 Jiajun Han
Jiajun Han Yaohua Fan
Yaohua Fan Peipei Wu1†
Peipei Wu1† Zifeng Huang
Zifeng Huang Xinrong Li
Xinrong Li Lijun Zhao
Lijun Zhao Meiling Zhu
Meiling Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 11 October 2021
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.743754
Parkinson’s disease dementia (PDD) is a common complication of Parkinson’s disease that seriously affects patients’ health and quality of life. At present, the process and pathological mechanisms of PDD remain controversial, which hinders the development of treatments. An increasing number of clinical studies have shown that alpha-synuclein (α-syn), tau, beta-amyloid (Aβ), and iron are closely associated with PDD severity. Thus, we inferred the vicious cycle that causes oxidative stress (OS), due to the synergistic effects of α-syn, tau, Aβ, and, iron, and which plays a pivotal role in the mechanism underlying PDD. First, iron-mediated reactive oxygen species (ROS) production can lead to neuronal protein accumulation (e.g., α-syn andAβ) and cytotoxicity. In addition, regulation of post-translational modification of α-syn by iron affects the aggregation or oligomer formation of α-syn. Iron promotes tau aggregation and neurofibrillary tangles (NFTs) formation. High levels of iron, α-syn, Aβ, tau, and NFTs can cause severe OS and neuroinflammation, which lead to cell death. Then, the increasing formation of α-syn, Aβ, and NFTs further increase iron levels, which promotes the spread of α-syn and Aβ in the central and peripheral nervous systems. Finally, iron-induced neurotoxicity promotes the activation of glycogen synthase kinase 3β (GSK3β) related pathways in the synaptic terminals, which in turn play an important role in the pathological synergistic effects of α-syn, tau and Aβ. Thus, as the central factor regulating this vicious cycle, GSK3β is a potential target for the prevention and treatment of PDD; this is worthy of future study.
Parkinson’s disease (PD) is a common neurodegenerative disease in middle-aged and older people. The main symptoms of PD include asymmetric resting tremor, bradykinesia, postural instability, and rigidity. Patients with PD often experience cognitive deficits in the course of the disease and may develop symptoms of dementia in the late stage of PD. At the initial diagnosis of PD, more than 20% of patients show cognitive impairment, and about 80% eventually develop Parkinson’s disease dementia (PDD; Aarsland et al., 2017; Goldman and Sieg, 2020). According to the Movement Disorder Society Task Force, the recommended criteria for PDD diagnosis include fulfilling the set of diagnostic criteria for PD, PD developed prior to the onset of dementia, PD associated with a decreased global cognitive efficiency, a cognitive deficiency that is severe enough to impair daily life, and impairment in more than one cognitive domain (Dubois et al., 2007). At present, the process and pathological mechanisms of PDD remain unclear. The primary pathological feature of PDD is the accumulation of misfolded alpha-synuclein (α-syn) aggregates that form intraneuronal Lewy bodies (LBs) that are predominantly localized to presynaptic terminals. Previous studies have reported that cortical, diffuse, or limbic LBs and Lewy neurites are closely associated with dementia in PD (Gomperts, 2016; Jellinger and Korczyn, 2018; Milán-Tomás et al., 2021), and cortical LBs and neuropathy in PDD were found to be more severe than those seen in PD. In addition to LBs, Alzheimer’s disease (AD)-type pathology is also closely associated with the pathological mechanism of PDD and is severe in more than 50% of patients with PDD (Irwin et al., 2012). A recent study has suggested that AD-type pathology, which is characterized by the superposition of beta-amyloid (Aβ) and hyperphosphorylated tau, plays a synergistic role in PDD (Irwin et al., 2012). Moreover, levels of tau, neurofibrillary tangles (NFTs), and Aβ plaques have been found to be positively correlated with cognitive impairment (Irwin et al., 2013). The combination of LBs and AD-type pathology is considered to have a strong pathological association with PDD (Irwin et al., 2013). Clinical studies have shown that PDD is associated with severe α-syn pathology in the hippocampus and entorhinal and occipitotemporal cortexes, and Aβ pathology and tau pathology in the striatum (Smith et al., 2019; Kouli et al., 2020; Howard et al., 2021). The combination of LBs, Aβ, and tau pathologies has a strong correlation with the pathology in patients with PDD (Compta et al., 2011). However, the underlying synergistic effects of α-syn, tau, and Aβ in PDD are still unclear.
Iron plays an important role in promoting nervous system development and maintaining neuronal function (Hsieh et al., 2020; Donker et al., 2021). Brain iron is essential for neurotransmission, myelination of neurons, DNA synthesis, gene expression, and mitochondrial respiration. Iron deficiency in the brain during childhood can lead to psychomotor disorders and delayed language and physical development (Thirupathi and Chang, 2019). In addition, the content of iron in the brain increases with age (Ashraf et al., 2018). Iron metabolism disorders have been associated with many neurodegenerative diseases, such as AD, PD, PDD, Friedreich’s ataxia, and multiple sclerosis (Costa-Mallen et al., 2017; Xuan et al., 2017; Chen et al., 2021; La Rosa et al., 2021; Tham et al., 2021). Several clinical studies have shown that iron accumulation in the substantia nigra (SN) and several basal ganglia structures is partly associated with cognitive decline in patients with PD and PDD (Rossi et al., 2014; Costa-Mallen et al., 2017; Genoud et al., 2017; Xuan et al., 2017; Gao et al., 2020). Excessive iron can induce oxidative stress by producing reactive oxygen species (ROS), especially hydroxyl free radicals (Ward et al., 2014; Thirupathi and Chang, 2019). Previous work has found that the specific insoluble amyloid plaques and NFTs in dementia contain high concentrations of iron, which are about three times more than those found in normal people (940 μM compared with 340 μM), and that the local accumulation of iron can lead to neuronal dysfunction (Roberts et al., 2012). Iron abnormalities are related to misfolding of Aβ produced by amyloid precursor protein (APP) and hyperphosphorylated tau (found in plaques and tangles; Derry et al., 2020). Abnormal iron metabolism and α-syn misfolding are prevalent in PD (Song and Xie, 2018), and there is also abnormal accumulation of Aβ and tau in PDD (Irwin et al., 2012). Therefore, exploring the interactions between Aβ, tau, α-syn, and iron could further our understanding of the pathological mechanism underlying PDD.
Aggregated α-syn, a landmark pathological characteristic of PDD, is encoded by the SNCA gene. The physiological role of α-syn is implicated in various cellular processes, such as maintaining the synaptic function, affecting neurotransmitter storage, and neurotransmitter release within the synapse (Nemani et al., 2010; Duce et al., 2017). SNCA gene duplication, triplication, or mutation aggravates the accumulation of misfolded α-syn, which can form insoluble fibrils through oligomerization (Hijaz and Volpicelli-Daley, 2020). In addition, α-syn aggregation has been found in cells treated with iron, which suggests that iron also plays a key role in inducing α-syn aggregation (Figure 1). Furthermore, iron-induced α-syn aggregation is dose-dependent, and the accumulation of iron in the SN during aging or LB disease may increase the aggregation rate of α-syn (Ostrerova-Golts et al., 2000).
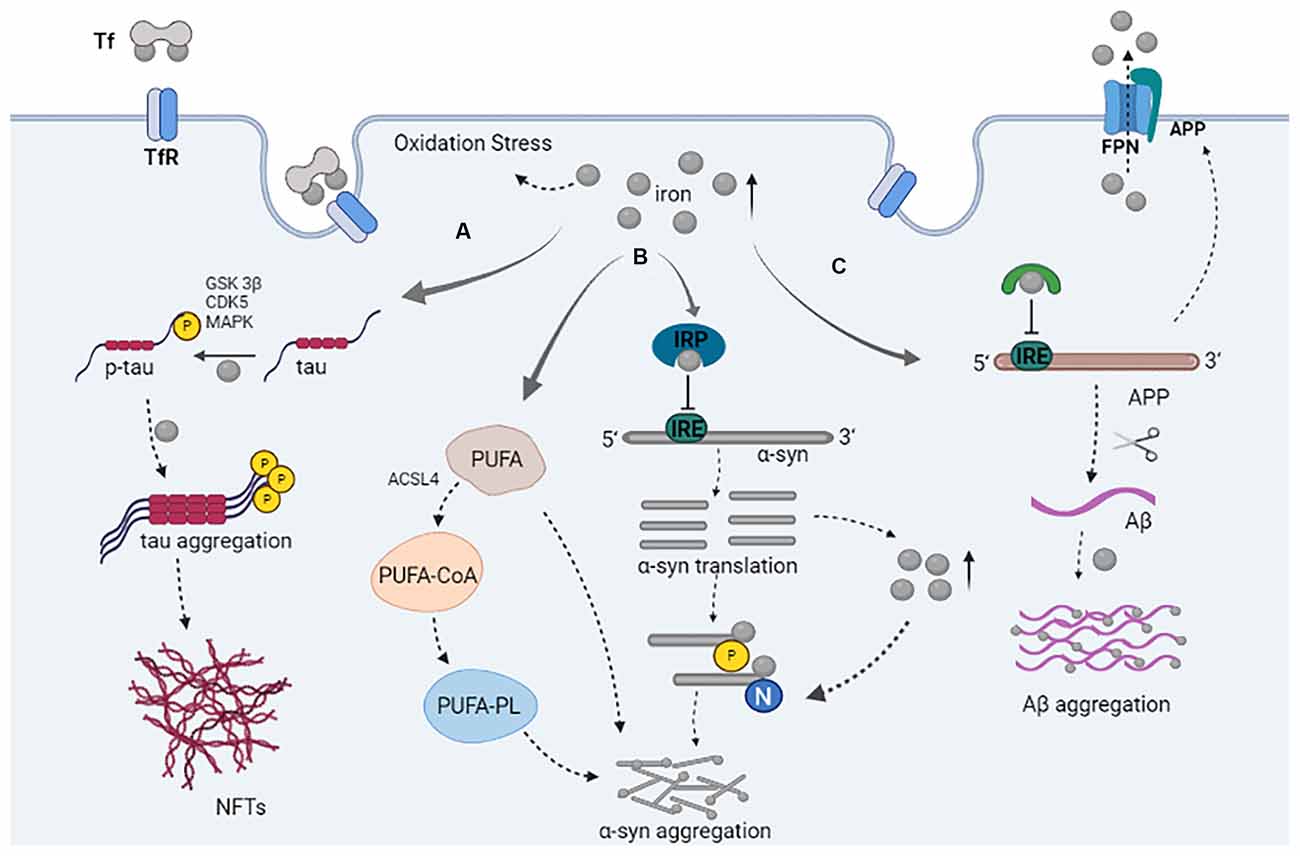
Figure 1. The role of iron in the pathology of tau, α-syn, and Aβ. (A) Iron and tau kinase (GSK3β, CDKs, MAPK) play a role in the phosphorylation of tau; iron promotes tau aggregation and NFTs formation. (B) The binding of IRP to iron inhibited its binding to IRE, resulting in increased expression of α-syn; α-syn can promote an increase in iron levels; iron participates in the post-translational modification of α-syn and promotes the aggregation of α-syn. Iron can promote the increase of PUFA; PUFA are conjugated to CoA by ACSL4 allowing PUFA-CoA to be incorporated into the phospholipids (PL); free or phospholipid-bound PUFAs promote the aggregation of α-syn. (C) The binding of IRP to iron inhibits its binding to IRE, which results in increased APP expression. The synergistic effect of APP and FPN causes iron outflow; Aβ produced from APP processing; iron promotes Aβ aggregation.
Using a focused monolayer fluorescence technique, previous work has found that Fe3+ significantly increased the aggregation of α-syn and induced the formation of large oligomers at low micromolar concentrations in vitro, due to direct interaction with amyloid fibrils (Kostka et al., 2008; Levin et al., 2011). A previous study demonstrated that even physiological concentrations of iron can form α-syn oligomer species at nanomolar protein concentrations (Nübling et al., 2014). In vitro experiments could already show that iron can bind to α-syn and is able to foster α-syn aggregation (Davies et al., 2011; Perfeito et al., 2014; Jansen van Rensburg et al., 2021), a recent study has found that iron enrichment occurring in particular areas may also affect α-syn spreading pathology (Dauer Née Joppe et al., 2021). Li et al. (2020) found that the accumulation of iron ions in the brains of patients with PD accelerates the prion-like proliferation of α-syn fibrils in a vicious cycle. Iron ions first promote the cellular internalization of the fibrils, and the internalized fibrils further aggregate to the natural α-syn seeds, and, finally, the mature fibrils are released into the extracellular space to induce further propagation (Li et al., 2020). The accumulation of both α-syn and iron in the SN establish the local conditions required for α-syn-mediated hydrogen peroxide formation and its conversion to hydroxyl radicals via the Fenton reaction, and this leads to the death of the vulnerable nigral neurons (Turnbull et al., 2001; Vasquez et al., 2017).
A recent study has found that iron can also promote the aggregation and transmission of α-syn by inhibiting autophagosome-lysosomal fusion (Xiao et al., 2018). The 5′ untranslated region of α-syn transcripts contains an iron response element (IRE; Zhou and Tan, 2017). Iron regulatory proteins (IRP) can bind to the IRE under iron deficiency conditions, thereby inhibiting protein synthesis, whereby increased iron levels cause IRP to bind to iron, which promotes the dissociation of IRP from the α-syn mRNA, leading to facilitation of the translation of the α-syn mRNA (Anderson et al., 2012; Zhou and Tan, 2017). Qu et al. (2019) demonstrated that rosmarinic acid might protect against iron-induced cellular neurotoxicity via the ability of the IRP/IRE system to inhibit the expression of α-syn and induce the degradation of α-syn. Increased iron levels lead to increased translation of the α-syn protein, and the over-expression of α-syn can promote the formation of aggregates by increasing the proportion of partially folded α-syn (Uversky, 2007). This further indicates that iron is a key contributor to α-syn aggregation.
In addition to directly promoting the aggregation of α-syn, iron can also indirectly promote the aggregation of α-syn through lipid metabolism. Given that iron is a cofactor to the family of lipoxygenase enzymes, an elevated labile iron pool catalyzes the formation of phospholipid hydroperoxides in the plasma membrane, which determines cellular sensitivity to ferroptosis (Mahoney-Sánchez et al., 2021). Free cytosolic polyunsaturated fatty acids (PUFA) are conjugated to coenzyme-A (CoA) by acyl-CoA synthetase long-chain family member 4 (ACSL4), which allows PUFA-CoA to be incorporated into the phospholipids in the plasma membrane (Doll et al., 2017). When exposed to free or phospholipid-bound PUFAs, α-syn will increase the propensity to aggregate and oligomerize (De Franceschi et al., 2009; Sánchez Campos et al., 2018).
There are many post-translational modifications of α-syn, including phosphorylation, acetylation, ubiquitination, oxidation, nitration, and truncation (Figure 1). Multiple post-translational modifications can change the structure of α-syn and regulate its physiological function. This may be related to aggregation or oligomer formation of α-syn (Barrett and Timothy Greenamyre, 2015). The following contents will focus on the relationship between nitration and phosphorylation of α-syn and iron, and iron-induced oxidative stress.
Phosphorylation is the most common and important protein modification associated with the pathology of α-syn. Several phosphorylation sites have been identified on α-syn, either at serine, tyrosine, or threonine, such as S129, S87, Y39, and Y125. Approximately 90% of α-syn deposited in LBs is phosphorylated at S129, while under normal physiological conditions, only 4% or less of α-syn was phosphorylated (Fujiwara et al., 2002; Anderson et al., 2006); this suggests that phosphorylation can affect α-syn aggregation. Phosphorylation typically occurs at S129. The involved kinases include polo-like kinase (PLKs), G protein-coupled receptor kinase (GRKs), casein kinase (CKs), and leucine-rich repeat kinase 2 (LRRK2; Qing et al., 2009; Oueslati, 2016; Rafael Guimarães et al., 2021). Iron can induce the up-regulation of α-syn and pS129 α-syn (S129 phosphorylation) through polo-like kinase 2 (PLK2) and casein kinase 2 (CK2; Wang et al., 2019). Fe3+ may promote pS129 α-syn oligomer formation, because Fe3+ not only prevents pS129 α-syn from combining with the membrane but also facilitates protein interactions via the additional negative charges introduced to the protein structure through phosphorylation (Nübling et al., 2014). It is also possible that iron plays a role in promoting the phosphorylation of S129. Electron microscope results have revealed that iron can change the morphology of α-syn fibrils and accelerate the aggregation of wild-type and mutant α-syn (Li et al., 2020). In addition to S129, there are other phosphorylation sites on the tyrosine residue of α-syn, such as Y125 and Y39 (Brahmachari et al., 2016; Zheng et al., 2019). The modified Y125 can increase the phosphorylation level of S129 through casein kinase 1 (CK1; Kosten et al., 2014), and a reduction of the Y125 modification can also reduce the phosphorylation level of S129, thereby reducing the abnormal aggregation of α-syn caused by phosphorylation of S129 (Fayyad et al., 2020). Antioxidants can completely block the iron-induced kinase up-regulation and α-syn phosphorylation, which indicates that there is a close association between these processes and oxidative stress (Wang et al., 2019).
α-Syn nitration modification is widespread in AD, PD, LB dementia, multiple system atrophy, and other neurodegenerative diseases (Chavarría and Souza, 2013). One study found that there were more nitrated α-syn-positive dopaminergic cell bodies in aged monkeys than in adult monkeys, and 20% of the α-syn positive dopaminergic neurons were co-immunoreactive with nitrated α-syn, which suggests that nitrated α-syn accumulated in SN neuronal bodies of aging primates (McCormack et al., 2012). Nitration often occurs at the four tyrosine residues of α-syn (Tyr-39, Tyr-125, Tyr-133, and Tyr-136; Burai et al., 2015). Nitration enhancement of α-syn Tyr-39 contributes to methamphetamine-induced dopaminergic toxicity (Qiao et al., 2019). Nitration of Tyr-125 contributes to the formation of α-syn dimers (Takahashi et al., 2002). Nitration of Tyr-39 and the formation of di-tyrosine involving this residue play a key role in the oligomerization of α-syn. The Tyr-39 mutation will lead to multiple di-tyrosine cross-linking and interfere with the formation of higher-order aggregates. Tyr-39 and Tyr-125 mutations also increase dimer and monomer levels (Burai et al., 2015), which leads to an increase in α-syn neurotoxicity.
Nitrated α-syn can induce adaptive immune responses and may intensify microglia activation (Reynolds et al., 2009). Activated microglia can induce oxidative stress in dopaminergic neurons, which leads to neuronal nitration and dopaminergic neuron death (Shavali et al., 2006; Yu et al., 2010). Recent studies have found that myeloperoxidase (MPO) is expressed in neurons of patients with PD and a humanized MPO-A53T mouse model of PD. MPO is co-located with neurons nitrated with α-syn, carbamylated with α-syn, and hypochlorous acid-modified with α-syn, and is associated with α-syn aggregation and motor damage. Furthermore, MPO inhibitors have beneficial effects in the treatment of neurodegenerative diseases such as PDD (Maki et al., 2019). Bibenzyl compound 20C, a Chinese medicinal extract, can inhibit the adaptive immune response mediated by nitration of α-syn (Wang et al., 2021), which may inhibit oxidative stress and neuronal death. Iron-induced oxidative stress is the most common harmful reaction, and excessive iron content is associated with an increase in nitration stress, which leads to increases in the tyrosine nitration level. There is extensive nitrative damage in neurodegeneration with brain iron accumulation type 1, intracellular nitrative insult will cause the accumulation of α-syn (Paxinou et al., 2001). In animals and patients with iron overload, excessive iron can promote the formation of nitrogen species (such as the hydroxyl radical, superoxide anion, and nitric oxide) that disrupt the redox balance of cells and lead to chronic nitrosative stress (Zhang et al., 2012). Therefore, iron may play an important regulatory role in α-syn nitration.
Many studies have investigated the role of iron in α-syn aggregation and the regulation of post-translational modification of α-syn. However, α-syn has also been found to play a role in iron homeostasis. α-Syn is considered to be an iron-binding protein that can bind to Fe, Fe3+, or Fe2+ (Peng et al., 2010; Davies et al., 2011). Fe3+ can also be reduced to Fe2+ by its iron reductase activity to increase the content of Fe2+ in cells (Brown, 2013; Ortega et al., 2016), and oxidative stress and neuronal degeneration can be induced by the Fenton reaction. Post-translational modification of α-syn can regulate iron transport, while N-terminal acetylation of α-syn can promote dynamic protein-mediated transferrin receptor (TfR) endosome transport and iron internalization. However, phosphorylated α-syn can reduce iron input through TfR endocytosis (Duce et al., 2017). In neurons exposed to excessive iron, the overexpression of α-syn leads to an increase in intracellular iron levels and the redistribution of iron from the cytoplasm to the perinuclear region in α-syn-rich inclusions (Ortega et al., 2016). This is further evidence of the binding between iron and α-syn.
Early study has found that when cells are exposed to dopamine alone, α-syn does not cause aggregation, but iron-treated cells induce aggregation. The overexpression of α-syn makes cells sensitive to iron-mediated toxicity. It is possible that the aggregation of α-syn increases the cellular iron content, resulting in increased oxidative toxicity (Ostrerova-Golts et al., 2000). Some data have also indicated that increased intracellular concentrations of α-syn, due to, for example, gene dose effects, autophagy injury, or proteasome dysfunction, may contribute to intracellular iron accumulation in neurons exposed to excessive iron, which in turn promotes further oligomerization and α-syn aggregation. In turn, α-syn oligomerization leads to an increase in intracellular iron levels (Ortega et al., 2016), which suggests that α-syn oligomerization and iron accumulation may be a mutual promotion process. Bi et al. (2020) found that α-syn could induce p38 mitogen-activated protein kinase (MAPK) activation to phosphorylate parkin at Ser131, inactivate parkin’s E3 ubiquitin ligase activity, further inhibit divalent metal transporter 1 (DMT1) ubiquitination, and increase the expression of DMT1 protein in α-syn-overexpressed SH-SY5Y cells and mutant human A53T α-syn transgenic mice. This process aggravates oxidative stress, interferes with iron homeostasis, and participates in the deposition of iron in the SN (Bi et al., 2020). Guo et al. (2021) have reported iron deposition in the SN–striatum system of monkeys after intranasal injection of exogenous α-synuclein preformed fibrils (α-syn PFFs). One potential mechanism underlying this effect is that α-syn-PFFs treatment triggers the accumulation of iron in microglia to initiate a neuroinflammatory reaction, which in turn induces a cascade reaction between iron deposition and microglia activation, producing hydrogen peroxide and hydroxyl radicals, and releasing pro-inflammatory factors; this, in turn, leads to neuroinflammatory α-syn accumulation and dopaminergic neuron degeneration (Guo et al., 2021).
Shekoohi et al. (2021) found that reduced expression of α-syn in retinal pigment epithelial (RPE) cells significantly reduced TfR1 and ferritin levels and reduced iron deposition compared to control RPE cells. However, overexpression of α-syn significantly increased the levels of these two proteins (Shekoohi et al., 2021). α-Syn can also damage ferritinophagy, which results in the accumulation of iron-rich ferritin in RPE cells (Baksi and Singh, 2017). In addition, other studies have shown that inhibition of α-syn expression by siRNA can prevent the effects of iron on mitochondria and cell death (Ganguly et al., 2020). Extracellular α-syn can mediate endoplasmic reticulum stress to regulate cellular iron-related proteins and further affect iron metabolism (Mi et al., 2021). Therefore, a high labile iron environment and α-syn aggregation could generate toxic dopamine reactive quinones and reactive species that induce the overproduction of ROS and mitochondrial dysfunction via mitochondrial respiratory complex I inhibition (Duce et al., 2017; Nonnekes et al., 2018; Zhou et al., 2019). Mitochondrial dysfunction results in increased ROS production, which may also contribute to lipid peroxidation in the plasma membrane.
NFTs formed by abnormal phosphorylation of tau protein are widely found in PDD and their levels are positively correlated with the severity of dementia. Tau dysfunction and misfolding may lead to iron deposition, which relies on iron-induced oxidative damage to produce toxicity, leading to neuron loss or ferroptosis (Dixon et al., 2012). Conversely, iron can induce tau phosphorylation and tau aggregation, promote the progression of dementia, increase Aβ aggregation and APP protein expression, promote the formation of NFTs, and finally lead to neuronal death (Xie et al., 2012; Spotorno et al., 2020; Figure 1). Fe3+ is associated with NFTs in dementia and progressive supranuclear palsy, and induces the accumulation of hyperphosphorylated tau. Reduction of Fe3+ to Fe2+ can reverse the aggregation of tau and dissolved tau species from the brain of patients with dementia (Yamamoto et al., 2002). It has been found that excessive intake of iron can induce hippocampal iron disorder in APP and presenilin 1 (PS1) double-transgenic mice and adult rats, and promote the overexpression of Aβ and phosphorylated tau (Chen et al., 2019). Metallic lithium can lead to iron accumulation by regulating tau expression. After lithium treatment, tau levels in mouse brains decreased, while iron in the SN and cortex increased. In neuron culture, lithium reduced the outflow of iron by reducing tau proteins. Tau protein and amyloid precursor gene-knockout mice were protected from lithium-induced iron elevation and neurotoxicity (Wood, 2016; Lei et al., 2017).
Tau kinase is also involved in the interaction between iron and tau. Bautista et al. (2016) found that tau hyperphosphorylation is one of the key steps in the formation of NFTs, and can be regulated by iron through the abnormal activation of tau kinases, such as GSK3β, cyclin-dependent kinase 5 (CDK5), and MAPK (Rao et al., 2020). Guo et al. (2013) demonstrated that high iron levels can induce tau phosphorylation at Thr205, Thr231, and Ser396 sites in the brains of APP/PS1 transgenic mice, while intranasal administration of deferoxamine (DFO) in APP/PS1 transgenic mice also reduced the activity of iron-induced CDK5 and GSK3β, which in turn inhibited tau phosphorylation. Therefore, iron and DFO antagonize and regulate tau phosphorylation in a CDK5- and GSK3β-dependent manner (Guo et al., 2013).
p-tau expression has been found to decrease after injection of an GSK3β inhibitor in MPTP mice (Hu et al., 2020). However, treatment with ebselen, a DMT1 inhibitor, has been found to inhibit CDK5 and GSK3β activity in ferrous-treated SH-SY5Y cells, which eventually reduces hyperphosphorylation of tau (Xie et al., 2012). APP can also interact with iron transporters on the neuron surface to regulate iron output. Tau loss leads to the accumulation of immature APP in the endoplasmic reticulum and prevents APP from being transported to the neuron surface, which leads to the toxic accumulation of iron (Lei et al., 2012; Bassil, 2013). Hephaestin (a transmembrane iron oxidase) has been recently identified as an active agent in the complex of APP and iron transporter proteins (Dlouhy et al., 2019). Ceruloplasmin also has the property of iron oxidase and can interact with iron transporters (Honarmand Ebrahimi et al., 2013).
Iron chelators reduce tau phosphorylation, and iron overload impairs the brain mitochondrial balance and increases brain oxidative stress, which leads to the loss of apoptotic dendrites and ultimate increases in AD-like lesions. Iron chelators, such as deferiprone (DFP) and DFO, can protect the brain following iron overload by reducing the accumulation of brain iron, the destruction of brain mitochondrial kinetics, and the loss of dendrites (Sripetchwandee et al., 2016). Age-dependent brain atrophy, iron accumulation, and SN neuron loss in tau-knockout mice with cognitive deficits and Parkinson’s syndrome can be prevented by oral administration of iron chelators (Xie et al., 2012). In addition, Wan et al. (2019) found that insulin signals were involved in iron-induced abnormal phosphorylation of tau. In primary cultured neurons, Fe2+ causes tau phosphorylation and reduces tyrosine phosphorylation of insulin receptor β, insulin signaling substrate 1, and phosphoinositide 3- kinase p85α. Insulin treatment also reduces tau hyperphosphorylation in neurons cultured with excessive iron, which is indicative of the existence of an insulin-dependent pathway (Wan et al., 2019). Homeostasis between tau and insulin signals can be restored by activating the insulin receptor, or by targeting the downstream pathway with protein tyrosine phosphatase 1B inhibitors, GLP-1 receptor agonists, or intranasal insulin administration (Gratuze et al., 2018). Stable insulin signals may facilitate the clearance of pathological forms of tau, which could be considered in potential treatments for PDD.
Accumulated Aβ and iron clearly play a central role in the pathology of PDD. First, extensive research has demonstrated that Aβ can bind to iron (Plascencia-Villa et al., 2016). The high affinity of Aβ for binding to iron and its ability to reduce iron levels lead to the formation of hydrogen peroxide, which causes oxidative damage (Foster et al., 2020). The ROS generated by iron-induced Aβ accumulation is also toxic to neurons (Liu et al., 2011). Animal models have shown that elevated brain iron levels lead to oxidative stress, promote Aβ toxicity and tau protein dysfunction, enhance neuronal cell death, and lead to neural degradation and cognitive dysfunction (Lane et al., 2018). In a study with patients with mild cognitive impairment, Ayton et al. (2017) found that individuals with high iron levels and a high amyloid load showed greater cognitive decline than individuals with only one of the pathological conditions. van Bergen et al. (2016) demonstrated that the progression of cognitive dysfunction was caused by cerebral iron deposition and spatial co-localization of Aβ plaques. Evidence has also suggested that iron accumulation in neurons containing NFTs and neurites adjacent to senile plaques is associated with cognitive decline (Ding et al., 2009; van Bergen et al., 2018). Low cerebrospinal fluid Aβ42 is also a predictor of future cognitive impairment in patients with PD (Lim et al., 2019). Redox-active metal ions such as iron can accelerate Aβ aggregation and induce ROS production (Ayton et al., 2017; Cheignon et al., 2018). In particular, iron can catalyze ROS production under acidic conditions via Fenton chemistry (Mai et al., 2017). And diffusely distributed Aβ spots were observed throughout the whole hippocampus of Young-High Fe mice (Chen et al., 2021). In addition, the interaction of iron with Aβ may result in the disorder of the iron balance (Qin et al., 2021).
Iron has not only been shown to bind to Aβ, but also to regulate APP production. Iron levels affect APP translation through iron-responsive elements in APP transcript 5′-UTR (Cho et al., 2010; Figure 1). Iron overload conditions can increase the protein expression of APP in the brain (Sripetchwandee et al., 2016). APP also plays a role in maintaining neuronal iron homeostasis. Cell surface APP stabilizes the iron export protein, ferroportin (FPN), and allows cellular iron to flow out through the porin (McCarthy et al., 2014; Venkataramani et al., 2018). Choi et al. (2021) demonstrated that treadmill exercise promoted α-secretase-dependent APP processing through low iron-induced enhancement of furin activity to reduce cognitive decline and Aβ-induced neuronal cell death, accompanied by a decrease in the levels of lipid peroxidation products and an increase in the ability of antioxidant defense enzymes. However, APP modified after translation, such as phosphorylation and glycosylation, can change its transport to the cell surface, and then affect its binding to FPN, which leads to imbalanced iron homeostasis (Tsatsanis et al., 2019). In addition, the intracellular processing of APP may impair iron output by destabilizing cell surface iron transporters. By contrast, non-amyloid processing of APP on the cell surface promotes the stability of iron transporters and thus reduces iron levels in neurons (Tsatsanis et al., 2020). Therefore, changes in intracellular transport that are related to changes in APP processing may lead to neuronal iron level increases and oxidative stress in dementia pathology.
A recent study has suggested that a high-iron diet boosted Aβ expression compared to mice fed normal diets. The iron concentration and ferritin in the hippocampus of APP/PS1 mice fed with a high-iron diet were significantly increased, the activity of superoxide dismutase was significantly decreased, and Aβ1–42 protein expression was higher. In addition, iron deposition and Aβ plaques were gathered in the hippocampal DG region, accompanied by cognitive dysfunction (Chen et al., 2019). It could be seen that an iron overload diet induced the disorder of iron in the mouse hippocampus and Aβ overexpression. Lipocalin 2 is involved in many physiological processes such as inflammation, iron metabolism, and cell death and may contribute to the pathophysiology of neurodegenerative diseases such as AD and PD. Ferroptosis is also involved in the pathogenesis of PD (Tian et al., 2020; Li et al., 2021; Si et al., 2021). Iron chelating agents such as DFO and DFO can inhibit the production of proteins such as Aβ-induced lipocalin 2 in primary cultured astrocytes, which may contribute to the protection of nerves (Dekens et al., 2020). Overexpression of hepcidin in astrocytes significantly improved Aβ 25–35 induced cellular damage in the cerebral cortex and hippocampus. It is possible that iron FPN1 acts on brain microvascular endothelial cells, which in turn reduce Aβ25–35-induced oxidative stress and apoptosis, and ultimately protects cells from damage (Zhang et al., 2020).
The increase in iron content in the brain may be related to the increase of hyperphosphorylated tau, Aβ, and α-syn (Lu et al., 2017). Additionally, p-tau, Aβ, and α-syn work together to promote the pathogenesis of PDD (Figure 1 and Table 1). In one study, APP transgenic mice exhibited severe hyperphosphorylated tau-positive neurites and synaptic dystrophy near amyloid plaques (Radde et al., 2006). Li et al. (2015) injected Aβ oligomers into the hippocampus of young and old tau gene knockout mice and found that tau ablation prevented Aβ-induced cognitive impairment, hippocampal neuron loss, and iron accumulation. Therefore, tau protein may be a downstream mediator of Aβ toxicity (Li et al., 2015). In addition, phosphorylation of tau can occur at many sites, and the hyperphosphorylation of tau protein at Thr181 has been associated with an increased expression of Aβ and APP proteins, as well as the progression of neurodegeneration (Spotorno et al., 2020). Tau-hyperphosphorylation of the Thr181 amino acid may be a specific biomarker of brain pathological states, such as AD (Saman et al., 2012). α-Syn and Aβ have also been found to have a certain synergistic effect. Bassil et al. (2020) injected α-syn PFFs into mice with abundant Aβ plaques. Aβ deposition significantly accelerated the pathogenesis of α-syn and spread throughout the whole brain. Notably, hyperphosphorylation of tau, partial neuronal loss, motor, and cognitive impairment were also noted in α-syn PFFs-injected 5xFAD mice. This may indicate the presence of a “pre-feedback” mechanism, whereby Aβ plaques enhance the dissemination and diffusion of endogenous α-syn following injection of PFFs (Bassil et al., 2020). Swirski et al. (2014) found a correlation between levels of S129-phosphorylated α-syn and Aβ. When SH-SY5Y cells transfected with the α-syn gene were exposed to aggregated Aβ42, the proportion of phosphorylated α-syn at S129 was significantly increased, which indicated an association between α-syn and Aβ (Swirski et al., 2014).

Table 1. Changes of α-syn, tau, Aβ, and iron in PDD.
In the synaptic terminals of adult and aged rats exposed to free iron, GSK3β phosphorylation has been reported to be enhanced and GSK3β-related signaling pathways activated (Uranga et al., 2009), while DFO treatment can attenuate the activation of GSK-3β (Zhang et al., 2015). Activated GSK3β signals may also play a role in the pathological synergy of tau, Aβ, and α-syn. First, the phosphorylation of tau by GSK-3β enhances metal ion-induced oligomer formation and co-aggregation with α-syn (Nübling et al., 2014). Overexpression of human P301L mutant tau promotes the phosphorylation and dimerization of endogenous α-syn by activating GSK-3β in rTg4510 mice (Takaichi et al., 2020). Tau not only promotes the synthesis and phosphorylation of α-syn, but also increases the insoluble form of α-syn and enhances its toxicity. Furthermore, α-syn promotes the hyperphosphorylation of GSK3β-dependent tau by reducing the phosphorylation of Ser9 and increasing the phosphorylation of Tyr216. Inhibition of GSK3β reduces the phosphorylation and accumulation of α-syn in PD pathology (Gąssowska et al., 2014). Yang et al. (2018) found that Aβ exposure induced GSK3β activity, while GSK3 proteins (including GSK3α and GSK3β) promoted Aβ production and stimulated apoptotic signals via Aβ. Both α-syn and Aβ overexpression were associated with increased levels of active GSK3β, which led an increased hyperphosphorylation of tau. Finally, inflammation associated with GSK3β activation was found to lead to apoptosis (Yang et al., 2018). Therefore, the synergistic effects among iron, tau, α-syn, Aβ, and GSK-3β are involved in the pathophysiology of PDD (Figure 2).
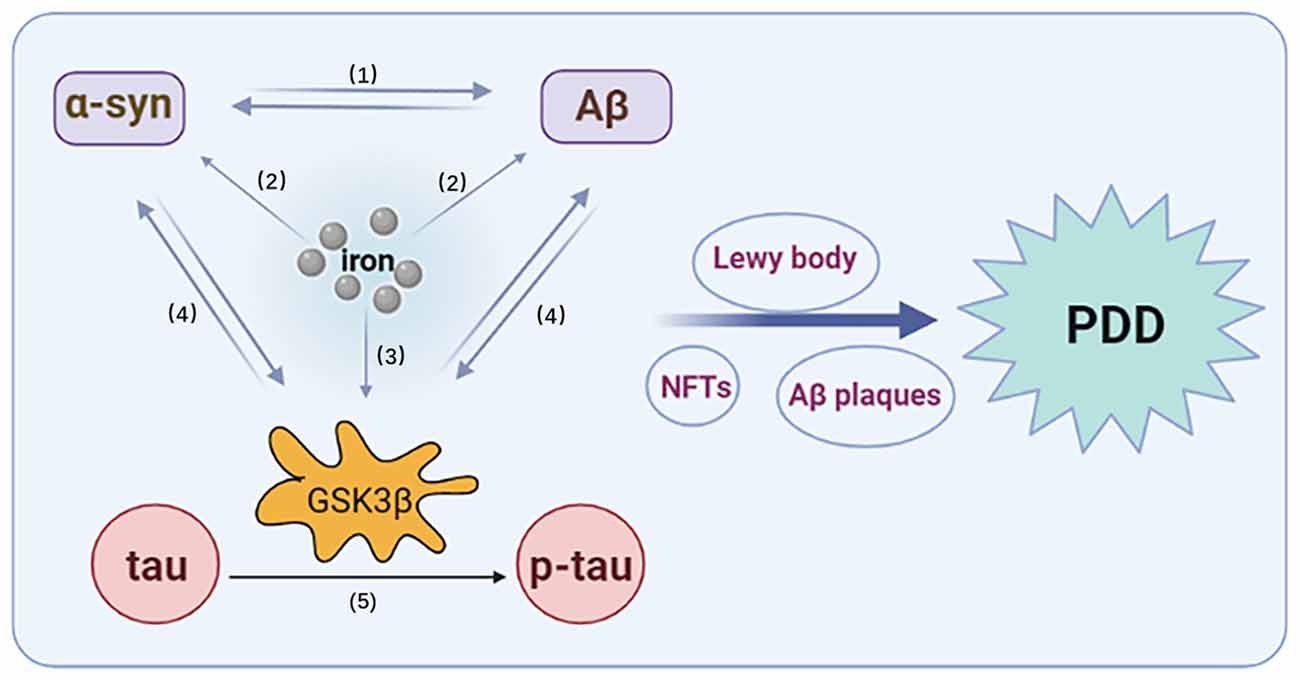
Figure 2. Interaction of tau, α-syn, Aβ, iron, and GSK3β. (1) The synergistic effect of α-syn and Aβ. (2) Iron promotes the overexpression of α-syn and Aβ. (3) Iron promotes the activation of GSK3β. (4) α-Syn and Aβ can promote an increase in GSK3β, which can, in turn, promote the expression of α-syn and Aβ. (5) GSK3β promotes the phosphorylation of tau.
Colom-Cadena et al. (2013) found that the density of MAPT·H1 haplotypes and amyloid deposits was related to the number of Aβ and α-syn deposits in the brains of patients with dementia with LBs. Aβ deposition has also been pathologically associated with synuclein PD and PDD cases. In addition, misfold-mediated heterotypic binding induced by the binding of α-syn and tau K18 has been reported to synergistically promote the formation of Aβ (Bhasne et al., 2018). Lu et al. (2020) found that two variants of monomers α-syn and tau (tau23 and K19) synergistically promoted amyloid fibril formation, causing them to co-aggregate in vitro. Nuclear magnetic resonance spectroscopy experiments showed that α-syn directly interacted with tau23 and K19 by virtue of its highly negatively charged carbon terminal. The C-terminal deletion effectively eliminated its association with tau23 and K19, and its synergistic effect of promoting fibrillation (Lu et al., 2020).
PDD is a common occurrence in PD and has a significant impact on the health and daily life of patients. Although most evidence has indicated an α-syn pathology in the development of PDD, there is a notable heterogeneity in symptomatology and timing of dementia between PDD cases. An increasing number of studies have shown that LBs, AD-type pathology, and iron dysmetabolism appear to contribute to the emergence of PDD (Smith et al., 2019; Kouli et al., 2020; Howard et al., 2021). It is possible that α-syn is transmitted from degenerating neurons into neighboring neurons and non-neuronal cells; indeed, α-syn aggregates have been found in the SN, the nucleus basalis of Meynert, cerebral cortex, locus coeruleus, cranial nerve motor nuclei, and locus coeruleus (Braak et al., 2006; Brundin and Melki, 2017). Astrocytes, which are the most abundant glial cells in the central nervous system, are responsible for the extracellular homeostasis of ions and neurotransmitters that are able to internalize extracellular α-syn (Lee et al., 2010, 2014). Moreover, Aβ has been found to induce an inflammatory response and iron accumulation in astrocytes, both in vivo and in vitro (Urrutia et al., 2013). Iron chelators inhibit the Aβ-induced production of lipocalin 2 in cultured astrocytes. Thus, astrocytes are a key participant in PDD pathogenesis and treatment (Zhu et al., 2019; Cui et al., 2020).
While some studies have suggested that the interaction of iron, α-syn, tau, and Aβ is a key pathological mechanism underlying PDD, the specific mechanism is not yet clear (Compta et al., 2011). According to a literature search, we found that iron can interact with α-syn, tau, and Aβ, and promote their aggregation. Iron indirectly affects disease-related proteins through oxidative stress or regulation of post-translational modifications. Conversely, α-syn, tau, and Aβ also affect iron metabolism and iron accumulation (Figure 1). Furthermore, α-syn and Aβ can promote an increase of GSK3β, which can, in turn, promote the expression of α-syn and Aβ, as well as the phosphorylation of tau (Figure 2). Thus, GSK3β plays a significant regulatory role in the synergistic effects of α-syn, tau, Aβ, and, iron, and further verification of its function in astrocytes is required. In addition, GSK3β may represent a potential biomarker of incipient cognitive decline in patients with PDD. Drug development for the prevention of GSK3β expression could also be considered in future research. For instance, immune-based therapies that target GSK3β could be an effective disease-modifying therapy in PDD. However, timing the initiation of this therapy is a critical and uncertain issue.
Recent studies on PD therapies have examined the role of the gut-brain axis, given the spread of LBs in the central nervous system and the peripheral nervous system, and particularly the enteric nervous system (Scheperjans et al., 2015; Surmeier et al., 2017; Diwakarla et al., 2021; Borah et al., 2021). Many studies have found that a high-iron diet can lead to iron disorders and overexpression of Aβ and phosphorylated tau, and iron chelating agents such as DFP and DFO can play a preventive and therapeutic role, as well as a neuroprotective role (Guo et al., 2013; Sripetchwandee et al., 2016; Rao et al., 2020). Moreover, Zederone could reduce the accumulation of amyloid plaques and improve cognitive capacity via the brain-gut axis. The above results suggest that targeted regulation of iron metabolism disorders via the brain-gut axis is a candidate treatment strategy to prevent PDD.
MZ and YF designed the article. JH, PW, ZH, XL, and LZ collected the article’s materials. JH and PW wrote the manuscript. MZ, YF, and YJ revised and polished the article. All authors contributed to the article and approved the submitted version.
This work was supported by the National Administration of Traditional Chinese Medicine, China (Grant No. GZY-FJS-2019-001 and GZY-FJS-2019-002), the Bao’an TCM Development Foundation (Grant No. 2020KJCX-KTYJ-130 and 2020KJCX-KTYJ-131), and the Natural Science Foundation of Shenzhen (Grant No. JCYJ20190807112405520).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ACSL4, Acyl-CoA synthetase long-chain family member 4; AD, Alzheimer’s disease; APP, Amyloid precursor protein; Aβ, Beta-amyloid; CDK5, Cyclin-dependent kinase 5; CK, Casein kinase; CK1, Casein kinase 1; CK2, Casein kinase 2; CoA, Coenzyme-A; DFO, Deferoxamine; DFP, Deferiprone; DMT1, Divalent metal transporter 1; FPN, Ferroportin; GRKs, G protein-coupled receptor kinase; GSK3β, Glycogen synthase kinase 3β; IRE, Iron response element; IRP, Iron regulatory proteins; LBD, Lewy body disease; LBs, Lewy bodies; LRRK2, Leucine-rich repeat kinase 2; MAPK, Mitogen-activated protein kinase; MPO, Myeloperoxidase; NFTs, Neurofibrillary tangles; PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; PLK2, Polo-like kinase 2; PLKs, Polo-like kinase; PS1, Presenilin 1; PUFA, Polyunsaturated fatty acids; ROS, Reactive oxygen species; RPE, Retinal pigment epithelial; SN, Substantia nigra; TfR, Transferrin receptor; α-syn, Alpha-synuclein; α-syn PFFs, α-synuclein preformed fibrils.
Aarsland, D., Creese, B., Politis, M., Chaudhuri, K. R., Ffytche, D. H., Weintraub, D., et al. (2017). Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 13, 217–231. doi: 10.1038/nrneurol.2017.27
Anderson, C. P., Shen, M., Eisenstein, R. S., and Leibold, E. A. (2012). Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483. doi: 10.1016/j.bbamcr.2012.05.010
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Ashraf, A., Clark, M., and So, P. W. (2018). The Aging of Iron Man. Front. Aging Neurosci. 10:65. doi: 10.3389/fnagi.2018.00065
Ayton, S., Fazlollahi, A., Bourgeat, P., Raniga, P., Ng, A., Lim, Y. Y., et al. (2017). Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain 140, 2112–2119. doi: 10.1093/brain/awx137
Ba, F., Zhou, Y., Zhou, J., and Chen, X. (2019). Repetitive transcranial magnetic stimulation protects mice against 6-OHDA-induced Parkinson’s disease symptoms by regulating brain amyloid β(1–42) level. Mol. Cell Biochem. 458, 71–78. doi: 10.1007/s11010-019-03531-w
Baksi, S., and Singh, N. (2017). α-Synuclein impairs ferritinophagy in the retinal pigment epithelium: implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 7:12843. doi: 10.1038/s41598-017-12862-x
Barrett, P. J., and Timothy Greenamyre, J. (2015). Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 1628, 247–253. doi: 10.1016/j.brainres.2015.06.002
Bassil, F., Brown, H. J., Pattabhiraman, S., Iwasyk, J. E., Maghames, C. M., Meymand, E. S., et al. (2020). Amyloid-Beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of lewy body disorders with Aβ pathology. Neuron 105, 260–275.e266. doi: 10.1016/j.neuron.2019.10.010
Bautista, E., Vergara, P., and Segovia, J. (2016). Iron-induced oxidative stress activates AKT and ERK1/2 and decreases Dyrk1B and PRMT1 in neuroblastoma SH-SY5Y cells. J. Trace Elem. Med. Biol. 34, 62–69. doi: 10.1016/j.jtemb.2015.11.005
Bhasne, K., Sebastian, S., Jain, N., and Mukhopadhyay, S. (2018). Synergistic amyloid switch triggered by early heterotypic oligomerization of intrinsically disordered α-synuclein and tau. J. Mol. Biol. 430, 2508–2520. doi: 10.1016/j.jmb.2018.04.020
Bi, M., Du, X., Jiao, Q., Liu, Z., and Jiang, H. (2020). α-synuclein regulates iron homeostasis via preventing Parkin-mediated DMT1 ubiquitylation in Parkinson’s disease models. ACS Chem. Neurosci. 11, 1682–1691. doi: 10.1021/acschemneuro.0c00196
Borah, S., Sarkar, P., and Sharma, H. K. (2021). Zederone improves the fecal microbial profile in dementia induced rat model: a first report. CNS Neurol. Disord. Drug Targets doi: 10.2174/1871527320666210827114227 [Online ahead of Print].
Braak, H., Bohl, J. R., Müller, C. M., Rüb, U., de Vos, R. A., and Del Tredici, K. (2006). Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 21, 2042–2051. doi: 10.1002/mds.21065
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to α-synuclein-induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Brown, D. R. (2013). α-synuclein as a ferrireductase. Biochem. Soc. Trans. 41, 1513–1517. doi: 10.1042/BST20130130
Brundin, P., and Melki, R. (2017). Prying into the prion hypothesis for Parkinson’s disease. J. Neurosci. 37, 9808–9818. doi: 10.1523/JNEUROSCI.1788-16.2017
Burai, R., Ait-Bouziad, N., Chiki, A., and Lashuel, H. A. (2015). Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and msutagenesis. J. Am. Chem. Soc. 137, 5041–5052. doi: 10.1021/ja5131726
Chavarría, C., and Souza, J. M. (2013). Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 533, 25–32. doi: 10.1016/j.abb.2013.02.009
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, M., Xu, E., Zeng, C., Zhu, W., Zheng, J., and Chen, H. (2021). High dietary iron has a greater impact on brain iron homeostasis and cognitive function in old compared with young C57BL/6J male mice. J. Nutr. 151, 2835–2842. doi: 10.1093/jn/nxab189
Chen, M., Zheng, J., Liu, G., Zeng, C., Xu, E., Zhu, W., et al. (2019). High dietary iron disrupts iron homeostasis and induces Amyloid-β and phospho-β expression in the hippocampus of adult wild-type and APP/PS1 transgenic mice. J. Nutr. 149, 2247–2254. doi: 10.1093/jn/nxz168
Cho, H. H., Cahill, C. M., Vanderburg, C. R., Scherzer, C. R., Wang, B., Huang, X., et al. (2010). Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 285, 31217–31232. doi: 10.1074/jbc.M110.149161
Choi, D. H., Choi, I. A., Lee, C. S., Yun, J. H., and Lee, J. (2019). The role of NOX4 in Parkinson’s disease with dementia. Int. J. Mol. Sci. 20:696. doi: 10.3390/ijms20030696
Choi, D. H., Kwon, K. C., Hwang, D. J., Koo, J. H., Um, H. S., Song, H. S., et al. (2021). Treadmill exercise alleviates brain iron dyshomeostasis accelerating neuronal Amyloid-β production, neuronal cell death and cognitive impairment in transgenic mice model of Alzheimer’s disease. Mol. Neurobiol. 58, 3208–3223. doi: 10.1007/s12035-021-02335-8
Clinton, L. K., Blurton-Jones, M., Myczek, K., Trojanowski, J. Q., and LaFerla, F. M. (2010). Synergistic interactions between Abeta, tau and alpha-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 30, 7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010
Colom-Cadena, M., Gelpi, E., Charif, S., Belbin, O., Blesa, R., Martí, M. J., et al. (2013). Confluence of α-synuclein, tau and β-amyloid pathologies in dementia with lewy bodies. J. Neuropathol. Exp. Neurol. 72, 1203–1212. doi: 10.1097/NEN.0000000000000018
Compta, Y., Parkkinen, L., O’Sullivan, S. S., Vandrovcova, J., Holton, J. L., Collins, C., et al. (2011). Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134, 1493–1505. doi: 10.1093/brain/awr031
Costa-Mallen, P., Gatenby, C., Friend, S., Maravilla, K. R., Hu, S. C., Cain, K. C., et al. (2017). Brain iron concentrations in regions of interest and relation with serum iron levels in Parkinson disease. J. Neurol. Sci. 378, 38–44. doi: 10.1016/j.jns.2017.04.035
Cui, J., Guo, X., Li, Q., Song, N., and Xie, J. (2020). Hepcidin-to-Ferritin ratio is decreased in astrocytes with extracellular alpha-synuclein and iron exposure. Front. Cell Neurosci. 14:47. doi: 10.3389/fncel.2020.00047
Dauer Née Joppe, K., Tatenhorst, L., Caldi Gomes, L., Zhang, S., Parvaz, M., Carboni, E., et al. (2021). Brain iron enrichment attenuates α-synuclein spreading after injection of preformed fibrils. J. Neurochem. doi: 10.1111/jnc.15461 [Online ahead of print].
Davies, P., Moualla, D., and Brown, D. R. (2011). Alpha-synuclein is a cellular ferrireductase. PLoS One 6:e15814. doi: 10.1371/journal.pone.0015814
De Franceschi, G., Frare, E., Bubacco, L., Mammi, S., Fontana, A., and de Laureto, P. P. (2009). Molecular insights into the interaction between alpha-synuclein and docosahexaenoic acid. J. Mol. Biol. 394, 94–107. doi: 10.1016/j.jmb.2009.09.008
Dekens, D. W., De Deyn, P. P., Sap, F., Eisel, U. L. M., and Naudé, P. J. W. (2020). Iron chelators inhibit amyloid-β-induced production of lipocalin 2 in cultured astrocytes. Neurochem. Int. 132:104607. doi: 10.1016/j.neuint.2019.104607
Derry, P. J., Hegde, M. L., Jackson, G. R., Kayed, R., Tour, J. M., Tsai, A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184:101716. doi: 10.1016/j.pneurobio.2019.101716
Ding, B., Chen, K. M., Ling, H. W., Sun, F., Li, X., Wan, T., et al. (2009). Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J. Magn. Reson. Imaging 29, 793–798. doi: 10.1002/jmri.21730
Diwakarla, S., McQuade, R. M., Constable, R., Artaiz, O., Lei, E., Barnham, K. J., et al. (2021). ATH434 reverses colorectal dysfunction in the A53T mouse model of Parkinson’s disease. J. Parkinsons Dis. doi: 10.3233/JPD-212731 [Online ahead of print].
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dlouhy, A. C., Bailey, D. K., Steimle, B. L., Parker, H. V., and Kosman, D. J. (2019). Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. J. Biol. Chem. 294, 4202–4214. doi: 10.1074/jbc.RA118.005142
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi: 10.1038/nchembio.2239
Donker, A. E., van der Staaij, H., and Swinkels, D. W. (2021). The critical roles of iron during the journey from fetus to adolescent: developmental aspects of iron homeostasis. Blood Rev. 100866. doi: 10.1016/j.blre.2021.100866 [Online ahead of print].
Dubois, B., Burn, D., Goetz, C., Aarsland, D., Brown, R. G., Broe, G. A., et al. (2007). Diagnostic procedures for Parkinson’s disease dementia: recommendations from the movement disorder society task force. Mov. Disord. 22, 2314–2324. doi: 10.1002/mds.21844
Duce, J. A., Wong, B. X., Durham, H., Devedjian, J. C., Smith, D. P., and Devos, D. (2017). Post translational changes to α-synuclein control iron and dopamine trafficking; a concept for neuron vulnerability in Parkinson’s disease. Mol. Neurodegener. 12:45. doi: 10.1186/s13024-017-0186-8
Fatima, A., Rahul, and Siddique, Y. H. (2019). Role of tangeritin against cognitive impairments in transgenic drosophila model of Parkinson’s disease. Neurosci. Lett. 705, 112–117. doi: 10.1016/j.neulet.2019.04.047
Fayyad, M., Erskine, D., Majbour, N. K., Vaikath, N. N., Ghanem, S. S., Sudhakaran, I. P., et al. (2020). Investigating the presence of doubly phosphorylated α-synuclein at tyrosine 125 and serine 129 in idiopathic lewy body diseases. Brain Pathol. 30, 831–843. doi: 10.1111/bpa.12845
Foster, C. M., Kennedy, K. M., Daugherty, A. M., and Rodrigue, K. M. (2020). Contribution of iron and Aβ to age differences in entorhinal and hippocampal subfield volume. Neurology 95, e2586–e2594. doi: 10.1212/WNL.0000000000010868
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Ganguly, U., Banerjee, A., Chakrabarti, S. S., Kaur, U., Sen, O., Cappai, R., et al. (2020). Interaction of α-synuclein and Parkin in iron toxicity on SH-SY5Y cells: implications in the pathogenesis of Parkinson’s disease. Biochem. J. 477, 1109–1122. doi: 10.1042/BCJ20190676
Gao, Y., Martínez-Cerdeño, V., Hogan, K. J., McLean, C. A., and Lockhart, P. J. (2020). Clinical and neuropathological features associated with loss of RAB39B. Mov. Disord. 35, 687–693. doi: 10.1002/mds.27951
Gąssowska, M., Czapski, G.A., Pająk, B., Cieślik, M., Lenkiewicz, A. M., and Adamczyk, A. (2014). Extracellular α-synuclein leads to microtubule destabilization via GSK-3β-dependent Tau phosphorylation in PC12 cells. PLoS One 9:e94259. doi: 10.1371/journal.pone.0094259
Genoud, S., Roberts, B. R., Gunn, A. P., Halliday, G. M., Lewis, S. J. G., Ball, H. J., et al. (2017). Subcellular compartmentalisation of copper, iron, manganese and zinc in the Parkinson’s disease brain. Metallomics 9, 1447–1455. doi: 10.1039/c7mt00244k
Gerson, J. E., Farmer, K. M., Henson, N., Castillo-Carranza, D. L., Carretero Murillo, M., Sengupta, U., et al. (2018). Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 13:13. doi: 10.1186/s13024-018-0245-9
Goldman, J. G., and Sieg, E. (2020). Cognitive impairment and dementia in Parkinson disease. Clin. Geriatr. Med. 36, 365–377. doi: 10.1016/j.cger.2020.01.001
Gomperts, S. N. (2016). Lewy body dementias: dementia with lewy bodies and Parkinson disease dementia. Continuum (Minneap Minn). 22, 435–463. doi: 10.1212/CON.0000000000000309
Gratuze, M., Joly-Amado, A., Vieau, D., Buée, L., and Blum, D. (2018). Mutual relationship between tau and central insulin signalling: consequences for AD and tauopathies? Neuroendocrinology 107, 181–195. doi: 10.1159/000487641
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Guo, J. J., Yue, F., Song, D. Y., Bousset, L., Liang, X., Tang, J., et al. (2021). Intranasal administration of α-synuclein preformed fibrils triggers microglial iron deposition in the substantia nigra of macaca fascicularis. Cell Death Dis. 12:81. doi: 10.1038/s41419-020-03369-x
Hall, H., Jewett, M., Landeck, N., Nilsson, N., Schagerlöf, U., Leanza, G., et al. (2013). Characterization of cognitive deficits in rats overexpressing human alpha-synuclein in the ventral tegmental area and medial septum using recombinant adeno-associated viral vectors. PLoS One 8:e64844. doi: 10.1371/journal.pone.0064844
Hijaz, B. A., and Volpicelli-Daley, L. A. (2020). Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 15:19. doi: 10.1186/s13024-020-00368-6
Honarmand Ebrahimi, K., Dienemann, C., Hoefgen, S., Than, M. E., Hagedoorn, P. L., and Hagen, W. R. (2013). The amyloid precursor protein (APP) does not have a ferroxidase site in its E2 domain. PLoS One 8:e72177. doi: 10.1371/journal.pone.0072177
Howard, E., Irwin, D. J., Rascovsky, K., Nevler, N., Shellikeri, S., Tropea, T. F., et al. (2021). Cognitive rofile and markers of Alzheimer disease-type pathology in patients with lewy body dementias. Neurology 96, e1855–e1864. doi: 10.1212/WNL.0000000000011699
Hsieh, H. Y., Chen, Y. C., Hsu, M. H., Yu, H. R., Su, C. H., Tain, Y. L., et al. (2020). Maternal iron deficiency programs offspring cognition and its relationship with gastrointestinal microbiota and metabolites. Int. J. Environ. Res. Public Health 17:6070. doi: 10.3390/ijerph17176070
Hu, S., Hu, M., Liu, J., Zhang, B., Zhang, Z., Zhou, F. H., et al. (2020). Phosphorylation of tau and α-synuclein induced neurodegeneration in MPTP mouse model of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 16, 651–663. doi: 10.2147/NDT.S235562
Irwin, D. J., Lee, V. M., and Trojanowski, J. Q. (2013). Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636. doi: 10.1038/nrn3549
Irwin, D. J., White, M. T., Toledo, J. B., Xie, S. X., Robinson, J. L., Van Deerlin, V., et al. (2012). Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 72, 587–598. doi: 10.1002/ana.23659
Jansen van Rensburg, Z., Abrahams, S., Bardien, S., and Kenyon, C. (2021). Toxic feedback loop involving iron, reactive oxygen species, α-synuclein and neuromelanin in Parkinson’s disease and intervention with turmeric. Mol. Neurobiol. doi: 10.1007/s12035-021-02516-5 [Online ahead of print].
Jellinger, K. A., and Korczyn, A. D. (2018). Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease. BMC Med. 16:34. doi: 10.1186/s12916-018-1016-8
Kosten, J., Binolfi, A., Stuiver, M., Verzini, S., Theillet, F. X., Bekei, B., et al. (2014). Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 5, 1203–1208. doi: 10.1021/cn5002254
Kostka, M., Högen, T., Danzer, K. M., Levin, J., Habeck, M., Wirth, A., et al. (2008). Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J. Biol. Chem. 283, 10992–11003. doi: 10.1074/jbc.M709634200
Kouli, A., Camacho, M., Allinson, K., and Williams-Gray, C. H. (2020). Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol. Commun. 8:211. doi: 10.1186/s40478-020-01083-5
La Rosa, P., Petrillo, S., Turchi, R., Berardinelli, F., Schirinzi, T., Vasco, G., et al. (2021). The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 38:101791. doi: 10.1016/j.redox.2020.101791
Lane, D. J. R., Ayton, S., and Bush, A. I. (2018). Iron and Alzheimer’s disease: an update on emerging mechanisms. J. Alzheimers Dis. 64, S379–s395. doi: 10.3233/JAD-179944
Lee, H. J., Bae, E. J., and Lee, S. J. (2014). Extracellular α—synuclein-a novel and crucial factor in lewy body diseases. Nat. Rev. Neurol. 10, 92–98. doi: 10.1038/nrneurol.2013.275
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.M109.081125
Lei, P., Ayton, S., Appukuttan, A. T., Moon, S., Duce, J. A., Volitakis, I., et al. (2017). Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry 22, 396–406. doi: 10.1038/mp.2016.96
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Levin, J., Högen, T., Hillmer, A. S., Bader, B., Schmidt, F., Kamp, F., et al. (2011). Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Parkinsons Dis. 1, 205–216. doi: 10.3233/JPD-2011-11040
Li, D. T. H., Hui, E. S., Chan, Q., Yao, N., Chua, S. E., McAlonan, G. M., et al. (2018). Quantitative susceptibility mapping as an indicator of subcortical and limbic iron abnormality in Parkinson’s disease with dementia. Neuroimage Clin. 20, 365–373. doi: 10.1016/j.nicl.2018.07.028
Li, X., Lei, P., Tuo, Q., Ayton, S., Li, Q. X., Moon, S., et al. (2015). Enduring elevations of hippocampal amyloid precursor protein and iron are features of β-amyloid toxicity and are mediated by tau. Neurotherapeutics 12, 862–873. doi: 10.1007/s13311-015-0378-2
Li, X., Si, W., Li, Z., Tian, Y., Liu, X., Ye, S., et al. (2021). miR-335 promotes ferroptosis by targeting ferritin heavy chain 1 in in vivo and in vitro models of Parkinson’s disease. Int. J. Mol. Med. 47:61. doi: 10.3892/ijmm.2021.4894
Li, Y., Yang, C., Wang, S., Yang, D., Zhang, Y., Xu, L., et al. (2020). Copper and iron ions accelerate the prion-like propagation of α-synuclein: a vicious cycle in Parkinson’s disease. Int. J. Biol. Macromol. 163, 562–573. doi: 10.1016/j.ijbiomac.2020.06.274
Lim, E. W., Aarsland, D., Ffytche, D., Taddei, R. N., van Wamelen, D. J., Wan, Y. M., et al. (2019). Amyloid-β and Parkinson’s disease. J. Neurol. 266, 2605–2619. doi: 10.1007/s00415-018-9100-8
Liu, B., Moloney, A., Meehan, S., Morris, K., Thomas, S. E., Serpell, L. C., et al. (2011). Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 286, 4248–4256. doi: 10.1074/jbc.M110.158980
Lu, L. N., Qian, Z. M., Wu, K. C., Yung, W. H., and Ke, Y. (2017). Expression of iron transporters and pathological hallmarks of Parkinson’s and Alzheimer’s diseases in the brain of young, adult and aged rats. Mol. Neurobiol. 54, 5213–5224. doi: 10.1007/s12035-016-0067-0
Lu, J., Zhang, S., Ma, X., Jia, C., Liu, Z., Huang, C., et al. (2020). Structural basis of the interplay between α-synuclein and Tau in regulating pathological amyloid aggregation. J. Biol. Chem. 295, 7470–7480. doi: 10.1074/jbc.RA119.012284
Magen, I., Fleming, S. M., Zhu, C., Garcia, E. C., Cardiff, K. M., Dinh, D., et al. (2012). Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease. Eur. J. Neurosci. 35, 870–882. doi: 10.1111/j.1460-9568.2012.08012.x
Mahoney-Sánchez, L., Bouchaoui, H., Ayton, S., Devos, D., Duce, J. A., and Devedjian, J. C. (2021). Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog. Neurobiol. 196:101890. doi: 10.1016/j.pneurobio.2020.101890
Mai, T. T., Hamaï, A., Hienzsch, A., Cañeque, T., Müller, S., Wicinski, J., et al. (2017). Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 9, 1025–1033. doi: 10.1038/nchem.2778
Maki, R. A., Holzer, M., Motamedchaboki, K., Malle, E., Masliah, E., Marsche, G., et al. (2019). Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 141, 115–140. doi: 10.1016/j.freeradbiomed.2019.05.033
McCarthy, R. C., Park, Y. H., and Kosman, D. J. (2014). sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. 15, 809–815. doi: 10.15252/embr.201338064
McCormack, A. L., Mak, S. K., and Di Monte, D. A. (2012). Increased α-synuclein phosphorylation and nitration in the aging primate substantia nigra. Cell Death Dis. 3:e315. doi: 10.1038/cddis.2012.50
Mi, X., Li, Q., Wen, X., Xie, J., Wang, Y., and Song, N. (2021). Extracellular α-synuclein modulates iron metabolism related proteins via endoplasmic reticulum stress in MES23.5 dopaminergic cells. Neurochem. Res. 46, 1502–1513. doi: 10.1007/s11064-021-03292-3
Milán-Tomás, Á., Fernández-Matarrubia, M., and Rodríguez-Oroz, M. C. (2021). Lewy body dementias: a coin with two sides. Behav. Sci. (Basel) 11:94. doi: 10.3390/bs11070094
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., et al. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. doi: 10.1016/j.neuron.2009.12.023
Niu, Y., Guo, X., Chen, Y., Wang, C. E., Gao, J., Yang, W., et al. (2015). Early Parkinson’s disease symptoms in α-synuclein transgenic monkeys. Hum. Mol. Genet. 24, 2308–2317. doi: 10.1093/hmg/ddu748
Nonnekes, J., Post, B., Tetrud, J. W., Langston, J. W., and Bloem, B. R. (2018). MPTP-induced parkinsonism: an historical case series. Lancet Neurol. 17, 300–301. doi: 10.1016/S1474-4422(18)30072-3
Nübling, G. S., Levin, J., Bader, B., Lorenzl, S., Hillmer, A., Högen, T., et al. (2014). Modelling Ser129 phosphorylation inhibits membrane binding of pore-forming alpha-synuclein oligomers. PLoS One 9:e98906. doi: 10.1371/journal.pone.0098906
Ortega, R., Carmona, A., Roudeau, S., Perrin, L., Dučić, T., Carboni, E., et al. (2016). α-Synuclein over-expression induces increased iron accumulation and redistribution in iron-exposed neurons. Mol. Neurobiol. 53, 1925–1934. doi: 10.1007/s12035-015-9146-x
Ostrerova-Golts, N., Petrucelli, L., Hardy, J., Lee, J. M., Farer, M., and Wolozin, B. (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000
Oueslati, A. (2016). Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade. J. Parkinsons Dis. 6, 39–51. doi: 10.3233/JPD-160779
Paxinou, E., Chen, Q., Weisse, M., Giasson, B. I., Norris, E. H., Rueter, S. M., et al. (2001). Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 21, 8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001
Peng, Y., Wang, C., Xu, H. H., Liu, Y. N., and Zhou, F. (2010). Binding of alpha-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 104, 365–370. doi: 10.1016/j.jinorgbio.2009.11.005
Perfeito, R., Lázaro, D. F., Outeiro, T. F., and Rego, A. C. (2014). Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell Neurosci. 62, 51–59. doi: 10.1016/j.mcn.2014.08.002
Plascencia-Villa, G., Ponce, A., Collingwood, J. F., Arellano-Jiménez, M. J., Zhu, X., Rogers, J. T., et al. (2016). High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci. Rep. 6:24873. doi: 10.1038/srep24873
Qiao, H. H., Zhu, L. N., Wang, Y., Hui, J. L., Xie, W. B., Liu, C., et al. (2019). Implications of alpha-synuclein nitration at tyrosine 39 in methamphetamine-induced neurotoxicity in vitro and in vivo. Neural Regen. Res. 14, 319–327. doi: 10.4103/1673-5374.244795
Qin, H., Cui, T., Liu, Z., Zhou, Y., Niu, J., Ren, J., et al. (2021). Engineering amyloid aggregation as a new way to eliminate cancer stem cells by the disruption of iron homeostasis. Nano Lett. 21, 7379–7387. doi: 10.1021/acs.nanolett.1c02734
Qing, H., Wong, W., McGeer, E. G., and McGeer, P. L. (2009). Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 387, 149–152. doi: 10.1016/j.bbrc.2009.06.142
Qu, L., Xu, H., Jia, W., Jiang, H., and Xie, J. (2019). Rosmarinic acid protects against MPTP-induced toxicity and inhibits iron-induced α-synuclein aggregation. Neuropharmacology 144, 291–300. doi: 10.1016/j.neuropharm.2018.09.042
Radde, R., Bolmont, T., Kaeser, S. A., Coomaraswamy, J., Lindau, D., Stoltze, L., et al. (2006). Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946. doi: 10.1038/sj.embor.7400784
Rafael Guimarães, T., Swanson, E., Kofler, J., and Thathiah, A. (2021). G protein-coupled receptor kinases are associated with Alzheimer’s disease pathology. Neuropathol. Appl. Neurobiol. doi: 10.1111/nan.12742 [Online ahead of print].
Rao, S. S., Portbury, S. D., Lago, L., Bush, A. I., and Adlard, P. A. (2020). The iron chelator deferiprone improves the phenotype in a mouse model of tauopathy. J. Alzheimers Dis. 78:1783. doi: 10.3233/JAD-200551
Reynolds, A. D., Stone, D. K., Mosley, R. L., and Gendelman, H. E. (2009). Nitrated alpha-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J. Immunol. 182, 4137–4149. doi: 10.4049/jimmunol.0803982
Roberts, B. R., Ryan, T. M., Bush, A. I., Masters, C. L., and Duce, J. A. (2012). The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. J. Neurochem. 120, 149–166. doi: 10.1111/j.1471-4159.2011.07500.x
Rossi, M. E., Ruottinen, H., Saunamäki, T., Elovaara, I., and Dastidar, P. (2014). Imaging brain iron and diffusion patterns: a follow-up study of Parkinson’s disease in the initial stages. Acad. Radiol. 21, 64–71. doi: 10.1016/j.acra.2013.09.018
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Sánchez Campos, S., Alza, N. P., and Salvador, G. A. (2018). Lipid metabolism alterations in the neuronal response to A53T α-synuclein and Fe-induced injury. Arch. Biochem. Biophys. 655, 43–54. doi: 10.1016/j.abb.2018.08.007
Scheperjans, F., Aho, V., Pereira, P. A., Koskinen, K., Paulin, L., Pekkonen, E., et al. (2015). Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358. doi: 10.1002/mds.26069
Shavali, S., Combs, C. K., and Ebadi, M. (2006). Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: relevance to Parkinson’s disease. Neurochem. Res. 31, 85–94. doi: 10.1007/s11064-005-9233-x
Shekoohi, S., Rajasekaran, S., Patel, D., Yang, S., Liu, W., Huang, S., et al. (2021). Knocking out alpha-synuclein in melanoma cells dysregulates cellular iron metabolism and suppresses tumor growth. Sci. Rep. 11:5267. doi: 10.1038/s41598-021-84443-y
Si, W., Huang, Z., Li, X., Zhao, L., Ji, Y., Li, H., et al. (2021). Super-enhancer-driven sorting Nexin 5 expression promotes dopaminergic neuronal ferroptosis in Parkinson’s disease models. Biochem. Biophys. Res. Commun. 567, 35–41. doi: 10.1016/j.bbrc.2021.06.024
Smith, C., Malek, N., Grosset, K., Cullen, B., Gentleman, S., and Grosset, D. G. (2019). Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 90, 1234–1243. doi: 10.1136/jnnp-2019-321111
Song, N., and Xie, J. (2018). Iron, dopamine and α-synuclein interactions in at-risk dopaminergic neurons in Parkinson’s disease. Neurosci. Bull. 34, 382–384. doi: 10.1007/s12264-018-0209-7
Spotorno, N., Acosta-Cabronero, J., Stomrud, E., Lampinen, B., Strandberg, O. T., van Westen, D., et al. (2020). Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 143, 1341–1349. doi: 10.1093/brain/awaa089
Sripetchwandee, J., Wongjaikam, S., Krintratun, W., Chattipakorn, N., and Chattipakorn, S. C. (2016). A combination of an iron chelator with an antioxidant effectively diminishes the dendritic loss, tau-hyperphosphorylation, amyloids-β accumulation and brain mitochondrial dynamic disruption in rats with chronic iron-overload. Neuroscience 332, 191–202. doi: 10.1016/j.neuroscience.2016.07.003
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Swirski, M., Miners, J. S., de Silva, R., Lashley, T., Ling, H., Holton, J., et al. (2014). Evaluating the relationship between amyloid-β and α-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimers Res. Ther. 6:77. doi: 10.1186/s13195-014-0077-y
Takahashi, T., Yamashita, H., Nakamura, T., Nagano, Y., and Nakamura, S. (2002). Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 938, 73–80. doi: 10.1016/s0006-8993(02)02498-8
Takaichi, Y., Chambers, J. K., Inoue, H., Ano, Y., Takashima, A., Nakayama, H., et al. (2020). Phosphorylation and oligomerization of α-synuclein associated with GSK-3β activation in the rTg4510 mouse model of tauopathy. Acta Neuropathol. Commun. 8:86. doi: 10.1186/s40478-020-00969-8
Tham, M., Frischer, J. M., Weigand, S. D., Fitz-Gibbon, P. D., Webb, S. M., Guo, Y., et al. (2021). Iron heterogeneity in early active multiple sclerosis lesions. Ann. Neurol. 89, 498–510. doi: 10.1002/ana.25974
Thirupathi, A., and Chang, Y. Z. (2019). Brain iron metabolism and CNS diseases. Adv. Exp. Med. Biol. 1173, 1–19. doi: 10.1007/978-981-13-9589-5_1
Thomas, G. E. C., Leyland, L. A., Schrag, A. E., Lees, A. J., Acosta-Cabronero, J., and Weil, R. S. (2020). Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 91, 418–425. doi: 10.1136/jnnp-2019-322042
Tian, Y., Lu, J., Hao, X., Li, H., Zhang, G., Liu, X., et al. (2020). FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson’s disease. Neurotherapeutics 17, 1796–1812. doi: 10.1007/s13311-020-00929-z
Tsatsanis, A., Dickens, S., Kwok, J. C. F., Wong, B. X., and Duce, J. A. (2019). Post translational modulation of β-amyloid precursor protein trafficking to the cell surface alters neuronal iron homeostasis. Neurochem. Res. 44, 1367–1374. doi: 10.1007/s11064-019-02747-y
Tsatsanis, A., Wong, B. X., Gunn, A. P., Ayton, S., Bush, A. I., Devos, D., et al. (2020). Amyloidogenic processing of Alzheimer’s disease β-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 25, 1958–1966. doi: 10.1038/s41380-020-0762-0
Turnbull, S., Tabner, B. J., El-Agnaf, O. M., Moore, S., Davies, Y., and Allsop, D. (2001). alpha-synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 30, 1163–1170. doi: 10.1016/s0891-5849(01)00513-5
Uranga, R. M., Giusto, N. M., and Salvador, G. A. (2009). Iron-induced oxidative injury differentially regulates PI3K/Akt/GSK3beta pathway in synaptic endings from adult and aged rats. Toxicol. Sci. 111, 331–344. doi: 10.1093/toxsci/kfp152
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Uversky, V. N. (2007). Neuropathology, biochemistry and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37. doi: 10.1111/j.1471-4159.2007.04764.x
van Bergen, J. M., Li, X., Hua, J., Schreiner, S. J., Steininger, S. C., Quevenco, F. C., et al. (2016). Colocalization of cerebral iron with amyloid beta in mild cognitive impairment. Sci. Rep. 6:35514. doi: 10.1038/srep35514
van Bergen, J. M. G., Li, X., Quevenco, F. C., Gietl, A. F., Treyer, V., Meyer, R., et al. (2018). Simultaneous quantitative susceptibility mapping and Flutemetamol-PET suggests local correlation of iron and β-amyloid as an indicator of cognitive performance at high age. Neuroimage 174, 308–316. doi: 10.1016/j.neuroimage.2018.03.021
Vasquez, V., Mitra, J., Hegde, P. M., Pandey, A., Sengupta, S., Mitra, S., et al. (2017). Chromatin-bound oxidized α-synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson’s disease. J. Alzheimers Dis. 60, S133–S150. doi: 10.3233/JAD-170342
Venkataramani, V., Doeppner, T. R., Willkommen, D., Cahill, C. M., Xin, Y., Ye, G., et al. (2018). Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 147, 831–848. doi: 10.1111/jnc.14580
Wan, W., Cao, L., Kalionis, B., Murthi, P., Xia, S., and Guan, Y. (2019). Iron deposition leads to hyperphosphorylation of tau and disruption of insulin signaling. Front. Neurol. 10:607. doi: 10.3389/fneur.2019.00607
Wang, R., Wang, Y., Qu, L., Chen, B., Jiang, H., Song, N., et al. (2019). Iron-induced oxidative stress contributes to α-synuclein phosphorylation and up-regulation via polo-like kinase 2 and casein kinase 2. Neurochem. Int. 125, 127–135. doi: 10.1016/j.neuint.2019.02.016
Wang, S., Han, Q. W., Zhou, T. T., Zhang, C. L., Zhu, C. G., Zhou, X., et al. (2021). A bibenzyl compound 20C protects rats against 6-OHDA-induced damage by regulating adaptive immunity associated molecules. Int. Immunopharmacol. 91:107269. doi: 10.1016/j.intimp.2020.107269
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/S1474-4422(14)70117-6
Wood, H. (2016). Neurodegenerative disease: Lithium promotes accumulation of brain iron via tau suppression. Nat. Rev. Neurol. 12, 492–493. doi: 10.1038/nrneurol.2016.112
Xiao, Y., Chen, X., Huang, S., Li, G., Mo, M., Zhang, L., et al. (2018). Iron promotes α-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J. Neurochem. 145, 34–50. doi: 10.1111/jnc.14312
Xie, L., Zheng, W., Xin, N., Xie, J. W., Wang, T., and Wang, Z. Y. (2012). Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 61, 334–340. doi: 10.1016/j.neuint.2012.05.016
Xuan, M., Guan, X., Gu, Q., Shen, Z., Yu, X., Qiu, T., et al. (2017). Different iron deposition patterns in early- and middle-late-onset Parkinson’s disease. Parkinsonism. Relat. Disord. 44, 23–27. doi: 10.1016/j.parkreldis.2017.08.013
Yamamoto, A., Shin, R. W., Hasegawa, K., Naiki, H., Sato, H., Yoshimasu, F., et al. (2002). Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x
Yang, L., Wang, H., Liu, L., and Xie, A. (2018). The role of insulin/IGF-1/PI3K/Akt/GSK3β signaling in Parkinson’s disease dementia. Front. Neurosci. 12:73. doi: 10.3389/fnins.2018.00073
Yang, L., Zhang, X., Li, S., Wang, H., Zhang, X., Liu, L., et al. (2020). Intranasal insulin ameliorates cognitive impairment in a rat model of Parkinson’s disease through Akt/GSK3β signaling pathway. Life Sci. 259:118159. doi: 10.1016/j.lfs.2020.118159
Yu, Z., Xu, X., Xiang, Z., Zhou, J., Zhang, Z., Hu, C., et al. (2010). Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS One 5:e9956. doi: 10.1371/journal.pone.0009956
Zhang, X.Y., Cao, J. B., Zhang, L. M., Li, Y. F., and Mi, W. D. (2015). Deferoxamine attenuates lipopolysaccharide-induced neuroinflammation and memory impairment in mice. J. Neuroinflammation 12:20. doi: 10.1186/s12974-015-0238-3
Zhang, X., Gou, Y. J., Zhang, Y., Li, J., Han, K., Xu, Y., et al. (2020). Hepcidin overexpression in astrocytes alters brain iron metabolism and protects against amyloid-β induced brain damage in mice. Cell Death Discov. 6:113. doi: 10.1038/s41420-020-00346-3
Zhang, Y., Huang, Y., Deng, X., Xu, Y., Gao, Z., and Li, H. (2012). Iron overload-induced rat liver injury: involvement of protein tyrosine nitration and the effect of baicalin. Eur. J. Pharmacol. 680, 95–101. doi: 10.1016/j.ejphar.2012.01.010
Zheng, W., Zhang, Z., Ye, Y., Wu, Q., Liu, M., and Li, C. (2019). Phosphorylation dependent α-synuclein degradation monitored by in-cell NMR. Chem. Commun. (Camb) 55, 11215–11218. doi: 10.1039/c9cc05662a
Zhou, Z. D., and Tan, E. K. (2017). Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegeneration 12:75. doi: 10.1186/s13024-017-0218-4
Zhou, Z. D., Xie, S. P., Saw, W. T., Ho, P. G. H., Wang, H., Lei, Z., et al. (2019). The therapeutic implications of tea polyphenols against dopamine (DA) neuron degeneration in Parkinson’s disease (PD). Cells 8:911. doi: 10.3390/cells8080911
Zhu, S., Wei, X., Yang, X., Huang, Z., Chang, Z., Xie, F., et al. (2019). Plasma lipoprotein-associated phospholipase A2 and superoxide dismutase are independent predicators of cognitive impairment in cerebral small vessel disease patients: diagnosis and assessment. Aging Dis. 10, 834–846. doi: 10.14336/AD.2019.0304
Keywords: beta-amyloid, tau, alpha-synuclein, iron, Parkinson’s disease dementia
Citation: Han J, Fan Y, Wu P, Huang Z, Li X, Zhao L, Ji Y and Zhu M (2021) Parkinson’s Disease Dementia: Synergistic Effects of Alpha-Synuclein, Tau, Beta-Amyloid, and Iron. Front. Aging Neurosci. 13:743754. doi: 10.3389/fnagi.2021.743754
Received: 19 July 2021; Accepted: 21 September 2021;
Published: 11 October 2021.
Edited by:
Ramesh Kandimalla, Indian Institute of Chemical Technology (CSIR), IndiaReviewed by:
Mohammad Mobashir, Karolinska Institutet (KI), SwedenCopyright © 2021 Han, Fan, Wu, Huang, Li, Zhao, Ji and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meiling Zhu, bWVpbGluZ3podTIwMjBAMTI2LmNvbQ==
† These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.