
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 10 December 2021
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.748388
 Afzal Misrani1,2,3†
Afzal Misrani1,2,3† Sidra Tabassum1,2,3†Qingwei Huo4†Sumaiya Tabassum2†
Sidra Tabassum1,2,3†Qingwei Huo4†Sumaiya Tabassum2† Jinxiang Jiang1†Adeel Ahmed2Xiangmao Chen2Jianwen Zhou2
Jinxiang Jiang1†Adeel Ahmed2Xiangmao Chen2Jianwen Zhou2 Jiajia Zhang2
Jiajia Zhang2 Sha Liu2Xiaoyi Feng2
Sha Liu2Xiaoyi Feng2 Cheng Long2,3*
Cheng Long2,3* Li Yang1*
Li Yang1*
Alzheimer’s disease (AD) is the most common neurodegenerative disorder worldwide. Mitochondrial dysfunction is thought to be an early event in the onset and progression of AD; however, the precise underlying mechanisms remain unclear. In this study, we investigated mitochondrial proteins involved in organelle dynamics, morphology and energy production in the medial prefrontal cortex (mPFC) and hippocampus (HIPP) of young (1∼2 months), adult (4∼5 months) and aged (9∼10, 12∼18 months) APP/PS1 mice. We observed increased levels of mitochondrial fission protein, Drp1, and decreased levels of ATP synthase subunit, ATP5A, leading to abnormal mitochondrial morphology, increased oxidative stress, glial activation, apoptosis, and altered neuronal morphology as early as 4∼5 months of age in APP/PS1 mice. Electrophysiological recordings revealed abnormal miniature excitatory postsynaptic current in the mPFC together with a minor connectivity change between the mPFC and HIPP, correlating with social deficits. These results suggest that abnormal mitochondrial dynamics, which worsen with disease progression, could be a biomarker of early-stage AD. Therapeutic interventions that improve mitochondrial function thus represent a promising approach for slowing the progression or delaying the onset of AD.
Alzheimer’s disease (AD) is a progressive, multifactorial, age-dependent, neurodegenerative disorder characterized by loss of memory, impairment of cognitive and non-cognitive functions, and changes in personality and behavior (Saez-Atienzar and Masliah, 2020). At the cellular and molecular levels, AD is associated with loss of neurons, neurofibrillary tangles, and amyloid β (Aβ) deposits in the cortex and hippocampus (Van Der Kant et al., 2020). However, several recent fundamental discoveries highlight important pathological roles for other critical cellular and molecular processes. Despite this, no disease-modifying treatment currently exists, and numerous phase 3 clinical trials targeting Aβ have failed to demonstrate benefit.
The mitochondrion, known as the powerhouse of the cell, is the organelle that produces the energy (in the form of adenosine triphosphate, ATP) necessary for the survival and optimal function of neurons (Spinelli and Haigis, 2018). Multiple studies suggest that mitochondrial ATP levels in affected brain regions of AD patients and mouse models are reduced (Reddy, 2008; Yao et al., 2009; Terni et al., 2010; Cha et al., 2015; Gauba et al., 2019). Neurons are especially vulnerable to mitochondrial dysfunction due to their high energy demand and dependence on the respiration for ATP generation. Mitochondrial dysfunction increases reactive oxygen species (ROS) production, leading to oxidative stress, neuroinflammation (Joshi et al., 2019), and subsequent neuronal damage in AD (Wang et al., 2020; Misrani et al., 2021). Despite evidence suggesting that mitochondrial dysfunction (Hauptmann et al., 2009; Wang et al., 2020) and abnormal synaptic transmission (Selkoe, 2002; Styr and Slutsky, 2018) are early events, earlier than the appearance of Aβ plaques in AD pathology, which comes first has not been evaluated side-by-side.
The prefrontal cortex (PFC) and hippocampus (HIPP) brain regions, which are critically involved in cognition and decision making (Spellman et al., 2015), are among the earliest areas to suffer impairment during AD progression (Grady et al., 2001; Wang et al., 2006). The PFC governs many higher-order executive tasks such as learning, memory (Warden and Miller, 2010), cognitive flexibility (Gruber et al., 2010), and emotional processing (Parfitt et al., 2017; Koush et al., 2019). Abnormal PFC activity, such as impaired executive functioning and working memory, is reported in AD patients (Satler and Tomaz, 2011; Stopford et al., 2012). The HIPP is widely studied in AD as this brain region is essential for forming new memories, and the progressive degeneration of neurons in the HIPP is responsible for the short-term memory loss that is a hallmark of AD (Selkoe, 2002; Wei et al., 2010). Moreover, reduced numbers of dendritic spines and altered synaptic transmission in PFC and HIPP are early events in AD (Wei et al., 2010; Styr and Slutsky, 2018; Ammassari-Teule, 2020). The neuronal projections from the HIPP to the PFC, referred to as the HIPP-PFC-circuit, play a critical role in cognitive, social, and emotional regulation. Furthermore, altered functional connectivity of the HIPP-PFC circuit occurs in AD, leading to cognitive impairment (Zaidel et al., 2012; Xue et al., 2019). Nevertheless, although synaptic dysfunction and altered connectivity of brain circuits are characteristics of AD (Selkoe, 2002; Styr and Slutsky, 2018; Xue et al., 2019), whether AD pathogenesis alters the HIPP-mPFC pathway in APP/PS1 mice is unclear.
In this paper, we used amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mice (Jankowsky et al., 2001) (1) to determine the earliest age at which mitochondrial alteration occurs in this mouse AD model; (2) to explore whether neuronal morphological and synaptic dysfunction co-occur with mitochondrial dysfunction; (3) to evaluate whether the earliest defects affect social behavior, which requires normal HIPP-mPFC activity. The results, which reveal severe defects in mitochondrial dynamics together with neuroinflammation in the brains of young APP/PS1 mice, may stimulate the development of new therapeutic strategies for AD.
Amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mice, derived from the B6C3-Tg (APPswe, PSEN1dE9) 85Dbo/J strain (JAX 004462), which expresses a chimeric mouse/human APP gene (APPswe) and human mutant PS1 (DeltaE9), were maintained at the animal house facility of the School of Life Sciences, Guangzhou University. Both male and female APPswe/PS1ΔE9 and their WT littermate mice were used in this study. The mice were genotyped by polymerase chain reaction (PCR) according to the Jackson Laboratory (JAX) protocol. Animals were housed in cages where mice could eat and drink freely, with a 12-h light-dark cycle. All animals involved in experiments were 1∼2, 4∼5, 9∼10, and 12∼18 months old unless otherwise indicated.
Mouse brains were rapidly dissected on ice, and mPFC and HIPP tissues were homogenized in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA pH 8.0, 1% SDS and protease inhibitors (Complete Mini; Roche)). After centrifugation at 4°C (14,000 rpm for 10 min), cellular debris was removed, and the supernatant was collected for western blotting. Tissue lysates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and separated proteins were transferred to nitrocellulose membranes. Membranes were then blocked with 5% defatted milk in Tris-buffered saline with Tween 20 (TBST) for 1 h and incubated overnight at 4°C with the following specific primary antibodies against Drp1 (Abcam, ab184247; dilution 1:1,000), Mfn1 (Abcam, ab57602; dilution 1:3,000), Mfn2 (Abcam, ab56889; dilution 1:3,000), OPA1 (Abcam, ab157457; dilution 1:2,000), ATP5A (Abcam, ab14748; dilution 1:3,000), PSD95 (Abcam, ab2723; dilution 1:3,000), GFAP (Thermofisher, 13-0300; dilution 1:1,000), Iba1 (Wako, 019-19741, dilution 1:1,000), nitrotyrosine (Santa Cruz: sc-32757; dilution 1:1,000), Cleaved caspase-3 (Abcam, ab13847; dilution 1:1,000) and Nrf2 (Abcam, ab137550; dilution 1:3,000). Anti γ-tubulin antibody (Sigma, T6557) was used as a loading control. After three washes with TBST, HRP-labeled secondary antibody (CWS, China) was added at room temperature for 1 h using 5% milk in TBST, followed by three additional washes with TBST. The Immobilon ECL western system (Millipore, United States) was then used to visualize the bands, which were quantified and analyzed with Gel-Pro Analysis software (Media Cybernetics, United States).
The mPFC and HIPP were fixed overnight in the 2.5 % (v/v) glutaraldehyde and then in 1 % (w/v) OsO4 for 1 h. The fixed slices were dehydrated in an ascending series of ethanol finishing with absolute alcohol and embedded in EPON resin. Ultra-thin sections (70 nm thick) were cut using (LEICA UC7/FC7 Ultramicrotome) from the tissue slices that were stained with uranyl acetate and lead citrate and then examined under a JEM 1400 plus Japan transmission electron microscope (TEM) as described previously (Moriguchi et al., 2019). The mitochondrial ultra-microstructure of pyramidal cells in the mPFC and HIPP was examined under magnification of × 23,000 and × 49,000. The number of mitochondria and mitochondrial aspect ratio (length/width) were calculated using ImageJ software.
The Golgi-Cox staining protocol followed a routine procedure (Zhang et al., 2020). All animals were coded before the Golgi-Cox method to blind the experimenter to the animal’s identity until the data analysis was completed. Briefly, animals were perfused transcardially with 4% paraformaldehyde (PFA). The brains were rapidly removed and stored in Golgi-Cox solution in the dark at room temperature (RT) for 14 days; the Golgi-Cox solution was refreshed every 48 h to remove sediments. These brains were then transferred into a 30% sucrose solution for 7 days, and the sucrose solution was changed every day. Brains were then embedded in paraffin wax, and 150 μm thick brain slices from mPFC and HIPP were cut using a microtome (Leica, Germany).
Medial prefrontal cortex and hippocampus pyramidal neurons were identified based on the following criteria: (1) location within the mPFC or HIPP; (2) staining of the intact neuron; (3) triangular-shaped soma and single axon; and(4) no direct contact with neighbouring neurons. All neurons were reconstructed under a light microscope (Nikon, Japan) using a 40× lens. Image J software was used to analyze dendritic length, the number of branches, and the morphological complexity of the cells.
Acute brain slices containing mPFC and HIPP (350 μm) were prepared according to routine procedures (Chen et al., 2017) from WT and APP/PS1 mice using a vibratome (VT 1000S, Leica, Germany) in an oxygenated ice-cold cutting solution containing (in mM), 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 11 D-glucose, 26.2 NaHCO3 (pH 7.2–7.4), saturated with 95% O2/5% CO2. Slices were kept in artificial cerebrospinal fluid (aCSF) containing (in mM) 140 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 11 D-glucose, 10 HEPES (pH 7.2–7.4), and gassed with 95% O2/5% CO2. Slices were then incubated for 1 h at 30–32°C before recording and then transferred to a submerged recording chamber where the temperature was held at 32 ± 0.5°C with an automatic temperature controller (TC-324B, Warner Instrument Corporation) with aCSF flow set at 2–3 ml/min.
To record miniature excitatory postsynaptic current (mEPSC) and miniature inhibitory postsynaptic current (mIPSC) from pyramidal neurons of the mPFC, the voltage was held at −60 and 0 mV, respectively. To block fast sodium channel activity and action potential, 1 μM TTX was added to the aCSF. The pipette was filled with the following internal solution (mM): 100 mM Cs-gluconate, 5 mM CsCl, 10 mM HEPES, 2 mM MgCl2, 1 mM CaCl2, 11 mM BAPTA, 4 mM ATP and 0.4 mM GTP (pH 7.3, adjusted with KOH) at an osmolality of 280–290 mOsm. In another experiment using a Drp1 inhibitor named mitochondrial division inhibitor (Mdivi-1) (Huang et al., 2015; Baek et al., 2017), we asked if neuronal functional alterations could be prevented. Therefore, we recorded mEPSC of pyramidal neurons of mPFC in 4∼5-month-old APP/PS1 mice, treated with DMSO (Vehicle) or Mdivi-1 (Mdivi-1). Mdivi-1 (Sigma Aldrich, 475856-10MG) was prepared as a stock solution (10 mM) and was diluted with DMSO to the final concentration immediately before use. Brain slices were incubated with either Mdivi-1 at a final concentration of 10 μmol/L or DMSO for 1 h followed by the recording of mEPSC. DMSO or Mdivi-1 containing aCSF was continuously perfused over slices during recording. Data were collected with a MultiClamp 700 B amplifier (Axon Instruments) and filtered during acquisition with a low pass filter set at 2 kHz using pCLAMP10 software (Molecular Devices, United States). The data were analyzed offline using Mini Analysis Program (Synaptosoft Inc., United States).
In vivo dual-site extracellular recordings were conducted as described (Engel et al., 2001). Mice were anesthetized with pentobarbital sodium (IP 80 mg/kg), then head-fixed in a stereotaxic apparatus (RWD Life Science) with body temperature maintained between 36 and 37°C. When necessary, a supplemental dose of anesthesia was given based on tail reflex. After a midline skin incision was made, two skull holes were drilled above the mPFC (1.98 mm anterior to the bregma, 0.5 mm lateral to the midline, 1.2 mm depth) and the CA1 subregion of the HIPP (−2.06 mm posterior to the bregma, −1.5 mm lateral to the midline, 1.0 mm depth) under a stereomicroscope (Sunny Optical Technology). Two glass microelectrodes for recording (filled with 0.5 M NaCl, with a resistance of 1.0–1.5 MΩ) were slowly inserted until the tips of the electrodes reached the mPFC and hippocampal CA1. Each recorded signal was amplified (1,000×) by an electrometer amplifier (Model 3000; A-M Systems) and digitized via a D/A converter (Micro 1401; Cambridge Electronic Design), then sent to data acquisition software (Spike2; Cambridge Electronic Design).
Local field potential data was analyzed offline in MATLAB 2012a (MathWorks) and spike2 (Chen et al., 2021). For processing the LFP, a Butterworth low pass filter (300 Hz) was applied to the raw recorded data. Synchronization was evaluated using cross-correlation analysis in line with our established protocol (Chen et al., 2019). Simultaneously recorded data were first low-passed using a third-order Butterworth filter in a phase-preserving manner and then subjected to normalized estimation of similarity. The maximal offset was set to ± 1 s for cross-correlation analysis. After the calculation, spectral coherence between the two LFPs from the mPFC and HIPP was analyzed under an FFT number of 512, and values were obtained in the range from 0 to 1, meaning non- (0) or completely (1) correlated in the frequency domain.
Mice were anesthetized by IP injection of 20% urethane (0.01 ml/g) and perfused transcardially with 0.9% saline and 4% paraformaldehyde (PFA) in PBS (0.01 M, pH 7.4). Immediately after perfusion, brains were dissected and post-fixed in 4% PFA for 24 h, then dehydrated by immersion in a 15% sucrose solution overnight followed by a 30% sucrose solution until the tissue no longer floated in the sucrose solution. Serial coronal/sagittal sections of mPFC and HIPP tissues were cut to a 30 μm thickness using a Leica CM30505 freezing microtome (Leica, Germany). The slices were then treated for membrane permeabilization with 0.5% Triton-X 100 and blocked with 5% BSA in 0.01 M PBS for 1.5 h at RT, followed by incubation with primary antibody against rabbit anti-Iba1 (Wako, 019-19741, 1:1,000) and anti-GFAP (Thermofisher, 13-0300, 1:1,000) overnight at 4°C in 1% BSA/PBS. The following day, slices were given three 10 min washes with PBS and incubated with secondary antibody (Invitrogen) at RT for 2 h followed by three additional washes with PBS for 10 min. Each section was imaged using a 20× lens under a fluorescence microscope (Nikon, Japan). Positive-cell counting was done using Image-Pro Plus (Media Cybernetics, United States) and Image J software (National Institutes of Health, Bethesda, MD, United States).
Social memory can be quantified by measuring the relative interaction durations with a novel and a familiar mouse under free-choice conditions (social discrimination test or SDT) as described previously (Okuyama et al., 2016). Briefly, the three-chambered apparatus is composed of a solid rectangular plexiglas container (60 × 44 × 40 cm3) separated into three sections (each 20 × 44 × 40 cm3) by plain Plexiglas walls containing openings. An inverted cage (10.5 cm high × 10.5 cm diameter bottom × 7.6 cm diameter top, 1 cm bar spacing) was placed in each of the two outer chambers (ZhengHua Instruments, China). During a habituation period, the mouse could freely explore the apparatus for 10 min. Subsequently, in the test phase, a cagemate mouse was placed in one social chamber, whereas a stranger non-cagemate mouse of the same age and sex as the familiar mouse was placed in another social chamber. Then, the experimental mouse was permitted to explore the entire apparatus for 10 min. Duration of sniffing by test mice of each cage was recorded, and the discrimination index was calculated as follows: Duration familiar, total sniffing duration for cagemate mouse; Duration novel, total sniffing duration for stranger non-cagemate mouse).
Prism 8.0 for Windows (GraphPad, United States) and OriginPro 2020 (OriginLab, United States) software were employed for graphing and statistical analysis. A two-sample t-test was used for statistical analyses between two-group comparisons. For the Golgi-Cox staining analysis, one-way ANOVA with a post hoc test was performed. Sholl analysis was performed using two-way repeated measure ANOVA; unless otherwise stated. p < 0.05 was considered statistically significant. The data are presented as mean ± SEM.
Mitochondria dynamics involve two specific, highly regulated opposing processes known as fission and fusion (Westermann, 2010; Kandimalla et al., 2016), which are fundamental aspects of mitochondrial biology and its quality control (Detmer and Chan, 2007). Mitochondrial fission requires the action of dynamin-1-like protein (Drp1), which is critical for mitochondrial division, size, shape and distribution throughout the neuron (Lee et al., 2016). In contrast, mitochondrial fusion requires the action of the mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) oligomeric complexes to tether the outer membranes of two fusing mitochondria (Chen et al., 2003). Inner membrane fusion is mediated by the inner membrane optic atrophy type 1 (OPA1) protein (Cipolat et al., 2004). Considering the crucial role of mitochondrial fission and fusion in neuronal function, and the occurrence of mitochondrial dysfunction in AD, we first sought to determine the earliest age at which any alteration in these fission/fusion proteins occurs in APP/PS1 mice. We evaluated mitochondrial fission, fusion, and ATP synthase protein levels in mPFC and HIPP of young 1∼2 month-old APP/PS1 mice. Our western blotting results of mPFC extracts revealed no significant difference in the levels of Drp1 (WT: 1 ± 0.31; APP/PS1: 0.87 ± 0.29; p = 0.792); Mfn1 (WT: 1 ± 0.32; APP/PS1: 0.56 ± 0.20; p = 0.321); Mfn2 (WT: 1 ± 0.35; APP/PS1: 0.89 ± 0.21; p = 0.815); OPA1 (WT: 1 ± 0.43; APP/PS1: 1.04 ± 0.32; p = 0.935); and ATP5A (WT: 1 ± 0.15; APP/PS1: 1.19 ± 0.30; p = 0.607; Supplementary Figures 1A, B). Consistent with the mPFC findings, we also found no significant difference in HIPP levels of Drp1 (WT: 1 ± 0.16; APP/PS1: 0.84 ± 0.17; p = 0.548); Mfn1 (WT: 1 ± 0.21; APP/PS1: 0.85 ± 0.05; p = 0.535); Mfn2 (WT: 1 ± 0.19; APP/PS1: 0.74 ± 0.11; p = 0.326); OPA1 (WT: 1 ± 0.14; APP/PS1: 0.88 ± 0.29; p = 0.741); and ATP5A (WT: 1 ± 0.19; APP/PS1: 0.90 ± 0.29; p = 0.801; Supplementary Figures 1C, D). Our data suggest that expression of mitochondrial fission/fusion proteins and ATP synthase is normal in the mPFC and HIPP of APP/PS1 mice at 1∼2 months of age.
Importantly, a balance of fission and fusion is crucial not only for mitochondrial morphology, but also for cell viability, synaptic function and neuronal morphology. We found identical fission/fusion and ATP synthase levels at 1∼2 months of age in APP/PS1 mice, so we asked whether alterations in these mitochondrial proteins occur later than this age. Considering that APP/PS1 mice exhibit a trend of increased soluble Aβ in cortex and HIPP at 4∼5 months-old (Jankowsky et al., 2004; Garcia-Alloza et al., 2006), we examined whether 4∼5 month-old APP/PS1 mice exhibit any alteration in these mitochondrial fission/fusion proteins. Western blotting was conducted in both mPFC and HIPP. In mPFC, we found significantly increased levels of fission protein Drp1 (WT: 1 ± 0.13; APP/PS1: 1.84 ± 0.24; p = 0.012), leaving levels of fusion proteins unchanged: Mfn1 (WT: 1 ± 0.12; APP/PS1: 0.94 ± 0.14; p = 0.782); Mfn2 (WT: 1 ± 0.16; APP/PS1: 1.18 ± 0.12; p = 0.384); OPA1 (WT: 1 ± 0.22; APP/PS1: 1.12 ± 0.22; p = 0.699). Notably, ATP5A levels decreased in APP/PS1 mice (WT: 1 ± 0.20; APP/PS1: 0.32 ± 0.08; p = 0.012; Figures 1A,B). The findings for HIPP reflected those for mPFC, revealing significantly increased levels of fission protein Drp1 (WT: 1 ± 0.19; APP/PS1: 2.10 ± 0.31; p = 0.013) and decreased ATP5A levels (WT: 1 ± 0.20; APP/PS1: 0.34 ± 0.10; p = 0.017) in APP/PS1 mice, while fusion protein levels were identical: Mfn1 (WT: 1 ± 0.20; APP/PS1: 0.94 ± 0.17; p = 0.852); Mfn2 (WT: 1 ± 0.32; APP/PS1: 0.75 ± 0.19; p = 0.525); OPA1 (WT: 1 ± 0.13; APP/PS1: 0.92 ± 0.20; p = 0.753; Figures 1C,D).
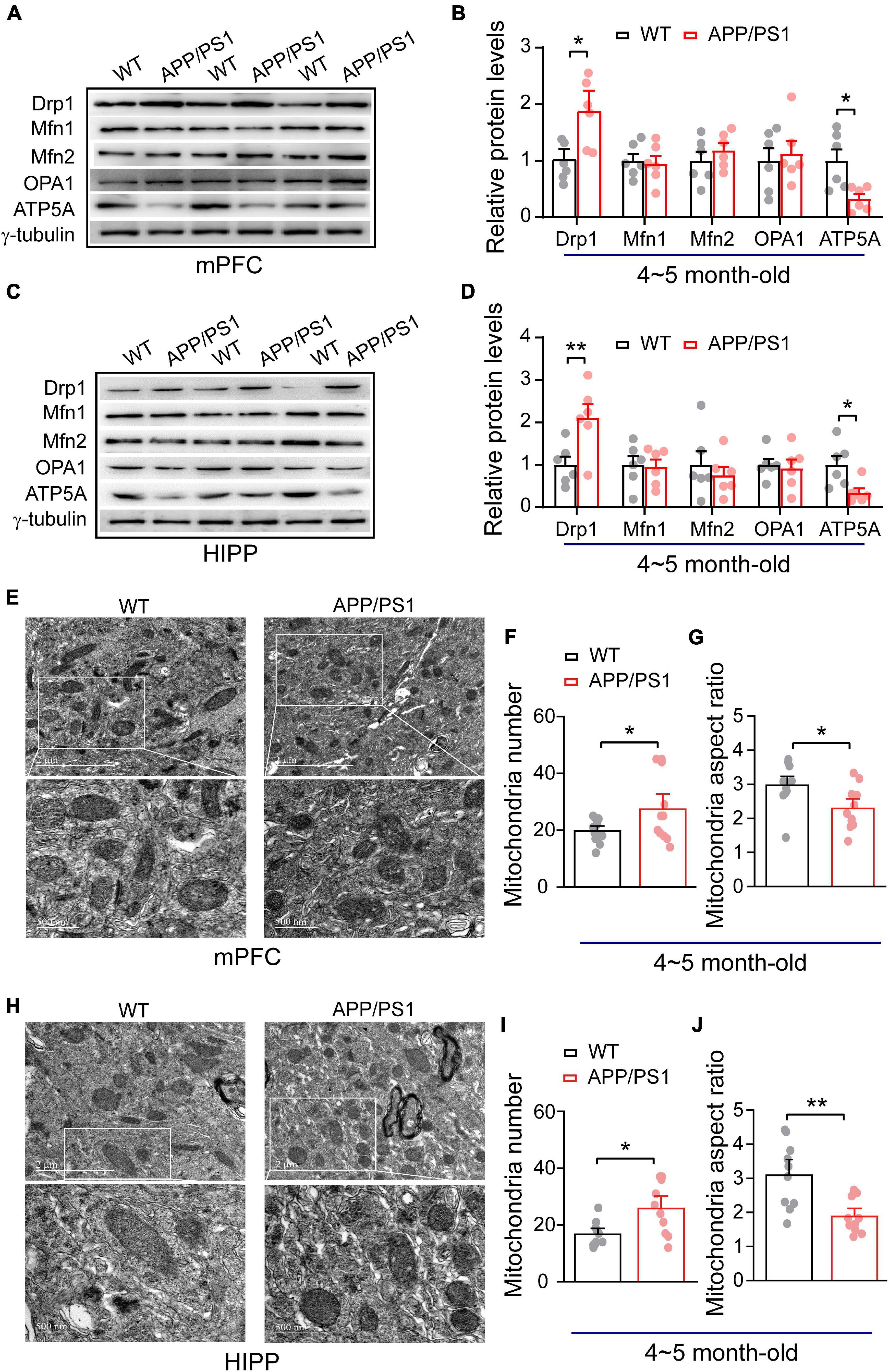
Figure 1. Alteration in mitochondrial fission, ATP synthase protein expression and mitochondrial morphology in the mPFC and HIPP of 4∼5-month-old APP/PS1 mice. (A) Representative immunoblotting images of mPFC extracts and (B) quantification of blots indicate significantly increased levels of Drp1 and decreased levels of ATP5A, whereas fusion proteins Mfn1, Mfn2, and OPA1 remain unchanged in APP/PS1 mice compared with age-matched WT mice (n = 6 mice per genotype; two repeats: two-sample t-test). (C) Representative immunoblotting images of hippocampus extracts and (D) quantification of blots indicate significantly increased levels of Drp1 and decreased levels of ATP5A, whereas fusion proteins Mfn1, Mfn2, and OPA1 remain unchanged in APP/PS1 mice compared with age-matched WT mice (n = 6 mice per genotype; two repeats; two-sample t-test). (E) Representative TEM images of mitochondrial morphology from mPFC of WT and APP/PS1 mice. (F) Increased mitochondrial number but (G) reduced mitochondrial aspect ratio in the mPFC of APP/PS1 mice than WT mice (n = 12 images for WT, 11 images for APP/PS1 mice; 3 mice per genotype: two-sample t-test). (H) Representative TEM images of mitochondrial morphology from HIPP of WT and APP/PS1 mice. (I) Increased mitochondrial number but (J) reduced mitochondrial aspect ratio in the HIPP of APP/PS1 mice than WT mice (n = 10 images for WT, 10 images for APP/PS1 mice; 3 mice per genotype: two-sample t-test). Each value represents the mean ± SEM, *p < 0.05, **p < 0.01.
Given that mitochondrial morphology is regulated by fission and fusion (Franco et al., 2016; Lee et al., 2016). We found increased mitochondrial fission (Drp1) in APP/PS1 mice; therefore, we asked if this could result in abnormal mitochondrial morphology. Using TEM, we have examined mitochondrial morphology in mPFC and HIPP of 4∼5 month-old APP/PS1 mice. Our data analysis of mPFC indicates an increased number of mitochondria (WT: 19.75 ± 1.14; APP/PS1: 27.36 ± 3.58; p = 0.047) and decreased mitochondria aspect ratio (WT: 2.96 ± 0.17; APP/PS1: 2.29 ± 0.18; p = 0.014; Figures 1F,G) in APP/PS1 mice. Similarly, HIPP also exhibits abnormal mitochondrial morphology in APP/PS1 mice, such as an increased number of mitochondria (WT: 16.7 ± 1.39; APP/PS1: 25.8 ± 2.93; p = 0.011); and decreased mitochondrial aspect ratio (WT: 3.08 ± 0.30; APP/PS1: 1.87 ± 0.16; p = 0.002; Figures 1I,J).
Aging is the primary risk factor for AD and is also associated with mitochondrial dysfunction, including impaired mitochondrial fission/fusion, biogenesis, and ATP production (Swerdlow et al., 2017; Hou et al., 2019). We next evaluated whether alterations in mitochondrial dynamics persist in older, i.e., 9∼10 month-old, APP/PS1 mice. Western blotting of mPFC extracts revealed an increased level of fission protein Drp1 (WT: 1 ± 0.06; APP/PS1: 2.16 ± 0.14; p < 0.001), but decreased levels of fusion proteins Mfn1 (WT: 1 ± 0.21; APP/PS1: 0.29 ± 0.11; p = 0.016) and OPA1 (WT: 1 ± 0.15; APP/PS1: 0.43 ± 0.06; p = 0.006), although no difference was found for Mfn2 (WT: 1 ± 0.06; APP/PS1: 0.83 ± 0.15; p = 0.361). Moreover, levels of ATP synthase protein ATP5A were decreased in mPFC (WT: 1 ± 0.11; APP/PS1: 0.40 ± 0.12; p = 0.005; Supplementary Figures 2A, B). A similar pattern was observed in HIPP, i.e., significantly increased levels of fission protein Drp1 (WT: 1 ± 0.20; APP/PS1: 3.51 ± 0.59; p = 0.002), but decreased levels of fusion proteins Mfn1 (WT: 1 ± 0.30; APP/PS1: 0.26 ± 0.06; p = 0.041), Mfn2 (WT: 1 ± 0.15; APP/PS1: 0.40 ± 0.11; p = 0.011) and OPA1 (WT: 1 ± 0.10; APP/PS1: 0.48 ± 0.04; p = 0.001), as well as decreased ATP5A levels (WT: 1 ± 0.13; APP/PS1: 0.49 ± 0.04; p = 0.004; Supplementary Figures 2C, D).
Given that aged APP/PS1 mice exhibit progressive development of Aβ plaques, which begins at 8 months of age (Garcia-Alloza et al., 2006), together with altered synaptic transmission and impaired memory (Mcclean and Holscher, 2014), we next asked whether aging worsened expression of mitochondrial proteins in AD model mice. Western blotting of proteins extracted from 12∼18 month-old APP/PS1 mPFC exhibited increased levels of fission protein Drp1 (WT: 1 ± 0.07; APP/PS1: 1.41 ± 0.04; p = 0.003), but decreased levels of fusion proteins Mfn1 (WT: 1 ± 0.12; APP/PS1: 0.38 ± 0.01; p = 0.002), Mfn2 (WT: 1 ± 0.05; APP/PS1: 0.66 ± 0.05; p < 0.001) and OPA1 (WT: 1 ± 0.08; APP/PS1: 0.57 ± 0.11; p = 0.010). Moreover, levels of ATP synthase subunit, ATP5A, also decreased (WT: 1 ± 0.04; APP/PS1: 0.67 ± 0.06; p = 0.002; Supplementary Figures 3A, B). HIPP, again, revealed significantly increased levels of fission protein Drp1 (WT: 1 ± 0.02; APP/PS1: 2.56. ± 0.14; p < 0.001), but decreased levels of fusion proteins Mfn1 (WT: 1 ± 0.03; APP/PS1: 0.66 ± 0.02; p < 0.001), Mfn2 (WT: 1 ± 0.02; APP/PS1: 0.69 ± 0.04; p < 0.001) and OPA1 (WT: 1 ± 0.01; APP/PS1: 0.61 ± 0.03; p < 0.001), as well as decreased levels of ATP synthase subunit, ATP5A (WT: 1 ± 0.06; APP/PS1: 0.65 ± 0.06; p = 0.002; Supplementary Figures 3C, D).
The above results suggest that 4∼5 months is the earliest age at which APP/PS1 mice exhibit alterations in proteins crucial for mitochondrial dynamics, abnormal mitochondrial morphology and energy metabolism, and that these effects persist in older mice (9∼10, 12∼18 month-old). We then focused on 4∼5 month-old APP/PS1 mice and age-matched WT controls in the following experiments unless otherwise indicated.
Dendritic morphology critically regulates the electrical properties of the neuron, with adult cortical neurons receiving around 15,000 synaptic inputs (Bianchi et al., 2013). To meet neuronal energy demands, normal mitochondrial fission and fusion are crucial as they enable mitochondrial transport within neurons from soma to dendrites and axons. Importantly, defects in mitochondrial function can lead to severe alteration in neuronal morphology, eventually resulting in the death of neurons (Sheng and Cai, 2012). The elevated Drp1 levels in 4∼5 month-old APP/PS1 mice, described above, make increased mitochondrial fragmentation likely, which in turn can alter neuronal morphology (Bertholet et al., 2016). We therefore examined the dendritic morphology of pyramidal neurons in the mPFC using Golgi-Cox analysis (Figure 2A) and found that 4∼5 month-old APP/PS1 mice had fewer intersections at a distance of 140–220 μm from the soma [F(1,84) = 485.113; p < 0.01; Figure 2B]. APP/PS1 mice also showed significantly reduced dendritic length [F(1,199) = 5.020; p = 0.026] and number of dendritic branches [F(1,199) = 4.620; p = 0.032], but no difference in the number of dendrites [F(1,199) = 0.743; p = 0.389] (Figures 2C–E).
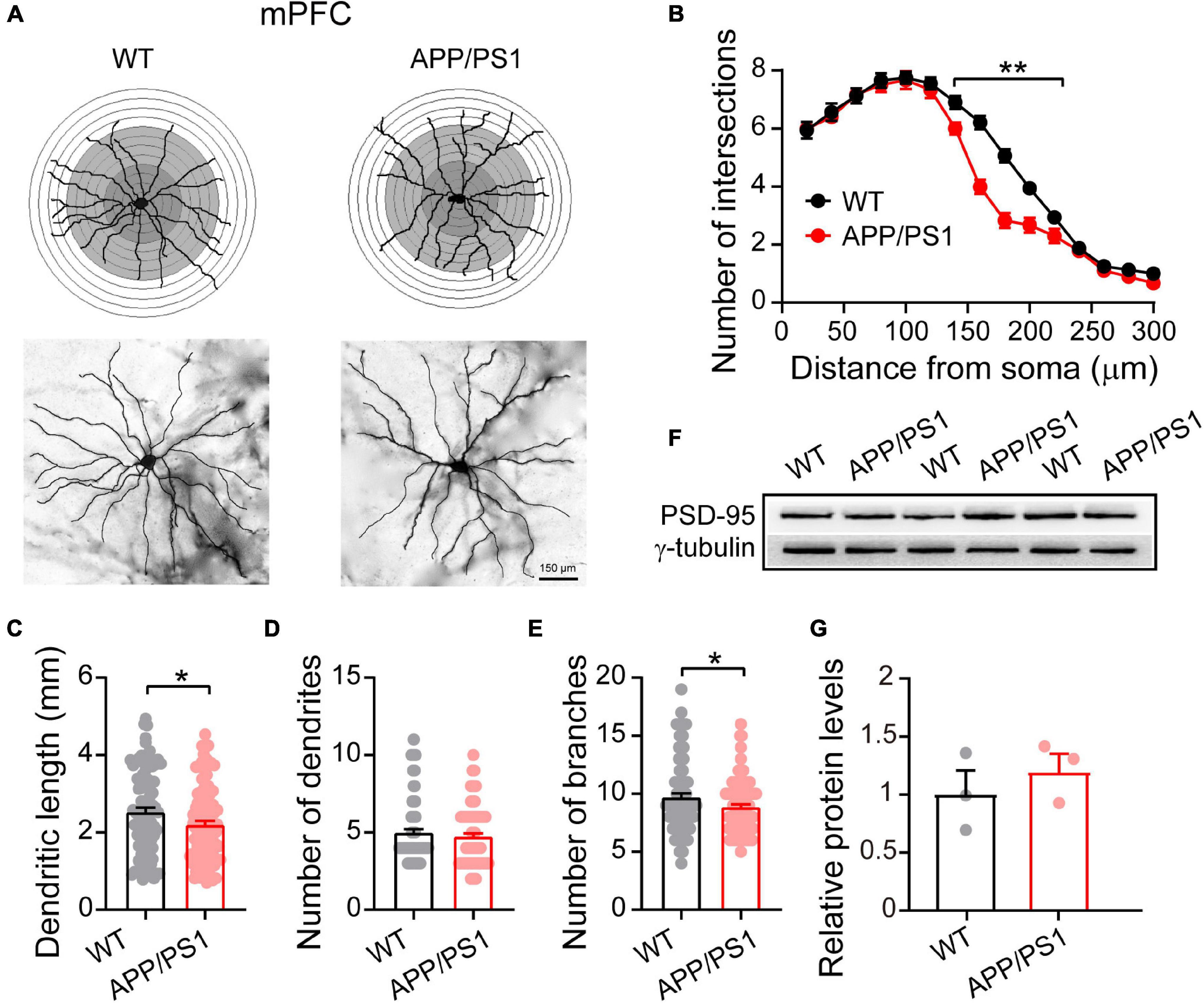
Figure 2. Altered dendritic morphology in the mPFC of 4∼5-month-old APP/PS1 mice. (A) Schematic photomicrographs of neurons with the allocation of dendrites between repeated 20 μm-spaced concentric rings. (B) APP/PS1 mice have fewer intersection points than age-matched WT mice (at a distance of 140–220 μm from the soma). (C–E) The dendrite length and number of branches of PFC neurons are significantly decreased in APP/PS1 mice compared with age-matched WT animals (n = 100 neurons from 4 animals per genotype; one-way ANOVA). (F) Representative immunoblotting images of PFC extracts and (G) quantification of blots indicate identical levels of PSD-95 in APP/PS1 mice compared with age-matched controls (n = 3 mice per genotype; two-sample t-test). Each value represents the mean ± SEM; *p < 0.05, **p < 0.01.
Having uncovered neuronal morphological alterations, we next analyzed the expression of PSD-95 in the mPFC. PSD-95 is an important postsynaptic scaffolding protein that plays a crucial role in dendritic remodeling and synaptic development (Sweet et al., 2011). However, the results revealed unchanged levels of PSD-95 in the mPFC of 4∼5 month-old APP/PS1 mice (WT: 1 ± 0.18; APP/PS1: 1.19 ± 0.14; p = 0.463; Figures 2F,G), suggesting that the changes in neuronal morphology were not due to abnormalities in PSD-95 expression.
Research suggests that HIPP shrinkage may be an early sign of AD, because it occurs years before memory loss, and other symptoms, appear (Fang et al., 2019). Our Golgi-Cox analysis of HIPP pyramidal neurons showed that 4∼5 month-old APP/PS1 mice had fewer intersections between 120 and 300 μm from the soma [F(1,98) = 439.813; p < 0.001; Figure 3B], and significantly reduced dendritic length [F(1,199) = 10.239; p = 0.001], number of dendrites [F(1,199) = 4.858; p = 0.028], and number of dendritic branches [F(1,199) = 4.683; p = 0.031] (Figures 3C–E). Interestingly, in contrast to the situation in mPFC, we observed decreased levels of PSD-95 in HIPP of 4∼5 month-old APP/PS1 mice (WT: 1 ± 0.13; APP/PS1: 0.46 ± 0.03; p = 0.020; Figures 3F,G).
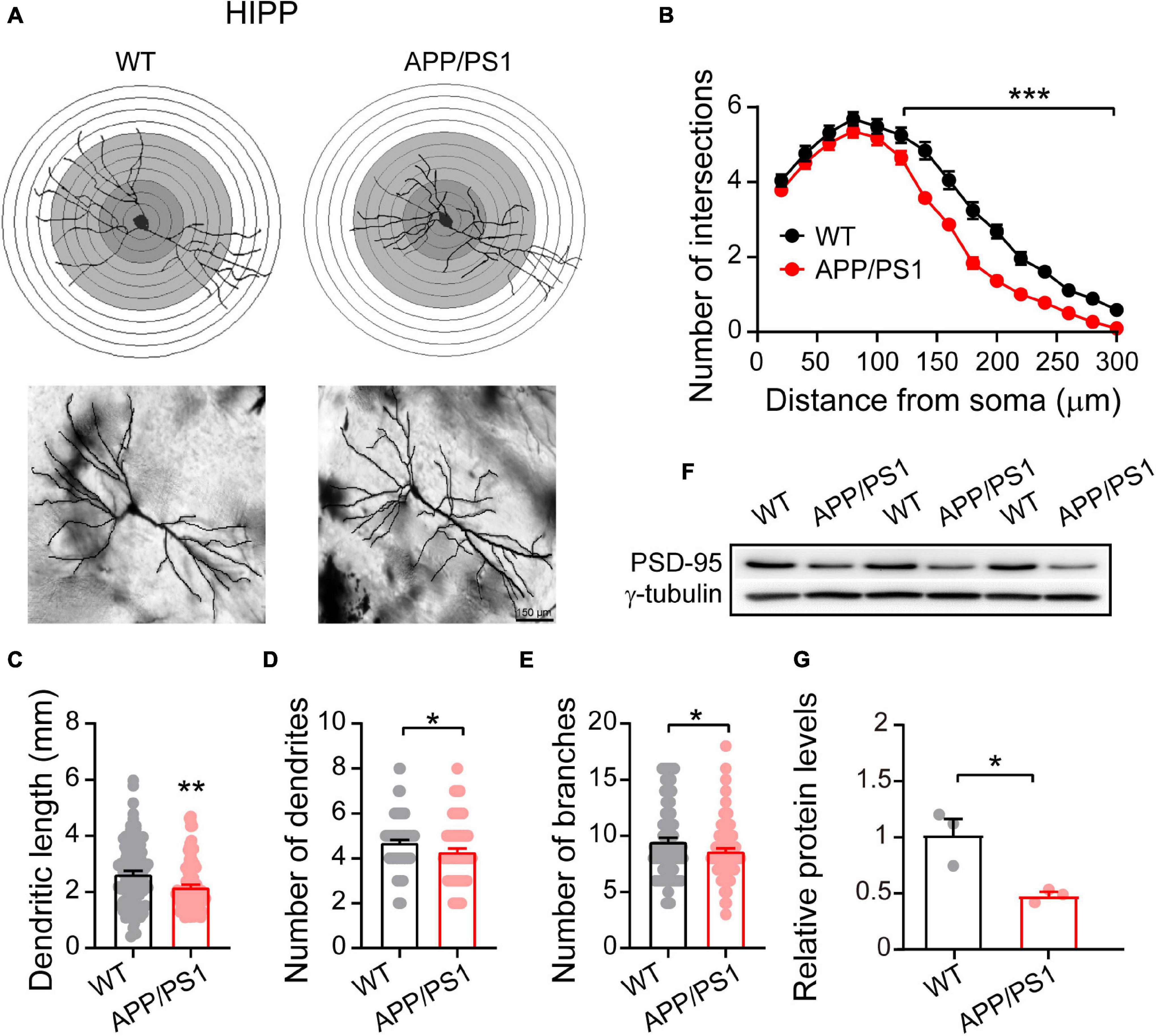
Figure 3. Altered dendritic morphology and decreased PSD-95 levels in the HIPP of 4∼5-month-old APP/PS1 mice. (A) Schematic photomicrographs of neurons with the allocation of dendrites between repeated 20 μm-spaced concentric rings. (B) APP/PS1 mice exhibit a reduced number of intersection points compared with age-matched WT (at a distance of 120–300 μm from the soma). (C–E) The dendrite length, number and branches of hippocampal neurons are significantly decreased in APP/PS1 mice compared with age-matched WT animals (n = 100 neurons from 4 animals per genotype; one-way ANOVA). (F) Representative immunoblotting images of HIPP extracts and (G) quantification of blots indicate significantly decreased levels of PSD-95 in APP/PS1 mice compared with age-matched WT mice (n = 3 mice per genotype; two-sample t-test). Each value represents the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001.
Mitochondria play an important role in presynaptic and postsynaptic neurotransmission through calcium buffering and a range of metabolic functions (Tang and Zucker, 1997; Devine and Kittler, 2018). Moreover, in neurons, fission uniquely facilitates the movement of mitochondria within axons and dendrites; disruptions of this movement due to alterations in the mitochondrial fission/fusion process specifically cause synaptic abnormalities and neuronal death (Shields et al., 2015; Devine and Kittler, 2018). Therefore, we determined whether mitochondrial dysfunction, as revealed by abnormal levels of mitochondrial proteins, might impair synaptic transmission in the mPFC and HIPP (Figure 4). Under the IR-DIC microscope, pyramidal neurons were identified by their typical triangular-shaped soma (Choy et al., 2018). The results showed unchanged mEPSC amplitude (WT: 9.80 ± 0.47 pA; APP/PS1: 9.43 ± 0.44 pA; p = 0.58; Figures 4B,D), but significantly decreased frequency (WT: 3.22 ± 0.43 Hz; APP/PS1: 1.95 ± 0.26 Hz; p = 0.0072; Figure 4E) in pyramidal neurons of mPFC in 4∼5-month-old APP/PS1 mice. In contrast, neither the amplitude nor the frequency of mIPSCs differed between the two groups (amplitude: WT: 8.22 ± 0.34 pA; APP/PS1: 7.73 ± 0.34 pA; p = 0.3179, frequency: WT: 2.35 ± 0.26 Hz; APP/PS1: 3.14 ± 0.3 Hz; p = 0.0512; Figures 4C,F,G). Moreover, mEPSC (n = 22 and 21 for WT and APP/PS1, respectively) and mIPSC (n = 23 and 22 for WT and APP/PS1, respectively) remained unchanged in the CA1 region of the HIPP in APP/PS1 compared to WT controls (data not shown).
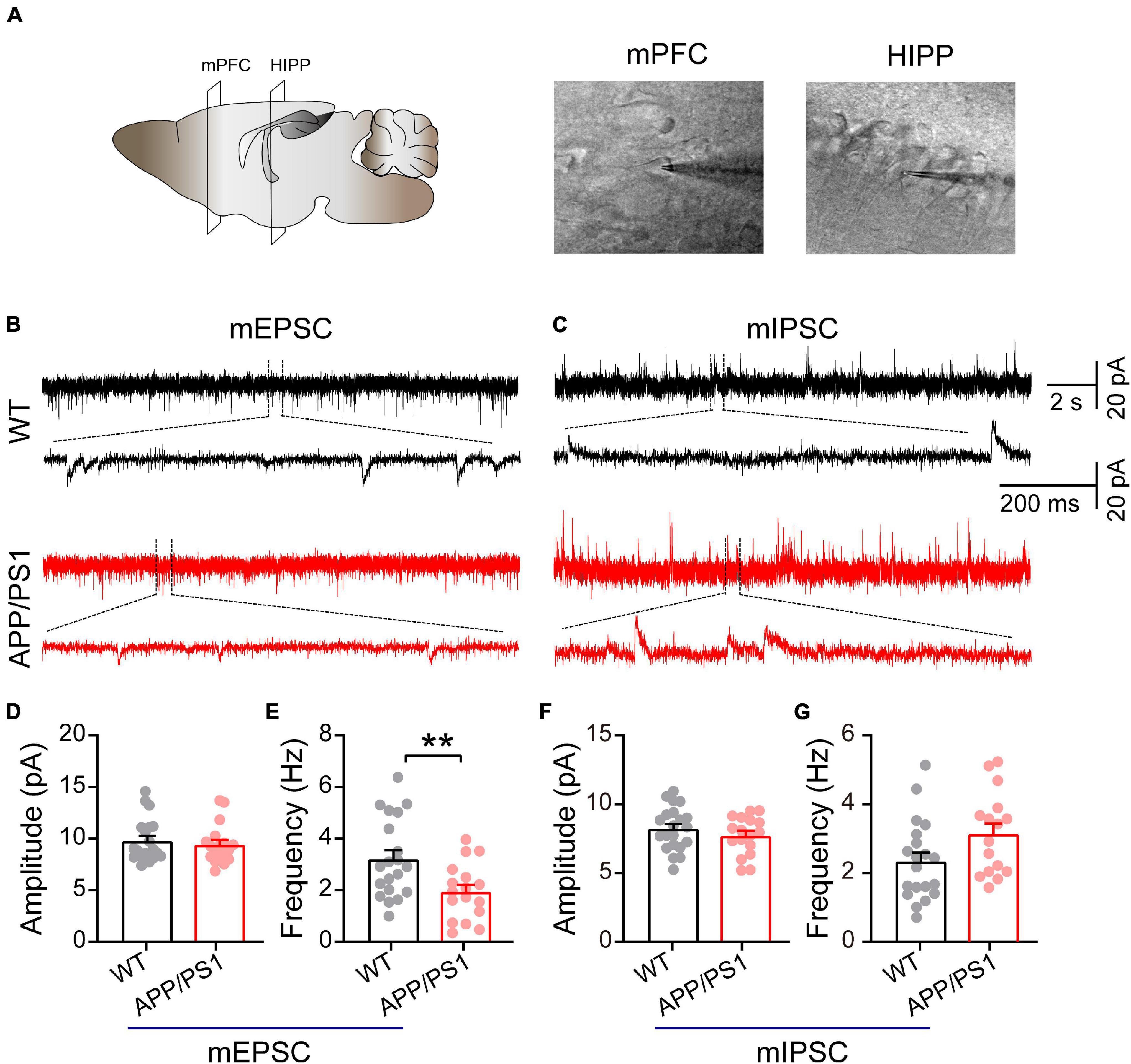
Figure 4. Decreased mEPSC frequency in pyramidal neurons of mPFC in 4∼5-month-old APP/PS1 mice. (A) Schematic of the mPFC and HIPP pyramidal neuron where whole-cell patch-clamp recording was performed. (B) Representative mEPSC and (C) mIPSC traces recorded in the mPFC. (D,E) Statistical analysis indicating significantly increased frequency but not amplitude of mEPSCs in APP/PS1 mice (frequency: p = 0.0072; amplitude: p = 0.5751; WT: n = 20 cells of 6 mice; APP/PS1: n = 17 cells of 6 mice). (F,G) Identical mIPSC frequency and amplitude in APP/PS1 mice (frequency: p = 0.0512; amplitude: p = 0.3179; WT: n = 21 cells of 6 mice; APP/PS1: n = 17 cells of 6 mice; two-sample t-test). Each value represents the mean ± SEM; **p < 0.01.
The direct HIPP-PFC pathway originates from the CA1 region of the HIPP and subiculum, selectively projecting to the prelimbic mPFC and orbitomedial frontal cortex (Thierry et al., 2000; Preston and Eichenbaum, 2013), and is critically involved in working memory and social interaction behavior (Sun et al., 2020). We thus evaluated HIPP-PFC connectivity by dual-site extracellular recordings in the mPFC and HIPP (Figure 5A). Although similar phase locking was observed in both brain areas of WT and APP/PS1 mice (Figure 5B), cross-correlation analysis revealed a significantly decreased correlation between mPFC and HIPP (Figures 5E,F), indicating reduced bidirectional communication and synchronization between the two areas in APP/PS1 mice.
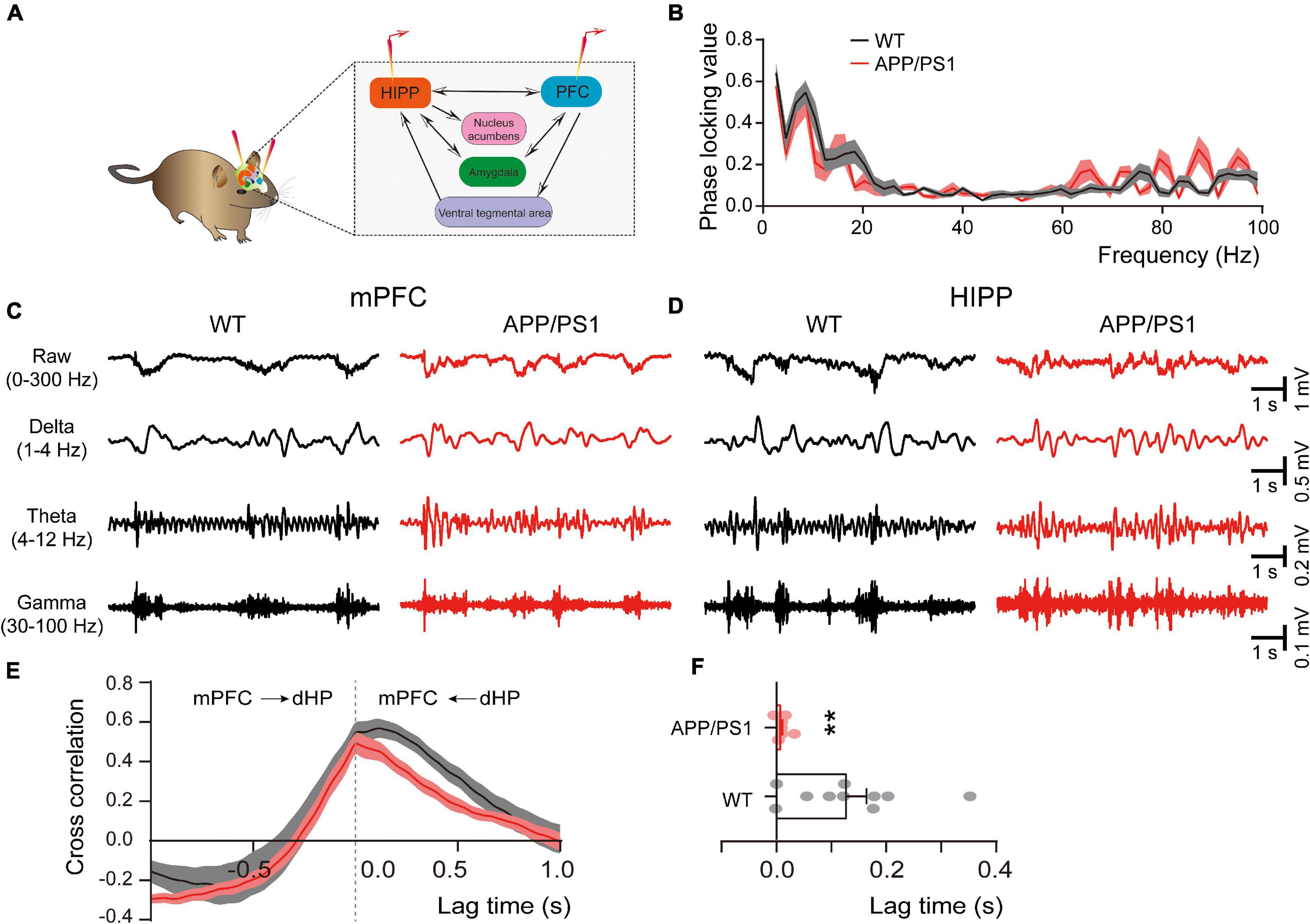
Figure 5. Altered connectivity between mPFC and HIPP of 4∼5 month-old APP/PS1 mice. (A) Schematic diagram showing in vivo dual-site LFP recording in the HIPP-mPFC. (B) The phase-locking curve between frequency ranges. (C,D) Representative traces of extracellular LFPs, as well as filtered delta, theta, and gamma oscillations, were recorded simultaneously in the mPFC and HIPP. (E) The LFP signals between mPFC and HIPP have approximately symmetrical cross-correlation values for positive (HIPP leading) and negative (mPFC leading) time lags in WT and APP/PS1, suggesting bidirectional communication between these two brain areas. (F) Quantification shows a significantly decreased cross-correlation value at positive time lag ranges between HIPP and mPFC in 4∼5 month-old APP/PS1 mice, indicating decreased synchronization between the two regions. WT: n = 10 mice; APP/PS1: n = 8 mice; two-sample t-test. Values represent the mean ± SEM, **p < 0.01.
The neuroinflammation ubiquitously observed in AD has emerged as a vital player in the progression of AD (Heneka et al., 2015; Da Mesquita et al., 2021; Leng and Edison, 2021). To examine whether astrogliosis occurs in 4∼5-month-old APP/PS1 mice, we conducted western blotting and immunofluorescent staining with anti-GFAP antibody, which is a standard marker of reactive astrocytes (Sofroniew, 2009). The staining and western blotting results revealed significantly enhanced levels of GFAP in both mPFC (WT: 1 ± 0.09; APP/PS1: 1.85 ± 0.32; p = 0.029; Figures 6A,G) and HIPP (WT: 1 ± 0.07; APP/PS1: 1.68 ± 0.15; p = 0.0013; Figures 7A,G) of APP/PS1 mice. Next, we asked if microglial activation, an indicator of neuroinflammation (Leng and Edison, 2021), occurs at this age in APP/PS1 mice. Utilizing anti-Iba1 antibody, widely used to detect active microglia under both normal and pathological conditions (Gheorghe et al., 2020), we observed a significantly increased number of Iba1-positive cells in mPFC (WT: 105.41 ± 4.22; APP/PS1: 119.29 ± 5.07; p = 0.042; Figure 6D) and HIPP (WT: 134.48 ± 6.05; APP/PS1: 151.36 ± 3.09; p = 0.017; Figure 7D) of APP/PS1 compared to WT mice. Moreover, significantly larger Iba1-labeled cell bodies occurred in the mPFC (WT: 26.76 ± 1.07; APP/PS1: 30.28 ± 1.28; p = 0.042; Figure 6E) and HIPP (WT: 34.14 ± 1.53; APP/PS1: 38.42 ± 0.78; p = 0.017; Figure 7E) of APP/PS1 mice. This microglial activation was further confirmed by western blotting (mPFC, WT: 1 ± 0.19; APP/PS1: 3.00 ± 0.30; p = 0.0014; Figures 6F,G) (HIPP, WT: 1 ± 0.09; APP/PS1: 1.59 ± 0.09; p = 0.005; Figures 7F,G), suggesting a remarkable enhancement of microglial activation, and likely subsequent neuroinflammation, at this age in AD mice.
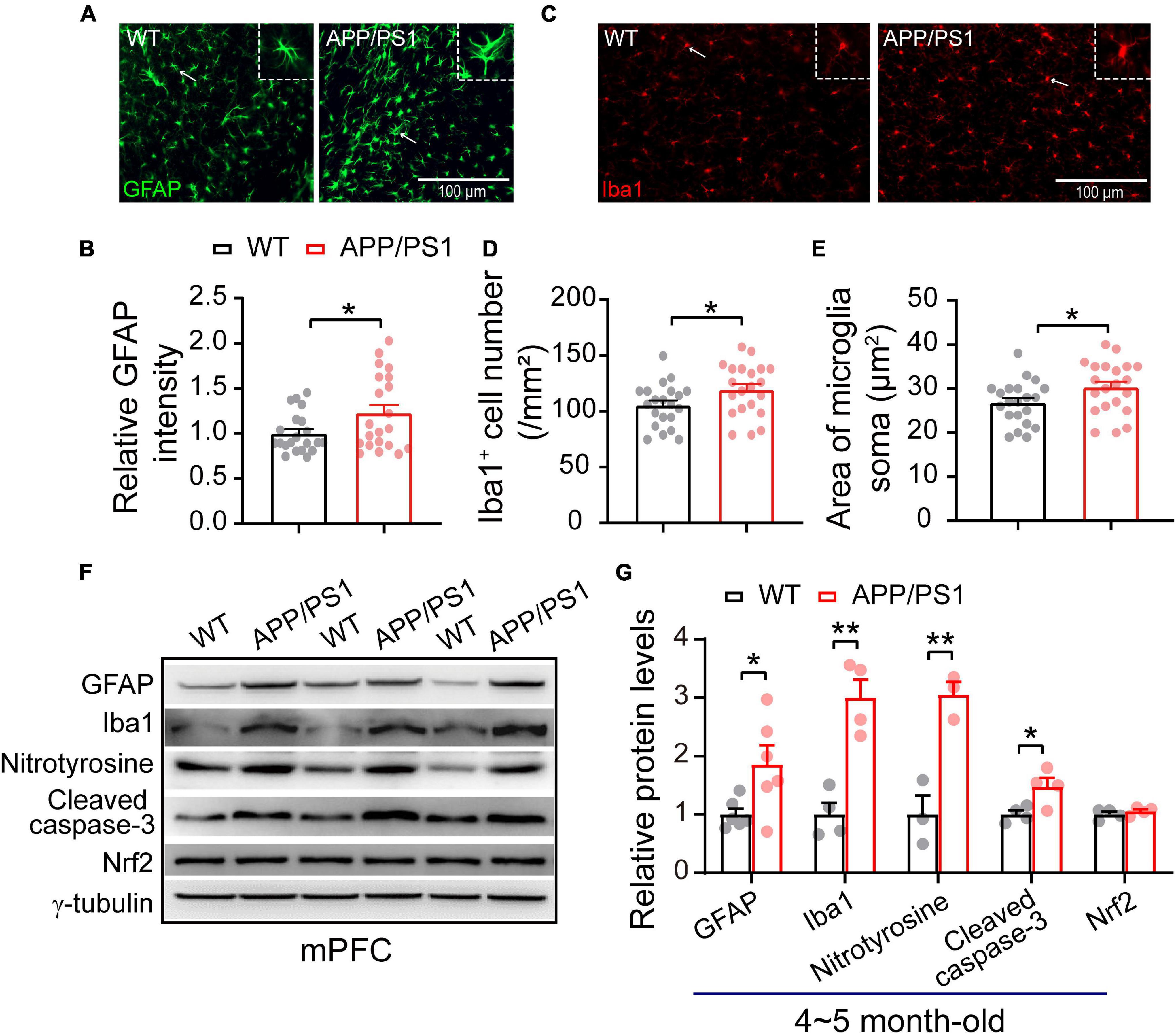
Figure 6. Increased neuroinflammation and oxidative stress in the mPFC of 4∼5 month-old APP/PS1 mice. (A) Immunofluorescent staining of GFAP in mPFC sections. (B) Higher levels of GFAP in the mPFC of APP/PS1 mice than in WT mice (7 slices/per mouse, 3 mice per genotype). (C) Immunofluorescent staining of Iba1 in mPFC sections. (D,E) Increased Iba1 + cell number and soma size, showing microglial activation, in the mPFC of APP/PS1 mice compared to WT mice (7 slices/per mouse, 3 mice per genotype). (F,G) Western blotting showing significantly increased levels of GFAP, Iba1, nitrotyrosine, and cleaved caspase-3 but unchanged Nrf2 in the mPFC of APP/PS1 mice compared to WT mice; n = 3–6 mice per genotype; two-sample t-test. Values represent the mean ± SEM, *p < 0.05, **p < 0.01.
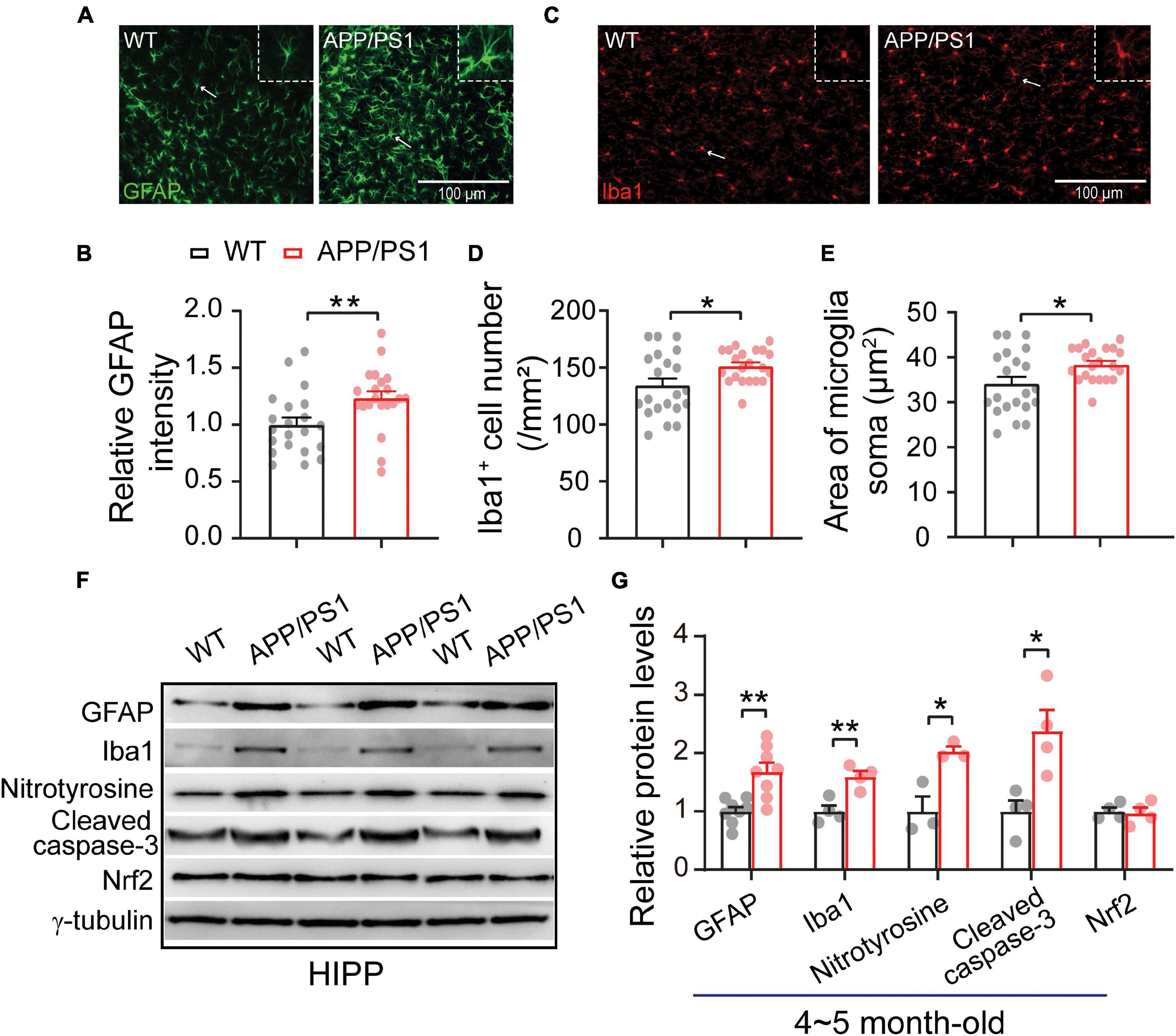
Figure 7. Increased neuroinflammation and oxidative stress in the HIPP of 4∼5 month-old APP/PS1 mice. (A) Immunofluorescent staining of GFAP in HIPP sections. (B) Increased levels of GFAP in the HIPP of APP/PS1 mice compared to WT mice (7 slices/per mouse, 3 mice per genotype). (C) Immunofluorescent staining of Iba1 in HIPP sections. (D,E) Increased Iba1 + cell number and soma size, showing microglial activation, in the HIPP of APP/PS1 mice compared to WT mice (7 slices/per mouse, 3 mice per genotype). (F,G) Western blotting showing significantly increased levels of GFAP, Iba1, nitrotyrosine, and cleaved caspase-3, but unchanged Nrf2 levels, in the HIPP of APP/PS1 mice compared to WT mice; n = 3–8 mice per genotype; two-sample t-test. Values represent the mean ± SEM, *p < 0.05, **p < 0.01.
Reciprocal interaction between neuroinflammation and dysfunctional mitochondria has been well documented (Van Horssen et al., 2019; Leng and Edison, 2021); these processes result in increased oxidative stress in the brain, leading to neuronal damage (Tonnies and Trushina, 2017; Misrani et al., 2021). Accordingly, we checked the expression of nitrotyrosine (a biomarker of oxidative stress) (Bandookwala and Sengupta, 2020) in the mPFC and HIPP of 4∼5-month-old APP/PS1 mice. Our results revealed a significantly increased level of nitrotyrosine in mPFC (WT: 1 ± 0.31; APP/PS1: 3.04 ± 0.21; p = 0.006) (Figures 6F,G) and in HIPP of APP/PS1 mice (WT: 1 ± 0.25; APP/PS1: 2.02 ± 0.08; p = 0.018) (Figures 7F,G). Cleaved caspase-3 induces synaptic dysfunction, neuronal loss and apoptosis in early AD (D’amelio et al., 2011). This caspase is responsible for the majority of proteolysis during apoptosis, and detection of cleaved caspase-3 is therefore considered a reliable marker for programmed cell death (Porter and Janicke, 1999; Lakhani et al., 2006). Neuroinflammation and oxidative stress can cause caspase-3 activation leading to apoptosis (Vince et al., 2018). We therefore tested levels of Cleaved caspase-3 and found these to be increased in mPFC (WT: 1 ± 0.06; APP/PS1: 1.47 ± 0.15; p = 0.029; Figures 6F,G) and in HIPP (WT: 1 ± 0.18; APP/PS1: 2.37 ± 0.36; p = 0.014; Figures 7F,G) of 4∼5-month-old APP/PS1 mice. However, levels of nuclear factor erythroid 2-related factor (Nrf2), which protects the brain from oxidative stress by upregulating the antioxidative defense pathway, inhibiting neuroinflammation, and maintaining protein homeostasis (Ahmed et al., 2017), remained unchanged in both mPFC (WT: 1 ± 0.04; APP/PS1: 1.05 ± 0.15; p = 0.36; Figures 6F,G) and HIPP (WT: 1 ± 0.06; APP/PS1: 0.96 ± 0.09; p = 0.079; Figures 7F,G).
Given that deficits in social communication, which requires normal HIPP and mPFC activity, occur in individuals with preclinical stage AD and mild cognitive impairment (MCI), we assessed the social interaction ability of 4∼5-month-old APP/PS1 and WT mice using a three-chamber social interaction test (Figure 8A). We found that APP/PS1 mice spent less time sniffing near the novel mouse chamber than WT mice (WT: 150.93 ± 15.44; APP/PS1: 110.49 ± 11.78; p = 0.041; Figures 8B,C). Moreover, data analysis of the discrimination index showed that APP/PS1 mice could not discriminate between familiar and novel mice as well as age-matched WT mice (WT: 27.16 ± 4.85; APP/PS1: 1.35 ± 5.26; p < 0.001; Figure 8D). Thus, although mice normally spend more time interacting with a novel mouse than a familiar one (Camats Perna and Engelmann, 2017), interaction time was decreased in APP/PS1 mice at the age tested.
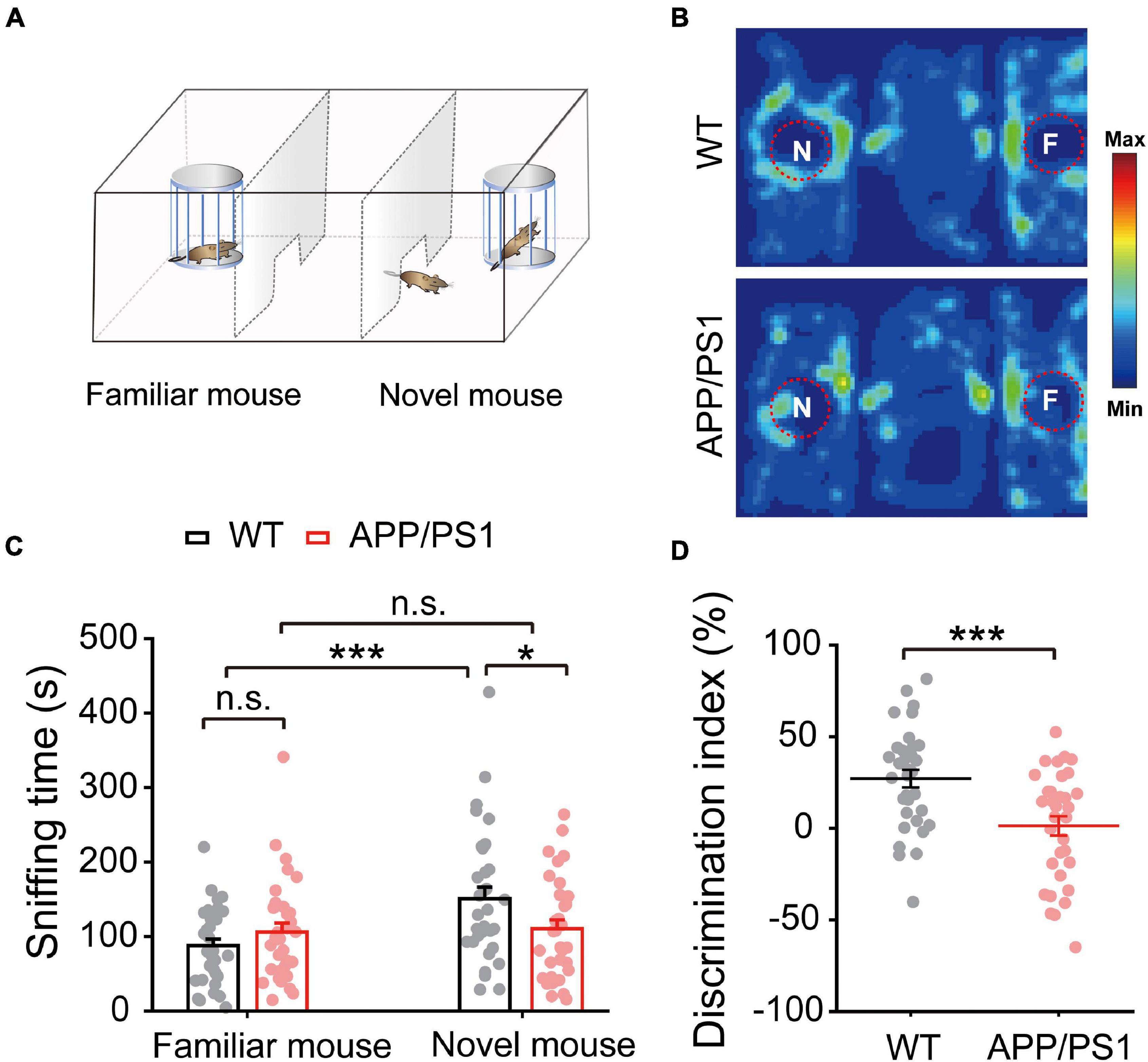
Figure 8. Impaired social interaction memory in 4∼5 month-old APP/PS1 mice. (A) Schematic diagram of the three-chamber social memory test setup, showing test mouse, familiar mouse chamber, and novel mouse chamber. (B) Representative heat map of a test mouse showing mouse activity during the test phase. (C) Time spent sniffing by test mouse in proximity to novel and familiar mice, showing that APP/PS1 mice spent less time than WT mice sniffing the novel mouse. (D) Decreased discrimination index of APP/PS1 mice compared to age-matched WT mice. Values represent mean ± SEM. WT: n = 33 mice; APP/PS1: n = 33 mice; one-way ANOVA, two-sample paired t-test. Values represent the mean ± SEM, *p < 0.05, ***p < 0.001.
It is becoming clear that the initial phase of AD begins years before the appearance of Aβ plaques and cognitive deficits (Frisoni et al., 2017; Styr and Slutsky, 2018). Recent research suggests that mitochondrial dysfunction (Hauptmann et al., 2009; Wang et al., 2020) and abnormal synaptic transmission (Selkoe, 2002; Styr and Slutsky, 2018) occur early at an early stage of disease progression, earlier than the emergence of histopathological or clinical abnormalities. Therefore, identifying the early mitochondrial and synaptic alterations at the prodromal phase of AD is of great importance for developing better diagnostic tools and more effective therapeutic interventions. Here, we show that, at 4∼5 months old (an age at which there are no obvious senile plaques) (Chen et al., 2021), APP/PS1 mice exhibit abnormal mitochondrial fission and morphology together with gliosis. These abnormalities are associated with neuroinflammation and oxidative stress (revealed by increased expression of nitrotyrosine), which may, in turn, cause activation of cleaved caspase-3 leading to apoptosis (Vince et al., 2018), as well as decreased levels of ATP5A (Terni et al., 2010). In combination, these alterations result in impairments of neuronal morphology, synaptic function and HIPP-mPFC network activity, which are associated with social interaction deficits (Figure 9). Together, the present study suggests that targeting mitochondrial dysfunction and neuroinflammation at an early stage of AD may slow down or prevent the pathogenesis of the disease.

Figure 9. Schematic illustration depicting early alterations in APP/PS1 mice. Increased Drp1 and decreased ATP5A levels in 4∼5 month-old APP/PS1 (Figure 1) accompanied by astroglial activation (Figure 6), suggesting mitochondrial dysfunction and neuroinflammation likely contribute to oxidative stress, apoptosis (Figures 6, 7), and neuronal morphological alterations (Figures 2, 3). These alterations together lead to impaired synaptic activity (Figure 4) and network activity between mPFC and HIPP (Figure 5), which is associated with social interaction deficits (Figure 8).
Postmortem brains of AD patients exhibit defects in mitochondrial morphology and dynamics, and energy metabolism (Devi et al., 2006; Pickett et al., 2018). An imbalance between mitochondrial fission and fusion (increased fission and decreased fusion), as well as impaired ATP metabolism, occurs in AD patients and mouse models (Wang et al., 2009; Manczak et al., 2011, 2019; Kandimalla et al., 2018). Moreover, mitochondrial dysfunction, correlating with increased fragmentation, which is a common theme of neurodegeneration mainly in AD and is considered to be a fundamental early marker in disease progression (Wang et al., 2017, 2020; Oliver and Reddy, 2019). Our results reveal increased levels of mitochondrial fission protein, Drp1, and decreased levels of ATP synthase as early as 4∼5 months of age in APP/PS1 mice, suggesting that mitochondrial dysfunction is an early marker of AD pathogenesis (Figure 1). Inhibition of Drp1 ameliorates mitochondrial and synaptic dysfunction in 7 month-old APP/PS1 and 3 month-old CRND8 APP transgenic AD mice (Baek et al., 2017; Wang et al., 2017), as well as in other neurodegenerative diseases (Filichia et al., 2016). It is worth noting that the mitochondrial fission and fusion process regulate mitochondrial shape, mobility and transport. For example, abnormalities in mitochondrial fission and fusion and consequent changes in mitochondrial morphology influence mitochondrial mobility and distribution (Chen and Chan, 2009). Exogenous Aβ or overexpression of amyloid precursor protein (APP) cause profound fragmentation and impair mitochondrial transport in neuronal cultures (Wang et al., 2009; Manczak et al., 2011) and Drosophila model (Iijima-Ando et al., 2009). Mitochondrial membrane ATP synthase (F1F0 ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the oxidative phosphorylation (OXPHOS). The dysfunction of ATP synthase leads to disrupted OXPHOS and progressive ATP depletion. Compromised mitochondrial OXPHOS constitutes a characteristic mitochondrial deficit in AD brains, resulting in lowered ATP production, increased oxidative stress, and eventually cell death (Terni et al., 2010; Du et al., 2012). Given that reduced ATP synthase is repeatedly reported in the AD brain, our results also represent an early alteration in the energy-producing enzyme in APP/PS1 mice. Moreover, we also found abnormal mitochondrial morphology (increased fragmentation, increased number of mitochondria and reduced mitochondrial aspect ratio) in 4∼5 months-old APP/PS1 mice (Figure 1). Mitochondrial morphology depends on the balance between mitochondrial fission and fusion, and increased Drp1-induced mitochondrial fission could result in increased fragmentation and abnormal mitochondrial morphology (Detmer and Chan, 2007). Our results are in line with the increased number of mitochondria with Ca2+ overload in the brain of APP/PS1 (Xu et al., 2018; Calvo-Rodriguez et al., 2020). Given that a balance between mitochondrial fission and fusion is necessary for optimal energy metabolism, our findings suggest an age-related increase in mitochondrial fission and abnormal energy metabolism in the brain of APP/PS1 mice. These effects persist in older mice: we also found increased mitochondrial fission and decreased fusion and ATP synthase subunit levels in the mPFC and HIPP of 9∼10 month-old (Supplementary Figure 2) and 12∼18 month-old APP/PS1 mice (Supplementary Figure 3).
Reduced dendritic branching and length are common in the HIPP and cortical pyramidal neurons of AD patients and animal models (Tsai et al., 2004; Grutzendler et al., 2007). The morphology of dendritic branches is highly variable, and dynamic structures are continuously formed and eliminated throughout life (Grutzendler et al., 2002), a process that depends on the balance between mitochondrial dynamics and oxidative stress. We show that 4∼5 month-old APP/PS1 mice exhibit abnormal dendritic complexity and relative shortening of dendrite length and branching in both mPFC and HIPP (Figures 2, 3). These alterations in dendritic morphology may be due to increased oxidative stress and increased fission, which is consistent with the notion that mitochondrial dysfunction increases oxidative stress production in the brain, leading to neuronal damage (Li et al., 2004). PSD-95 is crucial for the maintenance of neuronal morphology (Sweet et al., 2011), and our western blotting results showed significantly reduced levels of PSD-95 in the HIPP of 4∼5 month-old APP/PS1 mice, which may be attributed, at least in part, to increased oxidative stress (Ansari et al., 2008). However, our results show no change in PSD-95 expression in the mPFC of 4∼5 month-old APP/PS1 mice. Consistent with the notion that AD patients and animal models exhibit region-specific alterations. Similarly, AD mice exhibit region-specific and age-related alterations in metabolic signaling, synaptic marker protein synaptophysin (presynaptic) and synaptic loss (Rutten et al., 2005; Gonzalez-Dominguez et al., 2014).
Besides energy production, mitochondria play an essential role in buffering intracellular Ca2+, and consequently these organelles are involved in maintaining and regulating synaptic transmission (Devine and Kittler, 2018). We found decreased mEPSC frequency in mPFC of 4∼5 month-old APP/PS1 mice (Figure 4), suggesting that a loss of mitochondria from these regions may impair synaptic transmission due to altered energy metabolism and regulation of intracellular Ca2+. Thus, a decline in mitochondrial function can occur decades before a clinical diagnosis of AD and may serve as a biomarker of AD risk, as well as a therapeutic target for the preservation of synaptic function (Caldwell et al., 2015). It is worth noting that, although both mPFC and HIPP showed abnormal expression of mitochondrial proteins and dendritic morphology, mEPSC frequency was reduced only in the mPFC and not in the HIPP. This decrease in mEPSC frequency suggests an early presynaptic alteration in the mPFC pyramidal neurons of 4∼5 month-old APP/PS1 mice. It has been shown that 4∼5 month-old APP/PS1 mice do not exhibit any HIPP-dependent memory loss or electrophysiological alteration at this age (Tabassum et al., 2019; Chen et al., 2021), and, indeed, hippocampal abnormalities do not appear until after 6 months of age in these AD-model mice (Viana Da Silva et al., 2016). We reason that AD pathogenesis may cause age-dependent and region-specific alterations in neuronal activities (Gengler et al., 2010; Oyelami et al., 2016; Chen et al., 2021), and that synaptic transmission in the mPFC is more vulnerable to neurodegeneration. Because mitochondrial morphology and function are crucial for synaptic transmission and function (Tang and Zucker, 1997; Devine and Kittler, 2018), we next investigated potential mitochondria-dependent mechanisms of synaptic transmission in pyramidal neurons of 4∼5-month-old APP/PS1 mice. Although 4∼5-month-old APP/PS1 mice exhibit increased Drp1 and decreased frequency of mEPSC in mPFC, incubation of the brain slices with Drp1 inhibitor, Mdivi-1, did not reverse the decline of mEPSC frequency (Supplementary Figure 4). Worth noting that inhibition of Drp1-induced mitochondrial fission protected synaptic damage in AD mice (Huang et al., 2015; Baek et al., 2017) and cell lines (Reddy et al., 2017); however, chronic administration is recommended. It is likely that mitochondria are located inside an intracellular compartment, and the drug molecule requires exact dosage and physicochemical properties to achieve therapeutic effects.
Functional connectivity between distant brain structures is fundamental in coordinating neuronal communication during sensory processing. Altered brain connectivity between brain regions occurs in patients with MCI and AD (Chan et al., 2013; Zheng et al., 2019). In the present study, we demonstrate significantly reduced coupling between mPFC and HIPP in 4∼5 month-old APP/PS1 mice (Figure 5), indicating an early HIPP-mPFC network alteration in AD. Our results are in agreement with recent opinion that early network dysfunction (altered neuronal activity and synchrony) contributes to neurodegeneration and AD pathogenesis (Frere and Slutsky, 2018; Mondragon-Rodriguez et al., 2019). Although the exact causes and mechanisms of network alterations have not been defined, our results suggest that mitochondrial dysfunction, neuroinflammation and oxidative stress may underlie these neuronal activity deficits.
Emerging evidence suggests that neuroinflammation has a causal role in the progression and pathogenesis of AD. In the present study, we show an increase in reactive astrocytes and microglial activation in the mPFC and HIPP of 4∼5 month-old APP/PS1 mice (Figures 6, 7). Astrocytic and microglial activation have been observed at the pre-plaque stage in 3∼6 month-old animal models of AD (Furman et al., 2012; Hanzel et al., 2014; Leng and Edison, 2021), and a human neuroimaging study reported increased microglial activation in individuals with MCI (Okello et al., 2009). We thus speculate that gliosis associated with neuroinflammation at an early age inappropriately engulfs synapses, resulting in early synaptic and network abnormalities.
We demonstrated an early impairment of non-cognitive AD-like symptoms, i.e., deficits in social interest, interaction, and communication in 4∼5 month-old APP/PS1 mice (Figure 8), similar to previous findings of deficits in social memory in 3∼6 month-old APP/PS1 mice (Filali et al., 2011; Pietropaolo et al., 2012). Given the critical role of HIPP and mPFC in mediating social behavior (Phillips et al., 2019), the present study suggests that mitochondrial dysfunction, together with increased neuroinflammation, and abnormal synaptic and network activity in the mPFC and HIPP, may underlie the social deficits observed in 4∼5 month-old APP/PS1 mice.
In conclusion, the most provocative finding of the present study is that age-related alterations in mitochondrial dynamics, which could be attributed, at least in part, to a number of pathways damaged by soluble Aβ, such as impairment of oxidative phosphorylation, elevation of ROS production, and interaction of Aβ with mitochondrial proteins (Pagani and Eckert, 2011; Mossmann et al., 2014; Pinho et al., 2014), excessive gliosis, and increased oxidative stress are early indicators of AD pathogenesis, contributing to impaired HIPP-mPFC activity and social interaction deficits in 4∼5 month-old APP/PS1 mice. Given that pre-amyloid deposition in humans begins at least two decades before the signs and symptoms of AD appear (Villemagne et al., 2018), and that targeting the early pre-amyloid phase may be an effective paradigm for the prevention of AD (Uhlmann et al., 2020), therapeutic interventions that improve mitochondrial function and reduce neuroinflammation represent promising strategies for slowing the progression or delaying the onset of this incurable disease.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Guangzhou University and South China Normal University Institutional Review Boards.
AM: project initiation, experimental design, western blotting, TEM statistical analysis, and manuscript writing. SiT: western blotting, TEM analysis, statistical analysis, figure generation, and manuscript writing. QH: LFP recording data analysis and figure generation. SuT: Golgi cox staining and data analysis. JJ: behavior and analysis and TEM. AA: immunostaining and analysis. XC and JWZ: whole-cell recording. JJZ: western blotting. SL and XF: behavior and analysis. CL: supervision. LY: guiding the experiments, funding acquisition, and critical revision.
This study was supported by grants from the National Natural Science Foundation of China (32170950, 31970915, 31871170, and 31771219), the Guangdong Natural Science Foundation for Major Cultivation Project (2018B030336001), and the Guangdong Grant Key Technologies for Treatment of Brain Disorders (2018B030332001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.748388/full#supplementary-material
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017). Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 585–597. doi: 10.1016/j.bbadis.2016.11.005
Ammassari-Teule, M. (2020). Early-Occurring Dendritic Spines Alterations in Mouse Models of Alzheimer’s Disease Inform on Primary Causes of Neurodegeneration. Front. Synaptic. Neurosci. 12:566615. doi: 10.3389/fnsyn.2020.566615
Ansari, M. A., Roberts, K. N., and Scheff, S. W. (2008). Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 45, 443–452. doi: 10.1016/j.freeradbiomed.2008.04.038
Baek, S. H., Park, S. J., Jeong, J. I., Kim, S. H., Han, J., Kyung, J. W., et al. (2017). Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 37, 5099–5110. doi: 10.1523/JNEUROSCI.2385-16.2017
Bandookwala, M., and Sengupta, P. (2020). 3-Nitrotyrosine: a versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 130, 1047–1062. doi: 10.1080/00207454.2020.1713776
Bertholet, A. M., Delerue, T., Millet, A. M., Moulis, M. F., David, C., Daloyau, M., et al. (2016). Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 90, 3–19. doi: 10.1016/j.nbd.2015.10.011
Bianchi, S., Stimpson, C. D., Bauernfeind, A. L., Schapiro, S. J., Baze, W. B., Mcarthur, M. J., et al. (2013). Dendritic morphology of pyramidal neurons in the chimpanzee neocortex: regional specializations and comparison to humans. Cereb Cortex 23, 2429–2436. doi: 10.1093/cercor/bhs239
Caldwell, C. C., Yao, J., and Brinton, R. D. (2015). Targeting the prodromal stage of Alzheimer’s disease: bioenergetic and mitochondrial opportunities. Neurotherapeutics 12, 66–80. doi: 10.1007/s13311-014-0324-8
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 11:2146. doi: 10.1038/s41467-020-16074-2
Camats Perna, J., and Engelmann, M. (2017). Recognizing Others: Rodent’s Social Memories. Curr. Top. Behav. Neurosci. 30, 25–45. doi: 10.1007/7854_2015_413
Cha, M. Y., Cho, H. J., Kim, C., Jung, Y. O., Kang, M. J., Murray, M. E., et al. (2015). Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mole. Genet. 24, 6492–6504. doi: 10.1093/hmg/ddv358
Chan, H. L., Chu, J. H., Fung, H. C., Tsai, Y. T., Meng, L. F., Huang, C. C., et al. (2013). Brain connectivity of patients with Alzheimer’s disease by coherence and cross mutual information of electroencephalograms during photic stimulation. Med. Eng. Phys. 35, 241–252. doi: 10.1016/j.medengphy.2011.10.005
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, M., Chen, Y., Huo, Q., Wang, L., Tan, S., Misrani, A., et al. (2021). Enhancing GABAergic signaling ameliorates aberrant gamma oscillations of olfactory bulb in AD mouse models. Mol. Neurodegener. 16:14. doi: 10.1186/s13024-021-00434-7
Chen, M., Wang, J., Jiang, J., Zheng, X., Justice, N. J., Wang, K., et al. (2017). APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. Elife 2017:6. doi: 10.7554/eLife.20142
Chen, Y., Li, M., Zheng, Y., and Yang, L. (2019). Evaluation of Hemisphere Lateralization with Bilateral Local Field Potential Recording in Secondary Motor Cortex of Mice. J. Vis. Exp. 149:59310. doi: 10.3791/59310
Choy, J. M. C., Agahari, F. A., Li, L., and Stricker, C. (2018). Noradrenaline Increases mEPSC Frequency in Pyramidal Cells in Layer II of Rat Barrel Cortex via Calcium Release From Presynaptic Stores. Front. Cell Neurosci. 12:213. doi: 10.3389/fncel.2018.00213
Cipolat, S., Martins De Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U S A 101, 15927–15932. doi: 10.1073/pnas.0407043101
Da Mesquita, S., Papadopoulos, Z., Dykstra, T., Brase, L., Farias, F. G., Wall, M., et al. (2021). Meningeal lymphatics affect microglia responses and anti-Abeta immunotherapy. Nature 593, 255–260. doi: 10.1038/s41586-021-03489-0
D’amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14, 69–76. doi: 10.1038/nn.2709
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Du, H., Guo, L., and Yan, S. S. (2012). Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid Redox Signal 16, 1467–1475. doi: 10.1089/ars.2011.4277
Engel, A. K., Fries, P., and Singer, W. (2001). Dynamic predictions: oscillations and synchrony in top-down processing. Nat. Rev. Neurosci. 2, 704–716. doi: 10.1038/35094565
Fang, Y., Du, N., Xing, L., Duo, Y., and Zheng, L. (2019). Evaluation of hippocampal volume and serum brain-derived neurotrophic factor as potential diagnostic markers of conversion from amnestic mild cognitive impairment to Alzheimer disease: A STROBE-compliant article. Medicine 98:e16604. doi: 10.1097/MD.0000000000016604
Filali, M., Lalonde, R., and Rivest, S. (2011). Anomalies in social behaviors and exploratory activities in an APPswe/PS1 mouse model of Alzheimer’s disease. Physiol. Behav. 104, 880–885. doi: 10.1016/j.physbeh.2011.05.023
Filichia, E., Hoffer, B., Qi, X., and Luo, Y. (2016). Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016:6. doi: 10.1038/srep32656
Franco, A., Kitsis, R. N., Fleischer, J. A., Gavathiotis, E., Kornfeld, O. S., Gong, G., et al. (2016). Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540, 74–79. doi: 10.1038/nature20156
Frere, S., and Slutsky, I. (2018). Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 97, 32–58. doi: 10.1016/j.neuron.2017.11.028
Frisoni, G. B., Boccardi, M., Barkhof, F., Blennow, K., Cappa, S., Chiotis, K., et al. (2017). Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 16, 661–676. doi: 10.1016/S1474-4422(17)30159-X
Furman, J. L., Sama, D. M., Gant, J. C., Beckett, T. L., Murphy, M. P., Bachstetter, A. D., et al. (2012). Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., et al. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524. doi: 10.1016/j.nbd.2006.08.017
Gauba, E., Chen, H., Guo, L., and Du, H. (2019). Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 121, 138–147. doi: 10.1016/j.nbd.2018.09.020
Gengler, S., Hamilton, A., and Holscher, C. (2010). Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer’s disease is impaired in old but not young mice. PLoS One 5:e9764. doi: 10.1371/journal.pone.0009764
Gheorghe, R. O., Deftu, A., Filippi, A., Grosu, A., Bica-Popi, M., Chiritoiu, M., et al. (2020). Silencing the Cytoskeleton Protein Iba1 (Ionized Calcium Binding Adapter Protein 1) Interferes with BV2 Microglia Functioning. Cell Mol. Neurobiol. 40, 1011–1027. doi: 10.1007/s10571-020-00790-w
Gonzalez-Dominguez, R., Garcia-Barrera, T., Vitorica, J., and Gomez-Ariza, J. L. (2014). Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim. Biophys. Acta 1842, 2395–2402. doi: 10.1016/j.bbadis.2014.09.014
Grady, C. L., Furey, M. L., Pietrini, P., Horwitz, B., and Rapoport, S. I. (2001). Altered brain functional connectivity and impaired short-term memory in Alzheimer’s disease. Brain 124, 739–756. doi: 10.1093/brain/124.4.739
Gruber, A. J., Calhoon, G. G., Shusterman, I., Schoenbaum, G., Roesch, M. R., and O’donnell, P. (2010). More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J. Neurosci. 30, 17102–17110. doi: 10.1523/JNEUROSCI.4623-10.2010
Grutzendler, J., Helmin, K., Tsai, J., and Gan, W. B. (2007). Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1097, 30–39. doi: 10.1196/annals.1379.003
Grutzendler, J., Kasthuri, N., and Gan, W. B. (2002). Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. doi: 10.1038/nature01276
Hanzel, C. E., Pichet-Binette, A., Pimentel, L. S., Iulita, M. F., Allard, S., Ducatenzeiler, A., et al. (2014). Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 35, 2249–2262. doi: 10.1016/j.neurobiolaging.2014.03.026
Hauptmann, S., Scherping, I., Drose, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Huang, S., Wang, Y., Gan, X., Fang, D., Zhong, C., Wu, L., et al. (2015). Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes 64, 1728–1742. doi: 10.2337/db14-0758
Iijima-Ando, K., Hearn, S. A., Shenton, C., Gatt, A., Zhao, L., and Iijima, K. (2009). Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One 4:e8310. doi: 10.1371/journal.pone.0008310
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170. doi: 10.1093/hmg/ddh019
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/s1389-0344(01)00067-3
Joshi, A. U., Minhas, P. S., Liddelow, S. A., Haileselassie, B., Andreasson, K. I., and Dorn, G. W. II, et al. (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22, 1635–1648. doi: 10.1038/s41593-019-0486-0
Kandimalla, R., Manczak, M., Fry, D., Suneetha, Y., Sesaki, H., and Reddy, P. H. (2016). Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 4881–4897. doi: 10.1093/hmg/ddw312
Kandimalla, R., Manczak, M., Yin, X., Wang, R., and Reddy, P. H. (2018). Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 30–40. doi: 10.1093/hmg/ddx381
Koush, Y., Pichon, S., Eickhoff, S. B., Van De Ville, D., Vuilleumier, P., and Scharnowski, F. (2019). Brain networks for engaging oneself in positive-social emotion regulation. Neuroimage 189, 106–115. doi: 10.1016/j.neuroimage.2018.12.049
Lakhani, S. A., Masud, A., Kuida, K., Porter, G. A. Jr., Booth, C. J., Mehal, W. Z., et al. (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851. doi: 10.1126/science.1115035
Lee, J. E., Westrate, L. M., Wu, H., Page, C., and Voeltz, G. K. (2016). Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. doi: 10.1038/nature20555
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2019). Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 28, 177–199.
Mcclean, P. L., and Holscher, C. (2014). Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 76, 57–67. doi: 10.1016/j.neuropharm.2013.08.005
Misrani, A., Tabassum, S., and Yang, L. (2021). Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 13:617588.
Mondragon-Rodriguez, S., Gu, N., Fasano, C., Pena-Ortega, F., and Williams, S. (2019). Functional Connectivity between Hippocampus and Lateral Septum is Affected in Very Young Alzheimer’s Transgenic Mouse Model. Neuroscience 401, 96–105. doi: 10.1016/j.neuroscience.2018.12.042
Moriguchi, K., Jogahara, T., Oda, S., and Honda, M. (2019). Scanning transmission electron microscopic analysis of nitrogen generated by 3, 3’-diaminobenzidine-besed peroxidase reaction with resin ultrathin sections of rhinoceros parotid gland acinar cells. Microscopy 68, 111–121. doi: 10.1093/jmicro/dfy125
Mossmann, D., Vogtle, F. N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 20, 662–669. doi: 10.1016/j.cmet.2014.07.024
Okello, A., Edison, P., Archer, H. A., Turkheimer, F. E., Kennedy, J., Bullock, R., et al. (2009). Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72, 56–62. doi: 10.1212/01.wnl.0000338622.27876.0d
Okuyama, T., Kitamura, T., Roy, D. S., Itohara, S., and Tonegawa, S. (2016). Ventral CA1 neurons store social memory. Science 353, 1536–1541. doi: 10.1126/science.aaf7003
Oliver, D., and Reddy, P. H. (2019). Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019:8. doi: 10.3390/cells8090961
Oyelami, T., Bondt, A., Den Wyngaert, I. V., Hoorde, K. V., Hoskens, L., Shaban, H., et al. (2016). Age-dependent concomitant changes in synaptic dysfunction and GABAergic pathway in the APP/PS1 mouse model. Acta Neurobiol. Exp. 76, 282–293. doi: 10.21307/ane-2017-027
Pagani, L., and Eckert, A. (2011). Amyloid-Beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011:925050.
Parfitt, G. M., Nguyen, R., Bang, J. Y., Aqrabawi, A. J., Tran, M. M., Seo, D. K., et al. (2017). Bidirectional Control of Anxiety-Related Behaviors in Mice: Role of Inputs Arising from the Ventral Hippocampus to the Lateral Septum and Medial Prefrontal Cortex. Neuropsychopharmacology 42, 1715–1728. doi: 10.1038/npp.2017.56
Phillips, M. L., Robinson, H. A., and Pozzo-Miller, L. (2019). Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife 2019:8. doi: 10.7554/eLife.44182
Pickett, E. K., Rose, J., Mccrory, C., Mckenzie, C. A., King, D., Smith, C., et al. (2018). Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 136, 747–757. doi: 10.1007/s00401-018-1903-2
Pietropaolo, S., Delage, P., Lebreton, F., Crusio, W. E., and Cho, Y. H. (2012). Early development of social deficits in APP and APP-PS1 mice. Neurobiol. Aging 33, e1017–e1027. doi: 10.1016/j.neurobiolaging.2011.09.012
Pinho, C. M., Teixeira, P. F., and Glaser, E. (2014). Mitochondrial import and degradation of amyloid-beta peptide. Biochim. Biophys. Acta 1837, 1069–1074. doi: 10.1016/j.bbabio.2014.02.007
Porter, A. G., and Janicke, R. U. (1999). Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6, 99–104. doi: 10.1038/sj.cdd.4400476
Preston, A. R., and Eichenbaum, H. (2013). Interplay of hippocampus and prefrontal cortex in memory. Curr. Biol. 23, R764–R773.
Reddy, P. H. (2008). Mitochondrial medicine for aging and neurodegenerative diseases. Neuromolecular. Med. 10, 291–315. doi: 10.1007/s12017-008-8044-z
Reddy, P. H., Manczak, M., and Yin, X. (2017). Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimers Dis. 58, 147–162. doi: 10.3233/JAD-170051
Rutten, B. P., Van Der Kolk, N. M., Schafer, S., Van Zandvoort, M. A., Bayer, T. A., Steinbusch, H. W., et al. (2005). Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am. J. Pathol. 167, 161–173. doi: 10.1016/S0002-9440(10)62963-X
Saez-Atienzar, S., and Masliah, E. (2020). Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat. Rev. Neurosci. 21, 433–444. doi: 10.1038/s41583-020-0325-z
Satler, C., and Tomaz, C. (2011). Emotional working memory in Alzheimer’s disease patients. Dement Geriatr. Cogn. Dis. Extra 1, 124–138.
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shields, L. Y., Kim, H., Zhu, L., Haddad, D., Berthet, A., Pathak, D., et al. (2015). Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 6:e1725. doi: 10.1038/cddis.2015.94
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Spellman, T., Rigotti, M., Ahmari, S. E., Fusi, S., Gogos, J. A., and Gordon, J. A. (2015). Hippocampal-prefrontal input supports spatial encoding in working memory. Nature 522, 309–314. doi: 10.1038/nature14445
Spinelli, J. B., and Haigis, M. C. (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754. doi: 10.1038/s41556-018-0124-1
Stopford, C. L., Thompson, J. C., Neary, D., Richardson, A. M., and Snowden, J. S. (2012). Working memory, attention, and executive function in Alzheimer’s disease and frontotemporal dementia. Cortex 48, 429–446. doi: 10.1016/j.cortex.2010.12.002
Styr, B., and Slutsky, I. (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 21, 463–473. doi: 10.1038/s41593-018-0080-x
Sun, Q., Li, X., Li, A., Zhang, J., Ding, Z., Gong, H., et al. (2020). Ventral Hippocampal-Prefrontal Interaction Affects Social Behavior via Parvalbumin Positive Neurons in the Medial Prefrontal Cortex. iScience 23:100894. doi: 10.1016/j.isci.2020.100894
Sweet, E. S., Tseng, C. Y., and Firestein, B. L. (2011). To branch or not to branch: How PSD-95 regulates dendrites and spines. Bioarchitecture 1, 69–73. doi: 10.4161/bioa.1.2.15469
Swerdlow, R. H., Koppel, S., Weidling, I., Hayley, C., Ji, Y., and Wilkins, H. M. (2017). Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 146, 259–302. doi: 10.1016/bs.pmbts.2016.12.017
Tabassum, S., Misrani, A., Tang, B. L., Chen, J., Yang, L., and Long, C. (2019). Jujuboside A prevents sleep loss-induced disturbance of hippocampal neuronal excitability and memory impairment in young APP/PS1 mice. Sci. Rep. 9:4512. doi: 10.1038/s41598-019-41114-3
Tang, Y., and Zucker, R. S. (1997). Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18, 483–491. doi: 10.1016/s0896-6273(00)81248-9
Terni, B., Boada, J., Portero-Otin, M., Pamplona, R., and Ferrer, I. (2010). Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 20, 222–233. doi: 10.1111/j.1750-3639.2009.00266.x
Thierry, A. M., Gioanni, Y., Degenetais, E., and Glowinski, J. (2000). Hippocampo-prefrontal cortex pathway: anatomical and electrophysiological characteristics. Hippocampus 10, 411–419. doi: 10.1002/1098-1063(2000)10:4<411::AID-HIPO7>3.0.CO;2-A
Tonnies, E., and Trushina, E. (2017). Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 57, 1105–1121. doi: 10.3233/jad-161088
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. doi: 10.1038/nn1335
Uhlmann, R. E., Rother, C., Rasmussen, J., Schelle, J., Bergmann, C., Ullrich Gavilanes, E. M., et al. (2020). Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci. 23, 1580–1588. doi: 10.1038/s41593-020-00737-w
Van Der Kant, R., Goldstein, L. S. B., and Ossenkoppele, R. (2020). Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. doi: 10.1038/s41583-019-0240-3
Van Horssen, J., Van Schaik, P., and Witte, M. (2019). Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 710:132931. doi: 10.1016/j.neulet.2017.06.050
Viana Da Silva, S., Haberl, M. G., Zhang, P., Bethge, P., Lemos, C., et al. (2016). Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 7:11915. doi: 10.1038/ncomms11915
Villemagne, V. L., Dore, V., Burnham, S. C., Masters, C. L., and Rowe, C. C. (2018). Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 14, 225–236.
Vince, J. E., De Nardo, D., Gao, W., Vince, A. J., Hall, C., Mcarthur, K., et al. (2018). The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1beta Activation. Cell Rep. 25, 2339–2353e2334. doi: 10.1016/j.celrep.2018.10.103
Wang, L., Zang, Y., He, Y., Liang, M., Zhang, X., Tian, L., et al. (2006). Changes in hippocampal connectivity in the early stages of Alzheimer’s disease: evidence from resting state fMRI. Neuroimage 31, 496–504. doi: 10.1016/j.neuroimage.2005.12.033
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30. doi: 10.1186/s13024-020-00376-6
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Warden, M. R., and Miller, E. K. (2010). Task-dependent changes in short-term memory in the prefrontal cortex. J. Neurosci. 30, 15801–15810. doi: 10.1523/JNEUROSCI.1569-10.2010
Wei, W., Nguyen, L. N., Kessels, H. W., Hagiwara, H., Sisodia, S., and Malinow, R. (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196. doi: 10.1038/nn.2476
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
Xu, Y. J., Mei, Y., Qu, Z. L., Zhang, S. J., Zhao, W., Fang, J. S., et al. (2018). Ligustilide Ameliorates Memory Deficiency in APP/PS1 Transgenic Mice via Restoring Mitochondrial Dysfunction. Biomed. Res. Int. 2018:4606752. doi: 10.1155/2018/4606752
Xue, J., Guo, H., Gao, Y., Wang, X., Cui, H., Chen, Z., et al. (2019). Altered Directed Functional Connectivity of the Hippocampus in Mild Cognitive Impairment and Alzheimer’s Disease: A Resting-State fMRI Study. Front. Aging Neurosci. 11:326. doi: 10.3389/fnagi.2019.00326
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 14670–14675. doi: 10.1073/pnas.0903563106
Zaidel, L., Allen, G., Cullum, C. M., Briggs, R. W., Hynan, L. S., Weiner, M. F., et al. (2012). Donepezil effects on hippocampal and prefrontal functional connectivity in Alzheimer’s disease: preliminary report. J. Alzheimers Dis. 31(Suppl. 3), S221–S226. doi: 10.3233/JAD-2012-120709
Zhang, J. W., Tabassum, S., Jiang, J. X., and Long, C. (2020). Optimized Golgi-Cox Staining Validated in the Hippocampus of Spared Nerve Injury Mouse Model. Front. Neuroanat. 14, 585513. doi: 10.3389/fnana.2020.585513
Keywords: Alzheimer’s disease, hippocampus, medial prefrontal cortex, mitochondrial dynamics, social interaction
Citation: Misrani A, Tabassum S, Huo Q, Tabassum S, Jiang J, Ahmed A, Chen X, Zhou J, Zhang J, Liu S, Feng X, Long C and Yang L (2021) Mitochondrial Deficits With Neural and Social Damage in Early-Stage Alzheimer’s Disease Model Mice. Front. Aging Neurosci. 13:748388. doi: 10.3389/fnagi.2021.748388
Received: 27 July 2021; Accepted: 16 November 2021;
Published: 10 December 2021.
Edited by:
Nibaldo C. Inestrosa, Pontificia Universidad Católica de Chile, ChileCopyright © 2021 Misrani, Tabassum, Huo, Tabassum, Jiang, Ahmed, Chen, Zhou, Zhang, Liu, Feng, Long and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Long, bG9uZ2NoZW5nQG0uc2NudS5lZHUuY24=; Li Yang, eWFuZ19saUBnemh1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.