also observed to occur as secondary neuropathology in the nervous tissue of patients with different neurodegenerative diseases, including Parkinson’s disease (PD) and atypical parkinsonism. Mutations of TARDBP gene, mainly in exon 6 hotspot, have been reported to be causative of some forms of ALS and FTD, with clinical signs of parkinsonism observed in few mutation carriers.
Methods: Direct DNA sequencing of TARDBP exon 6 was performed in a large Italian cohort of 735 patients affected by PD (354 familial and 381 sporadic) and 142 affected by atypical parkinsonism, including 39 corticobasal syndrome (CBS) and 103 progressive sopranuclear palsy (PSP). Sequencing data from 1710 healthy, ethnically matched controls were already available.
Results: Four TARDBP missense variants (p.N267S, p. G294A, p.G295S, p.S393L) were identified in four patients with typical PD and in two individuals with atypical parkinsonism (1 CBS and 1 PSP). None of the detected mutations were found in healthy controls and only the variant p.N267S was previously described in association to idiopathic familial and sporadic PD and to CBS.
Conclusion: In this study we provide further insight into the clinical phenotypic heterogeneity associated with TARDBP mutations, which expands beyond the classical ALS and FTD diseases to include also PD and atypical parkinsonism, although with a low mutational frequency, varying considerably in different Caucasian populations. In addition, our study extends the spectrum of TARDBP pathogenetic mutations found in familial and sporadic PD.
TAR DNA-binding protein of 43 kDa (TDP-43), encoded by TARDBP gene, was originally described as the major component of ubiquitin-positive neuronal inclusions in nearly all (97%) amyotrophic lateral sclerosis (ALS) cases and in nearly 50% of patients with frontotemporal degeneration (FTLD-TDP), while most of the remaining cases have tau pathology (FTLD-tau). As such, ALS and frontotemporal dementia (FTD) are now recognized as opposite ends of the same disease continuum (Ling et al., 2013). Mutations of TARDBP gene were initially identified as causative of ALS, with a prevalence of 5% in familial and 1–2% in sporadic ALS cases (Sreedharan et al., 2008; Lattante et al., 2013), but ranging widely across populations from 0 to 12 and 0 to 5%, respectively (Gitcho et al., 2008; Guerreiro et al., 2008; Kabashi et al., 2008; Corrado et al., 2009; Gijselinck et al., 2009; Piaceri et al., 2012; Bertolin et al., 2014; Narain et al., 2018). Other studies detected TARDBP mutations also in FTD and ALS-FTD cases, although with a very low frequency (Benajiba et al., 2009; Borroni et al., 2009, 2010; Praline et al., 2012). Mutations in TARDBP gene predominantly map in exon 6 which encodes for the C-terminal region, a glycin-rich low complexity domain with a crucial role in protein-protein interaction, nucleo-cytoplasmic shuttling and aggregation propensity (Ticozzi et al., 2012). Interestingly, autoptic studies of ALS and FTD cases have shown a widespread distribution of TDP-43 aggregates through the whole central nervous system (CNS), suggesting a four-stage model of spreading of TDP-43 pathology with disease progression (Brettschneider et al., 2013). In stage 1, TDP-43 inclusions mainly occur in the projection neurons of the agranular motor cortex and lower motor neurons of brainstem and spinal cord, while in stage 2 they are observed in the reticular formation and parvocellular portion of the red nucleus. In stages 3 and 4, TDP-43 aggregates involve the pre-frontal areas, the striatum and the allocortical regions, providing a biological basis for the development of additional clinical manifestations observed in these patients. Indeed, although poorly reported in the past years, extrapyramidal features such as backward falls, retropulsion, bradykinesia, rigidity, and postural instability are now increasingly observed in ALS cases (Pasquini et al., 2022), being relevant for therapeutic treatment, rehabilitation and physiotherapy strategies. Nigrostriatal system dysfunction has been observed also by neuroimaging studies not only in ALS with parkinsonism (ALS-P) but also in ALS cases without clinical signs of parkinsonism, with substantia nigra hyperechogenicity reported by transcranial sonography in 67% of patients with sporadic ALS (Takahashi et al., 1993; Fathinia et al., 2013). In patients affected by the behavioral variant of FTD (bvFTD), parkinsonism represents a frequent clinical presentation, with symptoms like bradykinesia, parkinsonian gait, rigidity, and abnormal posture occurring before, during or after the onset of behavioral and cognitive changes (Baizabal-Carvallo and Jankovic, 2016). Significantly, the possibility of a common pathogenic mechanism affecting ALS/FTD and other neurodegenerative diseases has been suggested also by the finding of TDP-43 positive aggregates in the nervous tissue of patients suffering from alpha-synucleinopathies [Parkinson’s Disease (PD), dementia with Lewy Bodies (DLB), and multiple system atrophy (MSA)], tauopathies [progressive sopranuclear palsy (PSP) and corticobasal syndrome (CBS)] Alzheimer’s disease and Huntington’s disease (Chen-Plotkin et al., 2010).
Consistent with evidence indicating a broad role of TDP-43 in neuronal degeneration and with the co-existence of parkinsonism and ALS/FTD in few TARDBP mutation carriers, previous studies were conducted to investigate the role of TARDBP mutations in PD and other parkinsonisms (PSP and CBS). Three independent studies screened PD cohorts of Caucasian American (n = 463), French Canadian (n = 125), and Dutch (n = 429) origin, but failed to detect mutations (Kabashi et al., 2009; Ticozzi et al., 2011; van Blitterswijk et al., 2013). Nevertheless, the first direct link between TARDBP and PD was found in a cohort of PD patients of Sardinian descent, where the p.A382T variant was observed in 8 cases of sporadic PD cases and a family with atypical parkinsonism, with a reported mutational frequency of 2.5% (Quadri et al., 2011). This mutation is the most frequent TARDBP variant among Italian ALS cases and is especially common in Sardinia due to a founder effect, where it accounts for more than one third of all ALS cases (Chiò et al., 2011; Orrù et al., 2012; Borghero et al., 2014). Interestingly, parkinsonism has been reported as a clinical manifestation in some of these cases. Another report identified a single sporadic PD case in Southern Italy carrying the p.N267S mutation (Gagliardi et al., 2018), while a screening of a North American cohort found the same mutation in one familial PD case (Rayaprolu et al., 2013). Most ALS cases with TARDBP mutations manifesting parkinsonism were reported in the Italian population (Borghero et al., 2011; Mosca et al., 2012; Ticozzi et al., 2013; Pasquini et al., 2022) except for one Japanese pedigree. More recently, the first TARDBP mutation outside exon 6 manifesting with parkinsonism was identified in a Chinese ALS family (Chen et al., 2021).
Here, we describe the results of a mutational screening of TARDBP exon 6 in a large Italian cohort of patients with a clinical diagnosis of PD (n = 735) and atypical parkinsonism (n = 142).
The study cohort consisted of 735 patients with PD (443 males and 292 females, mean age at onset 57.8 ± 10.2 years) and 142 with atypical parkinsonism (77 males and 65 females, mean age at onset 65.1 ± 7.1 years). The latter group included 39 CBS and 103 PSP cases. All patients were individuals of Italian origin, who were consecutively recruited at Parkinson Institute, ASST Gaetano Pini-CTO and contributed to the Parkinson Institute Biobank.1 Clinical diagnosis was made by neurologists experienced in the field of movement disorders according to internationally recognized criteria (Postuma et al., 2015; Höglinger et al., 2017; Jabbari et al., 2020). For each patient the following information were collected: gender, age of onset, disease duration at last examination, clinical features. Within the study cohort, 354 PD patients and 12 individuals with atypical parkinsonism presented a positive family history (Supplementary Table 1), defined as at least one first- or second-degree relative having PD.
Publicly available genetic data from 1710 Italian individuals (non-Sardinians) without known neurological disorders previously screened for TARDBP exon 6 mutations were used as control data (Corrado et al., 2009; del Bo et al., 2009; Origone et al., 2010; Conforti et al., 2011; Gagliardi et al., 2018).
Genomic DNA was isolated from peripheral blood according to standard procedures. The TARDBP exon 6 and the exon-intron boundary regions were amplified by polymerase chain reaction (PCR) as previously described (Corrado et al., 2009). PCR products were purified enzimatically by using illustra™ ExoStar™ (GE Healthcare, Boston, MA, United States) and analyzed by direct sequencing using BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, United States) on 3500 Genetic Analyzer (Applied Biosystems, Waltham, MA, United States). All identified variants were confirmed by sequencing an independent PCR product. The following splicing analysis tools were used to assess the possible effects of identified variants on splicing regulatory sites: Human Splicing Finder, NNSplice, MaxEntScan, GeneSplicer, Splice site analysis-SSF, ESE Finder. Nucleotide numbering of TARDBP gene variants reflects cDNA numbering with + 1 corresponding to the A of the ATG translation initiation codon in reference sequence NM_007375.3. PD patients enrolled in this study were previously screened for major PD-related genes, including LRRK2, Parkin, PINK1, DJ1, and 45 were found to carry mutations in these genes.
Written informed consent for genetic studies was obtained from individuals recruited by the “Parkinson Institute Biobank,” member of the Telethon Network of Genetic Biobank. This study was approved by the ethical committee of IRCCS Istituto Auxologico Italiano (project DAMARE) and was performed in accordance with the 1964 Declaration of Helsinki and its later amendments. Pseudo-anonymized datasets analyzed for this study are archived on Zenodo (10.5281/zenodo.6985371) and will be shared upon reasonable request.
Genetic analysis of the mutational hotspot exon 6 of the TARDBP gene was performed in 735 patients with PD (354 familial and 381 sporadic) and 142 patients with atypical parkinsonism (39 CBS and 103 PSP). We identified six cases carrying TARDBP missense variants, all occurring in heterozygous state (Table 1 and Supplementary Figure 1).
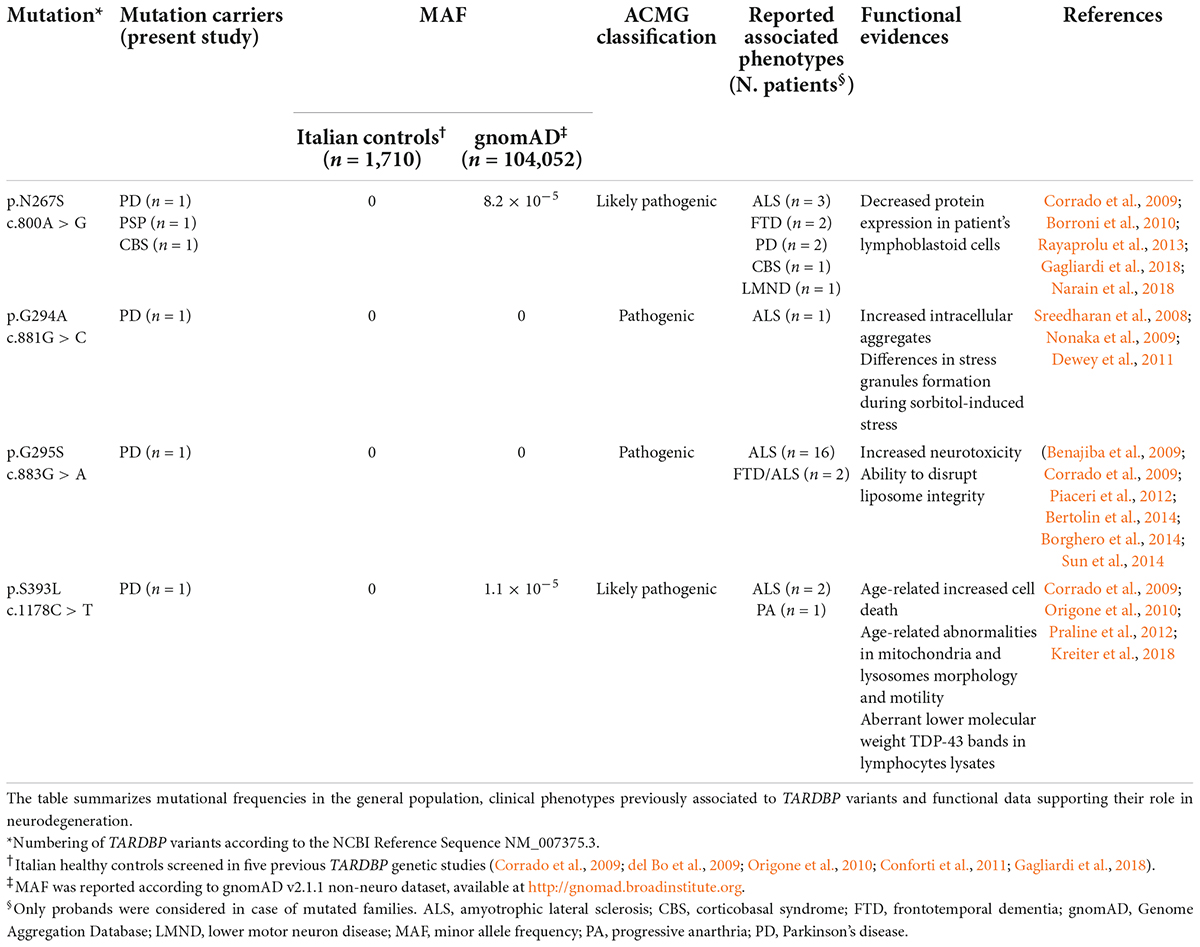
Table 1. TARDBP missense variants identified in our patient cohort.
In particular, four different missense mutations (c.800A > G, p.N267S; c.881G > C, p. G294A; c.883G > A, p.G295S; c.1178C > T, p.S393L) were observed in four individuals affected by typical PD (Table 1). Additionally, the mutation p.N267S was also detected in two patients with atypical parkinsonism (1 PSP and 1 CBS) (Table 1). Overall, we observed a mutational frequency for TARDBP of 0.7% (6/877) in our cohort of Italian parkinsonian patients. These variants were instead absent in 1710 Italian healthy controls, appear to be extremely rare in public sequencing databases and are classified as pathogenic or likely pathogenic according to the ACMG Criteria (Table 1).
Our mutational analysis also detected three other single nucleotide variants in three PD cases: an intronic variant (c.715-31C > T) and two synonymous variants, respectively p.Gly335Gly (c.1005T > A) and p.Ala341Ala (c.1023C > T), The intronic variant upstream exon 6 (c.715-31C > T) was not expected to induce significant splicing motif alterations by different bioinformatics predictive algorithms (Supplementary Table 2). According to the ESEfinder Tool, no impact on exonic splicing enhancers (ESEs) was predicted for the variant c.1005T > A, while the exonic variant c.1023C > T was predicted to disrupt the putative binding site for the SR protein SRSF1, although its biological effect on TDP-43 splicing activity remains unclear. All these variants are classified as likely benign according to the ACMG Criteria (Supplementary Table 2).
The clinical characteristics of patients carrying TARDBP missense mutations are reported in Table 2. Only patient #1149, harboring the p.N267S variant, reported a positive family history, having a brother affected by MSA but unfortunately DNA was not available for segregation analysis. The remaining mutation carriers were all sporadic patients. None of the TARDBP mutated patients carried additional mutations in other PD-associated genes.
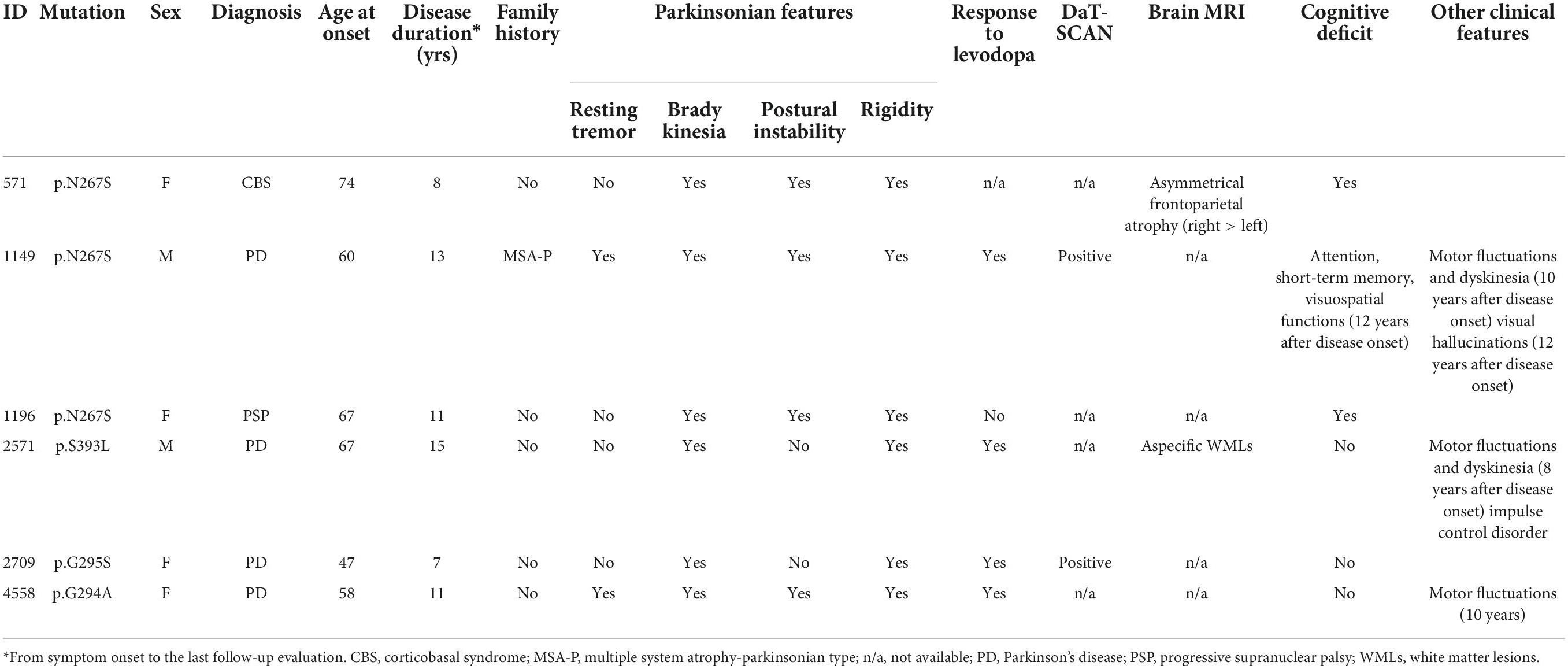
Table 2. Demographic and clinical characteristics of patients harboring TARDBP mutation in the study cohort.
Parkinsonisms are a group of clinically and pathologically heterogeneous neurodegenerative movement disorders, all characterized by the loss of dopaminergic nigrostriatal neurons and by the presence of typical motor symptoms of tremor, rigidity, bradykinesia, and postural instability. Some reports described a secondary TDP-43 pathology in parkinsonisms (7% of PD, 15.4% of CBS, 6% of PSP), although with a specific distinctive pattern of morphology, regional distribution and severity of TDP-43 aggregates compared to ALS/FTD TDP-43 proteinopathies.
In this study we sequenced exon 6 of TARDBP gene in a large cohort of Italian patients affected by different forms of parkinsonism and identified 4/735 (0.5%) PD and 2/142 (1.4%) atypical parkinsonism cases harboring pathogenic mutations. To date, 3,647 PD patients of Caucasian origin have been screened for TARDBP mutations by eight distinct genetic studies, including the present report (Kabashi et al., 2009; Quadri et al., 2011; Ticozzi et al., 2011; Cannas et al., 2013; Rayaprolu et al., 2013; van Blitterswijk et al., 2013; Gagliardi et al., 2018). With the exception of the founder p.A382T mutation in the Sardinian population, TARDBP variants have been reported to occur at very low rate (from 0 to 0.5%) in PD, with a heterogeneous frequency in different populations. TARDBP mutations have been associated with both familial and sporadic PD (literature data reviewed in Supplementary Table 3).
Regarding the TARDBP variants identified in our study, only the p.N267S mutation has been previously found in association with idiopathic familial and sporadic PD and to CBS (Huey et al., 2012; Rayaprolu et al., 2013; Gagliardi et al., 2018; Table 1 and Supplementary Table 3). The other three variants, p.G294A, p.G295S, and p.S393L, have been reported as causative mutations only in ALS or FTD cases with or without parkinsonism, but they have never been described in association with PD and PD-like phenotype. Interestingly, in our large dataset we did not identify p.A382T, which is the most common variant in the Italian population and accounts for 30% of ALS, 0.9–2.5% of PD, and up to 6% of atypical parkinsonisms, as well as 0.5–1.3% healthy controls in Sardinia (Chiò et al., 2011; Quadri et al., 2011; Cannas et al., 2013).
The TARDBP mutations identified in our cohort were instead absent in 1,710 Italian healthy controls, including 771 individuals with no reported history of neurological disorders that we previously screened (Corrado et al., 2009) and 939 subjects enrolled in other case-control studies (del Bo et al., 2009; Origone et al., 2010; Conforti et al., 2011; Gagliardi et al., 2018). Moreover, all these variants resulted to be extremely rare in public sequencing databases and are classified as pathogenic or likely pathogenic according to the ACMG Criteria, with functional data in support of their pathogenicity (Table 1).
The pathomechanisms associated to these specific TARDBP mutations are not fully understood, although functional studies and in silico predictions strongly support their pathogenicity (Nonaka et al., 2009; Dewey et al., 2011; Sun et al., 2014; Kreiter et al., 2018; Table 2). Almost all the mutations identified in our study lead to the creation/disruption of serine residues, with potential consequences on TDP-43 aggregation and function. As TDP-43 is involved in multiple steps of RNA processing, a dysregulation of this protein may impact on many RNA and protein targets. Interestingly, a link between TDP-43 and microtubule-associated protein tau and alfa-synuclein, the major components of the intraneuronal deposits in parkinsonism, has been proposed, although more studies are needed to investigate if TDP−43 and these proteins may mutually promote the reciprocal accumulation. There is evidence that TDP-43 may act as regulator of tau exon 10 splicing, which generates the two isoforms 3R-tau and 4R-tau, in different cell types and in transgenic mice (Gu et al., 2017a). A dysregulation of exon 10 inclusion, possibly induced by mutated TDP-43 protein, may therefore lead to an altered ratio of 3R-tau/4R-tau, similarly to the effect induced by pathological MAPT mutations in exon 10. Additionally, TDP-43 appears to bind the 3′-UTR of MAPT promoting mRNA instability (Gu et al., 2017b). Moreover, in double transgenic mice, the overexpression of wild-type TDP-43 potentiates mutant α-synuclein toxicity leading in turn to a significant loss of dopaminergic neurons compared to both single transgenic mice (Tian et al., 2011). Unfortunately, no brain autopsy material was available for any of the mutated patients in order to evaluate the neuropathological profile and to test the extent and type of TDP-43 pathology.
Despite the relatively small sample size of the atypical parkinsonism group, our findings suggest that TARDBP mutations may also be associated to PSP and CBS, being observed in 1.4% of cases. A previous study on 67 Sardinian patients identified the founder TARDBP variant p.A382T in four patients with atypical parkinsonisms (Cannas et al., 2013), while another report described the p.N267S mutation in. a single CBS case (Huey et al., 2012; Supplementary Table 3). Therefore, given the relatively small number of atypical parkinsonism cases screened so far, it will be interesting to study larger cohorts to determine the role of TARDBP mutations in the pathogenesis off CBS and PSP.
In conclusion, our study indicates that the clinical phenotypic presentation associated to TARDBP mutations, and in particular to the p.N267S variant, may expand beyond the classical ALS and FTD spectrum to include also PD and atypical parkinsonisms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Zenodo (doi: 10.5281/zenodo.6985371).
The studies involving human participants were reviewed and approved by the IRCCS Istituto Auxologico Italiano. The patients/participants provided their written informed consent to participate in this study.
CT, SG, GP, VS, and NT: study design. SG, AZ, and GP: data collection. CT, DC, FV, SP, DS, AR, and NT: genetic analysis. CT, SG, and NT: data analysis. CT, NC, SP, and NT: writing of first draft. SG and NT: supervision. All authors revised the manuscript for intellectual content.
Research funding was provided by the Italian Ministry of Health (Ricerca Corrente to IRCCS Istituto Auxologico Italiano, project DAMARE).
We thank the “Parkinson Institute Biobank,” member of the Telethon Network of Genetic Biobank funded by the TELETHON Italy (project no. GTB12001) and supported by the “Fondazione Grigioni per il Morbo di Parkinson.”
AR received research funding from AriSLA. VS received compensation for consulting services and/or speaking activities from AveXis, Cytokinetics, Italfarmaco, Liquidweb Srl, and Novartis Pharma AG. He receives or has received research support from the Italian Ministry of Health, AriSLA, and E-Rare Joint Transnational Call. He is on the Editorial Board of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, European Neurology, American Journal of Neurodegenerative Diseases, and Frontiers in Neurology. NT received compensation for consulting services and/or speaking activities from Amylyx Pharmaceuticals, Zambon Pharma AG, and Italfarmaco. He received research funding from the Italian Ministry of Health and AriSLA. He is Associate Editor for Frontiers in Aging Neuroscience.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1020948/full#supplementary-material
Baizabal-Carvallo, J. F., and Jankovic, J. (2016). Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 12, 175–185. doi: 10.1038/NRNEUROL.2016.14
Benajiba, L., Ber, I., Camuzat, A., Lacoste, M., Thomas-Anterion, C., Couratier, P., et al. (2009). TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 65, 470–473. doi: 10.1002/ANA.21612
Bertolin, C., D’Ascenzo, C., Querin, G., Gaiani, A., Boaretto, F., Salvoro, C., et al. (2014). Improving the knowledge of amyotrophic lateral sclerosis genetics: Novel SOD1 and FUS variants. Neurobiol. Aging 35, 1212.e7–1212.e10. doi: 10.1016/J.NEUROBIOLAGING.2013.10.093
Borghero, G., Floris, G., Cannas, A., Marrosu, M. G., Murru, M. R., Costantino, E., et al. (2011). A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol. Aging 32, 2327.e1–2327.e5. doi: 10.1016/j.neurobiolaging.2011.06.009
Borghero, G., Pugliatti, M., Marrosu, F., Marrosu, M. G., Murru, M. R., Floris, G., et al. (2014). Genetic architecture of ALS in Sardinia. Neurobiol. Aging 35, 2882.e7–2882.e12. doi: 10.1016/J.NEUROBIOLAGING.2014.07.012
Borroni, B., Archetti, S., del Bo, R., Papetti, A., Buratti, E., et al. (2010). TARDBP mutations in frontotemporal lobar degeneration: Frequency, clinical features, and disease course. Rejuvenation Res. 13, 509–517. doi: 10.1089/REJ.2010.1017
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S., et al. (2009). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30:E974–83. doi: 10.1002/humu.21100
Brettschneider, J., del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Cannas, A., Borghero, G., Floris, G. L., Solla, P., Chiò, A., Traynor, B. J., et al. (2013). The p.A382T TARDBP gene mutation in Sardinian patients affected by Parkinson’s disease and other degenerative parkinsonisms. Neurogenetics 14, 161–166. doi: 10.1007/s10048-013-0360-2
Chen, S., Zhou, R. L., Zhang, W., Che, C. H., Feng, S. Y., Huang, H. P., et al. (2021). Novel TARDBP missense mutation caused familial amyotrophic lateral sclerosis with frontotemporal dementia and parkinsonism. Neurobiol. Aging 107, 168–173. doi: 10.1016/J.NEUROBIOLAGING.2021.05.017
Chen-Plotkin, A. S., Lee, V. M. Y., and Trojanowski, J. Q. (2010). TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220. doi: 10.1038/nrneurol.2010.18
Chiò, A., Borghero, G., Pugliatti, M., Ticca, A., Calvo, A., Moglia, C., et al. (2011). Large proportion of amyotrophic lateral sclerosis cases in sardinia due to a single founder mutation of the TARDBP gene. Arch. Neurol. 68, 594–598. doi: 10.1001/archneurol.2010.352
Conforti, F. L., Sproviero, W., Simone, I. L., Mazzei, R., Valentino, P., Ungaro, C., et al. (2011). TARDBP gene mutations in south Italian patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 587–588. doi: 10.1136/jnnp.2009.198309
Corrado, L., Ratti, A., Gellera, C., Buratti, E., Castellotti, B., Carlomagno, Y., et al. (2009). High frequency of TARDBP gene mutations in italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 30, 688–694. doi: 10.1002/humu.20950
del Bo, R., Ghezzi, S., Corti, S., Pandolfo, M., Ranieri, M., et al. (2009). TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: Identification of two novel mutations. Eur. J. Neurol. 16, 727–32. doi: 10.1111/j.1468-1331.2009.02574.x
Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P., Good, S. K., et al. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell Biol. 31, 1098–1108. doi: 10.1128/MCB.01279-10
Fathinia, P., Hermann, A., Reuner, U., Kassubek, J., Storch, A., and Ludolph, A. C. (2013). Parkinson’s disease-like midbrain hyperechogenicity is frequent in amyotrophic lateral sclerosis. J. Neurol. 260, 454–457. doi: 10.1007/s00415-012-6654-8
Gagliardi, M., Arabia, G., Nisticò, R., Iannello, G., Procopio, R., Manfredini, L., et al. (2018). Mutational analysis of TARDBP gene in patients affected by Parkinson’s disease from Calabria. J. Neurol. Sci. 390, 209–211. doi: 10.1016/j.jns.2018.04.043
Gijselinck, I., Sleegers, K., Engelborghs, S., Robberecht, W., Martin, J. J., Vandenberghe, R., et al. (2009). Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol. Aging 30, 1329–1331. doi: 10.1016/j.neurobiolaging.2007.11.002
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ANA.21344
Gu, J., Chen, F., Iqbal, K., Gong, C. X., Wang, X., and Liu, F. (2017a). Transactive response stocktickerDNA-binding protein 43 (stocktickerTDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J. Biol. Chem. 292, 10600–10612. doi: 10.1074/jbc.M117.783498
Gu, J., Wu, F., Xu, W., Shi, J., Hu, W., Jin, N., et al. (2017b). TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 45, 6177–6193. doi: 10.1093/nar/gkx175
Guerreiro, R. J., Schymick, J. C., Crews, C., Singleton, A., Hardy, J., and Traynor, B. J. (2008). TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS One 3:e2450. doi: 10.1371/journal.pone.0002450
Höglinger, G. U., Respondek, G., Stamelou, M., Kurz, C., Josephs, K. A., Lang, A. E., et al. (2017). Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 32, 853–864. doi: 10.1002/MDS.26987
Huey, E. D., Ferrari, R., Moreno, J. H., Jensen, C., Morris, C. M., Potocnik, F., et al. (2012). FUS and TDP43 genetic variability in FTD and CBS. Neurobio. Aging 33, 1016.e9–17. doi: 10.1016/j.neurobiolaging.2011.08.004
Jabbari, E., Holland, N., Chelban, V., Jones, P. S., Lamb, R., Rawlinson, C., et al. (2020). Diagnosis Across the Spectrum of Progressive Supranuclear Palsy and Corticobasal Syndrome. JAMA Neurol. 77, 377–387. doi: 10.1001/JAMANEUROL.2019.4347
Kabashi, E., Daoud, H., Rivière, J. B., Valdamanis, P. N., Bourgouin, P., Provencher, P., et al. (2009). No TARDBP mutations in a french canadian population of patients with parkinson disease. Arch. Neurol. 66, 281–282. doi: 10.1001/archneurol.2008.568
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Gene. 40, 572–574. doi: 10.1038/ng.132
Kreiter, N., Pal, A., Lojewski, X., Corcia, P., Naujock, M., Reinhardt, P., et al. (2018). Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 115, 167–181. doi: 10.1016/J.NBD.2018.03.010
Lattante, S., Rouleau, G. A., and Kabashi, E. (2013). TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 34, 812–826. doi: 10.1002/humu.22319
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/J.NEURON.2013.07.033
Mosca, L., Lunetta, C., Tarlarini, C., Avemaria, F., Maestri, E., Melazzini, M., et al. (2012). Wide phenotypic spectrum of the TARDBP gene: Homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson’s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol. Aging 33, 1846.e1–4. doi: 10.1016/j.neurobiolaging.2012.01.108
Narain, P., Pandey, A., Gupta, S., Gomes, J., Bhatia, R., and Vivekanandan, P. (2018). Targeted next-generation sequencing reveals novel and rare variants in Indian patients with amyotrophic lateral sclerosis. Neurobiol. Aging 71, e9–265.e14. doi: 10.1016/J.NEUROBIOLAGING.2018.05.012
Nonaka, T., Kametani, F., Arai, T., Akiyama, H., and Hasegawa, M. (2009). Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364. doi: 10.1093/HMG/DDP275
Origone, P., Caponnetto, C., Bandettini Di Poggio, M., Ghiglione, E., Bellone, E., et al. (2010). Enlarging clinical spectrum of FALS with TARDBP gene mutations: S393L variant in an Italian family showing phenotypic variability and relevance for genetic counselling. Amyotroph. Lateral Scler. 11, 223–227. doi: 10.3109/17482960903165039
Orrù, S., Manolakos, E., Orrù, N., Kokotas, H., Mascia, V., Carcassi, C., et al. (2012). High frequency of the TARDBP p.Ala382Thr mutation in Sardinian patients with amyotrophic lateral sclerosis. Clin. Genet. 81, 172–178. doi: 10.1111/J.1399-0004.2011.01668.X
Pasquini, J., Trogu, F., Morelli, C., Poletti, B., Girotti, F., Peverelli, S., et al. (2022). Parkinsonian syndromes in motor neuron disease: A clinical study. Front. Aging Neurosci. 14:917706. doi: 10.3389/FNAGI.2022.917706
Piaceri, I., del Mastio, M., Tedde, A., Bagnoli, S., Latorraca, S., Massaro, F., et al. (2012). Clinical heterogeneity in Italian patients with amyotrophic lateral sclerosis. Clin. Genet. 82, 83–87. doi: 10.1111/J.1399-0004.2011.01726.X
Postuma, R. B., Berg, D., Stern, M., Poewe, W., Olanow, C. W., Oertel, W., et al. (2015). MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601. doi: 10.1002/MDS.26424
Praline, J., Vourc’H, P., Guennoc, A. M., Veyrat-Durebex, C., and Corcia, P. (2012). Co-occurrence of progressive anarthria with an S393L TARDBP mutation and ALS within a family. Amyotroph. Lateral Scler. 13, 155–157. doi: 10.3109/17482968.2011.598168
Quadri, M., Cossu, G., Saddi, V., Simons, E. J., Murgia, D., Melis, M., et al. (2011). Broadening the phenotype of TARDBP mutations: The TARDBP Ala382Thr mutation and Parkinson’s disease in Sardinia. Neurogenetics 12, 203–209. doi: 10.1007/s10048-011-0288-3
Rayaprolu, S., Fujioka, S., Traynor, S., Soto-Ortolaza, A. I., Petrucelli, L., Dickson, D. W., et al. (2013). TARDBP mutations in Parkinson’s disease. Parkinsonism Relat. Disord. 19, 312–315. doi: 10.1016/J.PARKRELDIS.2012.11.003
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Sun, C. S., Wang, C. Y. H., Chen, B. P. W., He, R. Y., Liu, G. C. H., Wang, C. H., et al. (2014). The influence of pathological mutations and proline substitutions in TDP-43 glycine-rich peptides on its amyloid properties and cellular toxicity. PLoS One 9:e103644. doi: 10.1371/JOURNAL.PONE.0103644
Takahashi, H., Snow, B., Bhatt, M. H., Peppard, R., Eisen, A., and Calne, D. B. (1993). Evidence for a dopaminergic deficit in sporadic amyotrophic lateral sclerosis on positron emission scanning. Lancet 342, 1016–1018. doi: 10.1016/0140-6736(93)92878-W
Tian, T., Huang, C., Tong, J., Yang, M., Zhou, H., and Xia, X. G. (2011). TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int. J. Biol. Sci. 7, 234–243. doi: 10.7150/ijbs.7.234
Ticozzi, N., LeClerc, A. L., van Blitterswijk, M., Keagle, P., McKenna-Yasek, D. M., Sapp, P. C., et al. (2011). Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol. Aging 32, 2096–20999. doi: 10.1016/j.neurobiolaging.2009.11.018
Ticozzi, N., Ratti, A., and Silani, V. (2012). Protein Aggregation and Defective RNA Metabolism as Mechanisms for Motor Neuron Damage. CNS Neurol. Disord. Drug Targets 9, 285–296. doi: 10.2174/187152710791292585
Ticozzi, N., Tiloca, C., Mencacci, N. E., Morelli, C., Doretti, A., Rusconi, D., et al. (2013). Oligoclonal bands in the cerebrospinal fluid of amyotrophic lateral sclerosis patients with disease-associated mutations. J. Neurol. 260, 85–92. doi: 10.1007/S00415-012-6589-0
Keywords: TARDBP, TDP-43, Parkinson’s disease, atypical parkinsonism, genetics
Citation: Tiloca C, Goldwurm S, Calcagno N, Verde F, Peverelli S, Calini D, Zecchinelli AL, Sangalli D, Ratti A, Pezzoli G, Silani V and Ticozzi N (2022) TARDBP mutations in a cohort of Italian patients with Parkinson’s disease and atypical parkinsonisms. Front. Aging Neurosci. 14:1020948. doi: 10.3389/fnagi.2022.1020948
Received: 16 August 2022; Accepted: 12 September 2022;
Published: 29 September 2022.
Edited by:
Shu G. Chen, Case Western Reserve University, United StatesReviewed by:
Emanuele Buratti, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2022 Tiloca, Goldwurm, Calcagno, Verde, Peverelli, Calini, Zecchinelli, Sangalli, Ratti, Pezzoli, Silani and Ticozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Ticozzi, bi50aWNvenppQGF1eG9sb2dpY28uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.