 Jie Liu
Jie Liu Zhen-Ni Guo1,2,3
Zhen-Ni Guo1,2,3 Xiu-Li Yan
Xiu-Li Yan Yi Yang
Yi Yang- 1Department of Neurology, Stroke Center & Clinical Trial and Research Center for Stroke, The First Hospital of Jilin University, Changchun, China
- 2China National Comprehensive Stroke Center, Changchun, China
- 3Jilin Provincial Key Laboratory of Cerebrovascular Disease, Changchun, China
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy that affects multiple systems including the muscle and heart. The mutant CTG expansion at the 3′-UTR of the DMPK gene causes the expression of toxic RNA that aggregate as nuclear foci. The foci then interfere with RNA-binding proteins, affecting hundreds of mis-spliced effector genes, leading to aberrant alternative splicing and loss of effector gene product functions, ultimately resulting in systemic disorders. In recent years, increasing clinical, imaging, and pathological evidence have indicated that DM1, though to a lesser extent, could also be recognized as true brain diseases, with more and more researchers dedicating to develop novel therapeutic tools dealing with it. In this review, we summarize the current advances in the pathogenesis and pathology of central nervous system (CNS) deficits in DM1, intervention measures currently being investigated are also highlighted, aiming to promote novel and cutting-edge therapeutic investigations.
Introduction
Myotonic dystrophies (DM1 and DM2) are inherited autosomal dominant skeletal muscle diseases that are characterized by progressive muscle weakness and myotonia, and involves multisystem engagement. DM1, also known as Steinert disease, is caused by the abnormal expansion of a CTG-trinucleotide repeat in the 3′-UTR of the dystrophia myotonic protein kinase (DMPK) gene (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992). In healthy individuals, there are approximately 5–38 CTG repeats in the DMPK gene, while DM1 patients harbor 50 to several thousands of repeats. CTG repeats tend to increase throughout aging. And as the number of repeats increase, disease severity escalates and age of onset decreases (Bird, 1993). As a progressively debilitating disease, DM1 tends to have an earlier onset and more severe phenotype from one generation to the next, while symptoms could be highly variable among patients (Udd and Krahe, 2012).
Conventionally, DM1 can be divided into five types: congenital, childhood-onset, juvenile-onset, adult-onset, and late-onset, with the adult-onset form being the most prevalent (De Antonio et al., 2016). In the past few years, several neurological symptoms, including cognitive impairment, behavioral impairment, and sensory-motor neural integration have attracted more and more attention, indicating cerebral involvement (Meola and Sansone, 2007; Meola and Cardani, 2015). These central nervous system (CNS) deficits among DM1 individuals significantly increase the disease burden, not only affecting neuropsychological domains but also decreasing the whole quality of life. A large clinical longitudinal study of DM1 patients by Miller et al. (2021) demonstrated that cognitive deficits, hypersomnolence, and apathy are critical brain symptoms of adult-onset DM1 caused by the underlying molecular mechanisms. In contrast, depression and anxiety are secondary coping symptoms with chronic physical and emotional stress (Miller et al., 2021). In addition, Simoncini et al. (2020) proved that the severity and progression rate of neurological impairments are highly variable over time, possibly attributed to underlying neuropathology (Mazzoli et al., 2020). The cognitive impairment in DM1 patients can be severe. This varies significantly with the phenotype, including the typical intellectual disability and lower intelligence quotient (IQ) levels in the congenital phenotype (Angeard et al., 2007, 2011; Echenne et al., 2008; Ekström et al., 2008; Douniol et al., 2012), the reading and spelling impairment, autistic behavior, attention deficits, deficiency in the speed of processing and severe difficulties in social interactions in childhood-onset phenotype (Steyaert et al., 1997), and dysfunctional personality, visuospatial deficits, unawareness of disease symptoms and signs, impaired facial expression and emotion recognition, and later apathy in the adult-onset phenotype (Meola et al., 2003; Winblad et al., 2006; Sansone et al., 2007; Takeda et al., 2009; Kobayakawa et al., 2010, 2012; Sistiaga et al., 2010; Jean et al., 2014; Gallais et al., 2015). Compared with the congenital groups, the childhood group shown greater cognitive and adaptive development (Lindeblad et al., 2019). Fatigue and sleep disorders including excessive daytime sleepiness (EDS) are also prominent complaints in DM1 patients (Steyaert et al., 1997; Quera Salva et al., 2006). Since fatigue involves both central and peripheral performances, the sense of tiredness is primarily caused by CNS dysfunction (Angelini and Tasca, 2012). Till now, a variety of neuropsychological tests have been developed to evaluate the CNS involvement in DM1 (Okkersen et al., 2017a; Simoncini et al., 2020). Tables 1, 2 briefly summarizes the critical CNS symptoms in DM1 and the recent findings about CNS involvement measured by neuropsychological tests in DM1, respectively (Meola and Sansone, 2007; Subramony et al., 2020). In addition, multiple neuroimaging studies relying on structural and functional explorations suggest a wide range of brain abnormalities in DM1 (Okkersen et al., 2017b; Minnerop et al., 2018; Angelini and Pinzan, 2019), mainly involving white matter (WM) abnormalities, widespread gray matter (GM) atrophy and hypometabolism in the frontal lobes. The primary studies focused on neuroimaging abnormalities in DM1 have been shown in Tables 3, 4. Furthermore, specific patterns of neuroimaging alterations and their correlations with other clinical parameters, such as clinical performances and neuropsychological test results have also been discovered, such as sleepiness might be associated with WM status in the superior longitudinal fasciculus and cingulum (Wozniak et al., 2014), and visuospatial impairment might be correlated with WM abnormalities and cortical atrophy (Cabada et al., 2017). Although the single structural or functional alterations seems to be critical for specific CNS dysfunctions, recent advance further revealed the abnormal functional connectivity patterns in DM1 brain and their effects on different personality traits (Serra et al., 2014, 2016a,b, 2020b). Besides, the main executive dysfunction and memory and visuo-spatial impairment were associated with the whole brain volume loss, but cannot be attributed to focal atrophy in any specific regions (Baldanzi et al., 2016b). Small but extensive WM damages in DM1 patients with normal-appearing WM beyond the signal changes detected with conventional MR imaging might be associated with the neuropsychological deficit (Anderson, 2013; Baldanzi et al., 2016b). This implying the critical effects of disrupted complex neuronal networks on cognitive impairments in DM1. Explorations on the complex neuronal networks and undetected lesions and their complex mechanisms contributing to clinical impairments might provide effective outcome measurements as well as effective therapeutic targets for DM1 CNS deficits. Despite a large amount of imaging and neuropsychological evidence of CNS involvement in DM1, the molecular mechanisms driving these deficits are largely unidentified, and targeted therapies aimed at ameliorating neurological deficits are scarce. This review summarizes the recent advances in the pathology and pathogenesis of CNS disorders in DM1 and the promising therapeutic strategies, hoping to provide new proposals for future investigations.

Table 1. CNS symptom in DM1.

Table 2. Neuropsychological tests in DM1.
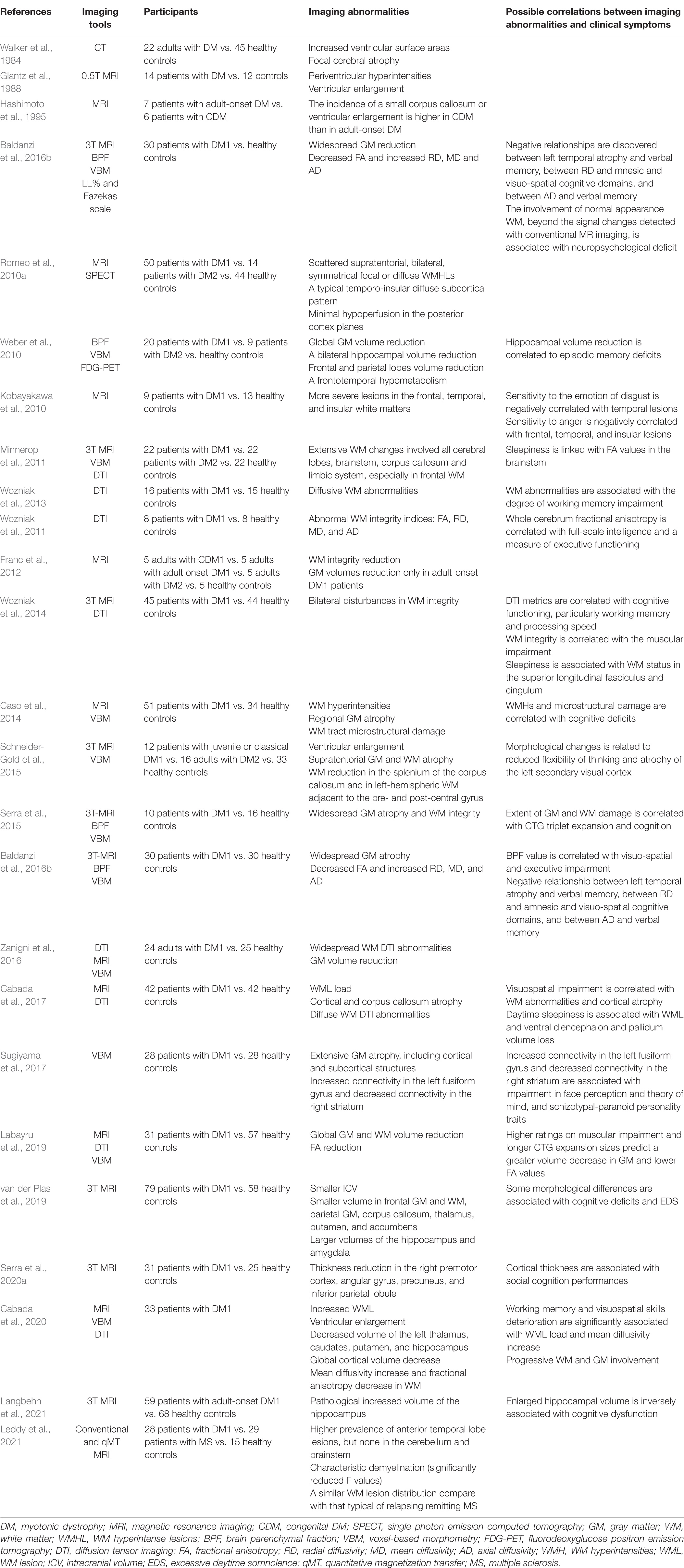
Table 3. Structural brain imaging in DM1.
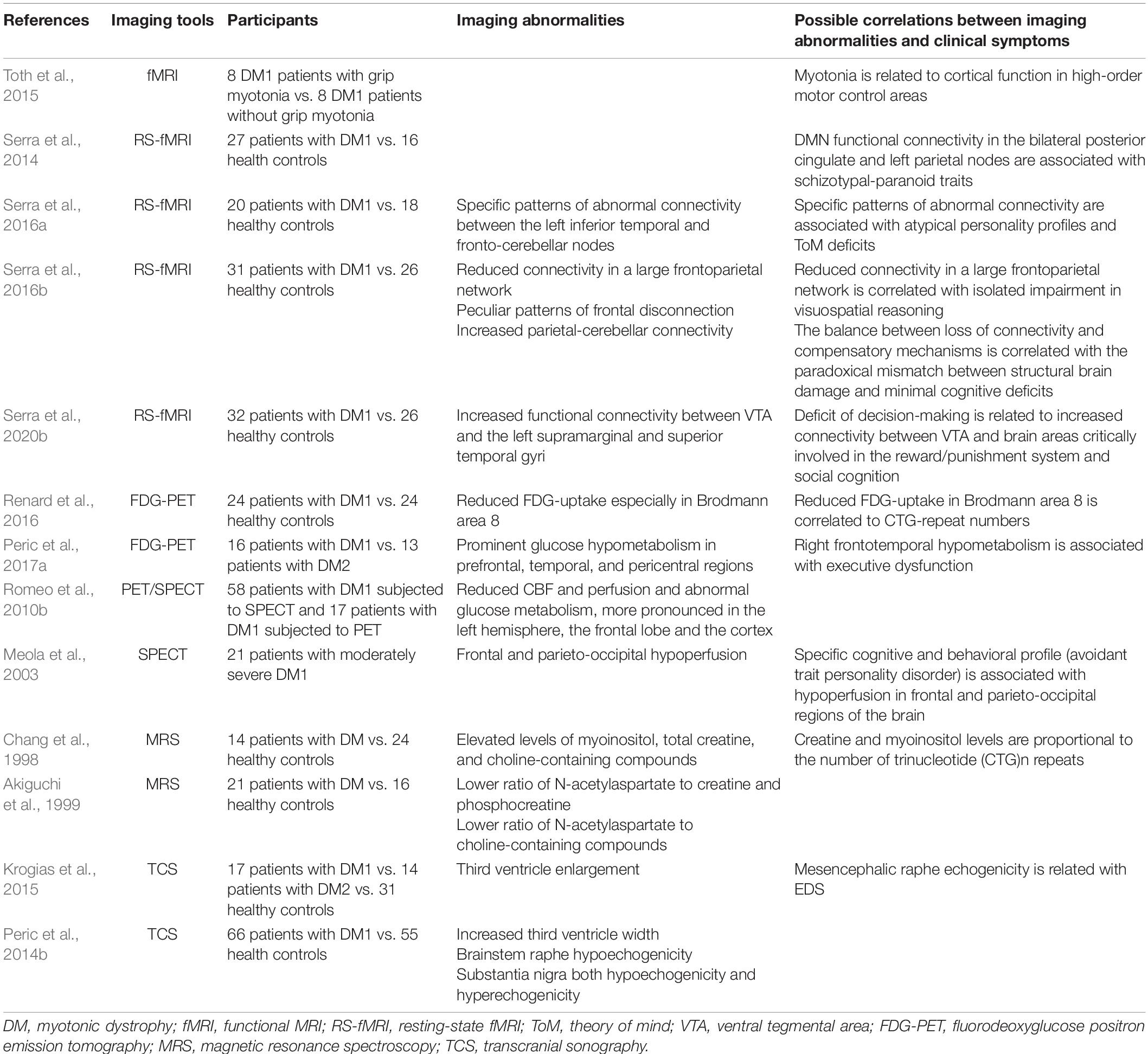
Table 4. Functional brain imaging in DM1.
Molecular Mechanisms
The pathogenesis of DM1 has been mainly attributed to the gain-of-function of toxic RNA (Pettersson et al., 2015). Specifically, the mutant CTG repeat at the 3′-UTR of the DMPK gene causes toxic RNA expression that accumulate in the nucleus called “nuclear foci,” which interferes with RNA-binding proteins (Miller et al., 2000; Fardaei et al., 2002), leading to the sequestration of muscleblind-like (MBNL) proteins and upregulation of CUGBP/Elav-like family (CELF) proteins. These alterations subsequently affected hundreds of mis-spliced effector genes, resulting in aberrant expression of embryonic splice isoforms and loss of these gene product functions, accounting for the multisystemic phenotype (Udd and Krahe, 2012; Chau and Kalsotra, 2015). In recent years, other factors, such as repeat-associated non-AUG (RAN) translation, which can contribute to the formation of toxic homopolymeric (e.g., polyQ) polypeptides, aberrant polyadenylation, activation of protein kinase C (PKC)-dependent signaling pathway, and microRNA deregulation have also been reported to play important roles in DM1 (Chau and Kalsotra, 2015). Regarding CNS, alternative splicing dysregulation has been frequently reported (Caillet-Boudin et al., 2014). Meanwhile, the widespread distributions of mutant DMPK mRNA accumulated in nuclear foci in neurons, astrocytes, oligodendrocytes, as well as in human DM1 induced pluripotent stem cell (iPSC)-derived neural stem cells (NSCs) have been reported (Jiang et al., 2004; Hernández-Hernández et al., 2013a; Xia et al., 2013; Sicot et al., 2017). In animal models, different splicing defects and their associated CNS symptoms were also studied (Figure 1). Except for these, emerging pathogenic events independent of splicing defects have also been discovered in the brain of DM1 patients (Marteyn et al., 2011; Hernández-Hernández et al., 2013b). Some CNS symptoms are non-linearly dependent on patient age and CTG repeat length, suggesting the complex and multifactorial mechanisms driving neurological deficits (Heatwole et al., 2012). This section concludes the current advance about CNS pathogenesis, hoping to better understand the complex nature of the DM1 CNS disorders.
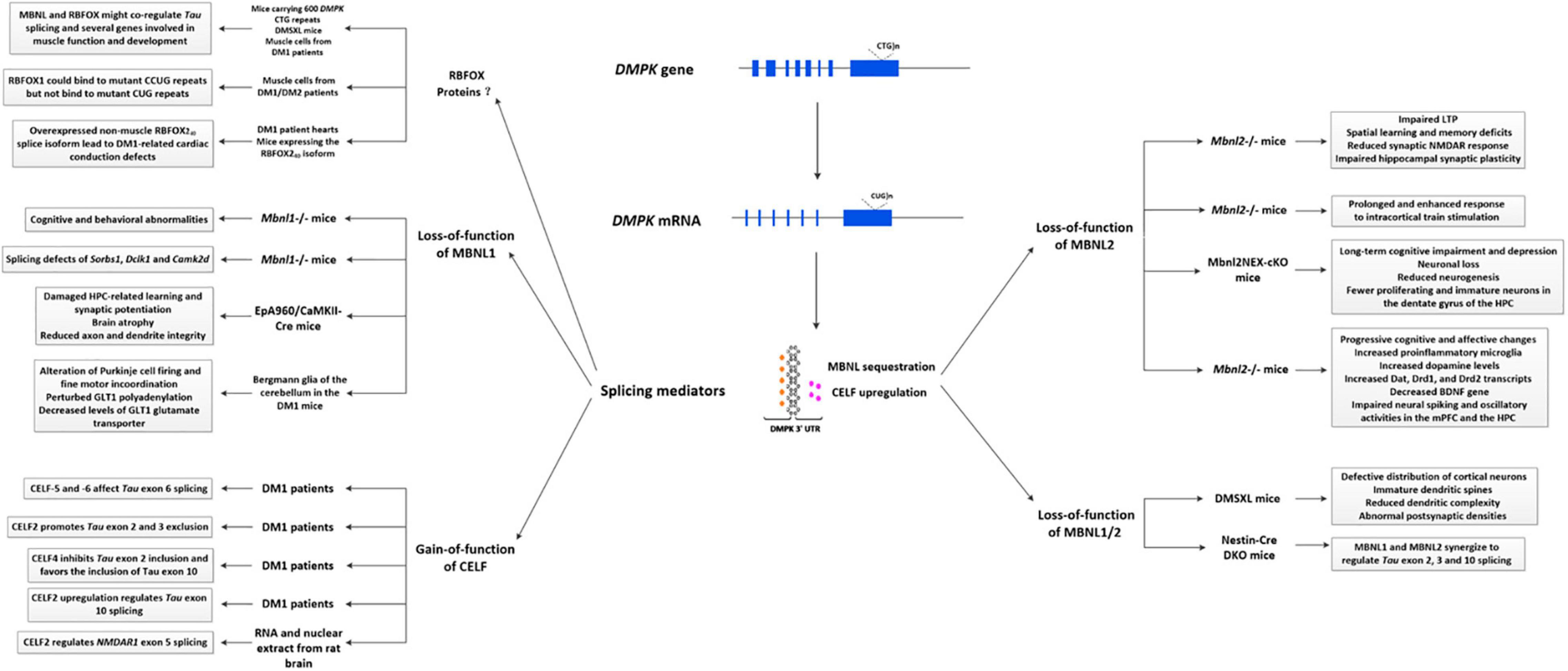
Figure 1. Primary splicing defects described in the DM1 brain. Splicing defects are generally considered the primary cause of DM1 pathology, similar to those in the brain. Specifically, the mutant DMPK gene transcripts into expanded CUG RNA, which folds into a hairpin-like structure in the nucleus called “nuclear foci.” These nuclear foci then bind and regulate RNA-binding proteins, affecting alternative splicing, eventually causing a wide range of pathogenic changes. Most splicing defects involved in brain pathology are mediated by the sequestration of MBNL proteins and upregulation of CELF proteins. In animal studies, it has been shown that loss-of-function of MBNL1 might be related to cognitive and behavioral abnormalities, learning abnormalities, alteration of Purkinje cell firing and fine motor incoordination, and perturbed GLT1 levels. Besides, it also contributes to alternative splicing alterations of several genes, such as Sorbs1, Dclk1, and Camk2d. Loss-of-function of MBNL2 could be associated with sleep alterations, spatial learning and memory deficits, impaired synaptic NMDAR response and hippocampal synaptic plasticity, neuronal loss, reduced neurogenesis, cognitive and affective changes and so on. Meanwhile, synergistic effects of MBNL1 and MBNL2 can regulate Tau exon 2, 3, and 10 splicing, while a combination loss of MBNL1/2 might be related to abnormal sleepiness, defective distribution of cortical neurons, immature dendritic spines, reduced dendritic complexity, and abnormal PSDs. In DM1 patients, it has been shown that gain-of-function of several CELF proteins can regulate Tau exon 2, 3, 6, and 10 splicing, and CELF2 can regulate NMDAR1 exon 5 splicing. A novel splicing regulator, RBFOX protein has recently been considered a new player in DM1 pathogenesis, via a competitive or cooperative interaction with MBNL1. Additionally, the non-muscle RBFOX240 splice isoform in DM1 heart may also contribute to cardiac conduction defects. DM1, myotonic dystrophy type 1; CELF, CUGBP/Elav-like family; MBNL, muscleblind-like; GLT1, glutamate transporter 1; LTP, long-term potentiation; mPFC, medial prefrontal cortex; HPC, hippocampus; BNDF, brain derived-neurotrophic factor.
Loss Function of Muscleblind-Like Proteins
Three MBNL paralogs are expressed in mammals, MBNL1, MBNL2, and MBNL3, which can be involved in regulating alternative splicing, mRNA stability, translation and so on (Charizanis et al., 2012; Wang et al., 2012). A large amount of previous studies have revealed the critical role of MBNL proteins in muscle or heart-related disorders, while in recent years, a close relationship between the loss-of-function of MBNL and DM1 brain-related phenotypes were also detected (Matynia et al., 2010; Charizanis et al., 2012; Goodwin et al., 2015), especially MBNL2. Nowadays, hundreds of dysregulated splicing factors, including in Cacna1d (McKinney et al., 2009), Tanc2 (Han et al., 2010), Ndrg4 (Yamamoto et al., 2011), and GRIN1 (Shimizu et al., 2000) have been detected in the brain of Mbnl 2–/– mice, most of which were similarly dysregulated in DM1 patients, indicating a critical role of the Mbnl2 loss in DM1 brain pathology (Charizanis et al., 2012). Further, Mbnl2–/– mice which are exposed to sleep deprivation also developed several CNS features including impaired long-term potentiation (LTP), deficits in spatial memory, reduced synaptic NMDAR response and impaired hippocampal synaptic plasticity (Charizanis et al., 2012), which were similar to the DM1 phenotype. In 2018, one study described prolonged and enhanced responsiveness to intracortical train stimulation in Mbnl2–/– mice, partially attributed to abnormal glutamate neurotransmission (Chen et al., 2018). Particularly, these abnormalities have also been observed in DM1 patients (Meola and Sansone, 2007; Takado et al., 2015).
Several studies also reported a potential role of MBNL1 protein in CNS disorders. For example, cognitive and behavioral abnormalities have been discovered in Mbnl1–/– mice (Matynia et al., 2010). In cultured primary hippocampal neurons and EpA960/CaMKII-Cre mice (a brain-specific DM1 model carrying 960 DMPK CTG repeats in the postnatal brain), expanded CUG repeats led to deubiquitination of cytoplasmic MBNL1, subsequent nuclear translocation, and morphological defects. These effects can be ameliorated by inhibiting the degradation of lysine 63-linked polyubiquitin chains or by promoting MBNL1 ubiquitination (Wang P. Y. et al., 2018). In 2020, cell studies shown that gain-of-function of MBNL1 could reverse the proliferation defect of skeletal muscle satellite cells in DM1 by inhibiting autophagy via the mTOR pathway (Song et al., 2020). Moreover, functional characterization of neuronal cells derived from human embryonic stem (ES) cells reported a reduced proliferative capacity and increased autophagy associated with alterations of the mTOR signaling pathway, while gain-of-function of MBNL1 rescued the phenotype (Denis et al., 2013), suggesting that MBNL1 loss might influence brain pathology by regulating the mTOR signaling pathway. Some important splicing defects have also been detected in the brain of Mbnl1–/– mice, in genes such as Sorbs1, Dclk1, and Camk2d (Suenaga et al., 2012). However, the extent of alternative splicing defects in the brain of Mbnl1–/– mice was much less than that observed in DM1. A number of alternative exons, such as GRIN1 exon 4, APP exon 7, and Tau exons 3 and 9, which have already been reported to be mis-spliced in the brains of DM1 patients, were unaltered in Mbnl1–/– mice, thus indicating a limited contribution of MBNL1 to DM1 CNS defects (Suenaga et al., 2012).
In addition, it’s worth noting that the combined loss of MBNL1 and MBNL2 has shown infant/immature structural phenotypes in mutant brains, similar to that of DM1 patients (Goodwin et al., 2015). Besides, single gene knockout may also contribute to the compensatory upregulation of the remaining Mbnl genes (Lee et al., 2013). In Mbnl1–/– mice, the expression of MBNL2 could be upregulated, which subsequently targets transcripts that are normally regulated by MBNL1 (Lee et al., 2013). These findings indicate a collaborative role of MBNL1 and MBNL2 involved in DM1 CNS.
Gain of CUGBP/Elav-Like Family Activities
The human CELF family has six members, all of which are involved in alternative splicing regulation (Ladd et al., 2001, 2004). Among these, the upregulations of CELF1 and CELF2 have been observed in the brain of DM1 patients (Dhaenens et al., 2011).
CELF1 has been identified as a (CUG)n repeat-binding protein (Timchenko et al., 1996). Unlike MBNL1, it is increased in DM1 patients mainly through PKC-mediated phosphorylation to stabilize the protein (Kuyumcu-Martinez et al., 2007), or through decreased levels of miR-23a/b (Kalsotra et al., 2014). In opposite, the increasing expression of CELF2 is not related to protein hyperphosphorylation, indicating other potential regulatory mechanisms. CELF1 and CELF2 can influence various transcripts (Ladd, 2013) in the DM1 brain, such as different exons of Tau and NMDAR1 exon 5 (Leroy et al., 2006a,b). Tau proteins can promote neurite outgrowth, organize axonal microtubules, and participate in kinesin-dependent axonal transport (Andreadis, 2012). NMDARs are key components of glutamate-mediated excitatory signaling, which can contribute to excitatory synaptic transmission and synaptic plasticity, thought to be the basis of learning and memory (Zorumski and Izumi, 2012). It has been discovered that four exons (2, 3, 6, and 10) of Tau isoforms could respond to one or more CELF proteins in DM1 (Leroy et al., 2006a,b; Dhaenens et al., 2011). CELF2 can regulate alternative exon 5 transcripts of NMDAR1 to change neuronal excitation in rat brain (Zhang et al., 2002), while CELF1 is inefficient. Aside from alternative splicing regulation, CELF proteins can also participate in regulating mRNA adenylation status, stability, and translation in various cell types (Dasgupta and Ladd, 2012), indicating its potential cytoplasmic functions in the brain.
A Novel Splicing Regulator—RBFOX Proteins
RBFOX proteins are sequence-specific RNA binding proteins which can regulate alternative splicing in multiple tissues, such as skeletal muscle, heart, and brain (Gehman et al., 2011; Singh et al., 2014; Conboy, 2017; Jacko et al., 2018). Nowadays, it has been discovered that RBFOX1 is involved in the regulation of synapses and autism-related genes in the cytoplasm of neurons (Wang et al., 2016). Klinck et al. (2014) reported that MBNL1 and RBFOX1 protein could co-regulate the splicing of a series genes involved in muscle function and development, some of which are also mis-spliced in DM1 tissues. And the decreased RBFOX1 may amplify the mis-splicing changes caused by the loss-of-function of MBNL1. At the same time, they found that the ectopic expression of RBFOX1 partially rescued the mis-splicing of Tau exon 2 in glioblastoma cells. Since the MBNL proteins have been shown to regulate the splicing of Tau exon 2 in DM1 brains, it is therefore interesting to postulate that cooperation of MBNL and RBFOX1 might regulate Tau splicing in DM1 brains. Nevertheless, Sellier’s researches proposed different results, that RBFOX1 could bind to expanded CUG RNA repeats, competing with MBNL1 and reducing the sequestration of MBNL1 in DM2 muscle cells, which suggest a partly competitive relationship between RBFOX1 and MBNL1 (Sellier et al., 2018). Misra et al. (2020) found a new non-muscle RBFOX240 splice isoform which is overexpressed in DM1 patient hearts. Particularly, mice expressing the RBFOX240 isoform in hearts also performed DM1-related cardiac conduction dysfunctions (Misra et al., 2020), which may due to its promotion of the production of pathogenic ion channel splice variants. All of these results provide a novel idea explaining the splicing dysregulation in DM1, though their possible roles in CNS dysfunctions still need further exploration.
Effector Genes Alterations Due to Splicing Defects
Nowadays, hundreds of spliced effector genes changes have been discovered in the DM1 brain (Jiang et al., 2004; de León and Cisneros, 2008; Suenaga et al., 2012; Hernández-Hernández et al., 2013a; Otero et al., 2021). Jiang et al. (2004) screened 45 exons (in 31 genes) spliced in the brain of DM1 patients, wherein four of them changed in the ratio of exon inclusion/exclusion splice products, including decreased inclusion of APP exon 7, Tau exons 2 and 10, and increased inclusion of NMDAR1 receptor exon 5. Interestingly, the sequences encoding APP exons 7 and 8 are excluded in neurons, but are included in astrocytes, indicating the splicing defects in astrocytes. In 2021, via detecting transcriptome alterations in frontal cortex of DM autopsy samples, Otero et al. (2021) reported 130 high-confidence splicing changes, which occur in ion channels, neurotransmitter receptors, and synaptic scaffolds, while mis-splicing of GRIP1 might change kinesin association. In frontal cortex samples, downregulated genes tend to express in neurons, while upregulated genes tended to express preferentially in endothelial and microglial, which suggest neuroinflammatory responses. In DMSXL mice (carrying ∼1,000–1,800 DMPK CTG repeats with multisystemic transgene expression), the mis-splicing patterns of GRIN1 exon 21, Ldb3 exon 11, and Mbnl2 exon 7 in frontal cortex, as well as GRIN1 exon 5, Ldb3 exon 11, APP exon 8, and Frx1 exons 15/16 in brainstem have been detected (Hernández-Hernández et al., 2013a), and GRIN1 and Tau mis-splicing appear to be involved in synaptic dysfunction. In the brain of Mbnl1–/– mice, 14 mis-spliced events have been observed using splicing-sensitive microarray, including Sorbs1 exons 6 and 25, Spag9 exon 31, Dclk1 exon 19, APP exon 7 and 8, GRIN1 exon 4, and so on (Suenaga et al., 2012).
Heterogeneity in Splicing Defects in Different Brain Regions
Recently, the heterogeneity of splicing defects in the DM1 brain also attracted increasing attention. In Mbnl knockdown mice, it has been identified that abnormal alternative splicing in the cerebellum are fewer than in other brain regions (Charizanis et al., 2012; Suenaga et al., 2012). The inclusion of Mbnl1 exon 5 and Mbnl2 exons 5 and 8 were higher in most brain areas, except in the cerebellum for Mbnl2 exons 5 and 8. Besides, RNA foci preferentially accumulate in the frontal cortex and certain areas of the brainstem of DM1 transgenic mice (Huguet et al., 2012; Hernández-Hernández et al., 2013a), and seem to be more abundant in cortical astrocytes than in neurons (Hernández-Hernández et al., 2013a). Using autopsied brain tissues of DM1 patients, researchers further observed varying degrees of mis-splicing among the cerebellar cell layers (Furuta et al., 2018). LASER capture microdissection revealed splicing defects in the molecular layer of the cerebellum, but not in the granular layer (Furuta et al., 2018). Similarly, one study reported that mis-splicing in WM is less apparent than in GM of the DM1 brain, which may be attributed to the inability to transfer the abnormal/fetal splicing isoform to the axon (Nishi et al., 2020). Future analysis of the mis-splicing diversity in the DM1 brain may favor a precise therapy targeting specific sites.
Translational RNA Differences
Though sharing with a common pathogenetic mechanism, alternative splicing defects, further findings suggested a new perspective influencing the performances of DM patients. According to the results of Salvatori et al. (2009) the molecular and biochemical differences of troponin T and the insulin receptor between DM1 and DM2 due to different translational patterns and distribution patterns might partially explain the apparent differences on their clinical phenotypes. This phenomenon promotes the exploration of DM1 pathogenesis at the translation level.
Somatic Expansion
Recently, an important aspect of the pathogenesis of DM1 regarding to somatic expansion has attracted increasing attention. It has been demonstrated that the length of modal allele in blood DNA samples of DM1 patients increased over time, driven primarily by the inherited progenitor allele length (ePAL), age-at-sampling, and age-at-onset (Morales et al., 2020). Since the DNA mismatch repair proteins MSH2, MSH3, and MSH6, are considered critical players in CTG repeat expansion, and their decreased expressions inhibit the expansion (Dragileva et al., 2009; Tomé et al., 2009), they provide a new insight into the mechanism of CTG repeat instability in DM1. Tomé et al. (2009) reported that MSH2 ATPase domain mutation could influence somatic instability in DM1 transgenic mice. Compared with fibroblasts, MSH2, MSH3, and MSH6, were highly expressed in DM1 patient-derived iPSC, accompanied with longer CTG repeat, while MSH2 silencing inhibited CTG repeat expansion (Du et al., 2013). Flower et al. (2019) reported that the altered MSH3 levels caused by MSH3 3a repeat allele decreased somatic expansion and changed the DM1 phenotype. Thus, regulation of MSH3 might represent a new roadmap for potential target therapy of DM1.
Cellular Processes Alterations
Synaptic Dysfunction
Synaptic protein dysfunction is an important feature demonstrated in the DM1 brain. RAB3A is an abundant synaptic vesicle protein that regulates neurotransmission by interacting with other synaptic proteins. The upregulation of RAB3A causes spontaneous exocytosis, and plays important roles in spatial learning, sleep control, and synaptic plasticity (Sudhof, 2004). Synapsin I (SYN1) can regulate synaptic vesicle release in a phosphorylation-dependent manner, and its hyperphosphorylation determines short-term synaptic plasticity alterations (Rosahl et al., 1993). Hernández-Hernández et al. (2013a) reported RAB3A upregulation and SYN1 hyperphosphorylation in DMSXL mice, transfected cells, and DM1 patient samples, which were related to altered spontaneous neurosecretion in cell culture, electrophysiological and behavioral deficits in mice, and possibly contributed to the neuropsychological manifestations in DM1 patients. Then, Jimenez-Marin et al. (2021) demonstrated that transcriptional signatures of synaptic vesicle genes at least partially explain the mechanisms of DM1 neurodegeneration. Nowadays, a close relationship between splicing regulator alterations and synaptic protein dysfunctions has been found, possibly contributing to DM1 neurological phenotypes (Hernández-Hernández et al., 2013b). For example, loss-of-function of MBNL1 in DMSXL mice brain mediated the upregulation of RAB3A levels and determined the length of neuronal dendrites and axons (Hernández-Hernández et al., 2013b). The overexpression of CELF1 or CELF2 in neuronal-like PC12 cells containing expanded CUG transcripts mediated SYN1 hyperphosphorylation (Hernández-Hernández et al., 2013b). And other splicing alterations, such as GRIN1 and Tau splicing defects, might also contribute to synaptic dysfunction in DM1 (Hernández-Hernández et al., 2013b). However, although influenced by different splicing regulators, these synaptic protein alterations seem to be independent of mis-splicing of their coding transcripts, suggesting that DM1 neuropathogenesis have far-reaching implications beyond the disruption of splicing programs.
In addition, other synaptic-related abnormalities have also been identified in the DM1 brain. Lee et al. (2019) found distribution defects of cortical neurons, abnormal dendritic morphology and complexity and postsynaptic densities in the Mbnl1/2–/– mice. A time-course study using EpA960/CaMKII-Cre mice revealed that hippocampus (HPC)-related learning and synaptic potentiation were damaged before structural alterations occurred in the brain, followed by brain atrophy associated with progressively reduced axon and dendrite integrity. Notably, cytoplasmic MBNL1 level on dendrites decreased before dendrite degeneration, whereas MBNL2 expression reduction and MBNL-mediated alternative splicing defects were evident after degeneration (Wang et al., 2017), suggesting that MBNL1 reduction might contribute to synaptic transmission dysfunction in the DM1 brain.
The Defective Neuroglial Interactions
A recent study shows that compared to neurons, cortical astrocytes contain more ribonuclear foci in DM1 transgenic mice (carrying 45 kb of human genomic DNA cloned from a DM1 patient) (Hernández-Hernández et al., 2013a). Meanwhile, RNA sequencing discovered more frequent splicing deficits in glia of DMSXL mice (Oude Ophuis et al., 2009; González-Barriga et al., 2021), suggesting a non-negligible presence of neuroglial damage in DM1. Glutamate transporter 1 (GLT1) is a glial-specific glutamate transporter that can recapture excitatory glutamate from the synaptic cleft and protect it from neurotoxicity caused by glutamate overstimulation (Bellamy, 2006). In the Bergmann glia of the cerebellum in DMSXL mice, scientists found glutamate excitotoxicity associated with decreased GLT1 (Sicot et al., 2017). In astrocytes of DMSXL mice, downregulation of GLT1 increased glutamate neurotoxicity and caused neuronal damage, while the upregulation of GLT1 corrected Purkinje cell firing and motor incoordination (Sicot et al., 2017). These studies indicated that the loss-of-function of GLT1 and defective neuroglial interactions might play critical roles in inducing DM1 brain disorders.
Nowadays, studies have identified that the expression of GLT1 is regulated by RNA transcription, splicing and stability, post-translational modifications, and protein activity (Kim et al., 2011). Specifically, loss-of-function of MBNL1 can perturb GLT1 polyadenylation, thus decreasing the levels of GLT1 glutamate transporter, while MBNL2 inactivation did not affect GLT1 levels, but contributed to the compensating increase in MBNL1 protein (Batra et al., 2014; Mohan et al., 2014). In the future, restoring GLT1 protein and glutamate neurotransmission by regulating MBNL proteins might be promising approaches to reverse the defective neuroglial interactions in DM1. Besides, in an inducible glial cell model of DM1 derived from human retinal Müller glial cells (MIO-M1) expressing 648 CUG repeats [MIO-M1 CTG(648)], scientists identified that the activation of inflammatory pathways and immune responses could partially explain DM1 CNS defects associated with defective glia (Azotla-Vilchis et al., 2021; González-Barriga et al., 2021). In glial cells of DMSXL mice, expanded CUG RNA affected preferentially differentiation-associated molecular events, which open new avenues in studying DM1 brain pathology with cell type resolution (González-Barriga et al., 2021).
Altered Brain Insulin Signaling
Previous studies have observed the insulin resistance (IR) phenotype in DM1 patients (Moxley et al., 1978), which could be attributed to the mis-splicing of the insulin receptor gene. While recently, altered insulin signaling in the brain has also been proposed, thus provides potential alternative explanations for the DM1 neuropathogenesis (Nieuwenhuis et al., 2019). For example, animals with impaired insulin receptor signaling have shown reduced motivation, which could translate to apathy in humans (Dagenhardt et al., 2017), a critical symptom in adult DM1 patients (Gallais et al., 2015). In patients with obsessive-compulsive disorder, altered brain insulin signaling has been observed (van de Vondervoort et al., 2016). Besides, cognitive deficits, especially visuospatial and verbal memory deficits (Kullmann et al., 2016), depressive symptoms (Minier et al., 2018; van der Velden et al., 2019) and decreased behavioral flexibility (van de Vondervoort et al., 2019), have also been shown to be associated with brain IR. Moreover, a recent review pointed out the effects of insulin signaling on tauopathy and Aβ metabolism (Gonçalves et al., 2019). And several neuroimaging studies have discovered a decrease in glucose uptake in the brain of DM1 patients (Fiorelli et al., 1992; Peric et al., 2017a), however, it remains unclear whether altered insulin signaling is involved.
Neurochemical Changes
A series of neurochemical changes were also found in DM1, which may account for specific symptoms. For example, the loss of serotonin (5-HT)-containing neurons in the dorsal raphe nucleus (DRN) and the superior central nucleus (SCN) of DM1 patients is associated with hypersomnia (Ono et al., 1998). And the alteration of serotonergic raphe structures might be involved in the pathogenesis of hypersomnia (Peric et al., 2014b; Krogias et al., 2015; Krogias and Walter, 2016). The hypoechogenicity of nucleus raphe might be correlated with EDS and depression in DM patients (Peric et al., 2014b). The extent of WM hyperintensities might be correlated with fatigue (Minnerop et al., 2011). Using conditional Mbnl2NEX-cKO mice (a tissue-specific knockout mouse model lacking the Mbnl2 gene in forebrain glutamatergic neurons), long-term cognitive impairment and depression were found (Ramon-Duaso et al., 2020). This might be associated with significant neuronal loss, reduced neurogenesis, and fewer proliferating and immature neurons in the dentate gyrus of the HPC. Additionally, in Mbnl2–/– mice, increased proinflammatory microglia, dopamine levels as well as Dat, Drd1, and Drd2 transcripts levels, decreased expression of the brain derived-neurotrophic factor (BDNF) gene, and impaired neural spiking and oscillatory activities in the medial prefrontal cortex (mPFC) and the HPC have been found, accompanied with progressive cognitive and affective changes (Ramon-Duaso et al., 2019). Similar abnormalities have also been observed in the mPFC and HPC of DM1 patients with severe depression and cognitive impairment (Romeo et al., 2010b). Currently, chronic treatment with methylphenidate (MPH) has been shown to reverse the behavioral abnormalities, reduce proinflammatory microglia and Dat level, and increase BDNF and Nrf2 mRNA expressions in Mbnl2–/– mice. This makes it a promising drug candidate to treat CNS dysfunctions in DM1 patients (Ramon-Duaso et al., 2019). However, this intervention also impaired glutamate uptake and increased glutamate levels in juvenile rats (Schmitz et al., 2016, 2017), querying whether MPH therapy would increase glutamate neurotoxicity and induce the defective neuroglial interactions.
In addition, dysregulation of cerebrospinal fluid (CSF) homeostasis was observed in early onset DM1. A study introducing DMPK CTG expansions into the mouse found that mis-splicing significantly affected brain choroid plexus epithelial cells (Nutter et al., 2019). Besides, increased levels of total-Tau, IgG, γ-globulin, and myelin basic protein (MBP), as well as decreased levels of Aß1-42 and orexin-A were found in the CSF of DM1 patients (Hirase and Araki, 1984; Martínez-Rodríguez et al., 2003; Winblad et al., 2008; Peric et al., 2014a). Orexins are hypothalamic peptides that play critical roles in sleep/wake regulation (Sakurai, 2014). A possible correlation between an altered CSF orexin-A levels and EDS in systemic lupus erythematosus (SLE) patients with hypothalamic lesions and patients with frontotemporal dementia (FTD) have been reported (Çoban et al., 2013; Suzuki et al., 2018). Notably, significantly lower orexin-A levels were also detected in the CSF of 6 DM1 patients affected by EDS (Martínez-Rodríguez et al., 2003), thus providing potential explanations for DM1 sleep disorders. Furthermore, in 2015, a decreased level of BDNF, a neurotrophin participate in learning and memory, was detected in the serum of DM1 patients. Since it can cross the brain-blood barrier (BBB), it might be considered a promising biomarker of CNS defects (Comim et al., 2015).
Neuropathological Defects in Early Developmental Processes
To determine the expression patterns of DMPK with age, Langbehn et al. (2021) analyzed the brain from 99 donors with DM1 ranging from 5 postconceptional weeks to 80 years old. They found that peak expression of wildtype DMPK coincides with a time of dynamic brain development, thus indicating that the abnormalities in DM1 brain DMPK expression may affect early brain development. Besides, direct injection of (CUG)91 repeat-containing mRNA into single-cell embryos of zebrafish induced CNS toxicity during early development, resulting in morphological abnormalities, behavioral abnormalities, and extensive transcriptional alterations, while co-injection of zebrafish Mbnl2 RNA suppressed this toxicity and reversed the associated behavioral and transcriptional abnormalities (Todd et al., 2014). In addition, defects in the genes involved in dysfunctional neurite outgrowth and synaptogenesis at the neuromuscular junction were also observed in neuronal progeny derived from DM1 mutant human ES cells, which are associated with the decreased expression of two genes that belong to the SLITRK family, SLITRK2 and SLITRK4 (Marteyn et al., 2011). Transfection of SLITRK2 and SLITRK4 into cultured DM1 cells restored neurite length to control levels. However, it is interesting that DMPK mutation and dysregulation of splicing by MBNL1 do not appear to be involved in SLITRK misexpression or neurite outgrowth, suggesting other new molecular mechanisms involved in DM1 abnormal neurodevelopment (Marteyn et al., 2011).
Pathological Features
Tau Pathology
In the DM1 brain, dysregulation of alternative splicing could lead to pathologic Tau proteins accumulations and the formation of neurofibrillary tangles (NFTs) (Caillet-Boudin et al., 2014), which are mainly located in the HPC, entorhinal cortex, and most of the temporal areas, called Tau pathology. Nowadays, Tau pathology has been confirmed a critical histopathological characteristic in the brain of DM1 patients (Caillet-Boudin et al., 2014), and could interfere with axonal transport and neurosecretion (Caillet-Boudin et al., 2014). Although there is no direct evidence, current research suggests that this pathology may be related to cognitive dysfunctions in DM1.
In the adult brain, Tau gene could encode six Tau isoforms through alternative splicing of exons 2, 3, and 10, whereas in DM1, all of these exons are absent, thus promoting fetal expression of the 3-repeat Tau isoform (Sergeant et al., 2001; Jiang et al., 2004). Interestingly, a recent study in a congenital DM1 patient with intellectual disability also suggested the existence of a 4-repeat tau dominant pathology (Mizuno et al., 2018). Originally, cell studies identified that long CUG repeats-mediated loss-of-function of MBNL1 could be responsible for the changes in Tau splicing (Dhaenens et al., 2008). While in 2014, researchers discovered that both MBNL1 and MBNL2 have an enhancer activity of Tau exon 2 inclusion, and only the interaction of MBNL1 and MBNL2 can fully reverse the splicing defect of Tau exon 2 induced by the mutant CUG repeats, similar to that observed in DM1 (Carpentier et al., 2014). Then in 2015, a further study examining the Tau splicing using Nestin-Cre DKO mice (a Mbnl1/2–/– mouse model) suggested that both MBNL1 and MBNL2 synergize to regulate Tau exon 2, 3, and 10 splicing (Goodwin et al., 2015). These findings proved that the regulation of MBNL1/2 and their interactions are highly essential in DM1 Tau pathology. Except MBNL proteins, CELF proteins also serve as potential regulators of Tau splicing. Four exons (2, 3, 6, and 10) of Tau isoforms have been discovered to respond to one or more CELF proteins (Leroy et al., 2006a,b). Among them, Tau exon 10 responds specifically to CELF2 upregulation (Dhaenens et al., 2011). Tau exon 6 splicing is regulated by CELF5 and CELF6 (Leroy et al., 2006b). Notably, the heterogeneous distribution of Tau protein in the DM1 brain has also been reported, which may affect its splicing regulation patterns.
Other than Tau pathology, several kinds of protein and nucleotide deposits have also been observed in the brain of DM1 patients (Weijs et al., 2021), including Lewy bodies (LBs), neuronal intranuclear eosinophilic inclusion bodies, intracytoplasmic inclusion bodies, increased Marinesco bodies, gliosis (Itoh et al., 2010; Jinnai et al., 2013), skein-like ubiquitin-positive inclusions and granulovacuolar degeneration (GVD), which suggest neurodegeneration in the DM1 brain (Itoh et al., 2010; Yamazaki et al., 2011; Nakamori et al., 2012).
RNAopathy and Spliceopathy
In the DM1 brain, the mutant DMPK RNA accumulates as nuclear foci in extensive areas, which contributes to abnormal alternative splicing (Miller et al., 2000; Fardaei et al., 2002). The nuclear foci are widely distributed throughout the brain of DM1 patients, including cortex, WM in subcortical and callosal areas, HPC, thalamus, brainstem, and cerebellum, and presented in various cell types, such as neurons, astrocytes, oligodendrocytes, Purkinje cells and human DM1 iPSC or ES-cell-derived NSCs (Jiang et al., 2004; Denis et al., 2013; Hernández-Hernández et al., 2013a). Their distributions varied from different genetic features, histological features and clinical features in each person (Jiang et al., 2004). Abnormal alternative splicing has been considered the critical pathogenesis of DM1. Changes in RNA-binding proteins, such as MBNL and CELF proteins, can lead to splicing defects of a variety of pre-mRNAs and misexpressions of different protein isoforms. Nowadays, the interaction between RNAopathy, spliceopathy, and Tau pathology have accounted for essential parts of DM1 neuropathology (Caillet-Boudin et al., 2014).
Other Pathological Features
Other neuropathological features of DM1 include neuronal loss in different areas, such as the superficial layer of the frontal and parietal cortices, the occipital cortex, the medullary arcuate nuclei, the anterior and dorsomedial thalamic nuclei, the midbrain, and pontine reticular formation, which may be related to cortical atrophy (Culebras et al., 1973; Ono et al., 1995, 1998; Mizukami et al., 1999). The cell loss of specific areas might also contribute to the cognitive and behavioral abnormalities in DM1 patients (Rosman and Kakulas, 1966; Ono et al., 1996; Mizukami et al., 1999). In the post mortem brain and spinal cord of DM1 patients with congenital or childhood onset with intellectual deficiency, heterotopic neurons have been found, suggesting abnormal neurodevelopment (Ogata et al., 1998). Degenerative WM changes were also reported in DM1 patients, including myelin and axonal loss, expansion of perivascular spaces, gliosis, and hyalinization of capillaries in deep and subcortical WM (Itoh et al., 2010). However, most studies exploring the microscopic brain pathology in DM1 patients, as well as their relationships with neuroimaging features and splicing changes are case reports or small-scale researches. Clearly, larger follow-up studies are eagerly needed to improve the understandings of pathological alterations in DM1.
Developmental or Neurodegenerative?
Multiple studies have identified a wide range of CNS alterations in DM1 patients. However, the varied characteristics between DM1 patients, such as CTG triplet expansion size, duration time, and age, significantly influence the cerebral performances. In several studies based on cross-sectional analyses, progressive GM loss and increased rate of cortex volume loss correlated with age have been reported (Weber et al., 2010; Minnerop et al., 2011; Caso et al., 2014; Serra et al., 2015; Zanigni et al., 2016; Cabada et al., 2017). A longitudinal study evaluating MRI in DM1 patients also revealed that the WM degeneration and ventricular enlargement progressed over time, though it varied between different individuals (Conforti et al., 2016). However, other studies did not find significant associations between WMHL and age or a significant increase of WML during disease progression (Meola and Sansone, 2007; Itoh et al., 2010; Bajrami et al., 2017; Cabada et al., 2017). Moreover, studies by Antonini et al. (2004) demonstrated a significantly reduced GM volume which was negatively correlated with age in DM1 patients, whereas WM volume was shown not to be correlated with cortical atrophy or age. Thus, further longitudinal evaluations are still need to assess spatiotemporal imaging changes. Regarding clinical features, Sansone et al. (2007) noted a progression in frontal cognitive impairment (attentional) in both DM1 and DM2 patients. Studies by Winblad and Gallais reported a cognitive decline in adult-onset DM1 is positively related to the earlier onset and longer duration of the disease (Winblad et al., 2016; Gallais et al., 2017). Moreover, a close relationship between CNS defects and CTG expansion size has also been discovered. For example, the impaired facial emotion recognition is significantly related to CTG repeat size (Winblad et al., 2006). The extent of GM and WM damage could be correlated with CTG expansion (Serra et al., 2015). And longer CTG expansion sizes could indicate a larger decrease in GM volume (Labayru et al., 2019). Further studies are needed to determine the progressive pattern of CNS dysfunctions.
Whether the progressive cerebral involvement in DM1 patients is due to a developmental or a neurodegenerative process is still an open question (Axford and Pearson, 2013). A variety of neurodegenerative pathological features, including NFTs, LBs and WM abnormalities, as well as a progressive cognitive decline reported by a limited number of longitudinal studies have been found in DM1 patients, which support that DM1 is in part a neurodegenerative process. However, another view supports that the progressive CNS dysfunction may be responsible for cognitive impairment, rather than neurodegenerative changes, since the distinct brain alteration patterns different from neurodegenerative features might also be associated with cognitive impairments in DM1 (Axford and Pearson, 2013; van der Plas et al., 2019; Labayru et al., 2020; Langbehn et al., 2021). A newly emerged opinion proposes that DM1 could be considered a progeroid disease (an early and accelerated aging process) (Sansone et al., 2007; Modoni et al., 2008; Caso et al., 2014; Winblad et al., 2016; Gallais et al., 2017; Solovyeva et al., 2021), as typical symptoms related to aging, such as cognitive decline, occur in the early years. However, a recent study shown an interesting contrast, that the presentation of simple tasks is hugely decreased while that of complex tasks are mostly retained in DM1, which is different from normal aging (Gallais et al., 2017). Further, it’s worth noting that in the congenital/childhood-onset DM1 patients, typical molecular and clinical deficits are observed during early developmental processes, which indicate an influence of DM1 on early brain development.
By the way, there are also some limited studies comparing the different characteristics between different phenotypes of DM1 (based on age-onset). In the congenital/childhood-onset DM1 patients, typical clinical deficits including intellectual disability and behavioral abnormalities are frequently reported, without further decline over these ages (Dhaenens et al., 2011; Caillet-Boudin et al., 2014; Fernandez-Gomez et al., 2019; Lindeblad et al., 2019). In addition, compared with adult-onset DM1 patients, verbal intelligence and memory was significantly deteriorated in juvenile-onset DM1 patients, reflecting a more pronounced developmental process in the juvenile type (Woo et al., 2019). However, in contrast, other studies demonstrated that adult-onset DM1 patients presented with more pronounced decrease of quality of life than juvenile-onset DM1 patients in almost all domains (Rakocevic-Stojanovic et al., 2014). A cross sectional study identified a significant cognitive impairment progression by aging in the majority of the cognitive domains in adult-onset DM1 patients (Baldanzi et al., 2016a), suggesting the existence of progressive degeneration. However, verbal memory abilities were relatively preserved, suggesting different changing patterns involved in memory and cognitive deficits (Baldanzi et al., 2016a). Regarding to neuroimaging observations, Caso et al. (2014) observed a more severe damage of GM in adult-onset DM1 patients than in juvenile-onset DM1 patients, supporting a degenerative origin of GM abnormalities. Conversely, the severe and distributed WM microstructural damage detected in both types might support a developmental change of microstructural WM damage. In 2020, the first longitudinal study of structural brain involvement in pediatric and adult/late-onset DM1 shown that the brain volume loss over time in both groups was not significant compared with their healthy controls, thus supporting the probable occurrence of the neurodevelopmental process. However, these findings cannot completely rule out the existence of the neurodegenerative process, since patients were not yet in their 60’s at follow-up (Labayru et al., 2020). In the future, additional studies with larger-sample and longitudinal observations are still needed to further clarify the feature of DM1 brain damages.
Therapeutic Strategies of DM1
Management of Neurological Defects
To date, there are no specific therapeutic agents available to reverse neurological defects in DM1. Modalities for management mainly rely on supportive care, including sleep hygiene improvement, cognitive−behavioral therapy, aerobic exercise training, and careful use of stimulant drugs. Sleep−related disorders have been recognized as primary symptoms of CNS involvement in DM1. Early recognition and treatment of sleep disorder breathing with nocturnal non-invasive mechanical ventilation have served as important countermeasures to deal with these problems (Pincherle et al., 2012). In a large clinical cohort study, scientists found that patients who insist on home mechanical ventilation for ≥5 h/24 h shown significantly higher survival rates than those who use it less (Seijger et al., 2021), and their tolerance and adherence were remarkably high. Besides, the biggest multicenter, randomized clinical trial in DM1 named the Observational Prolonged Trial in Myotonic Dystrophy Type 1 to Improve Quality of Life-Standards, a Target Identification Collaboration (OPTIMISTIC) revealed that cognitive behavioral therapy significantly improved the ability for activity and social participation at 10 months in severely fatigued patients with DM1 (NCT02118779) (Okkersen et al., 2018). Compared with usual care, cognitive behavioral therapy plus aerobic exercise training also increased the physical activity in DM1 patients (OPTIMISTIC) (van Engelen, 2015), indicating this therapy as a promising interventions for severe fatigue in DM1. Stimulant drugs also shown the potential to treat EDS. In 2007, the American Academy of Sleep Medicine (AASM) declared that MPH might be an effective tool for treating DM1-related EDS (Morgenthaler et al., 2007). However, a Cochrane review on well-designed psychostimulant trials in patients with DM1 and EDS pointed out the lack of evidence to support its routine use (Annane et al., 2002). Some clinical studies reported that modafinil might improve hypersomnia and fatigue without significantly increasing activity levels in DM1 (Hilton-Jones et al., 2012; Laberge et al., 2013). However, respiratory insufficiency (due to abnormal central drive and respiratory muscle weakness) and sleep fragmentation related to central or obstructive apnea must be excluded (Harper et al., 2002). Except for treating sleep-related disorders, in Mbnl2–/– mice, mirtazapine, a kind of antidepressant, has been discovered to reverse cognitive impairments and depression, as well as reduce microglia and neuronal loss (Ramon-Duaso et al., 2020). Moreover, metformin treatment reversed the metabolic and mitochondrial dysfunction and the accelerated aging process, such as impaired proliferation, in fibroblasts derived from DM1 patients (García-Puga et al., 2020). And its beneficial effects on muscle function have been confirmed in several clinical trials (Bassez et al., 2018). Since basic science reports have proposed the important role of abnormal insulin signaling in the brain (Nieuwenhuis et al., 2019), it indicates another possible therapeutic mechanism for metformin in DM1 brain. Furthermore, therapies via modulating glutamate levels and dopaminergic function have also emerged to attract more and more attention (Sicot et al., 2017; Serra et al., 2020b), which provide new insights for future DM1 CNS treatment.
Great progress of molecular therapies has been made in DM1, however, before we can move these treatments into clinical trials, there is also a great need to identify feasible outcome measures to evaluate the effectiveness of these therapies by characterizing the component of CNS deficits in DM1. Currently, the main outcome measures actually used to study CNS involvements in DM1 patients are represented by a variety of patient questionnaires and clinical neuropsychological tests which can assess cognitive and behavioral dysfunctions, and computerized neuroimaging techniques which can assess neuroimaging alterations (summarized in Tables 2–4). Besides, given the high heterogeneity of symptoms in DM1 patients, several patient-reported outcome measures are also developed, such as DM1 activity and participation scale for clinical use (DM1-ActivC), and the fatigue and daytime sleepiness scale (FDSS) (Hermans et al., 2013, 2015), which have shown valid measurements in DM1 populations (Angelini and Siciliano, 2020). Notably, due to the increasing risk and higher severity of COVID-19 in DM1 patients, and the difficulty in contacting a doctor during the COVID-19 epidemic, the Italian Association for Glycogenosis (AIG) developed telemedicine equipment, the AIGkit application (AIGkit app), which allows patients to receive constant remote monitoring by using dedicated questionnaires and getting personalized treatment (Symonds et al., 2017). A review by Simoncini et al. (2020) comprehensively summarized the clinical instruments available for cognitive and behavioral measures and neuroimaging assessment in DM1 brain. In the future, additional investigations are warranted to improve the reliability of these available measures, as well as to discover the new outcome markers.
Potential Therapeutic Strategies
With significant advances in understanding the molecular pathogenesis of DM1, several approaches targeting disease mechanisms have been developed, such as antisense oligonucleotide (ASO)-based therapy, small-molecule therapy, genome editing, non-coding RNAs (ncRNAs)-based therapy, and iPSC technique, which target different steps in the pathological process of DM1 (Mulders et al., 2010; Magaña and Cisneros, 2011). Focusing on the most upstream target should block the initiation of the toxic cascade and correct more defects in tissues. Since most therapeutic efforts of DM1 mainly focused on the myocardium and skeletal muscle, and only few studies described their effects on the neurological defects, we briefly introduce their roles in the treatment of DM1 and their therapeutic potential in CNS deficits, hoping to provide references for DM1 CNS treatment. Figure 2 specifically introduced the current management measures as well as promising therapeutic strategies expected to be applied to DM1 CNS disorders.
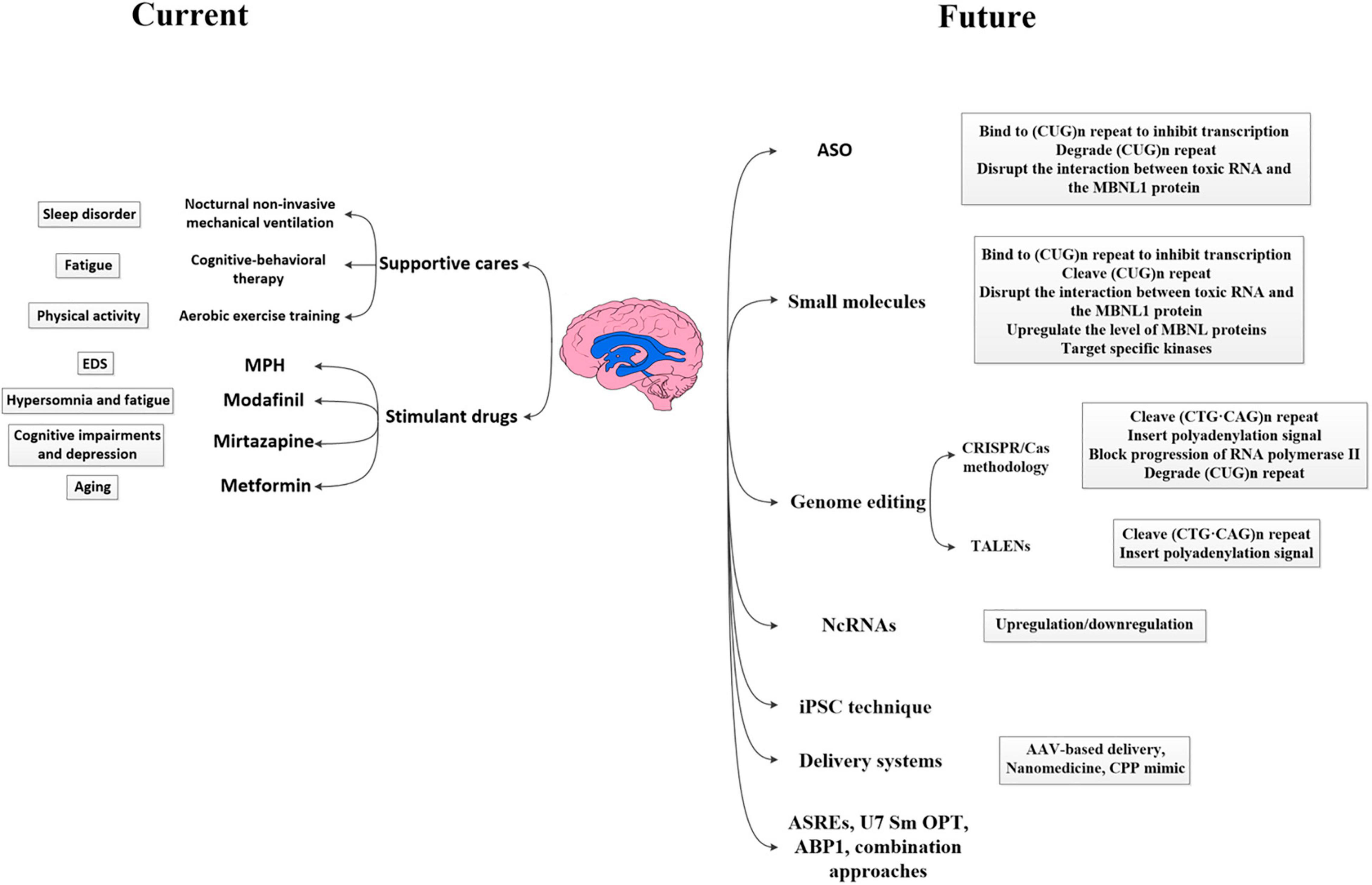
Figure 2. Current and future therapeutic strategies applied to DM1 CNS disorders. The therapeutic strategies for DM1 CNS disorders can be divided into available management measures and future promising strategies. Available modalities for management mainly rely on supportive care, including nocturnal non-invasive mechanical ventilation, cognitive–behavioral therapy, aerobic exercise training, and careful use of stimulant drugs such as MPH, modafinil, mirtazapine, and metformin. Future potential therapeutic strategies currently investigated include ASO-based therapy, small-molecule therapy, genome editing, ncRNAs-based therapy, iPSC technique and so on, which target different levels in the pathological process of DM1 (i.e., DNA level, RNA level, and protein level). Besides, the application of various effective delivery systems also improved the transportation efficiency and promoted the wide distribution of drugs. DM1, myotonic dystrophy type 1; MPH, methylphenidate; EDS, excessive daytime sleepiness; NcRNA, non-coding RNAs; iPSC, induced pluripotent stem cell; AAV, adeno-associated viral vector; ASREs, artificial site-specific RNA endonucleases; CPP, cell-penetrating peptide; ABP1, D-amino acid hexapeptide; ASO, antisense oligonucleotide; CRISPR-Cas9, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system 9; TALEN, transcription activator-like effector nucleases; MBNL, muscleblind-like.
Antisense Oligonucleotides
Antisense oligonucleotide is one of the important approaches which can target toxic RNA. It consists of a strand of nucleotides that can bind to a specific pre-mRNA/mRNA sequence and then alters protein synthesis through several mechanisms. In DM1, ASOs can interfere with the interaction between MBNL1 protein and toxic RNA, mainly by targeting the CUG repeat to reduce mutant transcripts, or by RNase-H-mediated degradation of expanded transcription (Klein et al., 2015). Other alterative mechanisms including inhibiting mRNA translation or altering RNA stability (Bennett and Swayze, 2010; Schoch and Miller, 2017).
Till now, various kinds of ASOs have shown efficacy in vitro and in vivo DM1 treatments. In 2003, scientists produced a retrovirus that expressed a 149-base pair (bp) antisense RNA, complementary to the (CUG)13 repeats and the proceeding 110-bp region (Furling et al., 2003). Injection of this ASO into human DM1 myoblasts significantly decreased toxic RNA and normalized CELF1 levels, eventually ameliorating the delay of muscle fusion and IR (Furling et al., 2003). However, this approach also reduced the level of normal DMPK transcripts and proteins. Since the unmodified ASOs are unstable and could easily be degraded, several chemical modifications to ASOs were developed to increase their stability and affinity for the target mRNA (Bennett et al., 2017; Khorkova and Wahlestedt, 2017). The first and the most widely used generation of modification was the phosphorothioate (PS) backbone modification (Bennett et al., 2017; Schoch and Miller, 2017), usually together with sugar modifications such as 2′-O-methyl (2′-O-Me) and 2′-O-methoxyethyl (2′-MOE). In 2009, a fully 2′-O-Me-PS-modified ASO, complementary to CUG repeats, called CAG7, was developed for DM1. Administration of this ASO in muscle tissue of DM500 mice (a DM1 model carrying > 300 DMPK CTG repeats) and HSALR20b mice (a DM1 model expressing human skeletal actin transcripts containing ∼250 DMPK CTG repeats) silenced the expression of mutant RNA and decreased the formation of nuclear foci in a selective and (CUG)n-length-dependent manner (Mulders et al., 2009). Subsequently, morpholino ASO was discovered, which can bind to the toxic RNA and inhibit its interactions with proteins as well as disrupt CUG-exp-MBNL1 complexes. CAG25 was the first morpholino ASO to be used in DM1. Injection of CAG25 into muscle fibers of HSALR mice by intramuscular injection followed by in vivo electroporation significantly reversed myotonia within 4–5 weeks, accompanied with the increased translation of the mutant RNA (Wheeler et al., 2009). Since morpholino ASO does not induce cleavage of target mRNA, they do not affect normal DMPK transcripts and proteins (Summerton, 1999).
To date, it is still a question whether DMPK knout-out will indeed cause DM1 phenotype. Some studies reported that both DMPK± and DMPK-/- mice shown abnormal cardiac conduction (Berul et al., 1999, 2000), and homozygous deletion also exhibited skeletal myopathy and muscle weakness (Reddy et al., 1996), while other findings countered that the administration of DMPK-targeting ASOs with heterozygous deletion did not influence the normal cardiac or muscle function in mice, thereby supporting the feasibility and safety of ASOs usage in DM1 (Carrell et al., 2016). In the future, more relevant experiments are needed to reach a conclusion.
Since nuclear-retained CUG repeats are sensitive to antisense silencing, and RNase H are essentially ubiquitous expressed in nuclear (Suzuki et al., 2010), the recruitment of diverse gapmer-based ASOs shown promising futures via mediating RNase H cleavage and decay of the target RNA (Walder and Walder, 1988; Wheeler et al., 2012; Nguyen and Yokota, 2020). In HSALR mice, subcutaneous injection of gapmer ASOs significantly degraded expanded CUG transcripts as well as lncRNAs in skeletal muscle, reversing MBNL1 sequestration, myotonia, and mis-splicing without apparent off-target effects (Schoch and Miller, 2017). This strategy is also more attractive since it’s highly specific to expanded CUG repeats compared with normal-size repeats, and could maintain effects for 1 year after the treatment. Besides, BNANC gapmers targeting the DMPK 3′ UTR specifically knockdown the expanded CUG RNA and reversed the mis-splicing and RNA foci accumulation without inducing caspase activation (Manning et al., 2017). Moreover, the combinative application of gapmer and CAG25 morpholino produced synergistic effects to reduce expanded CUG repeats by 80% and almost eliminate RNA foci in DM1 cell culture, and get a smaller CUG repeats reduction in skeletal muscle of DM1 mice by half (Lee et al., 2012). However, the competition between gapmer and CAG25 in targeting the CUG repeats might limit their effects.
Other chemical modifications of ASOs also significantly optimized its characteristics. For example, the establishment of modified human U7 small nuclear RNAs (hU7-snRNAs), which contain a poly-CAG antisense sequence targeting the mutant CUG repeats, specifically degraded toxic RNA transcripts without influencing the products of wild-type DMPK alleles (Francois et al., 2011). Since the instability of expanded CTG repeats is a critical characteristic of DM1, which could be enhanced by RNA repeats, recent work shown that early intervention with CAG-repeat ASOs can not only reduce RNA toxicity but also stabilize CTG:CAG repeats at subpathogenic lengths in both DM1 human cells and transgenic mice model (Nakamori et al., 2011). One study using a 2′-4′-constrained ethyl-modified (cEt) ASO (ISIS 486178) effectively decreased DMPK mRNA levels in multiple organs of DMSXL mice or cynomolgus monkeys (Pandey et al., 2015). This ASO also exhibits a high level of RNA binding affinity and in vivo potency, without any association with muscle or cardiac toxicity. Furthermore, the systemic treatment with ASO (ISIS 486178) targeted to the non-CUG sequence within the 3′-UTR of DMPK also specifically rescued DM1 phenotypes of myotonia and cardiac conduction defects in DM200 mice [a DM1 model carrying a GFP-DMPK 3′-UTR (CTG) 200 transgene] (Jauvin et al., 2017; Yadava et al., 2020). In order to overcome the low efficiency of ASOs due to its wide distribution, Klein et al. (2019) developed an arginine-rich 466 cell-penetrating peptide (CPP). Compared with previous ASO strategies, this Pip6a-conjugated morpholino phosphorodiamidate oligomer (PMO) significantly increased ASO delivery into striated muscles after systemic administration in HSALR mice. Only low-dose treatment with Pip6a-PMO-CAG sufficiently rescued splicing defects and myotonia in mice (Klein et al., 2019). Hsieh et al. (2018) designed a short miniPEG-γ peptide with terminal pyrenees, which also exhibited high affinity and sequence specificity to toxic RNA repeats and successfully disrupted the CUG-exp–MBNL1 complex. With the new discovery of the imbalance in the splice isoform profile of DMPK in DM1 (Groenen et al., 2000; Wansink et al., 2003), two chemically modified ASOs were developed to prompt exon skipping from mRNA and degrade the CUG repeat from pre-mRNA in fibroblasts of DM1 patients. Disappointingly, neither strategy was as successful as predicted, but they did improve the DM1-related molecular phenotypes (Stepniak-Konieczna et al., 2020). It’s worth noting that a newly selected ASO called ISIS-DMPKRx (ISIS 598769) gapmer-type candidate has been evaluated in a Phase 1/2a clinical trial for the treatment of DM1 (Lim et al., 2017), for it can bind to a specific 3′-UTR gene sequence outside the CUG repeat and degrade toxic RNA (NCT023412011). Though it presented great safety and tolerance, IONIS proposed an inefficient effect on the functional and biological endpoints set in the trial since no sufficient drug could reach the muscles.
Though a variety of modifications have improved pharmacokinetic and pharmacodynamic properties of ASOs, the systemic and tissue-targeted implementation of ASOs are still challenging due to their poor intracellular uptake in some tissues (particularly skeletal muscle, heart and brain) (Muntoni and Wood, 2011). To overcome this disadvantages, different administration routes were developed to increase the tissue specificity. Nowadays, the delivery of ASOs to a single muscle by intramuscular injection have been frequently used in DM1 pre-clinical trials. Given the multi-organ or systemic features of DM1, systemic delivery via intraperitoneal, subcutaneous, or intravenous administration results in a rapid and widespread absorption of ASOs to various peripheral tissues (Yin et al., 2008; Hua et al., 2011). Since the highly charged ASOs cannot cross the BBB (Smith et al., 2006), intraventricular or intrathecal injections were invented to directly introduce ASOs into the CSF or parenchyma, which highly increased the delivery efficiency and ensured adequate distribution of drugs in the CNS (Miller et al., 2013; Chiriboga et al., 2016). However, though intrathecal delivery is currently shown safe and well-tolerated, it’s relatively invasive compared with other administrations. Identification of easier ASO delivery routes to the CNS such as intranasal administration may be an important step to promote their translation to human clinical trials. In addition, assisted delivery systems such as CPPs (Lebleu et al., 2008; Lehto et al., 2012; Boisguérin et al., 2015), nanoparticles (Wang et al., 2015) and adeno-associated virus (AAV) vectors (Danos, 2008), also increase the efficacy of ASOs, which would be introduced below.
Small Molecules
Recently, small-molecule-related strategies have received increasing attention in the treatment of DM1. Compared to other strategies, small molecules have many benefits, including low manufacturing cost, better oral delivery with shorter half-lives, longer shelf lives than other biologics, and sufficient biodistribution to affect multiple systems. Since a number of small molecules are existing drugs that explored for new applications, it also reduces the development time and potential risks of toxicity. However, despite the multiple advantages, most small molecules currently established can only target downstream processes to reduce toxic RNA or alter protein levels but cannot correct gene mutations. Meanwhile, their instability in vivo also limit their uses.
The mechanisms of molecular therapies can be divided into four aspects: inhibiting transcription of mutant RNA, cleaving CUG repeats, disrupting the interaction between toxic RNA and the MBNL1 protein, or targeting downstream pathways (Mulders et al., 2010; López-Morató et al., 2018; Reddy et al., 2019a). Cell and animal studies have indicated that pentamidine and a series of methylene linker analogs could exert beneficial effects by binding the CTG repeat DNA to inhibit transcription (Coonrod et al., 2013). One study using a DM1 HeLa cell model screened out that multiple microtubule inhibitors can target the toxic CUG RNA to reduce r(CUG)480 levels and then rescue mis-splicing to some extent, wherein the clinical microtubule inhibitor colchicine could even make positive effects in HSALR mice and primary DM1 patient-derived cells (Reddy et al., 2019b). This strategy provides a new avenue for DM1 research and suggests an alternative method of repeat-selective screening. A new molecule, JM642, was reported to have the capacity to bind to the expanded r(CUG) repeat and disrupt ribonuclear foci in the C2C12 DM1 cells and HSALR mice, finally rescuing mis-splicing (Nakatani et al., 2020). Moreover, several potential therapeutic molecules focus on cleaving the aberrant CUG repeats from disease-affected cells. For example, cugamycin is a small molecule that has been confirmed to selectively bind expanded CUG repeat conjugated to a bleomycin A5-cleaving module to cleave expanded CUG repeat. And deglycobleomycin, an analog in which the carbohydrate domain of bleomycin A5 is removed, significantly improves its selectivity by reducing DNA damage as well as maintaining the cleave ability (Angelbello et al., 2020). The major small-molecule compounds identified in DM1 therapy have been summarized in Table 5.
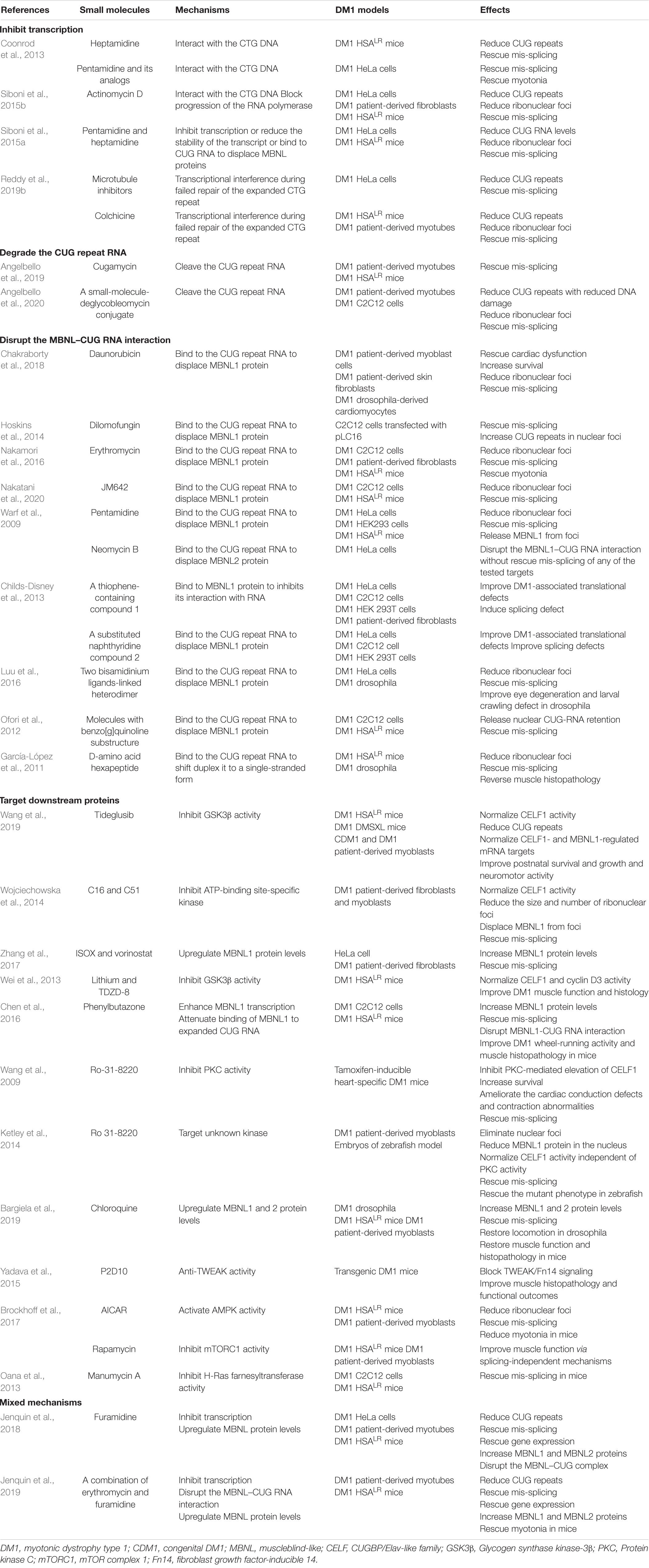
Table 5. Small molecules in DM1 treatments.
Some promising small molecules have even been tested in clinical trials. For example, AMO-02/tideglusib, a GSK-3β enzyme inhibitor, has been investigated for congenital DM by restoring the expression of CELF1 (Jones et al., 2012; Wang et al., 2019), which improved postnatal survival, weight, and neuromotor activity. Till now, a Phase II clinical trial of tideglusib on patients with congenital and juvenile-onset DM1 has already finished, and most participants presented with improved CNS and clinical neuromuscular performances (Horrigan et al., 2020). And a Phase II/III clinical trial on patients with congenital-onset DM1 is ongoing. In addition, MYD-0124 (erythromycin) and ERX-963 have been shown to bind to the CUG hairpin with high selectivity, reduce nuclear foci and reverse mis-splicing in DM1 vitro and vivo models. A Phase II clinical trial on adult patients with DM1 is currently underway to investigate the clinical effects of erythromycin after oral administration (Jenquin et al., 2019). Other molecules evaluated in clinical trials for specific disease symptoms (e.g., insulin resistance phenotype, myotonia, myalgia, or daytime sleepiness) include metformin, mexiletine, ranolazine, cannabinoids, pitolisant, Caffeine, and theobromine formulation MYODMTM (Kouki et al., 2005; Logigian et al., 2010; Laustriat et al., 2015; Bassez et al., 2018; Vita et al., 2019; Heatwole et al., 2021; reviewed in Pascual-Gilabert et al., 2021).
Genome Editing
Compared with ASO therapy, the development of genome editing provides an opportunity for permanent corrections of gene mutation, which mainly includes clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (Cas) methodology and transcription activator-like effector nucleases (TALENs) (Richard, 2015; Lee et al., 2016; Long et al., 2016; Nelson et al., 2017; Raaijmakers et al., 2019).
The CRISPR/Cas methodology can be used to target a specific genomic locus in the genome of eukaryotes (Knott and Doudna, 2018). Specifically, the Cas endonuclease is complexed with a small guide RNA (sgRNA) to target a specific genomic locus. Upon binding, the Cas protein generates a double-strand DNA (ds DNA) break by cleaving the DNA in both strands, thereby correcting the genetic defect (Raaijmakers et al., 2019). The main advantage of this strategy is to eliminate the disease defect at the DNA level, so the mutant transcripts and downstream dysregulations are not produced.
So far, the effects of CRISPR/Cas methodology have been validated. In 2017, one study reported that dual cleavage at either side of the CTG expansion can lead to complete and precise excision of the repeat tract from DMPK alleles in DM500 cells (myoblasts carrying 500 DMPK CTG repeats), myoblasts of DM1 patients, and unaffected individuals. And it also prevented damage to genes in the DM1 locus (van Agtmaal et al., 2017). Then, a CRISPR-Cas9 system from Staphylococcus aureus (Sa) was developed to cleave the CTG repeats in the human DMPK locus. A single intramuscular injection of recombinant AAV (rAAV) vectors expressing CRISPR-SaCas9 and selected sgRNAs has been shown to successfully delete the mutant CTG repeats in muscle fibers and reduce the RNA foci in myonuclei of DMSXL mice (Lo Scrudato et al., 2019).
Except for targeting the mutant DNA, RNA-targeting Cas9 (RCas9) systems which can bind to single-stranded RNA were also investigated (Strutt et al., 2018). Batra et al. (2017) found that through delivering a truncated RCas9 system in human DM1 cells, the toxic mRNAs were highly eliminated and aberrant splicing was corrected. This approach also has fewer side effects, since it does not affect normal transcripts. However, the delivery of RCas9 would gradually decay in the genome, which means the requirement of repeat treatments. Other scientists also proposed methods to prevent expanded transcription by inserting a homology-directed polyadenylation signal into the DMPK gene (Wang Y. et al., 2018), or recruiting catalytically deficient Cas9 (dCas9) to the repeat effectively to block progression of RNA polymerase II (Pinto et al., 2017). A recent review by Raaijmakers et al. (2019) carefully elaborated CRISPR/Cas-mediated approaches that target the causative mutation in the DNA and the RNA that cause DM1.
In recent years, some novel Cas-associated strategies have gradually emerged. For example, CRISPR-Cas13a is an RNA guided RNase. Zhang et al. (2020) using Leptotrichia shahii (Lsh) Cas13a in DM1 patient-derived myoblasts successfully degraded the expanded CUG RNA and reversed several important mis-splicing events. CRISPR/Cas9-associated base editing (BE) technology is a new therapy that do not rely on a dsDNA break at target sites but directly mediate the conversion of base pairs, and thus reduce the deletions or insertions (Komor et al., 2016; Gaudelli et al., 2017). However, the disability to generate precise edits beyond the allowed mutations might be a huge challenge.
Transcription activator-like effector nucleases is a newly discovered genome editing tool. It relies on modular transcription factors called transcription activator-like effectors, which enables the targeting of any specific DNA sequence (Sun and Zhao, 2013). Previous studies have reported that a dedicated TALEN can induce a dsDNA break into a CAG/CTG tri-nucleotide repeat in heterozygous yeast diploid cells, which shortened the repeat tract with nearly 100% efficacy and very high specificity (Richard et al., 2014). Currently, TALENs has shown great potential in the treatment of DM1. For example, TALENs application corrected the genetic defect of iPSCs, which contribute to the development of autologous stem cell therapy (Xia et al., 2015; Gao et al., 2016). By the way, TALENs seem to be the safest way to shorten trinucleotide repeats to non-pathological lengths, though more research is needed to combat possible off-target effects, immunogenicity to either the genome editing components or delivery particles and unpredictable DNA repair upon cleavage near the unstable repeat.
Non-coding RNAs
NcRNAs are RNA molecules that cannot be translated into proteins but are responsible for important regulatory events in cells (Fabbri et al., 2019). There are several types of ncRNAs, including microRNAs (miRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs). To date, global changes in ncRNA expression patterns in DM1 (Czubak et al., 2019a; López Castel et al., 2019; Voellenkle et al., 2019), as well as their key roles in contributing to DM pathogenesis have been presented (Perbellini et al., 2011; Wheeler et al., 2012; Gudde et al., 2016, 2017; Koutsoulidou et al., 2017; Czubak et al., 2019a,b; López Castel et al., 2019; Koehorst et al., 2020), which make them attractive biomarkers (Gambardella et al., 2010; Perbellini et al., 2011; Rau et al., 2011; Fernandez-Costa et al., 2013; Kalsotra et al., 2014; Perfetti et al., 2014, 2016; Koutsoulidou et al., 2015, 2017; Fritegotto et al., 2017; Koehorst et al., 2020; Pegoraro et al., 2020) and therapeutic targets in DM1 (Rau et al., 2011; Koutalianos et al., 2015; Zhang et al., 2016; Cerro-Herreros et al., 2018, 2020; López Castel et al., 2019; Sabater-Arcis et al., 2020). For example, muscle-specific miRNAs (myomiRNAs) like miR-1, miR-133a, miR-133b, and miR-206 as well as myostatin have been considered as attractive biomarkers of DM1 rehabilitation (Pegoraro et al., 2020). Besides, the therapeutic potential of miRNAs have been detected. Koutalianos et al. (2015) shown that in muscle cells from patients with congenital DM1, upregulation of the miR-206 expression by transfection with a miR-206 mimic into cells overexpressing CEFL1 induced myogenesis by inhibiting the expression of CELF1 and Twist-1. Replenishing of miR-7 with agomiR-7 reversed DM1 myoblast fusion defects and myotube growth, while blocking of miR-7 mediated by oligonucleotide worsen the outcomes (Sabater-Arcis et al., 2020). Silencing the regulatory dme-miR-277 and dme-miR-304 by miRNA sponge constructs successfully upregulated MBNL expression at the RNA and protein levels in a DM1 drosophila model, which then rescued mis-splicing and reduced muscle atrophy (Cerro-Herreros et al., 2016). This attempt evaluated miRNA sponge constructs as a powerful and attractive strategy to treat DM1 by blocking specific miRNAs. In recent years, RNA interference (RNAi) technology has attracted increased attention in DM1 (Bisset et al., 2015). Bisset et al. (2015) reported that miRNA-based RNAi hairpins delivered by rAAV vectors significantly downregulated mutant transcripts and reduced muscle pathology in HSALR mice. Besides, the intramuscular injection and electroporation of synthetic short interfering RNAs (siRNAs) significantly reduced toxic RNA transcripts and nuclear foci in HSALR mice (Sobczak et al., 2013). Antisense technology (antagomiRs) is a newly developed approach to block specific miRNA. Cerro-Herreros et al. (2020) reported that subcutaneous administration of antagomiR-23b in HSALR mice strongly increased the level of MBNL1 protein and reversed mis-splicing, grip strength, and myotonia in a dose-dependent manner. However, despite the huge potential of miRNA-based interventions, most of these attempts are still in preclinical phases because of their instability and delivery deficits.
Induced Pluripotent Stem Cell Technique
Nowadays, different differentiated cell models, such as neurons or muscle cells, have been used to investigate pathological mechanisms and to evaluate therapeutic strategies of DM1 before clinical validation (Larsen et al., 2011). However, these cell models are strongly limited due to the low DMPK transcript levels in affected cells and genetic background variation. Thus, there is a great need to generate alternative myogenic models that can be reliably used for in vitro disease modeling and/or drug screening purposes. It’s well-known that iPSCs are self-renewal and can differentiate into any cell type, including neurons and muscles cells. Since genome editing methodology could correct the genetic defect of iPSCs as mentioned before, the various phenotypes observed in DM1 can be subsequently reversed by correcting the lengths of CUG repeats in iPSCs and iPSC-derived cells, thereby offering a great translational platform for therapeutic development. Combining human iPSC lines and genome editing technology to create isogenic cell lines can also eliminate background genetic variation that might affect the expected results. In addition, reprogramming of somatic cells to iPSCs has been reported a valuable tool for disease modeling and drug discovery. Mondragon-Gonzalez and Perlingeiro (2018) established two DM1 iPSC lines from patient-derived fibroblasts, which then be differentiated into myotubes. The iPSC-derived myotubes highly recapitulate the molecular features of DM1 while ASO treatment successfully abolished RNA foci and rescued BIN1 mis-splicing. These results indeed confirmed DM1 iPSCs a kind of valuable alternative myogenic model to study DM1 pathogenesis and screen candidate drugs.
Assisted Delivery Systems
The inefficacy of conventional drug delivery and low bioavailability of drugs led to the rapid progress of assisted delivery systems in recent years. Currently, a number of delivery strategies have shown great potential in enhancing the efficacy of therapeutic molecules while minimizing their off-target effects, which can be broadly divided into four types: polymeric, peptide, lipid, and viral delivery systems, wherein AAV-based gene therapy has emerged to be a potent and promising therapeutic tool for DM1. Compared with other strategies, AAV vectors permanently change genetic defects and avoid repeated administrations. Given the multisystemic symptoms of DM1, they can also achieve systemic delivery (Gregorevic et al., 2004; Arruda et al., 2005). Kanadia et al. (2006) reported that upregulation of MBNL1 by AAV-mediated transfection to skeletal muscle effectively reversed mis-splicing and myotonia in HSALR mice. Besides, systemic AAV-delivered RNAi significantly improved disease phenotype in HSALR mice (Bisset et al., 2015). Currently, a possible AAV-delivered ASO, AT466 is being established to reduce the toxic RNA levels in cells derived from DM1 patients by RNA degradation, exon skipping or both (Audentes).1 Recent efforts on viral delivery to the CNS have also taken an exciting leap forward (Miller et al., 2012). The delivery of AAV9 via intravenous administration could traverse the BBB in both neonate and adult animals (van der Bent et al., 2018), which provides a promising tool to treat CNS disorders.
The hydrophilic nature as well as large size and high charge of peptides and proteins prevents their penetration across biological membranes. In order to achieve delivery of therapeutic peptide and protein into cells as well as across epithelial barriers and the BBB, a family of delivery vectors called CPPs has been developed, which shown the potential to traverse cellular membranes and promote the uptake of therapeutic peptides with lower toxicity. To date, CPPs have been applied for intracellular, transepithelial, and transendothelial delivery of various therapeutic cargos. Regarding DM1, one study using the CPP mimic as a scaffold to assemble the multivalent ligand construct significantly increased their binding affinity, which then contributed to phenotypic improvement in a DM1 drosophila model (Bai et al., 2016). Compared to unconjugated PMO, systemic administration of Pip6a-conjugated morpholino PMO remarkably enhanced ASO delivery into DM1 mice muscles. Besides, several CPPs have also been identified to mediate cargo delivery across the BBB and exerted protection in the brain (Kilic et al., 2003, 2004; Dietz et al., 2006). And the combination of CPPs and intranasal administration may further enhance CNS delivery (Kamei and Takeda-Morishita, 2015; McGowan et al., 2016; Khafagy et al., 2020; Akita et al., 2021). However, the BBB-specific CPPs remain not to be found and the mechanisms driving the transportation of CPPs are still unknown.
In order to overcome the instability, limited distribution, rapid degradation and toxicity of therapeutic molecules, nanomedicine has been rapidly developed as an effective drug delivery system in recent years, which greatly improved the efficiency and tissue compatibility of gene therapy, increased safety, and ensured systemic distribution (Koebis et al., 2013; Hermans et al., 2015; Lee et al., 2017; Amini et al., 2019), relying on their adjustable physicochemical properties to prevent toxicity and to carry specific biological molecules to target sites (Andreana et al., 2021). In HSALR mice, using bubble liposomes as delivery tools significantly improved the delivery efficiency of PMO into muscles, which then increased the expression of chloride channel 1 (Clcn1) protein in skeletal muscle and ameliorated the myotonia (Koebis et al., 2013). Besides, given the inefficiency of macromolecules to cross the BBB, the small nanocarriers-mediated transport, including active transport (e.g., receptor-mediated endocytosis) and facilitated diffusion, also provided a promising pathway for smooth BBB passage of large cargos and targeting various cells with intracellular localization specificity (Hernando et al., 2018; Di Filippo et al., 2021; Nehra et al., 2021; Song et al., 2021). Toward future directions, the combinative applications of these delivery vectors and administration routes would attracted more and more attentions since they provide numerous chances for the discovery of both safe and effective delivery strategies to avoid the side effects of any signal technique.
Other Potential Strategies
Artificial site-specific RNA endonucleases (ASREs) are newly discovered molecules specifically targeting mutant RNA accumulated in the nucleus. The results of Zhang et al. (2014) shown that ASRE treatment significantly decreased nuclear foci formation and reversed the mis-splicing of DM1-related genes with few side effects on wild-type alleles. U7 small nuclear ribonucleoproteins (snRNPs) are a specific type of snRNPs that do not participate in splicing mediation but is a key factor in the unique 3′ end processing of replication-dependent histone (RDH) pre-mRNAs. The modified U7 snRNP (U7 Sm OPT) has been used as a promising tool for gene therapy in splicing defects-associated diseases by targeting splicing to induce efficient skipping or inclusion of selected exons. It has multiple advantages, such as small size, good stability, ability to accumulate in the nucleus without toxicity and immunoreactivity, and low risk of transgene dysregulation. In addition, using U7 Sm OPT as a tool in gene therapy also ensures lifelong treatment. Gadgil and Raczyńska et al. (2021) demonstrated that incorporating ASO into the U7 Sm OPT successfully avoided repeated administration. Injection of U7 Sm OPT containing ASO with 15 CAG repeats in skeletal muscle cells isolated from DM1 patients resulted in long-time improvement in splicing and differentiation defects in a dose-dependent manner, without affecting the wild-type DMPK transcripts (Le Hir et al., 2013). The critical properties of U7 snRNP might deploy it as a new tool in gene therapy in the future. In a DM1 drosophila model, researchers screened a D-amino acid hexapeptide (ABP1) which can induce the CUG hairpin into a single-stranded conformation and bind the CUG RNA without displacing MBNL1. Compared to ASOs, this method avoids affecting endogenous transcripts. In fly eyes and muscles, overexpression of natural, L-amino acid ABP1 analogs reduced RNA toxicity. And in HSALR mice, ABP1 reversed muscle histopathology and partially rescued mis-splicing of MBNL1 targets (García-López et al., 2011). Notably, combination approaches such as different small molecules or gene therapies targeting different processes have also attracted increasing attention. This may produce greater benefits in disease modulation than simply additional effects. Importantly, this combination treatment might also reduce off-target effects.
Therapeutic Strategies Targeting Central Nervous System
The current advances in therapeutic strategies should also be applicable in principle to CNS since the pathogenesis of CNS deficits is similar to that of other organs, such as RNA toxicity and splicing defects. However, the existence of BBB hinders the delivery and distribution of drugs in the brain. Therefore, the ideal therapeutic agents need to be able to cross the BBB readily. Some improvements have been made, such as intracellular delivery of the therapeutic molecules to promote uptake, administering molecules intracerebroventricularly or intrathecally (Baughan et al., 2009; Geary et al., 2015), or regulating the molecule size and charge to achieve an efficient delivery to the brain. Recent advances also found that intranasal delivery efficiently bypasses the BBB and highly increases the CNS concentrations of drugs and is non-invasive. Besides, the combination of U7 methodology with highly efficient AAV-mediated delivery, receptor-mediated endocytosis of ASOs, and nanoparticles-, exosomes- or CPP-based delivery of large cargos also favor the BBB passage and the higher distributions of drugs in the CNS (Foust and Kaspar, 2009; Krupa et al., 2014; McGowan et al., 2015, 2016; Bai et al., 2016; Kristensen et al., 2016). In conclusion, all of these therapeutic agents, administration routes, and assisted delivery systems create numerous chances for the discovery of effective therapies for CNS disorders.
Several molecules applied for CNS treatment have shown positive effects in preclinical trials, which may provide new thoughts for DM1 CNS treatment. For example, in mice overexpressing APP, intracerebroventricular injections of PS-modified ASOs significantly reduced the expression of APP protein and improved learning and memory deficits (Kumar et al., 2000). In the Alzheimer’s disease (AD) mouse model, a designed 2′-O-Me-PS-modified ASO sustainly increased exon 19 splicing of apolipoprotein E receptor 2 (ApoER2) as well as restored synaptic function and learning and memory (Hinrich et al., 2016). Besides, an 2′MOE-modified ASO called IONIS MAPTRx (ISIS 814907) has been evaluated in a phase I/II study in patients with mild AD (NCT03186989), as it highly reduced the expression of tau protein through targeting MAPT mRNA. Hu et al. (2021) using a 20-mer RNase H-active gapmer ASO combined with a 3-month exercise training program in old HSALR mice reversed all measures of fatigue, though they did not detect the index of fatigue due to CNS dysfunctions. These findings may provide new thoughts for DM1 CNS treatment. Murlidharan et al. (2016) discovered a lab-derived AAV chimeric (AAV2g9), which has favorable CNS properties derived from both parental counterparts, AAV2 and AAV9. Administration of CRISPR/Cas9 with this synthetic AAV vector into the CSF minimized systemic leakage and reduced the sequestration and gene transfer in off-target organs.
The modification of stem cells also offers a new chance for CNS treatment. Previous studies have shown that NSC derived from ES cells or iPSCs could present critical features of DM1 (Marteyn et al., 2011; Denis et al., 2013; Xia and Ashizawa, 2015; Xia et al., 2015). In DM1 NSCs, insertion of poly A signals upstream of DMPK CTG repeats by TALEN-mediated homologous recombination significantly eliminated mutant transcripts and nuclear RNA foci, corrected mis-splicing, and ultimately reversed phenotypes (Xia et al., 2015; Gao et al., 2016). Despite the broad prospects of strategies in CNS treatment, there are still many challenges, such as lower efficiency and distribution to CNS and safety regarding immune response and gene therapy specificity. In addition, no studies have discussed the targeted brain cell types of different strategies and the corresponding alterations of brain cells after treatment. The resolution of these issues may provide a deeper understanding of the therapeutic mechanisms of drugs.
Limitations and Future Directions
Though great progresses have been made, there are still many limitations in current studies that hinder the understanding of the complex nature of CNS defects and the development of new treatments. Firstly, despite the current knowledge of the molecular basis underlying CNS defects, it is still unclear how these molecular alterations could be translated into specific pathological changes and clinical symptoms, nor do we know how these deficits progress over time. In the future, more longitudinal studies with large sample size are needed to fully understand the DM1 neuropathology and its progression. Secondly, although several cell and animal models have been developed to reproduce the DM1 gene mutation and pathogenesis similar to the human phenotype, such as MBNL-loss or CELF-overexpressing phenotypes, or in vitro and ex vivo alternatives which can assist animal experimentation, none of them completely recreate the multisystemic phenotypes of DM1, which block the in-depth assessment of DM1 defects and effective evaluations of feasible interventions. Thirdly, more applicable patient questionnaires and clinical neurocognitive evaluation protocols should be investigated to better identify the disease component and serve as potential markers to evaluate the effectiveness of therapeutic strategies. Fourthly, at the pharmacotherapeutic level, more efforts are needed to improve drug delivery efficiency, biodistribution and availability, and reduce toxicity. Meanwhile, the design of drugs for CNS treatment must be able to readily cross the BBB.
Conclusion
In this review, we primarily focus on CNS involvement in DM1, outlining the primary pathological alterations and pathogenesis underlying CNS. Then, we highlight promising therapeutic strategies for DM1. Excepting for some available drugs targeting for specific neurological impairments, the promising results of some biological molecules (i.e., ASO, small molecules, CRISPR/Cas9, ncRNA.) via targeting mutant DNA, RNA, or downstream proteins in preclinical models or even clinical trials have also provided relevant candidates for DM1 treatment. Particularly, a variety of re-purposed small molecules drugs have been evaluated in DM1, which present with low manufacturing cost, greater safety and stability in vivo treatment. Meanwhile, the development of screening and rational design technologies also promote the production of newly potent small molecules, which increases the availability of small molecule therapy in DM1 in the near future. In principle, these interventions should be applicable to the CNS, since strategies such as eliminating toxic RNAs, reducing the formation of nuclear foci, or restoring the levels of splicing-related factors are also effective in brain cells, however, additional researches are still needed to improve the understanding of DM1 progression and to transfer available therapeutic strategies and knowledge into actionable clinical applications.
Author Contributions
JL wrote the first draft of the manuscript. X-LY and Z-NG prepared the figures. SH and YY reviewed and edited the manuscript. All authors contributed to critical revision of the manuscript and approved the final manuscript for submission.
Funding
This work was supported by the National Natural Science Foundation of China (81971105) to Z-NG, the Program for JLU Science and Technology Innovative Research Team (2017TD-12), and the Jilin Provincial Key Laboratory (20190901005JC) to YY.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Akiguchi, I., Nakano, S., Shiino, A., Kimura, R., Inubushi, T., Handa, J., et al. (1999). Brain proton magnetic resonance spectroscopy and brain atrophy in myotonic dystrophy. Arch. Neurol. 56, 325–330. doi: 10.1001/archneur.56.3.325
Akita, T., Kimura, R., Akaguma, S., Nagai, M., Nakao, Y., Tsugane, M., et al. (2021). Usefulness of cell-penetrating peptides and penetration accelerating sequence for nose-to-brain delivery of glucagon-like peptide-2. J. Control. Release 335, 575–583. doi: 10.1016/j.jconrel.2021.06.007
Amini, M. A., Abbasi, A. Z., Cai, P., Lip, H., Gordijo, C. R., Li, J., et al. (2019). Combining tumor microenvironment modulating nanoparticles with doxorubicin to enhance chemotherapeutic efficacy and boost antitumor immunity. J. Natl. Cancer Institute 111, 399–408. doi: 10.1093/jnci/djy131
Anderson, P. J. (2013). Leader of the pack - neuropsychological assessment, 5th Edition, Muriel Lezak, Diane B. Howieson, Erin D. Bigler, & Daniel Tranel. J. Int. Neuropsychol. Soc. 19, 488–489. doi: 10.1017/S1355617713000337
Andreadis, A. (2012). Tau splicing and the intricacies of dementia. J. Cell. Physiol. 227, 1220–1225. doi: 10.1002/jcp.22842
Andreana, I., Repellin, M., Carton, F., Kryza, D., Briançon, S., Chazaud, B., et al. (2021). Nanomedicine for gene delivery and drug repurposing in the treatment of muscular dystrophies. Pharmaceutics 13:278. doi: 10.3390/pharmaceutics13020278
Angeard, N., Gargiulo, M., Jacquette, A., Radvanyi, H., Eymard, B., and Héron, D. (2007). Cognitive profile in childhood myotonic dystrophy type 1: is there a global impairment? Neuromusc. Disord. 17, 451–458. doi: 10.1016/j.nmd.2007.02.012
Angeard, N., Jacquette, A., Gargiulo, M., Radvanyi, H., Moutier, S., Eymard, B., et al. (2011). A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromusc. Disord. 21, 468–476. doi: 10.1016/j.nmd.2011.04.009
Angelbello, A. J., DeFeo, M. E., Glinkerman, C. M., Boger, D. L., and Disney, M. D. (2020). Precise targeted cleavage of a r(CUG) repeat expansion in cells by using a small-molecule-deglycobleomycin conjugate. ACS Chem. Biol. 15, 849–855. doi: 10.1021/acschembio.0c00036
Angelbello, A. J., Rzuczek, S. G., McKee, K. K., Chen, J. L., Olafson, H., Cameron, M. D., et al. (2019). Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U.S.A. 116, 7799–7804. doi: 10.1073/pnas.1901484116
Angelini, C., and Pinzan, E. (2019). Advances in imaging of brain abnormalities in neuromuscular disease. Ther. Adv. Neurol. Disord. 12:1756286419845567. doi: 10.1177/1756286419845567
Angelini, C., and Siciliano, G. (2020). Neuromuscular diseases and Covid-19: advices from scientific societies and early observations in Italy. Eur. J. Transl. Myol. 30:9032. doi: 10.4081/ejtm.2019.9032
Angelini, C., and Tasca, E. (2012). Fatigue in muscular dystrophies. Neuromusc. Disord. 22(Suppl. 3), S214–S220. doi: 10.1016/j.nmd.2012.10.010
Annane, D., Miller, R., and Barnes, P. (2002). Psychostimulants for hypersomnia (excessive daytime sleepiness) in myotonic dystrophy. Cochrane Database Syst. Rev. 4:Cd003218. doi: 10.1002/14651858.cd003218
Antonini, G., Mainero, C., Romano, A., Giubilei, F., Ceschin, V., Gragnani, F., et al. (2004). Cerebral atrophy in myotonic dystrophy: a voxel based morphometric study. J. Neurol. Neurosurg. Psychiatry 75, 1611–1613. doi: 10.1136/jnnp.2003.032417
Arruda, V. R., Stedman, H. H., Nichols, T. C., Haskins, M. E., Nicholson, M., Herzog, R. W., et al. (2005). Regional intravascular delivery of AAV-2-F.IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood 105, 3458–3464. doi: 10.1182/blood-2004-07-2908
Axford, M. M., and Pearson, C. E. (2013). Illuminating CNS and cognitive issues in myotonic dystrophy: workshop report. Neuromusc. Disord. 23, 370–374. doi: 10.1016/j.nmd.2013.01.003
Azotla-Vilchis, C. N., Sanchez-Celis, D., Agonizantes-Juárez, L. E., Suárez-Sánchez, R., Hernández-Hernández, J. M., Peña, J., et al. (2021). Transcriptome analysis reveals altered inflammatory pathway in an inducible glial cell model of myotonic dystrophy type 1. Biomolecules 11:159. doi: 10.3390/biom11020159
Bai, Y., Nguyen, L., Song, Z., Peng, S., Lee, J., Zheng, N., et al. (2016). Integrating display and delivery functionality with a cell penetrating peptide mimic as a scaffold for intracellular multivalent multitargeting. J. Am. Chem. Soc. 138, 9498–9507. doi: 10.1021/jacs.6b03697
Bajrami, A., Azman, F., Yayla, V., Cagirici, S., Keskinkiliç, C., and Sozer, N. (2017). MRI findings and cognitive functions in a small cohort of myotonic dystrophy type 1: retrospective analyses. Neuroradiol. J. 30, 23–27. doi: 10.1177/1971400916678223
Baldanzi, S., Cecchi, P., Fabbri, S., Pesaresi, I., Simoncini, C., Angelini, C., et al. (2016b). Relationship between neuropsychological impairment and grey and white matter changes in adult-onset myotonic dystrophy type 1. Neuroimage Clin. 12, 190–197. doi: 10.1016/j.nicl.2016.06.011
Baldanzi, S., Bevilacqua, F., Lorio, R., Volpi, L., Simoncini, C., Petrucci, A., et al. (2016a). Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J. Rare Dis. 11:34. doi: 10.1186/s13023-016-0417-z
Bargiela, A., Sabater-Arcis, M., Espinosa-Espinosa, J., Zulaica, M., Lopez de Munain, A., and Artero, R. (2019). Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. U.S.A. 116, 25203–25213. doi: 10.1073/pnas.1820297116
Bassez, G., Audureau, E., Hogrel, J. Y., Arrouasse, R., Baghdoyan, S., Bhugaloo, H., et al. (2018). Improved mobility with metformin in patients with myotonic dystrophy type 1: a randomized controlled trial. Brain 141, 2855–2865. doi: 10.1093/brain/awy231
Batra, R., Charizanis, K., Manchanda, M., Mohan, A., Li, M., Finn, D. J., et al. (2014). Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 56, 311–322. doi: 10.1016/j.molcel.2014.08.027
Batra, R., Nelles, D. A., Pirie, E., Blue, S. M., Marina, R. J., Wang, H., et al. (2017). Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell 170, 899.e10–912.e10. doi: 10.1016/j.cell.2017.07.010
Baughan, T. D., Dickson, A., Osman, E. Y., and Lorson, C. L. (2009). Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 18, 1600–1611. doi: 10.1093/hmg/ddp076
Bellamy, T. C. (2006). Interactions between Purkinje neurones and Bergmann glia. Cerebellum 5, 116–126. doi: 10.1080/14734220600724569
Bennett, C. F., Baker, B. F., Pham, N., Swayze, E., and Geary, R. S. (2017). Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 57, 81–105. doi: 10.1146/annurev-pharmtox-010716-104846
Bennett, C. F., and Swayze, E. E. (2010). RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259–293. doi: 10.1146/annurev.pharmtox.010909.105654
Bertrand, J. A., Jean, S., Laberge, L., Gagnon, C., Mathieu, J., Gagnon, J. F., et al. (2015). Psychological characteristics of patients with myotonic dystrophy type 1. Acta Neurol. Scand. 132, 49–58. doi: 10.1111/ane.12356
Berul, C. I., Maguire, C. T., Aronovitz, M. J., Greenwood, J., Miller, C., Gehrmann, J., et al. (1999). DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Investig. 103, R1–R7. doi: 10.1172/jci5346
Berul, C. I., Maguire, C. T., Gehrmann, J., and Reddy, S. (2000). Progressive atrioventricular conduction block in a mouse myotonic dystrophy model. J. Intervent. Cardiac Electrophysiol. 4, 351–358. doi: 10.1023/a:1009842114968
Bird, T. D. (1993). “Myotonic dystrophy type 1,” in Genereviews((r)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Bisset, D. R., Stepniak-Konieczna, E. A., Zavaljevski, M., Wei, J., Carter, G. T., Weiss, M. D., et al. (2015). Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum. Mol. Genet. 24, 4971–4983. doi: 10.1093/hmg/ddv219
Boisguérin, P., Deshayes, S., Gait, M. J., O’Donovan, L., Godfrey, C., Betts, C. A., et al. (2015). Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 87, 52–67. doi: 10.1016/j.addr.2015.02.008
Breton, É, Légaré, C., Overend, G., Guay, S. P., Monckton, D., Mathieu, J., et al. (2020). DNA methylation at the DMPK gene locus is associated with cognitive functions in myotonic dystrophy type 1. Epigenomics 12, 2051–2064. doi: 10.2217/epi-2020-0328
Brockhoff, M., Rion, N., Chojnowska, K., Wiktorowicz, T., Eickhorst, C., Erne, B., et al. (2017). Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J. Clin. Investig. 127, 549–563. doi: 10.1172/jci89616
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. doi: 10.1016/0092-8674(92)90154-5
Cabada, T., Díaz, J., Iridoy, M., López, P., Jericó, I., Lecumberri, P., et al. (2020). Longitudinal study in patients with myotonic dystrophy type 1: correlation of brain MRI abnormalities with cognitive performances. Neuroradiology 63, 1019–1029. doi: 10.1007/s00234-020-02611-9
Cabada, T., Díaz, J., Iridoy, M., López, P., Jericó, I., Lecumberri, P., et al. (2021). Longitudinal study in patients with myotonic dystrophy type 1: correlation of brain MRI abnormalities with cognitive performances. Neuroradiology 63, 1019–1029.
Cabada, T., Iridoy, M., Jericó, I., Lecumberri, P., Seijas, R., Gargallo, A., et al. (2017). Brain involvement in myotonic dystrophy type 1: a morphometric and diffusion tensor imaging study with neuropsychological correlation. Arch. Clin. Neuropsychol. 32, 401–412. doi: 10.1093/arclin/acx008
Caillet-Boudin, M. L., Fernandez-Gomez, F. J., Tran, H., Dhaenens, C. M., Buee, L., and Sergeant, N. (2014). Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front. Mol. Neurosci. 6:57. doi: 10.3389/fnmol.2013.00057
Carpentier, C., Ghanem, D., Fernandez-Gomez, F. J., Jumeau, F., Philippe, J. V., Freyermuth, F., et al. (2014). Tau exon 2 responsive elements deregulated in myotonic dystrophy type I are proximal to exon 2 and synergistically regulated by MBNL1 and MBNL2. Biochim. Biophys. Acta 1842, 654–664. doi: 10.1016/j.bbadis.2014.01.004
Carrell, S. T., Carrell, E. M., Auerbach, D., Pandey, S. K., Bennett, C. F., Dirksen, R. T., et al. (2016). Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 25, 4328–4338. doi: 10.1093/hmg/ddw266
Caso, F., Agosta, F., Peric, S., Rakočevid-Stojanovid, V., Copetti, M., Kostic, V. S., et al. (2014). Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PLoS One 9:e104697. doi: 10.1371/journal.pone.0104697
Cerro-Herreros, E., Fernandez-Costa, J. M., Sabater-Arcis, M., Llamusi, B., and Artero, R. (2016). Derepressing muscleblind expression by miRNA sponges ameliorates myotonic dystrophy-like phenotypes in Drosophila. Sci. Rep. 6:36230. doi: 10.1038/srep36230
Cerro-Herreros, E., González-Martínez, I., Moreno-Cervera, N., Overby, S., Pérez-Alonso, M., Llamusí, B., et al. (2020). Therapeutic potential of AntagomiR-23b for treating myotonic dystrophy. Mol. Ther. Nucleic Acids 21, 837–849. doi: 10.1016/j.omtn.2020.07.021
Cerro-Herreros, E., Sabater-Arcis, M., Fernandez-Costa, J. M., Moreno, N., Perez-Alonso, M., Llamusi, B., et al. (2018). miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 9:2482. doi: 10.1038/s41467-018-04892-4
Chakraborty, M., Sellier, C., Ney, M., Pascal, V., Charlet-Berguerand, N., Artero, R., et al. (2018). Daunorubicin reduces MBNL1 sequestration caused by CUG-repeat expansion and rescues cardiac dysfunctions in a Drosophila model of myotonic dystrophy. Dis. Models Mech. 11:dmm032557. doi: 10.1242/dmm.032557
Chang, L., Ernst, T., Osborn, D., Seltzer, W., Leonido-Yee, M., and Poland, R. E. (1998). Proton spectroscopy in myotonic dystrophy: correlations with CTG repeats. Arch. Neurol. 55, 305–311. doi: 10.1001/archneur.55.3.305
Charizanis, K., Lee, K. Y., Batra, R., Goodwin, M., Zhang, C., Yuan, Y., et al. (2012). Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75, 437–450. doi: 10.1016/j.neuron.2012.05.029
Chau, A., and Kalsotra, A. (2015). Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev. Dyn. 244, 377–390. doi: 10.1002/dvdy.24240
Chen, G., Carter, R. E., Cleary, J. D., Reid, T. S., Ranum, L. P., Swanson, M. S., et al. (2018). Altered levels of the splicing factor muscleblind modifies cerebral cortical function in mouse models of myotonic dystrophy. Neurobiol. Dis. 112, 35–48. doi: 10.1016/j.nbd.2018.01.003
Chen, G., Masuda, A., Konishi, H., Ohkawara, B., Ito, M., Kinoshita, M., et al. (2016). Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci. Rep. 6:25317. doi: 10.1038/srep25317
Childs-Disney, J. L., Stepniak-Konieczna, E., Tran, T., Yildirim, I., Park, H., Chen, C. Z., et al. (2013). Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat. Commun. 4:2044. doi: 10.1038/ncomms3044
Chiriboga, C. A., Swoboda, K. J., Darras, B. T., Iannaccone, S. T., Montes, J., De Vivo, D. C., et al. (2016). Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 86, 890–897. doi: 10.1212/wnl.0000000000002445
Çoban, A., Bilgiç, B., Lohmann, E., Küçükali, C., Benbir, G., Karadeniz, D., et al. (2013). Reduced orexin-A levels in frontotemporal dementia: possible association with sleep disturbance. Am. J. Alzheimers Dis. Dement. 28, 606–611. doi: 10.1177/1533317513494453
Comim, C. M., Mathia, G. B., Hoepers, A., Tuon, L., Kapczinski, F., Dal-Pizzol, F., et al. (2015). Neurotrophins, cytokines, oxidative parameters and funcionality in progressive muscular dystrophies. Anais Acad. Bras. Ciencias 87, 1809–1818. doi: 10.1590/0001-3765201520140508
Conboy, J. G. (2017). Developmental regulation of RNA processing by Rbfox proteins. Wiley Interdiscip. Rev. RNA 8:1398. doi: 10.1002/wrna.1398
Conforti, R., de Cristofaro, M., Cristofano, A., Brogna, B., Sardaro, A., Tedeschi, G., et al. (2016). Brain MRI abnormalities in the adult form of myotonic dystrophy type 1: a longitudinal case series study. Neuroradiol. J. 29, 36–45. doi: 10.1177/1971400915621325
Coonrod, L. A., Nakamori, M., Wang, W., Carrell, S., Hilton, C. L., Bodner, M. J., et al. (2013). Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 8, 2528–2537. doi: 10.1021/cb400431f
Culebras, A., Feldman, R. G., and Merk, F. B. (1973). Cytoplasmic inclusion bodies within neurons of the thalamus in myotonic dystrophy. A light and electron microscope study. J. Neurol. Sci. 19, 319–329. doi: 10.1016/0022-510x(73)90095-6
Czubak, K., Sedehizadeh, S., Kozlowski, P., and Wojciechowska, M. (2019a). An overview of circular RNAs and their implications in myotonic dystrophy. Int. J. Mol. Sci. 20:4385. doi: 10.3390/ijms20184385
Czubak, K., Taylor, K., Piasecka, A., Sobczak, K., Kozlowska, K., Philips, A., et al. (2019b). Global increase in circular RNA levels in myotonic dystrophy. Front. Genet. 10:649. doi: 10.3389/fgene.2019.00649
Dagenhardt, J., Trinh, A., Sumner, H., Scott, J., Aamodt, E., and Dwyer, D. S. (2017). Insulin signaling deficiency produces immobility in caenorhabditis elegans that models diminished motivation states in man and responds to antidepressants. Mol. Neuropsychiatry 3, 97–107. doi: 10.1159/000478049
Danos, O. (2008). AAV vectors for RNA-based modulation of gene expression. Gene Ther. 15, 864–869. doi: 10.1038/gt.2008.69
Dasgupta, T., and Ladd, A. N. (2012). The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 3, 104–121. doi: 10.1002/wrna.107
De Antonio, M., Dogan, C., Hamroun, D., Mati, M., Zerrouki, S., Eymard, B., et al. (2016). Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Revue Neurol. 172, 572–580. doi: 10.1016/j.neurol.2016.08.003
de León, M. B., and Cisneros, B. (2008). Myotonic dystrophy 1 in the nervous system: from the clinic to molecular mechanisms. J. Neurosci. Res. 86, 18–26. doi: 10.1002/jnr.21377
Denis, J. A., Gauthier, M., Rachdi, L., Aubert, S., Giraud-Triboult, K., Poydenot, P., et al. (2013). mTOR-dependent proliferation defect in human ES-derived neural stem cells affected by myotonic dystrophy type 1. J. Cell Sci. 126(Pt 8), 1763–1772. doi: 10.1242/jcs.116285
Dhaenens, C. M., Schraen-Maschke, S., Tran, H., Vingtdeux, V., Ghanem, D., Leroy, O., et al. (2008). Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Exp. Neurol. 210, 467–478. doi: 10.1016/j.expneurol.2007.11.020
Dhaenens, C. M., Tran, H., Frandemiche, M. L., Carpentier, C., Schraen-Maschke, S., Sistiaga, A., et al. (2011). Mis-splicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim. Biophys. Acta 1812, 732–742. doi: 10.1016/j.bbadis.2011.03.010
Di Filippo, L. D., Duarte, J., Fonseca-Santos, B., Tavares Júnior, A. G., Araújo, V., Roque Borda, C. A., et al. (2021). Mucoadhesive nanosystems for nose-to-brain drug delivery in the treatment of central nervous system diseases. Curr. Med. Chem. doi: 10.2174/0929867328666210813154019 [Epub ahead of print].
Dietz, G. P., Valbuena, P. C., Dietz, B., Meuer, K., Müeller, P., Weishaupt, J. H., et al. (2006). Application of a blood-brain-barrier-penetrating form of GDNF in a mouse model for Parkinson’s disease. Brain Res. 1082, 61–66. doi: 10.1016/j.brainres.2006.01.083
Douniol, M., Jacquette, A., Cohen, D., Bodeau, N., Rachidi, L., Angeard, N., et al. (2012). Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. 54, 905–911. doi: 10.1111/j.1469-8749.2012.04379.x
Dragileva, E., Hendricks, A., Teed, A., Gillis, T., Lopez, E. T., Friedberg, E. C., et al. (2009). Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 33, 37–47. doi: 10.1016/j.nbd.2008.09.014
Du, J., Campau, E., Soragni, E., Jespersen, C., and Gottesfeld, J. M. (2013). Length-dependent CTG⋅CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 22, 5276–5287. doi: 10.1093/hmg/ddt386
Echenne, B., Rideau, A., Roubertie, A., Sébire, G., Rivier, F., and Lemieux, B. (2008). Myotonic dystrophy type I in childhood Long-term evolution in patients surviving the neonatal period. Eur. J. Paediatr. Neurol. 12, 210–223. doi: 10.1016/j.ejpn.2007.07.014
Ekström, A. B., Hakenäs-Plate, L., Samuelsson, L., Tulinius, M., and Wentz, E. (2008). Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147b, 918–926. doi: 10.1002/ajmg.b.30698
Fabbri, M., Girnita, L., Varani, G., and Calin, G. A. (2019). Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 29, 1377–1388. doi: 10.1101/gr.247239.118
Fardaei, M., Rogers, M. T., Thorpe, H. M., Larkin, K., Hamshere, M. G., Harper, P. S., et al. (2002). Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11, 805–814. doi: 10.1093/hmg/11.7.805
Fernandez-Costa, J. M., Garcia-Lopez, A., Zuñiga, S., Fernandez-Pedrosa, V., Felipo-Benavent, A., Mata, M., et al. (2013). Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 22, 704–716. doi: 10.1093/hmg/dds478
Fernandez-Gomez, F., Tran, H., Dhaenens, C. M., Caillet-Boudin, M. L., Schraen-Maschke, S., Blum, D., et al. (2019). Myotonic dystrophy: an RNA toxic gain of function tauopathy? Adv. Exp. Med. Biol. 1184, 207–216. doi: 10.1007/978-981-32-9358-8_17
Fiorelli, M., Duboc, D., Mazoyer, B. M., Blin, J., Eymard, B., Fardeau, M., et al. (1992). Decreased cerebral glucose utilization in myotonic dystrophy. Neurology 42, 91–94. doi: 10.1212/wnl.42.1.91
Flower, M., Lomeikaite, V., Ciosi, M., Cumming, S., Morales, F., Lo, K., et al. (2019). MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 142, 1876–1886. doi: 10.1093/brain/awz115
Foust, K. D., and Kaspar, B. K. (2009). Over the barrier and through the blood: to CNS delivery we go. Cell Cycle 8, 4017–4018. doi: 10.4161/cc.8.24.10245
Franc, D. T., Muetzel, R. L., Robinson, P. R., Rodriguez, C. P., Dalton, J. C., Naughton, C. E., et al. (2012). Cerebral and muscle MRI abnormalities in myotonic dystrophy. Neuromusc. Disord. 22, 483–491. doi: 10.1016/j.nmd.2012.01.003
Francois, V., Klein, A. F., Beley, C., Jollet, A., Lemercier, C., Garcia, L., et al. (2011). Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat. Struct. Mol. Biol. 18, 85–87. doi: 10.1038/nsmb.1958
Fritegotto, C., Ferrati, C., Pegoraro, V., and Angelini, C. (2017). Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 38, 619–625. doi: 10.1007/s10072-017-2811-2
Fu, Y. H., Pizzuti, A., Fenwick, R. G. Jr., King, J., Rajnarayan, S., Dunne, P. W., et al. (1992). An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. doi: 10.1126/science.1546326
Furling, D., Doucet, G., Langlois, M. A., Timchenko, L., Belanger, E., Cossette, L., et al. (2003). Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther. 10, 795–802. doi: 10.1038/sj.gt.3301955
Furuta, M., Kimura, T., Nakamori, M., Matsumura, T., Fujimura, H., Jinnai, K., et al. (2018). Macroscopic and microscopic diversity of missplicing in the central nervous system of patients with myotonic dystrophy type 1. Neuroreport 29, 235–240. doi: 10.1097/wnr.0000000000000968
Gadgil, A., and Raczyńska, K. D. (2021). U7 snRNA: a tool for gene therapy. J. Gene Med. 23:e3321. doi: 10.1002/jgm.3321
Gallais, B., Gagnon, C., Mathieu, J., and Richer, L. (2017). Cognitive decline over time in adults with myotonic dystrophy type 1: a 9-year longitudinal study. Neuromusc. Disord. 27, 61–72. doi: 10.1016/j.nmd.2016.10.003
Gallais, B., Montreuil, M., Gargiulo, M., Eymard, B., Gagnon, C., and Laberge, L. (2015). Prevalence and correlates of apathy in myotonic dystrophy type 1. BMC Neurol. 15:148. doi: 10.1186/s12883-015-0401-6
Gambardella, S., Rinaldi, F., Lepore, S. M., Viola, A., Loro, E., Angelini, C., et al. (2010). Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med. 8:48. doi: 10.1186/1479-5876-8-48
Gao, Y., Guo, X., Santostefano, K., Wang, Y., Reid, T., Zeng, D., et al. (2016). Genome therapy of myotonic dystrophy type 1 ips cells for development of autologous stem cell therapy. Mol. Ther. 24, 1378–1387. doi: 10.1038/mt.2016.97
García-López, A., Llamusí, B., Orzáez, M., Pérez-Payá, E., and Artero, R. D. (2011). In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. U.S.A. 108, 11866–11871. doi: 10.1073/pnas.1018213108
García-Puga, M., Saenz-Antoñanzas, A., Fernández-Torrón, R., Munain, A. L., and Matheu, A. (2020). Myotonic Dystrophy type 1 cells display impaired metabolism and mitochondrial dysfunction that are reversed by metformin. Aging 12, 6260–6275. doi: 10.18632/aging.103022
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. doi: 10.1038/nature24644
Geary, R. S., Norris, D., Yu, R., and Bennett, C. F. (2015). Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 87, 46–51. doi: 10.1016/j.addr.2015.01.008
Gehman, L. T., Stoilov, P., Maguire, J., Damianov, A., Lin, C. H., Shiue, L., et al. (2011). The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 43, 706–711. doi: 10.1038/ng.841
Glantz, R. H., Wright, R. B., Huckman, M. S., Garron, D. C., and Siegel, I. M. (1988). Central nervous system magnetic resonance imaging findings in myotonic dystrophy. Arch. Neurol. 45, 36–37. doi: 10.1001/archneur.1988.00520250042017
Gonçalves, R. A., Wijesekara, N., Fraser, P. E., and De Felice, F. G. (2019). The link between tau and insulin signaling: implications for Alzheimer’s disease and other tauopathies. Front. Cell. Neurosci. 13:17. doi: 10.3389/fncel.2019.00017
González-Barriga, A., Lallemant, L., Dincã, D. M., Braz, S. O., Polvèche, H., Magneron, P., et al. (2021). Integrative cell type-specific multi-omics approaches reveal impaired programs of glial cell differentiation in mouse culture models of DM1. Front. Cell. Neurosci. 15:662035. doi: 10.3389/fncel.2021.662035
Goodwin, M., Mohan, A., Batra, R., Lee, K. Y., Charizanis, K., Fernández Gómez, F. J., et al. (2015). MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. 12, 1159–1168. doi: 10.1016/j.celrep.2015.07.029
Gregorevic, P., Blankinship, M. J., Allen, J. M., Crawford, R. W., Meuse, L., Miller, D. G., et al. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 10, 828–834. doi: 10.1038/nm1085
Groenen, P. J., Wansink, D. G., Coerwinkel, M., van den Broek, W., Jansen, G., and Wieringa, B. (2000). Constitutive and regulated modes of splicing produce six major myotonic dystrophy protein kinase (DMPK) isoforms with distinct properties. Hum. Mol. Genet. 9, 605–616. doi: 10.1093/hmg/9.4.605
Gudde, A., van Heeringen, S. J., de Oude, A. I., van Kessel, I. D. G., Estabrook, J., Wang, E. T., et al. (2017). Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. 14, 1374–1388. doi: 10.1080/15476286.2017.1279787
Gudde, A. E., González-Barriga, A., van den Broek, W. J., Wieringa, B., and Wansink, D. G. (2016). A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum. Mol. Genet. 25, 1648–1662. doi: 10.1093/hmg/ddw042
Han, S., Nam, J., Li, Y., Kim, S., Cho, S. H., Cho, Y. S., et al. (2010). Regulation of dendritic spines, spatial memory, and embryonic development by the TANC family of PSD-95-interacting proteins. J. Neurosci. 30, 15102–15112. doi: 10.1523/jneurosci.3128-10.2010
Harper, P. S., van Engelen, B. G., Eymard, B., Rogers, M., and Wilcox, D. (2002). 99th ENMC international workshop: myotonic dystrophy: present management, future therapy. 9-11 November 2001, Naarden, The Netherlands. Neuromusc. Disord. 12, 596–599. doi: 10.1016/s0960-8966(02)00020-2
Hashimoto, T., Tayama, M., Miyazaki, M., Murakawa, K., Kawai, H., Nishitani, H., et al. (1995). Neuroimaging study of myotonic dystrophy. II. MRI measurements of the brain. Brain Dev. 17, 28–32. doi: 10.1016/0387-7604(94)00097-h
Heatwole, C., Bode, R., Johnson, N., Quinn, C., Martens, W., McDermott, M. P., et al. (2012). Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 79, 348–357. doi: 10.1212/WNL.0b013e318260cbe6
Heatwole, C., Bode, R., Johnson, N. E., Dekdebrun, J., Dilek, N., Eichinger, K., et al. (2016). Myotonic dystrophy health index: correlations with clinical tests and patient function. Muscle Nerve 53, 183–190. doi: 10.1002/mus.24725
Heatwole, C., Johnson, N., Dekdebrun, J., Dilek, N., Eichinger, K., Hilbert, J., et al. (2018). Myotonic dystrophy patient preferences in patient-reported outcome measures. Muscle Nerve doi: 10.1002/mus.26066 [Epub ahead of print].
Heatwole, C., Luebbe, E., Rosero, S., Eichinger, K., Martens, W., Hilbert, J., et al. (2021). Mexiletine in myotonic dystrophy type 1: a randomized, double-blind, placebo-controlled trial. Neurology 96, e228–e240. doi: 10.1212/wnl.0000000000011002
Hermans, M. C., Hoeijmakers, J. G., Faber, C. G., and Merkies, I. S. (2015). Reconstructing the rasch-built myotonic dystrophy type 1 activity and participation scale. PLoS One 10:e0139944. doi: 10.1371/journal.pone.0139944
Hermans, M. C., Merkies, I. S., Laberge, L., Blom, E. W., Tennant, A., and Faber, C. G. (2013). Fatigue and daytime sleepiness scale in myotonic dystrophy type 1. Muscle Nerve 47, 89–95. doi: 10.1002/mus.23478
Hernández-Hernández, O., Guiraud-Dogan, C., Sicot, G., Huguet, A., Luilier, S., Steidl, E., et al. (2013a). Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 136(Pt 3), 957–970. doi: 10.1093/brain/aws367
Hernández-Hernández, O., Sicot, G., Dinca, D. M., Huguet, A., Nicole, A., Buée, L., et al. (2013b). Synaptic protein dysregulation in myotonic dystrophy type 1: disease neuropathogenesis beyond missplicing. Rare Dis. 1:e25553. doi: 10.4161/rdis.25553
Hernando, S., Herran, E., Figueiro-Silva, J., Pedraz, J. L., Igartua, M., Carro, E., et al. (2018). Intranasal administration of TAT-conjugated lipid nanocarriers loading GDNF for Parkinson’s disease. Mol. Neurobiol. 55, 145–155. doi: 10.1007/s12035-017-0728-7
Hilton-Jones, D., Bowler, M., Lochmueller, H., Longman, C., Petty, R., Roberts, M., et al. (2012). Modafinil for excessive daytime sleepiness in myotonic dystrophy type 1–the patients’ perspective. Neuromusc. Disord. 22, 597–603. doi: 10.1016/j.nmd.2012.02.005
Hinrich, A. J., Jodelka, F. M., Chang, J. L., Brutman, D., Bruno, A. M., Briggs, C. A., et al. (2016). Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol. Med. 8, 328–345. doi: 10.15252/emmm.201505846
Hirase, T., and Araki, S. (1984). Cerebrospinal fluid proteins in muscular dystrophy patients. Brain Dev. 6, 10–16. doi: 10.1016/s0387-7604(84)80003-0
Horrigan, J., Gomes, T. B., Snape, M., Nikolenko, N., McMorn, A., Evans, S., et al. (2020). A phase 2 study of AMO-02 (Tideglusib) in congenital and childhood-onset myotonic dystrophy type 1 (DM1). Pediatr. Neurol. 112, 84–93. doi: 10.1016/j.pediatrneurol.2020.08.001
Hoskins, J. W., Ofori, L. O., Chen, C. Z., Kumar, A., Sobczak, K., Nakamori, M., et al. (2014). Lomofungin and dilomofungin: inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res. 42, 6591–6602. doi: 10.1093/nar/gku275
Hsieh, W. C., Bahal, R., Thadke, S. A., Bhatt, K., Sobczak, K., Thornton, C., et al. (2018). Design of a “mini” nucleic acid probe for cooperative binding of an RNA-repeated transcript associated with myotonic dystrophy type 1. Biochemistry 57, 907–911. doi: 10.1021/acs.biochem.7b01239
Hu, N., Kim, E., Antoury, L., Li, J., González-Pérez, P., Rutkove, S. B., et al. (2021). Antisense oligonucleotide and adjuvant exercise therapy reverse fatigue in old mice with myotonic dystrophy. Mol. Ther. Nucleic Acids 23, 393–405. doi: 10.1016/j.omtn.2020.11.014
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F., et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
Huguet, A., Medja, F., Nicole, A., Vignaud, A., Guiraud-Dogan, C., Ferry, A., et al. (2012). Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8:e1003043. doi: 10.1371/journal.pgen.1003043
Itoh, K., Mitani, M., Kawamoto, K., Futamura, N., Funakawa, I., Jinnai, K., et al. (2010). Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with myotonic dystrophy type 1. Acta Histochem. Cytochem. 43, 149–156. doi: 10.1267/ahc.10019
Jacko, M., Weyn-Vanhentenryck, S. M., Smerdon, J. W., Yan, R., Feng, H., Williams, D. J., et al. (2018). Rbfox splicing factors promote neuronal maturation and axon initial segment assembly. Neuron 97, 853.e6–868.e6. doi: 10.1016/j.neuron.2018.01.020
Jauvin, D., Chretien, J., Pandey, S. K., Martineau, L., Revillod, L., Bassez, G., et al. (2017). Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol. Ther. Nucleic Acids 7, 465–474. doi: 10.1016/j.omtn.2017.05.007
Jean, S., Richer, L., Laberge, L., and Mathieu, J. (2014). Comparisons of intellectual capacities between mild and classic adult-onset phenotypes of myotonic dystrophy type 1 (DM1). Orphanet J. Rare Dis. 9:186. doi: 10.1186/s13023-014-0186-5
Jenquin, J. R., Coonrod, L. A., Silverglate, Q. A., Pellitier, N. A., Hale, M. A., Xia, G., et al. (2018). Furamidine rescues myotonic dystrophy type I associated mis-splicing through multiple mechanisms. ACS Chem. Biol. 13, 2708–2718. doi: 10.1021/acschembio.8b00646
Jenquin, J. R., Yang, H., Huigens, R. W. III, Nakamori, M., and Berglund, J. A. (2019). Combination treatment of erythromycin and furamidine provides additive and synergistic rescue of mis-splicing in myotonic dystrophy type 1 models. ACS Pharmacol. Transl. Sci. 2, 247–263. doi: 10.1021/acsptsci.9b00020
Jiang, H., Mankodi, A., Swanson, M. S., Moxley, R. T., and Thornton, C. A. (2004). Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079–3088. doi: 10.1093/hmg/ddh327
Jimenez-Marin, A., Diez, I., Labayru, G., Sistiaga, A., Caballero, M. C., Andres-Benito, P., et al. (2021). Transcriptional signatures of synaptic vesicle genes define myotonic dystrophy type I neurodegeneration. Neuropathol. Appl. Neurobiol. doi: 10.1111/nan.12725 [Epub ahead of print].
Jinnai, K., Mitani, M., Futamura, N., Kawamoto, K., Funakawa, I., and Itoh, K. (2013). Somatic instability of CTG repeats in the cerebellum of myotonic dystrophy type 1. Muscle Nerve 48, 105–108. doi: 10.1002/mus.23717
Jones, K., Wei, C., Iakova, P., Bugiardini, E., Schneider-Gold, C., Meola, G., et al. (2012). GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 122, 4461–4472. doi: 10.1172/jci64081
Kalsotra, A., Singh, R. K., Gurha, P., Ward, A. J., Creighton, C. J., and Cooper, T. A. (2014). The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 6, 336–345. doi: 10.1016/j.celrep.2013.12.025
Kamei, N., and Takeda-Morishita, M. (2015). Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J. Control. Release 197, 105–110. doi: 10.1016/j.jconrel.2014.11.004
Kanadia, R. N., Shin, J., Yuan, Y., Beattie, S. G., Wheeler, T. M., Thornton, C. A., et al. (2006). Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 103, 11748–11753. doi: 10.1073/pnas.0604970103
Ketley, A., Chen, C. Z., Li, X., Arya, S., Robinson, T. E., Granados-Riveron, J., et al. (2014). High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 23, 1551–1562. doi: 10.1093/hmg/ddt542
Khafagy, E. S., Kamei, N., Fujiwara, Y., Okumura, H., Yuasa, T., Kato, M., et al. (2020). Systemic and brain delivery of leptin via intranasal coadministration with cell-penetrating peptides and its therapeutic potential for obesity. J. Control. Release 319, 397–406. doi: 10.1016/j.jconrel.2020.01.016
Khorkova, O., and Wahlestedt, C. (2017). Oligonucleotide therapies for disorders of the nervous system. Nat. Biotechnol. 35, 249–263. doi: 10.1038/nbt.3784
Kilic, U., Kilic, E., Dietz, G. P., and Bähr, M. (2003). Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke 34, 1304–1310. doi: 10.1161/01.str.0000066869.45310.50
Kilic, U., Kilic, E., Dietz, G. P., and Bähr, M. (2004). The TAT protein transduction domain enhances the neuroprotective effect of glial-cell-line-derived neurotrophic factor after optic nerve transection. Neuro Degener. Dis. 1, 44–49. doi: 10.1159/000076669
Kim, K., Lee, S. G., Kegelman, T. P., Su, Z. Z., Das, S. K., Dash, R., et al. (2011). Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J. Cell. Physiol. 226, 2484–2493. doi: 10.1002/jcp.22609
Klein, A. F., Dastidar, S., Furling, D., and Chuah, M. K. (2015). Therapeutic approaches for dominant muscle diseases: highlight on myotonic dystrophy. Curr. Gene Ther. 15, 329–337. doi: 10.2174/1566523215666150630120537
Klein, A. F., Varela, M. A., Arandel, L., Holland, A., Naouar, N., Arzumanov, A., et al. (2019). Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 129, 4739–4744. doi: 10.1172/jci128205
Klinck, R., Fourrier, A., Thibault, P., Toutant, J., Durand, M., Lapointe, E., et al. (2014). RBFOX1 cooperates with MBNL1 to control splicing in muscle, including events altered in myotonic dystrophy type 1. PLoS One 9:e107324. doi: 10.1371/journal.pone.0107324
Knott, G. J., and Doudna, J. A. C. R. I. S. P. R. - (2018). Cas guides the future of genetic engineering. Science 361, 866–869. doi: 10.1126/science.aat5011
Kobayakawa, M., Tsuruya, N., and Kawamura, M. (2012). Theory of mind impairment in adult-onset myotonic dystrophy type 1. Neurosci. Res. 72, 341–346. doi: 10.1016/j.neures.2012.01.005
Kobayakawa, M., Tsuruya, N., Takeda, A., Suzuki, A., and Kawamura, M. (2010). Facial emotion recognition and cerebral white matter lesions in myotonic dystrophy type 1. J. Neurol. Sci. 290, 48–51. doi: 10.1016/j.jns.2009.11.011
Koebis, M., Kiyatake, T., Yamaura, H., Nagano, K., Higashihara, M., Sonoo, M., et al. (2013). Ultrasound-enhanced delivery of morpholino with Bubble liposomes ameliorates the myotonia of myotonic dystrophy model mice. Sci. Rep. 3:2242. doi: 10.1038/srep02242
Koehorst, E., Ballester-Lopez, A., Arechavala-Gomeza, V., Martínez-Piñeiro, A., and Nogales-Gadea, G. (2020). The biomarker potential of miRNAs in myotonic dystrophy type I. J. Clin. Med. 9:3939. doi: 10.3390/jcm9123939
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Kouki, T., Takasu, N., Nakachi, A., Tamanaha, T., Komiya, I., and Tawata, M. (2005). Low-dose metformin improves hyperglycaemia related to myotonic dystrophy. Diabetic Med. 22, 346–347. doi: 10.1111/j.1464-5491.2005.01432.x
Koutalianos, D., Koutsoulidou, A., Mastroyiannopoulos, N. P., Furling, D., and Phylactou, L. A. (2015). MyoD transcription factor induces myogenesis by inhibiting Twist-1 through miR-206. J. Cell Sci. 128, 3631–3645. doi: 10.1242/jcs.172288
Koutsoulidou, A., Kyriakides, T. C., Papadimas, G. K., Christou, Y., Kararizou, E., Papanicolaou, E. Z., et al. (2015). Elevated muscle-specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS One 10:e0125341. doi: 10.1371/journal.pone.0125341
Koutsoulidou, A., Photiades, M., Kyriakides, T. C., Georgiou, K., Prokopi, M., Kapnisis, K., et al. (2017). Identification of exosomal muscle-specific miRNAs in serum of myotonic dystrophy patients relating to muscle disease progress. Hum. Mol. Genet. 26, 3285–3302. doi: 10.1093/hmg/ddx212
Kristensen, M., Birch, D., and Mørck Nielsen, H. (2016). Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 17:185. doi: 10.3390/ijms17020185
Krogias, C., Bellenberg, B., Prehn, C., Schneider, R., Meves, S. H., Gold, R., et al. (2015). Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J. Neurol. 262, 365–374. doi: 10.1007/s00415-014-7566-6
Krogias, C., and Walter, U. (2016). Transcranial sonography findings in depression in association with psychiatric and neurologic diseases: a review. J. Neuroimaging 26, 257–263. doi: 10.1111/jon.12328
Krupa, P., Řehák, S., Diaz-Garcia, D., and Filip, S. (2014). Nanotechnology - new trends in the treatment of brain tumours. Acta Med. 57, 142–150. doi: 10.14712/18059694.2015.79
Kullmann, S., Heni, M., Hallschmid, M., Fritsche, A., Preissl, H., and Häring, H. U. (2016). Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 96, 1169–1209. doi: 10.1152/physrev.00032.2015
Kumar, V. B., Farr, S. A., Flood, J. F., Kamlesh, V., Franko, M., Banks, W. A., et al. (2000). Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 21, 1769–1775. doi: 10.1016/s0196-9781(00)00339-9
Kuyumcu-Martinez, N. M., Wang, G. S., and Cooper, T. A. (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78. doi: 10.1016/j.molcel.2007.07.027
Labayru, G., Arenzana, I., Aliri, J., Zulaica, M., López de Munain, A., and Sistiaga, A. A. (2018). Social cognition in myotonic dystrophy type 1: specific or secondary impairment? PLoS One 13:e0204227. doi: 10.1371/journal.pone.0204227
Labayru, G., Diez, I., Sepulcre, J., Fernández, E., Zulaica, M., Cortés, J. M., et al. (2019). Regional brain atrophy in gray and white matter is associated with cognitive impairment in Myotonic Dystrophy type 1. Neuroimage Clin. 24:102078. doi: 10.1016/j.nicl.2019.102078
Labayru, G., Jimenez-Marin, A., Fernández, E., Villanua, J., Zulaica, M., Cortes, J. M., et al. (2020). Neurodegeneration trajectory in pediatric and adult/late DM1: a follow-up MRI study across a decade. Ann. Clin. Transl. Neurol. 7, 1802–1815. doi: 10.1002/acn3.51163
Laberge, L., Gagnon, C., and Dauvilliers, Y. (2013). Daytime sleepiness and myotonic dystrophy. Curr. Neurol. Neurosci. Rep. 13:340. doi: 10.1007/s11910-013-0340-9
Ladd, A. N. (2013). CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol. Cell. Neurosci. 56, 456–464. doi: 10.1016/j.mcn.2012.12.003
Ladd, A. N., Charlet, N., and Cooper, T. A. (2001). The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol. 21, 1285–1296. doi: 10.1128/mcb.21.4.1285-1296.2001
Ladd, A. N., Nguyen, N. H., Malhotra, K., and Cooper, T. A. (2004). CELF6, a member of the CELF family of RNA-binding proteins, regulates muscle-specific splicing enhancer-dependent alternative splicing. J. Biol. Chem. 279, 17756–17764. doi: 10.1074/jbc.M310687200
Langbehn, K. E., van der Plas, E., Moser, D. J., Long, J. D., Gutmann, L., and Nopoulos, P. C. (2021). Cognitive function and its relationship with brain structure in myotonic dystrophy type 1. J. Neurosci. Res. 99, 190–199. doi: 10.1002/jnr.24595
Larsen, J., Pettersson, O. J., Jakobsen, M., Thomsen, R., Pedersen, C. B., Hertz, J. M., et al. (2011). Myoblasts generated by lentiviral mediated MyoD transduction of myotonic dystrophy type 1 (DM1) fibroblasts can be used for assays of therapeutic molecules. BMC Res. Notes 4:490. doi: 10.1186/1756-0500-4-490
Laustriat, D., Gide, J., Barrault, L., Chautard, E., Benoit, C., Auboeuf, D., et al. (2015). In vitro and In vivo modulation of alternative splicing by the biguanide metformin. Mol. Ther. Nucleic Acids 4:e262. doi: 10.1038/mtna.2015.35
Le Hir, M., Goyenvalle, A., Peccate, C., Précigout, G., Davies, K. E., Voit, T., et al. (2013). AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol. Ther. 21, 1551–1558. doi: 10.1038/mt.2013.121
Lebleu, B., Moulton, H. M., Abes, R., Ivanova, G. D., Abes, S., Stein, D. A., et al. (2008). Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 60, 517–529. doi: 10.1016/j.addr.2007.09.002
Leddy, S., Serra, L., Esposito, D., Vizzotto, C., Giulietti, G., Silvestri, G., et al. (2021). Lesion distribution and substrate of white matter damage in myotonic dystrophy type 1: comparison with multiple sclerosis. Neuroimage Clin. 29:102562. doi: 10.1016/j.nicl.2021.102562
Lee, H. B., Sundberg, B. N., Sigafoos, A. N., and Clark, K. J. (2016). Genome engineering with TALE and CRISPR systems in neuroscience. Front. Genet. 7:47. doi: 10.3389/fgene.2016.00047
Lee, J. E., Bennett, C. F., and Cooper, T. A. (2012). RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U.S.A. 109, 4221–4226. doi: 10.1073/pnas.1117019109
Lee, K., Conboy, M., Park, H. M., Jiang, F., Kim, H. J., Dewitt, M. A., et al. (2017). Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 1, 889–901. doi: 10.1038/s41551-017-0137-2
Lee, K. Y., Chang, H. C., Seah, C., and Lee, L. J. (2019). Deprivation of muscleblind-like proteins causes deficits in cortical neuron distribution and morphological changes in dendritic spines and postsynaptic densities. Front. Neuroanat. 13:75. doi: 10.3389/fnana.2019.00075
Lee, K. Y., Li, M., Manchanda, M., Batra, R., Charizanis, K., Mohan, A., et al. (2013). Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 5, 1887–1900. doi: 10.1002/emmm.201303275
Lehto, T., Kurrikoff, K., and Langel, Ü (2012). Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 9, 823–836. doi: 10.1517/17425247.2012.689285
Leroy, O., Dhaenens, C. M., Schraen-Maschke, S., Belarbi, K., Delacourte, A., Andreadis, A., et al. (2006a). ETR-3 represses Tau exons 2/3 inclusion, a splicing event abnormally enhanced in myotonic dystrophy type I. J. Neurosci. Res. 84, 852–859. doi: 10.1002/jnr.20980
Leroy, O., Wang, J., Maurage, C. A., Parent, M., Cooper, T., Buée, L., et al. (2006b). Brain-specific change in alternative splicing of Tau exon 6 in myotonic dystrophy type 1. Biochim. Biophys. Acta 1762, 460–467. doi: 10.1016/j.bbadis.2005.12.003
Lim, J. J., Derby, M. A., Zhang, Y., Deng, R., Larouche, R., Anderson, M., et al. (2017). A phase 1, randomized, double-blind, placebo-controlled, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of an anti-influenza B virus monoclonal antibody, MHAB5553A, in healthy volunteers. Antimicrob. Agents Chemother. 61:e00279-17. doi: 10.1128/aac.00279-17
Lindeblad, G., Kroksmark, A. K., and Ekström, A. B. (2019). Cognitive and adaptive functioning in congenital and childhood forms of myotonic dystrophy type 1: a longitudinal study. Dev. Med. Child Neurol. 61, 1214–1220. doi: 10.1111/dmcn.14161
Lo Scrudato, M., Poulard, K., Sourd, C., Tomé, S., Klein, A. F., Corre, G., et al. (2019). Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 27, 1372–1388. doi: 10.1016/j.ymthe.2019.05.021
Logigian, E. L., Martens, W. B., Moxley, RTt, McDermott, M. P., Dilek, N., Wiegner, A. W., et al. (2010). Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology 74, 1441–1448. doi: 10.1212/WNL.0b013e3181dc1a3a
Long, C., Amoasii, L., Bassel-Duby, R., and Olson, E. N. (2016). Genome editing of monogenic neuromuscular diseases: a systematic review. JAMA Neurol. 73, 1349–1355. doi: 10.1001/jamaneurol.2016.3388
López Castel, A., Overby, S. J., and Artero, R. (2019). MicroRNA-based therapeutic perspectives in myotonic dystrophy. Int. J. Mol. Sci. 20:5600. doi: 10.3390/ijms20225600
López-Morató, M., Brook, J. D., and Wojciechowska, M. (2018). Small molecules which improve pathogenesis of myotonic dystrophy type 1. Front. Neurol. 9:349. doi: 10.3389/fneur.2018.00349
Lopez-Titla, M. M., Chirino, A., Cruz Solis, S. V., Hernandez-Castillo, C. R., Diaz, R., Márquez-Quiroz, L. D. C., et al. (2021). Cognitive decline and white matter integrity degradation in myotonic dystrophy type I. J. Neuroimaging 31, 192–198. doi: 10.1111/jon.12786
Luu, L. M., Nguyen, L., Peng, S., Lee, J., Lee, H. Y., Wong, C. H., et al. (2016). A potent inhibitor of protein sequestration by expanded triplet (CUG) repeats that shows phenotypic improvements in a drosophila model of myotonic dystrophy. Chem. Med. Chem. 11, 1428–1435. doi: 10.1002/cmdc.201600081
Magaña, J. J., and Cisneros, B. (2011). Perspectives on gene therapy in myotonic dystrophy type 1. J. Neurosci. Res. 89, 275–285. doi: 10.1002/jnr.22551
Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., et al. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255, 1253–1255. doi: 10.1126/science.1546325
Manning, K. S., Rao, A. N., Castro, M., and Cooper, T. A. (2017). BNA (NC) gapmers revert splicing and reduce RNA foci with low toxicity in myotonic dystrophy cells. ACS Chem. Biol. 12, 2503–2509. doi: 10.1021/acschembio.7b00416
Marteyn, A., Maury, Y., Gauthier, M. M., Lecuyer, C., Vernet, R., Denis, J. A., et al. (2011). Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell 8, 434–444. doi: 10.1016/j.stem.2011.02.004
Martínez-Rodríguez, J. E., Lin, L., Iranzo, A., Genis, D., Martí, M. J., Santamaria, J., et al. (2003). Decreased hypocretin-1 (Orexin-A) levels in the cerebrospinal fluid of patients with myotonic dystrophy and excessive daytime sleepiness. Sleep 26, 287–290. doi: 10.1093/sleep/26.3.287
Matynia, A., Ng, C. H., Dansithong, W., Chiang, A., Silva, A. J., and Reddy, S. (2010). Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS One 5:e9857. doi: 10.1371/journal.pone.0009857
Mazzoli, M., Ariatti, A., Garuti, G. C., Agnoletto, V., Genovese, M., Gozzi, M., et al. (2020). Predictors of prognosis in type 1 myotonic dystrophy (DM1): longitudinal 18-years experience from a single center. Acta Myol. 39, 109–120. doi: 10.36185/2532-1900-015
McGowan, J. W., Bidwell, G. L. III, and Vig, P. J. (2015). Challenges and new strategies for therapeutic peptide delivery to the CNS. Ther. Deliv. 6, 841–853. doi: 10.4155/tde.15.30
McGowan, J. W., Shao, Q., Vig, P. J., and Bidwell, G. L. III (2016). Intranasal administration of elastin-like polypeptide for therapeutic delivery to the central nervous system. Drug Design Dev. Ther. 10, 2803–2813. doi: 10.2147/dddt.s106216
McKinney, B. C., Sze, W., Lee, B., and Murphy, G. G. (2009). Impaired long-term potentiation and enhanced neuronal excitability in the amygdala of Ca(V)1.3 knockout mice. Neurobiol. Learn. Mem. 92, 519–528. doi: 10.1016/j.nlm.2009.06.012
Meola, G., and Cardani, R. (2015). Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 1852, 594–606. doi: 10.1016/j.bbadis.2014.05.019
Meola, G., and Sansone, V. (2007). Cerebral involvement in myotonic dystrophies. Muscle Nerve 36, 294–306. doi: 10.1002/mus.20800
Meola, G., Sansone, V., Perani, D., Scarone, S., Cappa, S., Dragoni, C., et al. (2003). Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromusc. Disord. 13, 813–821. doi: 10.1016/s0960-8966(03)00137-8
Miller, J. N., Kruger, A., Moser, D. J., Gutmann, L., van der Plas, E., Koscik, T. R., et al. (2021). Cognitive deficits, apathy, and hypersomnolence represent the core brain symptoms of adult-onset myotonic dystrophy type 1. Front. Neurol. 12:700796. doi: 10.3389/fneur.2021.700796
Miller, J. W., Urbinati, C. R., Teng-Umnuay, P., Stenberg, M. G., Byrne, B. J., Thornton, C. A., et al. (2000). Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 19, 4439–4448. doi: 10.1093/emboj/19.17.4439
Miller, R. G., Mitchell, J. D., and Moore, D. H. (2012). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012:Cd001447. doi: 10.1002/14651858.CD001447.pub3
Miller, T. M., Pestronk, A., David, W., Rothstein, J., Simpson, E., Appel, S. H., et al. (2013). An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442. doi: 10.1016/s1474-4422(13)70061-9
Minier, L., Lignier, B., Bouvet, C., Gallais, B., and Camart, N. (2018). A review of psychopathology features, personality, and coping in myotonic dystrophy type 1. J. Neuromusc. Dis. 5, 279–294. doi: 10.3233/jnd-180310
Minnerop, M., Gliem, C., and Kornblum, C. (2018). Current progress in CNS imaging of myotonic dystrophy. Front. Neurol. 9:646. doi: 10.3389/fneur.2018.00646
Minnerop, M., Weber, B., Schoene-Bake, J. C., Roeske, S., Mirbach, S., Anspach, C., et al. (2011). The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 134(Pt 12), 3530–3546. doi: 10.1093/brain/awr299
Misra, C., Bangru, S., Lin, F., Lam, K., Koenig, S. N., Lubbers, E. R., et al. (2020). Aberrant expression of a non-muscle RBFOX2 isoform triggers cardiac conduction defects in myotonic dystrophy. Dev. Cell 52, 748.e6–763.e6. doi: 10.1016/j.devcel.2020.01.037
Mizukami, K., Sasaki, M., Baba, A., Suzuki, T., and Shiraishi, H. (1999). An autopsy case of myotonic dystrophy with mental disorders and various neuropathologic features. Psychiatry Clin. Neurosci. 53, 51–55. doi: 10.1046/j.1440-1819.1999.00470.x
Mizuno, Y., Maeda, N., Hamasaki, H., Arahata, H., Sasagasako, N., Honda, H., et al. (2018). Four-repeat tau dominant pathology in a congenital myotonic dystrophy type 1 patient with mental retardation. Brain Pathol. 28, 431–433. doi: 10.1111/bpa.12603
Modoni, A., Silvestri, G., Vita, M. G., Quaranta, D., Tonali, P. A., and Marra, C. (2008). Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow-up study. J. Neurol. 255, 1737–1742. doi: 10.1007/s00415-008-0017-5
Mohan, A., Goodwin, M., and Swanson, M. S. (2014). RNA-protein interactions in unstable microsatellite diseases. Brain Res. 1584, 3–14. doi: 10.1016/j.brainres.2014.03.039
Mondragon-Gonzalez, R., and Perlingeiro, R. C. R. (2018). Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: a tool for disease modeling and drug discovery. Dis. Models Mech. 11:dmm034728. doi: 10.1242/dmm.034728
Morales, F., Vásquez, M., Corrales, E., Vindas-Smith, R., Santamaría-Ulloa, C., Zhang, B., et al. (2020). Longitudinal increases in somatic mosaicism of the expanded CTG repeat in myotonic dystrophy type 1 are associated with variation in age-at-onset. Hum. Mol. Genet. 29, 2496–2507. doi: 10.1093/hmg/ddaa123
Morgenthaler, T. I., Kapur, V. K., Brown, T., Swick, T. J., Alessi, C., Aurora, R. N., et al. (2007). Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep 30, 1705–1711. doi: 10.1093/sleep/30.12.1705
Moxley, R. T. III, Griggs, R. C., Goldblatt, D., VanGelder, V., Herr, B. E., and Thiel, R. (1978). Decreased insulin sensitivity of forearm muscle in myotonic dystrophy. J. Clin. Investig. 62, 857–867. doi: 10.1172/jci109198
Mulders, S. A., van den Broek, W. J., Wheeler, T. M., Croes, H. J., van Kuik-Romeijn, P., de Kimpe, S. J., et al. (2009). Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 13915–13920. doi: 10.1073/pnas.0905780106
Mulders, S. A., van Engelen, B. G., Wieringa, B., and Wansink, D. G. (2010). Molecular therapy in myotonic dystrophy: focus on RNA gain-of-function. Hum. Mol. Genet. 19, R90–R97. doi: 10.1093/hmg/ddq161
Muntoni, F., and Wood, M. J. (2011). Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 10, 621–637. doi: 10.1038/nrd3459
Murlidharan, G., Sakamoto, K., Rao, L., Corriher, T., Wang, D., Gao, G., et al. (2016). CNS-restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV vector. Mol. Ther. Nucleic Acids 5:e338. doi: 10.1038/mtna.2016.49
Nakamori, M., Gourdon, G., and Thornton, C. A. (2011). Stabilization of expanded (CTG)•(CAG) repeats by antisense oligonucleotides. Mol. Ther. 19, 2222–2227. doi: 10.1038/mt.2011.191
Nakamori, M., Takahashi, T., Yamazaki, Y., Kurashige, T., Yamawaki, T., and Matsumoto, M. (2012). Cyclin-dependent kinase 5 immunoreactivity for granulovacuolar degeneration. Neuroreport 23, 867–872. doi: 10.1097/WNR.0b013e328358720b
Nakamori, M., Taylor, K., Mochizuki, H., Sobczak, K., and Takahashi, M. P. (2016). Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 3, 42–54. doi: 10.1002/acn3.271
Nakatani, K., Matsumoto, J., Nakamori, M., Okamoto, T., Murata, A., and Dohno, C. (2020). The dimeric form of 1,3-diaminoisoquinoline derivative rescued the mis-splicing of Atp2a1 and Clcn1 genes in myotonic dystrophy type 1 mouse model. Chemistry 26, 14305–14309. doi: 10.1002/chem.202001572
Nehra, M., Uthappa, U. T., Kumar, V., Kumar, R., Dixit, C., Dilbaghi, N., et al. (2021). Nanobiotechnology-assisted therapies to manage brain cancer in personalized manner. J. Control. Release 338, 224–243. doi: 10.1016/j.jconrel.2021.08.027
Nelson, C. E., Robinson-Hamm, J. N., and Gersbach, C. A. (2017). Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 13, 647–661. doi: 10.1038/nrneurol.2017.126
Nguyen, Q., and Yokota, T. (2020). Degradation of toxic RNA in myotonic dystrophy using gapmer antisense oligonucleotides. Methods Mol. Biol. 2176, 99–109. doi: 10.1007/978-1-0716-0771-8_7
Nieuwenhuis, S., Okkersen, K., Widomska, J., Blom, P., t Hoen, P. A. C., van Engelen, B., et al. (2019). Insulin signaling as a key moderator in myotonic dystrophy type 1. Front. Neurol. 10:1229. doi: 10.3389/fneur.2019.01229
Nishi, M., Kimura, T., Igeta, M., Furuta, M., Suenaga, K., Matsumura, T., et al. (2020). Differences in splicing defects between the grey and white matter in myotonic dystrophy type 1 patients. PLoS One 15:e0224912. doi: 10.1371/journal.pone.0224912
Nutter, C. A., Bubenik, J. L., Oliveira, R., Ivankovic, F., Sznajder, ŁJ., Kidd, B. M., et al. (2019). Cell-type-specific dysregulation of RNA alternative splicing in short tandem repeat mouse knockin models of myotonic dystrophy. Genes Dev. 33, 1635–1640. doi: 10.1101/gad.328963.119
Oana, K., Oma, Y., Suo, S., Takahashi, M. P., Nishino, I., Takeda, S., et al. (2013). Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci. Rep. 3:2142. doi: 10.1038/srep02142
Ofori, L. O., Hoskins, J., Nakamori, M., Thornton, C. A., and Miller, B. L. (2012). From dynamic combinatorial ‘hit’ to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res. 40, 6380–6390. doi: 10.1093/nar/gks298
Ogata, A., Terae, S., Fujita, M., and Tashiro, K. (1998). Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: an MRI and neuropathological study. Neuroradiology 40, 411–415. doi: 10.1007/s002340050613
Okkersen, K., Buskes, M., Groenewoud, J., Kessels, R. P. C., Knoop, H., van Engelen, B., et al. (2017a). The cognitive profile of myotonic dystrophy type 1: a systematic review and meta-analysis. Cortex 95, 143–155. doi: 10.1016/j.cortex.2017.08.008
Okkersen, K., Monckton, D. G., Le, N., Tuladhar, A. M., Raaphorst, J., and van Engelen, B. G. M. (2017b). Brain imaging in myotonic dystrophy type 1: a systematic review. Neurology 89, 960–969. doi: 10.1212/wnl.0000000000004300
Okkersen, K., Jimenez-Moreno, C., Wenninger, S., Daidj, F., Glennon, J., Cumming, S., et al. (2018). Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: a multicentre, single-blind, randomised trial. Lancet Neurol. 17, 671–680. doi: 10.1016/s1474-4422(18)30203-5
Ono, S., Kanda, F., Takahashi, K., Fukuoka, Y., Jinnai, K., Kurisaki, H., et al. (1995). Neuronal cell loss in the dorsal raphe nucleus and the superior central nucleus in myotonic dystrophy: a clinicopathological correlation. Acta Neuropathol. 89, 122–125. doi: 10.1007/bf00296355
Ono, S., Kanda, F., Takahashi, K., Fukuoka, Y., Jinnai, K., Kurisaki, H., et al. (1996). Neuronal loss in the medullary reticular formation in myotonic dystrophy: a clinicopathological study. Neurology 46, 228–231. doi: 10.1212/wnl.46.1.228
Ono, S., Takahashi, K., Jinnai, K., Kanda, F., Fukuoka, Y., Kurisaki, H., et al. (1998). Loss of serotonin-containing neurons in the raphe of patients with myotonic dystrophy: a quantitative immunohistochemical study and relation to hypersomnia. Neurology 50, 535–538. doi: 10.1212/wnl.50.2.535
Otero, B. A., Poukalov, K., Hildebrandt, R. P., Thornton, C. A., Jinnai, K., Fujimura, H., et al. (2021). Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 34:108634. doi: 10.1016/j.celrep.2020.108634
Oude Ophuis, R. J., Mulders, S. A., van Herpen, R. E., van de Vorstenbosch, R., Wieringa, B., and Wansink, D. G. (2009). DMPK protein isoforms are differentially expressed in myogenic and neural cell lineages. Muscle Nerve 40, 545–555. doi: 10.1002/mus.21352
Pandey, S. K., Wheeler, T. M., Justice, S. L., Kim, A., Younis, H. S., Gattis, D., et al. (2015). Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther. 355, 329–340. doi: 10.1124/jpet.115.226969
Pascual-Gilabert, M., López-Castel, A., and Artero, R. (2021). Myotonic dystrophy type 1 drug development: a pipeline toward the market. Drug Discov. Today 26, 1765–1772. doi: 10.1016/j.drudis.2021.03.024
Pegoraro, V., Cudia, P., Baba, A., and Angelini, C. (2020). MyomiRNAs and myostatin as physical rehabilitation biomarkers for myotonic dystrophy. Neurol. Sci. 41, 2953–2960. doi: 10.1007/s10072-020-04409-2
Perbellini, R., Greco, S., Sarra-Ferraris, G., Cardani, R., Capogrossi, M. C., Meola, G., et al. (2011). Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromusc. Disord. 21, 81–88. doi: 10.1016/j.nmd.2010.11.012
Perfetti, A., Greco, S., Bugiardini, E., Cardani, R., Gaia, P., Gaetano, C., et al. (2014). Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromusc. Disord. 24, 509–515. doi: 10.1016/j.nmd.2014.02.005
Perfetti, A., Greco, S., Cardani, R., Fossati, B., Cuomo, G., Valaperta, R., et al. (2016). Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 6:38174. doi: 10.1038/srep38174
Peric, S., Brajkovic, L., Belanovic, B., Ilic, V., Salak-Djokic, B., Basta, I., et al. (2017a). Brain positron emission tomography in patients with myotonic dystrophy type 1 and type 2. J. Neurol. Sci. 378, 187–192. doi: 10.1016/j.jns.2017.05.013
Peric, S., Heatwole, C., Durovic, E., Kacar, A., Nikolic, A., Basta, I., et al. (2017b). Prospective measurement of quality of life in myotonic dystrophy type 1. Acta Neurol. Scand. 136, 694–697. doi: 10.1111/ane.12788
Peric, S., Pavlovic, A., Ralic, V., Dobricic, V., Basta, I., Lavrnic, D., et al. (2014b). Transcranial sonography in patients with myotonic dystrophy type 1. Muscle Nerve 50, 278–282. doi: 10.1002/mus.24162
Peric, S., Mandic-Stojmenovic, G., Markovic, I., Stefanova, E., Ilic, V., Parojcic, A., et al. (2014a). Cerebrospinal fluid biomarkers of neurodegeneration in patients with juvenile and classic myotonic dystrophy type 1. Eur. J. Neurol. 21, 231–237. doi: 10.1111/ene.12237
Peric, S., Vujnic, M., Dobricic, V., Marjanovic, A., Basta, I., Novakovic, I., et al. (2016). Five-year study of quality of life in myotonic dystrophy. Acta Neurol. Scand. 134, 346–351. doi: 10.1111/ane.12549
Pettersson, O. J., Aagaard, L., Jensen, T. G., and Damgaard, C. K. (2015). Molecular mechanisms in DM1 - a focus on foci. Nucleic Acids Res. 43, 2433–2441. doi: 10.1093/nar/gkv029
Pincherle, A., Patruno, V., Raimondi, P., Moretti, S., Dominese, A., Martinelli-Boneschi, F., et al. (2012). Sleep breathing disorders in 40 Italian patients with Myotonic dystrophy type 1. Neuromusc. Disord. 22, 219–224. doi: 10.1016/j.nmd.2011.08.010
Pinto, B. S., Saxena, T., Oliveira, R., Méndez-Gómez, H. R., Cleary, J. D., Denes, L. T., et al. (2017). Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479.e5–490.e5. doi: 10.1016/j.molcel.2017.09.033
Quera Salva, M. A., Blumen, M., Jacquette, A., Durand, M. C., Andre, S., De Villiers, M., et al. (2006). Sleep disorders in childhood-onset myotonic dystrophy type 1. Neuromusc. Disord. 16, 564–570. doi: 10.1016/j.nmd.2006.06.007
Raaijmakers, R. H. L., Ripken, L., Ausems, C. R. M., and Wansink, D. G. (2019). CRISPR/Cas applications in myotonic dystrophy: expanding opportunities. Int. J. Mol. Sci. 20:3689. doi: 10.3390/ijms20153689
Rakocevic-Stojanovic, V., Peric, S., Madzarevic, R., Dobricic, V., Ralic, V., Ilic, V., et al. (2014). Significant impact of behavioral and cognitive impairment on quality of life in patients with myotonic dystrophy type 1. Clin. Neurol. Neurosurg. 126, 76–81. doi: 10.1016/j.clineuro.2014.08.021
Ramon-Duaso, C., Gener, T., Consegal, M., Fernández-Avilés, C., Gallego, J. J., Castarlenas, L., et al. (2019). Methylphenidate attenuates the cognitive and mood alterations observed in Mbnl2 knockout mice and reduces microglia overexpression. Cerebr. Cortex 29, 2978–2997. doi: 10.1093/cercor/bhy164
Ramon-Duaso, C., Rodríguez-Morató, J., Selma-Soriano, E., Fernández-Avilés, C., Artero, R., de la Torre, R., et al. (2020). Protective effects of mirtazapine in mice lacking the Mbnl2 gene in forebrain glutamatergic neurons: relevance for myotonic dystrophy 1. Neuropharmacology 170:108030. doi: 10.1016/j.neuropharm.2020.108030
Rau, F., Freyermuth, F., Fugier, C., Villemin, J. P., Fischer, M. C., Jost, B., et al. (2011). Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18, 840–845. doi: 10.1038/nsmb.2067
Reddy, K., Jenquin, J. R., Cleary, J. D., and Berglund, J. A. (2019a). Mitigating RNA toxicity in myotonic dystrophy using small molecules. Int. J. Mol. Sci. 20:4017. doi: 10.3390/ijms20164017
Reddy, K., Jenquin, J. R., McConnell, O. L., Cleary, J. D., Richardson, J. I., Pinto, B. S., et al. (2019b). A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. U.S.A. 116, 20991–21000. doi: 10.1073/pnas.1901893116
Reddy, S., Smith, D. B., Rich, M. M., Leferovich, J. M., Reilly, P., Davis, B. M., et al. (1996). Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 13, 325–335. doi: 10.1038/ng0796-325
Renard, D., Collombier, L., Castelli, C., Pouget, J. P., Kotzki, P. O., and Boudousq, V. (2016). In myotonic dystrophy type 1 reduced FDG-uptake on FDG-PET is most severe in Brodmann area 8. BMC Neurol. 16:100. doi: 10.1186/s12883-016-0630-3
Richard, G. F. (2015). Shortening trinucleotide repeats using highly specific endonucleases: a possible approach to gene therapy? Trends Genet. 31, 177–186. doi: 10.1016/j.tig.2015.02.003
Richard, G. F., Viterbo, D., Khanna, V., Mosbach, V., Castelain, L., and Dujon, B. (2014). Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS One 9:e95611. doi: 10.1371/journal.pone.0095611
Romeo, V., Pegoraro, E., Squarzanti, F., Sorarù, G., Ferrati, C., Ermani, M., et al. (2010b). Retrospective study on PET-SPECT imaging in a large cohort of myotonic dystrophy type 1 patients. Neurol. Sci. 31, 757–763. doi: 10.1007/s10072-010-0406-2
Romeo, V., Pegoraro, E., Ferrati, C., Squarzanti, F., Sorarù, G., Palmieri, A., et al. (2010a). Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J. Neurol. 257, 1246–1255. doi: 10.1007/s00415-010-5498-3
Rosahl, T. W., Geppert, M., Spillane, D., Herz, J., Hammer, R. E., Malenka, R. C., et al. (1993). Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell 75, 661–670. doi: 10.1016/0092-8674(93)90487-b
Rosman, N. P., and Kakulas, B. A. (1966). Mental deficiency associated with muscular dystrophy. A neuropathological study. Brain 89, 769–788. doi: 10.1093/brain/89.4.769
Sabater-Arcis, M., Bargiela, A., Furling, D., and Artero, R. (2020). miR-7 restores phenotypes in myotonic dystrophy muscle cells by repressing hyperactivated autophagy. Mol. Ther. Nucleic Acids 19, 278–292. doi: 10.1016/j.omtn.2019.11.012
Sakurai, T. (2014). The role of orexin in motivated behaviours. Nat. Rev. Neurosci. 15, 719–731. doi: 10.1038/nrn3837
Salvatori, S., Furlan, S., Fanin, M., Picard, A., Pastorello, E., Romeo, V., et al. (2009). Comparative transcriptional and biochemical studies in muscle of myotonic dystrophies (DM1 and DM2). Neurol. Sci. 30, 185–192. doi: 10.1007/s10072-009-0048-4
Sansone, V., Gandossini, S., Cotelli, M., Calabria, M., Zanetti, O., and Meola, G. (2007). Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol. Sci. 28, 9–15. doi: 10.1007/s10072-007-0742-z
Schmitz, F., Pierozan, P., Rodrigues, A. F., Biasibetti, H., Coelho, D. M., Mussulini, B. H., et al. (2016). Chronic treatment with a clinically relevant dose of methylphenidate increases glutamate levels in cerebrospinal fluid and impairs glutamatergic homeostasis in prefrontal cortex of juvenile rats. Mol. Neurobiol. 53, 2384–2396. doi: 10.1007/s12035-015-9219-x
Schmitz, F., Pierozan, P., Rodrigues, A. F., Biasibetti, H., Grings, M., Zanotto, B., et al. (2017). Methylphenidate decreases ATP levels and impairs glutamate uptake and Na(+),K(+)-ATPase activity in juvenile rat hippocampus. Mol. Neurobiol. 54, 7796–7807. doi: 10.1007/s12035-016-0289-1
Schneider-Gold, C., Bellenberg, B., Prehn, C., Krogias, C., Schneider, R., Klein, J., et al. (2015). Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One 10:e0130352. doi: 10.1371/journal.pone.0130352
Schoch, K. M., and Miller, T. M. (2017). Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. doi: 10.1016/j.neuron.2017.04.010
Seijger, C., Raaphorst, J., Vonk, J., van Engelen, B., Heijerman, H., Stigter, N., et al. (2021). New insights in adherence and survival in myotonic dystrophy patients using home mechanical ventilation. Respir. Int. Rev. Thor. Dis. 100, 154–163. doi: 10.1159/000511962
Sellier, C., Cerro-Herreros, E., Blatter, M., Freyermuth, F., Gaucherot, A., Ruffenach, F., et al. (2018). rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 9:2009. doi: 10.1038/s41467-018-04370-x
Sergeant, N., Sablonnière, B., Schraen-Maschke, S., Ghestem, A., Maurage, C. A., Wattez, A., et al. (2001). Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum. Mol. Genet. 10, 2143–2155. doi: 10.1093/hmg/10.19.2143
Serra, L., Scocchia, M., Meola, G., D’Amelio, M., Bruschini, M., Silvestri, G., et al. (2020b). Ventral tegmental area dysfunction affects decision-making in patients with myotonic dystrophy type-1. Cortex 128, 192–202. doi: 10.1016/j.cortex.2020.03.022
Serra, L., Bianchi, G., Bruschini, M., Giulietti, G., Domenico, C. D., Bonarota, S., et al. (2020a). Abnormal cortical thickness is associated with deficits in social cognition in patients with myotonic dystrophy type 1. Front. Neurol. 11:113. doi: 10.3389/fneur.2020.00113
Serra, L., Cercignani, M., Bruschini, M., Cipolotti, L., Mancini, M., Silvestri, G., et al. (2016a). “I know that you know that i know”: neural substrates associated with social cognition deficits in DM1 patients. PLoS One 11:e0156901. doi: 10.1371/journal.pone.0156901
Serra, L., Mancini, M., Silvestri, G., Petrucci, A., Masciullo, M., Spanò, B., et al. (2016b). Brain connectomics’ modification to clarify motor and nonmotor features of myotonic dystrophy type 1. Neural Plast. 2016:2696085. doi: 10.1155/2016/2696085
Serra, L., Petrucci, A., Spanò, B., Torso, M., Olivito, G., Lispi, L., et al. (2015). How genetics affects the brain to produce higher-level dysfunctions in myotonic dystrophy type 1. Funct. Neurol. 30, 21–31.
Serra, L., Silvestri, G., Petrucci, A., Basile, B., Masciullo, M., Makovac, E., et al. (2014). Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol. 71, 603–611. doi: 10.1001/jamaneurol.2014.130
Shimizu, E., Tang, Y. P., Rampon, C., and Tsien, J. Z. (2000). NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science 290, 1170–1174. doi: 10.1126/science.290.5494.1170
Siboni, R. B., Nakamori, M., Wagner, S. D., Struck, A. J., Coonrod, L. A., Harriott, S. A., et al. (2015b). Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 13, 2386–2394. doi: 10.1016/j.celrep.2015.11.028
Siboni, R. B., Bodner, M. J., Khalifa, M. M., Docter, A. G., Choi, J. Y., Nakamori, M., et al. (2015a). Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J. Med. Chem. 58, 5770–5780. doi: 10.1021/acs.jmedchem.5b00356
Sicot, G., Servais, L., Dinca, D. M., Leroy, A., Prigogine, C., Medja, F., et al. (2017). Downregulation of the glial GLT1 glutamate transporter and purkinje cell dysfunction in a mouse model of myotonic dystrophy. Cell Rep. 19, 2718–2729. doi: 10.1016/j.celrep.2017.06.006
Simoncini, C., Spadoni, G., Lai, E., Santoni, L., Angelini, C., Ricci, G., et al. (2020). Central nervous system involvement as outcome measure for clinical trials efficacy in myotonic dystrophy type 1. Front. Neurol. 11:624. doi: 10.3389/fneur.2020.00624
Singh, R. K., Xia, Z., Bland, C. S., Kalsotra, A., Scavuzzo, M. A., Curk, T., et al. (2014). Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol. Cell 55, 592–603. doi: 10.1016/j.molcel.2014.06.035
Sistiaga, A., Urreta, I., Jodar, M., Cobo, A. M., Emparanza, J., Otaegui, D., et al. (2010). Cognitive/personality pattern and triplet expansion size in adult myotonic dystrophy type 1 (DM1): CTG repeats, cognition and personality in DM1. Psychol. Med. 40, 487–495. doi: 10.1017/s0033291709990602
Smith, R. A., Miller, T. M., Yamanaka, K., Monia, B. P., Condon, T. P., Hung, G., et al. (2006). Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 116, 2290–2296. doi: 10.1172/jci25424
Sobczak, K., Wheeler, T. M., Wang, W., and Thornton, C. A. (2013). RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 21, 380–387. doi: 10.1038/mt.2012.222
Solovyeva, E. M., Ibebunjo, C., Utzinger, S., Eash, J. K., Dunbar, A., Naumann, U., et al. (2021). New insights into molecular changes in skeletal muscle aging and disease: differential alternative splicing and senescence. Mech. Age. Dev. 197:111510. doi: 10.1016/j.mad.2021.111510
Song, J., Lu, C., Leszek, J., and Zhang, J. (2021). Design and development of nanomaterial-based drug carriers to overcome the blood-brain barrier by using different transport mechanisms. Int. J. Mol. Sci. 22:10118. doi: 10.3390/ijms221810118
Song, K. Y., Guo, X. M., Wang, H. Q., Zhang, L., Huang, S. Y., Huo, Y. C., et al. (2020). MBNL1 reverses the proliferation defect of skeletal muscle satellite cells in myotonic dystrophy type 1 by inhibiting autophagy via the mTOR pathway. Cell Death Dis. 11:545. doi: 10.1038/s41419-020-02756-8
Stepniak-Konieczna, E., Konieczny, P., Cywoniuk, P., Dluzewska, J., and Sobczak, K. (2020). AON-induced splice-switching and DMPK pre-mRNA degradation as potential therapeutic approaches for myotonic dystrophy type 1. Nucleic Acids Research 48, 2531–2543. doi: 10.1093/nar/gkaa007
Steyaert, J., Umans, S., Willekens, D., Legius, E., Pijkels, E., de Die-Smulders, C., et al. (1997). A study of the cognitive and psychological profile in 16 children with congenital or juvenile myotonic dystrophy. Clin. Genet. 52, 135–141. doi: 10.1111/j.1399-0004.1997.tb02533.x
Strutt, S. C., Torrez, R. M., Kaya, E., Negrete, O. A., and Doudna, J. A. (2018). RNA-dependent RNA targeting by CRISPR-Cas9. eLife 7:e32724. doi: 10.7554/eLife.32724
Subramony, S. H., Wymer, J. P., Pinto, B. S., and Wang, E. T. (2020). Sleep disorders in myotonic dystrophies. Muscle Nerve 62, 309–320. doi: 10.1002/mus.26866
Sudhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Suenaga, K., Lee, K. Y., Nakamori, M., Tatsumi, Y., Takahashi, M. P., Fujimura, H., et al. (2012). Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One 7:e33218. doi: 10.1371/journal.pone.0033218
Sugiyama, A., Sone, D., Sato, N., Kimura, Y., Ota, M., Maikusa, N., et al. (2017). Brain gray matter structural network in myotonic dystrophy type 1. PLoS One 12:e0187343. doi: 10.1371/journal.pone.0187343
Summerton, J. (1999). Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta 1489, 141–158. doi: 10.1016/s0167-4781(99)00150-5
Sun, N., and Zhao, H. (2013). Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 110, 1811–1821. doi: 10.1002/bit.24890
Suzuki, K., Miyamoto, M., Miyamoto, T., Matsubara, T., Inoue, Y., Iijima, M., et al. (2018). Cerebrospinal fluid orexin-A levels in systemic lupus erythematosus patients presenting with excessive daytime sleepiness. Lupus 27, 1847–1853. doi: 10.1177/0961203318778767
Suzuki, Y., Holmes, J. B., Cerritelli, S. M., Sakhuja, K., Minczuk, M., Holt, I. J., et al. (2010). An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell. Biol. 30, 5123–5134. doi: 10.1128/mcb.00619-10
Symonds, T., Randall, J. A., and Campbell, P. (2017). Review of patient-reported outcome measures for use in myotonic dystrophy type 1 patients. Muscle Nerve 56, 86–92. doi: 10.1002/mus.25469
Takado, Y., Terajima, K., Ohkubo, M., Okamoto, K., Shimohata, T., Nishizawa, M., et al. (2015). Diffuse brain abnormalities in myotonic dystrophy type 1 detected by 3.0 T proton magnetic resonance spectroscopy. Eur. Neurol. 73, 247–256. doi: 10.1159/000371575
Takeda, A., Kobayakawa, M., Suzuki, A., Tsuruya, N., and Kawamura, M. (2009). Lowered sensitivity to facial emotions in myotonic dystrophy type 1. J. Neurol. Sci. 280, 35–39. doi: 10.1016/j.jns.2009.01.014
Timchenko, L. T., Miller, J. W., Timchenko, N. A., DeVore, D. R., Datar, K. V., Lin, L., et al. (1996). Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 24, 4407–4414. doi: 10.1093/nar/24.22.4407
Todd, P. K., Ackall, F. Y., Hur, J., Sharma, K., Paulson, H. L., and Dowling, J. J. (2014). Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis. Models Mech. 7, 143–155. doi: 10.1242/dmm.012427
Tomé, S., Holt, I., Edelmann, W., Morris, G. E., Munnich, A., Pearson, C. E., et al. (2009). MSH2 ATPase domain mutation affects CTG∗CAG repeat instability in transgenic mice. PLoS Genet. 5:e1000482. doi: 10.1371/journal.pgen.1000482
Toth, A., Lovadi, E., Komoly, S., Schwarcz, A., Orsi, G., Perlaki, G., et al. (2015). Cortical involvement during myotonia in myotonic dystrophy: an fMRI study. Acta Neurol. Scand. 132, 65–72. doi: 10.1111/ane.12360
Tremblay, M., Muslemani, S., Côté, I., Gagnon, C., Fortin, J., and Gallais, B. (2021). Accomplishment of instrumental activities of daily living and its relationship with cognitive functions in adults with myotonic dystrophy type 1 childhood phenotype: an exploratory study. BMC Psychol. 9:56. doi: 10.1186/s40359-021-00562-1
Udd, B., and Krahe, R. (2012). The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 11, 891–905. doi: 10.1016/s1474-4422(12)70204-1
van Agtmaal, E. L., André, L. M., Willemse, M., Cumming, S. A., van Kessel, I. D. G., van den Broek, W., et al. (2017). CRISPR/Cas9-induced (CTG⋅CAG)(n) repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 25, 24–43. doi: 10.1016/j.ymthe.2016.10.014
van de Vondervoort, I., Amiri, H., Bruchhage, M. M. K., Oomen, C. A., Rustogi, N., Cooper, J. D., et al. (2019). Converging evidence points towards a role of insulin signaling in regulating compulsive behavior. Transl. Psychiatry 9:225. doi: 10.1038/s41398-019-0559-6
van de Vondervoort, I., Poelmans, G., Aschrafi, A., Pauls, D. L., Buitelaar, J. K., Glennon, J. C., et al. (2016). An integrated molecular landscape implicates the regulation of dendritic spine formation through insulin-related signalling in obsessive-compulsive disorder. J. Psychiatry Neurosci. 41, 280–285. doi: 10.1503/jpn.140327
van der Bent, M. L., Paulino da Silva Filho, O., van Luijk, J., Brock, R., and Wansink, D. G. (2018). Assisted delivery of antisense therapeutics in animal models of heritable neurodegenerative and neuromuscular disorders: a systematic review and meta-analysis. Sci. Rep. 8:4181. doi: 10.1038/s41598-018-22316-7
van der Plas, E., Hamilton, M. J., Miller, J. N., Koscik, T. R., Long, J. D., Cumming, S., et al. (2019). Brain structural features of myotonic dystrophy type 1 and their relationship with CTG repeats. J. Neuromuscul. Dis. 6, 321–332. doi: 10.3233/jnd-190397
van der Velden, B. G., Okkersen, K., Kessels, R. P., Groenewoud, J., van Engelen, B., Knoop, H., et al. (2019). Affective symptoms and apathy in myotonic dystrophy type 1 a systematic review and meta-analysis. J. Affect. Disord. 250, 260–269. doi: 10.1016/j.jad.2019.03.036
van Engelen, B. (2015). Cognitive behaviour therapy plus aerobic exercise training to increase activity in patients with myotonic dystrophy type 1 (DM1) compared to usual care (OPTIMISTIC): study protocol for randomised controlled trial. Trials 16:224. doi: 10.1186/s13063-015-0737-7
Vita, G., Vita, G. L., Musumeci, O., Rodolico, C., and Messina, S. (2019). Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurol. Sci. 40, 671–681. doi: 10.1007/s10072-019-03764-z
Voellenkle, C., Perfetti, A., Carrara, M., Fuschi, P., Renna, L. V., Longo, M., et al. (2019). Dysregulation of circular RNAs in myotonic dystrophy type 1. Int. J. Mol. Sci. 20:1938. doi: 10.3390/ijms20081938
Walder, R. Y., and Walder, J. A. (1988). Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 85, 5011–5015. doi: 10.1073/pnas.85.14.5011
Walker, G. L., Rosser, R., Mastaglia, F. L., and Walton, J. N. (1984). Psychometric and cranial CT study in myotonic dystrophy. Clin. Exp. Neurol. 20, 161–167.
Wang, E. T., Cody, N. A., Jog, S., Biancolella, M., Wang, T. T., Treacy, D. J., et al. (2012). Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150, 710–724. doi: 10.1016/j.cell.2012.06.041
Wang, E. T., Taliaferro, J. M., Lee, J. A., Sudhakaran, I. P., Rossoll, W., Gross, C., et al. (2016). Dysregulation of mRNA localization and translation in genetic disease. J. Neurosci. 36, 11418–11426. doi: 10.1523/jneurosci.2352-16.2016
Wang, G. S., Kuyumcu-Martinez, M. N., Sarma, S., Mathur, N., Wehrens, X. H., and Cooper, T. A. (2009). PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Investig. 119, 3797–3806. doi: 10.1172/jci37976
Wang, M., Weng, W. C., Stock, L., Lindquist, D., Martinez, A., Gourdon, G., et al. (2019). Correction of glycogen synthase kinase 3β in myotonic dystrophy 1 reduces the mutant RNA and improves postnatal survival of DMSXL mice. Mol. Cell. Biol. 39:e00155-19. doi: 10.1128/mcb.00155-19
Wang, P. Y., Chang, K. T., Lin, Y. M., Kuo, T. Y., and Wang, G. S. (2018). Ubiquitination of MBNL1 Is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 22, 2294–2306. doi: 10.1016/j.celrep.2018.02.025
Wang, P. Y., Lin, Y. M., Wang, L. H., Kuo, T. Y., Cheng, S. J., and Wang, G. S. (2017). Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 26, 2247–2257. doi: 10.1093/hmg/ddx115
Wang, Y., Hao, L., Wang, H., Santostefano, K., Thapa, A., Cleary, J., et al. (2018). Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 26, 2617–2630. doi: 10.1016/j.ymthe.2018.09.003
Wang, Y., Miao, L., Satterlee, A., and Huang, L. (2015). Delivery of oligonucleotides with lipid nanoparticles. Adv. Drug Deliv. Rev. 87, 68–80. doi: 10.1016/j.addr.2015.02.007
Wansink, D. G., van Herpen, R. E., Coerwinkel-Driessen, M. M., Groenen, P. J., Hemmings, B. A., and Wieringa, B. (2003). Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol. Cell. Biol. 23, 5489–5501. doi: 10.1128/mcb.23.16.5489-5501.2003
Warf, M. B., Nakamori, M., Matthys, C. M., Thornton, C. A., and Berglund, J. A. (2009). Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 18551–18556. doi: 10.1073/pnas.0903234106
Weber, Y. G., Roebling, R., Kassubek, J., Hoffmann, S., Rosenbohm, A., Wolf, M., et al. (2010). Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 74, 1108–1117. doi: 10.1212/WNL.0b013e3181d8c35f
Wei, C., Jones, K., Timchenko, N. A., and Timchenko, L. (2013). GSK3β is a new therapeutic target for myotonic dystrophy type 1. Rare Dis. 1:e26555. doi: 10.4161/rdis.26555
Weijs, R., Okkersen, K., van Engelen, B., Küsters, B., Lammens, M., Aronica, E., et al. (2021). Human brain pathology in myotonic dystrophy type 1: a systematic review. Neuropathology 41, 3–20. doi: 10.1111/neup.12721
Wheeler, T. M., Leger, A. J., Pandey, S. K., MacLeod, A. R., Nakamori, M., Cheng, S. H., et al. (2012). Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115. doi: 10.1038/nature11362
Wheeler, T. M., Sobczak, K., Lueck, J. D., Osborne, R. J., Lin, X., Dirksen, R. T., et al. (2009). Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339. doi: 10.1126/science.1173110
Winblad, S., Hellström, P., Lindberg, C., and Hansen, S. (2006). Facial emotion recognition in myotonic dystrophy type 1 correlates with CTG repeat expansion. J. Neurol. Neurosurg. Psychiatry 77, 219–223. doi: 10.1136/jnnp.2005.070763
Winblad, S., Månsson, J. E., Blennow, K., Jensen, C., Samuelsson, L., and Lindberg, C. (2008). Cerebrospinal fluid tau and amyloid beta42 protein in patients with myotonic dystrophy type 1. Eur. J. Neurol. 15, 947–952. doi: 10.1111/j.1468-1331.2008.02217.x
Winblad, S., Samuelsson, L., Lindberg, C., and Meola, G. (2016). Cognition in myotonic dystrophy type 1: a 5-year follow-up study. Eur. J. Neurol. 23, 1471–1476. doi: 10.1111/ene.13062
Wojciechowska, M., Taylor, K., Sobczak, K., Napierala, M., and Krzyzosiak, W. J. (2014). Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 11, 742–754. doi: 10.4161/rna.28799
Woo, J., Lee, H. W., and Park, J. S. (2019). Differences in the pattern of cognitive impairments between juvenile and adult onset myotonic dystrophy type 1. J. Clin. Neurosci. 68, 92–96. doi: 10.1016/j.jocn.2019.07.029
Wozniak, J. R., Mueller, B. A., Bell, C. J., Muetzel, R. L., Lim, K. O., and Day, J. W. (2013). Diffusion tensor imaging reveals widespread white matter abnormalities in children and adolescents with myotonic dystrophy type 1. J. Neurol. 260, 1122–1131. doi: 10.1007/s00415-012-6771-4
Wozniak, J. R., Mueller, B. A., Lim, K. O., Hemmy, L. S., and Day, J. W. (2014). Tractography reveals diffuse white matter abnormalities in myotonic dystrophy type 1. J. Neurol. Sci. 341, 73–78. doi: 10.1016/j.jns.2014.04.005
Wozniak, J. R., Mueller, B. A., Ward, E. E., Lim, K. O., and Day, J. W. (2011). White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1: a diffusion tensor imaging study. Neuromusc. Disord. 21, 89–96. doi: 10.1016/j.nmd.2010.11.013
Xia, G., and Ashizawa, T. (2015). Dynamic changes of nuclear RNA foci in proliferating DM1 cells. Histochem. Cell Biol. 143, 557–564. doi: 10.1007/s00418-015-1315-5
Xia, G., Gao, Y., Jin, S., Subramony, S. H., Terada, N., Ranum, L. P., et al. (2015). Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33, 1829–1838. doi: 10.1002/stem.1970
Xia, G., Santostefano, K. E., Goodwin, M., Liu, J., Subramony, S. H., Swanson, M. S., et al. (2013). Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell. Reprogram. 15, 166–177. doi: 10.1089/cell.2012.0086
Yadava, R. S., Foff, E. P., Yu, Q., Gladman, J. T., Kim, Y. K., Bhatt, K. S., et al. (2015). TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum. Mol. Genet. 24, 2035–2048. doi: 10.1093/hmg/ddu617
Yadava, R. S., Yu, Q., Mandal, M., Rigo, F., Bennett, C. F., and Mahadevan, M. S. (2020). Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3′UTR RNA. Hum. Mol. Genet. 29, 1440–1453. doi: 10.1093/hmg/ddaa060
Yamamoto, H., Kokame, K., Okuda, T., Nakajo, Y., Yanamoto, H., and Miyata, T. (2011). NDRG4 protein-deficient mice exhibit spatial learning deficits and vulnerabilities to cerebral ischemia. J. Biol. Chem. 286, 26158–26165. doi: 10.1074/jbc.M111.256446
Yamazaki, Y., Matsubara, T., Takahashi, T., Kurashige, T., Dohi, E., Hiji, M., et al. (2011). Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PLoS One 6:e26996. doi: 10.1371/journal.pone.0026996
Yin, H., Moulton, H. M., Seow, Y., Boyd, C., Boutilier, J., Iverson, P., et al. (2008). Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 17, 3909–3918. doi: 10.1093/hmg/ddn293
Zanigni, S., Evangelisti, S., Giannoccaro, M. P., Oppi, F., Poda, R., Giorgio, A., et al. (2016). Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. Neuroimage Clin. 11, 678–685. doi: 10.1016/j.nicl.2016.04.012
Zhang, B. W., Cai, H. F., Wei, X. F., Sun, J. J., Lan, X. Y., Lei, C. Z., et al. (2016). miR-30-5p regulates muscle differentiation and alternative splicing of muscle-related genes by targeting MBNL. Int. J. Mol. Sci. 17:182. doi: 10.3390/ijms17020182
Zhang, F., Bodycombe, N. E., Haskell, K. M., Sun, Y. L., Wang, E. T., Morris, C. A., et al. (2017). A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 26, 3056–3068. doi: 10.1093/hmg/ddx190
Zhang, N., Bewick, B., Xia, G., Furling, D., and Ashizawa, T. (2020). A CRISPR-Cas13a based strategy that tracks and degrades toxic RNA in myotonic dystrophy type 1. Front. Genet. 11:594576. doi: 10.3389/fgene.2020.594576
Zhang, W., Liu, H., Han, K., and Grabowski, P. J. (2002). Region-specific alternative splicing in the nervous system: implications for regulation by the RNA-binding protein NAPOR. RNA 8, 671–685. doi: 10.1017/s1355838202027036
Zhang, W., Wang, Y., Dong, S., Choudhury, R., Jin, Y., and Wang, Z. (2014). Treatment of type 1 myotonic dystrophy by engineering site-specific RNA endonucleases that target (CUG)(n) repeats. Mol. Ther. 22, 312–320. doi: 10.1038/mt.2013.251
Keywords: myotonic dystrophy type 1, central nervous system, pathology, mechanism, treatment
Citation: Liu J, Guo Z-N, Yan X-L, Yang Y and Huang S (2021) Brain Pathogenesis and Potential Therapeutic Strategies in Myotonic Dystrophy Type 1. Front. Aging Neurosci. 13:755392. doi: 10.3389/fnagi.2021.755392
Received: 08 August 2021; Accepted: 20 October 2021;
Published: 15 November 2021.
Edited by:
Natalia Salvadores, Universidad Mayor, ChileReviewed by:
Stojan Z. Peric, University of Belgrade, SerbiaBulmaro Cisneros, Instituto Politécnico Nacional de México (CINVESTAV), Mexico
Corrado Italo Angelini, University of Padua, Italy
Copyright © 2021 Liu, Guo, Yan, Yang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuo Huang, Ym9yZWFzMDcyOUBqbHUuZWR1LmNu
†ORCID: Yi Yang, orcid.org/0000-0002-9729-8522