 Md. Mahiuddin Ahmed1,2,3†
Md. Mahiuddin Ahmed1,2,3† Noah R. Johnson1,2,3†
Noah R. Johnson1,2,3† Timothy D. Boyd1,2,4
Timothy D. Boyd1,2,4 Christina Coughlan1,2,3
Christina Coughlan1,2,3 Heidi J. Chial1,2,3
Heidi J. Chial1,2,3 Huntington Potter1,2,3*†
Huntington Potter1,2,3*†- 1Department of Neurology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2University of Colorado Alzheimer’s and Cognition Center, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 3Linda Crnic Institute for Down Syndrome, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 4Partner Therapeutics, Inc., Lexington, MA, United States
Innate immune system activation and inflammation are associated with and may contribute to clinical outcomes in people with Down syndrome (DS), neurodegenerative diseases such as Alzheimer’s disease (AD), and normal aging. In addition to serving as potential diagnostic biomarkers, innate immune system activation and inflammation may play a contributing or causal role in these conditions, leading to the hypothesis that effective therapies should seek to dampen their effects. However, recent intervention studies with the innate immune system activator granulocyte-macrophage colony-stimulating factor (GM-CSF) in animal models of DS, AD, and normal aging, and in an AD clinical trial suggest that activating the innate immune system and inflammation may instead be therapeutic. We consider evidence that DS, AD, and normal aging are accompanied by innate immune system activation and inflammation and discuss whether and when during the disease process it may be therapeutically beneficial to suppress or promote such activation.
Introduction
Down syndrome (DS), most often caused by triplication of human chromosome 21 (Hsa21), is the most common genetic cause of both intellectual disability (ID) and age-associated cognitive decline (Epstein, 1990; Chapman and Hesketh, 2000; Silverman, 2007), affecting 1 in 700–1,000 live births worldwide (Centers for Disease Control and Prevention (CDC), 2006; Irving et al., 2008; Loane et al., 2013). The amyloid precursor protein (APP) plays a major role in the pathophysiology of Alzheimer’s disease (AD), and because the APP gene resides on chromosome 21, its additional copy is primarily responsible for the fact that all people with DS develop AD brain pathology, including amyloid-β (Aβ) plaques and cerebral amyloid angiopathy, by age 40 (Epstein, 1990; Snyder et al., 2020). Additionally, adults with DS develop neurofibrillary tangles of hyperphosphorylated tau, oxidative stress, vascular abnormalities, and chronic neuroinflammation, which are pathologies also present in patients with AD or other neurodegenerative diseases (Wisniewski et al., 1985; Head et al., 2016; Snyder et al., 2020).
Despite substantial epidemiological, biochemical, and genetic evidence in support of the amyloid cascade hypothesis (Hardy, 2009), the AD pathogenic pathway can be modulated by other aspects of brain physiology, especially the innate immune system and neuroinflammation (Potter, 2001; El Khoury et al., 2007; Cribbs et al., 2012; Lambert et al., 2013; Bettcher et al., 2018; Barroeta-Espar et al., 2019; Taipa et al., 2019). Alois Alzheimer first suggested a potential role for inflammation in AD based on his observation of abnormal glial cells surrounding amyloid deposits (Alzheimer, 1907). The discovery that specific inflammatory proteins, such as the cytokine interleukin-1 (IL-1) and the inflammation/acute-phase protein α1-antichymotrypsin (ACT), were upregulated in the AD brain and were associated with amyloid deposits solidified these early clues (Abraham et al., 1988; Mrak and Griffin, 2001; McGeer et al., 2006).
Inflammation is a complex multifactorial process in both the central nervous system (CNS) and the periphery, the activity of which varies depending on the disease stage. Microglia are the primary cell type associated with the innate immune system and neuroinflammation in the brain, with growing evidence suggesting that other cells, including astrocytes, neurons, oligodendrocytes, and pericytes also play significant roles, and brain inflammation in age-associated AD differs from that in DS-associated AD (Perry and Gordon, 1988; Colton and Wilcock, 2010; Wilcock et al., 2015). Thus, neuroinflammation may play a pivotal role in the development of AD (Akiyama et al., 2000), but the underlying mechanisms driving this pathological manifestation and its association with DS remain poorly understood.
In this mini-review, we first discuss the evidence that innate immune system activation and inflammation characterize both the CNS and the periphery. We will then review data that challenge the view that inflammation is solely detrimental, and instead suggest that both suppression and activation of the innate immune system and neuroinflammation may be beneficial, depending on the stage of the disorder. Finally, we will consider several new therapeutic strategies for regulating neuroinflammation, including the immune-modulatory cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF), inhibitors of apolipoprotein E (apoE), and microglial depletion via drugs that target the colony-stimulating factor-1 receptor (CSF1R).
Neuroinflammatory Biomarkers in CSF and Plasma from AD and DS
Studies have found increased cerebrospinal fluid (CSF) levels of immune biomarkers in mild cognitive impairment (MCI) and AD patients (Tarkowski et al., 2003; Galimberti et al., 2006; Jesse et al., 2009; Buchhave et al., 2010; Westin et al., 2012; Kauwe et al., 2014; Counts et al., 2017; Whelan et al., 2019). Peripheral immune cells, such as neutrophils, monocytes, and lymphocytes, also produce (and respond to) inflammatory cytokines, which are significantly upregulated in the blood of AD patients and may also be derived from the CNS (Kim et al., 2008; Diniz et al., 2010; Morgan et al., 2019). Similar increases in blood levels of immune biomarkers are found in people with DS (Petersen and O’Bryant, 2019; Huggard et al., 2020a,b). One inflammation-based hypothesis is that people with DS are in a state of chronic abnormal inflammation, including features of auto-inflammation, across the lifespan that influences all phenotypes and disease risk. Specifically, proteomic analyses of plasma and brain tissue from people with DS revealed dysregulation of inflammatory protein expression, including increases in several pro-inflammatory cytokines, and decreases in numerous complement cascade components (Sullivan et al., 2016, 2017; Zhang et al., 2017). Another study revealed elevated levels of both pro-inflammatory and anti-inflammatory cytokines in plasma from children with DS (Huggard et al., 2020a,b). Activation of astrocytes and microglia, the secretion of inflammatory cytokines (e.g., IL-1, IL-6, and TNFα), and acute phase proteins are observed in both the brains and blood of people with DS, indicating an “inflammatory endophenotype” (Petersen and O’Bryant, 2019). In a cross-sectional analysis of people with DS, plasma glial fibrillary acidic protein (GFAP), a marker of astrogliosis, was found to increase starting in their mid-40s (Hendrix et al., 2021).
Timing and Effects of Innate Immune System Activation and Neuroinflammation in DS and AD
Epidemiological studies suggest that elevated immune biomarkers in the blood may be evident years prior to the manifestation of clinical symptoms of AD or AD-related dementias (ADRDs) in the typical population (Schmidt et al., 2002; Ridolfi et al., 2013; Leszek et al., 2016; Busse et al., 2017; Wendeln et al., 2018). Higher plasma levels of GFAP are correlated with lower measures of episodic memory and microstructural integrity in AD, MCI, and also in healthy aged donors (Bettcher et al., 2021). As discussed, people with DS also have increasing levels of plasma GFAP starting in their mid-40s (Hendrix et al., 2021). Thus, although initially thought to be a secondary effect of aberrant protein accumulation, changes in the innate immune system and neuroinflammation are now thought to be a core, early feature of both DS and AD that interface with, and may contribute to, clinical manifestations of cognitive disorders and decline (Lucin and Wyss-Coray, 2009; Heneka et al., 2015).
Hsa21 harbors numerous innate immune system and neuroinflammation-associated genes that are therefore triplicated in most people with DS (Table 1). Notably, four genes encoding interferon receptors reside on Hsa21, and interferon-related signaling is upregulated in people with DS (Sullivan et al., 2016; Araya et al., 2019; Powers et al., 2019). The inflammatory response microRNA miR-155 also resides on Hsa21 and is overexpressed in DS (Guo et al., 2019). Proteomic analyses have revealed a striking increase in both pro- and anti-inflammatory cytokines in plasma and brain tissue samples from people of all ages with DS (Sullivan et al., 2017; Zhang et al., 2017; Flores-Aguilar et al., 2020; Huggard et al., 2020b) and in mouse models of DS (Ahmed et al., 2012, 2013; Spellman et al., 2013; Block et al., 2015), which express homologs of Hsa21-encoded inflammation-related genes (Table 1). Together, these findings have led to the hypothesis that DS inherently results in chronic neuroinflammation, including auto-inflammation and astrogliosis (Sullivan et al., 2016; Rachubinski et al., 2019; Snyder et al., 2020). Although non-steroidal anti-inflammatory drugs (NSAIDs) were not therapeutic for AD, it is possible that more selective blockers of the innate immune system might be effective in AD and/or DS, for example, through inhibition of the TLR2-MyD88 interaction or of JAK-1 (Rangasamy et al., 2018; Rachubinski et al., 2019; Tuttle et al., 2020).

Table 1. A summary of inflammation-related genes located on HSA21.
Apolipoprotein E in Inflammation and Neurodegeneration in DS and AD
Inheritance of the APOE ε4 allele (APOE4) is the strongest risk factor for AD, besides age, with one copy of APOE4 leading to a three-fold increased risk of AD and two copies leading to a 15-fold increased risk of AD (Corder et al., 1993; Strittmatter et al., 1993; Strittmatter and Roses, 1995). Inheritance of the APOE4 allele also significantly increases the risk for cognitive decline and dementia in middle-aged people with DS (Rubinsztein et al., 1999; Deb et al., 2000), and it increases the risk of mortality by five-fold compared to non-APOE4 carriers (Zigman et al., 2005). Furthermore, brain APOE expression is significantly upregulated in people with DS compared to the typical population (Lockstone et al., 2007), which might exacerbate DS-associated AD.
Similar to the inflammation/acute-phase protein ACT, apoE is also associated with amyloid deposits (Wisniewski and Frangione, 1992; Wisniewski et al., 1993; Ma et al., 1994; Sanan et al., 1994; Wisniewski et al., 1994). Although not typically considered a neuroinflammatory molecule, APOE expression is upregulated by astrocytes and microglia early in the AD pathological process (Keren-Shaul et al., 2017; Krasemann et al., 2017; Kang et al., 2018; Rangaraju et al., 2018), and it also plays a pivotal role in modulating the neuroinflammatory cascade by both Aβ-dependent and Aβ-independent pathways (McGeer et al., 1997; Maezawa et al., 2006; Zhu et al., 2012; Cudaback et al., 2015; Shi et al., 2017; Lin et al., 2018).
Increasing evidence shows that apoE plays a role in amyloid formation by promoting the aggregation of the Aβ peptide to form insoluble filaments, thereby also inhibiting the clearance of Aβ from the brain (Potter and Wisniewski, 2012; Figure 1). For example, apoE4 catalyzes the formation of neurotoxic Aβ oligomers and fibrils (Ma et al., 1994, 1996; Sanan et al., 1994; Wisniewski et al., 1994; Castano et al., 1995; Golabek et al., 1996; Soto et al., 1996; Manelli et al., 2007; Cerf et al., 2011; Hashimoto et al., 2012; Koffie et al., 2012; Liu et al., 2017). Fortunately, this mechanistic pathway lends itself to therapeutic approaches targeting apoE using antibodies, antisense oligonucleotides, or structural correctors (Brodbeck et al., 2011; Chen et al., 2012; Liao et al., 2014; Huynh et al., 2017; Wang et al., 2018; Xiong et al., 2021), or by targeting the interaction between apoE and Aβ using peptide blockers or small molecule drugs (Ma et al., 1996; Sadowski et al., 2004; Pankiewicz et al., 2014; Johnson et al., 2021a). Recently, high-throughput screens from our lab have identified several Food and Drug Administration (FDA)-approved drugs, including the anti-depressant drug imipramine and the anti-psychotic drug olanzapine, that inhibit the apoE-Aβ interaction and appear to improve cognition in AD patients, and especially in APOE4 carriers, in our retrospective analyses of human clinical data (Johnson et al., 2021a).
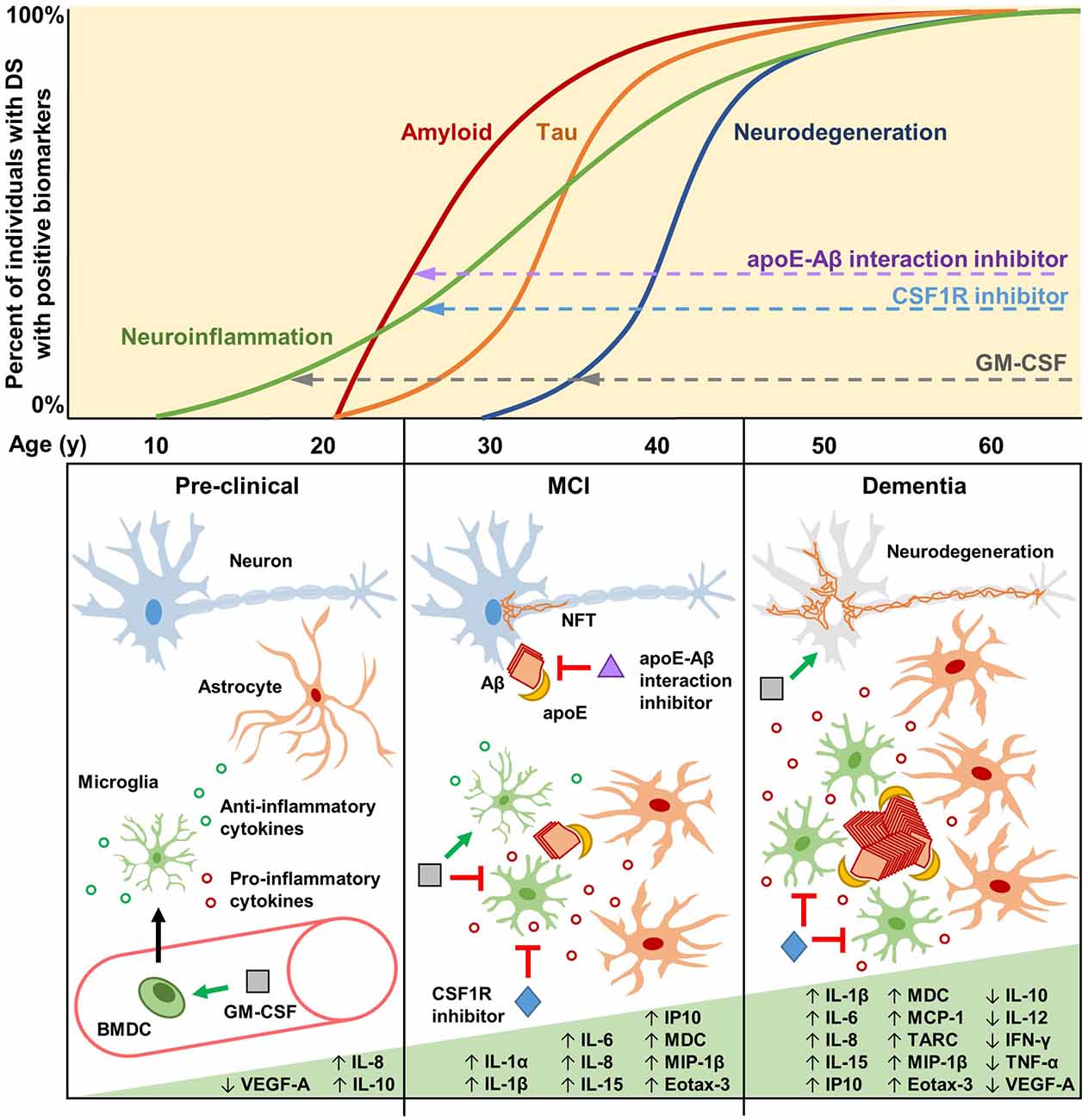
Figure 1. Biomarkers, cytokines, and potential therapeutic mechanisms for neuroinflammation in Down syndrome (DS) and Alzheimer’s disease (AD). Biomarkers of neuroinflammation, amyloid and tau pathology, and neurodegeneration increase over the lifetimes of the vast majority of individuals with DS. Neuroinflammatory biomarkers such as GFAP may begin increasing as early as the late teenage years and progressively increase throughout life (Hendrix et al., 2021). Changes in amyloid biomarkers are detectable after 20 years of age, with APOE genotype significantly contributing to risk (Rubinsztein et al., 1999; Deb et al., 2000), likely due to apoE-catalyzed amyloid-β (Aβ) polymerization during the early seeding stages of amyloid formation (Potter and Wisniewski, 2012). Tau pathology is detectable soon after amyloid forms, but it accumulates more slowly. By the age of 40, every individual with DS has the hallmark amyloid and tau neuropathology of AD. Biomarkers of neurodegeneration, such as changes in brain volume and glucose metabolism, are identifiable after 30 years of age, proceeding and generally correlating well with tau pathology. During the pre-clinical stage, GM-CSF may have therapeutic potential by modulating the immune/neuroinflammatory cascade in order to prevent and/or delay amyloidogenesis. GM-CSF stimulates bone marrow-derived cells (BMDCs) to mobilize and extravasate to the brain, or GM-CSF may enter the brain directly, where the resulting activated microglia modulate their cytokine expression and reactivity (Bhattacharya et al., 2015a; Abe et al., 2020). During the mild cognitive impairment (MCI) stage, preceding clinical dementia, Aβ aggregates begin to form, catalyzed by apoE, which then induces intraneuronal tau hyperphosphorylation and neurofibrillary tangle (NFT) formation. Therapeutic molecules that can inhibit the apoE-Aβ interaction (e.g., Sadowski et al., 2004; Pankiewicz et al., 2014; Johnson et al., 2021a) may significantly reduce amyloid deposition, thus preventing/reducing downstream pathologies. Activated microglia and astrocytes, characterized by increased soma size and shortened processes, cluster around amyloid plaques and increase the expression of interleukins and other pro-inflammatory cytokines. At this stage, GM-CSF may act to maintain microglia in a non-activated state, promote anti-inflammatory cytokine expression, and reduce pro-inflammatory cytokine expression (Ahmed et al., 2021; Potter et al., 2021). CSF1R inhibitors may also be beneficial at this stage to reduce the numbers of activated microglia while maintaining quiescent microglia that play important roles in immune surveillance and brain homeostasis (Johnson et al., 2021b). The clinical dementia stage is characterized by neurodegeneration due to widespread NFT formation and chronic neuroinflammation that persists through the mid-40s, 50s, and 60s, during which a majority of individuals with DS have clinical dementia. Some inflammatory markers are reported to decrease during this stage, possibly due to cellular exhaustion and degeneration (Flores-Aguilar et al., 2020). GM-CSF may therapeutically modify neurodegeneration via neuroprotective effects, while CSF1R inhibitors may reduce the numbers and effects of chronically activated microglia.
GM-CSF as a Neuroinflammatory Modulator in AD
Patients with rheumatoid arthritis (RA) have a three- to eight-fold reduced risk of developing AD, suggesting a potential role for inflammation and the innate immune system in AD (McGeer et al., 1996). Although the reduced AD risk in patients with RA was initially attributed to their frequent use of NSAIDs (McGeer et al., 1996), NSAID treatment showed no benefit in clinical trials of either AD or MCI patients (McGeer et al., 2006; ADAPT_FS_Research_Group, 2015).
As an alternative, we hypothesized that intrinsic factors associated with RA pathogenesis itself may underlie its AD protective effect(s). We identified GM-CSF as one such factor that is upregulated in the blood of RA patients and found that subcutaneous injection of GM-CSF for 20 days increased microglial activation, reduced amyloid pathology by more than 50%, and completely reversed the cognitive impairment of transgenic AD mice (Boyd et al., 2010), which has been replicated by other groups (Castellano et al., 2017; Kiyota et al., 2018). Treatment with recombinant human GM-CSF (sargramostim/Leukine®) was also associated with improved cognition in cancer patients undergoing hematopoietic stem cell transplantation (Jim et al., 2012). These findings led us to design and carry out a placebo-controlled, randomized, double-blind Phase II clinical trial in mild-to-moderate AD participants, which showed that subcutaneous injection of sargramostim (5 days/week for 3 weeks) was safe, associated with reduced plasma biomarkers of neuronal damage/neurodegeneration (i.e., total tau and UCH-L1), and improved cognition based on Mini-Mental State Examination (MMSE) scores (Potter et al., 2021). These findings suggest that during the early stages of AD, activation of the innate immune system may be beneficial, which has now led to a longer–term trial.
GM-CSF as an Amyloid-Independent Cognition Enhancer in DS and Normal Aging
People with DS have significant ID throughout their 60-year life expectancy (Bittles et al., 2007), with no treatments available. Several drugs have been tested in mouse models of DS, primarily in Ts65Dn mice, with some promising results showing the rescue of DS-related cognitive deficits. Unfortunately, no such drugs have shown significant benefits in clinical trials of people with DS (reviewed in Gardiner, 2015; Vacca et al., 2019).
Because GM-CSF treatment improved cognition and reduced amyloid in mouse models of AD, we investigated its effects in the Dp16 mouse model of DS. Our findings show that in both Dp16 mice and their wild-type (WT) littermates, GM-CSF treatment improves learning/memory in the radial arm water maze, a hippocampal-based task. In Dp16 mice, GM-CSF treatment also ameliorates the abnormal astrocyte morphology and aggregation and partially normalizes the levels of interneurons (Ahmed et al., 2021). Although GM-CSF treatment evidently improves learning/memory in mouse models of AD by removing amyloid plaques in the brain, it is noteworthy that WT mice and mouse models of DS, including Dp16 mice, do not develop AD amyloid pathology at any age. Therefore, GM-CSF treatment must lead to improved learning/memory in WT and Dp16 mice via an amyloid-independent mechanism(s), likely related to its pro-inflammatory or inflammation-modulating activity.
Despite its known pro-inflammatory properties, GM-CSF also affects multiple CNS processes that are consistent with, and provide some insights into, its unexpected beneficial effects on learning/memory in a mouse model of DS. Specifically, GM-CSF promotes recovery from neuronal damage or dysfunction in animal models of stroke (Schneider et al., 2007; Schäbitz et al., 2008; Kong et al., 2009; Theoret et al., 2016), traumatic brain injury (Shultz et al., 2014; Kelso et al., 2015), and acute retinal ganglion cell injury (Schallenberg et al., 2012; Legacy et al., 2013). GM-CSF can cross the blood-brain barrier (McLay et al., 1997) and is also produced within the brain, where numerous cell types express the GM-CSF receptor, including neurons, oligodendrocytes, microglia, astroglia, and endothelial cells, which would allow for both paracrine and autocrine signaling (Baldwin et al., 1993; Sawada et al., 1993).
Many studies have shown that both people with DS and typical aging adults exhibit an auto-inflammatory or “inflammaging” syndrome (Trollor et al., 2012; Frasca and Blomberg, 2016; Ashraf-Ganjouei et al., 2020; Serre-Miranda et al., 2020) that might predict that GM-CSF treatment would be detrimental. However, GM-CSF is not merely a pro-inflammatory molecule. A more accurate description is that GM-CSF modulates the innate immune system, especially in the setting of immune system dysregulation in the periphery and in the brain (Boyd et al., 2010; Bhattacharya et al., 2015a,b; Borriello et al., 2019). Indeed, GM-CSF treatment not only increases the levels of many cellular and cytokine biomarkers of inflammation in the blood of AD patients (e.g., neutrophils, monocytes, lymphocytes, IL-2, IL-6, and TNFα), but also reduces the levels of the inflammatory cytokine IL-8 and increases the levels of the typically anti-inflammatory cytokine IL-10 (Potter et al., 2021). Thus, GM-CSF has a much more complex physiological effect than simply being pro-inflammatory. Furthermore, suppressing the inflammation associated with DS in the periphery may be beneficial in the setting of certain acute disorders (Rachubinski et al., 2019). Thus, growing evidence highlights the complexity of the innate immune system in the context of inflammation in people with DS.
Notably, the fact that we observed improved memory in WT aging mice treated with GM-CSF (Boyd et al., 2010; Ahmed et al., 2021) suggests that GM-CSF has a cognition/memory enhancing activity that is independent of disease and suggests that modulation of the innate immune system may help prevent normal age-related memory decline. In contrast to previous expectations that inhibiting inflammation and the innate immune system would be the most effective therapy for co-morbidities of DS, the beneficial effects of GM-CSF on learning and memory may reflect its stimulating pro-inflammatory activity, its other physiological/cellular effects, or both (Figure 1).
Therapeutic Modulation of Microglial Numbers and Activation
It is becoming increasingly clear that microglia play multiple, and often disparate, roles at different stages of the innate immune and neuroinflammatory responses in DS and in AD. Accordingly, microglial reduction/depletion has also been investigated as a therapeutic approach to DS, AD, and other neurodegenerative diseases. For example, small molecule drugs targeting CSF1R, which is crucial for microglial proliferation and survival, have been repurposed from cancer indications (Cannarile et al., 2017) and used to modulate microglial levels in the CNS (Figure 1). Indeed, microglial reduction via CSF1R inhibition was found to rescue several cognitive deficits in Dp16 mice (Pinto et al., 2020). In AD mouse models, microglial depletion prior to amyloid deposition was shown to be critical for therapeutic efficacy (Sosna et al., 2018; Spangenberg et al., 2019; Son et al., 2020). Likewise, in mouse models of primary tauopathy, characterized by inclusions of the protein tau in neural cells (Kovacs, 2015), CSF1R inhibitors reduced pathological tau aggregation and subsequent neurodegeneration (Mancuso et al., 2019; Shi et al., 2019). However, recent evidence suggests that complete microglial depletion is neither necessary nor desirable for extending lifespan in tauopathy mice and that microglia resilient to CSF1R inhibition exist in a quiescent, non-activated state and may serve important roles in prevention and recovery from tau-induced neurodegeneration (Johnson et al., 2021b). Together, these studies underscore the importance of carefully considering microglial state and function over the disease course in order to appropriately balance microglial stimulation and repression therapeutically.
Conclusion
Collectively, the recent data indicate that targeted enhancement or inhibition of the innate immune system and inflammatory cytokines can effectively treat DS, AD, and normal aging. These findings provide compelling evidence that the long-held belief that inflammation and innate immune system activation primarily play negative roles in DS, AD, and normal aging must be reassessed. Although GM-CSF is the first modulatory cytokine to exhibit therapeutic potential in inflammation-associated disorders, it serves as a proof-of-principle, and other therapeutic molecules could use a similar approach. Furthermore, combination therapies that pair GM-CSF with small molecule inhibitors of apoE or CSF1R at appropriate neuroinflammatory stages may be particularly effective for treating people with DS and/or AD.
Author Contributions
All authors contributed to the design and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research support was provided by NIH grant R01 AG037942-01A1, the State of Colorado, the University of Colorado School of Medicine, the Linda Crnic Institute for Down Syndrome, Don and Sue Fisher, the Hewitt Family Foundation, Marcy and Bruce Benson, the Global Down Syndrome Foundation, and other generous philanthropists.
Conflict of Interest
HP and TB are two of the inventors on several U.S. patents owned by the University of South Florida, but not licensed. TB is an employee of Partner Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe, N., Nishihara, T., Yorozuya, T., and Tanaka, J. (2020). Microglia and macrophages in the pathological central and peripheral nervous systems. Cells 9:2132. doi: 10.3390/cells9092132
Abraham, C. R., Selkoe, D. J., and Potter, H. (1988). Immunochemical identification of the serine protease inhibitor α 1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell 52, 487–501. doi: 10.1016/0092-8674(88)90462-x
ADAPT_FS_Research_Group. (2015). Follow-up evaluation of cognitive function in the randomized Alzheimer’s disease anti-inflammatory prevention trial and its follow-up study. Alzheimers Dement. 11, 216.e1–225.e1. doi: 10.1016/j.jalz.2014.03.009
Ahmed, M. M., Dhanasekaran, A. R., Tong, S., Wiseman, F. K., Fisher, E. M., Tybulewicz, V. L., et al. (2013). Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum. Mol. Genet. 22, 1709–1724. doi: 10.1093/hmg/ddt017
Ahmed, M. M., Sturgeon, X., Ellison, M., Davisson, M. T., and Gardiner, K. J. (2012). Loss of correlations among proteins in brains of the Ts65Dn mouse model of down syndrome. J. Proteome Res. 11, 1251–1263. doi: 10.1021/pr2011582
Ahmed, M. M., Wang, A. C.-J., Elos, M., Chial, H. J., Sillau, S., Solano, D. A., et al. (2021). Pro-inflammatory cytokine GM-CSF improves learning/memory and brain pathology in Dp16 down syndrome mice and improves learning/memory in wild-type mice. bioRxiv [Preprint]. doi: 10.1101/2021.07.04.451073
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/s0197-4580(00)00124-x
Alzheimer, A. (1907). Uber eine eigenartige Erkrankung der Hirnride. Allg. Z. Psychiatr. Psych. Gerichtl. 64, 146–148.
Araya, P., Waugh, K. A., Sullivan, K. D., Núñez, N. G., Roselli, E., Smith, K. P., et al. (2019). Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. U S A 116, 24231–24241. doi: 10.1073/pnas.1908129116
Ashraf-Ganjouei, A., Moradi, K., Bagheri, S., and Aarabi, M. H. (2020). The association between systemic inflammation and cognitive performance in healthy adults. J. Neuroimmunol. 345:577272. doi: 10.1016/j.jneuroim.2020.577272
Baldwin, G. C., Benveniste, E. N., Chung, G. Y., Gasson, J. C., and Golde, D. W. (1993). Identification and characterization of a high-affinity granulocyte-macrophage colony-stimulating factor receptor on primary rat oligodendrocytes. Blood 82, 3279–3282. doi: 10.1182/blood.v82.11.3279.bloodjournal82113279
Barger, S. W., and Harmon, A. D. (1997). Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881. doi: 10.1038/42257
Barroeta-Espar, I., Weinstock, L. D., Perez-Nievas, B. G., Meltzer, A. C., Siao Tick Chong, M., Amaral, A. C., et al. (2019). Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 121, 327–337. doi: 10.1016/j.nbd.2018.10.009
Beecham, G. W., Hamilton, K., Naj, A. C., Martin, E. R., Huentelman, M., Myers, A. J., et al. (2014). Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 10:e1004606. doi: 10.1371/journal.pgen.1004606
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bettcher, B. M., Johnson, S. C., Fitch, R., Casaletto, K. B., Heffernan, K. S., Asthana, S., et al. (2018). Cerebrospinal fluid and plasma levels of inflammation differentially relate to CNS markers of Alzheimer’s disease pathology and neuronal damage. J. Alzheimers Dis. 62, 385–397. doi: 10.3233/JAD-170602
Bettcher, B. M., Olson, K. E., Carlson, N. E., McConnell, B. V., Boyd, T., Adame, V., et al. (2021). Astrogliosis and episodic memory in late life: higher GFAP is related to worse memory and white matter microstructure in healthy aging and Alzheimer’s disease. Neurobiol. Aging 103, 68–77. doi: 10.1016/j.neurobiolaging.2021.02.012
Beyer, K., Lao, J. I., Carrato, C., Rodriguez-Vila, A., Latorre, P., Mataro, M., et al. (2004). Cystathionine β synthase as a risk factor for Alzheimer disease. Curr. Alzheimer Res. 1, 127–133. doi: 10.2174/1567205043332243
Bhattacharya, P., Budnick, I., Singh, M., Thiruppathi, M., Alharshawi, K., Elshabrawy, H., et al. (2015a). Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: implications for immune therapy. J. Interferon Cytokine Res. 35, 585–599. doi: 10.1089/jir.2014.0149
Bhattacharya, P., Thiruppathi, M., Elshabrawy, H. A., Alharshawi, K., Kumar, P., and Prabhakar, B. S. (2015b). GM-CSF: an immune modulatory cytokine that can suppress autoimmunity. Cytokine 75, 261–271. doi: 10.1016/j.cyto.2015.05.030
Bittles, A. H., Bower, C., Hussain, R., and Glasson, E. J. (2007). The four ages of Down syndrome. Eur. J. Public Health 17, 221–225. doi: 10.1093/eurpub/ckl103
Block, A., Ahmed, M. M., Dhanasekaran, A. R., Tong, S., and Gardiner, K. J. (2015). Sex differences in protein expression in the mouse brain and their perturbations in a model of Down syndrome. Biol. Sex Differ. 6:24. doi: 10.1186/s13293-015-0043-9
Borriello, F., Galdiero, M. R., Varricchi, G., Loffredo, S., Spadaro, G., and Marone, G. (2019). Innate immune modulation by GM-CSF and IL-3 in health and disease. Int. J. Mol. Sci. 20:834. doi: 10.3390/ijms20040834
Boselli, D., Ragimbeau, J., Orlando, L., Cappello, P., Capello, M., Ambrogio, C., et al. (2010). Expression of IFNγR2 mutated in a dileucine internalization motif reinstates IFNγ signaling and apoptosis in human T lymphocytes. Immunol. Lett. 134, 17–25. doi: 10.1016/j.imlet.2010.08.005
Boyd, T. D., Bennett, S. P., Mori, T., Governatori, N., Runfeldt, M., Norden, M., et al. (2010). GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J. Alzheimers Dis. 21, 507–518. doi: 10.3233/JAD-2010-091471
Brodbeck, J., McGuire, J., Liu, Z., Meyer-Franke, A., Balestra, M. E., Jeong, D. E., et al. (2011). Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 286, 17217–17226. doi: 10.1074/jbc.M110.217380
Buchhave, P., Zetterberg, H., Blennow, K., Minthon, L., Janciauskiene, S., and Hansson, O. (2010). Soluble TNF receptors are associated with Aβ metabolism and conversion to dementia in subjects with mild cognitive impairment. Neurobiol. Aging 31, 1877–1884. doi: 10.1016/j.neurobiolaging.2008.10.012
Busse, M., Michler, E., von Hoff, F., Dobrowolny, H., Hartig, R., Frodl, T., et al. (2017). Alterations in the peripheral immune system in dementia. J. Alzheimers Dis. 58, 1303–1313. doi: 10.3233/JAD-161304
Cannarile, M. A., Weisser, M., Jacob, W., Jegg, A. M., Ries, C. H., and Ruttinger, D. (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5:53. doi: 10.1186/s40425-017-0257-y
Castano, E. M., Prelli, F., Wisniewski, T., Golabek, A., Kumar, R. A., Soto, C., et al. (1995). Fibrillogenesis in Alzheimer’s disease of amyloid β peptides and apolipoprotein E. Biochem. J. 306, 599–604. doi: 10.1042/bj3060599
Castellano, J. M., Mosher, K. I., Abbey, R. J., McBride, A. A., James, M. L., Berdnik, D., et al. (2017). Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492. doi: 10.1038/nature22067
Centers for Disease Control and Prevention (CDC). (2006). Improved national prevalence estimates for 18 selected major birth defects—United States, 1999–2001. MMWR Morb. Mortal. Wkly. Rep. 54, 1301–1305.
Cerf, E., Gustot, A., Goormaghtigh, E., Ruysschaert, J. M., and Raussens, V. (2011). High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 25, 1585–1595. doi: 10.1096/fj.10-175976
Chapman, R. S., and Hesketh, L. J. (2000). Behavioral phenotype of individuals with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 6, 84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P
Chen, H.-K., Liu, Z., Meyer-Franke, A., Brodbeck, J., Miranda, R. D., McGuire, J. G., et al. (2012). Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 287, 5253–5266. doi: 10.1074/jbc.M111.276162
Colton, C., and Wilcock, D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. doi: 10.2174/187152710791012053
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Counts, S. E., Ikonomovic, M. D., Mercado, N., Vega, I. E., and Mufson, E. J. (2017). Biomarkers for the early detection and progression of Alzheimer’s disease. Neurotherapeutics 14, 35–53. doi: 10.1007/s13311-016-0481-z
Cribbs, D. H., Berchtold, N. C., Perreau, V., Coleman, P. D., Rogers, J., Tenner, A. J., et al. (2012). Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflammation 9:179. doi: 10.1186/1742-2094-9-179
Cudaback, E., Yang, Y., Montine, T. J., and Keene, C. D. (2015). APOE genotype-dependent modulation of astrocyte chemokine CCL3 production. Glia 63, 51–65. doi: 10.1002/glia.22732
Danciger, E., Dafni, N., Bernstein, Y., Laver-Rudich, Z., Neer, A., and Groner, Y. (1986). Human Cu/Zn superoxide dismutase gene family: molecular structure and characterization of four Cu/Zn superoxide dismutase-related pseudogenes. Proc. Natl. Acad. Sci. U S A 83, 3619–3623. doi: 10.1073/pnas.83.11.3619
Deb, S., Braganza, J., Norton, N., Williams, H., Kehoe, P. G., Williams, J., et al. (2000). APOE epsilon 4 influences the manifestation of Alzheimer’s disease in adults with Down’s syndrome. Br. J. Psychiatry 176, 468–472. doi: 10.1192/bjp.176.5.468
Diniz, B. S., Teixeira, A. L., Ojopi, E. B., Talib, L. L., Mendonca, V. A., Gattaz, W. F., et al. (2010). Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 22, 1305–1311. doi: 10.3233/JAD-2010-100921
Donato, R., Cannon, B. R., Sorci, G., Riuzzi, F., Hsu, K., Weber, D. J., et al. (2013). Functions of S100 proteins. Curr. Mol. Med. 13, 24–57. doi: 10.2174/156652413804486214
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., et al. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438. doi: 10.1038/nm1555
Epstein, C. J. (1990). The consequences of chromosome imbalance. Am. J. Med. Genet. Suppl. 7, 31–37.
Flores-Aguilar, L., Iulita, M. F., Kovecses, O., Torres, M. D., Levi, S. M., Zhang, Y., et al. (2020). Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 143, 3653–3671. doi: 10.1093/brain/awaa326
Frasca, D., and Blomberg, B. B. (2016). Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17, 7–19. doi: 10.1007/s10522-015-9578-8
Galimberti, D., Fenoglio, C., Lovati, C., Venturelli, E., Guidi, I., Corra, B., et al. (2006). Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 27, 1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007
Ganesh, L., Yoshimoto, T., Moorthy, N. C., Akahata, W., Boehm, M., Nabel, E. G., et al. (2006). Protein methyltransferase 2 inhibits NF-kappaB function and promotes apoptosis. Mol. Cell. Biol. 26, 3864–3874. doi: 10.1128/MCB.26.10.3864-3874.2006
Gardiner, K. J. (2015). Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations. Drug Des. Devel. Ther. 9, 103–125. doi: 10.2147/DDDT.S51476
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291x(84)91209-9
Golabek, A. A., Soto, C., Vogel, T., and Wisniewski, T. (1996). The interaction between apolipoprotein E and Alzheimer’s amyloid β-peptide is dependent on β-peptide conformation. J. Biol. Chem. 271, 10602–10606. doi: 10.1074/jbc.271.18.10602
Guo, Y., Hong, W., Wang, X., Zhang, P., Körner, H., Tu, J., et al. (2019). MicroRNAs in microglia: how do MicroRNAs affect activation, inflammation, polarization of microglia and mediate the interaction between microglia and glioma? Front. Mol. Neurosci. 12:125. doi: 10.3389/fnmol.2019.00125
Guyant-Maréchal, L., Rovelet-Lecrux, A., Goumidi, L., Cousin, E., Hannequin, D., Raux, G., et al. (2007). Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology 68, 684–687. doi: 10.1212/01.wnl.0000255938.33739.46
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Head, E., Lott, I. T., Wilcock, D. M., and Lemere, C. A. (2016). Aging in down syndrome and the development of Alzheimer’s disease neuropathology. Curr. Alzheimer Res. 13, 18–29. doi: 10.2174/1567205012666151020114607
Hendrix, J. A., Airey, D. C., Britton, A., Burke, A. D., Capone, G. T., Chavez, R., et al. (2021). Cross-sectional exploration of plasma biomarkers of Alzheimer’s disease in down syndrome: early data from the longitudinal investigation for enhancing down syndrome research (LIFE-DSR) study. J. Clin. Med. 10:1907. doi: 10.3390/jcm10091907
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Huggard, D., Doherty, D. G., and Molloy, E. J. (2020a). Immune dysregulation in children with down syndrome. Front. Pediatr. 8:73. doi: 10.3389/fped.2020.00073
Huggard, D., Kelly, L., Ryan, E., McGrane, F., Lagan, N., Roche, E., et al. (2020b). Increased systemic inflammation in children with Down syndrome. Cytokine 127:154938. doi: 10.1016/j.cyto.2019.154938
Huynh, T.-P. V., Liao, F., Francis, C. M., Robinson, G. O., Serrano, J. R., Jiang, H., et al. (2017). Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron 96, P1013–1023.E4. doi: 10.1016/j.neuron.2017.11.014
Irving, C., Basu, A., Richmond, S., Burn, J., and Wren, C. (2008). Twenty-year trends in prevalence and survival of Down syndrome. Eur. J. Hum. Genet. 16, 1336–1340. doi: 10.1038/ejhg.2008.122
Jesse, S., Steinacker, P., Cepek, L., von Arnim, C. A., Tumani, H., Lehnert, S., et al. (2009). Glial fibrillary acidic protein and protein S-100B: different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. J. Alzheimers Dis. 17, 541–551. doi: 10.3233/JAD-2009-1075
Jim, H. S., Boyd, T. D., Booth-Jones, M., Pidala, J., and Potter, H. (2012). Granulocyte macrophage colony stimulating factor treatment is associated with improved cognition in cancer patients. Brain Disord. Ther. 1:1000101. doi: 10.4172/bdt.1000101
Johnson, N. R., Wang, A. C.-J., Coughlan, C., Sillau, S., Lucero, E., Viltz, L., et al. (2021a). Imipramine and olanzapine block apoE4-catalyzed polymerization of Aβ and show evidence of improving Alzheimer’s disease cognition. bioRxiv [Preprint]. doi: 10.1101/2021.03.28.437389
Johnson, N. R., Yuan, P., Lopez, T. P., Yue, W., Bond, A., Rivera, B. M., et al. (2021b). Sex-specific life extension in tauopathy mice by CSF1R inhibition causing selective microglial depletion and suppressed pathogenesis. bioRxiv [Preprint]. doi: 10.1101/2021.03.20.436288
Kang, S. S., Ebbert, M. T. W., Baker, K. E., Cook, C., Wang, X., Sens, J. P., et al. (2018). Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 215, 2235–2245. doi: 10.1084/jem.20180653
Kauwe, J. S., Bailey, M. H., Ridge, P. G., Perry, R., Wadsworth, M. E., Hoyt, K. L., et al. (2014). Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 10:e1004758. doi: 10.1371/journal.pgen.1004758
Kelso, M. L., Elliott, B. R., Haverland, N. A., Mosley, R. L., and Gendelman, H. E. (2015). Granulocyte-macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J. Neuroimmunol. 278, 162–173. doi: 10.1016/j.jneuroim.2014.11.002
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e7–1290.e7. doi: 10.1016/j.jneuroim.2014.11.002
Kim, S. H., Cohen, B., Novick, D., and Rubinstein, M. (1997). Mammalian type I interferon receptors consists of two subunits: IFNaR1 and IFNaR2. Gene 196, 279–286. doi: 10.1016/s0378-1119(97)00240-0
Kim, T.-S., Lim, H.-K., Lee, J. Y., Kim, D.-J., Park, S., Lee, C., et al. (2008). Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 436, 196–200. doi: 10.1016/j.neulet.2008.03.019
Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., et al. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23. doi: 10.1093/hmg/ddl437
Kiyota, T., Machhi, J., Lu, Y., Dyavarshetty, B., Nemati, M., Yokoyama, I., et al. (2018). Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J. Neuroimmunol. 319, 80–92. doi: 10.1016/j.jneuroim.2018.03.009
Koffie, R. M., Hashimoto, T., Tai, H.-C., Kay, K. R., Serrano-Pozo, A., Joyner, D., et al. (2012). Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 135, 2155–2168. doi: 10.1093/brain/aws127
Kong, T., Choi, J. K., Park, H., Choi, B. H., Snyder, B. J., Bukhari, S., et al. (2009). Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats. Laboratory investigation. J. Neurosurg. 111, 155–163. doi: 10.3171/2008.12.JNS08172
Kovacs, G. G. (2015). Invited review: neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23. doi: 10.1111/nan.12208
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, P566–581.E9. doi: 10.1016/j.immuni.2017.08.008
Kuhn, P.-H., Marjaux, E., Imhof, A., De Strooper, B., Haass, C., and Lichtenthaler, S. F. (2007). Regulated intramembrane proteolysis of the interleukin-1 receptor II by α-, β-, and γ-secretase. J. Biol. Chem. 282, 11982–11995. doi: 10.1074/jbc.M700356200
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Kuno, K., Kanada, N., Nakashima, E., Fujiki, F., Ichimura, F., and Matsushima, K. (1997). Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J. Biol. Chem. 272, 556–562. doi: 10.1074/jbc.272.1.556
Kurt, S., Tomatir, A. G., Tokgun, P. E., and Oncel, C. (2020). Altered expression of long non-coding RNAs in peripheral blood mononuclear cells of patients with Alzheimer’s disease. Mol. Neurobiol. 57, 5352–5361. doi: 10.1007/s12035-020-02106-x
Lambert, J.-C., Ferreira, S., Gussekloo, J., Christiansen, L., Brysbaert, G., Slagboom, E., et al. (2007). Evidence for the association of the S100β gene with low cognitive performance and dementia in the elderly. Mol. Psychiatry 12, 870–880. doi: 10.1038/sj.mp.4001974
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Latour, A., Gu, Y., Kassis, N., Daubigney, F., Colin, C., Gausseres, B., et al. (2019). LPS-induced inflammation abolishes the effect of DYRK1A on IkB stability in the brain of mice. Mol. Neurobiol. 56, 963–975. doi: 10.1007/s12035-018-1113-x
Lee, J. W., Kang, H. S., Lee, J. Y., Lee, E. J., Rhim, H., Yoon, J. H., et al. (2012). The transcription factor STAT2 enhances proteasomal degradation of RCAN1 through the ubiquitin E3 ligase FBW7. Biochem. Biophys. Res. Commun. 420, 404–410. doi: 10.1016/j.bbrc.2012.03.007
Legacy, J., Hanea, S., Theoret, J., and Smith, P. D. (2013). Granulocyte macrophage colony-stimulating factor promotes regeneration of retinal ganglion cells in vitro through a mammalian target of rapamycin-dependent mechanism. J. Neurosci. Res. 91, 771–779. doi: 10.1002/jnr.23205
Leszek, J., Barreto, G. E., Gasiorowski, K., Koutsouraki, E., Avila-Rodrigues, M., and Aliev, G. (2016). Inflammatory mechanisms and oxidative stress as key factors responsible for progression of neurodegeneration: role of brain innate immune system. CNS Neurol. Disord. Drug Targets 15, 329–336. doi: 10.2174/1871527315666160202125914
Li, Y., Wang, J., Sheng, J. G., Liu, L., Barger, S. W., Jones, R. A., et al. (1998). S100 β increases levels of β-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J. Neurochem. 71, 1421–1428. doi: 10.1046/j.1471-4159.1998.71041421.x
Liao, F., Hori, Y., Hudry, E., Bauer, A. Q., Jiang, H., Mahan, T. E., et al. (2014). Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. J. Neurosci. 34, 7281–7292. doi: 10.1523/JNEUROSCI.0646-14.2014
Lin, K. G., Tang, M., Guo, Y. B., Han, H. Y., and Lin, Y. H. (2011). Two polymorphisms of RCAN1 gene associated with Alzheimer’s disease in the Chinese Han population. East Asian Arch. Psychiatry 21, 79–84.
Lin, Y.-T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, P1141–1154.E7. doi: 10.1016/j.neuron.2018.05.008
Liu, L., Li, Y., Van Eldik, L. J., Griffin, W. S., and Barger, S. W. (2005). S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J. Neurochem. 92, 546–553. doi: 10.1111/j.1471-4159.2004.02909.x
Liu, C.-C., Zhao, N., Fu, Y., Wang, N., Linares, C., Tsai, C.-W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron 96, P1024–1032.E3. doi: 10.1016/j.neuron.2017.11.013
Loane, M., Morris, J. K., Addor, M. C., Arriola, L., Budd, J., Doray, B., et al. (2013). Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 21, 27–33. doi: 10.1038/ejhg.2012.9
Lockstone, H. E., Harris, L. W., Swatton, J. E., Wayland, M. T., Holland, A. J., and Bahn, S. (2007). Gene expression profiling in the adult Down syndrome brain. Genomics 90, 647–660. doi: 10.1016/j.ygeno.2007.08.005
Lucin, K. M., and Wyss-Coray, T. (2009). Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64, 110–122. doi: 10.1016/j.neuron.2009.08.039
Lv, H., Jia, L., and Jia, J. (2008). Promoter polymorphisms which modulate APP expression may increase susceptibility to Alzheimer’s disease. Neurobiol. Aging 29, 194–202. doi: 10.1016/j.neurobiolaging.2006.10.001
Ma, J., Brewer, H. B. Jr., and Potter, H. (1996). Alzheimer A β neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by A β-related peptides. Neurobiol. Aging 17, 773–780. doi: 10.1016/0197-4580(96)00112-1
Ma, J., Yee, A., Brewer, H. B. Jr., Das, S., and Potter, H. (1994). Amyloid-associated proteins α 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372, 92–94. doi: 10.1038/372092a0
Maezawa, I., Maeda, N., Montine, T. J., and Montine, K. S. (2006). Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J. Neuroinflammation 3:10. doi: 10.1186/1742-2094-3-10
Maher, K., Jeric Kokelj, B., Butinar, M., Mikhaylov, G., Mancek-Keber, M., Stoka, V., et al. (2014). A role for stefin B (cystatin B) in inflammation and endotoxemia. J. Biol. Chem. 289, 31736–31750. doi: 10.1074/jbc.M114.609396
Mancuso, R., Fryatt, G., Cleal, M., Obst, J., Pipi, E., Monzon-Sandoval, J., et al. (2019). CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 142, 3243–3264. doi: 10.1093/brain/awz241
Manelli, A. M., Bulfinch, L. C., Sullivan, P. M., and LaDu, M. J. (2007). Aβ42 neurotoxicity in primary co-cultures: effect of apoE isoform and Aβ conformation. Neurobiol. Aging 28, 1139–1147. doi: 10.1016/j.neurobiolaging.2006.05.024
McGeer, P. L., Rogers, J., and McGeer, E. G. (2006). Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimers Dis. 9, 271–276. doi: 10.3233/jad-2006-9s330
McGeer, P. L., Schulzer, M., and McGeer, E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47, 425–432. doi: 10.1212/wnl.47.2.425
McGeer, P. L., Walker, D. G., Pitas, R. E., Mahley, R. W., and McGeer, E. G. (1997). Apolipoprotein E4 (ApoE4) but not ApoE3 or ApoE2 potentiates β-amyloid protein activation of complement in vitro. Brain Res. 749, 135–138. doi: 10.1016/s0006-8993(96)01324-8
McLay, R. N., Kimura, M., Banks, W. A., and Kastin, A. J. (1997). Granulocyte-macrophage colony-stimulating factor crosses the blood—brain and blood—spinal cord barriers. Brain 120, 2083–2091. doi: 10.1093/brain/120.11.2083
Morgan, A. R., Touchard, S., Leckey, C., O’Hagan, C., Nevado-Holgado, A. J., Barkhof, F., et al. (2019). Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 15, 776–787. doi: 10.1016/j.jalz.2019.03.007
Mrak, R. E., and Griffin, W. S. (2001). Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 22, 903–908. doi: 10.1016/s0197-4580(01)00287-1
Myllykangas, L., Wavrant-De Vrieze, F., Polvikoski, T., Notkola, I. L., Sulkava, R., Niinisto, L., et al. (2005). Chromosome 21 BACE2 haplotype associates with Alzheimer’s disease: a two-stage study. J. Neurol. Sci. 236, 17–24. doi: 10.1016/j.jns.2005.04.008
Niu, L.-D., Xu, W., Li, J.-Q., Tan, C.-C., Cao, X.-P., Yu, J., et al. (2019). Genome-wide association study of cerebrospinal fluid neurofilament light levels in non-demented elders. Ann. Transl. Med. 7:657. doi: 10.21037/atm.2019.10.66
Nowotny, P., Simcock, X., Bertelsen, S., Hinrichs, A. L., Kauwe, J. S., Mayo, K., et al. (2007). Association studies testing for risk for late-onset Alzheimer’s disease with common variants in the β-amyloid precursor protein (APP). Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 469–474. doi: 10.1002/ajmg.b.30485
Pankiewicz, J. E., Guridi, M., Kim, J., Asuni, A. A., Sanchez, S., Sullivan, P. M., et al. (2014). Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol. Commun. 2:75. doi: 10.1186/s40478-014-0075-0
Patel, A., Rees, S. D., Kelly, M. A., Bain, S. C., Barnett, A. H., Thalitaya, D., et al. (2011). Association of variants within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in Alzheimer’s disease in subjects with Down syndrome. Neurosci. Lett. 487, 144–148. doi: 10.1016/j.neulet.2010.10.010
Patel, D., Zhang, X., Farrell, J. J., Lunetta, K. L., and Farrer, L. A. (2021). Set-based rare variant expression quantitative trait loci in blood and brain from alzheimer disease study participants. Genes 12:419. doi: 10.3390/genes12030419
Perry, V. H., and Gordon, S. (1988). Macrophages and microglia in the nervous system. Trends Neurosci. 11, 273–277. doi: 10.1016/0166-2236(88)90110-5
Petersen, M. E., and O’Bryant, S. E. (2019). Blood-based biomarkers for Down syndrome and Alzheimer’s disease: a systematic review. Dev. Neurobiol. 79, 699–710. doi: 10.1002/dneu.22714
Pinto, B., Morelli, G., Rastogi, M., Savardi, A., Fumagalli, A., Petretto, A., et al. (2020). Rescuing over-activated microglia restores cognitive performance in juvenile animals of the Dp(16) mouse model of down syndrome. Neuron 108, P887–904.E12. doi: 10.1016/j.neuron.2020.09.010
Potter, H. (2001). “Beyond β protein—the essential role of inflammation in Alzheimer amyloid formation,” in Inflammatory Events in Neurodegeneration, eds S. C. Bondy and A. Cambell (Shepparton, Australia: Prominent Press).
Potter, H., and Wisniewski, T. (2012). Apolipoprotein e: essential catalyst of the Alzheimer amyloid cascade. Int. J. Alzheimers Dis. 2012:489428. doi: 10.1155/2012/489428
Potter, H., Woodcock, J. H., Boyd, T. D., Coughlan, C. M., O’Shaughnessy, J. R., Borges, M. T., et al. (2021). Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimers Dement. 7:e12158. doi: 10.1002/trc2.12158
Powers, R. K., Culp-Hill, R., Ludwig, M. P., Smith, K. P., Waugh, K. A., Minter, R., et al. (2019). Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 10:4766. doi: 10.1038/s41467-019-12739-9
Rachubinski, A. L., Estrada, B. E., Norris, D., Dunnick, C. A., Boldrick, J. C., and Espinosa, J. M. (2019). Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep. 5, 365–367. doi: 10.1016/j.jdcr.2019.02.007
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 13:24. doi: 10.1186/s13024-018-0254-8
Rangasamy, S. B., Jana, M., Roy, A., Corbett, G. T., Kundu, M., Chandra, S., et al. (2018). Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J. Clin. Invest. 128, 4297–4312. doi: 10.1172/JCI96209
Ridolfi, E., Barone, C., Scarpini, E., and Galimberti, D. (2013). The role of the innate immune system in Alzheimer’s disease and frontotemporal lobar degeneration: an eye on microglia. Clin. Dev. Immunol. 2013:939786. doi: 10.1155/2013/939786
Rountree, R. B., Willis, C. R., Dinh, H., Blumberg, H., Bailey, K., Dean, C. Jr., et al. (2010). RIP4 regulates epidermal differentiation and cutaneous inflammation. J. Invest. Dermatol. 130, 102–112. doi: 10.1038/jid.2009.223
Rubinsztein, D. C., Hon, J., Stevens, F., Pyrah, I., Tysoe, C., Huppert, F. A., et al. (1999). Apo E genotypes and risk of dementia in Down syndrome. Am. J. Med. Genet. 88, 344–347. doi: 10.1002/(sici)1096-8628(19990820)88:4<344::aid-ajmg10>3.0.co;2-1
Sadowski, M., Pankiewicz, J., Scholtzova, H., Ripellino, J. A., Li, Y., Schmidt, S. D., et al. (2004). A synthetic peptide blocking the apolipoprotein E/β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. Am. J. Pathol. 165, 937–948. doi: 10.1016/s0002-9440(10)63355-x
Sanan, D. A., Weisgraber, K. H., Russell, S. J., Mahley, R. W., Huang, D., Saunders, A., et al. (1994). Apolipoprotein E associates with β amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Invest. 94, 860–869. doi: 10.1172/JCI117407
Sawada, M., Itoh, Y., Suzumura, A., and Marunouchi, T. (1993). Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci. Lett. 160, 131–134. doi: 10.1016/0304-3940(93)90396-3
Schäbitz, W. R., Krüger, C., Pitzer, C., Weber, D., Laage, R., Gassler, N., et al. (2008). A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J. Cereb. Blood Flow Metab. 28, 29–43. doi: 10.1038/sj.jcbfm.9600496
Schallenberg, M., Charalambous, P., and Thanos, S. (2012). GM-CSF protects rat photoreceptors from death by activating the SRC-dependent signalling and elevating anti-apoptotic factors and neurotrophins. Graefes Arch. Clin. Exp. Ophthalmol. 250, 699–712. doi: 10.1007/s00417-012-1932-9
Schmidt, R., Schmidt, H., Curb, J. D., Masaki, K., White, L. R., and Launer, L. J. (2002). Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann. Neurol. 52, 168–174. doi: 10.1002/ana.10265
Schneider, U. C., Schilling, L., Schroeck, H., Nebe, C. T., Vajkoczy, P., and Woitzik, J. (2007). Granulocyte-macrophage colony-stimulating factor-induced vessel growth restores cerebral blood supply after bilateral carotid artery occlusion. Stroke 38, 1320–1328. doi: 10.1161/01.STR.0000259707.43496.71
Sen, U., Givvimani, S., Abe, O. A., Lederer, E. D., and Tyagi, S. C. (2011). Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. Am. J. Physiol. Cell Physiol. 300, C155–C163. doi: 10.1152/ajpcell.00143.2010
Serre-Miranda, C., Roque, S., Santos, N. C., Costa, P., Sousa, N., Palha, J. A., et al. (2020). Cognition is associated with peripheral immune molecules in healthy older adults: a cross-sectional study. Front. Immunol. 11:2045. doi: 10.3389/fimmu.2020.02045
Shi, Y., Manis, M., Long, J., Wang, K., Sullivan, P. M., Remolina Serrano, J., et al. (2019). Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 216, 2546–2561. doi: 10.1084/jem.20190980
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shultz, S. R., Tan, X. L., Wright, D. K., Liu, S. J., Semple, B. D., Johnston, L., et al. (2014). Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J. Neurotrauma 31, 976–983. doi: 10.1089/neu.2013.3106
Silverman, W. (2007). Down syndrome: cognitive phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 13, 228–236. doi: 10.1002/mrdd.20156
Snyder, H. M., Bain, L. J., Brickman, A. M., Carrillo, M. C., Esbensen, A. J., Espinosa, J. M., et al. (2020). Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement. 16, 1065–1077. doi: 10.1002/alz.12112
Son, Y., Jeong, Y. J., Shin, N. R., Oh, S. J., Nam, K. R., Choi, H. D., et al. (2020). Inhibition of colony-stimulating factor 1 receptor by PLX3397 prevents amyloid β pathology and rescues dopaminergic signaling in aging 5xFAD mice. Int. J. Mol. Sci. 21:5553. doi: 10.3390/ijms21155553
Sosna, J., Philipp, S., Albay, R. III., Reyes-Ruiz, J. M., Baglietto-Vargas, D., LaFerla, F. M., et al. (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 13:11. doi: 10.1186/s13024-018-0244-x
Soto, C., Golabek, A., Wisniewski, T., and Castano, E. M. (1996). Alzheimer’s β-amyloid peptide is conformationally modified by apolipoprotein E in vitro. Neuroreport 7, 721–725. doi: 10.1097/00001756-199602290-00010
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. doi: 10.1038/s41467-019-11674-z
Spellman, C., Ahmed, M. M., Dubach, D., and Gardiner, K. J. (2013). Expression of trisomic proteins in Down syndrome model systems. Gene 512, 219–225. doi: 10.1016/j.gene.2012.10.051
Strittmatter, W. J., and Roses, A. D. (1995). Apolipoprotein E and Alzheimer disease. Proc. Natl. Acad. Sci. U S A 92, 4725–4727. doi: 10.1073/pnas.92.11.4725
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Subasinghe, W., Syed, I., and Kowluru, A. (2011). Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R12–R20. doi: 10.1152/ajpregu.00421.2010
Sullivan, K. D., Evans, D., Pandey, A., Hraha, T. H., Smith, K. P., Markham, N., et al. (2017). Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 7:14818. doi: 10.1038/s41598-017-13858-3
Sullivan, K. D., Lewis, H. C., Hill, A. A., Pandey, A., Jackson, L. P., Cabral, J. M., et al. (2016). Trisomy 21 consistently activates the interferon response. eLife 5:e16220. doi: 10.7554/eLife.16220
Taipa, R., das Neves, S. P., Sousa, A. L., Fernandes, J., Pinto, C., Correia, A. P., et al. (2019). Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 76, 125–132. doi: 10.1016/j.neurobiolaging.2018.12.019
Tan, M.-S., Yang, Y.-X., Xu, W., Wang, H.-F., Tan, L., Zuo, C.-T., et al. (2021). Associations of Alzheimer’s disease risk variants with gene expression, amyloidosis, tauopathy, and neurodegeneration. Alzheimers Res. Ther. 13:15. doi: 10.1186/s13195-020-00755-7
Tang, X., Sun, L., Wang, G., Chen, B., and Luo, F. (2018). RUNX1: a regulator of NF-kB signaling in pulmonary diseases. Curr. Protein Pept. Sci. 19, 172–178. doi: 10.2174/1389203718666171009111835
Tanzi, R. E., McClatchey, A. I., Lamperti, E. D., Villa-Komaroff, L., Gusella, J. F., and Neve, R. L. (1988). Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature 331, 528–530. doi: 10.1038/331528a0
Tarkowski, E., Andreasen, N., Tarkowski, A., and Blennow, K. (2003). Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205. doi: 10.1136/jnnp.74.9.1200
Theoret, J. K., Jadavji, N. M., Zhang, M., and Smith, P. D. (2016). Granulocyte macrophage colony-stimulating factor treatment results in recovery of motor function after white matter damage in mice. Eur. J. Neurosci. 43, 17–24. doi: 10.1111/ejn.13105
Trollor, J. N., Smith, E., Agars, E., Kuan, S. A., Baune, B. T., Campbell, L., et al. (2012). The association between systemic inflammation and cognitive performance in the elderly: the Sydney Memory and Ageing Study. Age 34, 1295–1308. doi: 10.1007/s11357-011-9301-x
Tuttle, K. D., Waugh, K. A., Araya, P., Minter, R., Orlicky, D. J., Ludwig, M., et al. (2020). JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of down syndrome. Cell Rep. 33:108407. doi: 10.1016/j.celrep.2020.108407
Vacca, R. A., Bawari, S., Valenti, D., Tewari, D., Nabavi, S. F., Shirooie, S., et al. (2019). Down syndrome: neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 98, 234–255. doi: 10.1016/j.neubiorev.2019.01.001
Wang, C., Najm, R., Xu, Q., Jeong, D. E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wendeln, A.-C., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. doi: 10.1038/s41586-018-0023-4
Westin, K., Buchhave, P., Nielsen, H., Minthon, L., Janciauskiene, S., and Hansson, O. (2012). CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One 7:e30525. doi: 10.1371/journal.pone.0030525
Whelan, C. D., Mattsson, N., Nagle, M. W., Vijayaraghavan, S., Hyde, C., Janelidze, S., et al. (2019). Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 7:169. doi: 10.1186/s40478-019-0795-2
Wilcock, D. M., and Griffin, W. S. (2013). Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation 10:84. doi: 10.1186/1742-2094-10-84
Wilcock, D. M., Hurban, J., Helman, A. M., Sudduth, T. L., McCarty, K. L., Beckett, T. L., et al. (2015). Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 36, 2468–2474. doi: 10.1016/j.neurobiolaging.2015.05.016
Wisniewski, T., Castano, E. M., Golabek, A., Vogel, T., and Frangione, B. (1994). Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am. J. Pathol. 145, 1030–1035.
Wisniewski, K. E., Dalton, A. J., McLachlan, C., Wen, G. Y., and Wisniewski, H. M. (1985). Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology 35, 957–961. doi: 10.1212/wnl.35.7.957
Wisniewski, T., and Frangione, B. (1992). Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 135, 235–238. doi: 10.1016/0304-3940(92)90444-c
Wisniewski, T., Golabek, A., Matsubara, E., Ghiso, J., and Frangione, B. (1993). Apolipoprotein E: binding to soluble Alzheimer’s β-amyloid. Biochem. Biophys. Res. Commun. 192, 359–365. doi: 10.1006/bbrc.1993.1423
Wojcik, A. J., Skaflen, M. D., Srinivasan, S., and Hedrick, C. C. (2008). A critical role for ABCG1 in macrophage inflammation and lung homeostasis. J. Immunol. 180, 4273–4282. doi: 10.4049/jimmunol.180.6.4273
Wollmer, M. A., Sleegers, K., Ingelsson, M., Zekanowski, C., Brouwers, N., Maruszak, A., et al. (2007). Association study of cholesterol-related genes in Alzheimer’s disease. Neurogenetics 8, 179–188. doi: 10.1007/s10048-007-0087-z
Xiong, M., Jiang, H., Serrano, J. R., Gonzales, E. R., Wang, C., Gratuze, M., et al. (2021). APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 13:eabd7522. doi: 10.1126/scitranslmed.abd7522
Yamanishi, Y., Boyle, D. L., Clark, M., Maki, R. A., Tortorella, M. D., Arner, E. C., et al. (2002). Expression and regulation of aggrecanase in arthritis: the role of TGF-β. J. Immunol. 168, 1405–1412. doi: 10.4049/jimmunol.168.3.1405
Yang, D.-S., Stavrides, P., Mohan, P. S., Kaushik, S., Kumar, A., Ohno, M., et al. (2011). Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 7, 788–789. doi: 10.4161/auto.7.7.15596
Yuen, S., Smith, J., Caruso, L., Balan, M., and Opavsky, M. A. (2011). The coxsackie-adenovirus receptor induces an inflammatory cardiomyopathy independent of viral infection. J. Mol. Cell. Cardiol. 50, 826–840. doi: 10.1016/j.yjmcc.2011.02.011
Zhang, Y., Che, M., Yuan, J., Yu, Y., Cao, C., Qin, X. Y., et al. (2017). Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget 8, 84489–84496. doi: 10.18632/oncotarget.21060
Zhu, Y., Nwabuisi-Heath, E., Dumanis, S. B., Tai, L. M., Yu, C., Rebeck, G. W., et al. (2012). APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569. doi: 10.1002/glia.22289
Keywords: innate immune system, inflammation, GM-CSF (granulocyte-macrophage colony-stimulating factor), Down syndrome, Alzheimer’s disease, apolipoprotein E, drug repurposing and discovery, amyloid-β
Citation: Ahmed MM, Johnson NR, Boyd TD, Coughlan C, Chial HJ and Potter H (2021) Innate Immune System Activation and Neuroinflammation in Down Syndrome and Neurodegeneration: Therapeutic Targets or Partners? Front. Aging Neurosci. 13:718426. doi: 10.3389/fnagi.2021.718426
Received: 31 May 2021; Accepted: 16 August 2021;
Published: 16 September 2021.
Edited by:
Elliott Jay Mufson, Barrow Neurological Institute (BNI), United StatesReviewed by:
Federico Paolini Paoletti, University of Perugia, ItalyMartin J. Sadowski, New York University, United States
Copyright © 2021 Ahmed, Johnson, Boyd, Coughlan, Chial and Potter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huntington Potter, aHVudGluZ3Rvbi5wb3R0ZXJAY3VhbnNjaHV0ei5lZHU=
† These authors have contributed equally to this work