
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 02 December 2022
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.1002138
 Wenmin Yi1,2,3,4,5
Wenmin Yi1,2,3,4,5 Fei Chen1,2,3,4,5Huiji Zhang1,2,3,4Peng Tang2,3Minghao Yuan1,2,3,4,5Jie Wen5,6Shengyuan Wang1,2,3,4,5
Fei Chen1,2,3,4,5Huiji Zhang1,2,3,4Peng Tang2,3Minghao Yuan1,2,3,4,5Jie Wen5,6Shengyuan Wang1,2,3,4,5 Zhiyou Cai1,2,3,4,5*
Zhiyou Cai1,2,3,4,5*Aging is an inevitable progressive decline in physiological organ function that increases the chance of disease and death. The renin–angiotensin system (RAS) is involved in the regulation of vasoconstriction, fluid homeostasis, cell growth, fibrosis, inflammation, and oxidative stress. In recent years, unprecedented advancement has been made in the RAS study, particularly with the observation that angiotensin II (Ang II), the central product of the RAS, plays a significant role in aging and chronic disease burden with aging. Binding to its receptors (Ang II type 1 receptor – AT1R in particular), Ang II acts as a mediator in the aging process by increasing free radical production and, consequently, mitochondrial dysfunction and telomere attrition. In this review, we examine the physiological function of the RAS and reactive oxygen species (ROS) sources in detail, highlighting how Ang II amplifies or drives mitochondrial dysfunction and telomere attrition underlying each hallmark of aging and contributes to the development of aging and age-linked diseases. Accordingly, the Ang II/AT1R pathway opens a new preventive and therapeutic direction for delaying aging and reducing the incidence of age-related diseases in the future.
During the past century, advances in modern medicine knowledge and improvements in examination technology have significantly augmented life expectancy (Kontis et al., 2017; Kalache et al., 2019). The global population, aged 60 years and above, is estimated to increase from 841 million in 2013 to approximately 2 billion in 2050 (Foreman et al., 2018). This demographic mile-stone will usually be accompanied by the high incidence of some diseases, such as cardiovascular disease, metabolic disorders, cancer, and neurodegenerative disorders (Lopez-Otin et al., 2013; Guzman-Castillo et al., 2017; Melzer et al., 2020), the incidence of which doubles every 5 years for people aged over 60 years. Aging is an irreversible and intricate process characterized by a generalized and time-dependent biological functional impairment, accompanied by genomic instability, telomere attrition, mitochondrial dysfunction, and cellular senescence (Lopez-Otin et al., 2013). Aging results in decreasing quality of life and has gradually become one of the major socioeconomic challenges of our era.
For the last few decades, the involvement of the renin–angiotensin system (RAS) in mediating vasoconstriction, ion entry and excretion, fibrosis, inflammatory and oxidative stress has been well documented (Benigni et al., 2009, 2010; Forrester et al., 2018). In addition to the circulating system, elements of the RAS are also found in diverse tissues of the brain, heart, and kidney, and contribute to the aging of these organs (Feng et al., 2011; Zablocki and Sadoshima, 2013; Forrester et al., 2018; Mogi, 2020; Labandeira-Garcia et al., 2021). Within the brain, different components of the RAS have been extensively studied in the context of neuroprotection and cognition (Jackson et al., 2018). Alterations in the brain RAS during aging may establish a link between impairment of autonomic reflex function and metabolic changes in aging (Diz et al., 2007). Further evidence for the relevance of the RAS in aging is derived from numerous experiments in vivo and in vitro, indicating that aging is accompanied by the increased activity of angiotensin II (Ang II), which is the major bioactive peptide of this system (Benigni et al., 2009; Salazar, 2018; Okuno et al., 2020). A prospective observational study (Benigni et al., 2009) effectively proved that the lifespan of mice with disrupted Ang II type 1 receptor (AT1R) was remarkably longer than that in the control group and showed that the mice lacking AT1R exhibited less oxidative damage, consequently indicating that Ang II-induced reactive oxygen species (ROS) by AT1R seemed to play a crucial part in the aging process. Considering that mitochondria are a key source of endogenous ROS (Cadenas, 2004; Balaban et al., 2005) and telomeres are particularly vulnerable to oxidative stress (Ahmed and Lingner, 2018; Barnes et al., 2019), in this article, we first describe the diverse components of the RAS as well as their physiological functions coupling with each other. Then, we provide an overview of the primary ROS sources and the mechanistic association of ROS with mitochondria and telomeres. This is followed by the discussion of the seemingly universal roles of mitochondrial dysfunction and telomere attrition in aging and how Ang II influences them, conducing to present new preventive strategies in fighting aging and age-associated diseases.
In 1898, Robert Tigerstedt and Per Gunnar Bergman discovered a powerful vasopressor in the renal cortex, named rennin (Tigerstedt and Bergman, 1898). Since then, the RAS has been widely researched. The RAS was initially considered a circulating hormonal system that played a major role in the regulation of body fluid homeostasis through the control of blood volume and peripheral resistance. It was generally thought that the RAS appeared only in the classic circulatory system (Kinoshita et al., 2009). Later on, however, the local or paracrine RAS was identified in various tissues, including the brain, heart, and kidney (Stoll et al., 1995; Vaughan et al., 1995; Lee et al., 2010; Salgado et al., 2010; Pan et al., 2014; Labandeira-Garcia et al., 2021). The elements of the RAS can be cleaved during circulation by a series of enzymes and delivered to and expressed in its numerous target tissues or cells. The local tissue RAS contains two forms: intracellular and extracellular (Zablocki and Sadoshima, 2011; Zhuo and Li, 2011; Chappell et al., 2014), which further increases the difficulty of studying the RAS. The synthesis of intracellular RAS components and different types of RAS receptors is observed in some cells, including vascular smooth muscle cells, fibroblasts, cardiomyocytes, renal cells, and neurons (Re and Cook, 2015; Li et al., 2018; Re, 2018; Escobales et al., 2019). Recently, researchers have paid more attention to the role of the local RAS in specific tissues, such as the brain, the heart, and the kidney (Dzau and Re, 1994). The local paracrine RAS in the brain has been associated with several brain disorders, including Parkinson's disease (PD) (Costa-Besada et al., 2018). Several studies have suggested that the circulating RAS and tissue paracrine RAS may act together in peripheral tissues and that the two forms of the RAS are deemed to work in a complementary way, especially in the heart and the kidney (Lee et al., 2010). For example, the tissue RAS in the kidney regulates, together with the circulating RAS, not only renal cell growth and production of glomerulosclerosis but also blood pressure (Kagami et al., 1994; Ruiz-Ortega and Egido, 1997).
The renin–angiotensin system comprises numerous peptides and enzymes, including renin, an acidic protease secreted by juxtaglomerular cells, angiotensinogen, and different types of angiotensin and their receptors (Figure 1). Angiotensinogen is cleaved by renin to form angiotensin I (Ang I), a decapeptide with weak biological activity. Ang I is subsequently hydrolyzed by angiotensin-converting enzyme (ACE) to form octapeptide Ang II, a hormone that has strong hemodynamic effects (Boehm and Nabel, 2002). Ang II can undergo further hydrolysis into angiotensin III (Ang III) and angiotensin IV (Ang IV) by other aminopeptidases in plasma and tissue. ACE is primarily membrane bound on the endothelium, mainly in the epididymis, testes, and lungs, and it is shed in the plasma. The ACE gene lies on human chromosome 17q23. Genetic analysis has uncovered that ACE I/D polymorphism, involving a 287 bp DNA repeat sequence that is inserted(I) or deleted (D) in intron 16 of chromosome 17, accounts for about 50% of the total phenotypic variance of ACE (Zhu et al., 2012). Studies on all the primary races have suggested that the ACE I/D polymorphism is associated with systemic lupus erythematosus, type 2 diabetes, and other related renal and cardiovascular disease (Huang, 1999; Sprovieri and Sens, 2005; Alsafar et al., 2015). Besides, it is also a credible means to select patients who may benefit the most from therapy with angiotensin receptor blocker (ARB) (Ruggenenti et al., 2008). Apart from ACE, chymase, a serine protease that exists in mast cells, endothelial cells, stromal cells of the heart (Urata et al., 1993), and in vascular smooth muscle cells of the kidney (Huang et al., 2003), can also convert Ang I to Ang II (Urata et al., 1993; Takai et al., 2012). Chymase-hydrolyzed Ang II generation has emerged as a substitute for ACE in cardiac, vascular, and renal tissue, particularly in disease conditions (Bacani and Frishman, 2006; Miyazaki and Takai, 2006).
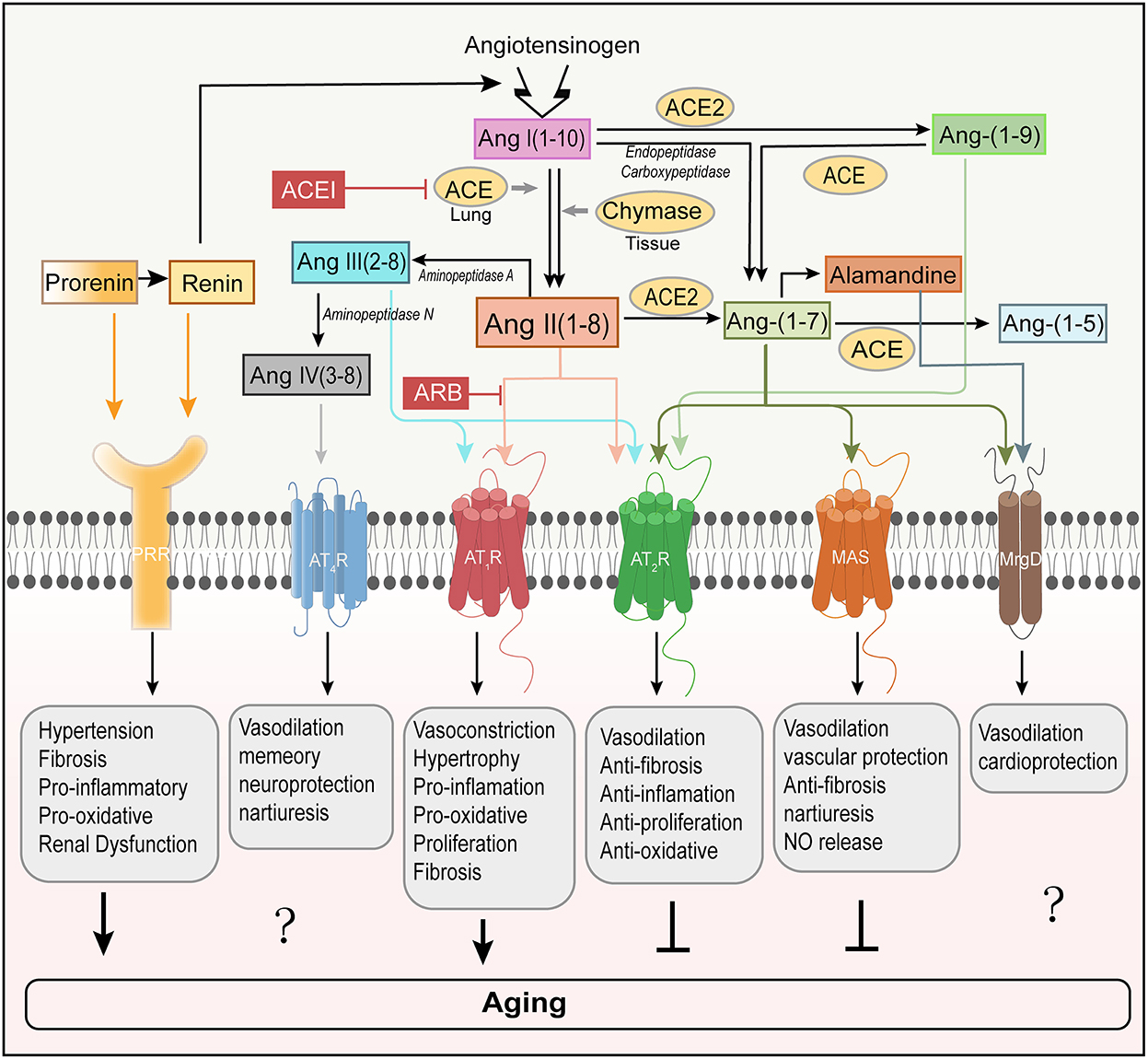
Figure 1. Involvement of RAS peptides and receptors in aging regulation. The classical RAS positively regulates the induction of aging by activating AT1R. In contrast, AT2R is regarded as a counterbalance to AT1R-mediated aging. PRR is a new member of the RAS receptors and mediates aging (Yoshida et al., 2019). Ang-(1–7) binds to MAS to counter-regulate of AT1R-mediated aging. However, the roles that Ang-(1–7) receptor MrgD and Ang IV receptor AT4R play in aging remain unclear. ACE, angiotensin-converting enzyme; ACE2, angiotensin converting enzyme 2; ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; AT1R, angiotensin type I receptor; AT2R, angiotensin type 2 receptor; AT4R, angiotensin type 4 receptor; MrgD, Mas-related-G protein-coupled receptor; PRR, (pro)renin receptor; NO, nitric oxide.
Recent research works have identified that ACE2, the first known human homolog ACE, is another carboxypeptidase and highly restricted to the heart, kidney, and testis in humans (Donoghue et al., 2000). As a counterbalance to ACE, ACE2 cleaves one aminoacid from Ang II, leading to the production of heptapeptide, angiotensin-(1–7)[Ang-(1–7)], which is generally considered to counter Ang II by activating Mas receptor (Mas) (Brand et al., 2002; Crackower et al., 2002; Ferrario and Chappell, 2004; Chamsi-Pasha et al., 2014; Romero et al., 2019). The Ang-(1–7)/Mas axis is downregulated with aging and may contribute to the aging-related susceptibility to neurodegeneration (Costa-Besada et al., 2018). Furthermore, ACE2 transforms Ang I into angiotensin-(1–9) [Ang-(1–9)] that, through the activation of angiotensin type 2 receptor (AT2R), may decrease blood pressure and protect the heart, blood vessels and possibly the kidney from adverse cardiovascular remodeling stimulated by hypertension or heart failure (Donoghue et al., 2000; Shariat-Madar and Schmaier, 2004; Ocaranza et al., 2014). Finally, these angiotensin family members can be further degraded into inactive peptide fragments, lowering the serum levels of endogenous vasodilators.
Ang II binds with high affinity to two pharmacologically distinct G protein-coupled receptors, the AT1R and the AT2R (Karnik et al., 2015) (Figure 1). Studies have identified that two isoforms can be found in rats and mice, dubbed AT1AR and AT1BR with 94% of aminoacid sequence identity and pharmacologically indistinguishable (Sasaki et al., 1991; de Gasparo et al., 1995). The AT1AR, the closest murine homolog to the single human AT1R, is predominantly expressed in organs including the brain (Burson et al., 1994). While AT1BR is primarily expressed in the anterior pituitary gland and adrenal zona glomerulosa (Oliverio and Coffman, 2000). Most of the classical actions of Ang II are mediated by AT1AR, such as an increase in blood pressure, aldosterone synthesis and release from the adrenal zona glomerulosa, and stimulation of the sympathetic nervous system (Davisson et al., 2000). Overactivation of tissue Ang II leads to proliferation, fibrosis, inflammatory response, and oxidative stress, which appear to be associated with aging in certain tissues. AT1BR controls blood pressure when AT1AR is in shortage. Besides, Ang II can also bind to AT2R, which generally exerts opposing effects on those mediated by AT1R (Forrester et al., 2018). Ang II activating the AT2R induces vasodilation of arteries through nitric oxide (NO) and cGMP (cyclic guanosine monophosphate) stimulation and improves resistance artery remodeling, proliferation, and inflammation and decreases fibrosis and oxidative stress. These functions are expressed in the adrenal medulla, uterus, ovary, and distinct brain regions (Jones et al., 2008; Matavelli and Siragy, 2015; Sumners et al., 2015). AT2R also provides a cardio-protective effect against ischemia-reperfusion injury and acute myocardial infarction and protects the kidney from fibrosis and ischemic injury (Schulman and Raij, 2008). Moreover, reduction of senescence markers induced by Ang II was observed in AT1AR-inactivated mice, and disruption of AT1AR promoted the lifespan of mice compared with controls, possibly through attenuation of oxidative stress and upregulation of the pro-survival genes in the kidney, suggesting the role of AT1AR in senescence (Benigni et al., 2009). In addition, during the past 20 years, significant improvements have been made in understanding the mechanism and function of new members. Ang-(1–7) acts on the Mas, one that signals to mediate the antagonistic effect of Ang II, leading to vasodilation, NO release, anti-fibrosis, antiproliferation, and anti-inflammation. As with the binding of Ang-(1–7) to the Mas, alamandine also promotes the hypotensive effect through the production of NO with Mas-related G protein-coupled receptor D (MrgD) (Lautner et al., 2013). Alamandine [Ala1-Ang-(1–7)] is endogenously synthesized from Ang-(1–7), and therefore, its serum level can be regarded as amounting to that of Ang-(1–7) (Carey, 2013). Alamandine can also be formed from Ala1-Ang II (angiotensin A) by ACE2 owing to decarboxylation of the Asp residue (Lautner et al., 2013). Ang III binds to and activates the AT1R and AT2R, while Ang IV binds to the angiotensin type 4 receptor (AT4R), causing vasodilation in the vascular beds of the brain, heart, and kidney, improving memory and increasing renal cortical blood flow and natriuresis. Finally, prorenin and renin both act at the (pro)renin receptor (PRR) to induce specific effects. These effects may be related to aging, therefore, summarizing the relationship between the RAS and aging process may be of great significance for providing new strategies for humans to delay aging in the future.
As discussed above, in general, we have already laid out the functions of Ang II. It is a key player in the induction of the oxidative stress toxicity process and the activation of redox-sensitive signaling cascades (Zhang et al., 2007; de Cavanagh et al., 2011). An abundance of evidence has demonstrated that Ang II can activate NAD(P)H oxidase (Nox) (Griendling et al., 1994; Hanna et al., 2002; Kimura et al., 2005) to produce ROS via binding to AT1R and stimulate mitochondrial ROS (mtROS) production (Doughan et al., 2008; Dai et al., 2011), leading to oxidative stress damage (Figure 2). ROS can damage macromolecules, such as proteins, DNA, and lipids (Cross et al., 1987; Young and Woodside, 2001; Schieber and Chandel, 2014; Silva et al., 2018), resulting in the accumulation of genetic and biological activity alterations, acceleration of mutagenesis, and eventually cell death (Pagan et al., 2022). Therefore, accretion of Ang II-mediated damage to cellular macromolecules is deemed as an underlying driving force in the aging process.
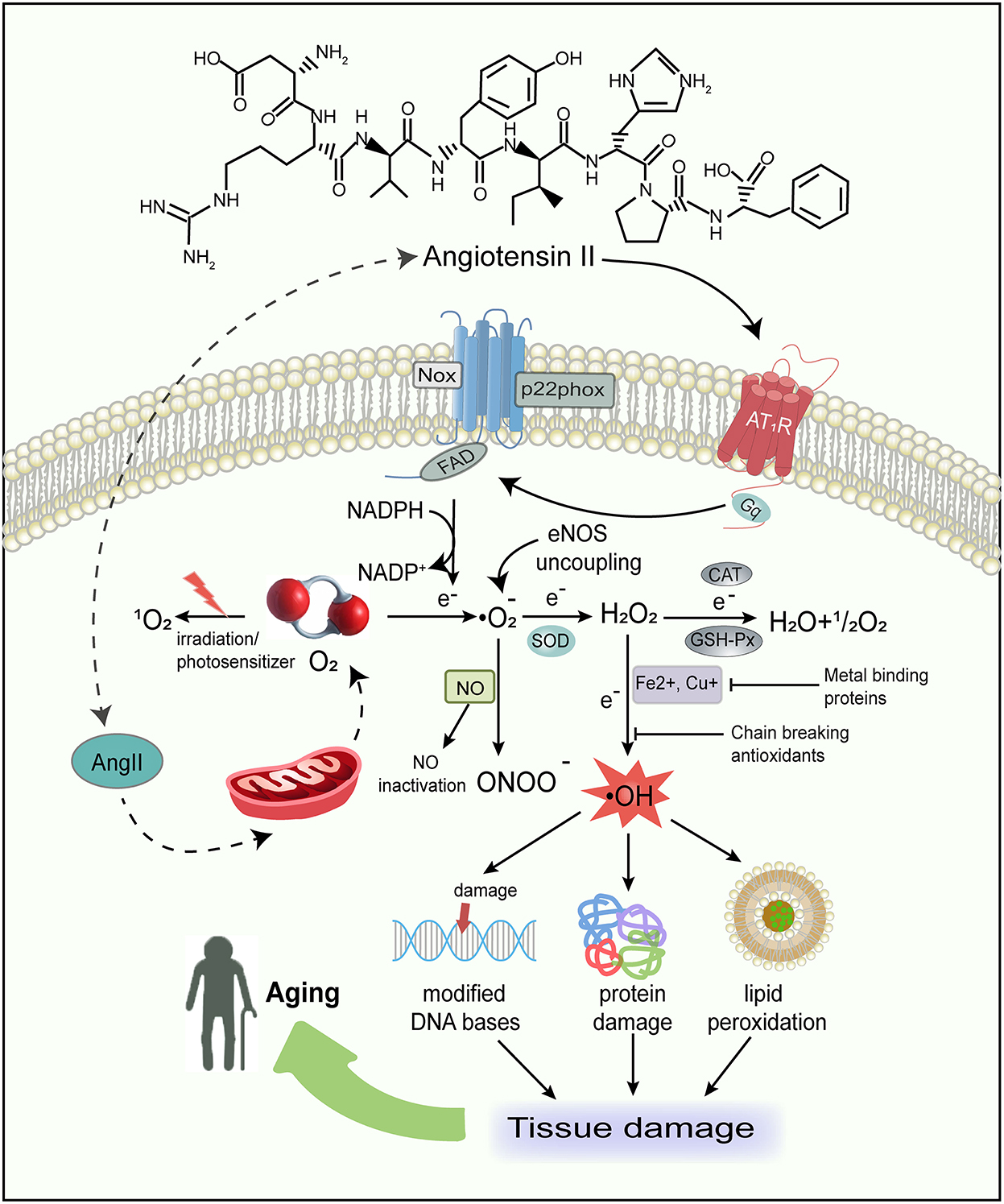
Figure 2. Schematic representation of major sources of free radicals in the body and the consequences of free radical damage. Ang II not only produces ROS by activating Nox and favoring eNOS uncoupling, but also has a direct action on mtROS production. The oxidative stress in turn can damage macromolecules, such as proteins, lipids, and DNA, therefore resulting in tissue damage and aging. ROS, reactive oxygen species; Nox, NAD(P)H oxidase; eNOS, endothelial NO synthase; mtROS, mitochondrial reactive oxygen species; O2, oxygen; , singlet oxygen; , superoxide; SOD, superoxide dismutase; H2O2, hydrogen peroxide; H2O, water; OH·, hydroxyl radical; NO, nitric oxide; ONOO-, peroxynitrite; CAT, catalase; GSH-Px, glutathione peroxidase; NADPH, nicotinamide adenine dinucleotide phosphate.
In addition, Ang II also enhances NO generation in the cytoplasm of different cell types such as endothelial, vascular smooth muscle, tubular epithelial, and fibroblast cells. Since the interaction of NO with superoxide () generates peroxynitrite (ONOO−) (Darley-Usmar et al., 1995; Fridovich, 1997; Pueyo et al., 1998), it can promote the production of both ROS and reactive nitrogen species (RNS), inhibit mitochondrial electron transport, and destroy DNA and cellular proteins (Radi et al., 1991). Furthermore, Ang II can promote endothelial NO synthase(eNOS) uncoupling, changing from NO to formation (Mollnau et al., 2002). Ang II-derived ROS, as significant intracellular second messengers, may activate many downstream signaling molecules such as mitogen-activated protein kinases (MAPK), tyrosine phosphatases, tyrosine kinases, and transcription factors (Madamanchi et al., 2005; Zhang et al., 2007). This suggests that ROS are highly related to regulating signal transduction pathways involved in cell growth, differentiation, apoptosis, and hypertrophy (Reckelhoff and Romero, 2003; Garrido and Griendling, 2009). We have previously reported that inhibition of MAPK induced by Ang II remarkably reduces senescence (Feresin et al., 2016), implying that Ang II-induced senescence is intricate and a combination of mechanisms may be involved.
The reactive oxygen species are generated as intermediates in the cellular reduction–oxidation reaction of oxygen, converting oxygen to water (Figure 2). In the presence of a free electron (e−), the univalent reduction of oxygen yields anion, hydrogen peroxide (H2O2), and hydroxyl radical (OH·) (Table 1). Interestingly, oxygen, a substance indispensable for human life activities, has detrimental effects on humans under certain conditions (Pham-Huy et al., 2008). The majority of the potentially destructive effects of oxygen are attributed to the formation and activation of ROS, which are inclined to transmit oxygen to other biomolecules.
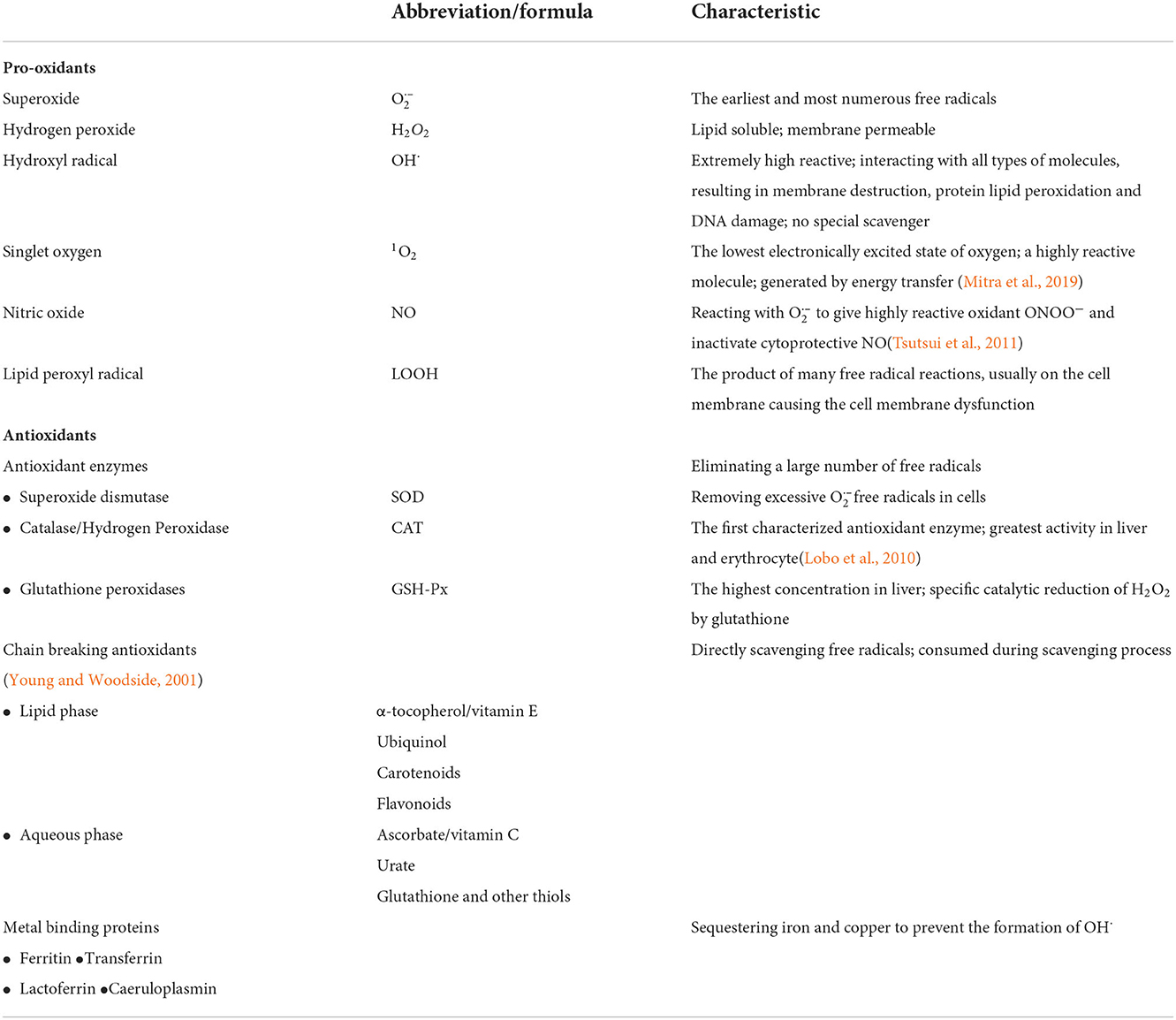
Table 1. ROS and the antioxidant defense mechanisms.
The reactive oxygen species play an important role in biological systems. Initially, ROS were considered as simple natural intermediate products involved in the molecular metabolism that can cause oxidative damage, various chronic diseases, and aging (Pham-Huy et al., 2008; Lobo et al., 2010; Rogov et al., 2021). However, with further research on free radicals in biology recently, they fulfill a more complicated and important physiological role than previously known, acting as vital intermediaries in cell signaling and homeostasis (Rogov et al., 2021).
The reactive oxygen species are highly diverse due to their respective dynamics and chemical properties, such as biological reactivity and the ability to cross membranes. The faster the elements in the ROS integrate with other molecules, the shorter the half-life. ROS can be charged or neutral, hydrophilic or hydrophobic, which dictates their capacities to cross biological membranes and move in aqueous or lipophilic environments. , which is considered the primary ROS, has an unpaired electron in its molecular outermost orbital. In an aerobic chemical reaction, free radicals grab electrons from other substances to form a stable state because electrons must come in pairs. So, the unpaired electron of imparts high oxidation reactivity and renders unstable and short-lived. can work not only as an oxidizing agent, in which is reduced to H2O2 but also as a reducing agent, where provides its unpaired electron for NO to form ONOO−, leading to NO quenching and the resultant decrease in NO availability. The disproportionation of to H2O2 can be spontaneous or catalyzed by superoxide dismutase (SOD). There are three forms of SOD in mammalian tissues: cytoplasmic copper/zinc SOD (SOD1), mitochondrial manganese SOD (MnSOD/SOD2), and extracellular SOD (SOD3) (Griendling et al., 1994). NO is regarded as an important protective factor against cardiac-cerebrovascular disease since it modulates vascular dilator tone and local cell growth, downregulates the expression of adhesion proteins, and prevents platelet aggregation, thrombosis in blood vessels and smooth muscle proliferation (Tousoulis et al., 2012). Although H2O2, mainly produced by spontaneous dismutation, is not a free radical, it is usually a member of the general ROS. H2O2 may directly destroy enzymes or proteins involving active thiol groups under a weak oxidation process (Young and Woodside, 2001). Also, it is a lipid-soluble molecule that can cross cell membranes directly, while can only go across cell membranes with the help of anion channels. Such a vital property allows H2O2 to diffuse over considerable distances from its site of production into other cells or cell compartments before decomposing to the highly reactive OH· (Halliwell and Gutteridge, 1990; Touyz, 2003). In general, upon the situation of excess anions, OH· is formed as a result of the decomposition of and H2O2 with transition metal (such as Fe2+ or Cu2+) catalyzed (Haber–Weiss reaction) (Koppenol, 2001). Although OH· has a very short half-life in vivo, it is the strongest oxidant among the oxygen free radicals (Zheng et al., 2015) and the most dangerous ROS. It is considered the ultimate mediator of most free radical-induced toxic effects (Lloyd et al., 1997). Owing to the presence of an unpaired electron, OH· could react at an extremely high rate with almost every type of biological molecule in living cells (del Río et al., 1992), especially with purines and pyrimidines in DNA, leading to cell death or mutation. All ROS discussed above exert tissue damage by inducing OH· formation. There are no antioxidants to eradicate OH· due to its extremely high reactivity.
Living organisms have evolved an extensive range of endogenous and exogenous antioxidant defense systems to protect cells against oxidative stress damage (Table 1). Free radicals-induced damage can be prevented by scavenging enzymes, as well as by other nonenzymatic antioxidants such as chain-breaking antioxidants, and transition metal-binding proteins (Figure 2). This can be realized by preventing the formation and scavenging of radicals or accelerating their decomposition (Finkel, 1998).
H2O2 is reduced to H2O and O2 by glutathione peroxidase (GSH-Px) and catalase. GSH-Px has higher affinity for H2O2 than catalase and is a vital antioxidant that not only prevents the formation of other more toxic free radicals, such as OH·, but also protects the cellular membrane from lipid peroxidation because glutathione contributes protons to the membrane lipids to keep them in a steady state. Furthermore, mice with GSH-Px overexpression were more resistant to myocardial oxidative stress and inhibited myocardial remodeling and failure in the heart of myocardial infarction, which might improve survival (Shiomi et al., 2004; Matsushima et al., 2006). Based on these lines of evidence, therapies aiming to interfere with oxidative stress may be a new avenue to prevent cardiac failure.
The rate and magnitude of oxidant formation and detoxification turn out to be balanced under normal physiological function. However, when the production of pro-oxidants overwhelms the body's antioxidative capacity, oxidative stress ensues (Lobo et al., 2010). As mentioned above, oxidative stress refers to the presence of excess free radicals in the nuclei and membranes of cells that attack lipids, nucleic acids, and proteins, triggering numerous chronic and degenerative illness diseases (Collin, 2019), such as PD (Trist et al., 2019) and Alzheimer disease (Tönnies and Trushina, 2017; Ionescu-Tucker and Cotman, 2021). Besides, it must be emphasized that cellular damage caused by oxidative stress is irreparable in that it induces irreversible macromolecular destruction, such as DNA or cellular membrane alteration or damage (Obrenovich et al., 2011). Hence, an external source of antioxidants should be applied prior to the development of oxidative stress.
The process of aging, a progressive functional decline and easier approach to death, has attracted boundless curiosity and deep exploration throughout human history. Although aging is a normal physiological process rather than a disease, it is recognized as the primary risk factor for all age-associated diseases (Rattan, 2014), such as cardiovascular disease, kidney failure, diabetes, cancer, and neurodegeneration, whose morbidity increases gradually along with aging (Brody and Grant, 2001; Lopez-Otin et al., 2013).
With the ever-increasing knowledge in molecular and cytology and continuous advances in technology, aging research has progressed unprecedentedly in recent years. Researchers surprisingly discovered that cancer and aging bear many similarities. They share mutual origins and are considered two different manifestations of the same potential process—the accumulation of cell damage. In addition, a lot of premature aging disorders result from the increased accumulation of DNA damage, such as Werner syndrome and Bloom syndrome (Burtner and Kennedy, 2010). In 2011, a landmark paper in the cancer field was published and enumerated 10 hallmarks of cancer (Hanahan and Weinberg, 2011); likewise, researchers have enumerated nine candidate hallmarks of aging, which is beneficial to conceptualizing the nature of age and its underlying mechanisms (Lopez-Otin et al., 2013).
The aging field first advanced a new concept in 1956, namely, the free radical theory of aging, with the publication of a significant article that proposed the basic chemical process of aging: the reaction between toxic ROS and cell cellular components (Harman, 1956). From then on, the free radical theory of aging was seriously questioned by many researchers. The identification of SOD in 1969 (McCord and Fridovich, 1969) and the following elucidation of cellular antioxidant defenses (Yu, 1994) gradually gave more credibility to the theory. There has been increasing evidence since then that the mechanism limiting longevity arises from ROS-induced damage (Harman, 1981; Barja, 2004). Subsequently, research in Britton Chance's lab found that oxygen in the mitochondrion is converted to H2O2 (Chance et al., 1979), which prompted Jaime Miquel to extend the free radical theory of aging and formulate in the early 1980s, the mitochondrial free radical theory of aging: oxidative stress damage to mitochondrial DNA (mtDNA) may result in mutation and replication arrest, and mitochondrial function impairment during aging (Miquel et al., 1980; Fleming et al., 1982). It became evident that the proposed theory initiated to link the accumulation of ROS with aging. ROS destroy the structure of cell membrane and protein function and mutate or delete genes through molecular peroxidation, which in turn critically influences the life span. Although substantial evidence that oxidative stress toxicity increases in aging, the theory is still seriously doubted by some scientists (Lapointe and Hekimi, 2010; Gladyshev, 2014). The question of the specific mechanism of how ROS act on aging remains unresolved.
Surprisingly, low ROS levels were likely to extend longevity in C. elegans (Palikaras et al., 2015). Similarly unexpected, elevating the levels of ROS in Drosophila via respiratory complex I could act as a signal to delay aging (Scialò et al., 2016), and finally, mice with overexpression of antioxidants did not present a prolonged lifespan (Pérez et al., 2009). These findings appear to contradict the destructive effects of ROS. In fact, in parallel studies of the detrimental effects of ROS, the area of the intercellular signal has experienced a rapid advance. Convincing evidence has accumulated that low levels of ROS may function as signaling molecules, improve systemic defense mechanisms by inducing an adaptive response (referred to as mitohormesis) to fight against cellular stress and damage with age, and eventually extend lifespan (Ristow and Schmeisser, 2014). Whereas, excessive ROS can defeat their original homeostatic goal and cause increased age-related damage and shortened lifespan (Hekimi et al., 2011; Gonzalez-Freire et al., 2015). Indeed, activating slight mitohormesis in Drosophila muscle significantly extends lifespan through systemically antagonizing insulin signaling, facilitating mitophagy, and regulating the mitochondrial unfolded protein response (Owusu-Ansah et al., 2013).
Under physiological conditions, Ang II-regulated ROS and RNS production and the subsequent promotion of oxidative stress are tightly controlled. On the contrary, under pathological conditions associated with the RAS overactivation, such as hypertension, atherosclerosis, myocardial infarction (Steinberg et al., 1989; Dhalla et al., 1996), diabetes (Rincon-Choles et al., 2002), and aging (Baylis et al., 1997; Thompson et al., 2000; Wang et al., 2003), the dysregulation of Ang II-dependent ROS generation may become a critical contributor to cell oxidation and connective tissue damage. Previous experiments showed that cell senescence was faster after exposure of human glomerular mesangial cells to Ang II (Feng et al., 2011). Recent research work has demonstrated that the pervasiveness of senescent vascular smooth muscle cells increases with age and can be accelerated by multiple Ang II-induced stress responses, including oxidative lesions, inflammation, and mitochondria dysfunction (Okuno et al., 2020).
In 1956, Harman proposed that ROS are the most significant molecular species involved in the aging process (Harman, 1956). Based on his theory, aging and age-associated degenerative diseases are attributed primarily to the deleterious side attacks of ROS. Hence, Ang II likely takes part in the aging process considering its capacity for mediating the generation and release of ROS. The upregulated activities of the RAS have been observed in aging mice kidneys. Here, AT1R was activated that accelerated ROS generation, as well as tubular senescence and renal fibrosis in D-galactose, establishing the aging model. These effects were blocked by treatment with AT1R blocker losartan (Miao et al., 2019). Recent advance in the field of cardiovascular diseases indicates that overactivation of Ang II is seriously harmful to cardiac function, including exacerbated fibrosis, oxidative lesion, and hypertrophy (Zablocki and Sadoshima, 2013; Hamilton et al., 2016). The later data showed that, in contrast to Ang II alone, mice administered with the combination of Ang II and L-NG-nitroarginine methyl ester (inhibition of NO synthesis) exerted a pernicious impact on mitochondria and exacerbated oxidative damage, leading to more deleterious heart failure (Hamilton et al., 2016). This result might present new therapeutic opportunities that modify mitochondrial damage and improve the redox state to prevent the incidence of heart failure. In animal models of PD, excessive brain AT1R stimulation enhanced oxidative stress and injured dopaminergic cells (Dominguez-Meijide et al., 2014), while ARB could enter the brain and decrease excessive brain inflammation and neuronal injury (Villapol and Saavedra, 2015). Clinical studies showed that ARB improved cognitive loss after stroke and aging. Since in brain cells, the intracellular RAS counteracts the oxidative stress induced by the extracellular/paracrine Ang II, ARB acting only on the extracellular or paracrine RAS may give cells better protection (Villar-Cheda et al., 2017). Overall, blocking the RAS using different antagonists, such as valsartan, candesartan, and telmisartan, as a possible strategy to reduce aging, would be advisable and would provide a more translational vision (Basso et al., 2005; Villapol and Saavedra, 2015).
Moreover, it has been shown that increasing ROS levels by exposing cells to exogenous H2O2 or enhancing the partial pressure of oxygen could induce human fibroblasts senescence with irreversible cell cycle arrest (von Zglinicki et al., 1995; Chen et al., 1998). Conversely, antioxidants, such as apocynin, kaempferol, and polyphenol extracts, markedly attenuated Ang II-induced oxidative damage and cellular senescence (Wang et al., 2013; Feresin et al., 2016; Du et al., 2019).
Mitochondria were described in the early 1890s as ubiquitous intracellular structures (Ernster and Schatz, 1981). Over the past century, with technological advances, several research studies elaborated on many comprehensive molecular details of mitochondria, including mitochondrial origin, construction, gene, metabolism, and signaling pathways (Son and Lee, 2021).
Mitochondria were once viewed as critical subcellular organelles dedicated to the generation of energy to keep the endergonic biochemical processes of cell life through the tricarboxylic acid cycle (TCA cycle). Now, they are regarded as remarkably multifunctional double membrane organelles that function as a crucial regulatory station for executing and coordinating essential cellular metabolism, ranging from calcium homeostasis (Nicholls, 2005), immune response (Pinegin et al., 2018), cell apoptosis (Brookes et al., 2000), tissue oxygen gradients (Thomas et al., 2001; Collins et al., 2012) to intracellular signaling (Cadenas, 2004). In the process of oxidative phosphorylation, mitochondria transfer electrons to oxygen through the electron transfer chain (ETC), producing the vast majority of cellular adenosine triphosphate (ATP). Although the mitochondrial ETC is a very efficient system, such an energetic process does have its downsides, and the ETC has been demonstrated to be somewhat “leaky”. During this process called “electron leak”, the “leaky” mitochondrial electron can spontaneously side react with some components of the respiratory chain to generate ROS, mainly from complexes I and III of the ETC (Cadenas and Davies, 2000; St-Pierre et al., 2002; Barja, 2014; Zhang et al., 2018) (Figure 3). For example, while the ubiquinone in the ETC cycle, from the states of quinone to semiquinone and to quinol, electrons tend to be passed directly to the oxygen molecule rather than to the next electron carrier in the respiratory chain. Furthermore, a few iron–sulfur clusters also generate toxic with oxygen during the respiratory chain (Cadenas and Davies, 2000). Apart from these ETC reactions in the mitochondrial inner membrane, another source of ROS is monoamine oxidases in the mitochondrial outer membrane which is not associated with the respiratory chain. The oxidative deamination of biogenic amines catalyzed by monoamine oxidases can directly reduce two-electron of to H2O2 (Hauptmann et al., 1996), which contributes to the modulation of ROS signaling and the maintenance of oxidative homeostasis within the mitochondrial matrix and cytosol. According to a putative vicious cycle, ROS derived from mitochondria can potentially feed back to the mitochondria forming more mtROS (Zorov et al., 2014). Therefore, mitochondria are commonly regarded as the major source of intracellular ROS under physiological conditions.
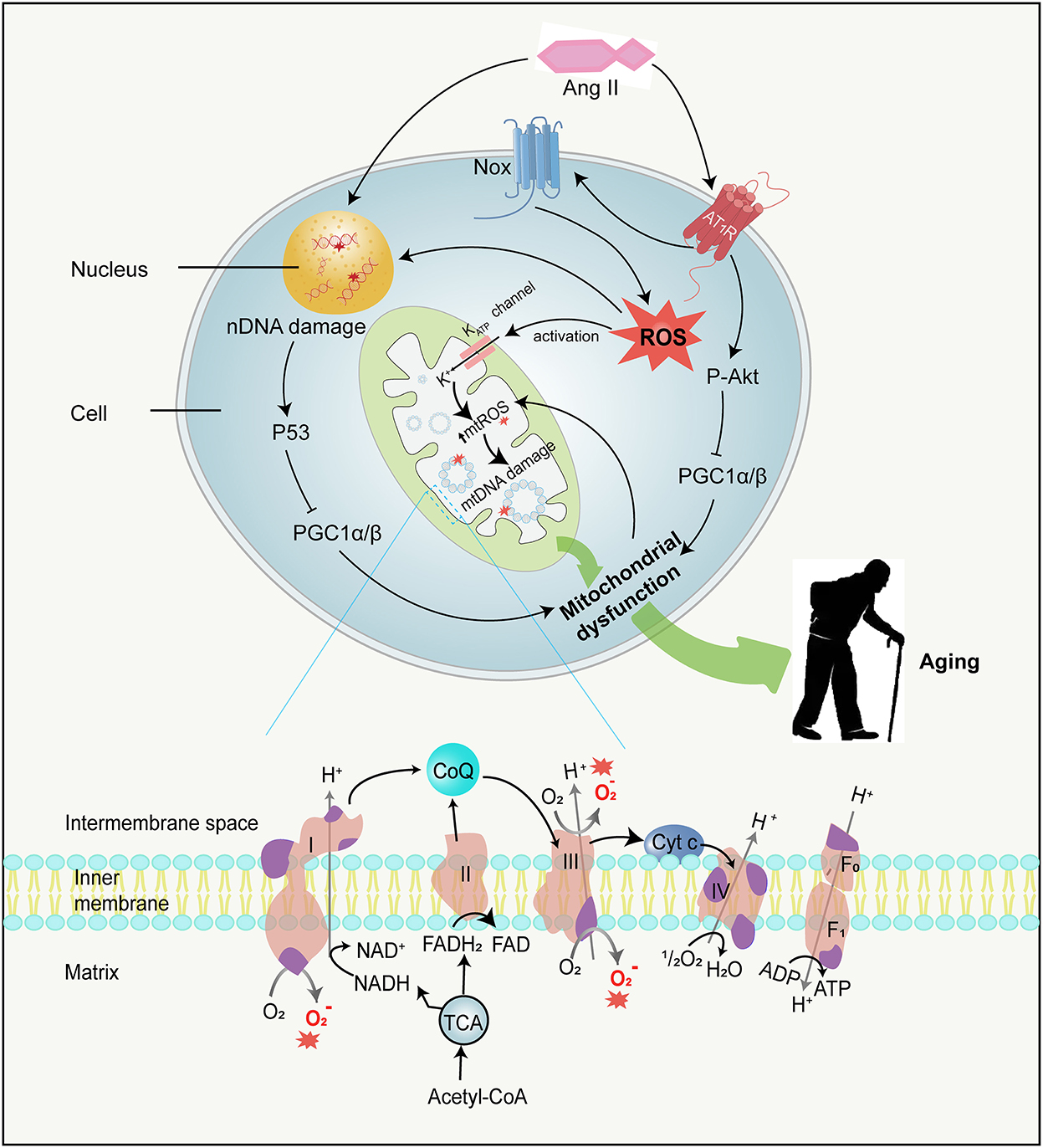
Figure 3. Ang II and mitochondrial dysfunction. Ang II can not only damage mtDNA by stimulating mtROS production in several ways but also damage nDNA directly. MtDNA mutations directly accelerate mitochondrial dysfunction, while nDNA damages result in mitochondrial dysfunction by activating P53 and subsequently inhibiting PGC-1αandPGC−1β, therefore contributing to aging. Besides, Ang II binding to AT1R activates Akt and in turn phosphorylates PGC-1α/β, leading to mitochondrial dysfunction. The mitochondrial ETC is composed of four respiratory complexes (1-IV) and ATP synthase(F0 − 1), which are all made up of proteins encoded by both mitochondrial (purple) and nuclear (pink) genes. MtROS (red asterisk) are primarily generated from complexes I and III in the ETC. mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PGC-1α and PGC-1β, peroxisome proliferator-activated receptor gamma coactivator 1-alpha and -beta; ETC, electron transfer chain; ATP, adenosine triphosphate; Cyt c, cytochrome c; ADP, adenosine diphosphate; NADH, nicotinamide adenine dinucleotide; FAD, flavin adenine dinucleotide; FADH2, flavine adenine dinucleotide reduced; CoQ, Coenzyme Q; TCA, tricarboxylic acid cycle.
Mitochondria consume more than 90% of cellular oxygen to generate energy to meet the needs of cellular activities. While most oxygen is converted into water, approximately 2% forms ROS (Boveris and Chance, 1973). However, mtROS, especially OH·, can react with almost every type of cell macromolecule and destroy their functions, thereby compromising mitochondrial integrity and function. Although proteins and phospholipids are turned over without perpetual damage, unrepaired ROS-induced nucleic acid damage can persistently exist and accrete over time. It is worth noting that both the mtDNA and nuclear DNA (nDNA) can be damaged by mtROS. In a previous study (Van Remmen et al., 2003), the MnSOD activity was reduced by about 50%, and the level of endogenously generated ROS was significantly increased in tissues of mice heterozygous for the SOD2 gene compared with wild-type mice. These heterozygous mice were also notably shown to exhibit elevated levels of 8-oxo-2-deoxyguanosine (8-oxoG) in both mtDNA and nDNA, which is both one of the most abundant mutations brought by oxidative conversion of guanosine and an important marker of oxidative stress. The increased 8-oxoG denoted a rising incidence of mtDNA and nDNA damage as well as a high rate of tumor formation in heterozygous SOD2 mice. Based on this finding, a conjecture that mitochondrial oxidative injury might be a considerable element of entire genomic instability started to attract the attention of researchers and was gradually embraced by them. The hypothesis is also supported by an experiment in irradiated human fibroblasts, in which exogenous ROS production leads to mitochondrial dysfunction, nDNA damage acceleration, and senescent phenotype through initiating signaling and forming a positive feedback loop (Passos et al., 2010). Similarly, mitochondrial oxidants can destroy nDNA (Balaban et al., 2005; Singhapol et al., 2013; Douiev et al., 2018), probably owing to the facts that several mtROS enhance oxidative lesions regarding the mitochondrial, cytosolic, and nuclear compartments (Van Remmen and Richardson, 2001), and that some ROS, e.g., H2O2, can spontaneously cross the mitochondrial membrane resulting in nuclear damage (Van Remmen et al., 2003). Although nDNA possesses repair mechanisms, the cell activates a series of signal transduction pathways if the damage is too large to repair (Figure 3). Damaged nDNA is further capable of promoting the phosphorylation of p53 (Balaban et al., 2005; Sahin and DePinho, 2012), which subsequently couples with the promoters of peroxisome proliferator-activated receptor gamma coactivator 1-alpha and –beta (PGC-1α and PGC-1β) and suppresses their expression (Sahin et al., 2011; Sahin and DePinho, 2012). PGC-1α and PGC-1β coordinate sophisticated metabolic responses including mitochondriogenesis, antioxidant defenses enhancement, and aliphatic acid oxidation improvement (Fernandez-Marcos and Auwerx, 2011; Feresin et al., 2016) and are regarded as the “master regulators” of mitochondria. The repression of both PGC-1α and PGC-1β finally affects entire mitochondria biogenesis, function, as well as homeostasis, thus leading to deficient ATP production and ROS enhancement (Scarpulla, 2011; Sahin and DePinho, 2012). In addition to mitochondrial dysfunction, decreased PGC-1α also reduces telomerase activity and increases DNA damage, resulting in telomere attrition and replicative senescence (Xiong et al., 2015). Unfortunately, the influence of p53 on mitochondriogenesis and subsequently caused cell pathophysiological mechanism is not overall clear. Additional research is needed to elucidate the exact mechanism of DNA damage in mitochondrial ETC dysfunction and the corresponding measures of prevention.
However, mtDNA is commonly considered to be more sensitive to oxygen radical attack, which can be ascribed to multiple factors. mtDNA is in the proximity of the reactive species generation site and exposed to a high concentration of reducing metabolites environment (Cadenas and Davies, 2000; Son and Lee, 2021); mtDNA lacks protective histones and has a relatively limited repair mechanism compared to nDNA (Linnane et al., 1989; Ames et al., 1995). As a corollary, the amount of oxidized bases in mtDNA is 10–20 times higher than that in nDNA(Richter et al., 1988; Adachi et al., 1995).
Importantly, over 900 proteins exist in mitochondria that are encoded by both nuclear and mitochondrial genomes. Although the majority of mitochondrial proteins are encoded by the nuclear genes, the mitochondrial genome is indispensable in the process of mitochondriogenesis, which is responsible for offering the templates for 13 essential proteins of ETC, and mitochondrial tRNAs and rRNAs transcription (Gomez-Cabrera et al., 2012; Lauri et al., 2014). Unlike wild-type mtDNA, mutated mtDNA is easier and faster to replicate, and in turn, contributes to its clonal expansion. If the rate of mutated mtDNA increases to 60%, insufficient respiration occurs in the cell and initiates a vicious spiral of progressively increasing mtDNA mutations (Lauri et al., 2014; Chinnery, 2015). Collectively, the extent of mtDNA base oxidation and deletion is increased with the H2O2 generated by inner (ETC) or outer (monoamine oxidase) membrane activities. Thus, mitochondria are, per se, targets of ROS/RNS-mediated damage, leading to the accumulation of mtDNA mutations, mitochondrial dysfunction, and therefore several diseases, such as cancer, mitochondrial diseases, aging, and age-related diseases (Chinnery, 2015; Yan et al., 2019; Fontana and Gahlon, 2020).
As cells and organisms age, proteasome activity descends, autophagy is inactivated, and mitochondrial function becomes perturbed, which emerges with the diminishing efficacy of the respiratory chain and ATP generation as well as the growing electron leakage and oxidative stress (Green et al., 2011; Theurey and Pizzo, 2018). Besides, it has been reported that in various organisms, oxidative injury to mitochondrial lipids, DNA, and proteins are highly augmented during the aging in some tissues (Van Remmen and Richardson, 2001). Interestingly, the mitochondria-targeted antioxidant mitoquinone can improve mitochondrial mass and diminish renal cell senescence and renal fibrosis in constituted aging models (Miao et al., 2019). Mutator mice overexpress catalase specifically in mitochondria, which decreases ROS-inflicted destruction and substantially ameliorates cardiac aging, evidenced by the important role of maintaining mitochondrial function in longevity (Dai et al., 2009, 2010). Oxidative stress is known to be the main reason for physiologic function decline in the aging process. Since mitochondria are a major site for ROS production, the relationship between mitochondrial function impairment and aging has long been attracting attention and suspicion. Anatomizing their relationship remains a key challenge in carrying out research work on aging.
Later, data greatly supports the importance of mtDNA damage in aging and age-related diseases. The first clear evidence comes from the observation and authentication of multisystem diseases caused by mtDNA mutations that partially phenocopy aging (Wallace, 2005). More convincing evidence that mtDNA point mutations or defects are a driving force behind the aging phenomenon derives from the creation of transgenic mice expressing a proofreading deficient mitochondrial DNA polymerase γ. These mutator mice show a higher rate than wild-type mice in mitochondrial mutation accumulation and an obvious early aging phenotype since the 25th week, such as osteoporosis progress, hair loss, and reduced fertility. Besides, a mosaic pattern related to cytochrome c oxidase (complex IV) deficiency, mitochondrial abnormalities, and enlargement is also observed in these transgenic mice. Finally, their life span averages 48 weeks (Trifunovic et al., 2004; Kujoth et al., 2005; Vermulst et al., 2008). Overall, the premature aging phenotype and short lifespan are associated with the accumulation of mtDNA mutations.
The continuous production of ROS and RNS by mitochondria provides evidence of the mitochondrial aging free radical theory, as an extension of the more general aging free radical theory (Miquel, 1998; Barja, 2014; Ziada et al., 2020). Based on the updated free radical theory of aging, oxidation of mitochondrial components destroys mitochondrial function primarily through mtROS, which in turn amplifies mtROS production and oxidizes macromolecule, resulting in deteriorating cellular and organ function. Afterward, evidence highly in favor of the mitochondrial version of the free radical aging theory is presented in a review (Barja, 2004) that generalizes two known features associating aging with oxidative stress damage by a comparative analysis of animals with different lifespans. Barja discovered that mtROS production was positively correlated with mtDNA destruction while negatively related to maximum longevity. Also, the unsaturated level of tissue fatty acid was linked inversely with maximum lifespan. Despite substantial evidence that mtDNA mutations are enhanced during aging, some recent findings have provided grounds for questioning the correlation between oxidative damage and mtDNA mutations. For instance, an experiment observed no increase in mtDNA transversion mutations caused by oxidative lesions, but an increase in mtDNA transition mutations within aging (Trifunovic et al., 2004; Kennedy et al., 2013). Therefore, it is inferred that rather than the direct function of oxidative stress, ROS may contribute to mtDNA mutations via altering mitochondrial polymerase γ, reducing its fidelity, and indirectly increasing somatic transition mutations (Kauppila et al., 2017; Ziada et al., 2020). ROS may as well affect mitochondrial biogenesis via signaling molecule pathway and, in turn, promote replication of existing mtDNA mutations (Ziada et al., 2020).
Recently, it has become increasingly apparent that the generation of ROS is connected with the reciprocal interaction between the RAS and mitochondria, the latter of which are major ROS sources. Several studies support the direct interactions between Ang II and nuclear and mitochondrial components. First, 125I-labeled Ang II was tested in cellular mitochondria and nuclei of rodents (Robertson and Khairallah, 1971; Sirett et al., 1977). Second, renin, angiotensinogen, and ACE were also found in intramitochondrial dense bodies of rat adrenal glands (Peters et al., 1996). In human and mouse cells, AT1R and AT2R were detected within the inner membrane of mitochondria and interacted with Ang II (Abadir et al., 2011); moreover, AT1R was also detected in cell nuclei (Booz et al., 1992; Eggena et al., 1993). Finally, studies showed that angiotensinogen was internalized by renal proximal tubule cells and trafficked to the nucleus and mitochondria. Studies also showed that the precursor was taken up by isolated mitochondria as well. Besides, Pendergrass et al. (2009) reported that AT1R sites in renal cells were coupled to ROS generation likely through NOX4.
Ang II is discovered to stimulate mtROS production (Doughan et al., 2008; Dai et al., 2011) (Figure 3), which thereby represses the mitochondrial respiratory chain and decreases the energy of ATP. A study has demonstrated that activation of the Ang system by the AT2R in mitochondria is accompanied by mitochondrial NO generation and is involved in the regulation of mitochondrial respiration (Abadir et al., 2011). Additionally, Ang II binding to AT1R could phosphorylate PGC-1α by activating Akt, which decreases the binding of transcription factor forkhead box O 1 (FoxO1) to catalase and Sirt1 promoters, reducing the expression of these target genes. Sirt1 deficiency further increases PGC-1α and FoxO1 acetylation, forming a negative feedback loop where mitochondrial dysfunction and cell senescence are accelerated (Xiong et al., 2010; Feresin et al., 2016; Salazar, 2018). Concomitantly, Ang II could also modulate the PKC(protein kinase C)/AMPK (adenosine monophosphate-activated protein kinase)/ULK1(unc-51 like autophagy activating kinase 1) axis and inhibit the autophagic flux to exacerbate lipid deposition and mitochondrial dysfunction (He et al., 2019).
It is necessary to elucidate the mechanism of how Ang II promotes the generation of mtROS. Some research has proved that ROS produced through Nox could facilitate the further generation of ROS via other channels. For instance, originated from Nox might uncouple NO synthase to form by oxidizing and degrading tetrahydyrobiopterin (BH4) (Laursen et al., 2001; Landmesser et al., 2003). Likewise, H2O2 can promote further ROS production via stimulating dehydrogenase to form xanthine oxidase (McNally et al., 2005). So, Ang II-mediated mtROS formation may also be triggered by Nox-originated ROS. The review of Zhang et al. (2007) supports this hypothesis and clarifies this mechanism in detail. Ang II activates Nox to increase cytosolic ROS levels, which in turn induces the opening of mitochondrial ATP-dependent potassium channels (mitoKATP) in the inner mitochondrial membrane, depolarizes mitochondrial membrane potential, and results in a burst of mtROS. Subsequently, mtROS are required for activation of downstream signaling cascades and autophagy (Yu et al., 2014), thus promoting cell apoptosis and senescence. Another piece of evidence that relates Ang II to mtROS presented by the experiment indicates that mitochondrial p66Shc deletion could protect from Ang II–inflicted myocardial hypertrophy (Graiani et al., 2005). Partly situated in the intermembrane space of mitochondria, p66Shc was thought to subtract electrons from cytochrome c and transfer them to oxygen to produce mtROS (de Cavanagh et al., 2015). Another fact worth mentioning is that the mtROS production stimulated by Ang II is related to vascular cell adhesion molecule-1. Ang II prompts the release of vascular cell adhesion molecule-1, which, like Ang II, can activate Nox and the consequent mtROS generation (Pérez et al., 2003; De Giusti et al., 2008). Ang II-mediated endothelial dysfunction has been reported in a variety of cardiovascular diseases (Dimmeler et al., 1997). Ang II, through attracting excessive expression of ROS in both cytoplasm and mitochondria, is involved in DNA damage, mitochondrial dysfunction, and inflammation, ultimately causing endothelial apoptosis (Caporali and Emanueli, 2011; Mendell and Olson, 2012; Zhang Y. et al., 2016; Li et al., 2019; Miyao et al., 2020). Later, it was shown that temporary Ang II stimulation promoted cell viability, including the ability of cell proliferation, migration, and angiogenesis (Li et al., 2019). While Ang II has long been considered a key regulator of endothelial apoptosis, the underlying mechanisms remain to be explored.
A study (Abadir et al., 2011) showed that mitochondrial AT1R increased and AT2R declined in mice kidneys along with age, and these consequences were combated by losartan. This may offer a novel way to decrease mitochondrial damage and chronic disease burden with aging. Moreover, apart from RAS activity enhancement, the quality and function of mitochondria were also destroyed with time in aging mice (Miao et al., 2019). This study has demonstrated that RAS signaling plays a primary role in the relationship between RAS-mediated age-associated renal fibrosis and mitochondrial dysfunction, and that inhibition of the signaling can efficiently attenuate renal fibrosis. Irbesartan, a typical ARB, considerably reversed mitochondrial dysfunction induced by Ang II by improving ATP production and mitochondrial potential and decreasing the amounts of ROS(He et al., 2019). In the future, thus, inhibition of RAS activity may be a feasible and practical therapy to ameliorate the progress of mitochondrial damage.
To sum up, an increasing number of studies show that mtROS are involved in the deleterious effects of Ang II, which are probably mediated by the activation of AT1R or direct interaction between Ang II and mitochondrial or nuclear components. Moreover, the existence of the RAS in human mitochondria contributes to understanding the interaction of mitochondria with aging and exposes underlying therapeutic targets to attenuate mitochondrial dysfunction and chronic disease during aging.
The intimate relationship between telomere attrition and cell senescence or aging has long been widely accepted (Allsopp et al., 1992; Kruk et al., 1995). With a growing understanding of the diverse molecular mechanisms of aging, the telomere is generally considered an instigator and amplifier of molecular loopbacks which pushes the aging process and degenerative diseases (Harley et al., 1990; Hayflick, 2000; Chakravarti et al., 2021) discovered a phenomenon, known as the Hayflick limit, that human fibroblasts stopped dividing after 40 to 60 times. Furthermore, telomere shortening occurred in parallel with the replicative lifespan of human fibroblasts, confirming telomere attrition could drive replicative senescence.
Telomeres are tandem repetitive 5′-TTAGGG-3′ nucleotide sequences at chromosome ends that contain proximal double-stranded and distal single-stranded regions. Telomeres combination with protegerin complex preserves the terminals from being regarded as DNA damage and therefore maintains chromosomal integrity and genomic stability (Figure 4A) (Chan and Blackburn, 2004). The Telomeric G-tail of single-stranded nucleotide is tucked into the duplex DNA to form a lariat-like structure named T loop. Then, the T loop forms a triple DNA strand secondary structure called a d-loop (Griffith et al., 1999; Doksani et al., 2013; Smith et al., 2020). Although telomeres are constitutionally unstable, vulnerable parts, they become stabilized by interacting with a distinctive so-called shelterin. Shelterin, a multiprotein complex, can disguise chromosome ends from DNA damage response (DDR) signals, prevent DNA repair systems from fusing ends by recombination or classical/alternative non-homologous end joining, and regulate telomerase access and activity at the end (Sfeir et al., 2009; Sfeir and de Lange, 2012). It comprises three protein subunits, namely telomeric-repeat-binding factor 1 and 2 (TRF1 and TRF2) and protection of telomeres 1 (POT1) that directly bind to DNA and three other subunits, namely TRF1-interacting nuclear protein 2 (TIN2), TIN2-interacting protein (TPP1), and repressor and activator protein 1 (RAP1), which regulate the interaction between the components (Figure 4B) (Martínez and Blasco, 2011; Shay and Wright, 2019). While TRF1 and TRF2 as homodimers both recognize the double-stranded DNA and effciently promote telomere replication, POT1 binds with high specificity and affinity to the single-stranded 3′ overhang (Schmutz and de Lange, 2016; Erdel et al., 2017). TIN2 bridges TRF1 and TRF2 by simultaneously interacting with the two proteins via independent domains and recruits the TPP1–POT1 complex (Hu et al., 2017; Kalathiya et al., 2018). RAP1 binds directly to TRF2 in order to localize at the telomere (Martinez et al., 2010). Thus, the higher-order structure of telomeres solves the end protection problem by covering telomeric DNA. Correspondingly, mutations or losses in the aforementioned constituents can impair the telomere–shelterin complex and the binding of telomerase-telomeric ends, resulting in telomere attrition and premature senescence (van Steensel et al., 1998).
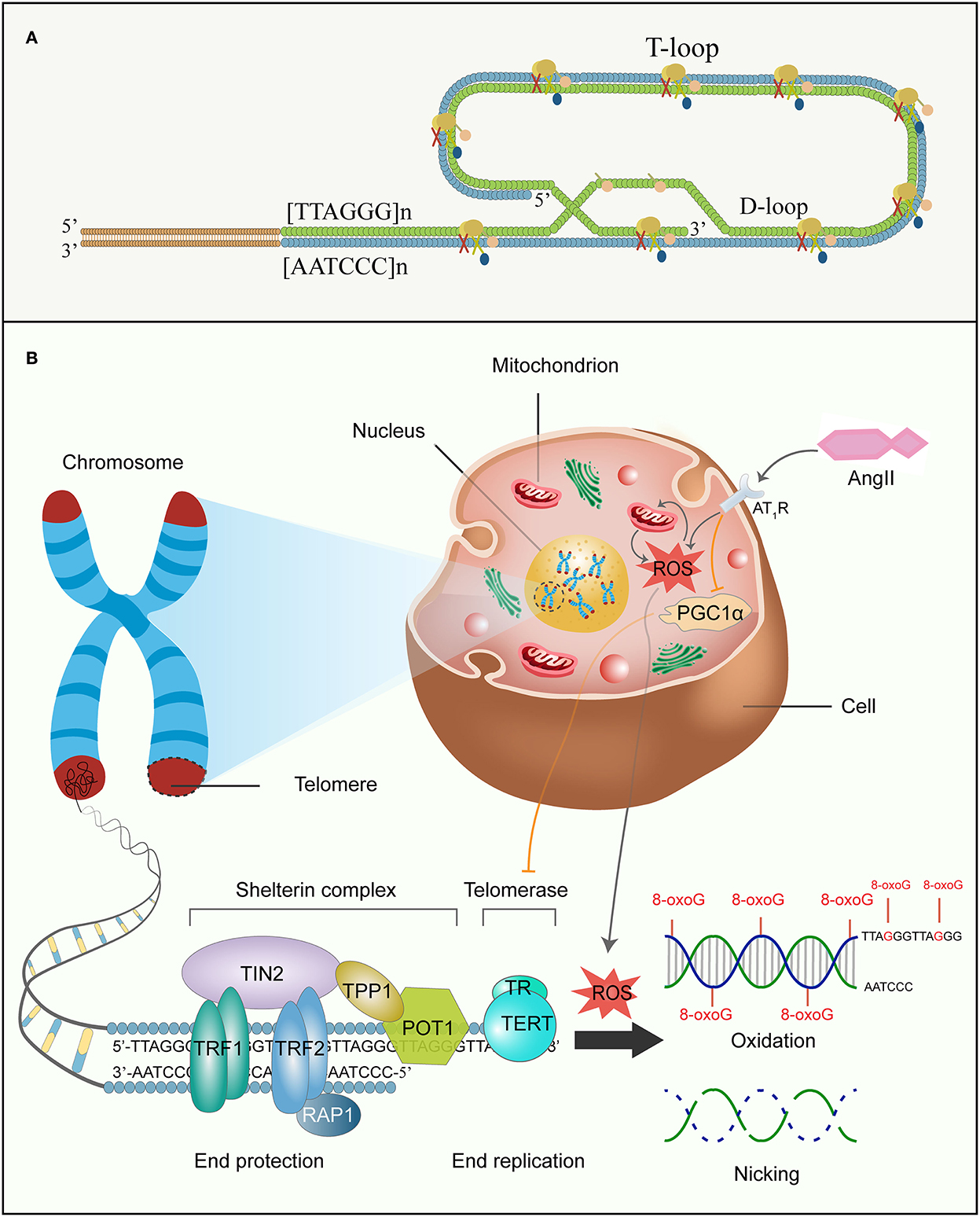
Figure 4. Ang II and telomere attrition. (A) Shelterin complex, structure of the D-loop and T-loop at the end of telomere. (B) Ang II, telomeres, and senescence. Telomeres are at the ends of linear chromosomes and combine with shelterin multiprotein complexes to escape from DDR. Shelterin complex comprises six diverse protein subunits: TRF1, TRF2, POT1, TIN2, TPP1, and RAP1, and are involved in chromosome end protection, while telomerase is made up of TERT and TR to function as end replication. However, the G-rich chain of telomeric DNA is sensitive to ROS induced by Ang II, and further accumulates 8-oxoG damages and forges high frequency of single-strand breakage in the DNA backbone. Concomitantly, Ang II also reduces telomerase activity by decreasing PGC-1α function. All these eventually accelerate replicative senescence. DDR, DNA damage response; TRF1 and TRF2, telomeric-repeat-binding factor 1 and 2; POT1, protection of telomeres 1; TIN2, TRF1-interacting nuclear protein 2; TPP1, TIN2-interacting protein; RAP1, repressor and activator protein 1; TERT, telomerase reverse transcriptase; TR, telomerase RNA; 8-oxoG, 8-oxo-2-deoxyguanosine.
Incomplete replication at chromosomal ends gives rise to gradual telomere attrition during every consecutive cell mitosis (O'Sullivan and Karlseder, 2010). On account of the unidirectional activity of replicative DNA polymerase, it lacks the ability to fully replicate the ends of linear DNA molecules. Consequently, each division would result in a progressive reduction in the chromosomal telomere length because of removing termini RNA primer for lagging-strand synthesis; finally, this shortens the coding sites of the DNA. To avoid this, when the length of somatic cell telomere is reduced to a critical level or is damaged, the region is considered to have double-stranded DNA breaks and activate P53 and P21, causing cell growth arrest and ultimately triggering replicative senescence (Olovnikov, 1973; d'Adda di Fagagna et al., 2003; Oganesian and Karlseder, 2009; Aunan et al., 2016). To a certain degree, some cells, including germ cells (Lee et al., 1998), cancer cells (Singhapol et al., 2013), hematopoietic cells (Colla et al., 2015), and embryonic stem cells of the skin(Lee et al., 1998) and gastrointestinal (Schepers et al., 2011), mitigate shortening through expressing holoenzyme telomerase, which repetitively adds successive 5′-TTAGGG-3′ to maintain telomere homeostasis and overcome the end replication problem (Barnes et al., 2019). Unfortunately, most human somatic cells are short of telomerase, and the critically shortened telomere is incapable of adequate shelterin binding parts, which leads to the chromosomal terminal being exposed to DDR and then the telomere being falsely considered as DNA breaks. The core telomerase is made up of a catalytic protein subunit termed telomerase reverse transcriptase (TERT), as well as a non-coding RNA subunit, termed telomerase RNA (TR) that involves the RNA template for telomere synthesis (Blackburn et al., 2006; Singhapol et al., 2013). Bodnar et al. have found that ectopic expression of TERT in normal human cells renders the cells with the ability to replicate immortally rather than induce malignant conversion (Bodnar et al., 1998). Importantly, numerous data have discovered a positive association of telomere length with age-linked diseases (Blasco, 2005) such as atherosclerosis (Hunt et al., 2015), atrial fibrillation (Carlquist et al., 2016) and osteoarthritis (Kuszel et al., 2015). Moreover, studies on long-lived families (Honig et al., 2015) as well as Ashkenazi centenarians and their offspring (Atzmon et al., 2010) also identified such association and uncovered that telomere length shortened with age and that those with longer telomeres had better overall health.
Further evidence in favor of the importance of telomeres in the human lifespan stems from the study of telomere maintenance-related gene mutations or losses in humans. For example, patients with dyskeratosis congenita carried gene TERT and TERC mutations, which resulted in shortened telomeres and reduced lifespan (Kirwan and Dokal, 2009). Other evidence comes from observation of patients with premature aging disorders, including Werner syndrome and ataxia telangiectasia (Martin, 2005). Interestingly, mutations in TERC and TERT alike had relevance to organ-restricted conditions, such as liver cirrhosis, idiopathic pulmonary fibrosis, and bone marrow failure syndromes (Armanios and Blackburn, 2012). Concomitantly, studies reported aplastic anemia and dyskeratosis congenita possessed mutations in the shelterin protein complex, particularly of TINF2 (Savage et al., 2008; Walne et al., 2008). Those diseases are featured decreased tissue regeneration and expedited aging (Armanios and Blackburn, 2012). Finally, an experiment proved that reactivation of the telomerase gene could reverse premature aging in telomerase-lack mice (Jaskelioff et al., 2011).
Aside from functioning as a maintenance telomere, however, telomerase, TERT, in particular, serves many non-telomere-linked functions including regulation of growth factors, gene expression, and cell multiplication (Smith et al., 2003; Li et al., 2005). Surprisingly, it has been reported that TERT is exported from the nucleus to the cytosol and then gets re-translocated to mitochondria where it protects mitochondrial function and increases resistance against oxidative stress (Ahmed et al., 2008; Haendeler et al., 2009). Similarly, a study has demonstrated that the TERT transferred to mitochondria specifically protects cells from nDNA damage and apoptosis by reducing mtROS levels (Singhapol et al., 2013). Nevertheless, what mechanism is behind the pro-survival function remains elusory and whether the mechanism is linked with telomere homeostasis or a non-telomeric function of TERT is not clear.
As is known, the maintenance of telomere homeostasis is a complex mechanism, and telomere shortening, as a biological hallmark of aging, occurs as a natural part of the aging process. However, at present, a robust body of research suggests that telomere length is subject to factors such as oxidative stress, inflammation, behavioral factors, and exposure to carcinogens or radiation (Starkweather et al., 2014; Patel et al., 2017; Barragán et al., 2021). Although plenty of environmental and genic factors are involved in expedited telomere attrition, oxidative stress is presumed to be the most important underlying mechanism (Figure 4B) (Barnes et al., 2019).
In recent years, increasing studies have reportedly shown that oxidative lesion and chronic inflammation either directly or indirectly promote telomere shortening in analyses of human different tissues, mice models, and cell cultures, particularly of vitro cultured cells (Reichert and Stier, 2017; Ahmed and Lingner, 2018; Barnes et al., 2019). Human correlative studies in a review summarized that six of eight population studies reported various oxidative stress markers remarkably related to shorter average telomere length in white blood cells (Reichert and Stier, 2017). Moreover, the majority of degenerative and inflammatory diseases with telomere attrition as mentioned above were parallels with elevated ROS (Zhang J. et al., 2016). Evidence from deficient nfkb1 subunit of the inflammatory gene nuclear factor κB (NF-κB) in mice models demonstrated that chronic inflammation-induced ROS accelerated aging through telomere dysfunction and cell senescence (Jurk et al., 2014). Finally, the mechanism of telomere attrition has been fully researched in vitro, clarifying that oxidative stress is a major factor in accelerating telomere loss in a dose-dependent manner (von Zglinicki, 2002; Richter and von Zglinicki, 2007). The later review indicated that oxidative stress or exposure to ROS-producing agents exasperated telomere loss in cultured normal human cells, whereas free-radical scavengers and antioxidants decelerated it and improved replicative lifespan (von Zglinicki, 2002).
There are several mechanisms to exposit how a high level of ROS accelerates telomere dysfunction, among which telomeric DNA damage is the most pervasive and conspicuous theory (Barnes et al., 2019). The following reasons explain why telomere is specifically susceptible to oxidative stress. First, on the account of high guanine content, telomeric DNA is particularly sensitive to oxidative damage and normally forms 8-oxoG, which participates in the acceleration of telomere shortening (von Zglinicki, 2002; Kawanishi and Oikawa, 2004). Oxidative stress can not only damage double stranded DNA but also lead to single-strand DNA breaks which result in incomplete replication of telomere in the next cellular division, consequently, causing more telomere loss (von Zglinicki et al., 2000). Second, due to the protective role of shelterin in telomeres, impaired telomeric DNA by ROS may not effectively activate DDR and make DNA repair proteins into telomeres. Thus, ROS-mediated telomeric regions may have low repair efficiency. Finally, the 3′ overhang of telomere and d-loop are single-stranded whose bases are not merely unprotected by hydrogen bonds unique to double-stranded, but cannot be repaired by base excision repair in the absence of the other complementary chain that offers a template for the impaired DNA re-synthesis (Ahmed and Lingner, 2018). Similarly, the G-quadruplex region, folded from telomeric G-tail, is a highly stable four-stranded helical structure and cannot be repaired via base excision repair as the 8-oxoG cannot be excised in quadruplex DNA (Zhou et al., 2013). While Neil3 and NEIL1 DNA glycosylases can remove 8-oxoG from quadruplex DNA, how to repair emerging abasic sites without the complementary stranded part is unclear at present (Zhou et al., 2013, 2015). When the ROS-induced 8-oxoG acts as a substrate for telomere extension, incorporated 8-oxoG influences telomerase activity and terminates 5'-TTAGGG-3' repeat sequences addition, other than perturbing the replication of telomeric DNA (Fouquerel et al., 2016). Strikingly, oxidative damage to telomeric DNA may likewise elongate telomere length by telomerase in certain conditions, nevertheless, present data from cell and animal models accommodate apparent evidence for the predominance of negative effects of ROS on telomere length in vivo (Ahmed and Lingner, 2018).
However, given that most of the evidence for the effects of oxidative damage on telomere function comes from cultured cells, oxidative damage has been questioned as a key factor in telomere shortening in vivo. A review summarized the limited relative experimental research in this regard and then inferred a conclusion that most experimental results partially or completely supported this view (Reichert and Stier, 2017). On top of that, due to the complexity of ROS that it can destroy multiple cellular components and change cell signaling and gene expression, whether the variations of telomeric length and completeness in oxidative lesion should be attributed to indirect factors or direct damage to the telomeres remains elusive (Barnes et al., 2019; Fouquerel et al., 2019). In the future, more experimental studies on different species are needed to find out the specific mechanism of the oxidative lesion in interfering with telomere length.
A previous study(Herbert et al., 2008) in vitro showed that the generation of ROS caused by Ang II binding to AT1R gave rise to DNA damage, and accordingly induced aging of human vascular smooth muscle cells. Ang II-induced ROS production contributes to cell senescence through two different pathways of DNA damage: stress-induced premature senescence independent of the telomere and accelerated replicative senescence dependent on the telomere. Interestingly, the type of vascular cell senescence appears to depend on the duration of Ang II stimulation (Min et al., 2009). Specifically, cells treated with Ang II for 24 h led to acute stress-induced premature senescence, while cells continually exposed to Ang II over a period of 30 days would accelerate telomere loss and further promote premature replicative senescence. Both senescence pathways were in a dose-dependent manner and ameliorated by catalase or losartan. Also, they shared mutual signaling pathways, i.e., increased p53-p21 axis expression and succeeding hypo-phosphorylation of Rb protein which regulates cell cycle arrest (Toussaint et al., 2000; Herbert et al., 2008). Feng et al. (2011) have demonstrated that Ang II-mediated oxidative stress accelerates senescence by telomere attrition. In this study, treatment of human glomerular mesangial cells with Ang II for 72 h exhibited marked telomere shortening, leading to cell growth arrest, probably via P53 and P21 overactivation. Equally, losartan prominently attenuated telomere attrition and cell senescence.
Similarly, two studies (Wilson et al., 2008; Atturu et al., 2010) have found reduced leukocyte telomere length in patients with abdominal aortic aneurysm, which is an age-related vascular disease and can be induced by Ang II via medial accretion of macrophages in aortic elastin degradation (Saraff et al., 2003). This case-control study (Atturu et al., 2010) collected data from 190 patients with abdominal aortic aneurysm and 183 controls and found that the average white cell telomere length in abdominal aortic aneurysm patients was significantly shorter compared to controls. Furthermore, in the murine model, TERT deficiency in bone marrow-derived macrophages alleviated Ang II-induced abdominal aortic aneurysm formation (Findeisen et al., 2011).
From molecular signaling pathways, Ang II-induced mitochondrial impairment and telomere attrition are closely related and interact with each other (Figure 5) (Sahin and Depinho, 2010; Chakravarti et al., 2021). Specifically, telomere attrition activates p53, inhibiting PGC1α/β expression, which triggers mitochondrial impairment and diminishes the expression of genes controlling oxidative defense. This signaling pathway of telomere/p53/PGC1α/β/mitochondria leads to escalated levels of mtROS generation and further aggravates guanosine base damage at telomeres. Besides, this increased mtROS activates the NF-κB, raising the expression of Nox, which lowers telomerase activity and expression and drives telomere shortening as well as replicative senescence (Sahin et al., 2011; Salazar et al., 2017; Salazar, 2018).
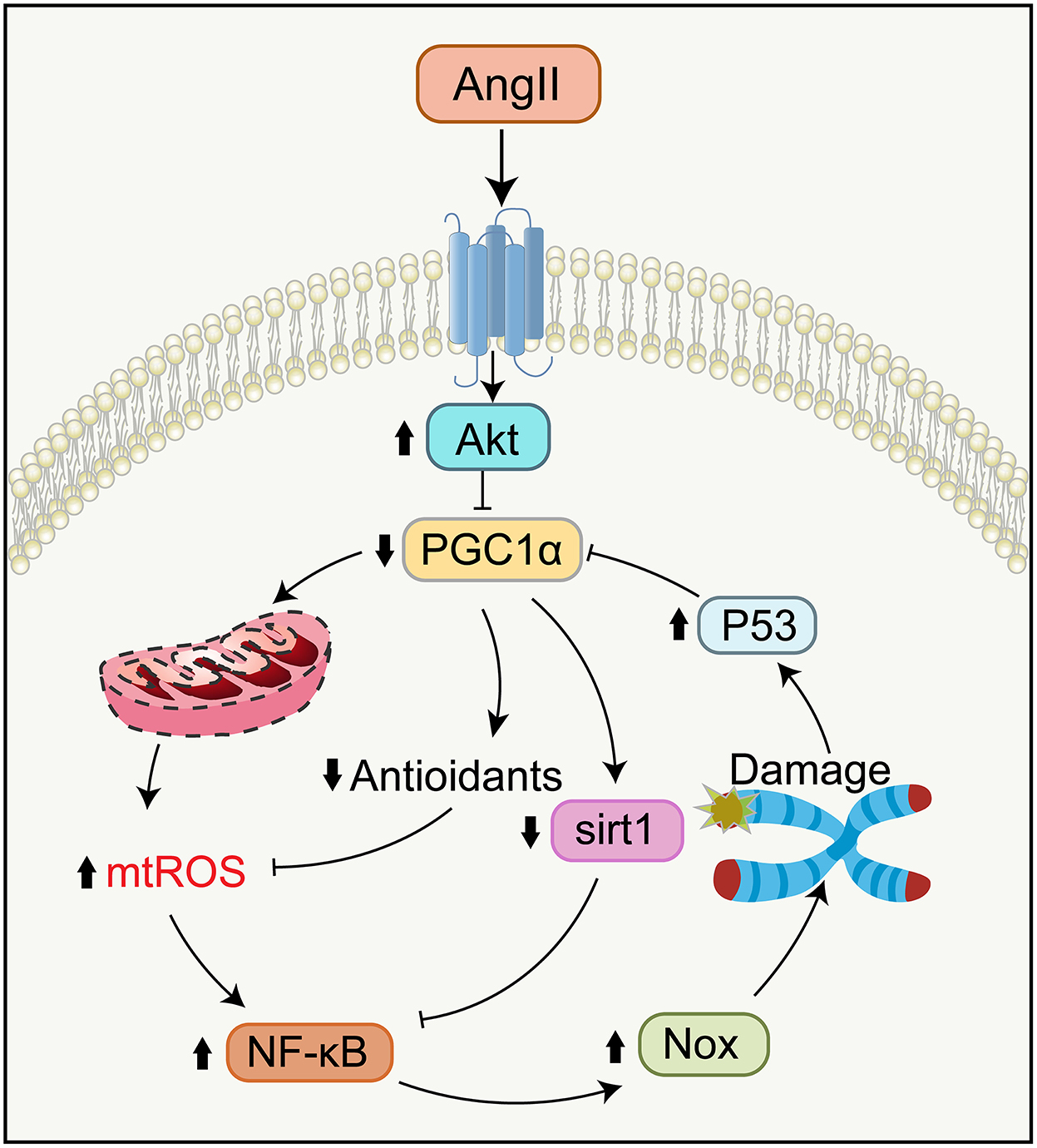
Figure 5. Signaling pathways in telomere attrition and mitochondrial dysfunction during aging. Ang II binding to AT1R reduces the function of PGC-1α via activating Akt, causing the compromises of Sirt1 activity and oxidative defense. Telomere attrition mediates mitochondrial impairment by the p53-dependent downregulation of PGC-1α. Impaired mitochondria generate more mtROS which leads to further telomere attrition and replicative senescence by NF-κB-dependent upregulation of Nox. NF-κB, nuclear factor κB.
Overall, Ang II-induced telomere attrition plays an important role in the aging of human cells. Despite many unanswered questions, these findings propose novel mechanisms to directly implicate Ang II in cell senescence and age-associated disorders.
The renin–angiotensin system is involved in a variety of mechanisms regarding tissue and cell senescence and lifespan. Ang II is a critical factor in such senescence, augmenting oxidative damage and inflammation through the AT1R. Accumulating evidence indicates that blocking the RAS or applying antioxidants can efficiently restrain organ dysfunction and aging caused by cellular senescence; accordingly, RAS inhibitors and antioxidants hold promise as an anti-aging drug. As detailed in this review, we describe the specific generation and constitution of Ang II-mediated ROS and their biological activities, followed by characterizing the theory of free radicals in aging that posits the gradual accretion of damage inflicted by ROS as an essential driver for aging. Some mechanisms have also been presented to interpret how raised ROS damage mitochondria, change telomeric homeostasis, and therefore lead to aging.
Indeed, these hallmarks during aging are intimately interconnected and develop a new molecular circuit of aging containing the interaction between telomere, p53, and mitochondria (Sahin and Depinho, 2010; Sahin and DePinho, 2012). Research work involving telomerase-deficient mice has uncovered that telomere dysfunction induces functional deterioration of mitochondria. Mitochondrial dysfunction is mediated by repressing PGC-1α/β expression through telomere attrition-induced activation of p53. Remarkably, damaged mitochondria can produce much more ROS and thereby forge a vicious cycle where telomeres may suffer further insult from oxidative stress. Furthermore, TERT can shuttle from the nucleus to mitochondria to protect the nucleus from oxidative damage-inflicted DNA damage and this protective activity is mediated by low levels of ROS. Importantly, mitochondrial dysfunction and fragmentation caused by Ang II via increasing PGC-1α acetylation also induce telomere attrition. Mechanistically, dysfunctional mitochondria generate mtROS, activating NF-κB dependent upregulation of Nox that reduces the activity of telomerase and causes telomere attrition and replicative senescence. Nevertheless, understanding their precise causal network is a huge and exciting challenge in future work as the hallmarks co-occur and interconnect with each other during aging.
ZC and WY designed and outlined the structure and contents of the review. WY and PT created the figures. All authors participated in the writing, revision of the manuscript, and approved the final manuscript.
This work was supported by the Construction Project of Capacity Improvement Plan for Chongqing Municipal Health Commission Affiliated Unit (2019NLTS001)-ZS03174, Operating Grant to Chongqing Key Laboratory of Neurodegenerative Diseases (1000013), and Plan for High-level Talent Introduction (2000055).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abadir, P. M., Foster, D. B., Crow, M., Cooke, C. A., Rucker, J. J., Jain, A., et al. (2011). Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. USA. 108, 14849–14854. doi: 10.1073/pnas.1101507108
Adachi, S., Zeisig, M., and Möller, L. (1995). Improvements in the analytical method for 8-hydroxydeoxyguanosine in nuclear DNA. Carcinogenesis. 16, 253–258. doi: 10.1093/carcin/16.2.253
Ahmed, S., Passos, J. F., Birket, M. J., Beckmann, T., Brings, S., Peters, H., et al. (2008). Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell. Sci. 121, 1046–1053. doi: 10.1242/jcs.019372
Ahmed, W., and Lingner, J. (2018). Impact of oxidative stress on telomere biology. Differentiation. 99, 21–27. doi: 10.1016/j.diff.2017.12.002
Allsopp, R. C., Vaziri, H., Patterson, C., Goldstein, S., Younglai, E. V., Futcher, A. B., et al. (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA. 89, 10114–10118. doi: 10.1073/pnas.89.21.10114
Alsafar, H., Hassoun, A., Almazrouei, S., Kamal, W., Almaini, M., Odama, U., et al. (2015). Association of angiotensin converting enzyme insertion-deletion polymorphism with hypertension in emiratis with type 2 diabetes mellitus and its interaction with obesity status. Dis. Markers. 2015, 536041. doi: 10.1155/2015/536041
Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1995). Mitochondrial decay in aging. Biochim. Biophys. Acta. 1271, 165–170. doi: 10.1016/0925-4439(95)00024-x
Armanios, M., and Blackburn, E. H. (2012). The telomere syndromes. Nat. Rev. Genet. 13, 693–704. doi: 10.1038/nrg3246
Atturu, G., Brouilette, S., Samani, N. J., London, N. J., Sayers, R. D., and Bown, M. J. (2010). Short leukocyte telomere length is associated with abdominal aortic aneurysm (AAA). Eur. J. Vasc. Endovasc. Surg. 39, 559–564. doi: 10.1016/j.ejvs.2010.01.013
Atzmon, G., Cho, M., Cawthon, R. M., Budagov, T., Katz, M., Yang, X., et al. (2010). Evolution in health and medicine Sackler colloquium: Genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proc. Natl. Acad. Sci. U S A. 107, 1710–1717. doi: 10.1073/pnas.0906191106
Aunan, J. R., Watson, M. M., Hagland, H. R., and Soreide, K. (2016). Molecular and biological hallmarks of ageing. Br. J. Surg. 103, e29–46. doi: 10.1002/bjs.10053
Bacani, C., and Frishman, W. J. C. (2006). Chymase: a new pharmacologic target in cardiovascular disease. Cardiol. Rev. 14, 187–193. doi: 10.1097/01.crd.0000195220.62533.c5
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell. 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Barja, G. (2004). Free radicals and aging. Trends Neurosci. 27, 595–600. doi: 10.1016/j.tins.2004.07.005
Barja, G. (2014). The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 127, 1–27. doi: 10.1016/B978-0-12-394625-6.00001-5
Barnes, R. P., Fouquerel, E., and Opresko, P. L. (2019). The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 177, 37–45. doi: 10.1016/j.mad.2018.03.013
Barragán, R., Ortega-Azorín, C., Sorl,í, J. V., Asensio, E. M., Coltell, O., St-Onge, M. P., et al. (2021). Effect of physical activity, smoking, and sleep on telomere length: a systematic review of observational and intervention studies. J Clin Med 11(1). doi: 10.3390/jcm11010076
Basso, N., Paglia, N., Stella, I., de Cavanagh, E. M., Ferder, L., del Rosario Lores Arnaiz, M., et al. (2005). Protective effect of the inhibition of the renin-angiotensin system on aging. Regul. Pept. 128, 247–252. doi: 10.1016/j.regpep.2004.12.027
Baylis, C., Engels, K., Hymel, A., and Navar, L. G. (1997). Plasma renin activity and metabolic clearance rate of angiotensin II in the unstressed aging rat. Mech. Ageing Dev. 97, 163–172. doi: 10.1016/s0047-6374(97)00053-5
Benigni, A., Cassis, P., and Remuzzi, G. (2010). Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med. 2, 247–257. doi: 10.1002/emmm.201000080
Benigni, A., Corna, D., Zoja, C., Sonzogni, A., Latini, R., Salio, M., et al. (2009). Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530. doi: 10.1172/JCI36703
Blackburn, E. H., Greider, C. W., and Szostak, J. W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12, 1133–1138. doi: 10.1038/nm1006-1133
Blasco, M. A. (2005). Telomeres and human disease: ageing, cancer and beyond. Nat. Rev. Genet. 6, 611–622. doi: 10.1038/nrg1656
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., et al. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science. 279, 349–352. doi: 10.1126/science.279.5349.349
Boehm, M., and Nabel, E. (2002). Angiotensin-converting enzyme 2–a new cardiac regulator. N. Engl. J. Med. 347, 1795–1797. doi: 10.1056/NEJMcibr022472
Booz, G. W., Conrad, K. M., Hess, A. L., Singer, H. A., and Baker, K. M. (1992). Angiotensin-II-binding sites on hepatocyte nuclei. Endocrinology. 130, 3641–3649. doi: 10.1210/endo.130.6.1597161
Boveris, A., and Chance, B. (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134, 707–716. doi: 10.1042/bj1340707
Brand, E., Chatelain, N., Paillard, F., Tiret, L., Visvikis, S., Lathrop, M., et al. (2002). Detection of putative functional angiotensinogen (AGT) gene variants controlling plasma AGT levels by combined segregation-linkage analysis. Eur. J. Hum. Genet. 10, 715–723. doi: 10.1038/sj.ejhg.5200874
Brody, J. A., and Grant, M. D. (2001). Age-associated diseases and conditions: implications for decreasing late life morbidity. Aging (Milano). 13, 64–67. doi: 10.1007/bf03351527
Brookes, P. S., Salinas, E. P., Darley-Usmar, K., Eiserich, J. P., Freeman, B. A., Darley-Usmar, V. M., et al. (2000). Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J. Biol. Chem. 275, 20474–20479. doi: 10.1074/jbc.M001077200
Burson, J. M., Aguilera, G., Gross, K. W., and Sigmund, C. D. (1994). Differential expression of angiotensin receptor 1A and 1B in mouse. Am. J. Physiol. 267, E260–267. doi: 10.1152/ajpendo.1994.267.2.E260
Burtner, C. R., and Kennedy, B. K. (2010). Progeria syndromes and ageing: what is the connection? Nat. Rev. Mol. Cell Biol. 11, 567–578. doi: 10.1038/nrm2944
Cadenas, E. (2004). Mitochondrial free radical production and cell signaling. Mol. Aspects Med. 25, 17–26. doi: 10.1016/j.mam.2004.02.005
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/s0891-5849(00)00317-8
Caporali, A., and Emanueli, C. J. (2011). MicroRNA regulation in angiogenesis. Vascul. Pharmacol. 55, 79–86. doi: 10.1016/j.vph.2011.06.006
Carey, R. M. (2013). Newly discovered components and actions of the renin-angiotensin system. Hypertension. 62, 818–822. doi: 10.1161/hypertensionaha.113.01111
Carlquist, J. F., Knight, S., Cawthon, R. M., Le, V. T., Jared Bunch, T., Horne, B. D., et al. (2016). Shortened telomere length is associated with paroxysmal atrial fibrillation among cardiovascular patients enrolled in the Intermountain Heart Collaborative Study. Heart Rhythm. 13, 21–27. doi: 10.1016/j.hrthm.2015.07.032
Chakravarti, D., LaBella, K. A., and DePinho, R. A. (2021). Telomeres: history, health, and hallmarks of aging. Cell. 184, 306–322. doi: 10.1016/j.cell.2020.12.028
Chamsi-Pasha, M., Shao, Z., and Tang, W. J. (2014). Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr. Heart Fail. Rep. 11, 58–63. doi: 10.1007/s11897-013-0178-0
Chan, S. R., and Blackburn, E. H. (2004). Telomeres and telomerase. Philos. Trans. R. Soc. Lond,. B,. Biol. Sci. 359, 109–121. doi: 10.1098/rstb.2003.1370
Chance, B., Sies, H., and Boveris, A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605. doi: 10.1152/physrev.1979.59.3.527
Chappell, M. C., Marshall, A. C., Alzayadneh, E. M., Shaltout, H. A., and Diz, D. I. (2014). Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front. Endocrinol. (Lausanne) 4, 201. doi: 10.3389/fendo.2013.00201
Chen, Q. M., Bartholomew, J. C., Campisi, J., Acosta, M., Reagan, J. D., and Ames, B. N. (1998). Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 332, 43–50. doi: 10.1042/bj3320043
Chinnery, P. F. (2015). Mitochondrial disease in adults: what's old and what's new? EMBO Mol. Med. 7, 1503–1512. doi: 10.15252/emmm.201505079
Colla, S., Ong, D. S., Ogoti, Y., Marchesini, M., Mistry, N. A., Clise-Dwyer, K., et al. (2015). Telomere dysfunction drives aberrant hematopoietic differentiation and myelodysplastic syndrome. Cancer Cell. 27, 644–657. doi: 10.1016/j.ccell.2015.04.007
Collin, F. (2019). Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 20, 10. doi: 10.3390/ijms20102407
Collins, S., Pi, J., and Yehuda-Shnaidman, E. (2012). Uncoupling and reactive oxygen species (ROS)–a double-edged sword for β-cell function? “Moderation in all things”. Best Pract. Res. Clin. Endocrinol. Metab. 26, 753–758. doi: 10.1016/j.beem.2012.08.002
Costa-Besada, M. A., Valenzuela, R., Garrido-Gil, P., Villar-Cheda, B., Parga, J. A., Lanciego, J. L., et al. (2018). Paracrine and intracrine angiotensin 1-7/mas receptor axis in the substantia nigra of rodents, monkeys, and humans. Mol. Neurobiol. 55, 5847–5867. doi: 10.1007/s12035-017-0805-y
Crackower, M., Sarao, R., Oudit, G., Yagil, C., Kozieradzki, I., Scanga, S., et al. (2002). Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 417, 822–828. doi: 10.1038/nature00786
Cross, C. E., Halliwell, B., Borish, E. T., Pryor, W. A., Ames, B. N., Saul, R. L., et al. (1987). Oxygen radicals and human disease. Ann. Intern. Med. 107, 526–545. doi: 10.7326/0003-4819-107-4-526
d'Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature. 426, 194–198. doi: 10.1038/nature02118
Dai, D. F., Chen, T., Wanagat, J., Laflamme, M., Marcinek, D. J., Emond, M. J., et al. (2010). Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 9, 536–544. doi: 10.1111/j.1474-9726.2010.00581.x
Dai, D. F., Johnson, S. C., Villarin, J. J., Chin, M. T., Nieves-Cintron, M., Chen, T., et al. (2011). Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 108, 837–846. doi: 10.1161/CIRCRESAHA.110.232306
Dai, D. F., Santana, L. F., Vermulst, M., Tomazela, D. M., Emond, M. J., MacCoss, M. J., et al. (2009). Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 119, 2789–2797. doi: 10.1161/circulationaha.108.822403
Darley-Usmar, V., Wiseman, H., and Halliwell, B. (1995). Nitric oxide and oxygen radicals: a question of balance. FEBS Lett. 369, 131–135. doi: 10.1016/0014-5793(95)00764-z
Davisson, R. L., Oliverio, M. I., Coffman, T. M., and Sigmund, C. D. (2000). Divergent functions of angiotensin II receptor isoforms in the brain. J. Clin. Invest. 106, 103–106. doi: 10.1172/jci10022
de Cavanagh, E. M., Inserra, F., and Ferder, L. (2011). Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc. Res. 89, 31–40. doi: 10.1093/cvr/cvq285
de Cavanagh, E. M., Inserra, F., and Ferder, L. (2015). Angiotensin II blockade: how its molecular targets may signal to mitochondria and slow aging. Coincidences with calorie restriction and mTOR inhibition. Am. J. Physiol. Heart Circ. Physiol. 309, H15–44. doi: 10.1152/ajpheart.00459.2014
de Gasparo, M., Husain, A., Alexander, W., Catt, K. J., Chiu, A. T., Drew, M., et al. (1995). Proposed update of angiotensin receptor nomenclature. Hypertension. 25, 924–927. doi: 10.1161/01.hyp.25.5.924
De Giusti, V. C., Correa, M. V., Villa-Abrille, M. C., Beltrano, C., Yeves, A. M., de Cingolani, G. E., et al. (2008). The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci. 83, 264–271. doi: 10.1016/j.lfs.2008.06.008
del Río, L. A., Sandalio, L. M., Palma, J. M., Bueno, P., and Corpas, F. J. (1992). Metabolism of oxygen radicals in peroxisomes and cellular implications. Free Radic. Biol. Med. 13, 557–580. doi: 10.1016/0891-5849(92)90150-f
Dhalla, A. K., Hill, M. F., and Singal, P. K. (1996). Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 28, 506–514. doi: 10.1016/0735-1097(96)00140-4
Dimmeler, S., Rippmann, V., Weiland, U., Haendeler, J., and Zeiher, A. M. (1997). Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ. Res. 81, 970–976. doi: 10.1161/01.res.81.6.970
Diz, D. I., Kasper, S. O., Sakima, A., and Ferrario, C. M. (2007). Aging and the brain renin-angiotensin system: insights from studies in transgenic rats. Cleve Clin. J. Med. 74, S95–98. doi: 10.3949/ccjm.74.suppl_1.s95
Doksani, Y., Wu, J. Y., de Lange, T., and Zhuang, X. (2013). Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 155, 345–356. doi: 10.1016/j.cell.2013.09.048
Dominguez-Meijide, A., Villar-Cheda, B., Garrido-Gil, P., Sierrra-Paredes, G., Guerra, M. J., and Labandeira-Garcia, J. L. (2014). Effect of chronic treatment with angiotensin type 1 receptor antagonists on striatal dopamine levels in normal rats and in a rat model of Parkinson's disease treated with L-DOPA. Neuropharmacology. 76, 156–168. doi: 10.1016/j.neuropharm.2013.07.016
Donoghue, M., Hsieh, F., Baronas, E., Godbout, K., Gosselin, M., Stagliano, N., et al. (2000). A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 87, E1–9. doi: 10.1161/01.res.87.5.e1
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi: 10.1161/CIRCRESAHA.107.162800
Douiev, L., Abu-Libdeh, B., and Saada, A. (2018). Cytochrome c oxidase deficiency, oxidative stress, possible antioxidant therapy and link to nuclear DNA damage. Eur. J. Hum. Genet. 26, 579–581. doi: 10.1038/s41431-017-0047-5
Du, Y., Han, J., Zhang, H., Xu, J., Jiang, L., and Ge, W. (2019). Kaempferol prevents against Ang II-induced cardiac remodeling through attenuating Ang II-induced inflammation and oxidative stress. J. Cardiovasc. Pharmacol. 74, 326–335. doi: 10.1097/fjc.0000000000000713
Dzau, V., and Re, R. J. C. (1994). Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation. 89, 493–498. doi: 10.1161/01.cir.89.1.493
Eggena, P., Zhu, J. H., Clegg, K., and Barrett, J. D. (1993). Nuclear angiotensin receptors induce transcription of renin and angiotensinogen mRNA. Hypertension. 22, 496–501. doi: 10.1161/01.hyp.22.4.496
Erdel, F., Kratz, K., Willcox, S., Griffith, J. D., Greene, E. C., and de Lange, T. (2017). Telomere recognition and assembly mechanism of mammalian shelterin. Cell Rep. 18, 41–53. doi: 10.1016/j.celrep.2016.12.005
Ernster, L., and Schatz, G. J. (1981). Mitochondria: a historical review. J. Cell Biol. 91, 227s−255s. doi: 10.1083/jcb.91.3.227s
Escobales, N., Nuñez, R., and Javadov, S. J. A. (2019). Mitochondrial angiotensin receptors and cardioprotective pathways. Am. J. Physiol. Heart Circ. Physiol. 316, H1426–H1438. doi: 10.1152/ajpheart.00772.2018
Feng, X., Wang, L., and Li, Y. (2011). Change of telomere length in angiotensin II-induced human glomerular mesangial cell senescence and the protective role of losartan. Mol. Med. Rep. 4, 255–260. doi: 10.3892/mmr.2011.436
Feresin, R. G., Huang, J., Klarich, D. S., Zhao, Y., Pourafshar, S., Arjmandi, B. H., et al. (2016). Blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in vascular smooth muscle cells. Food Funct. 7, 4175–4187. doi: 10.1039/c6fo00743k
Fernandez-Marcos, P. J., and Auwerx, J. (2011). Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884s−890. doi: 10.3945/ajcn.110.001917
Ferrario, C., and Chappell, M. J. C. (2004). Novel angiotensin peptides. Cell Mol. Life Sci. 61, 2720–2727. doi: 10.1007/s00018-004-4243-4
Findeisen, H. M., Gizard, F., Zhao, Y., Cohn, D., Heywood, E. B., Jones, K. L., et al. (2011). Telomerase deficiency in bone marrow-derived cells attenuates angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 31, 253–260. doi: 10.1161/ATVBAHA.110.218545
Finkel, T. (1998). Oxygen radicals and signaling. Curr. Opin. Cell Biol. 10, 248–253. doi: 10.1016/s0955-0674(98)80147-6
Fleming, J. E., Miquel, J., Cottrell, S. F., Yengoyan, L. S., and Economos, A. C. (1982). Is cell aging caused by respiration-dependent injury to the mitochondrial genome? Gerontology. 28, 44–53. doi: 10.1159/000212510
Fontana, G. A., and Gahlon, H. L. (2020). Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 48, 11244–11258. doi: 10.1093/nar/gkaa804
Foreman, K. J., Marquez, N., Dolgert, A., Fukutaki, K., Fullman, N., McGaughey, M., et al. (2018). Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. 392, 2052–2090. doi: 10.1016/s0140-6736(18)31694-5
Forrester, S. J., Booz, G. W., Sigmund, C. D., Coffman, T. M., Kawai, T., Rizzo, V., et al. (2018). Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98, 1627–1738. doi: 10.1152/physrev.00038.2017
Fouquerel, E., Barnes, R. P., Uttam, S., Watkins, S. C., Bruchez, M. P., and Opresko, P. L. (2019). Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol Cell 75(1) 117-130 e116. doi: 10.1016/j.molcel.2019.04.024
Fouquerel, E., Lormand, J., Bose, A., Lee, H. T., Kim, G. S., Li, J., et al. (2016). Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 23, 1092–1100. doi: 10.1038/nsmb.3319
Fridovich, I. (1997). Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J. Biol. Chem. 272, 18515–18517. doi: 10.1074/jbc.272.30.18515
Garrido, A. M., and Griendling, K. K. (2009). NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 302, 148–158. doi: 10.1016/j.mce.2008.11.003
Gladyshev, V. N. (2014). The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox. Signal. 20, 727–731. doi: 10.1089/ars.2013.5228
Gomez-Cabrera, M. C., Sanchis-Gomar, F., Garcia-Valles, R., Pareja-Galeano, H., Gambini, J., Borras, C., et al. (2012). Mitochondria as sources and targets of damage in cellular aging. Clin. Chem. Lab. Med. 50, 1287–1295. doi: 10.1515/cclm-2011-0795
Gonzalez-Freire, M., de Cabo, R., Bernier, M., Sollott, S. J., Fabbri, E., Navas, P., et al. (2015). Reconsidering the Role of Mitochondria in Aging. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1334–1342. doi: 10.1093/gerona/glv070
Graiani, G., Lagrasta, C., Migliaccio, E., Spillmann, F., Meloni, M., Madeddu, P., et al. (2005). Genetic deletion of the p66Shc adaptor protein protects from angiotensin II-induced myocardial damage. Hypertension 46, 433–440. doi: 10.1161/01.HYP.0000174986.73346.ba
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 333, 1109–1112. doi: 10.1126/science.1201940
Griendling, K. K., Minieri, C. A., Ollerenshaw, J. D., and Alexander, R. W. (1994). Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148. doi: 10.1161/01.res.74.6.1141
Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., et al. (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503–514. doi: 10.1016/s0092-8674(00)80760-6
Guzman-Castillo, M., Ahmadi-Abhari, S., Bandosz, P., Capewell, S., Steptoe, A., Singh-Manoux, A., et al. (2017). Forecasted trends in disability and life expectancy in England and Wales up to 2025: a modelling study. Lancet Public Health. 2, e307–e313. doi: 10.1016/s2468-2667(17)30091-9
Haendeler, J., Dröse, S., Büchner, N., Jakob, S., Altschmied, J., Goy, C., et al. (2009). Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler. Thromb. Vasc. Biol. 29, 929–935. doi: 10.1161/atvbaha.109.185546
Halliwell, B., and Gutteridge, J. M. (1990). Role of free radicals and catalytic metal ions in human disease: an overview. Meth. Enzymol. 186, 1–85. doi: 10.1016/0076-6879(90)86093-b
Hamilton, D. J., Zhang, A., Li, S., Cao, T. N., Smith, J. A., Vedula, I., et al. (2016). Combination of angiotensin II and l-NG-nitroarginine methyl ester exacerbates mitochondrial dysfunction and oxidative stress to cause heart failure. Am. J. Physiol. Heart Circ. Physiol. 310, H667–680. doi: 10.1152/ajpheart.00746.2015
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell. 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hanna, I. R., Taniyama, Y., Szöcs, K., Rocic, P., and Griendling, K. K. (2002). NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid. Redox Signal. 4, 899–914. doi: 10.1089/152308602762197443
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature. 345, 458–460. doi: 10.1038/345458a0
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Harman, D. (1981). The aging process. Proc. Natl. Acad. Sci. U S A. 78, 7124–7128. doi: 10.1073/pnas.78.11.7124
Hauptmann, N., Grimsby, J., Shih, J. C., and Cadenas, E. (1996). The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch. Biochem. Biophys. 335, 295–304. doi: 10.1006/abbi.1996.0510
Hayflick, L. (2000). The illusion of cell immortality. Br. J. Cancer 83, 841–846. doi: 10.1054/bjoc.2000.1296
He, J., Ding, J., Lai, Q., Wang, X., Li, A., and Liu, S. (2019). Irbesartan ameliorates lipid deposition by enhancing autophagy via PKC/AMPK/ULK1 axis in free fatty acid induced hepatocytes. Front. Physiol. 10, 681. doi: 10.3389/fphys.2019.00681
Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. doi: 10.1016/j.tcb.2011.06.008
Herbert, K. E., Mistry, Y., Hastings, R., Poolman, T., Niklason, L., and Williams, B. (2008). Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 102, 201–208. doi: 10.1161/CIRCRESAHA.107.158626
Honig, L. S., Kang, M. S., Cheng, R., Eckfeldt, J. H., Thyagarajan, B., Leiendecker-Foster, C., et al. (2015). Heritability of telomere length in a study of long-lived families. Neurobiol. Aging 36, 2785–2790. doi: 10.1016/j.neurobiolaging.2015.06.017
Hu, C., Rai, R., Huang, C., Broton, C., Long, J., Xu, Y., et al. (2017). Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. 27, 1485–1502. doi: 10.1038/cr.2017.144
Huang, J. (1999). Study of relationship between ACE gene I/D polymorphism and renal predisposition in SLE. J Clini. Int. Med. 16, 205–206
Huang, X., Chen, W., Truong, L., and Lan, H. (2003). Chymase is upregulated in diabetic nephropathy: implications for an alternative pathway of angiotensin II-mediated diabetic renal and vascular disease. J. Am. Soc. Nephrol. 14, 1738–1747. doi: 10.1097/01.asn.0000071512.93927.4e
Hunt, S. C., Kimura, M., Hopkins, P. N., Carr, J. J., Heiss, G., Province, M. A., et al. (2015). Leukocyte telomere length and coronary artery calcium. Am. J. Cardiol. 116, 214–218. doi: 10.1016/j.amjcard.2015.03.060
Ionescu-Tucker, A., and Cotman, C. W. (2021). Emerging roles of oxidative stress in brain aging and Alzheimer's disease. Neurobiol. Aging 107, 86–95. doi: 10.1016/j.neurobiolaging.2021.07.014
Jackson, L., Eldahshan, W., Fagan, S. C., and Ergul, A. (2018). Within the brain: the renin angiotensin system. Int. J. Mol. Sci. 19, 3. doi: 10.3390/ijms19030876
Jaskelioff, M., Muller, F. L., Paik, J. H., Thomas, E., Jiang, S., Adams, A. C., et al. (2011). Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106. doi: 10.1038/nature09603
Jones, E. S., Vinh, A., McCarthy, C. A., Gaspari, T. A., and Widdop, R. E. (2008). AT2 receptors: functional relevance in cardiovascular disease. Pharmacol. Ther. 120, 292–316. doi: 10.1016/j.pharmthera.2008.08.009
Jurk, D., Wilson, C., Passos, J. F., Oakley, F., Correia-Melo, C., Greaves, L., et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172. doi: 10.1038/ncomms5172
Kagami, S., Border, W., Miller, D., and Noble, N. J. T. J.,o.c.i. (1994). Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. 93, 2431–2437. doi: 10.1172/jci117251
Kalache, A., de Hoogh, A. I., Howlett, S. E., Kennedy, B., Eggersdorfer, M., Marsman, D. S., et al. (2019). Nutrition interventions for healthy ageing across the lifespan: a conference report. Eur J Nutr 58(Suppl 1), 1–11. doi: 10.1007/s00394-019-02027-z
Kalathiya, U., Padariya, M., and Baginski, M. (2018). The structurally similar TRFH domain of TRF1 and TRF2 dimers shows distinct behaviour towards TIN2. Arch. Biochem. Biophys. 642, 52–62. doi: 10.1016/j.abb.2018.02.005
Karnik, S. S., Unal, H., Kemp, J. R., Tirupula, K. C., Eguchi, S., Vanderheyden, P. M., et al. (2015). International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacol Rev 67, 754–819. doi: 10.1124/pr.114.010454
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N. G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kawanishi, S., and Oikawa, S. (2004). Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci. 1019, 278–284. doi: 10.1196/annals.1297.047
Kennedy, S. R., Salk, J. J., Schmitt, M. W., and Loeb, L. A. (2013). Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9, e1003794. doi: 10.1371/journal.pgen.1003794
Kimura, S., Zhang, G. X., Nishiyama, A., Shokoji, T., Yao, L., Fan, Y. Y., et al. (2005). Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 45, 860–866. doi: 10.1161/01.HYP.0000163462.98381.7f
Kinoshita, J., Fushida, S., Harada, S., Yagi, Y., Fujita, H., Kinami, S., et al. (2009). Local angiotensin II-generation in human gastric cancer: correlation with tumor progression through the activation of ERK1/2, NF-kappaB and survivin. Int. J. Oncol. 34, 1573–1582. doi: 10.3892/ijo_00000287
Kirwan, M., and Dokal, I. (2009). Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys. Acta. 1792, 371–379. doi: 10.1016/j.bbadis.2009.01.010
Kontis, V., Bennett, J. E., Mathers, C. D., Li, G., Foreman, K., and Ezzati, M. (2017). Future life expectancy in 35 industrialised countries: projections with a Bayesian model ensemble. Lancet 389, 1323–1335. doi: 10.1016/s0140-6736(16)32381-9
Koppenol, W. H. (2001). The Haber-Weiss cycle−70 years later. Redox Rep. 6, 229–234. doi: 10.1179/135100001101536373
Kruk, P. A., Rampino, N. J., and Bohr, V. A. (1995). DNA damage and repair in telomeres: relation to aging. Proc. Natl. Acad. Sci. USA. 92, 258–262. doi: 10.1073/pnas.92.1.258
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 309, 481–484. doi: 10.1126/science.1112125
Kuszel, L., Trzeciak, T., Richter, M., and Czarny-Ratajczak, M. (2015). Osteoarthritis and telomere shortening. J. Appl. Genet. 56, 169–176. doi: 10.1007/s13353-014-0251-8
Labandeira-Garcia, J. L., Valenzuela, R., Costa-Besada, M. A., Villar-Cheda, B., and Rodriguez-Perez, A. I. (2021). The intracellular renin-angiotensin system: Friend or foe. Some light from the dopaminergic neurons. Prog. Neurobiol. 199, 101919. doi: 10.1016/j.pneurobio.2020.101919
Landmesser, U., Dikalov, S., Price, S. R., McCann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi: 10.1172/jci14172
Lapointe, J., and Hekimi, S. (2010). When a theory of aging ages badly. Cell. Mol. Life Sci. 67, 1–8. doi: 10.1007/s00018-009-0138-8
Lauri, A., Pompilio, G., and Capogrossi, M. C. (2014). The mitochondrial genome in aging and senescence. Ageing Res. Rev. 18, 1–15. doi: 10.1016/j.arr.2014.07.001
Laursen, J. B., Somers, M., Kurz, S., McCann, L., Warnholtz, A., Freeman, B. A., et al. (2001). Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103, 1282–1288. doi: 10.1161/01.cir.103.9.1282
Lautner, R. Q., Villela, D. C., Fraga-Silva, R. A., Silva, N., Verano-Braga, T., Costa-Fraga, F., et al. (2013). Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ. Res. 112, 1104–1111. doi: 10.1161/circresaha.113.301077
Lee, H. W., Blasco, M. A., Gottlieb, G. J., Horner, J. W., Greider, C. W., and DePinho, R. A. (1998). Essential role of mouse telomerase in highly proliferative organs. Nature. 392, 569–574. doi: 10.1038/33345
Lee, S. H., Takahashi, R., Goto, T., and Oe, T. (2010). Mass spectrometric characterization of modifications to angiotensin II by lipid peroxidation products, 4-oxo-2(E)-nonenal and 4-hydroxy-2(E)-nonenal. Chem. Res. Toxicol. 23, 1771–1785. doi: 10.1021/tx100228q
Li, R., Mi, X., Yang, S., Yang, Y., Zhang, S., Hui, R., et al. (2019). Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Exp. Gerontol. 119, 212–220. doi: 10.1016/j.exger.2019.02.012
Li, S., Crothers, J., Haqq, C. M., and Blackburn, E. H. (2005). Cellular and gene expression responses involved in the rapid growth inhibition of human cancer cells by RNA interference-mediated depletion of telomerase RNA. J. Biol. Chem. 280, 23709–23717. doi: 10.1074/jbc.M502782200
Li, X., Zhu, D., Zheng, X., Zhang, J., and Zhuo, J. J. (2018). Intratubular and intracellular renin-angiotensin system in the kidney: a unifying perspective in blood pressure control. Clin. Sci. (Lond). 132, 1383–1401. doi: 10.1042/cs20180121
Linnane, A. W., Marzuki, S., Ozawa, T., and Tanaka, M. (1989). Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1, 642–645. doi: 10.1016/s0140-6736(89)92145-4
Lloyd, R. V., Hanna, P. M., and Mason, R. P. (1997). The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Radic. Biol. Med. 22, 885–888. doi: 10.1016/s0891-5849(96)00432-7
Lobo, V., Patil, A., Phatak, A., and Chandra, N. (2010). Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 4, 118–126. doi: 10.4103/0973-7847.70902
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell. 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Madamanchi, N. R., Vendrov, A., and Runge, M. S. (2005). Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 29–38. doi: 10.1161/01.ATV.0000150649.39934.13
Martin, G. M. (2005). Genetic modulation of senescent phenotypes in Homo sapiens. Cell. 120, 523–532. doi: 10.1016/j.cell.2005.01.031
Martínez, P., and Blasco, M. A. (2011). Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 11, 161–176. doi: 10.1038/nrc3025
Martinez, P., Thanasoula, M., Carlos, A. R., Gómez-López, G., Tejera, A. M., Schoeftner, S., et al. (2010). Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat. Cell Biol. 12, 768–780. doi: 10.1038/ncb2081
Matavelli, L. C., and Siragy, H. M. (2015). AT2 receptor activities and pathophysiological implications. J. Cardiovasc. Pharmacol. 65, 226–232. doi: 10.1097/fjc.0000000000000208
Matsushima, S., Kinugawa, S., Ide, T., Matsusaka, H., Inoue, N., Ohta, Y., et al. (2006). Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Am. J. Physiol. Heart Circ. Physiol. 291, H2237–2245. doi: 10.1152/ajpheart.00427.2006
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055
McNally, J. S., Saxena, A., Cai, H., Dikalov, S., and Harrison, D. G. (2005). Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler. Thromb. Vasc. Biol. 25, 1623–1628. doi: 10.1161/01.ATV.0000170827.16296.6e
Melzer, D., Pilling, L. C., and Ferrucci, L. (2020). The genetics of human ageing. Nat. Rev. Genet. 21, 88–101. doi: 10.1038/s41576-019-0183-6
Mendell, J. T., and Olson, E. N. (2012). MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187. doi: 10.1016/j.cell.2012.02.005
Miao, J., Liu, J., Niu, J., Zhang, Y., Shen, W., Luo, C., et al. (2019). Wnt/beta-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell 18, e13004. doi: 10.1111/acel.13004
Min, L. J., Mogi, M., Iwai, M., and Horiuchi, M. (2009). Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Res. Rev. 8, 113–121. doi: 10.1016/j.arr.2008.12.002
Miquel, J. (1998). An update on the oxygen stress-mitochondrial mutation theory of aging: genetic and evolutionary implications. Exp. Gerontol. 33, 113–126. doi: 10.1016/s0531-5565(97)00060-0
Miquel, J., Economos, A. C., Fleming, J., and Johnson, J. E. Jr. (1980). Mitochondrial role in cell aging. Exp. Gerontol. 15, 575–591. doi: 10.1016/0531-5565(80)90010-8
Mitra, S., Nguyen, L. N., Akter, M., Park, G., Choi, E. H., and Kaushik, N. K. (2019). Impact of ROS generated by chemical, physical, and plasma techniques on cancer attenuation. Cancers (Basel) 11(7). doi: 10.3390/cancers11071030
Miyao, M., Cicalese, S., Kawai, T., Cooper, H. A., Boyer, M. J., Elliott, K. J., et al. (2020). Involvement of senescence and mitochondrial fission in endothelial cell pro-inflammatory phenotype induced by angiotensin II. Int. J. Mol. Sci. 21(9). doi: 10.3390/ijms21093112
Miyazaki, M., and Takai, S. J. (2006). Tissue angiotensin II generating system by angiotensin-converting enzyme and chymase. 100, 391–397. doi: 10.1254/jphs.cpj06008x
Mogi, M. (2020). Effect of renin-angiotensin system on senescence. Geriatr. Gerontol. Int. 20, 520–525. doi: 10.1111/ggi.13927
Mollnau, H., Wendt, M., Szocs, K., Lassegue, B., Schulz, E., Oelze, M., et al. (2002). Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 90, E58–65. doi: 10.1161/01.res.0000012569.55432.02
Nicholls, D. J. (2005). Mitochondria and calcium signaling. Cell Calcium. 38, 311–317. doi: 10.1016/j.ceca.2005.06.011
Obrenovich, M. E., Li, Y., Parvathaneni, K., Yendluri, B. B., Palacios, H. H., Leszek, J., et al. (2011). Antioxidants in health, disease and aging. CNS Neurol. Disord. Drug Targets 10, 192–207. doi: 10.2174/187152711794480375
Ocaranza, M. P., Michea, L., Chiong, M., Lagos, C. F., Lavandero, S., and Jalil, J. E. (2014). Recent insights and therapeutic perspectives of angiotensin-(1-9) in the cardiovascular system. Clin. Sci. (Lond). 127, 549–557. doi: 10.1042/CS20130449
Oganesian, L., and Karlseder, J. (2009). Telomeric armor: the layers of end protection. J. Cell Sci. 122, 4013–4025. doi: 10.1242/jcs.050567
Okuno, K., Cicalese, S., Elliott, K. J., Kawai, T., Hashimoto, T., and Eguchi, S. (2020). Targeting molecular mechanism of vascular smooth muscle senescence induced by angiotensin II, a potential therapy via senolytics and senomorphics. Int. J. Mol. Sci. 21(18). doi: 10.3390/ijms21186579
Oliverio, M. I., and Coffman, T. M. (2000). Angiotensin II receptor physiology using gene targeting. News Physiol. Sci. 15, 171–175. doi: 10.1152/physiologyonline.2000.15.4.171
Olovnikov, A. M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41, 181–190. doi: 10.1016/0022-5193(73)90198-7
O'Sullivan, R. J., and Karlseder, J. (2010). Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 11, 171–181. doi: 10.1038/nrm2848
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 155, 699–712. doi: 10.1016/j.cell.2013.09.021
Pagan, L. U., Gomes, M. J., Gatto, M., Mota, G. A. F., Okoshi, K., and Okoshi, M. P. (2022). The Role of Oxidative Stress in the Aging Heart. Antioxidants (Basel). 11, 2. doi: 10.3390/antiox11020336
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Pan, H. W., Cui, Y. H., and Zeng, J. W. (2014). NF-κB mediates the survival of corneal myofibroblast induced by angiotensin II. Invest. Ophthalmol. Vis. Sci. 55, 4220–4228. doi: 10.1167/iovs.13-13735
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347. doi: 10.1038/msb.2010.5
Patel, C. J., Manrai, A. K., Corona, E., and Kohane, I. S. (2017). Systematic correlation of environmental exposure and physiological and self-reported behaviour factors with leukocyte telomere length. Int. J. Epidemiol. 46, 44–56. doi: 10.1093/ije/dyw043
Pendergrass, K. D., Gwathmey, T. M., Michalek, R. D., Grayson, J. M., and Chappell, M. C. (2009). The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem. Biophys. Res. Commun. 384, 149–154. doi: 10.1016/j.bbrc.2009.04.126
Pérez, N. G., Villa-Abrille, M. C., Aiello, E. A., Dulce, R. A., Cingolani, H. E., and Camilión de Hurtado, M. C. (2003). A low dose of angiotensin II increases inotropism through activation of reverse Na(+)/Ca(2+) exchange by endothelin release. Cardiovasc. Res. 60, 589–597. doi: 10.1016/j.cardiores.2003.09.004
Pérez, V. I., Van Remmen, H., Bokov, A., Epstein, C. J., Vijg, J., and Richardson, A. (2009). The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 8, 73–75. doi: 10.1111/j.1474-9726.2008.00449.x
Peters, J., Kränzlin, B., Schaeffer, S., Zimmer, J., Resch, S., Bachmann, S., et al. (1996). Presence of renin within intramitochondrial dense bodies of the rat adrenal cortex. Am. J. Physiol. 271, E439–450. doi: 10.1152/ajpendo.1996.271.3.E439
Pham-Huy, L. A., He, H., and Pham-Huy, C. (2008). Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 4, 89–96.
Pinegin, B., Vorobjeva, N., Pashenkov, M., and Chernyak, B. (2018). The role of mitochondrial ROS in antibacterial immunity. J. Cell. Physiol. 233, 3745–3754. doi: 10.1002/jcp.26117
Pueyo, M. E., Arnal, J. F., Rami, J., and Michel, J. B. (1998). Angiotensin II stimulates the production of NO and peroxynitrite in endothelial cells. Am. J. Physiol. 274, C214–220. doi: 10.1152/ajpcell.1998.274.1.C214
Radi, R., Beckman, J. S., Bush, K. M., and Freeman, B. A. (1991). Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 288, 481–487. doi: 10.1016/0003-9861(91)90224-7
Rattan, S. I. (2014). Aging is not a disease: implications for intervention. Aging Dis. 5, 196–202. doi: 10.14336/ad.2014.0500196
Re, R. N. (2018). Role of intracellular angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 314, H766–h771. doi: 10.1152/ajpheart.00632.2017
Re, R. N., and Cook, J. L. (2015). Studies of intracellular angiotensin II. Methods Mol. Biol. 1234, 1–8. doi: 10.1007/978-1-4939-1755-6_1
Reckelhoff, J. F., and Romero, J. C. (2003). Role of oxidative stress in angiotensin-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R893–912. doi: 10.1152/ajpregu.00491.2002
Reichert, S., and Stier, A. (2017). Does oxidative stress shorten telomeres in vivo? A review. Biol. Lett. 13, 12. doi: 10.1098/rsbl.2017.0463
Richter, C., Park, J. W., and Ames, B. N. (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA. 85, 6465–6467. doi: 10.1073/pnas.85.17.6465
Richter, T., and von Zglinicki, T. (2007). A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 42, 1039–1042. doi: 10.1016/j.exger.2007.08.005
Rincon-Choles, H., Kasinath, B. S., Gorin, Y., and Abboud, H. E. (2002). Angiotensin II and growth factors in the pathogenesis of diabetic nephropathy. Kidney Int. Suppl. 62, S8−11. doi: 10.1046/j.1523-1755.62.s82.3.x
Ristow, M., and Schmeisser, K. (2014). Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response. 12, 288–341. doi: 10.2203/dose-response.13-035.Ristow
Robertson, A. L. Jr., and Khairallah, P. A. (1971). Angiotensin II: rapid localization in nuclei of smooth and cardiac muscle. Science. 172, 1138–1139. doi: 10.1126/science.172.3988.1138
Rogov, A. G., Goleva, T. N., Epremyan, K. K., Kireev, I. I., and Zvyagilskaya, R. A. (2021). Propagation of mitochondria-derived reactive oxygen species within the dipodascus magnusii cells. Antioxidants (Basel). 10, 1. doi: 10.3390/antiox10010120
Romero, A., San Hipólito-Luengo, Á., Villalobos, L. A., Vallejo, S., Valencia, I., Michalska, P., et al. (2019). The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell. 18, e12913. doi: 10.1111/acel.12913
Ruggenenti, P., Bettinaglio, P., Pinares, F., and Remuzzi, G. J. (2008). Angiotensin converting enzyme insertion/deletion polymorphism and renoprotection in diabetic and nondiabetic nephropathies. Clin. J. Am. Soc. Nephrol. 3, 1511–1525. doi: 10.2215/cjn.04140907
Ruiz-Ortega, M., and Egido, J. J. (1997). Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int. 52, 1497–1510. doi: 10.1038/ki.1997.480
Sahin, E., Colla, S., Liesa, M., Moslehi, J., Müller, F. L., Guo, M., et al. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 470, 359–365. doi: 10.1038/nature09787
Sahin, E., and Depinho, R. A. (2010). Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 464, 520–528. doi: 10.1038/nature08982
Sahin, E., and DePinho, R. A. (2012). Axis of ageing: telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13, 397–404. doi: 10.1038/nrm3352
Salazar, G. (2018). NADPH oxidases and mitochondria in vascular senescence. Int. J. Mol. Sci. 19, 5. doi: 10.3390/ijms19051327
Salazar, G., Huang, J., Feresin, R. G., Zhao, Y., and Griendling, K. K. (2017). Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 108, 225–235. doi: 10.1016/j.freeradbiomed.2017.03.032
Salgado, D., Rocco, J., Silva, E., and Vincent, J. J. (2010). Modulation of the renin-angiotensin-aldosterone system in sepsis: a new therapeutic approach? 14, 11–20. doi: 10.1517/14728220903460332
Saraff, K., Babamusta, F., Cassis, L. A., and Daugherty, A. (2003). Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 1621–1626. doi: 10.1161/01.Atv.0000085631.76095.64
Sasaki, K., Yamano, Y., Bardhan, S., Iwai, N., Murray, J. J., Hasegawa, M., et al. (1991). Cloning and expression of a complementary DNA encoding a bovine adrenal angiotensin II type-1 receptor. Nature. 351, 230–233. doi: 10.1038/351230a0
Savage, S. A., Giri, N., Baerlocher, G. M., Orr, N., Lansdorp, P. M., and Alter, B. P. (2008). TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 82, 501–509. doi: 10.1016/j.ajhg.2007.10.004
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta. 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019
Schepers, A. G., Vries, R., van den Born, M., van de Wetering, M., and Clevers, H. (2011). Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J. 30, 1104–1109. doi: 10.1038/emboj.2011.26
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–462. doi: 10.1016/j.cub.2014.03.034
Schmutz, I., and de Lange, T. (2016). Shelterin. Curr. Biol. 26, R397–399. doi: 10.1016/j.cub.2016.01.056
Schulman, I. H., and Raij, L. (2008). The angiotensin II type 2 receptor: what is its clinical significance? Curr. Hypertens. Rep. 10, 188–193. doi: 10.1007/s11906-008-0036-8
Scialò, F., Sriram, A., Fernández-Ayala, D., Gubina, N., Lõhmus, M., Nelson, G., et al. (2016). Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734. doi: 10.1016/j.cmet.2016.03.009
Sfeir, A., and de Lange, T. (2012). Removal of shelterin reveals the telomere end-protection problem. Science. 336, 593–597. doi: 10.1126/science.1218498
Sfeir, A., Kosiyatrakul, S. T., Hockemeyer, D., MacRae, S. L., Karlseder, J., Schildkraut, C. L., et al. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 138, 90–103. doi: 10.1016/j.cell.2009.06.021
Shariat-Madar, Z., and Schmaier, A. J. (2004). The plasma kallikrein/kinin and renin angiotensin systems in blood pressure regulation in sepsis. J. Endotoxin Res. 10, 3–13. doi: 10.1179/096805104225003807
Shay, J. W., and Wright, W. E. (2019). Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 20, 299–309. doi: 10.1038/s41576-019-0099-1
Shiomi, T., Tsutsui, H., Matsusaka, H., Murakami, K., Hayashidani, S., Ikeuchi, M., et al. (2004). Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 109, 544–549. doi: 10.1161/01.Cir.0000109701.77059.E9
Silva, G. A. F., Nunes, R. A. L., Morale, M. G., Boccardo, E., Aguayo, F., and Termini, L. (2018). Oxidative stress: therapeutic approaches for cervical cancer treatment. Clinics (São Paulo). 73, e548s. doi: 10.6061/clinics/2018/e548s
Singhapol, C., Pal, D., Czapiewski, R., Porika, M., Nelson, G., and Saretzki, G. C. (2013). Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE. 8, e52989. doi: 10.1371/journal.pone.0052989
Sirett, N. E., McLean, A. S., Bray, J. J., and Hubbard, J. I. (1977). Distribution of angiotensin II receptors in rat brain. Brain Res. 122, 299–312. doi: 10.1016/0006-8993(77)90296-7
Smith, E. M., Pendlebury, D. F., and Nandakumar, J. (2020). Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 77, 61–79. doi: 10.1007/s00018-019-03369-x
Smith, L. L., Coller, H. A., and Roberts, J. M. (2003). Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 5, 474–479. doi: 10.1038/ncb985
Son, J. M., and Lee, C. (2021). Aging: All roads lead to mitochondria. Semin. Cell Dev. Biol. 116, 160–168. doi: 10.1016/j.semcdb.2021.02.006
Sprovieri, S., and Sens, Y. J. L. (2005). Polymorphisms of the renin-angiotensin system genes in Brazilian patients with lupus nephropathy. 14, 356–362. doi: 10.1191/0961203305lu2093oa
Starkweather, A. R., Alhaeeri, A. A., Montpetit, A., Brumelle, J., Filler, K., Montpetit, M., et al. (2014). An integrative review of factors associated with telomere length and implications for biobehavioral research. Nurs. Res. 63, 36–50. doi: 10.1097/nnr.0000000000000009
Steinberg, D., Parthasarathy, S., Carew, T. E., Khoo, J. C., and Witztum, J. L. (1989). Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 320, 915–924. doi: 10.1056/nejm198904063201407
Stoll, M., Steckelings, U., Paul, M., Bottari, S., Metzger, R., Unger, T. J. T. J., et al. (1995). The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin Invest. 95, 651–657. doi: 10.1172/jci117710
St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790. doi: 10.1074/jbc.M207217200
Sumners, C., de Kloet, A. D., Krause, E. G., Unger, T., and Steckelings, U. M. (2015). Angiotensin type 2 receptors: blood pressure regulation and end organ damage. Curr. Opin. Pharmacol. 21, 115–121. doi: 10.1016/j.coph.2015.01.004
Takai, S., Jin, D., and Miyazaki, M. J. (2012). Multiple mechanisms for the action of chymase inhibitors. J. Pharmacol. Sci. 118, 311–316. doi: 10.1254/jphs.11r11cp
Theurey, P., and Pizzo, P. (2018). The aging mitochondria. Genes (Basel). 9, 1. doi: 10.3390/genes9010022
Thomas, D. D., Liu, X., Kantrow, S. P., and Lancaster, J. R. (2001). The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA. 98, 355–360. doi: 10.1073/pnas.98.1.355
Thompson, M. M., Oyama, T. T., Kelly, F. J., Kennefick, T. M., and Anderson, S. (2000). Activity and responsiveness of the renin-angiotensin system in the aging rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1787–1794. doi: 10.1152/ajpregu.2000.279.5.R1787
Tigerstedt, R., and Bergman, P. Q. (1898). Niere and Kreislauf. Scand Arch Physiol Germany 8, 223–227.
Tönnies, E., and Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and alzheimer's disease. J. Alzheimers. Dis. 57, 1105–1121. doi: 10.3233/jad-161088
Tousoulis, D., Kampoli, A. M., Tentolouris, C., Papageorgiou, N., and Stefanadis, C. (2012). The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 10, 4–18. doi: 10.2174/157016112798829760
Toussaint, O., Medrano, E. E., and von Zglinicki, T. (2000). Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 35, 927–945. doi: 10.1016/s0531-5565(00)00180-7
Touyz, R. M. (2003). Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Rev. Cardiovasc. Ther. 1, 91–106. doi: 10.1586/14779072.1.1.91
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Trist, B. G., Hare, D. J., and Double, K. L. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease. Aging Cell 18, e13031. doi: 10.1111/acel.13031
Tsutsui, H., Kinugawa, S., and Matsushima, S. (2011). Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H2181–2190. doi: 10.1152/ajpheart.00554.2011
Urata, H., Boehm, K., Philip, A., Kinoshita, A., Gabrovsek, J., Bumpus, F., et al. (1993). Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. 91, 1269–1281. doi: 10.1172/jci116325
Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., et al. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 16, 29–37. doi: 10.1152/physiolgenomics.00122.2003
Van Remmen, H., and Richardson, A. (2001). Oxidative damage to mitochondria and aging. Exp. Gerontol. 36, 957–968. doi: 10.1016/s0531-5565(01)00093-6
van Steensel, B., Smogorzewska, A., and de Lange, T. (1998). TRF2 protects human telomeres from end-to-end fusions. Cell. 92, 401–413. doi: 10.1016/s0092-8674(00)80932-0
Vaughan, D., Lazos, S., and Tong, K. J. T. (1995). Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Invest. 95, 995–1001. doi: 10.1172/jci117809
Vermulst, M., Wanagat, J., Kujoth, G. C., Bielas, J. H., Rabinovitch, P. S., Prolla, T. A., et al. (2008). DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 40, 392–394. doi: 10.1038/ng.95
Villapol, S., and Saavedra, J. M. (2015). Neuroprotective effects of angiotensin receptor blockers. Am. J. Hypertens. 28, 289–299. doi: 10.1093/ajh/hpu197
Villar-Cheda, B., Costa-Besada, M. A., Valenzuela, R., Perez-Costas, E., Melendez-Ferro, M., and Labandeira-Garcia, J. L. (2017). The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 8, e3044. doi: 10.1038/cddis.2017.439
von Zglinicki, T. (2002). Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. doi: 10.1016/s0968-0004(02)02110-2
von Zglinicki, T., Pilger, R., and Sitte, N. (2000). Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 28, 64–74. doi: 10.1016/s0891-5849(99)00207-5
von Zglinicki, T., Saretzki, G., Döcke, W., and Lotze, C. (1995). Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp. Cell Res. 220, 186–193. doi: 10.1006/excr.1995.1305
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Walne, A. J., Vulliamy, T., Beswick, R., Kirwan, M., and Dokal, I. (2008). TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 112, 3594–3600. doi: 10.1182/blood-2008-05-153445
Wang, H. X., Yang, H., Han, Q. Y., Li, N., Jiang, X., Tian, C., et al. (2013). NADPH oxidases mediate a cellular “memory” of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic. Biol. Med. 65, 897–907. doi: 10.1016/j.freeradbiomed.2013.08.179
Wang, M., Takagi, G., Asai, K., Resuello, R. G., Natividad, F. F., Vatner, D. E., et al. (2003). Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41, 1308–1316. doi: 10.1161/01.Hyp.0000073843.56046.45
Wilson, W. R., Herbert, K. E., Mistry, Y., Stevens, S. E., Patel, H. R., Hastings, R. A., et al. (2008). Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur. Heart J. 29, 2689–2694. doi: 10.1093/eurheartj/ehn386
Xiong, S., Patrushev, N., Forouzandeh, F., Hilenski, L., and Alexander, R. W. (2015). PGC-1α Modulates telomere function and DNA damage in protecting against aging-related chronic diseases. Cell Rep. 12, 1391–1399. doi: 10.1016/j.celrep.2015.07.047
Xiong, S., Salazar, G., San Martin, A., Ahmad, M., Patrushev, N., Hilenski, L., et al. (2010). PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 285, 2474–2487. doi: 10.1074/jbc.M109.065235
Yan, C., Duanmu, X., Zeng, L., Liu, B., and Song, Z. (2019). Mitochondrial DNA: distribution, mutations, and elimination. Cells 8, 4. doi: 10.3390/cells8040379
Yoshida, N., Endo, J., Kinouchi, K., Kitakata, H., Moriyama, H., Kataoka, M., et al. (2019). (Pro)renin receptor accelerates development of sarcopenia via activation of Wnt/YAP signaling axis. Aging Cell. 18, e12991. doi: 10.1111/acel.12991
Young, I., and Woodside, J. J. (2001). Antioxidants in health and disease. J. Clin. Pathol. 54, 176–186. doi: 10.1136/jcp.54.3.176
Yu, B. P. (1994). Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 74, 139–162. doi: 10.1152/physrev.1994.74.1.139
Yu, K. Y., Wang, Y. P., Wang, L. H., Jian, Y., Zhao, X. D., Chen, J. W., et al. (2014). Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res. Cardiol. 109, 416. doi: 10.1007/s00395-014-0416-y
Zablocki, D., and Sadoshima, J. (2013). Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 19, 1095–1109. doi: 10.1089/ars.2012.4588
Zablocki, D., and Sadoshima, J. J. (2011). Knocking out angiotensin II in the heart. Curr. Hypertens. Rep. 13, 129–135. doi: 10.1007/s11906-011-0180-4
Zhang, G., Lu, X., Kimura, S., and Nishiyama, A. (2007). Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc. Res. 76, 204–212. doi: 10.1016/j.cardiores.2007.07.014
Zhang, H., Menzies, K. J., and Auwerx, J. (2018). The role of mitochondria in stem cell fate and aging. Development. 145, 8. doi: 10.1242/dev.143420
Zhang, J., Rane, G., Dai, X., Shanmugam, M. K., Arfuso, F., Samy, R. P., et al. (2016). Ageing and the telomere connection: an intimate relationship with inflammation. Ageing Res. Rev. 25, 55–69. doi: 10.1016/j.arr.2015.11.006
Zhang, Y., Zou, C., Yang, S., and Fu, J. (2016). P120 catenin attenuates the angiotensin II-induced apoptosis of human umbilical vein endothelial cells by suppressing the mitochondrial pathway. Int. J. Mol. Med. 37, 623–630. doi: 10.3892/ijmm.2016.2476
Zheng, X. Q., Wang, J. T., Liu, X. L., Sun, Y., Zheng, Y. J., Wang, X. J., et al. (2015). Effect of hydrolysis time on the physicochemical and functional properties of corn glutelin by Protamex hydrolysis. Food Chem. 172, 407–415. doi: 10.1016/j.foodchem.2014.09.080
Zhou, J., Fleming, A. M., Averill, A. M., Burrows, C. J., and Wallace, S. S. (2015). The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 43, 7171. doi: 10.1093/nar/gkv673
Zhou, J., Liu, M., Fleming, A. M., Burrows, C. J., and Wallace, S. S. (2013). Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 288, 27263–27272. doi: 10.1074/jbc.M113.479055
Zhu, M., Zhang, J., Nie, S., and Yan, W. J. (2012). Associations of ACE I/D, AGT M235T gene polymorphisms with pregnancy induced hypertension in Chinese population: a meta-analysis. J. Assist. Reprod. Genet. 29, 921–932. doi: 10.1007/s10815-012-9800-4
Zhuo, J. L., and Li, X. C. (2011). New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides. 32, 1551–1565. doi: 10.1016/j.peptides.2011.05.012
Ziada, A. S., Smith, M. R., and Cote, H. C. F. (2020). Updating the free radical theory of aging. Front Cell Dev Biol 8, 575645. doi: 10.3389/fcell.2020.575645
Keywords: aging, angiotensin II, reactive oxygen species, mitochondrial dysfunction, telomere attrition
Citation: Yi W, Chen F, Zhang H, Tang P, Yuan M, Wen J, Wang S and Cai Z (2022) Role of angiotensin II in aging. Front. Aging Neurosci. 14:1002138. doi: 10.3389/fnagi.2022.1002138
Received: 24 July 2022; Accepted: 08 November 2022;
Published: 02 December 2022.
Edited by:
Scott Miners, University of Bristol, United KingdomReviewed by:
Mark C. Chappell, Wake Forest University, United StatesCopyright © 2022 Yi, Chen, Zhang, Tang, Yuan, Wen, Wang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyou Cai, Y2FpemhpeW91QHVjYXMuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.