 Sarah Lecordier
Sarah Lecordier Daniel Manrique-Castano
Daniel Manrique-Castano Yara El Moghrabi
Yara El Moghrabi Ayman ElAli
Ayman ElAli- 1Neuroscience Axis, Research Center of CHU de Québec-Université Laval, Québec City, QC, Canada
- 2Department of Psychiatry and Neuroscience, Faculty of Medicine, Université Laval, Québec City, QC, Canada
Vascular dementia (VaD) constitutes the second most prevalent cause of dementia in the world after Alzheimer’s disease (AD). VaD regroups heterogeneous neurological conditions in which the decline of cognitive functions, including executive functions, is associated with structural and functional alterations in the cerebral vasculature. Among these cerebrovascular disorders, major stroke, and cerebral small vessel disease (cSVD) constitute the major risk factors for VaD. These conditions alter neurovascular functions leading to blood-brain barrier (BBB) deregulation, neurovascular coupling dysfunction, and inflammation. Accumulation of neurovascular impairments over time underlies the cognitive function decline associated with VaD. Furthermore, several vascular risk factors, such as hypertension, obesity, and diabetes have been shown to exacerbate neurovascular impairments and thus increase VaD prevalence. Importantly, air pollution constitutes an underestimated risk factor that triggers vascular dysfunction via inflammation and oxidative stress. The review summarizes the current knowledge related to the pathological mechanisms linking neurovascular impairments associated with stroke, cSVD, and vascular risk factors with a particular emphasis on air pollution, to VaD etiology and progression. Furthermore, the review discusses the major challenges to fully elucidate the pathobiology of VaD, as well as research directions to outline new therapeutic interventions.
Introduction
Dementia affects nearly 50 million people worldwide, and the World Health Organization (WHO) estimates that this number will triple by 2050 (Patterson, 2018). Dementia is a heterogeneous neurodegenerative pathology that encompasses Alzheimer’s disease (AD), vascular dementia (VaD), Lewy body dementia (LBD), frontotemporal dementia (FTD), and Parkinson’s disease (PD). Although the overall prevalence of dementia is higher in aging men, its severity is more pronounced in aging females, a disparity that might implicate sex hormones (Appelros et al., 2009; Podcasy and Epperson, 2016; Poorthuis et al., 2017). VaD comes just after AD as a main cause of dementia, accounting for approximately 15–20% of dementia cases in the Western countries and could reach up to 30% in Asia and developing countries (Rizzi et al., 2014). Vascular deficiencies are now considered relevant contributors to mixed dementia (MxD), which accounts for 25–35% of all dementia cases (Jellinger, 2007; Rosa et al., 2020).
The Vascular Impairment of Cognition Classification Consensus Study (VICCCS) defines VaD as “clinically significant deficits in at least one cognitive domain comprising sensation, perception, motor skills and construction, attention and concentration, memory, executive functioning, processing speed and language/verbal speed, that are of sufficient severity to cause severe disruption of activities of daily living” (Sachdev et al., 2006; Andrianopoulos et al., 2017; Skrobot et al., 2018). The cognitive functions are assessed through the Montreal Cognitive Assessment Test which evaluates five cognitive domains; executive function, attention, memory, language, and visuospatial function (Pendlebury et al., 2012; Skrobot et al., 2018; Iadecola et al., 2019). Diagnosis of VaD is divided into two major research fields; cognitive tests and neuroimaging. Indeed, the diagnosis does not rely only on memory impairments, but it is supported by the presence of diverse cognitive deficits accompanied by diagnostic imaging evidencing cerebrovascular abnormalities such as brain atrophy, white matter hyperintensities, infarcts, and hemorrhages. Accordingly, four different subtypes arise: post-stroke dementia (PSD) in which dementia appears 6 months after stroke, subcortical ischemic vascular dementia (SIVaD), multi-infarct dementia, and MxD (Skrobot et al., 2018). The cognitive deficits associated with VaD are caused by structural and functional vascular abnormalities that are exacerbated with age. These abnormalities promote the emergence of chronic alterations in the neurovascular functions that underlie the etiology of cognitive decline observed in VaD (Figure 1).
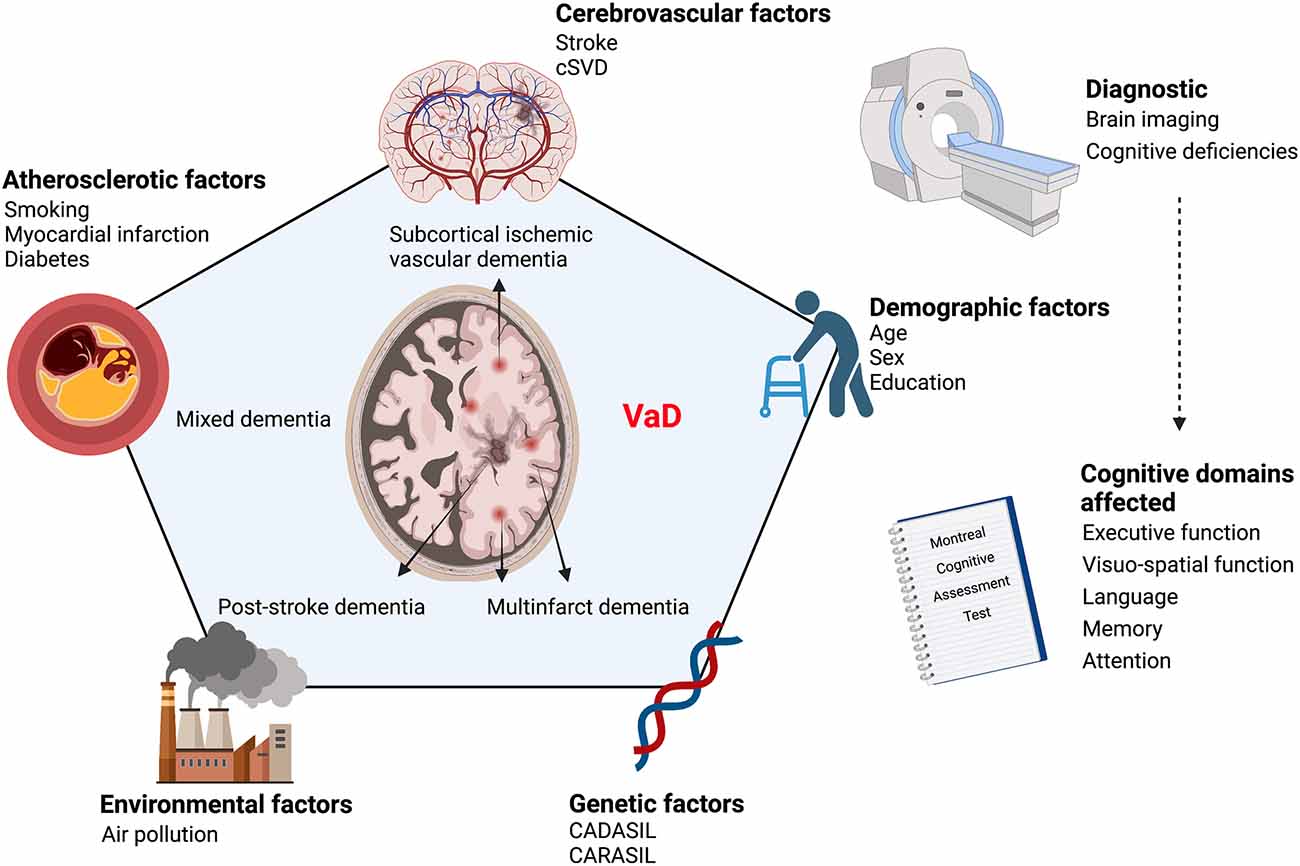
Figure 1. Scheme illustrating the continuum of risk factors that contribute to VaD etiology. VaD emerges as a conjunction of various risk factors affecting vascular homeostasis, namely cerebrovascular diseases, atherosclerosis, genetic and environmental factors. VaD diagnosis is based on the evaluation of cognitive deficiencies combined with neuroimaging to detect underlying vascular alterations. VaD, vascular dementia; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; cSVD, cerebral small vessel disease. Created with BioRender.com.
VaD is tightly associated with several risk factors that can be categorized into four groups, which comprise: (i) cerebrovascular disease-related factors; (ii) atherosclerotic factors, such as smoking, myocardial infarction, diabetes mellitus, and hyperlipidemia; (iii) demographic factors, such as age, biological sex and education; and (iv) genetic factors, such as the emergence of mutations leading to vascular encephalopathies (Ritchie and Lovestone, 2002; Gorelick, 2004). Noteworthy, these vascular risk factors are now being recognized as clinical risk factors for AD pathology (O’Brien and Markus, 2014; Figure 1).
The cerebrovascular disease-related factors include cerebral tissue loss volume, bilateral cerebral infarction, strategic infarction, and white matter disease (WMD; Ritchie and Lovestone, 2002; Gorelick, 2004). Furthermore, hypertension was shown to be associated with larger white matter and smaller brain volumes, silent or strategical subcortical or cortical infarcts, and loss of volume in the thalamus or temporal lobe that are critical for cognitive functions (Ritchie and Lovestone, 2002; Gorelick, 2004). While age remains a principal risk factor for dementia, the presence of familial dementia history, and the epsilon 4 allele of apolipoprotein E (ApoE)4 susceptibility gene were recognized as an important risk factor for VaD (Ritchie and Lovestone, 2002; Gorelick, 2004). In addition, variables such as the female sex, various types of infection of lipid concentrations, history of head injury, head circumference, hormone replacement therapy (HRT), as well as thyroid dysfunction and preceding history of depression could interact with ApoE genotype and hence increase the risk of dementia (Ritchie and Lovestone, 2002; Gorelick, 2004). The genetic factors include vascular encephalopathies such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), autosomal recessive cerebral arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), and potentially ApoE4 (Ritchie and Lovestone, 2002; Gorelick, 2004). Recently, environmental factors, namely air pollution, have been shown to constitute an important, yet underestimated, risk factor for dementia, inducing VaD and AD (Azarpazhooh and Hachinski, 2018; Béjot et al., 2018). Indeed, numerous studies have demonstrated that the elevated levels of air pollutants are directly linked to brain chronic inflammation and neurodegenerative diseases (Campbell et al., 2005; Schwartz et al., 2005; Calderón-Garcidueñas et al., 2007; Hartz et al., 2008; Block and Calderón-Garcidueñas, 2009; Mills et al., 2009; Rozemuller et al., 2012; Paul et al., 2019). Among air pollutants, ultrafine particles (UFPs) are particularly deleterious due to their ability to reach the brain where they act as inflammatory triggers and neurotoxins (Block and Calderón-Garcidueñas, 2009; Hameed et al., 2020).
As mentioned, VaD prevalence is strongly linked to cerebrovascular diseases, which essentially include stroke and cerebral small vessel disease (cSVD; Grinberg and Thal, 2010; Gorelick et al., 2011; Jellinger, 2013). Indeed, one patient in 10 has a stroke before developing a form of dementia that is not related to AD. In turn, cSVD was found in up to 62% of patients diagnosed with VaD, outlining a strong correlation between these pathologies (Gorelick et al., 2011; Venkat et al., 2015; van Veluw et al., 2017; Shih et al., 2018; Iadecola et al., 2019). Stroke and cSVD are characterized by the dysfunction of the neurovascular unit, which anatomically comprises sealed endothelial cells forming the blood-brain barrier (BBB), perivascular cells that include pericytes, and vascular smooth muscle cells (VSMCs), astrocytes, microglia, and neurons (Hermann and ElAli, 2012). The neurovascular unit integrates signals from the different neighboring cells to generate critical functions that include BBB maintenance, neurovascular coupling, vascular stability, and immunomodulation (Zlokovic, 2011; Hermann and ElAli, 2012). Neurovascular functions are impaired after stroke and cSVD leading to BBB dysfunction, neurovascular uncoupling, hypoperfusion, inflammation, and loss of neurons (Zlokovic, 2008, 2011; Guo and Lo, 2009; Moskowitz et al., 2010). These pathological events are at the origin of ischemic and hemorrhagic lesions, which strongly correlate with the cognitive deficits observed in VaD.
Despite being the second most common form of dementia after AD, little is known about the molecular and cellular mechanisms underlying the pathobiology of VaD. This gap in the literature is mainly due to disease heterogeneity in the clinical setup and the lack of an optimal experimental model that can accurately replicate most of the pathological events underlying the etiology and progression of the different forms of VaD. It is now established that accumulation of brain lesions over time mediated by neurovascular impairments constitutes a major contributor to the pathobiology of VaD in the elderly (Venkat et al., 2015; Corrada et al., 2016; Ince et al., 2017; Summers et al., 2017; van Veluw et al., 2017; Shih et al., 2018). The review summarizes the current knowledge related to the pathological mechanisms underlying the pathobiology of VaD with an emphasis on stroke, cSVD and risk factors with a focus on air pollution. We will discuss the challenges and research directions that might help in a better understanding of VaD pathobiology, thereby outlining new therapeutic interventions.
Major Risk Factor-Mediated Mechanisms Implicated in VaD Pathobiology
Stroke-Related Dementia
Ischemic or hemorrhagic strokes trigger major pathophysiological mechanisms that underlay VaD (Mijajlović et al., 2017). Indeed, epidemiological studies indicate that stroke history doubles the risk of dementia in the elderly (<65 years) and increases the incidence of early mortality (Savva and Stephan, 2010). Approximately 10% of patients exhibit signs of dementia before their first cerebrovascular accident, and another 10% manifest cognitive deficits soon after their first event (Desmond et al., 2002; Pendlebury and Rothwell, 2009). Particularly, recurrent stroke events raise the prevalence of dementia to 30%, constituting the most prominent causal factor of the disease (Pendlebury and Rothwell, 2009). Noteworthy, an examination of the association between stroke rates and dementia in the frame of the National Long-Term Care Survey (NLTCS) between 1984–2001 reported that the elevated incidence of post-stroke dementia is related to increased patient survival, due to clinical improvements in stroke management (Ukraintseva et al., 2006). Assessment of the neuropsychological parameters revealed that deterioration of the executive functions, abstraction, visual memory, and visuoconstruction constitute some of the most critical long-term cognitive disabilities observed in stroke patients (Sachdev et al., 2004). In contrast, praxis-gnosis, working memory, and language have been shown to be impacted to a lesser extent (Sachdev et al., 2004). The Sydney Stroke Study disclosed that in 50–85 years old patients diagnosed with VaD, the stroke volume and premorbid function were the most significant determinants of cognitive deterioration following the initial insult (Sachdev et al., 2006). It is remarkable to notice that in the last decade, stroke incidence increased by 23% in young adults aged between 35 and 50 years, especially because of the unhealthy lifestyle that meaningfully increased the rate of risk factors in this population, including smoking, hypertension, and obesity (Ekker et al., 2019; George, 2020). Due to the advances in acute stroke care, young stroke patients live longer and thus are at high risk of developing dementia at later stages (Pinter et al., 2019).
BBB Dysfunction
Hallmark of stroke pathophysiology (Yang et al., 2019), BBB dysfunction constitutes a pivotal factor implicated in the initiation and exacerbation of the cascade of events leading to dementia (Zlokovic, 2011; Sachdev et al., 2014; Noe et al., 2020). Following primary injury, BBB breakdown allows the uncontrolled infiltration into the brain of blood-borne molecules, including plasma proteins, metabolites, neurotoxic compounds, and peripheral immune cells that contribute to secondary injury progression via edema formation, neuroinflammation, and glial reactivity (Halder and Milner, 2019; Koizumi et al., 2019) that aggravate the initial neurological deficits (Khanna et al., 2014; Jiang et al., 2018). The experimental findings indicate that acute BBB impairment is widely mediated by early inflammatory mediators, such as cytokines and chemokines, as well as oxidative stress, including reactive oxygen species (ROS) and reactive nitrosative species (RNS; Yang et al., 2019). The action of these substrates is further potentiated by matricellular proteins, proteoglycans, and metalloproteinases (MMPs) secreted in the extracellular space (Jones and Bouvier, 2014). Experimental and clinical studies revealed that MMP-9 significantly contributes to long-term BBB breakdown in several brain disorders, namely stroke and neurodegenerative diseases (Barr et al., 2010; Montagne et al., 2017; Underly et al., 2017; Figure 2).
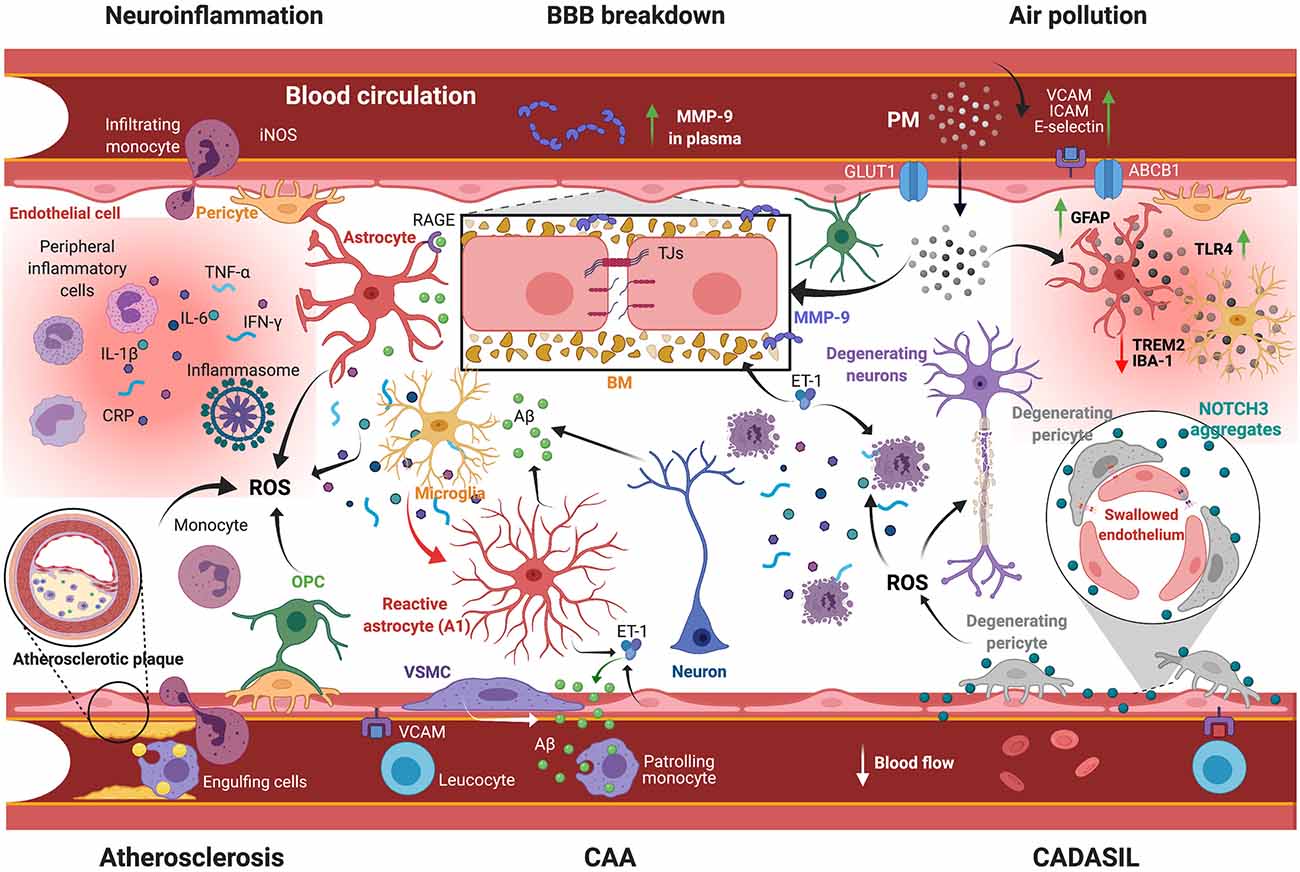
Figure 2. Scheme illustrating the mechanisms underlying VaD pathobiology. Several vascular risk factors are implicated in orchestrating pathological responses leading to VaD: (i) BBB breakdown involves the impairment of TJs and degradation of BM formed by ECM proteins via MMP-9 activity. Plasma MMP-9 levels constitute an effective prognostic marker for a poor neurological outcome; (ii) Post-stroke neuroinflammation comprises extravasation of peripheral immune cells and secretion of inflammatory mediators (e.g., CRP, TNF-α, IL-1β, IL-6, IFN-γ), as well as ROS generation and glial activation, accompanied by cerebral Aβ aggregation; (iii) Atherosclerosis comprises the accumulation of lipids and the calcification of immune cells into the intima, leading to vessel occlusion and hypoperfusion. This condition is associated with the generation of ROS that causes chronic inflammation; (iv) CAA is associated with the degeneration of VSMCs and vascular Aβ aggregation due to impaired clearance; (v) CADASIL is associated with NOTCH3 aggregation, causing endothelial cell swelling and pericyte degeneration and subsequently CBF impairment; and (vi) Exposure to air pollution, which implies PM infiltration into the brain, exacerbates BBB breakdown and neuroinflammation. VaD, vascular dementia; BBB, blood-brain barrier; TJs, tight junctions; BM, basement membrane; ECM, extracellular matrix proteins; MMP-9, matrix metalloproteinase-9; CRP, C-reactive protein; TNF-α, tumor necrosis factor-α; IL-1β/6, interleukin-1β/6; IFN-γ, interferon-γ; ROS, reactive oxygen species; Aβ, amyloid-β; CAA, cerebral amyloid angiopathy; VSMCs, vascular smooth muscle cell; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; NOTCH3, neurogenic locus notch homolog protein-3; CBF, cerebral blood flow; PM, particulate matter; GLUT1, glucose transporter-1; ABCB1, ATP binding cassette subfamily B member-1; VCAM, vascular cell adhesion protein; ICAM, intercellular adhesion molecule; TLR4, Toll like receptor-4; TREM2, triggering receptor expressed on myeloid cells-2; GFAP, glial fibrillary acidic protein; RAGE, receptor for advanced glycation endproducts; ET-1, endothelin-1; iNOS, inducible nitric oxide synthase; OPC, oligodendrocyte progenitor cell. Created with BioRender.com.
Upon ischemic stroke, MMP-9 is secreted by the cells forming the neurovascular unit via regulation of the extracellular signal-regulated kinase-(ERK)-1/2) and the signal transducer and activator of transcription (STAT)-3 pathways, leading to the degradation of basal lamina/extracellular matrix (ECM) proteins, and the recruitment and extravasation of peripheral immune cells (Nishikawa et al., 2018; Jäkel et al., 2020). Interestingly, human brain studies showed that MMP-9 is implicated in the degradation of type IV collagen at the basal lamina, resulting in hemorrhagic transformations (Rosell et al., 2006, 2008), enhanced leukocyte infiltration, and poor neurological outcomes (Kim et al., 2016). In parallel, studies employing MMP-9−/− mice showed that leukocyte-derived MMP-9 plays an essential role in mediating BBB dysfunction and is associated with elevated leukocyte transmigration that exacerbates the inflammatory signaling in the acute phase of stroke (Gidday et al., 2005). Furthermore, photothrombotic mouse models of cerebral ischemia have reported that BBB permeability at the level of the capillary is governed by pericytes exhibiting MMP-9 activation, which was later neutralized by specific MMP-9 inhibition (Underly et al., 2017). Elevated MMP-9 activity has been also reported to be associated with increased brain edema and IgG extravasation after ischemia in hyperlipidemic mice (ElAli et al., 2011). These observations indicate that hyperlipidemia exacerbates stroke-mediated BBB dysfunction, which could eventually aggravate dementia (Figure 2).
On an important note, stroke-induced MMP-9 expression has risen as an informative prognostic marker for a poor neurological outcome, increased mortality, and the emergence of typical signs of dementia (Zhong et al., 2017). Clinical investigations disclosed that high MMPs expression correlates with increased levels of albumin cerebrospinal fluid (CSF) in patients with vascular cognitive impairment (VCI) derived from SIVD, multiple strokes, and leukoaraiosis (Candelario-Jalil et al., 2011). In parallel, independently of the presence of vascular risk factors, elevated serum MMP-9 levels are associated with mild (25.6% of patients) and severe (27.4%) cognitive impairment 3 months following stroke according to the Mini-Mental State Examination and Montreal Cognitive Assessment (Zhong et al., 2018). Moreover, high MMP-9 levels in patients with cardioembolic stroke involving the middle cerebral artery (MCA) territory are related to large infarct volumes and poor behavioral scores based on the National Institutes of Health Stroke Scale (NIHSS; Montaner et al., 2001). Otherwise, the strong correlation between MMP-9 expression and the hyperintense acute reperfusion injury marker (HARM) has led to consider this protein as a revealing marker for BBB disruption (Barr et al., 2010). Furthermore, increased MMP-9 activity has been detected in the frontal and parietal cortex of postmortem human brains diagnosed with AD and patients exhibiting cognitive deficits (Bruno et al., 2009). MMP-9 enhanced expression is also notable in the CSF of AD patients, directly correlated with T-tau and P-tau levels (Stomrud et al., 2010). Importantly, a longitudinal study (4–10 years) enrolling AD and VaD patients revealed higher MMP-9 levels in the CSF of VaD patients compared to AD or controls (Adair et al., 2004). These findings imply that assessment of MMP-9 levels might constitute a potential strategy to distinguish between the different types of dementia.
Cell-based assays have demonstrated that exposure of pericytes to amyloid-β (Aβ)42 induced MMP-9 activity, which in turn ameliorated protein aggregation (Schultz et al., 2014). This outlines the presence of a direct pathological link between the molecular markers of dementia and MMP-9 activity, even though MMP-9 is an active substrate for Aβ degradation (Hernandez-Guillamon et al., 2015). Overall, current evidence suggests that MMP-9-mediated BBB dysfunction following stroke may constitute an early pathological mechanism that initiates the neurodegenerative cascades leading to cognitive deficits over time. Although MMP-9 inhibition has been proposed as a therapeutic strategy to attenuate BBB breakdown after cerebral ischemia (Dong et al., 2009; Chaturvedi and Kaczmarek, 2014), animal studies showed that MMP-9 is required for neurovascular remodeling and adaptation in the chronic phase after stroke (Zhao et al., 2006). Furthermore, MMP-9 is implicated in the clearance of various misfolded proteins involved in several neurodegenerative diseases, such as Aβ (Hernandez-Guillamon et al., 2015). The current knowledge implies that MMP-9 impact on BBB breakdown and neurodegeneration is time and context-dependent, which entails a contextualized modulation of protein expression/activity to preserve BBB integrity and attenuate cognitive deficits associated with stroke.
Post-stroke Neuroinflammation
Evidence obtained from AD studies outlined an important pathological link among chronic neuroinflammation, vascular damage, and cognitive decline in aged patients (Rhodin and Thomas, 2001; Kinney et al., 2018). Importantly, the neuroinflammatory responses associated with AD could be observed as well in other forms of dementia, including frontotemporal dementia (FTD; Bevan-Jones et al., 2020), PD (Caggiu et al., 2019), and VaD (Iadecola, 2013). Neuroinflammation plays a critical role in modulating tissue injury and repair after stroke. This process integrates various molecular and cellular mechanisms that comprise the release of inflammatory and oxidative stress mediators, glial reactivity, and peripheral immune cell activation and extravasation (Guruswamy and ElAli, 2017; Dzyubenko et al., 2018; Jayaraj et al., 2019). In this regard, uncontrolled microglial activation and the subsequent release of proinflammatory cytokines after stroke (Zhao et al., 2017) are strongly associated with demyelination and axonal loss (Sachdev et al., 2004).
It has been reported that in a rodent model of cerebral hypoperfusion, microglial activation via the complement (C)3-C3aR pathway, which is implicated in myelin phagocytosis, resulted in learning and memory deficits (Zhang et al., 2020). Interestingly, the cognitive impairments were attenuated by the genetic deletion of c3ar1 or via the administration of SB290157, a potent C3aR antagonist (Zhang et al., 2020). Similarly, experimental investigations using cerebral ischemia have shown that fingolimod (FTY720), a potent agonist of sphingosine 1 phosphate (S1P), induced a microglial anti-inflammatory phenotype (M2 phenotype) through the activation of STAT-3 signaling pathway (Qin et al., 2017). Modulating microglial activation to adopt a protective M2 phenotype resulted in enhanced oligodendrocytogenesis and white matter integrity, and reduced cognitive deficiencies associated with working memory (Qin et al., 2017). Induction of severe chronic cerebral hypoperfusion (SCCH) in APP/PS1 mice that overproduced Aβ accelerated spatial learning and memory decline in 4-month adult animals. This was correlated to the accumulation of parenchymal Aβ plaques in the hippocampus and diminished activity of the ERK-1/2 pathway. APP/PS1 mice subjected to SCCH had higher levels of patrolling monocytes in peripheral blood. Interestingly, this model revealed that SCCH reduces microglial interaction with Aβ plaques in the hippocampus, denoting a reduced capacity for Aβ clearing in the brain parenchyma (Bordeleau et al., 2016). Alternatively, the release of prostaglandins, which act as inflammatory mediators upon stroke, has been shown to be associated with exacerbated Aβ-mediated cognitive decline and impaired synaptic plasticity (Kotilinek et al., 2008; Figure 2).
Loss of white matter integrity by hyper-reactive ramified and amoeboid microglia was also found in the posterior cingulate cortex of post-mortem brains of patients diagnosed with Down syndrome who are at higher risk of developing AD neuropathology (Martini et al., 2020). Furthermore, microglia in post-mortem AD brains exhibit accelerated aging and transcriptional alterations associated with the isoforms of ApoE, a protein broadly related to both dementia and cardiovascular disease (Srinivasan et al., 2020). In this regard, findings from subarachnoid hemorrhage in mice indicate that ApoE mediates protective effects following injury by inducing M1 microglial quiescence (Pang et al., 2018), suggesting that adequate lipid metabolism modulates neuroinflammation. Functional human brain investigations using positron emission tomography (PET) coupled to 11C-PK11195, which is an in vivo marker of activated microglia, have unraveled a progressive microglial activation and neuroinflammation, which were correlated with long-term (14 to 16 months) cognitive decline in AD patients (Malpetti et al., 2020). In this regard, activated microglia exhibiting a pro-inflammatory neurotoxic phenotype (M1 phenotype) trigger the activation of pro-inflammatory astrocytes (A1 astrocytes) via tumor necrosis factor (TNF)-α, interleukin (IL)-1α, and C1q cytokines (Liddelow et al., 2017). In turn, A1 reactive astrocytes exacerbate oligodendrocyte and neuronal death (Liddelow et al., 2017).
In line with these findings, an exaggerated astrocyte reactivity has been related to dementia and cognitive decline (Jo et al., 2014; Csipo et al., 2020). The evidence suggests that morbid neuroinflammatory responses maintained by A1 reactive astrocytes could result in mediating brain injury or age-related neurodegeneration and cognitive deficits. For instance, hippocampal astrocytes are susceptible to the upregulation of inflammatory-related genes and pathways, such as C3 and C4b, C-X-C motif chemokine ligand (CXCL)-10, and the peptidase inhibitor serine protease inhibitor A3N (SERPINA3N; Clarke et al., 2018). Interestingly, in a mouse model of familial Danish dementia (FDD), abundant A1 reactive astrocytes were detected in the brain, which correlated with the appearance of cerebral amyloid angiopathy (CAA), a disease characterized by the deposition of Aβ within the cerebral vasculature and a major risk of VaD. In this context, an increased number of astrocytes was observed in perivascular zones, accompanied by numerous cell branches and enhanced glial fibrillary acid protein (GFAP) expression (Taylor et al., 2020; Figure 2).
Several approaches have demonstrated that the usage of anti-inflammatory strategies could attenuate cognitive deficits associated with dementia. For instance, activation of the cannabinoid receptor 2 (CB2R) using different agonists promoted memory restitution through the reduction of oxidative stress and mitochondrial dysfunction (Jayant and Sharma, 2016). In line with these findings, cell-based assays have shown that CB2R activation stimulated the microglial release of IL-10, a key anti-inflammatory cytokine, via activation of ERK1/2, c-Jun N-terminal kinase (JNK), and mitogen-activated protein kinases (MAPKs) pathways, accompanied by the inhibition of the nuclear factor-κB (NF-κB) pathway (Correa et al., 2010). Likewise, administration of the CB2R agonist paeoniflorin (PF) ameliorated memory and learning deficits in mice, accompanied by induction of M2 cells and the release of anti-inflammatory mediators, such as transforming growth factor (TGF)-β1, and IL-10, instead of pro-inflammatory ones, such as TNF-α, IL-1β, and IL-6. This phenotypic switch is driven by the enhanced activity of phosphoinositide-3-kinase (PI3K/AKT) anti-inflammatory pathway and the inhibition of the mammalian target of rapamycin (mTOR)/NF-κB pro-inflammatory signaling (Luo et al., 2018). Experimental findings indicate that the inhibition of mTOR attenuated cognitive deficits, which were accompanied by a restoration of M1/M2 microglia phenotypic switch following cerebral hypoperfusion (Chen et al., 2016).
Remarkably, in a rodent model of VaD, it has been shown that acupuncture could attenuate inflammation by reducing TNF-α and Toll-like receptor (TLR)4 expression in microglia, and suppressing the myeloid differentiation factor (MyD88)/NF-κB pathway (Wang et al., 2020). Furthermore, pharmacological administration of PLX5622, a potent inhibitor of colony stimulating factor-1 receptor (CSF-1R) that is required for microglial cell survival, improved short-term memory in a rodent model of induced hypertension. This effect was accompanied by controlled microglia reactivity and preservation of BBB integrity (Kerkhofs et al., 2020). It has also been demonstrated that depletion of microglia via CSF1R inhibition prevented Aβ plaque development in the hippocampus of a mouse model of AD (Spangenberg et al., 2019). Attenuation of microglial reactivity via blockage of CCL-5 signaling also preserved BBB integrity in the context of systemic inflammation (Haruwaka et al., 2019). Taken together, there is strong evidence associating chronic uncontrolled neuroinflammatory responses after stroke to the emergence of long-term cognitive disabilities and dementia, mediated essentially by microglial reactivity. Therefore, strategies aiming to modulate microglial response by stimulating a protective phenotype might constitute a potential approach to attenuate VaD occurrence after stroke.
Stroke-Mediated Proteinopathies
Accumulation of Aβ in the brain constitutes a hallmark of AD pathogenesis (Chen et al., 2017). Early studies using human post-mortem brains revealed that amyloid precursor protein (APP) is not implicated exclusively in AD pathology, and its expression is as well induced in the brain after stroke (Cochran et al., 1991; Jendroska et al., 1997). For instance, mutant mice overexpressing APP exhibited a substantial reduction of cerebral blood flow (CBF) accompanied by larger infarcts after stroke, suggesting that APP exacerbated ischemic injury by impairing structural and functional vascular integrity (Zhang et al., 1997). Moreover, it has been shown that cerebral ischemia promotes APP deposition in the lesion core, the perilesional regions, as well as in the white matter areas exhibiting myelin loss (Nihashi et al., 2001; Zhan et al., 2015).
Endothelin (ET)-1 is a powerful vasoconstrictor synthesized by endothelial cells and reactive astrocytes, which have been shown to be implicated in ischemic stroke pathobiology as well as Aβ deposition. Examination of post-mortem human brains showed strong ET-1 expression in reactive astrocytes surrounding Aβ plaques (Hung et al., 2015). Furthermore, ET-1 overexpression in the acute phase after stroke has been involved in BBB disruption, glial reactivity, and neuronal death. Indeed, mutant mice exhibiting astrocytic ET-1 overexpression (GET-1 mice) experience severe memory and spatial learning deficits, associated with the upregulation of cleaved caspase-3, TNF-α, and IL-1β (Thiel et al., 2014; Hung et al., 2015). Using cell-based assays, ET-1 overexpression in reactive astrocytes has been shown to amplify Aβ production (Hung et al., 2015). Aβ accretion contributes to the development of cognitive deficits by impairing the receptor for advanced glycation endproducts (RAGE)-mediated Aβ clearance, which exacerbates inflammation, oxidative stress, and neurodegeneration (Min et al., 2020). These findings indicate that ET-1 upregulation after ischemic stroke is tightly associated with Aβ production and deposition and has considerable effects on excitotoxicity and BBB integrity. Furthermore, comorbid models of Aβ toxicity and cerebral ischemia have reported that Aβ deposition exacerbates ischemic damage. This condition leads to ventricular enlargement and striatal atrophy, morphological alterations in microglia, increased production of inflammatory mediators and enhanced glial communication via the gap junction proteins connexin (CX)-43. These observations are particularly important as ventricular enlargement was associated with deposition of neurofibrillary tangles and Aβ plaques, directly implicated in the pathogenesis of various forms of dementia (Amtul et al., 2015). Furthermore, evidence indicates that the interactions among lipoprotein-associated triggering receptor expressed on myeloid cells (TREM) 2 and apolipoproteins are involved in modulating microglia-mediated Aβ phagocytosis (Yeh et al., 2016), thus suggesting that Aβ clearance is associated with lipid metabolism (Figure 2). Likewise, it has been shown that exogenous administration of Aβ triggers tau phosphorylation and magnifies learning and memory deficits in animals subjected to cerebral ischemia (Song et al., 2013).
Finally, the generation of RNS, including peroxynitrite, revealed by the formation of 3-nitrotyrosine (3-NT), is increased in perivascular astrocytes and microglial cells after Aβ42 injection, strongly correlating with BBB leakage (Ryu and McLarnon, 2006). These findings suggest that Aβ pathology could trigger the release of reactive nitrogen species in astrocytes, directly undermining cerebrovascular integrity. Moreover, it has been demonstrated that Aβ binding to RAGE induces ROS production leading to loss of the tight junction proteins claudin-5, occludin, and zonula occludens (ZO)-1, as well as deficient endothelial cell function (Carrano et al., 2011). Overall, this bidirectional pathological crosstalk implies that exacerbated cognitive decline strongly emerges when cerebral injury and Aβ toxicity occur comorbidly.
cSVD
cSVD comprises numerous pathologies impacting cerebral arteries, arterioles, venules, and capillaries, which are associated with diverse pathological and etiological processes (Østergaard et al., 2016; Staszewski et al., 2017; Li et al., 2018; Parkes et al., 2018). Six different types of cSVD are classified according to their etiology (Pantoni, 2010; Li et al., 2018). Atherosclerosis and sporadic and hereditary CAA are the most frequent forms. Recent reports outlined a significant increase in the number of genetic microangiopathies distinct from CAA such as CADASIL or Fabry’s disease (Razvi and Bone, 2006; Ballabio et al., 2007; Dichgans, 2007; Hara et al., 2009). Microangiopathies caused by inflammation or mediated by immunity are rare and characterized by the presence of inflammatory cells within the vasculature (Jennette and Falk, 1997), generally caused by mechanisms associated with systemic pathologies. Venous collagenosis is a pathologic thickening of the wall of veins and venules that are located near the lateral ventricles, thus leading to a smaller lumen and sometimes to an occlusion (Figure 2). Finally, post-radiation angiopathies are a side effect of cerebral irradiation that appears a few months to years after treatment. These angiopathies mainly affect small vessels of the white matter associated with fibrinoid necrosis, resulting in an increased thickness of the walls accompanied by a reduced diameter, which jointly could lead to a thrombotic occlusion (Dropcho, 1991).
cSVD Associated Parenchymal Pathology
cSVD refers to various and complex pathological and etiological processes. Therefore, the clinical manifestations depend on both the cause of the pathology and the affected brain territory. Among the most common symptoms are stroke-related manifestations, progressive cognitive deterioration, VaD, gait disturbance, sphincter dysfunction, and psychiatric disorders (van der Flier et al., 2005; Pantoni, 2010; Del Bene et al., 2013; Li et al., 2018; de Laat et al., 2011). cSVD is thought to constitute the major cause of vascular cognitive deficits and are responsible for up to 45% of dementia cases (Shi and Wardlaw, 2016; Li et al., 2018). Cognitive deficits are associated with impaired executive functions, decline in memory and attention, regression in verbal fluency, and delayed recall. These symptoms are accompanied by others that are not specific, including dizziness, trouble sleeping, tinnitus, and hearing loss. Moreover, neuropsychiatric symptoms can be observed, including hallucinations, agitation, depression, anxiety, disinhibition, apathy, irritability, and changes in appetite. Most of these manifestations are often accompanied by brain microbleeds. Cerebral microangiopathies are accountable for up to 20–30% of ischemic stroke as well as a considerable proportion of hemorrhage and encephalopathies caused by emboli, thrombosis, or stenosis of the vessel (Cai et al., 2015; Shi and Wardlaw, 2016; Regenhardt et al., 2019).
In addition to the cerebrovascular pathologies, cSVD exhibits a unique parenchymal pathology characterized by small subcortical infarcts, lacunar stroke, microbleeds, white matter hyperintensities (WMH), enlarged perivascular spaces, and brain atrophy detectable in imaging (van der Flier et al., 2005; de Laat et al., 2011; Del Bene et al., 2013; Wardlaw et al., 2013; Li et al., 2018; Regenhardt et al., 2018; Das et al., 2019). Small subcortical ischemic stroke is the result of severe tissue ischemia caused by the occlusion of a perforating arteriole. Patients either have typical stroke symptoms or a lesion visible only using neuroimaging approaches (Wardlaw et al., 2013; Li et al., 2018). Lesions can be anywhere in the brain and are round or ovoid and less than 20 mm in diameter (Smith et al., 2012; Brundel et al., 2012; van Veluw et al., 2017; Hartmann et al., 2018). They appear hyperintense in a diffusion-weighted image (DWI), hypointense on the map of apparent diffusion coefficients, and normal to hyperintense in fluid-attenuated inversion recovery (FLAIR)/T2 imaging (Okazaki et al., 2015; Potter et al., 2015; Li et al., 2018). DWI is the most sensitive technique currently used to detect ischemia a few hours after stroke onset. Recent infarcts will form a cavity characterized by morphological changes that include a reduction in volume and diameter within 90 days of the onset of the infarction (Moreau et al., 2012; Potter et al., 2015; Li et al., 2018). These infarcts can evolve in three different ways, namely lacuna, WMH without cavitation T2-weighted sequence, and finally, they could disappear without visible consequences in conventional magnetic resonance imaging (MRI). When a recent small subcortical ischemic stroke resolves into lacuna, it actually forms a fluid-filled cavity called a lacunar stroke and represents 40% of acute ischemic strokes. Lacunar insults are divided in two different categories; cavitated old infarcts and incomplete infarcts (Fisher, 1965; Lammie et al., 1998; Regenhardt et al., 2018). Old infarcts are pan-necrotic cavitation with scattered macrophages whereas incomplete infarcts are described as exhibiting loss of neurons and oligodendrocytes associated with invading CD68+ macrophages and reactive microglia in addition to reactive astrocytes that are found inside and around the lesion site (Merino and Hachinski, 2000; Brundel et al., 2012; Regenhardt et al., 2018).
The vascular damage can also develop into BBB leakage or cerebral microbleeds, which appear as small, round, and homogeneous hypointense foci on T2-weighted MRI and are mostly asymptomatic. They originate essentially from the rupture of a precapillary arteriole and are usually associated with vascular risk factor exposition or vascular Aβ deposition (Cordonnier et al., 2007; De Silva and Faraci, 2016; Shi and Wardlaw, 2016; Toth et al., 2017). The rupture is caused by various factors such as age, hypertension, cerebral ischemia, dementia, and cerebral amyloid angiopathy (CAA), and participates in cognitive deficits, dementia, and transient neurological deficits (Martinez-Ramirez et al., 2014; Shi and Wardlaw, 2016; Li et al., 2018). Microbleeds trigger proliferation and migration of microglia and astrocytes as well as monocyte recruitment (Liddelow et al., 2017). The immune cells release various inflammatory factors that impair neuronal function, as well as neurotransmitters that may be neurotoxic and interfere with neuronal circuitry to promote cognitive decline (Tancredi et al., 2000; Beattie et al., 2002; Rosidi et al., 2011; Donzis and Tronson, 2014).
The cerebral white matter is composed of myelinated axons, myelinating oligodendrocytes, oligodendrocyte precursor cells (OPCs), astrocytes, and microglia (Hase et al., 2018). WMH is common in older people and is a typical feature of cerebral microangiopathies, which are associated with BBB disruption, small white matter infarcts, glial activation, loss of oligodendrocytes, and demyelination caused by chronic diffuse hypoperfusion associated with a reduced CBF (Prins and Scheltens, 2015; Li et al., 2018). WMH is generally located within the white matter including the pons and brainstem but also in the deep gray matter. It is distributed symmetrically and bilaterally and appears hyperintense on FLAIR or T2 MRI. Importantly, WMH triples the risk of stroke, doubles the risk of dementia, and substantially increases the risk of death (Debette and Markus, 2010; Pantoni, 2010; Shi and Wardlaw, 2016). Symptoms develop insidiously and are associated essentially with cognitive impairments, dementia, and depression (Debette and Markus, 2010; Pantoni, 2010; Shi and Wardlaw, 2016).
The perivascular space is an extension of the subarachnoid space that surrounds the brain microvasculature. It is a liquid-filled space that cannot be detected by conventional imaging in a physiological context. When this space is widened, it often appears hyperintense on T2 MRI, hypointensity on T1 weighting, and sometimes hypointense on FLAIR (Aribisala et al., 2013; Shi and Wardlaw, 2016; Li et al., 2018). Finally, brain atrophy refers to a diminished brain volume on neuroimaging characterized by symmetrical or asymmetrical decreased total volume, increased ventricular volumes, enlarged superficial sulci, and decreased specific gray or white matter volumes (Mok et al., 2011). One main region affected is the hippocampus and is associated with cognitive decline (Muller et al., 2011; Jokinen et al., 2012).
Atherosclerosis
Atherosclerosis is an age-related condition that constitutes a major risk factor for cerebral microangiopathies. As its severity is increased by diabetes and hypertension, it is also called hypertensive microangiopathy (Tan et al., 2017; Li et al., 2018; Ter Telgte et al., 2018). The risk factors for atherosclerosis are also hyperlipidemia, smoking, and moderate to severe sleep apnea (Østergaard et al., 2016; Cannistraro et al., 2019). Atherosclerosis is characterized by a chronic inflammation associated with the deposition of low-density lipoproteins (LDL) within the vasculature, leading to its internalization by endothelial cells (Tabas et al., 2015), and resulting in the thickening and hardening of the arterial walls (Lusis, 2000; Shabir et al., 2018). Upon deposition, LDL undergoes oxidation by ROS to form oxidized (ox)-LDL, which further exacerbates the inflammatory response within the vasculature (Tabas et al., 2007). Indeed, by binding to vascular cell adhesion molecule (VCAM)-1 and P-selectin, monocytes can infiltrate the intima and differentiate into macrophages to engulf ox-LDL (Chistiakov et al., 2016). Macrophages, which are now called foam-cells due to the intracellular accumulation of lipids (Spann et al., 2012), accumulate and form stable fatty-streaks into the intima, and cells can calcify over time to slowly occlude the vessel (Alexander and Owens, 2012; Chistiakov et al., 2017). These pathological events occur in large to medium size arteries and lead to microbleeds, microinfarcts, as well as lipohyalinosis, characterized by the deposition of hyaline into the walls of connective tissue (Gorelick et al., 2011). This aspect is specific to the brain due to inflammation caused by ROS, ox-LDL, and gliosis involving astrocytes, OPCs, and microglia (Caplan, 2015). Lipohyalinosis fosters the infiltration of monocytes and T helper (TH)-1 lymphocytes that amplify the production of inflammatory mediators, such as TNF-α and interferon (INF)-γ (Stemme et al., 1995; Frostegård et al., 1999). Moreover, the assembly of inflammasomes can be promoted through the activation of nucleotide-binding oligomerization domain (NOD)-like receptor protein (NLRP)-3, stimulated by the formation of cholesterol crystals, caspase-1 and apoptosis-associated speck-like protein containing (ASC), caspase activation and recruitment domain (CARD; Weber and Noels, 2011). This results in IL-1β release, which in turn stimulates the release of IL-6 and C-reactive protein (CRP), implicated in the pathogenesis of atherosclerosis and thrombosis (Ridker et al., 2017; Figure 2).
Besides, the elevated levels of LDL combined with the low levels of high-density lipoprotein (HDL) play an important role in the pathogenesis of atherosclerosis and constitute as well a major risk factor for VaD (Hao and Friedman, 2014; Georgakis et al., 2020). Indeed, HDL exerts a protective role through its antioxidant properties and mediates beneficial effects on platelets and endothelial function, thus on coagulation and inflammation (Bandeali and Farmer, 2012). Furthermore, HDL interacts with triglyceride-rich lipoproteins, attenuating their deferential effects (Bandeali and Farmer, 2012). Moreover, it could contribute to the removal of cholesterol excess from the brain microvasculature through ApoE and heparin sulfate proteoglycans (Mulder and Terwel, 1998). HDL counteracts the inhibition of vessel relaxation caused by ox-LDL and decreases LDL peroxidation which affects cellular function and impairs membrane-bound receptors and enzymes (Braughler and Hall, 1992; Matsuda et al., 1993). Importantly, the impact of hyperlipidemia seems to be differentially modulated depending upon biological sex. Indeed, a recent study shows that females exhibit greater expression of genes related to neuroprotection in response to lipid stress compared to age-matching males (Nuthikattu et al., 2020). Finally, lipid derivatives are now being under the scope of researchers who are trying to unravel novel biomarkers to better understand and diagnose VaD pathology. Indeed, in an interesting recent study that aimed to discover lipid biomarkers in the context of VaD, it has been reported that patients with dementia exhibit low levels for ceramides, cholesterol esters, and phospholipids, and high levels of glycerides compared to controls (Liu et al., 2020). These observations indicate that lipid derivatives could indeed be used as novel diagnostic and prognostic biomarkers in VaD. However, more research is needed in this direction to validate the use of lipid derivatives as diagnostic and prognostic biomarkers in VaD.
Vascular damage caused by atherosclerosis can lead to microatheromas, microaneurysms, and even stenosis or obstruction of the vessel, impairing the mechanisms of blood flow autoregulation and leading to chronic cerebral hypoperfusion (Pantoni, 2010; Li et al., 2018). Importantly, occlusion of the cerebral arteries results in local ischemia or lacunar infarction (Kraft et al., 2017; Ter Telgte et al., 2018), while stenosis and hypoperfusion in the white matter cause incomplete ischemia lesions evidenced by neuroimaging as white matter hyperintensity (Rigsby et al., 2007). When the pathology affects the cerebral arterioles <50 μm in diameter, it is called small cerebrovascular atherosclerosis (Li et al., 2018).
Sporadic and Hereditary CAA
CAA is a chronic degenerative disease characterized by the loss of VSMCs and the accumulation into the vessel wall of eosinophilic hyaline material composed of soluble Aβ40 (Attems et al., 2011; Charidimou et al., 2017). CAA affects 50–60% of the elderly population affected by dementia, including 85–95% of AD cases (Sacco, 2000; Jellinger, 2002; Thal et al., 2003; Keage et al., 2009; Charidimou et al., 2017; Zhang et al., 2017; Li et al., 2018). The initial cognitive deficits associated with VaD could be explained by the particular sensitivity of the hippocampus and the cortex to CAA (Arvanitakis et al., 2011; Li et al., 2018). CAA is associated with changes in basal membrane (BM) composition and morphology that could predispose Aβ accumulation in the vessel even though the mechanism is not yet fully understood (Perlmutter et al., 1990, 1991; Su et al., 1992; Morris et al., 2014; Howe et al., 2020). Among the reported changes are BM thickening and degeneration, abnormal heparan sulfate proteoglycans (HSPGs) deposits, and irregular vasculature accompanied by increased collagen IV, fibronectin, agrin, and perlecan expression (Berzin et al., 2000; Farkas et al., 2000; Bourasset et al., 2009; Gama Sosa et al., 2010; Keable et al., 2016; Lepelletier et al., 2017; Magaki et al., 2018; Singh-Bains et al., 2019). Furthermore, vascular functional impairments are featured by BBB dysfunction caused by loss of endothelial cells, deregulation of mural cells mediated by oligomeric Aβ accumulation, as well as induction of astrocytosis with dystrophic endfeet surrounding BM Aβ deposits (Shimizu et al., 2009; de Jager et al., 2013; Giannoni et al., 2016; Yang et al., 2017; Magaki et al., 2018; Nortley et al., 2019). Two possible mechanisms for Aβ deposition have been proposed: (i) release of vascular Aβ from VSMCs directly into the vessel wall; and (ii) release of parenchymal Aβ by neurons which deposits afterward into the vessel wall (Davis et al., 2004; Herzig et al., 2004; Vidal et al., 2009; ElAli et al., 2013). In both cases, the protein accumulates due to a poor clearance towards the periphery (Davis et al., 2004; Herzig et al., 2004; Vidal et al., 2009; ElAli et al., 2013). Insufficient Aβ clearance can impair perivascular drainage pathways or diminish the ATP binding cassette subfamily B member-1 (ABCB1) and low-density lipoprotein receptor-related protein (LRP)1, a specialized endothelial-mediated active transport system implicated in Aβ mobilization from the brain into the blood circulation, namely (Deane et al., 2004; Herzig et al., 2006; Weller et al., 2008; Hawkes et al., 2011). Interestingly, LRP1 plays an important role in protecting against neurodegeneration. Indeed, LRP1 downregulation doesn’t only affect Aβ clearance but causes as well BBB breakdown through activation of MMP-9, thus leading to loss of neurons and cognitive deficits (Nikolakopoulou et al., 2021). Furthermore, recent evidence reveals that vascular Aβ could be engulfed and eliminated by circulating patrolling monocytes, which act as the housekeeper vascular homeostasis by surveying endothelial cells (Auffray et al., 2007; Carlin et al., 2013; Michaud et al., 2013; Thériault et al., 2015). In this regard, it has been demonstrated that patrolling monocytes located at the luminal wall internalize Aβ microaggregates that are diffusing from the parenchyma into the blood. Unfortunately, patrolling monocyte ability to phagocyte vascular Aβ in AD is defective, resulting in an overall increase of highly toxic Aβ40 and Aβ42 oligomers (Hallé et al., 2015; Gu et al., 2016). Moreover, chronic mild cerebral hypoperfusion impairs BBB functional properties and promotes the accumulation of circulating Aβ into the vessel wall, which initiates the cascade of parenchymal Aβ deposition (ElAli et al., 2013). Aβ accumulation and BM rearrangement trigger BBB breakdown, endorsing the formation of perivascular edema and the infiltration of toxic blood-derived substrates into the brain, which in turn contribute to the exacerbation of localized injuries and enlargement of the perivascular space (Holland et al., 2008; Hartz et al., 2012; Wardlaw et al., 2013; Li et al., 2018; Figure 2).
The overwhelming evidence is suggesting that the here mentioned vascular abnormalities leading to dementia reported in CAA occur as well in different forms of dementia, including AD and LBD (Salat et al., 2006; Okamoto et al., 2010; Soontornniyomkij et al., 2010; Arvanitakis et al., 2011; Love et al., 2014; Martinez-Ramirez et al., 2014; Reijmer et al., 2016; Li et al., 2018). This form of cSVD can be sporadic or of a genetic origin. For instance, a syndrome called hereditary brain hemorrhage with amyloidosis (HBHA) is associated with a mutation in the APP gene. This syndrome results in the deposition of misfolded amyloid fibrils in the walls of cerebral arterioles, which in turn activates a cascade of events leading to the development of CAA. The clinical phenotype develops between the ages of 45–65 years and is associated with intracerebral hemorrhages, WMH, multifocal lesions of a hemorrhagic and ischemic nature (Kamp et al., 2014; Marini et al., 2020). The presence of ApoE4 allele, which constitutes the main risk factor for AD, has also been demonstrated to constitute an important risk factor for this form of cSVD (Hermann and ElAli, 2012). ApoE4 is a lipid-binding protein which plays an important role in lipoprotein metabolism as well as transport of triglycerides and cholesterol (Hirsch-Reinshagen et al., 2009). It binds to LDL, very-low-density lipoprotein (VLDL) debris, and some HDL via LRP (Bu, 2009; Leduc et al., 2010). ApoE4 can form complexes with Aβ and impairs Aβ through LRP thus attenuating its clearance and subsequently leading to its accumulation in the brain (Cho et al., 2001; Verghese et al., 2013). Moreover, ApoE4 increases the formation of Aβ oligomers, which are now well established to constitute the most neurotoxic form of Aβ (Hashimoto et al., 2012; Youmans et al., 2012). Interestingly, human pericytes of the prefrontal cortex and hippocampus of ApoE4 carriers exhibit increased activation of nuclear factor of activated T-cells (NFAT), which might account for CAA occurrence (Blanchard et al., 2020; Figure 2).
Moreover, non-APP sources of CAA exist and are caused by mutations of the BRI2 [i.e., integral membrane protein (ITM)2B] gene, essentially a codon stop mutation. Indeed, processing of the mutated form of BRI2 protein leads to the generation of 34-mer amyloid Bri (ABri) and amyloid Dan (ADan) peptides that accumulate in the brain, either to the ABri amyloid subunit or the AD amyloidogenic fragment. ABri and ADan are responsible for the Familial British (FBD) and Danish (FDD) dementia characterized among other pathological features by severe CAA (Vidal et al., 1999, 2000; Yamada and Naiki, 2012).
Genetic cSVD
Mutations in specific genes constitute the third most common cause of cSVD, among which the mutation of the neurogenic locus notch homolog protein (NOTCH)3 gene is the better characterized (Cannistraro et al., 2019; Marini et al., 2020). NOTCH3 is a member of the transmembrane receptor NOTCH family, which is critically involved in developmental patterning, cell fate decisions, regulation of cell survival, and proliferation (Kopan and Ilagan, 2009; Bray, 2016; Baron, 2017; Hosseini-Alghaderi and Baron, 2020). During adulthood, NOTCH3 regulates stem cells and their lineages to promote tissue maintenance and repair. NOTCH3 is expressed by VSMCs and pericytes and plays a key role in regulating the crosstalk between the mural and endothelial cells. It controls the vascular tone and flow-mediated dilation via the modulation of the Ras homolog family member A (RHOA)/ Rho-associated protein kinase (ROCK) pathway in cerebral arteries (Joutel et al., 2000; Belin de Chantemèle et al., 2008; Li et al., 2009; Marini et al., 2020). However, the role of NOTCH3 is not restricted to the vasculature, since it is expressed in neural stem cells and is implicated in neuronal differentiation (Alunni et al., 2013; Kawai et al., 2017).
A mutation in the NOTCH 3 gene is responsible for CADASIL, the most common autosomal dominant inherited cSVD (Louvi et al., 2006; Di Donato et al., 2017; Hosseini-Alghaderi and Baron, 2020; Marini et al., 2020). NOTCH3 gene is affected essentially by missense mutations that lead to an odd number of cysteine residues located in the extracellular domain of the encoded receptor, and is associated with an early accumulation of the receptor’s extracellular domain containing aggregates in small vessels (Joutel et al., 2000, 2001; Monet-Leprêtre et al., 2013; Yamamoto et al., 2013). The function and activity of the NOTCH3 receptor are differently impacted by the mutations. However, the accumulation of extracellular domain containing aggregates in small vessels leads to mural cell degeneration via apoptosis or impaired proliferation (Joutel et al., 2000, 2001; Monet-Leprêtre et al., 2013; Yamamoto et al., 2013). Furthermore, the mutation itself causes profound morphological changes in pericytes, associated with dysfunctional mitochondria that could lead to oxidative and phosphorylation deficiencies, secondary lysosomes, and large cytoplasmic vesicles that result in cellular injury and autophagic apoptosis (de la Peña et al., 2001; Gu et al., 2012). This cascade of events cause neurovascular unit dysfunction characterized by detachment of astrocytic endfeet, destabilization of the vasculature, deregulation of vascular contractility, leakage of the BBB, and infiltration of toxic blood-born components into the brain parenchyma due to the decreased endothelial adherens junction protein, thus jointly resulting in diminished reactivity to CO2 (Ghosh et al., 2015; Figure 2).
Pericytes and endothelial cells are intimately interconnected through peg-and-socket junctions, which degenerate upon NOTCH3 mutations. For instance, endothelial cells exhibit degenerative features, such as selective death or swelling, causing vessel stenosis or occlusion (Dziewulska and Lewandowska, 2012). Deregulation of pericyte-endothelial cells crosstalk causes cerebrovascular dysfunction that comprises reduced vascular density and impaired CBF (Tuominen et al., 2004; Lacombe et al., 2005; Miao et al., 2006; Gu et al., 2012; De Guio et al., 2014; Liu X.-Y. et al., 2015; Ihara and Yamamoto, 2016; Ping et al., 2019). Moreover, chronic cerebral hypoperfusion resulting from cerebrovascular dysfunction exacerbates pericyte degeneration, reduces pericyte coverage for the capillaries, and subsequently increases BBB permeability leading to white matter impairments and neuronal loss (Ueno et al., 2002; Bell et al., 2010; Montagne et al., 2018; Liu et al., 2019; Nikolakopoulou et al., 2019). BBB breakdown allows the infiltration into the parenchyma of toxic blood-born metabolites that accumulate around the vasculature, thus inducing macrophage, microglia, and T-cells activation and recruitment, which jointly promote axonal degeneration (Davalos et al., 2012; Ryu et al., 2015). Neuronal loss is mainly provoked by the secretion of pro-inflammatory mediators and the generation of ROS and RNS by pericytes within the perivascular space, which further exacerbates leukocyte adhesion and infiltration as well as microglial cell activation (Matsumoto et al., 2018; Erdener and Dalkara, 2019). Finally, the structural lesions within the white matter are worsened by the release of pericyte-derived bone morphogenetic protein (BMP)-4, which promotes astrogliosis (Uemura et al., 2018, 2020). Evidence of these pathological events could be detected using imaging approaches that indicate WMH, ischemic manifestations, subcortical hemorrhages, and microbleeds. A new sensitive assay was recently developed allowing pericyte injury detection in the CSF, a new technology that could serve as a diagnostic tool for WMD (Sweeney et al., 2020).
CADASIL develops gradually over time and the earliest symptoms appear on average around 30 years of age, usually 10 years earlier in women than men, and are manifested as migraines with aura (Guey et al., 2016; Di Donato et al., 2017). The migraine could also manifest with atypical attacks with basilar, hemiplegic, or prolonged aura and a few patients can even develop very severe attacks leading to confusion, fever, meningitis, or even coma that can mimic encephalopathy (Schon et al., 2003; Vahedi et al., 2004; Ragno et al., 2013; Tan and Markus, 2016; Drazyk et al., 2019). Adults between the age of 20 and 65 years are subject to transient ischemic attacks and stroke (Lesnik Oberstein et al., 2001). CADASIL is associated as well with some psychiatric manifestations, which include mood disturbances, severe depression, and schizophrenia (Lågas and Juvonen, 2001; Valenti et al., 2008, 2011; Noh et al., 2014; Ho and Mondry, 2015; Di Donato et al., 2017). Finally, 40% of symptomatic cases report apathy which drastically impacts the quality of life of CADASIL patients (Reyes et al., 2009). The final stage of CADASIL progression is dementia, but cognitive decline starts years before (Brookes et al., 2016).
A recessive form of CADASIL exists under the name of CARASIL. This form of hereditary cSVD is caused by the mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene which has two major functions, degrading various substrates and inhibiting TGF-β1 signaling pathway that is involved in various processes namely angiogenesis and BBB formation via pericyte-endothelial cell crosstalk (Oka et al., 2004; Hara et al., 2009; Shiga et al., 2011; Akhtar-Schaefer et al., 2019; Kandasamy et al., 2020). HTRA1 is expressed in various brain cells comprising endothelial cells and VSMCs (De Luca et al., 2003; Oka et al., 2004; Campioni et al., 2010; Tennstaedt et al., 2012; Tiaden and Richards, 2013). Loss of HTRA1 function results in increased TGF-β1 availability and thereby signaling, leading to vascular fibrosis and extracellular matrix synthesis, which jointly cause microvascular degeneration, CBF reduction, and neurogenesis alterations (Wyss-Coray et al., 2000; Tarkowski et al., 2002; Gaertner et al., 2005; Yamamoto et al., 2011; Zhang et al., 2012; Martinez-Canabal et al., 2013; Beaufort et al., 2014; Friedrich et al., 2015). Moreover, the mutation has been shown to be associated with impaired pericyte proliferation, accumulation of protein within the vessel walls, MMPs activity, and BBB permeability (Joutel et al., 2016; Baron-Menguy et al., 2017; Ikawati et al., 2018; Marini et al., 2020). Cognitive decline begins early compared with CADASIL, as well as gait disturbances, lower back, pain and alopecia (Shiga et al., 2011; Marini et al., 2020).
Fabry’s disease is an X-inherited rare disorder that belongs to the family of lysosomal storage diseases and is caused by a mutation in the α-galactosidase (GAL)A gene that encodes for α-GAL enzyme that plays a key role in sphingolipid metabolism (El-Abassi et al., 2014). The mutation causes deficiencies in α-GAL activity that results in the accumulation of sphingolipids in various organs and tissues including the vessels (Rolfs et al., 2013). The cerebrovascular complications in Fabry’s disease arise from peripheral neuropathy and are associated with mild to severe headache, vertigo, transient ischemic attacks, ischemic stroke, intracerebral hemorrhage, and VaD (Okeda and Nisihara, 2008). Furthermore, deposition of toxic metabolites within the vasculature and VSMCs lead to ischemia, vessel stenosis, occlusion, and dilation with local changes in CBF (Shimotori et al., 2008). The disease is more severe in men compared to women and is often present with infantile neuropathy, gastrointestinal symptoms, corneal opacity, hearing loss, and angiokeratoma (Sims et al., 2009; Schiffmann, 2015; Marini et al., 2020). Brain structural damage and symptoms exacerbate with age.
Collagen IV, which exists as a heterodimer derived from the transcription of COL4A1 and COL4A2 genes, is an essential component of the vascular BM. Mutations in these two genes are associated with microangiopathies in several organs (Germain et al., 2019; Marini et al., 2020). More precisely, COL4A1 mutation is responsible for ocular, renal, muscular, and cerebral deficits (Vahedi and Alamowitch, 2011; Marini et al., 2020). Furthermore, cerebral microangiopathies have been shown to affect half of the carriers of this mutation. WMH, dilation of the perivascular spaces, lacunar infarctions, and microbleeds have been reported as well (Vahedi and Alamowitch, 2011). Pontine autosomal dominant microangiopathy and leukoencephalopathy (PADMAL) syndrome is a specific form of cerebral microangiopathies associated with the COL4A1 mutation (Verdura et al., 2016), and is associated with an overexpression of the gene with the absence of protein misfolding. Patients with this syndrome have dysarthria, ataxia, and stroke as well as mood disorders and dementia. On the other hand, COL4A2 mutation is associated with an increased prevalence of lacunar ischemic stroke and deep intracerebral hemorrhages (Verdura et al., 2016). In contrast to COL4A1 mutation, COL4A2 mutation impairs the trimerization of collagen IV due to defects in the α-helix structure, which cause BM instability, loss of vascular wall integrity, and increased BBB permeability, mediated by the intra- and extracellular accumulation of deficient collagen IV (Kuo et al., 2012; Meuwissen et al., 2015; Verdura et al., 2016; Zhang et al., 2017; Malik et al., 2018; Germain et al., 2019).
Forkhead Box C1 (FOXC1) is highly expressed in pericytes. Its expression plays an important role in controlling endothelial cell proliferation and vascular stability. FOXC1 mutation reproduces some of the events reported upon COL4A1 mutation, especially the ischemic infarctions, and cerebral microangiopathies that lead to WMH, which are visible in neuroimaging (French et al., 2014). The retinal vasculopathy with cerebral leukodystrophy (RVCL) syndrome includes three pathological conditions: (i) cerebral retinal vasculopathy (CRV); (ii) hereditary vascular retinopathy (HRV); and (iii) hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Patients with these syndromes possess a mutation in three prime repair exonuclease (TREX)-1 that encodes for a DNA exonuclease (Stam et al., 2016). This mutation causes a defect in apoptosis and INF signaling (Rice et al., 2015; Marini et al., 2020). All characteristics of the cerebral microangiopathies are found in RVCL patients who report the following symptoms: neurological deficits, migraines, cognitive deficits, psychiatric disorders, and seizures (Stam et al., 2016; Marini et al., 2020).
Environmental Risk Factors: Air Pollution
The increasing interaction with the environmental risk factors associated with human activities has a significant impact on health due to the exposure to various hazardous pollutants. Currently, environmental factors are directly implicated in the etiology and progression of diverse pathologies, including brain diseases. Indoor and outdoor air pollution is among the environmental factors that play a particularly important role in the deterioration of vascular health (Block and Calderón-Garcidueñas, 2009). Indeed, air pollution, which is defined as the release of an amalgam of pollutants into the atmosphere, has been reported to increase the prevalence of cardiovascular, cerebrovascular, and respiratory diseases, as well as cancer (Campbell et al., 2005; Schwartz et al., 2005; Calderón-Garcidueñas et al., 2007; Hartz et al., 2008; Block and Calderón-Garcidueñas, 2009; Mills et al., 2009; Rozemuller et al., 2012; Cho et al., 2018; Paul et al., 2019). Epidemiological studies have indicated that nearly one-third of the global stroke burden and about one-fifth of the global dementia burden, including VaD, are attributable to air pollution (Feigin et al., 2016; Azarpazhooh and Hachinski, 2018; Béjot et al., 2018). Furthermore, numerous studies have outlined a link between high levels of air pollutants, chronic brain inflammation, and neurodegeneration (Campbell et al., 2005; Schwartz et al., 2005; Calderón-Garcidueñas et al., 2007; Hartz et al., 2008; Block and Calderón-Garcidueñas, 2009; Mills et al., 2009; Rozemuller et al., 2012; Paul et al., 2019). These effects are mainly attributable to the exposure to fine particulate matter (PM), and more precisely to PM of 2.5 microns or less in diameter (PM2.5). Indeed, the experimental findings have indicated that UFPs could reach the brain through different routes, including the intranasal cavity, where they act as an inflammatory mediators, thus deregulating the function of cells forming the neurovascular unit (Oberdörster et al., 2004; Peters et al., 2006). In particular, exposure to PM leads to impaired olfactory function, one of the initial atypical symptoms that emerge in individuals affected by different forms of dementia (Campbell et al., 2005; Schwartz et al., 2005; Calderón-Garcidueñas et al., 2007; Hartz et al., 2008; Block and Calderón-Garcidueñas, 2009; Mills et al., 2009; Rozemuller et al., 2012; Paul et al., 2019). The correlation between air pollution and dementia, including VaD and AD, was highlighted in various epidemiological studies relying mostly on cohort studies in polluted regions (Åström et al., 2021). The Betula cohort revealed an association of dementia incidence, AD in particular, with traffic-related air pollution (TRAP; Oudin et al., 2019). Moreover, studies have found that exposure of the elderly to air pollution, notably PM10 and PM2.5 was associated with cognitive decline (Wu et al., 2015). In an interesting case-control study, it was reported that elevated long-term PM10 levels were associated with a significantly increased risk of AD and VaD prevalence in the elderly. A dose-response relationship between PM10 exposure and the risk of AD and VaD was reported (Wu et al., 2015).
Impact on Endothelial Functions
Various studies have investigated the impact of air pollutants, and more specifically PM, on endothelial functions. For instance, exposure of isolated rat brain capillaries to diesel exhaust particles (DEP) altered BBB function through oxidative stress generation and proinflammatory cytokine production, which jointly induced the expression of adhesion molecules that exacerbate infiltration of immune cells into the brain (Hartz et al., 2008). Exposure to DEP deregulated the expression of several transporters and receptors that are critically involved in BBB functionality namely ABCB1, multidrug-resistance associated proteins (MRP)1, MRP2, MRP4, breast cancer resistance protein (BCRP), glucose transporter (GLUT)1, and the metabolizing enzyme glutathione S-transferase (GST)π (Hartz et al., 2008). Several reports have demonstrated an increased BBB permeability following exposure to a mixed vehicular emission, translated essentially by deregulation of the TJs (Rojas et al., 2011; Oppenheim et al., 2013; Bernardi et al., 2021). For instance, human brain microvascular endothelial cells in culture exposed to nanoparticles of aluminum oxide exhibit reduced cell viability, altered mitochondrial function, increased oxidative stress, and diminished expression of the TJs proteins claudin-5 and occludin (Chen et al., 2008; Block and Calderón-Garcidueñas, 2009). Interestingly, epidemiological studies have outlined a strong correlation between air pollution and neuroinflammation in highly exposed residents (Calderón-Garcidueñas et al., 2008). Indeed, these studies reported an upregulation of some inflammatory markers, such as expression of cyclooxygenase (COX)-2 and IL-1β, as well as infiltration of immune cells into the olfactory bulb (OB), frontal cortex, substantia nigrae, and vagus nerves (Calderón-Garcidueñas et al., 2008). Importantly, most of the inflammatory responses were concentrated at the vasculature translated by activation of NF-κB pathway in brain endothelial cells, accompanied by oxidative stress, Aβ42 immunoreactivity, trafficking of inflammatory cells into the perivascular space, and an altered BBB (Calderón-Garcidueñas et al., 2008). In line with these observations, direct exposure of brain endothelial cells in culture to PM2.5 deregulated TJs and increased permeability and monocyte transmigration across the endothelial monolayer (Liu F. et al., 2015). In addition, exposure to both PM2.5 and PM10 induced the activation of endothelial cells accompanied by an enhanced adhesion of U937 monocytic cells to the endothelial monolayer (Montiel-Dávalos et al., 2007). Interestingly, exposure to PM2.5 also induced ICAM-1 expression, whereas exposure to PM10 induced expression of E-selectin and P-selectin (Montiel-Dávalos et al., 2007; Figure 2).
In vivo experiments in which mice were exposed to a mixed vehicular emission, a combination of gasoline and diesel engine exhausts, the animals exhibited altered BBB integrity through the deregulation of the TJs protein, namely claudin-5 and occludin (Oppenheim et al., 2013). This was accompanied by an augmentation of inducible nitric oxide synthase (iNOS) levels, an increase in the production of IL-1β in the parenchyma, and deregulation of ABCB1 transport activity (Oppenheim et al., 2013). Moreover, exposure to PM in vitro and in vivo has been shown to stimulate the re-localization of the TJs protein ZO-1 from the cell membrane and reduce its protein level (Wang et al., 2012). Importantly, PM mediated the intracellular mobilization of calcium (Ca2+) dependently upon ROS, activating calpain that is implicated in ZO-1 degradation and disruption of the endothelial barrier (Wang et al., 2012). Using a 3D human in vitro BBB model, indoor nanoscale particulate matter (INPM) was shown to translocate across the BBB and accentuate inflammation by inducing ROS (Li et al., 2020). This induction was followed by abnormal nuclear reactor factor (NRF)-2 expression and a disruption of the kelch ECH associating protein (KEAP)-1/antioxidant response elements (ARE) pathway which is involved in supporting cells to overcome stress (Li et al., 2020). Interestingly, exposure of rodents to urban PM increased the levels of ET-1 mRNA and reduced TNF-α mRNA levels in the cerebral hemisphere and the pituitary gland. These results suggest that the cerebrovascular effects of urban pollutants are associated with the modulation of gene expression involved in the regulation of vasoconstriction in the brain and pituitary gland (Benatti et al., 1993; Thomson et al., 2007).
Exposure to PM2.5 has been shown to increase the prevalence of carotid artery stenosis (CAS), a well-established risk factor for ischemic stroke, correlating with an increased BBB permeability (Newman et al., 2015; Szarmach et al., 2017). In line with these observations, a strong association between air pollution with systemic brain inflammation was revealed in children living in polluted areas, associated with short-term memory deficits, prefrontal WMH, and BBB disruption (Calderón-Garcidueñas et al., 2016). The same study reported a leaking vascular network, degeneration of pericytes, VSMCs, and endothelial cells, thickening of the BM, and reduced perivascular astroglial coverage in the prefrontal white matter of dog brains (Calderón-Garcidueñas et al., 2016). Exposure to PM2.5 has been demonstrated as well to accelerate atherosclerosis development through induction of vascular dysfunction as well as promotion of coagulopathies, which were accompanied by a strong inflammatory response and lipid abnormalities (Liang et al., 2020). Finally, exposure of ApoE-deficient mice to TRAP, mixed vehicle emissions, induced the cerebral expression of ICAM-1 and the release of pro-inflammatory mediators, such as TNF-α and IL-1β (Adivi et al., 2021).
Impact on the Dynamics of Astrocytes and Oligodendrocytes
Evidence indicates that astrocytes respond to PM in a context-dependent manner (Allen et al., 2014). For instance, early postnatal exposure to ambient UFPs decreased GFAP immunoreactivity in male subjects and increased GFAP expression as well as other neuroinflammation markers in females (Allen et al., 2014). Exposure of male and female rodents during gestation and early postnatal development to TRAP attenuated astrogliosis specifically in the dentate gyrus (DG) associated with reduced GFAP immunoreactivity, which remained unchanged in CA1 and CA3 regions (Patten et al., 2020). Previous findings have shown that maternal exposure to carbon black nanoparticles (CB-NP) induced astrogliosis in the cortex of rodents, affecting the interaction of the astrocyte endfeet with the endothelium and perivascular macrophages (Onoda et al., 2017). Interestingly, intranasal delivery of PM2.5 to male rodents subjected to ischemic stroke exacerbated astrocytic reactivity via GFAP activation and iNOS induction, aggravating post-stroke neurobehavioral impairments (Chen et al., 2016). In line with these results, rodents exposed to PM for a prolonged period exhibited altered neuronal and astrocytic functions via impairment of mitochondrial activity (Araújo et al., 2019). Likewise, exposure of rodents to natural air pollution sources, such as volcanic-derived particles, increased GFAP immunoreactivity in astrocytes (Camarinho et al., 2019; Navarro et al., 2021). Moreover, exposure of rodents to low doses of CB-NP induced endoplasmic reticulum (ER) stress in perivascular macrophages and reactive astrocytes, specifically around the vasculature of offspring animals, associated with the accumulation of β-sheet rich misfolded proteins (Onoda et al., 2020). Cell-based assays have shown that exposure of astrocytes to PM activated janus kinase (JAK)-2/STAT-3 and p38/JNK/ERK pathways in reactive astrocytes triggering iNOS induction and IL-1β production (Li et al., 2016). In this regard, PM has been reported to increase the expression and release of proinflammatory mediators through activation of the NF-κB signaling pathway (Li et al., 1999; Gómez-Budia et al., 2020).
Furthermore, exposure to UFPs altered adult OPCs turnover and survival of mature oligodendrocytes (OLs), accompanied by increased oxidative stress and decreased total antioxidant capacities (TAC) that impair the remyelination capacity of the brain (Kim J. Y. et al., 2020). Furthermore, prenatal exposure to concentrated ambient particles (CAPs), which were defined as UFPs, promoted a premature maturational shift in OLs in the corpus callosum (CC), followed by hypermyelination (Klocke et al., 2018). Interestingly, females showed significant alterations in oligodendrocytogenesis in the CC (Klocke et al., 2018). Finally, in a mouse model of lysolecithin-induced demyelination of the subcortical white matter, exposure to PM2.5 hampered remyelination and disrupted oligodendroglia differentiation (Parolisi et al., 2021).
Impact on Microglial Reactivity and Consequences on Neurons
In response to air pollution, microglia are activated through the upregulation of several proinflammatory mediators that affect neuronal function (Gómez-Budia et al., 2020). Microglia respond to air pollution by adopting an amoeboid shape in vitro (Cole et al., 2016; Roqué et al., 2016; Mumaw et al., 2017), as well as in vivo (Araújo et al., 2019). Indeed, mice exposed to DEP exhibited reduced adult neurogenesis through activation of microglia (Coburn et al., 2018). In contrast, attenuating microglial reactivity hampered neuroinflammation and oxidative stress (Coburn et al., 2018). Cell-based assays showed that exposure of BV-2 immortalized microglial cell to PM2.5 increased mRNA expression of various proinflammatory markers, namely IL-6, IL-1β, TNF-α, iNOS, COX-2, TREM2, and TLR2/4, while reducing mRNA expression of key anti-inflammatory markers, such as IL-10 and Arginase (ARG)-1 (Kim R.-E. et al., 2020). In parallel, exposure of rodents to DEP for a long period triggered the activation of microglia in the nuclei of the solitary tract (NTS), accompanied by ER dilation and mitochondrial vacuolization in the medulla, hence outlining structural alterations of the neuronal network (Chen et al., 2021). Interestingly, rodents exposed to DEP for a long period showed an induced ionized calcium-binding adaptor molecule (IBA)-1 expression in the brain (Levesque et al., 2011). Interestingly, microglial reactivity was accompanied by an increased mRNA expression of TNF-α, IL-6, and macrophage inflammatory protein (MCP)-1α in the midbrain, cortex, and OB, while IL-1β was expressed particularly in the midbrain (Levesque et al., 2011). Mice exposed to PM also triggered the induction of mRNA expression of TLR4, MyD88, TNF-α, and tumor necrosis factor receptor (TNFR)-2 in microglia (Woodward et al., 2017a). Activation of the latter was further confirmed through an increased intracellular expression of inflammatory mediators, such as COX-2, NF-κB, prostaglandin E2 (PGE2), and iNOS both in vitro and in vivo (Babadjouni et al., 2018; Chen et al., 2018). Using cell-based assays, TNF-α induction in microglia upon PM exposure inhibited neurite outgrowth (Cheng et al., 2016). Inhibition of NRF-2 activity prior to exposure of BV-2 microglial cells to PM2.5 attenuated cell viability, induced ROS generation, and stimulated NF-κB pathway, outlining NRF-2 role in mitigating PM deleterious effects (Chen et al., 2018).
Importantly, in vivo experiments showed that exposure to DEP in TREM2−/− mice accentuated IL-1β expression (Greve et al., 2020). PM has been demonstrated to impact neuron-glial crosstalk. Acute exposure of adult mice to PM2.5 increased the levels of lipoperoxidation and proinflammatory cytokines in the brain and activated microglia, accompanied by reduced neurogenesis in the subgranular zone (SGZ) and subventricular zone (SVZ; Bernardi et al., 2021). Neuronal cell cultures exhibited reduced viability upon exposure to PM2.5 associated with increased release of glutamate (Liu F. et al., 2015). Prior treatment of cells with the N-methyl-D-aspartate (NMDA) receptor mitigated PM2.5-mediated neuronal loss (Liu F. et al., 2015). Moreover, neuronal cultures displayed dopaminergic neurotoxicity upon exposure to DEP only in presence of microglia, which was associated with elevated levels of ROS (Block et al., 2004). Co-culture of neurons and microglia exposed to PM2.5 in presence of oligomeric oAβ exacerbated IL-1β and ROS release aggravating oAβ-induced neuronal injury and inflammation (Wang et al., 2018). PM have been shown to directly impact neuronal function. For instance, exposure of rodents to nano PM caused hippocampal neurite atrophy and decreased expression of myelin basic protein (MBP), accompanied by increased TNF-α mRNA expression (Woodward et al., 2017b). In this regard, epidemiological studies have outlined a strong correlation between the levels of PM in air and neuronal chromatolysis and satellitosis in exposed dogs, associated with cortical neurons degeneration, and neurofibrillary tangle formation (Calderón-Garcidueñas et al., 2002).
Interestingly, epidemiological studies comprising elderly women revealed that residence in places contaminated with high levels of fine PM increases the risks for global cognitive decline and all-cause of dementia by 81 and 92%, respectively, with stronger adverse effects in ApoE4 carriers (Cacciottolo et al., 2017). Experimental findings obtained from female AD mouse models (5xFAD; Familial AD) expressing either ApoE3 or ApoE4 mice that were exposed to urban nano PM for 15 weeks showed increased Aβ plaques and soluble Aβ oligomers, which were associated with neuronal changes in the hippocampus (Cacciottolo et al., 2017). These findings were confirmed in vitro upon exposure of neuroblastoma cells (N2a-APP/swe) to nano PM translated by an enhanced pro-amyloidogenic processing of the APP, explaining the elevated cerebral Aβ production (Cacciottolo et al., 2017). Furthermore, long-term exposure to ambient air pollution was found to be associated with rapid cognitive decline in aged adults, where ApoE4 carriers exhibited the fastest cognitive decline (Kulick et al., 2020; Figure 2).
Further in vivo experimental investigations implicating exposure to TRAP nano PM showed an increased production of Aβ peptides, associated with oxidative damage (Cacciottolo et al., 2020). Indeed, exposure of J20-APPswe mice, an AD mouse model, to nano PM for 150 h revealed exacerbated lipid oxidation and pro-amyloidogenic processing of APP in lipid raft fractions compared to controls (Cacciottolo et al., 2020). These observations were further confirmed in vitro using N2a-APPswe cells exposed to nano PM (Cacciottolo et al., 2020). Importantly, the link between air pollution and HDL was highlighted in the Multi-Ethnic Study of Atherosclerosis Air Pollution (MESA Air) study showing that exposure to air pollution was significantly associated with low levels of HDL (Bell et al., 2017). In this regard, ApoE-deficient mice exposed for 2 weeks to DEP, exhibited high systemic pro-oxidant effects associated with dysfunctional HDL (Yin et al., 2013). These observations would indicate that PM exposure could be responsible for the attenuated HDL protective effects against atherosclerosis (Yin et al., 2013).
Conclusion and Perspectives
Through this review, we aimed to highlight the structural and functional neurovascular alterations underlying VaD pathobiology. The epidemiological, clinical, and experimental investigations are indicating that the long-term outcomes of early pathophysiological events impacting neurovascular functions upon cerebrovascular disorders have major consequences on imitating a cascade of events that lead to VaD. Moreover, the recent findings are outlining air pollution as a major vascular risk factor that is directly implicated in promoting neurovascular impairments associated with VaD. Given the heterogeneity of cerebrovascular disorders, which include stroke, genetic and sporadic microangiopathies, combined with the diverse effects of environmental factors, a better understating of the short- and long-term remodeling processes at the neurovascular unit is urgently required to allow getting new insights into VaD pathobiology. Such knowledge will allow the identification of key “targetable” mechanisms for therapeutic purposes.
Recent findings are suggesting that BBB disruption occurs in some cases for a prolonged period beyond the acute and subacute phases after stroke (Bernardo-Castro et al., 2020). A prolonged, yet subtle, disruption of the BBB could trigger a cascade of events that lead to VaD. For instance, high expression levels of MMPs, the main mediator of BBB disruption, are significantly associated with cognitive deficits (Yang and Rosenberg, 2011). However, targeting MMPs is a double-edged sword as while preserving the BBB in the acute and sub-acute phase, its inhibition impairs neurovascular adaptation in the chronic phase, thus impeding brain plasticity (Yang and Rosenberg, 2011). More promising appears to be the inhibition of beta-site APP cleaving enzyme (BACE)1 that seems to protect endothelial cell integrity in the context of CADASIL and AD (Chacón-Quintero et al., 2021). In the same way, rodent models of aging suggest that the mitochondrial overexpression of catalase improves diminish vascular impairment and benefits neurovascular coupling (Csiszar et al., 2019).
On the other hand, uncontrolled post-stroke neuroinflammation, especially excessive microglial activation, is associated with cognitive deficits (Guruswamy and ElAli, 2017; Gefen et al., 2019; Jayaraj et al., 2019; Zhang et al., 2021). In this regard, strategies aiming to mitigate neuroinflammation via modulation of microglia could attenuate cognitive decline related to stroke (Guruswamy and ElAli, 2017; Dzyubenko et al., 2018; Jayaraj et al., 2019). In this regard, evidence suggests that the prominent microglial regulator, insulin-like growth factor (IGF)-1, reduces gliosis while preserving brain volume and myelination, as well as motor performance and memory when administered intranasally in aged mice (Farias Quipildor et al., 2019). Efficient immunomodulatory approaches capable of fine-tuning microglial activation are still to be developed.
Importantly, the emergence and turnover of stroke-mediated proteinopathies are associated with BBB disruption and neuroinflammation. Although the role of cSVD is critically important in the etiology of VaD, there is still a huge gap in the literature as to our understanding of the underlying mechanisms. This is due to the fact that the current knowledge is obtained either from genetic or non-clinically relevant sporadic animal models. Indeed, the majority of cSVD cases are sporadic and associated with diverse risk factors, as such, it is critically important to develop animal models that replicate some of the pathological features associated with VaD. However, the overwhelming findings indicate the loss-of-function on the local cerebrovascular network caused a central role in initiating a pathological cascade of events that lead to altered neuro-glial functions, and thus subsequently dementia (Rouhl et al., 2012; Shoamanesh et al., 2015; Fu and Yan, 2018; Li et al., 2018; Jian et al., 2020).
Air pollution has emerged as a significant risk factor for cerebrovascular and neurodegenerative disorders (Wu et al., 2015; Åström et al., 2021). It is becoming clear that exposure to PM, one of the most deleterious air pollutants, increases the risk of chronic neuroinflammation that leads to dementia (Wu et al., 2015; Åström et al., 2021). Cell-based assays have demonstrated that PM acts as a powerful inflammatory and oxidative stress mediator in various brain cell types. Interestingly, PM exposure seems to amplify the pathological responses underlying VaD pathobiology. Although the impact of PM on neurovascular functions is evidenced in vitro, little is known about its role in mediating neurovascular impairments in vivo. Future studies should consider investigating the consequences of PM exposure as a comorbid condition in the design of preclinical experiments.
As previously mentioned, besides PM, various modifiable vascular risk factors are recognized to impact VaD. For instance, stroke and cSVD share common risk factors such as hypertension, atherosclerosis, obesity, atrial fibrillation, diabetes, dyslipidemia, high homocysteine, metabolic syndrome, smoking, as well as cardiac and carotid arterial disease (Barnes and Yaffe, 2011; O’Brien and Thomas, 2015; Kalaria et al., 2016; Tariq and Barber, 2018). Given their central role, various preventive approaches have been developed and adopted to attenuate the impact of this triad of vascular on VaD. Such approaches are exemplified by the landmark multidomain Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), which has been shown to enhance all cognitive sub-domains through a multidomain lifestyle intervention that include dietary counseling, physical exercise, cognitive training, and vascular and metabolic risk monitoring for a period of 2 years (Ngandu et al., 2015; Kivipelto et al., 2020). Furthermore, knowing the detrimental role of high blood pressure in VaD prevalence, the Systolic Blood Pressure Intervention Trial (SPRINT) and its sub-study the Memory and Cognition in Decreased Hypertension (MIND) were established with an emphasis on investigating the consequences of lowering systolic blood pressure. Although the incidence of dementia was not improved, the trial reported a reduction of mild cognitive impairment (MCI) and MCI composite in subjects with lower blood pressure. Interestingly, these observations were associated with a reduction of WM lesion volumes (Kjeldsen et al., 2018; Peters et al., 2019). Despite that, each trial discloses important limitations, yet the impact of modifying main VaD risk factors was shown to allow the development of efficient interventions. Further research is needed for longer periods with a more representative population to lessen the limitations and improve the efficiency of interventions aiming to attenuate dementia prevalence.
Finally, a deep analysis of the current scientific knowledge outlines brain pericytes as a major effector in the pathobiology of VaD. Through their spatial localization, pericytes play a central role in integrating and processing signals from their milieu to generate critical neurovascular functions, which include BBB maintenance, CBF modulation, vascular stabilization, and immunomodulation (Zlokovic, 2011; Hermann and ElAli, 2012). Following a stroke, pericytes undergo cell death at the ischemic core and get activated in the peri-lesion site where they detach from the vasculature (Zlokovic, 2011; Hermann and ElAli, 2012). Importantly, it has been reported that even after successful recanalization, pericytes located at the peri-lesion site remain contracted impeding the capillary microcirculation, which leads to vascular constriction and chronic hypoperfusion (Dalkara, 2019). Moreover, microcirculation of the white matter was disrupted upon pericyte degeneration leading to the accumulation of toxic blood-derived fibrotic deposits within the vasculature, which promotes vascular fibrosis, accompanied by a reduction in the regional CBF (Montagne et al., 2018). These changes are supposed to be directly implicated in the pathogenesis of diffuse WMD associated with loss of oligodendrocytes and subsequently myelinated axons (Montagne et al., 2018). The findings indicate that pericyte degeneration plays a major role in the pathogenesis, and thereby therapies, of WMD associated with cSVD (Montagne et al., 2018). Furthermore, the generation of pericytes is translated by elevated levels of the soluble-platelet derived growth factor receptor (PDGFR)β (sPDGFRβ) in the CSF, strongly correlating with BBB breakdown, CBF reduction, and cognitive decline (Sweeney et al., 2020). Therefore, decoding the pericyte reactivity to stressors related to the vascular risk factors constitutes a promising avenue that might lead to achieving major breakthroughs in getting new mechanistic insights in the pathobiology of VaD and in developing novel therapeutic interventions.
Author Contributions
The authors contributed to the review as follows: SL wrote, edited, and finalized the draft as well as prepared the figures. DM-C wrote, edited, and finalized the draft as well as prepared the figures. YE wrote the section related to air pollution. AEA conceptualized, wrote, and finalized the draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC; #RGPIN-2017-06119), the Fonds de recherche du Québec—Santé (FRQS; #35170), Heart and Stroke Foundation of Canada (HSFC; #G-18-0022118), and the Canadian Institutes of Health Research (CIHR; #169062; all to AEA). AEA holds a CIHR Tier 2 Canada Research Chair in molecular and cellular neurovascular interactions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adair, J. C., Charlie, J., Dencoff, J. E., Kaye, J. A., Quinn, J. F., Camicioli, R. M., et al. (2004). Measurement of gelatinase B (MMP-9) in the cerebrospinal fluid of patients with vascular dementia and Alzheimer disease. Stroke 35, e159–e162. doi: 10.1161/01.STR.0000127420.10990.76
Adivi, A., Lucero, J., Simpson, N., McDonald, J. D., and Lund, A. K. (2021). Exposure to traffic-generated air pollution promotes alterations in the integrity of the brain microvasculature and inflammation in female ApoE−/− mice. Toxicol. Lett. 339, 39–50. doi: 10.1016/j.toxlet.2020.12.016
Akhtar-Schaefer, I., Reuten, R., Koch, M., Pietsch, M., and Langmann, T. (2019). AMD-associated HTRA1 variants do not influence TGF-β signaling in microglia. Adv. Exp. Med. Biol. 1185, 3–7. doi: 10.1007/978-3-030-27378-1_1
Alexander, M. R., and Owens, G. K. (2012). Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 74, 13–40. doi: 10.1146/annurev-physiol-012110-142315
Allen, J. L., Liu, X., Weston, D., Prince, L., Oberdörster, G., Finkelstein, J. N., et al. (2014). Developmental exposure to concentrated ambient ultrafine particulate matter air pollution in mice results in persistent and sex-dependent behavioral neurotoxicity and glial activation. Toxicol. Sci. 140, 160–178. doi: 10.1093/toxsci/kfu059
Alunni, A., Krecsmarik, M., Bosco, A., Galant, S., Pan, L., Moens, C. B., et al. (2013). Notch3 signaling gates cell cycle entry and limits neural stem cell amplification in the adult pallium. Development 140, 3335–3347. doi: 10.1242/dev.095018
Amtul, Z., Whitehead, S. N., Keeley, R. J., Bechberger, J., Fisher, A. L., McDonald, R. J., et al. (2015). Comorbid rat model of ischemia and β-amyloid toxicity: striatal and cortical degeneration. Brain Pathol. 25, 24–32. doi: 10.1111/bpa.12149
Andrianopoulos, V., Gloeckl, R., Vogiatzis, I., and Kenn, K. (2017). Cognitive impairment in COPD: should cognitive evaluation be part of respiratory assessment? Breathe 13, e1–e9. doi: 10.1183/20734735.001417
Appelros, P., Stegmayr, B., and Terént, A. (2009). Sex differences in stroke epidemiology: a systematic review. Stroke 40, 1082–1090. doi: 10.1161/STROKEAHA.108.540781
Araújo, J. E., Jorge, S., Santos, H. M., Chiechi, A., Galstyan, A., Lodeiro, C., et al. (2019). Proteomic changes driven by urban pollution suggest particulate matter as a deregulator of energy metabolism, mitochondrial activity, and oxidative pathways in the rat brain. Sci. Total Environ. 687, 839–848. doi: 10.1016/j.scitotenv.2019.06.102
Aribisala, B. S., Valdés Hernández, M. C., Royle, N. A., Morris, Z., Muñoz Maniega, S., Bastin, M. E., et al. (2013). Brain atrophy associations with white matter lesions in the ageing brain: the Lothian Birth Cohort 1936. Eur. Radiol. 23, 1084–1092. doi: 10.1007/s00330-012-2677-x
Arvanitakis, Z., Leurgans, S. E., Wang, Z., Wilson, R. S., Bennett, D. A., and Schneider, J. A. (2011). Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327. doi: 10.1002/ana.22112
Åström, D. O., Adolfsson, R., Segersson, D., Forsberg, B., and Oudin, A. (2021). Local contrasts in concentration of ambient particulate air pollution (PM2.5) and incidence of Alzheimer’s disease and dementia: results from the betula cohort in northern sweden. J. Alzheimers Dis. 81, 83–85. doi: 10.3233/JAD-201538
Attems, J., Jellinger, K., Thal, D. R., and Van Nostrand, W. (2011). Review: sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 37, 75–93. doi: 10.1111/j.1365-2990.2010.01137.x
Auffray, C., Fogg, D., Garfa, M., Elain, G., Join-Lambert, O., Kayal, S., et al. (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670. doi: 10.1126/science.1142883
Azarpazhooh, M. R., and Hachinski, V. (2018). Air pollution: a silent common killer for stroke and dementia. Int. J. Stroke 13, 667–668. doi: 10.1177/1747493018784476
Babadjouni, R., Patel, A., Liu, Q., Shkirkova, K., Lamorie-Foote, K., Connor, M., et al. (2018). Nanoparticulate matter exposure results in neuroinflammatory changes in the corpus callosum. PLoS One 13:e0206934. doi: 10.1371/journal.pone.0206934
Ballabio, E., Bersano, A., Bresolin, N., and Candelise, L. (2007). Monogenic vessel diseases related to ischemic stroke: a clinical approach. J. Cereb. Blood Flow Metab. 27, 1649–1662. doi: 10.1038/sj.jcbfm.9600520
Bandeali, S., and Farmer, J. (2012). High-density lipoprotein and atherosclerosis: the role of antioxidant activity. Curr. Atheroscler. Rep. 14, 101–107. doi: 10.1007/s11883-012-0235-2
Barnes, D. E., and Yaffe, K. (2011). The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 10, 819–828. doi: 10.1016/S1474-4422(11)70072-2
Baron, M. (2017). Combining genetic and biophysical approaches to probe the structure and function relationships of the notch receptor. Mol. Membr. Biol. 34, 33–49. doi: 10.1080/09687688.2018.1503742
Baron-Menguy, C., Domenga-Denier, V., Ghezali, L., Faraci, F. M., and Joutel, A. (2017). Increased notch3 activity mediates pathological changes in structure of cerebral arteries. Hypertension 69, 60–70. doi: 10.1161/HYPERTENSIONAHA.116.08015
Barr, T. L., Latour, L. L., Lee, K.-Y., Schaewe, T. J., Luby, M., Chang, G. S., et al. (2010). Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke 41, e123–e128. doi: 10.1161/STROKEAHA.109.570515
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNFα. Science 295, 2282–2285. doi: 10.1126/science.1067859
Beaufort, N., Scharrer, E., Kremmer, E., Lux, V., Ehrmann, M., Huber, R., et al. (2014). Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc. Natl. Acad. Sci. U S A 111, 16496–16501. doi: 10.1073/pnas.1418087111
Béjot, Y., Reis, J., Giroud, M., and Feigin, V. (2018). A review of epidemiological research on stroke and dementia and exposure to air pollution. Int. J. Stroke 13, 687–695. doi: 10.1177/1747493018772800
Belin de Chantemèle, E. J., Retailleau, K., Pinaud, F., Vessières, E., Bocquet, A., Guihot, A. L., et al. (2008). Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler. Thromb. Vasc. Biol. 28, 2216–2224. doi: 10.1161/ATVBAHA.108.171751
Bell, G., Mora, S., Greenland, P., Tsai, M., Gill, E., and Kaufman, J. D. (2017). Association of air pollution exposures with high-density lipoprotein cholesterol and particle number: the multi-ethnic study of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 37, 976–982. doi: 10.1161/ATVBAHA.116.308193
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Benatti, L., Bonecchi, L., Cozzi, L., and Sarmientos, P. (1993). Two preproendothelin 1 mRNAs transcribed by alternative promoters. J. Clin. Invest. 91, 1149–1156. doi: 10.1172/JCI116274
Bernardi, R. B., Zanchi, A. C. T., Damaceno-Rodrigues, N. R., Veras, M. M., Saldiva, P. H. N., Barros, H. M. T., et al. (2021). The impact of chronic exposure to air pollution over oxidative stress parameters and brain histology. Environ. Sci. Pollut. Res. Int. doi: 10.1007/s11356-021-14023-0 [Epub ahead of print].
Bernardo-Castro, S., Sousa, J. A., Brás, A., Cecília, C., Rodrigues, B., Almendra, L., et al. (2020). Pathophysiology of blood-brain barrier permeability throughout the different stages of ischemic stroke and its implication on hemorrhagic transformation and recovery. Front. Neurol. 11:594672. doi: 10.3389/fneur.2020.594672
Berzin, T. M., Zipser, B. D., Rafii, M. S., Kuo-Leblanc, V., Yancopoulos, G. D., Glass, D. J., et al. (2000). Agrin and microvascular damage in Alzheimer’s disease. Neurobiol. Aging 21, 349–355. doi: 10.1016/s0197-4580(00)00121-4
Bevan-Jones, W. R., Cope, T. E., Jones, P. S., Kaalund, S. S., Passamonti, L., Allinson, K., et al. (2020). Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain 143, 1010–1026. doi: 10.1093/brain/awaa033
Blanchard, J. W., Bula, M., Davila-Velderrain, J., Akay, L. A., Zhu, L., Frank, A., et al. (2020). Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 26, 952–963. doi: 10.1038/s41591-020-0886-4
Block, M. L., and Calderón-Garcidueñas, L. (2009). Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 32, 506–516. doi: 10.1016/j.tins.2009.05.009
Block, M. L., Wu, X., Pei, Z., Li, G., Wang, T., Qin, L., et al. (2004). Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 18, 1618–1620. doi: 10.1096/fj.04-1945fje
Bordeleau, M., ElAli, A., and Rivest, S. (2016). Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice. Oncotarget 7, 11864–11880. doi: 10.18632/oncotarget.7689
Bourasset, F., Ouellet, M., Tremblay, C., Julien, C., Do, T. M., Oddo, S., et al. (2009). Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer’s disease. Neuropharmacology 56, 808–813. doi: 10.1016/j.neuropharm.2009.01.006
Braughler, J. M., and Hall, E. D. (1992). Involvement of lipid peroxidation in CNS injury. J. Neurotrauma 9, S1–S7.
Bray, S. J. (2016). Notch signalling in context. Nat. Rev. Mol. Cell Biol. 17, 722–735. doi: 10.1038/nrm.2016.94
Brookes, R. L., Hollocks, M. J., Tan, R. Y. Y., Morris, R. G., and Markus, H. S. (2016). Brief screening of vascular cognitive impairment in patients with cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy without dementia. Stroke 47, 2482–2487. doi: 10.1161/STROKEAHA.116.013761
Brundel, M., de Bresser, J., van Dillen, J. J., Kappelle, L. J., and Biessels, G. J. (2012). Cerebral microinfarcts: a systematic review of neuropathological studies. J. Cereb. Blood Flow Metab. 32, 425–436. doi: 10.1038/jcbfm.2011.200
Bruno, M. A., Mufson, E. J., Wuu, J., and Cuello, A. C. (2009). Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 68, 1309–1318. doi: 10.1097/NEN.0b013e3181c22569
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Cacciottolo, M., Morgan, T. E., Saffari, A. A., Shirmohammadi, F., Forman, H. J., Sioutas, C., et al. (2020). Traffic-related air pollutants (TRAP-PM) promote neuronal amyloidogenesis through oxidative damage to lipid rafts. Free Radic. Biol. Med. 147, 242–251. doi: 10.1016/j.freeradbiomed.2019.12.023
Cacciottolo, M., Wang, X., Driscoll, I., Woodward, N., Saffari, A., Reyes, J., et al. (2017). Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 7:e1022. doi: 10.1038/tp.2016.280
Caggiu, E., Arru, G., Hosseini, S., Niegowska, M., Sechi, G., Zarbo, I. R., et al. (2019). Inflammation, infectious triggers, and parkinson’s disease. Front. Neurol. 10:122. doi: 10.3389/fneur.2019.00122
Cai, Z., Wang, C., He, W., Tu, H., Tang, Z., Xiao, M., et al. (2015). Cerebral small vessel disease and Alzheimer’s disease. Clin. Interv. Aging 10, 1695–1704. doi: 10.2147/CIA.S90871
Calderón-Garcidueñas, L., Azzarelli, B., Acuna, H., Garcia, R., Gambling, T. M., Osnaya, N., et al. (2002). Air pollution and brain damage. Toxicol. Pathol. 30, 373–389. doi: 10.1080/01926230252929954
Calderón-Garcidueñas, L., Reynoso-Robles, R., Vargas-Martínez, J., Gómez-Maqueo-Chew, A., Pérez-Guillé, B., Mukherjee, P. S., et al. (2016). Prefrontal white matter pathology in air pollution exposed Mexico City young urbanites and their potential impact on neurovascular unit dysfunction and the development of Alzheimer’s disease. Environ. Res. 146, 404–417. doi: 10.1016/j.envres.2015.12.031
Calderón-Garcidueñas, L., Solt, A. C., Henríquez-Roldán, C., Torres-Jardón, R., Nuse, B., Herritt, L., et al. (2008). Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid β-42 and α-synuclein in children and young adults. Toxicol. Pathol. 36, 289–310. doi: 10.1177/0192623307313011
Calderón-Garcidueñas, L., Vincent, R., Mora-Tiscareño, A., Franco-Lira, M., Henríquez-Roldán, C., Barragán-Mejía, G., et al. (2007). Elevated plasma endothelin-1 and pulmonary arterial pressure in children exposed to air pollution. Environ. Health Perspect. 115, 1248–1253. doi: 10.1289/ehp.9641
Camarinho, R., Garcia, P. V., Choi, H., and Rodrigues, A. S. (2019). Overproduction of TNF-α and lung structural remodelling due to chronic exposure to volcanogenic air pollution. Chemosphere 222, 227–234. doi: 10.1016/j.chemosphere.2019.01.138
Campbell, A., Oldham, M., Becaria, A., Bondy, S. C., Meacher, D., Sioutas, C., et al. (2005). Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology 26, 133–140. doi: 10.1016/j.neuro.2004.08.003
Campioni, M., Severino, A., Manente, L., Tuduce, I. L., Toldo, S., Caraglia, M., et al. (2010). The serine protease HtrA1 specifically interacts and degrades the tuberous sclerosis complex 2 protein. Mol. Cancer Res. 8, 1248–1260. doi: 10.1158/1541-7786.MCR-09-0473
Candelario-Jalil, E., Thompson, J., Taheri, S., Grossetete, M., Adair, J. C., Edmonds, E., et al. (2011). Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke 42, 1345–1350. doi: 10.1161/STROKEAHA.110.600825
Cannistraro, R. J., Badi, M., Eidelman, B. H., Dickson, D. W., Middlebrooks, E. H., and Meschia, J. F. (2019). CNS small vessel disease: a clinical review. Neurology 92, 1146–1156. doi: 10.1212/WNL.0000000000007654
Caplan, L. R. (2015). Lacunar infarction and small vessel disease: pathology and pathophysiology. J. Stroke 17, 2–6. doi: 10.5853/jos.2015.17.1.2
Carlin, L. M., Stamatiades, E. G., Auffray, C., Hanna, R. N., Glover, L., Vizcay-Barrena, G., et al. (2013). Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153, 362–375. doi: 10.1016/j.cell.2013.03.010
Carrano, A., Hoozemans, J. J. M., van der Vies, S. M., Rozemuller, A. J. M., van Horssen, J., and de Vries, H. E. (2011). Amyloid β induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Chacón-Quintero, M. V., Pineda-López, L. G., Villegas-Lanau, C. A., Posada-Duque, R., and Cardona-Gómez, G. P. (2021). β-secretase 1 underlies reactive astrocytes and endothelial disruption in neurodegeneration. Front. Cell. Neurosci. 15:656832. doi: 10.3389/fncel.2021.656832
Charidimou, A., Boulouis, G., Gurol, M. E., Ayata, C., Bacskai, B. J., Frosch, M. P., et al. (2017). Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140, 1829–1850. doi: 10.1093/brain/awx047
Chaturvedi, M., and Kaczmarek, L. (2014). Mmp-9 inhibition: a therapeutic strategy in ischemic stroke. Mol. Neurobiol. 49, 563–573. doi: 10.1007/s12035-013-8538-z
Chen, Z., Chen, F., Fang, Z., Zhao, H., Zhan, C., Li, C., et al. (2021). Glial activation and inflammation in the NTS in a rat model after exposure to diesel exhaust particles. Environ. Toxicol. Pharmacol. 83:103584. doi: 10.1016/j.etap.2021.103584
Chen, X., Liu, S., Zhang, W., Wu, C., Liu, H., Zhang, F., et al. (2018). Nrf2 deficiency exacerbates PM2.5-induced olfactory bulb injury. Biochem. Biophys. Res. Commun. 505, 1154–1160. doi: 10.1016/j.bbrc.2018.10.057
Chen, G.-F., Xu, T.-H., Yan, Y., Zhou, Y.-R., Jiang, Y., Melcher, K., et al. (2017). Amyloid β: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chen, L., Yokel, R. A., Hennig, B., and Toborek, M. (2008). Manufactured aluminum oxide nanoparticles decrease expression of tight junction proteins in brain vasculature. J. Neuroimmune Pharmacol. 3, 286–295. doi: 10.1007/s11481-008-9131-5
Chen, L., Zhang, Y., Li, D., Zhang, N., Liu, R., Han, B., et al. (2016). Everolimus (RAD001) ameliorates vascular cognitive impairment by regulating microglial function via the mTORC1 signaling pathway. J. Neuroimmunol. 299, 164–171. doi: 10.1016/j.jneuroim.2016.09.008
Cheng, H., Davis, D. A., Hasheminassab, S., Sioutas, C., Morgan, T. E., and Finch, C. E. (2016). Urban traffic-derived nanoparticulate matter reduces neurite outgrowth via TNFα in vitro. J. Neuroinflammation 13:19. doi: 10.1186/s12974-016-0480-3
Chistiakov, D. A., Bobryshev, Y. V., and Orekhov, A. N. (2016). Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 20, 17–28. doi: 10.1111/jcmm.12689
Chistiakov, D. A., Myasoedova, V. A., Melnichenko, A. A., Grechko, A. V., and Orekhov, A. N. (2017). Calcifying matrix vesicles and atherosclerosis. Biomed Res. Int. 2017:7463590. doi: 10.1155/2017/7463590
Cho, C.-C., Hsieh, W.-Y., Tsai, C.-H., Chen, C.-Y., Chang, H.-F., and Lin, C.-S. (2018). In vitro and in vivo experimental studies of PM2.5 on disease progression. Int. J. Environ. Res. Public Health 15:1380. doi: 10.3390/ijerph15071380
Cho, H. S., Hyman, B. T., Greenberg, S. M., and Rebeck, G. W. (2001). Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Aβ aggregation. J. Neuropathol. Exp. Neurol. 60, 342–349. doi: 10.1093/jnen/60.4.342
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U S A 115, E1896–E1905. doi: 10.1073/pnas.1800165115
Coburn, J. L., Cole, T. B., Dao, K. T., and Costa, L. G. (2018). Acute exposure to diesel exhaust impairs adult neurogenesis in mice: prominence in males and protective effect of pioglitazone. Arch. Toxicol. 92, 1815–1829. doi: 10.1007/s00204-018-2180-5
Cochran, E., Bacci, B., Chen, Y., Patton, A., Gambetti, P., and Autilio-Gambetti, L. (1991). Amyloid precursor protein and ubiquitin immunoreactivity in dystrophic axons is not unique to Alzheimer’s disease. Am. J. Pathol. 139, 485–489.
Cole, T. B., Coburn, J., Dao, K., Roqué, P., Chang, Y.-C., Kalia, V., et al. (2016). Sex and genetic differences in the effects of acute diesel exhaust exposure on inflammation and oxidative stress in mouse brain. Toxicology 374, 1–9. doi: 10.1016/j.tox.2016.11.010
Cordonnier, C., Al-Shahi Salman, R., and Wardlaw, J. (2007). Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain 130, 1988–2003. doi: 10.1093/brain/awl387
Corrada, M. M., Sonnen, J. A., Kim, R. C., and Kawas, C. H. (2016). Microinfarcts are common and strongly related to dementia in the oldest-old: the 90+ study. Alzheimers Dement. 12, 900–908. doi: 10.1016/j.jalz.2016.04.006
Correa, F., Hernangómez, M., Mestre, L., Loría, F., Spagnolo, A., Docagne, F., et al. (2010). Anandamide enhances IL-10 production in activated microglia by targeting CB(2) receptors: roles of ERK1/2, JNK, and NF-κB. Glia 58, 135–147. doi: 10.1002/glia.20907
Csipo, T., Lipecz, A., Ashpole, N. M., Balasubramanian, P., and Tarantini, S. (2020). Astrocyte senescence contributes to cognitive decline. Geroscience 42, 51–55. doi: 10.1007/s11357-019-00140-9
Csiszar, A., Yabluchanskiy, A., Ungvari, A., Ungvari, Z., and Tarantini, S. (2019). Overexpression of catalase targeted to mitochondria improves neurovascular coupling responses in aged mice. Geroscience 41, 609–617. doi: 10.1007/s11357-019-00111-0
Dalkara, T. (2019). Pericytes: a novel target to improve success of recanalization therapies. Stroke 50, 2985–2991. doi: 10.1161/STROKEAHA.118.023590
Das, A. S., Regenhardt, R. W., Feske, S. K., and Gurol, M. E. (2019). Treatment approaches to lacunar stroke. J. Stroke Cerebrovasc. Dis. 28, 2055–2078. doi: 10.1016/j.jstrokecerebrovasdis.2019.05.004
Davalos, D., Ryu, J. K., Merlini, M., Baeten, K. M., Le Moan, N., Petersen, M. A., et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3:1227. doi: 10.1038/ncomms2230
Davis, J., Xu, F., Deane, R., Romanov, G., Previti, M. L., Zeigler, K., et al. (2004). Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J. Biol. Chem. 279, 20296–20306. doi: 10.1074/jbc.M312946200
De Guio, F., Vignaud, A., Ropele, S., Duering, M., Duchesnay, E., Chabriat, H., et al. (2014). Loss of venous integrity in cerebral small vessel disease: a 7-T MRI study in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke 45, 2124–2126. doi: 10.1161/STROKEAHA.114.005726
de Jager, M., van der Wildt, B., Schul, E., Bol, J. G. J. M., van Duinen, S. G., Drukarch, B., et al. (2013). Tissue transglutaminase colocalizes with extracellular matrix proteins in cerebral amyloid angiopathy. Neurobiol. Aging 34, 1159–1169. doi: 10.1016/j.neurobiolaging.2012.10.005
de la Peña, P., Bornstein, B., del Hoyo, P., Fernández-Moreno, M. A., Martín, M. A., Campos, Y., et al. (2001). Mitochondrial dysfunction associated with a mutation in the Notch3 gene in a CADASIL family. Neurology 57, 1235–1238. doi: 10.1212/wnl.57.7.1235
de Laat, K. F., Tuladhar, A. M., van Norden, A. G. W., Norris, D. G., Zwiers, M. P., and de Leeuw, F.-E. (2011). Loss of white matter integrity is associated with gait disorders in cerebral small vessel disease. Brain 134, 73–83. doi: 10.1093/brain/awq343
De Luca, A., De Falco, M., Severino, A., Campioni, M., Santini, D., Baldi, F., et al. (2003). Distribution of the serine protease HtrA1 in normal human tissues. J. Histochem. Cytochem. 51, 1279–1284. doi: 10.1177/002215540305101004
De Silva, T. M., and Faraci, F. M. (2016). Microvascular dysfunction and cognitive impairment. Cell. Mol. Neurobiol. 36, 241–258. doi: 10.1007/s10571-015-0308-1
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
Debette, S., and Markus, H. S. (2010). The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 341:c3666. doi: 10.1136/bmj.c3666
Del Bene, A., Makin, S. D. J., Doubal, F. N., Inzitari, D., and Wardlaw, J. M. (2013). Variation in risk factors for recent small subcortical infarcts with infarct size, shape, and location. Stroke 44, 3000–3006. doi: 10.1161/STROKEAHA.113.002227
Desmond, D. W., Moroney, J. T., Sano, M., and Stern, Y. (2002). Incidence of dementia after ischemic stroke: results of a longitudinal study. Stroke 33, 2254–2260. doi: 10.1161/01.str.0000028235.91778.95
Di Donato, I., Bianchi, S., De Stefano, N., Dichgans, M., Dotti, M. T., Duering, M., et al. (2017). Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med. 15:41. doi: 10.1186/s12916-017-0778-8
Dichgans, M. (2007). Genetics of ischaemic stroke. Lancet Neurol. 6, 149–161. doi: 10.1016/S1474-4422(07)70028-5
Dong, X., Song, Y.-N., Liu, W.-G., and Guo, X.-L. (2009). Mmp-9, a potential target for cerebral ischemic treatment. Curr. Neuropharmacol. 7, 269–275. doi: 10.2174/157015909790031157
Donzis, E. J., and Tronson, N. C. (2014). Modulation of learning and memory by cytokines: signaling mechanisms and long term consequences. Neurobiol. Learn. Mem. 115, 68–77. doi: 10.1016/j.nlm.2014.08.008
Drazyk, A. M., Tan, R. Y. Y., Tay, J., Traylor, M., Das, T., and Markus, H. S. (2019). Encephalopathy in a large cohort of british cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients. Stroke 50, 283–290. doi: 10.1161/STROKEAHA.118.023661
Dropcho, E. J. (1991). Central nervous system injury by therapeutic irradiation. Neurol. Clin. 9, 969–988. doi: 10.1016/s0733-8619(18)30260-3
Dziewulska, D., and Lewandowska, E. (2012). Pericytes as a new target for pathological processes in CADASIL. Neuropathology 32, 515–521. doi: 10.1111/j.1440-1789.2011.01290.x
Dzyubenko, E., Manrique-Castano, D., Kleinschnitz, C., Faissner, A., and Hermann, D. M. (2018). Role of immune responses for extracellular matrix remodeling in the ischemic brain. Ther. Adv. Neurol. Disord. 11:1756286418818092. doi: 10.1177/1756286418818092
Ekker, M. S., Verhoeven, J. I., Vaartjes, I., van Nieuwenhuizen, K. M., Klijn, C. J. M., and de Leeuw, F.-E. (2019). Stroke incidence in young adults according to age, subtype, sex, and time trends. Neurology 92, e2444–e2454. doi: 10.1212/WNL.0000000000007533
El-Abassi, R., Singhal, D., and England, J. D. (2014). Fabry’s disease. J. Neurol. Sci. 344, 5–19. doi: 10.1016/j.jns.2014.06.029
ElAli, A., Doeppner, T. R., Zechariah, A., and Hermann, D. M. (2011). Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9 and RhoA overactivation. Stroke 42, 3238–3244. doi: 10.1161/STROKEAHA.111.615559
ElAli, A., Thériault, P., Préfontaine, P., and Rivest, S. (2013). Mild chronic cerebral hypoperfusion induces neurovascular dysfunction, triggering peripheral β-amyloid brain entry and aggregation. Acta Neuropathol. Commun. 1:75. doi: 10.1186/2051-5960-1-75
Erdener, S. E., and Dalkara, T. (2019). Small vessels are a big problem in neurodegeneration and neuroprotection. Front. Neurol. 10:889. doi: 10.3389/fneur.2019.00889
Farias Quipildor, G. E., Mao, K., Hu, Z., Novaj, A., Cui, M.-H., Gulinello, M., et al. (2019). Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience 41, 185–208. doi: 10.1007/s11357-019-00065-3
Farkas, E., De Jong, G. I., de Vos, R. A., Jansen Steur, E. N., and Luiten, P. G. (2000). Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 100, 395–402. doi: 10.1007/s004010000195
Feigin, V. L., Roth, G. A., Naghavi, M., Parmar, P., Krishnamurthi, R., Chugh, S., et al. (2016). Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 15, 913–924. doi: 10.1016/S1474-4422(16)30073-4
Fisher, C. M. (1965). Lacunes: small, deep cerebral infarcts. Neurology 15, 774–784. doi: 10.1212/wnl.15.8.774
French, C. R., Seshadri, S., Destefano, A. L., Fornage, M., Arnold, C. R., Gage, P. J., et al. (2014). Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Invest. 124, 4877–4881. doi: 10.1172/JCI75109
Friedrich, U., Datta, S., Schubert, T., Plössl, K., Schneider, M., Grassmann, F., et al. (2015). Synonymous variants in HTRA1 implicated in AMD susceptibility impair its capacity to regulate TGF-β signaling. Hum. Mol. Genet. 24, 6361–6373. doi: 10.1093/hmg/ddv346
Frostegård, J., Ulfgren, A. K., Nyberg, P., Hedin, U., Swedenborg, J., Andersson, U., et al. (1999). Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 145, 33–43. doi: 10.1016/s0021-9150(99)00011-8
Fu, Y., and Yan, Y. (2018). Emerging role of immunity in cerebral small vessel disease. Front. Immunol. 9:67. doi: 10.3389/fimmu.2018.00067
Gaertner, R. F., Wyss-Coray, T., Von Euw, D., Lesné, S., Vivien, D., and Lacombe, P. (2005). Reduced brain tissue perfusion in TGF-β 1 transgenic mice showing Alzheimer’s disease-like cerebrovascular abnormalities. Neurobiol. Dis. 19, 38–46. doi: 10.1016/j.nbd.2004.11.008
Gama Sosa, M. A., Gasperi, R. D., Rocher, A. B., Wang, A. C.-J., Janssen, W. G. M., Flores, T., et al. (2010). Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am. J. Pathol. 176, 353–368. doi: 10.2353/ajpath.2010.090482
Gefen, T., Kim, G., Bolbolan, K., Geoly, A., Ohm, D., Oboudiyat, C., et al. (2019). Activated microglia in cortical white matter across cognitive aging trajectories. Front. Aging Neurosci. 11:94. doi: 10.3389/fnagi.2019.00094
Georgakis, M. K., Malik, R., Anderson, C. D., Parhofer, K. G., Hopewell, J. C., and Dichgans, M. (2020). Genetic determinants of blood lipids and cerebral small vessel disease: role of high-density lipoprotein cholesterol. Brain 143, 597–610. doi: 10.1093/brain/awz413
George, M. G. (2020). Risk factors for ischemic stroke in younger adults: a focused update. Stroke 51, 729–735. doi: 10.1161/STROKEAHA.119.024156
Germain, D. P., Elliott, P. M., Falissard, B., Fomin, V. V., Hilz, M. J., Jovanovic, A., et al. (2019). The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 19:100454. doi: 10.1016/j.ymgmr.2019.100454
Ghosh, M., Balbi, M., Hellal, F., Dichgans, M., Lindauer, U., and Plesnila, N. (2015). Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann. Neurol. 78, 887–900. doi: 10.1002/ana.24512
Giannoni, P., Arango-Lievano, M., Neves, I. D., Rousset, M.-C., Baranger, K., Rivera, S., et al. (2016). Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol. Dis. 88, 107–117. doi: 10.1016/j.nbd.2016.01.001
Gidday, J. M., Gasche, Y. G., Copin, J.-C., Shah, A. R., Perez, R. S., Shapiro, S. D., et al. (2005). Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 289, H558–H568. doi: 10.1152/ajpheart.01275.2004
Gómez-Budia, M., Konttinen, H., Saveleva, L., Korhonen, P., Jalava, P. I., Kanninen, K. M., et al. (2020). Glial smog: interplay between air pollution and astrocyte-microglia interactions. Neurochem. Int. 136:104715. doi: 10.1016/j.neuint.2020.104715
Gorelick, P. B. (2004). Risk factors for vascular dementia and Alzheimer disease. Stroke 35, 2620–2622. doi: 10.1161/01.STR.0000143318.70292.47
Gorelick, P. B., Scuteri, A., Black, S. E., DeCarli, C., Greenberg, S. M., Iadecola, C., et al. (2011). Vascular contributions to cognitive impairment and dementia. Stroke 42, 2672–2713. doi: 10.1161/STR.0b013e3182299496
Greve, H. J., Mumaw, C. L., Messenger, E. J., Kodavanti, P. R. S., Royland, J. L., Kodavanti, U. P., et al. (2020). Diesel exhaust impairs TREM2 to dysregulate neuroinflammation. J. Neuroinflammation 17:351. doi: 10.1186/s12974-020-02017-7
Grinberg, L. T., and Thal, D. R. (2010). Vascular pathology in the aged human brain. Acta Neuropathol. 119, 277–290. doi: 10.1007/s00401-010-0652-7
Gu, B. J., Huang, X., Ou, A., Rembach, A., Fowler, C., Avula, P. K., et al. (2016). Innate phagocytosis by peripheral blood monocytes is altered in Alzheimer’s disease. Acta Neuropathol. 132, 377–389. doi: 10.1007/s00401-016-1596-3
Gu, X., Liu, X.-Y., Fagan, A., Gonzalez-Toledo, M. E., and Zhao, L.-R. (2012). Ultrastructural changes in cerebral capillary pericytes in aged Notch3 mutant transgenic mice. Ultrastruct. Pathol. 36, 48–55. doi: 10.3109/01913123.2011.620220
Guey, S., Mawet, J., Hervé, D., Duering, M., Godin, O., Jouvent, E., et al. (2016). Prevalence and characteristics of migraine in CADASIL. Cephalalgia 36, 1038–1047. doi: 10.1177/0333102415620909
Guo, S., and Lo, E. H. (2009). Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke 40, S4–S7. doi: 10.1161/STROKEAHA.108.534388
Guruswamy, R., and ElAli, A. (2017). Complex roles of microglial cells in ischemic stroke pathobiology: new insights and future directions. Int. J. Mol. Sci. 18:496. doi: 10.3390/ijms18030496
Halder, S. K., and Milner, R. (2019). A critical role for microglia in maintaining vascular integrity in the hypoxic spinal cord. Proc. Natl. Acad. Sci. U S A 116, 26029–26037. doi: 10.1073/pnas.1912178116
Hallé, M., Tribout-Jover, P., Lanteigne, A.-M., Boulais, J., St-Jean, J. R., Jodoin, R., et al. (2015). Methods to monitor monocytes-mediated amyloid-β uptake and phagocytosis in the context of adjuvanted immunotherapies. J. Immunol. Methods 424, 64–79. doi: 10.1016/j.jim.2015.05.002
Hameed, S., Zhao, J., and Zare, R. N. (2020). Ambient PM particles reach mouse brain, generate ultrastructural hallmarks of neuroinflammation, and stimulate amyloid deposition, tangles, and plaque formation. Talanta Open 2:100013. doi: 10.1016/j.talo.2020.100013
Hao, W., and Friedman, A. (2014). The LDL-HDL profile determines the risk of atherosclerosis: a mathematical model. PLoS One 9:e90497. doi: 10.1371/journal.pone.0090497
Hara, K., Shiga, A., Fukutake, T., Nozaki, H., Miyashita, A., Yokoseki, A., et al. (2009). Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 360, 1729–1739. doi: 10.1056/NEJMoa0801560
Hartmann, D. A., Hyacinth, H. I., Liao, F.-F., and Shih, A. Y. (2018). Does pathology of small venules contribute to cerebral microinfarcts and dementia? J. Neurochem. 144, 517–526. doi: 10.1111/jnc.14228
Hartz, A. M. S., Bauer, B., Block, M. L., Hong, J.-S., and Miller, D. S. (2008). Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 22, 2723–2733. doi: 10.1096/fj.08-106997
Hartz, A. M. S., Bauer, B., Soldner, E. L. B., Wolf, A., Boy, S., Backhaus, R., et al. (2012). Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 43, 514–523. doi: 10.1161/STROKEAHA.111.627562
Haruwaka, K., Ikegami, A., Tachibana, Y., Ohno, N., Konishi, H., Hashimoto, A., et al. (2019). Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 10:5816. doi: 10.1038/s41467-019-13812-z
Hase, Y., Horsburgh, K., Ihara, M., and Kalaria, R. N. (2018). White matter degeneration in vascular and other ageing-related dementias. J. Neurochem. 144, 617–633. doi: 10.1111/jnc.14271
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Hawkes, C. A., Härtig, W., Kacza, J., Schliebs, R., Weller, R. O., Nicoll, J. A., et al. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 121, 431–443. doi: 10.1007/s00401-011-0801-7
Hermann, D. M., and ElAli, A. (2012). The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci. Signal. 5:re4. doi: 10.1126/scisignal.2002886
Hernandez-Guillamon, M., Mawhirt, S., Blais, S., Montaner, J., Neubert, T. A., Rostagno, A., et al. (2015). Sequential amyloid-β degradation by the matrix metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 290, 15078–15091. doi: 10.1074/jbc.M114.610931
Herzig, M. C., Van Nostrand, W. E., and Jucker, M. (2006). Mechanism of cerebral β-amyloid angiopathy: murine and cellular models. Brain Pathol. 16, 40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x
Herzig, M. C., Winkler, D. T., Burgermeister, P., Pfeifer, M., Kohler, E., Schmidt, S. D., et al. (2004). Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 7, 954–960. doi: 10.1038/nn1302
Hirsch-Reinshagen, V., Burgess, B. L., and Wellington, C. L. (2009). Why lipids are important for Alzheimer disease? Mol. Cell. Biochem. 326, 121–129. doi: 10.1007/s11010-008-0012-2
Ho, C. S. H., and Mondry, A. (2015). CADASIL presenting as schizophreniform organic psychosis. Gen. Hosp. Psychiatry 37, 273.e11–273.e13. doi: 10.1016/j.genhosppsych.2015.02.006
Holland, C. M., Smith, E. E., Csapo, I., Gurol, M. E., Brylka, D. A., Killiany, R. J., et al. (2008). Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 39, 1127–1133. doi: 10.1161/STROKEAHA.107.497438
Hosseini-Alghaderi, S., and Baron, M. (2020). Notch3 in development, health and disease. Biomolecules 10:485. doi: 10.3390/biom10030485
Howe, M. D., McCullough, L. D., and Urayama, A. (2020). The role of basement membranes in cerebral amyloid angiopathy. Front. Physiol. 11:601320. doi: 10.3389/fphys.2020.601320
Hung, V. K. L., Yeung, P. K. K., Lai, A. K. W., Ho, M. C. Y., Lo, A. C. Y., Chan, K. C., et al. (2015). Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. J. Cereb. Blood Flow Metab. 35, 1687–1696. doi: 10.1038/jcbfm.2015.109
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Iadecola, C., Duering, M., Hachinski, V., Joutel, A., Pendlebury, S. T., Schneider, J. A., et al. (2019). Vascular cognitive impairment and dementia: JACC scientific expert panel. J. Am. Coll. Cardiol. 73, 3326–3344. doi: 10.1016/j.jacc.2019.04.034
Ihara, M., and Yamamoto, Y. (2016). Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 47, 554–560. doi: 10.1161/STROKEAHA.115.009627
Ikawati, M., Kawaichi, M., and Oka, C. (2018). Loss of HtrA1 serine protease induces synthetic modulation of aortic vascular smooth muscle cells. PLoS One 13:e0196628. doi: 10.1371/journal.pone.0196628
Ince, P. G., Minett, T., Forster, G., Brayne, C., Wharton, S. B., and Medical Research Council Cognitive Function and Ageing Neuropathology Study. (2017). Microinfarcts in an older population-representative brain donor cohort (MRC CFAS): prevalence, relation to dementia, and mobility and implications for the evaluation of cerebral Small Vessel Disease. Neuropathol. Appl. Neurobiol. 43, 409–418. doi: 10.1111/nan.12363
Jäkel, L., Kuiperij, H. B., Gerding, L. P., Custers, E. E. M., van den Berg, E., Jolink, W. M. T., et al. (2020). Disturbed balance in the expression of MMP9 and TIMP3 in cerebral amyloid angiopathy-related intracerebral haemorrhage. Acta Neuropathol. Commun. 8:99. doi: 10.1186/s40478-020-00972-z
Jayant, S., and Sharma, B. (2016). Selective modulator of cannabinoid receptor type 2 reduces memory impairment and infarct size during cerebral hypoperfusion and vascular dementia. Curr. Neurovasc. Res. 13, 289–302. doi: 10.2174/1567202613666160902102007
Jayaraj, R. L., Azimullah, S., Beiram, R., Jalal, F. Y., and Rosenberg, G. A. (2019). Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation 16:142. doi: 10.1186/s12974-019-1516-2
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm. 109, 813–836. doi: 10.1007/s007020200068
Jellinger, K. A. (2007). The enigma of mixed dementia. Alzheimers Dement. 3, 40–53. doi: 10.1016/j.jalz.2006.09.002
Jellinger, K. A. (2013). Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front. Aging Neurosci. 5:17. doi: 10.3389/fnagi.2013.00017
Jendroska, K., Hoffmann, O. M., and Patt, S. (1997). Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. N Y Acad. Sci. 826, 401–405. doi: 10.1111/j.1749-6632.1997.tb48492.x
Jennette, J. C., and Falk, R. J. (1997). Small-vessel vasculitis. N. Engl. J. Med. 337, 1512–1523. doi: 10.1056/NEJM199711203372106
Jian, B., Hu, M., Cai, W., Zhang, B., and Lu, Z. (2020). Update of immunosenescence in cerebral small vessel disease. Front. Immunol. 11:585655. doi: 10.3389/fimmu.2020.585655
Jiang, X., Andjelkovic, A. V., Zhu, L., Yang, T., Bennett, M. V. L., Chen, J., et al. (2018). Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 163–164, 144–171. doi: 10.1016/j.pneurobio.2017.10.001
Jo, W. K., Law, A. C. K., and Chung, S. K. (2014). The neglected co-star in the dementia drama: the putative roles of astrocytes in the pathogeneses of major neurocognitive disorders. Mol. Psychiatry 19, 159–167. doi: 10.1038/mp.2013.171
Jokinen, H., Lipsanen, J., Schmidt, R., Fazekas, F., Gouw, A. A., van der Flier, W. M., et al. (2012). Brain atrophy accelerates cognitive decline in cerebral small vessel disease: the LADIS study. Neurology 78, 1785–1792. doi: 10.1212/WNL.0b013e3182583070
Jones, E. V., and Bouvier, D. S. (2014). Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. 2014:321209. doi: 10.1155/2014/321209
Joutel, A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., et al. (2000). The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105, 597–605. doi: 10.1172/JCI8047
Joutel, A., Favrole, P., Labauge, P., Chabriat, H., Lescoat, C., Andreux, F., et al. (2001). Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet 358, 2049–2051. doi: 10.1016/S0140-6736(01)07142-2
Joutel, A., Haddad, I., Ratelade, J., and Nelson, M. T. (2016). Perturbations of the cerebrovascular matrisome: a convergent mechanism in small vessel disease of the brain? J. Cereb. Blood Flow Metab. 36, 143–157. doi: 10.1038/jcbfm.2015.62
Kalaria, R. N., Akinyemi, R., and Ihara, M. (2016). Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 1862, 915–925. doi: 10.1016/j.bbadis.2016.01.015
Kamp, J. A., Moursel, L. G., Haan, J., Terwindt, G. M., Lesnik Oberstein, S. A. M. J., van Duinen, S. G., et al. (2014). Amyloid β in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Rev. Neurosci. 25, 641–651. doi: 10.1515/revneuro-2014-0008
Kandasamy, M., Anusuyadevi, M., Aigner, K. M., Unger, M. S., Kniewallner, K. M., de Sousa, D. M. B., et al. (2020). TGF-β signaling: a therapeutic target to reinstate regenerative plasticity in vascular dementia? Aging Dis. 11, 828–850. doi: 10.14336/AD.2020.0222
Kawai, H., Kawaguchi, D., Kuebrich, B. D., Kitamoto, T., Yamaguchi, M., Gotoh, Y., et al. (2017). Area-specific regulation of quiescent neural stem cells by Notch3 in the adult mouse subependymal zone. J. Neurosci. 37, 11867–11880. doi: 10.1523/JNEUROSCI.0001-17.2017
Keable, A., Fenna, K., Yuen, H. M., Johnston, D. A., Smyth, N. R., Smith, C., et al. (2016). Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta 1862, 1037–1046. doi: 10.1016/j.bbadis.2015.08.024
Keage, H. A. D., Carare, R. O., Friedland, R. P., Ince, P. G., Love, S., Nicoll, J. A., et al. (2009). Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 9:3. doi: 10.1186/1471-2377-9-3
Kerkhofs, D., van Hagen, B. T., Milanova, I. V., Schell, K. J., van Essen, H., Wijnands, E., et al. (2020). Pharmacological depletion of microglia and perivascular macrophages prevents vascular cognitive impairment in Ang II-induced hypertension. Theranostics 10, 9512–9527. doi: 10.7150/thno.44394
Khanna, A., Kahle, K. T., Walcott, B. P., Gerzanich, V., and Simard, J. M. (2014). Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl. Stroke Res. 5, 3–16. doi: 10.1007/s12975-013-0307-9
Kim, J. Y., Kim, J.-H., Kim, Y.-D., and Seo, J. H. (2020). High vulnerability of oligodendrocytes to oxidative stress induced by ultrafine urban particles. Antioxidants 10:4. doi: 10.3390/antiox10010004
Kim, J. Y., Park, J., Chang, J. Y., Kim, S.-H., and Lee, J. E. (2016). Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp. Neurobiol. 25, 241–251. doi: 10.5607/en.2016.25.5.241
Kim, R.-E., Shin, C. Y., Han, S.-H., and Kwon, K. J. (2020). Astaxanthin suppresses PM2.5-induced neuroinflammation by regulating Akt phosphorylation in BV-2 microglial cells. Int. J. Mol. Sci. 21:7227. doi: 10.3390/ijms21197227
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Kivipelto, M., Mangialasche, F., Snyder, H. M., Allegri, R., Andrieu, S., Arai, H., et al. (2020). World-wide FINGERS network: a global approach to risk reduction and prevention of dementia. Alzheimers Dement. 16, 1078–1094. doi: 10.1002/alz.12123
Kjeldsen, S. E., Narkiewicz, K., Burnier, M., and Oparil, S. (2018). Intensive blood pressure lowering prevents mild cognitive impairment and possible dementia and slows development of white matter lesions in brain: the SPRINT Memory and Cognition IN Decreased Hypertension (SPRINT MIND) study. Blood Press. 27, 247–248. doi: 10.1080/08037051.2018.1507621
Klocke, C., Allen, J. L., Sobolewski, M., Blum, J. L., Zelikoff, J. T., and Cory-Slechta, D. A. (2018). Exposure to fine and ultrafine particulate matter during gestation alters postnatal oligodendrocyte maturation, proliferation capacity and myelination. Neurotoxicology 65, 196–206. doi: 10.1016/j.neuro.2017.10.004
Koizumi, T., Taguchi, K., Mizuta, I., Toba, H., Ohigashi, M., Onishi, O., et al. (2019). Transiently proliferating perivascular microglia harbor M1 type and precede cerebrovascular changes in a chronic hypertension model. J. Neuroinflammation 16:79. doi: 10.1186/s12974-019-1467-7
Kopan, R., and Ilagan, M. X. G. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. doi: 10.1016/j.cell.2009.03.045
Kotilinek, L. A., Westerman, M. A., Wang, Q., Panizzon, K., Lim, G. P., Simonyi, A., et al. (2008). Cyclooxygenase-2 inhibition improves amyloid-β-mediated suppression of memory and synaptic plasticity. Brain 131, 651–664. doi: 10.1093/brain/awn008
Kraft, P., Schuhmann, M. K., Garz, C., Jandke, S., Urlaub, D., Mencl, S., et al. (2017). Hypercholesterolemia induced cerebral small vessel disease. PLoS One 12:e0182822. doi: 10.1371/journal.pone.0182822
Kulick, E. R., Elkind, M. S. V., Boehme, A. K., Joyce, N. R., Schupf, N., Kaufman, J. D., et al. (2020). Long-term exposure to ambient air pollution, APOE-ε4 status and cognitive decline in a cohort of older adults in northern Manhattan. Environ. Int. 136:105440. doi: 10.1016/j.envint.2019.105440
Kuo, D. S., Labelle-Dumais, C., and Gould, D. B. (2012). COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum. Mol. Genet. 21, R97–R110. doi: 10.1093/hmg/dds346
Lacombe, P., Oligo, C., Domenga, V., Tournier-Lasserve, E., and Joutel, A. (2005). Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy. Stroke 36, 1053–1058. doi: 10.1161/01.STR.0000163080.82766.eb
Lågas, P. A., and Juvonen, V. (2001). Schizophrenia in a patient with cerebral autosomally dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL disease). Nord. J. Psychiatry 55, 41–42. doi: 10.1080/080394801750093724
Lammie, G. A., Brannan, F., and Wardlaw, J. M. (1998). Incomplete lacunar infarction (Type Ib lacunes). Acta Neuropathol. 96, 163–171. doi: 10.1007/s004010050877
Leduc, V., Jasmin-Bélanger, S., and Poirier, J. (2010). APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 16, 469–477. doi: 10.1016/j.molmed.2010.07.008
Lepelletier, F.-X., Mann, D. M. A., Robinson, A. C., Pinteaux, E., and Boutin, H. (2017). Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 43, 167–182. doi: 10.1111/nan.12295
Lesnik Oberstein, S. A., van den Boom, R., van Buchem, M. A., van Houwelingen, H. C., Bakker, E., Vollebregt, E., et al. (2001). Cerebral microbleeds in CADASIL. Neurology 57, 1066–1070. doi: 10.1212/wnl.57.6.1066
Levesque, S., Taetzsch, T., Lull, M. E., Kodavanti, U., Stadler, K., Wagner, A., et al. (2011). Diesel exhaust activates and primes microglia: air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 119, 1149–1155. doi: 10.1289/ehp.1002986
Li, Z. W., Chu, W., Hu, Y., Delhase, M., Deerinck, T., Ellisman, M., et al. (1999). The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med. 189, 1839–1845. doi: 10.1084/jem.189.11.1839
Li, Q., Yang, Y., Reis, C., Tao, T., Li, W., Li, X., et al. (2018). Cerebral small vessel disease. Cell Transplant. 27, 1711–1722. doi: 10.1177/0963689718795148
Li, X., Zhang, X., Leathers, R., Makino, A., Huang, C., Parsa, P., et al. (2009). Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 15, 1289–1297. doi: 10.1038/nm.2021
Li, Y., Liu, Y., Hu, C., Chang, Q., Deng, Q., Yang, X., et al. (2020). Study of the neurotoxicity of indoor airborne nanoparticles based on a 3D human blood-brain barrier chip. Environ. Int. 143:105598. doi: 10.1016/j.envint.2020.105598
Li, T., Zhao, J., Ge, J., Yang, J., Song, X., Wang, C., et al. (2016). Particulate matter facilitates C6 glioma cells activation and the release of inflammatory factors through MAPK and JAK2/STAT3 pathways. Neurochem. Res. 41, 1969–1981. doi: 10.1007/s11064-016-1908-y
Liang, S., Zhang, J., Ning, R., Du, Z., Liu, J., Batibawa, J. W., et al. (2020). The critical role of endothelial function in fine particulate matter-induced atherosclerosis. Part. Fibre Toxicol. 17:61. doi: 10.1186/s12989-020-00391-x
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liu, Y., Chan, D. K. Y., Thalamuthu, A., Wen, W., Jiang, J., Paradise, M., et al. (2020). Plasma lipidomic biomarker analysis reveals distinct lipid changes in vascular dementia. Comput. Struct. Biotechnol. J. 18, 1613–1624. doi: 10.1016/j.csbj.2020.06.001
Liu, F., Huang, Y., Zhang, F., Chen, Q., Wu, B., Rui, W., et al. (2015). Macrophages treated with particulate matter PM2.5 induce selective neurotoxicity through glutaminase-mediated glutamate generation. J. Neurochem. 134, 315–326. doi: 10.1111/jnc.13135
Liu, X.-Y., Gonzalez-Toledo, M. E., Fagan, A., Duan, W.-M., Liu, Y., Zhang, S., et al. (2015). Stem cell factor and granulocyte colony-stimulating factor exhibit therapeutic effects in a mouse model of CADASIL. Neurobiol. Dis. 73, 189–203. doi: 10.1016/j.nbd.2014.09.006
Liu, Q., Radwanski, R., Babadjouni, R., Patel, A., Hodis, D. M., Baumbacher, P., et al. (2019). Experimental chronic cerebral hypoperfusion results in decreased pericyte coverage and increased blood-brain barrier permeability in the corpus callosum. J. Cereb. Blood Flow Metab. 39, 240–250. doi: 10.1177/0271678X17743670
Louvi, A., Arboleda-Velasquez, J. F., and Artavanis-Tsakonas, S. (2006). CADASIL: a critical look at a Notch disease. Dev. Neurosci. 28, 5–12. doi: 10.1159/000090748
Love, S., Chalmers, K., Ince, P., Esiri, M., Attems, J., Jellinger, K., et al. (2014). Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am. J. Neurodegener. Dis. 3, 19–32.
Luo, X.-Q., Li, A., Yang, X., Xiao, X., Hu, R., Wang, T.-W., et al. (2018). Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin. Med. 13:14. doi: 10.1186/s13020-018-0173-1
Magaki, S., Tang, Z., Tung, S., Williams, C. K., Lo, D., Yong, W. H., et al. (2018). The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 70, 70–77. doi: 10.1016/j.neurobiolaging.2018.06.004
Malik, R., Rannikmäe, K., Traylor, M., Georgakis, M. K., Sargurupremraj, M., Markus, H. S., et al. (2018). Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Ann. Neurol. 84, 934–939. doi: 10.1002/ana.25369
Malpetti, M., Kievit, R. A., Passamonti, L., Jones, P. S., Tsvetanov, K. A., Rittman, T., et al. (2020). Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 143, 1588–1602. doi: 10.1093/brain/awaa088
Marini, S., Anderson, C. D., and Rosand, J. (2020). Genetics of cerebral small vessel disease. Stroke 51, 12–20. doi: 10.1161/STROKEAHA.119.024151
Martinez-Canabal, A., Wheeler, A. L., Sarkis, D., Lerch, J. P., Lu, W.-Y., Buckwalter, M. S., et al. (2013). Chronic over-expression of TGFβ1 alters hippocampal structure and causes learning deficits. Hippocampus 23, 1198–1211. doi: 10.1002/hipo.22159
Martinez-Ramirez, S., Greenberg, S. M., and Viswanathan, A. (2014). Cerebral microbleeds: overview and implications in cognitive impairment. Alzheimers Res. Ther. 6:33. doi: 10.1186/alzrt263
Martini, A. C., Helman, A. M., McCarty, K. L., Lott, I. T., Doran, E., Schmitt, F. A., et al. (2020). Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement. 12:e12113. doi: 10.1002/dad2.12113
Matsuda, Y., Hirata, K., Inoue, N., Suematsu, M., Kawashima, S., Akita, H., et al. (1993). High density lipoprotein reverses inhibitory effect of oxidized low density lipoprotein on endothelium-dependent arterial relaxation. Circ. Res. 72, 1103–1109. doi: 10.1161/01.res.72.5.1103
Matsumoto, J., Dohgu, S., Takata, F., Machida, T., Bölükbaşi Hatip, F. F., Hatip-Al-Khatib, I., et al. (2018). TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res. 1692, 34–44. doi: 10.1016/j.brainres.2018.04.023
Merino, J. G., and Hachinski, V. (2000). Leukoaraiosis: reifying rarefaction. Arch. Neurol. 57, 925–926. doi: 10.1001/archneur.57.7.925
Meuwissen, M. E. C., Halley, D. J. J., Smit, L. S., Lequin, M. H., Cobben, J. M., de Coo, R., et al. (2015). The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet. Med. 17, 843–853. doi: 10.1038/gim.2014.210
Miao, Q., Paloneva, T., Tuisku, S., Roine, S., Poyhonen, M., Viitanen, M., et al. (2006). Arterioles of the lenticular nucleus in CADASIL. Stroke 37, 2242–2247. doi: 10.1161/01.STR.0000236838.84150.c2
Michaud, J.-P., Bellavance, M.-A., Préfontaine, P., and Rivest, S. (2013). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid β. Cell Rep. 5, 646–653. doi: 10.1016/j.celrep.2013.10.010
Mijajlović, M. D., Pavlović, A., Brainin, M., Heiss, W.-D., Quinn, T. J., Ihle-Hansen, H. B., et al. (2017). Post-stroke dementia—a comprehensive review. BMC Med. 15:11. doi: 10.1186/s12916-017-0779-7
Mills, N. L., Donaldson, K., Hadoke, P. W., Boon, N. A., MacNee, W., Cassee, F. R., et al. (2009). Adverse cardiovascular effects of air pollution. Nat. Clin. Pract. Cardiovasc. Med. 6, 36–44. doi: 10.1038/ncpcardio1399
Min, L.-J., Iwanami, J., Shudou, M., Bai, H.-Y., Shan, B.-S., Higaki, A., et al. (2020). Deterioration of cognitive function after transient cerebral ischemia with amyloid-β infusion-possible amelioration of cognitive function by AT2 receptor activation. J. Neuroinflammation 17:106. doi: 10.1186/s12974-020-01775-8
Mok, V., Wong, K. K., Xiong, Y., Wong, A., Schmidt, R., Chu, W., et al. (2011). Cortical and frontal atrophy are associated with cognitive impairment in age-related confluent white-matter lesion. J. Neurol. Neurosurg. Psychiatry 82, 52–57. doi: 10.1136/jnnp.2009.201665
Monet-Leprêtre, M., Haddad, I., Baron-Menguy, C., Fouillot-Panchal, M., Riani, M., Domenga-Denier, V., et al. (2013). Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain 136, 1830–1845. doi: 10.1093/brain/awt092
Montagne, A., Nikolakopoulou, A. M., Zhao, Z., Sagare, A. P., Si, G., Lazic, D., et al. (2018). Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 24, 326–337. doi: 10.1038/nm.4482
Montagne, A., Zhao, Z., and Zlokovic, B. V. (2017). Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J. Exp. Med. 214, 3151–3169. doi: 10.1084/jem.20171406
Montaner, J., Alvarez-Sabín, J., Molina, C., Anglés, A., Abilleira, S., Arenillas, J., et al. (2001). Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke 32, 1759–1766. doi: 10.1161/01.str.32.8.1759
Montiel-Dávalos, A., Alfaro-Moreno, E., and López-Marure, R. (2007). PM2.5 and PM10 induce the expression of adhesion molecules and the adhesion of monocytic cells to human umbilical vein endothelial cells. Inhal. Toxicol. 19, 91–98. doi: 10.1080/08958370701495212
Moreau, F., Patel, S., Lauzon, M. L., McCreary, C. R., Goyal, M., Frayne, R., et al. (2012). Cavitation after acute symptomatic lacunar stroke depends on time, location, and MRI sequence. Stroke 43, 1837–1842. doi: 10.1161/STROKEAHA.111.647859
Morris, A. W. J., Carare, R. O., Schreiber, S., and Hawkes, C. A. (2014). The cerebrovascular basement membrane: role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:251. doi: 10.3389/fnagi.2014.00251
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Mulder, M., and Terwel, D. (1998). Possible link between lipid metabolism and cerebral amyloid angiopathy in Alzheimer’s disease: a role for high-density lipoproteins? Haemostasis 28, 174–194. doi: 10.1159/000022429
Muller, M., Appelman, A. P. A., van der Graaf, Y., Vincken, K. L., Mali, W. P. T. M., and Geerlings, M. I. (2011). Brain atrophy and cognition: interaction with cerebrovascular pathology? Neurobiol. Aging 32, 885–893. doi: 10.1016/j.neurobiolaging.2009.05.005
Mumaw, C. L., Surace, M., Levesque, S., Kodavanti, U. P., Kodavanti, P. R. S., Royland, J. E., et al. (2017). Atypical microglial response to biodiesel exhaust in healthy and hypertensive rats. Neurotoxicology 59, 155–163. doi: 10.1016/j.neuro.2016.10.012
Navarro, A., García, M., Rodrigues, A. S., Garcia, P. V., Camarinho, R., and Segovia, Y. (2021). Reactive astrogliosis in the dentate gyrus of mice exposed to active volcanic environments. J. Toxicol. Environ. Health Part A 84, 213–226. doi: 10.1080/15287394.2020.1850381
Newman, J. D., Thurston, G. D., Cromar, K., Guo, Y., Rockman, C. B., Fisher, E. A., et al. (2015). Particulate air pollution and carotid artery stenosis. J. Am. Coll. Cardiol. 65, 1150–1151. doi: 10.1016/j.jacc.2014.12.052
Ngandu, T., Lehtisalo, J., Solomon, A., Levälahti, E., Ahtiluoto, S., Antikainen, R., et al. (2015). A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385, 2255–2263. doi: 10.1016/S0140-6736(15)60461-5
Nihashi, T., Inao, S., Kajita, Y., Kawai, T., Sugimoto, T., Niwa, M., et al. (2001). Expression and distribution of β amyloid precursor protein and β amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 143, 287–295. doi: 10.1007/s007010170109
Nikolakopoulou, A. M., Montagne, A., Kisler, K., Dai, Z., Wang, Y., Huuskonen, M. T., et al. (2019). Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 22, 1089–1098. doi: 10.1038/s41593-019-0434-z
Nikolakopoulou, A. M., Wang, Y., Ma, Q., Sagare, A. P., Montagne, A., Huuskonen, M. T., et al. (2021). Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J. Exp. Med. 218:e20202207. doi: 10.1084/jem.20202207
Nishikawa, H., Liu, L., Nakano, F., Kawakita, F., Kanamaru, H., Nakatsuka, Y., et al. (2018). Modified citrus pectin prevents blood-brain barrier disruption in mouse subarachnoid hemorrhage by inhibiting galectin-3. Stroke 49, 2743–2751. doi: 10.1161/STROKEAHA.118.021757
Noe, C. R., Noe-Letschnig, M., Handschuh, P., Noe, C. A., and Lanzenberger, R. (2020). Dysfunction of the blood-brain barrier-a key step in neurodegeneration and dementia. Front. Aging Neurosci. 12:185. doi: 10.3389/fnagi.2020.00185
Noh, S.-M., Chung, S. J., Kim, K.-K., Kang, D.-W., Lim, Y.-M., Kwon, S. U., et al. (2014). Emotional disturbance in CADASIL: its impact on quality of life and caregiver burden. Cerebrovasc. Dis. 37, 188–194. doi: 10.1159/000357798
Nortley, R., Korte, N., Izquierdo, P., Hirunpattarasilp, C., Mishra, A., Jaunmuktane, Z., et al. (2019). Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365:eaav9518. doi: 10.1126/science.aav9518
Nuthikattu, S., Milenkovic, D., Rutledge, J. C., and Villablanca, A. C. (2020). Sex-dependent molecular mechanisms of lipotoxic injury in brain microvasculature: implications for dementia. Int. J. Mol. Sci. 21:8146. doi: 10.3390/ijms21218146
O’Brien, J. T., and Markus, H. S. (2014). Vascular risk factors and Alzheimer’s disease. BMC Med. 12:218. doi: 10.1186/s12916-014-0218-y
O’Brien, J. T., and Thomas, A. (2015). Vascular dementia. Lancet 386, 1698–1706. doi: 10.1016/S0140-6736(15)00463-8
Oberdörster, G., Sharp, Z., Atudorei, V., Elder, A., Gelein, R., Kreyling, W., et al. (2004). Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol. 16, 437–445. doi: 10.1080/08958370490439597
Oka, C., Tsujimoto, R., Kajikawa, M., Koshiba-Takeuchi, K., Ina, J., Yano, M., et al. (2004). HtrA1 serine protease inhibits signaling mediated by Tgfβ family proteins. Development 131, 1041–1053. doi: 10.1242/dev.00999
Okamoto, Y., Ihara, M., Tomimoto, H., Taylor Kimberly, W., and Greenberg, S. M. (2010). Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 74:93; author reply 93. doi: 10.1212/WNL.0b013e3181c77627
Okazaki, S., Hornberger, E., Griebe, M., Gass, A., Hennerici, M. G., and Szabo, K. (2015). MRI characteristics of the evolution of supratentorial recent small subcortical infarcts. Front. Neurol. 6:118. doi: 10.3389/fneur.2015.00118
Okeda, R., and Nisihara, M. (2008). An autopsy case of Fabry disease with neuropathological investigation of the pathogenesis of associated dementia. Neuropathology 28, 532–540. doi: 10.1111/j.1440-1789.2008.00883.x
Onoda, A., Kawasaki, T., Tsukiyama, K., Takeda, K., and Umezawa, M. (2020). Carbon nanoparticles induce endoplasmic reticulum stress around blood vessels with accumulation of misfolded proteins in the developing brain of offspring. Sci. Rep. 10:10028. doi: 10.1038/s41598-020-66744-w
Onoda, A., Takeda, K., and Umezawa, M. (2017). Dose-dependent induction of astrocyte activation and reactive astrogliosis in mouse brain following maternal exposure to carbon black nanoparticle. Part. Fibre Toxicol. 14:4. doi: 10.1186/s12989-017-0184-6
Oppenheim, H. A., Lucero, J., Guyot, A.-C., Herbert, L. M., McDonald, J. D., Mabondzo, A., et al. (2013). Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part. Fibre Toxicol. 10:62. doi: 10.1186/1743-8977-10-62
Østergaard, L., Engedal, T. S., Moreton, F., Hansen, M. B., Wardlaw, J. M., Dalkara, T., et al. (2016). Cerebral small vessel disease: capillary pathways to stroke and cognitive decline. J. Cereb. Blood Flow Metab. 36, 302–325. doi: 10.1177/0271678X15606723
Oudin, A., Andersson, J., Sundström, A., Nordin Adolfsson, A., Oudin Åström, D., Adolfsson, R., et al. (2019). Traffic-related air pollution as a risk factor for dementia: no clear modifying effects of APOEε4 in the betula cohort. J. Alzheimers Dis. 71, 733–740. doi: 10.3233/JAD-181037
Patterson, C. (2018). World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. London, UK: Alzheimer’s Disease International.
Pang, J., Peng, J., Matei, N., Yang, P., Kuai, L., Wu, Y., et al. (2018). Apolipoprotein E exerts a whole-brain protective property by promoting M1? Microglia quiescence after experimental subarachnoid hemorrhage in mice. Transl. Stroke Res. 9, 654–668. doi: 10.1007/s12975-018-0665-4
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi: 10.1016/S1474-4422(10)70104-6
Parkes, I., Chintawar, S., and Cader, M. Z. (2018). Neurovascular dysfunction in dementia—human cellular models and molecular mechanisms. Clin. Sci. 132, 399–418. doi: 10.1042/CS20160720
Parolisi, R., Montarolo, F., Pini, A., Rovelli, S., Cattaneo, A., Bertolotto, A., et al. (2021). Exposure to fine particulate matter (PM2.5) hampers myelin repair in a mouse model of white matter demyelination. Neurochem. Int. 145:104991. doi: 10.1016/j.neuint.2021.104991
Patten, K. T., González, E. A., Valenzuela, A., Berg, E., Wallis, C., Garbow, J. R., et al. (2020). Effects of early life exposure to traffic-related air pollution on brain development in juvenile Sprague-Dawley rats. Transl. Psychiatry 10:166. doi: 10.1038/s41398-020-0845-3
Paul, K. C., Haan, M., Mayeda, E. R., and Ritz, B. R. (2019). Ambient air pollution, noise and late-life cognitive decline and dementia risk. Annu. Rev. Public Health 40, 203–220. doi: 10.1146/annurev-publhealth-040218-044058
Pendlebury, S. T., Mariz, J., Bull, L., Mehta, Z., and Rothwell, P. M. (2012). MoCA, ACE-R, and MMSE versus the national institute of neurological disorders and stroke-canadian stroke network vascular cognitive impairment harmonization standards neuropsychological battery after TIA and stroke. Stroke 43, 464–469. doi: 10.1161/STROKEAHA.111.633586
Pendlebury, S. T., and Rothwell, P. M. (2009). Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018. doi: 10.1016/S1474-4422(09)70236-4
Perlmutter, L. S., Barrón, E., Saperia, D., and Chui, H. C. (1991). Association between vascular basement membrane components and the lesions of Alzheimer’s disease. J. Neurosci. Res. 30, 673–681. doi: 10.1002/jnr.490300411
Perlmutter, L. S., Chui, H. C., Saperia, D., and Athanikar, J. (1990). Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 508, 13–19. doi: 10.1016/0006-8993(90)91111-s
Peters, A., Veronesi, B., Calderón-Garcidueñas, L., Gehr, P., Chen, L. C., Geiser, M., et al. (2006). Translocation and potential neurological effects of fine and ultrafine particles a critical update. Part. Fibre Toxicol. 3:13. doi: 10.1186/1743-8977-3-13
Peters, R., Warwick, J., Anstey, K. J., and Anderson, C. S. (2019). Blood pressure and dementia: what the SPRINT-MIND trial adds and what we still need to know. Neurology 92, 1017–1018. doi: 10.1212/WNL.0000000000007543
Ping, S., Qiu, X., Kyle, M., Hughes, K., Longo, J., and Zhao, L.-R. (2019). Stem cell factor and granulocyte colony-stimulating factor promote brain repair and improve cognitive function through VEGF-A in a mouse model of CADASIL. Neurobiol. Dis. 132:104561. doi: 10.1016/j.nbd.2019.104561
Pinter, D., Enzinger, C., Gattringer, T., Eppinger, S., Niederkorn, K., Horner, S., et al. (2019). Prevalence and short-term changes of cognitive dysfunction in young ischaemic stroke patients. Eur. J. Neurol. 26, 727–732. doi: 10.1111/ene.13879
Podcasy, J. L., and Epperson, C. N. (2016). Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 18, 437–446. doi: 10.31887/DCNS.2016.18.4/cepperson
Poorthuis, M. H. F., Algra, A. M., Algra, A., Kappelle, L. J., and Klijn, C. J. M. (2017). Female- and male-specific risk factors for stroke: a systematic review and meta-analysis. JAMA Neurol. 74, 75–81. doi: 10.1001/jamaneurol.2016.3482
Potter, G. M., Doubal, F. N., Jackson, C. A., Chappell, F. M., Sudlow, C. L., Dennis, M. S., et al. (2015). Enlarged perivascular spaces and cerebral small vessel disease. Int. J. Stroke 10, 376–381. doi: 10.1111/ijs.12054
Prins, N. D., and Scheltens, P. (2015). White matter hyperintensities, cognitive impairment and dementia: an update. Nat. Rev. Neurol. 11, 157–165. doi: 10.1038/nrneurol.2015.10
Qin, C., Fan, W.-H., Liu, Q., Shang, K., Murugan, M., Wu, L.-J., et al. (2017). Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke 48, 3336–3346. doi: 10.1161/STROKEAHA.117.018505
Ragno, M., Pianese, L., Morroni, M., Cacchiò, G., Manca, A., Di Marzio, F., et al. (2013). “CADASIL coma” in an Italian homozygous CADASIL patient: comparison with clinical and MRI findings in age-matched heterozygous patients with the same G528C NOTCH3 mutation. Neurol. Sci. 34, 1947–1953. doi: 10.1007/s10072-013-1418-5
Razvi, S. S. M., and Bone, I. (2006). Single gene disorders causing ischaemic stroke. J. Neurol. 253, 685–700. doi: 10.1007/s00415-006-0048-8
Regenhardt, R. W., Das, A. S., Lo, E. H., and Caplan, L. R. (2018). Advances in understanding the pathophysiology of lacunar stroke: a review. JAMA Neurol. 75, 1273–1281. doi: 10.1001/jamaneurol.2018.1073
Regenhardt, R. W., Das, A. S., Ohtomo, R., Lo, E. H., Ayata, C., and Gurol, M. E. (2019). Pathophysiology of lacunar stroke: history’s mysteries and modern interpretations. J. Stroke Cerebrovasc. Dis. 28, 2079–2097. doi: 10.1016/j.jstrokecerebrovasdis.2019.05.006
Reijmer, Y. D., van Veluw, S. J., and Greenberg, S. M. (2016). Ischemic brain injury in cerebral amyloid angiopathy. J. Cereb. Blood Flow Metab. 36, 40–54. doi: 10.1038/jcbfm.2015.88
Reyes, S., Viswanathan, A., Godin, O., Dufouil, C., Benisty, S., Hernandez, K., et al. (2009). Apathy: a major symptom in CADASIL. Neurology 72, 905–910. doi: 10.1212/01.wnl.0000344166.03470.f8
Rhodin, J. A., and Thomas, T. O. M. (2001). A vascular connection to alzheimer’s disease. Microcirculation 8, 207–220. doi: 10.1038/sj/mn/7800086
Rice, G. I., Rodero, M. P., and Crow, Y. J. (2015). Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 35, 235–243. doi: 10.1007/s10875-015-0147-3
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi: 10.1056/NEJMoa1707914
Rigsby, C. S., Pollock, D. M., and Dorrance, A. M. (2007). Spironolactone improves structure and increases tone in the cerebral vasculature of male spontaneously hypertensive stroke-prone rats. Microvasc. Res. 73, 198–205. doi: 10.1016/j.mvr.2006.12.001
Ritchie, K., and Lovestone, S. (2002). The dementias. Lancet 360, 1759–1766. doi: 10.1016/S0140-6736(02)11667-9
Rizzi, L., Rosset, I., and Roriz-Cruz, M. (2014). Global epidemiology of dementia: Alzheimer’s and vascular types. Biomed Res. Int. 2014:908915. doi: 10.1155/2014/908915
Rojas, H., Ritter, C., and Pizzol, F. D. (2011). Mechanisms of dysfunction of the blood-brain barrier in critically ill patients: emphasis on the role of matrix metalloproteinases. Rev. Bras. Ter. Intensiva 23, 222–227. doi: 10.1590/S0103-507X2011000200016
Rolfs, A., Fazekas, F., Grittner, U., Dichgans, M., Martus, P., Holzhausen, M., et al. (2013). Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke 44, 340–349. doi: 10.1161/STROKEAHA.112.663708
Roqué, P. J., Dao, K., and Costa, L. G. (2016). Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology 56, 204–214. doi: 10.1016/j.neuro.2016.08.006
Rosa, G., Giannotti, C., Martella, L., Massa, F., Serafini, G., Pardini, M., et al. (2020). Brain aging, cardiovascular diseases, mixed dementia, and frailty in the oldest old: from brain phenotype to clinical expression. J. Alzheimers Dis. 75, 1083–1103. doi: 10.3233/JAD-191075
Rosell, A., Cuadrado, E., Ortega-Aznar, A., Hernández-Guillamon, M., Lo, E. H., and Montaner, J. (2008). MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39, 1121–1126. doi: 10.1161/STROKEAHA.107.500868
Rosell, A., Ortega-Aznar, A., Alvarez-Sabín, J., Fernández-Cadenas, I., Ribó, M., Molina, C. A., et al. (2006). Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke 37, 1399–1406. doi: 10.1161/01.STR.0000223001.06264.af
Rosidi, N. L., Zhou, J., Pattanaik, S., Wang, P., Jin, W., Brophy, M., et al. (2011). Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One 6:e26612. doi: 10.1371/journal.pone.0026612
Rouhl, R. P. W., Damoiseaux, J. G. M. C., Lodder, J., Theunissen, R. O. M. F. I. H., Knottnerus, I. L. H., Staals, J., et al. (2012). Vascular inflammation in cerebral small vessel disease. Neurobiol. Aging 33, 1800–1806. doi: 10.1016/j.neurobiolaging.2011.04.008
Rozemuller, A. J. M., Jansen, C., Carrano, A., van Haastert, E. S., Hondius, D., van der Vies, S. M., et al. (2012). Neuroinflammation and common mechanism in Alzheimer’s disease and prion amyloidosis: amyloid-associated proteins, neuroinflammation and neurofibrillary degeneration. Neurodegener. Dis. 10, 301–304. doi: 10.1159/000335380
Ryu, J. K., and McLarnon, J. G. (2006). Minocycline or iNOS inhibition block 3-nitrotyrosine increases and blood-brain barrier leakiness in amyloid β-peptide-injected rat hippocampus. Exp. Neurol. 198, 552–557. doi: 10.1016/j.expneurol.2005.12.016
Ryu, J. K., Petersen, M. A., Murray, S. G., Baeten, K. M., Meyer-Franke, A., Chan, J. P., et al. (2015). Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat. Commun. 6:8164. doi: 10.1038/ncomms9164
Sacco, R. L. (2000). Lobar intracerebral hemorrhage. N. Engl. J. Med. 342, 276–279. doi: 10.1056/NEJM200001273420410
Sachdev, P. S., Blacker, D., Blazer, D. G., Ganguli, M., Jeste, D. V., Paulsen, J. S., et al. (2014). Classifying neurocognitive disorders: the DSM-5 approach. Nat. Rev. Neurol. 10, 634–642. doi: 10.1038/nrneurol.2014.181
Sachdev, P. S., Brodaty, H., Valenzuela, M. J., Lorentz, L., Looi, J. C. L., Berman, K., et al. (2006). Clinical determinants of dementia and mild cognitive impairment following ischaemic stroke: the Sydney Stroke Study. Dement. Geriatr. Cogn. Disord. 21, 275–283. doi: 10.1159/000091434
Sachdev, P. S., Brodaty, H., Valenzuela, M. J., Lorentz, L., Looi, J. C. L., Wen, W., et al. (2004). The neuropsychological profile of vascular cognitive impairment in stroke and TIA patients. Neurology 62, 912–919. doi: 10.1212/01.wnl.0000115108.65264.4b
Salat, D. H., Smith, E. E., Tuch, D. S., Benner, T., Pappu, V., Schwab, K. M., et al. (2006). White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke 37, 1759–1764. doi: 10.1161/01.STR.0000227328.86353.a7
Savva, G. M., Stephan, B. C. M., and Alzheimer’s Society Vascular Dementia Systematic Review Group. (2010). Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke 41, e41–e46. doi: 10.1161/STROKEAHA.109.559880
Schiffmann, R. (2015). Fabry disease. Handb. Clin. Neurol. 132, 231–248. doi: 10.1016/B978-0-444-62702-5.00017-2
Schon, F., Martin, R. J., Prevett, M., Clough, C., Enevoldson, T. P., and Markus, H. S. (2003). “CADASIL coma”: an underdiagnosed acute encephalopathy. J. Neurol. Neurosurg. Psychiatry 74, 249–252. doi: 10.1136/jnnp.74.2.249
Schultz, N., Nielsen, H. M., Minthon, L., and Wennström, M. (2014). Involvement of matrix metalloproteinase-9 in amyloid-β 1–42-induced shedding of the pericyte proteoglycan NG2. J. Neuropathol. Exp. Neurol. 73, 684–692. doi: 10.1097/NEN.0000000000000084
Schwartz, J., Litonjua, A., Suh, H., Verrier, M., Zanobetti, A., Syring, M., et al. (2005). Traffic related pollution and heart rate variability in a panel of elderly subjects. Thorax 60, 455–461. doi: 10.1136/thx.2004.024836
Shabir, O., Berwick, J., and Francis, S. E. (2018). Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 19:62. doi: 10.1186/s12868-018-0465-5
Shi, Y., and Wardlaw, J. M. (2016). Update on cerebral small vessel disease: a dynamic whole-brain disease. Stroke Vasc. Neurol. 1, 83–92. doi: 10.1136/svn-2016-000035
Shiga, A., Nozaki, H., Yokoseki, A., Nihonmatsu, M., Kawata, H., Kato, T., et al. (2011). Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-β1 via cleavage of proTGF-β1. Hum. Mol. Genet. 20, 1800–1810. doi: 10.1093/hmg/ddr063
Shih, A. Y., Hyacinth, H. I., Hartmann, D. A., and van Veluw, S. J. (2018). Rodent models of cerebral microinfarct and microhemorrhage. Stroke 49, 803–810. doi: 10.1161/STROKEAHA.117.016995
Shimizu, H., Ghazizadeh, M., Sato, S., Oguro, T., and Kawanami, O. (2009). Interaction between β-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer’s disease. J. Clin. Neurosci. 16, 277–282. doi: 10.1016/j.jocn.2008.04.009
Shimotori, M., Maruyama, H., Nakamura, G., Suyama, T., Sakamoto, F., Itoh, M., et al. (2008). Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 29:331. doi: 10.1002/humu.9520
Shoamanesh, A., Preis, S. R., Beiser, A. S., Vasan, R. S., Benjamin, E. J., Kase, C. S., et al. (2015). Inflammatory biomarkers, cerebral microbleeds and small vessel disease: framingham heart study. Neurology 84, 825–832. doi: 10.1212/WNL.0000000000001279
Sims, K., Politei, J., Banikazemi, M., and Lee, P. (2009). Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry registry. Stroke 40, 788–794. doi: 10.1161/STROKEAHA.108.526293
Singh-Bains, M. K., Linke, V., Austria, M. D. R., Tan, A. Y. S., Scotter, E. L., Mehrabi, N. F., et al. (2019). Altered microglia and neurovasculature in the Alzheimer’s disease cerebellum. Neurobiol. Dis. 132:104589. doi: 10.1016/j.nbd.2019.104589
Skrobot, O. A., Black, S. E., Chen, C., DeCarli, C., Erkinjuntti, T., Ford, G. A., et al. (2018). Progress toward standardized diagnosis of vascular cognitive impairment: guidelines from the vascular impairment of cognition classification consensus study. Alzheimers Dement. 14, 280–292. doi: 10.1016/j.jalz.2017.09.007
Smith, E. E., Schneider, J. A., Wardlaw, J. M., and Greenberg, S. M. (2012). Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 11, 272–282. doi: 10.1016/S1474-4422(11)70307-6
Song, B., Ao, Q., Niu, Y., Shen, Q., Zuo, H., Zhang, X., et al. (2013). Amyloid β-peptide worsens cognitive impairment following cerebral ischemia-reperfusion injury. Neural Regen. Res. 8, 2449–2457. doi: 10.3969/j.issn.1673-5374.2013.26.006
Soontornniyomkij, V., Lynch, M. D., Mermash, S., Pomakian, J., Badkoobehi, H., Clare, R., et al. (2010). Cerebral microinfarcts associated with severe cerebral β-amyloid angiopathy. Brain Pathol. 20, 459–467. doi: 10.1111/j.1750-3639.2009.00322.x
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. doi: 10.1038/s41467-019-11674-z
Spann, N. J., Garmire, L. X., McDonald, J. G., Myers, D. S., Milne, S. B., Shibata, N., et al. (2012). Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151, 138–152. doi: 10.1016/j.cell.2012.06.054
Srinivasan, K., Friedman, B. A., Etxeberria, A., Huntley, M. A., van der Brug, M. P., Foreman, O., et al. (2020). Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 31:107843. doi: 10.1016/j.celrep.2020.107843
Stam, A. H., Kothari, P. H., Shaikh, A., Gschwendter, A., Jen, J. C., Hodgkinson, S., et al. (2016). Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain 139, 2909–2922. doi: 10.1093/brain/aww217
Staszewski, J., Piusińska-Macoch, R., Brodacki, B., Skrobowska, E., Macek, K., and Stepień, A. (2017). Risk of vascular events in different manifestations of cerebral small vessel disease: a 2-year follow-up study with a control group. Heliyon 3:e00455. doi: 10.1016/j.heliyon.2017.e00455
Stemme, S., Faber, B., Holm, J., Wiklund, O., Witztum, J. L., and Hansson, G. K. (1995). T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. U S A 92, 3893–3897. doi: 10.1073/pnas.92.9.3893
Stomrud, E., Björkqvist, M., Janciauskiene, S., Minthon, L., and Hansson, O. (2010). Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2:20. doi: 10.1186/alzrt44
Su, J. H., Cummings, B. J., and Cotman, C. W. (1992). Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience 51, 801–813. doi: 10.1016/0306-4522(92)90521-3
Summers, P. M., Hartmann, D. A., Hui, E. S., Nie, X., Deardorff, R. L., McKinnon, E. T., et al. (2017). Functional deficits induced by cortical microinfarcts. J. Cereb. Blood Flow Metab. 37, 3599–3614. doi: 10.1177/0271678X16685573
Sweeney, M. D., Sagare, A. P., Pachicano, M., Harrington, M. G., Joe, E., Chui, H. C., et al. (2020). A novel sensitive assay for detection of a biomarker of pericyte injury in cerebrospinal fluid. Alzheimers Dement. 16, 821–830. doi: 10.1002/alz.12061
Szarmach, A., Halena, G., Kaszubowski, M., Piskunowicz, M., Studniarek, M., Lass, P., et al. (2017). Carotid artery stenting and blood-brain barrier permeability in subjects with chronic carotid artery stenosis. Int. J. Mol. Sci. 18:1008. doi: 10.3390/ijms18051008
Tabas, I., García-Cardeña, G., and Owens, G. K. (2015). Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 209, 13–22. doi: 10.1083/jcb.201412052
Tabas, I., Williams, K. J., and Borén, J. (2007). Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116, 1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890
Tan, R. Y. Y., and Markus, H. S. (2016). CADASIL: migraine, encephalopathy, stroke and their inter-relationships. PLoS One 11:e0157613. doi: 10.1371/journal.pone.0157613
Tan, R., Traylor, M., Rutten-Jacobs, L., and Markus, H. (2017). New insights into mechanisms of small vessel disease stroke from genetics. Clin. Sci. 131, 515–531. doi: 10.1042/CS20160825
Tancredi, V., D’Antuono, M., Cafè, C., Giovedì, S., Buè, M. C., D’Arcangelo, G., et al. (2000). The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J. Neurochem. 75, 634–643. doi: 10.1046/j.1471-4159.2000.0750634.x
Tariq, S., and Barber, P. A. (2018). Dementia risk and prevention by targeting modifiable vascular risk factors. J. Neurochem. 144, 565–581. doi: 10.1111/jnc.14132
Tarkowski, E., Issa, R., Sjögren, M., Wallin, A., Blennow, K., Tarkowski, A., et al. (2002). Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 23, 237–243. doi: 10.1016/s0197-4580(01)00285-8
Taylor, X., Cisternas, P., You, Y., You, Y., Xiang, S., Marambio, Y., et al. (2020). A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflammation 17:223. doi: 10.1186/s12974-020-01900-7
Tennstaedt, A., Pöpsel, S., Truebestein, L., Hauske, P., Brockmann, A., Schmidt, N., et al. (2012). Human high temperature requirement serine protease A1 (HTRA1) degrades tau protein aggregates. J. Biol. Chem. 287, 20931–20941. doi: 10.1074/jbc.M111.316232
Ter Telgte, A., van Leijsen, E. M. C., Wiegertjes, K., Klijn, C. J. M., Tuladhar, A. M., and de Leeuw, F.-E. (2018). Cerebral small vessel disease: from a focal to a global perspective. Nat. Rev. Neurol. 14, 387–398. doi: 10.1038/s41582-018-0014-y
Thal, D. R., Ghebremedhin, E., Orantes, M., and Wiestler, O. D. (2003). Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropathol. Exp. Neurol. 62, 1287–1301. doi: 10.1093/jnen/62.12.1287
Thériault, P., ElAli, A., and Rivest, S. (2015). The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther. 7:41. doi: 10.1186/s13195-015-0125-2
Thiel, A., Cechetto, D. F., Heiss, W.-D., Hachinski, V., and Whitehead, S. N. (2014). Amyloid burden, neuroinflammation and links to cognitive decline after ischemic stroke. Stroke 45, 2825–2829. doi: 10.1161/STROKEAHA.114.004285
Thomson, E. M., Kumarathasan, P., Calderón-Garcidueñas, L., and Vincent, R. (2007). Air pollution alters brain and pituitary endothelin-1 and inducible nitric oxide synthase gene expression. Environ. Res. 105, 224–233. doi: 10.1016/j.envres.2007.06.005
Tiaden, A. N., and Richards, P. J. (2013). The emerging roles of HTRA1 in musculoskeletal disease. Am. J. Pathol. 182, 1482–1488. doi: 10.1016/j.ajpath.2013.02.003
Toth, P., Tarantini, S., Csiszar, A., and Ungvari, Z. (2017). Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment and neurovascular uncoupling in aging. Am. J. Physiol. Heart Circ. Physiol. 312, H1–H20. doi: 10.1152/ajpheart.00581.2016
Tuominen, S., Miao, Q., Kurki, T., Tuisku, S., Pöyhönen, M., Kalimo, H., et al. (2004). Positron emission tomography examination of cerebral blood flow and glucose metabolism in young CADASIL patients. Stroke 35, 1063–1067. doi: 10.1161/01.STR.0000124124.69842.2d
Uemura, M. T., Ihara, M., Maki, T., Nakagomi, T., Kaji, S., Uemura, K., et al. (2018). Pericyte-derived bone morphogenetic protein 4 underlies white matter damage after chronic hypoperfusion. Brain Pathol. 28, 521–535. doi: 10.1111/bpa.12523
Uemura, M. T., Maki, T., Ihara, M., Lee, V. M. Y., and Trojanowski, J. Q. (2020). Brain microvascular pericytes in vascular cognitive impairment and dementia. Front. Aging Neurosci. 12:80. doi: 10.3389/fnagi.2020.00080
Ueno, M., Tomimoto, H., Akiguchi, I., Wakita, H., and Sakamoto, H. (2002). Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 22, 97–104. doi: 10.1097/00004647-200201000-00012
Ukraintseva, S., Sloan, F., Arbeev, K., and Yashin, A. (2006). Increasing rates of dementia at time of declining mortality from stroke. Stroke 37, 1155–1159. doi: 10.1161/01.STR.0000217971.88034.e9
Underly, R. G., Levy, M., Hartmann, D. A., Grant, R. I., Watson, A. N., and Shih, A. Y. (2017). Pericytes as inducers of rapid, matrix metalloproteinase-9-dependent capillary damage during ischemia. J. Neurosci. 37, 129–140. doi: 10.1523/JNEUROSCI.2891-16.2016
Vahedi, K., and Alamowitch, S. (2011). Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr. Opin. Neurol. 24, 63–68. doi: 10.1097/WCO.0b013e32834232c6
Vahedi, K., Chabriat, H., Levy, C., Joutel, A., Tournier-Lasserve, E., and Bousser, M.-G. (2004). Migraine with aura and brain magnetic resonance imaging abnormalities in patients with CADASIL. Arch. Neurol. 61, 1237–1240. doi: 10.1001/archneur.61.8.1237
Valenti, R., Pescini, F., Antonini, S., Castellini, G., Poggesi, A., Bianchi, S., et al. (2011). Major depression and bipolar disorders in CADASIL: a study using the DSM-IV semi-structured interview. Acta Neurol. Scand. 124, 390–395. doi: 10.1111/j.1600-0404.2011.01512.x
Valenti, R., Poggesi, A., Pescini, F., Inzitari, D., and Pantoni, L. (2008). Psychiatric disturbances in CADASIL: a brief review. Acta Neurol. Scand. 118, 291–295. doi: 10.1111/j.1600-0404.2008.01015.x
van der Flier, W. M., van Straaten, E. C. W., Barkhof, F., Verdelho, A., Madureira, S., Pantoni, L., et al. (2005). Small vessel disease and general cognitive function in nondisabled elderly: the LADIS study. Stroke 36, 2116–2120. doi: 10.1161/01.STR.0000179092.59909.42
van Veluw, S. J., Shih, A. Y., Smith, E. E., Chen, C., Schneider, J. A., Wardlaw, J. M., et al. (2017). Detection, risk factors and functional consequences of cerebral microinfarcts. Lancet Neurol. 16, 730–740. doi: 10.1016/S1474-4422(17)30196-5
Venkat, P., Chopp, M., and Chen, J. (2015). Models and mechanisms of vascular dementia. Exp. Neurol. 272, 97–108. doi: 10.1016/j.expneurol.2015.05.006
Verdura, E., Hervé, D., Bergametti, F., Jacquet, C., Morvan, T., Prieto-Morin, C., et al. (2016). Disruption of a miR-29 binding site leading to COL4A1 upregulation causes pontine autosomal dominant microangiopathy with leukoencephalopathy. Ann. Neurol. 80, 741–753. doi: 10.1002/ana.24782
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U S A 110, E1807–E1816. doi: 10.1073/pnas.1220484110
Vidal, R., Barbeito, A. G., Miravalle, L., and Ghetti, B. (2009). Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol. 19, 58–68. doi: 10.1111/j.1750-3639.2008.00164.x
Vidal, R., Frangione, B., Rostagno, A., Mead, S., Révész, T., Plant, G., et al. (1999). A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399, 776–781. doi: 10.1038/21637
Vidal, R., Revesz, T., Rostagno, A., Kim, E., Holton, J. L., Bek, T., et al. (2000). A decamer duplication in the 3’ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl. Acad. Sci. U S A 97, 4920–4925. doi: 10.1073/pnas.080076097
Wang, B.-R., Shi, J.-Q., Ge, N.-N., Ou, Z., Tian, Y.-Y., Jiang, T., et al. (2018). PM2.5 exposure aggravates oligomeric amyloid β-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer’s disease. J. Neuroinflammation 15:132. doi: 10.1186/s12974-018-1178-5
Wang, T., Wang, L., Moreno-Vinasco, L., Lang, G. D., Siegler, J. H., Mathew, B., et al. (2012). Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 9:35. doi: 10.1186/1743-8977-9-35
Wang, L., Yang, J.-W., Lin, L.-T., Huang, J., Wang, X.-R., Su, X.-T., et al. (2020). Acupuncture attenuates inflammation in microglia of vascular dementia rats by inhibiting miR-93-mediated TLR4/MyD88/NF-κB signaling pathway. Oxid. Med. Cell. Longev. 2020:8253904. doi: 10.1155/2020/8253904
Wardlaw, J. M., Smith, E., Biessels, G. J., Cordonnier, C., Fazekas, F., Frayne, R., et al. (2013). Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838. doi: 10.1016/S1474-4422(13)70124-8
Weber, C., and Noels, H. (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422. doi: 10.1038/nm.2538
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
Woodward, N. C., Levine, M. C., Haghani, A., Shirmohammadi, F., Saffari, A., Sioutas, C., et al. (2017a). Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J. Neuroinflammation 14:84. doi: 10.1186/s12974-017-0858-x
Woodward, N. C., Pakbin, P., Saffari, A., Shirmohammadi, F., Haghani, A., Sioutas, C., et al. (2017b). Traffic-related air pollution impact on mouse brain accelerates myelin and neuritic aging changes with specificity for CA1 neurons. Neurobiol. Aging 53, 48–58. doi: 10.1016/j.neurobiolaging.2017.01.007
Wu, Y.-C., Lin, Y.-C., Yu, H.-L., Chen, J.-H., Chen, T.-F., Sun, Y., et al. (2015). Association between air pollutants and dementia risk in the elderly. Alzheimers Dement. 1, 220–228. doi: 10.1016/j.dadm.2014.11.015
Wyss-Coray, T., Lin, C., Sanan, D. A., Mucke, L., and Masliah, E. (2000). Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 156, 139–150. doi: 10.1016/s0002-9440(10)64713-x
Yamada, M., and Naiki, H. (2012). Cerebral amyloid angiopathy. Prog. Mol. Biol. Transl. Sci. 107, 41–78. doi: 10.1016/B978-0-12-385883-2.00006-0
Yamamoto, Y., Craggs, L., Baumann, M., Kalimo, H., and Kalaria, R. N. (2011). Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol. Appl. Neurobiol. 37, 94–113. doi: 10.1111/j.1365-2990.2010.01147.x
Yamamoto, Y., Craggs, L. J. L., Watanabe, A., Booth, T., Attems, J., Low, R. W. C., et al. (2013). Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J. Neuropathol. Exp. Neurol. 72, 416–431. doi: 10.1097/NEN.0b013e31829020b5
Yang, C., Hawkins, K. E., Doré, S., and Candelario-Jalil, E. (2019). Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 316, C135–C153. doi: 10.1152/ajpcell.00136.2018
Yang, Y., and Rosenberg, G. A. (2011). Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42, 3323–3328. doi: 10.1161/STROKEAHA.110.608257
Yang, J., Zhang, R., Shi, C., Mao, C., Yang, Z., Suo, Z., et al. (2017). AQP4 association with amyloid deposition and astrocyte pathology in the Tg-arcswe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 57, 157–169. doi: 10.3233/JAD-160957
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ and thereby facilitates uptake of amyloid-β by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yin, F., Lawal, A., Ricks, J., Fox, J. R., Larson, T., Navab, M., et al. (2013). Diesel exhaust induces systemic lipid peroxidation and development of dysfunctional pro-oxidant and pro-inflammatory high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 33, 1153–1161. doi: 10.1161/ATVBAHA.112.300552
Youmans, K. L., Tai, L. M., Nwabuisi-Heath, E., Jungbauer, L., Kanekiyo, T., Gan, M., et al. (2012). APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287, 41774–41786. doi: 10.1074/jbc.M112.407957
Zhan, X., Cox, C., Ander, B. P., Liu, D., Stamova, B., Jin, L.-W., et al. (2015). Inflammation combined with ischemia produces myelin injury and plaque-like aggregates of myelin, amyloid-β and AβPP in adult rat brain. J. Alzheimers Dis. 46, 507–523. doi: 10.3233/JAD-143072
Zhang, F., Eckman, C., Younkin, S., Hsiao, K. K., and Iadecola, C. (1997). Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J. Neurosci. 17, 7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997
Zhang, D., Li, S., Hou, L., Jing, L., Ruan, Z., Peng, B., et al. (2021). Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J. Neuroinflammation 18:4. doi: 10.1186/s12974-020-02065-z
Zhang, L., Lim, S. L., Du, H., Zhang, M., Kozak, I., Hannum, G., et al. (2012). High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-β family member growth differentiation factor 6. J. Biol. Chem. 287, 1520–1526. doi: 10.1074/jbc.M111.275990
Zhang, L.-Y., Pan, J., Mamtilahun, M., Zhu, Y., Wang, L., Venkatesh, A., et al. (2020). Microglia exacerbate white matter injury via complement C3/C3aR pathway after hypoperfusion. Theranostics 10, 74–90. doi: 10.7150/thno.35841
Zhang, C. E., Wong, S. M., van de Haar, H. J., Staals, J., Jansen, J. F. A., Jeukens, C. R. L. P. N., et al. (2017). Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology 88, 426–432. doi: 10.1212/WNL.0000000000003556
Zhong, C., Bu, X., Xu, T., Guo, L., Wang, X., Zhang, J., et al. (2018). Serum matrix metalloproteinase-9 and cognitive impairment after acute ischemic stroke. J. Am. Heart Assoc. 7:e007776. doi: 10.1161/JAHA.117.007776
Zhong, C., Yang, J., Xu, T., Xu, T., Peng, Y., Wang, A., et al. (2017). Serum matrix metalloproteinase-9 levels and prognosis of acute ischemic stroke. Neurology 89, 805–812. doi: 10.1212/WNL.0000000000004257
Zhao, S.-C., Ma, L.-S., Chu, Z.-H., Xu, H., Wu, W.-Q., and Liu, F. (2017). Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 38, 445–458. doi: 10.1038/aps.2016.162
Zhao, B.-Q., Wang, S., Kim, H.-Y., Storrie, H., Rosen, B. R., Mooney, D. J., et al. (2006). Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 12, 441–445. doi: 10.1038/nm1387
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Keywords: vascular dementia (VaD), stroke, cerebral small vessel disease (cSVD), neurovascular abnormalities, blood-brain barrier, neuroinflammation, air pollution
Citation: Lecordier S, Manrique-Castano D, El Moghrabi Y and ElAli A (2021) Neurovascular Alterations in Vascular Dementia: Emphasis on Risk Factors. Front. Aging Neurosci. 13:727590. doi: 10.3389/fnagi.2021.727590
Received: 19 June 2021; Accepted: 05 August 2021;
Published: 10 September 2021.
Edited by:
Shereen Nizari, University College London, United KingdomReviewed by:
Scott Edward Counts, Michigan State University, United StatesMarcus O. W. Grimm, Saarland University, Germany
Copyright © 2021 Lecordier, Manrique-Castano, El Moghrabi and ElAli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ayman ElAli, YXltYW4uZWwtYWxpQGNyY2h1ZGVxdWViZWMudWxhdmFsLmNh