 M. I. Holubiec
M. I. Holubiec M. Gellert
M. Gellert E. M. Hanschmann
E. M. Hanschmann- 1IBioBA-MPSP Instituto de Investigación en Biomedicina de Buenos Aires, Partner Institute of the Max Planck Society, Buenos Aires, Argentina
- 2Institute for Medical Biochemistry and Molecular Biology, University Medicine Greifwald, University Greifswald, Greifswald, Germany
- 3Independent Researcher Essen, Germany
Reduction and oxidation reactions are essential for biochemical processes. They are part of metabolic pathways and signal transduction. Reactive oxygen species (ROS) as second messengers and oxidative modifications of cysteinyl (Cys) residues are key to transduce and translate intracellular and intercellular signals. Dysregulation of cellular redox signaling is known as oxidative distress, which has been linked to various pathologies, including neurodegeneration. Alzheimer's disease (AD) is a neurodegenerative pathology linked to both, abnormal amyloid precursor protein (APP) processing, generating Aβ peptide, and Tau hyperphosphorylation and aggregation. Signs of oxidative distress in AD include: increase of ROS (H2O2, O2•−), decrease of the levels or activities of antioxidant enzymes, abnormal oxidation of macromolecules related to elevated Aβ production, and changes in mitochondrial homeostasis linked to Tau phosphorylation. Interestingly, Cys residues present in APP form disulfide bonds that are important for intermolecular interactions and might be involved in the aggregation of Aβ. Moreover, two Cys residues in some Tau isoforms have been shown to be essential for Tau stabilization and its interaction with microtubules. Future research will show the complexities of Tau, its interactome, and the role that Cys residues play in the progression of AD. The specific modification of cysteinyl residues in redox signaling is also tightly connected to the regulation of various metabolic pathways. Many of these pathways have been found to be altered in AD, even at very early stages. In order to analyze the complex changes and underlying mechanisms, several AD models have been developed, including animal models, 2D and 3D cell culture, and ex-vivo studies of patient samples. The use of these models along with innovative, new redox analysis techniques are key to further understand the importance of the redox component in Alzheimer's disease and the identification of new therapeutic targets in the future.
Redox signaling and metabolism
The transport of electrons within reduction and oxidation (redox) reactions is important for biochemical processes within cells. Cells possess electron carriers in the form of co-enzymes and proteins that fulfill different functions. Co-enzymes include the electron acceptors flavin adenine dinucleotide (FAD/FADH), flavin mononucleotide (FMN/FMNH), as well as nicotinamide adenine dinucleotide (NAD+/NADH) and the electron donor nicotinamide adenine dinucleotide phosphate (NADP+/NADPH). Proteins include cytochromes (containing heme), flavoproteins (containing FAD or FMN), iron-sulfur proteins (containing iron) and proteins of the Thioredoxin (Trx) family (containing Cys or Sec residue(s) within their active site motifs). Redox reactions are often part of metabolic pathways and play a role in catabolic and anabolic reactions. Therefore, they are needed for the break-down of molecules and the release of energy, as well as for the biosynthesis of complex molecules including amino acids/proteins, fatty acids/lipids and nucleotides/nucleic acids [reviewed in Hosios and Vander Heiden (2018)].
These biomolecules are known to be prone to modification and even inactivation by oxidation. In fact, first introduced in 1985, the term oxidative stress (today known as oxidative distress) has long linked non-physiological levels of reactive oxygen species (ROS) to oxidation of biomolecules and cellular structures, gene mutations, protein inactivation and aggregation and eventually clinical pathologies (Hanschmann, 2013; Sies, 2015). These reactive species can be produced exogenously or endogenously. Endogenous sources include the production by enzymes such as NADPH oxidase [NOX; generates superoxide (O2•−)], superoxide dismutases [SOD; generates hydrogen peroxide (H2O2) or NO-synthase (NOS; generates nitric oxide (NO))], and generation as by-products by enzymes or aerobic metabolism (e.g. in the respiratory chain) (Hanschmann, 2013). Organelles such as mitochondria contain specific redox networks that are linked to their specific physiological relevance (Riemer et al., 2015). Mitochondria are a major source of O2•− and H2O2. Interestingly, mitochondrial subcompartments have distinct functions and are equipped with different sets of enzymes and redox networks. Today we appreciate the absence of a redox equilibrium and the relevance of reactive species for signal transduction and cell physiology, i.e., oxidative eustress (Hanschmann, 2013). Numerous specialized transporters and shuttles link redox metabolism to other compartments. In fact, a substrate or protein is transported in a specific redox state to another compartment, where it undergoes oxidative modification (Hosios and Vander Heiden, 2018). Many key enzymes of metabolic pathways can be regulated via specific and reversible oxidative modifications that act as thiol switches (Brandes et al., 2009; López-Grueso et al., 2019; Gao et al., 2020). They are part of redox signaling circuits and depend on i) second messengers like H2O2, hydrogen sulfide (H2S), and NO, and ii) the catalysis by enzymes of the Trx family (including thioredoxins, glutaredoxins, and peroxiredoxins) (Hanschmann, 2013).
Redox in Alzheimer's disease
Alzheimer's disease (AD) is a neurodegenerative pathology that has both neurological and cognitive effects (Knopman et al., 2021). AD is first evidenced in two main brain areas: the entorhinal cortex (EC) and the hippocampus (Leng et al., 2021). Like other neurodegenerative diseases, AD presents with selective vulnerability (Leng et al., 2021). Recent studies state that early dysregulation of signaling pathways in the EC include alterations in the redox signaling and neuroinflammation (Olajide et al., 2021). Redox changes and alterations of mitochondrial homeostasis seem to be of utmost importance for the early development of the AD pathology in the EC. Different neuronal populations present distinct sensitivities toward oxidative damage (Wang and Michaelis, 2010), e.g., damage to macromolecules in the early stages of AD was detected in EC neurons, which are particularly sensitive to oxidative damage (Terni et al., 2010). Furthermore, distinct neuronal populations in the hippocampus respond differently to increased oxidative conditions, particularly the CA1 neurons are greatly affected (Wang et al., 2005; Olajide et al., 2021). Redox changes, such as an increase in H2O2, RNA oxidation observed in samples from patients, and mitochondrial dysfunction, that was evidenced as an increase of lipid peroxidation of the α subunit of the mitochondrial ATP-synthase, seem to be of utmost importance for the early development of the AD pathology in the EC (Nunomura et al., 2001; Terni et al., 2010; Olajide et al., 2021).
The most known hallmarks of AD in patients are neurofibrillary tangles (NFT) and amyloid beta (Aβ) deposits. NFTs are mainly conformed by phosphorylated Tau proteins that aggregate in intracellular tangles. Aβ deposits are extracellular aggregates generated from the Aβ-peptide (Figure 1A). These are produced from the proteolysis of the amyloid precursor protein (APP) that takes place through the action of two different secretases (β- and γ-), generating different peptides among which Aβ-40 and Aβ-42 are of utmost relevance in AD (Figures 1A,B). An increase of positive β-secretase levels has been observed in the presence of high levels of peroxides (Tamagno et al., 2008). APP cleavage by α-secretase initiates the non-amyloidogenic pathway, the primary α-secretase is a metalloprotease called ADAM10 that is also involved in the regulation of redox related proteins, such as Trx1 cleavage and generation of the secreted Trx80 (Kuhn et al., 2010; Gil-Bea et al., 2012; Swomley et al., 2014), as well as being subject to activation through disulfide isomerization depending on oxidative conditions (Atapattu et al., 2016). Tau is a microtubule-associated protein (MAP) that, along with other proteins, dynamically interacts with neuronal microtubules, stabilizing them under physiological conditions (Maccioni and Cambiazo, 1995; Avila et al., 2004; Pîrşcoveanu et al., 2017), allowing dynamic changes in the extremes of these elements (Peña-Ortega et al., 2022). Under certain circumstances Tau undergoes posttranslational modifications that may result in the generation of oligomers and aggregates in neurons (Avila et al., 2004). The toxicity of Aβ and Tau aggregates has been thoroughly studied during the last years and was linked to changes in metabolism and the production of ROS, such as , H2O2 and •OH (Carrillo-Mora et al., 2014; Yan and Wang, 2014; Cheignon et al., 2017). However, these metabolic and redox changes could be related to other consequences of AD. For instance, it has been proven that microglial activation leads to inflammation and an increase of ROS such as produced by NOX (Simpson and Oliver, 2020). NOX is a group of seven enzymes that particularly produce upon activation. Two isoforms (NOX2 and 4) have been identified in microglial cells and they are activated upon neuroinflammation or neurodegeneration (Simpson and Oliver, 2020). ROS, such as H2O2 or , are important second messengers and as such modulators of the immune response by activating signaling pathways in microglia, e.g., H2O2 activates NFκB signaling (Kim et al., 2008).
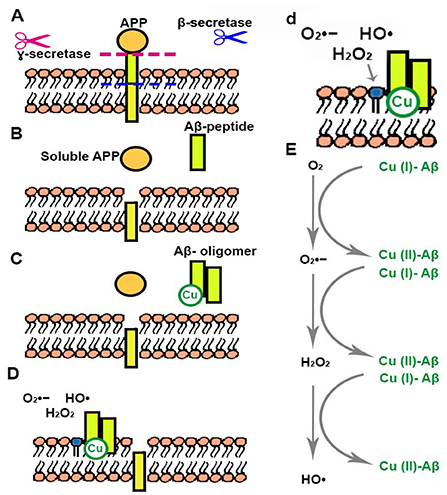
Figure 1. Generation of Aβ peptides and Cu related ROS production. (A) Aβ peptide is produced from the proteolysis of APP that takes place through the action of β- and γ-secretases. (B) Aβ peptide and soluble APP are released. (C) Aβ peptide forms oligomers that can interact with Cu. (D,d) Aβ oligomer-Cu complexes are incorporated into the cellular membrane generating changes in nearby phospholipids. (E) Aβ oligomer-Cu complexes can be oxidized in the presence of O2, generating different ROS.
Other AD related cellular changes include astrogliosis, dystrophic neurites and synapse and neuronal loss (Selkoe, 2001). The relation between neuronal survival and redox regulation has been thoroughly studied and reviewed in recent years (Satoh and Lipton, 2007; Sabens Liedhegner et al., 2012; Sultana et al., 2013; Yin et al., 2014; Sbodio et al., 2019). For instance, an increase of ROS gives rise to cysteine modifications. Reversible protein mixed disulfides with glutathione (GSH) may be the most common steady-state derivative, since GSH is abundant in the cell. Thus, S-glutathionylation may act in redox signal transduction and glutaredoxins (Grxs) serve not only as reducing enzymes upon neuroinflammation but are also involved in the catalysis of (de-)glutathionylation (Sabens Liedhegner et al., 2012; Hanschmann, 2013). Grx1 and Grx2 are expressed in the brain (Aon-Bertolino et al., 2010; Godoy et al., 2011). It has been shown that neurons that present different levels of Grx also show a different viability response in AD. Immunohistochemical analysis showed lower Grx1 and 2 levels in neurons presenting signs of neurodegeneration compared to healthy neurons (Akterin et al., 2006; Arodin et al., 2014). Furthermore, higher levels of Grx1 and Trx1 have been found in plasma and cerebrospinal fluid of AD patients (Arodin et al., 2014). Interestingly, there are no apparent changes in Trx1 levels in the brain. However, there is a decrease in Trx2 immunostaining in the axons of the CA1 hippocampal area in AD patients.
Uric acid, bilirubin, lycopene, α and β carotene, vitamin A, vitamin C, and vitamin E, present decreased levels in patients with AD (Foy et al., 1999; Kim et al., 2006). Even though initial works have shown that some antioxidant enzymes present a greater activity in cells from AD patients (Zemlan et al., 1989), it has recently been shown that many antioxidant enzymes, such as SOD2, catalase and glutathione peroxidase, present lower activities in AD (Marcus et al., 1998; Omar et al., 1999; Wang et al., 2014). A study performed in patients has shown that low GSH levels in plasma lead to a lower risk of AD development (Charisis et al., 2021). Furthermore, a study performed in mice showed that the increase of SOD levels could diminish some of the characteristic symptoms of AD including learning and memory deficits (Massaad et al., 2009).
All in all, the pathophysiology of AD goes beyond Aβ aggregates and Tau neurofibrils and it is linked to a complex network of redox regulation and signaling that should be the focus of further investigation.
Sporadic and familial Alzheimer's disease
There are two major forms of AD that present particular characteristics albeit the same symptoms (Piaceri et al., 2013). Sporadic AD has a late onset and the etiology of the disease is not clear. Environmental and genetic causes have been pinpointed as key to this form of AD. It is generally known that AD is tightly related to advanced age, this being one of the main risk factors in sporadic AD (Katzman, 1988; Riedel et al., 2016; Armstrong, 2019). The relation between oxidative damage and aging has been previously addressed (Muller et al., 2007; Sanz and Stefanatos, 2008). This relation has received some criticism since there are many examples that contradict the fact that a higher production of free radicals is directly related to longevity. However, subsequent and complex studies have pointed to senescence pathways as key players in the relation between aging and redox regulation (Chandrasekaran et al., 2017). Mitochondrial metabolism, bioenergetics and redox signaling are of utmost importance in neurodegeneration and the progression of AD (Yap et al., 2009; Yin et al., 2014). Some areas of the brain, such as the hippocampus, are more susceptible to lower cell excitability, changes in synaptic plasticity and diminished synaptic transmission (Kumar et al., 2018). It was shown that increased oxidative conditions in neurons can be an early marker of Alzheimer's disease and, through molecular signaling, it may contribute to impaired synaptic function (Foster et al., 2017).
Familial AD (FA) occurs in many patients at an earlier onset and has a clear etiology. It is caused by mutations in three specific genes: APP (Amyloid Beta Precursor Protein), PSEN1 (Presenlin1) and PSEN2 (Presenilin2) (Piaceri et al., 2013). FA was firstly associated with mutations in APP and has been shown to increase Aβ-42 levels compared to Aβ-40 (Finckh et al., 2005; Munter et al., 2010). Autosomal FA may account for ~0.5% of AD cases (Finckh et al., 2005). Although most of the mutations causing FA are known to generate an increase in Aβ-42 levels, some mutations that occur within the APP GxxxG motif can cause a decrease in this form of the peptide (Munter et al., 2010). Mutations in APP, PSEN1 and PSEN2 are responsible for the increase in Aβ-peptide production in patients and cause the same symptoms and cellular changes as described for sporadic AD (Finckh et al., 2005). Thus, the study of the impact of these specific mutations can shed light on the etiology of AD (Selkoe, 2001).
Aβ and APP, and their role in redox regulation in AD
Mutations in APP, PSEN1 or PSEN2 have been shown to lead to increased production of Aβ peptide (Gaitonde et al., 1975; Sultana et al., 2009), and these are closely linked to FA. Regarding presenilin, changes in different Cys residues present in PSEN1 have been related to the onset of FA. Of these two, Cys92 located at the N-terminus is highly conserved and it has been shown that its mutation leads to an increase of Aβ-42 in HEK293 cells (Zhang et al., 2000; Tandon and Fraser, 2002). Mutations in PSEN2 are not so common in FA or AD patients and the described mutations do not include changes in Cys residues (Tandon and Fraser, 2002). Nevertheless, mutations near a Cys residue can also affect protein regulation and function, and these should be taken into further consideration. It is worth noting that APP presents a Cu binding site and that it has been shown that incubation of APP with Cu (II) results in the reduction to Cu (I) and the oxidation of Cys144 in APP (Multhaup et al., 1996; Ruiz et al., 1999; Barnham et al., 2003a; Kong et al., 2008). Binding Cu leads to oxidative modification of APP. Moreover, the reduction of Cu can give rise to the formation of ROS, such as or hydroxyl radicals (•OH) (Ruiz et al., 1999). The production of ROS by Cu-APP can be linked to theories that state that AD arises well before the formation of Aβ and that modifications in APP are the main cause (Multhaup et al., 1996). In APP full length, Cys144 forms a disulfide bond with an additional Cys residue in the protein, that could belong to a Cys-rich domain present at the N-terminus of APP, but was not clearly identified (Multhaup et al., 1996; Zhang et al., 2021). Peptides with only Cys144 form intermolecular disulfide bonds creating dimers (Multhaup et al., 1996). Zhang et al. described a rare mutation in APP, near the Cys-rich domain, that generates an increase in Aβ production related to a fast exit of APP from the endoplasmatic reticulum (ER) (Zhang et al., 2021). Interestingly, the palmitoylation of Cys 186 and 187 was shown to be of importance for APP release from the ER, leading to more amyloidogenic processing and Aβ production (Bhattacharyya et al., 2013). Furthermore, this Cys cluster appears to be important for APP dimerization, which decreases APP localization to the plasma membrane, thus diminishing amyloidogenic processing (Ciuculescu et al., 2005; Baumkötter et al., 2014). These studies point to a clear relation between APP (and its processing) and redox regulation, ranging from the presence of highly conserved Cys residues in APP and PSEN1 to the increased generation of different ROS (, •OH, H2O2).
There are many studies linking Aβ with oxidation and toxicity, explaining how mutations that produce larger amounts of this peptide can lead to an early onset development of AD (Barnham et al., 2003b; Everett et al., 2014; Cheignon et al., 2017; Elsworthy et al., 2021). Increased oxidation of macromolecules, such as nucleic acids, proteins and lipids, has been related to higher levels of Aβ-40 and Aβ1-42 in AD hippocampus and cortex (Butterfield and Lauderback, 2002). It has been shown that Aβ peptide can reduce metal ions such as iron (Fe) and copper (Cu) generating •OH radicals through the Fenton reaction (Sbodio et al., 2019). Aβ aggregation leads to the generation of β-sheet rich structures composed of oligomeric species that are reorganized into protofibrils and fibrils (Figure 1C) (Finder and Glockshuber, 2007; Pham et al., 2010; Forloni et al., 2016; Cheignon et al., 2017). Aβ oligomers can impair Cys uptake and GSH synthesis through excitatory amino acid transporter 3 (EAAT3) inhibition (Hodgson et al., 2013). This transporter plays a critical role in neuronal redox regulation, and its depletion can promote oxidative distress and neurodegeneration (Hodgson et al., 2013). However, the most accepted hypothesis to this day states that Aβ oligomers that accumulate in the central nervous system are of utmost toxicity for cells since they can interact with lipids and permeabilize cellular membranes (Figures 1D,d), leading to cell dysfunction, and neurodegeneration (Cheignon et al., 2017). The permeabilization of membranes by Aβ-42 oligomers has been reported as a common component of amyloid toxicity making the disruption of neuronal membrane biomolecules a common feature of AD (Figures 1D,d) (Schubert et al., 1995; Glabe, 2006). The hydrophobicity of some of the aggregates allows them to incorporate into the lipid bilayer initiating the production of ROS, mainly O2•−, •OH, and H2O2 leading to the oxidation of lipids and membrane proteins (Figures 1D,E) (Fraser et al., 1993; Stege and Bosman, 1999; Schmidt et al., 2009; Butterfield et al., 2013; Evangelisti et al., 2013; Swomley et al., 2014; Yan and Wang, 2014). Cu is capable of cycling between two redox states, which makes it a redox-active ion (Drommi et al., 2021). It has been suggested that the high levels of Cu present in senile plaques are related to covalent crosslinking of Aβ. Cu interaction with Aβ in these structures could be responsible for the formation and stabilization of oligomers and aggregates (Figures 1D,d) (Atwood et al., 2004; Chassaing et al., 2012; Drommi et al., 2021). Since Cu can be incompletely oxidized it can lead to the formation of ROS such as O2•−, •OH, H2O2, increasing oxidation and damage to macromolecules (Figure 1E) (Drommi et al., 2021). Recent studies suggest a mechanism mediated by methionine-35 (Met35) of Aβ-42 in the process of ROS production and oxidation (Swomley et al., 2014; Friedemann et al., 2015). Briefly, Aβ-42 oligomers re-enter the bilayer acquiring an alpha-helical structure in which Met35 would interact with the carbonyl of ilein-31 allowing the priming of the electrons in the methionine (Friedemann et al., 2015). In this hydrophobic environment the primed Met stabilizes as sulfuranyl free radical that is able to oxidize thiols or ascorbate and increases and H2O2 levels (Schöneich, 2002; Friedemann et al., 2015). In addition to the oxidation of Met35, tyrosine can also be oxidized in Aβ peptides, to form a dityrosine covalent dimer (Al-Hilaly et al., 2013). Different studies have shown that aggregates present this dityrosine dimer, linking this oxidation to the formation of neurofibrils (Atwood et al., 2004; Al-Hilaly et al., 2013). Furthermore, it has been shown that lack of methionine sulfoxide reductase A-1 (MsrA-1), that specifically reduces oxidized methionines in protein targets, produces a decrease in amyloid aggregates in a Caenorhabditis elegans model, and tips the balance toward an increase of oligomeric aggregates, correlating with an increased synaptic dysfunction (Minniti et al., 2015). On this note, the mainstream ideas on Aβ aggregation suggest that amyloid aggregates present less toxicity than oligomeric ones (Uddin et al., 2020; Koike et al., 2021). However, the impact of Aβ oxidation on its aggregation is still controversial. Some studies state that oxidation of Met35 impedes amyloid aggregate formation (Hou et al., 2013; Pilkington et al., 2019), while others propose that it increases aggregation of the peptide (Maiti et al., 2021).
All in all, Aβ plays a key role in the generation of increased levels of oxidation. However, the pathways through which this occurs are still not clear and further studies focused on the molecular production of specific ROS seems to be essential.
Mitochondrial dysfunction in AD
Mitochondria function in ATP generation, a process known as oxidative phosphorylation (Dikalov, 2011). During this process electrons, supplied by NADP or succinate, are transferred through different complexes of the respiratory chain (Dikalov, 2011). Here, production occurs at the ubiquinol oxidation center of complex III and the FAD site of complex II. Also, both and H2O2 are produced at the 2-oxoacid dehydrogenase complexes (Wang et al., 2014; Larosa and Remacle, 2018). Previous reviews delve into mitochondrial altered dynamics and morphology in AD (Wang et al., 2009a; Misrani et al., 2021). Mitochondrial dysfunction has been linked to Aβ and Tau (Meng et al., 1990; Calkins and Reddy, 2011; Manczak and Reddy, 2012; Manczak et al., 2016). Interestingly, alterations and functional changes in mitochondria appear before the formation of Aβ plaques or Tau neurofibrils (Calkins et al., 2011; Correia et al., 2016). Briefly, changes in mitochondrial fission and fusion occur in AD, and some findings suggest that this dynamical balance is tipped toward less fission in AD (Wang et al., 2008a). On the contrary, several analyses suggest that there is an increase of mitochondrial fragmentation in AD (Figure 2A), that have been ascribed to Aβ toxicity (Wang et al., 2008b, 2009b, 2017). These alterations in the fusion/fission balance are clearly related to mitochondrial morphological changes that have been observed in AD (Collins, 1990; Wang et al., 2008a, 2009b; Flannery and Trushina, 2019). Importantly, during the early stages of AD there is evidence of changes in mitochondrial axonal transport (Figure 2B) (Correia et al., 2016; Flannery and Trushina, 2019). Changes in mitochondrial transport could be the cause of early axonal degeneration and the beginning of neuronal loss (Calkins et al., 2011; Guo et al., 2013; Correia et al., 2016). There is a decrease in mitochondrial DNA coupled with mitochondrial DNA damage, mediated by 8-oxo-7,8-dihydro-29-deoxyguanosine (8-OhdG) accumulation in patients with AD (Figure 2C) (de la Monte et al., 2000; Brown et al., 2001). This points to lower expression of mitochondrial proteins, particularly some components of the respiratory chain, such as cytochrome oxidase (COX) in complex IV (Brown et al., 2001). The activity of several mitochondrial enzymes, such as the pyruvate dehydrogenase complex, the α-ketoglutarate dehydrogenase complex and the respiratory complex IV, is reduced in AD (Parker et al., 1994; Fišar et al., 2019). However, different works have reported somewhat contrary results regarding mitochondrial protein expression (Silva et al., 2012). Some studies have shown an increase in COX levels (Hirai et al., 2001) while others have evidenced a decrease of this enzyme (Nagy et al., 1999; de la Monte et al., 2000). Manczak et al. suggested that an increase in these protein levels could be a compensatory mechanism since they found that in the same samples that showed increased expressions of COX 1 and 2 there was decreased downregulation of the subunit 3 in complex III and of ATPase 6 and 8 in complex V (Manczak et al., 2004). A transcriptomic study using neurons from donor patients suffering from AD, coupled with western blot analysis, showed that there is a lower expression of genes related to the mitochondrial electron transport chain. This study suggests a relation between lower cerebral metabolic rates and the reduction in the expression of genes encoding for the electron transport chain (Liang et al., 2008).
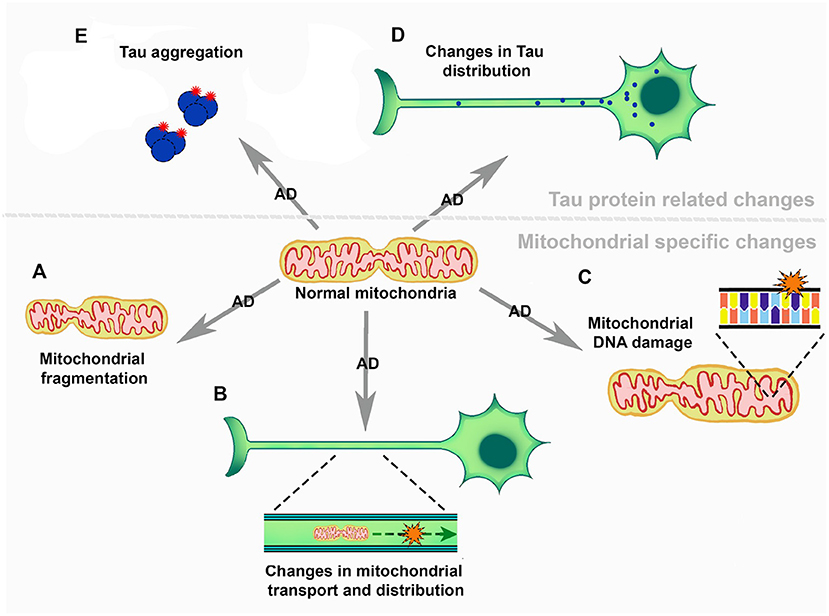
Figure 2. The role of mitochondria in the development and progression of Alzheimer's disease. (A) Mitochondrial fragmentation. (B) Mitochondrial transport and distribution across neurons. (C) Mitochondrial DNA damage. (D) Changes in Tau distribution and (E) Tau aggregation.
There are several mitochondrial alterations in AD, ranging from fragmentation to mitochondrial DNA damage (Figure 2). It is known that the mitochondria play a key role in redox metabolism and regulation, and present an association to neurodegenerative diseases such as AD. However, the studies focused on this topic are scarce and they are not particularly focused on mitochondrial homeostasis and redox metabolism in AD.
Tau toxicity, and its role in oxidation and redox regulation
Tau is a complex protein with intrinsically disordered regions, that presents 6 different isoforms in the CNS, two of these depend on the inclusion and exclusion of exon 10, generating 3R Tau and 4R Tau isoforms (Bachmann et al., 2021). In healthy human brains 3R/4R Tau levels are present at a 50/50 ratio (Chen et al., 2010). Mutations in the Microtubule Associated Protein Tau (MAPT) gene, which encodes the Tau protein, are known to be related to a number of neurodegenerative diseases (Guo et al., 2017; Cherry et al., 2021; Esteras et al., 2022). Mutations altering exon 10 splicing can increase the levels of 4R or 3R Tau which are also related to several neurodegenerative diseases, e.g., fronto temporal dementia and progressive supranuclear palsy (Chen et al., 2010; Qian and Liu, 2014; Bachmann et al., 2021; Stamelou et al., 2021). NMDA activation can generate ROS, particularly O2•− (Brennan et al., 2009; Girouard et al., 2009; Esteras et al., 2022). Inversely, NMDA receptors can be modulated by the cellular redox state, altering synaptic functions in AD (Bodhinathan et al., 2010; Guidi et al., 2015). Esteras et al. (2022) demonstrated that 4R Tau generates changes in NMDA receptor activity and that these changes can be modulated by mitochondrial antioxidants, such as MitoQ. This drug mimics the endogenous mitochondrial antioxidant coenzyme Q10 activity and augments the enzyme capacity (Tauskela, 2007). Similarly, hyperphosphorylated Tau has been shown to interact with the mitochondrial Dynamin-related protein 1 (Drp1), affecting mitochondrial fission and fusion. Decreased levels of Drp1 protect against mitochondrial alterations generated by Tau (Manczak and Reddy, 2012; Kandimalla et al., 2016). Cofilin-1 is a redox regulated protein that plays a key role in mitochondrial function (Kang and Woo, 2019). Upon oxidation, intermolecular disulfide bonds form and Cofilin loses its affinity for actin. Cofilin then translocates to the mitochondria, where it generates a drop in mitochondrial membrane potential, and cytochrome c release through promotion of the opening of the permeability transition pore (Kang and Woo, 2019). A very recent and interesting work has shown that a cluster of mitochondria belonging to the axon initial segment, and with particular characteristics and little motility, is important for Tau sorting (Figure 2D) (Tjiang and Zempel, 2022). It has been shown that ROS produced in mitochondria, such as H2O2, can generate Tau oligomer formation in a mouse model (Figure 2E) (Du et al., 2022). Moreover, inhibition of GSH synthesis in a cell model produced an increase in tau phosphorylation, a first step toward Tau oligomer formation (Su et al., 2010). Also, Tau 4R presents two Cys residues that seem to be important for Tau pathology development in a Drosophila Taupathy model (Prifti et al., 2021). These Cys residues seem to be important for Tau stability and its interaction with microtubules, and Cys-322 can affect tau aggregation (Prifti et al., 2021). Cys-322 as well as Cys-291 appear to be important for the polymerization of human Tau (Bhattacharya et al., 2001; Chen et al., 2018). Other works in Dropsophila melanogaster have shown that the substitution of these Cys diminishes 4R Tau toxicity and they contribute to Tau accumulation under oxidative conditions (Saito et al., 2021).
There are few studies linking modifications in Tau to the onset of AD. Some works mention Lys modifications, such as succinylation (in Tau, APP, and mitochondrial proteins) (Yang et al., 2022), tau acetylation (Min et al., 2010) that, in fact, can be linked to redox regulation (Lucke-Wold et al., 2017) and APP acetylation (Bai et al., 2022). The presence of Cys residues that are important for Tau polymerization led us to believe that redox regulation and signaling of and via this protein are extremely important in AD, since one of the clearest hallmarks of the disease is Tau aggregation into neurofibrils. Furthermore, modifications in Tau aggregation and localization are related to mitochondrial homeostasis. This link should be further explored in the future.
It is important to mention that Tau is a complex protein that does not only bind microtubules. Recent studies have found that both tau and p-tau present multiple interaction partners, some of which are located in the mitochondria and are related to redox homeostasis (Drummond et al., 2020; Sinsky et al., 2020; Jiménez, 2022; Tracy et al., 2022). Drummond et al. analyzed the interactome of p-tau inclusions dissected from AD patient brains. It is interesting to note that among the p-tau interaction partners, mitochondrial proteins such as Cytochrome c oxidase subunit 5B and Aldehyde dehydrogenase were found (Drummond et al., 2020). Moreover, an interaction with Prx5, a member of the Thioredoxin family proteins, was found (Drummond et al., 2020) clearly linking tau and its phosphorylated form not only to mitochondria but also to redox regulation in AD. Tracy et al. (2022) used a very clever approach with multiple tau mutations that cause fronto temporal dementia, showing that these mutations affect tau interaction with several mitochondrial proteins and impair metabolism and bioenergetics. Tau has not only been shown to interact with other proteins but also with DNA, making it less susceptible to peroxidation (Wei et al., 2008). A list of redox-relevant Tau interaction partners can be found in Table 1.
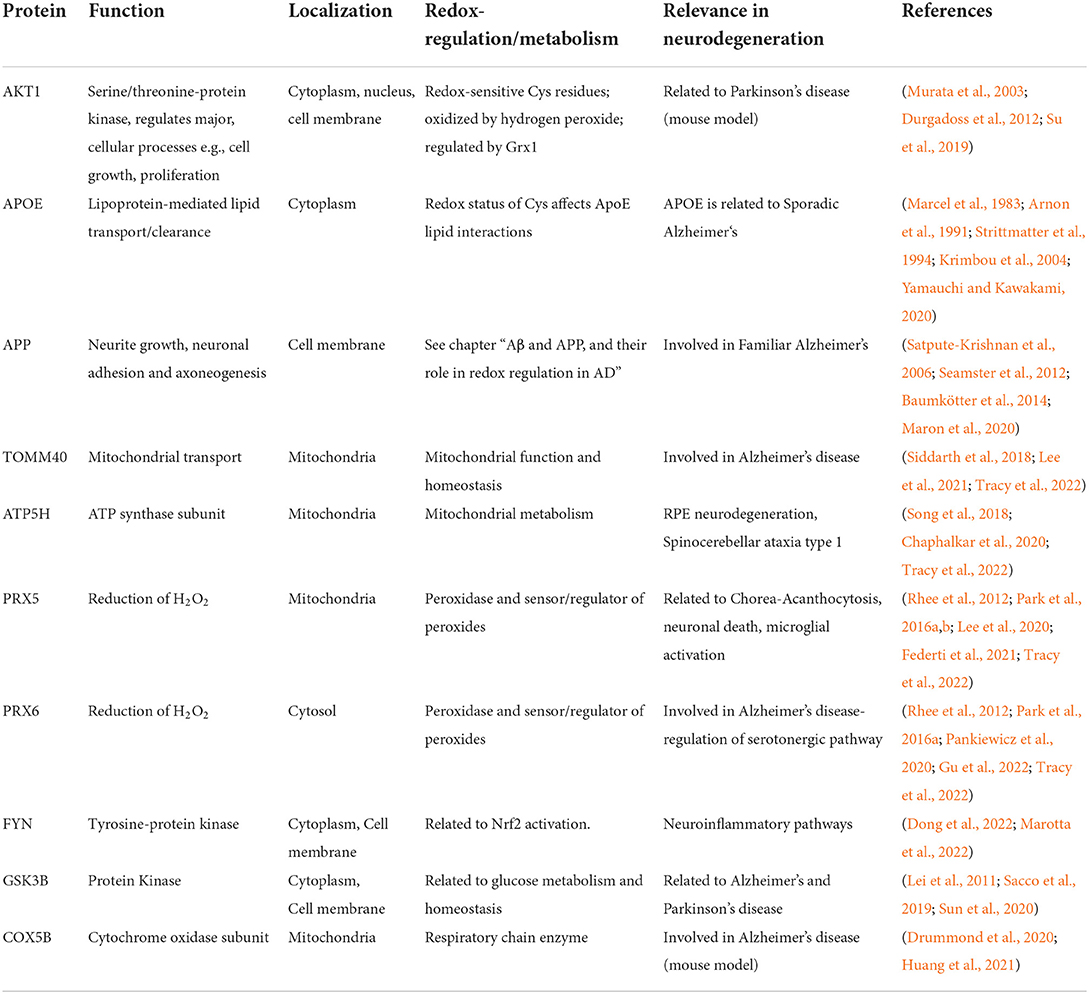
Table 1. List of redox regulated Tau interaction partners, their function(s), localization, as well as a link to redox regulation and metabolism.
Redox metabolism in AD
Several metabolic alterations have been described in different AD models (Toledo et al., 2017). A general overview about metabolic pathways in AD-related pathologies, i.e., lipid, glucose, tryptophan, purin, vitamin, and metal ion metabolism, and the involvement of redox signaling in these pathways is given in (Chen et al., 2020). Glucose metabolism in the CNS has been found to be impaired in AD patients in early stages of the disease (Chen and Zhong, 2013). It has been shown that there is a correlation between AD and its severity with elevated glucose concentrations in the CNS, reduced glycolytic flux, and lower levels of GLUT1 and GLUT3 (An et al., 2018; Han et al., 2021). Besides impaired glucose uptake, a rise in inactive pyruvate dehydrogenase (PDH) and increased aerobic glycolysis and ketone body metabolism were observed in different AD models and patients (Han et al., 2021). Redox active α-lipoic acid, which is one of the co-factors of pyruvate dehydrogenase, may have neuroprotective effects. Research on the molecular effects and therapeutic potential of α-lipoic acid was recently summarized in (Kaur et al., 2021). Interestingly, under oxidative conditions, such as high H2O2 concentrations, the α-lipoic acid in α-ketoglutarate dehydrogenase, an enzyme similar to the PDH, the activity of which is also decreased in AD, is reversibly glutathionylated thereby protecting the sulfur-containing compound from modification e.g., by 4-hydroxynonenal (Applegate et al., 2008). Impaired pyruvate metabolism leads to the deficiency of acetyl CoA as an energy source, also causing a decrease in acetylcholin synthesis (Kaur et al., 2021). Although Tau acetylation has been linked to AD, there is only little evidence of a connection between this modification and metabolic changes. Shin et al. described that increased NAD levels decrease Tau acetylation and neurodegeneration (Shin et al., 2021). A recently published acetylome analysis in AD brains has shown that there are acetylation changes mainly in mitochondrial proteins related to ATP synthesis and proton transport (Sun et al., 2021). In addition, mitochondrial homeostasis is altered in AD (see “Mitochondrial dysfunction in AD”). Changes in the succinylation of Lys residues in samples from AD patients occur mainly in the mitochondria and these changes have been linked to mitochondrial metabolic dysfunction (Yang et al., 2022). However, Glucose is not only most important as energy source but is substrate to the pentose phosphate pathway (PPP). Already in 1999 it was discovered, that the key enzyme of the PPP, the glucose-6-phosphate dehydrogenase, is upregulated in AD (Russell et al., 1999). Both parts of the PPP, the oxidative and non-oxidative part, are active in neurons even though to different degrees depending on the cell type (Brekke et al., 2012). The reducing equivalents generated in the PPP are essential for the function of redox enzymes including the Trx and Grx/GSH systems.
Besides glucose metabolism and mitochondrial dysfunction, the impaired ability of a controlled protein metabolism plays an important role in AD. The downregulation of proteasome subunits as well as the inhibition of the proteasome activity, e.g., by Tau polymers, leads to the accumulation not only of damaged and missfolded proteins but also hyperphosphorylated Tau (Graham and Liu, 2017). Additionally, the lysosomal degradation of proteins and organelles is impaired through a byproduct of lipid peroxydation (4-hydroxynonenal), leading to the accumulation of autophagosomes and polyubiqinated proteins (Zhang et al., 2017). It was also suggested that this loss of protein degradation via the proteasome occurs already at the earliest stages of AD (Cecarini et al., 2007). Lipid peroxidation products such as hydroxynonenal and malondialdehyde may be the result of an impaired iron metabolism thereby inducing neurodegeneration (Stockwell et al., 2017). There is evidence that treatment with mesenchymal stem cell leads to a decrease in malondialdeyde and phosphorylated tau aggregates (Abozaid et al., 2022). Elevated iron levels may be one source for hydroxyl radicals as it reacts easily with H2O2 in the Fenton reaction. A number of features found in neurodegenerative diseases including AD are consistent with iron induced cell death, i.e., ferroptosis (Stockwell et al., 2017; Wang et al., 2022). The importance of iron metabolism and ferroptosis in neurodegeneration is summarized e.g., in (Stockwell et al., 2017; Li et al., 2022; Wang et al., 2022). Next to high iron levels, glutathione is significantly decreased in the hippocampus of AD patients (Mandal et al., 2022). At the same time the amount of S-glutathionylated proteins is elevated in AD brains already at early stages and especially in the hippocampus (Zhang et al., 2012). For an overview of alterations observed in AD from a molecular level to their effects on the organism see Figure 3.
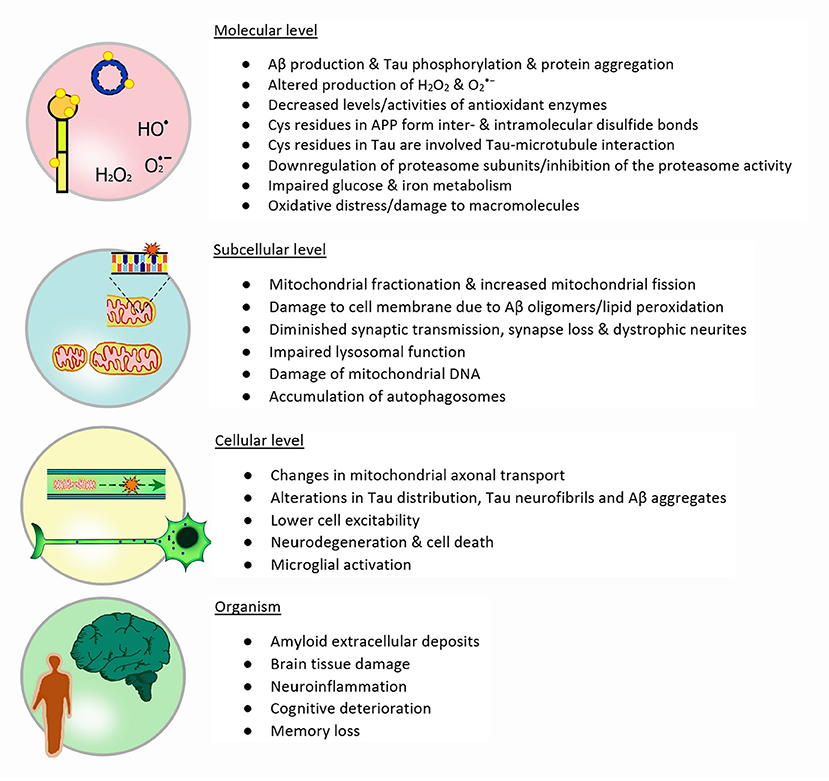
Figure 3. Redox metabolism in AD: The illustration depicts the four different levels of redox and metabolic changes in Alzheimer's disease: Molecules involved and their importance, different subcellular and functional changes, cellular alterations and finally the impact on the whole organism and particularly in patients. Each stage presents features that are a direct or indirect consequence of events described in the previous level.
Redox tools for in vivo analysis of Alzheimer's disease–Technical obstacles and limitations
Suitable experimental models, including animal models, organoids, and cell culture models, are the key to gaining a better understanding of the pathogenesis of diseases such as Alzheimer's disease as well as to identify new potential therapeutic approaches (Drummond and Wisniewski, 2017; Esquerda-Canals et al., 2017; Papaspyropoulos et al., 2020). Transgenic mice constitute the animal model in AD research that is used the most. Overviews of the most common transgenic mouse and rat models of AD with indicated mutations are summarized in (Drummond and Wisniewski, 2017; Esquerda-Canals et al., 2017). The use of non-human primates has the benefit that they share a 100% sequence homology with human Aβ. Rhesus monkeys are the most practical primate model with an age-related Aβ pathology (Drummond and Wisniewski, 2017). Other species with age-related AD associated pathologies are dogs and the Octodon degu but their use has practical and scientific limitations, i.e., long lifespans and variable pathologies (Drummond and Wisniewski, 2017). Another model that should be used more widely is the zebra fish, which can easily be modified by the morpholino technique (Newman et al., 2011, 2014; Caramillo and Echevarria, 2017; Drummond and Wisniewski, 2017). Analysis of human patient samples revealed several mutations and biomarkers that were indicative of the AD pathology (Bondi et al., 2017). Therefore, especially cerebrospinal fluid and blood are investigated to identify peripheral oxidatively modified proteins potentially involved in AD progression (Di Domenico et al., 2011). However, as summarized in (Drummond and Wisniewski, 2017), there is currently no model available replicating all features of human AD. As discussed above, Alzheimer's disease results from multiple pathogenic mechanisms and dysfunctions, one of which is the dysregulation of the redox homeostasis (von Bernhardi and Eugenín, 2012; Conrad et al., 2013). Brain organoids are a good approach since they are a model that presents a human proteomic and genomic background, with different cell types (neurons and astrocytes) organized in tissular structures. Nevertheless, they still lack some very important characteristics, such as all the cell types present in the human brain (Bi et al., 2021).
The in vivo analysis of redox modifications is very challenging due to the variety of possible and mostly transient modifications (Hanschmann, 2013). Proteome analysis using 2D-gel electrophoresis followed by mass spectrometry (MS) and/or computer-based analysis allows to compare samples and thereby to identify isoforms, mutants, stable protein interactions, and posttranslational modifications (Sultana et al., 2006a,b). To identify specifically oxidized proteins in human AD brain tissue the 2D-gel electrophoresis can be coupled to an immunochemical detection of protein carbonyl and nitrated proteins followed by mass spectrometry (Butterfield et al., 2006). However, the limitation of this method are membrane proteins, low abundant proteins, and proteins with high trypsin content, which cannot be solubilized or detected, respectively (Sultana et al., 2006b). An alternative are non-SDS-PAGE based methods coupled with MS analysis that are increasingly used. Lennicke et al. summarized gel-based and non-gel-based methods in general for the identification and enrichment of redox-modified proteins by redox proteomics in (Lennicke et al., 2016).
Monitoring the redox state in vivo requires specific biocompatible probes for different reactive species and, since their transient concentrations are usually very low, high sensitivity. Specific electron paramagnetic resonance (EPR) probes (aminoacyl radicals) are in vivo reporters to analyze the redox state of the brain and the blood-brain barrier integrity in a 5xFAD mouse model of AD (Vesković et al., 2021). A near-infrared emissive iridium probe is sensitive and selective toward peroxynitrite/glutathione redox cycles (Wu et al., 2021). Available small molecule near-infrared fluorescent probes are used to detect Tau, Aβ, and reactive species in vivo, e.g., CRANAD-61 and CRANAD-5 detect ROS at micro- and macro-levels in APP/PS1 mice (Fang et al., 2019). Numerous fluorescent probes for detection of varying redox active species (ROS/RNS/RSS) in vivo/in cellulo are summarized in (Lü, 2017) and (Wu et al., 2019). Luminescent lanthanide complexes selectively detect i.e., O2•−, •OH, 1O2, and H2O2 in a μM to nM range (Galaup et al., 2021). Disadvantages of probes for the in vivo detection of redox reactive compounds may include artifactual amplification of the intensity by the formation of intermediate radicals, light sensitivity leading to artifactual H2O2 generation, reaction with other reactive species, and interference/dependency of the reaction on enzymatic activity (Kalyanaraman et al., 2012).
New tools to address the difficult analysis of redox modifications are being developed and optimized. Redox sensors measuring real time dynamic changes of e.g., H2O2 production in different compartments of the cell, are a powerful tool to analyze the impact of AD induced changes in the cellular redox state in 2D and 3D cell culture and animal models. However, the data analysis of redox modifications and measurements of ROS need to be very conscientious since commercially available kits bear challenges and limitations that too easily lead to misinterpretations (Murphy et al., 2022). In the future it will not only be important to identify the exact nature of reactive species of ROS/RNS/RSS but also the sources, as they could pose potential targets for new therapeutic approaches.
Conclusion and future challenges
The emergence of novel experimental models, such as 3-dimensional cell cultures and complex transgenic animals, coupled with new imaging techniques and redox sensors promise great advances in the study of redox regulation in AD.
Many of the studies revised in the present work focus on the action of the Aβ peptide and the oligomers or plaques over the formation of different ROS and an increased oxidation of macromolecules. However, the central role of this peptide in AD has been questioned during the last two decades, changing the focus to other factors such as APP, Tau, its phosphorylation and the formation of neurofibrils. In the present bibliographical research, we were able to find scant works that put their whole attention on these proteins and their probable relation to redox regulation. It has been shown that APP presents different redox states and that these could be related to dimerization of the protein or its subproducts. Furthermore, 4R Tau possesses two Cys residues that are key for its aggregation. This protein has also been linked to mitochondrial changes, and this is of most importance, since many authors have found a clear relation between mitochondrial modifications, the production of O2•− and H2O2, metabolic changes and the onset of AD. Thus, further investigations that focus not only on Aβ peptide, but on its precursor protein and Tau are of most interest in the field. Additionally, further investigations on alterations of metabolic pathways and redox metabolism are necessary to fully understand their interplay and importance in the development and progression of AD. Even small, post translational changes such as oxidative modifications regulate the function of biomolecules and can have a strong impact on the onset and progression of diseases linked to neurodegeneration and neuroinflammation. However, we found that many studies use unspecific markers and kits to analyze ROS and the oxidation of macromolecules. These tools can be quite misleading, and they give incomplete and inconsistent information. Future research will aim to understand redox regulation of specific proteins and pathways in neurodegenerative diseases, through specific second messengers, that need to be identified with certain detail. Thus, we believe that the use of novel, advanced and specific techniques promise a continuous progress in the untangling of the relations of AD and redox signaling and dys/regulation.
More research is needed to characterize the physiological function and the pathological impact of thiol switches in key proteins linked to AD, such as Aβ, Tau, and the interaction partners that regulate their function.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
We acknowledge the financial support to EMH from the Deutsche Forschungsgemeinschaft (Ha 8334/2-2) within the cooperative DFG program SPP1710: Dynamics of Thiol-based Redox Switches in Cellular Physiology). We acknowledge support for the Article Processing Charge by the German Research Foundation and the Open Access Publication Fund of the University of Greifswald.
Acknowledgments
We thank Tomas Falzone and Cayetana Arnaiz Yepez for their most helpful input, pointing to a number of important works that were included in the present review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abozaid, O. A. R., Sallam, M. W., and Ahmed, E. S. A. (2022). Mesenchymal stem cells modulate SIRT1/MiR-134/GSK3β signaling pathway in a rat model of Alzheimer's disease. J. Prev. Alzheimers Dis. 9, 458–468. doi: 10.14283/jpad.2022.26
Akterin, S., Cowburn, R. F., Miranda-Vizuete, A., Jiménez, A., Bogdanovic, N., Winblad, B., et al. (2006). Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 13, 1454–1465. doi: 10.1038/sj.cdd.4401818
Al-Hilaly, Y. K., Williams, T. L., Stewart-Parker, M., Ford, L., Skaria, E., Cole, M., et al. (2013). A central role for dityrosine crosslinking of Amyloid-β in Alzheimer's disease. Acta Neuropathol. Commun. 1, 83. doi: 10.1186/2051-5960-1-83
An, Y., Varma, V. R., Varma, S., Casanova, R., Dammer, E., Pletnikova, O., et al. (2018). Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers. Dement. 14, 318–329. doi: 10.1016/j.jalz.2017.09.011
Aon-Bertolino, M. L., Romero, J. I., Galeano, P., Holubiec, M., Badorrey, M. S., Saraceno, G. E., et al. (2010). Thioredoxin and glutaredoxin system proteins-immunolocalization in the rat central nervous system. Biochim. Biophys. Acta. 1810, 93–110. doi: 10.1016/j.bbagen.2010.06.011
Applegate, M. A. B., Humphries, K. M., and Szweda, L. I. (2008). Reversible inhibition of α-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 47, 473–478. doi: 10.1021/bi7017464
Armstrong, R. A. (2019). Risk factors for Alzheimer's disease. Folia Neuropathol. 57, 87–105. doi: 10.5114/fn.2019.85929
Arnon, R., Sehayek, E., Vogel, T., and Eisenberg, S. (1991). Effects of exogenous apo E-3 and of cholesterol-enriched meals on the cellular metabolism of human chylomicrons and their remnants. Biochim. Biophys. Acta. 1085, 336–342. doi: 10.1016/0005-2760(91)90138-8
Arodin, L., Lamparter, H., Karlsson, H., Nennesmo, I., Björnstedt, M., Schröder, J., et al. (2014). Alteration of thioredoxin and glutaredoxin in the progression of Alzheimer's disease. J. Alzheimers. Dis. 39, 787–797. doi: 10.3233/JAD-131814
Atapattu, L., Saha, N., Chheang, C., Eissman, M. F., Xu, K., Vail, M. E., et al. (2016). An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth. J. Exp. Med. 213, 1741–1757. doi: 10.1084/jem.20151095
Atwood, C. S., Perry, G., Zeng, H., Kato, Y., Jones, W. D., Ling, K. Q., et al. (2004). Copper mediates dityrosine cross-linking of Alzheimer's amyloid-beta. Biochemistry. 43, 560–568. doi: 10.1021/bi0358824
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Bachmann, S., Bell, M., Klimek, J., and Zempel, H. (2021). Differential effects of the six human TAU isoforms: somatic retention of 2N-TAU and increased microtubule number induced by 4R-TAU. Front. Neurosci. 15, 643115. doi: 10.3389/fnins.2021.643115
Bai, N., Li, N., Cheng, R., Guan, Y., Zhao, X., Song, Z., et al. (2022). Inhibition of SIRT2 promotes APP acetylation and ameliorates cognitive impairment in APP/PS1 transgenic mice. Cell Rep. 40, 111062. doi: 10.1016/j.celrep.2022.111062
Barnham, K. J., Ciccotosto, G. D., Tickler, A. K., Ali, F. E., Smith, D. G., Williamson, N. A., et al. (2003b). Neurotoxic, Redox-competent Alzheimer's β-Amyloid Is Released from Lipid Membrane by Methionine Oxidation*. J. Biol. Chem. 278, 42959–42965. doi: 10.1074/jbc.M305494200
Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., et al. (2003a). Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407. doi: 10.1074/jbc.M300629200
Baumkötter, F., Schmidt, N., Vargas, C., Schilling, S., Weber, R., Wagner, K., et al. (2014). Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 34, 11159–11172. doi: 10.1523/JNEUROSCI.0180-14.2014
Bhattacharya, K., Rank, K. B., Evans, D. B., and Sharma, S. K. (2001). Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun. 285, 20–26. doi: 10.1006/bbrc.2001.5116
Bhattacharyya, R., Barren, C., and Kovacs, D. M. (2013). Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 33, 11169–11183. doi: 10.1523/JNEUROSCI.4704-12.2013
Bi, F. C., Yang, X. H., Cheng, X. Y., Deng, W. B., Guo, X. L., Yang, H., et al. (2021). Optimization of cerebral organoids: a more qualified model for Alzheimer's disease research. Transl. Neurodegener. 10, 27. doi: 10.1186/s40035-021-00252-3
Bodhinathan, K., Kumar, A., and Foster, T. C. (2010). Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30, 1914–1924. doi: 10.1523/JNEUROSCI.5485-09.2010
Bondi, M. W., Edmonds, E. C., and Salmon, D. P. (2017). Alzheimer's disease: past, present, and future. J. Int. Neuropsychol. Soc. 23, 818–831. doi: 10.1017/S135561771700100X
Brandes, N., Schmitt, S., and Jakob, U. (2009). Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11, 997–1014. doi: 10.1089/ars.2008.2285
Brekke, E. M. F., Walls, A. B., Schousboe, A., Waagepetersen, H. S., and Sonnewald, U. (2012). Quantitative importance of the pentose phosphate pathway determined by incorporation of 13C from [2-13C]- and [3-13C]glucose into TCA cycle intermediates and neurotransmitter amino acids in functionally intact neurons. J. Cereb. Blood Flow Metab. 32, 1788–1799. doi: 10.1038/jcbfm.2012.85
Brennan, A. M., Suh, S. W., Won, S. J., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brown, A. M., Sheu, R. K., Mohs, R., Haroutunian, V., and Blass, J. P. (2001). Correlation of the clinical severity of Alzheimer's disease with an aberration in mitochondrial DNA (mtDNA). J. Mol. Neurosci. 16, 41–48. doi: 10.1385/JMN:16:1:41
Butterfield, D. A., and Lauderback, C. M. (2002). Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress1, 2 1Guest Editors: Mark A, Smith and George Perry 2This article is part of a series of reviews on “Causes and Consequences of Oxidative Stress in Alzheimer's Disease.” The full list of papers may be found on the homepage of the journal. Free Radic. Biol. Med. 32, 1050–1060. doi: 10.1016/S0891-5849(02)00794-3
Butterfield, D. A., Perluigi, M., and Sultana, R. (2006). Oxidative stress in Alzheimer's disease brain: new insights from redox proteomics. Eur. J. Pharmacol. 545, 39–50. doi: 10.1016/j.ejphar.2006.06.026
Butterfield, D. A., Swomley, A. M., and Sultana, R. (2013). Amyloid β-Peptide (1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxid. Redox Signal. 19, 823–835. doi: 10.1089/ars.2012.5027
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Calkins, M. J., and Reddy, P. H. (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim. Biophys. Acta. 1812, 507–513. doi: 10.1016/j.bbadis.2011.01.007
Caramillo, E. M., and Echevarria, D. J. (2017). Alzheimer's disease in the zebrafish: where can we take it? Behav. Pharmacol. 28, 179–186. doi: 10.1097/FBP.0000000000000284
Carrillo-Mora, P., Luna, R., and Colín-Barenque, L. (2014). Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev. 2014:795375. doi: 10.1155/2014/795375
Cecarini, V., Ding, Q., and Keller, J. N. (2007). Oxidative inactivation of the proteasome in Alzheimer's disease. Free Radic. Res. 41, 673–680. doi: 10.1080/10715760701286159
Chandrasekaran, A., Idelchik, D. P. S. M., and Melendez, J. A. (2017). Redox control of senescence and age-related disease. Redox Biol. 11, 91–102. doi: 10.1016/j.redox.2016.11.005
Chaphalkar, R. M., Stankowska, D. L., He, S., Kodati, B., Phillips, N., Prah, J., et al. (2020). Endothelin-1 mediated decrease in mitochondrial gene expression and bioenergetics contribute to neurodegeneration of retinal ganglion cells. Sci. Rep. 10, 3571. doi: 10.1038/s41598-020-60558-6
Charisis, S., Ntanasi, E., Yannakoulia, M., Anastasiou, C. A., Kosmidis, M. H., Dardiotis, E., et al. (2021). Plasma GSH levels and Alzheimer's disease. A prospective approach.: Results from the HELIAD study. Free Radic. Biol Med. 162, 274–282. doi: 10.1016/j.freeradbiomed.2020.10.027
Chassaing, S., Collin, F., Dorlet, P., Gout, J., Hureau, C., and Faller, P. (2012). Copper and heme-mediated abeta toxicity: redox chemistry, abeta oxidations and anti-ROS compounds. Curr. Top. Med. Chem. 12, 2573–2595. doi: 10.2174/1568026611212220011
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2017). Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, H., Liu, S., Li, S., Chen, J., Ni, J., and Liu, Q. (2018). Blocking the thiol at cysteine-322 destabilizes tau protein and prevents its oligomer formation. ACS Chem. Neurosci. 9, 1560–1565. doi: 10.1021/acschemneuro.8b00003
Chen, S., Townsend, K., Goldberg, T. E., Davies, P., and Conejero-Goldberg, C. (2010). MAPT isoforms: differential transcriptional profiles related to 3R and 4R splice variants. J. Alzheimers. Dis. 22, 1313–1329. doi: 10.3233/JAD-2010-101155
Chen, Y. Y., Wang, M. C., Wang, Y. N., Hu, H. H., Liu, Q. Q., Liu, H. J., et al. (2020). Redox signaling and Alzheimer's disease: from pathomechanism insights to biomarker discovery and therapy strategy. Biomarker Res. 8, 42. doi: 10.1186/s40364-020-00218-z
Chen, Z., and Zhong, C. (2013). Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 108, 21–43. doi: 10.1016/j.pneurobio.2013.06.004
Cherry, J. D., Esnault, C. D., Baucom, Z. H., Tripodis, Y., Huber, B. R., Alvarez, V. E., et al. (2021). Tau isoforms are differentially expressed across the hippocampus in chronic traumatic encephalopathy and Alzheimer's disease. Acta Neuropathol. Commun. 9, 86. doi: 10.1186/s40478-021-01189-4
Ciuculescu, E.-D., Mekmouche, Y., and Faller, P. (2005). Metal-binding properties of the peptide APP170-188: a model of the ZnII-binding site of amyloid precursor protein (APP). Chemistry. 11, 903–909. doi: 10.1002/chem.200400786
Collins, P. F. (1990). The challenge of change. N. Z. Dent. J. 86, 84–85. doi: 10.1097/00152193-199004000-00024
Conrad, M., Schick, J., and Angeli, J. P. F. (2013). Glutathione and thioredoxin dependent systems in neurodegenerative disease: what can be learned from reverse genetics in mice. Neurochem. Int. 62, 738–749. doi: 10.1016/j.neuint.2013.01.010
Correia, S. C., Perry, G., and Moreira, P. I. (2016). Mitochondrial traffic jams in Alzheimer's disease - pinpointing the roadblocks. Biochim. Biophys. Acta. 1862, 1909–1917. doi: 10.1016/j.bbadis.2016.07.010
de la Monte, S. M., Luong, T., Neely, T. R., Robinson, D., and Wands, J. R. (2000). Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab. Invest. 80, 1323–1335. doi: 10.1038/labinvest.3780140
Di Domenico, F., Coccia, R., Butterfield, D. A., and Perluigi, M. (2011). Circulating biomarkers of protein oxidation for Alzheimer disease: Expectations within limits. Biochim. Biophys. Acta 1814, 1785–1795. doi: 10.1016/j.bbapap.2011.10.001
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Dong, D., Zhang, Y., He, H., Zhu, Y., and Ou, H. (2022). Alpinetin inhibits macrophage infiltration and atherosclerosis by improving the thiol redox state: Requirement of GSk3β/Fyn-dependent Nrf2 activation. FASEB J. 36, e22261. doi: 10.1096/fj.202101567R
Drommi, M., Rulmont, C., Esmieu, C., and Hureau, C. (2021). Hybrid bis-histidine phenanthroline-based ligands to lessen Aβ-bound Cu ROS production: an illustration of Cu(I) significance. Molecules. 26, 7630. doi: 10.3390/molecules26247630
Drummond, E., Pires, G., MacMurray, C., Askenazi, M., Nayak, S., Bourdon, M., et al. (2020). Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain. 143, 2803–2817. doi: 10.1093/brain/awaa223
Drummond, E., and Wisniewski, T. (2017). Alzheimer's disease: experimental models and reality. Acta Neuropathol. 133, 155–175. doi: 10.1007/s00401-016-1662-x
Du, F., Yu, Q., and Yan, S. S. (2022). Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer's disease. Hum Mol Genet, ddab363. doi: 10.1093/hmg/ddab363
Durgadoss, L., Nidadavolu, P., Valli, R. K., Saeed, U., Mishra, M., Seth, P., et al. (2012). Redox modification of Akt mediated by the dopaminergic neurotoxin MPTP, in mouse midbrain, leads to down-regulation of pAkt. FASEB J. 26, 1473–1483. doi: 10.1096/fj.11-194100
Elsworthy, R. J., King, M. C., Grainger, A., Fisher, E., Crowe, J. A., Alqattan, S., et al. (2021). Amyloid-β precursor protein processing and oxidative stress are altered in human iPSC-derived neuron and astrocyte co-cultures carrying presenillin-1 gene mutations following spontaneous differentiation. Mol. Cell. Neurosci. 114, 103631. doi: 10.1016/j.mcn.2021.103631
Esquerda-Canals, G., Montoliu-Gaya, L., Güell-Bosch, J., and Villegas, S. (2017). Mouse models of Alzheimer's disease. J. Alzheimers. Dis. 57, 1171–1183. doi: 10.3233/JAD-170045
Esteras, N., Kopach, O., Maiolino, M., Lariccia, V., Amoroso, S., Qamar, S., et al. (2022). Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimers. Dement. 18, 318–338. doi: 10.1002/alz.12394
Evangelisti, E., Wright, D., Zampagni, M., Cascella, R., Fiorillo, C., Bagnoli, S., et al. (2013). Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer's disease. Curr. Alzheimer Res. 10, 143–153. doi: 10.2174/1567205011310020004
Everett, J., Céspedes, E., Shelford, L. R., Exley, C., Collingwood, J. F., Dobson, J., et al. (2014). Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer's disease Peptide β-Amyloid. Inorg. Chem. 53, 2803–2809. doi: 10.1021/ic402406g
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA, 116, 2672–2680. doi: 10.1073/pnas.1821022116
Federti, E., Matte, A., Riccardi, V., Peikert, K., Alper, S. L., Danek, A., et al. (2021). Adaptative Up-regulation of PRX2 and PRX5 expression characterizes brain from a mouse model of chorea-acanthocytosis. Antioxidants (Basel). 11, 76. doi: 10.3390/antiox11010076
Finckh, U., Kuschel, C., Anagnostouli, M., Patsouris, E., Pantes, G. V., Gatzonis, S., et al. (2005). Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 6, 85–89. doi: 10.1007/s10048-005-0211-x
Finder, V. H., and Glockshuber, R. (2007). Amyloid-β aggregation. NDD 4, 13–27. doi: 10.1159/000100355
Fišar, Z., Hansíková, H., KríŽová, J., Jirák, R., Kitzlerová, E., Zvěrová, M., et al. (2019). Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer's disease and depressive disorder. Mitochondrion. 48, 67–77. doi: 10.1016/j.mito.2019.07.013
Flannery, P. J., and Trushina, E. (2019). Mitochondrial dynamics and transport in Alzheimer's disease. Mol. Cell. Neurosci. 98, 109–120. doi: 10.1016/j.mcn.2019.06.009
Forloni, G., Artuso, V., La Vitola, P., and Balducci, C. (2016). Oligomeropathies and pathogenesis of Alzheimer and Parkinson's diseases. Mov. Disord. 31, 771–781. doi: 10.1002/mds.26624
Foster, T. C., Kyritsopoulos, C., and Kumar, A. (2017). Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer's disease. Behav. Brain Res. 322, 223–232. doi: 10.1016/j.bbr.2016.05.012
Foy, C. J., Passmore, A. P., Vahidassr, M. D., Young, I. S., and Lawson, J. T. (1999). Plasma chain-breaking antioxidants in Alzheimer's disease, vascular dementia and Parkinson's disease. QJ M 92, 39–45. doi: 10.1093/qjmed/92.1.39
Fraser, P. E., Lévesque, L., and McLachlan, D. R. (1993). Biochemistry of Alzheimer's disease amyloid plaques. Clin. Biochem. 26, 339–349. doi: 10.1016/0009-9120(93)90110-R
Friedemann, M., Helk, E., Tiiman, A., Zovo, K., Palumaa, P., and Tõugu, V. (2015). Effect of methionine-35 oxidation on the aggregation of amyloid-β peptide. Biochem. Biophys. Rep. 3, 94–99. doi: 10.1016/j.bbrep.2015.07.017
Gaitonde, M. K., Fayein, N. A., and Johnson, A. L. (1975). Decreased metabolism in vivo of glucose into amino acids of the brain of thiamine-deficient rats after treatment with pyrithiamine. J. Neurochem. 24, 1215–1223. doi: 10.1111/j.1471-4159.1975.tb03901.x
Galaup, C., Picard, C., Couderc, F., Gilard, V., and Collin, F. (2021). Luminescent lanthanide complexes for reactive oxygen species biosensing and possible application in Alzheimer's diseases. FEBS J., doi: 10.1111/febs.15859
Gao, X. H., Li, L., Parisien, M., Wu, J., Bederman, I., Gao, Z., et al. (2020). Discovery of a redox thiol switch: implications for cellular energy metabolism. Mol. Cell. Proteomics. 19, 852–870. doi: 10.1074/mcp.RA119.001910
Gil-Bea, F., Akterin, S., Persson, T., Mateos, L., Sandebring, A., Avila-Cariño, J., et al. (2012). Thioredoxin-80 is a product of alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer's disease brain. EMBO Mol. Med. 4, 1097–1111. doi: 10.1002/emmm.201201462
Girouard, H., Wang, G., Gallo, E. F., Anrather, J., Zhou, P., Pickel, V. M., et al. (2009). NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 29, 2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009
Glabe, C. G. (2006). Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging. 27, 570–575. doi: 10.1016/j.neurobiolaging.2005.04.017
Godoy, J. R., Funke, M., Ackermann, W., Haunhorst, P., Oesteritz, S., Capani, F., et al. (2011). Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse. Biochim. Biophys. Acta. 1810, 2–92. doi: 10.1016/j.bbagen.2010.05.006
Graham, S. H., and Liu, H. (2017). Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res. Rev. 34, 30–38. doi: 10.1016/j.arr.2016.09.011
Gu, S. M., Yu, E., Kim, Y. E., Yoon, S. S., Lee, D., Hong, J. T., et al. (2022). Peroxiredoxin 6 overexpression induces anxiolytic and depression-like behaviors by regulating the serotonergic pathway in mice. Biomol. Ther. (Seoul). 30, 334–339. doi: 10.4062/biomolther.2021.169
Guidi, M., Kumar, A., and Foster, T. C. (2015). Impaired attention and synaptic senescence of the prefrontal cortex involves redox regulation of NMDA receptors. J. Neurosci. 35, 3966–3977. doi: 10.1523/JNEUROSCI.3523-14.2015
Guo, L., Du, H., Yan, S., Wu, X., McKhann, G. M., Chen, J. X., et al. (2013). Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer's neurons. PLoS ONE. 8, e54914. doi: 10.1371/journal.pone.0054914
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Han, R., Liang, J., and Zhou, B. (2021). Glucose metabolic dysfunction in neurodegenerative diseases—new mechanistic insights and the potential of hypoxia as a prospective therapy targeting metabolic reprogramming. Int. J. Mol. Sci. 22, 5887. doi: 10.3390/ijms22115887
Hanschmann, E. M. (2013). Thioredoxins, glutaredoxins, and peroxiredoxins— molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605. doi: 10.1089/ars.2012.4599
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 21, 3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001
Hodgson, N., Trivedi, M., Muratore, C., Li, S., and Deth, R. (2013). Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers. Dis. 36, 197–209. doi: 10.3233/JAD-130101
Hosios, A. M., and Vander Heiden, M. G. (2018). The redox requirements of proliferating mammalian cells. J. Biol. Chem. 293, 7490–7498. doi: 10.1074/jbc.TM117.000239
Hou, L., Lee, H. G., Han, F., Tedesco, J. M., Perry, G., Smith, M. A., et al. (2013). Modification of amyloid-β1-42 fibril structure by methionine-35 oxidation. J. Alzheimers. Dis. 37, 9–18. doi: 10.3233/JAD-122389
Huang, X., Yang, J., Huang, X., Zhang, Z., Liu, J., Zou, L., et al. (2021). Tetramethylpyrazine improves cognitive impairment and modifies the hippocampal proteome in two mouse models of Alzheimer's disease. Front Cell Dev Biol. 9, 632843. doi: 10.3389/fcell.2021.632843
Jiménez, J. S. (2022). Macromolecular structures and proteins interacting with the microtubule associated tau protein. Neuroscience. doi: 10.1016/j.neuroscience.2022.05.023
Kalyanaraman, B., Darley-Usmar, V., Davies, K. J. A., Dennery, P. A., Forman, H. J., Grisham, M. B., et al. (2012). Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6. doi: 10.1016/j.freeradbiomed.2011.09.030
Kandimalla, R., Manczak, M., Fry, D., Suneetha, Y., Sesaki, H., and Reddy, P. H. (2016). Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum. Mol. Genet. 25, 4881–4897. doi: 10.1093/hmg/ddw312
Kang, D. E., and Woo, J. A. (2019). Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer's disease. J. Alzheimers. Dis. 72, S131–S144. doi: 10.3233/JAD-190585
Katzman, R. (1988). Alzheimer's disease as an age-dependent disorder. Ciba Found. Symp. 134, 69–85. doi: 10.1002/9780470513583.ch6
Kaur, D., Behl, T., Sehgal, A., Singh, S., Sharma, N., Chigurupati, S., et al. (2021). Life Sciences. 284, 119899. doi: 10.1016/j.lfs.2021.119899
Kim, J. H., Na, H. J., Kim, C. K., Kim, J. Y., Ha, K. S., Lee, H., et al. (2008). The non-provitamin A carotenoid, lutein, inhibits NF-kappaB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-kappaB-inducing kinase pathways: role of H(2)O(2) in NF-kappaB activation. Free Radic. Biol. Med. 45, 885–896. doi: 10.1016/j.freeradbiomed.2008.06.019
Kim, T. S., Pae, C. U., Yoon, S. J., Jang, W. Y., Lee, N. J., Kim, J. J., et al. (2006). Decreased plasma antioxidants in patients with Alzheimer's disease. Int. J. Geriatr. Psychiatry. 21, 344–348. doi: 10.1002/gps.1469
Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers. 7, 1–21. doi: 10.1038/s41572-021-00269-y
Koike, H., Iguchi, Y., Sahashi, K., and Katsuno, M. (2021). Significance of oligomeric and fibrillar species in amyloidosis: insights into pathophysiology and treatment. Molecules. 26, 5091. doi: 10.3390/molecules26165091
Kong, G. K. W., Miles, L. A., Crespi, G. A. N., Morton, C. J., Ng, H. L., Barnham, K. J., et al. (2008). Copper binding to the Alzheimer's disease amyloid precursor protein. Eur. Biophys. J. 37, 269–279. doi: 10.1007/s00249-007-0234-3
Krimbou, L., Denis, M., Haidar, B., Carrier, M., Marcil, M., and Genest, J. (2004). Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J. Lipid Res. 45, 839–848. doi: 10.1194/jlr.M300418-JLR200
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., et al. (2010). ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032. doi: 10.1038/emboj.2010.167
Kumar, A., Yegla, B., and Foster, T. C. (2018). Redox signaling in neurotransmission and cognition during aging. Antioxid. Redox Signal. 28, 1724–1745. doi: 10.1089/ars.2017.7111
Larosa, V., and Remacle, C. (2018). Insights into the respiratory chain and oxidative stress. Biosci Rep. 38, BSR20171492. doi: 10.1042/BSR20171492
Lee, D. G., Kam, M. K., Lee, S. R., Lee, H. J., and Lee, D. S. (2020). Peroxiredoxin 5 deficiency exacerbates iron overload-induced neuronal death via ER-mediated mitochondrial fission in mouse hippocampus. Cell Death Dis. 11, 204. doi: 10.1038/s41419-020-2402-7
Lee, E. G., Chen, S., Leong, L., Tulloch, J., and Yu, C. E. (2021). TOMM40 RNA transcription in Alzheimer's disease brain, and its implication in mitochondrial dysfunction. Genes (Basel). 12, 871. doi: 10.3390/genes12060871
Lei, P., Ayton, S., Bush, A. I., and Adlard, P. A. (2011). GSK-3 in neurodegenerative diseases. Int. J. Alzheimers. Dis. 2011, 189246. doi: 10.4061/2011/189246
Leng, K., Li, E., Eser, R., Piergies, A., Sit, R., Tan, M., et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat. Neurosci. 24, 276–287. doi: 10.1038/s41593-020-00764-7
Lennicke, C., Rahn, J., Heimer, N., Lichtenfels, R., Wessjohann, L. A., Seliger, B., et al. (2016). Redox proteomics: Methods for the identification and enrichment of redox-modified proteins and their applications. Proteomics. 16, 197–213. doi: 10.1002/pmic.201500268
Li, J., Jia, B., Cheng, Y., Song, Y., Li, Q., and Luo, C. (2022). Targeting molecular mediators of ferroptosis and oxidative stress for neurological disorders. Oxid. Med. Cell. Longev. 2022, 3999083. doi: 10.1155/2022/3999083
Liang, W. S., Reiman, E. M., Valla, J., Dunckley, T., Beach, T. G., Grover, A., et al. (2008). Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA. 105, 4441–4446. doi: 10.1073/pnas.0709259105
López-Grueso, M. J., González-Ojeda, R., Requejo-Aguilar, R., McDonagh, B., Fuentes-Almagro, C. A., Muntan,é, J., et al. (2019). Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 21, 101049. doi: 10.1016/j.redox.2018.11.007
Lü, R. (2017). Reaction-based small-molecule fluorescent probes for dynamic detection of ROS and transient redox changes in living cells and small animals. J. Mol. Cell. Cardiol. 110, 96–108. doi: 10.1016/j.yjmcc.2017.07.008
Lucke-Wold, B., Seidel, K., Udo, R., Omalu, B., Ornstein, M., Nolan, R., et al. (2017). Role of tau acetylation in alzheimer's disease and chronic traumatic encephalopathy: the way forward for successful treatment. J. Neurol. Neurosurg. 4, 140.
Maccioni, R. B., and Cambiazo, V. (1995). Role of microtubule-associated proteins in the control of microtubule assembly. Physiol. Rev. 75, 835–864. doi: 10.1152/physrev.1995.75.4.835
Maiti, P., Piacentini, R., Ripoli, C., Grassi, C., and Bitan, G. (2021). Correction: Surprising toxicity and assembly behaviour of amyloid β-protein oxidized to sulfone. Biochem. J. 478, 4151. doi: 10.1042/BJ20101391_COR
Manczak, M., Kandimalla, R., Fry, D., Sesaki, H., and Reddy, P. H. (2016). Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum. Mol. Genet. 25, 5148–5166. doi: 10.1093/hmg/ddw330
Manczak, M., Park, B. S., Jung, Y., and Reddy, P. H. (2004). Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 5, 147–162. doi: 10.1385/NMM:5:2:147
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Mandal, P. K., Goel, A., Bush, A. I., Punjabi, K., Joon, S., Mishra, R., et al. (2022). Hippocampal glutathione depletion with enhanced iron level in patients with mild cognitive impairment and Alzheimer's disease compared with healthy elderly participants. Brain Commun. 4, fcac215. doi: 10.1093/braincomms/fcac215
Marcel, Y. L., Vezina, C., and Milne, R. W. (1983). Cholesteryl ester and apolipoprotein E transfer between human high density lipoproteins and chylomicrons. Biochim. Biophys. Acta. 750, 411–417. doi: 10.1016/0005-2760(83)90047-4
Marcus, D. L., Thomas, C., Rodriguez, C., Simberkoff, K., Tsai, J. S., Strafaci, J. A., et al. (1998). Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp. Neurol. 150, 40–44. doi: 10.1006/exnr.1997.6750
Maron, R., Armony, G., Tsoory, M., Wilchek, M., Frenkel, D., and Arnon, R. (2020). Peptide interference with APP and tau association: relevance to Alzheimer's disease amelioration. Int. J. Mol. Sci. 21, E3270. doi: 10.3390/ijms21093270
Marotta, G., Basagni, F., Rosini, M., and Minarini, A. (2022). Role of fyn kinase inhibitors in switching neuroinflammatory pathways. Curr. Med. Chem. 29, 4738–4755. doi: 10.2174/0929867329666211221153719
Massaad, C. A., Washington, T. M., Pautler, R. G., and Klann, E. (2009). Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 106, 13576–13581. doi: 10.1073/pnas.0902714106
Meng, Y. X., Jiang, H. Y., Chen, A. J., Lu, F. Y., Yang, H., Zhang, M. Y., et al. (1990). Hemostatic changes in women using a monthly injectable contraceptive for one year. Contraception. 42, 455–466. doi: 10.1016/0010-7824(90)90052-W
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Minniti, A. N., Arrazola, M. S., Bravo-Zehnder, M., Ramos, F., Inestrosa, N. C., and Aldunate, R. (2015). The protein oxidation repair enzyme methionine sulfoxide reductase a modulates Aβ aggregation and toxicity in vivo. Antioxid. Redox Signal. 22, 48–62. doi: 10.1089/ars.2013.5803
Misrani, A., Tabassum, S., and Yang, L. (2021). Mitochondrial dysfunction and oxidative stress in Alzheimer's disease. Front. Aging Neurosci. 13, 617588. doi: 10.3389/fnagi.2021.617588
Muller, F. L., Lustgarten, M. S., Jang, Y., Richardson, A., and Van Remmen, H. (2007). Trends in oxidative aging theories. Free Radic. Biol. Med. 43, 477–503. doi: 10.1016/j.freeradbiomed.2007.03.034
Multhaup, G., Schlicksupp, A., Hesse, L., Beher, D., Ruppert, T., Masters, C. L., et al. (1996). The amyloid precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I). Science. 271, 1406–1409. doi: 10.1126/science.271.5254.1406
Munter, L. M., Botev, A., Richter, L., Hildebrand, P. W., Althoff, V., Weise, C., et al. (2010). Aberrant Amyloid Precursor Protein (APP) processing in hereditary forms of alzheimer disease caused by APP familial alzheimer disease mutations can be rescued by mutations in the APP GxxxG Motif. J. Biol. Chem. 285, 21636–21643. doi: 10.1074/jbc.M109.088005
Murata, H., Ihara, Y., Nakamura, H., Yodoi, J., Sumikawa, K., and Kondo, T. (2003). Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 278, 50226–50233. doi: 10.1074/jbc.M310171200
Murphy, M. P., Bayir, H., Belousov, V., Chang, C. J., Davies, K. J. A., Davies, M. J., et al. (2022). Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab. 4, 651–662. doi: 10.1038/s42255-022-00591-z
Nagy, Z., Esiri, M. M., LeGris, M., and Matthews, P. M. (1999). Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 97, 346–354. doi: 10.1007/s004010050997
Newman, M., Ebrahimie, E., and Lardelli, M. (2014). Using the zebrafish model for Alzheimer's disease research. Front. Genet. 5, 189. doi: 10.3389/fgene.2014.00189
Newman, M., Verdile, G., Martins, R. N., and Lardelli, M. (2011). Zebrafish as a tool in Alzheimer's disease research. Biochim. Biophys. Acta. 1812, 346–352. doi: 10.1016/j.bbadis.2010.09.012
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Olajide, O. J., Suvanto, M. E., and Chapman, C. A. (2021). Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer's disease. Biol. Open. 10, bio056796. doi: 10.1242/bio.056796
Omar, R. A., Chyan, Y. J., Andorn, A. C., Poeggeler, B., Robakis, N. K., and Pappolla, M. A. (1999). Increased Expression but Reduced Activity of Antioxidant Enzymes in Alzheimer's Disease. J. Alzheimers Dis. 1, 139–145. doi: 10.3233/JAD-1999-1301
Pankiewicz, J. E., Diaz, J. R., Martá-Ariza, M., Lizińczyk, A. M., Franco, L. A., and Sadowski, M. J. (2020). Peroxiredoxin 6 mediates protective function of astrocytes in Aβ proteostasis. Mol. Neurodegener. 15, 50. doi: 10.1186/s13024-020-00401-8
Papaspyropoulos, A., Tsolaki, M., Foroglou, N., and Pantazaki, A. A. (2020). Modeling and targeting Alzheimer's disease with organoids. Front. Pharmacol. 11, 396. doi: 10.3389/fphar.2020.00396
Park, J., Choi, H., Kim, B., Chae, U., Lee, D. G., Lee, S. R., et al. (2016b). Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca2+/calcineurin-Drp1-dependent mitochondrial fission. Free Radic. Biol. Med. 99, 392–404. doi: 10.1016/j.freeradbiomed.2016.08.030
Park, M. H., Jo, M., Kim, Y. R., Lee, C. K., and Hong, J. T. (2016a). Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 163, 1–23. doi: 10.1016/j.pharmthera.2016.03.018
Parker, W. D., Parks, J., Filley, C. M., and Kleinschmidt-DeMasters, B. K. (1994). Electron transport chain defects in Alzheimer's disease brain. Neurology. 44, 1090–1096. doi: 10.1212/WNL.44.6.1090
Peña-Ortega, F., Robles-Gómez, Á. A., and Xolalpa-Cueva, L. (2022). Microtubules as regulators of neural network shape and function: focus on excitability, plasticity and memory. Cells. 11, 923. doi: 10.3390/cells11060923
Pham, E., Crews, L., Ubhi, K., Hansen, L., Adame, A., Cartier, A., et al. (2010). Progressive accumulation of amyloid-β oligomers in Alzheimer's disease and APP transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 277, 3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer's disease. Front. Biosci. Elite. 5, 167–177. doi: 10.2741/E605
Pilkington, A. W., Donohoe, G. C., Akhmedov, N. G., Ferrebee, T., Valentine, S. J., and Legleiter, J. (2019). Hydrogen Peroxide Modifies Aβ-Membrane Interactions with Implications for Aβ40 Aggregation. Biochemistry. 58, 2893–2905. doi: 10.1021/acs.biochem.9b00233
Pîrşcoveanu, D. F. V., Pirici, I., Tudorică, V., Bălşeanu, T. A., Albu, V. C., Bondari, S., et al. (2017). Tau protein in neurodegenerative diseases - a review. Rom. J. Morphol. Embryol. 58, 1141–1150.
Prifti, E., Tsakiri, E. N., Vourkou, E., Stamatakis, G., Samiotaki, M., and Papanikolopoulou, K. (2021). The two cysteines of tau protein are functionally distinct and contribute differentially to its pathogenicity in vivo. J. Neurosci. 41, 797–810. doi: 10.1523/JNEUROSCI.1920-20.2020
Qian, W., and Liu, F. (2014). Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 30, 367–377. doi: 10.1007/s12264-013-1411-2
Rhee, S. G., Woo, H. A., Kil, I. S., and Bae, S. H. (2012). Peroxiredoxin Functions as a Peroxidase and a Regulator and Sensor of Local Peroxides*. J. Biol. Chem. 287, 4403–4410. doi: 10.1074/jbc.R111.283432
Riedel, B. C., Thompson, P. M., and Brinton, R. D. (2016). Age, APOE and sex: triad of risk of Alzheimer's disease. J. Steroid Biochem. Mol. Biol. 160, 134–147. doi: 10.1016/j.jsbmb.2016.03.012
Riemer, J., Schwarzländer, M., Conrad, M., and Herrmann, J. M. (2015). Thiol switches in mitochondria: operation and physiological relevance. Biol. Chem. 396, 465–482. doi: 10.1515/hsz-2014-0293
Ruiz, F. H., González, M., Bodini, M., Opazo, C., and Inestrosa, N. C. (1999). Cysteine 144 is a key residue in the copper reduction by the beta-amyloid precursor protein. J. Neurochem. 73, 1288–1292. doi: 10.1046/j.1471-4159.1999.0731288.x
Russell, R. L., Siedlak, S. L., Raina, A. K., Bautista, J. M., Smith, M. A., and Perry, G. (1999). Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 370, 236–239. doi: 10.1006/abbi.1999.1404
Sabens Liedhegner, E. A., Gao, X. H., and Mieyal, J. J. (2012). Mechanisms of altered redox regulation in neurodegenerative diseases–focus on S–glutathionylation. Antioxid. Redox Signal. 16, 543–566. doi: 10.1089/ars.2011.4119
Sacco, F., Seelig, A., Humphrey, S. J., Krahmer, N., Volta, F., Reggio, A., et al. (2019). Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab. 29, 1422–1432.e3. doi: 10.1016/j.cmet.2019.02.012
Saito, T., Chiku, T., Oka, M., Wada-Kakuda, S., Nobuhara, M., Oba, T., et al. (2021). Disulfide bond formation in microtubule-associated tau protein promotes tau accumulation and toxicity in vivo. Hum. Mol. Genet. 30, 1955–1967. doi: 10.1093/hmg/ddab162
Sanz, A., and Stefanatos, R. K. A. (2008). The mitochondrial free radical theory of aging: a critical view. Curr. Aging Sci. 1, 10–21. doi: 10.2174/1874609810801010010
Satoh, T., and Lipton, S. A. (2007). Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 30, 37–45. doi: 10.1016/j.tins.2006.11.004
Satpute-Krishnan, P., DeGiorgis, J. A., Conley, M. P., Jang, M., and Bearer, E. L. (2006). A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 103, 16532–16537. doi: 10.1073/pnas.0607527103
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2019). Redox Mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 30, 1450–1499. doi: 10.1089/ars.2017.7321
Schmidt, M., Sachse, C., Richter, W., Xu, C., Fändrich, M., and Grigorieff, N. (2009). Comparison of Alzheimer Aβ(1–40) and Aβ(1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. USA. 106, 19813–19818. doi: 10.1073/pnas.0905007106
Schöneich, C. (2002). Redox processes of methionine relevant to beta-amyloid oxidation and Alzheimer's disease. Arch. Biochem. Biophys. 397, 370–376. doi: 10.1006/abbi.2001.2621
Schubert, D., Behl, C., Lesley, R., Brack, A., Dargusch, R., Sagara, Y., et al. (1995). Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. USA. 92, 1989–1993. doi: 10.1073/pnas.92.6.1989
Seamster, P. E., Loewenberg, M., Pascal, J., Chauviere, A., Gonzales, A., Cristini, V., et al. (2012). Quantitative measurements and modeling of cargo-motor interactions during fast transport in the living axon. Phys. Biol. 9, 055005. doi: 10.1088/1478-3975/9/5/055005
Selkoe, D. J. (2001). Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Shin, M. K., Vázquez-Rosa, E., Koh, Y., Dhar, M., Chaubey, K., Cintrón-Pérez, C. J., et al. (2021). Reducing acetylated tau is neuroprotective in brain injury. Cell. 184, 2715–2732.e23. doi: 10.1016/j.cell.2021.03.032
Siddarth, P., Burggren, A. C., Merrill, D. A., Ercoli, L. M., Mahmood, Z., Barrio, J. R., et al. (2018). Longer TOMM40 poly-T variants associated with higher FDDNP-PET medial temporal tau and amyloid binding. PLoS ONE. 13, e0208358. doi: 10.1371/journal.pone.0208358
Sies, H. (2015). Oxidative stress: a concept in redox biology and medicine. Redox Biol. 4, 180–183. doi: 10.1016/j.redox.2015.01.002
Silva, D. F., Selfridge, J. E., Lu, J. E. L., Cardoso, S. M., and Swerdlow, R. H. (2012). Mitochondrial abnormalities in Alzheimer's disease: possible targets for therapeutic intervention. Adv. Pharmacol. 64, 83–126. doi: 10.1016/B978-0-12-394816-8.00003-9
Simpson, D. S. A., and Oliver, P. L. (2020). ROS Generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants (Basel). 9, 743. doi: 10.3390/antiox9080743
Sinsky, J., Majerova, P., Kovac, A., Kotlyar, M., Jurisica, I., and Hanes, J. (2020). Physiological Tau Interactome in Brain and Its Link to Tauopathies. J. Proteome Res. 19, 2429–2442. doi: 10.1021/acs.jproteome.0c00137
Song, K. H., Kim, J. H., Lee, Y. H., Bae, H. C., Lee, H. J., Woo, S. R., et al. (2018). Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance. J. Clin. Invest. 128, 4098–4114. doi: 10.1172/JCI96804
Stamelou, M., Respondek, G., Giagkou, N., Whitwell, J. L., Kovacs, G. G., and Höglinger, G. U. (2021). Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat. Rev. Neurol. 17, 601–620. doi: 10.1038/s41582-021-00541-5
Stege, G. J. J., and Bosman, C. G. M. G. J. (1999). The biochemistry of Alzheimer's disease. Drugs Aging. 14, 437–446. doi: 10.2165/00002512-199914060-00004
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Strittmatter, W. J., Saunders, A. M., Goedert, M., Weisgraber, K. H., Dong, L. M., Jakes, R., et al. (1994). Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA. 91, 11183–11186. doi: 10.1073/pnas.91.23.11183
Su, B., Wang, X., Lee, H. G., Tabaton, M., Perry, G., Smith, M. A., et al. (2010). Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 468, 267–271. doi: 10.1016/j.neulet.2009.11.010
Su, Z., Burchfield, J. G., Yang, P., Humphrey, S. J., Yang, G., Francis, D., et al. (2019). Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 10, 5486. doi: 10.1038/s41467-019-13114-4
Sultana, R., Boyd-Kimball, D., Poon, H. F., Cai, J., Pierce, W. M., Klein, J. B., et al. (2006a). Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging. 27, 1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021
Sultana, R., Perluigi, M., and Butterfield, D. A. (2006b). Redox proteomics identification of oxidatively modified proteins in Alzheimer's disease brain and in vivo and in vitro models of AD centered around Aβ(1–42). J. Chromatogr. B 833, 3–11. doi: 10.1016/j.jchromb.2005.09.024
Sultana, R., Perluigi, M., and Butterfield, D. A. (2009). Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 118, 131. doi: 10.1007/s00401-009-0517-0
Sultana, R., Perluigi, M., and Butterfield, D. A. (2013). Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169. doi: 10.1016/j.freeradbiomed.2012.09.027
Sun, L., Bhawal, R., Xu, H., Chen, H., Anderson, E. T., Haroutunian, V., et al. (2021). The human brain acetylome reveals that decreased acetylation of mitochondrial proteins associates with Alzheimer's disease. J. Neurochem. 158, 282–296. doi: 10.1111/jnc.15377
Sun, Y., Ma, C., Sun, H., Wang, H., Peng, W., Zhou, Z., et al. (2020). Metabolism: a novel shared link between diabetes mellitus and Alzheimer's disease. J. Diabetes Res. 2020, 4981814. doi: 10.1155/2020/4981814
Swomley, A. M., Förster, S., Keeney, J. T., Triplett, J., Zhang, Z., Sultana, R., et al. (2014). Abeta, oxidative stress in alzheimer disease: evidence based on proteomics studies. Biochim. Biophys. Acta. 1842, 1248–1257. doi: 10.1016/j.bbadis.2013.09.015
Tamagno, E., Guglielmotto, M., Aragno, M., Borghi, R., Autelli, R., Giliberto, L., et al. (2008). Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 104, 683–695. doi: 10.1111/j.1471-4159.2007.05072.x
Tandon, A., and Fraser, P. (2002). The presenilins. Genome Biol. 3, reviews3014. doi: 10.1186/gb-2002-3-11-reviews3014
Terni, B., Boada, J., Portero-Otin, M., Pamplona, R., and Ferrer, I. (2010). Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer's disease pathology. Brain Pathol. 20, 222–233. doi: 10.1111/j.1750-3639.2009.00266.x
Tjiang, N., and Zempel, H. (2022). A mitochondria cluster at the proximal axon initial segment controls axodendritic TAU trafficking in rodent primary and human iPSC-derived neurons. Cell. Mol. Life Sci. 79, 120. doi: 10.1007/s00018-022-04150-3
Toledo, J. B., Arnold, M., Kastenmüller, G., Chang, R., Baillie, R. A., Han, X., et al. (2017). Alzheimer's disease neuroimaging initiative and the Alzheimer disease metabolomics consortium, metabolic network failures in alzheimer's disease: a biochemical road map. Alzheimers. Dement. 13, 965–984. doi: 10.1016/j.jalz.2017.01.020
Tracy, T. E., Madero-Pérez, J., Swaney, D. L., Chang, T. S., Moritz, M., Konrad, C., et al. (2022). Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 185, 712–728.e14. doi: 10.1016/j.cell.2021.12.041
Uddin, M. S., Kabir, M. T., Rahman, M. S., Behl, T., Jeandet, P., Ashraf, G. M., et al. (2020). Revisiting the amyloid cascade hypothesis: from Anti-Aβ therapeutics to auspicious new ways for Alzheimer's disease. Int. J. Mol. Sci. 21, E5858. doi: 10.3390/ijms21165858
Vesković, A., Nakarada, ., Pavićević, A., Prokić, B., Perović, M., Kanazir, S., et al. (2021). In vivo/ex vivo EPR investigation of the brain redox status and blood–brain barrier integrity in the 5xFAD mouse model of Alzheimer's disease. Curr. Alzheimer Res. 18, 25–34. doi: 10.2174/1567205018666210324121156
von Bernhardi, R., and Eugenín, J. (2012). Alzheimer's disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 16, 974–1031. doi: 10.1089/ars.2011.4082
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer's disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, X., and Michaelis, E. K. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2, 12. doi: 10.3389/fnagi.2010.00012
Wang, X., Pal, R., Chen, X. W., Limpeanchob, N., Kumar, K. N., and Michaelis, E. K. (2005). High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Brain Res. Mol. Brain Res. 140, 120–126. doi: 10.1016/j.molbrainres.2005.07.018
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008a). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am. J. Pathol. 173, 470–482. doi: 10.2353/ajpath.2008.071208
Wang, X., Su, B., Lee, H., Li, X., Perry, G., Smith, M. A., et al. (2009b). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008b). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA. 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009a). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J. Neurochem. 109 Suppl 1, 153–159. doi: 10.1111/j.1471-4159.2009.05867.x
Wang, X., Wang, W., Li, L., Perry, G., Lee, H., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim. Biophys. Acta. 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Wang, Y., Tang, B., Zhu, J., Yu, J., Hui, J., Xia, S., et al. (2022). Emerging mechanisms and targeted therapy of ferroptosis in neurological diseases and neuro-oncology. Int. J. Biol. Sci. 18, 4260–4274. doi: 10.7150/ijbs.72251
Wei, Y., Qu, M. H., Wang, X. S., Chen, L., Wang, D. L., Liu, Y., et al. (2008). Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE. 3, e2600. doi: 10.1371/journal.pone.0002600
Wu, L., Sedgwick, A. C., Sun, X., Bull, S. D., He, X. P., and James, T. D. (2019). Reaction-based fluorescent probes for the detection and imaging of reactive oxygen, nitrogen, and sulfur species. Acc. Chem. Res. 52, 2582–2597. doi: 10.1021/acs.accounts.9b00302
Wu, W., Liao, X., Chen, Y., Ji, L., and Chao, H. (2021). Mitochondria-targeting and reversible near-infrared emissive iridium(III) probe for in vivo ONOO-/GSH redox cycles monitoring. Anal. Chem. 93, 8062–8070. doi: 10.1021/acs.analchem.1c01409
Yamauchi, K., and Kawakami, Y. (2020). The redox status of cysteine thiol residues of apolipoprotein E impacts on its lipid interactions. Biol. Chem. 401, 617–627. doi: 10.1515/hsz-2019-0414
Yan, Y., and Wang, C. (2014). Protection mechanisms against Aβ42 aggregation. Curr. Alzheimer Res. 5, 548–554. doi: 10.2174/156720508786898460
Yang, Y., Tapias, V., Acosta, D., Xu, H., Chen, H., Bhawal, R., et al. (2022). Altered succinylation of mitochondrial proteins, APP and tau in Alzheimer's disease. Nat. Commun. 13, 159. doi: 10.1038/s41467-021-27572-2
Yap, L.-P., Garcia, J. V., Han, D., and Cadenas, E. (2009). The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 61, 1283–1298. doi: 10.1016/j.addr.2009.07.015
Yin, F., Boveris, A., and Cadenas, E. (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 20, 353–371. doi: 10.1089/ars.2012.4774
Zemlan, F. P., Thienhaus, O. J., and Bosmann, H. B. (1989). Superoxide dismutase activity in Alzheimer's disease: possible mechanism for paired helical filament formation. Brain Res. 476, 160–162. doi: 10.1016/0006-8993(89)91550-3
Zhang, C., Rodriguez, C., Spaulding, J., Aw, T. Y., and Feng, J. (2012). Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer's disease. J. Alzheimers. Dis. 28, 655–666. doi: 10.3233/JAD-2011-111244
Zhang, D. M., Levitan, D., Yu, G., Nishimura, M., Chen, F., Tandon, A., et al. (2000). Mutation of the conserved N-terminal cysteine (Cys92) of human presenilin 1 causes increased A beta42 secretion in mammalian cells but impaired Notch/lin-12 signalling in C. elegans. Neuroreport. 11, 3227–3230. doi: 10.1097/00001756-200009280-00035
Zhang, S., Eitan, E., and Mattson, M. P. (2017). Early involvement of lysosome dysfunction in the degeneration of cerebral cortical neurons caused by the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 140, 941–954. doi: 10.1111/jnc.13957
Keywords: redox signaling, redox metabolism, Alzheimer's disease, neurodegeneration, Tau, APP
Citation: Holubiec MI, Gellert M and Hanschmann EM (2022) Redox signaling and metabolism in Alzheimer's disease. Front. Aging Neurosci. 14:1003721. doi: 10.3389/fnagi.2022.1003721
Received: 26 July 2022; Accepted: 14 October 2022;
Published: 03 November 2022.
Edited by:
Ramesh Kandimalla, Indian Institute of Chemical Technology (CSIR), IndiaReviewed by:
Haritha Kunhiraman, Emory University, United StatesRuchika Bhawal, Cornell University, United States
Liviu Mirica, University of Illinois at Urbana-Champaign, United States
Copyright © 2022 Holubiec, Gellert and Hanschmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Gellert, Z2VsbGVydG1AdW5pLWdyZWlmc3dhbGQuZGU=; M. I. Holubiec, bWhvbHViaWVjQGZtZWQudWJhLmFy; E. M. Hanschmann, ZXZhX2hhbnNjaG1hbm5Ad2ViLmRl