 Nad’a Majerníková
Nad’a Majerníková Wilfred F. A. den Dunnen
Wilfred F. A. den Dunnen Amalia M. Dolga
Amalia M. Dolga- 1Research School of Behavioural and Cognitive Neuroscience, University of Groningen, Groningen, Netherlands
- 2Department of Pathology and Medical Biology, University Medical Centre Groningen, University of Groningen, Groningen, Netherlands
- 3Department of Molecular Pharmacology, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, Netherlands
- 4Research Institute Brain and Cognition, Molecular Neuroscience and Aging Research (MOLAR), University Medical Centre Groningen, Groningen, Netherlands
Alzheimer’s disease (AD), the most common form of dementia, currently affects 40–50 million people worldwide. Despite the extensive research into amyloid β (Aβ) deposition and tau protein hyperphosphorylation (p-tau), an effective treatment to stop or slow down the progression of neurodegeneration is missing. Emerging evidence suggests that ferroptosis, an iron-dependent and lipid peroxidation-driven type of programmed cell death, contributes to neurodegeneration in AD. Therefore, how to intervene against ferroptosis in the context of AD has become one of the questions addressed by studies aiming to develop novel therapeutic strategies. However, the underlying molecular mechanism of ferroptosis in AD, when ferroptosis occurs in the disease course, and which ferroptosis-related genes are differentially expressed in AD remains to be established. In this review, we summarize the current knowledge on cell mechanisms involved in ferroptosis, we discuss how these processes relate to AD, and we analyze which ferroptosis-related genes are differentially expressed in AD brain dependant on cell type, disease progression and gender. In addition, we point out the existing targets for therapeutic options to prevent ferroptosis in AD. Future studies should focus on developing new tools able to demonstrate where and when cells undergo ferroptosis in AD brain and build more translatable AD models for identifying anti-ferroptotic agents able to slow down neurodegeneration.
Introduction
Alzheimer’s disease (AD) is the most prevalent age-related neurodegenerative disorder, affecting over 44 million people worldwide (Gaugler et al., 2016). In AD, formation of amyloid β (Aβ) plaques and neurofibrillary tangles (NFTs) are associated with progressive cortical and hippocampal neuronal dysfunction and death (Dugger and Dickson, 2017). Many cell death mechanisms have been studied in AD pathology. The aggregation of Aβ was linked with caspase-9 and caspase-3-dependant apoptosis in neurons (Obulesu and Lakshmi, 2014), autophagy deficiency (Li and Sun, 2017), necrosis (Tanaka et al., 2020) and microglia-dependant activation of inflammasome pathway (Heneka et al., 2018). Despite extensive research into main hallmarks and molecular pathways of cell death in AD, many degenerative processes cannot be explained by these mechanisms alone, resulting in failure of over 200 AD drugs trials aiming at these targets over the past decade (Yiannopoulou et al., 2019).
In addition to apoptosis and necrosis, ferroptosis, an iron dependent and lipid-peroxidation driven cell death (Dixon, 2017), seems to be associated with AD (Hambright et al., 2017). Ferroptosis, the process increasing with aging (Zhou et al., 2020), is morphologically, genetically, and biochemically different from other types of cell death (Dixon et al., 2012). Its hallmarks, such as increased iron levels and oxidative stress, have been long noted in the AD brain (Praticò et al., 2001; Praticò and Sung, 2004; Castellani et al., 2007; Derry et al., 2020). It has been shown that formation of Aβ plaques and NFTs is related to iron overload in AD models and post mortem tissue (Yamamoto et al., 2002; Peters et al., 2018). Moreover, iron levels positively correlate with cognitive decline in human subjects (Ayton et al., 2017), and glutathione peroxidase (GPx4, also known as GPX4), the critical regulator of ferroptosis, is protective in AD mice model (Yoo et al., 2010).
Human genome-wide association studies (GWAS) support these results by showing a relation between the risk of developing AD and GPX4 polymorphism (Karch et al., 2016; da Rocha et al., 2018). Moreover, PSEN1/2 mutations identified in Alzheimer patients affected the hypoxic response in mouse embryonic fibroblasts by regulating hypoxia inducible factor-1α (HIF-1α), a driver of vulnerability to ferroptosis in cancer (Kaufmann et al., 2013; Zou et al., 2019). These results suggest that higher risk of developing AD is associated with deregulation of ferroptosis-related proteins, and thus ferroptosis inhibitors may have a therapeutic potential in AD (Weiland et al., 2019). However, the underlying mechanism of ferroptosis in AD, and whether ferroptosis happens at the onset, during or as a consequence of AD remains to be established.
Our aim is to examine the potential of ferroptosis inhibition as a therapeutic strategy for AD. We will first recapitulate ferroptosis pathway and its relation to AD, identify which ferroptosis-related genes are differentially expressed in AD and lastly, discuss the therapeutic options to prevent ferroptosis in AD.
Processes Involved in the Underlying Pathway of Ferroptosis
Ferroptosis mechanism can be divided into three parts: (1) iron homeostasis, (2) glutathione (GSH) metabolism and (3) oxidative stress and lipid peroxidation (Figure 1). Disruption of one or more of these mechanisms can induce lipid peroxidation-driven ferroptotic cell death.
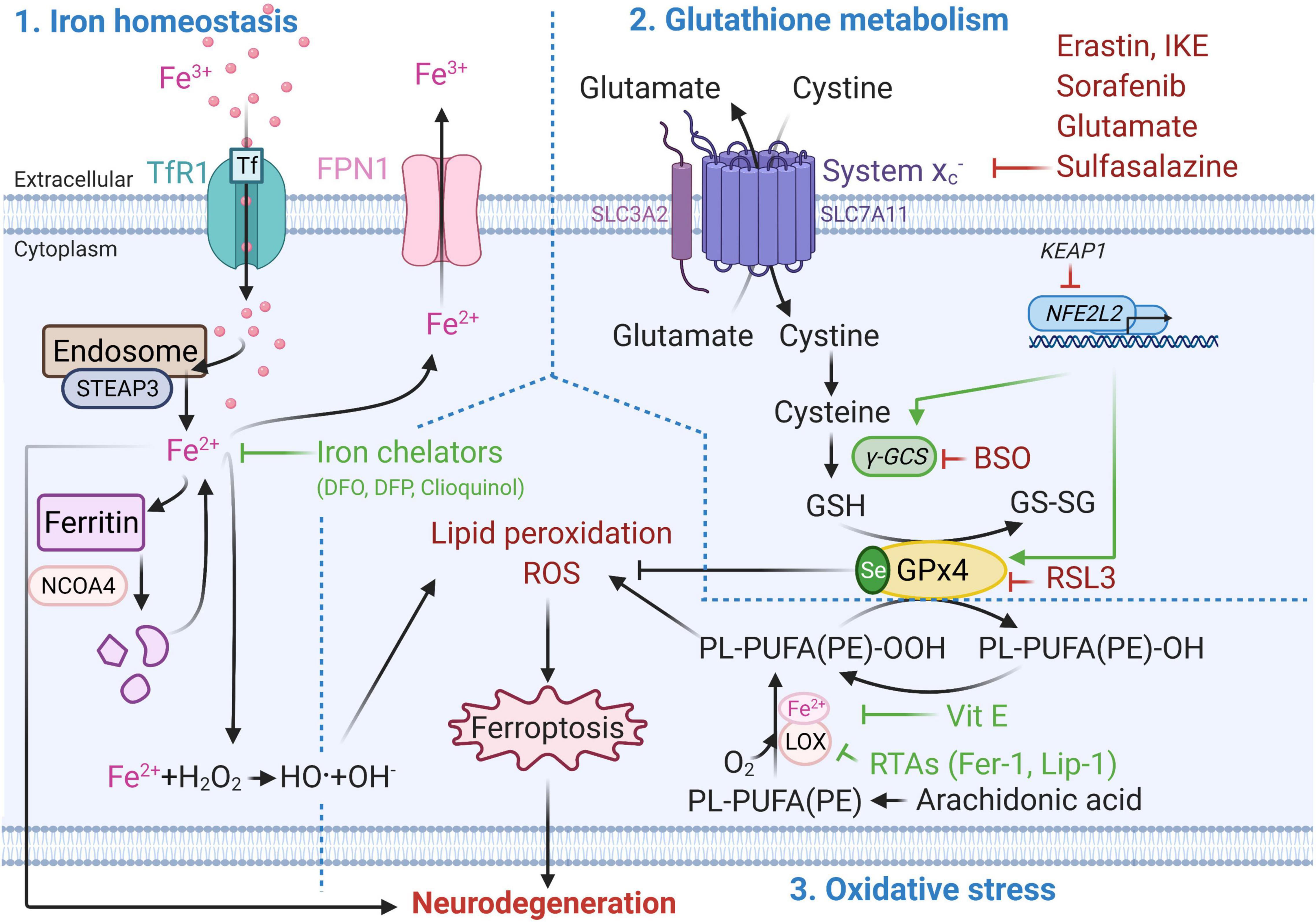
Figure 1. Molecular mechanisms of ferroptotic cell death. Metabolic pathways such as iron metabolism (left), cysteine and glutathione metabolism (top right), and polyunsaturated fatty acid metabolism (bottom right) play an essential role in the ferroptotic pathway. Well established ferroptosis inducers and inhibitors and their mode of action are depicted in red and green respectively. BSO, Buthionine sulphoximine; DFO, Deferoxamine; DFP, Deferiprone; Fe2 +, Ferrous iron; Fe3 +, Ferric iron; FPN1, Ferroportin; GPx4, Glutathione peroxidase 4; GSH, Glutathione (reduced glutathione form); GS-SG, Glutathione disulfide (oxidized glutathione form); Keap1, Kelch-like ECH-associated protein 1, LOX, Lipoxygenase; NCOA4, Nuclear receptor co-activator 4; NFE2L2, nuclear factor E2 related factor 2 encoding for Nrf2; PL-PUFA(PE)-OH, Polyunsaturated-fatty-acids (phosphatidylethanolamine)-reduced; PL-PUFA(PE)-OOH, Polyunsaturated-fatty-acid-containing-phospholipid hydroperoxides; ROS, Reactive oxygen species; RSL3, (1S,3R)-methyl-2-(2-chloroacetyl)-2,3,4,9-tetrahydro-1-[4-(methoxycarbonyl)phenyl]-1H-pyrido [3,4-b]indole-3-carboxylic acid; RTAs, Radical-trapping antioxidants; Se, Selenocysteine; STEAP3, Six-Transmembrane Epithelial Antigen Of Prostate 3; xCT subunit of system xc–, Glutamate/cystine antiporter system; TfR1, Transferrin 1 receptor; Vit E, Vitamin E; γ-GCS, Gamma-glutamylcysteine synthetase. This figure was created using Biorender.
Iron Homeostasis
Iron homeostasis plays a key role in ferroptosis (Yan and Zhang, 2020). Iron can enter the cell via transferrin 1 receptor (TfR1, also known as TFR1) and be reduced from ferric (Fe3+) to ferrous (Fe2+) form via metalloreductase STEAP3 in the endosome (Zhang et al., 2012). In this form, iron can be stored in ferritin, or exported from the cell via ferroportin (FPN1) (Chang, 2019). Ferritin degradation via the nuclear receptor coactivator 4 (NCOA4) contributes to ferroptosis by increasing the free intracellular iron levels (Hou et al., 2016). Excessive intracellular iron accumulation can lead to generation of reactive oxygen species (ROS) and oxidative stress via the Fenton reaction (Ward et al., 2014). Iron accumulation-induced ROS, such as superoxide anion (O2-•) and hydroxyl radical (•OH), possess an unpaired electron at their outer orbit which allows them to react with all cellular components including proteins, lipids and nucleic acid. This results in lipid peroxidation, oxidative damage to membranes and other lipid-containing molecules, and ultimately to cellular damage and ferroptotic cell death (Aprioku, 2013).
Glutathione Metabolism
On the other hand, inhibition of glutamate/cystine antiporter (system xc–, xc-, with xCT as the functional subunit of system xc–) and depletion of GSH cause inactivation of GPx4, the critical antioxidant enzyme and regulator of ferroptosis (Seibt et al., 2019). This can lead to ferroptotic cell death through increased lipid peroxidation and accumulation of ROS (Wang et al., 2020). GPx4 reduces hydroperoxides of polyunsaturated-fatty-acid-containing-phospholipids (PL-PUFA(PE)-OOH) to polyunsaturated-fatty-acids (phosphatidylethanol amine)-reduced (PL-PUFA(PE)-OH) (Seibt et al., 2019). GPx4 uses GSH as a reducing substrate and converts it into oxidized form, also referred to as glutathione disulphide (GS-SG) (Cozza et al., 2017). Apart from nuclear factor erythroid 2-related factor 2 (Nrf2, coded by NFE2L2 gene) (Habib et al., 2015), the xCT mRNA can be positively regulated by the activation of transcription factor 4 (ATF4) under oxidative stress (Sato et al., 2004), while its negative regulation by p53 results in cysteine deprivation and increased susceptibility to ferroptosis (Jiang et al., 2015).
Oxidative Stress and Lipid Peroxidation
Oxidative stress occurs due to the imbalance between generation of free radicals and the ability to neutralize or eliminate them through antioxidants (Birben et al., 2012). One of the main drivers of ferroptosis is ROS-mediated lipid peroxidation, which can result in oxidative stress (Kuang et al., 2020). Inhibition of GPx4 and decrease in GSH levels lead to activation of 12/15-lipoxygenase (12/15-LOX, which is the protein product of the ALOX15 gene). The association of Fe2+ with lipoxygenases (LOX, a dioxygenase containing non-heme iron) can lead to oxygenation of polyunsaturated-fatty-acids (PUFA), such as arachidonic acid present in phospholipids, and trigger lipid peroxidation-induced ferroptosis (Kagan et al., 2017). The LOX nomenclature is defined by the specific site of their oxygenation product: in humans there are six LOX isoforms 15-LOX-1, 15-LOX-2, 12-LOX-1, 12-LOX-2, E3-LOX, and 5-LOX, of which 12/15-LOX (15-LOX) are the most abundant. 12/15-LOX are considered as one of the key regulators of ferroptotic cell death (Yang et al., 2016; Kagan et al., 2017). Although, this has been supported by the findings that pharmacological inhibition of 15-LOX-1 exerts a cytoprotective effect (Seiler et al., 2008; Eleftheriadis et al., 2016), some off-target effects of lipoxygenase inhibitors have also been reported (Shah et al., 2018).
In addition to iron accumulation-induced generation of ROS, mitochondria also contribute to ROS production. Electrons leak from complex I and III of the electron transport chain (ETC) located on the inner membrane of mitochondria (Zhao et al., 2019). This can result in the formation of ROS such as O2-• and hydrogen peroxides (H2O2), and potentially can lead to loss of mitochondrial membrane potential (ΔΨm) (Gao et al., 2019). Reduced ΔΨm was associated with ferroptosis and involves different regulatory mechanisms than apoptosis (Kuang et al., 2020). GSH depletion-induced activation of 12/15-LOX can increase cytosolic Ca2+ via both (1) the import from the extracellular compartment and (2) release from mitochondria and endoplasmic reticulum (Maher et al., 2018). Decrease in GSH levels can also lead to dysregulation of Ca2+ transport in and out of mitochondria by voltage dependant anion channels (VDAC) and mitochondrial Ca2+ uniporter (MCU) (Zorov et al., 2014; DeHart et al., 2018). This results in mitochondrial Ca2+ overload and collapse of the mitochondrial function which activates Ca2+-dependant proteases (Zorov et al., 2014; DeHart et al., 2018; Marmolejo-Garza and Dolga, 2021). Consequently, ROS-induced transactivation of BH3 interacting-domain death agonist (BID) to mitochondria and Ca2+overload-induced translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus causes the cell to die (Neitemeier et al., 2017). This caspase-independent process is accompanied by mitochondrial fragmentation and enlarged cristae (Dixon et al., 2012). The rescue of mitochondria (Jelinek et al., 2018), decrease of mitochondria-associated endoplasmic reticulum membranes (MAMs) interaction (Guo et al., 2019) and small conductance calcium-activated potassium (KCa2/SK) channel activation have the potential to protect from ferroptotic cell death (Krabbendam et al., 2020).
Contributions of Ferroptosis to Alzheimer’s Disease
Iron Homeostasis
Advanced age is associated with iron dysregulation affecting most of our organs (Xu et al., 2012; Picca et al., 2019). Many studies show that iron dysregulation can also contribute to AD pathology (Bush, 2013; Nuñez and Chana-Cuevas, 2018). With aging, iron deposits in the brain (Acosta-Cabronero et al., 2016), which can increase the formation of Aβ plaques (Becerril-Ortega et al., 2014) and tau hyperphosphorylation in AD transgenic mouse brain (Guo et al., 2013). Imaging and histological experiments support this by showing increased iron deposition in AD-specific brain regions (Altamura and Muckenthaler, 2009; Bush, 2013; Apostolakis and Kypraiou, 2017; Lee and Lee, 2019). Magnetic resonance imaging (MRI) studies revealed increased iron levels in the putamen, pulvinar thalamus, red nucleus, hippocampus, and temporal cortex of AD patients (Langkammer et al., 2014). Later, quantitative susceptibility mapping showed higher iron levels in caudate and putamen nucleus of AD patients than in controls. Interestingly, the increased iron level in the left caudate nucleus correlated with the degree of cognitive impairment (Du et al., 2018). Finally, higher iron levels in the frontal cortex were associated with AD severity (Bulk et al., 2018b). This evidence suggested that iron contributes to AD pathology and presented an important avenue for therapy development (Masaldan et al., 2019).
Glutathione Metabolism
Ferroptosis can be induced by compounds interfering with system xc–, such as erastin, which induces cysteine deprivation, GSH depletion, endoplasmic reticulum stress, and cell death (Dixon et al., 2012, 2014; Sato et al., 2018). System xc– can also be inhibited by adding small concentrations of sorafenib (Lachaier et al., 2014), glutamate (Jiang et al., 2020) and sulfasalazine (Yu et al., 2019) to the extracellular compartment. Inhibition of gamma-glutamylcysteine synthetase (γ-GCS) by buthionine sulphoximine (BSO) results in GSH depletion and can lead to ferroptosis (Griffith, 1982). Irreversible and direct inhibition of GPx4 by the (1S,3R)-RSL3 (RSL3), causes the production of polyunsaturated-fatty-acid-containing-phospholipid hydroperoxides, which leads to lipid peroxidation and ferroptotic cell death (Liang et al., 2019). In addition to pharmacological compounds, genetic modifications targeting regulators of the system xc– can induce ferroptosis. The Gpx4BI-KO mouse was generated by a conditional deletion of Gpx4 in forebrain neurons by administration of tamoxifen. In this mouse model, 75–85% decrease of Gpx4 was shown to induce hippocampal neuronal loss, lipid peroxidation, neuroinflammation and spatial learning deficits (Hambright et al., 2017). Similarly, the knockout of Gpx1, facilitated memory impairment induced by β-Amyloid in mice (Joo et al., 2020). The Western blot analysis of AD post mortem brain tissue revealed enhanced expression of the light-chain subunit of the xCT (Ashraf et al., 2020). These results suggest that impaired GSH metabolism might play a role in ferroptosis during AD pathology (Ashraf et al., 2020).
Oxidative Stress and Lipid Peroxidation
The brain is the most vulnerable organ to oxidative stress. It represents only 2% of the body but uses 20% of the total oxygen supply (Sokoloff, 1999). Oxidative stress plays a key role in AD pathology by initiating the generation and enhancing of both Aβ plaques and hyperphosphorylation of Tau (p-Tau) (Huang et al., 2016; Nassireslami et al., 2016). Oxidative stress can be enhanced in AD via metal accumulation. In addition to iron, the Aβ precursor protein (APP) has a high affinity to binding zinc and copper at the N terminal metal-binding sites (Barnham et al., 2003). Additionally, high concentrations of these metals were also found in Aβ plaques in mouse and human brain (Plascencia-Villa et al., 2016; James et al., 2017). As copper is the potent mediator of •OH, and the binding of zinc leads to production of toxic Aβ and further uncontrolled zinc release, these metals can contribute to the increase of oxidative stress in AD (Strozyk et al., 2009). Post mortem tissue from AD patients shows higher levels of oxidized bases in the frontal, parietal and temporal lobes compared to control subjects (Wang et al., 2005), which correlates with imbalanced levels of copper, zinc and iron (Deibel et al., 1996). Other studies have shown higher level of lipid peroxidation, in diseased regions of AD brain compared to controls (Montine et al., 1998; Lovell et al., 2001; Bradley-Whitman and Lovell, 2015). These results support that oxidative stress might be an important factor contributing to the development and progression of AD (Zhao and Zhao, 2013).
Differential Expression of Ferroptosis-Related Genes in Alzheimer’s Disease
Many AD differentially expressed genes (DEGs) have been identified in animal and human studies. Using available RNAseq datasets of AD mouse models, AD patients and age-matched controls, we analyzed which of the 44 ferroptosis-related genes are differentially expressed in AD (Supplementary Table 1). To this end, we analyzed the expression of ferroptosis-related genes in one mouse [Alzmap (Chen et al., 2020)] and three human datasets of AD-DEGs [scREAD (Mathys et al., 2019), ACTA (Gerrits et al., 2021), AMPA-AD (Wan et al., 2020)]. All four datasets were available to the public and compared the gene expression between cell types, stages of disease progression and gender.
We first used the Alzmap gene retrieving function to make a qualitative assessment of the expression of three representative ferroptosis-related genes. We included (i) Gpx4, as it can suppress phospholipid peroxidation, an important process during ferroptosis, (ii) Gss, as it can facilitate the production of GSH, and (iii) Acsl4 for its role in supporting the incorporation of long PUFAs into lipid membranes, a process associated with ferroptosis (Figure 2). We choose t-distributed stochastic neighbor embedding (TSNE) statistical method to visualize the representative genes in a high-dimensional dataset (Figure 2). However, Alzmap website offers other modes of analysis and visualization tools such as the principal component analysis (PCA) and uniform manifold approximation and projections for dimension reduction (UMAP). The distribution and visualization of the chosen genes might render different output since these methods of visualization and reduction tools are based on specific clustering algorithms, i.e., unsupervised linear dimensionality reduction and data visualization technique for very high dimensional data for PCA, while t-SNE is based on a non-linear statistical method, calculating the similarity probability score in a low dimensional space. Therefore, visualization of genes could appear to render various outcomes. The alterations observed in the ferroptosis-related genes generated by Alzmap are purely based on a qualitative assessment. These data can be freely accessible on the https://alzmap.org/website.
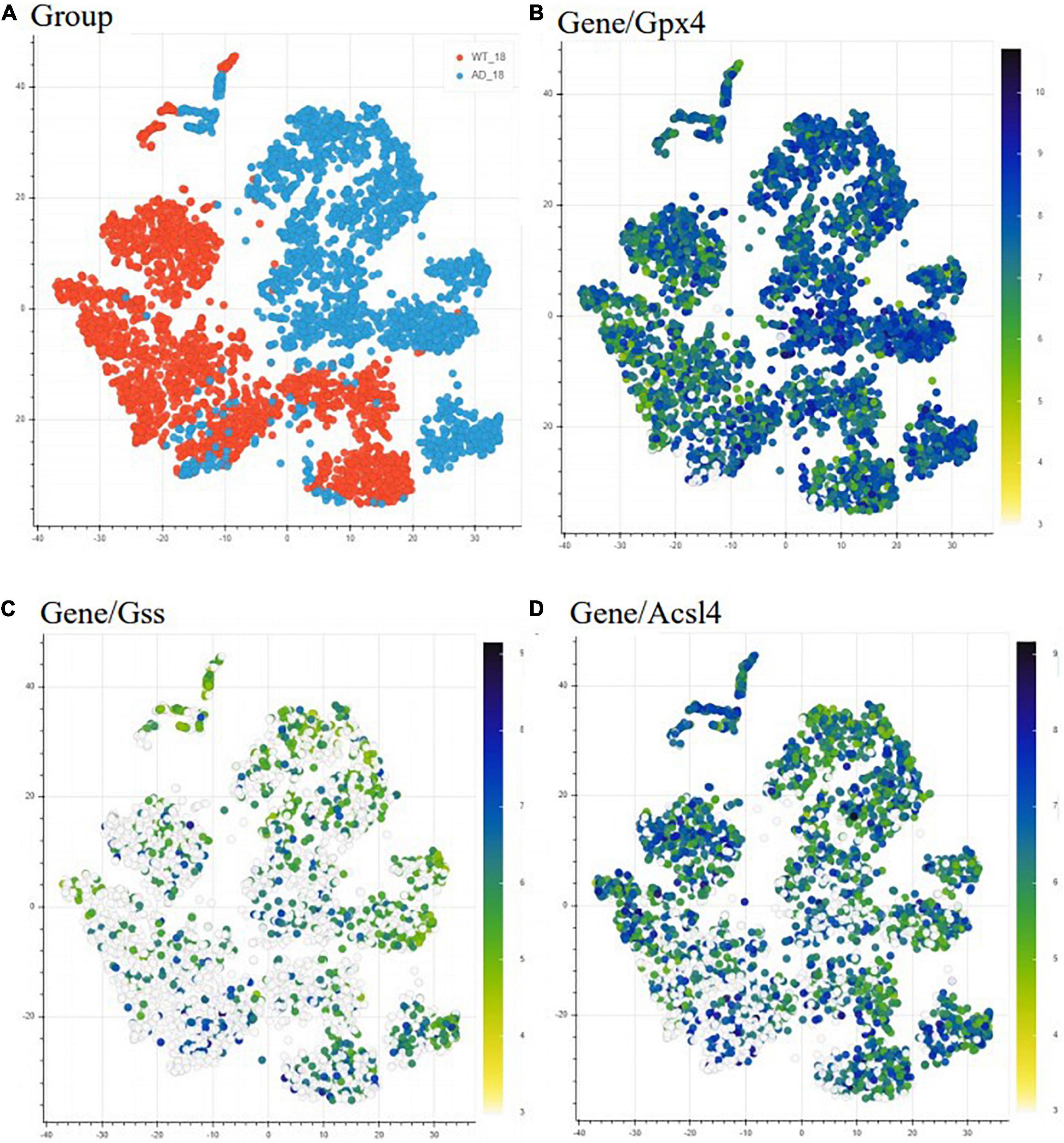
Figure 2. Differential expression of ferroptosis related genes in AD mice model compared to WT mice. Heatmap representing the difference in expression of ferroptosis-related genes between 18 months old WT (orange) and AD mice (light blue) (A). The heatmap depicts gene expression from low/white to high/dark blue. Each point indicates one spatial transcriptomic spot defining one tissue domain on the slide. Glutathione peroxidase 4 (Gpx4) is upregulated with pathology (B), while glutathione synthase (Gss) (C) and acyl-CoA synthetase long-chain family member (Acsl4) (D) are downregulated with the pathology. This data is freely accessible online, Alzmap (Chen et al., 2020).
In the Alzmap study, one left and one right hemisphere was collected for each experimental group and analyzed according to the spatial transcriptomic manual (Stockholm, Sweden) (Ståhl et al., 2016) using Fiji groovy script package (Chen et al., 2020). Our analysis revealed Gpx4 upregulation and Gss and Acsl4 downregulation in AppNL-G-F knock-in AD mice compared to WT mice. Although this analysis shows that these ferroptosis-related genes are differentially expressed in AppNL-G-F knock-in AD mice, it is known that downregulation of Gpx4 and upregulation of Acsl4 can induce ferroptosis (Dixon et al., 2012). Our observation from the TSNE analysis can be explained by cells trying to increase resistance against ferroptosis by increasing the generation of antioxidants (from the observation of increased Gpx4) and depleting the substrates for lipid peroxidation (as Acsl4 gene expression was found decreased) (Stockwell et al., 2017).
In the second study containing the scREAD dataset (Mathys et al., 2019), 48 participants were divided into early and late stage groups based on nine clinical pathological traits. Data was acquired by single-nucleus RNA sequencing (snRNAseq)-based differential expression analysis and assessed by Wilcoxon rank-sum test and false discovery rate (FDR) multiple-testing correction (Mathys et al., 2019). Our analysis revealed that ferroptosis-related genes in excitatory neurons from human brains are mostly downregulated at an early clinical stage of AD, while they are upregulated at a later clinical stage of the disease relative to early stage (Table 1). The same was observed with inhibitory neurons, astrocytes and glia cells. For instance, genes which are important for ferroptosis resistance [e.g., ACSL3, ferritin heavy chain (FTH1), GPX4, GSS and voltage-dependent anion channel 2 and 3 (VDAC2/3)] are downregulated in an early stage of AD pathology but upregulated at later AD stage. This could imply that ferroptosis already happens at early stages of the diseases. The shift from downregulation to upregulation at later stages can be explained by cells trying to compensate and rescue the ferroptotic cell death by increasing the expression of antioxidant proteins and enzymes. Furthermore, the observation that neurons show a higher number of ferroptosis DEGs in AD than astrocytes and oligodendrocytes suggests that ferroptosis affects neurons and glia cells differently (Kim et al., 2021). Although it seems from this dataset that ferroptosis gene expression changes primarily in neurons, it might be because glia cells were not primarily sorted out in this study. Therefore, next we analyzed a dataset that specifically looked at glia cells.
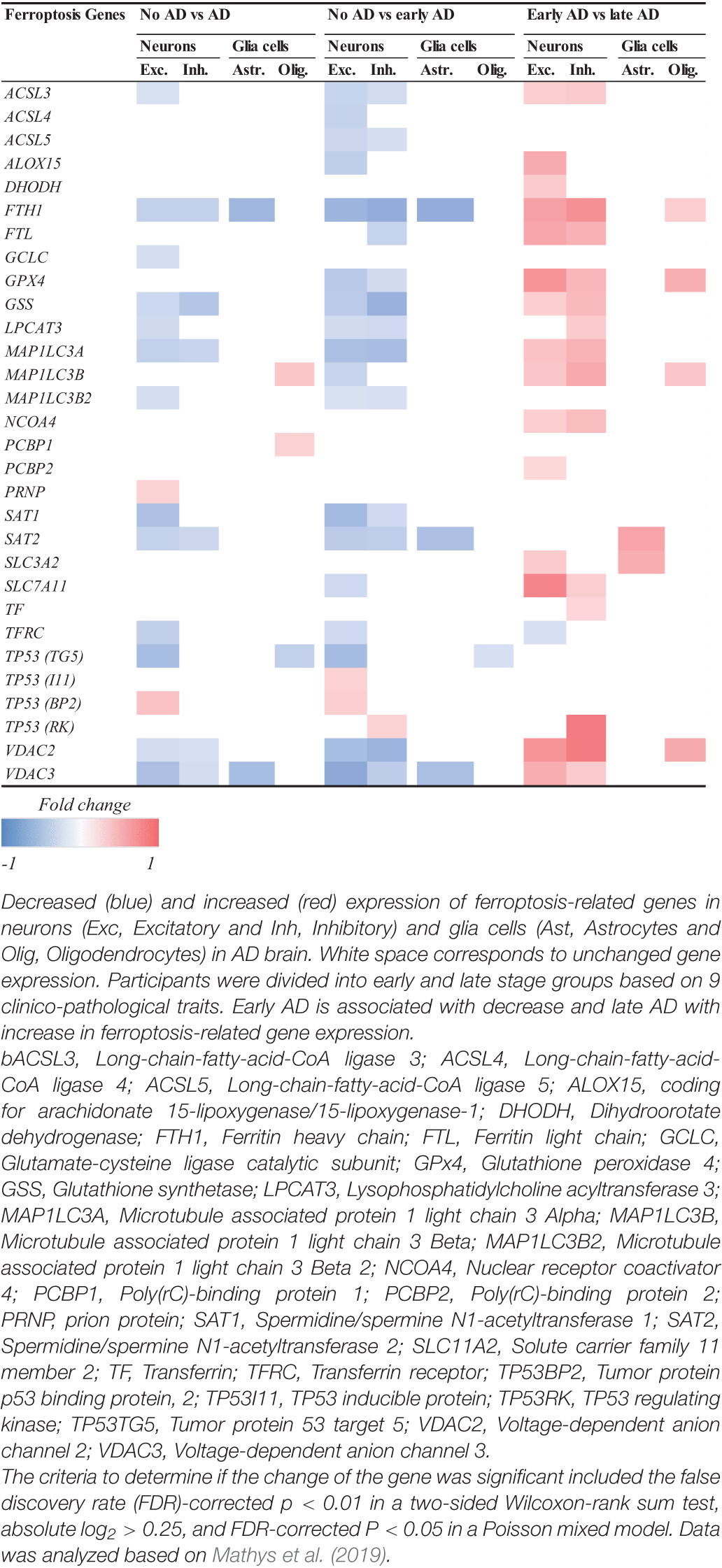
Table 1. Log2-fold change of ferroptosis-related DEGs related to AD.
To further investigate how ferroptosis could affect glia cells in AD, we looked at the difference in expression of ferroptosis-related genes in microglia between control and AD brains containing only amyloid-β plaques in the occipital cortex (OC) and both amyloid-β and tau pathology in the occipitotemporal cortex (OTC) (Gerrits et al., 2021). In this study, the differential expression analysis was performed using a logistic regression and adjusted p-value below 0.05 was used to determine the significance (Gerrits et al., 2021). Microglia belonging to different subclusters (homeostatic, Aβ-related = AD1 and tau-related = AD2) showed changes in the expression of ferroptosis-related genes between AD and control subjects (Table 2). Microglia affected by Aβ pathology alone, or the combination of Aβ and tau pathology showed more DEGs than cells in the homeostatic subcluster. Microglia in the Aβ-related subcluster showed increase in the expression of ferroptosis-related genes, while microglia in tau pathology-related subcluster showed decrease in the expression of these genes. As the presence of tau pathology in OC is typical for later stages of the diseases, these results could suggest that there seem to be a difference between the expression of ferroptosis-related genes between early and late stages of AD. However, whether glia cells die via ferroptotic cell death at later stages of AD should be investigated further.
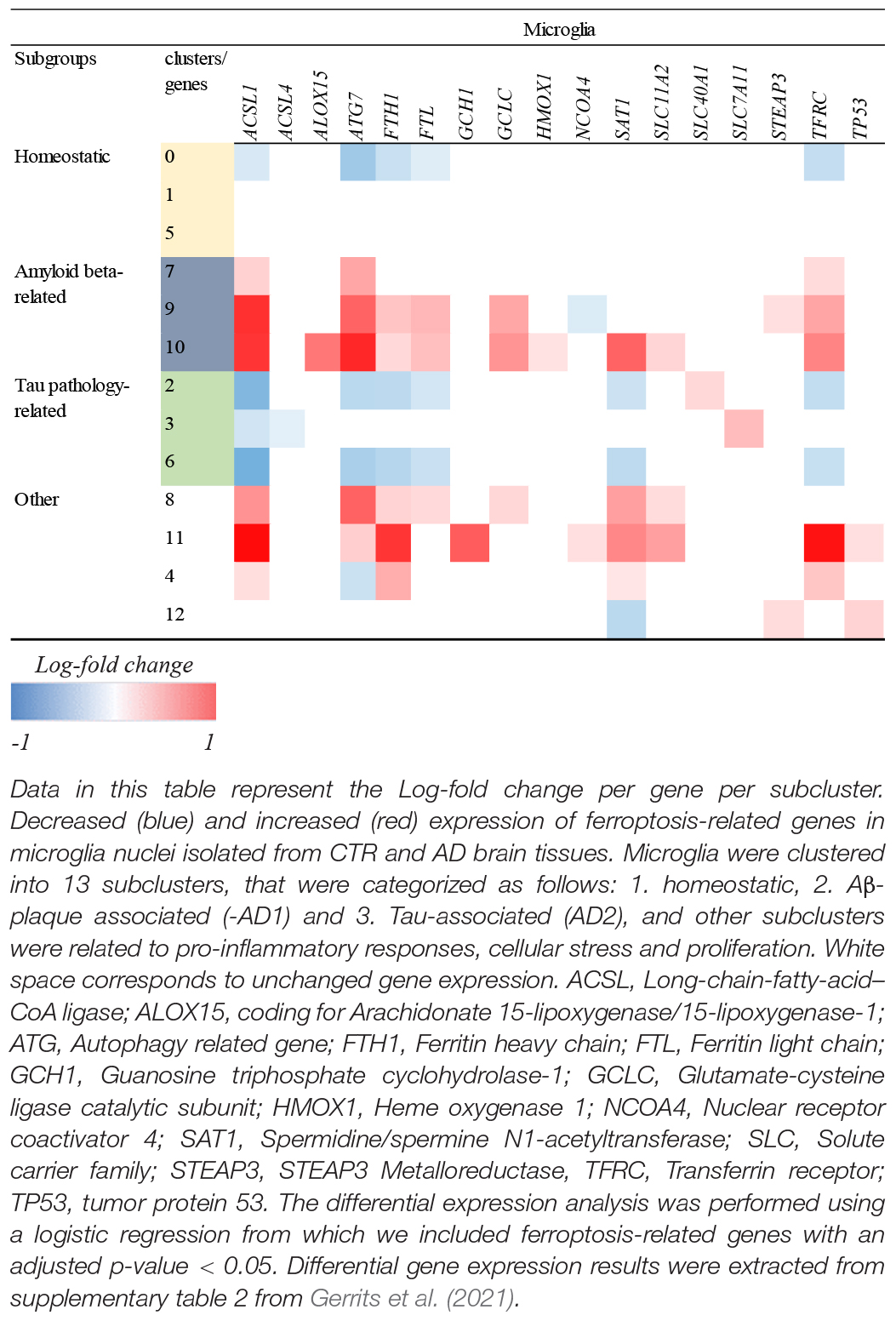
Table 2. Log-fold change of ferroptosis-related DEGs in glia cells in AD.
Previous analysis of the whole brain human DEGs in AD revealed more AD-DEGs in women than men (Wan et al., 2020). To see whether this is also specifically true for ferroptosis-related genes, we analyzed the 44 ferroptosis-related genes in the AMPA-AD dataset where AD-DEGs were compared between genders (Table 3). The sample size included 478 AD (female: 318, male: 160) and 300 control (female: 148, male: 152) cases on which sex-stratified meta-analysis (Wan et al., 2020). Our analysis revealed three downregulated genes in both men and women while only GSS was downregulated in both. Only one gene, Cytochrome B-245 Beta Chain (CYBB), was upregulated in men while eleven genes were upregulated in women (Table 3). The analysis of the dataset available in this study indicates that like AD-DEGs, ferroptosis-related genes seem to be more differentially expressed in women than men. Finally, nine of the 44 ferroptosis-related genes were not differentially expressed in any of the analyzed datasets (Supplementary Table 1).
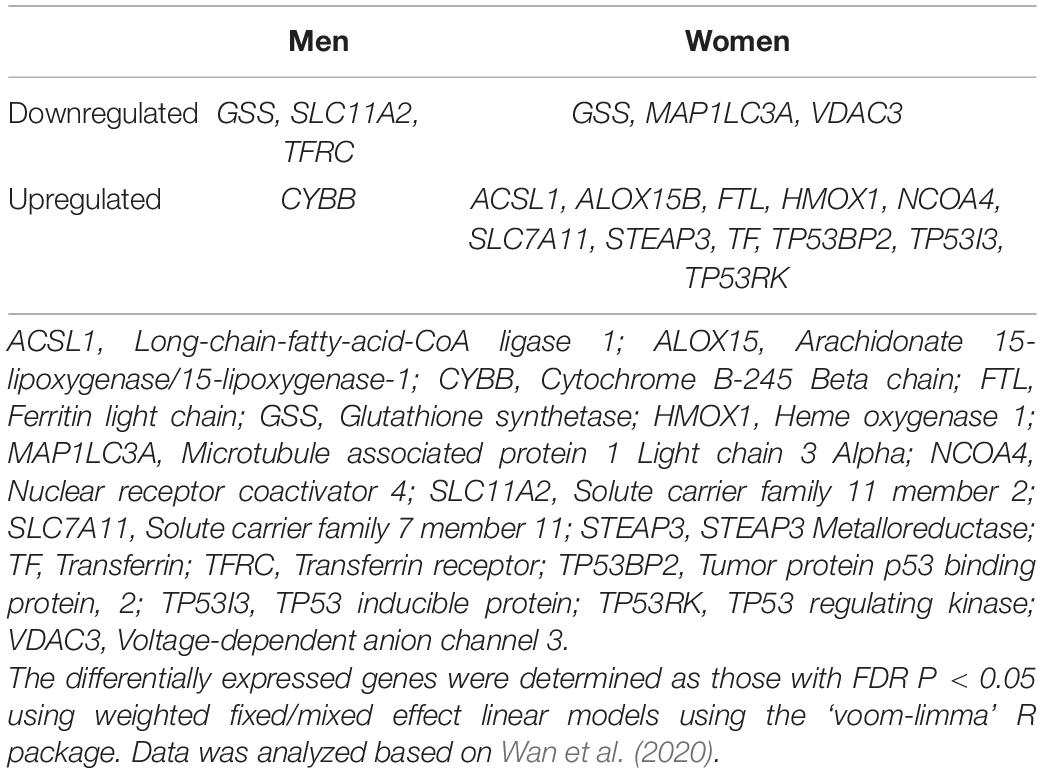
Table 3. Comparison of ferroptosis-related DEGs in AD between genders.
Inhibition of Ferroptosis to Treat Alzheimer’s Disease
An increasing amount of literature suggests that anti-ferroptotic therapies may be efficient in AD (Ashraf et al., 2020; Li et al., 2020; Table 4).
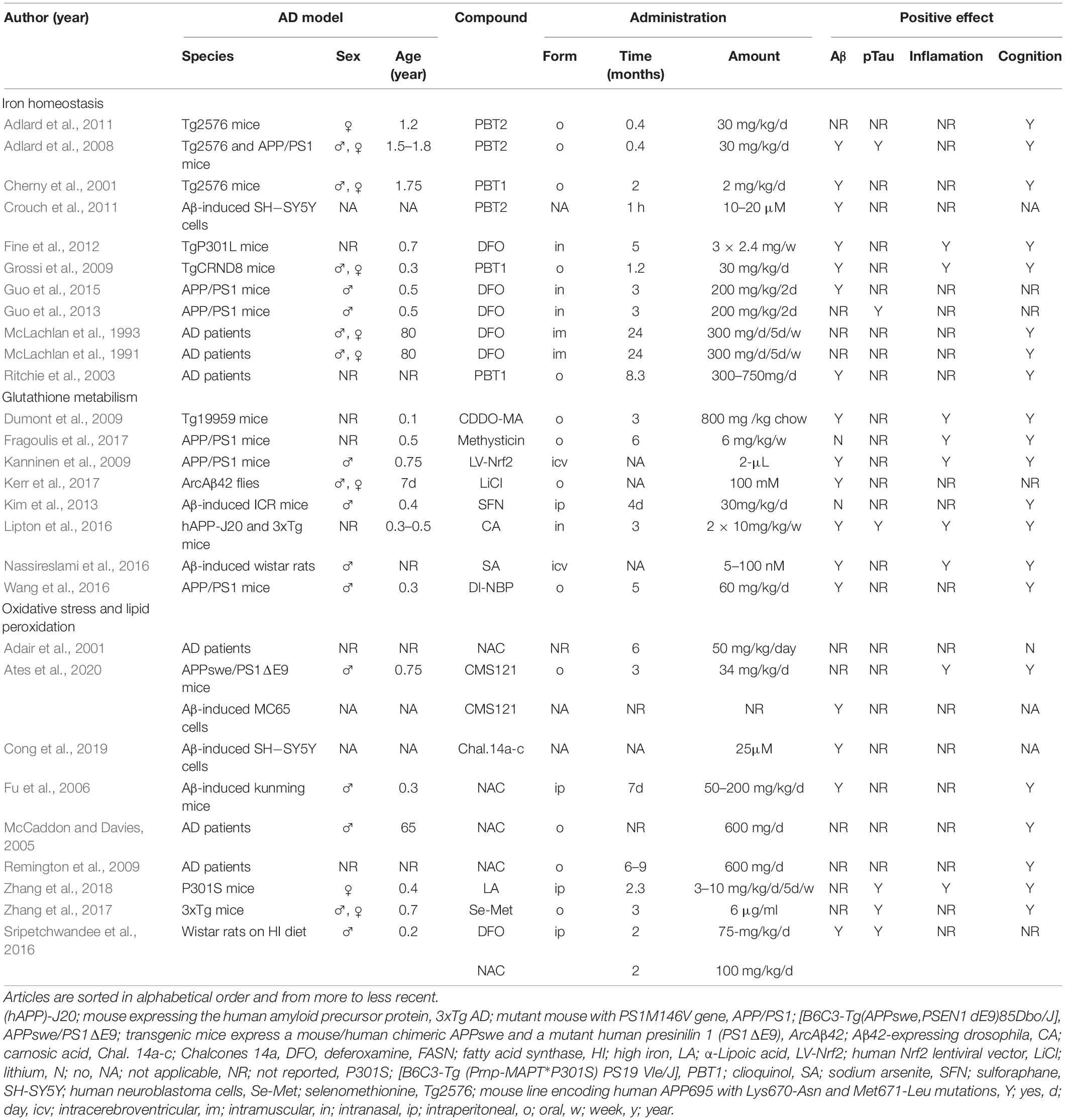
Table 4. Characteristics of included articles assessing therapeutic options to prevent ferroptosis in AD stratified by mechanisms involved in ferroptosis.
Iron Homeostasis
Our transcriptomic analysis revealed that FTH1, component responsible for iron storage, is differentially expressed in early and late stages of AD. Furthermore, excessive iron deposition in specific brain areas contributes to AD pathology (Antharam et al., 2012; Moon et al., 2016). Therefore, an increased interest in the development of therapeutic strategies targeting iron has emerged in the past years. In animal models, DFO treatment decreased AD hallmarks, iron overload, iron-induced kinase activity [cyclin-dependent kinase 5 (CDK5), glycogen synthase kinase 3β (GSK3β)], mitochondrial dysfunction, synaptic loss, and neuronal damage (Fine et al., 2012; Guo et al., 2013, 2015; Sripetchwandee et al., 2016). DFO increased expression of transferrin receptor (TfR1) and brain-derived neurotrophic factor (BDNF), leading to reduced iron-induced memory deficits in rodents (Fine et al., 2012; C. Guo et al., 2013, 2015; Sripetchwandee et al., 2016). In a clinical trial, DFO slowed down the progression of AD in patients (McLachlan et al., 1991, 1993). However, the dosing regimens need to be standardized before DFO could be implemented in the clinical setting (Farr and Xiong, 2021). In addition, to reduce DFO-related cytotoxicity and prolong its presence into circulation, new DFO component-containing nanogels were proposed as promising alternatives for iron-chelation in AD (Wang et al., 2018). Besides AD, DFO alone or co/treatment with ferrostatin (Fer-1, inhibitor of lipid peroxidation) also improved α-synuclein-induced pathology in a PD animal model (Febbraro et al., 2013). PBT1, a drug inhibiting zinc and copper ions from binding to Aβ, reduced Aβ deposition, attenuated astrogliosis and prevented memory impairment in AD animal models AD (Cherny et al., 2001; Grossi et al., 2009). In pilot-phase 2 clinical trial, PBT1 reduced Aβ plasma levels and, when looked specifically on severely affected AD patients, PBT1 was able to slow down the clinical decline (Ritchie et al., 2003). PBT2, a second-generation 8-hydroxyquinoline analog produced as a successor to clioquinol, induced GSK3β phosphorylation and prevented formation of Aβ in neuroblastoma SH-SY5Y cells (Crouch et al., 2011). In animal models of AD, PBT2 induced Aβ plaque degradation, decreased p-tau, rescued decreased spine density, increased brain-levels of BDNF and improved cognitive performance (Adlard et al., 2008, 2011). PBT2 was also assessed in a phase 2 clinical trial, where it lead to reduced levels of Aβ in cerebrospinal fluid and improved executive function compared to placebo (Lannfelt et al., 2008). However, PBT2 did not show any significant effect on cognition. Currently, deferiprone (DFP), a compound that alleviates symptoms related to PD pathology (Devos et al., 2014; Grolez et al., 2015; Gutbier et al., 2020), is evaluated a in phase 2 randomized placebo-controlled clinical trial with AD patients (NCT03234686). As previously reported, iron chelators can attenuate symptoms and slow down the progression of AD, which shows the potential for novel therapeutic approaches (Nuñez and Chana-Cuevas, 2018).
Glutathione Metabolism
The revealed differential gene expression of GPX4 and GSS suggests that modifying the expression or/and the activity of these gene-encoded proteins might be beneficial to treat AD. The expression of GPx4 can be directly upregulated by α-Lipoic acid (LA) (Zhang et al., 2018). LA treatment on P301S Tau transgenic mice enhanced the activity of system xc–, GPx4, superoxide dismutase 1 (Sod1), CDK5, GSK3β, TfR1 and FPN1 (Zhang et al., 2018). LA reduced the hippocampal levels of glial fibrillary acidic protein (GFAP), tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), as well as the calcium (Ca2+) content, p-tau, calpain1 levels, and synaptic loss. As a result, these processes led to enhanced memory function (Zhang et al., 2018). Apart from LA, GPx4 can be activated in an indirect manner through Nrf2. Nrf2 plays an important role in neurodegeneration and ferroptosis by regulating a wide range of genes (Song and Long, 2020). In addition to the activation of GPx4 and GSH synthesis (Dodson et al., 2019), it can also affect the activity of glucose-6-phosphate dehydrogenase, GSH reductase, glutamate-cysteine ligase modifier subunit (GLCM), solute carrier family 7 member 11 (SLC7A11) and others as previously summarized by Song and Long (2020). Nrf2 can be upregulated using a human lentiviral vector or compounds such as sodium arsenite, triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid-methylamide (CDDO-MA), dl-3-n-butylphthalide (DI-NBP), kavalactone methysticin, carnosic acid (CA) and sulforaphane (SFN). Nrf2 upregulation increased heme oxygenase-1 (HMOX1) levels and decreased AD hallmarks, hippocampal inflammation, oxidative stress, and Aβ-induced memory deficits in AD mouse models (Dumont et al., 2009; Kanninen et al., 2009; Kim et al., 2013; Lipton et al., 2016; Nassireslami et al., 2016; Wang et al., 2016; Fragoulis et al., 2017). Finally, genetic downregulation of Kelch-like ECH-associated protein 1 (Keap1), the negative regulator of Nrf2, in ArcAβ42 flies, activated Nrf2, induced Aβ42 degradation, prevented neuronal toxicity in response to Aβ42 peptide, rescued neuronal-specific motor defects and increased life span (Kerr et al., 2017).
Altogether, these results suggest that inhibition of ferroptosis by targeting GSH metabolism is an important avenue for the development of new therapies for AD (Ashraf et al., 2020).
Oxidative Stress and Lipid Peroxidation
Lipid peroxidation represents an important hallmark of AD (Sultana et al., 2013), which was also supported by the observed differential expression of ACSL3 and 4 in the course of the pathology (Tables 1, 2). In many studies, oxidative stress was targeted to reduce neuronal damage and alleviate symptoms related to AD pathology. Anti-ferroptotic compounds that reduce oxidative stress include liproxstatin 1 (Lip-1) (inhibitor of ROS and lipid peroxidation), chalcones 14a-c (inhibitor of Aβ and lipid peroxidation), Selenomethionine (Se-Met) (inhibitor of lipid peroxidation), CMC121 (fatty acid synthase inhibitor), N-acetylcysteine (NAC) (free radical scavenger), Vitamin E (Vit E) and PD146176 (15-LOX-1 inhibitor). Studies using in vitro and in vivo models of AD have shown that targeting oxidative stress has a positive effect on neural degeneration, inflammation, Aβ1-42 aggregation, p-tau formation, GSH levels, iron overload, mitochondrial function, motor dysfunction and learning and memory (Fu et al., 2006; Sripetchwandee et al., 2016; Hambright et al., 2017; Zhang et al., 2017; Cong et al., 2019; Ates et al., 2020). In concordance with these results, clinical trials have shown that NAC and co-treatment of NAC, Vit E and Se-Met improved behavioral symptoms, general well-being, and neuropsychiatric and cognitive scores of AD patients (Adair et al., 2001; McCaddon and Davies, 2005; Remington et al., 2009). Although Vitamin E treatment had no beneficial effect on patients with mild cognitive impairment (Marder, 2005), it was able to improve symptoms related to other neurodegenerative diseases such as PD (Taghizadeh et al., 2017) and cerebellar ataxia (Gabsi et al., 2001). Considering the lack of adverse events of these antioxidants, ferroptosis inhibition by targeting oxidative stress is a new promising therapeutic strategy for AD.
Discussion
Improved understanding of underlying mechanisms of ferroptosis in AD may lead to the development and application of anti-ferroptotic strategies to slow down or prevent AD progression (Han et al., 2020). Iron accumulation (Bulk et al., 2018a), lipid peroxidation (Majerníková et al., 2020) and mitochondrial dysfunction (Horowitz and Greenamyre, 2010), the main hallmarks of ferroptosis, are observed early in AD pathology, suggesting that targeting ferroptosis in AD may lead to the prevention of symptoms manifestation such as cognitive decline at advanced stages of AD.
Our analysis of DEGs in AD revealed that differential expression of ferroptosis-related genes in AD affects mostly neurons and that the changes observed in glia cells could be related to both tau phosphorylation and Aβ accumulation. This may explain the difference in the expression of ferroptotic markers between early (Aβ) and late (Aβ + p-tau) stages of AD. Even though this review has shed more light on the role of different brain cell types in ferroptosis during AD, whether ferroptosis in glia cells is related to later stages of the pathology should be investigated further.
While it is known that AD brain shows ferroptosis characteristics, it is unknown what is the causal relationship between AD and ferroptosis. Plasma ferritin increases with increasing age and Aβ deposition. Recent work on the inhibition of lipid peroxidation and iron accumulation in C. elegans revealed extended life- and health-span independently of other mechanisms (Jenkins et al., 2020). This evidence suggests that ferroptosis may be an age-related as well as disease-related process (Goozee et al., 2018; Larric et al., 2020). Therefore, ferroptosis inhibition may not only lead to slowing down the neurodegeneration but also contribute to longer health-span (Larric et al., 2020).
Iron dysregulation aggravates formation and aggregation of both Aβ and p-tau protein forming plaques and NFT respectively (Derry et al., 2020). Even though the link between ferroptosis and Aβ has been extensively studied, much less is known about its role in NFT formation. Therefore, future studies should try to investigate the role of ferroptosis in hyperphosphorylation of tau protein and formation of fibrillary tangles independently of Aβ pathology. This could be achieved by comparing the characteristics of ferroptotic cell death in AD with patients with primary age-related tauopathy (PART) (Crary et al., 2014).
Further research should also address the effect of ferroptosis on the interactions between different cell types in AD context. Although cell-cell interactions are dysregulated in AD brain (Henstridge et al., 2019), this feature of AD is often overlooked in in vitro studies. The brain-on-a-chip platform using induced pluripotent stem cells (iPSCs) -derived neurons and glia from AD patients could allow a high throughput screening of the effect of anti-ferroptotic drugs in AD, while mimicking the cell-cell interactions in AD context (Trombetta-Lima et al., 2021). Moreover, this model is easily reproducible and thanks to the use of iPSCs from AD patients, also more translatable to humans compared to well-established animal models.
Conclusion
This review summarizes the evidence supporting the important role of ferroptosis in AD pathology and presents what is known about the targets for its inhibition for a potential treatment. Ferroptosis-related genes are differentially expressed in AD, supporting our hypothesis that ferroptosis inhibition could slow down the AD progression and memory decline, however, many questions remain unanswered. Developing new AD models allowing us to study how ferroptosis effects cell-cell interaction is needed to understand the causal relationship and timing of ferroptosis in AD. Future efforts should be directed toward developing detection techniques of ferroptosis in vivo and organizing large, randomized clinical trials of anti-ferroptotic drugs in early and late stages of AD progression.
Author Contributions
NM, AD, and WD designed the theme of the manuscript. NM contributed by writing all the sections and creating all tables and figures. AD and WD conducted critical revisions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
NM received a De Cock research grant and a fellowship from the Behavioural and Cognitive Neuroscience Graduate School, University Medical Centre Groningen. AD is the recipient of an Alzheimer Nederland grant (WE.03- 2018-04, Netherlands), and a Rosalind Franklin Fellowship co-funded by the European Union and the University of Groningen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Alejandro Marmolejo-Garza for the support on the transcriptomic analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.745046/full#supplementary-material
References
Acosta-Cabronero, J., Betts, M. J., Cardenas-Blanco, A., Yang, S., and Nestor, P. J. (2016). In vivo MRI mapping of brain iron deposition across the adult lifespan. J. Neurosci. 36, 364–374. doi: 10.1523/JNEUROSCI.1907-15.2016
Adair, J. C., Knoefel, J. E., and Morgan, N. (2001). Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 57, 1515–1517. doi: 10.1212/WNL.57.8.1515
Adlard, P. A., Bica, L., White, A. R., Nurjono, M., Filiz, G., Crouch, P. J., et al. (2011). Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS One 6:e17669. doi: 10.1371/journal.pone.0017669
Adlard, P. A., Cherny, R. A., Finkelstein, D. I., Gautier, E., Robb, E., Cortes, M., et al. (2008). Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 59, 43–55. doi: 10.1016/j.neuron.2008.06.018
Altamura, S., and Muckenthaler, M. U. (2009). Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimer’s Dis. 16, 879–895. doi: 10.3233/JAD-2009-1010
Antharam, V., Collingwood, J. F., Bullivant, J. P., Davidson, M. R., Chandra, S., Mikhaylova, A., et al. (2012). High field magnetic resonance microscopy of the human hippocampus in Alzheimer’s disease: Quantitative imaging and correlation with iron. NeuroImage 59, 1249–1260. doi: 10.1016/j.neuroimage.2011.08.019
Apostolakis, S., and Kypraiou, A. M. (2017). Iron in neurodegenerative disorders: Being in the wrong place at the wrong time? Rev. Neurosci. 28, 893–911. doi: 10.1515/revneuro-2017-0020
Aprioku, J. S. (2013). Pharmacology of free radicals and the impact of reactive oxygen species on the testis. J. Reproduct. Infertil. 14, 158–172.
Ashraf, A., Jeandriens, J., Parkes, H. G., and So, P. W. (2020). Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol. 32:101494. doi: 10.1016/j.redox.2020.101494
Ates, G., Goldberg, J., Currais, A., and Maher, P. (2020). CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol. 36:101648. doi: 10.1016/j.redox.2020.101648
Ayton, S., Fazlollahi, A., Bourgeat, P., Raniga, P., Ng, A., Lim, Y. Y., et al. (2017). Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain 140, 2112–2119. doi: 10.1093/brain/awx137
Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., et al. (2003). Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407. doi: 10.1074/jbc.M300629200
Becerril-Ortega, J., Bordji, K., Fréret, T., Rush, T., and Buisson, A. (2014). Iron overload accelerates neuronal amyloid-β production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol. Aging 35, 2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S., and Kalayci, O. (2012). Oxidative stress and antioxidant defense. World Allergy Organizat. J. 5, 9–19. doi: 10.1097/WOX.0b013e3182439613
Bradley-Whitman, M. A., and Lovell, M. A. (2015). Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch. Toxicol. 89, 1035–1044. doi: 10.1007/s00204-015-1517-6
Bulk, M., Kenkhuis, B., Van Der Graaf, L. M., Goeman, J. J., Natté, R., and Van Der Weerd, L. (2018b). Postmortem T2*-Weighted MRI Imaging of Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 65, 1125–1137. doi: 10.3233/JAD-180317
Bulk, M., Abdelmoula, W. M., Nabuurs, R. J. A., van der Graaf, L. M., Mulders, C. W. H., Mulder, A. A., et al. (2018a). Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer’s disease. Neurobiol. Aging 62, 231–242. doi: 10.1016/j.neurobiolaging.2017.10.017
Bush, A. I. (2013). The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 33(Suppl. 1), S277–S281. doi: 10.3233/JAD-2012-129011
Castellani, R. J., Moreira, P. I., Liu, G., Dobson, J., Perry, G., Smith, M. A., et al. (2007). Iron: The redox-active center of oxidative stress in Alzheimer disease. Neurochem. Res. 32, 1640–1645. doi: 10.1007/s11064-007-9360-7
Chang, Y. (2019). Cellulat iron metabolism and regulation. Brain Iron Metabol. CNS Dis. 1173, 21–32. doi: 10.1007/978-981-13-9589-5_2
Chen, W. T., Lu, A., Craessaerts, K., Pavie, B., Sala Frigerio, C., Corthout, N., et al. (2020). Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 182, 976.e–991.e. doi: 10.1016/j.cell.2020.06.038
Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., McLean, C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676. doi: 10.1016/S0896-6273(01)00317-8
Cong, L., Dong, X., Wang, Y., Deng, Y., Li, B., and Dai, R. (2019). On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 166, 11–21. doi: 10.1016/j.ejmech.2019.01.039
Cozza, G., Rossetto, M., Bosello-Travain, V., Maiorino, M., Roveri, A., Toppo, S., et al. (2017). Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 112, 1–11. doi: 10.1016/j.freeradbiomed.2017.07.010
Crary, J. F., Trojanowski, J. Q., Schneider, J. A., Abisambra, J. F., Abner, E. L., Alafuzoff, I., et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. doi: 10.1007/s00401-014-1349-0
Crouch, P. J., Savva, M. S., Hung, L. W., Donnelly, P. S., Mot, A. I., Parker, S. J., et al. (2011). The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 119, 220–230. doi: 10.1111/j.1471-4159.2011.07402.x
da Rocha, T. J., Silva Alves, M., Guisso, C. C., de Andrade, F. M., Camozzato, A., de Oliveira, A. A., et al. (2018). Association of GPX1 and GPX4 polymorphisms with episodic memory and Alzheimer’s disease. Neurosci. Lett. 666, 32–37. doi: 10.1016/j.neulet.2017.12.026
DeHart, D. N., Fang, D., Heslop, K., Li, L., Lemasters, J. J., and Maldonado, E. N. (2018). Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 148, 155–162. doi: 10.1016/j.bcp.2017.12.022
Deibel, M. A., Ehmann, W. D., and Markesbery, W. R. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 143, 137–142. doi: 10.1016/S0022-510X(96)00203-1
Derry, P. J., Hegde, M. L., Jackson, G. R., Kayed, R., Tour, J. M., Tsai, A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184:101716. doi: 10.1016/j.pneurobio.2019.101716
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
Dixon, S. J. (2017). Ferroptosis: bug or feature? Immunol. Rev. 277, 150–157. doi: 10.1111/imr.12533
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D., Welsch, M., Skouta, R., Lee, E., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. ELife 3:e02523. doi: 10.7554/eLife.02523
Dodson, M., Castro-Portuguez, R., and Zhang, D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23:101107. doi: 10.1016/j.redox.2019.101107
Du, L., Zhao, Z., Cui, A., Zhu, Y., Zhang, L., Liu, J., et al. (2018). Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chemical Neurosci. 9, 1849–1857. doi: 10.1021/acschemneuro.8b00194
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Dumont, M., Wille, E., Calingasan, N. Y., Tampellini, D., Williams, C., Gouras, G. K., et al. (2009). Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 109, 502–512. doi: 10.1111/j.1471-4159.2009.05970.x
Eleftheriadis, N., Poelman, H., Leus, N. G. J., Honrath, B., Neochoritis, C. G., Dolga, A., et al. (2016). Design of a novel thiophene inhibitor of 15-lipoxygenase-1 with both anti-inflammatory and neuroprotective properties. Eur. J. Med. Chem. 122, 786–801. doi: 10.1016/j.ejmech.2016.07.010
Farr, A. C., and Xiong, M. P. (2021). Challenges and Opportunities of Deferoxamine Delivery for Treatment of Alzheimer’s Disease, Parkinson’s Disease, and Intracerebral Hemorrhage. Mol. Pharmaceut. 18, 593–609. doi: 10.1021/acs.molpharmaceut.0c00474
Febbraro, F., Andersen, K. J., Sanchez-Guajardo, V., Tentillier, N., and Romero-Ramos, M. (2013). Chronic intranasal deferoxamine ameliorates motor defects and pathology in the α-synuclein rAAV Parkinson’s model. Exp. Neurol. 247, 45–58. doi: 10.1016/j.expneurol.2013.03.017
Fine, J. M., Baillargeon, A. M., Renner, D. B., Hoerster, N. S., Tokarev, J., Colton, S., et al. (2012). Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3β in P301L tau transgenic mice. Exp. Brain Res. 219, 381–390. doi: 10.1007/s00221-012-3101-0
Fragoulis, A., Siegl, S., Fendt, M., Jansen, S., Soppa, U., Brandenburg, L. O., et al. (2017). Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol. 12, 843–853. doi: 10.1016/j.redox.2017.04.024
Fu, A. L., Dong, Z. H., and Sun, M. J. (2006). Protective effect of N-acetyl-l-cysteine on amyloid β-peptide-induced learning and memory deficits in mice. Brain Res. 1109, 201–206. doi: 10.1016/j.brainres.2006.06.042
Gabsi, S., Gouider-Khouja, N., Belal, S., Fki, M., Kefi, M., Turki, I., et al. (2001). Effect of vitamin E supplementation in patients with ataxia with vitamin E deficiency. Eur. J. Neurol. 8, 477–481. doi: 10.1046/j.1468-1331.2001.00273.x
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of Mitochondria in Ferroptosis. Mol. Cell 73, 354.e–363.e. doi: 10.1016/j.molcel.2018.10.042
Gaugler, J., James, B., Johnson, T., Scholz, K., and Weuve, J. (2016). 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2, 459–509. doi: 10.1016/j.jalz.2016.03.001
Gerrits, E., Brouwer, N., Kooistra, S. M., Woodbury, M. E., Vermeiren, Y., Lambourne, M., et al. (2021). Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696. doi: 10.1007/s00401-021-02263-w
Goozee, K., Chatterjee, P., James, I., Shen, K., Sohrabi, H. R., Asih, P. R., et al. (2018). Elevated plasma ferritin in elderly individuals with high neocortical amyloid-β load. Mol. Psychiatry 23, 1807–1812. doi: 10.1038/mp.2017.146
Griffith, O. W. (1982). Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 257, 13704–13712.
Grolez, G., Moreau, C., Sablonnière, B., Garçon, G., Devedjian, J. C., Meguig, S., et al. (2015). Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 6:74. doi: 10.1186/s12883-015-0331-3
Grossi, C., Francese, S., Casini, A., Rosi, M. C., Luccarini, I., Fiorentini, A., et al. (2009). Clioquinol decreases amyloid-β burden and reduces working memory impairment in a transgenic mouse model of alzheimer’s disease. J. Alzheimer’s Dis. 17, 423–440. doi: 10.3233/JAD-2009-1063
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Guo, C., Zhang, Y. X., Wang, T., Zhong, M. L., Yang, Z. H., Hao, L. J., et al. (2015). Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 7:104. doi: 10.3389/fnagi.2015.00104
Guo, X., Lin, H., Liu, J., and Yao, P. (2019). Quercetin Protects Hepatocyte from Ferroptosis by Depressing Mitochondria-reticulum Interaction Through PERK Downregulation in Alcoholic Liver (P06-056-19). Curr. Dev. Nutrit. 2019:19. doi: 10.1093/cdn/nzz031.p06-056-19
Gutbier, S., Kyriakou, S., Schildknecht, S., Ückert, A. K., Brüll, M., Lewis, F., et al. (2020). Design and evaluation of bi-functional iron chelators for protection of dopaminergic neurons from toxicants. Arch. Toxicol. 94, 3105–3123. doi: 10.1007/s00204-020-02826-y
Habib, E., Linher-Melville, K., Lin, H. X., and Singh, G. (2015). Expression of xCT and activity of system xc- are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 5, 33–42. doi: 10.1016/j.redox.2015.03.003
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Han, C., Liu, Y., Dai, R., Ismail, N., Su, W., and Li, B. (2020). Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 11:239. doi: 10.3389/fphar.2020.00239
Heneka, M. T., McManus, R. M., and Latz, E. (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621. doi: 10.1038/s41583-018-0055-7
Henstridge, C. M., Hyman, B. T., and Spires-Jones, T. L. (2019). Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 20, 94–108. doi: 10.1038/s41583-018-0113-1
Horowitz, M. P., and Greenamyre, J. T. (2010). Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimer’s Dis. 20(Suppl. 2), S551–S568. doi: 10.3233/JAD-2010-100354
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi: 10.1080/15548627.2016.1187366
Huang, W. J., Zhang, X., and Chen, W. W. (2016). Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 4, 519–522. doi: 10.3892/br.2016.630
James, S. A., Churches, Q. I., De Jonge, M. D., Birchall, I. E., Streltsov, V., McColl, G., et al. (2017). Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem. Neurosci. 8, 629–637. doi: 10.1021/acschemneuro.6b00362
Jelinek, A., Heyder, L., Daude, M., Plessner, M., Krippner, S., Grosse, R., et al. (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 117, 45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
Jenkins, N. L., James, S. A., Salim, A., Sumardy, F., Speed, T. P., Conrad, M., et al. (2020). Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging caenorhabditis elegans. ELife 9:e56580. doi: 10.7554/eLife.56580
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi: 10.1038/nature14344
Jiang, T., Cheng, H., Su, J., Wang, X., Wang, Q., Chu, J., et al. (2020). Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. Vitro 2020:104715. doi: 10.1016/j.tiv.2019.104715
Joo, E., Yoon, S., Chung, H., Sharma, N., Trong, B., Sung, N., et al. (2020). Glutathione Peroxidase - 1 Knockout Facilitates Memory Impairment Induced by β - Amyloid (1 – 42) in Mice via Inhibition of PKC βII - Mediated ERK Signaling; Application with Glutathione Peroxidase - 1 Gene - Encoded Adenovirus Vector. Neurochem. Res. 2020:0123456789. doi: 10.1007/s11064-020-03147-3
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P. F., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Kanninen, K., Heikkinen, R., Malm, T., Rolova, T., Kuhmonen, S., Leinonen, H., et al. (2009). Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 106, 16505–16510. doi: 10.1073/pnas.0908397106
Karch, C. M., Ezerskiy, L. A., Bertelsen, S., Goate, A. M., Albert, M. S., Albin, R. L., et al. (2016). Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 loci. PLoS One 11:e0148717. doi: 10.1371/journal.pone.0148717
Kaufmann, M. R., Barth, S., Konietzko, U., Wu, B., Egger, S., Kunze, R., et al. (2013). Dysregulation of hypoxia-inducible factor by presenilin/γ-secretase loss-of-function mutations. J. Neurosci. 33, 1915–1926. doi: 10.1523/JNEUROSCI.3402-12.2013
Kerr, F., Sofola-Adesakin, O., Ivanov, D. K., Gatliff, J., Gomez Perez-Nievas, B., Bertrand, H. C., et al. (2017). Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 13:e1006593. doi: 10.1371/journal.pgen.1006593
Kim, H. V., Kim, H. Y., Ehrlich, H. Y., Choi, S. Y., Kim, D. J., and Kim, Y. S. (2013). Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 20, 7–12. doi: 10.3109/13506129.2012.751367
Kim, S., Kim, Y., Kim, S. E., and An, J. (2021). Ferroptosis-Related Genes in Neurodevelopment and Central Nervous System. Biology 10:35. doi: 10.3390/biology10010035
Krabbendam, I. E., Honrath, B., Dilberger, B., Iannetti, E. F., Branicky, R. S., Meyer, T., et al. (2020). SK channel-mediated metabolic escape to glycolysis inhibits ferroptosis and supports stress resistance in C. elegans. Cell Death Dis. 11:263. doi: 10.1038/s41419-020-2458-4
Kuang, F., Liu, J., Tang, D., and Kang, R. (2020). Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020:1–10. doi: 10.3389/fcell.2020.586578
Lachaier, E., Louandre, C., Godin, C., Saidak, Z., Baert, M., Diouf, M., et al. (2014). Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 34, 6417–6422.
Langkammer, C., Ropele, S., Pirpamer, L., Fazekas, F., and Schmidt, R. (2014). MRI for iron mapping in Alzheimer’s disease. Neurodegenerat. Dis. 13, 189–191. doi: 10.1159/000353756
Lannfelt, L., Blennow, K., Zetterberg, H., Batsman, S., Ames, D., Harrison, J., et al. (2008). Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786. doi: 10.1016/S1474-4422(08)70167-4
Larric, J. W., Larric, J. W., and Mendelsoh, A. R. (2020). Contribution of Ferroptosis to Aging and Frailty. Rejuvenat. Res. 23, 434–438. doi: 10.1089/rej.2020.2390
Lee, J. H., and Lee, M. S. (2019). Brain iron accumulation in atypical parkinsonian syndromes: In vivo MRI evidences for distinctive patterns. Front. Neurol. 10:74. doi: 10.3389/fneur.2019.00074
Li, J., Cao, F., Yin, H. I, Huang, Z. J., Lin, Z. T., Mao, N., et al. (2020). Ferroptosis: past, present and future. Cell Death Dis. 11:2. doi: 10.1038/s41419-020-2298-2
Li, Q., and Sun, M. (2017). The role of autophagy in Alzheimer’s disease. J. Syst. Integrat. Neurosci. 3, 1–6. doi: 10.15761/jsin.1000172
Liang, C., Zhang, X., Yang, M., and Dong, X. (2019). Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 31:e1904197. doi: 10.1002/adma.201904197
Lipton, S. A., Rezaie, T., Nutter, A., Lopez, K. M., Parker, J., Kosaka, K., et al. (2016). Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 7:389. doi: 10.1038/cddis.2016.389
Lovell, M. A., Xie, C., and Markesbery, W. R. (2001). Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging 22, 187–194. doi: 10.1016/S0197-4580(00)00235-9
Maher, P., van Leyen, K., Dey, P. N., Honrath, B., Dolga, A., and Methner, A. (2018). The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 70, 47–55. doi: 10.1016/j.ceca.2017.05.007
Majerníková, N., Jia, J., and Andrea, Y. (2020). CuATSM PET to diagnose age - related diseases: a systematic literature review. Clin. Translat. Imaging 8, 449–460. doi: 10.1007/s40336-020-00394-w
Marder, K. (2005). Vitamin E and donepezil for the treatment of mild cognitive impairment. Curr. Neurol. Neurosci. Rep. 5, 337–338. doi: 10.1007/s11910-005-0056-6
Marmolejo-Garza, A., and Dolga, A. M. (2021). PEG out through the pores with the help of ESCRTIII. Cell Calcium 97:102422. doi: 10.1016/j.ceca.2021.102422
Masaldan, S., Belaidi, A. A., Ayton, S., and Bush, A. I. (2019). Cellular senescence and iron dyshomeostasis in alzheimer’s disease. Pharmaceuticals 12:93. doi: 10.3390/ph12020093
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. doi: 10.1038/s41586-019-1195-2
McCaddon, A., and Davies, G. (2005). Co-administration of N-acetylcysteine, vitamin B12 and folate in cognitively impaired hyperhomocysteinaemic patients. Int. J. Geriatr. Psychiatry 20, 998–1000. doi: 10.1002/gps.1376
McLachlan, D. R. C., Kruck, T. P. A., Kalow, W., Andrews, D. F., Dalton, A. J., Bell, M. Y., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-B
McLachlan, D. R., Smith, W. L., and Kruck, T. P. (1993). Desferrioxamine and alzheimer’s disease: Video home behavior assessment of clinical course and measures of brain aluminum. Therapeut. Drug Monitor. 15, 602–607. doi: 10.1097/00007691-199312000-00027
Montine, T. J., Markesbery, W. R., Morrow, J. D., and Roberts, L. J. (1998). Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer’s disease. Ann. Neurol. 44, 410–413. doi: 10.1002/ana.410440322
Moon, Y., Han, S. H., and Moon, W. J. (2016). Patterns of Brain Iron Accumulation in Vascular Dementia and Alzheimer’s Dementia Using Quantitative Susceptibility Mapping Imaging. J. Alzheimer’s Dis. 51, 737–745. doi: 10.3233/JAD-151037
Nassireslami, E., Nikbin, P., Amini, E., Payandemehr, B., Shaerzadeh, F., Khodagholi, F., et al. (2016). How sodium arsenite improve amyloid β-induced memory deficit? Physiol. Behav. 163, 97–106. doi: 10.1016/j.physbeh.2016.04.046
Neitemeier, S., Jelinek, A., Laino, V., Hoffmann, L., Eisenbach, I., Eying, R., et al. (2017). BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570. doi: 10.1016/j.redox.2017.03.007
Nuñez, M. T., and Chana-Cuevas, P. (2018). New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 11:109. doi: 10.3390/ph11040109
Obulesu, M., and Lakshmi, M. J. (2014). Apoptosis in Alzheimer’s Disease: An Understanding of the Physiology, Pathology and Therapeutic Avenues. Neurochem. Res. 39, 2301–2312. doi: 10.1007/s11064-014-1454-4
Peters, D. G., Pollack, A. N., Cheng, K. C., Sun, D., Saido, T., Haaf, M. P., et al. (2018). Dietary lipophilic iron alters amyloidogenesis and microglial morphology in Alzheimer’s disease knock-in APP mice. Metallomics 10, 426–443. doi: 10.1039/c8mt00004b
Picca, A., Mankowski, R. T., Kamenov, G., Anton, S. D., Manini, T. M., Buford, T. W., et al. (2019). Advanced Age Is Associated with Iron Dyshomeostasis and Mitochondrial DNA Damage in Human Skeletal Muscle. Cells 8:1525. doi: 10.3390/cells8121525
Plascencia-Villa, G., Ponce, A., Collingwood, J. F., Josefina Arellano-Jiménez, M., Zhu, X., Rogers, J. T., et al. (2016). High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci. Rep. 6, 1–12. doi: 10.1038/srep24873
Praticò, D., and Sung, S. (2004). Lipid Peroxidation and Oxidative imbalance: Early functional events in Alzheimer’s disease. J. Alzheimer’s Dis. 6, 171–175. doi: 10.3233/JAD-2004-6209
Praticò, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. Y. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of alzheimer amyloidosis. J. Neurosci. 21, 4183–4187. doi: 10.1523/jneurosci.21-12-04183.2001
Remington, R., Chan, A., Paskavitz, J., and Shea, T. B. (2009). Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage alzheimer’s disease: A placebo-controlled pilot study. Am. J. Alzheimer’s Dis. Dement. 24, 27–33. doi: 10.1177/1533317508325094
Ritchie, C. W., Bush, A. I., Mackinnon, A., Macfarlane, S., Mastwyk, M., MacGregor, L., et al. (2003). Metal-Protein Attenuation with Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease: A Pilot Phase 2 Clinical Trial. Arch. Neurol. 60, 1685–1691. doi: 10.1001/archneur.60.12.1685
Sato, H., Nomura, S., Maebara, K., Sato, K., Tamba, M., and Bannai, S. (2004). Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 325, 109–116. doi: 10.1016/j.bbrc.2004.10.009
Sato, M., Kusumi, R., Hamashima, S., Kobayashi, S., Sasaki, S., Komiyama, Y., et al. (2018). The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 8:968. doi: 10.1038/s41598-018-19213-4
Seibt, T. M., Proneth, B., and Conrad, M. (2019). Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 133, 144–152. doi: 10.1016/j.freeradbiomed.2018.09.014
Seiler, A., Schneider, M., Förster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metabol. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Shah, R., Shchepinov, M. S., and Pratt, D. A. (2018). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Central Sci. 4, 387–396. doi: 10.1021/acscentsci.7b00589
Sokoloff, L. (1999). Energetics of functional activation in neural tissues. Neurochem. Res. 24, 321–329. doi: 10.1023/A:1022534709672
Song, X., and Long, D. (2020). Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 14:1–15. doi: 10.3389/fnins.2020.00267
Sripetchwandee, J., Wongjaikam, S., Krintratun, W., Chattipakorn, N., and Chattipakorn, S. C. (2016). A combination of an iron chelator with an antioxidant effectively diminishes the dendritic loss, tau-hyperphosphorylation, amyloids-β accumulation and brain mitochondrial dynamic disruption in rats with chronic iron-overload. Neuroscience 332, 191–202. doi: 10.1016/j.neuroscience.2016.07.003
Ståhl, P. L., Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. F., Magnusson, J., et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. doi: 10.1126/science.aaf2403
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Strozyk, D., Launer, L. J., Adlard, P. A., Cherny, R. A., Tsatsanis, A., Volitakis, I., et al. (2009). Zinc and copper modulate Alzheimer Aβ levels in human cerebrospinal fluid. Neurobiol. Aging 30, 1069–1077. doi: 10.1016/j.neurobiolaging.2007.10.012
Sultana, R., Perluigi, M., and Butterfield, D. A. (2013). Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169. doi: 10.1016/j.freeradbiomed.2012.09.027
Taghizadeh, M., Tamtaji, O. R., Dadgostar, E., Daneshvar Kakhaki, R., Bahmani, F., Abolhassani, J., et al. (2017). The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 108, 183–189. doi: 10.1016/j.neuint.2017.03.014
Tanaka, H., Homma, H., Fujita, K., Kondo, K., Yamada, S., Jin, X., et al. (2020). YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun. 11:507. doi: 10.1038/s41467-020-14353-6
Trombetta-Lima, M., Sabogal-Guáqueta, A. M., and Dolga, A. M. (2021). Mitochondrial dysfunction in neurodegenerative diseases: A focus on iPSC-derived neuronal models. Cell Calcium 94:102362. doi: 10.1016/j.ceca.2021.102362
Wan, Y. W., Al-Ouran, R., Mangleburg, C. G., Perumal, T. M., Lee, T. V., Allison, K., et al. (2020). Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep. 32:107908. doi: 10.1016/j.celrep.2020.107908
Wang, C. Y., Wang, Z. Y., Xie, J. W., Wang, T., Wang, X., Xu, Y., et al. (2016). Dl-3-n-butylphthalide-induced upregulation of antioxidant defense is involved in the enhancement of cross talk between CREB and Nrf2 in an Alzheimer’s disease mouse model. Neurobiol. Aging 38, 32–46. doi: 10.1016/j.neurobiolaging.2015.10.024
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
Wang, L., Liu, Y., Du, T., Yang, H., Lei, L., Guo, M., et al. (2020). ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Different. 27, 662–675. doi: 10.1038/s41418-019-0380-z
Wang, Y., Liu, Z., Lin, T. M., Chanana, S., and Xiong, M. P. (2018). Nanogel-DFO conjugates as a model to investigate pharmacokinetics, biodistribution, and iron chelation in vivo. Int. J. Pharmaceut. 538, 79–86. doi: 10.1016/j.ijpharm.2018.01.004
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/S1474-4422(14)70117-6
Weiland, A., Wang, Y., Wu, W., Lan, X., Han, X., Li, Q., et al. (2019). Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 56, 4880–4893. doi: 10.1007/s12035-018-1403-3
Xu, J., Jia, Z., Knutson, M. D., and Leeuwenburgh, C. (2012). Impaired iron status in aging research. Int. J. Mol. Sci. 13, 2368–2386. doi: 10.3390/ijms13022368
Yamamoto, A., Shin, R. W., Hasegawa, K., Naiki, H., Sato, H., Yoshimasu, F., et al. (2002). Iron (III) induces aggregation of hyperphosphorylated τ and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2002.01061.x
Yan, N., and Zhang, J. J. (2020). Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 13:1443. doi: 10.3389/fnins.2019.01443
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U S A. 113, E4966–E4975. doi: 10.1073/pnas.1603244113
Yiannopoulou, K. G., Anastasiou, A. I., Zachariou, V., and Pelidou, S. H. (2019). Reasons for failed trials of disease-modifying treatments for alzheimer disease and their contribution in recent research. Biomedicines 7:97. doi: 10.3390/biomedicines7040097
Yoo, M. H., Gu, X., Xu, X. M., Kim, J. Y., Carlson, B. A., Patterson, A. D., et al. (2010). Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in alzheimer’s disease. Antioxid. Redox Signal. 12, 819–827. doi: 10.1089/ars.2009.2891
Yu, H., Yang, C., Jian, L., Guo, S., Chen, R., Li, K., et al. (2019). Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 42, 826–838. doi: 10.3892/or.2019.7189
Zhang, F., Tao, Y., Zhang, Z., Guo, X., An, P., Shen, Y., et al. (2012). Metalloreductase steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica 97, 1826–1835. doi: 10.3324/haematol.2012.063974
Zhang, Y. H., Wang, D. W., Xu, S. F., Zhang, S., Fan, Y. G., Yang, Y. Y., et al. (2018). α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. doi: 10.1016/j.redox.2017.11.001
Zhang, Z. H., Wu, Q. Y., Zheng, R., Chen, C., Chen, Y., Liu, Q., et al. (2017). Selenomethionine mitigates cognitive decline by targeting both tau hyperphosphorylation and autophagic clearance in an Alzheimer’s disease mouse model. J. Neurosci. 37, 2449–2462. doi: 10.1523/JNEUROSCI.3229-16.2017
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44, 3–15. doi: 10.3892/ijmm.2019.4188
Zhao, Y., and Zhao, B. (2013). Oxidative stress and the pathogenesis of alzheimer’s disease. Oxidat. Med. Cell. Longev. 14, 450–464. doi: 10.1155/2013/316523
Zhou, R. P., Chen, Y., Wei, X., Yu, B., Xiong, Z. G., Lu, C., et al. (2020). Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 10, 11976–11997. doi: 10.7150/thno.50663
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950. doi: 10.1152/physrev.00026.2013
Keywords: neurodegeneration, iron dysregulation, glutathione, lipid peroxidation, amyloid β
Citation: Majerníková N, den Dunnen WFA and Dolga AM (2021) The Potential of Ferroptosis-Targeting Therapies for Alzheimer’s Disease: From Mechanism to Transcriptomic Analysis. Front. Aging Neurosci. 13:745046. doi: 10.3389/fnagi.2021.745046
Received: 21 July 2021; Accepted: 18 November 2021;
Published: 20 December 2021.
Edited by:
Anne Eckert, University Psychiatric Clinic Basel, SwitzerlandReviewed by:
Mitsuru Shinohara, National Center for Geriatrics and Gerontology (NCGG), JapanJeff Liddell, The University of Melbourne, Australia
Copyright © 2021 Majerníková, den Dunnen and Dolga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amalia M. Dolga, YS5tLmRvbGdhQHJ1Zy5ubA==