
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 23 June 2021
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.650740
This article is part of the Research TopicBlood Biomarkers of Neurodegenerative DiseasesView all 26 articles
 Mohammad Reza Asadi1,2
Mohammad Reza Asadi1,2 Marziyeh Sadat Moslehian2
Marziyeh Sadat Moslehian2 Hani Sabaie2Abbas Jalaiei2
Hani Sabaie2Abbas Jalaiei2 Soudeh Ghafouri-Fard3
Soudeh Ghafouri-Fard3 Mohammad Taheri4*Maryam Rezazadeh1,2*
Mohammad Taheri4*Maryam Rezazadeh1,2*Cytoplasmic ribonucleoproteins called stress granules (SGs) are considered as one of the main cellular solutions against stress. Their temporary presence ends with stress relief. Any factor such as chronic stress or mutations in the structure of the components of SGs that lead to their permanent presence can affect their interactions with pathological aggregations and increase the degenerative effects. SGs involved in RNA mechanisms are important factors in the pathophysiology of neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), frontotemporal degeneration (FTD), and Alzheimer's diseases (AD). Although many studies have been performed in the field of SGs and neurodegenerative disorders, so far, no systematic studies have been executed in this field. The purpose of this study is to provide a comprehensive perspective of all studies about the role of SGs in the pathogenesis of neurodegenerative disorders with a focus on the protein ingredients of these granules. This scoping review is based on a six-stage methodology structure and the PRISMA guideline. A systematic search of seven databases for qualified articles was conducted until December 2020. Publications were screened independently by two reviewers and quantitative and qualitative analysis was performed on the extracted data. Bioinformatics analysis was used to plot the network and predict interprotein interactions. In addition, GO analysis was performed. A total of 48 articles were identified that comply the inclusion criteria. Most studies on neurodegenerative diseases have been conducted on ALS, AD, and FTD using human post mortem tissues. Human derived cell line studies have been used only in ALS. A total 29 genes of protein components of SGs have been studied, the most important of which are TDP-43, TIA-1, PABP-1. Bioinformatics studies have predicted 15 proteins to interact with the protein components of SGs, which may be the constituents of SGs. Understanding the interactions between SGs and pathological aggregations in neurodegenerative diseases can provide new targets for treatment of these disorders.
Cell function is divided between the organelles sited inside the cell. Based on the presence of lipid membrane, organelles can be divided into main two groups. Nucleus, mitochondria, endoplasmic reticulum, and Golgi apparatus are the major membranous organelles. Membraneless organelles are ribosomes (Turi et al., 2019), stress granules (Arrigo et al., 1988), p-body (Sheth and Parker, 2003), and nucleolus (Shaw and Jordan, 1995) that are formed during a process called the liquid-liquid phase separation phase (Marnik and Updike, 2019). The existence of stress at the cellular level can lead to a variety of responses, including global translational inhibition, leading to the formation of stress granules (SG) (Aulas et al., 2018). SGs, membraneless ribonucleoproteins containing mRNA, are cytoplasmic accumulations being stopped at the initiation of translation and disappear after the end of stress induction (Boncella et al., 2020). These stresses in mammalian cells include viral infections (biotic stress), induction of redox stress with sodium arsenite, heat and UV radiation which are environmental stress conditions (Kedersha et al., 2013). Three groups can form the protein component of stress granules: RNA-binding proteins, translation initiation factors, and non-RNA-binding proteins (Cao et al., 2020; Samadian et al., 2021). Stoppage at the critical stage of translation initiation due to biotic or environmental stress leads to the isolation of the translating polysomes resulting in the creation of a huge reservoir of RNA and related proteins that build and increase the number of SGs. On the other hand, relieving stress and increasing translated mRNAs is associated with disassembly and reduction in the number of these granules (Panas et al., 2016; Marnik and Updike, 2019).
There are two mechanisms for stopping translation initiation at the cellular level. Phosphorylation of the α subunit of eIF2 transcription initiation factor and prevention of the eIF-4F complex assembly. eIF2 is present in the ternary complex and is responsible for transferring the initiator tRNA to the pre-initiation complex at the 5'-ends of mRNAs. The result of eIF2 phosphorylation is reduction in its binding to GTP and loss of its ability to transfer the initiator tRNA to ribosomes for start codon recognition. Four stress associated kinases (HRI, PERK, PKR, GCN2) have the ability to phosphorylate the α subunit in the eIF2 factor (Aulas et al., 2017; Wolozin and Ivanov, 2019). eIF-4F complex is responsible for detecting the structure of the cap at the 5' mRNA end, and assembly of this complex is controlled by the PI3K-mTOR kinase cascade. eIF4E is inactivated in phosphorylated form and this phosphorylation is performed by mTOR, a member of the phosphatidylinositol 3-kinase-related kinase family of protein kinases (Mitra et al., 2015). eIF4E in active mode prevents the eIF-4F complex assembly and the translation process halts at the initiation point (Gingras et al., 1999; Aulas et al., 2017; Wolozin and Ivanov, 2019).
No specific function can be considered for SGs, but they can be considered as a “decision point” (Wolozin and Ivanov, 2019) for the fate of mRNA trapped in its structure. SG can define the fate of an mRNA that could be subjected to storage, degeneration, or re-initiation of translation. The probability of mRNA being in the SGs structure is sequence-independent but is directly related to the low translatability and the increase in the length of encoded region as well as the UTR region in the mRNA (Khong et al., 2017). Since proteins and RNAs with important roles can be included in SGs, this would affect biological interactions (Arimoto-Matsuzaki et al., 2008).
Traces of SGs have been found in many diseases, such as cancer (Gao et al., 2019), neurodegenerative disorders (Wolozin, 2012) and autoimmune conditions (McCormick and Khaperskyy, 2017). Neurodegenerative diseases include as amyotrophic lateral sclerosis (ALS) (Baron et al., 2013), Alzheimer's disease (AD) (Ash et al., 2014), and multiple sclerosis (MS) (Salapa et al., 2018). The neurodegeneration process involves atrophy and loss of neuronal activity (Wolozin and Ivanov, 2019). In neurodegenerative diseases, many mutations and misfolding events have been identified in this protein components of SGs, which can lead to the accumulation of abnormal proteins and the formation of SGs. Pathological symptoms appear when the presence of SGs becomes permanent due to an increase in their formation resulting from chronic stress (Brown et al., 2020) and a decrease in their deletion due to mutations in genes involved in the process of autophagy (Chitiprolu et al., 2018; Brown et al., 2020).
So far, many studies have been done on the nature of SGs, their components, structures and their pathological characteristics in various neurodegenerative diseases, and useful results have been obtained. In this study, we tried to establish a strong correlation between clinical evidence and genetic characteristics in neurodegenerative diseases in the form of a scoping review study by summarizing all human clinical studies and human-derived cell lines in the field of SGs.
The writing strategy of this article is based on the methodology proposed by Arksey and O'Malley (2005). This strategy was later improved by Levac et al. (2010) and Colquhoun et al. (2014). In this review, 5 steps of the 6-step framework have been followed which includes (1) Development of research questions, (2) Search strategy, (3) Study eligibility criteria, (4) Data extraction, (5) Collating, summarizing and reporting the results. The sixth step, consultation, is optional and is not included in this article. Also, in order to observe the principles of clarity and transparency in writing the article, the Preferred Reporting Items for Systematic Reviews and Meta-Analyses Extension for Scoping Reviews (PRISMA-ScR) Checklist has been used well (Tricco et al., 2018).
To survey, summarize and discuss the studies on SGs in human and human-derived cell lines in neurodegenerative diseases, our review was guided by the following questions:
1. What exactly have been done on SGs in humans with neurodegenerative disease?
2. Exactly what studies have been done on SGs in human-derived cell lines in neurodegenerative disease?
The publications were searched using seven databases: PubMed, Scopus, Cochrane, Google Scholar, Embase, Web of Science, and ProQuest, based on the search method of each database and was not limited to the date, language, subject or type of publication. Also, review articles published in this field were reviewed to reduce the possibility of losing related articles. “Neurodegeneration” keyword was the medical subject heading (MeSH) used in search strategy if available. PubMed and Embase related search strategies are shown in Appendix I. The last search was conducted on December 8, 2020. The references were managed using EndNote X8.1.
Studies in neurodegenerative diseases in relation to SGs in humans or in human-derived cell lines were screened from publications obtained during the search process. All types of publications were reviewed, including journal articles, conference presentations, conference abstracts and reports. Non-English articles with English abstracts were also included. Screening was done in two stages. At first, both researchers (MRA, MSM) screened articles separately based on title and abstract, according to the inclusion criteria mentioned above. In the next step, full texts of the selected articles were investigated to measure its relevance to the research question. Finally, appropriate articles were selected based on the eligibility criteria. Any contradiction was resolved in agreement with the opinion of the third person.
Two separate charts were designed for human samples and human-derived cell lines in Microsoft Excel to help extracting the data. Chart related to human sample articles included author's name, year of publication, diagnosed neurodegenerative disease (number of patients), age and country, sample and method of analysis, protein component of SGs and major findings. The chart related to human-derived cell lines articles included the author's name, year of publication, origin of cell lines, age at biopsy, country, sample, method of analysis, mutation, and major findings. Two researchers (MRA, MSM) separately extracted data from articles based on charts.
Quantitative and qualitative analysis was performed on the data obtained from the publications. In the quantitative analysis section, a descriptive numerical summary of the extent, nature and distribution of the studies were reviewed. In the qualitative analysis section, based on the research question mentioned earlier, a narrative review was performed on the available information with affirmation on the broader context suggested by Levac et al. (2010).
Two disease-protein interaction networks were designed using Cytoscape v3.8.0 software (Shannon et al., 2003) based on data extracted from articles. One was the network of neurodegenerative diseases with protein components of SGs and their interactions according to the results of human studies and the other was the protein components of SGs studied in human-derived cell lines from ALS. Therefore, the gene/proteins found from the literature search were used as input. The output was the interaction network between these proteins. Gene ontology (GO) analysis was performed using Enrichr's web-based tools and services (Kuleshov et al., 2016) on the genes of SGs protein components in neurodegenerative diseases and ALS. Protein-protein interaction was predicted using the string-db cytoscape plugin (Doncheva et al., 2019) on the data extracted from the articles. The analyzed graph of GO was ranked based on p < 0.05.
Keyword-based searches in various databases yielded 1,087 related records. Also, 7 records from other sources were added to the results. Of these, 758 duplicates were identified. After excluding the duplicates, the number of articles with potential to be related to the research question reached 329. A total of 262 articles were excluded from the study by screening titles and abstracts and 61 articles remained. By reviewing the full text of the remaining articles, 49 articles were included in the study, of which 5 articles were conference abstracts. The process of selecting eligible studies is detailed in the flowchart in Figure 1. Table 1 provides an overview of the included articles.
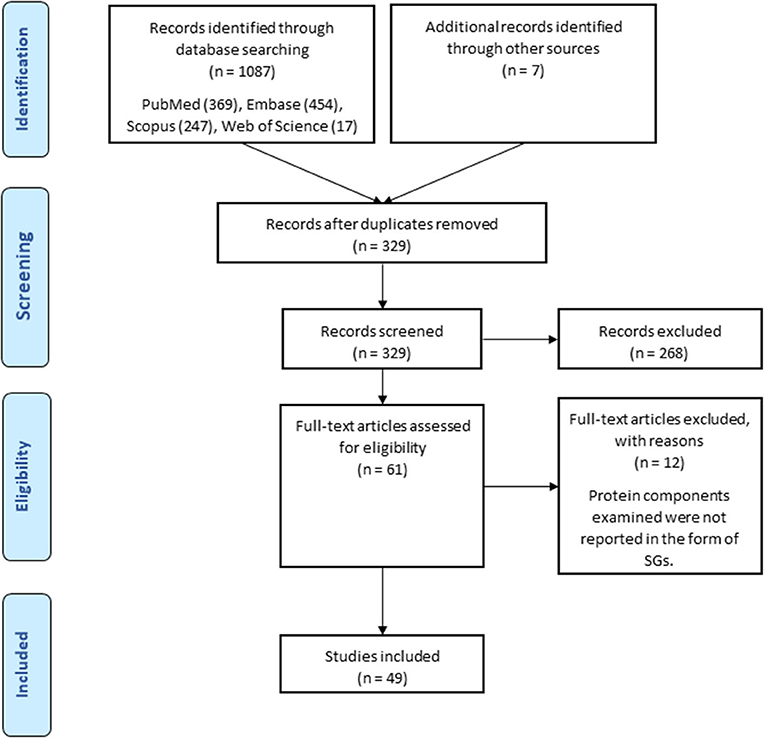
Figure 1. Search strategy flow chart based on the PRISMA flow diagram.

Table 1. Detailed characteristics of included publications.
The studies were published from 2008 to 2020. The total number of neurodegenerative cases in this study is 258. These studies were conducted ALS (Fujita et al., 2008; Colombrita et al., 2009; Volkening et al., 2009; Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Farg et al., 2013, 2014; McGurk et al., 2014; Cohen et al., 2015; Lim et al., 2016; Manghera et al., 2016; Dreser et al., 2017; Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017; Bennett et al., 2018; Chen and Cohen, 2019; Mann et al., 2019; Montalbano et al., 2020; Vassileff et al., 2020), ALS/FTD (Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017), ALS/frontotemporal lobar degeneration (FTLD) (Mann et al., 2019), AD (Castellani et al., 2011; Vanderweyde et al., 2012; Ivanov et al., 2016; Maziuk et al., 2018; Silva et al., 2019; Montalbano et al., 2020; Younas et al., 2020), MS (Salapa et al., 2018, 2020; Levin et al., 2020), FTD (McGurk et al., 2014; Montalbano et al., 2020), FTDP-17 (Vanderweyde et al., 2012), FTLD-u (Fujita et al., 2008; Dormann et al., 2010; Liu-Yesucevitz et al., 2010), FTLD with TDP-43 inclusions (FTLD-TDP) (Dormann et al., 2010; Bentmann et al., 2012; Cohen et al., 2015), FTLD-FUS (Hock et al., 2018), neuronal intermediate filament inclusion disease (NIFID) (Dormann et al., 2010), basophilic inclusion body disease (BIBD) (Dormann et al., 2010; McGurk et al., 2014) and motor neuron disease (MND) (Fujita et al., 2008), which are summarized in Table 2. Cases in this study include both sexes, but because the gender of the cases was not mentioned in many studies, it was not indexed in Table 2. The samples used in the studies were brain, spinal cord and blood, of which twelve studies used only brain (Castellani et al., 2011; Vanderweyde et al., 2012; Manghera et al., 2016; Hock et al., 2018; Maziuk et al., 2018; Salapa et al., 2018, 2020; Silva et al., 2019; Levin et al., 2020; Montalbano et al., 2020; Vassileff et al., 2020; Younas et al., 2020), seven studies used only spinal cord (Colombrita et al., 2009; Farg et al., 2013, 2014; McGurk et al., 2014; Dreser et al., 2017; Mackenzie et al., 2017; Chen and Cohen, 2019), ten studies used both tissues (Fujita et al., 2008; Volkening et al., 2009; Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Cohen et al., 2015; Lim et al., 2016; Hirsch-Reinshagen et al., 2017; Bennett et al., 2018; Mann et al., 2019) and one study used blood samples (Ivanov et al., 2016). The analysis methods of most studies were immunohistochemistry (IHC), but immunoprecipitation (IP) (Volkening et al., 2009; Farg et al., 2013; Montalbano et al., 2020), immunofluorescent microscope imager (Ivanov et al., 2016), RNA-FISH (Mann et al., 2019), mass spectrometry (Vassileff et al., 2020) and confocal microscopy (Volkening et al., 2009; Montalbano et al., 2020) were also used. The protein component of the SGs examined in the studies is also summarized in Figure 2. The included studies have been conducted in the United States, Canada, Australia, Germany, the Netherlands, Japan, Italy, Russia, South Korea, China and the United Kingdom. Based on the bioinformatics studies using the string-db Cytoscape plugin, 15 new genes were predicted to interact with the genes extracted from these studies, including HNRNPC, HNRNPDL, KHDRBS1, HNRNPH1, EDC4, DCP1A, ELAVL1, MATR3, TAF15, HNRNPF, PTBP1, CAPRIN1, PABPC1, KHSRP, AND ILF3.
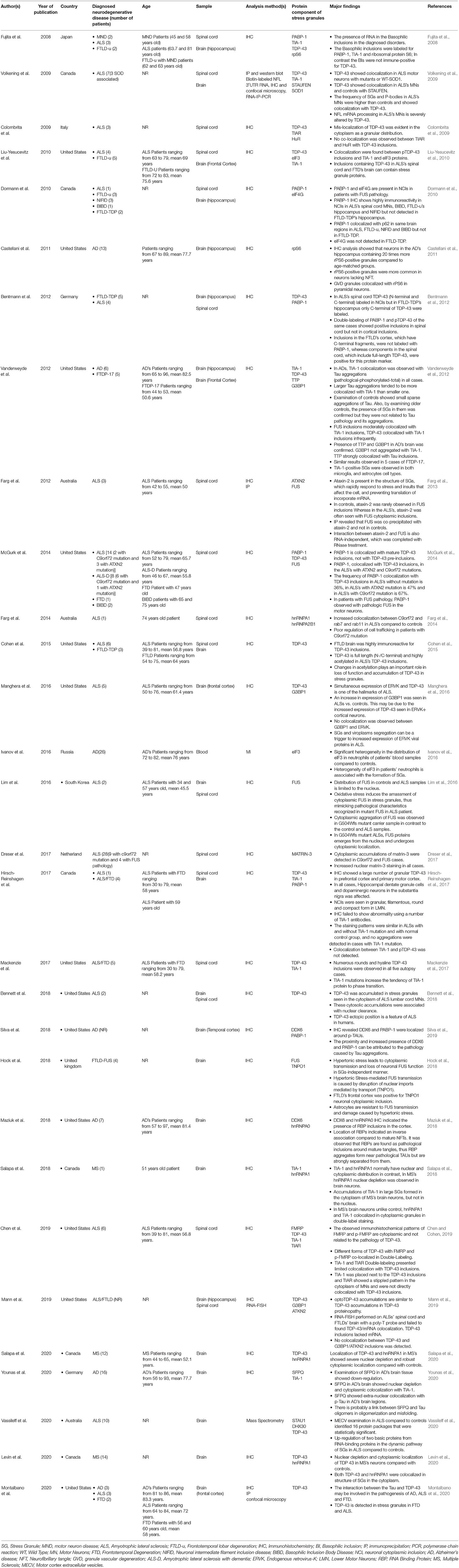
Table 2. Stress Granules and neurodegenerative disorders in human samples.
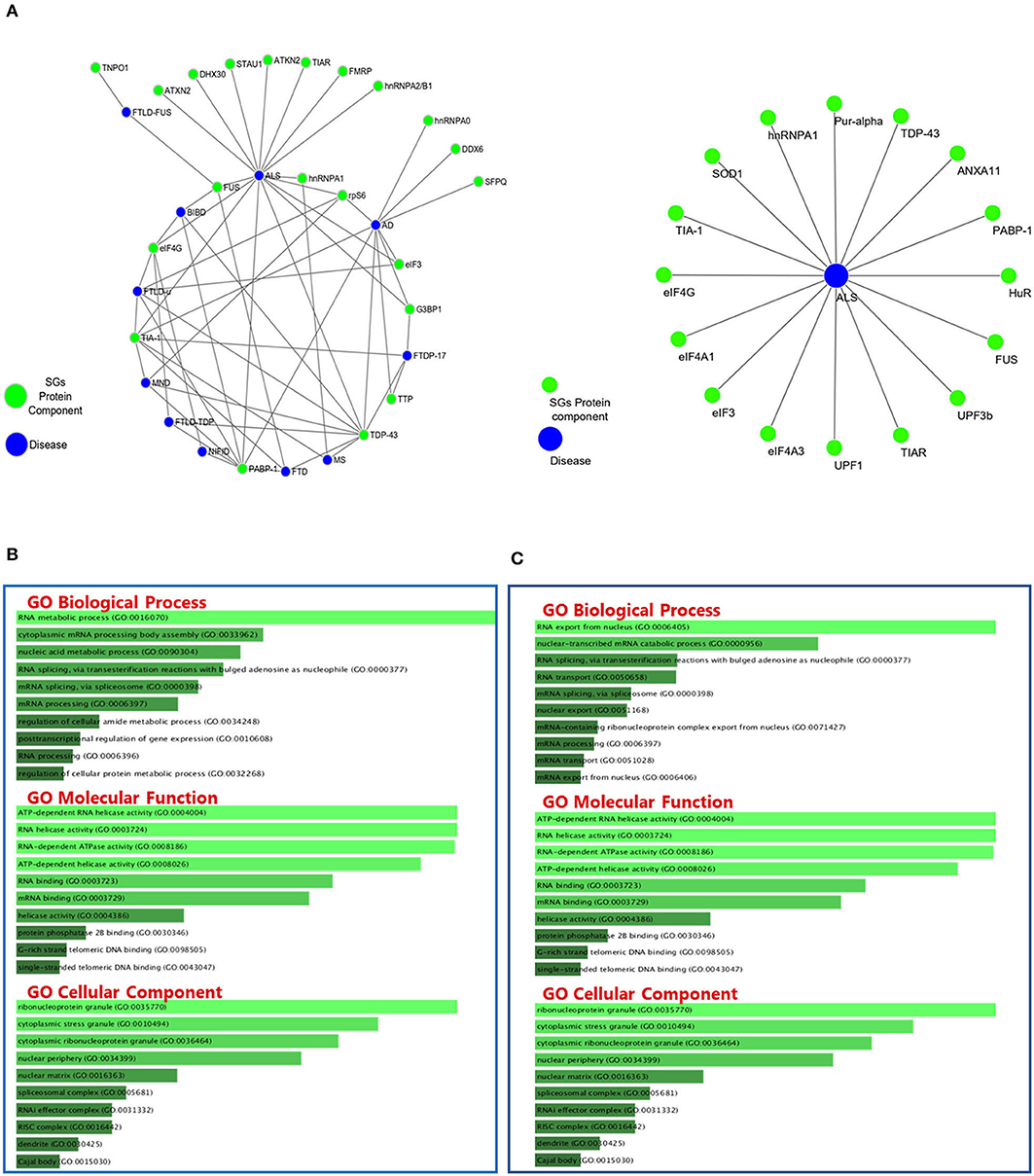
Figure 2. Protein-Protein Interactions and Top 10 GO analysis of target genes in SGs protein components in neurodegenerative disease. The network of neurodegenerative diseases with protein components of SGs and their interactions. (A) Gene ontology analysis of the genes in Table 2 (B) and Table 3 (C) has been performed. The length of each bar represents the degree of significance in that particular category sorted by p-value. Note that the lower the color intensity of the bars, the greater the relationship with that category.
The studies were published from 2016 to 2020. In this ward, only ALS patients were sampled, whose numbers were around 50 as their number has not been reported in 5 studies. The average age of the people reported in this section is 47.6 years. Samples used in the studies included human skin fibroblasts in 18 studies, hair follicle cells (Japtok et al., 2015) and human B-lymphoblastoid cells (Daigle et al., 2016) each in one study. The protein component of SGs examined in these studies includes G3BP1, TIA-1, hnRNPA1, TDP-43, PABP-1, FUS, TIAR, eIF4A3, eIF4A1, eIF3, eIF4G, UPF1, UPF3b, HuR, ANXA11, Pur-alpha, which are also summarized in Figure 3. The cell lines used in this study carry missense mutations in SOD1 (Gal et al., 2016; Rajpurohit et al., 2020), TARDBP (Orrù et al., 2016; Loginov et al., 2018; Ratti et al., 2020), FUS (Japtok et al., 2015; Lenzi et al., 2015; Daigle et al., 2016; Ichiyanagi et al., 2016; Lim et al., 2016; Lo Bello et al., 2017; Kamelgarn et al., 2018; Arenas et al., 2020), TDP-43 (Kreiter et al., 2018; Loginov et al., 2018; Feneberg et al., 2020) and ANXA11 (Gieseler et al., 2019) genes and frameshift mutations in FUS (Japtok et al., 2015; Lim et al., 2016) gene and hexanucleotide repeat expansion mutations in C9ORF72 gene (Dafinca et al., 2016; Loginov et al., 2018; Ratti et al., 2020). Studies were conducted in the United States, Italy, Germany, France, Japan, South Korea, India and the United Kingdom.
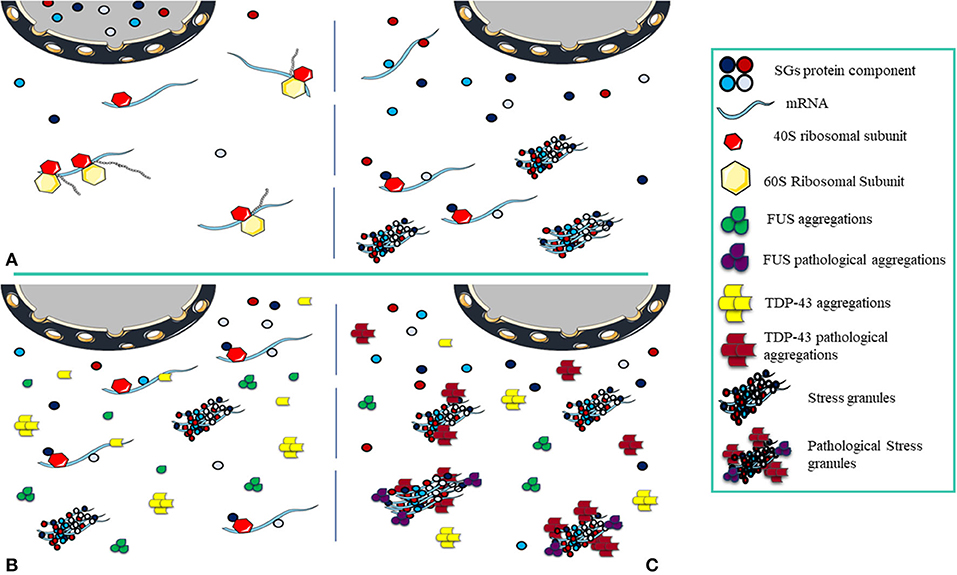
Figure 3. Pathological Stress Granules Formation. (A) The cell is in equilibrium, and as stress arrives, nuclear depletion of SGs components occurs, the translation process halts and the structure of transient SGs is formed. Once the stress is relieved, the cell returns to normal and SGs disassemble. (B) The cell is exposed to acute stress, the components of SGs and protein aggregations (FUS and TDP-43) localize to the cytoplasm. In this condition, FUS and TDP-43 aggregations have not become pathological and do not interact with SGs, the number of SGs decreases as stress is relieved. (C) Under chronic stress, FUS and TDP-43 aggregations become pathological and in interaction with SGs, under the constant presence, pathological effects appear.
Inhibition of translation due to stress is the main factor for SGs assembly. The size of the SGs can vary from 100 to 1,000 nm (Kedersha et al., 1999). SG assembly needs inhibition of the assembly of polysomes so that only the 40S ribosome subunit remains attached. Non-translated mRNAs are attached to other RNA binding proteins at the nucleation phase in the SGs formation. These proteins at the nucleation phase, also known as SG nucleators, are generally multi-domain proteins in which the IDR “intrinsically disordered region” and RBD domains are prominent (Kedersha et al., 2005) and facilitate the assembly of SGs (Gilks et al., 2004). The nucleation phase continues with the formation of a repository of mRNPs in situ, where it is accompanied by interactions between RNA-RNA and RNA-protein to create a stable form of SG cores. Increasing the interactions with the growth of these cores brings the concentration to the critical level, initiates the phase changes (Kedersha et al., 2005; Molliex et al., 2015; Patel et al., 2015) and creates the primary biphasic core/shell structures. The composition of biphasic SGs creates a larger mature assembly during the microtubule-dependent process (Wheeler et al., 2016). The opposite point of assembling SGs is disassembly. In general, disassembly involves the return of non-translated mRNAs trapped in the SG structures to the translation process and is caused by a variety of factors including chaperones in the stress relief phase (Mazroui et al., 2007), microtubules (Loschi et al., 2009), autophagy mechanisms (Dormann et al., 2010), and post-translational modifications (Stoecklin et al., 2004). Assembling and disassembling of SGs are in equilibrium with polysomes (Kedersha et al., 2000). Since biotic or environmental stress stops translation and increases the number of SGs, relieving stress and increasing the number of translated mRNAs is associated with return to equilibrium and disassembly of SGs (Panas et al., 2016; Marnik and Updike, 2019). When the condition progresses toward disequilibrium, by increasing the assembly of SGs and decreasing the clearance, pathogenic processes would be evolved (Wolozin, 2012; Chen and Liu, 2017) (Figure 3).
This pathogenesis links SGs to a wide range of neurodegenerative diseases including ALS (Colombrita et al., 2009; Volkening et al., 2009; Farg et al., 2013; Manghera et al., 2016; Dreser et al., 2017; Chen and Cohen, 2019), AD (Castellani et al., 2011; Vanderweyde et al., 2012; Ivanov et al., 2016; Maziuk et al., 2018; Silva et al., 2019; Younas et al., 2020), FTD (Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017; Hock et al., 2018; Montalbano et al., 2020), and FTDP (Vanderweyde et al., 2012). Many of the protein components of SGs such as TIA-1 (Fujita et al., 2008; Volkening et al., 2009; Liu-Yesucevitz et al., 2010; Vanderweyde et al., 2012; Dafinca et al., 2016; Gal et al., 2016; Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017; Salapa et al., 2018; Chen and Cohen, 2019; Younas et al., 2020), PABP-1 (Fujita et al., 2008; Dormann et al., 2010; Bentmann et al., 2012; McGurk et al., 2014; Dafinca et al., 2016; Gal et al., 2016; Hirsch-Reinshagen et al., 2017; Kamelgarn et al., 2018; Silva et al., 2019), G3BP1 (Vanderweyde et al., 2012; Daigle et al., 2016; Gal et al., 2016; Ichiyanagi et al., 2016; Lim et al., 2016; Manghera et al., 2016; Orrù et al., 2016; Loginov et al., 2018; Mann et al., 2019), eIF4G (Dormann et al., 2010; Lim et al., 2016; Kamelgarn et al., 2018), TTP (Vanderweyde et al., 2012) and TIAR (Colombrita et al., 2009; Lenzi et al., 2015; Lo Bello et al., 2017; Codron et al., 2018; Loginov et al., 2018; Chen and Cohen, 2019; Ratti et al., 2020) have been studied in neurodegenerative diseases. IHC has revealed the colocalization of pathological accumulations of proteins such as TDP-43 (Fujita et al., 2008; Colombrita et al., 2009; Volkening et al., 2009; Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Vanderweyde et al., 2012; McGurk et al., 2014; Gal et al., 2016; Manghera et al., 2016; Orrù et al., 2016; Chen and Liu, 2017; Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017; Bennett et al., 2018; Kreiter et al., 2018; Loginov et al., 2018; Chen and Cohen, 2019; Mann et al., 2019; Feneberg et al., 2020; Levin et al., 2020; Montalbano et al., 2020; Ratti et al., 2020; Salapa et al., 2020; Vassileff et al., 2020) or FUS (Farg et al., 2013; McGurk et al., 2014; Japtok et al., 2015; Lenzi et al., 2015; Daigle et al., 2016; Lim et al., 2016; Lo Bello et al., 2017; Hock et al., 2018; Kamelgarn et al., 2018; Arenas et al., 2020) with SGs in neurodegeneration, indicating a strong association of SGs with the pathogenic mechanisms. In this study, by reviewing all studies on SGs and neurodegenerative diseases in humans, we tried to answer the question that SGs can act as nests or sources for these pathological aggregations, or disruption and mutations in the main components of these accumulations can disrupt the balance of SGs.
ALS is a neurodegenerative disease specific to motor neurons (MNs), and with progressive loss of upper MNs (UMN) in the motor cortex of the brain and lower MNs (LMN) in the brain stem and the spinal cord (Robberecht and Philips, 2013), muscle weakness and atrophy appear (Hardiman et al., 2017). The main pathological signature of ALS is the presence of inclusion bodies in the cytoplasmic region of MNs. The key component of these inclusions in 95% of cases is TDP-43 in the form of hyperphosphorylated, Ubiquitinated, and truncated (Neumann et al., 2006; Nonaka et al., 2016). FUS and SOD1 are other proteins that can be involved in the formation of these inclusions (Volkening et al., 2009; Farg et al., 2013). TDP-43 and FUS are RNA-binding proteins that are often nuclear localized but can commute between the nucleus and the cytoplasm (Kapeli et al., 2017). The nuclear activity of these proteins is summarized in transcription, pre-mRNA splicing and processing non-coding RNAs (Ederle and Dormann, 2017). In the cytoplasm, these proteins can contribute in the regulation of mRNA stability, mRNA transport, translation, autophagy, and stress response and LLPS (Birsa et al., 2020). SOD1 is a superoxide dismutase enzyme. Mutations in this gene can result in these accumulation (Volkening et al., 2009). Based on bioinformatics studies and overlap in the function of these proteins in pathological aggregations and impairment of mRNA mechanisms, these proteins can be considered as pathological factors in these disorders (Liu-Yesucevitz et al., 2010; Wolozin, 2012; Ramaswami et al., 2013). One of the symptoms of impaired mRNA mechanisms is the disequilibrium of SGs. Numerous studies have been performed on the presence of SGs and their association with pathological aggregations in humans using post mortem tissue (brain, spinal cord) and blood, which are summarized in Table 2. To investigate the presence of SGs in the target tissue, SG markers were used, which include TIA-1, G3BP1, PABP-1, TIAR, HuR, and TTP. Studies of human-derived cell lines have been mostly performed using autopsy skin fibroblasts and differentiation into motor (Dimos et al., 2008). These studies have allowed the study of mutations in the TDP-43, FUS, SOD1, and C9orf72 genes and their effects on cell and other components of pathological aggregations and SGs, which are summarized in Table 3.

Table 3. Stress Granules and ALS in human derived cell lines.
TAR DNA-binding protein 43 (TDP-43), product of the TARDBP gene, plays the largest role in the formation of pathological aggregations. Most of the changes observed in this protein in aggregations include phosphorylation (Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Cohen et al., 2015; Hirsch-Reinshagen et al., 2017; Ratti et al., 2020), acetylation (Cohen et al., 2015), and cleavage at N/C terminals (Bentmann et al., 2012; Cohen et al., 2015). General observations suggest that hyperphosphorylation and acetylation may predispose TDP-43 to accumulation. TDP-43 has been studied in both full-length and cleaved (Cohen et al., 2015) forms in aggregations in spinal cord (McGurk et al., 2014; Chen and Cohen, 2019) and brain (Liu-Yesucevitz et al., 2010; Manghera et al., 2016; Hirsch-Reinshagen et al., 2017; Vassileff et al., 2020). In studies of colocalization of TDP-43 with SGs, it is most often colocalized with TIA-1, PABP-1, G3BP1, TIAR, and HuR, which are markers for the presence of SGs in cell and are the core proteins in nucleation phase in the assembly of SGs (Kedersha et al., 2005). In addition, in MNs differentiated from fibroblasts, the effect of TDP43 on nucleation phase and interaction with G3BP1 in CORE formation has been determined (Orrù et al., 2016). TDP-43 is colocalized with STAUFEN and FMRP, which play important roles in mRNA mechanisms. STAUFEN (Volkening et al., 2009; Vassileff et al., 2020), encoded by the STAU1 gene, is involved in the transport of mRNA to different subcellular compartments and organelles (Thomas et al., 2005). FMRP is involved in the nucleocytoplasmic shuttling and dendritic localization of mRNA (Antar et al., 2005). So far, the effects of TDP-43 mutants including S393L (Kreiter et al., 2018), G294V (Kreiter et al., 2018), M337V (Loginov et al., 2018; Feneberg et al., 2020), I383T (Loginov et al., 2018) and TARDBP-A382T (Orrù et al., 2016; Loginov et al., 2018; Ratti et al., 2020) mutations in human-derived cell line studies have been considered. S393L, G294V missense mutations can cause neurodegeneration in MNs in a TDP-43 accumulation independent manner (Kreiter et al., 2018). The TDP-43 M337V mutant can increase the assembly of SGs by interfering with the function of eIF4A1 and endoplasmic reticulum chaperone Grp78 (Feneberg et al., 2020). In contrast, TARDBP-A382T mutation due to loss of TDP-43 function significantly has reduced the number of SGs in cells (Orrù et al., 2016).
FUS (fused in sarcoma) is present in pathological aggregations with a lower percentage than TDP-43 and its association and effects on SGs have been studied. The mechanism of FUS toxicity is not fully understood, but due to its cytoplasmic localization, loss of nuclear activity and acquisition of cytoplasmic function might be involved (Kino et al., 2015). Studies in post-mortem tissue have shown the association of FUS aggregations with SGs through colocalization with ATAXIN2 (Farg et al., 2013) and PABP-1 (McGurk et al., 2014). FUS aggregations are directly affected by the type of mutation, benign and malignant (Japtok et al., 2015). The amount of mislocalization and recruitment in the structure of SGs is related to the type of mutation (Lenzi et al., 2015). FUS in the cell carrying the P525L mutation has more cytoplasmic localization than in control, and when exposed to stress, this localization increases leading to nuclear depletion (Lim et al., 2016). After stress relief, the mutant carrier cell needs more time to return to normal than control. SGs are more numerous in this cell and have a longer persistence, which indicates the direct effect of FUS mutations on SGs (Lo Bello et al., 2017). Post-transcription modifications, such as acetylation in lysine 510, which is located in the NLS sequence, disrupts the interaction between FUS and transportin1, causing its cytoplasmic mislocalization, which is more common in the pathogenesis of ALS than in controls (Arenas et al., 2020). The localization of SOD1 has been confirmed in both mutant and WT form with TDP-43 accumulations in spinal cord motor neurons (Volkening et al., 2009). Mutations that occur in SOD1 can also affect the dynamics of SGs. The mutant types SOD1-L144F (Gal et al., 2016) and SOD1-L39R (Rajpurohit et al., 2020) colocalize with G3BP1 in fibroblast-derived motor neurons, whereas SOD1-WT does not colocalize with SGs (Gal et al., 2016; Rajpurohit et al., 2020).
Hexanucleotide repeat expansion (HRE) is another common mutation in ALS that is associated with an increase in the number of G4C2 repeats in the C9orf72 gene. The number of repeats in normal people are between 20 and 30, but in people with mutations, the number of repeats increases to hundreds (Khan et al., 2012). Three mechanisms explain the effect of C9orf72 mutations on SGs.
1. C9orf72-related RNA transcripts accumulate in the nucleus and cytoplasm causing sequestration of RNA-binding proteins, including proteins involved in SG dynamics (Rossi et al., 2015; Dafinca et al., 2016). Colocalization of PABP-1 with TDP-43 inclusions was higher in ALS patients with C9orf72 mutation (67%) than in patients with ATXN2 mutation (47%) and patients without any mutation (36%) (McGurk et al., 2014).
2. The effect of mutations on C9orf72 protein function and the destruction of interactions with other proteins is another proposed mechanism. DENNL72 is another name for C9orf72, which stands for differentially expressed in normal and neoplastic cells and One of the molecular roles envisaged for it is acting as a guanine nucleotide exchange factor (GEF) (Levine et al., 2013). The interaction of C9orf72 as GEF with Rab proteins (Tang, 2016) is involved in autophagy and cellular trafficking mechanisms such as Rab7 and Rab11 (Farg et al., 2014) and disruption of these pathways can lead to cytoplasmic accumulations of TDP-43 (Ratti et al., 2020) and decreased clearance of SGs (Monahan et al., 2016).
3. The transcript of the mutant C9orf72 gene containing GGGGCC repeats can be translated during the non-ATG translation mechanism and produce 5 different types of dipeptide repeats (DPRs) (Mori et al., 2013). Among these, arginine-containing dipeptide repeats can interact with a number of SG protein components that have the IDR domain. DPRs containing glycine and proline also play a role in the assembly of SGs by inhibiting the translation and phosphorylation of eIF2a and G3BP1 (Lee et al., 2016).
FTD is one of the most common types of dementia that affects people under the age of 65 (Bird et al., 1999) and represents a diverse range of subtypes with neurodegenerative disorders such as FTLD (Faber, 1999). Due to common characteristics in clinical observations, ALS and FTD now form a broad continuum of neurodegeneration that can occur in an individual or a family. Like ALS, studies on post-mortem tissue, specifically the brain of FTD patients, have revealed the association and development of the disease with SGs (Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017). Most pathological mechanisms and aggregations such as TDP43, FUS, and C9orf72 mutations overlap with ALS. But FTD has some distinctive features. No colocalization was observed between PABP-1 and eIF4G with cytoplasmic neuronal inclusions in FTLD-TDP brain (Dormann et al., 2010), and acetylated and full-length pTDP-43 have no effects in the pathogenesis of FTLD-TDP (Cohen et al., 2015).
AD is a chronic neurodegenerative disease that is accompanied by death of neurons and loss of synapses in the cerebral cortex and certain subcortical regions of the brain (Tiraboschi et al., 2004; Burns and Iliffe, 2009). The most common form of dementia is AD, which occurs with abnormal structures (Wang et al., 2018), extracellular senile plaques, composed mainly of small proteins called Aβ42 (Bate et al., 2006), and intraneuronal neurofibrillary tangles, which are the result of accumulations of hyperphosphorylated Tau proteins (Bancher et al., 1989). Tau proteins are a group of six protein isoforms produced by alternative splicing of the MAPT (microtubule-associated protein Tau) gene (Goedert et al., 1989). The main function of Tau proteins is to maintain the stability of microtubules in axons due to their high expression in CNS neurons (Haritani et al., 1994), which are highly soluble in the cytoplasm and contribute to the dynamic and functions of microtubules (Arendt et al., 2016). Tau undergoes many post-translational modifications, including hyperphosphorylation (Grundke-Iqbal et al., 1986), acetylation (Min et al., 2010), C-terminal truncation (Zhao et al., 2016), and n-glycosylation (Wang et al., 1996), which can play a role in regulating its localization and functions. Usually, Tau is non-phosphorylated in interaction with microtubules in the axons. When stress is applied, it is phosphorylated near the microtubule binding domain and loses its ability to bind to microtubules (Trinczek et al., 1995). The hyperphosphorylated form of Tau is seen in all six isoforms in neurofibrillary tangles (Hernández and Avila, 2007). Hyperphosphorylation increases the affinity of Tau proteins for each other and binds together to form oligomers and misfolded Taus. Oligomers bind to other units to form Tau deposits, which are the Elements of NFTs that bind together to form NFTs (Shafiei et al., 2017).
Tau can act as a negative regulator of protein translation by binding to ribosomes and reducing protein synthesis (Meier et al., 2016). Stopping translation and providing RNA-binding proteins to mRNA are key elements in the formation of SGs. TIA-1 and TTP are among the core nucleation proteins. The size of Tau aggregations is directly related to TIA-1 colocalization. The larger the Tau aggregations, the greater the rate of TIA-1 colocalization (Vanderweyde et al., 2012). PABP-1 and DDX6 are also proteins found near Tau aggregations in the temporal cortex neurons of AD (Silva et al., 2019). The association of aggregations between RNA-binding proteins and the formation of SGs with Tau aggregates has not been well-studied. Tau aggregations might promote the dynamic equilibrium of SGs toward further assembly or to SGs disequilibrium pave the way for the accumulation of Tau proteins. The formation of RBPs pathological aggregations close to the Tau pathological aggregates supports this hypothesis (Maziuk et al., 2018). Tau proteins have interaction with rps6, which is a component of the 40s ribosome complex, and affects translation inhibition (Koren et al., 2019). Notably, hippocampal neurons in AD patients have more positive rps6 granules than controls (Castellani et al., 2011). Moreover, the colocalization of rps6 with TIA-1 and PABP-1 in basophilic inclusions may indicate one of the main shared mechanisms between Tau aggregations and stress granules (Fujita et al., 2008). The association of TDP-43 oligomers with Tau aggregations has also been suggested as one of the interactions involved in the pathogenesis of Tau (Montalbano et al., 2020).
MS is a demyelinating disease in which the cover of nerve cells in the brain and spinal cord is damaged (Noyes and Weinstock-Guttman, 2013). The causative mechanism of the disease is summarized in the destruction of myelin sheath by the immune system and defects in myelin-producing cells (Nakahara et al., 2011). The disease has three main features, including the formation of lesions in the CNS, inflammation and destruction of the myelin sheath (Compston and Coles, 2008). Neurodegeneration is also an important feature in the pathology of MS (Frohman et al., 2006; Lassmann and van Horssen, 2011), but so far, no specific mechanism has been proposed for it. The impact of SGs on neurodegeneration and neurodegenerative diseases has been fully discussed so far. Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) is a major component in the formation of SGs (Guil et al., 2006) and is discussed as a factor in the pathogenesis of autoimmune mediated CNS neurological diseases and as a link between SGs and autoimmune neurological diseases (Douglas et al., 2016). The interaction between TIA-1 and hnRNPA1 in cytoplasmic granules and its nuclear depletion in MS patients is significant. The aggregations of TIA-1 in the structure of large SGs in the cytoplasm can be a link between degeneration in neurons and MS (Salapa et al., 2018). Nuclear depletion of TDP-43 and interaction with hnRNPA1 and colocalization of both in the structure of SGs also emphasizes the importance of SGs in MS (Levin et al., 2020; Salapa et al., 2020).
GO classifies relationships between genes by annotating and categorizing them into three levels: biological process, molecular function, and cellular component. Biological process describes the cellular or physiological role performed on a larger scale by a gene in relation to other genes. The molecular function describes the molecular activity of the desired gene, and the cellular component determines where the gene product executes its function (The Gene Ontology Consortium., 2019). GO analysis was performed on the list of genes associated with SGs in neurodegenerative diseases (Figure 3A) and the list of genes associated with SGs in ALS disease was extracted from human derived cell lines studies (Figure 3C) using Enrichr's web-based tools and services (Kuleshov et al., 2016). Most of the proteins in the structure of SGs are RBPs (Kedersha et al., 2005), and GO analysis confirms this. According to the GO biological process, the connections that can be made in one biological pathway with other proteins are more involved in the RNA metabolic pathways and, as expected, all of these proteins act in conjunction with RNAs, including cytoplasmic mRNA, body assembly, and RNA splicing. Further use of these proteins in the structure of ribonucleoprotein granules, cytoplasmic SGs and cytoplasmic ribonucleoprotein granules was confirmed by GO cellular component analysis. In ALS, the biological association of these proteins with other proteins, based on the GO biological process, significantly confirmed their role in RNA export from nucleus. This was expected, given that these proteins are mostly shuttling between nucleus and cytoplasm. Gene prediction was performed on the gene list extracted in neurodegenerative disease in Table 3 by string-db cytoscape plugin (Doncheva et al., 2019). GO biological process showed these proteins interact with other proteins in p-bodies, cytoplasmic ribonucleoprotein granules and cytoplasmic SGs. Overall, bioinformatics analysis determined the association of these proteins and genes with RNA-related mechanisms that are specifically involved in the formation and assembly of ribonucleoprotein granules. Their functional position is also shared between nucleus and cytoplasm and is an evidence to trafficking and shuttling of SGs protein components between them (Figure 4).
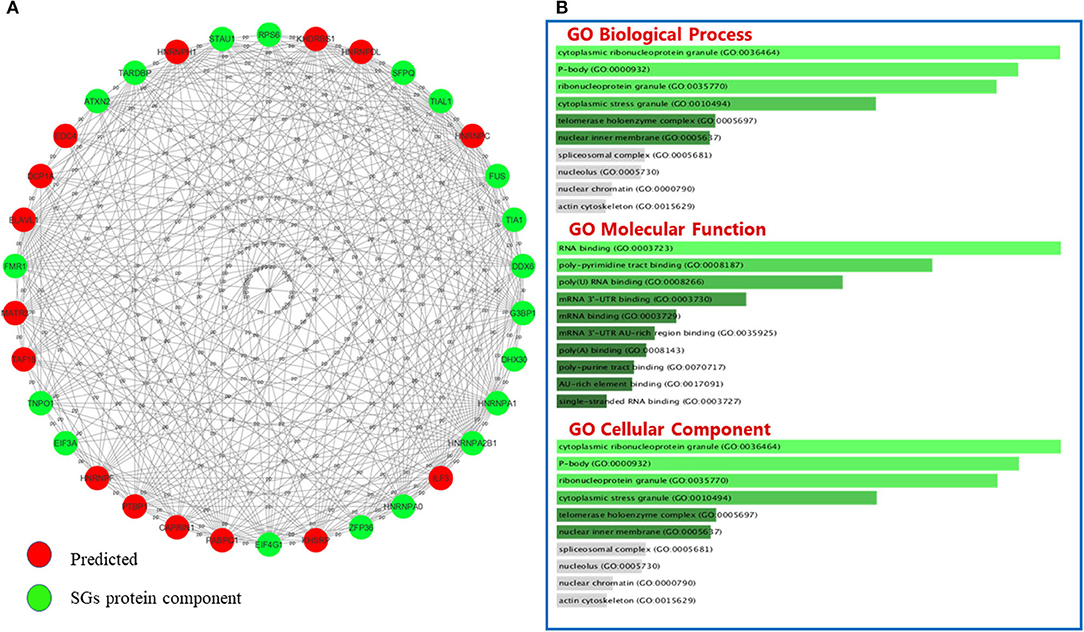
Figure 4. The interactions between SGs protein components and predicted proteins and GO analysis on predicted ones in neurodegenerative disease. (A) Fifteen new proteins were predicted by interaction with SGs protein components using the Cytoscape string-db plugin on data extracted from articles. (B) Fifteen new proteins were predicted by interaction with SGs protein components using the Cytoscape string-db plugin on data extracted from articles. GO analysis was performed on the predicted proteins in three biological processes: molecular function and cellular component. The length of each bar indicates the importance in that particular category sorted by p-value. Note that the lower the color intensity of the bars, the greater the relationship with that category.
Equilibrium is the most important point in SGs. SGs are in the nature of the cell and are considered as cell solutions to stress. They are temporary constructions and when the stress is relieved, they are disassembled and reduced in number, and the cell condition returns to normal. If under any circumstances the presence of SGs becomes permanent and leads to disequilibrium, in interaction with pathological aggregations such as TDP and FUS aggregations, they can lead to pathological conditions such as neuron degeneration. SGs have been studied in many neurodegenerative diseases in humans, including ALS, FTD, AD, and MS. These studies have indicated common features in SG biology among neurodegenerative diseases. At least 15 proteins have been predicted to interact with the protein components of SGs in mentioned neurodegenerative disorders. Therefore, it seems that SG biology is common between these disorders. Yet, some components of SGs might be specific to these disorders. Based on the rarity of comparative analysis between these disorders, it is not possible to male conclusive interpretations in this regard. We have tried to provide a comprehensive summary of these studies and an overview of SGs in neurodegenerative diseases. To conclude, more studies can be done in diseases such as AD and MS and the association of the Tau protein with other protein components of SGs or the association of inflammatory pathways with the formation of SGs could also be assessed.
MT, SG-F, and MR wrote the manuscript and contributed in study design. MA, MS, HS, and AJ contributed in the data collection, designed the tables and figures. All authors approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.650740/full#supplementary-material
SGs, Stress granules; ALS, amyotrophic lateral sclerosis; FTD, frontotemporal degeneration; AD, Alzheimer's diseases; MS, multiple sclerosis; PRISMA-ScR, Preferred Reporting Items for Systematic Reviews and Meta-Analyses Extension for Scoping Reviews; MeSH, medical subject heading; GO, Gene ontology; FTLD, frontotemporal lobar degeneration; NIFID, neuronal intermediate filament inclusion disease; BIBD, basophilic inclusion body disease; MND, motor neuron disease; IHC, immunohistochemistry; IP, immunoprecipitation; MNs, motor neurons; LMN, lower MNs; TDP-43, TAR DNA-binding protein 43; HRE, Hexanucleotide repeat expansion; GEF, guanine nucleotide exchange factor; hnRNPA1, Heterogeneous nuclear ribonucleoprotein A1.
Antar, L. N., Dictenberg, J. B., Plociniak, M., Afroz, R., and Bassell, G. J. (2005). Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350–359. doi: 10.1111/j.1601-183X.2005.00128.x
Arenas, A., Chen, J., Kuang, L., Barnett, K., Kasarskis, E., Gal, J., et al. (2020). Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum. Mol. Genet. 29, 2684–2697. doi: 10.1093/hmg/ddaa159
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126(Pt 3), 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Arimoto-Matsuzaki, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H., and Takekawa, M. (2008). Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332. doi: 10.1038/ncb1791
Arksey, H., and O'Malley, L. (2005). Scoping studies: towards a methodological framework. Int. J. Soc. Res. Methodol. 8, 19–32. doi: 10.1080/1364557032000119616
Arrigo, A. P., Suhan, J. P., and Welch, W. J. (1988). Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol. Cell. Biol. 8, 5059–5071. doi: 10.1128/MCB.8.12.5059
Ash, P. E. A., Vanderweyde, T. E., Youmans, K. L., Apicco, D. J., and Wolozin, B. (2014). Pathological stress granules in Alzheimer's disease. Brain Res. 1584, 52–58. doi: 10.1016/j.brainres.2014.05.052
Aulas, A., Fay, M. M., Lyons, S. M., Achorn, C. A., Kedersha, N., Anderson, P., et al. (2017). Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 130:927. doi: 10.1242/jcs.199240
Aulas, A., Lyons, S. M., Fay, M. M., Anderson, P., and Ivanov, P. (2018). Nitric oxide triggers the assembly of “type II” stress granules linked to decreased cell viability. Cell Death Dis. 9:1129. doi: 10.1038/s41419-018-1173-x
Bancher, C., Brunner, C., Lassmann, H., Budka, H., Jellinger, K., Wiche, G., et al. (1989). Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 477, 90–99. doi: 10.1016/0006-8993(89)91396-6
Baron, D. M., Kaushansky, L. J., Ward, C. L., Sama, R. R. K., Chian, R. J., Boggio, K. J., et al. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegen. 8:30. doi: 10.1186/1750-1326-8-30
Bate, C., Kempster, S., Last, V., and Williams, A. (2006). Interferon-gamma increases neuronal death in response to amyloid-beta1-42. J. Neuroinflamm. 3:7. doi: 10.1186/1742-2094-3-7
Bennett, C. L., Dastidar, S. G., Ling, S. C., Malik, B., Ashe, T., Wadhwa, M., et al. (2018). Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 136, 425–443. doi: 10.1007/s00401-018-1852-9
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.M111.328757
Bird, T. D., Nochlin, D., Poorkaj, P., Cherrier, M., Kaye, J., Payami, H., et al. (1999). A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122, 741–756. doi: 10.1093/brain/122.4.741
Birsa, N., Bentham, M. P., and Fratta, P. (2020). Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 99, 193–201. doi: 10.1016/j.semcdb.2019.05.023
Boncella, A. E., Shattuck, J. E., Cascarina, S. M., Paul, K. R., Baer, M. H., Fomicheva, A., et al. (2020). Composition-based prediction and rational manipulation of prion-like domain recruitment to stress granules. Proc. Natl. Acad. Sci. U.S.A. 117, 5826–5835. doi: 10.1073/pnas.1912723117
Brown, D. G., Shorter, J., and Wobst, H. J. (2020). Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorgan. Med. Chem. Lett. 30:126942. doi: 10.1016/j.bmcl.2019.126942
Cao, X., Jin, X., and Liu, B. (2020). The involvement of stress granules in aging and aging-associated diseases. Aging Cell 19:e13136. doi: 10.1111/acel.13136
Castellani, R. J., Gupta, Y., Sheng, B., Siedlak, S. L., Harris, P. L., Coller, J. M., et al. (2011). A novel origin for granulovacuolar degeneration in aging and Alzheimer's disease: parallels to stress granules. Lab. Invest. 91, 1777–1786. doi: 10.1038/labinvest.2011.149
Chen, L., and Liu, B. (2017). Relationships between stress granules, oxidative stress, and neurodegenerative diseases. Oxidat. Med. Cell. Long. 2017:1809592. doi: 10.1155/2017/1809592
Chen, Y., and Cohen, T. J. (2019). Aggregation of the nucleic acid-binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation. J. Biol. Chem. 294, 3696–3706. doi: 10.1074/jbc.RA118.006351
Chitiprolu, M., Jagow, C., Tremblay, V., Bondy-Chorney, E., Paris, G., Savard, A., et al. (2018). A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 9:2794. doi: 10.1038/s41467-018-05273-7
Codron, P., Cassereau, J., Vourc'h, P., Veyrat-Durebex, C., Blasco, H., Kane, S., et al. (2018). Primary fibroblasts derived from sporadic amyotrophic lateral sclerosis patients do not show ALS cytological lesions. Amyotroph. Lat. Scl. Frontotemp. Degener. 19, 446–456. doi: 10.1080/21678421.2018.1431787
Cohen, T. J., Hwang, A. W., Restrepo, C. R., Yuan, C. X., Trojanowski, J. Q., and Lee, V. M. Y. (2015). An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6:5845. doi: 10.1038/ncomms6845
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
Colquhoun, H. L., Levac, D., O'Brien, K. K., Straus, S., Tricco, A. C., Perrier, L., et al. (2014). Scoping reviews: time for clarity in definition, methods, and reporting. J. Clin. Epidemiol. 67, 1291–1294. doi: 10.1016/j.jclinepi.2014.03.013
Compston, A., and Coles, A. (2008). Multiple sclerosis. Lancet 372, 1502–1517. doi: 10.1016/S0140-6736(08)61620-7
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells. 34, 2063–2078. doi: 10.1002/stem.2388
Daigle, J. G., Krishnamurthy, K., Ramesh, N., Casci, I., Monaghan, J., McAvoy, K., et al. (2016). Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 131, 605–620. doi: 10.1007/s00401-015-1530-0
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. (2008) 321, 1218–1221. doi: 10.1126/science.1158799
Doncheva, N. T., Morris, J. H., Gorodkin, J., and Jensen, L. J. (2019). Cytoscape stringapp: network analysis and visualization of proteomics data. J. Proteome Res. 18, 623–632. doi: 10.1021/acs.jproteome.8b00702
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Douglas, J. N., Gardner, L. A., Salapa, H. E., Lalor, S. J., Lee, S., Segal, B. M., et al. (2016). Antibodies to the RNA-binding protein hnRNP A1 contribute to neurodegeneration in a model of central nervous system autoimmune inflammatory disease. J. Neuroinflamm. 13:178. doi: 10.1186/s12974-016-0647-y
Dreser, A., Vollrath, J. T., Sechi, A., Johann, S., Roos, A., Yamoah, A., et al. (2017). The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Diff. 24, 1655–1671. doi: 10.1038/cdd.2017.88
Ederle, H., and Dormann, D. (2017). TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 591, 1489–1507. doi: 10.1002/1873-3468.12646
Faber, R. (1999). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 53:1159. doi: 10.1212/WNL.53.5.1158-b
Farg, M. A., Soo, K. Y., Warraich, S. T., Sundaramoorthy, V., Blair, I. P., and Atkin, J. D. (2013). Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 717–728. doi: 10.1093/hmg/dds479
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Feneberg, E., Gordon, D., Thompson, A., Finelli, M., Dafinca, R., Candalija Iserte, A., et al. (2020). An ALS-linked mutation in TDP-43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol. Dis. 144:105050. doi: 10.1016/j.nbd.2020.105050
Frohman, E. M., Racke, M. K., and Raine, C. S. (2006). Multiple sclerosis–the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955. doi: 10.1056/NEJMra052130
Fujita, K., Ito, H., Nakano, S., Kinoshita, Y., Wate, R., and Kusaka, H. (2008). Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol. 116, 439–445. doi: 10.1007/s00401-008-0415-x
Gal, J., Kuang, L., Barnett, K. R., Zhu, B. Z., Shissler, S. C., Korotkov, K. V., et al. (2016). ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 132, 563–576. doi: 10.1007/s00401-016-1601-x
Gao, X., Jiang, L., Gong, Y., Chen, X., Ying, M., Zhu, H., et al. (2019). Stress granule: a promising target for cancer treatment. Br. J. Pharmacol. 176, 4421–4433. doi: 10.1111/bph.14790
Gieseler, A., Hillert, R., Krusche, A., and Zacher, K. H. (2019). Theme 5 human cell biology and pathology. Amyotroph. Lat. Scl. Frontotemp. Degen. 20, 188–205. doi: 10.1080/21678421.2019.1646993
Gilks, N., Kedersha, N., Ayodele, M., Shen, L., Stoecklin, G., Dember, L. M., et al. (2004). Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell. 15, 5383–5398. doi: 10.1091/mbc.e04-08-0715
Gingras, A. C., Raught, B., and Sonenberg, N. (1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68, 913–963. doi: 10.1146/annurev.biochem.68.1.913
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Guil, S., Long, J. C., and Cáceres, J. F. (2006). hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 26, 5744–5758. doi: 10.1128/MCB.00224-06
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3:17085. doi: 10.1038/nrdp.2017.85
Haritani, M., Spencer, Y. I., and Wells, G. A. H. (1994). Hydrated autoclave pretreatment enhancement of prion protein immunoreactivity in formalin-fixed bovine spongiform encephalopathy-affected brain. Acta Neuropathol. 87, 86–90. doi: 10.1007/BF00386258
Hernández, F., and Avila, J. (2007). Tauopathies. Cell. Mol. Life Sci. 64, 2219–2233. doi: 10.1007/s00018-007-7220-x
Hirsch-Reinshagen, V., Pottier, C., Nicholson, A. M., Baker, M., Hsiung, G. R., Krieger, C., et al. (2017). Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol. Commun. 5:96. doi: 10.1186/s40478-017-0493-x
Hock, E. M., Maniecka, Z., Hruska-Plochan, M., Reber, S., Laferrière, F., Sahadevan, M. K. S., et al. (2018). Hypertonic stress causes cytoplasmic translocation of neuronal, but not astrocytic, FUS due to impaired transportin function. Cell Rep. 24, 987–1000.e7. doi: 10.1016/j.celrep.2018.06.094
Ichiyanagi, N., Fujimori, K., Yano, M., Ishihara-Fujisaki, C., Sone, T., Akiyama, T., et al. (2016). Establishment of in vitro FUS-associated familial amyotrophic lateral sclerosis model using human induced pluripotent stem cells. Stem Cell Rep. 6, 496–510. doi: 10.1016/j.stemcr.2016.02.011
Ivanov, P. A., Mikhaylova, N. M., and Klyushnik, T. P. (2016). Distribution of translation initiation factor eIF3 in neutrophils in Alzheimer disease. Biochem. Suppl. A Memb. Cell Biol. 10, 328–332. doi: 10.1134/S1990747816030053
Japtok, J., Lojewski, X., Naumann, M., Klingenstein, M., Reinhardt, P., Sterneckert, J., et al. (2015). Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 82, 420–429. doi: 10.1016/j.nbd.2015.07.017
Kamelgarn, M., Chen, J., Kuang, L., Jin, H., Kasarskis, E. J., and Zhu, H. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U.S.A. 115, E11904–E11913. doi: 10.1073/pnas.1810413115
Kapeli, K., Martinez, F. J., and Yeo, G. W. (2017). Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 136, 1193–1214. doi: 10.1007/s00439-017-1830-7
Kedersha, N., Cho, M. R., Li, W., Yacono, P. W., Chen, S., Gilks, N., et al. (2000). Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell. Biol. 151, 1257–1268. doi: 10.1083/jcb.151.6.1257
Kedersha, N., Ivanov, P., and Anderson, P. (2013). Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 38, 494–506. doi: 10.1016/j.tibs.2013.07.004
Kedersha, N., Stoecklin, G., Ayodele, M., Yacono, P., Lykke-Andersen, J., Fritzler, M. J., et al. (2005). Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell. Biol. 169, 871–884. doi: 10.1083/jcb.200502088
Kedersha, N. L., Gupta, M., Li, W., Miller, I., and Anderson, P. (1999). RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell. Biol. 147, 1431–1442. doi: 10.1083/jcb.147.7.1431
Khan, B. K., Yokoyama, J. S., Takada, L. T., Sha, S. J., Rutherford, N. J., Fong, J. C., et al. (2012). Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J. Neurol. Neurosurg. Psychiatry 83, 358–364. doi: 10.1136/jnnp-2011-301883
Khong, A., Matheny, T., Jain, S., Mitchell, S. F., Wheeler, J. R., and Parker, R. (2017). The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol. Cell. 68, 808–20.e5. doi: 10.1016/j.molcel.2017.10.015
Kino, Y., Washizu, C., Kurosawa, M., Yamada, M., Miyazaki, H., Akagi, T., et al. (2015). FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 3:24. doi: 10.1186/s40478-015-0202-6
Koren, S. A., Hamm, M. J., Meier, S. E., Weiss, B. E., Nation, G. K., Chishti, E. A., et al. (2019). Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 137, 571–583. doi: 10.1007/s00401-019-01970-9
Kreiter, N., Pal, A., Lojewski, X., Corcia, P., Naujock, M., Reinhardt, P., et al. (2018). Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 115, 167–181. doi: 10.1016/j.nbd.2018.03.010
Kuleshov, M. V., Jones, M. R., Rouillard, A. D., Fernandez, N. F., Duan, Q., Wang, Z., et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. doi: 10.1093/nar/gkw377
Lassmann, H., and van Horssen, J. (2011). The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 585, 3715–3723. doi: 10.1016/j.febslet.2011.08.004
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e17. doi: 10.1016/j.cell.2016.10.002
Lenzi, J., De Santis, R., de Turris, V., Morlando, M., Laneve, P., Calvo, A., et al. (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Models Mech. 8, 755–766. doi: 10.1242/dmm.020099
Levac, D., Colquhoun, H., and O'Brien, K. K. (2010). Scoping studies: advancing the methodology. Implement. Sci. 5:69. doi: 10.1186/1748-5908-5-69
Levin, M., Salapa, H., Libner, C., Hutchinson, C., and Popescu, B. (2020). A role for dysfunctional RNA binding proteins in the pathogenesis of neurodegeneration in MS (2669). Neurology 94(Suppl. 15):2669.
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Lim, S. M., Choi, W. J., Oh, K. W., Xue, Y., Choi, J. Y., Kim, S. H., et al. (2016). Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol Neurodegener. 11:8. doi: 10.1186/s13024-016-0075-6
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE. 5:e13250. doi: 10.1371/journal.pone.0013250
Lo Bello, M., Di Fini, F., Notaro, A., Spataro, R., Conforti, F. L., and La Bella, V. (2017). ALS-related mutant FUS protein is mislocalized to cytoplasm and is recruited into stress granules of fibroblasts from asymptomatic FUS P525L mutation carriers. Neuro Degen. Dis. 17, 292–303. doi: 10.1159/000480085
Loginov, V. I., Burdennyy, A. M., Filippova, E. A., Pronina, I. V., Kazubskaya, T. P., Kushlinsky, D. N., et al. (2018). [Hypermethylation of miR-107, miR-130b, miR-203a, miR-1258 genes associated with ovarian cancer development and metastasis]. Molekul. Biol. 52, 801–809. doi: 10.1134/S0026893318050102
Loschi, M., Leishman, C. C., Berardone, N., and Boccaccio, G. L. (2009). Dynein and kinesin regulate stress-granule and P-body dynamics. J. Cell. Sci. 122(Pt 21), 3973–3982. doi: 10.1242/jcs.051383
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Manghera, M., Ferguson-Parry, J., and Douville, R. N. (2016). TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol. Dis. 94, 226–236. doi: 10.1016/j.nbd.2016.06.017
Mann, J. R., Gleixner, A. M., Mauna, J. C., Gomes, E., DeChellis-Marks, M. R., Needham, P. G., et al. (2019). RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8. doi: 10.1016/j.neuron.2019.01.048
Marnik, E. A., and Updike, D. L. (2019). Membraneless organelles: P granules in Caenorhabditis elegans. Traffic 20, 373–379. doi: 10.1111/tra.12644
Maziuk, B. F., Apicco, D. J., Cruz, A. L., Jiang, L., Ash, P. E. A., da Rocha, E. L., et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 6:71. doi: 10.1186/s40478-018-0574-5
Mazroui, R., Marco, S., Kaufman, R., and Gallouzi, I. E. (2007). Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell. 18, 2603–2618. doi: 10.1091/mbc.e06-12-1079
McCormick, C., and Khaperskyy, D. A. (2017). Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 17:647. doi: 10.1038/nri.2017.63
McGurk, L., Lee, V. M., Trojanowksi, J. Q., Van Deerlin, V. M., Lee, E. B., and Bonini, N. M. (2014). Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 73, 837–845. doi: 10.1097/NEN.0000000000000102
Meier, S., Bell, M., Lyons, D. N., Rodriguez-Rivera, J., Ingram, A., Fontaine, S. N., et al. (2016). Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J. Neurosci. 36, 1001–1007. doi: 10.1523/JNEUROSCI.3029-15.2016
Min, S. W., Cho, S.-H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Mitra, A., Luna, J. I., Marusina, A. I., Merleev, A., Kundu-Raychaudhuri, S., Fiorentino, D., et al. (2015). Dual mTOR inhibition is required to prevent TGF-β-mediated fibrosis: implications for scleroderma. J. Invest. Dermatol. 135, 2873–2876. doi: 10.1038/jid.2015.252
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. doi: 10.1016/j.cell.2015.09.015
Monahan, Z., Shewmaker, F., and Pandey, U. B. (2016). Stress granules at the intersection of autophagy and ALS. Brain. Res. 1649(Pt B), 189–200. doi: 10.1016/j.brainres.2016.05.022
Montalbano, M., McAllen, S., Cascio, F. L., Sengupta, U., Garcia, S., Bhatt, N., et al. (2020). TDP-43 and tau oligomers in alzheimer's disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol. Dis. 146:105130. doi: 10.1016/j.nbd.2020.105130
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Nakahara, J., Maeda, M., Aiso, S., and Suzuki, N. (2011). Current concepts in multiple sclerosis: autoimmunity versus oligodendrogliopathy. Clin. Rev. Allergy Immunol. 42, 26–34. doi: 10.1007/s12016-011-8287-6
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nonaka, T., Suzuki, G., Tanaka, Y., Kametani, F., Hirai, S., Okado, H., et al. (2016). Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 291, 5473–5483. doi: 10.1074/jbc.M115.695379
Noyes, K., and Weinstock-Guttman, B. (2013). Impact of diagnosis and early treatment on the course of multiple sclerosis. Am. J. Manag. care. 19(Suppl. 17), s321–s331. doi: 10.1155/2013/713627
Orrù, S., Coni, P., Floris, A., Littera, R., Carcassi, C., Sogos, V., et al. (2016). Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 25, 4473–4483. doi: 10.1093/hmg/ddw276
Panas, M. D., Ivanov, P., and Anderson, P. (2016). Mechanistic insights into mammalian stress granule dynamics. J. Cell. Biol. 215, 313–323. doi: 10.1083/jcb.201609081
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS Accelerated By Disease Mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Rajpurohit, C. S., Kumar, V., Cheffer, A., Oliveira, D., Ulrich, H., Okamoto, O. K., et al. (2020). Mechanistic insights of astrocyte-mediated hyperactive autophagy and loss of motor neuron function in SOD1(L39R) linked amyotrophic lateral sclerosis. Mol. Neurobiol. 57, 4117–4133. doi: 10.1007/s12035-020-02006-0
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Ratti, A., Gumina, V., Lenzi, P., Bossolasco, P., Fulceri, F., Volpe, C., et al. (2020). Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 145:105051. doi: 10.1016/j.nbd.2020.105051
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rossi, S., Serrano, A., Gerbino, V., Giorgi, A., Di Francesco, L., Nencini, M., et al. (2015). Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell. Sci. 128, 1787–1799. doi: 10.1242/jcs.165332
Salapa, H. E., Hutchinson, C., Popescu, B. F., and Levin, M. C. (2020). Neuronal RNA-binding protein dysfunction in multiple sclerosis cortex. Anna. Clin. Transl. Neurol. 7, 1214–1224. doi: 10.1002/acn3.51103
Salapa, H. E., Johnson, C., Hutchinson, C., Popescu, B. F., and Levin, M. C. (2018). Dysfunctional RNA binding proteins and stress granules in multiple sclerosis. J. Neuroimmunol. 324, 149–156. doi: 10.1016/j.jneuroim.2018.08.015
Samadian, M., Gholipour, M., Hajiesmaeili, M., Taheri, M., and Ghafouri-Fard, S. (2021). The eminent role of microRNAs in the pathogenesis of Alzheimer's disease. Front Aging Neurosci. 13:107. doi: 10.3389/fnagi.2021.641080
Shafiei, S. S., Guerrero-Muñoz, M. J., and Castillo-Carranza, D. L. (2017). Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci. 9:83. doi: 10.3389/fnagi.2017.00083
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shaw, P. J., and Jordan, E. G. (1995). The nucleolus. Annu. Rev. Cell Dev. Biol. 11, 93–121. doi: 10.1146/annurev.cb.11.110195.000521
Sheth, U., and Parker, R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808. doi: 10.1126/science.1082320
Silva, J. M., Rodrigues, S., Sampaio-Marques, B., Gomes, P., Neves-Carvalho, A., Dioli, C., et al. (2019). Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Diff. 26, 1411–1427. doi: 10.1038/s41418-018-0217-1
Stoecklin, G., Stubbs, T., Kedersha, N., Wax, S., Rigby, W. F. C., Blackwell, T. K., et al. (2004). MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23, 1313–1324. doi: 10.1038/sj.emboj.7600163
Tang, B. L. (2016). C9orf72's interaction with rab GTPases-modulation of membrane traffic and autophagy. Front. Cell. Neurosci. 10:228. doi: 10.3389/fncel.2016.00228
The Gene Ontology Consortium. (2019). The Gene Ontology Resource: 20 Years and still GOing strong. Nucleic Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
Theme 4 Human cell biology and pathology. (2018). Amyotroph. Lat. Scl. Frontotemp. Degen. 19, 154–177. doi: 10.1080/21678421.2018.1510571
Thomas, M. G., Martinez Tosar, L. J., Loschi, M., Pasquini, J. M., Correale, J., Kindler, S., et al. (2005). Staufen recruitment into stress granules does not affect early mRNA transport in oligodendrocytes. Mol. Biol. Cell. 16, 405–420. doi: 10.1091/mbc.e04-06-0516
Tiraboschi, P., Hansen, L., Thal, L. J., and Corey-Bloom, J. (2004). The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62, 1984–1989. doi: 10.1212/01.WNL.0000129697.01779.0A
Tricco, A. C., Lillie, E., Zarin, W., O'Brien, K. K., Colquhoun, H., Levac, D., et al. (2018). PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann. Intern. Med. 169, 467–473. doi: 10.7326/M18-0850
Trinczek, B., Biernat, J., Baumann, K., Mandelkow, E. M., and Mandelkow, E. (1995). Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell. 6, 1887–1902. doi: 10.1091/mbc.6.12.1887
Turi, Z., Lacey, M., Mistrik, M., and Moudry, P. (2019). Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging 11, 2512–2540. doi: 10.18632/aging.101922
Vanderweyde, T., Yu, H., Varnum, M., Liu-Yesucevitz, L., Citro, A., Ikezu, T., et al. (2012). Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 32, 8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012
Vassileff, N., Vella, L. J., Rajapaksha, H., Shambrook, M., Kenari, A. N., McLean, C., et al. (2020). Revealing the proteome of motor cortex derived extracellular vesicles isolated from amyotrophic lateral sclerosis human postmortem tissues. Cells 9:1709. doi: 10.3390/cells9071709
Volkening, K., Leystra-Lantz, C., Yang, W., Jaffee, H., and Strong, M. J. (2009). Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 1305, 168–182. doi: 10.1016/j.brainres.2009.09.105
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (1996). Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat. Med. 2, 871–875. doi: 10.1038/nm0896-871
Wang, Z.-H., Liu, P., Liu, X., Yu, S. P., Wang, J.-Z., and Ye, K. (2018). Delta-secretase (AEP) mediates tau-splicing imbalance and accelerates cognitive decline in tauopathies. J. Exp. Med. 215, 3038–3056. doi: 10.1084/jem.20180539
Wheeler, J. R., Matheny, T., Jain, S., Abrisch, R., and Parker, R. (2016). Distinct stages in stress granule assembly and disassembly. Elife. 5:e18413. doi: 10.7554/eLife.18413
Wolozin, B. (2012). Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegen. 7:56. doi: 10.1186/1750-1326-7-56
Wolozin, B., and Ivanov, P. (2019). Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666. doi: 10.1038/s41583-019-0222-5
Younas, N., Zafar, S., Shafiq, M., Noor, A., Siegert, A., Arora, A. S., et al. (2020). SFPQ and Tau: critical factors contributing to rapid progression of Alzheimer's disease. Acta Neuropathol. 140, 317–339. doi: 10.1007/s00401-020-02178-y
Keywords: stress granules, pathological aggregations, neurodegenerative disorders, amyotrophic lateral sclerosis, Alzheimer's, TDP-43, TIA-1, PABP-1
Citation: Asadi MR, Sadat Moslehian M, Sabaie H, Jalaiei A, Ghafouri-Fard S, Taheri M and Rezazadeh M (2021) Stress Granules and Neurodegenerative Disorders: A Scoping Review. Front. Aging Neurosci. 13:650740. doi: 10.3389/fnagi.2021.650740
Received: 07 January 2021; Accepted: 17 May 2021;
Published: 23 June 2021.
Edited by:
Thomas K. Karikari, University of Gothenburg, SwedenReviewed by:
Manuela Basso, University of Trento, ItalyCopyright © 2021 Asadi, Sadat Moslehian, Sabaie, Jalaiei, Ghafouri-Fard, Taheri and Rezazadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Taheri, bW9oYW1tYWRfODIzQHlhaG9vLmNvbQ==; Maryam Rezazadeh, cmV6YXphZGVobUB0YnptZWQuYWMuaXI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.