 Estibaliz Santiago-Mujika1
Estibaliz Santiago-Mujika1 Ruth Luthi-Carter1
Ruth Luthi-Carter1 Flaviano Giorgini2
Flaviano Giorgini2 Raj N. Kalaria3
Raj N. Kalaria3 Elizabeta B. Mukaetova-Ladinska1,4*
Elizabeta B. Mukaetova-Ladinska1,4*- 1Department of Neuroscience, Behavior and Psychology, University of Leicester, Leicester, United Kingdom
- 2Department of Genetics and Genome Biology, University of Leicester, Leicester, United Kingdom
- 3Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom
- 4Evington Centre, Leicester General Hospital, Leicester, United Kingdom
Alzheimer’s disease (AD) and vascular dementia (VaD) are the two most common forms of dementia in older people. Although these two dementia types differ in their etiology, they share many pathophysiological and morphological features, including neuronal loss, which is associated with the microtubule (MT) destabilization. Stabilization of MTs is achieved in different ways: through interactions with MT binding proteins (MTBP) or by posttranslational modifications (PTMs) of tubulin. Polyglutamylation and tyrosination are two foremost PTMs that regulate the interaction between MTs and MTBPs, and play, therefore, a role in neurodegeneration. In this review, we summarize key information on tubulin PTMs in relation to AD and VaD and address the importance of studying further the tubulin code to reveal sites of potential intervention in development of novel and effective dementia therapy.
Alzheimer’s Disease and Vascular Dementia
Alzheimer’s disease (AD) and vascular dementia (VaD) are two of the most common types of dementia, representing ∼85% of all dementia cases (Qiu et al., 2009; Iadecola, 2013). In some cases, patients show mixed dementia, where they combine features of AD with ischemic lesions (Iadecola, 2013). Although they have been thoroughly studied, there is no effective treatment to halt or reverse these diseases.
AD is the most common form of dementia and it accounts for more than 70% of all clinically diagnosed dementias (Qiu et al., 2009). Although memory is the main impaired feature, language difficulties, emotional and behavioral changes are also present (Steinberg et al., 2008). The neuropathological hallmarks are amyloid plaques and neurofibrillary tangles (NFTs) (Šimić et al., 2016). Additional neuropathological AD features include dystrophic neurites, astrogliosis, neuronal loss, and cortical atrophy (McKhann et al., 2011; Serrano-Pozo et al., 2011).
Amyloid plaques are extracellular aggregates of insoluble 40 and 42 amyloid-β (Aβ) peptide, that although present in healthy people, are significantly increased in AD patients. On the other hand, NFTs are intraneuronal aggregates of hyperphosphorylated and/or truncated and misfolded tau. Contrary to amyloid plaques, there is a correlation between the burden of NFTs and disease progression, as well as AD clinical symptoms (Giannakopoulos et al., 2009; Suemoto et al., 2017).
VaD is the second most common form of dementia, accounting for ∼15% of all cases. In spite of the number of patients diagnosed and the annual costs for this syndrome being highly significant, VaD has not been as thoroughly studied as AD (Iadecola, 2013).
There are four types of VaD: post-stroke dementia, subcortical ischemic vascular dementia, multi-infarct dementia and mixed dementia (Kalaria, 2018; Skrobot et al., 2018). VaD is considered a heterogeneous group of brain disorders since it can be caused by several cerebrovascular pathologies (Iadecola, 2013), such as atherosclerosis, small vessel disease or cerebral amyloid angiopathy (CAA), which is the accumulation of Aβ in vessel walls (Serrano-Pozo et al., 2011). These diseases, in turn, can lead to different cerebrovascular lesions, i.e., ischemic or hemorrhagic infarct, white matter lesions or hemorrhages (McAleese et al., 2016; Smith, 2017; Kalaria, 2018). Although in different ways, these lesions ultimately reduce the blood flow to the brain, and consequently the oxygen supply. The lack of oxygen in the brain is what ultimately leads to vascular dementia (Figure 1).
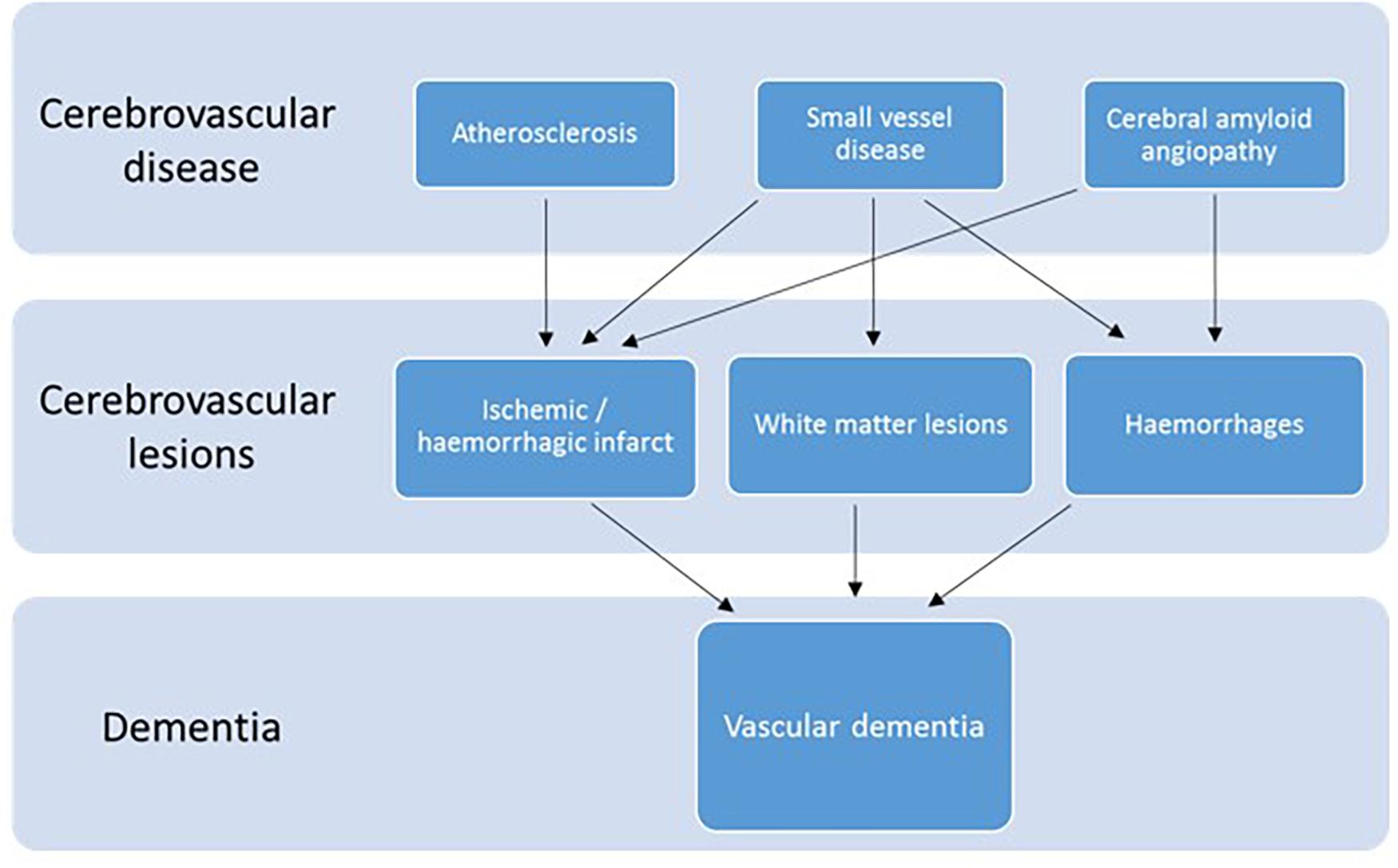
Figure 1. Diagram of the cerebrovascular diseases and lesions that lead to VaD. Modified after (McAleese et al., 2016).
Although AD and VaD differ in their etiology, they share risk factors, such as age, obesity, the apolipoprotein E4 allele (ApoE ε4 allele) and hypercholesterolemia (Akinyemi et al., 2013). Furthermore, they also share similarities in their pathophysiology (Table 1; Kalaria and Ballard, 1999), i.e., a tendency of Aβ42 to be significantly higher in the temporal lobe (Lewis et al., 2006), or a neuronal cell volume loss (Gemmell et al., 2012). In fact, neuronal and synaptic loss are the best predictors of cognitive decline in neurodegenerative diseases (Coleman et al., 2004; Andrade-Moraes et al., 2013; Theofilas et al., 2018). In one study on 14 brains from 80-year-old women, a novel technique called isotropic fractionator was used to determine the absolute cellular composition of brain regions. The study showed there was ∼50% reduction of total neuronal cell numbers in the hippocampus of AD subjects (Andrade-Moraes et al., 2013).

Table 1. Pathophysiological similarities between AD and VaD.
Not only is neuronal cell death a hallmark of neurodegenerative diseases, but in AD it is also correlated with the severity of the disease (Bussière et al., 2003; Arendt et al., 2015; Martínez-Pinilla et al., 2016; Chi et al., 2018). The same has been found in VaD, where both reduced neuronal volume (Gemmell et al., 2012) and cell counts (Jellinger, 2013) are associated with the severity of cognitive impairment in VaD (Kril et al., 2002; Gemmell et al., 2012).
Although there are several descriptions of mechanisms of cell death (Galluzzi et al., 2018), the major ones linked to neurodegenerative diseases are apoptosis and necrosis (Chi et al., 2018). Apoptosis is a controlled process where there are no spillages of the cell contents to the surrounding, whereas necrosis consists of an uncontrolled cell death, induced by external injury, such as hypoxia or inflammation. Ultimately, the cell membrane breaks and the cellular contents are spilled (D’Arcy, 2019). A new type of controlled necrosis has been recently described, called necroptosis (Degterev et al., 2005). Although similar to necrosis, necroptosis can be activated by death receptors such as TNFR1, which leads to the activation of RIPK1 (Yuan et al., 2019). Furthermore, RIPK1 promotes neuroinflammation, another feature of neurodegenerative diseases (Yuan et al., 2019). Necroptosis has been reported to be activated in AD human brains and positively correlated with Braak stages (Caccamo et al., 2017), with changes in the pathways of apoptosis, autophagy and necrosis depending on the stage of AD (Telegina et al., 2019). Necroptosis has also been reported in the hippocampus of one case of VaD due to ischemic injury. In this case, the necroptosis was related to inflammation and increased cytokines, such as TNF-α and IL-1β (Belkhelfa et al., 2018).
In apoptosis, the cytoskeleton undergoes significant morphological changes (Bonfoco et al., 1995) due to a disruption of MTs (Liepins and Bustamante, 1994; Figure 2A). Colchicine, an alkaloid plant extract with a therapeutic use in coronary artery diseases, inflammatory and fibrotic conditions, is a drug that disrupts the MTs by fragmenting the tubulin heterodimers and, consequently, leading to apoptosis. Induced apoptosis could be, thus, prevented by the addition of taxol, a drug that stabilizes the MTs (Bonfoco et al., 1995). Although many of the MT-stabilizing drugs, including taxol, do not cross the blood-brain barrier, recent advances of their nanosuspension delivery (Fan et al., 2021) or nasal administration of paclitaxel (Cross et al., 2021) appear to effectively overcome the brain blood barrier and accomplish neuronal cell-targeted drug delivery, thus, offering a potential for novel dementia therapeutic opportunities.
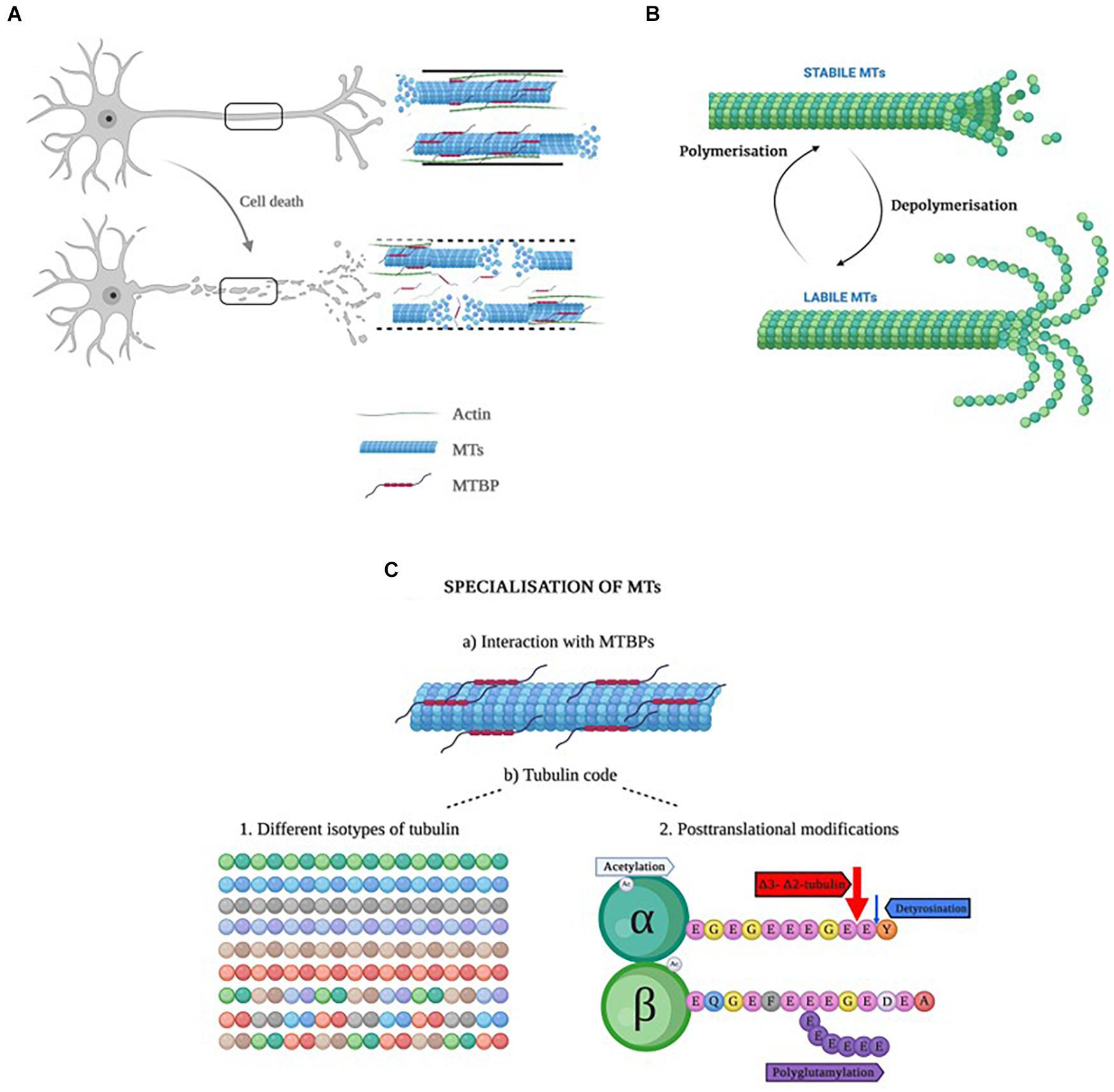
Figure 2. (A) Representation of a healthy neuron with a healthy cytoskeleton (top), and a neuron in apoptosis with increased ratio of labile to stabile MTs. (B) Representation of the dynamic instability process in microtubules. When MTs are highly dynamic, they are called labile, whereas when the dynamics are slow, they are named stable MTs (Li and Black, 1996). The important aspect of dynamic instability is the presence of stochastic changes between a growth and a shrinkage phase and vice versa. These two phases can occur in parallel in the cells. The dynamic instability is energy-dependent and driven by GTP hydrolysis. (C) Schematic representation of the specialization of MTs. Created with BioRender.com.
Protein aggregates in neurodegenerative diseases also contribute to neuronal loss (Chi et al., 2018). The localization as well as the composition of the aggregates differ from disease to disease [i.e., intraneuronal aggregates of tau protein in AD (Šimić et al., 2016) or aggregations of α-synuclein in Parkinson’s disease (Peng et al., 2018)]. In AD, tau protein undergoes posttranslational modifications (i.e., hyperphosphorylation) and/or truncation (Wischik et al., 1995), which is observed in NFTs. It is not clear, however, whether the phosphorylation occurs before the aggregation or after the formation of NFTs. Similarly, the debate whether tau aggregates or soluble tau oligomers are the toxic species is ongoing. Nonetheless, the NFTs may contribute to the activation of neuronal apoptotic mechanisms in AD (Nixon and Yang, 2011; Chi et al., 2018; Liu et al., 2020). Even though there are many papers in the literature that describe tau as a MT stabilizer which detaches from MTs when hyperphosphorylated (Chi et al., 2018), recent studies might call this statement into question (Baas and Qiang, 2019). Nonetheless, the fact that phosphorylation of tau hinders tubulin and MT assembly remains (Savastano et al., 2021). Furthermore, a study showed that tau protein is only bound to MTs for a very short time of 40 ms. The authors described that tau protein presented a “kiss and hop” interaction with tubulin molecules (Janning et al., 2014).
Not only have the NFTs been related to MT destabilization, but also Aβ. In fact, Fifre et al. (2006) described the possible role of Aβ in the degradation of different MAPs. Gevorkian et al. (2008) detected MT disruption in primary neurons exposed to Aβ as a result of the binding of Aβ with MAP1. In addition, modifications of MTs can also influence the ability of neurons to cope with Aβ neurotoxicity. Namely, MTs disruption leads to increased levels of cytotoxicity caused by the Aβ1–42 exposure, and vice versa, the stabilized MTs delay the toxicity caused by Aβ1–42 (Shamitko-Klingensmith et al., 2016). Similarly, in isolated neuronal cultures, Aβ oligomers cause tau-dependent MT breakdown mediated by spastin, an MT-severing enzyme (Zempel et al., 2013).
When analyzing VaD, it has been found that there is a loss neuronal cell volume in the dorsolateral prefrontal cortex (DLPFC) of patients with VaD when compared to controls (Mukaetova-Ladinska et al., 2009). Another study reported a selective loss of total tau protein in the temporal lobe of subjects with VaD that did not correlate with NFT, senile plaques or amyloid beta (Mukaetova-Ladinska et al., 2015). In a study by Gallart-Palau et al. (2015) where the levels of α1-tubulin and βII-tubulin isotypes were measured in VaD subjects and controls, no differences were observed in the temporal lobe. It is worth mentioning that the same study showed the tubulin proteins were significantly deamidated in VaD patients when compared to controls. Furthermore, these changes were not due to neurofibrillary pathology or any other lesion in VaD, i.e., visible infarcts (Gallart-Palau et al., 2015). Similarly, in a work done with human brain homogenates from VaD and AD patients, Mukaetova-Ladinska’s lab found a tendency for reduced levels of tubulin in the temporal lobe of VaD patients (Li et al., 2014), raising the question of whether changes in tubulin, i.e., posttranslational modifications, could be related to the observed loss of tau and neuronal volume.
Although there is no accumulation of NFTs in VaD, the temporal lobe appears to have a significant loss of soluble tau protein (Mukaetova-Ladinska et al., 2015), and this may result in the decreased neuronal cell volume followed by neuronal cell loss and apoptosis (Gemmell et al., 2012). The tubulin loss additionally may be result of: (1) neuronal death secondary to ischemic injury, (2) the process of diaschisis, (3) axonal injury in white matter and reduction of white matter volume, and (4) reduced dendritic arborization due to cell atrophy. Furthermore, these early changes in the interaction between microtubule associated proteins and tubulin may denote the neurobiological crossroad of further clinical and neurobiological progression to either AD or VaD. Having a better understanding of MTs and tubulin changes in the brain might lead to the development of an effective form of therapy.
Microtubules
The cytoskeleton is a complex network of filaments that gives the cell its shape and mechanical resistance. In neurons, the cytoskeleton is formed by MTs, microfilaments and neurofilaments (Penazzi et al., 2016). Out of the three, MTs are the main protein filaments of the cytoskeleton and they are abundant in the cells (Horio and Murata, 2014; Burbaeva et al., 2020). They constitute approximately 10% of the total protein concentration in the brain (Cleveland et al., 1980).
MTs are involved in a variety of functions, such as cell motility, transport, cell shape and polarity, and mitosis (Mandelkow and Mandelkow, 1995). They are also necessary for synaptic plasticity, and their stability is essential for the physiological functioning of neurons (Carnwath et al., 2018). Cytoskeletal defects and altered MT-mediated processes are indeed linked to neurodevelopmental disorders, such as severe lissencephaly due to a mutation in the tubulin α1A gene (Kumar et al., 2010), or autosomal dominant disorders of axon guidance due to mutations in the TUBB3 gene (Tischfield et al., 2010).
A fundamental characteristic of MTs is that they are dynamic structures, which means they are able to alter their organization in order to adapt to changes in cellular shape (Goodson and Jonasson, 2018). This process, named “dynamic instability” (Horio and Murata, 2014), is considered an intrinsic property consisting on two opposed processes: polymerization or growth, and depolymerization or shrinkage, as seen in Figure 2B. The dynamic instability derives from the tubulin’s GTPase activity (Hyman et al., 1995; Nogales and Wang, 2006). Both polymerization and depolymerization can happen at the same time in the cell: whereas polymerization occurs in a temperature and concentration-dependent manner, depolymerization occurs randomly (Horio and Murata, 2014). Interestingly, this process occurs stochastically within the same filament, depending on whether MTs are bound to GTP or GDP, and both growing and shrinking MTs can be present in the same cell at any one given time. Thanks to this dynamic instability, fibroblasts are able to migrate and neurons extend their axon and dendrites (Mitchison and Kirschner, 1984; Brouhard, 2015).
MTs are hollow cylinders formed by heterodimers of α- and β-tubulin (Bryan and Wilson, 1971), with an approximate ratio of 1:1 (Alvarez et al., 1998). Both α- and β-tubulin monomers are very similar; they are ∼40% identical and 63% homologous (Nogales, 2013). Each tubulin monomer is composed of 450 amino acid residues which differ slightly from one another (Ludueńa et al., 1977; Sullivan and Cleveland, 1986), and a molecular mass of ∼50 kDa (Alvarez et al., 1998; Nogales et al., 1998). The monomers present some structural differences (Table 2). Due to the different functions MTs perform, they need to be specialized, and this specialization is achieved in two different ways (Gadadhar et al., 2017): (a) by interacting with microtubule binding proteins (MTBPs), or (b) by the tubulin code (Figure 2C), a highly important event in that it not only controls specific cellular functions, but it also has a role in human pathologies (Gadadhar et al., 2017). Moreover, the tubulin code is a mechanism by which MTs regulate themselves in yet another two different ways (Gadadhar et al., 2017):

Table 2. Structural differences between α- and β-tubulin (modified after Nogales et al., 1998; Nogales, 2013).
1- Expression of different isotypes of α- and β-tubulin: both α- and β-tubulin have different isotypes that are encoded by different genes. In humans, there are 7α- and 8β-tubulin isotypes which differ in their C-terminal sequence and present tissue specificity (Ludueña, 1998; Fukushima et al., 2009; Ludueña, 2013). For example, α-tubulin Class II is mostly found in the testis, whereas α-tubulin III is found in brain and muscle (Ludueña, 2013). On the other hand, β-tubulin III is present only in neurons (Katsetos et al., 2003) whereas β tubulin Class VI is present mainly in hematopoietic cells (Murphy et al., 1987). Brains express α 1A-, α 1B-, α 1C-, α 4A-, and α8-tubulin, and β 2A-, β 3-, β 4-, and β5-tubulin (Fukushima et al., 2009). β-tubulin isotypes are evolutionary very well conserved and significantly better understood than α-tubulin isotypes (Ludueña, 2013). The level of expression of the isotypes in each specific tissue is very important since high βI-tubulin and low βII-tubulin levels have been described in breast cancer (Hasegawa et al., 2003; Ohishi et al., 2007). Furthermore, each isotype presents a specific function. Whereas βII is involved in neurite formation, βIII protects the MTs from reactive oxygen species (ROS) (Ludueña, 2013).
2- Posttranslational modifications (PTMs) of tubulin (Table 3): there are more than ten modifications that can occur at any given time (Fukushima et al., 2009; Janke, 2014; Gadadhar et al., 2017). More often than not, PTMs are found on stable long-lived MTs, such as neuronal, axonemal, and centriolar MTs (Janke, 2014). In modifying the tubulin, PTMs also modify the binding affinity of tubulin to MTBPs, such as tau, as it is observed by an increase in polyglutamylated tubulin (Eddé et al., 1990).
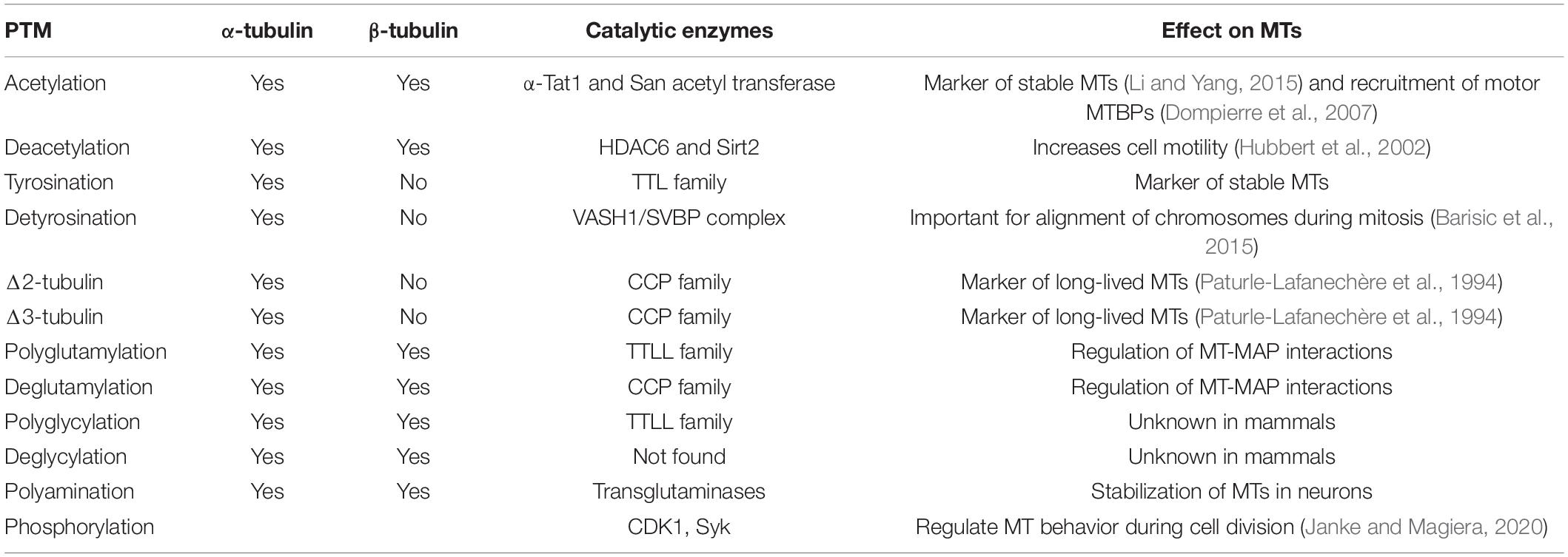
Table 3. Summary of main PTMs, the catalytic enzymes taking part in the reaction and the effects of PTMs on MTs/cells.
Interaction With Microtubule Binding Proteins
Any protein that binds to the MTs in a specified manner is considered a MTBP, and they are functionally categorized as stabilizers, destabilizers, capping proteins or bundler/cross-linkers (Goodson and Jonasson, 2018). Probably the most well-known MTBPs are the ones that comprise the MT associated protein (MAP) family, which includes tau protein and MAP2, the major MAPs in the brain (Melková et al., 2019), as well as MAP6, previously known as STOP.
1. Tau: this protein is enriched in axonal MTs, although it is also present in dendrites (Melková et al., 2019). Due to alternative splicing, there are six different tau isoforms, which differ in the number of MT-binding domain repeats (Fischer and Baas, 2020). Five of those isoforms weight around 40 kDa, and present multiple phosphorylation sites. When hyperphosphorylated, these form the characteristic NFTs observed in AD (Himmler et al., 1989), which hinder tubulin assembly (Savastano et al., 2021). On the other hand, the sixth isoform termed “Big Tau,” mainly expressed in the peripheral nervous system, weighs 100 kDa and does not present many phosphorylation sites; protecting it from aggregation into tangles (Fischer and Baas, 2020). In addition, the speculated function of big tau is the stabilization of mature axonal cytoskeleton (Oblinger et al., 1991).
2. MAP2: present in cell bodies and dendrites, MAP2 is considered a MT stabilizer (Goodson and Jonasson, 2018) as it increases MT rigidity (Felgner et al., 1997). Similar to tau protein, MAP2 is accumulated into granules, which leads to neurotoxicity and neuronal loss (Xie and Miyasaka, 2016). Loss of MAP2 has been widely linked to AD in human samples, as well as in cellular and animal models (Mukaetova-Ladinska et al., 2009; Baazaoui et al., 2017; Kandimalla et al., 2018; Manczak et al., 2018; Reddy et al., 2018; Beggiato et al., 2020; Liang et al., 2020).
3. MAP6: formerly known as STOP (stable tubule only polypeptide), it is a genuine MT stabilizer found predominantly in the axon, along with detyrosinated and acetylated tubulin; two PTMs present in stable MTs (Slaughter and Black, 2003).
Posttranslational Modifications of Tubulin
Tubulin can undergo more than 10 different PTMs, some of them occurring solely on the tubulin, such as polyglutamylation. The major modifications that have been linked to neurodegeneration are acetylation, detyrosination, and polyglutamylation. In the following pages, we will discuss the tubulin PTMs and what it is known thus far about their role in neurodegenerative diseases.
1. Acetylation and Deacetylation: this modification is different from the others in that it happens in the lumen of MTs (Gadadhar et al., 2017). Acetylation can take place either on lysine 40 (K40) on α-tubulin (L’Hernault and Rosenbaum, 1985), where it is linked to stable MTs (Westermann and Weber, 2003; Wloga and Gaertig, 2010; Janke and Bulinski, 2011), or on K252 on β-tubulin (Janke, 2014), where it might slow down the incorporation of tubulin into MTs (Chu et al., 2011). The enzymes that catalyze the reactions are α-Tat1 (specific acetylation of tubulin) and San acetyl transferase (Akella et al., 2010; Janke, 2014), and HDAC6 and Sirt2 (involved in the deacetylation of tubulin and other substrates).
In a functional level, acetylated tubulin is implicated in intracellular trafficking, endoplasmic reticulum (ER) localization and ER-mitochondria interactions, as well as the regulation of MT dynamics (Daire et al., 2009). It has been observed that in most NFT bearing neurons there is a decrease in acetylated α-tubulin (Brion et al., 2001), a marker of stable MTs (Janke and Bulinski, 2011). Similarly, deacetylation is associated with dysfunctional MT-mediated axonal transport in neurodegenerative diseases, such as AD, Parkinson’s disease (PD) and Huntington’s disease (HD) (Li and Yang, 2015; Fernández-Barrera et al., 2018). In fact, HDAC6 (histone deacetylase 6, a multidomain cytosolic enzyme with α-tubulin deacetylase activity) is increased in AD (Zhang et al., 2013) and HD (Ferrante et al., 2003), and dysregulation of HDACs also take part in ischemic strokes (Fang et al., 2020).
2. Detyrosination and tyrosination: α-tubulin isotypes present a tyrosine residue at the end of the C-terminal sequence that β-tubulin isotypes lack; thus, rendering this modification specific to α-tubulin (Prota et al., 2013; Janke and Magiera, 2020). The tyrosine residue can be reversibly removed by the VASH1/SVBP complex (Aillaud et al., 2017) generating detyrosinated tubulin. Retyrosination of the tubulin, however, happens thanks to the tubulin tyrosine ligase (TTL) (Janke, 2014). These two modifications are spatially and temporally regulated. Whereas tyrosination is mostly found in the growing end of the axons in developing neurons (Ahmad et al., 1993; Sferra et al., 2020), detyrosination is mainly found in the dendrites of mature neurons, and it is considered a marker of stable MTs (Sferra et al., 2020).
Detyrosination regulates MT-MAP interactions whereas tyrosination has a role in spindle orientation and growth cone guidance in neuronal pathfinding (Magiera and Janke, 2014). In the context of AD, one study has suggested that the levels of tyrosinated levels are increased (Zhang et al., 2015).
3. Δ 2-tubulin and Δ 3-tubulin: these terms are used when after detyrosination, additional C-terminal glutamate residues are eliminated from the tubulin sequence, which means that retyrosination is not possible (Janke, 2014; Gadadhar et al., 2017). Because of the lack of retyrosination, it is believed that one of the functions of Δ2-tubulin is to lock the tubulin in the non-tyrosinatable status or to reduce sites for polyglutamylation or polyglycylation (Magiera and Janke, 2014). It is estimated that 35% of α-tubulin in the brain corresponds to Δ2-tubulin (Paturle-Lafanechère et al., 1994). The enzymes that catalyze these reactions belong to the cytosolic carboxypeptidases (CCPs) family (Janke, 2014; Gadadhar et al., 2017). A study showed that there were increased levels of glutamylated Δ2-tubulin in the hippocampi of post-mortem patients of AD (Vu et al., 2017).
4. Polyglutamylation: first reported in 1990, it involves the addition of 1–12 glutamate units into a glutamic acid residue in the C-terminal region of either α- or β-tubulin (Eddé et al., 1990). In the brain tissue, there is an average of 3–6 glutamate residues on each tail, with as many as 11 and 7 residues detected on the α- and β-tubulin tails, respectively (Redeker, 2010). The polyglutamylation catalyzing enzymes belong to the TTL like (TTLL) family (Rogowski et al., 2010; Wloga and Gaertig, 2010) whereas the deglutamylases belong to the CCP family (Kimura et al., 2010). There are 8 members in the TTLL family that present different affinities for α- and β-tubulin, and different preferences for either initiating or elongating the glutamylation reaction, as shown in Table 4 (Yu et al., 2015). As for the CCP family, there are six members. CCP1, -2, -3, -4, and -6 shorten polyglutamylated chains, whereas CCP5 is the only monodeglutamylase found until date (Rogowski et al., 2010; Berezniuk et al., 2013).

Table 4. Family members of the TTLL family and their specific role in glutamylation.
Polyglutamylation is abundant in neurons, centrioles, basal bodies, axonemes of cilia and flagella, as well as in the mitotic spindle (Redeker, 2010), and it is implicated in the regulation of MT and MAP electrostatic interactions, since polyglutamylation affects the charges of the tubulin tails (Janke, 2014).
Maintaining the correct levels of polyglutamylation is essential. Several defects have been associated with either depletion or overexpression of the enzymes catalyzing glutamylation: TTLL1 knockout mice show respiratory problems (Ikegami et al., 2010), and TTLL6 knock out zebrafish show defective assembly of olfactory cilia (Pathak et al., 2007), whereas overexpression of TTLL6 causes ciliary defects in Tetrahymena (Suryavanshi et al., 2010). Another example comes from the Purkinje cell degeneration (pcd) mouse model, which lacks the CCP1 enzyme (Rogowski et al., 2010). These mice show overexpression of polyglutamylated tubulin and degeneration of Purkinje cells in the cerebellum (Magiera et al., 2018). Thus, balanced tubulin glutamylation levels are important for neuronal function and survival.
The excess of TTLL1-mediated polyglutamylation is the cause of cerebellar neurodegeneration in the pcd mouse model, and this neurodegeneration could be rescued upon depletion of TTLL1 (Magiera et al., 2018). The excessive polyglutamylation also impairs neuronal transport (Magiera and Janke, 2014), with the mutations of CCP1 in humans leading to childhood-onset neurodegeneration (Shashi et al., 2018). Of interest, in people with Alzheimer’s disease (AD), there is a reduction of α-tubulin and a tendency for increased polyglutamylation in the hippocampus when compared to age-matched controls (Zhang et al., 2015). It has already been shown that if tubulin is polyglutamylated with four or more glutamyl residues, the affinity to bind tau decreases (Eddé et al., 1990). However, little is known about MT-MAP interaction in neurodegenerative diseases and the implications this lower affinity has.
5. Polyglycylation: it refers to the generation of side chains of glycine residues within the C-terminal tails of α- and β-tubulin (Redeker et al., 1994). It happens at the same site as glutamylation and it is also catalyzed by TTLL enzymes (Rogowski et al., 2010). This PTM is restricted to cilia and flagella and the extent of the modification correlates with the length of the axonemes (Rogowski et al., 2010; Wloga and Gaertig, 2010). To date, there is very little known about the function of this modification in mammals. However, a link between the downregulation of TTLL3 and colon cancer has been described in humans (Rocha et al., 2014). Furthermore, it has been observed that polyglycylation in humans is not possible since the enzyme responsible for the elongation, TTLL10, is inactivated (Janke, 2014). There is little to no knowledge about the role of polyglycylation in neurodegeneration.
6. Polyamination: for this modification, glutamine 15 (Q15) is considered to be the primary modification site, and the reaction is catalyzed by transglutaminases (Song et al., 2013). The reaction consists on the addition of amines to either α- or β-tubulin, adding positive charges to the acidic tubulin (Janke and Magiera, 2020). This PTM is irreversible and it most likely stabilizes microtubule subpopulations in neurons (Janke, 2014).
7. Phopshorylation: the Cdk1 enzyme catalyzes the phosphorylation on Serine 172 (S172) of β-tubulin (Fourest-Lieuvin et al., 2006), and it seems that this modification might be implicated in microtubule dynamics during cell division (Janke, 2014). Another tyrosine kinase known as Syk has been shown to phosphorylate an unidentified residue of α-tubulin (Peters et al., 1996).
8. Others: tubulin can also undergo palmitoylation (Caron, 1997), ubiquitination (Ren et al., 2003), glycosylation (Walgren et al., 2003), arginylation (Wong et al., 2007), methylation (Xiao et al., 2010), and sumoylation (Rosas-Acosta et al., 2005). However, there is not much information on these processes and how they affect disease pathogenesis.
Therapeutic Drugs Targeting Microtubule Stability
Microtubule Stabilizers
Several studies with MT stabilizers have been carried out as possible treatment for AD and other tauopathies:
1. Epothilone D (BM2-241027): this is a brain penetrant microtubule stabilizing agent able to polymerize tubulin and inhibit its depolymerization (Cheng and Huang, 2018). In a study following treatment with this drug, MT dynamicity decreased, whilst cognition improved, as shown in the Morris Water Maze task (Barten et al., 2012). Another study in cortical neurons put in display the importance of the dose, since it can affect the mitochondrial transport (Clark et al., 2020). Studies have also shown that BM2-241027 reduces axonal dysfunction, neurotoxicity, cognitive deficits, and AD-like pathology in PS19 aged tau transgenic mice (Zhang et al., 2012). Although in animal studies Epothilone D rescued working and spatial memory deficits in aged tau transgenic mice (reviewed in Yu et al., 2021), this drug was discontinued following a Phase 1 clinical study in 2013, with no information regarding drug’s effectiveness and side effects in humans (Bristol-Myers Squibb, 2013).
2. TPI-287: this CNS penetrating taxane was used in another clinical study to assess its efficiency as an AD treatment, progressive supranuclear palsy and corticobasal syndrome. Unfortunately, it was discontinued due to safety, i.e., severe anaphylactoid reactions (Tsai et al., 2020).
3. Paclitaxel: it is the generic name of taxol, a very common MT stabilizing drug approved by the Food and Drug Administration for the treatment of several types of cancer (Weaver, 2014). However, they evoke axonal degeneration (Gornstein and Schwarz, 2014). This neurotoxicity prevents it from being used in neurodegenerative diseases. However, its’ newer nasal formulation has been successfully used in transgenic animals. Cross et al. (2019) showed that intranasal paclitaxel administered once daily, prevented injury-induced memory deficits in mice. Furthermore, there was reduced evidence of axonal injury and synaptic loss (Cross et al., 2019). The same group has also shown that a transgenic mouse model of AD presented less tau-containing neurons in the CA1 and had improved memory after paclitaxel treatment (Cross et al., 2021). On the other hand, Lehrer and Rheinstein recently proposed that transdermal patches over the cervical spine could revolutionize drug therapy for AD, and may avoid the systemic side effects of the paclitaxel, such as anemia, leukopenia or peripheral neuropathy (Lehrer and Rheinstein, 2019).
4. NAP (Davunetide): this agent protects MTs against degradation induced by numerous MT disrupting agents, rendering it a possible potent drug against neurodegenerative diseases. The clinical trials performed in humans with tauopathies had no positive outcome (Ivashko-Pachima and Gozes, 2021). In a phase II double-blind randomized controlled trial, NAP showed cognitive and functional improvement in mild cognitive impairment (MCI) patients, after a 12-week intranasal NAP administration (reviewed by Yu et al. (2021). By the time this review is being written, clinical trials on MCI, progressive supranuclear palsy and schizophrenia have all been discontinued, and the one for frontotemporal dementia is inactive (as per alzforum webpage 20.09.2021).
As observed in these three examples, MT stabilizers have severe side effects (Chiorazzi et al., 2009; Brandt and Bakota, 2017), and although they might help in neurodegenerative diseases, they require very fine-tuning to avoid toxicity.
Drugs Targeting Tubulin Posttranslational Modifications
The only tubulin PTM that has been targeted thus far has been acetylation. There are several histone deacetylases (HDACs) which deacetylase α-tubulin, such as HDAC6 and SIRT2 (Hubbert et al., 2002; North et al., 2003). There are studied that show prevention of cognitive decline upon inhibition of SIRT2 (Diaz-Perdigon et al., 2020). Positive effects were also observed with HDAC6 inhibitors (Selenica et al., 2014). Vorinostat is an HDAC inhibitor which tolerable doses is being studied in patients with mild AD (clinical trial identifier: NCT03056495).
For a more detailed review on cytoskeleton-targeted drugs, refer to Eira et al. (2016).
Drugs Targeting Necroptosis May Stabilize Microtubule
Necrostatin-1 (Nec-1) is an anti-necroptotic molecule that directly targets Aβ and tau proteins, alleviates brain cell death and ameliorates cognitive impairment in AD models (Yang et al., 2017). Via targeting and reducing both Aβ oligomers and hyperphosphorylation of tau protein, it may also improve the MT stabilization and may serve an important role in the development of preventive approach for AD. These findings come from an animal APP/PS1 model (Yang et al., 2017), and have been recently extended to a vascular animal model with bilateral common carotid artery stenosis. Namely, in the latter vascular animal model, Nec-1 improved animal behavior and enhanced the inhibitory effect of environment enrichment on inflammation response (Zhang et al., 2019).
Another necroptosis inhibitor, necrosulfonamide (NSA), has also been investigated in a rat model of AD. Administration of NSA intraperitoneally for 6 weeks alleviated phosphorylated tau protein and Aβ accumulation, and regulated the high hippocampal expression of tumor necrosis factor-alpha (TNF-α), β-site amyloid precursor protein cleaving enzyme 1 (BACE1), glycogen synthase kinase-3β (GSK-3β), and acetylcholinesterase (Motawi et al., 2020). NSA has been identified as a novel promising anti-AD treatment via targeting necroptosis, and possibly indirectly contributing to MT stabilization in AD.
All the evidence for the necroptotic agents in AD treatment comes from animal studies, and no human clinical trials are being conducted Similarly, the evidence of these agents upon the MT network, as a result of their effect upon hyperphosphorylated tau protein and Aβ, is still missing.
Conclusion
AD and VaD represent almost 85% of all clinically diagnosed dementia cases, percentage that raises if we take mixed dementia into account, and neuronal loss is a hallmark in all of them. The neuronal loss is not only the best predictor for cognitive decline, but in AD and VaD it also correlates with the severity of the disease, as well as with Braak stages (Buchanan et al., 2020). On a morphological level, neuronal loss results in significant changes in the cytoskeleton of the cells, and consequently, in the MTs. Stability of MTs is, thus, essential for the physiological functioning of neurons.
Tau and MAP2 are MTBP involved in regulating the dynamics and assembly of MTs. In AD, tau is hyperphosphorylated and leads to neuronal loss. Polyglutamylation also regulates the interaction between tau and tubulin, where the more polyglutamylated the tubulin is, the less affinity it has to binding tau, ultimately leading to destabilization of MTs (Eddé et al., 1990). Although this was shown already in 1990, we still don’t know what exactly happens to MT-MTBP interactions in neurodegenerative diseases. Understanding the interplay between tubulin, its PTMs and the binding affinity to MTBPs may help determine if there is a specific event that can prevent or stop neurodegeneration.
Maintaining the levels of tubulin polyglutamylation has proven to be essential for neuronal survival. In fact, it has been shown that there is a tendency for increased α-tubulin polyglutamylation in AD brains when compared to controls (Zhang et al., 2015). However, the number of subjects in this study was relatively low, and a larger number of brain samples should be studied. In addition, tyrosinated levels of tubulin were also measured, with the same outcome. Increasing the number of subjects in these studies is very important. Determining whether polyglutamylation and tyrosination play a role in AD and VaD could have clinical relevance that might lead to the development of new and efficient dementia treatments. It may be possible that tubulin plays a more important role than we know in these diseases, and that its stabilization and PTMs could have a big impact on the severity and progression of the diseases.
Even though there is no increase of phosphorylated tau protein in VaD patients, there is a decrease in the levels of total tau protein (Mukaetova-Ladinska et al., 2015). It is possible that changes in tubulin, such as an increase in polyglutamylation, may lead to a decreased affinity for tau, changes in MT dynamics and increasing the ratio of shrinkage to growth. This could be one plausible explanation for the observed neuronal loss in VaD (Gemmell et al., 2012). It is worth mentioning that some authors state tau works through a “gain of toxicity” mechanism, and that tau-lowering levels might be beneficial in AD and other tauopathies (Goedert, 2016). However, we do not speculate that a decrease in tau could lead to MT destabilization, but that the opposite might happen: tubulin PTMs could lead to molecular changes in tau that, ultimately, could take part in the destabilization of MTs. Even though there is no much literature linking VaD with axonal transport disruption, there are a couple of studies that do. One of them suggests that compromised axonal transport might play an important role in a type of VaD (Binswanger’s disease) (Akiguchi et al., 1997). The other one states that since white matter is often affected in VaD, likely both demyelination and focal axonal injury occurs in VaD (Ihara et al., 2010). Therefore, it is fundamental for in-depth studying of VaD to understand the molecular mechanisms as well as the MT-MTBP interactions to aid the pharmacological treatment of VaD.
It is unclear whether MT abnormalities have a causal and early role in the disease process or represent a common end point downstream of the neurodegenerative cascade. Their presence in absence of overt Aβ and tau pathology, as it is the case in VaD, would argue that they may be an early change, though the MT destabilization as seen in AD argues for the opposite. Understanding the chronology of this triad of tightly woven molecular events will provide a new window of opportunity to arrest the dementia process in early, preclinical stages. It is intriguing to speculate that the chronology of events may differ among different dementia syndromes, and thus drive the design of novel dementia-specific therapies.
Restoring the MT network and the balance between labile and stable MTs might be fundamental to stop the progression of cell death and neurodegeneration. We believe that regulation of MTs by the different PTMs might be key on determining if and with how much affinity does tubulin bind to MTBPs. These changes might not affect the cells immediately, but they might accumulate over time and have a progressively negative impact on the brain. What if tubulin is the key link that is missing? What if the accumulation of proteins and gradual synaptic loss comes from a previous gradual accumulation of tubulin PTMs? What if these accumulations or sudden increase in some of the PTMs lead to a strong enough cell destabilization that produces the neuronal loss in neurodegenerative diseases? These are only some of the questions that may help guide the development of the new generation anti-dementia treatments.
Author Contributions
ES-M wrote the article. RL-C, FG, RK, and EM-L contributed to the discussion, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This financial support for this research has been provided by a Ph.D. grant by the University of Leicester (ES-M/EBM-L).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, F. J., Pienkowski, T. P., and Baas, P. W. (1993). Regional differences in microtubule dynamics in the axon. J. Neurosci. 13, 856–866. doi: 10.1523/jneurosci.13-02-00856.1993
Aillaud, C., Bosc, C., Peris, L., Bosson, A., Heemeryck, P., Van Dijk, J., et al. (2017). Vasohibins/SVBP are tubulin carboxypeptidases (TCPs) that regulate neuron differentiation. Science 358, 1448–1453. doi: 10.1126/science.aao4165
Akella, J. S., Wloga, D., Kim, J., Starostina, N. G., Lyons-Abbott, S., Morrissette, N. S., et al. (2010). MEC-17 is an alpha-tubulin acetyltransferase. Nature 467, 218–222. doi: 10.1038/nature09324
Akiguchi, I., Tomimoto, H., Suenaga, T., Wakita, H., and Budka, H. (1997). Alterations in glia and axons in the brains of Binswanger’s disease patients. Stroke 28, 1423–1429. doi: 10.1161/01.str.28.7.1423
Akinyemi, R. O., Mukaetova-Ladinska, E. B., Attems, J., Ihara, M., and Kalaria, R. N. (2013). Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr. Alzheimer Res. 10, 642–653. doi: 10.2174/15672050113109990037
Alvarez, P., Smith, A., Fleming, J., and Solomon, F. (1998). Modulation of tubulin polypeptide ratios by the yeast protein Pac10p. Genetics 149, 857–864.
Andrade-Moraes, C. H., Oliveira-Pinto, A. V., Castro-Fonseca, E., da Silva, C. G., Guimarães, D. M., Szczupak, D., et al. (2013). Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136(Pt. 12), 3738–3752. doi: 10.1093/brain/awt273
Arendt, T., Brückner, M. K., Morawski, M., Jäger, C., and Gertz, H. J. (2015). Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol. Commun. 3:10. doi: 10.1186/s40478-015-0187-1
Baas, P. W., and Qiang, L. (2019). Tau: it’s not what you think. Trends Cell Biol. 29, 452–461. doi: 10.1016/j.tcb.2019.02.007
Baazaoui, N., Flory, M., and Iqbal, K. (2017). Synaptic compensation as a probable cause of prolonged mild cognitive impairment in Alzheimer’s disease: implications from a transgenic mouse model of the disease. J. Alzheimers Dis. 56, 1385–1401. doi: 10.3233/jad-160845
Barisic, M., Silva e Sousa, R., Tripathy, S. K., Magiera, M. M., Zaytsev, A. V., Pereira, A. L., et al. (2015). Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 348, 799–803. doi: 10.1126/science.aaa5175
Barten, D. M., Fanara, P., Andorfer, C., Hoque, N., Wong, P. Y., Husted, K. H., et al. (2012). Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J. Neurosci. 32, 7137–7145. doi: 10.1523/jneurosci.0188-12.2012
Beggiato, S., Cassano, T., Ferraro, L., and Tomasini, M. C. (2020). Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer’s disease. Life Sci. 257:118037. doi: 10.1016/j.lfs.2020.118037
Belkhelfa, M., Beder, N., Mouhoub, D., Amri, M., Hayet, R., Tighilt, N., et al. (2018). The involvement of neuroinflammation and necroptosis in the hippocampus during vascular dementia. J. Neuroimmunol. 320, 48–57. doi: 10.1016/j.jneuroim.2018.04.004
Berezniuk, I., Lyons, P. J., Sironi, J. J., Xiao, H., Setou, M., Angeletti, R. H., et al. (2013). Cytosolic carboxypeptidase 5 removes α- and γ-linked glutamates from tubulin. J. Biol. Chem. 288, 30445–30453. doi: 10.1074/jbc.M113.497917
Bonfoco, E., Ceccatelli, S., Manzo, L., and Nicotera, P. (1995). Colchicine induces apoptosis in cerebellar granule cells. Exp. Cell Res. 218, 189–200. doi: 10.1006/excr.1995.1147
Brandt, R., and Bakota, L. (2017). Microtubule dynamics and the neurodegenerative triad of Alzheimer’s disease: the hidden connection. J. Neurochem. 143, 409–417. doi: 10.1111/jnc.14011
Brion, J. P., Anderton, B. H., Authelet, M., Dayanandan, R., Leroy, K., Lovestone, S., et al. (2001). Neurofibrillary tangles and tau phosphorylation. Biochem. Soc. Symp. 67, 81–88. doi: 10.1042/bss0670081
Bristol-Myers Squibb (2013). Therapeutics: Epothilone D. Available online at: https://www.alzforum.org/therapeutics/epothilone-d
Brouhard, G. J. (2015). Dynamic instability 30 years later: complexities in microtubule growth and catastrophe. Mol. Biol. Cell 26, 1207–1210. doi: 10.1091/mbc.E13-10-0594
Bryan, J., and Wilson, L. (1971). Are cytoplasmic microtubules heteropolymers? Proc. Natl. Acad. Sci. U.S.A. 68, 1762–1766. doi: 10.1073/pnas.68.8.1762
Buchanan, H., Mackay, M., Palmer, K., Tothová, K., Katsur, M., Platt, B., et al. (2020). Synaptic loss, ER stress and neuro-inflammation emerge late in the lateral temporal cortex and associate with progressive tau pathology in Alzheimer’s disease. Mol. Neurobiol. 57, 3258–3272. doi: 10.1007/s12035-020-01950-1
Burbaeva, G. S., Androsova, L. V., and Savushkina, O. K. (2020). Binding of colchicine to tubulin in the brain structures in normal conditions and in schizophrenia. Neurochem. J. 14, 235–238. doi: 10.1134/S1819712420010067
Bussière, T., Giannakopoulos, P., Bouras, C., Perl, D. P., Morrison, J. H., and Hof, P. R. (2003). Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: stereologic analysis of prefrontal cortex area 9. J. Comp. Neurol. 463, 281–302. doi: 10.1002/cne.10760
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246. doi: 10.1038/nn.4608
Carnwath, T., Mohammed, R., and Tsiang, D. (2018). The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 14, 1685–1695. doi: 10.2147/ndt.S166322
Caron, J. M. (1997). Posttranslational modification of tubulin by palmitoylation: I. in vivo and cell-free studies. Mol. Biol. Cell 8, 621–636. doi: 10.1091/mbc.8.4.621
Cheng, H., and Huang, G. (2018). Synthesis and activity of epothilone D. Curr. Drug Targets 19, 1866–1870. doi: 10.2174/1389450119666180803122118
Chi, H., Chang, H. Y., and Sang, T. K. (2018). Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 19:3082. doi: 10.3390/ijms19103082
Chiorazzi, A., Nicolini, G., Canta, A., Oggioni, N., Rigolio, R., Cossa, G., et al. (2009). Experimental epothilone B neurotoxicity: results of in vitro and in vivo studies. Neurobiol. Dis. 35, 270–277. doi: 10.1016/j.nbd.2009.05.006
Chu, C. W., Hou, F., Zhang, J., Phu, L., Loktev, A. V., Kirkpatrick, D. S., et al. (2011). A novel acetylation of β-tubulin by san modulates microtubule polymerization via down-regulating tubulin incorporation. Mol. Biol. Cell 22, 448–456. doi: 10.1091/mbc.E10-03-0203
Clark, J. A., Chuckowree, J. A., Dyer, M. S., Dickson, T. C., and Blizzard, C. A. (2020). Epothilone D alters normal growth, viability and microtubule dependent intracellular functions of cortical neurons in vitro. Sci. Rep. 10:918. doi: 10.1038/s41598-020-57718-z
Cleveland, D. W., Lopata, M. A., MacDonald, R. J., Cowan, N. J., Rutter, W. J., and Kirschner, M. W. (1980). Number and evolutionary conservation of alpha- and beta-tubulin and cytoplasmic beta- and gamma-actin genes using specific cloned cDNA probes. Cell 20, 95–105. doi: 10.1016/0092-8674(80)90238-x
Coleman, P., Federoff, H., and Kurlan, R. (2004). A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63, 1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a
Cross, D. J., Huber, B. R., Silverman, M. A., Cline, M. M., Gill, T. B., Cross, C. G., et al. (2021). Intranasal paclitaxel alters Alzheimer’s disease phenotypic features in 3xTg-AD mice. J. Alzheimers Dis. 83, 379–394. doi: 10.3233/jad-210109
Cross, D. J., Meabon, J. S., Cline, M. M., Richards, T. L., Stump, A. J., Cross, C. G., et al. (2019). Paclitaxel reduces brain injury from repeated head trauma in mice. J. Alzheimers Dis. 67, 859–874. doi: 10.3233/jad-180871
Daire, V., Giustiniani, J., Leroy-Gori, I., Quesnoit, M., Drevensek, S., Dimitrov, A., et al. (2009). Kinesin-1 regulates microtubule dynamics via a c-Jun N-terminal kinase-dependent mechanism. J. Biol. Chem. 284, 31992–32001. doi: 10.1074/jbc.M109.007906
D’Arcy, M. S. (2019). Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 43, 582–592. doi: 10.1002/cbin.11137
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Diaz-Perdigon, T., Belloch, F. B., Ricobaraza, A., Elboray, E. E., Suzuki, T., Tordera, R. M., et al. (2020). Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacology 45, 347–357. doi: 10.1038/s41386-019-0503-8
Dompierre, J. P., Godin, J. D., Charrin, B. C., Cordelières, F. P., King, S. J., Humbert, S., et al. (2007). Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583. doi: 10.1523/jneurosci.0037-07.2007
Eddé, B., Rossier, J., Le Caer, J. P., Desbruyères, E., Gros, F., and Denoulet, P. (1990). Posttranslational glutamylation of alpha-tubulin. Science 247, 83–85. doi: 10.1126/science.1967194
Eira, J., Silva, C. S., Sousa, M. M., and Liz, M. A. (2016). The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 141, 61–82. doi: 10.1016/j.pneurobio.2016.04.007
Fan, Y., Cui, Y., Hao, W., Chen, M., Liu, Q., Wang, Y., et al. (2021). Carrier-free highly drug-loaded biomimetic nanosuspensions encapsulated by cancer cell membrane based on homology and active targeting for the treatment of glioma. Bioact. Mater. 6, 4402–4414. doi: 10.1016/j.bioactmat.2021.04.027
Fang, Y. C., Chan, L., Liou, J. P., Tu, Y. K., Lai, M. J., Chen, C. I., et al. (2020). HDAC inhibitor protects chronic cerebral hypoperfusion and oxygen-glucose deprivation injuries via H3K14 and H4K5 acetylation-mediated BDNF expression. J. Cell Mol. Med. 24, 6966–6977. doi: 10.1111/jcmm.15358
Felgner, H., Frank, R., Biernat, J., Mandelkow, E. M., Mandelkow, E., Ludin, B., et al. (1997). Domains of neuronal microtubule-associated proteins and flexural rigidity of microtubules. J. Cell Biol. 138, 1067–1075. doi: 10.1083/jcb.138.5.1067
Fernández-Barrera, J., Correas, I., and Alonso, M. A. (2018). Age-related neuropathies and tubulin acetylation. Aging 10, 524–525. doi: 10.18632/aging.101432
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., et al. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23, 9418–9427. doi: 10.1523/jneurosci.23-28-09418.2003
Fifre, A., Sponne, I., Koziel, V., Kriem, B., Yen Potin, F. T., Bihain, B. E., et al. (2006). Microtubule-associated protein MAP1A, MAP1B, and MAP2 proteolysis during soluble amyloid beta-peptide-induced neuronal apoptosis. Synergistic involvement of calpain and caspase-3. J. Biol. Chem. 281, 229–240. doi: 10.1074/jbc.M507378200
Fischer, I., and Baas, P. W. (2020). Resurrecting the mysteries of big tau. Trends Neurosci. 43, 493–504. doi: 10.1016/j.tins.2020.04.007
Fourest-Lieuvin, A., Peris, L., Gache, V., Garcia-Saez, I., Juillan-Binard, C., Lantez, V., et al. (2006). Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol. Biol. Cell 17, 1041–1050. doi: 10.1091/mbc.e05-07-0621
Fukushima, N., Furuta, D., Hidaka, Y., Moriyama, R., and Tsujiuchi, T. (2009). Post-translational modifications of tubulin in the nervous system. J. Neurochem. 109, 683–693. doi: 10.1111/j.1471-4159.2009.06013.x
Gadadhar, S., Bodakuntla, S., Natarajan, K., and Janke, C. (2017). The tubulin code at a glance. J. Cell Sci. 130, 1347–1353. doi: 10.1242/jcs.199471
Gallart-Palau, X., Serra, A., Qian, J., Chen, C. P., Kalaria, R. N., and Sze, S. K. (2015). Temporal lobe proteins implicated in synaptic failure exhibit differential expression and deamidation in vascular dementia. Neurochem Int 80, 87–98. doi: 10.1016/j.neuint.2014.12.002
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Gemmell, E., Bosomworth, H., Allan, L., Hall, R., Khundakar, A., Oakley, A. E., et al. (2012). Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 43, 808–814. doi: 10.1161/strokeaha.111.636498
Gevorkian, G., Gonzalez-Noriega, A., Acero, G., Ordoñez, J., Michalak, C., Munguia, M. E., et al. (2008). Amyloid-beta peptide binds to microtubule-associated protein 1B (MAP1B). Neurochem. Int. 52, 1030–1036. doi: 10.1016/j.neuint.2007.10.020
Giannakopoulos, P., Gold, G., von Gunten, A., Hof, P. R., and Bouras, C. (2009). Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 24:20–29. doi: 10.1159/000197881
Goedert, M. (2016). The ordered assembly of tau is the gain-of-toxic function that causes human tauopathies. Alzheimers Dement. 12, 1040–1050. doi: 10.1016/j.jalz.2016.09.001
Goodson, H. V., and Jonasson, E. M. (2018). Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 10:a022608. doi: 10.1101/cshperspect.a022608
Gornstein, E., and Schwarz, T. L. (2014). The paradox of paclitaxel neurotoxicity: mechanisms and unanswered questions. Neuropharmacology 76(Pt. A), 175–183. doi: 10.1016/j.neuropharm.2013.08.016
Govindpani, K., McNamara, L. G., Smith, N. R., Vinnakota, C., Waldvogel, H. J., Faull, R. L., et al. (2019). Vascular Dysfunction in Alzheimer’s disease: a prelude to the pathological process or a consequence of it? J. Clin. Med. 8:651. doi: 10.3390/jcm8050651
Hasegawa, S., Miyoshi, Y., Egawa, C., Ishitobi, M., Taguchi, T., Tamaki, Y., et al. (2003). Prediction of response to docetaxel by quantitative analysis of class I and III beta-tubulin isotype mRNA expression in human breast cancers. Clin. Cancer Res. 9, 2992–2997.
Himmler, A., Drechsel, D., Kirschner, M. W., and Martin, D. W. Jr. (1989). Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell Biol. 9, 1381–1388. doi: 10.1128/mcb.9.4.1381
Horio, T., and Murata, T. (2014). The role of dynamic instability in microtubule organization. Front. Plant Sci. 5:511. doi: 10.3389/fpls.2014.00511
Hubbert, C., Guardiola, A., Shao, R., Kawaguchi, Y., Ito, A., Nixon, A., et al. (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. doi: 10.1038/417455a
Hyman, A. A., Chrétien, D., Arnal, I., and Wade, R. H. (1995). Structural changes accompanying GTP hydrolysis in microtubules: information from a slowly hydrolyzable analogue guanylyl-(alpha,beta)-methylene-diphosphonate. J. Cell Biol. 128, 117–125. doi: 10.1083/jcb.128.1.117
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Ihara, M., Polvikoski, T. M., Hall, R., Slade, J. Y., Perry, R. H., Oakley, A. E., et al. (2010). Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol. 119, 579–589. doi: 10.1007/s00401-009-0635-8
Ikegami, K., Sato, S., Nakamura, K., Ostrowski, L. E., and Setou, M. (2010). Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proc. Natl. Acad. Sci. U.S.A. 107, 10490–10495. doi: 10.1073/pnas.1002128107
Ivashko-Pachima, Y., and Gozes, I. (2021). Activity-dependent neuroprotective protein (ADNP)-end-binding protein (EB) interactions regulate microtubule dynamics toward protection against tauopathy. Prog. Mol. Biol. Transl. Sci. 177, 65–90. doi: 10.1016/bs.pmbts.2020.07.008
Janke, C. (2014). The tubulin code: molecular components, readout mechanisms, and functions. J. Cell Biol. 206, 461–472. doi: 10.1083/jcb.201406055
Janke, C., and Bulinski, J. C. (2011). Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773–786. doi: 10.1038/nrm3227
Janke, C., and Magiera, M. M. (2020). The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 21, 307–326. doi: 10.1038/s41580-020-0214-3
Janning, D., Igaev, M., Sündermann, F., Brühmann, J., Beutel, O., Heinisch, J. J., et al. (2014). Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell 25, 3541–3551. doi: 10.1091/mbc.E14-06-1099
Jellinger, K. A. (2013). Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front. Aging Neurosci. 5:17. doi: 10.3389/fnagi.2013.00017
Kalaria, R. N. (2018). The pathology and pathophysiology of vascular dementia. Neuropharmacology 134(Pt. B), 226–239. doi: 10.1016/j.neuropharm.2017.12.030
Kalaria, R. N., and Ballard, C. (1999). Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis. Assoc. Disord. 13(Suppl. 3), S115–S123. doi: 10.1097/00002093-199912003-00017
Kandimalla, R., Manczak, M., Yin, X., Wang, R., and Reddy, P. H. (2018). Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 30–40. doi: 10.1093/hmg/ddx381
Katsetos, C. D., Legido, A., Perentes, E., and Mörk, S. J. (2003). Class III beta-tubulin isotype: a key cytoskeletal protein at the crossroads of developmental neurobiology and tumor neuropathology. J. Child Neurol. 18, 851–866;discussion 867. doi: 10.1177/088307380301801205
Kimura, Y., Kurabe, N., Ikegami, K., Tsutsumi, K., Konishi, Y., Kaplan, O. I., et al. (2010). Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 285, 22936–22941. doi: 10.1074/jbc.C110.128280
Kril, J. J., Patel, S., Harding, A. J., and Halliday, G. M. (2002). Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss. J. Neurol. Neurosurg. Psychiatry 72, 747–751. doi: 10.1136/jnnp.72.6.747
Kumar, R. A., Pilz, D. T., Babatz, T. D., Cushion, T. D., Harvey, K., Topf, M., et al. (2010). TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 19, 2817–2827. doi: 10.1093/hmg/ddq182
Lehrer, S., and Rheinstein, P. H. (2019). Transspinal delivery of drugs by transdermal patch back-of-neck for Alzheimer’s disease: a new route of administration. Discov. Med. 27, 37–43.
Lewis, H., Beher, D., Cookson, N., Oakley, A., Piggott, M., Morris, C. M., et al. (2006). Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 32, 103–118. doi: 10.1111/j.1365-2990.2006.00696.x
L’Hernault, S. W., and Rosenbaum, J. L. (1985). Chlamydomonas alpha-tubulin is posttranslationally modified by acetylation on the epsilon-amino group of a lysine. Biochemistry 24, 473–478. doi: 10.1021/bi00323a034
Li, Y., and Black, M. M. (1996). Microtubule assembly and turnover in growing axons. J. Neurosci. 16, 531–544. doi: 10.1523/JNEUROSCI.16-02-00531.1996
Li, L., and Yang, X. J. (2015). Tubulin acetylation: responsible enzymes, biological functions and human diseases. Cell Mol. Life Sci. 72, 4237–4255. doi: 10.1007/s00018-015-2000-5
Li, M., Mukaetova-Ladinska, E. B., and Kalaria, R. N. (2014). Tau Protein Changes in Vascular Dementia. Master thesis.
Liang, S., Zheng, Y., Lei, L., Deng, X., Ai, J., Li, Y., et al. (2020). Corydalis edulis total alkaloids (CETA) ameliorates cognitive dysfunction in rat model of Alzheimer disease through regulation of the antioxidant stress and MAP2/NF-κB. J. Ethnopharmacol. 251:112540. doi: 10.1016/j.jep.2019.112540
Liepins, A., and Bustamante, J. O. (1994). Cell injury and apoptosis. Scanning Microsc. 8, 631–641;discussion 641–633.
Liu, M., Sui, D., Dexheimer, T., Hovde, S., Deng, X., Wang, K. W., et al. (2020). Hyperphosphorylation renders tau prone to aggregate and to cause cell death. Mol. Neurobiol. 57, 4704–4719. doi: 10.1007/s12035-020-02034-w
Ludueña, R. F. (1998). Multiple forms of tubulin: different gene products and covalent modifications. Int. Rev. Cytol. 178, 207–275. doi: 10.1016/s0074-7696(08)62138-5
Ludueña, R. F. (2013). A hypothesis on the origin and evolution of tubulin. Int. Rev. Cell Mol. Biol. 302, 41–185. doi: 10.1016/b978-0-12-407699-0.00002-9
Ludueńa, R. F., Shooter, E. M., and Wilson, L. (1977). Structure of the tubulin dimer. J. Biol. Chem. 252, 7006–7014.
Magiera, M. M., and Janke, C. (2014). Post-translational modifications of tubulin. Curr. Biol. 24, R351–R354. doi: 10.1016/j.cub.2014.03.032
Magiera, M. M., Bodakuntla, S., Žiak, J., Lacomme, S., Marques Sousa, P., Leboucher, S., et al. (2018). Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37:e100440. doi: 10.15252/embj.2018100440
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2018). Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 1332–1342. doi: 10.1093/hmg/ddy042
Mandelkow, E., and Mandelkow, E. M. (1995). Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 7, 72–81. doi: 10.1016/0955-0674(95)80047-6
Martínez-Pinilla, E., Ordóñez, C., Del Valle, E., Navarro, A., and Tolivia, J. (2016). Regional and gender study of neuronal density in brain during aging and in Alzheimer’s disease. Front. Aging Neurosci. 8:213. doi: 10.3389/fnagi.2016.00213
McAleese, K. E., Alafuzoff, I., Charidimou, A., De Reuck, J., Grinberg, L. T., Hainsworth, A. H., et al. (2016). Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med. 14:129. doi: 10.1186/s12916-016-0676-5
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Melková, K., Zapletal, V., Narasimhan, S., Jansen, S., Hritz, J., Škrabana, R., et al. (2019). Structure and functions of microtubule associated proteins tau and MAP2c: similarities and differences. Biomolecules 9:105. doi: 10.3390/biom9030105
Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. doi: 10.1038/312237a0
Motawi, T. M. K., Abdel-Nasser, Z. M., and Shahin, N. N. (2020). Ameliorative effect of necrosulfonamide in a rat model of Alzheimer’s disease: targeting mixed lineage kinase domain-like protein-mediated necroptosis. ACS Chem. Neurosci. 11, 3386–3397. doi: 10.1021/acschemneuro.0c00516
Mukaetova-Ladinska, E. B., Abdel-All, Z., Mugica, E. S., Li, M., Craggs, L. J., Oakley, A. E., et al. (2015). Tau proteins in the temporal and frontal cortices in patients with vascular dementia. J. Neuropathol. Exp. Neurol. 74, 148–157. doi: 10.1097/nen.0000000000000157
Mukaetova-Ladinska, E. B., Harrington, C. R., Roth, M., and Wischik, C. M. (1993). Biochemical and anatomical redistribution of tau protein in Alzheimer’s disease. Am. J. Pathol. 143, 565–578.
Mukaetova-Ladinska, E. B., Xuereb, J. H., Garcia-Sierra, F., Hurt, J., Gertz, H. J., Hills, R., et al. (2009). Lewy body variant of Alzheimer’s disease: selective neocortical loss of t-SNARE proteins and loss of MAP2 and alpha-synuclein in medial temporal lobe. ScientificWorldJournal 9, 1463–1475. doi: 10.1100/tsw.2009.151
Murphy, D. B., Wallis, K. T., Machlin, P. S., Ratrie, H. III, and Cleveland, D. W. (1987). The sequence and expression of the divergent beta-tubulin in chicken erythrocytes. J. Biol. Chem. 262, 14305–14312.
Nixon, R. A., and Yang, D. S. (2011). Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol. Dis. 43, 38–45. doi: 10.1016/j.nbd.2011.01.021
Nogales, E. (2013). “Tubulin and Its Isoforms,” in Encyclopedia of Biological Chemistry, 2nd Edn, eds W. J. Lennarz and M. D. Lane (Waltham, MA: Academic Press), 450–453.
Nogales, E., and Wang, H. W. (2006). Structural intermediates in microtubule assembly and disassembly: how and why? Curr. Opin. Cell Biol. 18, 179–184. doi: 10.1016/j.ceb.2006.02.009
Nogales, E., Wolf, S. G., and Downing, K. H. (1998). Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391, 199–203. doi: 10.1038/34465
North, B. J., Marshall, B. L., Borra, M. T., Denu, J. M., and Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444. doi: 10.1016/s1097-2765(03)00038-8
Oblinger, M. M., Argasinski, A., Wong, J., and Kosik, K. S. (1991). Tau gene expression in rat sensory neurons during development and regeneration. J. Neurosci. 11, 2453–2459. doi: 10.1523/jneurosci.11-08-02453.1991
Ohishi, Y., Oda, Y., Basaki, Y., Kobayashi, H., Wake, N., Kuwano, M., et al. (2007). Expression of beta-tubulin isotypes in human primary ovarian carcinoma. Gynecol. Oncol. 105, 586–592. doi: 10.1016/j.ygyno.2007.01.044
Pathak, N., Obara, T., Mangos, S., Liu, Y., and Drummond, I. A. (2007). The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol. Biol. Cell 18, 4353–4364. doi: 10.1091/mbc.e07-06-0537
Paturle-Lafanechère, L., Manier, M., Trigault, N., Pirollet, F., Mazarguil, H., and Job, D. (1994). Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J. Cell Sci. 107(Pt. 6), 1529–1543.
Penazzi, L., Bakota, L., and Brandt, R. (2016). Microtubule dynamics in neuronal development, plasticity, and neurodegeneration. Int. Rev. Cell Mol. Biol. 321, 89–169. doi: 10.1016/bs.ircmb.2015.09.004
Peng, C., Gathagan, R. J., and Lee, V. M. (2018). Distinct α-Synuclein strains and implications for heterogeneity among α-Synucleinopathies. Neurobiol. Dis. 109(Pt. B), 209–218. doi: 10.1016/j.nbd.2017.07.018
Peters, J. D., Furlong, M. T., Asai, D. J., Harrison, M. L., and Geahlen, R. L. (1996). Syk, activated by cross-linking the B-cell antigen receptor, localizes to the cytosol where it interacts with and phosphorylates alpha-tubulin on tyrosine. J. Biol. Chem. 271, 4755–4762. doi: 10.1074/jbc.271.9.4755
Prota, A. E., Magiera, M. M., Kuijpers, M., Bargsten, K., Frey, D., Wieser, M., et al. (2013). Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 200, 259–270. doi: 10.1083/jcb.201211017
Qiu, C., Kivipelto, M., and von Strauss, E. (2009). Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 11, 111–128.
Reddy, P. H., Yin, X., Manczak, M., Kumar, S., Pradeepkiran, J. A., Vijayan, M., et al. (2018). Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 27, 2502–2516. doi: 10.1093/hmg/ddy154
Redeker, V. (2010). Mass spectrometry analysis of C-terminal posttranslational modifications of tubulins. Methods Cell Biol. 95, 77–103. doi: 10.1016/s0091-679x(10)95006-1
Redeker, V., Levilliers, N., Schmitter, J. M., Le Caer, J. P., Rossier, J., Adoutte, A., et al. (1994). Polyglycylation of tubulin: a posttranslational modification in axonemal microtubules. Science 266, 1688–1691. doi: 10.1126/science.7992051
Ren, Y., Zhao, J., and Feng, J. (2003). Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci 23, 3316–3324. doi: 10.1523/jneurosci.23-08-03316.2003
Rocha, C., Papon, L., Cacheux, W., Marques Sousa, P., Lascano, V., Tort, O., et al. (2014). Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon. EMBO J. 33, 2247–2260. doi: 10.15252/embj.201488466
Rogowski, K., van Dijk, J., Magiera, M. M., Bosc, C., Deloulme, J. C., Bosson, A., et al. (2010). A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143, 564–578. doi: 10.1016/j.cell.2010.10.014
Rosas-Acosta, G., Russell, W. K., Deyrieux, A., Russell, D. H., and Wilson, V. G. (2005). A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol. Cell Proteom. 4, 56–72. doi: 10.1074/mcp.M400149-MCP200
Savastano, A., Flores, D., Kadavath, H., Biernat, J., Mandelkow, E., and Zweckstetter, M. (2021). Disease-associated tau phosphorylation hinders tubulin assembly within tau condensates. Angew. Chem. Int. Edn. Engl. 60, 726–730. doi: 10.1002/anie.202011157
Selenica, M. L., Benner, L., Housley, S. B., Manchec, B., Lee, D. C., Nash, K. R., et al. (2014). Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 6:12. doi: 10.1186/alzrt241
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Sferra, A., Nicita, F., and Bertini, E. (2020). Microtubule dysfunction: a common feature of neurodegenerative diseases. Int. J. Mol. Sci. 21:7354. doi: 10.3390/ijms21197354
Shamitko-Klingensmith, N., Boyd, J. W., and Legleiter, J. (2016). Microtubule modification influences cellular response to amyloid-β exposure. AIMS Biophys. 3, 261–285. doi: 10.3934/biophy.2016.2.261
Shashi, V., Magiera, M. M., Klein, D., Zaki, M., Schoch, K., Rudnik-Schöneborn, S., et al. (2018). Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 37:e100540. doi: 10.15252/embj.2018100540
Šimić, G., Babić Leko, M., Wray, S., Harrington, C., Delalle, I., Jovanov-Milošević, N., et al. (2016). Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6:6. doi: 10.3390/biom6010006
Skrobot, O. A., Black, S. E., Chen, C., DeCarli, C., Erkinjuntti, T., Ford, G. A., et al. (2018). Progress toward standardized diagnosis of vascular cognitive impairment: guidelines from the vascular impairment of cognition classification consensus study. Alzheimers Dement. 14, 280–292. doi: 10.1016/j.jalz.2017.09.007
Slaughter, T., and Black, M. M. (2003). STOP (stable-tubule-only-polypeptide) is preferentially associated with the stable domain of axonal microtubules. J. Neurocytol. 32, 399–413. doi: 10.1023/b:Neur.0000011334.70648.87
Smith, E. E. (2017). Clinical presentations and epidemiology of vascular dementia. Clin. Sci. 131, 1059–1068. doi: 10.1042/cs20160607
Song, Y., Kirkpatrick, L. L., Schilling, A. B., Helseth, D. L., Chabot, N., Keillor, J. W., et al. (2013). Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron 78, 109–123. doi: 10.1016/j.neuron.2013.01.036
Steinberg, M., Shao, H., Zandi, P., Lyketsos, C. G., Welsh-Bohmer, K. A., Norton, M. C., et al. (2008). Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the cache county study. Int. J. Geriatr. Psychiatry 23, 170–177. doi: 10.1002/gps.1858
Suemoto, C. K., Ferretti-Rebustini, R. E., Rodriguez, R. D., Leite, R. E., Soterio, L., Brucki, S. M., et al. (2017). Neuropathological diagnoses and clinical correlates in older adults in Brazil: a cross-sectional study. PLoS Med. 14:e1002267. doi: 10.1371/journal.pmed.1002267
Sullivan, K. F., and Cleveland, D. W. (1986). Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proc. Natl. Acad. Sci. U.S.A. 83, 4327–4331. doi: 10.1073/pnas.83.12.4327
Suryavanshi, S., Eddé, B., Fox, L. A., Guerrero, S., Hard, R., Hennessey, T., et al. (2010). Tubulin glutamylation regulates ciliary motility by altering inner dynein arm activity. Curr. Biol. 20, 435–440. doi: 10.1016/j.cub.2009.12.062
Telegina, D. V., Suvorov, G. K., Kozhevnikova, O. S., and Kolosova, N. G. (2019). Mechanisms of neuronal death in the cerebral cortex during aging and development of Alzheimer’s disease-like pathology in rats. Int. J. Mol. Sci. 20:5632. doi: 10.3390/ijms20225632
Theofilas, P., Ehrenberg, A. J., Nguy, A., Thackrey, J. M., Dunlop, S., Mejia, M. B., et al. (2018). Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol. Aging 61, 1–12. doi: 10.1016/j.neurobiolaging.2017.09.007
Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87. doi: 10.1016/j.cell.2009.12.011
Tsai, R. M., Miller, Z., Koestler, M., Rojas, J. C., Ljubenkov, P. A., Rosen, H. J., et al. (2020). Reactions to multiple ascending doses of the microtubule stabilizer TPI-287 in patients with alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol. 77, 215–224. doi: 10.1001/jamaneurol.2019.3812
Vu, H. T., Akatsu, H., Hashizume, Y., Setou, M., and Ikegami, K. (2017). Increase in α-tubulin modifications in the neuronal processes of hippocampal neurons in both kainic acid-induced epileptic seizure and Alzheimer’s disease. Sci. Rep. 7:40205. doi: 10.1038/srep40205
Walgren, J. L., Vincent, T. S., Schey, K. L., and Buse, M. G. (2003). High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am. J. Physiol. Endocrinol. Metab. 284, E424–E434. doi: 10.1152/ajpendo.00382.2002
Weaver, B. A. (2014). How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 25, 2677–2681. doi: 10.1091/mbc.E14-04-0916
Westermann, S., and Weber, K. (2003). Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 4, 938–947. doi: 10.1038/nrm1260
Wischik, C. M., Edwards, P. C., Lai, R. Y., Gertz, H. N., Xuereb, J. H., Paykel, E. S., et al. (1995). Quantitative analysis of tau protein in paired helical filament preparations: implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer’s disease. Neurobiol. Aging 16, 409–417;discussion 418–431. doi: 10.1016/0197-4580(95)97327-d
Wloga, D., and Gaertig, J. (2010). Post-translational modifications of microtubules. J. Cell Sci. 123(Pt. 20), 3447–3455. doi: 10.1242/jcs.063727
Wong, C. C., Xu, T., Rai, R., Bailey, A. O., Yates, J. R. III, Wolf, Y. I., et al. (2007). Global analysis of posttranslational protein arginylation. PLoS Biol. 5:e258. doi: 10.1371/journal.pbio.0050258
Xiao, H., El Bissati, K., Verdier-Pinard, P., Burd, B., Zhang, H., Kim, K., et al. (2010). Post-translational modifications to Toxoplasma gondii alpha- and beta-tubulins include novel C-terminal methylation. J. Proteome Res. 9, 359–372. doi: 10.1021/pr900699a
Xie, C., and Miyasaka, T. (2016). The role of the carboxyl-terminal sequence of tau and MAP2 in the pathogenesis of dementia. Front. Mol. Neurosci. 9:158. doi: 10.3389/fnmol.2016.00158
Yang, S. H., Lee, D. K., Shin, J., Lee, S., Baek, S., Kim, J., et al. (2017). Nec-1 alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS1 mice. EMBO Mol. Med. 9, 61–77. doi: 10.15252/emmm.201606566
Yu, I., Garnham, C. P., and Roll-Mecak, A. (2015). Writing and reading the tubulin code. J. Biol. Chem. 290, 17163–17172. doi: 10.1074/jbc.R115.637447
Yu, T. W., Lane, H. Y., and Lin, C. H. (2021). Novel therapeutic approaches for Alzheimer’s disease: an updated review. Int. J. Mol. Sci. 22:8208. doi: 10.3390/ijms22158208
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33. doi: 10.1038/s41583-018-0093-1
Zempel, H., Luedtke, J., Kumar, Y., Biernat, J., Dawson, H., Mandelkow, E., et al. (2013). Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937. doi: 10.1038/emboj.2013.207
Zhang, B., Carroll, J., Trojanowski, J. Q., Yao, Y., Iba, M., Potuzak, J. S., et al. (2012). The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 32, 3601–3611. doi: 10.1523/jneurosci.4922-11.2012
Zhang, F., Su, B., Wang, C., Siedlak, S. L., Mondragon-Rodriguez, S., Lee, H. G., et al. (2015). Posttranslational modifications of α-tubulin in alzheimer disease. Transl. Neurodegener. 4:9. doi: 10.1186/s40035-015-0030-4
Zhang, L., Sheng, S., and Qin, C. (2013). The role of HDAC6 in Alzheimer’s disease. J. Alzheimers Dis. 33, 283–295. doi: 10.3233/jad-2012-120727
Keywords: vascular dementia, Alzheimer’s disease, tubulin, posttranslational modification, tubulin code
Citation: Santiago-Mujika E, Luthi-Carter R, Giorgini F, Kalaria RN and Mukaetova-Ladinska EB (2021) Tubulin and Tubulin Posttranslational Modifications in Alzheimer’s Disease and Vascular Dementia. Front. Aging Neurosci. 13:730107. doi: 10.3389/fnagi.2021.730107
Received: 24 June 2021; Accepted: 04 October 2021;
Published: 29 October 2021.
Edited by:
Natalia Salvadores, Universidad Mayor, ChileReviewed by:
Cathryn Louise Haigh, Rocky Mountain Laboratories (NIAID), United StatesRoland Brandt, University of Osnabrück, Germany
Copyright © 2021 Santiago-Mujika, Luthi-Carter, Giorgini, Kalaria and Mukaetova-Ladinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeta B. Mukaetova-Ladinska, ZW1sMTJAbGUuYWMudWs=