
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 06 October 2020
Sec. Alzheimer's Disease and Related Dementias
Volume 12 - 2020 | https://doi.org/10.3389/fnagi.2020.577996
 Alejandro Martín-Belmonte1
Alejandro Martín-Belmonte1 Carolina Aguado1
Carolina Aguado1 Rocío Alfaro-Ruíz1
Rocío Alfaro-Ruíz1 Makoto Itakura2
Makoto Itakura2 Ana Esther Moreno-Martínez1
Ana Esther Moreno-Martínez1 Luis de la Ossa3
Luis de la Ossa3 Elek Molnár4
Elek Molnár4 Yugo Fukazawa5
Yugo Fukazawa5 Rafael Luján1*
Rafael Luján1*Synapse loss occurs early in Alzheimer’s disease (AD) patients and animal models. Alterations at synaptic level are a major morphological correlate of the memory deficits and related symptoms of AD. Given the predominant roles of synaptic AMPA receptors (AMPARs) in excitatory synaptic transmission in the brain, changes in their dynamic regulation are also implicated in the pathophysiology of AD. Here, we used immunolocalization techniques to analyze the expression and subcellular distribution of AMPARs in the hippocampal region of APP/PS1 mouse model of AD. Immunoblots and histoblots revealed that the total amount of AMPARs and their regional expression pattern in the hippocampus was similar in APP/PS1 mice and in age-matched wild type mice. At the ultrastructural level, two synapse populations were examined using SDS-digested freeze-fracture replica labeling in the stratum radiatum in mice: (i) on spines of CA1 pyramidal cells; and (ii) on randomly found dendritic shafts of CA1 interneurons. While 1- and 6-months-old APP/PS1 mice exhibited no change, we observed a significant reduction at 12 months in AMPAR density at synapses in both pyramidal cells and interneurons, compared to wild-type. This reduction of AMPARs in dendritic spines was accompanied by a significant increase in AMPAR subunit proteins identified in intracellular compartments. Our data demonstrate an age-dependent reduction of synaptic AMPARs in APP/PS1 mice, which may contribute to impaired learning and memory at later stages of AD.
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder characterized by progressive cognitive deficit and memory loss. Characteristic pathological features of AD are amyloid-β (Aβ) plaques and neurofibrillary tangles of aggregated hyperphosphorylated tau proteins in the brain (Bloom, 2014). Whilst this pathology is widespread throughout the brain, the entorhinal cortex and hippocampus are the most affected areas in AD (Hyman et al., 1984). In the hippocampus, CA1 pyramidal cells are the neurons most vulnerable to neurodegeneration, with a prominent loss of dendritic spines (West et al., 2000, 2004) and eventual cell death, but interneurons are also targeted in AD (Villette and Dutar, 2017). Soluble Aβ oligomers reduce the number of dendritic spines and impair glutamatergic synaptic transmission (Mucke and Selkoe, 2012).
Glutamatergic synaptic neurotransmission is regulated by the abundance and molecular composition of the glutamate receptor subtypes expressed at synapses by specific neuron types. The main targets of glutamatergic input in the hippocampus are the dendritic spines of pyramidal cells or granule cells and the dendritic shafts of interneurons (Spruston, 2008). Ionotropic glutamate receptors mediate the fast component of glutamatergic responses, mostly consisting of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) receptors (Hollmann and Heinemann, 1994; Traynelis et al., 2010). AMPA receptors (AMPARs) are tetrameric complexes composed of homomeric or heteromeric combinations of four (GluA1–4) subunits, encoded by distinct genes, GRIA1–GRIA4 (Traynelis et al., 2010). The majority of AMPARs in the hippocampus consist of heteromeric combinations of GluA1, GluA2, and GluA3 subunits (Keinänen et al., 1990). Immunoelectron microscopy using post-embedding immunogold labeling has been used to investigate AMPARs at excitatory synapses in the hippocampus in normal brains (Nusser et al., 1998; Petralia et al., 1999; Takumi et al., 1999; Racca et al., 2000). However, until now, these high-resolution techniques have not been applied for the identification of AD-related pathological changes in AMPAR numbers and densities.
Given the central role of AMPARs in learning and memory, their disfunction likely contribute to synaptic and memory deficits associated with AD. Consistent with this idea, previous studies have shown that Aβ can contribute to the down-regulation of synaptic transmission as a consequence of AMPAR internalization (Almeida et al., 2005; Hsieh et al., 2006; Gu et al., 2009; but see Whitcomb et al., 2015). Previous studies using autoradiography, in situ hybridization, immunoblots or immunohistochemical techniques produced conflicting results regarding the expression of AMPARs in AD brains. Some studies have shown that the distributions of subunits (GluA1, GluA2/3, GluA4) in the hippocampus of AD brains are similar to control brains (Hyman et al., 1994). In contrast, other studies suggested that GluA2 and GluA2/3 are reduced, but GluA1 is unchanged (Carter et al., 2004). Furthermore, earlier studies also proposed a reduction in GluA1 levels (Pellegrini-Giampietro et al., 1994), or consistent decrease in all four AMPAR subunits (Ikonomovic et al., 1995, 1997; Aronica et al., 1997; Thorns et al., 1997; Wakabayashi et al., 1999). However, these changes were not investigated at the level of individual neurons or synapses.
At present, it is not clear whether AMPARs are altered at all excitatory synapses in the hippocampus of AD brains. They may also be selectively disrupted at specific postsynaptic sites or neuron types. To clarify these possibilities in the hippocampal CA1 region, we employed immunoblots, histoblots, and high-resolution quantitative immunocytochemical techniques, with specific focus on the hippocampal regions of APP/PS1 mice. Here we provide compelling new evidence for a reduction in synaptic AMPARs in pyramidal cells and interneurons of the hippocampal CA1 region in the APP/PS1 AD mouse model, with a parallel increase in intracellular AMPAR population.
Male APP/PS1 mice (RRID:IMSR_MMRRC:034832) were obtained from the Jackson Laboratory1 and expressed Mo/Hu APP695swe construct in conjunction with the exon-9-deleted variant of human presenilin 1 [Tg(APPswe,PSEN1dE9)85Dbo/Mmjax] (Jankowsky et al., 2001, 2004). The “control” wild type (WT) mice were age-matched littermates without the transgene. The following ages were selected for analysis: (i) no sign of pathology (1 month), (ii) first signs of Aβ deposition (6 months) (Jankowsky et al., 2004), and (iii) onset of memory deficits with severe synapse loss and widespread Aβ deposition (12 months) (Garcia-Alloza et al., 2006; Gimbel et al., 2010). For all ages and genotypes, mice were used as follows for the experiments: Immunoblot (4), Histoblot (4), SDS-digested freeze-fracture replica labeling (SDS-FRL) (4) and pre-embedding immunogold experiments (3). All mice were maintained at the Animal House Facility of the University of Castilla-La Mancha (Albacete, Spain) in cages of two or more mice, on a 12 h light/12 h dark cycle at 24°C and received food and water ad libitum. Care and handling of animals prior to and during experimental procedures were in accordance with Spanish (RD 1201/2015) and European Union regulations (86/609/EC), and all protocols and methodologies were approved by the local Animal Care and Use Committee.
For immunoblotting and histoblotting, animals were deeply anesthetized by intraperitoneal injection of ketamine/xylazine 1:1 (ketamine, 100 mg/Kg; xylazine, 10 mg/Kg), the hippocampus was dissected, frozen rapidly in liquid nitrogen and stored at −80°C. For immunohistochemistry experiments at both the light microscopic and electron microscopic level, using the pre-embedding immunogold technique, animals were firstly deeply anesthetized by intraperitoneal injection of ketamine-xylazine 1:1 (ketamine, 100 mg/Kg; xylazine, 10 mg/Kg) and then transcardially perfused with ice-cold fixative containing 4% (w/v) paraformaldehyde with 0.05% (v/v) glutaraldehyde in 0.1 M phosphate buffer (PB, pH 7.4) for 15 min. After perfusion, brains were removed and immersed in the same fixative for 2 h or overnight at 4°C. Tissue blocks were washed thoroughly in 0.1 M PB. Coronal sections (60 μm thickness) were cut using a Vibratome (Leica V1000). For SDS-FRL, see below.
For SDS-FRL, we used a new in-house generated rabbit anti-GluA1–4 (pan-AMPA) receptor polyclonal antibody (D160) (Supplementary Figure 1), whose preparation and purification were carried out by following and updating protocols used to prepare a previous in-house rabbit pan-AMPAR antibody (Nusser et al., 1998; Pickard et al., 2000). In brief, the antibody was raised against a glutathione S-transferase (GST) fusion protein that contained the 58 extracellular amino-acid residues (724–781) that preceded the last membrane-spanning segment of GluR1flop [GSTGluA1flop(724–781)] and affinity purified with a maltose-binding protein fusion protein with the identical amino acid residues used for immunization. The aa sequence is highly conserved in GluA1–4 and therefore the obtained antibody is expected to react with all AMPAR subunit proteins (Pickard et al., 2000; Supplementary Figure 1).
For histoblot and immunoblots, we used a guinea pig pan-GluA1–4 receptor antibody (GP-Af580; aa. 717–754 of mouse AMPA; Frontier Institute Co., Japan). The antibody characteristics have been described previously (Fukaya et al., 2006). For the pre-embedding immunogold technique, we used a polyclonal rabbit antibody anti-GluA2/3 (AB1506; Chemicon, Temecula, CA, United States). These antibodies were raised against synthetic peptides derived from intracellular C-terminal sequences of the GluA2 and GluA3 subunits (Wenthold et al., 1992). A monoclonal anti-α-tubulin antibody (DM1A; ref CP06) was obtained from Millipore (Millipore Corporation, Burlington, MA, United States). For double-SDS-FRL, we used a mouse monoclonal antibody against the GluN1 subunit of NMDA receptor (MAB363, Millipore Bioscience Research Reagents). The characteristics and specificity of GluN1 was characterized previously (Siegel et al., 1994; Iwasato et al., 2000; Masugi-Tokita et al., 2007; Szabadits et al., 2011). While the guinea pig AMPAR antibody worked well for histoblots and immunoblots, it produced relatively weak labeling in SDS-FRL. Therefore, we used rabbit anti-GluA1–4 antibodies for SDS-FRL.
The following secondary antibodies were used: goat anti-mouse IgG-horseradish peroxidase (1: 2,000; Santa Cruz Biotechnology, Santa Cruz, CA, United States), goat anti-rabbit IgG-horseradish peroxidase (1: 15,000; Pierce, Rockford, United States), alkaline phosphatase (AP)-goat anti-mouse IgG (H + L) and AP-goat anti-rabbit IgG (H + L) (1: 5,000; Invitrogen, Paisley, United Kingdom), anti-rabbit IgG conjugated to 10 nm gold particles and anti-mouse IgG conjugated to 5 nm gold particles (1: 100; British Biocell International, Cardiff, United Kingdom).
Hippocampi were homogenized in 50 mM Tris Base, pH 7.4, and protease inhibitor cocktail (Thermo Fisher Scientific, Pierce, Rockford, United States) with a motorized pestle (Sigma-Aldrich). The homogenized tissue was initially centrifuged 10 min at 1,000 × g at 4°C and the supernatant was further centrifuged 30 min at 12,000 × g (Centrifuge 5415R, Eppendorf, Hamburg, Germany) at 4°C. The resulting pellet, containing the membrane extracts, was resuspended in 50 mM Tris-Cl, pH 7.4, and protease inhibitor cocktail (Thermo Fisher Scientific, Pierce, Rockford, United States). The protein content of each membrane extract was determined by the BCA protein assay kit (Thermo Fisher Scientific). Twenty five micrograms of membrane protein was loaded onto sodium dodecyl sulfate polyacrylamide (7.5%) gels (SDS/PAGE) in sample buffer [0.05 M Tris pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 0.05% (v/v) β-mercaptoethanol, and 0.001% (w/v) bromophenol blue]. The proteins were transferred to PVDF membranes using a semidry transfer system, followed by immunolabeling with anti-GluA1–4 (1:1,000) and anti-α-tubulin (1:1,000) antibodies. Protein bands were visualized and detected after application of a mouse IgG kappa binding protein coupled to horseradish peroxidase (1:2,000 dilution) and using the enhanced chemiluminescence (ECL) blotting detection kit (SuperSignal West Dura Extended Duration Substrate, Pierce, Rockford, United States). Blots were captured and quantified by densitometry using a LAS4000 MINI (Fujifilm, Japan).
The regional distribution of AMPARs was analyzed in rodent brains, using the histoblot technique (Molnár, 2016; Aguado and Luján, 2019). Briefly, horizontal cryostat sections (10 μm) from mouse brain were overlayed with nitrocellulose membranes moistened with 48 mM Tris-base, 39 mM glycine, 2% (w/v) sodium dodecyl sulfate and 20% (v/v) methanol for 15 min at room temperature (∼20°C). After blocking in 5% (w/v) non-fat dry milk in phosphate-buffered saline for 1 h, nitrocellulose membranes were treated with DNase I (5 U/mL), washed and incubated in 2% (w/v) sodium dodecyl sulfate and 100 mM β-mercaptoethanol in 100 mM Tris–HCl (pH 7.0) for 60 min at 45°C to remove any remaining tissue residues. After extensive washing, the blots were reacted with affinity-purified anti-GluA1–4 antibodies (0.5 mg/mL) in blocking solution overnight at 4°C. The bound primary antibodies were detected with alkaline phosphatase-conjugated anti-rabbit IgG secondary antibodies (Molnár, 2016; Aguado and Luján, 2019). To compare the expression levels of AMPARs between the wild type and APP/PS1 mice and at all ages, all nitrocellulose membranes were processed in parallel, and with the same incubation time for each reagent was used for the antibody. Digital images were acquired by scanning the nitrocellulose membranes using a desktop scanner (HP Scanjet 8300). Image analysis and processing were performed using the Adobe Photoshop software (Adobe Systems, San Jose, CA, United States) as described previously (Martín-Belmonte et al., 2019). For both, a series of primary and secondary antibody dilutions and incubation times were used to optimize the experimental conditions for the linear sensitivity range of all reactions and to confirm that all labeling was below saturation levels.
Immunohistochemical reactions at the electron microscopic level were carried out using the pre-embedding immunogold and the SDS-FRL methods as described earlier (Tanaka et al., 2005; Luján et al., 2018).
Briefly, free-floating sections obtained from WT and APP/PS1 and three ages (1, 6, and 12-months) were incubated in parallel in 10% (v/v) NGS diluted in TBS. Sections were then incubated in anti-GluA2/3 antibodies (3–5 μg/mL diluted in TBS containing 1% (v/v) NGS), followed by incubation in goat anti-rabbit IgG coupled to 1.4 nm gold (Nanoprobes Inc., Stony Brook, NY, United States). Sections were post-fixed in 1% (v/v) glutaraldehyde and washed in double-distilled water, followed by silver enhancement of the gold particles with an HQ Silver kit (Nanoprobes Inc.). Sections were then treated with osmium tetroxide [1% (w/v) in 0.1 M phosphate buffer], block-stained with uranyl acetate, dehydrated in graded series of ethanol and flat-embedded on glass slides in Durcupan (Fluka) resin. Regions of interest were cut at 70–90 nm using an ultramicrotome (Reichert Ultracut E, Leica, Austria) and collected on single slot pioloform-coated copper grids. Ultrastructural analyses were performed in a JEOL-1010 electron microscope.
Animals were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 25 mM PBS for 1 min, followed by perfusion with ice-cold fixative containing 2% (w/v) paraformaldehyde in 0.1 M phosphate buffer (PB) for 12 min. After perfusion, brains were immediately removed from the skull, and then the hippocampi were dissected and cut into coronal slices (130 μm) using a Microslicer (Dosaka, Kyoto, Japan) in 0.1 M PB. Next, hippocampal slices containing the CA1 region were trimmed out of the slices, and immersed in graded glycerol of 10–30% (v/v) in 0.1 M PB at 4°C overnight. Slices were frozen using a high-pressure freezing machine (HPM010, BAL-TEC, Balzers). Slices were then fractured into two parts at −120°C and replicated by carbon deposition (5 nm thick), platinum (60° unidirectional from horizontal level, 2 nm), and carbon (15–20 nm) in a freeze-fracture replica machine (BAF060, BAL-TEC, Balzers). Replicas were transferred to 2.5% (w/v) SDS and 20% (w/v) sucrose in 15 mM Tris-Cl buffer (pH 8.3) for 18 h at 80°C with shaking to dissolve tissue debris. The replicas were washed three times in 50 mM Tris-buffered saline (TBS, pH 7.4), containing 0.05% (w/v) bovine serum albumin (BSA), and then blocked with 5% (w/v) BSA in the washing buffer for 1 h at room temperature. Next, the replicas were washed and reacted with the following primary antibodies: (1) pan-GluA1–4 antibody raised in rabbit (7.3 μg/mL), or (2) mixture of the pan-GluA1–4 and mouse monoclonal antibody against the GluN1 subunit of NMDA receptor (10 μg/mL; Millipore Bioscience Research Reagents), diluted in 25 mM TBS containing 1% (w/v) BSA overnight at 15°C. Following three washes in 0.05% BSA in TBS and blocking in 5% (w/v) BSA/TBS, replicas were incubated in goat anti-rabbit (for pan-GluA1–4) IgGs coupled to 5 nm gold particles (1:30; British BioCell Research Laboratories) diluted in 25 mM TBS containing 5% (w/v) BSA overnight at room temperature. In the double-labeling protocols, replicas were first reacted with the pan-GluA1–4 antibody and then anti-rabbit secondary antibody conjugated to 10 nm gold particle, followed by incubation with the GluN1 antibody and then anti-mouse secondary antibody conjugated to 5 nm gold particle. When the primary antibody was omitted, no immunoreactivity was observed. After immunogold labeling, the replicas were immediately rinsed three times with 0.05% BSA in TBS, washed twice with distilled water, and picked up onto grids coated with pioloform (Agar Scientific, Stansted, Essex, United Kingdom) and examined with an electron microscope (Hitach H-7650) equipped with a digital camera (Quemesa, EM SIS).
The labeled replicas were examined using a transmission electron microscope (JEOL-1010) and images captured at magnifications of 80,000× or 100,000×. All antibodies used in this study were visualized by immunoparticles on the exoplasmic face (E-face), consistent with the extracellular location of their epitopes. Non-specific background labeling for anti-AMPAR was estimated by counting immunogold particles on the protoplasmic face (P-face) surfaces in wild type mice. This value was on average 2.3 immunoparticles/μm2, and was not subtracted from values for specific labeling given the low value. Digitized images were then modified for brightness and contrast using Adobe PhotoShop CS5 (Mountain View, CA, United States) to optimize them for quantitative analysis.
We determined the number of AMPAR immunoparticles at excitatory synapses present in dendritic spines of CA1 pyramidal cells in the stratum radiatum and in the dendritic shafts of interneurons located in the stratum radiatum of the CA1 region of the hippocampus, in both wild type and APP/PS1 mice and at all three ages. For this aim, we used the software GPDQ (Gold Particle Detection and Quantification) developed recently to perform automated and semi-automated detection of gold particles present in a given compartment of neurons (Luján et al., 2018). The vast majority of the spines in stratum radiatum arise from pyramidal cells, thus we refer to them as pyramidal cell spines. Dendritic shafts receiving multiple excitatory and inhibitory synapses are considered to originate from interneurons. For identification of neuronal compartment in the SDS-FRL samples, oblique dendrites were identified based on their small diameter and the presence of at least one emerging spine from the dendritic shaft. Dendritic spines were considered as such if: (i) they emerged from a dendritic shaft, or (ii) they opposed an axon terminal. Dendritic spines are smaller in size compared to dendritic shafts of interneurons. Given these differences in size, excitatory synapses in spines are normally observed with a concave shape, while in interneurons they have a more flattened morphology. Axon terminals were identified based on: (i) the presence of an active zone (AZ) facing a postsynaptic density (PSD), recognized by an accumulation of intramembrane particles (IMPs), on the opposing exoplasmic-face (E-face) of a spine or dendrite; or (ii) the presence of synaptic vesicles on their cross-fractured portions. Non-specific background labeling was measured on P-face structures surrounding the measured E-faces.
Quantitative analysis of immunogold labeling for AMPAR subunits was performed on excitatory postsynaptic specializations indicated by the presence of intramembrane particle (IMP) clusters on the exoplasmic face (E-face) (Harris and Landis, 1986). One of the advantages of the SDS-FRL technique is that the whole synaptic specialization of excitatory synapses is immediately visible over the surface of neurons. The outline of postsynaptic specialization (IMP clusters) is manually demarcated by connecting the outermost IMP particles, and the area of synaptic sites is measured using the software GPDQ. Immunogold particles for AMPARs were regarded as synaptic labeling if they were within demarcated IMP clusters and those located in the immediate vicinity within 30 nm from the edge of the IMP clusters, i.e., the potential distance between the immunogold particles and antigens. The number of immunogold particles was counted in both complete and incomplete (partially fractured) postsynaptic membrane specialization. An incomplete postsynaptic membrane specialization was considered as such when it was partially cut by a fracture, but contained at least 30 IMPs. AMPAR-positive excitatory synapses that were co-labeled with more than two immunoparticles for GluN1 were included as synaptic IMP clusters in the analysis to make sure we were dealing with glutamatergic synaptic contacts. As densities of immunogold labeling for the pan-GluA1–4 antibody obtained from complete and incomplete synapses were not significantly different, they were pooled. The density of the immunoparticles for AMPARs in each synaptic site was calculated by dividing the number of the immunoparticles by the area of the demarcated IMP clusters. Measurements were performed in three animals, and results were pooled due to the fact that the density for immunogold particles was not significantly different in the different animals. Then, we calculated the average density across synapses. Immunoparticle densities are presented as mean ± SEM.
Finally, it is worth mentioning that neuronal loss is only observed adjacent to plaques in the APP/PS1 mouse model2. Accordingly, our quantitative analysis was performed in Aβ plaque-free regions of the hippocampus to avoid the destroyed tissue in dystrophic neurites adjacent to Aβ plaques (Alonso-Nanclares et al., 2013), and also carried out systematically on identified excitatory synapses, which were similar in numbers (n) and total surface areas between the two genotypes. Thus, the density values expressed as immunoparticles/μm2 in the APP/PS1 mice at 12 months of age represent genuine reduction of AMPARs in different types of CA1 excitatory synapses, regardless of any possible neuronal and/or synaptic loss.
To test method specificity in the procedures for electron microscopy, the primary antibodies were either omitted or replaced with 5% (v/v) normal serum of the species of the primary antibody, resulting in total loss of the signal. For the pre-embedding technique, labeling patterns were also compared with those obtained by calbindin (polyclonal rabbit anti-calbindin D-9k CB9; Swant, Marly, Switzerland); only the antibodies against GluA1–4 consistently labeled excitatory synapses.
To avoid observer bias, we performed blinded experiments for immunoblots and immohistochemistry prior to data analysis. Statistical analyses were performed using GraphPad Prism (San Diego, CA, United States) and data were presented as mean ± SEM unless indicated otherwise. Statistical significance was defined as p < 0.05. The statistical evaluation of the immunoblots was performed using the Student-Fisher t-test, with Levene’s test for homogeneity of variance and Shapiro-Wilks normality test. The application of Levene’s test indicated that variances are equal, and the application of Shapiro-Wilks test indicated that distributions were normal. To compute SEM error bars, five blots were measured from each animal. The statistical evaluation of the immunogold densities in the mouse model was performed using the two-way ANOVA test and Bonferroni post-hoc test. Correlations were assessed using Pearson’s correlation test. Finally, statistical evaluation of the frequency of immunogold measured with pre-embedding and frequency of co-localization measured with immunofluorescence were performed using student-t test with Holm-Sidak correction, following the application of Shapiro-Wilks test indicating that distributions were normal.
Using the guinea pig anti-GluA1–4 antibody in conventional histoblots (Aguado and Luján, 2019), we determined in first place whether the expression of AMPAR was altered in the brain of APP/PS1 mice at different ages: 1 month (Figures 1A–C), 6 months (Figures 1D–F), and 12 months (Figures 1G–I). The overall expression patterns of GluA1–4 was very similar in wild type and APP/PS1 mice (Figures 1A–I). In wild type mice at all three ages, the strongest GluA1–4 immunoreactivity was found in the neuropil layers of the hippocampal formation, in the superficial layers of neocortex, and in the cerebellar molecular layer (Figures 1A,D,G). Moderate labeling was found in deeper layers of the neocortex, in the caudate putamen and thalamus (Figures 1A,D,G). This expression pattern was very similar in the brain of APP/PS1 mice at all ages (Figures 1B,E,H). Quantitative analyses performed to compare the protein densities for all ages revealed that GluA1–4 immunoreactivities were similar between wild type and APP/PS1 mice (Figures 1C,F,I).

Figure 1. Regional expression of AMPARs in the brain in wild type and APP/PS1 mice. (A–I) Expression of GluA1–4 proteins was visualized using histoblots of horizontal brain sections at 1, 6, and 12 months of age in wild type and APP/PS1 mice, using an affinity-purified anti-GluA1–4 antibody. AMPAR expression in different brain regions was determined by densitometric analysis of the scanned histoblots (C,F,I). The highest level of AMPAR expression was detected in the hippocampus (Hp), followed by the cerebellum (Cb), cortex (Cx), and septum (Sp). Lower expression levels were detected in the caudate putamen (CPu) and the thalamus (Th). Densitometric analysis showed no differences in AMPAR expression in APP/PS1 mice compared to age-matched wild type controls. Error bars indicate SEM. Scale bars: 0.2 cm.
Given that one of the most vulnerable brain regions affected in AD is the hippocampus (West et al., 2000, 2004), we next focused on this brain region to explore the laminar expression pattern of AMPARs. GluA1–4 is widely expressed in all hippocampal regions and dendritic layers at all ages of both wild type and APP/PS1 mice (Figures 2A–I). In the CA1 and CA3 regions of wild type mice, at all ages, GluA1–4 expression was moderate to strong, with the strata oriens and lacunosum-moleculare showing the lowest and the stratum radiatum showing the highest expression levels (Figures 2A,D,G). The stratum lucidum in the CA3 region showed weak expression level throughout (Figures 2A,D,G). In the dentate gyrus of wild type mice, GluA1–4 immunolabeling was weak in the hilus and moderate in the molecular layer (Figures 2A,D,G). The stratum pyramidale of the CA1 and CA3 regions and the granule cell layer of the dentate gyrus showed the weakest GluA1–4 immunolabeling (Figures 2A,D,G). The labeling pattern of wild type mice was similar to that of APP/PS1 mice at all ages (Figures 2B,E,H). Quantitative analyses of immunoreactivities performed at the three ages confirmed that the expression levels of GluA1–4 in all subfields and dendritic layers analyzed was unchanged in APP/PS1 mice compared to age-matched wild type controls (Figures 2C,F,I).
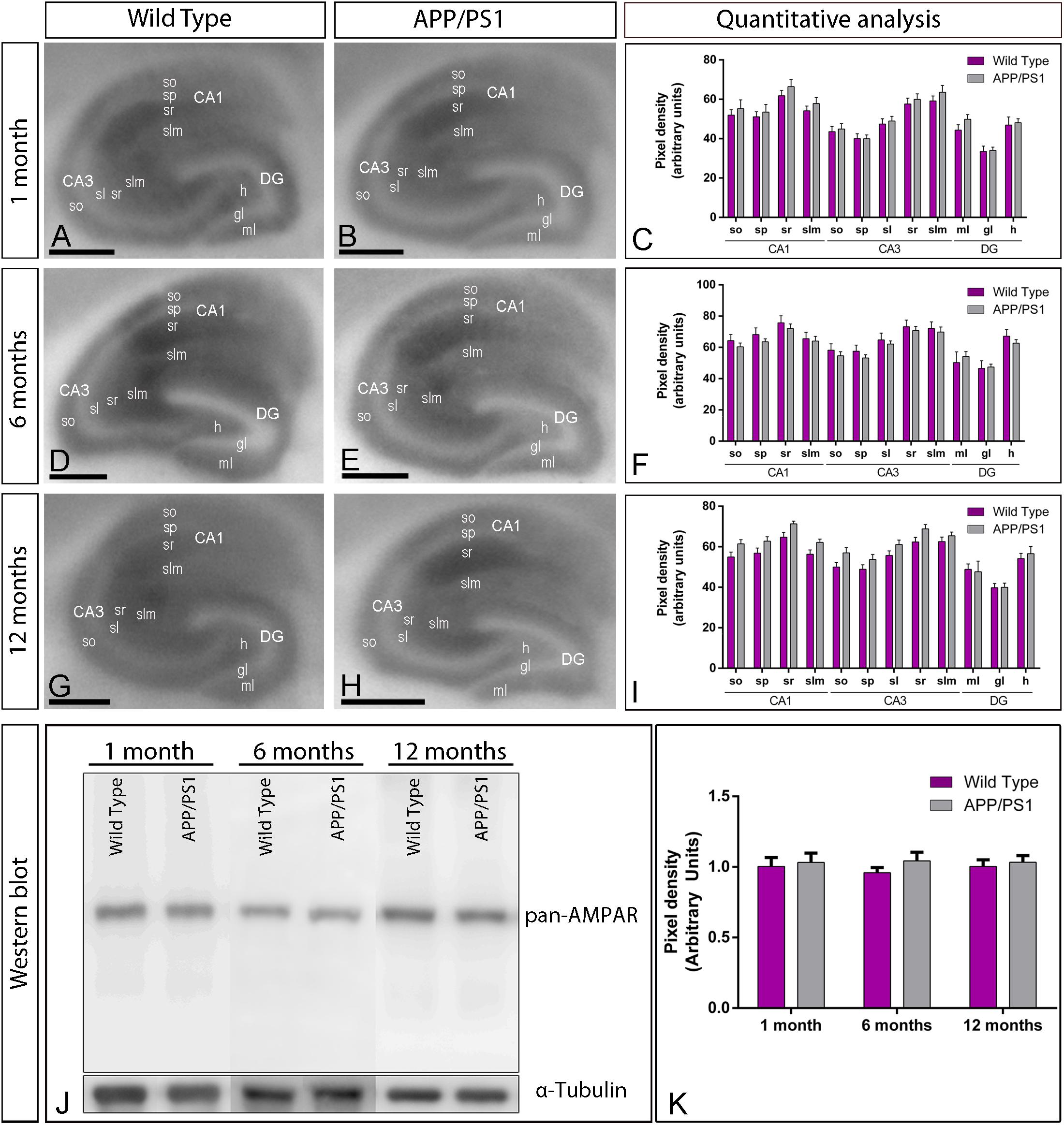
Figure 2. Hippocampal expression and distribution of AMPARs in wild type and APP/PS1 mice. (A–I) The expression of GluA1–4 proteins was visualized in histoblots of horizontal brain sections at 1, 6, and 12 months of age in wild type and APP/PS1 mice using an affinity-purified anti-GluA1–4 antibody. The level of AMPAR immunoreactivities in different hippocampal subfields and dendritic layers were determined by densitometric analysis of scanned histoblots. AMPAR expression was moderate to strong in all dendritic layers of the CA1 and CA3 region and dentate gyrus, with the stratum radiatum (sr) of CA1 and CA3 showing the highest expression level. Densitometric analysis showed no differences in AMPAR expression in APP/PS1 mice compared to age-matched wild type controls. Error bars indicate SEM. Scale bars: 0.05 cm. (J,K) Immunoblots showing the expression of the AMPAR protein in the hippocampus at 1, 6, and 12 months of age in wild type and APP/PS1 mice. Crude membrane preparations were probed with the anti-GluA1–4 antibody, which recognized a band with estimated molecular mass of 100 kDa. The developed immunoblots were scanned and densitometric measurements from five independent experiments (n = 4 brains) were averaged to compare the protein densities for each age and experimental group. AMPAR immunoreactivities were normalized to the α-tubulin content of each sample and expressed as pixel density. No differences were detected in APP/PS1 mice compared to age-matched wild type controls. Error bars indicate SEM.
We further evaluated the expression of GluA1–4 in the hippocampus using immunoblots of membrane fractions, which reaffirmed the data obtained using histoblots. The guinea pig anti-GluA1–4 antibody revealed one immunoreactive band with estimated molecular mass of ∼105 kDa (Figure 2J). The levels of AMPAR proteins were unchanged in APP/PS1 compared to age-matched wild type mice (Figures 2J,K). Overall, these results indicate no detectable changes in overall expression of AMPAR subunit proteins in APP/PS1 mice brains in any of the investigated brain regions.
Using the SDS-FRL method, we analyzed the distribution and densities of AMPARs at excitatory synapses in the CA1 region of hippocampal sections obtained from 1, 6, and 12-months of age wild type and APP/PS1 mice with an antibody against highly conserved extracellular amino acid residues of GluA1–4 (pan-AMPA). We showed that this antibody reacts selectively with all AMPAR subunits GluA1–4. Using the SDS-FRL technique, clusters of IMPs on the E-face represent the postsynaptic membrane specialization (PSDs) of glutamatergic synapses (Tarusawa et al., 2009). Many IMP clusters were labeled with anti-GluA1–4-linked immunogold particles in the CA1 region of the hippocampus.
We initially performed the ultrastructural analysis at 1 month (Figure 3) and 6 months of age (Figure 4), both in wild type and APP/PS1 mice. In the four experimental groups, immunoparticles for AMPARs were distributed over the entire postsynaptic membrane specializations of spines and interneurons with no apparent clustering (Figures 3A–F, 4A–E). The number of functional AMPARs in individual IMP clusters was plotted against the synaptic area and found to be proportional (Figures 3G, 4F). The density of labeling varied between synapses both in dendritic spines and interneurons (Table 1), but we found no differences between APP/PS1 and WT mice (Figures 3H,I, 4G,H). Overall, these results indicate no changes in the synaptic localization of AMPAR subunit proteins in APP/PS1 mice brains up until 6 months of age.
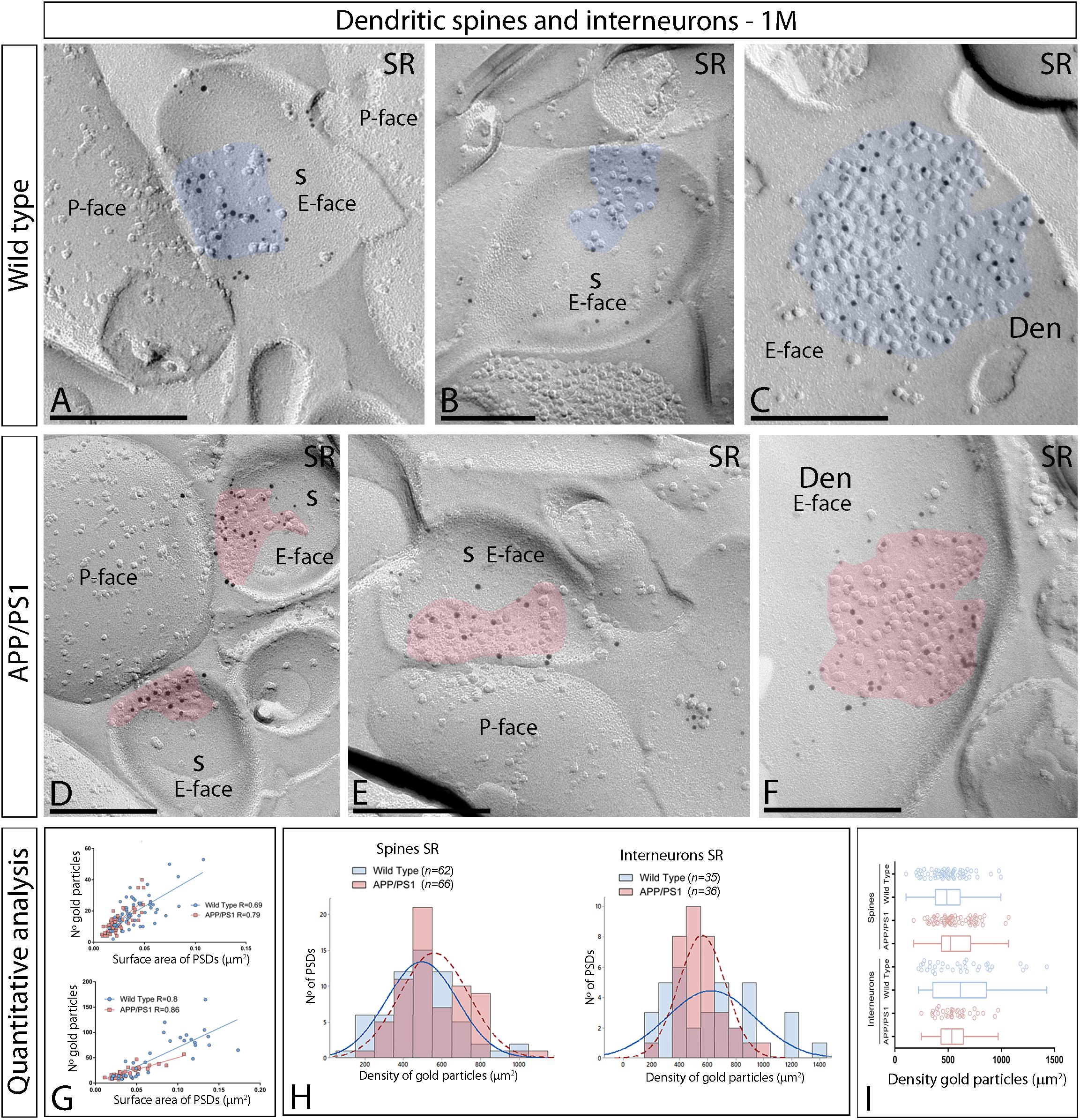
Figure 3. Synaptic AMPARs in dendritic spines and interneurons of APP/PS1 mice at 1 month. (A–F) Electron micrographs of the hippocampus showing immunoparticles for AMPARs at excitatory synaptic sites of dendritic spines of pyramidal cells and shafts of interneurons in the stratum radiatum of the CA1 region, as detected using the SDS-FRL technique. Postsynaptic membrane specializations (IMP clusters, pseudo colored in blue for wild type and in red for APP/PS1 to aid visualization) show strong immunoreactivity for AMPARs (10 nm gold particles), both in the wild type and in the APP/PS1 mice. Scale bars: (A–F), 200 nm. (G) Scatter plots showing the correlation between surface areas of postsynaptic membrane specializations and numbers of gold particles labeling AMPARs in wild type and APP/PS1 mice. (H) Histograms showing the distribution of densities of gold particles that label AMPARs of individual postsynaptic membrane specializations in the stratum radiatum of the hippocampal CA1 region in wild type and APP/PS1 mice. (I) Quantitative analysis showing mean densities of AMPARs in excitatory synapses in spines and interneurons in the stratum radiatum in wild type and APP/PS1 mice. No differences were detected in densities of AMPAR immunoparticles in the spines (WT = 496 ± 23 immunoparticles/μm2; APP = 560 ± 22 immunoparticles/μm2) or interneurons (WT = 626 ± 53 immunoparticles/μm2; APP = 562 ± 29 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, p > 0.05).
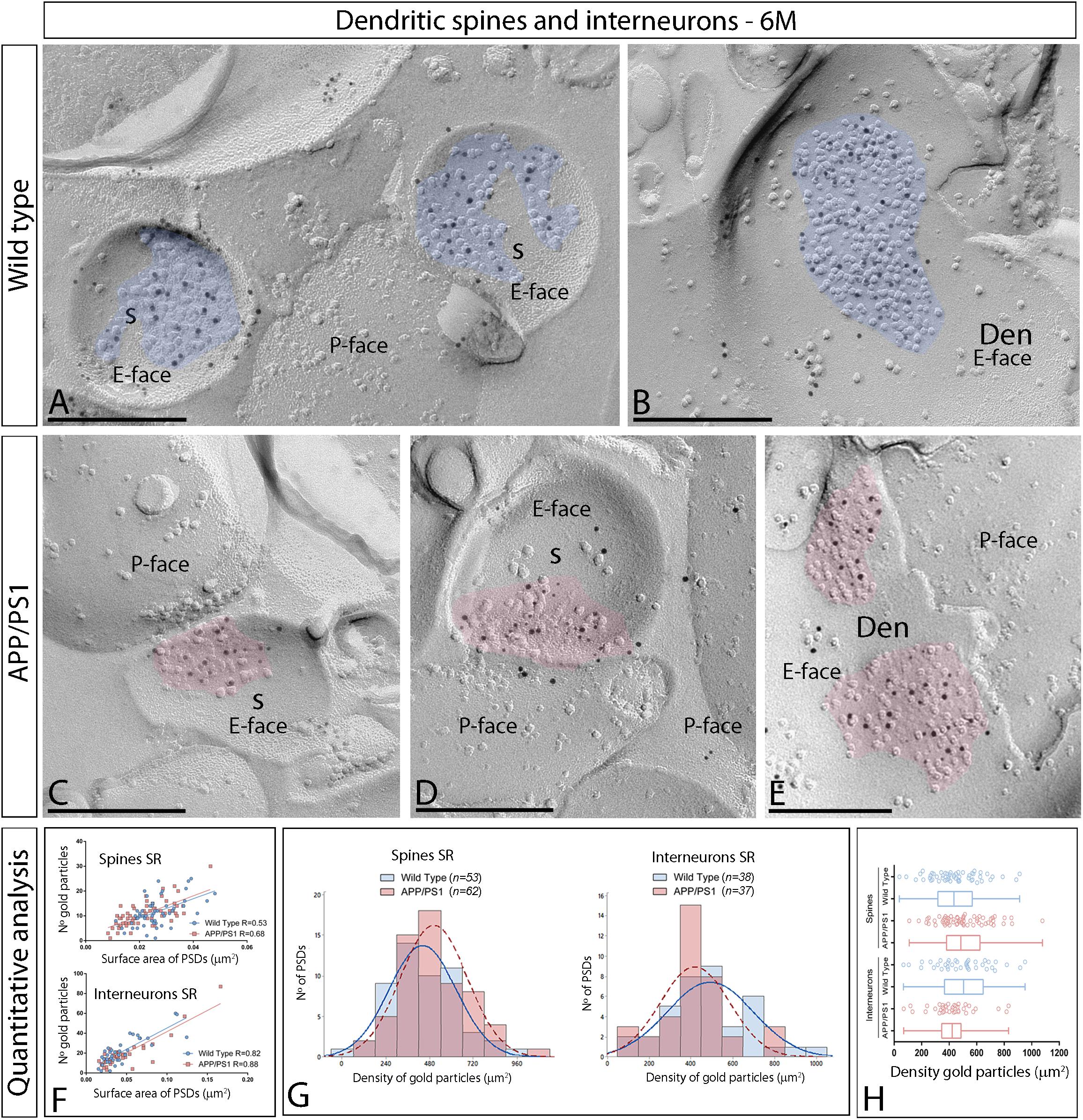
Figure 4. Synaptic AMPARs in dendritic spines and interneurons of APP/PS1 mice at 6 months of age. (A–F) Electron micrographs of the hippocampus showing immunoparticles for AMPARs at excitatory synaptic sites of dendritic spines of pyramidal cells and shafts of interneurons in the CA1 stratum radiatum, as detected using the SDS-FRL technique at 6 months of age. Postsynaptic membrane specializations (IMP clusters, pseudo colored in blue for wild type and in red for APP/PS1 to aid visualization) show strong immunoreactivity for GluA1–4 (10 nm gold particles) both in the wild type and in the APP/PS1 mice. Scale bars: (A–E), 200 nm. (F) Scatter plots showing the correlation between surface areas of postsynaptic membrane specializations and numbers of gold particles labeling AMPARs in wild type and APP/PS1 mice. (G) Histograms showing the distribution of densities of gold particles that label AMPARs of individual postsynaptic membrane specializations in the hippocampal CA1 region in wild type and APP/PS1 mice. (H) Quantitative analysis showing mean densities of AMPARs in excitatory synapses in spines and interneurons in the stratum radiatum in wild type and APP/PS1 mice. No differences were detected in densities of AMPAR immunoparticles in the spines (WT = 443 ± 25 immunoparticles/μm2; APP = 505 ± 23 immunoparticles/μm2) or interneurons (WT = 494 ± 33 immunoparticles/μm2; APP = 421 ± 27 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, p > 0.05).
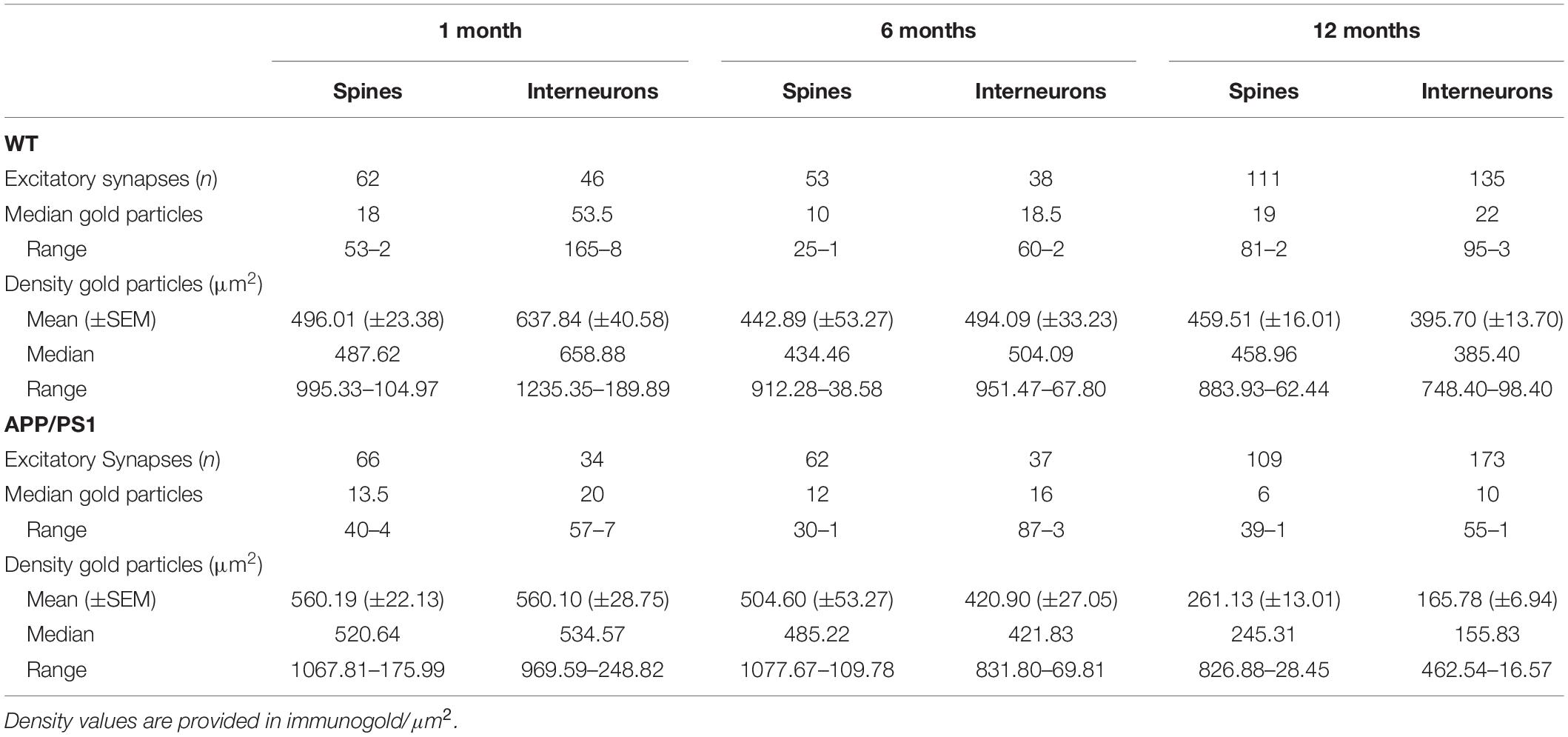
Table 1. Number and density of gold particles for pan-AMPAR at different excitatory synapses in the CA1 stratum radiatum.
In the APP/PS1 mouse model of AD severe pathological damage appears at 12 months of age (Garcia-Alloza et al., 2006). In wild type mice, immunoparticles for AMPARs in dendritic spines followed a similar synaptic pattern as at earlier ages, observed over the postsynaptic membrane specializations (Figures 5A–C). In contrast, fewer AMPAR immunoparticles were detected in excitatory synapses of dendritic spines in APP/PS1 mice (Figures 5D–F). The number of gold particles labeling AMPARs was variable (Table 1), but proportional to the synaptic area both in wild type (R = 0.74) and APP/PS1 (R = 0.66) mice (p < 0.0001, Pearson’s correlation test) (Figure 5G). Quantitative analyses revealed a reduction in the number and density of AMPARs in excitatory synapses of dendritic spines. The number of AMPAR immunoparticles distributed in excitatory synapses (n = 111) varied in the range of 2–81 with a median value of 19, but in APP/PS1 dendritic spines (n = 109) in the range of 1–39 with a median value of 6 (Figure 5H and Table 1). Although the density of labeling varied between synapses (Figure 5I), we found a significant reduction in AMPAR levels in APP/PS1 synapses (median = 245 immunoparticles/μm2) compared to age-matched wild type controls (median = 459 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, ****p < 0.0001) (Figure 5I and Table 1).
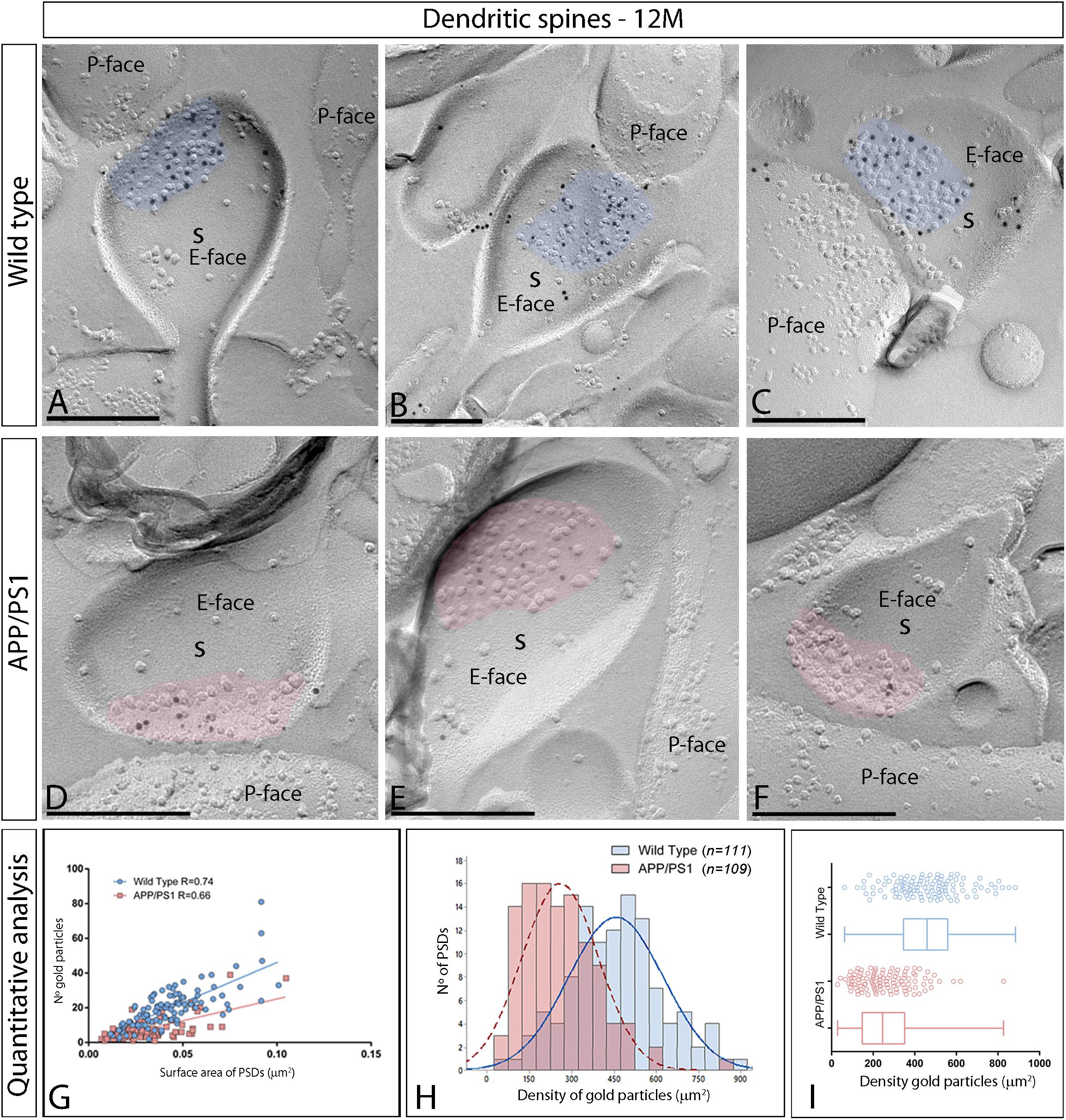
Figure 5. Reduced density of synaptic AMPARs in dendritic spines of APP/PS1 mice at 12 months. (A–F) Electron micrographs of the hippocampus showing immunoparticles for AMPARs at excitatory synaptic sites of dendritic spines of pyramidal cells in the CA1 stratum radiatum, as detected using the SDS-FRL technique. Postsynaptic membrane specializations (IMP clusters, pseudo colored in blue for wild type and in red for APP/PS1 to aid visualization) show strong immunoreactivity for AMPARs (10 nm gold particles) in the wild type, while they show weaker immunoreactivity in the APP/PS1. Scale bars: (A–F), 200 nm. (G) Scatter plots showing the correlation between surface areas of postsynaptic membrane specializations and numbers of gold particles labeling AMPARs in wild type and APP/PS1 mice. (H) Histograms showing the distribution of densities of gold particles that label AMPARs of individual postsynaptic membrane specializations in the hippocampal CA1 region in wild type and APP/PS1 mice. (I) Quantitative analysis showing mean densities of AMPARs in excitatory synapses in spines in wild type and APP/PS1 mice. A significant reduction in the densities of AMPAR immunoparticles were detected in dendritic spines of APP/PS1 mice (261 ± 13 immunoparticles/μm2) compared to age matched wild type (459 ± 16 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, p < 0.0001).
Next, we performed ultrastructural analyses to determine whether synaptic AMPARs are also altered in excitatory synapses on interneurons (Figure 6). Similarly to 1 and 6 month old mice, immunoparticles for AMPARs in interneurons at 12 months of age were observed over the postsynaptic membrane specializations in wild type mice (Figures 6A–C), but were consistently fewer in APP/PS1 mice (Figures 6D–F). Although the number of gold particles labeling AMPARs was variable (Table 1), it was proportional to the synaptic area (Figure 6G). In wild type mice, immunoparticles distributed in excitatory synapses (n = 135) varied in the range of 3–95 with a median value of 22, but in APP/PS1 dendritic spines (n = 173) in the range of 1–55 with a median value of 10 (Figure 6H and Table 1). When density of AMPAR labeling was analyzed, there was a significant reduction of AMPARs in APP/PS1 synapses (median = 156 immunoparticles/μm2) compared to age-matched wild type (median = 385 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, ****p < 0.0001) (Figure 6I and Table 1). In summary, these results indicate a significant decrease in the synaptic localization of AMPAR subunit proteins in APP/PS1 mouse brains at 12 months of age.
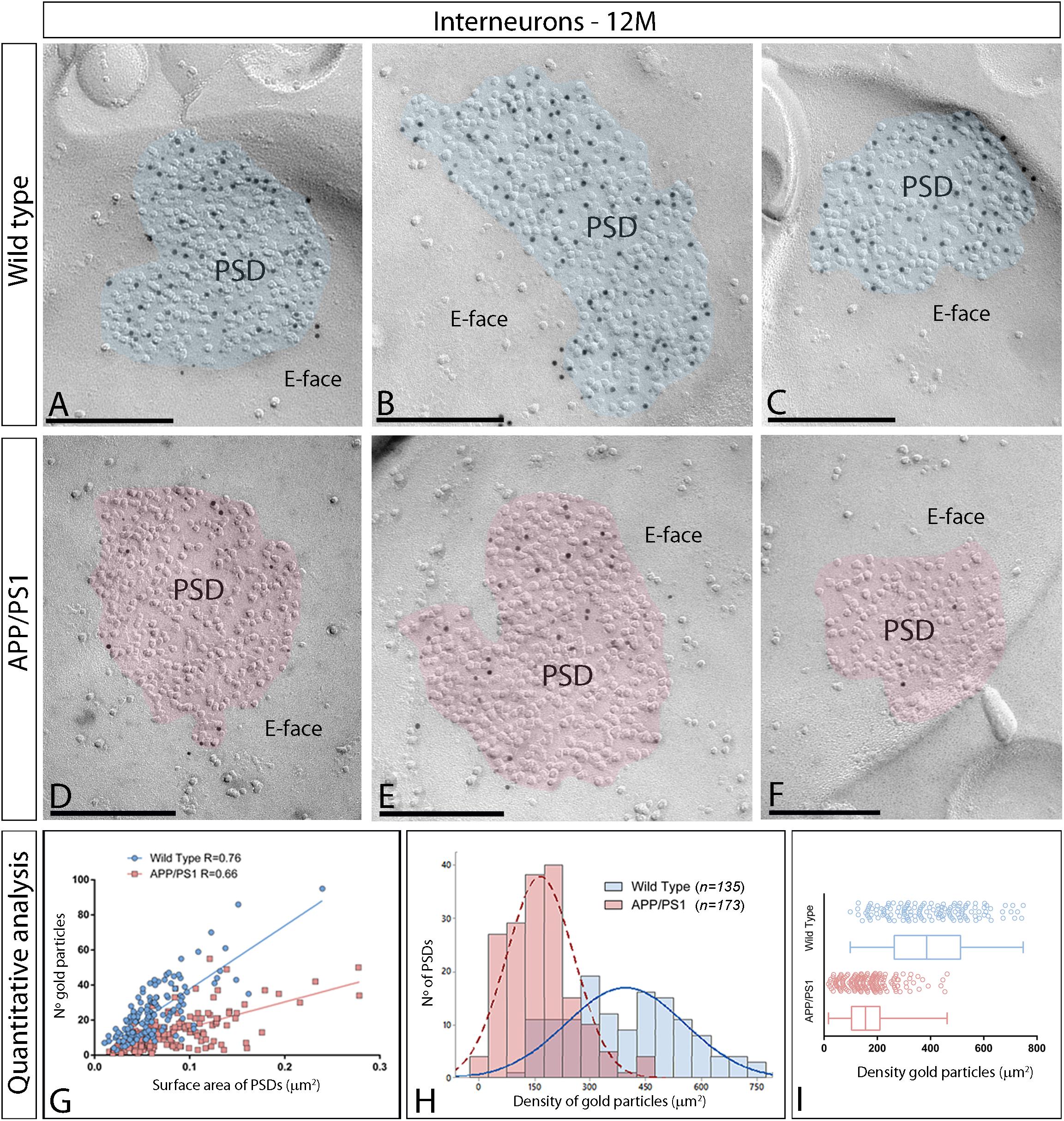
Figure 6. Density of synaptic AMPARs in interneurons of APP/PS1 mice at 12 months. (A–F) Electron micrographs of the hippocampus showing immunoparticles for AMPARs at synaptic sites of interneurons in the stratum radiatum of the CA1 region, as detected using the SDS-FRL technique. Postsynaptic membrane specializations (IMP clusters, pseudo colored in blue for wild type and in red for APP/PS1 to aid visualization) show strong immunoreactivity for AMPARs (10 nm gold particles) in the wild type, while they show weaker immunoreactivity in the APP/PS1. Scale bars: (A–F), 200 nm. (I) Quantitative analysis showing mean densities of AMPARs in excitatory synapses in spines in wild type and APP/PS1 mice. A significant reduction in the densities of AMPAR immunoparticles were detected in synaptic specialization of interneurons in APP/PS1 mice (166 ± 7 immunoparticles/μm2) compared to age matched wild type (396 ± 14 immunoparticles/μm2) (Two-way ANOVA test and Bonferroni post-hoc test, p < 0.0001). (H) Histograms showing the distribution of densities of gold particles that label AMPARs in SDS-FRL replicas of individual postsynaptic membrane specializations in the hippocampal CA1 region in wild type and APP/PS1 mice. (G) Scatter plots showing the correlation between surface areas of postsynaptic membrane specializations and numbers of gold particles labeling AMPARs in wild type and APP/PS1 mice.
We recently described using the same mouse model that whilst the GABAB1 receptor protein expression is not altered, its density is reduced in the plasma membrane and increased at cytoplasmic sites in CA1 pyramidal cells (Martín-Belmonte et al., 2019). To explore whether AMPARs undergo a similar redistribution, their subcellular localization was investigated at 12 months using the quantitative pre-embedding immunogold techniques on tissue blocks taken from the stratum radiatum of the CA1 area (Figure 7). The anti-GluA1–4 antibody recognizes extracellular domains of AMPAR subunits, and this prevents the determination of their precise localization, because when two neuronal compartments are close to each other in the neuropil, the origin of immunoparticles cannot always be established unambiguously. Therefore, we used an anti-GluA2/3 antibody raised against the intracellular C-terminal domain of AMPAR subunits, which are exposed on the surface of intracellular compartments and identifiable using immunolabeling (Bernard et al., 1997).
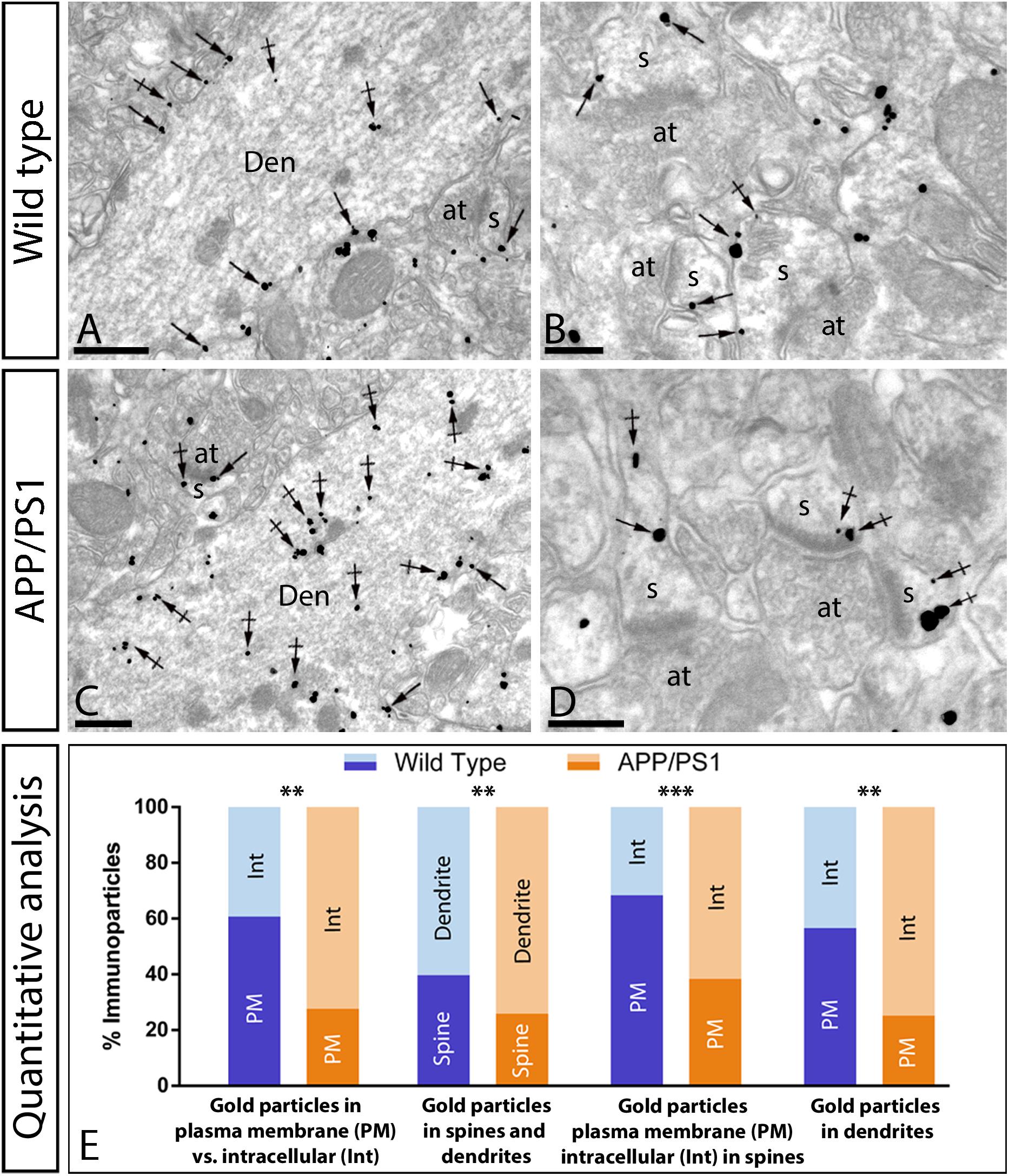
Figure 7. Increased intracellular distribution of GluA2/3 receptors in APP/PS1 mice at 12 months. Electron micrographs showing immunoparticles for GluA2/3 receptors in pyramidal cells of the CA1 region in wild type and APP/PS1 mice, as detected using a pre-embedding immunogold technique. (A,B) In wild type mice, immunoparticles for GluA2/3 were found along the extrasynaptic plasma membrane (arrows) and intracellular sites (crossed arrows) of dendritic shafts (Den) and spines (s) contacted by axon terminals (at). (C,D) In APP/PS1 mice, immunoparticles for GluA2/3 were more frequently detected at intracellular sites (crossed arrows) and few immunoparticles along the plasma membrane (arrows). (E) Quantitative analysis showing that immunoparticles for GluA2/3 receptors were less frequently observed along the extrasynaptic plasma membrane of dendrites and spines of CA1 pyramidal cells in APP/PS1 mice, but instead they more frequently detected at intracellular sites in pyramidal cells. (Student-T test and Holm-Sidak correction, **p < 0.01; ***p < 0.001) Scale bars: (A,B,D), 1 μm; (C), 0.5 μm.
In both wild type and APP/PS1 mice, immunoreactivity for GluA2/3 was detected in dendritic spines and shafts of CA1 pyramidal cells (Figures 7A–D), along their extrasynaptic plasma membrane and also associated at intracellular sites. In wild type mice, most immunoparticles for GluA2/3 were detected along the extrasynaptic plasma membranes of pyramidal cells (Figures 7A,B). In the APP/PS1 mice, however, immunoparticles for AMPARs were more frequently detected intracellularly (Figures 7C,D). This change in localization was demonstrated using quantitative analyses (Figure 7E), which showed significant differences in the total amount of plasma membrane-associated vs. intracellular AMPAR labeling in CA1 pyramidal cells (Plasma membrane: 61 ± 1.9% in wild type and 28 ± 3.6% in APP/PS1; Intracellular: 39 ± 1.9% in wild type and 72 ± 3.6% in APP/PS1; ∗∗p < 0.01) (Student-T test and Holm-Sidak correction). This change took place both in dendritic spines (Plasma membrane: 68 ± 0.4% in wild type and 38 ± 2.4% in APP/PS1; Intracellular: 32 ± 0.4% in wild type and 62 ± 2.4% in APP/PS1; ∗∗∗p < 0.001) and dendritic shafts (Plasma membrane: 57 ± 2.6% in wild type and 25 ± 3.5% in APP/PS1; Intracellular: 43 ± 2.6% in wild type and 75 ± 3.5% in APP/PS1; ∗∗p < 0.01) (Student’s T-test and Holm-Sidak correction) (Figure 7E). Given the low expression of GluA2/3 in interneurons compared to pyramidal cells, quantitative analysis was not feasible. To establish the types of intracellular compartments in which AMPARs are present, we performed double labeling immunofluorescence with different marker proteins (Rab4, Rab5 and EEA1), and found no differences in the co-localization pattern in the APP/PS1 mice compared to age-matched wild type controls (Supplementary Figure 3).
In AD, hippocampal dysfunction is produced by synaptic failure (Li and Selkoe, 2020), which leads to memory deficits. Changes in AMPARs are involved in Aβ-related synaptic dysfunction and pathophysiology of AD (reviewed by Keifer and Zheng, 2010; Palop and Mucke, 2010; Whitehead et al., 2017). Here, using biochemical and immunocytochemical techniques we examined the expression of AMPARs, and found no change in GluA1–4 protein expression in the APP/PS1 transgenic mouse model of AD. However, using sensitive immunoelectron microscopic techniques to analyze CA1 excitatory synapses in these mice, we identified a reduction of synaptic AMPARs in two different excitatory synapses and a parallel increase in GluA2/3 subunits at intracellular sites. These differences in the density of AMPARs were prominent at 12 months, but not detectable at earlier ages in the APP/PS1 mouse model. These changes likely to be responsible for pathological events in AD.
The expression of AMPARs has been extensively studied in the brain of rodents (Boulter et al., 1990; Keinänen et al., 1990; Petralia and Wenthold, 1992; Sato et al., 1993), and there is substantial knowledge relating to their distribution and regulation during development and aging (Jurado, 2018). In the present study, we have shown by two different techniques that expression levels of AMPARs proteins are not significantly different in APP/PS1 mice with the progressive increase of Aβ levels (Alonso-Nanclares et al., 2013) at different ages in the hippocampus. Our data differs from other studies using different mouse models of AD reporting down-regulation of the protein for AMPAR subunits (Snyder et al., 2005; Baglietto-Vargas et al., 2018; Li et al., 2019).
Previous studies using different techniques in human tissue yielded inconsistent results (Harrison et al., 1990; Dewar et al., 1991; Hyman et al., 1994; Pellegrini-Giampietro et al., 1994; Ikonomovic et al., 1995, 1997; Aronica et al., 1997; Thorns et al., 1997; Wakabayashi et al., 1999; Carter et al., 2004; Ginsberg et al., 2012). Our study identified a clear reduction in AMPAR protein levels in the hippocampus of AD patients (see Supplementary Figure 2), consistent with the reported down-regulation in the expression of GluA1 and GluA2 in AD (Martín-Belmonte et al., 2019). This may be due to the use of different techniques, post-mortem delays, brain weight and the age of the subjects, duration of tissue storage, stage of AD or the variability in tissue sampling.
The discrepancy observed between protein expression levels in the mouse model and in human tissue could be the result of differences in brain organization or development of the disease between humans and mice. It is also possible that such differences are due to the fact that the APP/PS1 is an incomplete model of AD. For instance, this model does not develop neurofibrillary tangles, a typical pathologic alteration observed in AD human patients (Irizarry et al., 1997). Alternatively, 12 months old in the APP/PS1 model could be a relatively early stage of pathological evolution to be compared with the stages of AD in human patients with severe AD neuropathology. Despite the unaltered AMPAR protein expression levels in APP/PS1 mice, we explored the possibility that they may still undergo changes in the synaptic localization vs. intracellular sites with AD progression, as also recently described for GABAB receptors (Martín-Belmonte et al., 2019).
While previous studies used the post-embedding immunogold technique to investigate numbers and densities of AMPARs in the hippocampus (Nusser et al., 1998; Petralia et al., 1999; Takumi et al., 1999; Racca et al., 2000), we utilized the highly sensitive SDS-FRL method, with nearly one gold particle-one functional channel sensitivity and proving to be an ideal tool to study the high-resolution subcellular localization of surface-localized molecules (Tanaka et al., 2005). Using this technique, we were able to unravel the distribution of AMPARs in specific CA1 excitatory synapses obtaining data about their density in hippocampal samples displaying AD pathology, at a level of detail and sensitivity never previously attained.
The efficacy of fast glutamatergic neurotransmission in hippocampal neurons depends on the type and number of ionotropic receptors in synapses, particularly AMPARs. Our analysis revealed a great variability in the AMPAR content at individual synapses, similarly to previous reports using post-embedding immunogold techniques (Nusser et al., 1998; Petralia et al., 1999; Takumi et al., 1999; Racca et al., 2000). Consistent with these studies, the number of AMPAR immunoparticles correlated with synaptic area in all experimental groups, but we observed an age-dependent reduction in the density AMPARs in Schaffer collateral synapses in APP/PS1 mice. We detected significant decrease in the synaptic localization of AMPARs and subsequent increase at cytoplasmic sites in CA1 pyramidal cell spines and shafts at 12 months of age, but not earlier.
One important aspect to consider is that the size of individual postsynaptic membrane specializations can affect synaptic transmission. Previous studies have shown that CA1 dendritic spines have PSDs with size around 0.04–0.07 μm2 (Schikorski and Stevens, 1997; Shepherd and Harris, 1998). In our work, the average size of PSDs that we found in the stratum radiatum of CA1 pyramidal cells in wild type mice at the three ages fell in that range (for instance, 0.046 ± 0.018 μm2 at 12 months), and was similar to the size observed previously with similar techniques (Antal et al., 2008). The smaller average size PSDs we encountered in APP/PS1 mice at 12 months is in agreement with previous studies in the same animal model and same age showing a higher proportion of spines with a small head volume in the stratum radiatum of the CA1 region (Merino-Serrais et al., 2011). The size of dendritic spines and their PSD are key determinants of synaptic efficacy via control of the distribution of AMPARs (Nusser et al., 1998; Takumi et al., 1999; Matsuzaki et al., 2001).
Interestingly, internalization of AMPARs is required for the induction of LTD (Beattie et al., 2000), a form of synaptic plasticity significantly enhanced in animal models of AD (Shankar et al., 2008). In addition, APP and soluble oligomeric Aβ can induce the removal of surface AMPARs at excitatory synapses, leading to synaptic depression and inhibition of LTP (Walsh et al., 2002; Klyubin et al., 2005; Hsieh et al., 2006). In relation to the effect of the observed unbalance between synaptic and intracellular alterations, the reduction of surface AMPARs and its abnormal trafficking may contribute to the memory deficits in the AD mouse model. This may reflect changes in synaptic plasticity, and this contribute to the memory deficits in the AD mouse model used in this study (Trinchese et al., 2004).
Most studies of AD brains have concentrated on excitatory neuronal functions, but compelling studies also implicate impaired inhibition in the pathogenesis (Palop and Mucke, 2010; Verret et al., 2012; Hazra et al., 2013; Martinez-Losa et al., 2018). Our results demonstrate a significant reduction in the synaptic localization of AMPARs in excitatory synapses onto dendritic shafts of interneurons in the APP/PS1 mouse model, consistent with past evidence that interneurons and the oscillatory network activities they regulate are altered in AD (Palop and Mucke, 2016). Given the random fractures in our replica samples (Masugi-Tokita and Shigemoto, 2007), dendritic shafts of interneurons could not be followed back to the parent cell body. Therefore, we could not exclude the possibility that dendritic shafts establishing excitatory synapses with different AMPAR contents represent dendrites originating from distinct interneurons or just the same interneuron receiving distinct inputs. Technical limitations prevented unequivocal identification of the source of dendritic shafts, but since the region of stratum radiatum we analyzed was close to pyramidal cell bodies, these dendrites are likely to arise from PV-positive cells (Nyíri et al., 2003), which represent basket and axo-axonic cells whose axon arborizations target the soma and the axon initial segment of principal cells, respectively (Freund and Buzsáki, 1996; Klausberger and Somogyi, 2008).
Upon post mortem examination, the hippocampi of AD patients show a reduction in the number of PV-expressing interneurons, as well as other subpopulations, in the hippocampus (Brion and Résibois, 1994; Brady and Mufson, 1997). The significant reduction of synaptic AMPARs described here in interneurons would lead to an aberrant weakening of excitatory synapses, thus contributing to hyperexcitability and deficits in synchrony. Interestingly, weakening of synapses due to the reduction of AMPAR currents has been involved in the impairment of network oscillatory activity (Caputi et al., 2012), which is known to be essential for learning and memory. This too contributes to cognitive abnormalities in AD (Verret et al., 2012; Palop and Mucke, 2016). Therefore, improving the function of interneurons may be of therapeutic benefit in AD.
In summary, this study is the first to report that reduction of AMPARs takes place in excitatory synapses established on both pyramidal cells and interneurons in APP/PS1 mice. Reductions in the receptors at synapses in both pyramidal cells and interneurons are likely to contribute to the memory deficits associated with AD. A deeper understanding of the subcellular localization of AMPARs in the neurobiology of AD may elucidate further mechanisms of the disease.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by the Brain Bank of the Institute of Neuropathology of the University Hospital Bellvitge IDIBELL (HUB-ICO-IDIBELL-Biobank). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Comité Ético de Experimentación Animal (CEEA) UCLM.
RL and YF designed the project. MI developed the rabbit anti-GluA1–4 (D160, pan-AMPA) receptor polyclonal antibody. AM-B, RL, and YF performed SDS-FRL immunoelectron microscopy. AM-B, CA, and AM-M performed histoblot analysis. AM-B performed western blots. AM-B, RA-R, and AM-M performed pre-embedding immunoelectron microscopy and its quantitative analysis. LO developed in-house software and performed computational analysis. EM provided reagents, training in the histoblot method, and feedback on the analysis. AM-B, RA-R, CA, and RL analyzed the data. RL wrote the manuscript. All authors read and approved the final manuscript, had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
This research has received funding from the European Union’s Horizon 2020 Framework Programme for Research and Innovation under the Specific Grant Agreement No. 785907 (Human Brain Project SGA2) to RL. This work was supported by grants from the Spanish Ministerio de Economía y Competitividad (RTI2018-095812-B-I00) and Junta de Comunidades de Castilla-La Mancha (SBPLY/17/180501/000229) to RL, and Life Science Innovation Center (Research and Education Program for Life Science) at University of Fukui and JSPS KAKENHI Grant Nos. 16H04662, 17K19446, and 18H05120 to YF. EM’s research was supported by the Biotechnology and Biological Sciences Research Council (Grant BB/J015938/1) and Innovate UK (UK Research and Innovation) Knowledge Transfer Partnership Grant (Ref. No: 12333).
EM is Scientific Advisory Board member of Hello Bio (http://www.hellobio.com).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Antonio Hernández-Martínez of the Nursing School of UCLM at Ciudad Real for assistance in the statistical analysis, Miss Diane Latawiec for the English revision of the manuscript, Takako Maegawa and Hitoshi Takagi of the Life Science Research Laboratory at the University of Fukui for technical assistance in SDS-FRL experiments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2020.577996/full#supplementary-material
Aguado, C., and Luján, R. (2019). “The histoblot technique: a reliable approach to analyse expression profile of proteins and to predict their molecular association,” in Neuromethods, Ed. W. Wolfgang (Berlin: Springer), 65–88. doi: 10.1007/978-1-4939-8985-0
Almeida, C. G., Tampellini, D., Takahashi, R. H., Greengard, P., Lin, M. T., Snyder, E. M., et al. (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 20, 187–198. doi: 10.1016/j.nbd.2005.02.008
Alonso-Nanclares, L., Merino-Serrais, P., Gonzalez, S., and Defelipe, J. (2013). Synaptic changes in the dentate gyrus of APP/PS1 transgenic mice revealed by electron microscopy. J. Neuropathol. Exp. Neurol. 72, 386–395. doi: 10.1097/NEN.0b013e31828d41ec
Antal, M., Fukazawa, Y., Eördögh, M., Muszil, D., Molnár, E., Itakura, M., et al. (2008). Numbers, densities, and colocalization of AMPA- and NMDA-type glutamate receptors at individual synapses in the superficial spinal dorsal horn of rats. J. Neurosci. 28, 9692–9701. doi: 10.1523/JNEUROSCI.1551-08.2008
Aronica, E., Dickson, D., Kress, Y., Morrison, J., and Zukin, R. (1997). Non-plaque dystrophic dendrites in Alzheimer hippocampus: a new pathological structure revealed by glutamate receptor immunocytochemistry. Neuroscience 82, 979–991.
Baglietto-Vargas, D., Prieto, G. A., Limon, A., Forner, S., Rodriguez-Ortiz, C. J., Ikemura, K., et al. (2018). Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell 17, 1–14. doi: 10.1111/acel.12791
Beattie, E. C., Carroll, R. C., Yu, X., Morishita, W., Yasuda, H., Von Zastrow, M., et al. (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3, 1291–1300. doi: 10.1038/81823
Bernard, V., Somogyi, P., and Bolam, J. P. (1997). Cellular, subcellular, and subsynaptic distribution of AMPA-type glutamate receptor subunits in the neostriatum of the rat. J. Neurosci. 17, 819–833. doi: 10.1523/jneurosci.17-02-00819.1997
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Boulter, J., Hollmann, M., O’Shea-Greenfield, A., Hartley, M., Deneris, E., Maron, C., et al. (1990). Molecular cloning and functional expression of glutamate receptor subunit genes. Science 249, 1033–1037. doi: 10.1126/science.2168579
Brady, D. R., and Mufson, E. J. (1997). Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 80, 1113–1125. doi: 10.1016/S0306-4522(97)00068-7
Brion, J. P., and Résibois, A. (1994). A subset of calretinin-positive neurons are abnormal in Alzheimer’s disease. Acta Neuropathol. 88, 33–43. doi: 10.1007/BF00294357
Caputi, A., Fuchs, E. C., Allen, K., Magueresse, C., and Monyer, H. (2012). Selective reduction of AMPA currents onto hippocampal interneurons impairs network oscillatory activity. PLoS One 7:e37318. doi: 10.1371/journal.pone.0037318
Carter, T. L., Rissman, R. A., Mishizen-Eberz, A. J., Wolfe, B. B., Hamilton, R. L., Gandy, S., et al. (2004). Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer’s disease patients according to Braak stage. Exp. Neurol. 187, 299–309. doi: 10.1016/j.expneurol.2003.12.010
Dewar, D., Chalmers, D. T., Graham, D. I., and McCulloch, J. (1991). Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: an autoradiographic study of the hippocampus. Brain Res. 553, 58–64. doi: 10.1016/0006-8993(91)90230-S
Freund, T. F., and Buzsáki, G. (1996). Interneurons of the hippocampus. Hippocampus 6, 347–470. doi: 10.1002/(SICI)1098-106319966:4<347::AID-HIPO1<3.0.CO;2-I
Fukaya, M., Tsujita, M., Yamazaki, M., Kushiya, E., Abe, M., Akashi, K., et al. (2006). Abundant distribution of TARP γ-8 in synaptic and extrasynaptic surface of hippocampal neurons and its major role in AMPA receptor expression on spines and dendrites. Eur. J. Neurosci. 24, 2177–2190. doi: 10.1111/j.1460-9568.2006.05081.x
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., et al. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524. doi: 10.1016/j.nbd.2006.08.017
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Laurén, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Ginsberg, S. D., Alldred, M. J., and Che, S. (2012). Gene expression levels assessed by CA1 pyramidal neuron and regional hippocampal dissections in Alzheimer’s disease. Neurobiol. Dis. 45, 99–107. doi: 10.1016/j.nbd.2011.07.013
Gu, Z., Liu, W., and Yan, Z. (2009). β-Amyloid Impairs AMPA Receptor Trafficking and Function by Reducing Ca 2+ /Calmodulin-dependent Protein Kinase II Synaptic Distribution. J. Biol. Chem. 284, 10639–10649. doi: 10.1074/jbc.M806508200
Harris, K. M., and Landis, D. M. D. (1986). Membrane structure at synaptic junctions in area CA1 of the rat hippocampus. Neuroscience 19, 857–872. doi: 10.1016/0306-4522(86)90304-0
Harrison, P. J., Bartont, A. J., Najlerahim, A., and Pearson, R. C. (1990). Distribution of a kainate/AMPA receptor mRNA in normal and Alzheimer brain. Neuroreport 1, 149–152. doi: 10.1097/00001756-199010000-00017
Hazra, A., Gu, F., Aulakh, A., Berridge, C., Eriksen, J. L., and Žiburkus, J. (2013). Inhibitory Neuron and Hippocampal Circuit Dysfunction in an Aged Mouse Model of Alzheimer’s Disease. PLoS One 8:e64318. doi: 10.1371/journal.pone.0064318
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108. doi: 10.1146/annurev.neuro.17.1.31
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Aβ-Induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Hyman, B., Van Hoesen, G., Damasio, A., and Barnes, C. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170. doi: 10.1126/science.6474172
Hyman, B. T., Penney, J. B., Blackstone, C. D., and Young, A. B. (1994). Localization of non–N-methyl-D-aspartate glutamate receptors in normal and Alzheimer hippocampal formation. Ann. Neurol. 35, 31–37. doi: 10.1002/ana.410350106
Ikonomovic, M. D., Mizukami, K., Davies, P., Hamilton, R., Sheffield, R., and Armstrong, D. M. (1997). The Loss of GluR2(3) immunoreactivity precedes neurofibrillary tangle formation in the entorhinal cortex and hippocampus of alzheimer brains. J. Neuropathol. Exp. Neurol. 56, 1018–1027. doi: 10.1097/00005072-199709000-00007
Ikonomovic, M. D., Sheffield, R., and Armstrong, D. M. (1995). AMPA-Selective glutamate receptor subtype immunoreactivity in the hippocampal dentate gyrus of patients with Alzheimer disease. Mol. Chem. Neuropathol. 28, 59–64. doi: 10.1007/BF02815205
Irizarry, M. C., Mcnamara, M., Fedorchak, K., Hsiao, K., and Hyman, B. T. (1997). APPSW Transgenic Mice Develop Age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 56, 965–973. doi: 10.1097/00005072-199709000-00002
Iwasato, T., Datwani, A., Wolf, A. M., Nishiyama, H., Taguchi, Y., Tonegawa, S., et al. (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406, 726–731. doi: 10.1038/35021059
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170. doi: 10.1093/hmg/ddh019
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/S1389-0344(01)00067-3
Jurado, S. (2018). AMPA receptor trafficking in natural and pathological aging. Front. Mol. Neurosci. 10:446. doi: 10.3389/fnmol.2017.00446
Keifer, J., and Zheng, Z. (2010). AMPA receptor trafficking and learning. Eur. J. Neurosci. 32, 269–277. doi: 10.1111/j.1460-9568.2010.07339.x
Keinänen, K., Wisden, W., Sommer, B., Werner, P., Herb, A., Verdoorn, T. A., et al. (1990). A family of AMPA-selective glutamate receptors. Science 249, 556–560. doi: 10.1126/science.2166337
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Klyubin, I., Walsh, D. M., Lemere, C. A., Cullen, W. K., Shankar, G. M., Betts, V., et al. (2005). Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 11, 556–561. doi: 10.1038/nm1234
Li, N., Li, Y., Li, L. J., Zhu, K., Zheng, Y., and Wang, X. M. (2019). Glutamate receptor delocalization in postsynaptic membrane and reduced hippocampal synaptic plasticity in the early stage of Alzheimer’s disease. Neural Regen. Res. 14, 1037–1045. doi: 10.4103/1673-5374.250625
Li, S., and Selkoe, D. J. (2020). A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Ab oligomers from Alzheimer brain. J. Neurochem. 154, 583–597. doi: 10.1111/jnc.15007
Luján, R., Aguado, C., Ciruela, F., Cózar, J., Kleindienst, D., de la Ossa, L., et al. (2018). Differential association of GABA B receptors with their effector ion channels in Purkinje cells. Brain Struct. Funct. 223, 1565–1587. doi: 10.1007/s00429-017-1568-y
Martín-Belmonte, A., Aguado, C., Alfaro-Ruíz, R., Moreno-Martínez, A. E., de la Ossa, L., Martínez-Hernández, J., et al. (2019). Reduction in the neuronal surface of post- and pre-synaptic GABA B receptors in the hippocampus in a mouse model of Alzheimer’s disease. Brain Pathol. 30, 554–575. doi: 10.1111/bpa.12802
Martinez-Losa, M., Tracy, T. E., Ma, K., Verret, L., Clemente-Perez, A., Khan, A. S., et al. (2018). Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98, 75–89.e5. doi: 10.1016/j.neuron.2018.02.029
Masugi-Tokita, M., and Shigemoto, R. (2007). High-resolution quantitative visualization of glutamate and GABA receptors at central synapses. Curr. Opin. Neurobiol. 17, 387–393. doi: 10.1016/j.conb.2007.04.012
Masugi-Tokita, M., Tarusawa, E., Watanabe, M., Molnár, E., Fujimoto, K., and Shigemoto, R. (2007). Number and density of AMPA receptors in individual synapses in the rat cerebellum as revealed by SDS-digested freeze-fracture replica labeling. J. Neurosci. 27, 2135–2144. doi: 10.1523/JNEUROSCI.2861-06.2007
Matsuzaki, M., Ellis-Davies, G. C. R., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092. doi: 10.1038/nn736
Merino-Serrais, P., Knafo, S., Alonso-Nanclares, L., Fernaud-Espinosa, I., and Defelipe, J. (2011). Layer-specific alterations to CA1 dendritic spines in a mouse model of Alzheimer’s disease. Hippocampus 21, 1037–1044. doi: 10.1002/hipo.20861
Molnár, E. (2016). “Analysis of the expression profile and regional distribution of neurotransmitter receptors and ion channels in the central nervous system using histoblots,” in Receptor and Ion Channel Detection in the Brain, eds F. Ciruela and R. Luján (Berlin: Springer), 157–170. doi: 10.1007/978-1-4939-3064-7_12
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, 1–17. doi: 10.1101/cshperspect.a006338
Nusser, Z., Lujan, R., Laube, G., Roberts, J. D. B., Molnar, E., and Somogyi, P. (1998). Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21, 545–559. doi: 10.1016/S0896-6273(00)80565-6
Nyíri, G., Stephenson, F. A., Freund, T. F., and Somogyi, P. (2003). Large variability in synaptic N-methyl-D-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience 119, 347–363. doi: 10.1016/S0306-4522(03)00157-X
Palop, J. J., and Mucke, L. (2010). Amyloid-B -induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Palop, J. J., and Mucke, L. (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792. doi: 10.1038/nrn.2016.141
Pellegrini-Giampietro, D. E., Bennett, M. V. L., and Zukin, R. S. (1994). Ampa/kainate receptor gene expression in normal and alzheimer’s disease hippocampus. Neuroscience 61, 41–49. doi: 10.1016/0306-4522(94)90058-2
Petralia, R. S., Esteban, J. A., Wang, Y. X., Partridge, J. G., Zhao, H. M., Wenthold, R. J., et al. (1999). Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat. Neurosci. 2, 31–36. doi: 10.1038/4532
Petralia, R. S., and Wenthold, R. J. (1992). Light and electron immunocytochemical localization of AMPA-selective glutamate receptors in the rat brain. J. Comp. Neurol. 318, 329–354. doi: 10.1002/cne.903180309
Pickard, L., Noel, J., Henley, J. M., Collingridge, G. L., and Molnar, E. (2000). Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J. Neurosci. 20, 7922–7931. doi: 10.1523/jneurosci.20-21-07922.2000
Racca, C., Stephenson, F. A., Streit, P., Roberts, J. D. B., and Somogyi, P. (2000). NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J. Neurosci. 20, 2512–2522. doi: 10.1523/jneurosci.20-07-02512.2000
Sato, K., Kiyama, H., and Tohyama, M. (1993). The differential expression patterns of messenger RNAs encoding non-N-methyl-d-aspartate glutamate receptor subunits (GluR1-4) in the rat brain. Neuroscience 52, 515–539. doi: 10.1016/0306-4522(93)90403-3
Schikorski, T., and Stevens, C. F. (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 17, 5858–5867.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shepherd, G. M. G., and Harris, K. M. (1998). Three-dimensional structure and composition of CA3→CA1 Axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 18, 8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998
Siegel, S. J., Brose, N., Janssen, W. G., Gasic, G. P., Jahn, R., Heinemann, S. F., et al. (1994). Regional, cellular, and ultrastructural distribution of N-methyl-D- aspartate receptor subunit 1 in monkey hippocampus. Proc. Natl. Acad. Sci. U.S.A. 91, 564–568. doi: 10.1073/pnas.91.2.564
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 8, 1051–1058. doi: 10.1038/nn1503
Spruston, N. (2008). Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221. doi: 10.1038/nrn2286
Szabadits, E., Cserép, C., Szonyi, A., Fukazawa, Y., Shigemoto, R., Watanabe, M., et al. (2011). NMDA receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J. Neurosci. 31, 5893–5904. doi: 10.1523/JNEUROSCI.5938-10.2011
Takumi, Y., Ramírez-León, V., Laake, P., Rinvik, E., and Ottersen, O. P. (1999). Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat. Neurosci. 2, 618–624. doi: 10.1038/10172
Tanaka, J. I., Matsuzaki, M., Tarusawa, E., Momiyama, A., Molnar, E., Kasai, H., et al. (2005). Number and density of AMPA receptors in single synapses in immature cerebellum. J. Neurosci. 25, 799–807. doi: 10.1523/JNEUROSCI.4256-04.2005
Tarusawa, E., Matsui, K., Budisantoso, T., Molnár, E., Watanabe, M., Matsui, M., et al. (2009). Input-specific intrasynaptic arrangements of ionotropic glutamate receptors and their impact on postsynaptic responses. J. Neurosci. 29, 12896–12908. doi: 10.1523/JNEUROSCI.6160-08.2009
Thorns, V., Mallory, M., Hansen, L., and Masliah, E. (1997). Alterations in glutamate receptor 2/3 subunits and amyloid precursor protein expression during the course of Alzheimer’s disease and Lewy body variant. Acta Neuropathol. 94, 539–548. doi: 10.1007/s004010050748
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Trinchese, F., Liu, S., Battaglia, F., Walter, S., Mathews, P. M., and Arancio, O. (2004). Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814. doi: 10.1002/ana.20101
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M. I., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Villette, V., and Dutar, P. (2017). GABAergic Microcircuits in Alzheimer’s Disease Models. Curr. Alzheimer Res. 14, 30–39. doi: 10.2174/1567205013666160819125757
Wakabayashi, K., Narisawa-Saito, M., Iwakura, Y., Arai, T., Ikeda, K., Takahashi, H., et al. (1999). Phenotypic down-regulation of glutamate receptor subunit GluR1 in Alzheimer’s disease. Neurobiol. Aging 20, 287–295. doi: 10.1016/S0197-4580(99)00035-4
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wenthold, R. J., Yokotani, N., Doi, K., and Wada, K. (1992). Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies: evidence for a hetero-oligomeric structure in rat brain. J. Biol. Chem. 267, 501–507.
West, M. J., Kawas, C. H., Martin, L. J., and Troncoso, J. C. (2000). The CA1 Region of the Human Hippocampus Is a Hot Spot in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 908, 255–259. doi: 10.1111/j.1749-6632.2000.tb06652.x
West, M. J., Kawas, C. H., Stewart, W. F., Rudow, G. L., and Troncoso, J. C. (2004). Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol. Aging 25, 1205–1212. doi: 10.1016/j.neurobiolaging.2003.12.005
Whitcomb, D. J., Hogg, E. L., Regan, P., Piers, T., Narayan, P., Whitehead, G., et al. (2015). Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 5:10934. doi: 10.1038/srep10934
Keywords: AD mouse model, hippocampus, electron microscopy, AMPA receptors, Alzheimer’s disease
Citation: Martín-Belmonte A, Aguado C, Alfaro-Ruíz R, Itakura M, Moreno-Martínez AE, de la Ossa L, Molnár E, Fukazawa Y and Luján R (2020) Age-Dependent Shift of AMPA Receptors From Synapses to Intracellular Compartments in Alzheimer’s Disease: Immunocytochemical Analysis of the CA1 Hippocampal Region in APP/PS1 Transgenic Mouse Model. Front. Aging Neurosci. 12:577996. doi: 10.3389/fnagi.2020.577996
Received: 30 June 2020; Accepted: 14 September 2020;
Published: 06 October 2020.
Edited by:
Mercè Pallàs, University of Barcelona, SpainReviewed by:
Richard J. Weinberg, University of North Carolina at Chapel Hill, United StatesCopyright © 2020 Martín-Belmonte, Aguado, Alfaro-Ruíz, Itakura, Moreno-Martínez, de la Ossa, Molnár, Fukazawa and Luján. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Luján, UmFmYWVsLkx1amFuQHVjbG0uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.