
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Aging Neurosci. , 29 June 2021
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.671296
This article is part of the Research Topic Genes and Aging: From Bench Side to Bed side View all 10 articles
 Yi-Hui Liu1,2†
Yi-Hui Liu1,2† Hai-Feng Zhang2†Jie-Yuan Jin1†
Hai-Feng Zhang2†Jie-Yuan Jin1† Yan-Qiu Wei2Chen-Yu Wang1
Yan-Qiu Wei2Chen-Yu Wang1 Liang-Liang Fan1,3,4*
Liang-Liang Fan1,3,4* Lv Liu1,3*
Lv Liu1,3*Leukodystrophies are a heterogeneous group of inherited disorders with highly variable clinical manifestations and pathogenetic backgrounds. At present, variants in more than 20 genes have been described and may be responsible for different types of leukodystrophies. Members of the phospholipase D family of enzymes catalyze the hydrolysis of membrane phospholipids. Meanwhile, phospholipase D3 (PLD3) has also been found to exhibit single stranded DNA (ssDNA) acid 5′ exonuclease activity. Variants in phospholipase D3 (PLD3) may increase the risk of Alzheimer's disease and spinocerebellar ataxia, but this hypothesis has not been fully confirmed. In this study, we identified a novel homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 in a consanguineous family with white matter lesions, hearing and vision loss, and kidney disease by whole exome sequencing. Real-time PCR revealed that the novel mutation may lead to non-sense-mediated messenger RNA (mRNA) decay. This may be the first case report on the homozygous mutation of PLD3 in patients worldwide. Our studies indicated that homozygous mutation of PLD3 may result in a novel leukoencephalopathy syndrome with white matter lesions, hearing and vision loss, and kidney disease.
Leukoencephalopathy (LE) is a structural alteration of the cerebral white matter in which myelin suffers the most damage (Kohler et al., 2018). Leukodystrophies can be broadly subdivided into hypomyelinating leukodystrophies, which are characterized by primary deficits in myelin development, and demyelinating leukodystrophies, where myelin develops normally but subsequently undergoes progressive disruption (Vanderver et al., 2015). At present, ~20 distinct disorders are defined as adulthood leukodystrophies (Tillema and Renaud, 2012; Kohler et al., 2018), such as Pelizaeus–Merzbacher disease, adult polyglucosan body disease, and X-linked adrenoleukodystrophy.
The human PLD3 gene, which encodes a single-pass type II membrane protein with two phospholipase D (PLD) phosphodiesterase domains, is located on chromosome 19q13.2 and consists of 13 exons spanning 32 kb. As a member of the phospholipase D family of enzymes that catalyze the hydrolysis of membrane phospholipids, PLD3 has been proven to be involved in the processing of amyloid-beta precursor protein (Fazzari et al., 2017). Recently, two proteins from the PLD family, namely, phospholipase D3 (PLD3) and phospholipase D4 (PLD4), were found to exhibit single stranded DNA (ssDNA) acid 5′ exonuclease activity (Gavin et al., 2018; Cappel et al., 2020). Previous studies revealed that a heterozygous mutation of PLD3 might increase the risk of Alzheimer's disease and spinocerebellar ataxia (Wang et al., 2015; Nibbeling et al., 2017). However, the effect of homozygous mutations in PLD3 is still not clear.
In this study, we identified a novel homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 by whole exome sequencing in a patient from a consanguineous family with white matter lesions, hearing and vision loss, and kidney disease.
This study was carried out in accordance with the guidelines of the institutional ethics committee of the Affiliated Hospital of Yangzhou University in China. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
We enrolled a consanguineous family from the Han-Chinese population (Figure 1A). The proband (IV-1), a 57-year-old woman, was admitted to our hospital due to sudden onset of lightheadedness and vertigo accompanied by nausea and vomiting. Magnetic resonance imaging (MRI) testing detected brain lesions in the proband. T2 image showed high intensity of white matter and thalamus, high intensity of brain stem and thalamus, high intensity of bilateral white matter, high intensity of bilateral thalamus, and high intensity of brain stem and bilateral cerebellum (Figure 1B). The Mini-Mental State Examination (MMSE) suggested a normal cognitive state (score, 27). Except for several episodes of lightheadedness and vertigo, the patient did not present with other motor or cognitive impairment during the hospital stay. A medical history investigation found that the proband suffered from sudden hearing and vision loss. The computed tomography (CT) image of the proband indicated tapering of bilateral optic nerves (Figure 1C). Eye examination revealed normal eye movement but a significant low vision (left, 0.3; right, 0.1). Pure-tone audiometry (PTA) showed severe hearing loss (Figure 1D). In addition, the patient also suffered from chronic kidney disease (CKD) and was diagnosed with focal segmental glomerulosclerosis by renal biopsy in another hospital 10 years ago (Figure 1E). The body mass index of the proband was 21.7, and the blood pressure was 121/77 mmHg. Blood lipid and glucose levels, as well as cerebrovascular CT of the proband, did not display any abnormalities. Upon further interviews on the family history, we found that her parents married consanguineously. The parents (III-1 and III-2) and her brother (IV-3) presented with normal vision, hearing, and kidney function. Brain MRI of the proband's parents and her brother also showed no obvious lesions (Supplementary Figure 1).
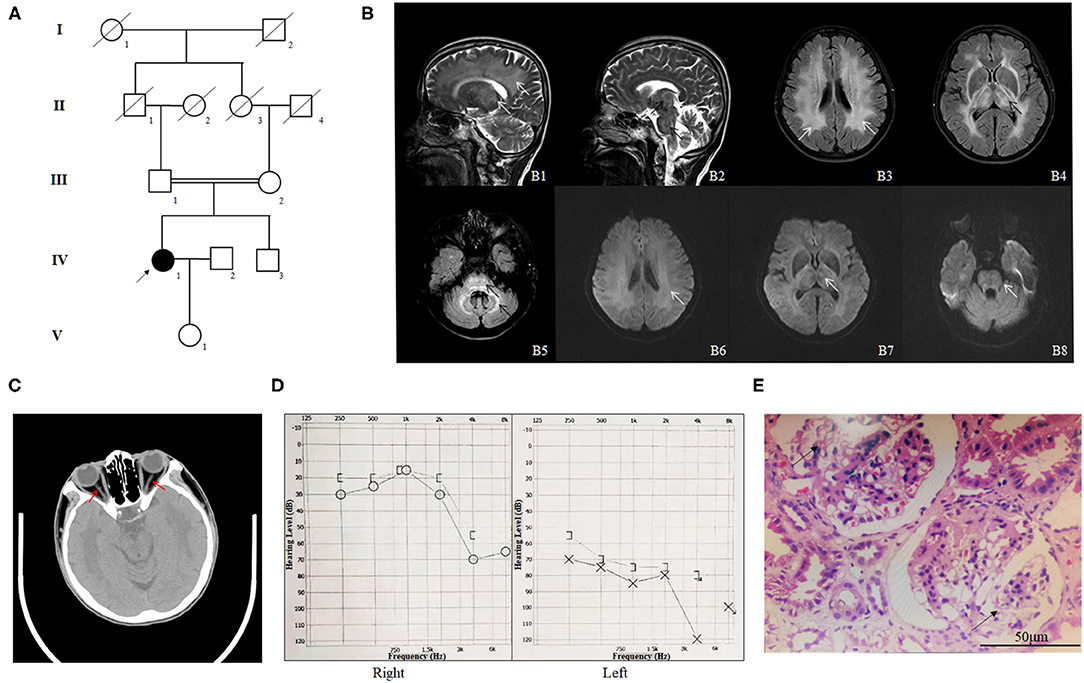
Figure 1. The clinical profile of the family. (A) Pedigree of the phospholipase D3 (PLD3)-deficient family. The pedigree chart shows five generations of the family. Roman numerals refer to generations. Circles refer to female subjects. Squares refer to male subjects. Solid symbols refer to affected subjects. Crossed-out symbols refer to deceased subjects. The arrow indicates the proband. (B) The MR images of the proband. T2 image shows high intensity of white matter and thalamus (B1, white arrows), high intensity of brain stem and thalamus (B2, black arrows), high intensity of bilateral white matter (B3, white arrows), high intensity of bilateral thalamus (B4, black arrows), and high intensity of brain stem and bilateral cerebellum (B5, black arrows). B6–8 are the diffusion weighted image (DWI) corresponding to B3–5 and shows slightly higher intensity. (C) The CT image of the proband indicating tapering of bilateral optic nerves (red arrows). (D) Pure-tone audiometry (PTA) results of the proband. (E) HE staining of the renal biopsy specimen of the proband (IV-1); the arrows indicate glomerulosclerosis.
We then performed whole exome sequencing in an effort to identify the genetic lesions responsible for the disease phenotype of the proband. The central part of the whole exome sequencing was provided by the Novogene Bioinformatics Institute (Beijing, China). The exomes were captured using Agilent SureSelect Human All Exon V6 kits (Agilent Technologies, Sta Clara, CA, USA), and high-throughput sequencing was performed using Illumina HiSeq X-10 (Illumina, San Diego, CA, USA). The necessary bioinformatics analyses, including reads, mapping, variant detection, filtering, and annotation, were also carried out by Novogene Bioinformatics Institute as previously described (Fan et al., 2019).
The strategies of data filtering are as follows (Wang et al., 2020): (a) non-synonymous single-nucleotide polymorphisms (SNPs) or frameshift-causing INDELs with an alternative allele frequency >0.05 in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500), dbSNP152 (http://www.ncbi.nlm.nih.gov/projects/SNP/index.html), the 1000 Genomes project (http://www.1000genomes.org/), the ExAC database (http://exac.broadinstitute.org), or in-house exome databases of Novogene (2500 exomes) were excluded; (b) the filtered single nucleotide variants (SNVs) and INDELs, predicted to be damaging by SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/) were remained; (c) all the homozygous mutations were retained; and (d) cosegregation analysis was conducted in the family.
After data filtering and American College of Medical Genetics and Genomics (ACMG) guideline assessment (Richards et al., 2015), only a novel homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 identified in the proband met the likely pathogenic criteria (Supplementary Table 1). The novel non-sense mutation, resulting in a premature stop codon in exon 5 of the PLD3 gene, was validated in the proband in a homozygous form and existed in the proband's parents and her brother in a heterozygous form (Figure 2A). We then decided to determine whether the novel variant of PLD3 is sensitive to non-sense-mediated messenger RNA (mRNA) decay. According to the GTEx database, PLD3 is expressed in white blood cells. Hence, we isolated the total RNA from peripheral white blood cells derived from three groups or donors (five healthy controls, three heterozygote carriers, and one homozygote patient). After synthesizing complementary DNA (cDNA), real-time PCR found that the mRNA level of PLD3 in heterozygous carriers (III-1, III-2, and IV-3) was decreased by ~43% compared with that in healthy controls (five healthy people without PLD3 mutations), and the expression of PLD3 in homozygous patients (IV-1, repeated three times) was reduced by ~90% compared to that in healthy controls (Figure 2B), which indicated that the non-sense mutation may have led to non-sense-mediated mRNA decay. Concurrently, we isolated the total proteins from white blood cells obtained from two healthy people, two heterozygote carriers (III-1 and IV-3), and the proband (IV-1). Western blot analysis found that the expression of PLD3 in heterozygote group was decreased dramatically compared to healthy controls, and PLD3 was almost absent in the proband (IV-1) (Figure 2C).
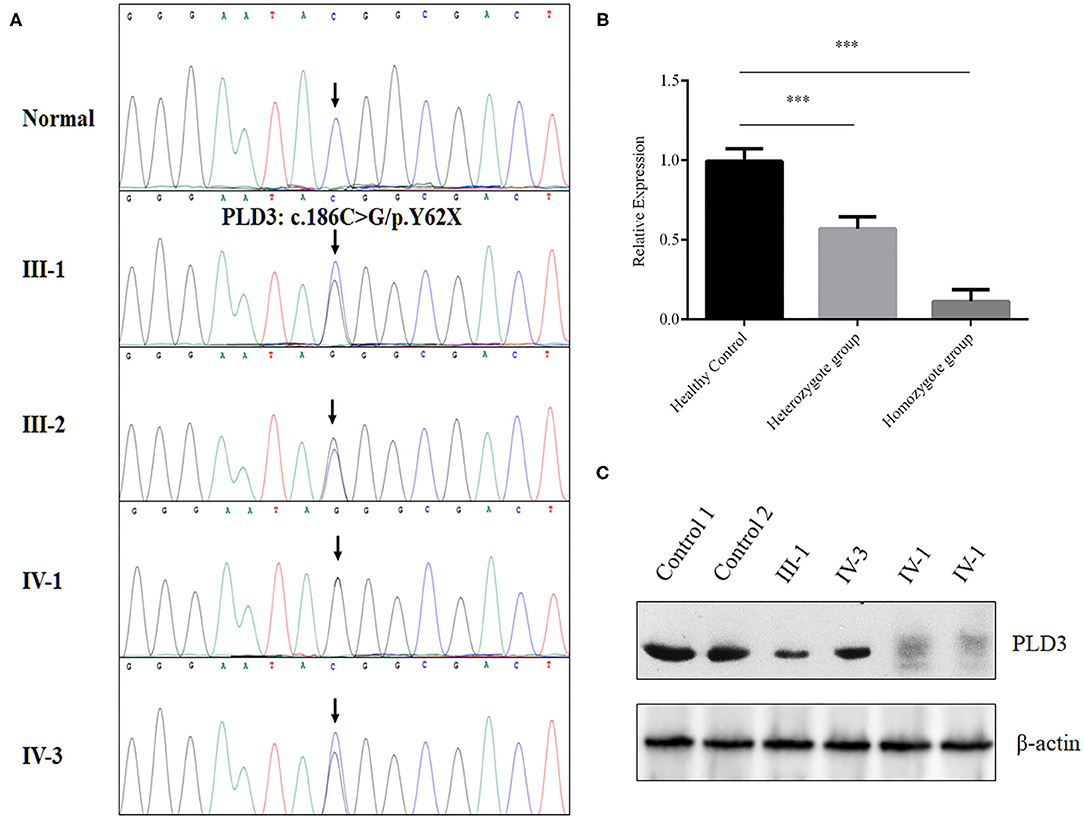
Figure 2. Genetic analysis of the family. (A) Sanger sequencing analysis of the PLD3 gene in the patients, their family members, and the controls. (B) Real-time PCR determined the messenger RNA (mRNA) levels of phospholipase D3 (PLD3) in white blood cells from healthy controls (five samples), heterozygotes (III-1, III-2, and IV-3), and a homozygote (IV-1, repeated three times). ***p < 0.01. (C) Western blot analysis of the expression of PLD3 in white blood cells from healthy controls (two samples), the heterozygote group (III-1 and IV-3), and the homozygote group (IV-1, repeated two times).
In this study, a novel homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 was identified in a consanguineous family with leukoencephalopathy syndrome by whole exome sequencing. Furthermore, real-time PCR and Western blot assays confirmed that the homozygous mutation might have led to non-sense-mediated mRNA decay and resulted in loss of function in PLD3. PLD3 is highly expressed in neurons and may regulate early neuronal development in the central nervous system (Wang et al., 2015). Meanwhile, studies found that the function of PLD3 was to catalyze the hydrolysis of membrane phospholipids. In leukoencephalopathy, neurons are damaged, and myelin is also disrupted (Kohler et al., 2018). Myelin, a phospholipids, might also be regulated by PLD3. Recently, PLD3 was found to exhibit ssDNA acid 5′ exonuclease activity (Gavin et al., 2018; Cappel et al., 2020), which was also associated with brain diseases such as Alzheimer's disease and spinocerebellar ataxia (SCA) (Gavin et al., 2018). Hence, in our case, the pathological changes in the patient who presented with high intensity white matter may have resulted from PLD3 deficiency.
Mutations in PLD3 may also lead to endoplasmic reticulum (ER) stress and reduced phospholipase activity (Nibbeling et al., 2017). Further studies revealed that PLD3 mutation can impair O-glycosylation at pT271 in PLD3, which is essential for normalizing antioxidative phospholipid levels and protecting the brain (Demirev et al., 2019). In addition, variants in PLD3 can reduce PLD3 activity and affect amyloid-β levels in a cellular model of Alzheimer's disease, possibly via the autophagy-dependent mTOR signaling pathway (Tan et al., 2019). Recently, PLD3 has also been found to play a crucial role in regulating inflammatory cytokine responses (Gavin et al., 2018). Macrophages from PLD3-deficient mice had exaggerated TLR9 responses (Gavin et al., 2018). Here, in our study, the patient who carried a homozygous mutation of PLD3 presented with white matter lesions, hearing and vision loss, and focal segmental glomerulosclerosis. We speculated that the homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 may lead to PLD3 deficiency, which may induce ER stress and reduce phospholipase and exonuclease activities in neurons, as well as loss of O-glycosylation at pT271 of PLD3, ultimately damaging neuros in the central nervous system and optic and vestibulocochlear nerves. Simultaneously, PLD3 deficiency may also induce inflammatory cytokine responses in the kidney. Hence, the proband presented with phenotypes not only in the nervous system but also in the kidney. Our studies indicated that homozygous mutation of PLD3 may result in a novel leukoencephalopathy syndrome including white matter lesions, hearing and vision loss, and kidney disease.
Previous genetic studies revealed that variants in PLD3 may increase the risk for late-onset Alzheimer's disease (van der Lee et al., 2015; Tan et al., 2018). However, studies in Belgium found that rare variants in PLD3 do not raise the risk for early-onset Alzheimer's disease (Cacace et al., 2015). Subsequently, Nibbeling et al. identified novel genes (FAT2, PLD3, KIF26B, EP300, and FAT1) in autosomal dominant SCA patients by whole exome sequencing. Functional studies revealed that PLD3 is located in the ER and that the missense mutation p. Leu308Pro of PLD3 may lead to loss of function, which can induce ER stress and reduce phospholipase activity in COS-7 cells (Nibbeling et al., 2017). However, Gonzalez et al. discovered that PLD3 was located in lysosomes but not in the ER and acted as a 5′ exonuclease in HeLa cells. In addition, they also found that loss of PLD3 did not disrupt lipid catabolism and that PLD3 knockout mice did not present cerebellar ataxia phenotypes, which challenged the interpretation of PLD3 mutations as the causative SCA46 gene (Gonzalez et al., 2018). Hence, the identification of additional patients carrying PLD3 mutations will further strengthen the role of PLD3 in brain disease.
In our study, four heterozygous mutation carriers (III-1, III-2, IV-3, and V-1) showed normal physical features, which indicated that heterozygous non-sense mutation of PLD3 might not be the responsible genetic lesion of SCA and Alzheimer's disease. However, homozygous non-sense mutation of PLD3 can lead to white matter lesions, which may develop into Alzheimer's disease in the future. Certainly, we cannot exclude the genetic heterogeneity and incomplete appearance of PLD3. In fact, we enrolled almost 181 patients with white matter lesions in the past 5 years, but we only detected one homozygous mutation of PLD3 in this family. This may be the first case report on a homozygous mutation of PLD3 in patients with leukoencephalopathy syndrome. Our study may reveal a relationship between leukoencephalopathy syndrome and PLD3 homozygous mutation in patients.
In summary, we identified a novel homozygous mutation (NM_012268.3: c.186C>G/ p.Y62X) of PLD3 in a consanguineous family with white matter lesions, hearing and vision loss, and kidney disease. This may be the first case report on a homozygous mutation of PLD3 in patients worldwide. Our study also provided new insights into function of PLD3 in human diseases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://ncbi.nlm.nih.gov/, PRJNA723675.
The studies involving human participants were reviewed and approved by Affiliated Hospital of Yangzhou University in China. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Y-HL and H-FZ enrolled the samples and performed the Sanger sequencing. J-YJ performed the real-time PCR and Western blot. Y-QW and C-YW enrolled the clinical data. L-LF and LL revised the manuscript and support the project. Y-HL, H-FZ, and J-YJ wrote the draft. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (82000079 and 82000427) and Hunan Province Natural Science Foundation (2020JJ5785).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all subjects for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.671296/full#supplementary-material
Cacace, R., Van den Bossche, T., Engelborghs, S., Geerts, N., Laureys, A., Dillen, L., et al. (2015). Rare variants in PLD3 do not affect risk for early-onset Alzheimer disease in a European consortium cohort. Hum. Mutat. 36, 1226–1235. doi: 10.1002/humu.22908
Cappel, C., Gonzalez, A. C., and Damme, M. (2020). Quantification and characterization of the 5'exonuclease activity of the lysosomal nuclease PLD3 by a novel cell-based assay. J. Biol. Chem. 296:100152. doi: 10.1074/jbc.RA120.015867
Demirev, A. V., Song, H. L., Cho, M. H., Cho, K., Peak, J. J., Yoo, H. J., et al. (2019). V232M substitution restricts a distinct O-glycosylation of PLD3 and its neuroprotective function. Neurobiol. Dis. 129, 182–194. doi: 10.1016/j.nbd.2019.05.015
Fan, L. L., Ding, D. B., Huang, H., Chen, Y. Q., Jin, J. Y., Xia, K., et al. (2019). A de novo mutation of SMYD1 (p.F272L) is responsible for hypertrophic cardiomyopathy in a Chinese patient. Clin. Chem. Lab. Med. 57, 532–539. doi: 10.1515/cclm-2018-0578
Fazzari, P., Horre, K., Arranz, A. M., Frigerio, C. S., Saito, T., Saido, T. C., et al. (2017). PLD3 gene and processing of APP. Nature 541, E1–E2. doi: 10.1038/nature21030
Gavin, A. L., Huang, D., Huber, C., Martensson, A., Tardif, V., Skog, P. D., et al. (2018). PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat. Immunol. 19, 942–953. doi: 10.1038/s41590-018-0179-y
Gonzalez, A. C., Stroobants, S., Reisdorf, P., Gavin, A. L., Nemazee, D., Schwudke, D., et al. (2018). PLD3 and spinocerebellar ataxia. Brain 141:e78. doi: 10.1093/brain/awy258
Kohler, W., Curiel, J., and Vanderver, A. (2018). Adulthood leukodystrophies. Nat. Rev. Neurol. 14, 94–105. doi: 10.1038/nrneurol.2017.175
Nibbeling, E. A. R., Duarri, A., Verschuuren-Bemelmans, C. C., Fokkens, M. R., Karjalainen, J. M., Smeets, C., et al. (2017). Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 140, 2860–2878. doi: 10.1093/brain/awx251
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Tan, M., Li, J., Ma, F., Zhang, X., Zhao, Q., and Cao, X. (2019). PLD3 rare variants identified in late-onset Alzheimer's disease affect amyloid-beta levels in cellular model. Front. Neurosci. 13, 116. doi: 10.3389/fnins.2019.00116
Tan, M. S., Zhu, J. X., Cao, X. P., Yu, J. T., and Tan, L. (2018). Rare variants in PLD3 increase risk for Alzheimer's disease in Han Chinese. J. Alzheimers. Dis. 64, 55–59. doi: 10.3233/JAD-180205
Tillema, J. M., and Renaud, D. L. (2012). Leukoencephalopathies in adulthood. Semin. Neurol. 32, 85–94. doi: 10.1055/s-0032-1306391
van der Lee, S. J., Holstege, H., Wong, T. H., Jakobsdottir, J., Bis, J. C., Chouraki, V., et al. (2015). PLD3 variants in population studies. Nature 520, E2–3. doi: 10.1038/nature14038
Vanderver, A., Prust, M., Tonduti, D., Mochel, F., Hussey, H. M., Helman, G., et al. (2015). Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 114, 494–500. doi: 10.1016/j.ymgme.2015.01.006
Wang, C. Y., Chen, Y. Q., Jin, J. Y., Du, R., Fan, L. L., and Xiang, R. (2020). A novel nonsense mutation of ABCA8 in a Han-Chinese family with ASCVD leads to the reduction of HDL-c levels. Front. Genet. 11:755. doi: 10.3389/fgene.2020.00755
Keywords: leukoencephalopathy, white matter lesions, hearing and vision loss, PLD3, Homozygous mutation, chronic kidney disease
Citation: Liu Y-H, Zhang H-F, Jin J-Y, Wei Y-Q, Wang C-Y, Fan L-L and Liu L (2021) Case Report: A Homozygous Mutation (p.Y62X) of Phospholipase D3 May Lead to a New Leukoencephalopathy Syndrome. Front. Aging Neurosci. 13:671296. doi: 10.3389/fnagi.2021.671296
Received: 23 February 2021; Accepted: 31 May 2021;
Published: 29 June 2021.
Edited by:
Chenju Yi, Sun Yat-sen University, ChinaReviewed by:
Raj Kamal Srivastava, Indira Gandhi National Tribal University, IndiaCopyright © 2021 Liu, Zhang, Jin, Wei, Wang, Fan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang-Liang Fan, c3dmYW5saWFuZ2xpYW5nQGNzdS5lZHUuY24=; Lv Liu, ZG9jbGl1bHZAY3N1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.