 Mingchao Shi
Mingchao Shi Chunrong Li
Chunrong Li Xiaoping Tian
Xiaoping Tian Fengna Chu
Fengna Chu Jie Zhu
Jie Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 22 July 2021
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.685863
Alzheimer’s disease as the most common age-related dementia affects more than 40 million people in the world, representing a global public health priority. However, the pathogenesis of Alzheimer’s disease (AD) is complex, and it remains unclear. Over the past decades, all efforts made in the treatments of AD, with targeting the pathogenic amyloid β (Aβ), neurofibrillary tangles, and misfolded tau protein, were failed. Recently, many studies have hinted that infection, and chronic inflammation that caused by infection are crucial risk factors for AD development and progress. In the review, we analyzed the role of infections caused by bacteria, viruses, and other pathogens in the pathogenesis of AD and its animal models, and explored the therapeutic possibility with anti-infections for AD. However, based on the published data, it is still difficult to determine their causal relationship between infection and AD due to contradictory results. We think that the role of infection in the pathogenesis of AD should not be ignored, even though infection does not necessarily cause AD, it may act as an accelerator in AD at least. It is essential to conduct the longitudinal studies and randomized controlled trials in humans, which can determine the role of infection in AD and clarify the links between infection and the pathological features of AD. Finding targeting infection drugs and identifying the time window for applying antibacterial or antiviral intervention may be more promising for future clinical therapeutic strategies in AD.
- The infections caused by various pathogens have been evidenced to involve in the pathogenesis of AD; however, it is still controversial. Therefore, in-depth study of the causal relationship between infections and AD is very necessary.
- The infections may be one of the crucial pathogenic factors in development of AD through inducing neuroinflammation and aggravating the pathological changes, promoting the progression of AD.
- The treatments with anti-infections may slow down AD progression, but it needs to identify the time window for applying antibacterial or antiviral intervention, which might be a beneficial to AD patients.
Alzheimer’s disease is the most prevalent form of age-related dementia in the elderly population, and it has reported that about 40 million people suffered from Alzheimer’s disease (AD) in the world in 2017 (Levy et al., 2017), representing a global public health priority. The main pathological features of AD consist of amyloid-β (Aβ) plaque deposits, neurofibrillary tangles formed by hyperphosphorylated tau protein, and neuroinflammation, which are current major theory of the pathogenesis of AD (Angelucci et al., 2019). Despite a lot of intensive studies in the past, the pathogenesis and the etiology of AD remain largely unknown. Previous treatment of AD based on targeting the pathogenic Aβ or tau was unsuccessful, speculating that the pathogenesis of AD should be multifactorial and is more complex than a simple pathogenic Aβ or tau would suggest. Recently, several groups have demonstrated that infections or chronic inflammation caused by infectious agents are strongly involved in the pathogenesis of AD, which reignited interest in the infectious theory that was largely disregarded by AD research (Ashraf et al., 2019; Beydoun et al., 2020; Haditsch et al., 2020; Schnaider et al., 2020), since the role of infection in the development and progress of AD has long been debated and is still uncertain (Allnutt et al., 2020; Ou et al., 2020). Currently, the crucial role of infectious agents in AD-associated inflammatory pathways and antimicrobial properties of Aβ has been evidenced (Ashraf et al., 2019; Itzhaki et al., 2020).
Evidence based on the published studies showed that bacteria, such as Helicobacter pylori (H. pylori) (Malaguarnera et al., 2004; Kountouras et al., 2006; Albaret et al., 2020; Doulberis et al., 2020), and Porphyromonas gingivalis (P. gingivalis) (Singhrao et al., 2015; Haditsch et al., 2020), viruses, such as human immunodeficiency virus (HIV) (Lieberman, 2000; Smits et al., 2000; de Almeida et al., 2020), herpes simplex virus type 1 (HSV-1) (Cribbs et al., 2000; La Rosa et al., 2019; Linard et al., 2020), human herpes virus-6 (HHV-6) (Lin et al., 2002; Carbone et al., 2014), HHV-7 (Readhead et al., 2018), Epstein–Barr virus (EBV) (Xu and Gaskin, 1997; Gate et al., 2020), the novel coronavirus 2019 (COVID-19) virus that broke out in the end of 2019 (Ferini-Strambi and Salsone, 2020; Ou et al., 2020), Chlamydia pneumoniae (Arking et al., 1999; Itzhaki et al., 2020; Sundar et al., 2020; Woods et al., 2020), and Borrelia burgdorferi (Itzhaki et al., 2020), as well as fungi, such as Saccharomyces cerevisiae, Malassezia globosa, Candida albicans (C. albicans), and Cryptococcus neoformans (C. neoformans) (Alonso et al., 2014a, 2015; Pisa et al., 2015a; Carter, 2017; Santiago-Tirado et al., 2017), etc., pathogens, and the prion protein (PrP) (Snow et al., 1989; Come et al., 1993; Zhang B. et al., 2020), may be associated with development and progress of AD. Generally, it has a great possibility that infectious pathogens invading the brain may play a triggering role in AD development, supporting the existence of a direct or indirect nexus between infections/chronic inflammation caused by infections and AD.
In the review, we analyze the role and the underlying mechanisms of these infectious agents in the pathogenesis of AD in order to evidence targeting these infectious pathogens as a novel therapeutic manipulation in AD.
It has been evidenced and is beyond doubt that aging results in increasing the severity and susceptibility to infections and diminishes ability to fight infection in aged people (Kim et al., 2015). The main reason is due to immunosenescence, which manifests as obviously decreasing immunoglobulin (Ig) diversity associated with declined number of mature B cells and circulating antibodies, weakening their response capacity to new antigens and impairing antibody function, including lowering affinities and opsonizing abilities during aging (Cicin-Sain et al., 2012; Derhovanessian et al., 2013). Also aged memory T helper (Th) 1-type autoreactivity results in age-related immune hyporeactivity in aged people and AD, whereas autoreactive memory Th2 cells could promote tumorigenesis (Pawelec, 2017; Seledtsov and von Delwig, 2020; Thomas et al., 2020), which is related to a harmful clinical outcome always (Pawelec, 2017; Dugan et al., 2020). In short, immunosenescence leads to immune dysfunction in aged people and AD (Pawelec, 2017; Dugan et al., 2020).
To date, the published results have displayed the possibility that infection causes AD by creating inflammation (Soscia et al., 2010; Hur et al., 2020). Infections might cause a chronic, low-grade, sterile, and systemic inflammatory condition during aging called as “inflammaging” that has been recognized as a crucial pathogenic factor in the pathogenesis of AD. The features of inflammaging consist of enhancement of microglial activation and pro-inflammatory molecule production (Ponnappan and Ponnappan, 2011; Fulop et al., 2018), which deteriorates inflammation in aging people and AD (Abbas et al., 2002; Lambert et al., 2009).
There is growing evidence that reactive gliosis and microglial activation in AD brain, as the features of inflammation response, are the histological hallmarks and important factors in initial AD pathology (Salter and Stevens, 2017; Yeh et al., 2017; Zhang X. et al., 2020). Activated microglia in the central nervous system (CNS) are heterogeneous and categorized into two types: pro-inflammatory and anti-inflammatory microglia (Tang and Le, 2016; Yu et al., 2019), which play a dual role as either cytotoxic or neuroprotective roles, depending on the microglial phenotype and at the different stages of AD. The pro-inflammatory microglia can enhance inflammation and promote Aβ accumulation, inducing neural cell death and worsening disease through producing inflammatory mediators (Fakhoury, 2018; Zhang X. et al., 2020). Oppositely, Aβ plaques can be phagocytized by the anti-inflammatory microglia, which clean and degrade Aβ by increased phagocytic and lysosomal activity, as well as restriction of the inflammatory response (Kamphuis et al., 2016; Dubbelaar et al., 2018). We speculated that microglial over-activation or malfunction may be the basis of AD pathogenesis, which aggravates inflammation and accelerates the development of AD.
A large number of research results in the past showed that elevated levels of inflammatory cytokines/chemokines and mediators were associated with infections and inflammation in patients with AD and its animal models. Serum levels of pro-inflammatory cytokines/chemokines, including interleukin (IL)-6 (IL-6), CXCL2, and IL-1β, were higher in cognitive-impaired patients with brain amyloidosis than those without brain amyloidosis and controls; however, a significantly declined anti-inflammatory cytokine IL-10 was seen in the patients with brain amyloidosis compared with patients without brain amyloidosis (Cattaneo et al., 2017), implying pro-inflammatory cytokines/chemokines, contributing to brain amyloidosis and cognitive impairment. Additionally, it was observed a close relationship between higher levels of pro-inflammatory cytokines IL-1β and CXCL2 with bacterial infections (Cattaneo et al., 2017).
Similarly, the infections by P. gingivalis increased productions of inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), IL-6, and IL-1β, as well as complement factors, nitric oxide (NO) and reactive oxygen species, which augmented Aβ deposits and activated complement system, causing inflammation, brain tissue degeneration, and cognitive impairment, consistent with the damage in AD (Boje and Arora, 1992; Floyd, 1999; Liccardo et al., 2020; Costa et al., 2021). Additionally, high levels of TNF-α, IL-1β, IL-6, IL-12, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in plasma increased gut permeability in aging macaques, which was associated with leaky gut, inflammatory phenotype, and dysbiosis of gut microbiota, leading to AD and neurological disorders through a gut-brain axis pathway (Schneider et al., 2019; Sochocka et al., 2019).
Moreover, interferon (IFN) as an inflammatory boosting factor in the cultured brain cells enhanced levels of Aβ and IFN-induced transmembrane protein 3 (IFITM3) that can bind to γ-secretase, raising Aβ production, and act as a mediator in the process of inflammation and Aβ formation. When viruses invade into the brain, IFITM3 is activated and exercises its function (Hur et al., 2020). Furthermore, IFN-stimulated gene (ISG) signature was found in the brains of AD murine models and of wild-type mice, stimulated with generic nucleic acid-containing amyloid fibrils. Most convincingly, activated ISG-expressing microglia only surrounded nucleic acid+ Aβ plaques accumulated in an age-related way. In AD models, injection of IFN-β into brain caused microglial activation and blockading IFN receptor reduced microgliosis and synapse loss. The same phenomenon with activated ISG-expressing microglia with nucleic acid-containing neuritic plaques was observed in postmortem brains of AD. An IFN pathway in AD was obviously upregulated and clearly related to AD severity and inflammation (Devanand et al., 2020).
Previously, our study displayed that major inflammatory cytokines, IL-12, and IFN-γ in brain of transgenic mice APPSWE (Tg2576) that overexpress the human Aβ precursor protein gene as an AD mouse model were associated with Aβ plaque formation and microglial and astrocyte activation (Abbas et al., 2002). In the brains of patients with AD and its animal models, infection stimulating microglia and/or microglial activation by peripheral cytokines caused robust production of free radicals with another wave of pro-inflammatory cytokines, which, in turn, led to inflammation and massive neuronal damage (Nakamura et al., 2021; Wiatrak and Balon, 2021).
Inflammation, playing a central role in the pathogenesis of AD, has reached consensus, while inflammatory mediators caused neuroinflammation through the damaged blood-brain barrier (BBB) entering to CNS. Thereby, BBB disruption was one of the determinant factors in development of AD, which has been evidenced by genetic, proteomic, and epidemiological study (Carter, 2017). So far, diverse pathogens have been detected in AD brains, indicating that infections may be related to the pathogenesis of AD. Disruption of BBB facilitated most pathogens to enter brain, which appeared an early pathological character of AD (van de Haar et al., 2016; Lecler et al., 2017). Siderosis and microbleeds in brain were also the features associated with BBB disruption in patients with (Yates et al., 2014; Inoue et al., 2016), since bacteria relied on the availability of free iron to assist in their successful colonization in AD.
It is noteworthy that infections not only cause acute diseases but also lurk in the body for a long period, leading to a chronic, low-grade, sterile inflammation and tissues damage, which is a key pathogenic factor in neurodegenerative diseases, such as AD (Chitnis and Weiner, 2017; Stephenson et al., 2018).
The infectious hypothesis of AD pathogenesis was highly overlooked previously due to lack of evidence. Recently, the infectious hypothesis of AD has been more and more recognized and accepted by the broad scientific community, since there are many new overpowering pieces of evidence of a close relationship between the infections by various pathogenic microbes and development of AD (Malaguarnera et al., 2004; Singhrao et al., 2015; Beydoun et al., 2020; de Almeida et al., 2020; Doulberis et al., 2020; Ferini-Strambi and Salsone, 2020; Gate et al., 2020; Haditsch et al., 2020; Linard et al., 2020). It is often seen that patients with mild cognitive impairment (MCI) or early dementia, suffering from infection, manifested a speedy decline in cognitive function and could not completely recover after therapy of infections. It is possible that persistent infection worsens gradually cognitive function (Itzhaki et al., 2020). It has been proved that P. gingivalis infiltration was found in autopsy specimens from brains and in cerebrospinal fluid (CSF) of patients with AD (Laugisch et al., 2018; Ryder, 2020). Moreover, it has been observed that the etiology and pathologies of AD may be related to the infections by several bacteria, viruses, and fungi, as well as other pathogens, which support that infections are a risk factor and play a crucial role in AD (Snow et al., 1989; Come et al., 1993; Xu and Gaskin, 1997; Cribbs et al., 2000; Lieberman, 2000; Smits et al., 2000; Lin et al., 2002; Malaguarnera et al., 2004; Kountouras et al., 2006; Alonso et al., 2014b, 2015; Carbone et al., 2014; Singhrao et al., 2015; La Rosa et al., 2019; Albaret et al., 2020; de Almeida et al., 2020; Doulberis et al., 2020; Gate et al., 2020; Haditsch et al., 2020; Linard et al., 2020; Zhang B. et al., 2020).
Bacterial infections, mainly referring to the infections with P. gingivalis and H. pylori, have been shown to be linked to AD, which is definitive evidence, proving the infection associated with AD so far (Malaguarnera et al., 2004; Kountouras et al., 2006; Laugisch et al., 2018; Beydoun et al., 2020; Liccardo et al., 2020; Ou et al., 2020).
Porphyromonas gingivalis and other periodontal pathogens can penetrate the bloodstream and enter the brain, inducing a low-grade systemic inflammation that negatively affects brain function (Liccardo et al., 2020). These periodontal pathogens were found in periodontium, serum, and CSF of AD. However, periodontal pathogens could not trigger development of AD in the demented patients younger than 70 years old (Laugisch et al., 2018). The antibodies to periodontal bacteria, including Aggregatibacter actinomycetemcomitans, P. gingivalis, Campylobacter rectus, Treponema denticola, Fusobacterium nucleatum, Tannerella forsythia, and Prevotella intermedia, were enhanced obviously in the patients before cognitive impairment, indicating that periodontal bacteria are key risk factors in AD occurrence (Sparks Stein et al., 2012). The infected neurons by P. gingivalis appeared the similar signs to AD neuropathological changes, for example, increased in the phospho-tau/tau ratio, synapse loss, etc. (Haditsch et al., 2020), supporting the existence of a direct nexus between periodontitis and AD. The gingipains, a class of P. gingivalis proteases, are related to neuronal damage, tau tangles, and Aβ deposit in the brains of patients with AD (Ryder, 2020). Additionally, the brain of the patients with AD can be infected by oral pathogens and their toxic products. During the period of the disease progress, patients with AD are unable to adequately maintain oral hygiene, resulting in oral infections and periodontitis; in turn, it deteriorated AD forming a vicious circle (Liccardo et al., 2020; Sadrameli et al., 2020). A diminished level of salivary lactoferrin, a credible biomarker for Aβ accumulation in AD brains, has been found more recently, because a lower level of salivary lactoferrin altered the oral microbiota, which was an outcome of poor oral hygiene and dry mouth in the elderly and promoted oral bacteria to grow and to spread elsewhere in the body (Nie et al., 2019). Oral bacteria could grow locally and followed the infected inflammatory monocytes/macrophages in the mouth, spreading elsewhere in the body through the bloodstream or these bacteria penetrated the bloodstream directly to reach various parts of the body, in which the activated cathepsin (Cat) B (CatB)/nuclear factor kappa-B (NF-κB) signaling pathway by bacteria may contribute to the spreading process (Nie et al., 2019). The pathogens of periodontal disease causing chronic inflammatory responses impact significantly on the development of AD. The evidence showed that microbial infections and chronic inflammation in the brain may be involved in the pathogenesis of AD (Angelucci et al., 2019; Ashraf et al., 2019; Beydoun et al., 2020; Haditsch et al., 2020).
In mice, oral infections with P. gingivalis showed P. gingivalis infiltration into brain, accompanying with various neuropathological features of AD and exacerbated disease (Nie et al., 2019). P. gingivalis as an oral pathogen can entry into apolipoprotein (APOE) knockout (KO) (equivalent to APOE 4 genotype) brain of mice to induce complement activation (Poole et al., 2015). Although there are various means of entry for periodontal bacteria to access brain (Riviere et al., 2002), P. gingivalis entered APOE KO brain of mice mainly through blood circulation and circumventricular organs (Velsko et al., 2014; Poole et al., 2015; Ilievski et al., 2018). Also P. gingivalis was able to access APOE KO brain of mice via other routes, including a permeable BBB affected by P. gingivalis or the choroid plexus, containing a rich blood supply and CSF (Riviere et al., 2002). Particularly, it is very easy for P. gingivalis accessing APOE KO brain of mice due to APOE deficiency, leading to a BBB leakage (Hafezi-Moghadam et al., 2007). Treatment with the compounds that inhibit P. gingivalis or its toxins in infected mice reduced the abundance of P. gingivalis DNA in brain and ameliorated the pathologies and the neurotoxic effects of P. gingivalis infection, which implies that control of P. gingivalis infection can affect the pathological condition of AD (Matsushita et al., 2020; Olsen and Singhrao, 2020), and the inhibitors of P. gingivalis provided a potential approach to treat both periodontitis and AD (Ryder, 2020). In addition, chronic systemic P. gingivalis infection led to the accumulation of Aβ in the brain, the most significant pathological hallmark of AD, in middle-aged mice, and also induced Aβ accumulation in inflammatory monocytes/macrophages through activating CatB/NF-κB signaling, indicating that monocytes/macrophages act as a circulating pool of Aβ in patients with periodontitis (Nie et al., 2019). It is worth noting that repeated oral infection by P. gingivalis caused neurodegeneration and an extracellular Aβ42 deposit in brain of young adult wild-type (WT) mice, which displayed that low-grade chronic periodontal pathogen infection led to occurrence and development of AD-like neuropathology (Ilievski et al., 2018). These results from cross-sectional and longitudinal studies showed that periodontitis is closely associated with cognitive impairment and AD.
The exact molecular mechanisms behind the infections by P. gingivalis and periodontal disease contributing the pathogenesis of AD remain unclear; however, it has been accepted, although not confirmed completely, that oral pathogens can infect and shift to the brain, inducing a low-grade local and systemic inflammation and affecting brain functions.
Helicobacter pylori infections are a common gastrointestinal disease, leading to gastrointestinal dysfunction always. AD and cognitive decline, as well as other neurodegenerative diseases, are often associated with gastrointestinal dysfunction (Zhuang et al., 2018; Sochocka et al., 2019; Ticinesi et al., 2019). Levels of IgG and IgA antibodies to H. pylori were detected in 30 patients with AD, 30 patients with vascular dementia (VaD), and 30 control subjects, respectively, in a study (Malaguarnera et al., 2004). The results exhibited that significant high H. pylori-IgG levels were seen in AD when compared with normal controls but lower than those of patients with VaD. H. pylori-IgA levels were equally enhanced in patients with VaD and AD, which found a relationship between H. pylori infection and AD (Malaguarnera et al., 2004). Another study investigated the association between H. pylori infection and AD by using histology for diagnosis of H. pylori in total of 50 patients with AD and 30 iron deficiency anemic controls without AD. The results showed high histologic prevalence of H. pylori with 88% in AD and 46.7% in controls; the comparison between the two groups was very significant (p < 0.001) (Kountouras et al., 2006). More recently, a systematic review article has reported that the most common investigated microbiota was H. pylori in 241 published papers on a connection between AD and gastrointestinal microbiota, and displayed an association between AD and H. pylori (Doulberis et al., 2020). The risk of H. pylori infections in AD and Parkinson’s disease (PD) from the obtained data revealed increase in the risk of developing AD (Fu et al., 2020), indicating AD clearly associated with intestinal disorders, especially intestinal infections by H. pylori (Fu et al., 2020). H. pylori infection was associated with a low mini-mental state examination (MMSE) score in patients with AD, possibly via triggering the inflammatory mediator productions (Roubaud-Baudron et al., 2012) and increasing levels of Aβ40 and Aβ42 in serum of patients with AD (Bu et al., 2015). In vitro, the neuroblastoma cell cultures treated by H. pylori filtrate induced tau hyperphosphorylation, which was similar to AD tau pathological changes (Wang et al., 2015). Infected aged C57BL/6 mice with H. pylori or Helicobacter felis or left uninfected for 18 months appeared severe gastritis and increased neuroinflammation in infected groups (H. felis > H. pylori), but without brain Aβ deposition or systemic inflammation (Albaret et al., 2020).
The onset of AD and all-cause dementia can be changed by coinfection with H. pylori and the groups of periodontal pathogens, including P. gingivalis, which was evidenced via investigating 1,431 participants (≥65 years), with a mean follow-up of 10–11 years for AD and an all-cause dementia incidence (Beydoun et al., 2020). Epidemiological and clinical studies have shown that infections with H. pylori increased the risk of developing AD. However, the questions are whether this association is due to the ubiquity of H. pylori, or whether it promotes neuroinflammation in AD. The relationship between H. pylori and AD, if confirmed by additional studies with a large sample of patients, might have important clinical and therapeutic implications.
Alzheimer’s disease is a multifactorial disease caused by both environmental factors and a genetic predisposition.
With the discovery of bacteria, viruses, and other pathogens in the brain of patients with AD, more and more evidence shows that AD is caused by microbial infection and inflammation (Jamieson et al., 1991; Kountouras et al., 2006; Readhead et al., 2018). Besides bacteria, the infections with viruses also contribute to the pathogenesis of AD. The viruses linked to development of AD include HIV, HSV-1, HHV-6, HHV-7, EBV, hepatitis C, and COVID-19, as well as Torque teno virus (TTV), which are gradually being recognized as important factors affecting the development of AD.
In the patients with acquired immune deficiency syndrome (AIDS), infected by HIV, manifested cognitive deficits or dementia as the age-related complications, following patients living longer after treatments. Aβ deposits have been observed in patients with AIDS, which may be associated with HIV infection-induced neuroinflammation (Canet et al., 2018; Hategan et al., 2019; Kodidela et al., 2019). Recently, it has been found that the connection between HIV and AD could be extracellular vesicles (EVs)/exosomes, because they are able to package and transport the toxic proteins in both AD and HIV (Aβ/tau and gp120/tat, respectively) (Kodidela et al., 2019). Elevated Aβ in neuron-derived exosomes from the plasma of patients with AIDS showed neuron-to-neuron exosomal transport of Aβ and tau also contributing to AD-like pathology in the patients with AIDS (Kodidela et al., 2019).
Human immunodeficiency virus passing BBB causes neuronal dysfunction, leading to cognitive impairment. In the process of HIV infections, many factors involved in AD pathology, such as members of Aβ and tau pathways, are regulated by HIV infection, resulting in the accumulation of Aβ plaques or neurofibrillary tangles (NFT) in brains, indicating the similarities and convergences of the two pathologies in AD- and HIV-infected brains. HIV and its proteins, such as trans-activator of transcription (Tat) and envelope glycoprotein gp120, can modulate cellular pathways to deteriorate AD (Canet et al., 2018). HIV-Tat interacts also with Aβ and tau via novel a protein-protein coaction and induces tau phosphorylation through a cascade of cellular processes to form NFT, eventually resulting in AD, despite adequate antiretroviral therapy (Hategan et al., 2019). HIV-Tat may accelerate AD-like pathology in patients with HIV. There is a double risk in the older patients infected with HIV, those who may suffer from both HIV-associated neurocognitive disorder (HAND) and AD, which has been evidenced by investigating neuroimaging and neuropsychological performance in 61 older adults with HAND, 53 with MCI of a type of AD and 89 healthy controls cross-sectional over 20 months (Matsushita et al., 2020).
HIV-associated neurocognitive disorder as a cognitive disorder is similar to AD, and its pathogenesis involves the effects of HIV and its proteins, such as Tat and gp120 by destroying neurons and initiating neuroinflammation contributing to the deposition of Aβ and leading to dementia. HIV infection is associated with increased Aβ deposits in brain, a feature of AD (Fulop et al., 2019). Thus, the questions arise whether HAND may lead to AD or is it a form of AD? These questions are needed to further explore and study.
A study that has followed more than 8,000 people with HSV infection and 25,000 healthy controls in Taiwan for 10 years found that increased risk of developing AD in patients with HSV was 2.5-fold higher than that in controls (Tzeng et al., 2018). Recent study has emerged that anti-herpetic drugs play a protective role in dementia through declining the risk of dementia in patients with HSV infections (Tzeng et al., 2018).
Infection with HSV-1 is very common, and more than half of the world population hide it in their bodies. As early as 1990, HSV-1 was observed in postmortem AD brains (Jamieson et al., 1991), which evidenced the relationship between HSV-1 infection and AD for the first time. However, someone questioned that HSV-1 or other microbes were able to leak into brains easily after death due to tissue of brain breaks down; therefore, the pathogenesis of AD basis on postmortem material was an assumption (Jamieson et al., 1991).
A study on evaluating the impact of HSV-1 infection on the incidence of AD in the light of APOE4 status, a genetic susceptibility factor for AD, has been conducted via estimating 10-year risk of AD associated with anti-HSV antibodies in 1,037 subjects by Cox models (Linard et al., 2020). The results showed that frequency of HSV-1 reactivation was very high in APOE4 carriers manifested as raised an HSV-1 IgG antibody accompanied by increased risk of AD (Linard et al., 2020), suggesting a role of HSV-1 in the pathogenesis of AD in populations with a genetic susceptible factor, the APOE4 allele.
There is growing evidence that HSV-1 is gradually recognized as an important factor impacting the occurrence and development of AD. However, the mechanism linking HSV-1 and AD remains unclear. New research finds that HSV-1 manipulates the activity of cofilin-1 involved in the plastic of dendritic spines and axon regeneration of neurons to ensure HSV-1-infecting neurons efficiently. Dysfunction of cofilin-1 was observed in patients with AD and its animal models. The cognitive impairment in AD models can be improved by administration of inhibitors of cofilin-1 (Wang et al., 2020). HSV-1 infection-causing dysregulation of cofilin-1 led to AD finally probably. Pathologically, Aβ and hyperphosphorylated tau accumulations were observed in the neurons infected by HSV-1 and bacterial ligands; viral and bacterial DNA and RNA can increase the expression of pro-inflammatory molecules (Ashraf et al., 2019). To this, very strong evidence is that, after HSV-1 was injected into the brains of young 5xFAD mice, transgenic mice of AD, and of normal mice in 3 weeks, the brains of transgenic mice were covered fully with Aβ plaques (Eimer et al., 2018).
The role of HHV-6 in AD has been controversial, and recent studies have renewed the debate again. Previously, it was reported that HHV-6 existed in 10% of healthy brains in humans and mice; therefore, humans were not susceptible to HHV-6 infection (Eimer et al., 2018). But more recent study has demonstrated that adding HHV-6 to the human neural cell cultures that simulated pathologies of AD led to Aβ-plaques and tau tangles accumulation after 2 days compared with the cultures without HHV-6 after cultures for 6 weeks (Eimer et al., 2018), hitting that HHV-6 infection-accelerated Aβ-plaques and tau tangles accumulation, the two main pathological features of AD.
Human herpes virus-6 as neurotropic virus can dysfunction autophagy and activate endoplasmic reticulum (ER) stress/unfolded protein response (UPR) in vitro cell lines (Romeo et al., 2020), which is necessary for cell homeostasis, particularly in postmitotic cells, such as neuronal cells. Autophagy dysregulation and abnormal UPR activation are always observed in AD and other neurodegenerative disorders. A recent study has evidenced that primary neurons infected by HHV-6A decreased obviously autophagy, increased Aβ deposition, and activated ER stress/UPR, promoting tau protein hyper-phosphorylation. The levels of HHV-6A and HHV-7 were higher in AD than in controls by detecting the snippets of genes or proteins specific to herpes (Readhead et al., 2018; Romeo et al., 2020). These results showed HHV-6A infection playing a role in AD (Romeo et al., 2020).
However, a recent experimental result has negated the above conclusion and considered no association between HHV-6 and AD through screening for HHV-6 across three independent AD brain repositories, using (1) RNA sequencing (RNA-seq) datasets and (2) DNA samples extracted from AD and non-AD control brains. These direct methods of viral detection did not support HHV-6 playing a role in AD (Allnutt et al., 2020). The proving causality between HHV-6 and AD has been elusive, and more studies are needed to conduct in the future. Fortunately, valacyclovir, an antiviral drug started to treat mild AD with HSV antibodies together in 2017 and will be completed in 2022. If the clinical trial is successful, it will provide a proof that HHV can increase cognitive impairment and participate in the development of AD.
Currently, the global epidemic of COVID-19 infection has potential implications for the field of AD, because COVID-19 caused severe respiratory symptoms in people aged 60 years and older; it has important implications for patients with AD. However, the exact impact of COVID-19 infection on AD is still uncertain due to lack of long-term follow-up and research. According to the published data, the preliminary conclusions concerning the effects of COVID-19 virus infections on AD are inconsistent (Brown et al., 2020; Fotuhi et al., 2020; Li et al., 2020; Naughton et al., 2020; Yu et al., 2020). Whether COVID-19 increases the risk and the incidence rate of future dementia is still to be further studied.
The clinical evidence showed that COVID-19 infection can cause acute neurological symptoms, such as a headache, confusion, epilepsy, delirium, and encephalopathy, as well as loss of taste and smell, etc., it also develops future diseases as long-term consequences of COVID-19 infection, such as depression, insomnia, cognitive decline, accelerated aging, PD, or AD in the future (Ferini-Strambi and Salsone, 2020; Fotuhi et al., 2020). It has been corroborated the presence of COVID-19 in the brain tissue and CSF of the infected patients by electron microscopy, immunohistochemistry, and real-time reverse transcription-PCR (Netland et al., 2008; Baig et al., 2020). COVID-19 infection can induce neuroinflammation, a prominent characteristic in AD pathology, and immune response in brain, which accelerates brain inflammatory neurodegeneration, and the elderly people are more susceptible to severe outcomes after COVID-19 infection. The patients with type 2 diabetes increased a risk for development of AD after COVID-19 infection (Bostanciklioglu, 2020; Fotuhi et al., 2020; Naughton et al., 2020). The epidemic has restricted activities and aggravated AD as a previous study showed movement restriction exacerbating AD symptoms (Lin et al., 2015). Higher rates of deaths of COVID-19 infection have been reported in older patients, who had comorbidities, such as hypertension, cardiovascular disease (CVD), and dementia (Vallamkondu et al., 2020). However, a report from Wuhan city of China showed, when compared with patients with non-AD, patients with AD had a better prognosis after COVID-19 infection due to the patients with AD had full access to health care (Li et al., 2020). In addition, a study that tested 13,338 cases for COVID-19 in United Kingdom showed that the patients with AD had highest risk of COVID-19 infection and mortality than normal elderly. However, the patients with PD enhanced risk of COVID-19 infection, but not mortality from COVID-19, suggesting that there was different susceptibility to COVID-19 in patients with neurodegenerative disorders (Yu et al., 2020). Patients carrying APOEε4 infected with COVID-19 had obvious odor recognition deficits when compared with those not carrying APOEε4, speculating that COVID-19 infection may increase the risk of dementia in APOEε4 carriers in the future (Ferini-Strambi and Salsone, 2020). Generally, the impact of COVID-19 on AD is age rather than neuropathology itself, whereas neuropathology rather than age may be critical in neuroimmune diseases, such as multiple sclerosis (Ferini-Strambi and Salsone, 2020).
Epstein–Barr virus has been reported to associate with AD pathogenesis, and it was presented as two separate EBV antigens in CSF of patients with AD (Gate et al., 2020). EBV-transformed B cell lines from the peripheral blood of the patients with AD produced anti-Aβ antibodies in Aβ plaques of AD (Xu and Gaskin, 1997). Besides the descripted infectious pathogens as the above, recently, some researchers have demonstrated that the respiratory bacteria Chlamydia pneumoniae may be also implicated in AD, because the infection with Chlamydia pneumoniae has been associated with dementia, cortical atrophy, and pathological and biological hallmarks of AD (Ashraf et al., 2019). However, the results remain still controversial (Taylor et al., 2002; Boelen et al., 2007; Woods et al., 2020). Therefore, further studies are required for substantiation of causality and for the discovery of promising novel therapies.
These data further demonstrated that infections and their causes of chronic inflammation may be associated with developing AD. Nevertheless, it is still elusive whether immune dysfunction in AD brains reactivates latent viruses to prompt neurodegeneration or these viruses themselves trigger the neurodegenerative process in AD. Even though the microorganisms do not necessarily cause AD, they may act as accelerators in AD at least. This association does not yet prove a causal link, but these results call to conduct more studies on the microbial infection-AD hypothesis to determine whether therapeutic strategy targets infectious pathogens, which can slow down AD progress and is viable in the future (Ou et al., 2020; Schnaider et al., 2020).
Nonetheless, the infection hypothesis has been challenged in the pathogenesis of AD. The special issue is lack of experimental and preclinical evidence, showing that pathogens can cause directly neuropathological changes and cognitive impairment, which are the main characteristics of AD. Several studies have demonstrated that the transmissibility of AD neuropathology or cognitive impairment could not be observed after injection of AD brain homogenate directly into the brain of primates (Ou et al., 2020; Schnaider et al., 2020). There is still insufficient evidence to prove the existence of invaded intact virions or viral proteins in AD brain (Lellouch-Tubiana et al., 2010). Thus, infection associated with the etiology of AD may be unconvincing (Ou et al., 2020; Schnaider et al., 2020). Additionally, there was no significant difference between patients with AD and the controls for tested Chlamydia pneumoniae infection, which may conclude Chlamydia pneumoniae infection did not play a role in the etiology of AD (Campbell et al., 1998).
After providing extensive evidence by several research groups, the fungi infections are considered to be causative agents and risk factor in AD unequivocally on the basis of detected diverse fungi in AD brains, such as Saccharomyces cerevisiae, Malassezia globosa, C. albicans, C. neoformans, Malassezia restricta, Cladosporium, Neosartorya hiratsukae, and several other Candida species (Alonso et al., 2014a,b, 2017; Pisa et al., 2015a,b; Carter, 2017). Another piece of irrefutable evidence is that fungal DNA and proteins were found in CSF and peripheral blood of patients with AD (Alonso et al., 2014b, 2015). Besides, there were fungal components in postmortem brain tissues of patients with AD, and several peptides clearly corresponding to fungal proteins were observed by proteomics analysis (Alonso et al., 2014a; Pisa et al., 2015b). Strikingly, yeast-like cells and hyphal structures in AD brain were observed directly, which were related to neural cells both intra- and extracellularly (Pisa et al., 2015a,b). Additionally, increased levels of chitinase, a component of fungi and an inflammatory marker, were detected in blood and CSF of patients with AD, even before the onset of AD, which is a further support for the viewpoint that mycoses are present in AD (Rosén et al., 2014; Melah et al., 2016).
In patients with AD, Alternaria, Botrytis, Candida, and Malassezia were prevalent fungal genera analyzed by the group of Alonso (Alonso et al., 2018). Fungi detected from frontal cortex samples of patients with AD emerged clustered together and differed from the controls (Alonso et al., 2018). It is worth noting that coinfections with fungi and bacteria together were found in patients with AD, implying polymicrobial infections existing in AD (Carter, 2017; Alonso et al., 2018).
The cell surface amyloid-like structures in the pathogenic fungi are for adhesion and biofilm formation. An in vitro study, binding of the human constitutive plasma protein serum amyloid P component (SAP) to the surface amyloid of fungi inhibited immune response to fungi and diminished the phagocytosis of fungi by macrophages, there by SAP binding to fungi may help fungi infections in AD (Behrens et al., 2019).
In short, these observations support the hypothesis that the existence of fungal infections in patients with AD may be associated with the pathogenesis of AD (Pisa et al., 2015b; Alonso et al., 2017, 2018). However, the same fungi could not detect in all AD brains, or in different laboratories; therefore, further studies are required to confirm certain fungi in patients with AD.
Accumulation of the pathological prion protein, scrapie prion protein (PrPSc), results in the fatal prion diseases. Cellular prion protein (PrPC) is also involved in other neurological disorders, including AD (Snow et al., 1989; Come et al., 1993; Zhang B. et al., 2020). There are protein misfolding and aggregation in both AD and prion diseases, which are underlying mechanisms of the diseases (Kodidela et al., 2019). It is clear that PrPC plays a crucial role in AD development. Infections led to Aβ deposition and increased PrPC and then caused high affinity binding between Aβ oligomers and PrPC, finally activated the Fyn signaling cascade, resulting in neuron death in brain of AD (Guerrero-Muñoz et al., 2014; Kodidela et al., 2019; Zhang B. et al., 2020).
Cellular prion protein can act as a neurotoxic protein, and Aβ oligomer in AD has been found as a ligand of PrPC that is critical to Aβ42-mediated autophagy in neurons. However, the role of PrPC as an Aβ42-binding protein is not clear (Onodera, 2017). The role of miRNA-146a among all studied microRNAs is age-related and associated with human inflammatory neurodegenerative disease, including AD and prion diseases (Lukiw, 2020). Infection and inflammation hypothesis of AD pathology is summarized in Figure 1.
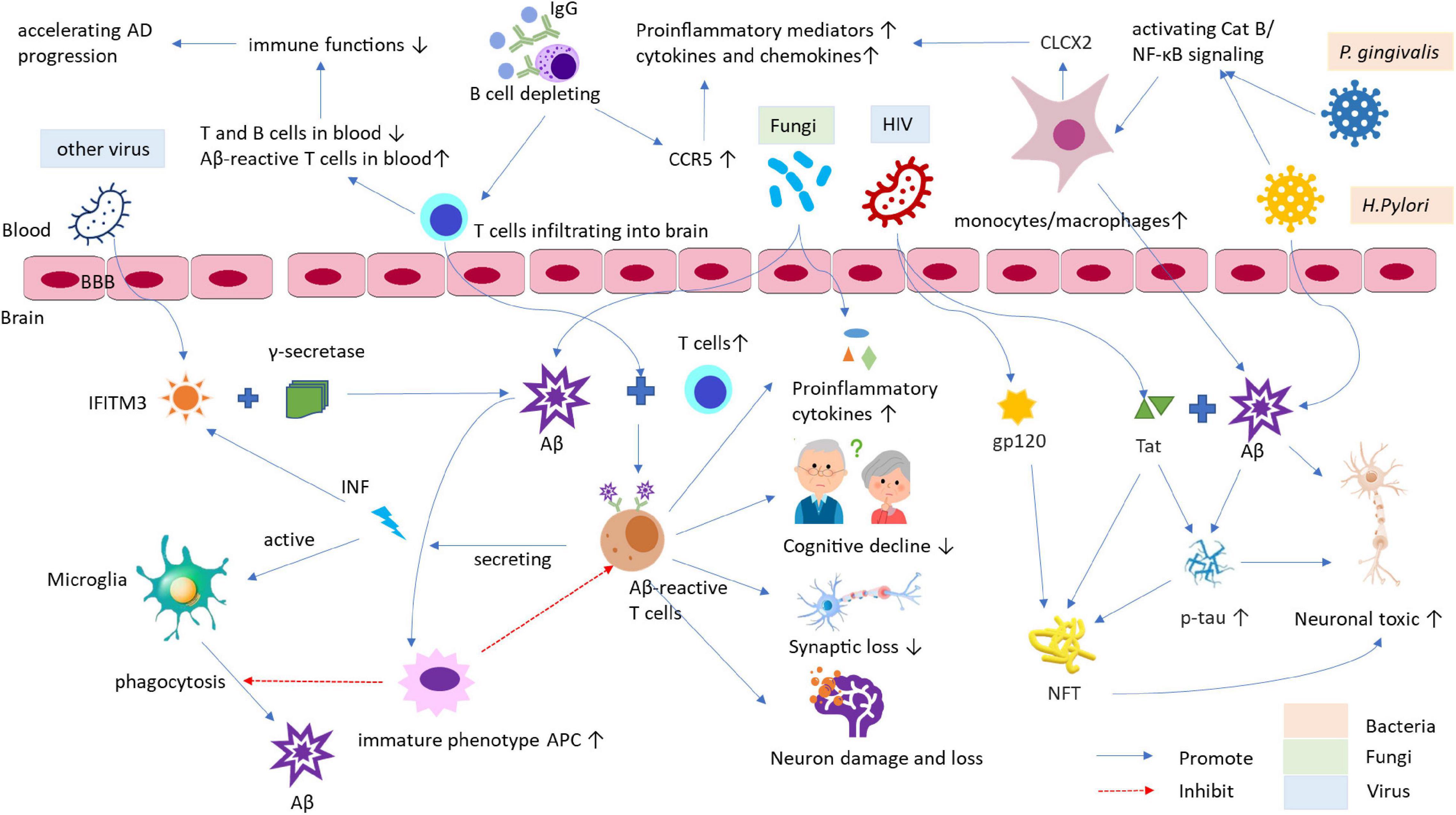
Figure 1. The role of infection and inflammation in the pathogenesis of AD and its animal models. The infections by bacteria, viruses, and other pathogens, and it caused chronic inflammation may be the crucial risk factors for AD development and progress. These pathogens can access into the brain through increased permeability of BBB. Afterward, the viruses active IFITM3 can bind to γ-secretase raising Aβ accumulation and deposition in brain and act as a mediator in the process of inflammation and Aβ formation. The bacteria, such as P. gingivalis and H. Pylori, can induce Aβ accumulation in inflammatory monocytes/macrophages through Cat B/NF-κB signaling. Activated monocytes/macrophages secrete CXCL2 can increase productions of proinflammatory mediators and cytokines/chemokines, leading to the pathological changes being similar to those of AD, for example, the elevated phospho-tau/tau ratio and synapse loss, etc. HIV and its proteins, such as Tat and envelope glycoprotein gp120, can modulate cellular pathways to deteriorate AD. HIV-Tat can interact with Aβ and tau via novel protein-protein coaction and induces tau phosphorylation through a cascade of cellular processes to form NFT, eventually resulting in neuronal toxicity. Additionally, fungi can also enter the brain to enhance levels of proinflammatory cytokines and bind to SAP to diminish the phagocytosis of macrophages, which cause Aβ accumulation, exacerbating AD. Furthermore, infection and inflammation can decline immune function and response contributing the pathogenesis of AD. AD, Alzheimer’s disease; APC, antigen-presenting cell; Aβ, β-amyloid; BBB, blood-brain barrier; Cat, Cathepsin; CCR5, C chemokine receptor type 5; CNS, central nervous system; CXCL2, chemokine ligand 2; HIV, human immunodeficiency virus; IFITM3, IFN-induced transmembrane protein 3; IFN, interferon; NFT, neurofibrillary tangles; NF-κB, nuclear factor kappa B; p-tau, phospho-tau; SAP, serum amyloid P component; Tat, trans-activator of transcription.
Alzheimer’s disease has been considered as a systemic disease related to infections, and the inflammatory-infectious hypothesis of its pathogenesis becomes more significant (Bronzuoli et al., 2016). The microbes and their products in the brain causing chronic inflammation are an important predisposing factor in neuroinflammation and neurodegeneration in AD (Cattaneo et al., 2017; Ashraf et al., 2019). Thus, a proper combination of anti-inflammatory, antibiotic, and antiviral therapeutics might potentially prevent the development and progression of AD, even though there was much disappointment with anti-AD drug discovery previously, it is still promising and possible to find a new-treatments basis on infections impacting on AD.
The causal relationship between infection and AD remains uncertain and controversial; however, there is a possibility of reversed causality. Generally, patients with AD have poorer general health, and their ability and response of innate and adaptive immune are low, resulting in more likely to have infections (Shi et al., 2021). That is, infection may trigger AD, which, in turn, is more susceptible to infection, forming a vicious circle and aggravating the disease.
The main effects of antibiotic therapies are eliminating and avoiding bacterial colonization in human body via their directly killing bacteria or indirectly changing the composition of gut microbiota that have been evidenced to associate with AD pathogenesis (Angelucci et al., 2019). Therapies with antibiotics could alter behavior and brain chemistry in both humans and animals (Fröhlich et al., 2016; Neuman et al., 2018). Antibiotic therapy lowered obviously amounts of microglia and astrocyte accumulation around Aβ plaques in the hippocampus, and reduced insoluble Aβ plaques in aged APPSWE/PS1DeltaE9 transgenic mice of the AD model, indicating that antibiotic therapy was effective in AD mice (Minter et al., 2017). However, the therapy has not yet widely investigated in patients with AD, because it is unknown whether there are safe antibiotics available (Panza et al., 2019). Since the patients with AD infected with H. pylori, receiving antibiotic treatment, displayed neuropsychiatric symptoms, such as anxiety, psychosis, and delirium in two studies (Loeb et al., 2004; Molloy et al., 2013), these neuropsychiatric symptoms did not appear in the general subjects (Neufeld et al., 2017). Similarly, the harmful effects of antibiotics on APP/PS1 transgenic mice manifested as enhanced neuroinflammation and pro-inflammatory cytokine levels and deteriorated disease (Minter et al., 2016). Ampicillin increased rat serum corticosterone related to memory dysfunctions and a lowered brain-derived neurotrophic factor (BDNF) in the hippocampus, the features of AD pathology, and deteriorated the anxiety-like behavior and impaired spatial memory in rats (Fröhlich et al., 2016). The clinical and experimental studies highlight that the results with antibiotic treatment in AD are controversial. Also, there were some adverse consequences after antibiotics treatments. It is currently difficult to determine the effect of antibiotics on AD. Therefore, the future studies on therapeutic trials with antibiotics in AD are required.
Activated IFN type I, a viral infection inhibitor, and stimulated gene (ISG)-expressing microglia were found in Aβ plaques in an age-dependent manner in AD mice. Injection of recombinant IFN-β into mouse brain caused complement C3-dependent synapse loss in vivo. In turn, a blockade of the IFN receptor lightened microgliosis and synapse loss in AD mice. ISG-expressing microglia were also detected in postmortem brains of patients with AD, indicating that the IFN pathway was markedly upregulated in AD and close related to disease severity and complement activation. Therefore, IFN critically promotes neuropathogenic processes (Roy et al., 2020). Valacyclovir, as an antivirus drug, has been conducted for the clinical trial in 130 patients with mild AD (65 patients in valacyclovir and in placebo groups, respectively) in a randomized, double-blind, 78-week phase II to decide valacyclovir to apply in treatment for AD via its anti-HSV effect (Devanand et al., 2020). There is no doubt when completion of the trial and the effect of valacyclovir on AD might be clear. The anti-infection therapies in AD animal models and patients are summarized in Table 1.
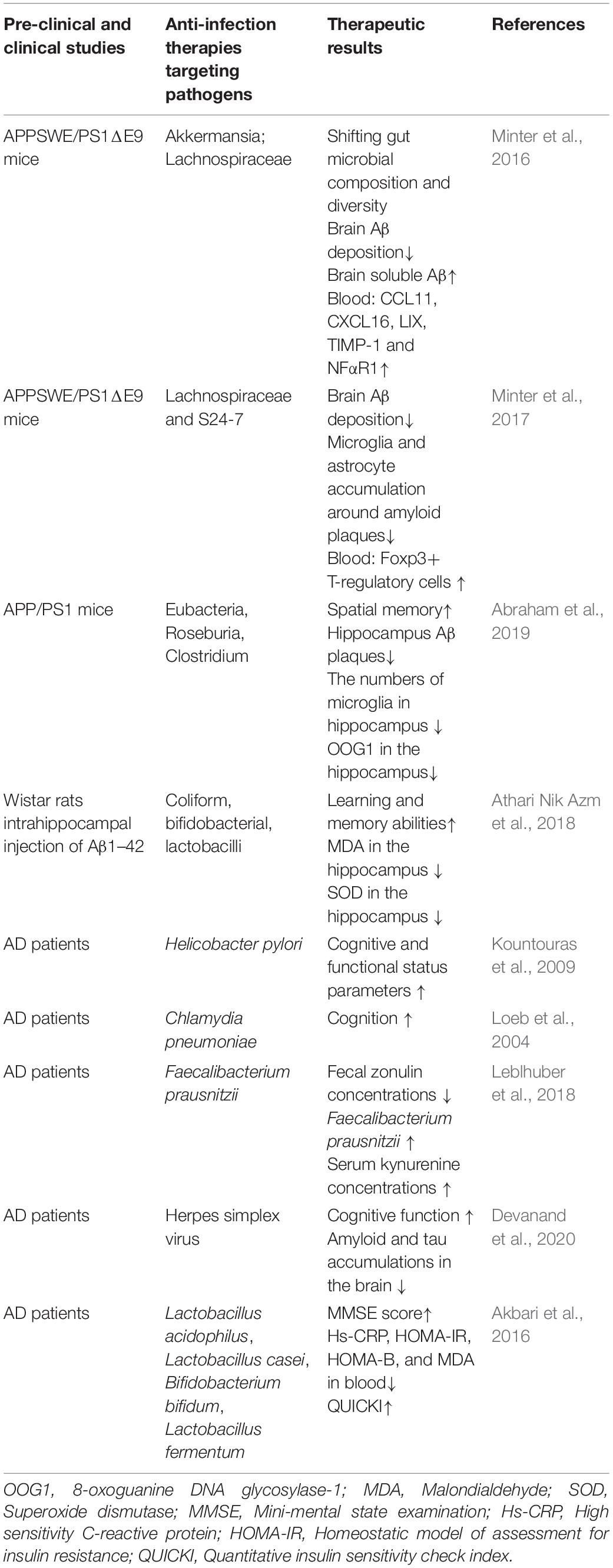
Table 1. Anti-infection therapies in AD animal models and patients.
Up to now, no study has confirmed clearly that anti-microbe treatment in AD is beneficial and effective, which is needed to evidence in the future studies.
Infections mainly caused by P. gingivalis, H. pylori, HIV, and HSV-1, and chronic inflammation caused by infections as the most likely etiologies contribute to the pathogenesis of AD. It is linked to rising pro-inflammatory and lower anti-inflammatory response, and to promote Aβ plaques deposits and hyperphosphorylated tau protein formation in brain of AD, which damages brain health and causes the pathological changes in AD. Infection may be a key pathogenic factor in development and progression of AD, which has been evidenced through the experimental and preclinical studies currently. However, the results obtained are controversial, and it is still difficult to determine their causal relationship between infection and AD so far. By listing a large number of results and in-depth discussions in the present review, we should pay attention to the pathogenic role of infection in AD; albeit infection may not necessarily cause AD directly, it is a trigger or promoter in development of AD definitely. The further study and randomized controlled trials in humans are essential to define the impact of infection on AD. Discovery of novel anti-infectious compounds and determining the time window application of antibacterial or antiviral intervention are more hopeful for the future clinical treatment strategy of AD.
MS and CL prepared the manuscript. XT and FC provided views and revised the manuscript. JZ designed the framework of the manuscript and prepared and finalized the manuscript. All authors agreed to approved the final manuscript.
This work was supported by The First Hospital, Jilin University, Changchun of China and Sanming Project of Medicine in Shenzhen city (SZSM201801014), Guangdong Province of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AD, Alzheimer’s disease; A β, amyloid β; P. gingivalis, Porphyromonas gingivalis; H. pylori, Helicobacter pylori; C. albicans, Candida albicans; C. neoformans, Cryptococcus neoformans; HIV, human immunodeficiency virus; HSV-1, herpes simplex virus type 1; HHV-6, human herpes virus-6; EBV, Epstein–Barr virus; COVID-19, coronavirus 2019; CNS, central nervous system; IL, interleukin; TNF- α, tumor necrosis factor-alpha; NO, nitric oxide; Th, T helper; CSF, cerebrospinal fluid; APOE, apolipoprotein; KO, knockout; Cat, cathepsin; NF- κ B, nuclear factor kappa-B; WT, wild type; VaD, vascular dementia; PD, Parkinson’s disease; MMSE, mini-mental state examination; TTV, Torque teno virus; AIDS, acquired immune deficiency syndrome; EVs, extracellular vesicles; BBB, blood-brain barrier; NFT, neurofibrillary tangles; Tat, transcription; HAND, HIV-associated neurocognitive disorder; MCI, mild cognitive impairment; ER, endoplasmic reticulum; UPR, unfolded protein response; RNA-seq, RNA sequencing; CVD, cardiovascular disease; PrPSc, prion protein scrapie; PrPC, cellular prion protein; BDNF, brain-derived neurotrophic factor; IFN, type I interferon; ISG, interferon-stimulated gene.
Abbas, N., Bednar, I., Mix, E., Marie, S., Paterson, D., Ljungberg, A., et al. (2002). Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. 126, 50–57. doi: 10.1016/s0165-5728(02)00050-4
Abraham, D., Feher, J., Scuderi, G. L., Szabo, D., Dobolyi, A., Cservenak, M., et al. (2019). Exercise and probiotics attenuate the development of Alzheimer’s disease in transgenic mice: Role of microbiome. Exp. Gerontol. 115, 122–131. doi: 10.1016/j.exger.2018.12.005
Akbari, E., Asemi, Z., Daneshvar Kakhaki, R., Bahmani, F., Kouchaki, E., Tamtaji, O. R., et al. (2016). Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 8:256. doi: 10.3389/fnagi.2016.00256
Albaret, G., Sifré, E., Floch, P., Laye, S., Aubert, A., Dubus, P., et al. (2020). Alzheimer’s Disease and Helicobacter pylori Infection: Inflammation from Stomach to Brain? J. Alzheimers Dis. 73, 801–809. doi: 10.3233/jad-190496
Allnutt, M. A., Johnson, K., Bennett, D. A., Connor, S. M., Troncoso, J. C., Pletnikova, O., et al. (2020). Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 105, 1027–1035.e1022. doi: 10.1016/j.neuron.2019.12.031
Alonso, R., Pisa, D., Aguado, B., and Carrasco, L. (2017). Identification of Fungal Species in Brain Tissue from Alzheimer’s Disease by Next-Generation Sequencing. J. Alzheimers Dis. 58, 55–67. doi: 10.3233/jad-170058
Alonso, R., Pisa, D., Fernández-Fernández, A. M., and Carrasco, L. (2018). Infection of Fungi and Bacteria in Brain Tissue From Elderly Persons and Patients With Alzheimer’s Disease. Front. Aging Neurosci. 10:159. doi: 10.3389/fnagi.2018.00159
Alonso, R., Pisa, D., Marina, A. I., Morato, E., Rábano, A., and Carrasco, L. (2014a). Fungal infection in patients with Alzheimer’s disease. J. Alzheimers Dis. 41, 301–311. doi: 10.3233/jad-132681
Alonso, R., Pisa, D., Rábano, A., and Carrasco, L. (2014b). Alzheimer’s disease and disseminated mycoses. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1125–1132. doi: 10.1007/s10096-013-2045-z
Alonso, R., Pisa, D., Rábano, A., Rodal, I., and Carrasco, L. (2015). Cerebrospinal Fluid from Alzheimer’s Disease Patients Contains Fungal Proteins and DNA. J. Alzheimers Dis. 47, 873–876. doi: 10.3233/jad-150382
Angelucci, F., Cechova, K., Amlerova, J., and Hort, J. (2019). Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinfl. 16:108. doi: 10.1186/s12974-019-1494-4
Arking, E. J., Appelt, D. M., Abrams, J. T., Kolbe, S., Hudson, A. P., and Balin, B. J. (1999). Ultrastructural Analysis of Chlamydia Pneumoniae in the Alzheimer’s Brain. Pathogenesis 1, 201–211.
Ashraf, G. M., Tarasov, V. V., Makhmutova, A., Chubarev, V. N., Avila-Rodriguez, M., Bachurin, S. O., et al. (2019). The Possibility of an Infectious Etiology of Alzheimer Disease. Mol. Neurobiol. 56, 4479–4491. doi: 10.1007/s12035-018-1388-y
Athari Nik Azm, S., Djazayeri, A., Safa, M., Azami, K., Ahmadvand, B., et al. (2018). Lactobacilli and bifidobacteria ameliorate memory and learning deficits and oxidative stress in β-amyloid (1-42) injected rats. Appl. Physiol. Nutr. Metabol. 43, 718–726. doi: 10.1139/apnm-2017-0648
Baig, A. M., Khaleeq, A., Ali, U., and Syeda, H. (2020). Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 11, 995–998. doi: 10.1021/acschemneuro.0c00122
Behrens, N. E., Lipke, P. N., Pilling, D., Gomer, R. H., and Klotz, S. A. (2019). Serum Amyloid P Component Binds Fungal Surface Amyloid and Decreases Human Macrophage Phagocytosis and Secretion of Inflammatory Cytokines. mBio 10, e00218–19. doi: 10.1128/mBio.00218-19
Beydoun, M. A., Beydoun, H. A., Weiss, J., Hossain, S., El-Hajj, Z. W., and Zonderman, A. B. (2020). Helicobacter pylori, periodontal pathogens, and their interactive association with incident all-cause and Alzheimer’s disease dementia in a large national survey. Mol. Psychiatry doi: 10.1038/s41380-020-0736-2
Boelen, E., Stassen, F. R. M., van der Ven, A. J. A. M., Lemmens, M. A. M., Steinbusch, H. P. J., Bruggeman, C. A., et al. (2007). Detection of amyloid beta aggregates in the brain of BALB/c mice after Chlamydia pneumoniae infection. Acta Neuropathol. 114, 255–261.
Boje, K. M., and Arora, P. K. (1992). Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 587, 250–256. doi: 10.1016/0006-8993(92)91004-x
Bostanciklioglu, M. (2020). Severe acute respiratory syndrome coronavirus 2 is penetrating to dementia research. Curr. Neurovasc. Res. 17, 342–343. doi: 10.2174/1567202617666200522220509
Bronzuoli, M. R., Iacomino, A., Steardo, L., and Scuderi, C. (2016). Targeting neuroinflammation in Alzheimer’s disease. J. inflamm. Res. 9, 199–208.
Brown, E. E., Kumar, S., Rajji, T. K., Pollock, B. G., and Mulsant, B. H. (2020). Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am. J. Geriatr. Psychiatry 28, 712–721. doi: 10.1016/j.jagp.2020.04.010
Bu, X. L., Yao, X. Q., Jiao, S. S., Zeng, F., Liu, Y. H., Xiang, Y., et al. (2015). A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 22, 1519–1525. doi: 10.1111/ene.12477
Campbell, L. A., Cho-Chou, K., and Grayston, J. T. (1998). Chlamydia pneumoniae and cardiovascular disease. Emerg. Infect. Dis. 4, 571–579.
Canet, G., Dias, C., Gabelle, A., Simonin, Y., Gosselet, F., Marchi, N., et al. (2018). HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell Neurosci. 12:307. doi: 10.3389/fncel.2018.00307
Carbone, I., Lazzarotto, T., Ianni, M., Porcellini, E., Forti, P., Masliah, E., et al. (2014). Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol. Aging 35, 122–129. doi: 10.1016/j.neurobiolaging.2013.06.024
Carter, C. J. (2017). Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 1, 125–157. doi: 10.3233/adr-170017
Cattaneo, A., Cattane, N., Galluzzi, S., Provasi, S., Lopizzo, N., Festari, C., et al. (2017). Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68. doi: 10.1016/j.neurobiolaging.2016.08.019
Chitnis, T., and Weiner, H. L. (2017). CNS inflammation and neurodegeneration. J. Clin. Invest. 127, 3577–3587. doi: 10.1172/jci90609
Cicin-Sain, L., Brien, J. D., Uhrlaub, J. L., Drabig, A., Marandu, T. F., and Nikolich-Zugich, J. (2012). Cytomegalovirus Infection Impairs Immune Responses and Accentuates T-cell Pool Changes Observed in Mice with Aging. PLoS Pathog. 8:e1002849. doi: 10.1371/journal.ppat.1002849
Come, J. H., Fraser, P. E., and Lansbury, P. T. Jr. (1993). A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc. Natl. Acad. Sci. U.S.A. 90, 5959–5963. doi: 10.1073/pnas.90.13.5959
Costa, M. J. F., de Araújo, I. D. T., da Rocha, Alves, L., da Silva, R. L., and Dos Santos, et al. (2021). Relationship of Porphyromonas gingivalis and Alzheimer’s disease: a systematic review of pre-clinical studies. Clin. Oral. Investig. 25, 797–806. doi: 10.1007/s00784-020-03764-w
Cribbs, D. H., Azizeh, B. Y., Cotman, C. W., and LaFerla, F. M. (2000). Fibril formation and neurotoxicity by a herpes simplex virus glycoprotein B fragment with homology to the Alzheimer’s A beta peptide. Biochemistry 39, 5988–5994. doi: 10.1021/bi000029f
de Almeida, S. M., Ribeiro, C. E., Rotta, I., Letendre, S., Potter, M., Tang, B., et al. (2020). Blood amyloid-β protein isoforms are affected by HIV-1 in a subtype-dependent pattern. J. Neurovirol. 26, 3–13. doi: 10.1007/s13365-019-00783-6
Derhovanessian, E., Theeten, H., Hähnel, K., Van Damme, P., Cools, N., and Pawelec, G. (2013). Cytomegalovirus-associated accumulation of late-differentiated CD4 T-cells correlates with poor humoral response to influenza vaccination. Vaccine 31, 685–690. doi: 10.1016/j.vaccine.2012.11.041
Devanand, D. P., Andrews, H., Kreisl, W. C., Razlighi, Q., Gershon, A., Stern, Y., et al. (2020). Antiviral therapy: Valacyclovir Treatment of Alzheimer’s Disease (VALAD) Trial: protocol for a randomised, double-blind,placebo-controlled, treatment trial. BMJ Open 10:e032112. doi: 10.1136/bmjopen-2019-032112
Doulberis, M., Kotronis, G., Gialamprinou, D., Polyzos, S. A., Papaefthymiou, A., Katsinelos, P., et al. (2020). Alzheimer’s disease and gastrointestinal microbiota; impact of Helicobacter pylori infection involvement. Int. J. Neurosci. 131, 1–19. doi: 10.1080/00207454.2020.1738432
Dubbelaar, M. L., Kracht, L., Eggen, B. J. L., and Boddeke, E. (2018). The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 9:1753. doi: 10.3389/fimmu.2018.01753
Dugan, H. L., Henry, C., and Wilson, P. C. (2020). Aging and influenza vaccine-induced immunity. Cell. Immunol. 348:103998. doi: 10.1016/j.cellimm.2019.103998
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Rodriguez, A. S., Mitchell, T., Washicosky, K. J., et al. (2018). Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 100, 1527–1532. doi: 10.1016/j.neuron.2018.11.043
Fakhoury, M. (2018). Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 16, 508–518. doi: 10.2174/1570159x15666170720095240
Ferini-Strambi, L., and Salsone, M. (2020). COVID-19 and neurological disorders: are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 268, 409–419. doi: 10.1007/s00415-020-10070-8
Floyd, R. A. (1999). Neuroinflammatory processes are important in neurodegenerative diseases: an hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Radic. Biol. Med. 26, 1346–1355. doi: 10.1016/s0891-5849(98)00293-7
Fotuhi, M., Mian, A., Meysami, S., and Raji, C. A. (2020). Neurobiology of COVID-19. J. Alzheimers Dis. 76, 3–19. doi: 10.3233/jad-200581
Fröhlich, E. E., Farzi, A., Mayerhofer, R., Reichmann, F., Jačan, A., Wagner, B., et al. (2016). Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain 56, 140–155. doi: 10.1016/j.bbi.2016.02.020
Fu, P., Gao, M., and Yung, K. K. L. (2020). Association of Intestinal Disorders with Parkinson’s Disease and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. ACS Chem. Neurosci. 11, 395–405. doi: 10.1021/acschemneuro.9b00607
Fulop, T., Witkowski, J. M., Larbi, A., Khalil, A., Herbein, G., and Frost, E. H. (2019). Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J. Neurovirol. 25, 634–647. doi: 10.1007/s13365-019-00732-3
Fulop, T., Witkowski, J. M., Olivieri, F., and Larbi, A. (2018). The integration of inflammaging in age-related diseases. Sem. Immunol. 40, 17–35. doi: 10.1016/j.smim.2018.09.003
Gate, D., Saligrama, N., Leventhal, O., Yang, A. C., Unger, M. S., Middeldorp, J., et al. (2020). Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404. doi: 10.1038/s41586-019-1895-7
Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., Krishnamurthy, S., Paulucci-Holthauzen, A. A., Sengupta, U., Lasagna-Reeves, C. A., et al. (2014). Amyloid-β oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol. Dis. 71, 14–23. doi: 10.1016/j.nbd.2014.08.008
Haditsch, U., Roth, T., Rodriguez, L., Hancock, S., Cecere, T., Nguyen, M., et al. (2020). Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains. J. Alzheimers Dis. 75, 1361–1376. doi: 10.3233/jad-200393
Hafezi-Moghadam, A., Thomas, K. L., and Wagner, D. D. (2007). ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am. J. Physiol. Cell Physiol. 292, C1256–C1262. doi: 10.1152/ajpcell.00563.2005
Hategan, A., Masliah, E., and Nath, A. (2019). HIV and Alzheimer’s disease: complex interactions of HIV-Tat with amyloid β peptide and Tau protein. J. Neurovirol. 25, 648–660. doi: 10.1007/s13365-019-00736-z
Hur, J. Y., Frost, G. R., Wu, X., Crump, C., Pan, S. J., Wong, E., et al. (2020). The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 586, 735–740. doi: 10.1038/s41586-020-2681-2
Ilievski, V., Zuchowska, P. K., Green, S. J., Toth, P. T., Ragozzino, M. E., Le, K., et al. (2018). Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13:e0204941. doi: 10.1371/journal.pone.0204941
Inoue, Y., Nakajima, M., Uetani, H., Hirai, T., Ueda, M., Kitajima, M., et al. (2016). Diagnostic Significance of Cortical Superficial Siderosis for Alzheimer Disease in Patients with Cognitive Impairment. AJNR 37, 223–227. doi: 10.3174/ajnr.A4496
Itzhaki, R. F., Golde, T. E., Heneka, M. T., and Readhead, B. (2020). Do infections have a role in the pathogenesis of Alzheimer disease? Nat. Rev. Neurol. 16, 193–197. doi: 10.1038/s41582-020-0323-9
Jamieson, G. A., Maitland, N. J., Wilcock, G. K., Craske, J., and Itzhaki, R. F. (1991). Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 33, 224–227. doi: 10.1002/jmv.1890330403
Kamphuis, W., Kooijman, L., Schetters, S., Orre, M., and Hol, E. M. (2016). Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 1862, 1847–1860. doi: 10.1016/j.bbadis.2016.07.007
Kim, J., Kim, A. R., and Shin, E.-C. (2015). Cytomegalovirus Infection and Memory T Cell Inflation. Immune Network 15, 186–190. doi: 10.4110/in.2015.15.4.186
Kodidela, S., Gerth, K., Haque, S., Gong, Y., Ismael, S., Singh, A., et al. (2019). Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells 8:9. doi: 10.3390/cells8090968
Kountouras, J., Boziki, M., Gavalas, E., Zavos, C., Grigoriadis, N., Deretzi, G., et al. (2009). Eradication of Helicobacter pylori may be beneficial in the management of Alzheimer’s disease. J. Neurol. 256, 758–767. doi: 10.1007/s00415-009-5011-z
Kountouras, J., Tsolaki, M., Gavalas, E., Boziki, M., Zavos, C., Karatzoglou, P., et al. (2006). Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology 66, 938–940. doi: 10.1212/01.wnl.0000203644.68059.5f
La Rosa, F., Agostini, S., Bianchi, A., Nemni, R., Piancone, F., Marventano, I., et al. (2019). Herpes simplex virus-1 (HSV-1) infection induces a potent but ineffective IFN-λ production in immune cells of AD and PD patients. J. Transl. Med. 17:286. doi: 10.1186/s12967-019-2034-9
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Laugisch, O., Johnen, A., Maldonado, A., Ehmke, B., Bürgin, W., Olsen, I., et al. (2018). Periodontal Pathogens and Associated Intrathecal Antibodies in Early Stages of Alzheimer’s Disease. JAD 66, 105–114. doi: 10.3233/JAD-180620
Leblhuber, F., Steiner, K., Schuetz, B., Fuchs, D., and Gostner, J. M. (2018). Probiotic Supplementation in Patients with Alzheimer’s Dementia - An Explorative Intervention Study. Curr. Alzheimer Res. 15, 1106–1113. doi: 10.2174/1389200219666180813144834
Lecler, A., Fournier, L., Diard-Detoeuf, C., and Balvay, D. (2017). Blood-Brain Barrier Leakage in Early Alzheimer Disease. Radiology 282, 923–925. doi: 10.1148/radiol.2017162578
Lellouch-Tubiana, A., Fohlen, M., Robain, O., and Rozenberg, F. (2010). Immunocytochemical characterization of long−term persistent immune activation in human brain after herpes simplex encephalitis. Neuropathol. Appl. Neurobiol. 26, 285–294.
Levy, M., Kolodziejczyk, A. A., Thaiss, C. A., and Elinav, E. (2017). Dysbiosis and the immune system. Nat. Rev. Immunol. 17, 219–232. doi: 10.1038/nri.2017.7
Li, J., Long, X., Huang, H., Tang, J., Zhu, C., Hu, S., et al. (2020). Resilience of Alzheimer’s Disease to COVID-19. J. Alzheimers Dis. 77, 67–73. doi: 10.3233/jad-200649
Liccardo, D., Marzano, F., Carraturo, F., Guida, M., Femminella, G. D., Bencivenga, L., et al. (2020). Potential Bidirectional Relationship Between Periodontitis and Alzheimer’s Disease. Front. Physiol. 11:683. doi: 10.3389/fphys.2020.00683
Lieberman, R. (2000). HIV in older Americans: an epidemiologic perspective. J. Midwifery Womens Health 45, 176–182. doi: 10.1016/s1526-9523(00)00002-7
Lin, T.-W., Shih, Y.-H., Chen, S.-J., Lien, C.-H., Chang, C.-Y., Huang, T.-Y., et al. (2015). Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol. Learn. Memory 118, 189–197. doi: 10.1016/j.nlm.2014.12.005
Lin, W. R., Wozniak, M. A., Cooper, R. J., Wilcock, G. K., and Itzhaki, R. F. (2002). Herpesviruses in brain and Alzheimer’s disease. J. Pathol. 197, 395–402. doi: 10.1002/path.1127
Linard, M., Letenneur, L., Garrigue, I., Doize, A., Dartigues, J. F., and Helmer, C. (2020). Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimers Dement 16, 200–208. doi: 10.1002/alz.12008
Loeb, M. B., Molloy, D. W., Smieja, M., Standish, T., Goldsmith, C. H., Mahony, J., et al. (2004). A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 52, 381–387.
Lukiw, W. J. (2020). microRNA-146a Signaling in Alzheimer’s Disease (AD) and Prion Disease (PrD). Front. Neurol. 11:462. doi: 10.3389/fneur.2020.00462
Malaguarnera, M., Bella, R., Alagona, G., Ferri, R., Carnemolla, A., and Pennisi, G. (2004). Helicobacter pylori and Alzheimer’s disease: a possible link. Eur. J. Intern. Med. 15, 381–386. doi: 10.1016/j.ejim.2004.05.008
Matsushita, K., Yamada-Furukawa, M., Kurosawa, M., and Shikama, Y. (2020). Periodontal Disease and Periodontal Disease-Related Bacteria Involved in the Pathogenesis of Alzheimer’s Disease. J. inflamm. Res. 13, 275–283. doi: 10.2147/JIR.S255309
Melah, K. E., Lu, S. Y., Hoscheidt, S. M., Alexander, A. L., Adluru, N., Destiche, D. J., et al. (2016). Cerebrospinal Fluid Markers of Alzheimer’s Disease Pathology and Microglial Activation are Associated with Altered White Matter Microstructure in Asymptomatic Adults at Risk for Alzheimer’s Disease. J. Alzheimers Dis. 50, 873–886. doi: 10.3233/jad-150897
Minter, M. R., Hinterleitner, R., Meisel, M., Zhang, C., Leone, V., Zhang, X., et al. (2017). Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APP/PS1 murine model of Alzheimer’s disease. Scientific Rep. 7:10411. doi: 10.1038/s41598-017-11047-w
Minter, M. R., Zhang, C., Leone, V., Ringus, D. L., Zhang, X., Oyler-Castrillo, P., et al. (2016). Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Scientific Rep. 6:30028. doi: 10.1038/srep30028
Molloy, D. W., Standish, T. I., Zhou, Q., and Guyatt, G. (2013). A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: the DARAD trial. Int. J. Geriatr. Psych. 28, 463–470. doi: 10.1002/gps.3846
Nakamura, T., Oh, C. K., Liao, L., Zhang, X., Lopez, K. M., Gibbs, D., et al. (2021). Noncanonical transnitrosylation network contributes to synapse loss in Alzheimer’s disease. Science 371:6526. doi: 10.1126/science.aaw0843
Naughton, S. X., Raval, U., and Pasinetti, G. M. (2020). Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies. J. Alzheimers Dis. 76, 21–25. doi: 10.3233/jad-200537
Netland, J., Meyerholz, D. K., Moore, S., Cassell, M., and Perlman, S. (2008). Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 82, 7264–7275. doi: 10.1128/JVI.00737-08
Neufeld, N. H., Mohamed, N. S., Grujich, N., and Shulman, K. (2017). Acute Neuropsychiatric Symptoms Associated With Antibiotic Treatment of Helicobacter Pylori Infections: A Review. J. Psychiatr. Pract. 23, 25–35. doi: 10.1097/PRA.0000000000000205
Neuman, H., Forsythe, P., Uzan, A., Avni, O., and Koren, O. (2018). Antibiotics in early life: dysbiosis and the damage done. FEMS Microbiol. Rev. 42, 489–499. doi: 10.1093/femsre/fuy018
Nie, R., Wu, Z., Ni, J., Zeng, F., Yu, W., Zhang, Y., et al. (2019). Porphyromonas gingivalis Infection Induces Amyloid-β Accumulation in Monocytes/Macrophages. JAD 72, 479–494. doi: 10.3233/JAD-190298
Olsen, I., and Singhrao, S. K. (2020). infection may contribute to systemic and intracerebral amyloid-beta: implications for Alzheimer’s disease onset. Exp. Rev. Anti Infect. Ther. 18, 1063–1066. doi: 10.1080/14787210.2020.1792292
Onodera, T. (2017). Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc. Jap. Acad. Ser B 93, 155–173. doi: 10.2183/pjab.93.010
Ou, Y.-N., Zhu, J.-X., Hou, X.-H., Shen, X.-N., Xu, W., Dong, Q., et al. (2020). Associations of Infectious Agents with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. JAD 75, 299–309. doi: 10.3233/JAD-191337
Panza, F., Lozupone, M., Solfrizzi, V., Watling, M., and Imbimbo, B. P. (2019). Time to test antibacterial therapy in Alzheimer’s disease. Brain 142, 2905–2929. doi: 10.1093/brain/awz244
Pawelec, G. (2017). Immunosenescence and cancer. Biogerontology 18, 717–721. doi: 10.1007/s10522-017-9682-z
Pisa, D., Alonso, R., Juarranz, A., Rábano, A., and Carrasco, L. (2015a). Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J. Alzheimers Dis. 43, 613–624. doi: 10.3233/jad-141386
Pisa, D., Alonso, R., Rábano, A., Rodal, I., and Carrasco, L. (2015b). Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 5:15015. doi: 10.1038/srep15015
Ponnappan, S., and Ponnappan, U. (2011). Aging and Immune Function: Molecular Mechanisms to Interventions. Antioxid. Redox Signal. 14, 1551–1585. doi: 10.1089/ars.2010.3228
Poole, S., Singhrao, S. K., Chukkapalli, S., Rivera, M., Velsko, I., Kesavalu, L., et al. (2015). Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J. Alzheimers Dis. 43, 67–80. doi: 10.3233/jad-140315
Readhead, B., Haure-Mirande, J. V., Funk, C. C., Richards, M. A., Shannon, P., Haroutunian, V., et al. (2018). Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 99, 64–82.e67. doi: 10.1016/j.neuron.2018.05.023
Riviere, G. R., Riviere, K. H., and Smith, K. S. (2002). Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 17, 113–118. doi: 10.1046/j.0902-0055.2001.00100.x
Romeo, M. A., Gilardini Montani, M. S., Gaeta, A., D’Orazi, G., Faggioni, A., and Cirone, M. (2020). HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta. Mol. Basis Dis. 1866:165647. doi: 10.1016/j.bbadis.2019.165647
Rosén, C., Andersson, C. H., Andreasson, U., Molinuevo, J. L., Bjerke, M., Rami, L., et al. (2014). Increased Levels of Chitotriosidase and YKL-40 in Cerebrospinal Fluid from Patients with Alzheimer’s Disease. Dement Geriatr. Cogn. Dis. Extra 4, 297–304. doi: 10.1159/000362164
Roubaud-Baudron, C., Krolak-Salmon, P., Quadrio, I., Mégraud, F., and Salles, N. (2012). Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: preliminary results. Neurobiol. Aging 33, 1009.e11–9. doi: 10.1016/j.neurobiolaging.2011.10.021
Roy, E. R., Wang, B., Wan, Y.-W., Chiu, G., Cole, A., Yin, Z., et al. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 130, 1912–1930. doi: 10.1172/JCI133737
Ryder, M. I. (2020). Porphyromonas gingivalis and Alzheimer disease: Recent findings and potential therapies. J. Periodontol. 91, S45–S49. doi: 10.1002/JPER.20-0104
Sadrameli, M., Bathini, P., and Alberi, L. (2020). Linking mechanisms of periodontitis to Alzheimer’s disease. Curr. Opin. Neurol. 33, 230–238. doi: 10.1097/WCO.0000000000000797
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Santiago-Tirado, F. H., Onken, M. D., Cooper, J. A., Klein, R. S., and Doering, T. L. (2017). Trojan Horse Transit Contributes to Blood-Brain Barrier Crossing of a Eukaryotic Pathogen. mBio 8, e02183–16. doi: 10.1128/mBio.02183-16
Schnaider, L., Arnon, Z. A., and Gazit, E. (2020). Reevaluating the Microbial Infection Link to Alzheimer’s Disease. J. Alzheimers Dis. 73, 59–62. doi: 10.3233/jad-190765
Schneider, S., Wright, C. M., and Heuckeroth, R. O. (2019). Unexpected Roles for the Second Brain: Enteric Nervous System as Master Regulator of Bowel Function. Annu. Rev. Physiol. 81, 235–259. doi: 10.1146/annurev-physiol-021317-121515
Seledtsov, V. I., and von Delwig, A. A. (2020). Immune memory limits human longevity: the role of memory ÑD4+ T cells in age-related immune abnormalities. Expert Rev. Vacc. 19, 209–215. doi: 10.1080/14760584.2020.1745638
Shi, M., Chu, F., Tian, X., Aerqin, Q., Zhu, F., and Zhu, J. (2021). Role of Adaptive Immune and Impacts of Risk Factors on Adaptive Immune in Alzheimer’s Disease: Are Immunotherapies Effective or Off-Target? Neuroscientist 177:073858420987224. doi: 10.1177/1073858420987224
Singhrao, S. K., Harding, A., Poole, S., Kesavalu, L., and Crean, S. (2015). Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer’s Disease. Mediat. Inflamm. 2015:137357. doi: 10.1155/2015/137357
Smits, H. A., Boven, L. A., Pereira, C. F., Verhoef, J., and Nottet, H. S. (2000). Role of macrophage activation in the pathogenesis of Alzheimer’s disease and human immunodeficiency virus type 1-associated dementia. Eur. J. Clin. Invest. 30, 526–535. doi: 10.1046/j.1365-2362.2000.00661.x
Snow, A. D., Kisilevsky, R., Willmer, J., Prusiner, S. B., and DeArmond, S. J. (1989). Sulfated glycosaminoglycans in amyloid plaques of prion diseases. Acta Neuropathol. 77, 337–342. doi: 10.1007/bf00687367
Sochocka, M., Donskow-Lysoniewska, K., Diniz, B. S., Kurpas, D., Brzozowska, E., and Leszek, J. (2019). The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease-a Critical Review. Mol. Neurobiol. 56, 1841–1851. doi: 10.1007/s12035-018-1188-4
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Sparks Stein, P., Steffen, M. J., Smith, C., Jicha, G., Ebersole, J. L., Abner, E., et al. (2012). Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dementia 8, 196–203. doi: 10.1016/j.jalz.2011.04.006
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219. doi: 10.1111/imm.12922
Sundar, S., Battistoni, C., McNulty, R., Morales, F., Gorky, J., Foley, H., et al. (2020). An agent-based model to investigate microbial initiation of Alzheimer’s via the olfactory system. Theoret. Biol. Med. Model. 17:5. doi: 10.1186/s12976-020-00123-w
Tang, Y., and Le, W. (2016). Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Taylor, G. S., Vipond, I. B., Paul, I. D., Matthews, S., Wilcock, G. K., and Caul, E. O. (2002). Failure to correlate C. pneumoniae with late onset Alzheimer’s disease. Neurology 59, 142–143.
Thomas, R., Wang, W., and Su, D.-M. (2020). Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 17:2. doi: 10.1186/s12979-020-0173-8
Ticinesi, A., Nouvenne, A., Tana, C., Prati, B., and Meschi, T. (2019). Gut Microbiota and Microbiota-Related Metabolites as Possible Biomarkers of Cognitive Aging. Adv. Exp. Med. Biol, 1178, 129–154. doi: 10.1007/978-3-030-25650-0_8
Tzeng, N. S., Chung, C. H., Lin, F. H., Chiang, C. P., Yeh, C. B., Huang, S. Y., et al. (2018). Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 15, 417–429. doi: 10.1007/s13311-018-0611-x
Vallamkondu, J., John, A., Wani, W. Y., Ramadevi, S. P., Jella, K. K., Reddy, P. H., et al. (2020). SARS-CoV-2 pathophysiology and assessment of coronaviruses in CNS diseases with a focus on therapeutic targets. Biochim. Biophys. Acta 1866:165889. doi: 10.1016/j.bbadis.2020.165889
van de Haar, H. J., Burgmans, S., Jansen, J. F., van Osch, M. J., van Buchem, M. A., Muller, M., et al. (2016). Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 281, 527–535. doi: 10.1148/radiol.2016152244
Velsko, I. M., Chukkapalli, S. S., Rivera, M. F., Lee, J. Y., Chen, H., Zheng, D., et al. (2014). Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS One 9:e97811. doi: 10.1371/journal.pone.0097811
Wang, X.-L., Zeng, J., Yang, Y., Xiong, Y., Zhang, Z.-H., Qiu, M., et al. (2015). Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3β. JAD 43, 153–165. doi: 10.3233/JAD-140198
Wang, Y., Song, X., Wang, Y., Huang, L., Luo, W., Li, F., et al. (2020). Dysregulation of cofilin-1 activity-the missing link between herpes simplex virus type-1 infection and Alzheimer’s disease. Critical Rev. Microbiol 2020:32715819. doi: 10.1080/1040841X.2020.1794789
Wiatrak, B., and Balon, K. (2021). Protective Activity of Aβ on Cell Cultures (PC12 and THP-1 after Differentiation) Preincubated with Lipopolysaccharide (LPS). Mol. Neurobiol. 58, 1453–1464. doi: 10.1007/s12035-020-02204-w
Woods, J. J., Skelding, K. A., Martin, K. L., Aryal, R., Sontag, E., Johnstone, D. M., et al. (2020). Assessment of evidence for or against contributions of Chlamydia pneumoniae infections to Alzheimer’s disease etiology. Brain 83, 22–32. doi: 10.1016/j.bbi.2019.10.014
Xu, S., and Gaskin, F. (1997). Increased incidence of anti-beta-amyloid autoantibodies secreted by Epstein-Barr virus transformed B cell lines from patients with Alzheimer’s disease. Mech. Ageing Dev. 94, 213–222. doi: 10.1016/s0047-6374(96)01861-1
Yates, P. A., Desmond, P. M., Phal, P. M., Steward, C., Szoeke, C., Salvado, O., et al. (2014). Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 82, 1266–1273. doi: 10.1212/wnl.0000000000000285
Yeh, F. L., Hansen, D. V., and Sheng, M. (2017). TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 23, 512–533. doi: 10.1016/j.molmed.2017.03.008
Yu, T., Yu, H., Zhang, B., Wang, D., Li, B., Zhu, J., et al. (2019). Promising Neuroprotective Function for M2 Microglia in Kainic Acid-Induced Neurotoxicity Via the Down-Regulation of NF-κB and Caspase 3 Signaling Pathways. Neuroscience 406, 86–96. doi: 10.1016/j.neuroscience.2019.03.002
Yu, Y., Travaglio, M., Popovic, R., Leal, N., and Martins, L. M. (2020). Alzheimer’s and Parkinson’s diseases predict different COVID-19 outcomes, a UK Biobank study. Geriatrics 6:10. doi: 10.3390/geriatrics6010010
Zhang, B., Shen, P., Yin, X., Dai, Y., Ding, M., and Cui, L. (2020). Expression and functions of cellular prion proteins in immunocytes. Scand. J. Immunol. 91:e12854. doi: 10.1111/sji.12854
Zhang, X., Wang, D., Zhang, B., Zhu, J., Zhou, Z., and Cui, L. (2020). Regulation of microglia by glutamate and its signal pathway in neurodegenerative diseases. Drug Discov. Today 25, 1074–1085. doi: 10.1016/j.drudis.2020.04.001
Keywords: Alzheimer’s disease, infection, inflammation, treatment, microbiota
Citation: Shi M, Li C, Tian X, Chu F and Zhu J (2021) Can Control Infections Slow Down the Progression of Alzheimer’s Disease? Talking About the Role of Infections in Alzheimer’s Disease. Front. Aging Neurosci. 13:685863. doi: 10.3389/fnagi.2021.685863
Received: 25 March 2021; Accepted: 24 June 2021;
Published: 22 July 2021.
Edited by:
Rodrigo Morales, University of Texas Health Science Center at Houston, United StatesReviewed by:
Tatiana Barichello, University of Texas Health Science Center at Houston, United StatesCopyright © 2021 Shi, Li, Tian, Chu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Zhu, anpodWhzQHlhaG9vLmNvbQ==; amllLnpodUBraS5zZQ==; anpodUBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.