 Nelson de Oliveira Manzanza
Nelson de Oliveira Manzanza Lucia Sedlackova2
Lucia Sedlackova2 Raj N. Kalaria
Raj N. Kalaria- 1Translational and Clinical Research Institute, Campus for Ageing and Vitality, Newcastle University, Newcastle upon Tyne, United Kingdom
- 2Biosciences Institute, Campus for Ageing and Vitality, Newcastle University, Newcastle upon Tyne, United Kingdom
Lewy Body Disorders (LBDs) lie within the spectrum of age-related neurodegenerative diseases now frequently categorized as the synucleinopathies. LBDs are considered to be among the second most common form of neurodegenerative dementias after Alzheimer's disease. They are progressive conditions with variable clinical symptoms embodied within specific cognitive and behavioral disorders. There are currently no effective treatments for LBDs. LBDs are histopathologically characterized by the presence of abnormal neuronal inclusions commonly known as Lewy Bodies (LBs) and extracellular Lewy Neurites (LNs). The inclusions predominantly comprise aggregates of alpha-synuclein (aSyn). It has been proposed that post-translational modifications (PTMs) such as aSyn phosphorylation, ubiquitination SUMOylation, Nitration, o-GlcNacylation, and Truncation play important roles in the formation of toxic forms of the protein, which consequently facilitates the formation of these inclusions. This review focuses on the role of different PTMs in aSyn in the pathogenesis of LBDs. We highlight how these PTMs interact with aSyn to promote misfolding and aggregation and interplay with cell membranes leading to the potential functional and pathogenic consequences detected so far, and their involvement in the development of LBDs.
Highlights
- aSyn aggregation and transformation into LBs and LNs underlie progression of LBDs and other synucleinopathies.
- The failure of proteostasis network is linked to maintain biologically active aSyn.
- Post-translational modifications (PTMs) contribute to aSyn pathogenicity.
- aSyn PTMs include ubiquitination, nitration, acetylation, SUMOylation, glutathionylation, Glycosylation, and phosphorylation.
- PTMs interventions or modifications may reduce accumulation of LBs and prevent LBDs.
Introduction
Lewy body dementias (LBDs) are progressive neurodegenerative disorders characterized by the presence of abnormal intra-neuronal deposits commonly called Lewy bodies (LBs) and deposition of extracellular Lewy neurites (LNs) (Figure 1) (McKeith et al., 2017; Outeiro et al., 2019). LBDs are currently regarded to encompass dementia with Lewy Bodies (DLB), Parkinson's disease (PD), and Parkinson's disease with dementia (PDD). They are the second most prevalent types of neurodegenerative dementia, after Alzheimer's disease (AD), accounting for ~22% of all dementias in older people. The incidence of LBDs is associated with advancing age as it increasingly manifests in individuals over 65 years of age (Spillantini et al., 1998; Braak et al., 1999; McKeith et al., 2003, 2004, 2017; Yang et al., 2018; Outeiro et al., 2019). The clinical and pathological profiles of the LBDs demonstrate considerable overlap among the subtypes. Patients diagnosed with DLB often present with parkinsonism associated with brainstem and midbrain pathology and many PD cases present with dementia owing to pathological involvement of the limbic and cortical structures progressing to PDD (Figure 1) (Jellinger and Korczyn, 2018). The differential diagnosis between PDD and DLB is achieved using the temporal profile during onset of disease such as the appearance of cognitive symptoms after the presentation of Parkinsonian features. Thus, if the onset of dementia occurs at least 1 year after the onset of initial PD related symptoms, the diagnosis is consistent with PDD. In contrast, if the onset of dementia begins within 1 year of the development of parkinsonism, then the profile is consistent with DLB (Spillantini et al., 1997, 1998; Gurd et al., 2000; McKeith, 2006; Goedert et al., 2013; McKeith et al., 2017). If dementia develops in the context of established PD, then it is consistent with PDD (McKeith et al., 2017). Generally, PDD develops after ~10 years of the onset of PD (Aarsland et al., 2003; McKeith et al., 2017). However, the neuropsychological profile of PDD is not phenotypically very different compared with DLB (Aarsland et al., 2003). Despite progress in our understanding and identification of the variable symptoms, which may be indicative of either PD or DLB disease progression, a definitive clinical diagnosis remains difficult to achieve. At present, a confirmed diagnosis is only provided through post-mortem examination by the detection of intraneuronal inclusions, consistent with LB pathology found in the neuronal soma and LN pathology within the neuronal processes.
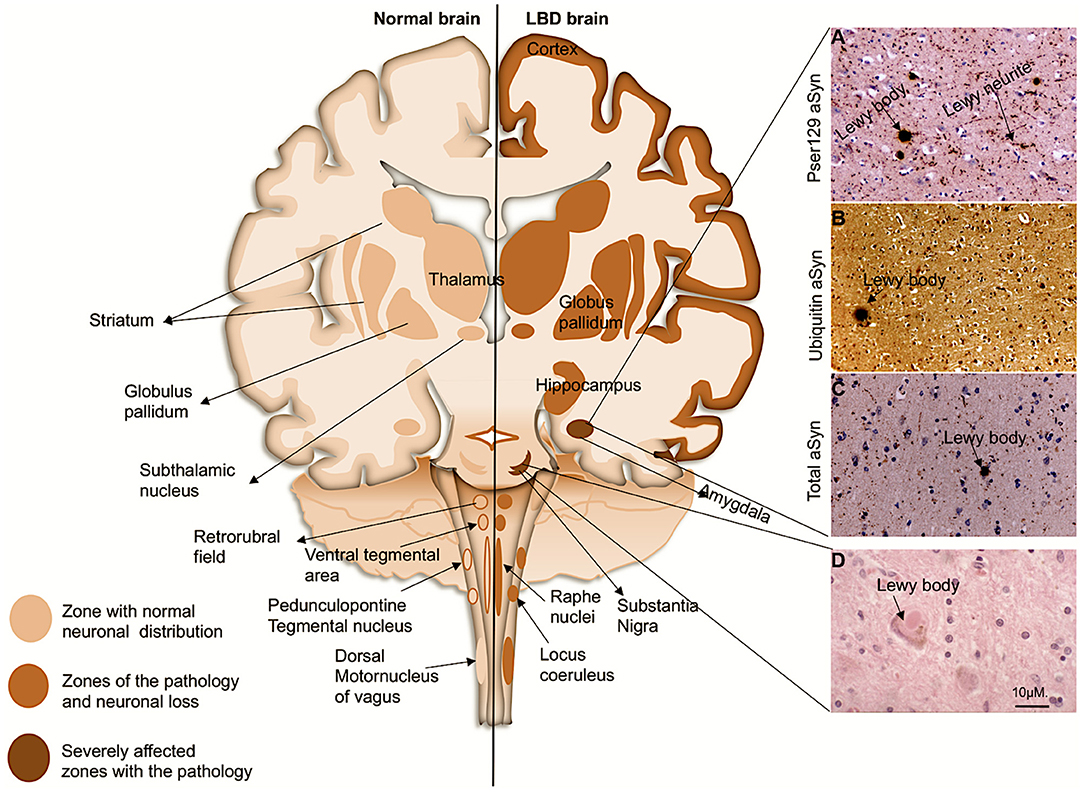
Figure 1. Coronal schematic of the brain, depicting normal structures and in LBD subjects. Shading shows the distribution of aSyn in LBs/LNs in cortical and subcortical regions. Subcortical regions are associated with PD, and as the disease progresses to cortical regions it corresponds with DLB and PDD. (Right, A–D): LB pathology (arrows) revealed in the amygdala of a patient with DLB with antibody to pSer129 aSyn (A), ubiquitin (B), and whole aSyn counterstained with haematoxylin. (D), LB with a halo typically found in the substantia nigra. Magnification bar: 10μM.
Upon light microscopy, LBs are characteristically seen as neuronal inclusions usually eosinophilic round bodies surrounded by a halo (Figure 1). LBs form in subsets of large or long projection neurons which seem to be prone to such accumulation particularly monoaminergic neurons in the brainstem and midbrain (Halliday et al., 1990; Braak et al., 2004). However, in the limbic system and the neocortical areas, LBs are most frequently found in glutamatergic pyramidal neurons; LBs have also been reported in cholinergic neurons of the basal forebrain (Wakabayashi et al., 1995; Marui et al., 2003; Dugger and Dickson, 2010; Bernstein et al., 2011). LBs and LNs are not exclusive to LBDs, as they also occur in other neurodegenerative diseases each defined by different regional and cellular distributions and collectively termed as the synucleinopathies (Spillantini et al., 1997, 1998; Mattila et al., 2000; Goedert et al., 2013).
LBs and LNs were originally immunohistochemically identified by antibodies to ubiquitin. However, it is now standard to demonstrate them with antibodies to alpha-synuclein (aSyn) (Yoshimoto et al., 1995; Spillantini et al., 1998). aSyn protein is the most abundant constituent of the inclusions (Spillantini et al., 1997, 1998; Baba et al., 1998). More recent studies have shown that LBs and LNs also comprise many cellular components including dysmorphic organelles, vesicular structures and membranes (Spillantini et al., 1997, 1998; Braak et al., 1999; Shults, 2006; Shahmoradian et al., 2018). aSyn protein was first identified in 1988, as a small intracellular protein, and it was named so because of its localization in the nuclear envelopes and synaptic vesicles of the cells (Maroteaux et al., 1988). aSyn belongs to a family of three homologous synuclein proteins, which include β-synuclein and γ-synuclein (Nakajo et al., 1990; Jakes et al., 1994; Buchman et al., 1998; Lavedan et al., 1998; Wales et al., 2013). Subsequently, Saitoh and colleagues discovered that aSyn was associated with amyloid plaques of Alzheimer's disease (AD) patients (Kalaria, 1997) and called it the non-amyloid beta component (NAC) (Ueda et al., 1993). aSyn further became a major focus in LBDs upon discovery of point mutations in the aSyn gene in familial forms of PD (Polymeropoulos et al., 1997; Nemani et al., 2010). Point mutations within the aSyn encoding gene (SNCA) leading to formation of LBs and LNs has largely confirmed its pathogenic role in LBDs (Polymeropoulos et al., 1997; Bussell and Eliezer, 2001; Kruger et al., 2001; Zarranz et al., 2004; Lesage et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014).
Although the exact mechanism that triggers the aggregation of aSyn and the consequent inclusion formation is not clearly understood it is reported that the influence of mutations and post-translational modifications (PTMs) add to the protein pathogenicity. It is thought that the PTMs play important roles in impairing the native state of the protein, which consequently induces protein misfolding and aggregation (Villar-Pique et al., 2016). Thus, it is important to assess the disease associated PTMs of aSyn, as it may enable elucidation of potential processes involved in its pathological aggregation (Jellinger and Korczyn, 2018; Moors et al., 2018). In addition, it may help to elucidate the observed anatomical and distribution patterns of LBs seen within the LBDs, as well as provide better insights in strategies for treatments. This review outlines the involvement of different PTMs of aSyn described in LBDs, with particular focus on aSyn phosphorylation, ubiquitination SUMOylation, Nitration, o-GlcNacylation, and Truncation (Figure 2). Our aim is to provide insight into how the PTMs affect aSyn to misfold and aggregate, leading to the potential functional and pathogenic consequences so far detected, that lead to the development of LBDs.
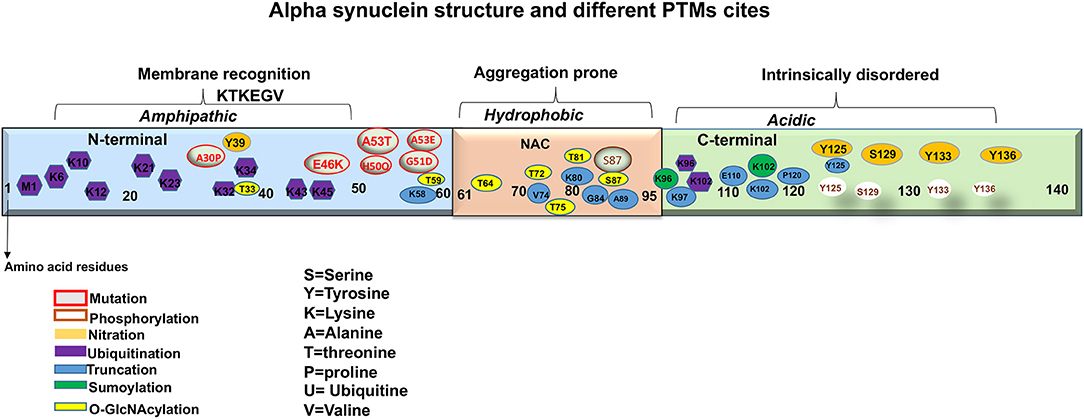
Figure 2. A representation of aSyn structure and main sites of mutations and post-translational modifications (PTMs).
aSyn Structure
aSyn is ubiquitously expressed in the CNS comprising ~1% of total cytosolic neuronal protein (Kahle, 2008). It is a relatively small protein with molecular weight of 14.5 kDa, nearly 4 times as large as the amyloid-β peptide. Synucleins are predominantly neuronal proteins, most abundant in presynaptic terminals (Maroteaux et al., 1988; Ueda et al., 1993). aSyn is a product of the synuclein alpha (SNCA) gene, located at position 21 of the long arm of chromosome 4. The gene encodes 140 highly charged amino acid (aa) residues, which do not adopt a defined structure in an aqueous solution (Polymeropoulos et al., 1997; Wales et al., 2013). aSyn consists of three distinct domains: The N-terminal, the Central and C terminal domains (Figure 2). The N-terminal domain or the amino terminus, (aa 1-60) contains four 11-dimer repeats that contain a KTKEGV consensus sequence. This region is predisposed to fold into alpha helices and has been hypothesized to be linked with the lipid binding capacity of the protein (Polymeropoulos et al., 1997; Marui et al., 2004; Wales et al., 2013). The Central domain, also called the NAC (aa 61-95) is particularly hydrophobic and amyloidogenic (Maroteaux et al., 1988; Ueda et al., 1993). Studies have reported that this domain is associated with aSyn aggregation when it adopts a beta-sheet structure. Moreover, identified mutations that are linked with synucleinopathies are also found in this region (Figure 2) (Polymeropoulos et al., 1997; Lesage et al., 2013; Proukakis et al., 2013). Finally, the C terminal domain or the carboxyl terminus (aa 96-140), provides flexibility to the polypeptide, because of its abundance in proline residues, that are known to affect the secondary structure of the region (Maroteaux et al., 1988; Ueda et al., 1993). In addition, this region is highly acidic, intrinsically unstructured and is thought to be the target of diverse PTMs (Figure 2) (Davidson et al., 1998; Bussell and Eliezer, 2001, 2003). Furthermore, this region is also involved in protein-protein interactions, polyamine binding, protection from protein aggregation and modulation of membrane binding properties of aSyn (Crowther et al., 1998; Nielsen et al., 2001; Park et al., 2002; Fernandez et al., 2004; Brown, 2007; Bodner et al., 2009; Oueslati et al., 2010; Sevcsik et al., 2011).
aSyn Function
The precise function of aSyn remains the focus of intense debate and research. The predominant cytoplasmic expression of aSyn and its specific transport to presynaptic terminals, suggests a regulatory function within the synapse (Burre, 2015). Modulation of aSyn expression by either overexpression, knock-down or knockout, alters synaptic vesicle trafficking, and maintenance, as well as neurotransmitter compartmentalisation, storage, release, recycling, and changes in synaptic activity and plasticity (Lashuel et al., 2012; Burre et al., 2013; Allen Reish and Standaert, 2015). Given the extensive nature of the synaptic parameters affected, it is plausible that aSyn may act as an accessory/adaptor protein for these processes. aSyn is thought to empower a number of better-defined synaptic proteins. aSyn supports N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins in their role, potentially acting with some level of functional redundancy with Cysteine Stirring Protein alpha (CSP-α) 1- (Goda, 1997; Gerst, 1999; Burre et al., 2010; Lautenschlager et al., 2017; Lou et al., 2017; Hawk et al., 2019; Agliardi et al., 2021). aSyn is thought to assist in the formation of SNARE complexes via the direct binding of synaptobrevin 2. Such facilitation of SNARE complex formation ultimately influences the dynamics of synaptic vesicle cycling during neurotransmitter release (Table 1) (Burre et al., 2010). An interesting study by Fortin et al. (2005), supports this, as it demonstrated that during neural activity, fluorescently labeled aSyn retracts from synaptic vesicles and then returns progressively to the vesicles (Fortin et al., 2005). Nevertheless, despite these research efforts the full extent of aSyn function(s) remains poorly understood.
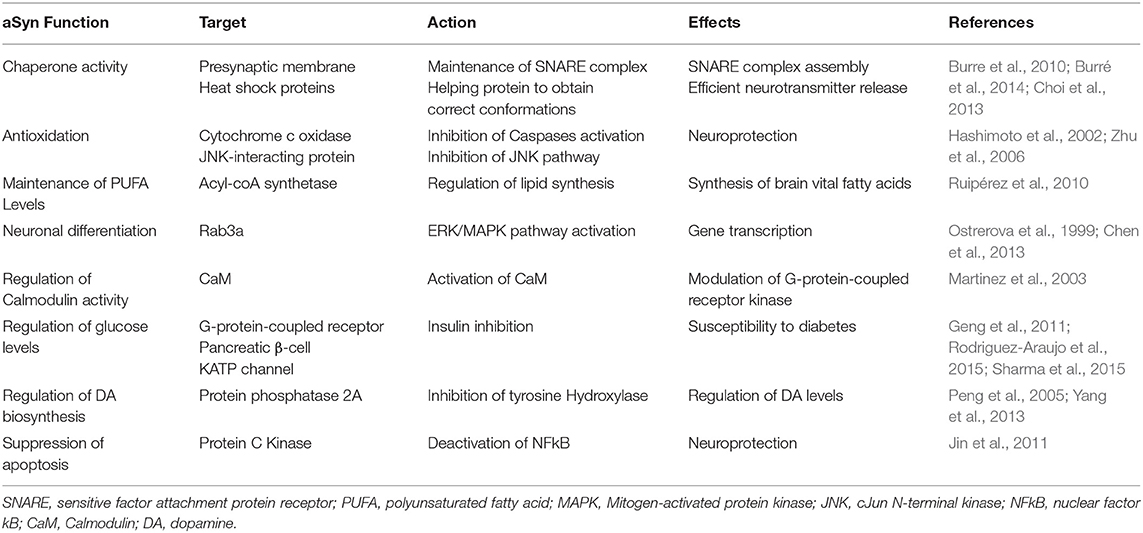
Table 1. Summary of aSyn function, targets, action, and effects.
aSyn Aggregation and Its Role in Cellular Dysfunction
aSyn aggregates are also the main constituents of LBs and LNs. Aggregates are also present in mixed neuropathological cases where both LB pathology and atypical AD with amyloid-β and Tau pathology are present (Spillantini et al., 1997, 1998). Multiple lines of evidence shows that unstructured soluble monomers of aSyn are transformed into oligomers, which then aggregate to form mature insoluble fibrils and form LBs (Harper et al., 1997; Walsh et al., 1997; Lambert et al., 1998; Miake et al., 2002). In vitro studies demonstrated that the aggregation process occurs in three different stages, named the lag, elongation and stationary phases (Buell et al., 2014). In the lag phase, the aggregation occurs through key structural changes within the protein resulting in the formation of misfolded monomers and aggregation, culminating in the formation of oligomers. In the elongation phase, an exponential growth of oligomers takes place, to form non-soluble fibrils achieved by the continual addition of monomers. In the stationary phase, the consumption of monomeric aggregates culminates into reduction of the fibril growth rate and formation of LBs (Invernizzi et al., 2012; Buell et al., 2014). However, this sequence of events appears to be much more complex in vivo as different oligomeric forms may be involved in the process (Fairfoul et al., 2016; Shahnawaz et al., 2017).
Whilst it remains unclear whether the end-stage products of aggregation (LBs and LNs) are the most disease relevant toxic forms of aSyn, it is widely assumed that the process of aggregation is critical and perhaps its intermediates such as prefibrils or fibrillary oligomers formed during such a process are the toxic elements (Periquet et al., 2007). This is supported by the fact that, aSyn and β-synuclein fragments which do not contain the NAC domain, were found to be resistant to aggregation (Periquet et al., 2007). Moreover, aSyn oligomers and aggregated fibrils were found to exhibit toxicity even before forming LBs/LNs, and these aggregation precursors have been associated with progressive motor impairments and neuronal cell death (Periquet et al., 2007). Cell culture and in vivo studies in animal models have shown that aggregated fibrils cause further aggregation of aSyn monomers that consequently translocate between cells in prion-like manner (Wood et al., 1999; Desplats et al., 2009; Hansen et al., 2011). This finding indicates that aSyn pathogenic cascade, may begin even before LBs are formed, causing functional alterations before the structural changes are evident. Furthermore, aggregated aSyn was shown to affect various cellular pathways, impairing the adaptation of the neurons to endoplasmic reticulum (ER) stress, inducing dysfunction of the proteasome degradation process, mitochondria functioning, and leading to membrane damage as well as loss of synapses (Figure 3) (Wood et al., 1999; Desplats et al., 2009; Hansen et al., 2011).
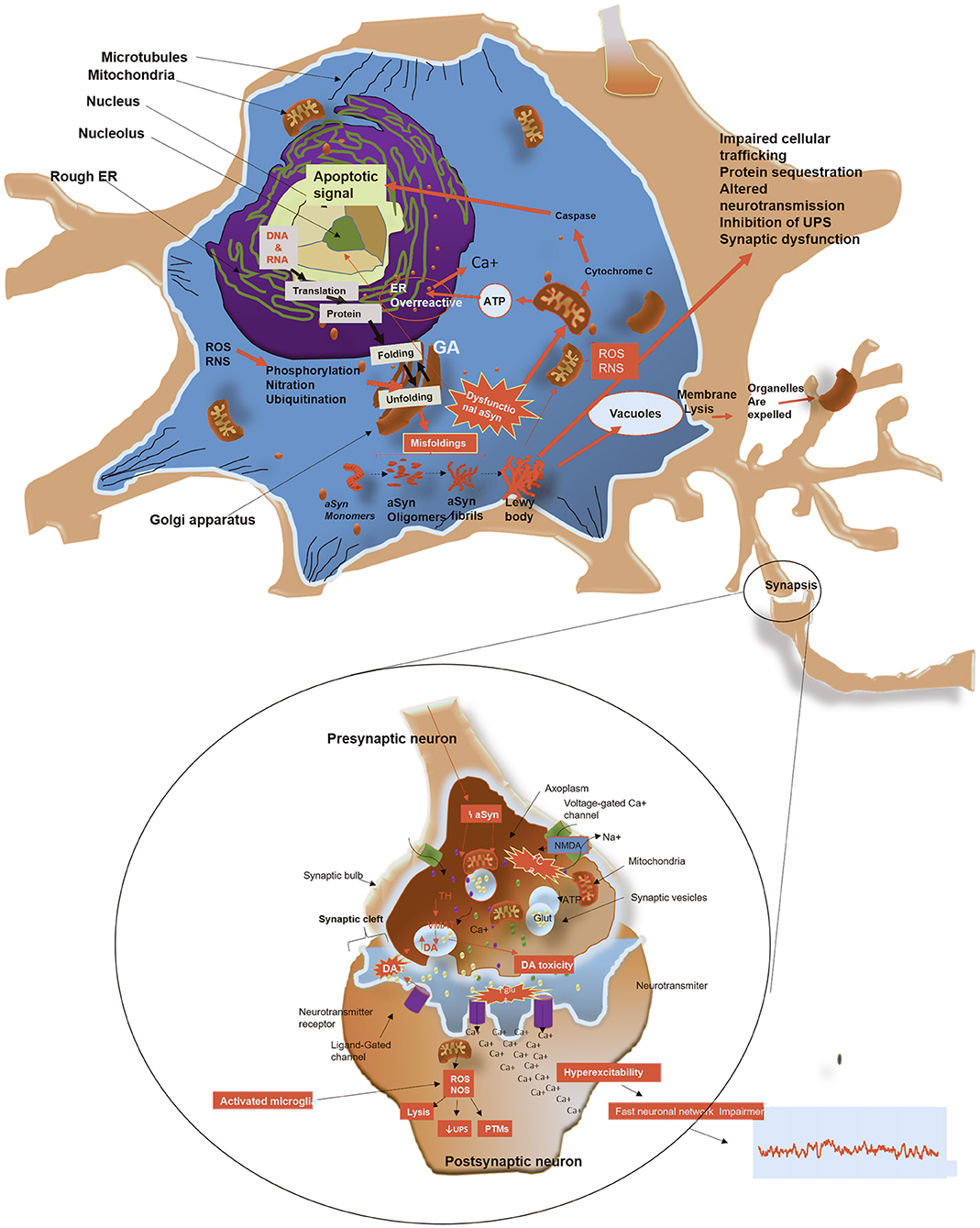
Figure 3. Schematic representation of potential mechanisms by which aSyn form aggregates, leads to toxicity and causes cell death. Monomeric aSyn assembles to form oligomers and producing mature fibrils. (1) LBs and LNs formation: fibrillary forms of aSyn are segregated into protein inclusions, which contain dysmorphic organelles, vesicular structures and various other cellular proteins, depleting cells of these vital components. (2) Mitochondrial impairment: Altered aSyn translocate to the mitochondria, causing oxidative stress by increasing production of reactive oxygen and nitrogen species with consequent impairment of energy production. Damage to mitochondria leads to release of Cytochrome C. This is in turn activates caspases, which translocate to the nucleus to initiate an apoptotic response. (3) ER dysfunction and protein trafficking inhibition: depletion of ATP due to mitochondrial dysfunction, causes ER overactivity leading to increased release of calcium. This further causes blockage of protein trafficking from ER to Golgi, leading to Golgi fragmentation. (4) Membrane pore formation: putative toxic forms of aSyn may penetrate cellular membranes, altering its permeability, causing excess of calcium and other ions within the cytosol. (5) Impaired autophagy: adhesion of aSyn to the membrane of lysosomes alters autophagy mediated by chaperones, resulting in aggregation of substrates and impairment of the proteasome system. (6) Release of toxic aSyn and other cellular components into extracellular space: toxic aSyn may be passively released in the extracellular space by dying neurons and consequently taken by adjacent neurons, resulting in seeding aggregation and synaptic impairments. (7) Defects in axonal transport: toxic aSyn causes Tau hyper phosphorylation, which inhibit the modulation of microtubule assembly, with consequent detrimental effects on cellular transport, increasing aggregation of toxic substances in the cytosol. (8) Synaptic terminal dysfunction: toxic aSyn alters distribution of proteins in the synaptic terminal, reduces synaptic vesicle release, which leads to changes of the synaptic protein composition and hyperexcitability. (9) Impairment in dopamine (DA) metabolism: aggregation of aSyn impairs metabolism of DA, which induces ROS. Figure was constructed using several references (Hampton, 2000; Lindersson et al., 2004; Danzer et al., 2007; Li W. et al., 2007; Prots et al., 2013; Shahmoradian et al., 2018).
Dysfunction of the Ubiquitin-Dependent Proteasome Degradation System
It is expected that in the course of time, large quantities of misfolded aSyn aggregates, and accumulates within the neurons, consequently leading to inclusion formation. In physiological conditions as other proteins, aSyn should be eliminated before forming aggregates. This process normally occurs via different proteolytic pathways within cellular compartments, such as the lysosomal pathway or the ubiquitin dependent proteasome degradation system also known as the ubiquitin proteasome system (UPS), which is a major protein degradation pathway targeting misfolded and unwanted proteins (Hyttinen et al., 2014). UPS is mediated by a tri-enzymatic reaction that leads to the mono-ubiquitination or poly-ubiquitination of the target protein on lysine residues. Poly-ubiquitination is associated with protein degradation by the 29S proteasome and require substrate recognition by 19S proteasome subunit prior to degradation by the proteolytic core of the 20S proteasome complex (Tanaka and Chiba, 1998; Tanaka, 2009).
aSyn is found to be highly ubiquitinated in LBs (Lindersson et al., 2004). It has been indicated that, the accumulation of these proteins marks aSyn for degradation. Major components of the proteolytic 20S proteasome are widely present in LBs/LNs (Lindersson et al., 2004). Thus, it can be considered that these proteins have been sequestered from their physiological role to be degraded and/or eliminated. In addition or even preceding the incorporation of the 20S proteasomes into LBs/LNs, its degradation capacity may be impaired as result of the direct binding of aSyn oligomers (Nielsen et al., 2001; Tanaka, 2009; Emmanouilidou et al., 2010). This was proven in a study which showed that, aSyn oligomers prominently impair proteasomal activity in PC12 cells expressing mutant A53T (Zondler et al., 2017). Thus, supporting the notion that in pathologic cellular environments aSyn cannot be degraded by the proteasome system, and thus contributing to selective vulnerability of cells incorporating LBD pathology. The selective vulnerability of the substantia nigra to proteasome inhibition in PD patients, is thought to occur as a result of proteasomal inhibition by the oligomeric species of aSyn (McNaught and Jenner, 2001; McNaught et al., 2001; Olanow and McNaught, 2006). On the other hand, when proteasomal activity is enhanced, as it was demonstrated with a potent proteasome activator such as Tetramethylpyrazine Analog T-006, aSyn is efficiently degraded not only by the UPS but also in autophagy independent manner (Zhou et al., 2019). Other studies have demonstrated that the inhibition of UPS by aSyn was reversed with the supplementation of Congo red, which is an aSyn oligomerization inhibitor (Emmanouilidou et al., 2010). Thus, the selective vulnerability of aSyn to form aggregates is greatly enhanced by the failures of ubiquitin dependent clearance mechanisms.
Mitochondrial Dysfunction and Membrane Damage
Although, aSyn is abundant in the cytosol, accumulating evidence suggests the presence of endogenous or overexpressed aSyn within mitochondria (Li W. W. et al., 2007; Nakamura et al., 2008; Liu et al., 2009; Loeb et al., 2010). This is thought to lead to aSyn related mitochondrial toxicity. Some studies proposed that aSyn binds to and inhibits the function of a subunit of the mitochondrial protein import system, such as TOM20 (Wiedemann et al., 2004; Wurm et al., 2011; Di Maio et al., 2016). On the other hand, mitochondrial damage can lead to oligomerization and aggregation of aSyn, thus revealing bidirectional toxicity (Betarbet et al., 2000; Cannon et al., 2009).
Moreover, aSyn shows high affinity with voltage-dependent anion channel (VDAC), which is an outer mitochondrial membrane channel that regulates flux of small hydrophilic molecules and calcium in and out of the mitochondria (Lu et al., 2013). Rostovtseva et al. (2015) showed that, aSyn monomers reversibly inhibit VDAC in a voltage dependent manner, creating a high influx of calcium into the mitochondria (Rostovtseva et al., 2015). The high influx of calcium into the mitochondria is thought to be facilitated by aSyn oligomers, which consequently propels mitochondrial swelling, that occurs as a result of depolarisation, and retention of exogenous calcium inside the mitochondrial matrix (Luth et al., 2014). Another study revealed that, overexpression and subsequent oligomerization of aSyn led to mitochondrial fragmentation in dopaminergic SH-SY5Y cell line (Plotegher et al., 2014). Mitochondrial fragmentation was also found to be induced by mutant forms of aSyn, and this is thought to be associated with excessive mitophagy within cells (Choubey et al., 2011; Nakamura et al., 2011). In addition, toxic aSyn species are thought to cause downregulation of complex I in the electron transport chain of mitochondria (Nakamura et al., 2008; Loeb et al., 2010). The effects of aSyn on mitochondria, particularly the inhibition of complex I, induces the production of reactive oxygen species (ROS) and oxidative stress which leads to the impairments of mitochondrial function, with consequent upregulation of apoptotic signals within the cell (Figure 3) (Hsu et al., 2000; Ryan et al., 2015). This suggests loss of mitochondria functions as a key part of a pathogenic process of LBDs.
Cell stability relies on the integrity of its membranes which function as a barrier between intracellular and extracellular environments, controlling the movement of the metabolites. When incubated with natural and synthetic phospholipids or other lipid bilayer membranes in vitro, aSyn rapidly formed aggregates and oligomeric species (Haque et al., 2010; Grey et al., 2011). In similitude to the mitochondria membrane, extracellular oligomeric aSyn species were shown to form pores on the cell membranes. This is thought to facilitate the influx of excessive exogenous calcium within the cytosol, which is a process that often precedes cellular disruption and death (Danzer et al., 2007). In addition, unfolded monomeric and oligomeric species of aSyn are thought to cause calcium dysregulation by interacting with the calcium membrane signaling in a site-specific manner (Angelova et al., 2016). Interestingly, only oligomeric species are capable of inducing cell death. It would be interesting to study if other pre-aggregated and/or aggregated forms have the same effects. Contrastingly, protecting the membrane with new compounds such as NPT100-18A, which can displace aSyn from the lipid bilayer membrane, reduces toxicity of oligomeric aSyn (Wrasidlo et al., 2016). Supporting this, endosulfine-alpha, a compound that selectively binds to membrane-associated aSyn, was found to block the formation of toxic aggregated forms within the cell and reduce the death of dopaminergic neurons in PD (Ysselstein et al., 2017). Thus, demonstrating that abnormal interaction between toxic aSyn with the membranes is linked with the impairment of the cellular membrane function in LBDs.
aSyn and Synaptic Membrane Dysfunction
Synaptic dysfunction is an early event in the evolution of LBDs, as well as in other neurodegenerative diseases (Schulz-Schaeffer, 2010). Considering the physiological role of aSyn at the synapse, it is plausible that overexpressed and aggregated aSyn induces synaptic dysfunction and neurotoxicity. In contrast to physiological aSyn, which facilitates formation of the SNARE complex, and control movement of synaptic vesicles and release of neurotransmitters, oligomeric aSyn, acting via an association with synaptobrevin, inhibits SNARE complex formation. Thus preventing the fusion of synaptic vesicles with the plasma membranes and the subsequent release of neurotransmitters (Choi et al., 2013, 2018; Burré et al., 2018; Masaracchia et al., 2018; Man et al., 2021). Moreover, in COS-7 cells, aSyn overexpression was shown to cause disruption of the Golgi apparatus, the site of synaptic vesicle formation, and thus led to synaptic dysfunction by the depletion of synaptic vesicles (Gosavi et al., 2002). In addition, oligomeric species of aSyn were found to cause reduction in neurotransmitter release by inducing premature intracellular rupture of synaptic vesicles (Danzer et al., 2007). In a similar manner, aSyn oligomers promote increased calcium influx due to cell membrane permeabilization with consequent result of increased neuronal excitotoxicity (Danzer et al., 2007) (Figure 3). Furthermore, aSyn oligomers decreased the stability of microtubules (MT), which are fundamental for axonal transport and the regulation of neuronal homeostasis (Goldstein et al., 2008; Prots et al., 2013).
In vitro studies demonstrated that the interaction of aSyn with 3,4-dihydroxyphenylacetaldehyde (DOPA), a toxic product of dopamine degradation, promotes aSyn oligomerization (Burke, 2003). The consequence of this interaction is the permeabilization of cholesterol-rich lipid membranes, including synaptic vesicles (Burke, 2003). Altogether, these studies suggest a synergistic effect of DOPA formation and aSyn toxicity and may explain the selective vulnerability of dopaminergic neurons to the rupture of synaptic vesicles (Burke, 2003; Plotegher et al., 2017; Lima et al., 2019). Overall, the main effects of aggregated and oligomeric forms of aSyn are mediated by the reduction of neurotransmitter release, inhibition of synaptic vesicle recycling, enlargement of synaptic vesicle, loss of presynaptic proteins, and redistribution of the SNARE proteins (Chung et al., 2009; Garcia-Reitbock et al., 2010; Nemani et al., 2010). These are key pathogenic features that may underlie the synaptic impairment observed in LBDs.
Endoplasmic Reticulum Stress and Membranes
Endoplasmic reticulum (ER) is the cellular site of protein synthesis, folding, modification, and release to the secretory pathway. Perturbation at any stage of ER-mediated proteostasis leads to ER stress (Hampton, 2000). ER stress is thought to occur early in the development of aSyn pathology. This hypothesis was tested in a study using A53T aSyn mutant expressing PC12 cell lines, which reported a lower effect of aSyn neurotoxicity in cells treated with Salubrinal, a pharmacological inhibitor of ER stress (Smith et al., 2005). In support of this, a study using SH-SY5Y cells showed that ER stress was linked with overexpressed aSyn (Castillo-Carranza et al., 2012). Oligomeric species of aSyn are also associated with increased levels of chronic ER stress transgenic mouse models of synucleinopathy (Colla et al., 2012; Colla, 2019). Moreover, Rab1, which is a protein that regulates the membrane trafficking, was found to be associated with cytoplasmic aSyn inclusions. Its role in aSyn-mediated neurotoxicity was demonstrated by a study using a yeast model of synucleinopathy by showed showing its synergic effects in blocking ER-Golgi vesicular trafficking (Cooper et al., 2006). Thus, the ER may be a primary target of aSyn toxicity.
Neurons may also release toxic aSyn species into the extracellular space (Qiao et al., 2012). These could be taken up by microglia (Figure 4) to prevent further ER stress and toxicity (Park et al., 2008). aSyn synergistically interacts with Toll-like receptor2 (TLR2), which is a potent microglia activator (Qiao et al., 2012; Kim et al., 2013; Stivers et al., 2017).
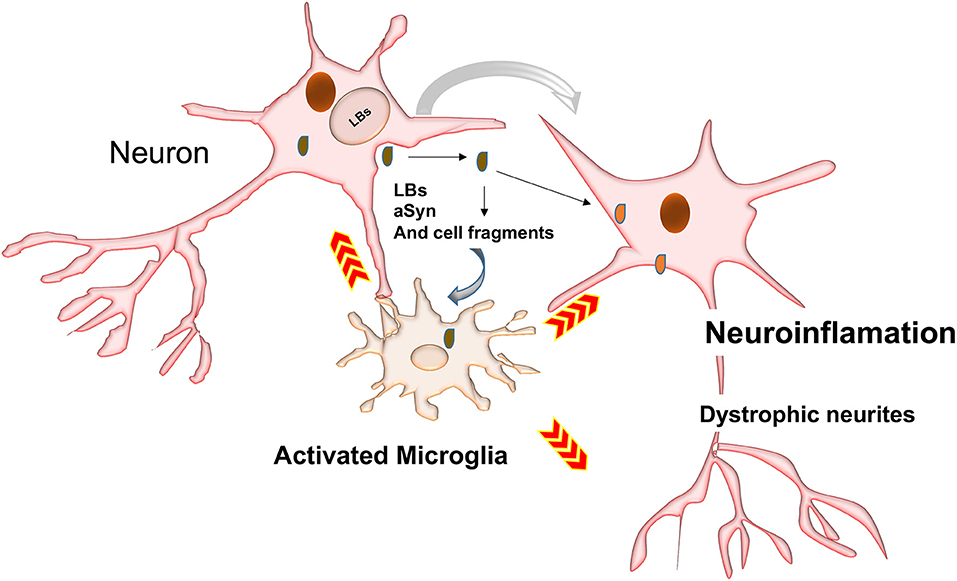
Figure 4. Possible mechanisms involved in activation of phagocytosis and inflammatory responses. LBs may be passively secreted in the extracellular environment following cell death by cell-to-cell transfer. Inclusions and their toxic intermediates may activate astrocytes and glia cells surrounding neurons, resulting in the release of pro-inflammatory cytokines and ROS, which could damage neighboring neurons.
Post-translational Modifications of aSyn
It is likely that loss of the dynamic equilibrium between the soluble, membrane-associated and aggregated forms of aSyn leads to aSyn toxicity and LBD pathology. Exactly how this phenomenon occurs, remains to be determined, and to address this, it is important to know first, what occurs before aSyn forms aggregates. We posit that the remarkable number of PTMs that is reported to occur in aSyn may underlie such disequilibrium. For a relatively small molecule, a large number of PTMs are reported to occur in the protein (Figure 2). PTMs are chemical modifications of amino acid residues that carry a potential to modify protein structure and regulate protein localization, activity, binding affinity, and degradation (Beck-Sickinger and Mörl, 2006; Prabakaran et al., 2012; Schmid et al., 2013). aSyn PTMs, such as ubiquitination, nitration (Oueslati et al., 2010), acetylation (Fauvet et al., 2012; Maltsev et al., 2012; Theillet et al., 2016), truncation (Li et al., 2005; Beyer and Ariza, 2013), SUMOylation (Dorval and Fraser, 2006; Wilkinson and Henley, 2010; Krumova et al., 2011), glutathionylation (Kim et al., 2012; Zhang et al., 2018), Glycosylation (Spiro, 2002), fatty acid acylation (Robinson et al., 1970; Resh, 2016), and phosphorylation, have been reported to occur in aSyn, and more will probably come in the future. To date, these PTMs have been studied in isolation and of all them, the most studied is aSyn phosphorylation at serine 129 (pSer129) (Oueslati et al., 2010). Nevertheless, the precise roles of these PTMs in LBD pathogenesis are the focus of much research, as this is still not well-understood (see Table 2).
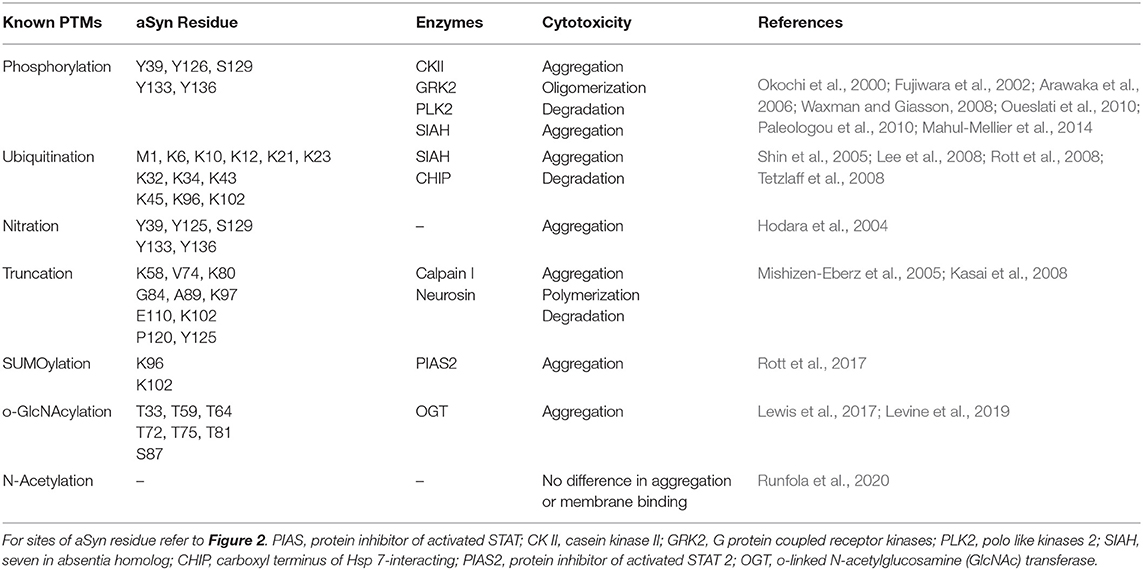
Table 2. Known post-translational modifications in αSyn, sites, enzymes, and effects.
aSyn Phosphorylation
Phosphorylation is a form of reversible PTM that plays an essential role in the regulation of physiological and pathological functions of proteins involved in pathways including but not limited to differentiation, cellular metabolism, gene expression, cell cycle progression, intercellular communication, cellular motility, migration, and cytoskeletal arrangements (Manning et al., 2002a,b). Under physiological conditions, the level of aSyn phosphorylation is very low, and the presence of phosphorylated residues such as tyrosine (pY39, 125) and serine (pSer87) are almost absent (Okochi et al., 2000; Fujiwara et al., 2002; Anderson et al., 2012). In contrast, phosphorylation of threonine (pT125, 133, 135), serine (pSer87, pSer129), and tyrosine (pY125, 133, 136) was detected in pathologically aggregated aSyn. Most of the phosphorylated residues are localized in C terminus of the protein, which is believed to be involved in aSyn pathology, except pSer87 (Chen et al., 2009, 2010; Venda et al., 2010; Xu et al., 2015). The most abundant phosphorylation PTM occurs at pSer129, which is now considered an important marker for synucleinopathies (Figure 2) (Okochi et al., 2000; Fujiwara et al., 2002; Takahashi et al., 2003; Anderson et al., 2006). In addition, this type of aSyn phosphorylation is found to be widespread within the LBs and LNs in corresponding regions of the brain affected in LBDs (Takahashi et al., 2003; Zhou et al., 2011; Walker et al., 2013).
aSyn phosphorylation is also linked to the formation of the toxic species of aSyn, leading to the rapid formation LBs (Kahle et al., 2000a,b; Okochi et al., 2000; Fujiwara et al., 2002; Anderson et al., 2006). In PD, over 90% of endogenous aSyn is found to be phosphorylated at Ser129; in direct contrast to only 4% aSyn phosphorylation detected in healthy individuals (Kahle et al., 2000b; Fujiwara et al., 2002). Similarly, pSer129 was found in post-mortem brain in LBDs subjects and in SH-SY5Y cells that overexpress wild-type aSyn (Swirski et al., 2014). A recent study demonstrated behavioral deficits associated to widespread pSer129 aSyn in brains of wild-type d transgenic mice that overexpress aSyn under Thy1-promoter (Gabrielyan et al., 2021), as pSer129 phospho-memetic reported slower/inhibited aggregation kinetic than wild-type aSyn, despite its prominence in LBs and the preferential phosphorylation of fibrillar aSyn. It has been, suggested that pSer129 is a late-stage event in aggregation leading to the fibrillization of aSyn, and pSer129 is deemed to be crucial for the regulation of disease progression (Paleologou et al., 2010; Waxman and Giasson, 2011; Wales et al., 2013). Other studies suggest different forms of phosphorylation upregulate each other. For instance, both pSer87 and pSer129 were found to be present in LBs aggregates and enhance aSyn toxic membrane interactions in synucleinopathies (Paleologou et al., 2010). Moreover, the binding of pSer129 with vesicular trafficking, cytoskeletal proteins and other phosphorylated linked enzymes, is thought to reinforce the important interrelationships among these pathways in the pathogenesis of LBDs, by interfering with functions of these proteins as well as utilizing such proteins as co-aggregates (McFarland et al., 2009). In addition, proteasomal inhibition (previously described to be a result of aSyn toxicity), was reported to increase the toxicity of aSyn pSer129, as well as its toxicity in dopaminergic neurons in PD. This suggests there is more rapid degradation mechanism of phosphorylated aSyn under physiological conditions (Chau et al., 2009; Oueslati et al., 2013; Mahul-Mellier et al., 2014; Arawaka et al., 2017). Hence, it can be surmised that phosphorylation of aSyn, particularly at residue 129, may be a key factor in the formation of LBs and the development of LBD.
Kinases Involved in aSyn Phosphorylation
In all cellular environments, including the extracellular matrix, phosphorylation process is enzymatically balanced by protein kinases and phosphatases (Bononi et al., 2011; Datta and Ganapathy, 2017). Several kinases are known to be involved in aSyn Ser129 phosphorylation, including casein kinases (CK1, CK2) (Okochi et al., 2000; Ishii et al., 2007; Takahashi et al., 2007; Waxman and Giasson, 2008, 2011), polo like kinase 2 (PLK2, also known as serum inducible kinase (SNK) (Inglis et al., 2009), and G protein-coupled receptor kinases (GRK1, GRK2, GRK5, and GRK6) (Pronin et al., 2000; Arawaka et al., 2006; Sakamoto et al., 2009). The co-localization of GRK5 and aSyn was observed in LBs/LNs inclusions in post-mortem brains of PD patients (Arawaka et al., 2006). However, this was not corroborated in DLB (Arawaka et al., 2006). Moreover, GRK5 knockdown in SH-SY5Y cells failed to completely suppress aSyn phosphorylation (Liu et al., 2010). On the other hand, CK1 and CK2 were found to induce aSyn phosphorylation in mammalian cells, in a yeast model and in rat primary cortical neurons (Okochi et al., 2000; Ishii et al., 2007; Waxman and Giasson, 2008; Zabrocki et al., 2008). Furthermore, oxidative stress increases aSyn phosphorylation mediated by CK2 and its autologues, resulting in an intensification in the formation of LBs/LNs inclusions in SH-SY5Y cells (Pronin et al., 2000). Similarly, PLKs, a family of kinases known to be essential for cellular response to carcinogenesis, stress response, and cell cycle regulation (Ng et al., 2006), were found to participate in aSyn Pser129 (Inglis et al., 2009; Mbefo et al., 2010). Although yet to be confirmed by others, Leucine-rich repeat kinase 2 (LRRK2), an enzyme involved in the pathogenesis of PD, was reported to directly interact with and phosphorylate aSyn (Table 2) (Qing et al., 2009).
aSyn Phosphorylation and Oxidative Stress
Increasing evidence indicates an association between aSyn aggregation and oxidative stress. It has been suggested that the latter has a direct causal relationship with aSyn phosphorylation (Chau et al., 2009). In SH-SY5Y cell line, treatment with low doses of rotenone, an electron transport chain complex I inhibitor that causes an increase in reactive oxygen species (ROS) production, led to increased levels of aSyn pSer129 (Sugeno et al., 2008). Similarly, an in vitro study of a synucleinopathy model demonstrated that aSyn phosphorylation increases when exposed to rotenone and ferrous iron (Perfeito et al., 2014). The increase in aSyn phosphorylation was also observed in SH-SY5Y cells exposed to exogenous toxins such as 6-hydroxidopamine, epoxomicin (a proteasome inhibitor), and paraquat (an environmental toxin that causes oxidative stress) (Chau et al., 2009; Ganapathy et al., 2016). Perfeito et al. (2014), hypothesized that stimuli that promote mitochondrial dysfunction and the formation of ROS are associated with mutant A53T aSyn pSer129. They found correlation between rotenone and aSyn pSer129 in the formation of reactive oxygen species which may underlie neuronal degeneration in LBDs (Perfeito et al., 2014). Thus, biochemical changes linked with LBDs pathogenesis such as oxidative stress, mitochondrial complex I dysfunction, and proteasome impairments, could underlie changes in aSyn phosphorylation (Lee and Trojanowski, 2006; Lashuel et al., 2012; Robson et al., 2018).
Subcellular Distribution of Phosphorylated aSyn
aSyn is a predominantly cytosolic protein but is also found in subcellular organelles such as mitochondria. Whether aSyn localizes within the nucleus is still under debate. Notwithstanding, studies performed in rat and mouse models of synucleinopathy showed that phosphorylated aSyn localized in the nucleus of DA neurons in hydroxydopamine treated rats (Yamada et al., 2004; Wakamatsu et al., 2007). Moreover, pSer129 was shown to modulate the interchange of aSyn between the nucleus and cytoplasm in human neuroglioma cells (Outeiro et al., 2008). This is supported the finding that the translocation dynamics of aSyn between the cytosol and the nucleus was altered upon co-expression of aSyn with various kinases (reviewed in Goncalves and Outeiro, 2013). Correspondingly, cytoplasmic and nuclear shuttling of aSyn was also found to be modulated by GRK5, PLK2, and PLK3 (Table 2) (Monti et al., 2010). In addition, the phospho-mimicking Ser129A and Ser129D were found to localize in the nucleus and increase cellular toxicity (Ganapathy et al., 2016). Pathological insults such as oxidative stress that induce increased aSyn phosphorylation have shown to promote nuclear aSyn localization (Monti et al., 2010; Siddiqui et al., 2012; Ganapathy et al., 2016). Although, we still do not know the exact function of aSyn within the nucleus, it is believed that once aSyn translocates to the nucleus, it acts as a transcriptional regulator via interacting with PGC-1alpha and histones, thus enhancing the toxicity of aSyn in the nucleus (Goers et al., 2003; Kontopoulos et al., 2006; Vasquez et al., 2017; Schaser et al., 2019).
Phosphorylated aSyn as Biomarker in Synucleinopathies
The efficiency of phosphorylated aSyn as a potential biomarker is being widely explored in the diagnosis of LBDs. Quantification of total aSyn in the cerebrospinal fluid (CSF) in patients diagnosed with synucleinopathies and related disorders was proposed as a potential biomarker particularly in differentiating early stages of other related pathologies (Mollenhauer et al., 2008; Hong et al., 2010). However, recent study reported that in LBDs, aSyn pSer129 was found present in the human plasma but not in the CSF (Cariulo et al., 2019). Nevertheless, according to recent discoveries phosphorylated aSyn, improves the performance of aSyn as biomarker on other tissues. For instance, aSyn pSer129 in the retina was proposed as a biomarker for PD, as it was found to accumulate in parallel with aSyn pSer129 in the brain (Ortuño-Lizarán et al., 2018). Moreover, skin biopsies performed in PD subjects, has proven to be a reliable tool for the detection of aSyn pSer129 to discriminate LBDs from controls (Tang et al., 2020). Another important diagnostic test that could also be used for LBDs is the detection of phosphorylated aSyn in the peripheral nervous system, which revealed aggregation of phosphorylated aSyn in large and small fibers (Wang et al., 2012; Doppler et al., 2014; Stewart et al., 2015). Furthermore, aggregation of phosphorylated aSyn was detected in the gastrointestinal tract of PD subjects (Pouclet et al., 2012; Hilton et al., 2014). Although, Foulds et al. (2013), showed that the total number of aSyn in the blood plasma of PD patients were similar to the findings in non-PD controls. The levels of aSyn pSer129 were remarkably higher in PD patients compared to non-PD controls (Foulds et al., 2013; Cariulo et al., 2019). A more recent study has reported aSyn pSer129 in red blood cells, could be used as a potential biomarker, as it was found to be higher in patients with synucleinopathy (Xu et al., 2015; Cariulo et al., 2019; Li et al., 2020). Thus, phosphsphorylated aSyn is perhaps a more applicable candidate for the development of biomarkers for the diagnosis of LBDs.
aSyn Ubiquitination
Intraneuronal compartments are highly specialized to facilitate many operations such as cell cycle, transcription, transduction, antigen presentation, protein degradation, and apoptosis (Hershko and Ciechanover, 1998). Many of these processes require ubiquitin, which is a small protein with 76 amino acid residue that is ubiquitously expressed in the nervous system (Varshavsky, 2001). Ubiquitination, is a PTMs that regulates protein abundance inside the neurons, in a process by which ubiquitin is covalently conjugated to a substrate protein, in the presence of ubiquitin activating enzyme (E1-UBA), ubiquitin conjugating enzyme (E2-UBC) and a ubiquitin ligase (E3-UBL) (Stone, 2016). The process terminates with the formation of an amide bond between ubiquitin and lysine or in other instances with cysteine, serine or threonine residues of the substrate protein (Wang et al., 2007; Vosper et al., 2009). When the conjugation culminates with the single molecule of ubiquitin attached to the residue protein, the process is referred to as mono-ubiquitination. If the process is repeated, the addition of several ubiquitin molecules leads to the formation of a poly-ubiquitin chain. Poly-ubiquitination leads to protein degradation, primarily by the UPS, and it sometimes uses macro-autophagy as an alternative pathway linked to degradation of long-lived ubiquitinated proteins, protein aggregates and organelles (Wang and Pickart, 2005; Hochstrasser, 2006; Li W. et al., 2007; Deshaies and Joazeiro, 2009; Maspero et al., 2011).
Ubiquitinated aSyn is often present in the LBs and LNs inclusions. In fact, multiple lines of evidence have reported the presence of mono-ubiquitinated aSyn as a predominant species in LBs and LNs (Lowe et al., 1990; Wakabayashi et al., 2000; Hasegawa et al., 2002; Tofaris et al., 2003; Anderson et al., 2006). Although, mono-ubiquitination mechanisms that underlie the modulation of aSyn aggregation are still unknown, a study on isolated LBs and LNs from PD patients detected the presence of the seven in absentia homolog (SIAH), an E3 ubiquitin ligase enzyme, previously reported to interact with and ubiquitinated aSyn (Liani et al., 2004; Engelender, 2008; Lee et al., 2008). De facto, lysine 12, 21, and 23 residues, the common targets of aSyn mono ubiquitination, are also ubiquitinated by SIAH (Anderson et al., 2006). This suggests SIAH is involved in the ubiquitination of aSyn, contributing to LBD pathology. However, this remains to be confirmed because some studies have found that only a small fraction of aSyn is ubiquitinated in LBs and LNs (Hasegawa et al., 2002).
In LBDs, neurons are heavily laden with proteasome subunits and ubiquitinated structures, suggesting their involvement and impairments in their degradation (Kuzuhara et al., 1988; Bentea et al., 2017; Lehtonen et al., 2019). Although UPS and the autophagy-lysosomal pathways constitute two major degradation pathways which manage the elimination of misfolded, aggregated, and damaged neuronal proteins (Seglen et al., 1996; Ciechanover and Kwon, 2015; Bustamante et al., 2018; Blasiak et al., 2019), the UPS, chaperone-mediated autophagy (CMA), and macroautophagy all appear to be involved in the proteolytic degradation of aSyn. CMA is proposed to be the main mechanism involved in aSyn clearance (Bennett et al., 1999; Paxinou et al., 2001; Webb et al., 2003).
aSyn SUMOylation
aSyn SUMOylation occurs when a lysine residue of aSyn is covalently linked with a small ubiquitin-like modifier (SUMO) protein. SUMO proteins share structural and mechanistic similarity with ubiquitin (Jentsch and Pyrowolakis, 2000; Dohmen, 2004). Like ubiquitin, SUMO forms covalent links with its substrates through isopeptide bonds between glycine and lysine residues, using similar enzymes to those that mediate the conjugation of ubiquitin (Jentsch and Pyrowolakis, 2000; Dohmen, 2004). This process is believed to be associated with the pathological formation of proteinaceous inclusions within neurons (Dohmen, 2004). In fact, abnormal aSyn SUMOylation was detected in many neurodegenerative diseases such as LBDs, Huntington's disease, AD, amyotrophic lateral sclerosis (Figure 4) (Fei et al., 2006; Kim et al., 2011; Krumova et al., 2011; O'Rourke et al., 2013; Luo et al., 2014; Ochaba et al., 2016). aSyn is conjugated to SUMO at lysine residues (K6, K10, K12, K21, K23, K34, K45, K60, K80, K96, and K102) (Dorval and Fraser, 2006; Kim et al., 2011; Krumova et al., 2011; Coelho-Silva et al., 2017; Iyer and Claessens, 2019). Two studies have demonstrated that aSyn SUMOylation is a signal for its proteasome-mediated degradation and proteasome inhibition led to aggregation of SUMOylated aSyn (Table 2) (Kim et al., 2011; Oh et al., 2011). Additionally, aSyn SUMOylation levels were increased in LBs/LNs in LBD cases (Rott et al., 2017).
It has been demonstrated that E3 SUMO-protein ligase Protein inhibitor of activated STATS (PIAS2) SUMOylates aSyn, thus promoting aSyn aggregation, and an increase in the levels of extracellular aSyn, preceding the formation of the proteinaceous inclusions (Kunadt et al., 2015). It is believed that the increase of extracellular aSyn is a consequence of SUMOylation build up in the endosomal complex, which is linked with aSyn efflux (Kunadt et al., 2015). This process was also demonstrated to occur in the absence of proteasome inhibitors (Kunadt et al., 2015; Rott et al., 2017). SUMOylation is thought to impair the process of mono/poly ubiquitination of aSyn mediated by SIAH and Nedd4 (Liani et al., 2004; Rott et al., 2008, 2017). Two ligases found to be involved in aSyn are SUMOylation, E3 SUMO-protein ligase PIAS, and E3 SUMO-protein ligase PIAS2, as their increase was markedly detected in PD brains (Rott et al., 2017). It is thought that PIAS2 promotes SUMOylation of aSyn, thus inhibiting the action of SIAH and Nedd4 ubiquitin ligases, leading to a decrease of aSyn Ubiquitination, causing aSyn aggregation and LBs formation (Rott et al., 2017). Therefore, it is suggested that these enzymes upregulate aSyn SUMOylation, which promotes overexpression of the protein, thus increasing its aggregation and toxicity.
aSyn Nitration
Protein nitration is a nitrosative stress related PTM that affects the tyrosine (Tyr) residues (Radi, 2004; Bartesaghi and Radi, 2018). aSyn is nitrated at its C terminal in residues Y125, Y133, Y136, and at N terminal residue Y39 (Figure 2) (Giasson et al., 2000; Souza et al., 2000). These Tyrosine residues are found to be necessary for aSyn aggregation, especially under oxidative stress conditions (Souza et al., 2000). Indeed, it is believed that aSyn exposure to nitrating agents leads to the formation of covalently stable oligomers through the oxidation of tyrosine to o,o'-dityrosine (Souza et al., 2000). Although, high rates of oligomerization were observed upon Tyr39 nitration, studies have shown that Tyr125 is more relevant to aSyn dimer formation (Takahashi et al., 2002; Olteanu and Pielak, 2004; Yu et al., 2004; Danielson et al., 2011). Moreover, fibril formation is increased by both monomeric and dimeric-nitrated aSyn (Hodara et al., 2004).
aSyn Nitration-Mediated Interaction With Membranes and Oxidative Stress
The association of aSyn with membranes is thought to lead to peroxidation of polyunsaturated fatty acids, as membrane lipid peroxidation can be triggered by ROS and RNOS. aSyn incubation with 4-hydroxy-2-nonenal (4HNE), a lipid peroxidation marker, leads to the formation of aSyn-HNE adducts (Trostchansky et al., 2006; Qin et al., 2007). Some studies have suggested that the interaction of aSyn with membrane phospholipids and polyunsaturated fatty acids is necessary for the formation of aSyn oligomers (Perrin et al., 2001; Assayag et al., 2007; Beyer and Ariza, 2013).
aSyn also promotes nitric oxides (NO) activity at the plasma membrane, which leads to NOS production. This excess of NOS is thought to inhibit the electron transport chain, thus increasing the production of superoxide (Lashuel et al., 2002). This creates a vicious cycle that facilitates nitration and oxidation that potentiate cellular dysfunction and loss of viability (Radi et al., 2002; Radi, 2004). Likewise, aSyn oxidation and nitration impedes the autophagy-mediated degradation, therefore inhibiting the degradation of nitrated aSyn (Martinez-Vicente et al., 2008). This resistance to degradation of nitrated aSyn promotes its intracellular increase in half life and consequently its concentration, which boosts the toxicity of the protein, stimulating neuronal degeneration (Chavarria and Souza, 2013).
aSyn o-GlcNAcylation
O-beta-N-acetyl-D-glucosaminyl transferase (O-GlcNAc/ OGT) is an enzyme that catalyzes the glycosylation of the serine and threonine aa residues in a dynamic that is opposed by O-GlcNAcase (OGA) (Hart et al., 2007). O-GlcNAc of aSyn was detected on multiple threonine residues (33, 34, 54, 59, 64, 72, 75, 81, and 87) of aSyn extracted from human tissues of individuals with LBD and mouse models of synucleinopathies (Wang et al., 2009, 2010, 2017; Alfaro et al., 2012; Morris et al., 2015). Although, glycation exerts a large inhibitory effect on unmodified aSyn, it was, however, found to promote aggregation of pathological aSyn and increase the toxicity of the protein (Levine et al., 2019). Moreover, in vivo and in vitro studies performed in Drosophila and in A53T mouse model have shown that glycation potentiates the toxicity of aSyn by affecting its N-terminal domain, impairing the clearance of aSyn. This is thought to decrease the membrane binding properties of aSyn and enhancing the aggregation of its oligomeric species, altogether resulting in the impairment of synaptic transmission (Table 2) (Vicente Miranda et al., 2017). In DLB, aSyn o-GlcNAcylation was found to induce the formation and aggregation of its oligomeric forms (Zhang et al., 2017). Conversely, other studies revealed that, use of glycation inhibitors decreased aSyn aggregation, restored aSyn clearance, and abated clinical phenotype of the disease (Vicente Miranda et al., 2017).
aSyn Truncation
Truncated forms of aSyn are ubiquitous in LBs inclusions. This is thought to be associated with abnormal proteolysis of aSyn at the C terminus of the protein (Tofaris et al., 2003, 2006; Periquet et al., 2007). aSyn C-terminal truncation is thought to occur through the lysosomal-associated pathways (Murray et al., 2003; Sevlever et al., 2008; Tsujimura et al., 2015; Sorrentino et al., 2018). Nevertheless, the mechanism by which truncated aSyn contributes to the formation of LBs and LNs inclusions remains to be elucidated. However, it is believed, that the amount of truncated aSyn species represents about 15% of all aSyn in LBs/LNs inclusions, and they may act as seeds that enhance the aggregation of aSyn to form the LBs/LNs under physiological pH (van der Wateren et al., 2018; Zhang et al., 2019). In vivo studies using transgenic mice and Drosophila that overexpress αSyn, also demonstrated that truncated aSyn is associated with increased aggregation of aSyn and neurotoxicity (Tofaris et al., 2003, 2006; Periquet et al., 2007). Interestingly, truncated aSyn was found to be important for the prion-like propagation property of toxic aSyn, which is believed to be at the root of the phenotypic diversity of the synucleinopathies (Terada et al., 2018). Indeed, in brains of DLB subjects the full-length as well as truncated and insoluble aggregates of aSyn are evident in LBs and LNs (Baba et al., 1998).
Furthermore, enzymes such as calpin1, cathepsin D, matrix metalloproteinase 3, and neurosin are involved in aSyn truncation (Table 2) (Iwata et al., 2003; Mishizen-Eberz et al., 2003; Dufty et al., 2007; Sevlever et al., 2008; Choi et al., 2011; Weber et al., 2019). aSyn is thought to be a substrate of calpain 1, a neural calcium-activated protease. Calpain 1 cleaves wild-type aSyn at amino acids 57 and 73, 74, and 83 located within the NAC region. This process promotes aggregation, therefore calpain 1 is thought to generate aggregation-prone forms of aSyn promoting fibril formation (Mishizen-Eberz et al., 2003). Similarly, neurosin, a protease that is ubiquitously expressed in brain cells and cleaves aSyn after aa 80 also increases the propensity of aSyn to form aggregates (Iwata et al., 2003; Kasai et al., 2008). These observations strongly support the involvement of truncated aSyn in the formation LBs/LNs and selective neuronal degeneration seen in DLB.
Conclusions
aSyn aggregation and transformation into LBs and LNs underlie the onset and progression of LBDs and related synucleinopathies. It represents a failure in proteostasis network directly linked to maintain biologically active aSyn and reduce toxic forms of the protein. Multiple lines of evidence suggest PTMs are key in aSyn aggregation and toxicity across the synucleinopathies. The most studied PTMs associated with the LBDs are phosphorylation, ubiquitination SUMOylation, Nitration, o-GlcNacylation, and truncation. Under normal physiological conditions PTMs are known regulators of protein biology which is important for localization and function. However, recent findings demonstrate that they are associated with toxic species of aSyn with the consequent formation of LBs and LNs, disrupting cellular homeostasis leading to neuronal cell death.
Further understanding of the exact role of PTMs in LBDs is warranted. To date, few reports have largely focused on PD. Most studies reported thus far, are concentrated on non-acetylated aSyn, which needs more validation. It is therefore difficult to assess the full modulation of PTMs with the existing models of PTMs. Studies on specific PTMs using purified forms of aSyn are also essential, as this would permit more detailed study of each PTM to uncover the mechanisms of how PTMs modify aggregation and form LBDs. It is still unclear what role aSyn PTMs play in the propagation of LBD pathology. Nonetheless, the reports so far of the biological relevance of aSyn PTMs, has highlighted their importance in the pathogenesis of disease. This could lead to development of interventions aimed at reducing the accumulation of LBs and slowing or preventing LBDs. PTMs represent a more selective potential therapeutic target that addresses LB accumulation and therefore more specific for DLB, rather than simply be duplicative of AD or PD directed treatments.
Author Contributions
NM: established concept, searched references (~300 articles from PubMed, PNAS databases, selecting 270), curation of data, writing original draft, and editing several version. LS: reviewing and editing drafts. RK: reviewing references and editing several drafts of the article. All authors contributed to the article and approved the submitted version.
Funding
NM was supported by a Ph.D. Studentship grant from INAGBE-ANGOLA, Bairro Talatona. Rua do MAT, complexo administrativo, classicos de talatona, Edificio 4, second floor, Luanda Angola. RK's work was supported by grants from the UK Medical Research Council (MRC, G0500247), Newcastle Center for Brain Aging and Vitality (BBSRC, EPSRC, ESRC and MRC, LLHW). The NBTR was funded by a grant from the UK MRC (G0400074) with further support from the Newcastle NIHR Biomedical Research Center in Aging and Age-Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust, and a grant from the Alzheimer's Society and ART as part of the Brains for Dementia Research Project and BrainNet II Europe, Germany.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Professor Stephen Atwood for proofreading, Dr. Viktor Korolchuk, Dr. David Koss, and Professor Tiago F. Outeiro for initiating key discussions in this research topic.
Abbreviations
DLB, Dementia with Lewy Bodies; LBDs, Lewy body disorders; LBs, Lewy bodies; LNs, Lewy neurites; PD, Parkinson's disease; PDD, Parkinson's disease dementia; ETC, electron transport chain; PTMs, post-translational modifications; aSyn, Alpha-synuclein; AA, amino acid; CSP, α-Cysteine Stirring Protein alpha; SNARE, Soluble NSF attachment receptor; UPS, Ubiquitin proteasome system; VDAC, voltage-dependent anion channel; ROS, reactive oxygen species; RNOS, reactive nitrogen species; MT, Microtubules; ER, Endoplasmic reticulum; TLR2, Toll-like receptor2.
References
Aarsland, D., Andersen, K., Larsen, J. P., Lolk, A., and Kragh-Sorensen, P. (2003). Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch. Neurol. 60, 387–392. doi: 10.1001/archneur.60.3.387
Agliardi, C., Meloni, M., Guerini, F. R., Zanzottera, M., Bolognesi, E., Baglio, F., et al. (2021). Oligomeric alpha-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson's disease. Neurobiol. Dis. 148:105185. doi: 10.1016/j.nbd.2020.105185
Alfaro, J. F., Gong, C. X., Monroe, M. E., Aldrich, J. T., Clauss, T. R., Purvine, S. O., et al. (2012). Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. U.S.A. 109, 7280–7285. doi: 10.1073/pnas.1200425109
Allen Reish, H. E., and Standaert, D. G. (2015). Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons Dis. 5, 1–19. doi: 10.3233/JPD-140491
Anderson, E. J., Katunga, L. A., and Willis, M. S. (2012). Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 39, 179–193. doi: 10.1111/j.1440-1681.2011.05641.x
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Angelova, P. R., Ludtmann, M. H., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Arawaka, S., Sato, H., Sasaki, A., Koyama, S., and Kato, T. (2017). Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 5:48. doi: 10.1186/s40478-017-0452-6
Arawaka, S., Wada, M., Goto, S., Karube, H., Sakamoto, M., Ren, C. H., et al. (2006). The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J. Neurosci. 26, 9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006
Assayag, K., Yakunin, E., Loeb, V., Selkoe, D. J., and Sharon, R. (2007). Polyunsaturated fatty acids induce alpha-synuclein-related pathogenic changes in neuronal cells. Am. J. Pathol. 171, 2000–2011. doi: 10.2353/ajpath.2007.070373
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bartesaghi, S., and Radi, R. (2018). Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 14, 618–625. doi: 10.1016/j.redox.2017.09.009
Beck-Sickinger, A. G., and Mörl, K. (2006). Posttranslational modification of proteins. expanding nature's inventory. by Christopher T. Walsh. Angew. Chem. Int. Ed. 45, 1020–1020. doi: 10.1002/anie.200585363
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bentea, E., Verbruggen, L., and Massie, A. (2017). The proteasome inhibition model of Parkinson's disease. J. Parkinson's Dis. 7, 31–63. doi: 10.3233/JPD-160921
Bernstein, H.-G., Johnson, M., Perry, R. H., LeBeau, F. E. N., Dobrowolny, H., Bogerts, B., et al. (2011). Partial loss of parvalbumin-containing hippocampal interneurons in dementia with Lewy bodies. Neuropathology 31, 1–10. doi: 10.1111/j.1440-1789.2010.01117.x
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Beyer, K., and Ariza, A. (2013). alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 47, 509–524. doi: 10.1007/s12035-012-8330-5
Blasiak, J., Pawlowska, E., Szczepanska, J., and Kaarniranta, K. (2019). Interplay between autophagy and the ubiquitin-proteasome system and its role in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci. 20:210. doi: 10.3390/ijms20010210
Bodner, C. R., Dobson, C. M., and Bax, A. (2009). Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 390, 775–790. doi: 10.1016/j.jmb.2009.05.066
Bononi, A., Agnoletto, C., De Marchi, E., Marchi, S., Patergnani, S., Bonora, M., et al. (2011). Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011:329098. doi: 10.4061/2011/329098
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H., and Del Tredici, K. (2004). Stages in the development of Parkinson's disease-related pathology. Cell. Tissue Res. 318, 121–134. doi: 10.1007/s00441-004-0956-9
Braak, H., Sandmann-Keil, D., Gai, W., and Braak, E. (1999). Extensive axonal Lewy neurites in Parkinson's disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neurosci. Lett. 265, 67–69. doi: 10.1016/S0304-3940(99)00208-6
Brown, D. R. (2007). Interactions between metals and alpha-synuclein–function or artefact? FEBS J. 274, 3766–3774. doi: 10.1111/j.1742-4658.2007.05917.x
Buchman, V. L., Hunter, H. J., Pinon, L. G., Thompson, J., Privalova, E. M., Ninkina, N. N., et al. (1998). Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system. J. Neurosci. 18, 9335–9341. doi: 10.1523/JNEUROSCI.18-22-09335.1998
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P. J., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676. doi: 10.1073/pnas.1315346111
Burke, W. J. (2003). 3,4-dihydroxyphenylacetaldehyde: a potential target for neuroprotective therapy in Parkinson's disease. Curr. Drug. Targets CNS Neurol. Disord. 2, 143–148. doi: 10.2174/1568007033482913
Burre, J. (2015). The synaptic function of alpha-synuclein. J. Parkinsons Dis. 5, 699–713. doi: 10.3233/JPD-150642
Burré, J., Sharma, M., and Südhof, T. C. (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Burré, J., Sharma, M., and Südhof, T. C. (2018). Cell biology and pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 8:a024091. doi: 10.1101/cshperspect.a024091
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Burre, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Sudhof, T. C. (2013). Properties of native brain alpha-synuclein. Nature 498, E4–6; discussion E6-7. doi: 10.1038/nature12125
Bussell, R. Jr., and Eliezer, D. (2001). Residual structure and dynamics in Parkinson's disease-associated mutants of alpha-synuclein. J. Biol. Chem. 276, 45996–46003. doi: 10.1074/jbc.M106777200
Bussell, R. Jr., and Eliezer, D. (2003). A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 329, 763–778. doi: 10.1016/S0022-2836(03)00520-5
Bustamante, H. A., González, A. E., Cerda-Troncoso, C., Shaughnessy, R., Otth, C., Soza, A., et al. (2018). Interplay between the autophagy-lysosomal pathway and the ubiquitin-proteasome system: a target for therapeutic development in Alzheimer's disease. Front. Cell. Neurosci. 12:126. doi: 10.3389/fncel.2018.00126
Cannon, J. R., Tapias, V., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson's disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
Cariulo, C., Martufi, P., Verani, M., Azzollini, L., Bruni, G., Weiss, A., et al. (2019). Phospho-S129 alpha-synuclein is present in human plasma but not in cerebrospinal fluid as determined by an ultrasensitive immunoassay. Front. Neurosci. 13:889. doi: 10.3389/fnins.2019.00889
Castillo-Carranza, D. L., Zhang, Y., Guerrero-Munoz, M. J., Kayed, R., Rincon-Limas, D. E., and Fernandez-Funez, P. (2012). Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem. Res. 37, 1707–1717. doi: 10.1007/s11064-012-0780-7
Chau, K. Y., Ching, H. L., Schapira, A. H., and Cooper, J. M. (2009). Relationship between alpha synuclein phosphorylation, proteasomal inhibition and cell death: relevance to Parkinson's disease pathogenesis. J. Neurochem. 110, 1005–1013. doi: 10.1111/j.1471-4159.2009.06191.x
Chavarria, C., and Souza, J. M. (2013). Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 533, 25–32. doi: 10.1016/j.abb.2013.02.009
Chen, L., Periquet, M., Wang, X., Negro, A., McLean, P. J., Hyman, B. T., et al. (2009). Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 119, 3257–3265. doi: 10.1172/JCI39088
Chen, R. H. C., Wislet-Gendebien, S., Samuel, F., Visanji, N. P., Zhang, G., Marsilio, D., et al. (2013). α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 288, 7438–7449. doi: 10.1074/jbc.M112.439497
Chen, S. C.-C., Chen, F.-C., and Li, W.-H. (2010). Phosphorylated and nonphosphorylated serine and threonine residues evolve at different rates in mammals. Mol. Biol. Evol. 27, 2548–2554. doi: 10.1093/molbev/msq142
Choi, B. K., Choi, M. G., Kim, J. Y., Yang, Y., Lai, Y., Kweon, D. H., et al. (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U.S.A. 110, 4087–4092. doi: 10.1073/pnas.1218424110
Choi, D. H., Kim, Y. J., Kim, Y. G., Joh, T. H., Beal, M. F., and Kim, Y. S. (2011). Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J. Biol. Chem. 286, 14168–14177. doi: 10.1074/jbc.M111.222430
Choi, M. G., Kim, M. J., Kim, D. G., Yu, R., Jang, Y. N., and Oh, W. J. (2018). Sequestration of synaptic proteins by alpha-synuclein aggregates leading to neurotoxicity is inhibited by small peptide. PLoS ONE 13:e0195339. doi: 10.1371/journal.pone.0195339
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.M110.132514
Chung, C. Y., Koprich, J. B., Siddiqi, H., and Isacson, O. (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 29, 3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009
Ciechanover, A., and Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47:e147. doi: 10.1038/emm.2014.117
Coelho-Silva, L., Stephens Gary, J., and Cimarosti, H. (2017). SUMOylation and calcium signalling: potential roles in the brain and beyond. Neuronal. Signal. 1:NS20160010. doi: 10.1042/NS20160010
Colla, E. (2019). Linking the endoplasmic reticulum to Parkinson's disease and alpha-synucleinopathy. Front. Neurosci. 13, 560. doi: 10.3389/fnins.2019.00560
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328. doi: 10.1126/science.1129462
Crowther, R. A., Jakes, R., Spillantini, M. G., and Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 436, 309–312. doi: 10.1016/S0014-5793(98)01146-6
Danielson, S. R., Held, J. M., Oo, M., Riley, R., Gibson, B. W., and Andersen, J. K. (2011). Quantitative mapping of reversible mitochondrial Complex I cysteine oxidation in a Parkinson disease mouse model. J. Biol. Chem. 286, 7601–7608. doi: 10.1074/jbc.M110.190108
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
Datta, I., and Ganapathy, K. (2017). “Modification of α-Synuclein by Phosphorylation: A Pivotal Event in the Cellular Pathogenesis of Parkinson's Disease.” Protein Phosphorylation, Claude Prigent. IntechOpen. doi: 10.5772/intechopen.70405
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
Deshaies, R. J., and Joazeiro, C. A. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. doi: 10.1146/annurev.biochem.78.101807.093809
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci. Transl. Med. 8:342ra378. doi: 10.1126/scitranslmed.aaf3634
Dohmen, R. J. (2004). SUMO protein modification. Biochim. Biophys. Acta 1695, 113–131. doi: 10.1016/j.bbamcr.2004.09.021
Doppler, K., Ebert, S., Uceyler, N., Trenkwalder, C., Ebentheuer, J., Volkmann, J., et al. (2014). Cutaneous neuropathy in Parkinson's disease: a window into brain pathology. Acta Neuropathol. 128, 99–109. doi: 10.1007/s00401-014-1284-0
Dorval, V., and Fraser, P. E. (2006). Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 281, 9919–9924. doi: 10.1074/jbc.M510127200
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., et al. (2007). Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 170, 1725–1738. doi: 10.2353/ajpath.2007.061232
Dugger, B. N., and Dickson, D. W. (2010). Cell type specific sequestration of choline acetyltransferase and tyrosine hydroxylase within Lewy bodies. Acta Neuropathol. 120, 633–639. doi: 10.1007/s00401-010-0739-1
Emmanouilidou, E., Stefanis, L., and Vekrellis, K. (2010). Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 31, 953–968. doi: 10.1016/j.neurobiolaging.2008.07.008
Engelender, S. (2008). Ubiquitination of α-synuclein and autophagy in Parkinson's disease. Autophagy 4, 372–374. doi: 10.4161/auto.5604
Fairfoul, G., McGuire, L. I., Pal, S., Ironside, J. W., Neumann, J., Christie, S., et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 3, 812–818. doi: 10.1002/acn3.338
Fauvet, B., Mbefo, M. K., Fares, M. B., Desobry, C., Michael, S., Ardah, M. T., et al. (2012). alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. doi: 10.1074/jbc.M111.318949
Fei, E., Jia, N., Yan, M., Ying, Z., Sun, Q., Wang, H., et al. (2006). SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 347, 406–412. doi: 10.1016/j.bbrc.2006.06.092
Fernandez, C. O., Hoyer, W., Zweckstetter, M., Jares-Erijman, E. A., Subramaniam, V., Griesinger, C., et al. (2004). NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. Embo J. 23, 2039–2046. doi: 10.1038/sj.emboj.7600211
Fortin, D. L., Nemani, V. M., Voglmaier, S. M., Anthony, M. D., Ryan, T. A., and Edwards, R. H. (2005). Neural activity controls the synaptic accumulation of alpha-synuclein. J. Neurosci. 25, 10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005
Foulds, P. G., Diggle, P., Mitchell, J. D., Parker, A., Hasegawa, M., Masuda-Suzukake, M., et al. (2013). A longitudinal study on alpha-synuclein in blood plasma as a biomarker for Parkinson's disease. Sci. Rep. 3:2540. doi: 10.1038/srep02540
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gabrielyan, L., Liang, H., Minalyan, A., Hatami, A., John, V., and Wang, L. (2021). Behavioral deficits and brain α-Synuclein and phosphorylated serine-129 α-Synuclein in male and female mice overexpressing human α-Synuclein. J. Alzheimer's Dis. 79, 875–893. doi: 10.3233/JAD-200983
Ganapathy, K., Datta, I., Sowmithra, S., Joshi, P., and Bhonde, R. (2016). Influence of 6-hydroxydopamine toxicity on alpha-synuclein phosphorylation, resting vesicle expression, and vesicular dopamine release. J. Cell Biochem. 117, 2719–2736. doi: 10.1002/jcb.25570
Garcia-Reitbock, P., Anichtchik, O., Bellucci, A., Iovino, M., Ballini, C., Fineberg, E., et al. (2010). SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain 133, 2032–2044. doi: 10.1093/brain/awq132
Geng, X., Lou, H., Wang, J., Li, L., Swanson, A. L., Sun, M., et al. (2011). α-Synuclein binds the K(ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Endocrinol. Metab. 300, E276–286. doi: 10.1152/ajpendo.00262.2010
Gerst, J. E. (1999). SNAREs and SNARE regulators in membrane fusion and exocytosis. Cell Mol. Life Sci. 55, 707–734. doi: 10.1007/s000180050328
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Goda, Y. (1997). SNAREs and regulated vesicle exocytosis. Proc. Natl. Acad. Sci. U.S.A. 94, 769–772. doi: 10.1073/pnas.94.3.769
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Goers, J., Manning-Bog, A. B., McCormack, A. L., Millett, I. S., Doniach, S., Di Monte, D. A., et al. (2003). Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42, 8465–8471. doi: 10.1021/bi0341152
Goldstein, A. Y., Wang, X., and Schwarz, T. L. (2008). Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 18, 495–503. doi: 10.1016/j.conb.2008.10.003
Goncalves, S., and Outeiro, T. F. (2013). Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol. Neurobiol. 47, 1081–1092. doi: 10.1007/s12035-013-8406-x
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Grey, M., Linse, S., Nilsson, H., Brundin, P., and Sparr, E. (2011). Membrane interaction of alpha-synuclein in different aggregation states. J. Parkinsons Dis. 1, 359–371. doi: 10.3233/JPD-2011-11067
Gurd, J. M., Herzberg, L., Joachim, C., Marshall, J. C., Jobst, K., McShane, R. H., et al. (2000). Dementia with Lewy bodies: a pure case. Brain Cogn. 44, 307–323. doi: 10.1006/brcg.1999.1124
Halliday, G. M., Li, Y. W., Blumbergs, P. C., Joh, T. H., Cotton, R. G., Howe, P. R., et al. (1990). Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann. Neurol. 27, 373–385. doi: 10.1002/ana.410270405
Hampton, R. Y. (2000). ER stress response: getting the UPR hand on misfolded proteins. Curr. Biol. 10, R518–R521. doi: 10.1016/S0960-9822(00)00583-2
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Haque, F., Pandey, A. P., Cambrea, L. R., Rochet, J.-C., and Hovis, J. S. (2010). Adsorption of alpha-synuclein on lipid bilayers: modulating the structure and stability of protein assemblies. J. Phys. Chem. B 114, 4070–4081. doi: 10.1021/jp1006704
Harper, J. D., Lieber, C. M., and Lansbury, P. T. Jr. (1997). Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem. Biol. 4, 951–959. doi: 10.1016/S1074-5521(97)90303-3
Hart, G. W., Housley, M. P., and Slawson, C. (2007). Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446:1017. doi: 10.1038/nature05815
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M., et al. (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hashimoto, M., Hsu, L. J., Rockenstein, E., Takenouchi, T., Mallory, M., and Masliah, E. (2002). alpha-Synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J. Biol. Chem. 277, 11465–11472. doi: 10.1074/jbc.M111428200
Hawk, B. J. D., Khounlo, R., and Shin, Y. K. (2019). Alpha-synuclein continues to enhance SNARE-dependent vesicle docking at exorbitant concentrations. Front. Neurosci. 13:216. doi: 10.3389/fnins.2019.00216
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hilton, D., Stephens, M., Kirk, L., Edwards, P., Potter, R., Zajicek, J., et al. (2014). Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson's disease. Acta Neuropathol. 127, 235–241. doi: 10.1007/s00401-013-1214-6
Hochstrasser, M. (2006). Lingering mysteries of ubiquitin-chain assembly. Cell 124, 27–34. doi: 10.1016/j.cell.2005.12.025
Hodara, R., Norris, E. H., Giasson, B. I., Mishizen-Eberz, A. J., Lynch, D. R., Lee, V. M., et al. (2004). Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753. doi: 10.1074/jbc.M408906200
Hong, Z., Shi, M., Chung, K. A., Quinn, J. F., Peskind, E. R., Galasko, D., et al. (2010). DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 133, 713–726. doi: 10.1093/brain/awq008
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., et al. (2000). alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410. doi: 10.1016/S0002-9440(10)64553-1
Hyttinen, J. M. T., Amadio, M., Viiri, J., Pascale, A., Salminen, A., and Kaarniranta, K. (2014). Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 18, 16–28. doi: 10.1016/j.arr.2014.07.002
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S. S., Schobel, S., Frigon, N. L., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602. doi: 10.1074/jbc.C800206200
Invernizzi, G., Papaleo, E., Sabate, R., and Ventura, S. (2012). Protein aggregation: mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 44, 1541–1554. doi: 10.1016/j.biocel.2012.05.023
Ishii, A., Nonaka, T., Taniguchi, S., Saito, T., Arai, T., Mann, D., et al. (2007). Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 581, 4711–4717. doi: 10.1016/j.febslet.2007.08.067
Iwata, A., Maruyama, M., Akagi, T., Hashikawa, T., Kanazawa, I., Tsuji, S., et al. (2003). Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 12, 2625–2635. doi: 10.1093/hmg/ddg283
Iyer, A., and Claessens, M. (2019). Disruptive membrane interactions of alpha-synuclein aggregates. Biochim. Biophys. Acta Proteins Proteom. 1867, 468–482. doi: 10.1016/j.bbapap.2018.10.006
Jakes, R., Spillantini, M. G., and Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32. doi: 10.1016/0014-5793(94)00395-5
Jellinger, K. A., and Korczyn, A. D. (2018). Are dementia with Lewy bodies and Parkinson's disease dementia the same disease? BMC Med. 16:34. doi: 10.1186/s12916-018-1016-8
Jentsch, S., and Pyrowolakis, G. (2000). Ubiquitin and its kin: how close are the family ties? Trends Cell Biol. 10, 335–342. doi: 10.1016/S0962-8924(00)01785-2
Jin, H., Kanthasamy, A., Ghosh, A., Yang, Y., Anantharam, V., and Kanthasamy, A. G. (2011). α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 31, 2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011
Kahle, P. J. (2008). alpha-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol. 115, 87–95. doi: 10.1007/s00401-007-0302-x
Kahle, P. J., Neumann, M., Ozmen, L., and Haass, C. (2000a). Physiology and pathophysiology of alpha-synuclein. cell culture and transgenic animal models based on a Parkinson's disease-associated protein. Ann. N. Y. Acad. Sci. 920, 33–41. doi: 10.1111/j.1749-6632.2000.tb06902.x
Kahle, P. J., Neumann, M., Ozmen, L., Müller, V., Jacobsen, H., Schindzielorz, A., et al. (2000b). Subcellular localization of wild-type and Parkinson's disease-associated mutant α-synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000
Kasai, T., Tokuda, T., Yamaguchi, N., Watanabe, Y., Kametani, F., Nakagawa, M., et al. (2008). Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56. doi: 10.1016/j.neulet.2008.02.057
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4:1562. doi: 10.1038/ncomms2534
Kim, K., Kim, S. H., Kim, J., Kim, H., and Yim, J. (2012). Glutathione s-transferase omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J. Biol. Chem. 287, 6628–6641. doi: 10.1074/jbc.M111.291179
Kim, Y. M., Jang, W. H., Quezado, M. M., Oh, Y., Chung, K. C., Junn, E., et al. (2011). Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. J. Neurol. Sci. 307, 157–161. doi: 10.1016/j.jns.2011.04.015
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Kruger, R., Kuhn, W., Leenders, K. L., Sprengelmeyer, R., Muller, T., Woitalla, D., et al. (2001). Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology 56, 1355–1362. doi: 10.1212/WNL.56.10.1355
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Kunadt, M., Eckermann, K., Stuendl, A., Gong, J., Russo, B., Strauss, K., et al. (2015). Extracellular vesicle sorting of alpha-Synuclein is regulated by sumoylation. Acta Neuropathol. 129, 695–713. doi: 10.1007/s00401-015-1408-1
Kuzuhara, S., Mori, H., Izumiyama, N., Yoshimura, M., and Ihara, Y. (1988). Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 75, 345–353. doi: 10.1007/BF00687787
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2012). The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., et al. (2002). Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102. doi: 10.1016/S0022-2836(02)00735-0
Lautenschlager, J., Kaminski, C. F., and Kaminski Schierle, G. S. (2017). alpha-Synuclein - regulator of exocytosis, endocytosis, or both? Trends Cell Biol. 27, 468–479. doi: 10.1016/j.tcb.2017.02.002
Lavedan, C., Leroy, E., Dehejia, A., Buchholtz, S., Dutra, A., Nussbaum, R. L., et al. (1998). Identification, localization and characterization of the human γ-synuclein gene. Hum. Genet. 103, 106–112. doi: 10.1007/s004390050792
Lee, J. T., Wheeler, T. C., Li, L., and Chin, L. S. (2008). Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 17, 906–917. doi: 10.1093/hmg/ddm363
Lee, V. M., and Trojanowski, J. Q. (2006). Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52, 33–38. doi: 10.1016/j.neuron.2006.09.026
Lehtonen, Š., Sonninen, T.-M., Wojciechowski, S., Goldsteins, G., and Koistinaho, J. (2019). Dysfunction of cellular proteostasis in Parkinson's disease. Front. Neurosci. 13:457. doi: 10.3389/fnins.2019.00457
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honore, A., Rozas, N., et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Levine, P. M., Galesic, A., Balana, A. T., Mahul-Mellier, A. L., Navarro, M. X., De Leon, C. A., et al. (2019). alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 116, 1511–1519. doi: 10.1073/pnas.1808845116
Lewis, Y. E., Galesic, A., Levine, P. M., De Leon, C. A., Lamiri, N., Brennan, C. K., et al. (2017). O-GlcNAcylation of α-Synuclein at Serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 12, 1020–1027. doi: 10.1021/acschembio.7b00113
Li, W., Tu, D., Brunger, A. T., and Ye, Y. (2007). A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 446, 333–337. doi: 10.1038/nature05542
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167. doi: 10.1073/pnas.0406976102
Li, W. W., Yang, R., Guo, J. C., Ren, H. M., Zha, X. L., Cheng, J. S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Li, X.-Y., Yang, W., Li, X., Li, X.-R., Li, W., Song, Q., et al. (2020). phosphorylated alpha-synuclein in red blood cells as a potential diagnostic biomarker for multiple system atrophy: a pilot study. Parkinson's Dis. 2020:8740419. doi: 10.1155/2020/8740419
Liani, E., Eyal, A., Avraham, E., Shemer, R., Szargel, R., Berg, D., et al. (2004). Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 5500–5505. doi: 10.1073/pnas.0401081101
Lima, V. A., do Nascimento, L. A., Eliezer, D., and Follmer, C. (2019). Role of Parkinson's disease-linked mutations and N-Terminal acetylation on the oligomerization of alpha-synuclein induced by 3,4-Dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. 10, 690–703. doi: 10.1021/acschemneuro.8b00498
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. doi: 10.1074/jbc.M306390200
Liu, G., Zhang, C., Yin, J., Li, X., Cheng, F., Li, Y., et al. (2009). alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 454, 187–192. doi: 10.1016/j.neulet.2009.02.056
Liu, P., Wang, X., Gao, N., Zhu, H., Dai, X., Xu, Y., et al. (2010). G protein-coupled receptor kinase 5, overexpressed in the alpha-synuclein up-regulation model of Parkinson's disease, regulates bcl-2 expression. Brain Res. 1307, 134–141. doi: 10.1016/j.brainres.2009.10.036
Loeb, V., Yakunin, E., Saada, A., and Sharon, R. (2010). The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J. Biol. Chem. 285, 7334–7343. doi: 10.1074/jbc.M109.061051
Lou, X., Kim, J., Hawk, B. J., and Shin, Y. K. (2017). alpha-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 474, 2039–2049. doi: 10.1042/BCJ20170200
Lowe, J., McDermott, H., Landon, M., Mayer, R. J., and Wilkinson, K. D. (1990). Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 161, 153–160. doi: 10.1002/path.1711610210
Lu, L., Zhang, C., Cai, Q., Lu, Q., Duan, C., Zhu, Y., et al. (2013). Voltage-dependent anion channel involved in the α-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim. Biophys. Sin. 45, 170–178. doi: 10.1093/abbs/gms114
Luo, H.-B., Xia, Y.-Y., Shu, X.-J., Liu, Z.-C., Feng, Y., Liu, X.-H., et al. (2014). SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA. 111, 16586–16591. doi: 10.1073/pnas.1417548111
Luth, E. S., Stavrovskaya, I. G., Bartels, T., Kristal, B. S., and Selkoe, D. J. (2014). Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507. doi: 10.1074/jbc.M113.545749
Mahul-Mellier, A. L., Fauvet, B., Gysbers, A., Dikiy, I., Oueslati, A., Georgeon, S., et al. (2014). c-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum. Mol. Genet. 23, 2858–2879. doi: 10.1093/hmg/ddt674
Maltsev, A. S., Ying, J., and Bax, A. (2012). Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 51, 5004–5013. doi: 10.1021/bi300642h
Man, W. K., Tahirbegi, B., Vrettas, M. D., Preet, S., Ying, L., Vendruscolo, M., et al. (2021). The docking of synaptic vesicles on the presynaptic membrane induced by alpha-synuclein is modulated by lipid composition. Nat. Commun. 12:927. doi: 10.1038/s41467-021-21027-4
Manning, G., Plowman, G. D., Hunter, T., and Sudarsanam, S. (2002a). Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520. doi: 10.1016/S0968-0004(02)02179-5
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002b). The protein kinase complement of the human genome. Science 298, 1912–1934. doi: 10.1126/science.1075762
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Martinez, J., Moeller, I., Erdjument-Bromage, H., Tempst, P., and Lauring, B. (2003). Parkinson's disease-associated alpha-synuclein is a calmodulin substrate. J. Biol. Chem. 278, 17379–17387. doi: 10.1074/jbc.M209020200
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Marui, W., Iseki, E., Kato, M., Akatsu, H., and Kosaka, K. (2004). Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer's disease. Acta Neuropathol. 108, 121–128. doi: 10.1007/s00401-004-0869-4
Marui, W., Iseki, E., Kato, M., and Kosaka, K. (2003). Degeneration of tyrosine hydroxylase-immunoreactive neurons in the cerebral cortex and hippocampus of patients with dementia with Lewy bodies. Neurosci. Lett. 340, 185–188. doi: 10.1016/S0304-3940(03)00101-0
Masaracchia, C., Hnida, M., Gerhardt, E., Lopes da Fonseca, T., Villar-Pique, A., Branco, T., et al. (2018). Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol. Commun. 6:79. doi: 10.1186/s40478-018-0578-1
Maspero, E., Mari, S., Valentini, E., Musacchio, A., Fish, A., Pasqualato, S., et al. (2011). Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 12, 342–349. doi: 10.1038/embor.2011.21
Mattila, P. M., Rinne, J. O., Helenius, H., Dickson, D. W., and Roytta, M. (2000). Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol. 100, 285–290. doi: 10.1007/s004019900168
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., et al. (2010). Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822. doi: 10.1074/jbc.M109.081950
McFarland, N. R., Fan, Z., Xu, K., Schwarzschild, M. A., Feany, M. B., Hyman, B. T., et al. (2009). Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524. doi: 10.1097/NEN.0b013e3181a24b53
McKeith, I., Mintzer, J., Aarsland, D., Burn, D., Chiu, H., Cohen-Mansfield, J., et al. (2004). Dementia with Lewy bodies. Lancet Neurol. 3, 19–28. doi: 10.1016/S1474-4422(03)00619-7
McKeith, I. G. (2006). Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J. Alzheimers Dis. 9, 417–423. doi: 10.3233/JAD-2006-9S347
McKeith, I. G., Boeve, B. F., Dickson, D. W., Halliday, G., Taylor, J.-P., Weintraub, D., et al. (2017). Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89, 88–100. doi: 10.1212/WNL.0000000000004058
McKeith, I. G., Burn, D. J., Ballard, C. G., Collerton, D., Jaros, E., Morris, C. M., et al. (2003). Dementia with Lewy bodies. Semin. Clin. Neuropsychiatry 8, 46–57. doi: 10.1053/scnp.2003.50006
McNaught, K. S., and Jenner, P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci. Lett. 297, 191–194. doi: 10.1016/S0304-3940(00)01701-8
McNaught, K. S., Olanow, C. W., Halliwell, B., Isacson, O., and Jenner, P. (2001). Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat. Rev. Neurosci. 2, 589–594. doi: 10.1038/35086067
Miake, H., Mizusawa, H., Iwatsubo, T., and Hasegawa, M. (2002). Biochemical characterization of the core structure of alpha-synuclein filaments. J. Biol. Chem. 277, 19213–19219. doi: 10.1074/jbc.M110551200
Mishizen-Eberz, A. J., Guttmann, R. P., Giasson, B. I., Day, G. A. 3rd, Hodara, R., Ischiropoulos, H., et al. (2003). Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 86, 836–847. doi: 10.1046/j.1471-4159.2003.01878.x
Mishizen-Eberz, A. J., Norris, E. H., Giasson, B. I., Hodara, R., Ischiropoulos, H., Lee, V. M., et al. (2005). Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry 44, 7818–7829. doi: 10.1021/bi047846q
Mollenhauer, B., Cullen, V., Kahn, I., Krastins, B., Outeiro, T. F., Pepivani, I., et al. (2008). Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp. Neurol. 213, 315–325. doi: 10.1016/j.expneurol.2008.06.004
Monti, B., Gatta, V., Piretti, F., Raffaelli, S. S., Virgili, M., and Contestabile, A. (2010). Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of α-Synuclein. Neurotox. Res. 17, 130–141. doi: 10.1007/s12640-009-9090-5
Moors, T. E., Maat, C. A., Niedieker, D., Mona, D., Petersen, D., Timmermans-Huisman, E., et al. (2018). Detailed structural orchestration of Lewy pathology in Parkinson's disease as revealed by 3D multicolor STED microscopy. bioRxiv 470476. doi: 10.1101/470476
Morris, M., Sanchez, P. E., Verret, L., Beagle, A. J., Guo, W., Dubal, D., et al. (2015). Network dysfunction in α-synuclein transgenic mice and human Lewy body dementia. Ann. Clin. Transl. Neurol. 2, 1012–1028. doi: 10.1002/acn3.257
Murray, I. V., Giasson, B. I., Quinn, S. M., Koppaka, V., Axelsen, P. H., Ischiropoulos, H., et al. (2003). Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42, 8530–8540. doi: 10.1021/bi027363r
Nakajo, S., Omata, K., Aiuchi, T., Shibayama, T., Okahashi, I., Ochiai, H., et al. (1990). Purification and characterization of a novel brain-specific 14-kDa protein. J. Neurochem. 55, 2031–2038. doi: 10.1111/j.1471-4159.1990.tb05792.x
Nakamura, K., Nemani, V. M., Azarbal, F., Skibinski, G., Levy, J. M., Egami, K., et al. (2011). Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 286, 20710–20726. doi: 10.1074/jbc.M110.213538
Nakamura, K., Nemani, V. M., Wallender, E. K., Kaehlcke, K., Ott, M., and Edwards, R. H. (2008). Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 28, 12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., et al. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. doi: 10.1016/j.neuron.2009.12.023
Ng, S. S., Papadopoulou, K., and McInerny, C. J. (2006). Regulation of gene expression and cell division by Polo-like kinases. Curr. Genet. 50, 73–80. doi: 10.1007/s00294-006-0077-y
Nielsen, M. S., Vorum, H., Lindersson, E., and Jensen, P. H. (2001). Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 276, 22680–22684. doi: 10.1074/jbc.M101181200
Ochaba, J., Monteys, A. M., O'Rourke, J. G., Reidling, J. C., Steffan, J. S., Davidson, B. L., et al. (2016). PIAS1 regulates mutant huntingtin accumulation and huntington's disease-associated phenotypes in vivo. Neuron 90, 507–520. doi: 10.1016/j.neuron.2016.03.016
Oh, Y., Kim, Y. M., Mouradian, M. M., and Chung, K. C. (2011). Human Polycomb protein 2 promotes alpha-synuclein aggregate formation through covalent SUMOylation. Brain Res. 1381, 78–89. doi: 10.1016/j.brainres.2011.01.039
Okochi, M., Walter, J., Koyama, A., Nakajo, S., Baba, M., Iwatsubo, T., et al. (2000). Constitutive phosphorylation of the Parkinson's disease associated alpha-synuclein. J. Biol. Chem. 275, 390–397. doi: 10.1074/jbc.275.1.390
Olanow, C. W., and McNaught, K. S. (2006). Ubiquitin-proteasome system and Parkinson's disease. Mov. Disord. 21, 1806–1823. doi: 10.1002/mds.21013
Olteanu, A., and Pielak, G. J. (2004). Peroxidative aggregation of alpha-synuclein requires tyrosines. Protein Sci. 13, 2852–2856. doi: 10.1110/ps.04947204
O'Rourke, J. G., Gareau, J. R., Ochaba, J., Song, W., Rasko, T., Reverter, D., et al. (2013). SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell Rep. 4, 362–375. doi: 10.1016/j.celrep.2013.06.034
Ortuño-Lizarán, I., Beach, T. G., Serrano, G. E., Walker, D. G., Adler, C. H., and Cuenca, N. (2018). Phosphorylated α-synuclein in the retina is a biomarker of Parkinson's disease pathology severity. Mov. Disord. 33, 1315–1324. doi: 10.1002/mds.27392
Ostrerova, N., Petrucelli, L., Farrer, M., Mehta, N., Choi, P., Hardy, J., et al. (1999). alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 19, 5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999
Oueslati, A., Fournier, M., and Lashuel, H. A. (2010). “Chapter 7 - Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies,” in Progress in Brain Research, Vol. 183, eds. A. Björklund and M. A. Cenci (Elsevier), 115–145. doi: 10.1016/S0079-6123(10)83007-9
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–3954. doi: 10.1073/pnas.1309991110
Outeiro, T. F., Koss, D. J., Erskine, D., Walker, L., Kurzawa-Akanbi, M., Burn, D., et al. (2019). Dementia with Lewy bodies: an update and outlook. Mol. Neurodegener. 14:5. doi: 10.1186/s13024-019-0306-8
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., et al. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE 3:e1867. doi: 10.1371/annotation/9282f173-df82-4b70-9120-b4e62b3dacb1
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Park, J. Y., Paik, S. R., Jou, I., and Park, S. M. (2008). Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson's disease. Glia 56, 1215–1223. doi: 10.1002/glia.20691
Park, S. M., Jung, H. Y., Kim, T. D., Park, J. H., Yang, C. H., and Kim, J. (2002). Distinct roles of the N-terminal-binding domain and the C-terminal-solubilizing domain of alpha-synuclein, a molecular chaperone. J. Biol. Chem. 277, 28512–28520. doi: 10.1074/jbc.M111971200
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol. Aging 35, 2180.e2181–2185. doi: 10.1016/j.neurobiolaging.2014.03.024
Paxinou, E., Chen, Q., Weisse, M., Giasson, B. I., Norris, E. H., Rueter, S. M., et al. (2001). Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 21, 8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001
Peng, X., Tehranian, R., Dietrich, P., Stefanis, L., and Perez, R. G. (2005). Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118, 3523–3530. doi: 10.1242/jcs.02481
Perfeito, R., Lázaro, D. F., Outeiro, T. F., and Rego, A. C. (2014). Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 62, 51–59. doi: 10.1016/j.mcn.2014.08.002
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Perrin, R. J., Woods, W. S., Clayton, D. F., and George, J. M. (2001). Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 276, 41958–41962. doi: 10.1074/jbc.M105022200
Plotegher, N., Berti, G., Ferrari, E., Tessari, I., Zanetti, M., Lunelli, L., et al. (2017). DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 7:40699. doi: 10.1038/srep40699
Plotegher, N., Gratton, E., and Bubacco, L. (2014). Number and Brightness analysis of alpha-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim. Biophys. Acta 1840, 2014–2024. doi: 10.1016/j.bbagen.2014.02.013
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pouclet, H., Lebouvier, T., Coron, E., Neunlist, M., and Derkinderen, P. (2012). Lewy pathology in gastric and duodenal biopsies in Parkinson's disease. Mov. Disord. 27:708. doi: 10.1002/mds.24993
Prabakaran, S., Lippens, G., Steen, H., and Gunawardena, J. (2012). Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 565–583. doi: 10.1002/wsbm.1185
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522. doi: 10.1074/jbc.M003542200
Prots, I., Veber, V., Brey, S., Campioni, S., Buder, K., Riek, R., et al. (2013). alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754. doi: 10.1074/jbc.M113.451815
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Qiao, S., Luo, J. H., and Jin, J. H. (2012). [Role of microglial activation induced by alpha-synuclein in pathogenesis of Parkinson's disease]. Zhejiang Da Xue Xue Bao Yi Xue Ban. 41, 210–214. doi: 10.1093/jmcb/mjr011
Qin, Z., Hu, D., Han, S., Reaney, S. H., Di Monte, D. A., and Fink, A. L. (2007). Effect of 4-Hydroxy-2-nonenal modification on α-Synuclein aggregation. J. Biol. Chem. 282, 5862–5870. doi: 10.1074/jbc.M608126200
Qing, H., Wong, W., McGeer, E. G., and McGeer, P. L. (2009). Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 387, 149–152. doi: 10.1016/j.bbrc.2009.06.142
Radi, R. (2004). Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 101, 4003–4008. doi: 10.1073/pnas.0307446101
Radi, R., Cassina, A., Hodara, R., Quijano, C., and Castro, L. (2002). Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 33, 1451–1464. doi: 10.1016/S0891-5849(02)01111-5
Resh, M. D. (2016). Fatty acylation of proteins: the long and the short of it. Prog. Lipid. Res. 63, 120–131. doi: 10.1016/j.plipres.2016.05.002
Robinson, A. B., McKerrow, J. H., and Cary, P. (1970). Controlled deamidation of peptides and proteins: an experimental hazard and a possible biological timer. Proc. Natl. Acad. Sci. U.S.A. 66, 753–757. doi: 10.1073/pnas.66.3.753
Robson, E., Tweedy, C., Manzanza, N., Taylor, J. P., Atkinson, P., Randall, F., et al. (2018). Impaired fast network oscillations and mitochondrial dysfunction in a mouse model of alpha-synucleinopathy (A30P). Neuroscience 377, 161–173. doi: 10.1016/j.neuroscience.2018.02.032
Rodriguez-Araujo, G., Nakagami, H., Takami, Y., Katsuya, T., Akasaka, H., Saitoh, S., et al. (2015). Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci. Rep. 5:12081. doi: 10.1038/srep12081
Rostovtseva, T. K., Gurnev, P. A., Protchenko, O., Hoogerheide, D. P., Yap, T. L., Philpott, C. C., et al. (2015). alpha-Synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 290, 18467–18477. doi: 10.1074/jbc.M115.641746
Rott, R., Szargel, R., Haskin, J., Shani, V., Shainskaya, A., Manov, I., et al. (2008). Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328. doi: 10.1074/jbc.M704809200
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA. 114, 13176–13181. doi: 10.1073/pnas.1704351114
Ruipérez, V., Darios, F., and Davletov, B. (2010). Alpha-synuclein, lipids and Parkinson's disease. Prog. Lipid. Res. 49, 420–428. doi: 10.1016/j.plipres.2010.05.004
Runfola, M., De Simone, A., Vendruscolo, M., Dobson, C. M., and Fusco, G. (2020). The N-terminal acetylation of α-Synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep. 10:204. doi: 10.1038/s41598-019-57023-4
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Sakamoto, M., Arawaka, S., Hara, S., Sato, H., Cui, C., Machiya, Y., et al. (2009). Contribution of endogenous G-protein-coupled receptor kinases to Ser129 phosphorylation of alpha-synuclein in HEK293 cells. Biochem. Biophys. Res. Commun. 384, 378–382. doi: 10.1016/j.bbrc.2009.04.130
Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S. W., et al. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9:10919. doi: 10.1038/s41598-019-47227-z
Schmid, A. W., Fauvet, B., Moniatte, M., and Lashuel, H. A. (2013). Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell. Proteomics 12, 3543–3558. doi: 10.1074/mcp.R113.032730
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 120, 131–143. doi: 10.1007/s00401-010-0711-0
Seglen, P. O., Berg, T. O., Blankson, H., Fengsrud, M., Holen, I., and Strømhaug, P. E. (1996). “Structural aspects of autophagy,” in Intracellular Protein Catabolism, eds K. Suzuki and J.S. Bond (Boston, MA: Springer US), 103–111. doi: 10.1007/978-1-4613-0335-0_12
Sevcsik, E., Trexler, A. J., Dunn, J. M., and Rhoades, E. (2011). Allostery in a disordered protein: oxidative modifications to alpha-synuclein act distally to regulate membrane binding. J. Am. Chem. Soc. 133, 7152–7158. doi: 10.1021/ja2009554
Sevlever, D., Jiang, P., and Yen, S. H. (2008). Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687. doi: 10.1021/bi800699v
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Graff-Meyer, A., Hench, J., Moors, T., et al. (2018). Lewy pathology in Parkinson's disease consists of a crowded organellar, membranous medley. bioRxiv 137976. doi: 10.1101/137976
Shahnawaz, M., Tokuda, T., Waragai, M., Mendez, N., Ishii, R., Trenkwalder, C., et al. (2017). Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 74, 163–172. doi: 10.1001/jamaneurol.2016.4547
Sharma, S. K., Chorell, E., Steneberg, P., Vernersson-Lindahl, E., Edlund, H., and Wittung-Stafshede, P. (2015). Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 5:12531. doi: 10.1038/srep12531
Shin, Y., Klucken, J., Patterson, C., Hyman, B. T., and McLean, P. J. (2005). The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 280, 23727–23734. doi: 10.1074/jbc.M503326200
Shults, C. W. (2006). Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 103, 1661–1668. doi: 10.1073/pnas.0509567103
Siddiqui, A., Chinta, S. J., Mallajosyula, J. K., Rajagopolan, S., Hanson, I., Rane, A., et al. (2012). Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson's disease. Free Radic. Biol. Med. 53, 993–1003. doi: 10.1016/j.freeradbiomed.2012.05.024
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811. doi: 10.1093/hmg/ddi396
Sorrentino, Z. A., Vijayaraghavan, N., Gorion, K.-M., Riffe, C. J., Strang, K. H., Caldwell, J., et al. (2018). Physiological carboxy-truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 293, 18914–18932. doi: 10.1074/jbc.RA118.005603
Souza, J. M., Giasson, B. I., Chen, Q., Lee, V. M., and Ischiropoulos, H. (2000). Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 275, 18344–18349. doi: 10.1074/jbc.M000206200
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Spiro, R. G. (2002). Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R−56R. doi: 10.1093/glycob/12.4.43R
Stewart, T., Sossi, V., Aasly, J. O., Wszolek, Z. K., Uitti, R. J., Hasegawa, K., et al. (2015). Phosphorylated α-synuclein in Parkinson's disease: correlation depends on disease severity. Acta Neuropathol. Commun. 3:7. doi: 10.1186/s40478-015-0185-3
Stivers, N. S., Pelisch, N., Orem, B. C., Williams, J., Nally, J. M., and Stirling, D. P. (2017). The toll-like receptor 2 agonist Pam3CSK4 is neuroprotective after spinal cord injury. Exp. Neurol. 294, 1–11. doi: 10.1016/j.expneurol.2017.04.012
Stone, S. L. (2016). “Chapter 26 - ubiquitination of plant transcription factors,” in Plant Transcription Factors, ed D. H. Gonzalez. (Boston, MA: Academic Press), 395–409. doi: 10.1016/B978-0-12-800854-6.00026-9
Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., et al. (2008). Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 283, 23179–23188. doi: 10.1074/jbc.M802223200
Swirski, M., Miners, J. S., de Silva, R., Lashley, T., Ling, H., Holton, J., et al. (2014). Evaluating the relationship between amyloid-β and α-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson's disease. Alzheimer's Res. Therap. 6:77. doi: 10.1186/s13195-014-0077-y
Takahashi, M., Kanuka, H., Fujiwara, H., Koyama, A., Hasegawa, M., Miura, M., et al. (2003). Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 336, 155–158. doi: 10.1016/S0304-3940(02)01258-2
Takahashi, M., Ko, L. W., Kulathingal, J., Jiang, P., Sevlever, D., and Yen, S. H. (2007). Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur. J. Neurosci. 26, 863–874. doi: 10.1111/j.1460-9568.2007.05736.x
Takahashi, T., Yamashita, H., Nakamura, T., Nagano, Y., and Nakamura, S. (2002). Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 938, 73–80. doi: 10.1016/S0006-8993(02)02498-8
Tanaka, K. (2009). The proteasome: overview of structure and functions. Proc. JPN. Acad. Ser. B,. Phys. Biol. Sci. 85, 12–36. doi: 10.2183/pjab.85.12
Tanaka, K., and Chiba, T. (1998). The proteasome: a protein-destroying machine. Genes Cells 3, 499–510. doi: 10.1046/j.1365-2443.1998.00207.x
Tang, A., Rajan, S., Penzlin, A. I., Galvis, V., Freeman, R., and Gibbons, C. (2020). Cutaneous phosphorylated α-synuclein as a biomarker for the α-synucleinopathies: a meta-analysis (1178). Neurology 94:1178. doi: 10.1212/WNL.0000000000011941
Terada, M., Suzuki, G., Nonaka, T., Kametani, F., Tamaoka, A., and Hasegawa, M. (2018). The effect of truncation on prion-like properties of alpha-synuclein. J. Biol. Chem. 293, 13910–13920. doi: 10.1074/jbc.RA118.001862
Tetzlaff, J. E., Putcha, P., Outeiro, T. F., Ivanov, A., Berezovska, O., Hyman, B. T., et al. (2008). CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem. 283, 17962–17968. doi: 10.1074/jbc.M802283200
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O'Connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., and Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. doi: 10.1074/jbc.M308041200
Trostchansky, A., Lind, S., Hodara, R., Oe, T., Blair, I. A., Ischiropoulos, H., et al. (2006). Interaction with phospholipids modulates alpha-synuclein nitration and lipid-protein adduct formation. Biochem. J. 393, 343–349. doi: 10.1042/BJ20051277
Tsujimura, A., Taguchi, K., Watanabe, Y., Tatebe, H., Tokuda, T., Mizuno, T., et al. (2015). Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous alpha-synuclein fibrils. Neurobiol. Dis. 73, 244–253. doi: 10.1016/j.nbd.2014.10.011
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
van der Wateren, I. M., Knowles, T. P. J., Buell, A. K., Dobson, C. M., and Galvagnion, C. (2018). C-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH. Chem. Sci. 9, 5506–5516. doi: 10.1039/C8SC01109E
Varshavsky, A. (2001). “Ubiquitin,” in Encyclopedia of Genetics, eds S. Brenner and J. H. Miller (New York, NY: Academic Press), 2091–2093. doi: 10.1006/rwgn.2001.1348
Vasquez, V., Mitra, J., Hegde, P. M., Pandey, A., Sengupta, S., Mitra, S., et al. (2017). Chromatin-bound oxidized α-Synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson's disease. J. Alzheimer's Dis. 60, S133–S150. doi: 10.3233/JAD-170342
Venda, L. L., Cragg, S. J., Buchman, V. L., and Wade-Martins, R. (2010). alpha-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 33, 559–568. doi: 10.1016/j.tins.2010.09.004
Vicente Miranda, H., Szego, E. M., Oliveira, L. M. A., Breda, C., Darendelioglu, E., de Oliveira, R. M., et al. (2017). Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 140, 1399–1419. doi: 10.1093/brain/awx056
Villar-Pique, A., Lopes da Fonseca, T., and Outeiro, T. F. (2016). Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J. Neurochem. 139(Suppl. 1), 240–255. doi: 10.1111/jnc.13249
Vosper, J. M. D., McDowell, G. S., Hindley, C. J., Fiore-Heriche, C. S., Kucerova, R., Horan, I., et al. (2009). Ubiquitylation on canonical and non-canonical sites targets the transcription factor neurogenin for ubiquitin-mediated proteolysis. J. Biol. Chem. 284, 15458–15468. doi: 10.1074/jbc.M809366200
Wakabayashi, K., Engelender, S., Yoshimoto, M., Tsuji, S., Ross, C. A., and Takahashi, H. (2000). Synphilin-1 is present in Lewy bodies in Parkinson's disease. Ann. Neurol. 47, 521–523. doi: 10.1002/1531-8249(200004)47:4<521::AID-ANA18=3.0.CO;2-B
Wakabayashi, K., Hansen, L. A., and Masliah, E. (1995). Cortical Lewy body-containing neurons are pyramidal cells: laser confocal imaging of double-immunolabeled sections with anti-ubiquitin and SMI32. Acta Neuropathol. 89, 404–408. doi: 10.1007/BF00307643
Wakamatsu, M., Ishii, A., Ukai, Y., Sakagami, J., Iwata, S., Ono, M., et al. (2007). Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. J. Neurosci. Res. 85, 1819–1825. doi: 10.1002/jnr.21310
Wales, P., Pinho, R., Lazaro, D. F., and Outeiro, T. F. (2013). Limelight on alpha-synuclein: pathological and mechanistic implications in neurodegeneration. J. Parkinsons Dis. 3, 415–459. doi: 10.3233/JPD-130216
Walker, D. G., Lue, L. F., Adler, C. H., Shill, H. A., Caviness, J. N., Sabbagh, M. N., et al. (2013). Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 240, 190–204. doi: 10.1016/j.expneurol.2012.11.020
Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M., and Teplow, D. B. (1997). Amyloid beta-protein fibrillogenesis. detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372. doi: 10.1074/jbc.272.35.22364
Wang, M., and Pickart, C. M. (2005). Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. Embo J. 24, 4324–4333. doi: 10.1038/sj.emboj.7600895
Wang, S., Yang, F., Petyuk, V. A., Shukla, A. K., Monroe, M. E., Gritsenko, M. A., et al. (2017). Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer's disease. J. Pathol. 243, 78–88. doi: 10.1002/path.4929
Wang, X., Herr, R. A., Chua, W. J., Lybarger, L., Wiertz, E. J., and Hansen, T. H. (2007). Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624. doi: 10.1083/jcb.200611063
Wang, Y., Shi, M., Chung, K. A., Zabetian, C. P., Leverenz, J. B., Berg, D., et al. (2012). Phosphorylated α-Synuclein in Parkinson's disease. Sci. Transl. Med. 4:121ra120-121ra120. doi: 10.1126/scitranslmed.3002566
Wang, Z., Park, K., Comer, F., Hsieh-Wilson, L. C., Saudek, C. D., and Hart, G. W. (2009). Site-specific GlcNAcylation of human erythrocyte proteins: potential biomarker(s) for diabetes. Diabetes 58, 309–317. doi: 10.2337/db08-0994
Wang, Z., Udeshi, N. D., O'Malley, M., Shabanowitz, J., Hunt, D. F., and Hart, G. W. (2010). Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell Proteom. 9, 153–160. doi: 10.1074/mcp.M900268-MCP200
Waxman, E. A., and Giasson, B. I. (2008). Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 67, 402–416. doi: 10.1097/NEN.0b013e3186fc995
Waxman, E. A., and Giasson, B. I. (2011). Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J. Neurosci. Res. 89, 231–247. doi: 10.1002/jnr.22537
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Weber, J. J., Pereira Sena, P., Singer, E., and Nguyen, H. P. (2019). Killing two angry birds with one stone: autophagy activation by inhibiting calpains in neurodegenerative diseases and beyond. Biomed. Res. Int. 2019:4741252. doi: 10.1155/2019/4741252
Wiedemann, N., Frazier, A. E., and Pfanner, N. (2004). The protein import machinery of mitochondria. J. Biol. Chem. 279, 14473–14476. doi: 10.1074/jbc.R400003200
Wilkinson, K. A., and Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145. doi: 10.1042/BJ20100158
Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M., and Biere, A. L. (1999). alpha-synuclein fibrillogenesis is nucleation-dependent. implications for the pathogenesis of Parkinson's disease. J. Biol. Chem. 274, 19509–19512. doi: 10.1074/jbc.274.28.19509
Wrasidlo, W., Tsigelny, I. F., Price, D. L., Dutta, G., Rockenstein, E., Schwarz, T. C., et al. (2016). A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson's disease. Brain 139, 3217–3236. doi: 10.1093/brain/aww238
Wurm, C. A., Neumann, D., Lauterbach, M. A., Harke, B., Egner, A., Hell, S. W., et al. (2011). Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc. Natl. Acad. Sci. U.S.A. 108, 13546–13551. doi: 10.1073/pnas.1107553108
Xu, Y., Deng, Y., and Qing, H. (2015). The phosphorylation of α-synuclein: development and implication for the mechanism and therapy of the Parkinson's disease. J. Neurochem. 135, 4–18. doi: 10.1111/jnc.13234
Yamada, M., Iwatsubo, T., Mizuno, Y., and Mochizuki, H. (2004). Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J. Neurochem. 91, 451–461. doi: 10.1111/j.1471-4159.2004.02728.x
Yang, S. K., Chen, W., Su, C. H., and Liu, C. H. (2018). Incidence and comorbidity of dementia with lewy bodies: a population-based Cohort study. Behav. Neurol. 2018:7631951. doi: 10.1155/2018/7631951
Yang, W., Wang, X., Duan, C., Lu, L., and Yang, H. (2013). Alpha-synuclein overexpression increases phospho-protein phosphatase 2A levels via formation of calmodulin/Src complex. Neurochem. Int. 63, 180–194. doi: 10.1016/j.neuint.2013.06.010
Yoshimoto, M., Iwai, A., Kang, D., Otero, D. A., Xia, Y., and Saitoh, T. (1995). NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc. Natl. Acad. Sci. U.S.A. 92, 9141–9145. doi: 10.1073/pnas.92.20.9141
Ysselstein, D., Dehay, B., Costantino, I. M., McCabe, G. P., Frosch, M. P., George, J. M., et al. (2017). Endosulfine-alpha inhibits membrane-induced α-synuclein aggregation and protects against α-synuclein neurotoxicity. Acta Neuropathol. Commun. 5:3. doi: 10.1186/s40478-016-0403-7
Yu, S., Zuo, X., Li, Y., Zhang, C., Zhou, M., Zhang, Y. A., et al. (2004). Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 367, 34–39. doi: 10.1016/j.neulet.2004.05.118
Zabrocki, P., Bastiaens, I., Delay, C., Bammens, T., Ghillebert, R., Pellens, K., et al. (2008). Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780. doi: 10.1016/j.bbamcr.2008.06.010
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, J., Lei, H., Chen, Y., Ma, Y. T., Jiang, F., Tan, J., et al. (2017). Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci. Lett. 655, 90–94. doi: 10.1016/j.neulet.2017.06.034
Zhang, J., Li, X., and Li, J.-D. (2019). The Roles of Post-translational Modifications on α-Synuclein in the pathogenesis of Parkinson's diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Zhang, J., Ye, Z. W., Singh, S., Townsend, D. M., and Tew, K. D. (2018). An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 120, 204–216. doi: 10.1016/j.freeradbiomed.2018.03.038
Zhou, H., Shao, M., Guo, B., Li, C., Lu, Y., Yang, X., et al. (2019). Tetramethylpyrazine analogue T-006 promotes the clearance of alpha-synuclein by enhancing proteasome activity in Parkinson's disease models. Neurotherapeutics 16, 1225–1236. doi: 10.1007/s13311-019-00759-8
Zhou, J., Broe, M., Huang, Y., Anderson, J. P., Gai, W. P., Milward, E. A., et al. (2011). Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson's disease. Acta Neuropathol. 121, 695–704. doi: 10.1007/s00401-011-0815-1
Zhu, M., Qin, Z. J., Hu, D., Munishkina, L. A., and Fink, A. L. (2006). Alpha-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry 45, 8135–8142. doi: 10.1021/bi052584t
Keywords: alpha synuclein, neuronal membrane, Lewy body disorders, post-translational modifications, dementia
Citation: Manzanza NdO, Sedlackova L and Kalaria RN (2021) Alpha-Synuclein Post-translational Modifications: Implications for Pathogenesis of Lewy Body Disorders. Front. Aging Neurosci. 13:690293. doi: 10.3389/fnagi.2021.690293
Received: 02 April 2021; Accepted: 24 May 2021;
Published: 25 June 2021.
Edited by:
Jiajie Diao, University of Cincinnati, United StatesReviewed by:
Carlo Scialò, International School for Advanced Studies (SISSA), ItalyMassimiliano Mirabella, Catholic University of the Sacred Heart, Rome, Italy
Copyright © 2021 Manzanza, Sedlackova and Kalaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nelson de Oliveira Manzanza, bi5kLm8ubWFuemFuemEyQG5ld2Nhc3RsZS5hYy51aw==