
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 08 February 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.813544
 Gayathri Srinivasan
Gayathri Srinivasan David A. Brafman*
David A. Brafman*Numerous epidemiological studies have demonstrated that individuals who have sustained a traumatic brain injury (TBI) have an elevated risk for developing Alzheimer’s disease and Alzheimer’s-related dementias (AD/ADRD). Despite these connections, the underlying mechanisms by which TBI induces AD-related pathology, neuronal dysfunction, and cognitive decline have yet to be elucidated. In this review, we will discuss the various in vivo and in vitro models that are being employed to provide more definite mechanistic relationships between TBI-induced mechanical injury and AD-related phenotypes. In particular, we will highlight the strengths and weaknesses of each of these model systems as it relates to advancing the understanding of the mechanisms that lead to TBI-induced AD onset and progression as well as providing platforms to evaluate potential therapies. Finally, we will discuss how emerging methods including the use of human induced pluripotent stem cell (hiPSC)-derived cultures and genome engineering technologies can be employed to generate better models of TBI-induced AD.
Alzheimer’s disease (AD) affects over 6 million individuals in the U.S. and has a direct cost estimated in excess of $355 billion/year (Alzheimer’s Association, 2021). Although a vast majority of late onset AD (LOAD) cases are sporadic, numerous genetic and environmental risk factors have been identified that contribute to lifetime risk of developing the disease. In this vein, studies over the past few decades have also implicated traumatic brain injury (TBI) in the onset and progression of neurodegenerative diseases later in life (Schofield et al., 1997; Chen et al., 2007; Gardner et al., 2018). Indeed, it has been generally accepted that in addition to the primary mechanical damage inflicted, TBI sets in place various secondary injury mechanisms with acute and long-term effects. In addition, several studies have shown that a large-percentage of TBI patients not only suffer from short-term cognitive impairment but also experience long-term cognitive decline similar to that observed in AD patients (Rabinowitz and Levin, 2014). Furthermore, pathologies associated with AD have been observed in post-mortem brain tissue of TBI patients (Ikonomovic et al., 2004).
Despite these associations between a TBI and AD onset, the mechanisms by which a TBI induces or augments AD-related neurodegenerative processes are unclear. In this review, we will first discuss the various epidemiological and clinical evidence that exists for TBI-induced AD. Next, we will describe the various potential mechanisms for TBI-induced AD that have been suggested by these studies. Finally, we will discuss the emergence of in vitro models of TBI and speculate about how these can be used to further elucidate the mechanisms by which TBI induces AD-related pathology, neuronal dysfunction, and cognitive decline.
Because cognitive impairment often follows a TBI, the onset of AD following head trauma has been an active area of investigation. In this section, we will review the recent epidemiological, clinical, and animal-based studies that have investigated this relationship between TBI and AD. In addition, we will discuss the limitations of these studies as it relates to providing a definitive mechanistic link between TBI and age-related progression of AD.
Broadly speaking, epidemiological studies investigating the link between TBI and AD have been divided in their conclusions with some demonstrating an increased risk of AD post-TBI while others have not identified a significant association (Lye and Shores, 2000; Dams-O’Connor et al., 2016). With respect to the studies that support the link between TBI and AD, it has been reported that TBI approximately doubles the likelihood of an individual developing AD (Fleminger et al., 2003). In support of these studies, it has been suggested that after age and Apolipoprotein (E) genotype, TBI is the strongest risk factor associated with non-familial, sporadic forms of AD (Daviglus et al., 2011; Armstrong, 2019). Likewise, TBI has also been reported to accelerate the age of onset of AD (Nemetz et al., 1999; Schaffert et al., 2018). However, it should be noted that additional studies have suggested that several factors might modulate this risk of TBI-induced AD. For example, the severity of injury has a significant effect on the likelihood of development of AD with the reported risk increasing only twofold with moderate TBI but over fourfold with severe TBI (Guo et al., 2000; Plassman et al., 2000). Likewise, some studies report an increased AD risk only in TBI cases with loss of consciousness (Schofield et al., 1997; Guo et al., 2000).
It also has been suggested that the risk of AD after a TBI might be sex-dependent with a significant association only found in men (Fleminger et al., 2003; Weiner et al., 2017). This is interesting given that women have a higher risk of developing AD in the absence of head injury (Viña and Lloret, 2010; Laws et al., 2018; Guo et al., 2022). It has been suggested that female hormones such as estrogen and progesterone exert protective effects post-TBI by modulating anti-inflammatory and antioxidant processes (Vagnerova et al., 2008; Brotfain et al., 2016; Ma et al., 2019). In the same vein, these same sex-related hormones may alter AD-related mechanisms post-injury as well. However, others have suggested that sex-based differences in clinical outcomes after TBI might be independent of biological-related mechanisms but rather due to sex differences in injury type or treatment post-TBI (Ma et al., 2019; reviewed here Mollayeva et al., 2018). Moving forward, establishing definitive correlations between sex and TBI-induced AD will require more extensive, well-controlled studies (Ma et al., 2019).
Finally, as is the case with many epidemiological studies, there are several limitations that should be noted. For example, many of the studies cited are retrospective in nature, introducing recall bias due to which the association between TBI and AD might be significant (Weiner et al., 2017). Moreover, many of these studies suffer from the well-documented limitations of diagnosing AD using only neurophysiological profiling or cognitive examination (Tarawneh and Holtzman, 2012; Dubois et al., 2016; Weiner et al., 2017). Perhaps because of these limitations, some prospective studies have failed to find significant association between TBI and AD (Katzman et al., 1989; Launer et al., 1999; Mehta et al., 1999; Lindsay et al., 2002) even in cases with loss of consciousness (Crane et al., 2016).
Clinicopathological studies have demonstrated a link between TBI and the development of AD-related pathologies such as the formation of amyloid beta (Aβ) plaques and neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau (p-tau). While amyloid precursor protein (APP) accumulation is a well-known marker for diffuse axonal injury (DAI) (Gentleman et al., 1993), a major pathology of TBI, evidence for Aβ accumulation post-injury first appeared in studies by Clinton et al. (1991) and Roberts et al. (1991) where Aβ deposits were observed in one-third of short term survivors of TBI within days of the injury. Moreover, these deposits were observed up to a few years after the injury in one-third of TBI survivors (Roberts et al., 1994). Similarly, Aβ42 containing diffuse Aβ deposits have been observed acutely after injury (Gentleman et al., 1997). While Aβ plaques similar to those observed in AD patients have been reported in a few long-term survivors of TBI (Johnson et al., 2012), some studies report a lack of dense mature plaques in short-term (Ikonomovic et al., 2004) as well as long-term survivors of TBI (Chen et al., 2009). It has been suggested that clearance of Aβ deposits post injury by the enzyme neprilysin might explain this lack of Aβ plaques in long-term TBI survivors (Chen et al., 2009). Similarly, although the enzymes involved in the amyloidogenic processing of APP such PS1 and BACE1 were found to colocalize with Aβ in damaged axons, plaque formation was not observed even years after the injury (Uryu et al., 2007). Overall, although the evidence for Aβ plaques post-injury is unclear, these studies demonstrate increased amyloid burden in TBI patients.
Repetitive mild TBI in in athletes (Stern et al., 2019), boxers (McCrory et al., 2007) and veterans (McKee et al., 2013) has been identified as a risk factor for the development of a tauopathy, known as chronic traumatic encephalopathy (CTE). On the other hand, the effect of a single TBI on tau pathology is yet to be understood (Collins-Praino and Corrigan, 2017). In fact, NFTs have been only rarely, if at all, reported in acute survivors of TBI (Smith et al., 2003; Ikonomovic et al., 2004) although alterations in tau immunoreactivity even in the absence of NFTs have been observed and warrant further studies. For instance, after injury, accumulation of cis-p tau, the conformation of p-tau prone to aggregation and found in NFTs in AD patients, has been reported (Albayram et al., 2017). In contrast to studies focused on acute effects of injury, NFTs, tau deposition and white matter degeneration have been reported in long-term survivors of TBI as indicated by immunohistochemical studies (Johnson et al., 2012, 2013). Tau deposits in long-term survivors of TBI have also been correlated with onset of neuropsychiatric conditions later in life (Takahata et al., 2019).
Overall, while clinical studies have provided important evidence for the connection between TBI and subsequent AD onset, the majority of these studies are focused on the examination of post-mortem tissue which only provide an endpoint of the disease. As such, it is difficult to establish temporal relationships between the initial TBI and formation of AD-related pathologies. Recently, progress has been made in the use of biomarkers and advanced imaging techniques to determine such connections. For example, amyloid PET imaging in long-term survivors of TBI revealed amyloid pathology in patients after brain trauma albeit in a pattern different from those observed in AD patients (Scott et al., 2016). As it relates to tauopathy, PET scans have revealed tau deposition in long-term survivors of TBI (Gorgoraptis et al., 2019). Along similar lines, cleaved tau in cerebrospinal fluid (CSF) and total tau in interstitial fluid (ISF) have been observed to increase post-injury and have been suggested as biomarkers for TBI (Gabbita et al., 2005; Marklund et al., 2009).
Because of the limitations of epidemiological and clinical studies, animal models have been employed to identify potential mechanistic links between TBI and the development of AD. Broadly speaking, these studies have employed various AD transgenic mouse models (e.g., 3xTg, APP/PS1, Tg2576, PS19, APP knockin) in the context of numerous injury systems [e.g., controlled cortical impact (CCI), midline fluid percussion injury (mFPI), lateral fluid percussion injury (LFPI), closed head impact model of engineered rotational acceleration (CHIMERA)]. These studies are too numerous to review here but have been summarized in Table 1. Instead, we highlight some of the key studies that demonstrate a link between TBI and AD.
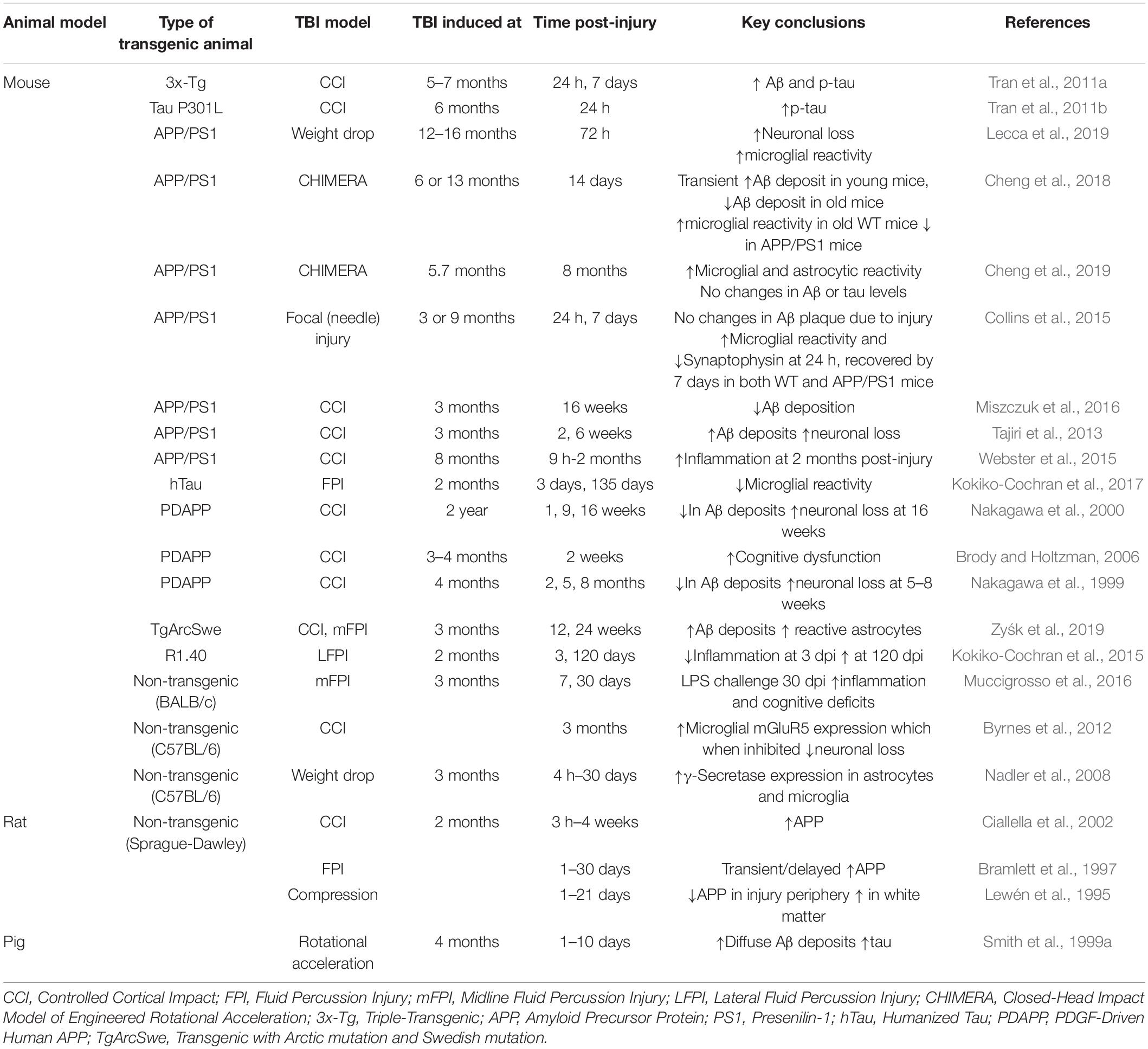
Table 1. Summary of AD phenotypes observed in animal models of injury.
With respect to amyloid-related phenotypes, increased APP immunoreactivity, a marker of axonal damage has been observed in the white matter in rats acutely (Ciallella et al., 2002) as well as chronically (Pierce et al., 1998; Acosta et al., 2017) (up to 1 year) post-injury. It is important to note that the temporal and spatial pattern of APP immunoreactivity in the gray matter, however, seem to vary depending on the mode of injury as well as its severity (Bramlett et al., 1997). For instance, in a rat CCI model, increased APP immunoreactivity was observed in gray matter 6 months post-injury (Acosta et al., 2017) whereas in an FPI model, the immunoreactivity increased transiently in the striatum but remained elevated at the site of injury up to 1 month (Bramlett et al., 1997). Similarly, in a rat mFPI model APP immunoreactivity was observed to increase acutely in the hippocampus but decrease by 7 days post-injury which could be attributed to the cell death observed at this time point (Murakami et al., 1998). Several transgenic mouse models of AD that express human APP, such as 3x-Tg and APP/PS1 have shown acute intra-axonal Aβ accumulation (Tran et al., 2011b). APP/PS1 mice have also shown to accumulate extracellular Aβ deposits (Tajiri et al., 2013), but not Aβ plaques due to focal injury (Collins et al., 2015). Critically, age at injury also appears to affect the extent of these deposits (Nakagawa et al., 2000; Cheng et al., 2018). Despite increased APP expression, only increased Aβ (Hoshino et al., 1998) but not plaques have been reported in rodent models most likely due to the differences in rodent and human Aβ species. Diffuse Aβ plaques, however, were observed in a pig model with gyrencephalic brains similar to humans rather than lissencephalic brains as present in rodents upon DAI (Smith et al., 1999a).
As it relates to tauopathies, while repetitive injury models have been utilized to understand tau pathologies after injury, the effect of a single injury is yet to be elucidated. Presence of tau oligomers as well as tau phosphorylation has been reported in rat FPI (Hoshino et al., 1998; Hawkins et al., 2013), CCI (Acosta et al., 2017) and mouse blast injury (Huber et al., 2013) models as well as in tau transgenic mouse models (Tran et al., 2011b). Similar to studies that suggest an Aβ independent tau pathology in AD (reviewed here (van der Kant et al., 2020), tau phosphorylation due to injury in 3xTg mice has also been suggested to be independent of Aβ pathology (Tran et al., 2011a).
Despite the utility of animal studies, the inherent complexities and multi-cellular nature of the in vivo environment have made it difficult to make definitive mechanistic links between TBI-induced cellular injury and AD-related phenotypes as well as tease apart cell-autonomous vs. cell-non autonomous aspects of such relationships. Similarly, because animal models do not recapitulate all aspects of the human disease (Duff and Suleman, 2004; Seok et al., 2013; Warren et al., 2015), it is questionable to what degree these findings in animal models will translate to the human condition. In the future, the use of complimentary human-based systems will be necessary to confirm findings made using animal models.
Despite the limitations of epidemiological, clinical, and animal-based studies, these investigations have suggested some potential mechanisms by which TBI can lead to AD onset and age-related progression. In this section, we will summarize some of these hypothesized mechanisms (Figure 1).
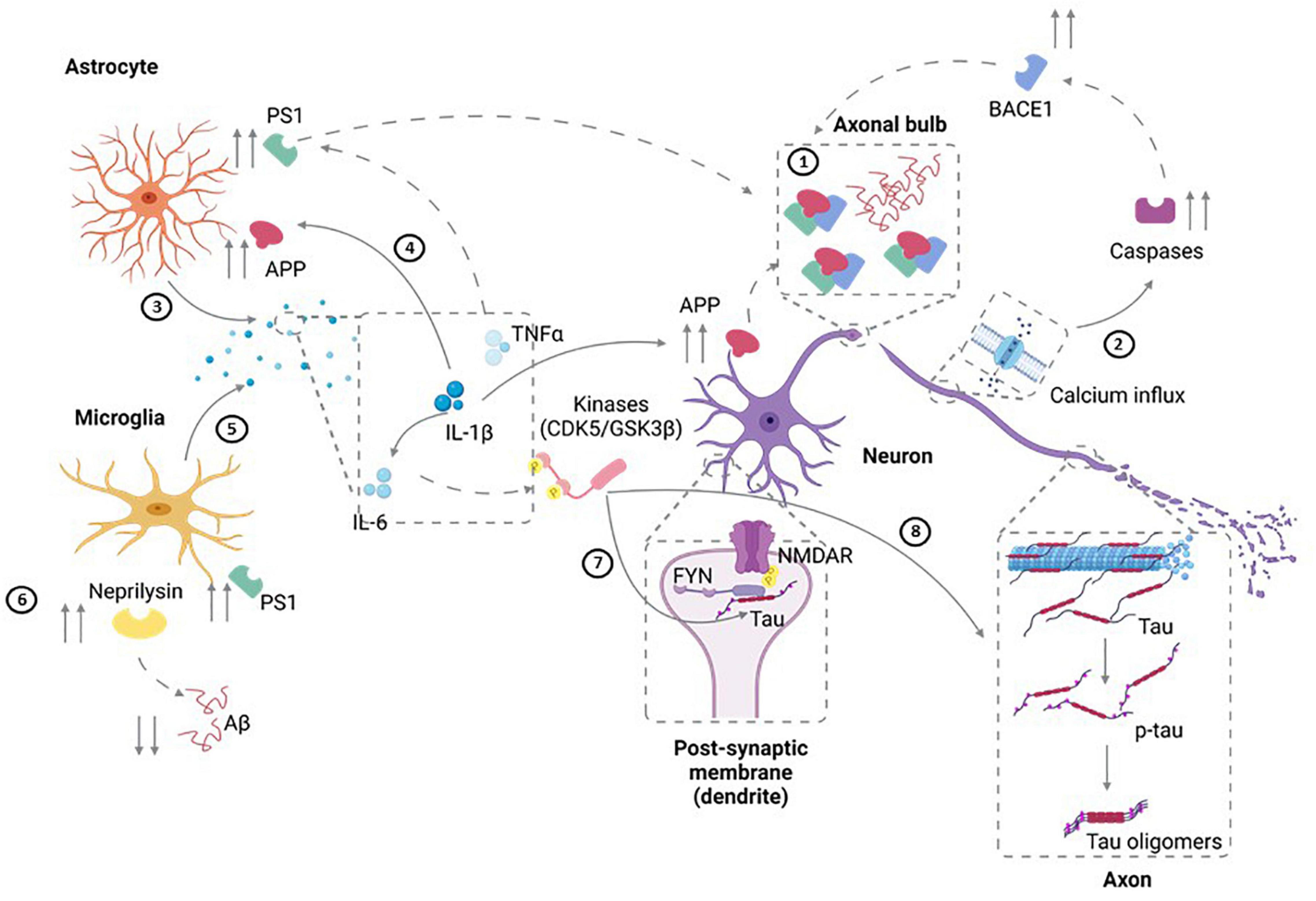
Figure 1. Potential Mechanisms for TBI-Induced AD. ➀ Diffuse axonal injury that results from TBI can lead to upregulation of APP as well as increased amyloidogenic processing of APP to pathogenic Aβ40 and Aβ42. In addition, axonal degeneration could also lead to the extracellular release of Aβ. ➁ Injury can also result in calcium influx in neurons. In turn, this can result in activation of caspases that induce the amyloidogenic processing of APP through elevated BACE1 availability. ➂ Injury-induced cytokine release (IL-1) by astrocytes can also result in ➃ the upregulation of APP expression and an associated increase in its amyloidogenic progressing through upregulation of BACE and PS1. ➄ Along similar lines, microglial activation and production of IL-6, IL-1 and TNFα can increase APP transcription, upregulation of PS1, and increased amyloidogenic APP processing. ➅ TBI also can modulate Aβ clearance by neprilysin generated by microglia leading to aberrant clearance of Aβ. ➆ Cell injury can also elevate FYN tyrosine kinase activity leading to increased tau phosphorylation as well as activation of the NMDAR subunit NR2B, thereby modulating synaptic plasticity and increasing excitotoxic vulnerability. ➇ IL-6 upregulation post-injury can also lead to tau hyperphosphorylation through elevated MAPK-p38 signaling. Figure was generated with the assistance of Biorender.
The amyloid cascade hypothesis posits that Aβ deposition is central to the pathogenesis of AD and might be due to either the increased production of amyloidogenic Aβ or due its aberrant clearance (reviewed here Selkoe and Hardy, 2016). Aβ peptides are generated from APP which is a transmembrane protein with an extracellular N-terminus and intracellular C-terminus. APP can be cleaved by the amyloidogenic pathway leading to Aβ peptide formation or by the non-amyloidogenic pathway. In the non-amyloidogenic pathway, α-secretase cleaves APP to form soluble APPα (sAPPα) and a membrane-bound C-terminal fragment called C83 (Kojro and Fahrenholz, 2005; LaFerla et al., 2007). In the amyloidogenic pathway, β-secretase cleaves APP to form soluble APPβ (sAPPβ) and a 99 amino acid C-terminal fragment called C99 which is further cleaved by y-secretase to form Aβ peptides of varying lengths and a membrane-bound APP intracellular domain (AICD). γ-secretase cleaves C83 formed by α-secretase cleavage to generate 3kDa protein called p3 (Haass et al., 1993) and a membrane-bound AICD. AICD formed by γ-secretase cleavage of C99 could be internalized and further cleaved by caspases to form toxic species (C31). Aβ peptides, predominantly Aβ42 are prone to aggregation (Kim and Hecht, 2005) and might form amyloid fibrils that accumulate to form senile plaques (Jarrett et al., 1993; Younkin, 1998). The exact species that is toxic to neurons is still unknown, although, several in vitro studies point toward Aβ oligomers being neurotoxic (Lambert et al., 1998; Hartley et al., 1999).
Diffuse axonal injury observed in TBI is marked by APP upregulation. Increased levels of Aβ40 as well as of Aβ42 and of Aβ deposits as a result of amyloidogenic processing of APP have been observed in animal models post injury (Hoshino et al., 1998; Smith et al., 1999a; Tran et al., 2011a; Tajiri et al., 2013). Various mechanisms of Aβ generation and accumulation have been posited-persistent axonal damage after a single injury might be a continual source of Aβ formation. Degeneration of these axons could release Aβ into the extracellular space. The accumulation of APP, BACE1 and PS-1 might also be due to the impaired axonal transport observed in DAI as suggested by a rodent peripheral nervous system (PNS) injury model (Cribbs et al., 1996; Chen et al., 2004). In fact, targeting these enzymes including BACE and γ-secretase has been shown to reduce neuronal cell loss post-injury (Loane et al., 2009).
APP expression could also be upregulated upon cytokine induction (IL-1) by neurons and glia observed post-injury (Goldgaber et al., 1989; Ciallella et al., 2002). Oxidative stress-induced upregulation of BACE and γ-secretase could also be speculated to contribute to the amyloidogenic processing of APP post-TBI (Tamagno et al., 2002). Activation of caspases due to calcium influx in neurons could also contribute to the amyloidogenic cleavage of APP potentially through increasing BACE availability (Walker et al., 2012). On the other hand, it is unclear if upregulation of APP post-injury has neuroprotective effects with cognitive outcomes declining upon knocking out APP (Corrigan et al., 2012a) or if it has no effect at all (Murai et al., 1998).
Excitotoxicity is a major secondary injury mechanism in TBI. N-methyl-D-aspartate receptor (NMDAR) activation post-injury has been shown to shift APP processing to the amyloidogenic pathway from non-amyloidogenic, potentially contributing to increased Aβ (Lesné et al., 2005). Upregulation of Aβ clearing enzyme neprilysin has also been observed in TBI patients who didn’t display Aβ plaque pathology indicating the possibility of Aβ clearance by neprilysin (Chen et al., 2009) and potentially an imbalance between the generation and clearance of Aβ in plaque formation. Recent studies indicate that promoting the non-amyloidogenic processing of accumulated APP by soluble APP alpha (sAPPα) administration (Corrigan et al., 2012b), increasing AB CA1 levels (Loane et al., 2011) and inhibiting c-JNK (Rehman et al., 2018) reduced Aβ pathology further suggesting a role for Aβ accumulation in AD pathogenesis post-TBI.
Tau is a microtubule associated protein expressed in neurons whose primary function is to stabilize microtubules. It has six isoforms characterized by the number of tubulin binding domains (R) in the C-terminal and the presence of one or two or the absence of a 29-amino acid insert (N). Tau undergoes phosphorylation in at least 80 different sites, such as Thr231, Ser404 that allow it to detach from microtubules (Buée et al., 2000). Its phosphorylation depends upon the activity of various kinases such as MAPK (Drewes et al., 1992), CDK5 (Baumann et al., 1993) and GSK3β (Mandelkow et al., 1992) and phosphatases such as PP2A (Wang et al., 2013). Pathological hyperphosphorylation of tau leads to its oligomerization and formation of paired helical fragments that form the core of neurofibrillary tangles in AD. Furthermore, tau pathology has also been observed to spread across cells in a prion-like manner (Clavaguera et al., 2015).
Tau localizes mainly in axons, although low levels are present in dendrites as well (Ittner et al., 2010). Tau has been shown to associate with FYN tyrosine kinase in dendrites allowing FYN to phosphorylate and activate the NMDAR subunit NR2B, thereby regulating synaptic plasticity as well as potentially rendering neurons vulnerable to excitotoxicity (Salter and Kalia, 2004; Ittner and Götz, 2011). This has been speculated to contribute to Aβ induced toxicity in neurons in AD with FYN kinase inhibition suggested as a potential therapeutic for AD (Nygaard et al., 2014). FYN inhibition in a repetitive injury model has been shown to reduce tau phosphorylation and memory deficits (Tang et al., 2020) indicating a potential route for tau phosphorylation and pathology post-injury (Schumann et al., 2008; Rubenstein et al., 2017).
Tau pathology has been observed to spread to the contralateral hemisphere post-injury in a tau transgenic mouse model (Edwards et al., 2019). Prion-like transmission of tau pathology has also been observed in uninjured rodents upon injection of tau oligomers or brain homogenates from rodent models of injury (Gerson et al., 2016; Zanier et al., 2018). A reduction in phosphatases observed in rat injury models could also contribute to tau hyperphosphorylation (Arun et al., 2015). Additionally, tau phosphorylation post- injury has been suggested to alter the microglia and macrophage mediated inflammatory response hinting at a link between tau pathology and inflammation post-injury (Kokiko-Cochran et al., 2017).
Chronic inflammation is a major component of AD pathology (Kinney et al., 2018). Recent GWAS studies in fact identify a number of immune responsive genes as risk factors for AD (Efthymiou and Goate, 2017). Inflammation has been shown to exacerbate Aβ and tau pathology as well as play a key role in neurodegeneration (Barroeta-Espar et al., 2019; Griciuc and Tanzi, 2021). While the inflammatory response to TBI is mediated by a number of cell types, we will focus on the microglial and astrocytic contribution.
Studies indicate that microglia phagocytose Aβ at the earlier stages of the disease contributing to its clearance but remain chronically activated marked by a decrease in their phagocytic capacity (Hickman et al., 2008; Yang et al., 2011). Such prolonged activation is linked to a chronic inflammatory response marked by impaired Aβ clearance, secretion of cytokines and reactive oxygen species (ROS) (Meda et al., 1995; McDonald et al., 1997). However, since markers for microglial activation have not been clearly established, there is some controversy on the nature of microglia surrounding Aβ plaques in AD post-mortem brains with reports of activated/reactive and of senescent microglia being present (Streit et al., 2009).
In brain trauma, microglia respond to injury by acutely elevating pro- and anti-inflammatory cytokine levels and by phagocytosing Aβ generated as a result of injury; in fact, microglia containing Aβ have been found in TBI patients (Chen et al., 2009; Mannix and Whalen, 2012). Acute release of pro-inflammatory cytokines have been shown to have neuroprotective effects (Penkowa et al., 2000). However, chronic activation of microglia has been observed in long-term survivors of TBI (Johnson et al., 2013) as well as in animal models of injury (Loane et al., 2014a) with the existence of primed microglia with exacerbated responses to immune challenges post-injury observed in a rodent model (Muccigrosso et al., 2016). Persistent microglial activation is hypothesized to contribute to neurodegeneration post-injury (Kokiko-Cochran et al., 2015). Upregulation of NADPH oxidase (NOX) enzymes that generate ROS post-injury (Loane et al., 2014a) could alter microglial activation by polarizing them to an M1-like state promoting neurodegeneration (Kumar et al., 2016).
Microglial activation and secretion of IL-6, IL-1, and TNFα by microglia and astrocytes have been suggested to increase APP transcription and y-secretase activity leading to amyloidogenic processing of APP (Sheng et al., 2003; Lee et al., 2008; Breunig et al., 2013). Additionally, upregulation of PS-1 in astrocytes and microglia has been observed in an animal model of injury (Nadler et al., 2008), suggesting glial role in Aβ processing. Moreover, Aβ accumulation post-injury has been shown to modulate microglial activation, with chronic activation observed in AD transgenic but not wild type (WT) mice after injury (Kokiko-Cochran et al., 2015). Upregulation of cytokines such as IL-6 (Kumar et al., 2015) reported after TBI could also lead to tau hyperphosphorylation, potentially through MAPK-p38 signaling (Quintanilla et al., 2004).
Additionally, metabotropic glutamate receptor activation in microglia has been suggested to decrease microglial activation as well as neuronal loss in a mouse model of injury (Byrnes et al., 2012; Loane et al., 2014b). However, the timing of inhibiting microglial activation might be important since a recent study suggests that inhibition of chronic microglial activation increases neurodegeneration post-injury in TBI patients (Scott et al., 2018).
Work with various model systems has allowed for the identification of several potential mechanisms by which TBI exerts its risk modifying effects as it relates to AD onset. As discussed previously each of these model systems has numerous limitations which make it difficult to dissect the mechanistic links between TBI and AD. As such, accessible in vitro models are needed to complement these existing model systems. In this section, we will review currently existing in vitro TBI models as it relates to mode of injury (e.g., transection, compression, stretch, blast, shear, microfluidic) and cell types (e.g., primary tissue, dissociated primary cells, immortalized cells lines, pluripotent cells) employed (Table 2). In addition, we will speculate how these models can be utilized in the future to study TBI-induced AD.
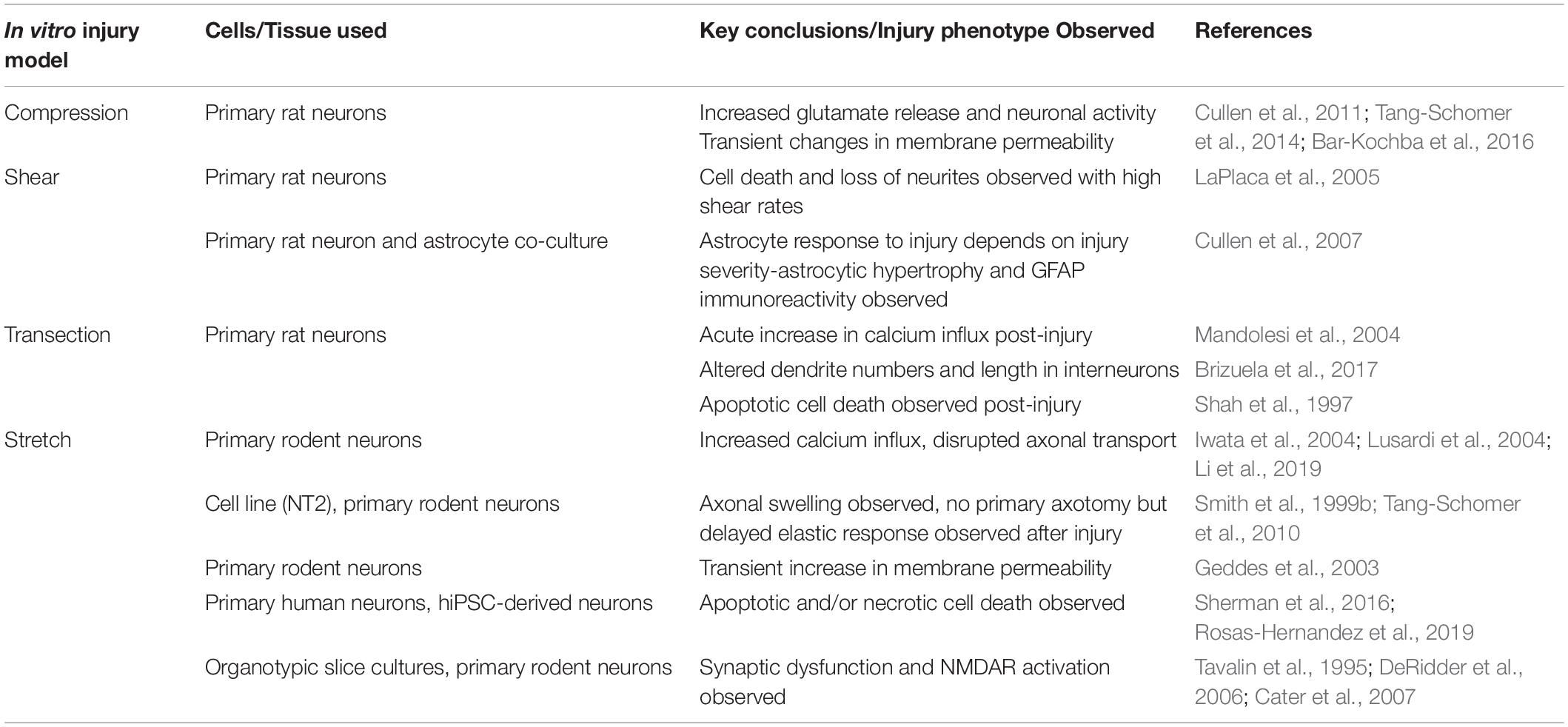
Table 2. Summary of in vitro injury model studies with key injury phenotypes observed in these models highlighted.
Due to the heterogeneous nature of the type of injury that is induced in animal models, correlations between injury intensity and frequency with cellular phenotypes have been difficult to precisely ascertain (DeWitt et al., 2018). In this vein, various in vitro injury paradigms have been engineered which eliminate the complexities associated with in vivo experiments by minimizing confounding variables and facilitating a more direct investigation of the effects of mechanical insult. Here, we will discuss how each of these modes of injury recapitulate various aspects of a TBI. In addition, the limitations of each of these injury paradigms will be summarized.
Transection models involve utilizing stylets (Tecoma et al., 1989), blades (Chuckowree and Vickers, 2003) or rotating scribes (Mukhin et al., 1997) to introduce cuts in cultures, typically transecting axons. Although simple, the injury caused by these models are not often reproducible or controllable in terms of the biomechanical force involved. Moreover, they mimic primary axotomy in TBI, which occurs only in a small proportion of all TBI cases. Nonetheless, these models have lent insight into axonal regeneration post-injury (Chuckowree and Vickers, 2003), apoptotic cell death in mixed cultures (Shah et al., 1997) and calcium influx (Mandolesi et al., 2004) or proteomic changes (Lööv et al., 2013) following injury.
Compression injury models involve weight drop or pistons to compress 3-D, organoid or organotypic slice cultures (Tang-Schomer et al., 2014). Closed loop models allowing for reproducible injury have been developed (Cullen et al., 2011; Bar-Kochba et al., 2016). Additionally, these systems allow for live cell imaging. Importantly, controlling the piston size allows for the injury of various regions of the culture and study the effects in surrounding regions. However, the strain field is difficult to characterize with techniques to do so only being developed recently (Bar-Kochba et al., 2016).
Stretch injury models involve culturing neurons on a flexible, deformable substrate (e.g., silicone) and stretching the substrate along one (uniaxial; Pfister et al., 2003) or both (biaxial; Morrison et al., 2006) directions using pneumatic systems. These models recapitulate DAI pathology and have shown axonal varicosities, axonal transport disruption and apoptotic cell death as observed in animal models or in clinical studies (DeRidder et al., 2006; Monnerie et al., 2010; Tang-Schomer et al., 2012). Additionally, these models allow for the culture of organotypic slice cultures, although typically 2-D cultures are utilized in these models. Moreover, these systems can be combined with multi-electrode array (MEA) systems to allow for the monitoring of electrophysiological changes post-injury (Kang et al., 2014). A disadvantage of these systems is that the applied strain is not uniform throughout the substrate and is not well characterized (Morrison et al., 2011).
Blast injury devices employ high pressure waves using compressed gas delivered via a shock-tube (Effgen et al., 2012) or using focused ultrasound (Lai et al., 2020) to mimic shock waves, particularly a transient increase in pressure due to a blast in 3-D cultures or organotypic slice cultures (Effgen et al., 2014; Zander et al., 2017). These have been used to study blood brain barrier disruption (Hue et al., 2013) and electrophysiological changes post-injury (Vogel et al., 2015). One limitation of these systems, though, is that the construction of these devices might be cost-prohibitive.
Shear stress models introduce shear stress either by moving one portion of the culture rapidly while its parallel surface is held fixed (LaPlaca et al., 2005) or by utilizing fluid shear stress by placing a rotating plate over cultures (LaPlaca and Thibault, 1997). Use of 3-D cultures in these models allows for complex-co-culture systems (Cullen et al., 2007). Critically, deformation due to shear stress is a major component of TBI and, thus, these systems uniquely mimic that aspect of the in vivo injury.
More recently, microfluidic platforms to injure cells are also being employed to study axonal transport (Magdesian et al., 2012), hyperexcitability (Nagendran and Taylor, 2019), and mitochondrial damage (Dollé et al., 2014) post-injury. These devices typically culture cells in a microfluidic device patterned such that the neuronal cell bodies are restricted to one compartment and the axons are directed along a channel; some models also allow for culturing organotypic slice cultures on flexible substrates. Axotomy or injury is introduced by vacuum aspiration (Taylor et al., 2005), stretching the flexible substrate (Dollé et al., 2013) or compressing cultures using polydimethylsiloxane (PDMS) pads (Hosmane et al., 2011). Microfluidic platforms allow us to restrict the injury to specific regions of the cell (Shrirao et al., 2018). However, these systems require complex fabrication techniques as the pneumatic systems that often drive the compression/stretch require precise connections with the microfluidic chambers.
Various cell and tissue types exist for the in vitro modeling of TBI. These cell types offer several advantages over their in vivo counterparts. For example, these cell types allow for a higher degree of control as specific cell types can be interrogated individually or combined at reproducibly defined ratios. In this vein, analysis of such cultures allows for the dissection of TBI-induced AD related phenotypes that are exerted through cell-autonomous vs. –non-autonomous mechanisms. In addition, injury with in vitro cell preparations allows for a greater ease-of-use than in vivo models which require technically complex surgical procedures. Finally, these in vitro cell types are amenable to a greater level of genetic and pharmacological manipulation than in vivo models. Despite these advantages, each of these cell preparations have inherent limitations that we will highlight in this section.
Acute preparations of rodent tissue (∼400 μm or less in thickness) can be isolated and subjected to injury within a few hours of isolation. These slices could be isolated from animals irrespective of their age, thereby allowing us to utilize mature cells in in vitro models. Additionally, these preparations preserve the native architecture of the tissue. However, the isolation procedure in itself could confound the injury response (Morrison et al., 2011).
On the other hand, organotypic slice cultures, isolated typically from rodent hippocampus are cultured for days before being used in an injury platform (often used in blast, stretch and compression models). Similar to acute preparations, these cultures retain the 3-D architecture and complexity of in vivo tissues. However, these slice cultures are generally isolated from younger animals, slowing down the maturation of the cells when cultured.
Primary cells enzymatically isolated from rodent tissue are used in several in vitro models (Shah et al., 1997; LaPlaca et al., 2005; Cullen et al., 2011; Bar-Kochba et al., 2016). These allow us to study cell-type specific responses to TBI as well as allow for complex co-culture models. Depending on the method, the isolation procedure introduces some mechanical damage prior to the in vitro injury. This is specifically an issue in case of cultures that utilize microglia that are transcriptionally different when isolated and cultured. Additionally, these cells are typically isolated from embryonic tissue and require long periods before maturation (Morrison et al., 2011). In addition, primary cell sources rapidly lose disease phenotypes upon ex vivo culture and are not amenable to genetic modification.
Various immortalized cell lines of various subtypes (e.g., neuronal-like, microglial, endothelial) and origins (e.g., human, mouse, rat) have been used extensively in conjunction with in vitro injury models (Smith et al., 1999b; Triyoso and Good, 1999; Pfister et al., 2003; Salvador et al., 2018; Li et al., 2019; Yin et al., 2020). The main advantage of these cell lines is their accessibility and extensive characterization. However, one major limitation of immortalized cells is that they might not represent the phenotypic maturity of their in vivo counterparts or display the same functional properties (Gordon et al., 2013; Carter and Shieh, 2015). In addition, immortalized cell lines often have abnormal karyotypes with unknown dosage at key disease-relevant genes (Ouellette et al., 2000).
Advances in cellular reprogramming have enabled the generation of in vitro central nervous system (CNS) disease-in-a-dish models that can be used to investigate the molecular mechanisms of disease origins as well as interrogate potential therapeutic interventions (Goldstein et al., 2015; Ghaffari et al., 2018). In particular, human induced pluripotent stem cells (hiPSCs), which can be derived from the reprogramming of somatic cells, can differentiate into all of the neural lineages and supporting cell types that comprise the CNS (Hong and Do, 2019). As such, hiPSC-derived cell types have been recently used in stretch and blast models of injury (Sherman et al., 2016; Lai et al., 2020), providing the ability to study TBI in a more relevant human system. However, hiPSC-derived cells resemble fetal neurons in nature, require prolonged differentiations to generate certain cell types and exhibit a lot of variability across clones and differentiations (Dolmetsch and Geschwind, 2011).
Although in vitro models have been used extensively to interrogate the effects of TBI on neural cell phenotypes, only recently have these systems been applied to investigate the molecular and cellular mechanisms that might induce the onset of AD post-TBI. For example, Wu et al. (2020) used mouse hippocampal slice cultures in conjunction with a weight drop model to interrogate the TBI-induced AD-related pathologies. Interestingly, the authors observed that injury induced a marked increase in APP cleavage. Mechanistically, the authors determined that injury induces increased delta-secretase (AEP) expression which mediates APP fragmentation and subsequent neuronal cell death.
Several studies have also used in vitro platforms to investigate the potential mechanisms by which cell injury can lead to tau-related pathologies. For example, in one such study a stretch model employing rodent hippocampal cells was used to investigate the effect of injury on tauopathy (Braun et al., 2020). This study revealed that mechanical stretching of cultured neurons resulted in tau mislocalization to dendritic spines which results in subsequent synaptic dysfunction. Critically, the authors identified a strong relationship between injury dynamics and the extent of tau mislocalization. Finally, through the use of pharmacological inhibitors the authors showed that the injury-induced synaptic deficits due to tau hyperphosphorylation were mediated likely by GSK3β and CDK5, kinases that phosphorylate tau and whose expression has been observed to be upregulated in AD brains (Yamaguchi et al., 1996; Blalock et al., 2004).
Collectively, these studies set a strong precedent for using in vitro models for the identification of possible mechanisms linking TBI and AD. In the future, given the utility of neurons, astrocytes, and microglia differentiated from patient derived hiPSCs in various culture formats, these cells could be utilized in in vitro injury models to investigate the mechanisms of TBI-induced AD. In one such study, a high-intensity focused ultrasound was used to induce mechanical injury in 3-D hiPSC-derived cortical organoids. Remarkably, injured organoids displayed increased levels of pathologically associated phosphorylated tau (Lai et al., 2020). In addition, injury disrupted nucleocytoplasmic transport in a manner similar to that observed in AD (Eftekharzadeh et al., 2018). Finally, a more recent study that used hiPSC-derived neurons in the context of a stretch-based model demonstrated that injury reduced APP axonal transport as well increased the accumulation of axonal amyloidogenic fragments (Chaves et al., 2021). Moving forward, given that phenotypes such as elevated Aβ peptides and tau hyperphosphorylation are readily observed in AD hiPSC lines (Israel et al., 2012; Kim et al., 2015; Jones et al., 2017), future studies could employ these cell lines to investigate the effect of cell injury on the induction or augmentation of AD-related molecular, biochemical, and cellular changes.
There is emerging evidence that sex can have a significant influence on TBI risk and related clinical outcomes (Gupte et al., 2019; Ma et al., 2019). The potential genetic, biochemical, and environmental causes for these sex-specific differences have been reviewed elsewhere (Gupte et al., 2019; Ma et al., 2019). On the other hand, there is a paucity of research related to investigating the mechanisms by which TBI-induced AD can be influenced by sex. Interestingly, while AD is more prevalent among females, the clinical outcomes associated with TBI appear to be worse in males (Viña and Lloret, 2010; Laws et al., 2018; Gupte et al., 2019; Ma et al., 2019; Guo et al., 2022). Thus, future studies that employ in vivo and in vitro models might be able to identify the molecular underpinnings of sex-based differences. Indeed, several studies have used hiPSC-based models of other diseases to identify the role of sex in disease onset and progression (Huo et al., 2019; Lock et al., 2021; Paci et al., 2021). In the same regard, moving forward similar study designs could be employed to determine the presence and mechanisms of sex-based differences in TBI-induced AD.
In addition to age at injury and injury severity, the pathological consequence of TBI can be influenced by a variety of genetic factors (Wilson and Montgomery, 2007; Dardiotis et al., 2010; Bennett et al., 2016; Kurowski et al., 2019). As it relates to TBI-induced AD, polymorphisms in Apolipoprotein E (APOE) appear to be the most prominent risk-modifying genetic factor. APOE is a cholesterol transport lipoprotein that is primarily secreted by astroglial cells in the CNS. Broadly speaking, APOE has three main isoforms (E2, E3, and E4) of which APOE4 has been reported to increase the risk as well as decrease the median age of AD onset whereas APOE2 has been demonstrated to mitigate the onset and age-related progression of AD (Bales et al., 1997; Castellano et al., 2011). The role of APOE in modulating AD-related phenotypes has been reviewed extensively elsewhere (Liu et al., 2013). Briefly, APOE isoforms have been shown to differentially clear Aβ and affect its aggregation (Bales et al., 1997; Castellano et al., 2011) as well as modulate tau phosphorylation and immune responses in AD (LaDu et al., 2001; Brecht et al., 2004).
Despite the strong evidence linking APOE isoforms to modulation of AD onset and age-related progression, the role of APOE polymorphism in influencing AD-related outcomes post-TBI remains unclear. For example, several studies have indicated that clinical outcomes worsen in APOE4 individuals post-injury with Aβ deposition being observed more frequently (Nicoll et al., 1995; Friedman et al., 1999). Confirmatory work with a transgenic AD mouse model demonstrated that in ApoE4 animals injury induced Aβ plaque formation whereas only diffuse Aβ deposits were observed in ApoE3 or ApoE knockout mice indicating a role for ApoE4 in Aβ aggregation (Hartman et al., 2002). On the other hand, in a 3xTg AD mouse model crossed with human ApoE (ApoE2, 3 and 4 isoforms), increased APP immunoreactivity, but similar levels Aβ40 or Aβ42, were observed acutely after injury in 3xTg-ApoE4 mice compared to mice with other ApoE genotypes (Bennett et al., 2013). However, injury outcomes have been reported to worsen with time in human ApoE4 expressing mice so evaluation of Aβ and tau pathology at further time points are required (Sabo et al., 2000). Interestingly, a recent transcriptomic study addressing this question observed no ApoE isoform specific changes in the transcriptome post-injury in human ApoE expressing mice (Castranio et al., 2017). However, the isoform specific effects may also be brain-region dependent (Ezra et al., 2003) with gene expression changes observed in an isoform-dependent manner to a larger extent in the hippocampus (Crawford et al., 2009). Some epidemiological studies have found no additional risk in developing AD conferred by ApoE4 after TBI with one study indicating age-dependent effects of ApoE in determining injury outcome (Teasdale et al., 2005) suggesting the need for further studies elucidating the role of ApoE in determining injury outcome as well as in mediating neurodegenerative processes post-injury (O’Meara et al., 1997; Chamelian et al., 2004).
As it relates to the use of in vitro models, recent studies have demonstrated the utility of hiPSC-based platforms combined with powerful gene editing techniques such as CRISPR/Cas9 in investigating the contribution of genetic risk factors to disease onset and progression. With respect to APOE, isogenic hiPSCs to investigate the mechanisms by which APOE4 increases and APOE2 decreases AD risk (Lin et al., 2018; Wang et al., 2018; Brookhouser et al., 2021; Martens et al., 2021; Sienski et al., 2021) can be used in conjunction with in vitro injury models to determine the combinatorial effect of APOE isoforms and cell injury on the manifestation of AD-related phenotypes. In addition, numerous genome-wide association studies (GWAS) studies have identified several risk factors associated with altered probability of AD onset (Bettens et al., 2010). In fact, some of these genes, such as BDNF, IL-1, and p53, have also been identified to influence clinical outcomes post-TBI (Dardiotis et al., 2010; Cortes and Pera, 2021; Zeiler et al., 2021). In addition, many of the genetic risk factors that affect clinical outcomes post-TBI play critical roles in pathways (e.g., inflammation, microglia activation, neurotransmitter synthesis, synaptic formation) that are dysregulated in AD (Dardiotis et al., 2010; Cortes and Pera, 2021; Zeiler et al., 2021). Moving forward, hiPSC-based isogenic models employed with in vitro injury systems can be used to investigate the influence of these additional genetic risk factors on the development of AD-related pathologies post-injury.
While there is some conflicting evidence for TBI-induced AD in epidemiological studies, clinical studies and animal models suggest a strong link between the two. Pathologies observed in AD including Aβ deposition, hyperphosphorylated tau and persistent inflammation have been observed in a fraction of TBI patients as well as in animal models. The mechanisms underlying the development of such pathologies are yet to be elucidated—injury induced amyloidogenic processing of APP, dysregulation of kinases phosphorylating tau and chronic inflammation mediated by microglia are some of the major avenues currently being explored. While transgenic rodent models have lent valuable insight into these mechanisms, several in vitro models developed to mimic aspects of traumatic injury could be leveraged to further probe the link between TBI and AD. Nonetheless, as the famous statistician George Box stated “All models are wrong, but some are useful.” Thus, moving forward researchers could utilize the complementary strengths of in vivo and in vitro systems to address the underlying causes of TBI-induced AD and identify potentially novel therapeutic targets.
GS and DB collected, analyzed, and interpreted current literature, and wrote the manuscript. Both authors contributed to the article and approved the submitted version.
This study was funded by the National Institutes of Health-National Institute on Aging (5R21AG056706).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acosta, S. A., Tajiri, N., Sanberg, P. R., Kaneko, Y., and Borlongan, C. V. (2017). Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J. Cell. Physiol. 232, 665–677. doi: 10.1002/jcp.25629
Albayram, O., Kondo, A., Mannix, R., Smith, C., Tsai, C.-Y., Li, C., et al. (2017). Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 8:1000. doi: 10.1038/s41467-017-01068-4
Alzheimer’s Association (2021). Alzheimer’s Disease Facts and Figures. Chicago, IL: Alzheimer’s Association.
Arun, P., Oguntayo, S., Albert, S. V., Gist, I., Wang, Y., Nambiar, M. P., et al. (2015). Acute decrease in alkaline phosphatase after brain injury: a potential mechanism for tauopathy. Neurosci. Lett. 609, 152–158. doi: 10.1016/j.neulet.2015.10.036
Bales, K. R., Verina, T., Dodel, R. C., Du, Y., Altstiel, L., Bender, M., et al. (1997). Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat. Genet. 17, 263–264. doi: 10.1038/ng1197-263
Bar-Kochba, E., Scimone, M. T., Estrada, J. B., and Franck, C. (2016). Strain and rate-dependent neuronal injury in a 3D in vitro compression model of traumatic brain injury. Sci. Rep. 6:30550. doi: 10.1038/srep30550
Barroeta-Espar, I., Weinstock, L. D., Perez-Nievas, B. G., Meltzer, A. C., Siao Tick Chong, M., Amaral, A. C., et al. (2019). Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 121, 327–337. doi: 10.1016/j.nbd.2018.10.009
Baumann, K., Mandelkow, E.-M., Biernat, J., Piwnica-Worms, H., and Mandelkow, E. (1993). Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 336, 417–424. doi: 10.1016/0014-5793(93)80849-p
Bennett, E. R., Reuter-Rice, K., and Laskowitz, D. T. (2016). “Genetic influences in traumatic brain injury,” in Translational Research in Traumatic Brain Injury, eds D. Laskowitz and G. Grant (Boca Raton, FL: CRC Press/Taylor and Francis Group).
Bennett, R. E., Esparza, T. J., Lewis, H. A., Kim, E., Donald, C. L. M., Sullivan, P. M., et al. (2013). Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-A immunoreactivity after traumatic brain injury in 3xTG-AD mice. J. Neuropathol. Exp. Neurol. 72:8. doi: 10.1097/NEN.0b013e31828e24ab
Bettens, K., Sleegers, K., and Van Broeckhoven, C. (2010). Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum. Mol. Genet. 19, R4–R11. doi: 10.1093/hmg/ddq142
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178. doi: 10.1073/pnas.0308512100
Bramlett, H. M., Kraydieh, S., Green, E. J., and Dietrich, W. D. (1997). Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in Rats. J Neuropathol. Exp. Neurol. 56, 1132–1141. doi: 10.1097/00005072-199710000-00007
Braun, N. J., Yao, K. R., Alford, P. W., and Liao, D. (2020). Mechanical injuries of neurons induce tau mislocalization to dendritic spines and tau-dependent synaptic dysfunction. Proc. Natl. Acad. Sci. U.S.A. 117, 29069–29079. doi: 10.1073/pnas.2008306117
Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G.-Q., Xu, Q., et al. (2004). Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004
Breunig, J., Guillot-Sestier, M.-V., and Town, T. (2013). Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 5:26.
Brizuela, M., Blizzard, C. A., Chuckowree, J. A., Pitman, K. A., Young, K. M., and Dickson, T. (2017). Mild traumatic brain injury leads to decreased inhibition and a differential response of calretinin positive interneurons in the injured cortex. J. Neurotrauma 34, 2504–2517. doi: 10.1089/neu.2017.4977
Brody, D. L., and Holtzman, D. M. (2006). Morris water maze search strategy analysis in PDAPP mice before and after experimental traumatic brain injury. Exp. Neurol. 197, 330–340. doi: 10.1016/j.expneurol.2005.10.020
Brookhouser, N., Raman, S., Frisch, C., Srinivasan, G., and Brafman, D. A. (2021). APOE2 mitigates disease-related phenotypes in an isogenic hiPSC-based model of Alzheimer’s disease. Mol. Psychiatry 26, 5715–5732. doi: 10.1038/s41380-021-01076-3
Brotfain, E., Gruenbaum, S. E., Boyko, M., Kutz, R., Zlotnik, A., and Klein, M. (2016). Neuroprotection by estrogen and progesterone in traumatic brain injury and spinal cord injury. Curr. Neuropharmacol. 14, 641–653. doi: 10.2174/1570159x14666160309123554
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 33, 95–130. doi: 10.1016/s0165-0173(00)00019-9
Byrnes, K. R., Loane, D. J., Stoica, B. A., Zhang, J., and Faden, A. I. (2012). Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflamm. 9:43. doi: 10.1186/1742-2094-9-43
Carter, M., and Shieh, J. (2015). “Chapter 14 – cell culture techniques,” in Guide to Research Techniques in Neuroscience (Second Edition), eds M. Carter and J. Shieh (Cambridge, MA: Academic Press), 295–310. doi: 10.1016/B978-0-12-800511-8.00014-9
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Castranio, E. L., Mounier, A., Wolfe, C. M., Nam, K. N., Fitz, N. F., Letronne, F., et al. (2017). Gene co-expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol. Dis. 105, 1–14. doi: 10.1016/j.nbd.2017.05.006
Cater, H. L., Gitterman, D., Davis, S. M., Benham, C. D., Morrison, B., and Sundstrom, L. E. (2007). Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J. Neurochem. 101, 434–447. doi: 10.1111/j.1471-4159.2006.04379.x
Chamelian, L., Reis, M., and Feinstein, A. (2004). Six-month recovery from mild to moderate traumatic brain injury: the role of APOE-ε4 allele. Brain 127, 2621–2628. doi: 10.1093/brain/awh296
Chaves, R. S., Tran, M., Holder, A. R., Balcer, A. M., Dickey, A. M., Roberts, E. A., et al. (2021). Amyloidogenic processing of amyloid precursor protein drives stretch-induced disruption of axonal transport in hiPSC-derived neurons. J. Neurosci. 41, 10034–10053. doi: 10.1523/JNEUROSCI.2553-20.2021
Chen, H., Richard, M., Sandler, D. P., Umbach, D. M., and Kamel, F. (2007). Head injury and amyotrophic lateral sclerosis. Am. J. Epidemiol. 166, 810–816.
Chen, X.-H., Johnson, V. E., Uryu, K., Trojanowski, J. Q., and Smith, D. H. A. (2009). Lack of amyloid β plaques despite persistent accumulation of amyloid β in axons of long-term survivors of traumatic brain Injury. Brain Pathol. 19, 214–223. doi: 10.1111/j.1750-3639.2008.00176.x
Chen, X.-H., Siman, R., Iwata, A., Meaney, D. F., Trojanowski, J. Q., and Smith, D. H. (2004). Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371. doi: 10.1016/s0002-9440(10)63303-2
Cheng, W. H., Martens, K. M., Bashir, A., Cheung, H., Stukas, S., Gibbs, E., et al. (2019). CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimer’s Res. Ther. 11:6. doi: 10.1186/s13195-018-0461-0
Cheng, W. H., Stukas, S., Martens, K. M., Namjoshi, D. R., Button, E. B., Wilkinson, A., et al. (2018). Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp. Neurol. 301, 26–38. doi: 10.1016/j.expneurol.2017.12.007
Chuckowree, J. A., and Vickers, J. C. (2003). Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron axons In Vitro. J. Neurosci. 23, 3715–3725. doi: 10.1523/JNEUROSCI.23-09-03715.2003
Ciallella, J. R., Ikonomovic, M. D., Paljug, W. R., Wilbur, Y. I., Dixon, C. E., Kochanek, P. M., et al. (2002). Changes in expression of amyloid precursor protein and interleukin-1β after experimental traumatic brain injury in rats. J. Neurotrauma 19, 1555–1567. doi: 10.1089/089771502762300229
Clavaguera, F., Hench, J., Goedert, M., and Tolnay, M. (2015). Invited review: prion-like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 41, 47–58. doi: 10.1111/nan.12197
Clinton, J., Ambler, M. W., and Roberts, G. W. (1991). Post–traumatic Alzheimer’s disease: preponderance of a single plaque type. Neuropathol. Appl. Neurobiol. 17, 69–74. doi: 10.1111/j.1365-2990.1991.tb00695.x
Collins, J. M., King, A. E., Woodhouse, A., Kirkcaldie, M. T. K., and Vickers, J. C. (2015). The effect of focal brain injury on beta-amyloid plaque deposition, inflammation and synapses in the APP/PS1 mouse model of Alzheimer’s disease. Exp. Neurol. 267, 219–229. doi: 10.1016/j.expneurol.2015.02.034
Collins-Praino, L. E., and Corrigan, F. (2017). Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav. Immunity 60, 369–382. doi: 10.1016/j.bbi.2016.09.027
Corrigan, F., Vink, R., Blumbergs, P. C., Masters, C. L., Cappai, R., and van den Heuvel, C. (2012a). Characterisation of the effect of knockout of the amyloid precursor protein on outcome following mild traumatic brain injury. Brain Res. 1451, 87–99. doi: 10.1016/j.brainres.2012.02.045
Corrigan, F., Vink, R., Blumbergs, P. C., Masters, C. L., Cappai, R., and Heuvel, C. (2012b). sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J. Neurochem. 122, 208–220. doi: 10.1111/j.1471-4159.2012.07761.x
Cortes, D., and Pera, M. F. (2021). The genetic basis of inter-individual variation in recovery from traumatic brain injury. Npj Regen. Med. 6, 1–9. doi: 10.1038/s41536-020-00114-y
Crane, P. K., Gibbons, L. E., Dams-O’Connor, K., Trittschuh, E., Leverenz, J. B., Keene, C. D., et al. (2016). Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. 73, 1062–1069. doi: 10.1001/jamaneurol.2016.1948
Crawford, F., Wood, M., Ferguson, S., Mathura, V., Gupta, P., Humphrey, J., et al. (2009). Apolipoprotein E-genotype dependent hippocampal and cortical responses to traumatic brain injury. Neuroscience 159, 1349–1362. doi: 10.1016/j.neuroscience.2009.01.033
Cribbs, D. H., Chen, L., Cotman, C. W., and LaFerla, F. M. (1996). Injury induces presenilin-1 gene expression in mouse brain. Neuroreport 7, 1773–1776. doi: 10.1097/00001756-199607290-00016
Cullen, D. K., Simon, C. M., and LaPlaca, M. C. (2007). Strain rate-dependent induction of reactive astrogliosis and cell death in three-dimensional neuronal–astrocytic co-cultures. Brain Res. 1158, 103–115. doi: 10.1016/j.brainres.2007.04.070
Cullen, D. K., Vernekar, V. N., and LaPlaca, M. C. (2011). Trauma-induced plasmalemma disruptions in three-dimensional neural cultures are dependent on strain modality and rate. J. Neurotrauma 28, 2219–2233. doi: 10.1089/neu.2011.1841
Dams-O’Connor, K., Guetta, G., Hahn-Ketter, A. E., and Fedor, A. (2016). Traumatic brain injury as a risk factor for Alzheimer’s disease: current knowledge and future directions. Neurodegener. Dis. Manag. 6, 417–429. doi: 10.2217/nmt-2016-0017
Dardiotis, E., Fountas, K. N., Dardioti, M., Xiromerisiou, G., Kapsalaki, E., Tasiou, A., et al. (2010). Genetic association studies in patients with traumatic brain injury. Neurosurgical Focus 28:E9. doi: 10.3171/2009.10.FOCUS09215
Daviglus, M. L., Plassman, B. L., Pirzada, A., Bell, C. C., Bowen, P. E., Burke, J. R., et al. (2011). Risk Factors and preventive interventions for Alzheimer disease: state of the science. Arch. Neurol. 68, 1185–1190. doi: 10.1001/archneurol.2011.100
DeRidder, M. N., Simon, M. J., Siman, R., Auberson, Y. P., Raghupathi, R., and Meaney, D. F. (2006). Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol. Dis. 22, 165–176. doi: 10.1016/j.nbd.2005.10.011
DeWitt, D. S., Hawkins, B. E., Dixon, C. E., Kochanek, P. M., Armstead, W., Bass, C. R., et al. (2018). Pre-clinical testing of therapies for traumatic brain injury. J. Neurotrauma 35, 2737–2754. doi: 10.1089/neu.2018.5778
Dollé, J.-P., Iii, B. M., Schloss, S., and Yarmush, L. (2013). An organotypic uniaxial strain model using microfluidics. Lab Chip 13, 432–442. doi: 10.1039/c2lc41063j
Dollé, J.-P., Morrison, B., Schloss, R. S., and Yarmush, M. L. (2014). Brain-on-a-chip microsystem for investigating traumatic brain injury: axon diameter and mitochondrial membrane changes play a significant role in axonal response to strain injuries. Technology 02, 106–117. doi: 10.1142/S2339547814500095
Dolmetsch, R., and Geschwind, D. H. (2011). The human brain in a dish: the promise of iPSC-derived neurons. Cell 145, 831–834. doi: 10.1016/j.cell.2011.05.034
Drewes, G., Lichtenberg-Kraag, B., Döring, F., Mandelkow, E. M., Biernat, J., Goris, J., et al. (1992). Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. The EMBO Journal 11, 2131–2138.
Dubois, B., Padovani, A., Scheltens, P., Rossi, A., and Dell’Agnello, G. (2016). Timely diagnosis for Alzheimer’s disease: a literature review on benefits and challenges. J. Alzheimer’s Dis. 49, 617–631. doi: 10.3233/JAD-150692
Duff, K., and Suleman, F. (2004). Transgenic mouse models of Alzheimer’s disease: how useful have they been for therapeutic development? Brief. Funct. Genomics 3, 47–59. doi: 10.1093/bfgp/3.1.47
Edwards, G., Zhao, J., Dash, P. K., Soto, C., and Moreno-Gonzalez, I. (2019). Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma 37, 80–92. doi: 10.1089/neu.2018.6348
Effgen, G. B., Hue, C. D., Vogel, E. I., Panzer, M. B., Meaney, D. F., and Bass, C. (2012). A multiscale approach to blast neurotrauma modeling: part ii: methodology for inducing blast injury to in vitro models. Front. Neurol. 3:23. doi: 10.3389/fneur.2012.00023
Effgen, G. B., Vogel, E. W., Lynch, K. A., Lobel, A., Hue, C. D., Meaney, D. F., et al. (2014). Isolated primary blast alters neuronal function with minimal cell death in organotypic hippocampal slice cultures. J. Neurotrauma 31, 1202–1210. doi: 10.1089/neu.2013.3227
Eftekharzadeh, B., Daigle, J. G., Kapinos, L. E., Coyne, A., Schiantarelli, J., Carlomagno, Y., et al. (2018). Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99, 925–940.e7.
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Ezra, Y., Oron, L., Moskovich, L., Roses, A. D., Beni, S. M., Shohami, E., et al. (2003). Apolipoprotein e4 decreases whereas apolipoprotein e3 increases the level of secreted amyloid precursor protein after closed head injury. Neuroscience 121, 315–325. doi: 10.1016/s0306-4522(03)00436-6
Fleminger, S., Oliver, D. L., Lovestone, S., Rabe-Hesketh, S., and Giora, A. (2003). Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 74, 857–862. doi: 10.1136/jnnp.74.7.857
Friedman, G., Froom, P., Sazbon, L., Grinblatt, I., Shochina, M., Tsenter, J., et al. (1999). Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology 52, 244–248. doi: 10.1212/wnl.52.2.244
Gabbita, S. P., Scheff, S. W., Menard, R. M., Roberts, K., Fugaccia, I., and Zemlan, F. P. (2005). Cleaved-Tau: a biomarker of neuronal damage after traumatic brain injury. J. Neurotrauma 22, 83–94. doi: 10.1089/neu.2005.22.83
Gardner, R. C., Byers, A. L., Barnes, D. E., Li, Y., Boscardin, J., and Yaffe, K. (2018). Mild TBI and risk of Parkinson disease: a chronic effects of neurotrauma consortium study. Neurology 90, e1771–e1779. doi: 10.1212/WNL.0000000000005522
Geddes, D. M., Cargill, R. S., and LaPlaca, M. C. (2003). Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J. Neurotrauma 20, 1039–1049. doi: 10.1089/089771503770195885
Gentleman, S. M., Greenberg, B. D., Savage, M. J., Noori, M., Newman, S. J., Roberts, G. W., et al. (1997). β42 is the predominant form of amyloid b-protein in the brains of short-term survivors of head injury. Neuroreport 8, 1519–1522. doi: 10.1097/00001756-199704140-00039
Gentleman, S. M., Nash, M. J., Sweeting, C. J., Graham, D. I., and Roberts, G. W. (1993). β-Amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci. Lett. 160, 139–144.
Gerson, J., Castillo-Carranza, D. L., Sengupta, U., Bodani, R., Prough, D. S., DeWitt, D. S., et al. (2016). Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in htau mice. J. Neurotrauma 33, 2034–2043. doi: 10.1089/neu.2015.4262
Ghaffari, L. T., Starr, A., Nelson, A. T., and Sattler, R. (2018). Representing diversity in the dish: using patient-derived in vitro models to recreate the heterogeneity of neurological disease. Front. Neurosci. 12:56. doi: 10.3389/fnins.2018.00056
Goldgaber, D., Harris, H. W., Hla, T., Maciag, T., Donnelly, R. J., Jacobsen, J. S., et al. (1989). Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 86, 7606–7610. doi: 10.1073/pnas.86.19.7606
Goldstein, L. S. B., Reyna, S., and Woodruff, G. (2015). Probing the secrets of Alzheimer’s disease using human-induced pluripotent stem cell technology. Neurotherapeutics 12, 121–125. doi: 10.1007/s13311-014-0326-6
Gordon, J., Amini, S., and White, M. K. (2013). “General overview of neuronal cell culture,” in Neuronal Cell Culture: Methods and Protocols, eds S. Amini and M. K. White (Totowa, NJ: Humana Press), 1–8. doi: 10.1007/978-1-62703-640-5_1
Gorgoraptis, N., Li, L. M., Whittington, A., Zimmerman, K. A., Maclean, L. M., McLeod, C., et al. (2019). In vivo detection of cerebral tau pathology in long-term survivors of traumatic brain injury. Sci. Transl. Med. 11, eaaw1993. doi: 10.1126/scitranslmed.aaw1993
Griciuc, A., and Tanzi, R. E. (2021). The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 34, 228–236. doi: 10.1097/WCO.0000000000000911
Guo, L., Zhong, M. B., Zhang, L., Zhang, B., and Cai, D. (2022). Sex differences in Alzheimer’s disease: insights from the multiomics landscape. Biological Psychiatry 91, 61–71. doi: 10.1016/j.biopsych.2021.02.968
Guo, Z., Cupples, L. A., Kurz, A., Auerbach, S. H., Volicer, L., Chui, H., et al. (2000). Head injury and the risk of AD in the MIRAGE study. Neurology 54, 1316–1323. doi: 10.1212/wnl.54.6.1316
Gupte, R. P., Brooks, W. M., Vukas, R. R., Pierce, J. D., and Harris, J. L. (2019). Sex differences in traumatic brain injury: what we know and what we should know. J. Neurotrauma 36, 3063–3091. doi: 10.1089/neu.2018.6171
Haass, C., Hung, A. Y., Schlossmacher, M. G., Teplow, D. B., and Selkoe, D. J. (1993). beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 268, 3021–3024.
Hartley, D. M., Walsh, D. M., Ye, C. P., Diehl, T., Vasquez, S., Vassilev, P. M., et al. (1999). Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999
Hartman, R. E., Laurer, H., Longhi, L., Bales, K. R., Paul, S. M., McIntosh, T. K., et al. (2002). Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer’s disease. J. Neurosci. 22, 10083–10087. doi: 10.1523/JNEUROSCI.22-23-10083.2002
Hawkins, B. E., Krishnamurthy, S., Castillo-Carranza, D. L., Sengupta, U., Prough, D. S., Jackson, G. R., et al. (2013). Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible Link between traumatic brain injury and sporadic tauopathies. J. Biol. Chem. 288, 17042–17050. doi: 10.1074/jbc.M113.472746
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hong, Y. J., and Do, J. T. (2019). Neural lineage differentiation from pluripotent stem cells to mimic human brain tissues. Front. Bioeng. Biotechnol. 7:400. doi: 10.3389/fbioe.2019.00400
Hoshino, S., Tamaoka, A., Takahashi, M., Kobayashi, S., Furukawa, T., Oaki, Y., et al. (1998). Emergence of immunoreactivities for phosphorylated tau and amyloid-β protein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9, 1879–1883. doi: 10.1097/00001756-199806010-00039
Hosmane, S., Fournier, A., Wright, R., Rajbhandari, L., Siddique, R., Hong Yang, I., et al. (2011). Valve-based microfluidic compression platform: single axon injury and regrowth. Lab Chip 11, 3888–3895. doi: 10.1039/c1lc20549h
Huber, B. R., Meabon, J. S., Martin, T. J., Mourad, P. D., Bennett, R., Kraemer, B. C., et al. (2013). Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J. Alzheimer’s Dis. 37, 309–323. doi: 10.3233/JAD-130182
Hue, C. D., Cao, S., Haider, S. F., Vo, K. V., Effgen, G. B., Vogel, E., et al. (2013). Blood-brain barrier dysfunction after primary blast injury in vitro. J. Neurotrauma 30, 1652–1663. doi: 10.1089/neu.2012.2773
Huo, J., Wei, F., Cai, C., Lyn-Cook, B., and Pang, L. (2019). Sex-related differences in drug-induced QT prolongation and torsades de pointes: a new model system with human iPSC-CMs. Toxicol. Sci. 167, 360–374.
Ikonomovic, M. D., Uryu, K., Abrahamson, E. E., Ciallella, J. R., Trojanowski, J. Q., Lee, V. M.-Y., et al. (2004). Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190, 192–203. doi: 10.1016/j.expneurol.2004.06.011
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Ittner, L. M., and Götz, J. (2011). Amyloid-β and tau — a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 67–72.
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Iwata, A., Stys, P. K., Wolf, J. A., Chen, X.-H., Taylor, A. G., Meaney, D. F., et al. (2004). Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J. Neurosci. 24:4605. doi: 10.1523/JNEUROSCI.0515-03.2004
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. doi: 10.1093/brain/aws322
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans: long-term AD-like pathology after single TBI. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Jones, V. C., Atkinson-Dell, R., Verkhratsky, A., and Mohamet, L. (2017). Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 8:e2696.
Kang, W. H., Cao, W., Graudejus, O., Patel, T. P., Wagner, S., Meaney, D. F., et al. (2014). Alterations in hippocampal network activity after in vitro traumatic brain injury. J. Neurotrauma 32, 1011–1019. doi: 10.1089/neu.2014.3667
Katzman, R., Aronson, M., Fuld, P., Kawas, C., Brown, T., Morgenstern, H., et al. (1989). Development of dementing illnesses in an 80-year-old volunteer cohort. Ann. Neurol. 25, 317–324. doi: 10.1002/ana.410250402
Kim, W., and Hecht, M. H. (2005). Sequence determinants of enhanced amyloidogenicity of alzheimer Aβ42 peptide relative to Aβ40. J. Biol. Chem. 280, 35069–35076.
Kim, Y. H., Choi, S. H., D’Avanzo, C., Hebisch, M., Sliwinski, C., Bylykbashi, E., et al. (2015). A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat. Protoc. 10, 985–1006. doi: 10.1038/nprot.2015.065
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Intervent. 4, 575–590.
Kojro, E., and Fahrenholz, F. (2005). “The non-amyloidogenic pathway: structure and function of α-secretases,” in Alzheimer’s Disease: Cellular and Molecular Aspects of Amyloid β, eds J. R. Harris and F. Fahrenholz (Berlin: Springer US), 105–127. doi: 10.1007/0-387-23226-5_5
Kokiko-Cochran, O. N., Saber, M., Puntambekar, S., Bemiller, S. M., Katsumoto, A., Lee, Y.-S., et al. (2017). Traumatic brain injury in hTau model mice: enhanced acute macrophage response and altered long-term recovery. J. Neurotrauma 35, 73–84. doi: 10.1089/neu.2017.5203
Kokiko-Cochran, O., Ransohoff, L., Veenstra, M., Lee, S., Saber, M., Sikora, M., et al. (2015). Altered neuroinflammation and behavior after traumatic brain injury in a mouse model of Alzheimer’s disease. J. Neurotrauma 33, 625–640. doi: 10.1089/neu.2015.3970
Kumar, A., Barrett, J. P., Alvarez-Croda, D.-M., Stoica, B. A., Faden, A. I., and Loane, D. J. (2016). NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immunity 58, 291–309. doi: 10.1016/j.bbi.2016.07.158
Kumar, R. G., Diamond, M. L., Boles, J. A., Berger, R. P., Tisherman, S. A., Kochanek, P. M., et al. (2015). Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav. Immunity 45, 253–262. doi: 10.1016/j.bbi.2014.12.021
Kurowski, B. G., Treble-Barna, A., Pilipenko, V., Wade, S. L., Yeates, K. O., Taylor, H. G., et al. (2019). Genetic influences on behavioral outcomes after childhood TBI: a novel systems biology-informed approach. Frontiers in Genetics 10:481. doi: 10.3389/fgene.2019.00481
LaDu, M. J., Shah, J. A., Reardon, C. A., Getz, G. S., Bu, G., Hu, J., et al. (2001). Apolipoprotein E and apolipoprotein E receptors modulate Aβ-induced glial neuroinflammatory responses. Neurochem. Int. 39, 427–434. doi: 10.1016/s0197-0186(01)00050-x
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509.
Lai, J. D., Berlind, J. E., Fricklas, G., Maria, N. S., Jacobs, R., Yu, V., et al. (2020). A model of traumatic brain injury using human iPSC-derived cortical brain organoids. bioRxiv [Preprint]. doi: 10.1101/2020.07.05.180299
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
LaPlaca, M. C., and Thibault, L. E. (1997). Anin vitro traumatic injury model to examine the response of neurons to a hydrodynamically-induced deformation. Ann. Biomed. Eng. 25, 665–677. doi: 10.1007/BF02684844
LaPlaca, M. C., Cullen, D. K., McLoughlin, J. J., and Cargill, R. S. (2005). High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. J. Biomech. 38, 1093–1105. doi: 10.1016/j.jbiomech.2004.05.032
Launer, L. J., Andersen, K., Dewey, M. E., Letenneur, L., Ott, A., Amaducci, L. A., et al. (1999). Rates and risk factors for dementia and Alzheimer’s disease: results from EURODEM pooled analyses. Neurology 52:78. doi: 10.1212/wnl.52.1.78
Laws, K. R., Irvine, K., and Gale, T. M. (2018). Sex differences in Alzheimer’s disease. Curr. Opin. Psychiatry 31, 133–139.
Lecca, D., Bader, M., Tweedie, D., Hoffman, A. F., Jung, Y. J., Hsueh, S.-C., et al. (2019). (-)-Phenserine and the prevention of pre-programmed cell death and neuroinflammation in mild traumatic brain injury and Alzheimer’s disease challenged mice. Neurobiol. Dis. 130:104528. doi: 10.1016/j.nbd.2019.104528
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 5:37. doi: 10.1186/1742-2094-5-37
Lesné, S., Ali, C., Gabriel, C., Croci, N., MacKenzie, E. T., Glabe, C. G., et al. (2005). Receptor activation inhibits α-secretase and promotes neuronal amyloid-β production. J. Neurosci. 25, 9367–9377.
Lewén, A., Li, G., Nilsson, P., Olsson, Y., and Hillered, L. (1995). Traumatic brain injury in rat produces changes of beta-amyloid precursor protein immunoreactivity. Neuroreport 6, 357–360. doi: 10.1097/00001756-199501000-00032
Li, Y., Li, C., Gan, C., Zhao, K., Chen, J., Song, J., et al. (2019). A precise, controllable in vitro model for diffuse axonal injury through uniaxial stretch injury. Front. Neurosci. 13:1063. doi: 10.3389/fnins.2019.01063
Lin, Y.-T., Seo, J., Gao, F., Feldman, H. M., Wen, H.-L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7.
Lindsay, J., Laurin, D., Verreault, R., Hébert, R., Helliwell, B., Hill, G. B., et al. (2002). Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am. J. Epidemiol. 156, 445–453. doi: 10.1093/aje/kwf074
Liu, C.-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms, and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Loane, D. J., Kumar, A., Stoica, B. A., Cabatbat, R., and Faden, A. I. (2014a). Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 73, 14–29. doi: 10.1097/NEN.0000000000000021
Loane, D. J., Pocivavsek, A., Moussa, C. E.-H., Thompson, R., Matsuoka, Y., Faden, A. I., et al. (2009). Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 15, 377–379. doi: 10.1038/nm.1940
Loane, D. J., Stoica, B. A., Tchantchou, F., Kumar, A., Barrett, J. P., Akintola, T., et al. (2014b). Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics 11, 857–869. doi: 10.1007/s13311-014-0298-6
Loane, D. J., Washington, P. M., Vardanian, L., Pocivavsek, A., Hoe, H.-S., Duff, K. E., et al. (2011). Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J. Neurotrauma 28, 225–236. doi: 10.1089/neu.2010.1595
Lock, R., Al Asafen, H., Fleischer, S., Tamargo, M., Zhao, Y., Radisic, M., et al. (2021). A framework for developing sex-specific engineered heart models. Nat. Rev. Mater. 1–19. doi: 10.1038/s41578-021-00381-1
Lööv, C., Shevchenko, G., Geeyarpuram Nadadhur, A., Clausen, F., Hillered, L., Wetterhall, M., et al. (2013). Identification of injury specific proteins in a cell culture model of traumatic brain injury. PLoS One 8:e55983. doi: 10.1371/journal.pone.0055983
Lusardi, T. A., Wolf, J. A., Putt, M. E., Smith, D. H., and Meaney, D. F. (2004). Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J. Neurotrauma 21, 61–72. doi: 10.1089/089771504772695959
Lye, T. C., and Shores, E. A. (2000). Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol. Rev. 10, 115–129. doi: 10.1023/a:1009068804787
Ma, C., Wu, X., Shen, X., Yang, Y., Chen, Z., Sun, X., et al. (2019). Sex differences in traumatic brain injury: a multi-dimensional exploration in genes, hormones, cells, individuals, and society. Chin. Neurosurg. J. 5:24. doi: 10.1186/s41016-019-0173-8
Magdesian, M. H., Sanchez, F. S., Lopez, M., Thostrup, P., Durisic, N., Belkaid, W., et al. (2012). Atomic force microscopy reveals important differences in axonal resistance to injury. Biophys. J. 103, 405–414. doi: 10.1016/j.bpj.2012.07.003
Mandelkow, E.-M., Drewes, G., Biernat, J., Gustke, N., Lint, J. V., Vandenheede, J. R., et al. (1992). Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 314, 315–321. doi: 10.1016/0014-5793(92)81496-9
Mandolesi, G., Madeddu, F., Bozzi, Y., Maffei, L., and Ratto, G. M. (2004). Acute physiological response of mammalian central neurons to axotomy: ionic regulation and electrical activity. FASEB J. 18, 1934–1936. doi: 10.1096/fj.04-1805fje
Mannix, R. C., and Whalen, M. J. (2012). Traumatic brain injury, microglia, and beta amyloid. Int. J. Alzheimer’s Dis. 2012:e608732. doi: 10.1155/2012/608732
Marklund, N., Blennow, K., Zetterberg, H., Ronne-Engström, E., Enblad, P., and Hillered, L. (2009). Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury: Clinical article. J. Neurosurg. 110, 1227–1237. doi: 10.3171/2008.9.JNS08584
Martens, Y. A., Xu, S., Tait, R., Li, G., Zhao, X. C., Lu, W., et al. (2021). Generation and validation of APOE knockout human iPSC-derived cerebral organoids. STAR Protoc. 2:100571. doi: 10.1016/j.xpro.2021.100571
McCrory, P., Zazryn, T., and Cameron, P. (2007). The Evidence for Chronic Traumatic Encephalopathy in boxing. Sports Med. 37, 467–476. doi: 10.2165/00007256-200737060-00001
McDonald, D. R., Brunden, K. R., and Landreth, G. E. (1997). Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J. Neurosci. 17:2284. doi: 10.1523/JNEUROSCI.17-07-02284.1997
McKee, A. C., Stein, T. D., Nowinski, C. J., Stern, R. A., Daneshvar, D. H., Alvarez, V. E., et al. (2013). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64. doi: 10.1093/brain/aws307
Meda, L., Cassatella, M. A., Szendrei, G. I., Otvos, L., Baron, P., Villalba, M., et al. (1995). Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 374, 647–650.
Mehta, K. M., Ott, A., Kalmijn, S., Slooter, A. J. C., van Duijn, C. M., Hofman, A., et al. (1999). Head trauma and risk of dementia and Alzheimer’s disease. Neurology 53:1959. doi: 10.1212/wnl.53.9.1959
Miszczuk, D., Dębski, K. J., Tanila, H., Lukasiuk, K., and Pitkänen, A. (2016). Traumatic brain injury increases the expression of Nos1, Aβ clearance, and epileptogenesis in APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. 53, 7010–7027. doi: 10.1007/s12035-015-9578-3
Mollayeva, T., Mollayeva, S., and Colantonio, A. (2018). Traumatic brain injury: sex, gender and intersecting vulnerabilities. Nat. Rev. Neurol. 14, 711–722. doi: 10.1038/s41582-018-0091-y
Monnerie, H., Tang-Schomer, M. D., Iwata, A., Smith, D. H., Kim, H. A., and Le Roux, P. D. (2010). Dendritic alterations after dynamic axonal stretch injury in vitro. Exp. Neurol. 224, 415–423. doi: 10.1016/j.expneurol.2010.05.001
Morrison, B., Cater, H. L., Benham, C. D., and Sundstrom, L. E. (2006). An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J. Neurosci. Methods 150, 192–201. doi: 10.1016/j.jneumeth.2005.06.014
Morrison, B., Elkin, B. S., Dollé, J.-P., and Yarmush, M. L. (2011). In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 13, 91–126.
Muccigrosso, M. M., Ford, J., Benner, B., Moussa, D., Burnsides, C., Fenn, A. M., et al. (2016). Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav. Immunity 54, 95–109. doi: 10.1016/j.bbi.2016.01.009
Mukhin, A. G., Ivanova, S. A., Knoblach, S. M., and Faden, A. I. (1997). New in vitro model of traumatic neuronal injury: evaluation of secondary injury and glutamate receptor-mediated neurotoxicity. J. Neurotrauma 14, 651–663. doi: 10.1089/neu.1997.14.651
Murai, H., Pierce, J. E. S., Raghupathi, R., Smith, D. H., Saatman, K. E., Trojanowski, J. Q., et al. (1998). Twofold overexpression of human β-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J. Comp. Neurol. 392, 428–438.
Murakami, N., Yamaki, T., Iwamoto, Y., Sakakibara, T., Kobori, N., Fushiki, S., et al. (1998). Experimental brain injury induces expression of amyloid precursor protein, which may be related to neuronal loss in the hippocampus. J. Neurotrauma 15, 993–1003. doi: 10.1089/neu.1998.15.993
Nadler, Y., Alexandrovich, A., Grigoriadis, N., Hartmann, T., Rao, K. S. J., Shohami, E., et al. (2008). Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56, 552–567. doi: 10.1002/glia.20638
Nagendran, T., and Taylor, A. M. (2019). Unique axon-to-soma signaling pathways mediate dendritic spine loss and hyper-excitability post-axotomy. Front. Cell. Neurosci. 13:431. doi: 10.3389/fncel.2019.00431
Nakagawa, Y., Nakamura, M., Mcintosh, T. K., Rodriguez, A., Berlin, J. A., Smith, D. H., et al. (1999). Traumatic brain injury in young, amyloid-β peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Aβ deposition during aging. J. Comp. Neurol. 411, 390–398.
Nakagawa, Y., Reed, L., Nakamura, M., McIntosh, T. K., Smith, D. H., Saatman, K. E., et al. (2000). Brain Trauma in aged transgenic mice induces regression of established Aβ deposits. Exp. Neurol. 163, 244–252. doi: 10.1006/exnr.2000.7375
Nemetz, P. N., Leibson, C., Naessens, J. M., Beard, M., Kokmen, E., Annegers, J. F., et al. (1999). Traumatic brain injury and time to onset of alzheimer’s disease: a population-based study. Am. J. Epidemiol. 149, 32–40. doi: 10.1093/oxfordjournals.aje.a009724
Nicoll, J. A. R., Roberts, G. W., and Graham, D. I. (1995). Apolipoprotein E ε4 allele is associated with deposition of amyloid β-protein following head injury. Nat. Med. 1, 135–137.
Nygaard, H. B., van Dyck, C. H., and Strittmatter, S. M. (2014). Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alz. Res. Ther. 6:8. doi: 10.1186/alzrt238
O’Meara, E. S., Kukull, W. A., Sheppard, L., Bowen, J. D., McCormick, W. C., Teri, L., et al. (1997). Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am. J. Epidemiol. 146, 373–384. doi: 10.1093/oxfordjournals.aje.a009290
Ouellette, M. M., McDaniel, L. D., Wright, W. E., Shay, J. W., and Schultz, R. A. (2000). The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 9, 403–411. doi: 10.1093/hmg/9.3.403
Paci, M., Penttinen, K., Pekkanen-Mattila, M., and Koivumäki, J. T. (2021). Arrhythmia mechanisms in human induced pluripotent stem cell–derived cardiomyocytes. J. Cardiovasc. Pharmacol. 77, 300–316. doi: 10.1097/FJC.0000000000000972
Penkowa, M., Giralt, M., Carrasco, J., Hadberg, H., and Hidalgo, J. (2000). Impaired inflammatory response and increased oxidative stress and neurodegeneration after brain injury in interleukin-6-deficient mice. Glia 32, 271–285. doi: 10.1002/1098-1136(200012)32:3<271::aid-glia70>3.0.co;2-5
Pfister, B. J., Weihs, T. P., Betenbaugh, M., and Bao, G. (2003). An in vitro uniaxial stretch model for axonal injury. Ann. Biomed. Eng. 31, 589–598.
Pierce, J. E. S., Smith, D. H., Trojanowski, J. Q., and McIntosh, T. K. (1998). Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 87, 359–369. doi: 10.1016/s0306-4522(98)00142-0
Plassman, B. L., Havlik, R. J., Steffens, D. C., Helms, M. J., Newman, T. N., Drosdick, D., et al. (2000). Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 55, 1158–1166. doi: 10.1212/wnl.55.8.1158
Quintanilla, R. A., Orellana, D. I., González-Billault, C., and Maccioni, R. B. (2004). Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 295, 245–257. doi: 10.1016/j.yexcr.2004.01.002
Rabinowitz, A. R., and Levin, H. S. (2014). Cognitive sequelae of traumatic brain injury. Psychiatric Clin. North Am. 37, 1–11.
Rehman, S. U., Ahmad, A., Yoon, G.-H., Khan, M., Abid, M. N., and Kim, M. O. (2018). Inhibition of c-Jun N-terminal kinase protects against brain damage and improves learning and memory after traumatic brain injury in adult mice. Cereb. Cortex 28, 2854–2872. doi: 10.1093/cercor/bhx164
Roberts, G. W., Gentleman, S. M., Lynch, A., and Graham, D. I. (1991). beta A4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423. doi: 10.1016/0140-6736(91)92724-g
Roberts, G. W., Gentleman, S. M., Lynch, A., Murray, L., Landon, M., and Graham, D. I. (1994). Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425. doi: 10.1136/jnnp.57.4.419
Rosas-Hernandez, H., Burks, S. M., Cuevas, E., and Ali, S. F. (2019). Stretch-induced deformation as a model to study dopaminergic dysfunction in traumatic brain injury. Neurochem. Res. 44, 2546–2555. doi: 10.1007/s11064-019-02872-8
Rubenstein, R., Chang, B., Grinkina, N., Drummond, E., Davies, P., Ruditzky, M., et al. (2017). Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathol. Commun. 5:30. doi: 10.1186/s40478-017-0435-7
Sabo, T., Lomnitski, L., Nyska, A., Beni, S., Maronpot, R. R., Shohami, E., et al. (2000). Susceptibility of transgenic mice expressing human apolipoprotein E to closed head injury: The allele E3 is neuroprotective whereas E4 increases fatalities. Neuroscience 101, 879–884. doi: 10.1016/s0306-4522(00)00438-3
Salter, M. W., and Kalia, L. V. (2004). Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328. doi: 10.1038/nrn1368
Salvador, E., Burek, M., and Förster, C. Y. (2018). “An in vitro model of traumatic brain injury,” in Traumatic and Ischemic Injury: Methods and Protocols, ed. B. Tharakan (Berlin: Springer), 219–227. doi: 10.1007/978-1-4939-7526-6_17
Schaffert, J., LoBue, C., White, C. L., Chiang, H.-S., Didehbani, N., Lacritz, L., et al. (2018). Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer’s disease. Neuropsychology 32, 410–416. doi: 10.1037/neu0000423
Schofield, P. W., Tang, M., Marder, K., Bell, K., Dooneief, G., Chun, M., et al. (1997). Alzheimer’s disease after remote head injury: an incidence study. J. Neurol. Neurosurg. Psychiatry 62, 119–124. doi: 10.1136/jnnp.62.2.119
Schumann, J., Alexandrovich, G. A., Biegon, A., and Yaka, R. (2008). Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J. Neurotrauma 25, 945–957. doi: 10.1089/neu.2008.0521
Scott, G., Ramlackhansingh, A. F., Edison, P., Hellyer, P., Cole, J., Veronese, M., et al. (2016). Amyloid pathology and axonal injury after brain trauma. Neurology 86, 821–828. doi: 10.1212/WNL.0000000000002413
Scott, G., Zetterberg, H., Jolly, A., Cole, J. H., De Simoni, S., Jenkins, P. O., et al. (2018). Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 141, 459–471. doi: 10.1093/brain/awx339
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Seok, J., Warren, H. S., Cuenca, A. G., Mindrinos, M. N., Baker, H. V., Xu, W., et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U.S.A. 110:3507.
Shah, P. T., Yoon, K.-W., Xu, X. M., and Broder, L. D. (1997). Apoptosis mediates cell death following traumatic injury in rat hippocampal neurons. Neuroscience 79, 999–1004. doi: 10.1016/s0306-4522(97)00013-4
Sheng, J. G., Bora, S. H., Xu, G., Borchelt, D. R., Price, D. L., and Koliatsos, V. E. (2003). Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. doi: 10.1016/s0969-9961(03)00069-x
Sherman, S. A., Phillips, J. K., Costa, J. T., Cho, F. S., Oungoulian, S. R., and Finan, J. D. (2016). Stretch Injury of human induced pluripotent stem cell derived neurons in a 96 well format. Sci. Rep. 6:34097.
Shrirao, A. B., Kung, F. H., Omelchenko, A., Schloss, R. S., Boustany, N. N., Zahn, J. D., et al. (2018). Microfluidic platforms for the study of neuronal injury in vitro. Biotechnol. Bioeng. 115, 815–830. doi: 10.1002/bit.26519
Sienski, G., Narayan, P., Bonner, J. M., Kory, N., Boland, S., Arczewska, A. A., et al. (2021). APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 13:eaaz4564. doi: 10.1126/scitranslmed.aaz4564
Smith, C., Graham, D. I., Murray, L. S., and Nicoll, J. A. R. (2003). Tau immunohistochemistry in acute brain injury. Neuropathol. Appl. Neurobiol. 29, 496–502. doi: 10.1046/j.1365-2990.2003.00488.x
Smith, D. H., Chen, X. H., Nonaka, M., Trojanowski, J., Lee, V. Y., Saatman, K. E., et al. (1999a). Accumulation of amyloid β and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J. Neuropathol. Exp. Neurol. 58, 982–992. doi: 10.1097/00005072-199909000-00008
Smith, D. H., Wolf, J. A., Lusardi, T. A., Lee, V. M.-Y., and Meaney, D. F. (1999b). High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J. Neurosci. 19, 4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999
Stern, R. A., Adler, C. H., Chen, K., Navitsky, M., Luo, J., Dodick, D. W., et al. (2019). Tau positron-emission tomography in former national football league players. N. Engl. J. Med. 380, 1716–1725. doi: 10.1056/NEJMoa1900757
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Tajiri, N., Kellogg, S. L., Shimizu, T., Arendash, G. W., and Borlongan, C. V. (2013). Traumatic brain injury precipitates cognitive impairment and extracellular Aβ aggregation in Alzheimer’s disease transgenic mice. PLoS One 8:e78851. doi: 10.1371/journal.pone.0078851
Takahata, K., Kimura, Y., Sahara, N., Koga, S., Shimada, H., Ichise, M., et al. (2019). PET-detectable tau pathology correlates with long-term neuropsychiatric outcomes in patients with traumatic brain injury. Brain 142, 3265–3279. doi: 10.1093/brain/awz238
Tamagno, E., Bardini, P., Obbili, A., Vitali, A., Borghi, R., Zaccheo, D., et al. (2002). Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 10, 279–288. doi: 10.1006/nbdi.2002.0515
Tang, S. J., Fesharaki-Zadeh, A., Takahashi, H., Nies, S. H., Smith, L. M., Luo, A., et al. (2020). Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol. Commun. 8:96. doi: 10.1186/s40478-020-00976-9
Tang-Schomer, M. D., Johnson, V. E., Baas, P. W., Stewart, W., and Smith, D. H. (2012). Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp. Neurol. 233, 364–372. doi: 10.1016/j.expneurol.2011.10.030
Tang-Schomer, M. D., Patel, A. R., Baas, P. W., and Smith, D. H. (2010). Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 24, 1401–1410. doi: 10.1096/fj.09-142844
Tang-Schomer, M. D., White, J. D., Tien, L. W., Schmitt, L. I., Valentin, T. M., Graziano, D. J., et al. (2014). Bioengineered functional brain-like cortical tissue. Proc. Natl. Acad. Sci. U.S.A. 111, 13811–13816. doi: 10.1073/pnas.1324214111
Tarawneh, R., and Holtzman, D. M. (2012). The clinical problem of symptomatic Alzheimer’s disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2:a006148.
Tavalin, S. J., Ellis, E. F., and Satin, L. S. (1995). Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J. Neurophysiol. 74, 2767–2773.
Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W., and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605. doi: 10.1038/nmeth777
Teasdale, G. M., Murray, G. D., and Nicoll, J. A. R. (2005). The association between APOE ε4, age and outcome after head injury: a prospective cohort study. Brain 128, 2556–2561.
Tecoma, E. S., Monyer, H., Goldberg, M. P., and Choi, D. W. (1989). Traumatic neuronal injury in vitro is attenuated by NMDA antagonists. Neuron 2, 1541–1545. doi: 10.1016/0896-6273(89)90042-1
Tran, H. T., LaFerla, F. M., Holtzman, D. M., and Brody, D. L. (2011a). Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid- accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 31, 9513–9525. doi: 10.1523/JNEUROSCI.0858-11.2011
Tran, H. T., Sanchez, L., Esparza, T. J., and Brody, D. L. (2011b). Distinct temporal and anatomical distributions of amyloid-β and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 6:e25475. doi: 10.1371/journal.pone.0025475
Triyoso, D. H., and Good, T. A. (1999). Pulsatile shear stress leads to DNA fragmentation in human SH-SY5Y neuroblastoma cell line. J. Physiol. 515, 355–365. doi: 10.1111/j.1469-7793.1999.355ac.x
Uryu, K., Chen, X.-H., Martinez, D., Browne, K. D., Johnson, V. E., Graham, D. I., et al. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192. doi: 10.1016/j.expneurol.2007.06.018
Vagnerova, K., Koerner, I. P., and Hurn, P. D. (2008). Gender and the Injured Brain. Anesthesia Analgesia 107, 201–214.
van der Kant, R., Goldstein, L. S. B., and Ossenkoppele, R. (2020). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35.
Viña, J., and Lloret, A. (2010). Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-β peptide. J. Alzheimer’s Dis. 20, S527–S533. doi: 10.3233/JAD-2010-100501
Vogel, E. W., Effgen, G. B., Patel, T. P., Meaney, D. F., and Bass, C. R. (2015). Isolated primary blast inhibits long-term potentiation in organotypic hippocampal slice cultures. J. Neurotrauma 33, 652–661. doi: 10.1089/neu.2015.4045
Walker, K. R., Kang, E. L., Whalen, M. J., Shen, Y., and Tesco, G. (2012). Depletion of GGA1 and GGA3 Mediates Postinjury Elevation of BACE1. J. Neurosci. 32, 10423–10437. doi: 10.1523/JNEUROSCI.5491-11.2012
Wang, C., Najm, R., Xu, Q., Jeong, D., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nature Medicine 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wang, J.-Z., Xia, Y.-Y., Grundke-Iqbal, I., and Iqbal, K. (2013). Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimer’s Dis. 33, S123–S139.
Warren, H. S., Tompkins, R. G., Moldawer, L. L., Seok, J., Xu, W., Mindrinos, M. N., et al. (2015). Mice are not men. Proc. Natl. Acad. Sci. U.S.A. 112, E345–E345.
Webster, S. J., Van Eldik, L. J., Watterson, D. M., and Bachstetter, A. D. (2015). Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J. Neurosci. 35, 6554–6569. doi: 10.1523/JNEUROSCI.0291-15.2015
Weiner, M. W., Crane, P. K., Montine, T. J., Bennett, D. A., and Veitch, D. P. (2017). Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology 89:1923. doi: 10.1212/WNL.0000000000004608
Wilson, M., and Montgomery, H. (2007). Impact of genetic factors on outcome from brain injury. Br. J. Anaesthesia 99, 43–48. doi: 10.1093/bja/aem142
Wu, Z., Wang, Z.-H., Liu, X., Zhang, Z., Gu, X., Yu, S. P., et al. (2020). Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer’s disease pathology. Prog. Neurobiol. 185:101730. doi: 10.1016/j.pneurobio.2019.101730
Yamaguchi, H., Ishiguro, K., Uchida, T., Takashima, A., Lemere, C. A., and Imahori, K. (1996). Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3β and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 92, 232–241. doi: 10.1007/s004010050513
Yang, C.-N., Shiao, Y.-J., Shie, F.-S., Guo, B.-S., Chen, P.-H., Cho, C.-Y., et al. (2011). Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol. Dis. 42, 221–230.
Yin, Z., Han, Z., Hu, T., Zhang, S., Ge, X., Huang, S., et al. (2020). Neuron-derived exosomes with high miR-21-5p expression promoted polarization of M1 microglia in culture. Brain Behav. Immunity 83, 270–282.
Zander, N. E., Piehler, T., Hogberg, H., and Pamies, D. (2017). Explosive blast loading on human 3D aggregate minibrains. Cell Mol. Neurobiol. 37, 1331–1334. doi: 10.1007/s10571-017-0463-7
Zanier, E. R., Bertani, I., Sammali, E., Pischiutta, F., Chiaravalloti, M. A., Vegliante, G., et al. (2018). Induction of a transmissible tau pathology by traumatic brain injury. Brain 141, 2685–2699.
Zeiler, F. A., McFadyen, C., Newcombe, V. F. J., Synnot, A., Donoghue, E. L., Ripatti, S., et al. (2021). Genetic influences on patient-oriented outcomes in traumatic brain injury: a living systematic review of non-apolipoprotein E single-nucleotide polymorphisms. J. Neurotrauma 38, 1107–1123. doi: 10.1089/neu.2017.5583
Keywords: traumatic brain injury, Alzheimer’s disease, in vivo models, in vitro models, pluripotent stem cells, genome engineering
Citation: Srinivasan G and Brafman DA (2022) The Emergence of Model Systems to Investigate the Link Between Traumatic Brain Injury and Alzheimer’s Disease. Front. Aging Neurosci. 13:813544. doi: 10.3389/fnagi.2021.813544
Received: 11 November 2021; Accepted: 20 December 2021;
Published: 08 February 2022.
Edited by:
Yi Li, Cornell University, United StatesReviewed by:
Jean Kim, Baylor College of Medicine, United StatesCopyright © 2022 Srinivasan and Brafman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Brafman, RGF2aWQuQnJhZm1hbkBhc3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.