 Gilbert Ho1
Gilbert Ho1 Yoshiki Takamatsu2
Yoshiki Takamatsu2 Ryoko Wada2
Ryoko Wada2 Shuei Sugama3
Shuei Sugama3 Masaaki Waragai1
Masaaki Waragai1 Takato Takenouchi4Eliezer Masliah5
Takato Takenouchi4Eliezer Masliah5 Makoto Hashimoto2*
Makoto Hashimoto2*- 1PCND Neuroscience Research Institute, Poway, CA, United States
- 2Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
- 3Department of Physiology, Nippon Medical School, Tokyo, Japan
- 4Institute of Agrobiological Sciences, National Agriculture and Food Research Organization, Tsukuba, Japan
- 5Division of Neurosciences, National Institute on Aging, National Institutes of Health, Bethesda, MD, United States
Type 2 diabetes mellitus (T2DM) has been clearlylinked to oxidative stress and amylin amyloidosis in pancreatic β-cells. Yet despite extensive investigation, the biological significance of this is not fully understood. Recently, we proposed that Alzheimer’s disease (AD)-relevant amyloidogenic proteins (APs), such as amyloid-β (Aβ) and tau, might be involved in evolvability against diverse stressors in the brain. Given the analogous cellular stress environments shared by both T2DM and AD, the objective of this study is to explore T2DM pathogenesis from the viewpoint of amyloidogenic evolvability. Similar to AD-related APs, protofibrillar amylin might confer resistance against the multiple stressors in β-cells and be transmitted to offspring to deliver stress information, in the absence of which, type 1 DM (T1DM) in offspring might develop. On the contrary, T2DM may be manifested through an antagonistic pleiotropy mechanism during parental aging. Such evolvability-associated processes might be affected by parental diabetic conditions, including T1DM and T2DM. Furthermore, the T2DM-mediated increase in AD risk during aging might be attributed to an interaction of amylin with AD-related APs through evolvability, in which amylin protofibrillar formation presumably caused by adiponectin (APN) resistance could increase protofibril formation of AD-related APs in evolvability and subsequently lead to T2DM promotion of AD through antagonistic pleiotropy in aging. This suggests that targeting APN combined with an anti-T2DM agent might be therapeutic against neurodegeneration. Collectively, T1DM and T2DM might be linked through amylin evolvability, and a better understanding of amyloidogenic evolvability might also reveal clues to therapeutic interventions for AD comorbid with T2DM.
Introduction
Accumulating evidence suggests that type 2 diabetes mellitus (T2DM) may drive the pathogenesis of various nervous system disorders, including ischemia, depression, and neurodegenerative conditions (Atlantis et al., 2014; Takamatsu et al., 2017). In Alzheimer’s disease (AD), T2DM may increase the risk of mild cognitive impairment and subsequent progression to dementia (Watts et al., 2013; Ng et al., 2016). Additionally, T2DM has been linked to other neurodegenerative disorders, including Parkinson’s disease (PD) and Huntington’s disease (HD; Aviles-Olmos et al., 2013; Montojo et al., 2017). Yet despite a plethora of such observations, the precise mechanistic underpinning of the comorbidity between T2DM and neurodegeneration remains elusive.
Our recent work suggests that the evolvability of amyloidogenic proteins (APs) relevant to neurodegeneration, such as β-amyloid (Aβ) in AD, α-synuclein (αS) in PD, and huntingtin in HD, might be physiologically important (Hashimoto et al., 2018a,b,c; Takamatsu et al., 2018). More specifically, APs might act as vehicles to transgenerationally deliver information regarding diverse biological stressors to cope with such forthcoming stressors in offspring’s brain (Hashimoto et al., 2018a). On the contrary, evolvability might also cause aging-associated neurodegenerative disease through the antagonistic pleiotropy mechanism during the course of parental aging (Hashimoto et al., 2018b). Since evolvability is critical for reproduction, neurodegenerative diseases in aging have evaded “weeding out” by natural selection during evolution.
Given analogous pathology between T2DM and neurodegenerative disorders in terms of amyloidosis-associated stressors (Abedini and Schmidt, 2013; Singh et al., 2015), common mechanisms might form the basis for both groups of disorders. In this context, the main objective of this article is to discuss the mechanisms by which T2DM might increase AD risk from the viewpoint of amyloidogenic evolvability. We hypothesize that amylin protofibrils might confer resistance against multiple stressors in β-cells in the pancreas, which might be transgenerationally transmitted via the germ line to offspring, the absence of which might lead to type 1 DM (T1DM), and where T2DM in parents might promote amylin evolvability, while T1DM in parents might increase T1DM in offspring. Furthermore, we assume that the comorbidity of AD and T2DM in aging might be attributed to cross seeding of APs, which may stimulate evolvability. Yet the increased risk of AD from T2DM during aging might be due to upstream amylin activity relative to other more prominent APs, such as Aβ and tau, in evolvability, where the adiponectin (APN) paradox might be important. Collectively, DM and AD might be connected through amyloid evolvability, which might shed light on novel therapeutic strategies against such comorbid disorders.
Evolvability of Amyloidogenic Proteins Relevant to Type 2 Diabetes Mellitus
Since T2DM is associated with increased biological stressors and formation of amyloid-like fibrils of amylin in pancreatic β cells, we propose that amylin evolvability might be involved in the pathogenesis of T2DM.
Amylin and Insulin Are Amyloidogenic Proteins
Amylin, also called islet amyloid polypeptide, is a 37-residue peptide that is co-secreted with insulin by pancreatic β-cells (Mietlicki-Baase, 2016). The normal functions of amylin include inhibition of glucagon secretion and reduction of the gastric emptying rate, effects that contribute to the maintenance of postprandial glucose homeostasis (Mietlicki-Baase, 2016). Amylin is prone to aggregate, leading to the formation of amyloid fibrils with characteristic β-sheet structures (Jaikaran and Clark, 2001). Consistent with this, amylin is the major component of the islet amyloid found in the pancreas in T2DM, suggesting that amylin may play a significant role in the pathogenesis of T2DM (Luca et al., 2007). In addition, insulin, a 51-residue protein that is composed of a dimer of an A- and a B-chain linked by disulfide bonds (Brange and Langkjoer, 1993), also forms amyloid fibrils. Insulin, and its related peptide, insulin-like growth factor-1 (IGF-1), are essential to multiple physiological processes, including cell proliferation, differentiation, and survival, in addition to the regulation of energy storage and glucose metabolism (Stewart and Rotwein, 1996; Dadon et al., 2012). In contrast to amylin, the biological role of insulin fibril formation remains unclear. Therefore, our article will henceforth refer exclusively to amylin in this context.
Evolvability of Amylin
Because APs such as amylin are intrinsically disordered proteins composed of heterogeneous structures (Moore et al., 2018), APs might be involved in resistance against diverse stressors (Takamatsu et al., 2018). In addition, amylin toxicity in pancreatic β-cells has been well described in the pathogenesis of T2DM (Bharadwaj et al., 2017). Collectively, this may be comparable with the hormesis conferred by amyloidogenic evolvability (Hashimoto et al., 2018a). Because evolvability is defined as the capacity of a population of organisms to generate not only genetic diversity, but also adaptive genetic diversity, often overriding natural selection (Kirschner and Gerhart, 1998), both hormesis and heredity are critical steps in evolvability. In humans, AP protofibrils that encode information regarding biological and environmental stressors might be transmitted to offspring via germ lines (Hashimoto et al., 2018a). Although monomers of APs are unstable due to their intrinsically disordered nature (Takamatsu et al., 2018), formation of protofibrils might confer greater stability, which might provide an advantage for the transgenerational transmission of APs to offspring (Takamatsu et al., 2018). The presence of circulatory amylin is consistent with the transgenerational transmission via germ cells, although little is known about the expression of amylin in gonadal tissues. Yet given that amyloid fibrils are abundantly expressed in the semen (Roan et al., 2017), further exploration of this is definitely warranted. Taken together, our concept proposes that amylin protofibrils may convey stress information from parental pancreatic β-cells to an offspring’s pancreas. By virtue of amylin evolvability, offspring can then better cope with the forthcoming stressors, reducing the risk of developing T1DM (Figure 1A). Indeed, it was previously shown that amylin-knockout mice developed a more severe form of alloxan-induced diabetes (Mulder et al., 2000), which is consistent with our hypothesis that amylin protofibrils might be involved in evolvability of β-cells in the pancreas.
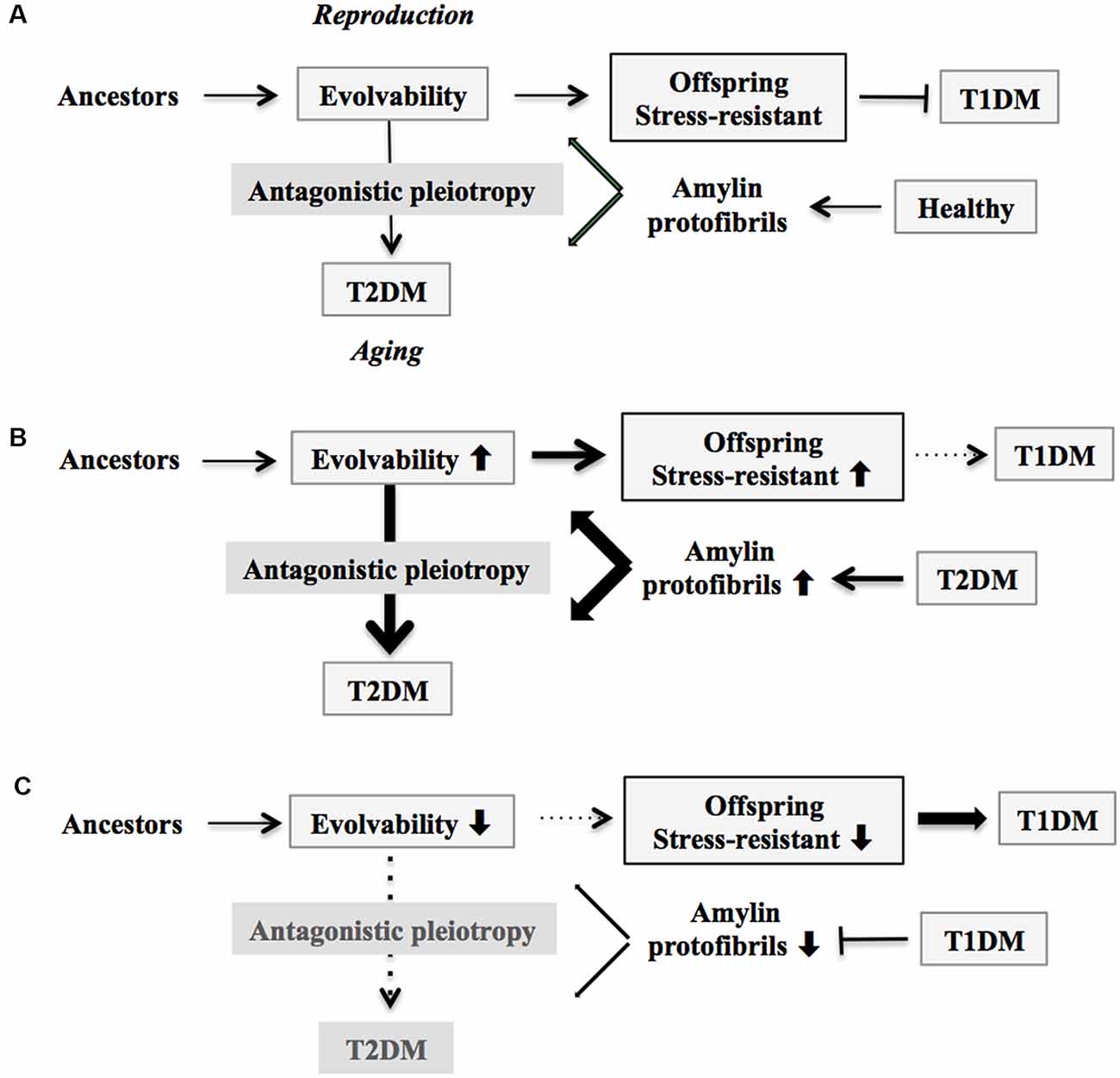
Figure 1. Schematic of disease manifestation caused by alterations in amylin evolvability. (A) Amylin protofibrils might confer resistance against multiple stressors in parental β-cells in the pancreas under the healthy conditions and are transgenerationally transmitted to offspring during reproduction to deliver the stress information. By virtue of this, the β-cells in the pancreas in offspring can cope with forthcoming multiple stressors in β-cells in the pancreas. Thus, amylin evolvability is an epigenetic phenomenon that is beneficial in evolution. However, amylin protofibrils might lead to type 2 diabetes mellitus (T2DM) during parental aging through the antagonistic pleiotropy mechanism. (B) Increased evolvability of amylin protofibrils by various causes, such as T2DM (thick bold line), may result in an efficient delivery of information about stressors for offspring, leading to reduced frequency of T1DM in offspring (thin dot line) and increased frequency of T2DM in parents (thick bold line). (C) In contrast, inefficient delivery of the information of stresses due to decrease of amylin evolvability caused by type 1 diabetes mellitus (T1DM) in parents may result in the increased frequency of T1DM in offspring (thick bold line) and the decreased frequency of T2DM in parents (thin dot line).
On the other hand, T2DM might manifest in later life through the mechanism of antagonistic pleiotropy during parental aging (Figure 1A). Briefly, according to the antagonistic pleiotropy hypothesis, a prominent theory of aging proposed by G. C. Williams a half-century ago, certain genes whose functions are beneficial during reproductive stages may in turn exert adverse effects later in aging (Williams, 1957). Such a view may explain why T2DM, a primarily post-reproductive and biologically disadvantageous disorder, has emerged and persisted across evolution. Yet it is recognized that T2DM is a complex disorder further modulated by interaction of a combination of susceptible genes and perhaps lifestyle factors such as exercise and diet.
Connecting Type 1 Diabetes Mellitus and Type 2 Diabetes Mellitus Through Amyloidogenic Evolvability
Although it is generally believed that T1DM occurs during youth and may be etiologically distinct from T2DM, which emerges during later adulthood, we alternatively propose that the two types of DM might actually be closely linked through amyloidogenic evolvability.
The Conventional View of Type 1 Diabetes Mellitus and Type 2 Diabetes Mellitus Pathogenesis
In T1DM, which accounts for 5–10% of all diabetes cases and occurs in approximately 0.3% of young individuals, amassed evidence suggests that various etiologies, such as autoimmune dysfunction and viral infection, might be pathogenetically involved (Daneman, 2006; Menke et al., 2013). Furthermore, recent genome-wide association studies have shown that the major susceptibility for the T1DM locus maps to the HLA class II genes at 6p21, which accounts for up to 30~50% of genetic risk of T1DM, with minor contributions to disease risk from several other non-MHC loci (Steck and Rewers, 2011). On the contrary, the more common T2DM, estimated to be ~7% of the general population, in contrast to early-onset T1DM, most often begins in those over the age of 65 years (although some early-onset T2DM cases are reported; Deshpande et al., 2008). Despite similar pathology in terms of dysfunctional pancreatic β cells, T1DM and T2DM are understood to be etiologically different entities.
Equilibrium Between Type 1 Diabetes Mellitus in Offspring and Parental Type 2 Diabetes Mellitus Through Amylin Evolvability
Based on our evolvability concept, stress information from parental/adult pancreatic β-cells could be delivered to their offspring through amylin protofibrils. The resulting pancreatic β-cells in offspring should therefore become more resistant against such stressors (Figure 1A).
According to our view, the greater the stress information from parental pancreatic β-cells that is delivered through amylin protofibrils to offspring through increased evolvability, the more resistant the pancreatic β-cells in offspring become against stressors, and the less prone they are to developing T1DM. This could result from various factors, including amylin missense mutation (Akter et al., 2016), T2DM in both parents, and maternal gestational diabetes mellitus (GDM). Consistent with our view, it was previously shown that parental history of T2DM is associated with a later onset of T1DM, the metabolic syndrome, and a metabolic profile related to insulin resistance (Thorn et al., 2009). Because of the action of antagonistic pleiotropy, T2DM is expressed more prominently during aging (Figure 1B).
Yet if the parents are afflicted with T1DM, then reduced evolvability associated with the down-regulation of amylin protofibrils might result in the delivery of less stress information to offspring, and as a consequence, T1DM risk may dramatically increase in offspring (Figure 1C). Supporting this, T1DM is preferentially transmitted from parents to offspring, although the mechanism of gender difference is obscure (Guo and Tuomilehto, 2002). Of interest, the inverse relationship between T1DM in offspring and parental T2DM is reminiscent of the concept of transgenerational equilibrium previously described between parental AD and schizophrenia in offspring related to amyloid evolvability (Takamatsu et al., 2019), raising a possibility that certain chronic disorders in offspring and their aging-associated amyloidogenic disorder counterparts in parents may exist in an inverse relationship through amyloidogenic evolvability.
Are There any Roles for Gestational Diabetes Mellitus in Amylin Evolvability?
The third diabetic subtype, GDM, occurs among pregnant women without a previous history of DM (Kampmann et al., 2015). GDM, with a prevalence of ~39% of pregnancies, correlates with increased maternal obesity (Carpenter, 2007; Egan et al., 2017). GDM shows a complex etiology, with genetic and environmental factors implicated across mechanistic and epidemiological studies. Compared with T1DM and T2DM, there have been fewer studies on the etiological basis of GDM, because of its transient nature and resolution upon delivery of the infant. Thus, a possible etiologic role for GDM might also relate to evolvability. Given that GDM is associated with increased insulin resistance (Catalano, 2014), GDM might be involved in transmitting amylin protofibrils encoding stress information from parental pancreatic β-cells to offspring in order to mitigate the risk of T1DM in offspring. On the other hand, symptomatic T2DM might emerge later during parental aging. If this notion can be verified, GDM may become central to linking T1DM in offspring and parental T2DM through amylin evolvability. Yet, presently, there is no evidence that amylin can cross the transplacental barrier, which is generally believed to be permeable to only small molecules (Berveiller et al., 2016). Therefore, it would be fair to conclude that GDM might be unlikely to promote amylin evolvability. As will be described later for T2DM, GDM might represent an antagonistic pleiotropy of amylin evolvability. Consistently, after the occurrence of GDM, there is a higher likelihood of developing subsequent maternal T2DM, as well as possible abnormal cardiometabolic phenotypes in offspring (Carpenter, 2007; Kawasaki et al., 2018).
Moreover, pregnant women often experience memory dysfunction through the course of their pregnancy (John et al., 2018), which is generally attributed to elevated hormone levels affecting the brain, although the precise mechanism is unknown. Similar to GDM, gestational dementia might be relevant to amyloid evolvability, perhaps involving Aβ and tau. To the best of our knowledge, limited information is currently available regarding changes in brain APs during pregnancy.
Comorbidity of Alzheimer’s Disease and Type 2 Diabetes Mellitus
Supposing that both T2DM and AD are antagonistic phenomena derived from amyloidogenic evolvability during reproduction, it follows that comorbidity of T2DM with AD in aging might be attributed to the synergistic interaction of amylin with APs, including Aβ and tau, in regulating evolvability during reproduction.
The Conventional View of the Comorbidity of Alzheimer’s Disease and Type 2 Diabetes Mellitus
With regard to the mechanisms underlying increased neurodegeneration related to T2DM, numerous studies have shown that both T2DM and AD are associated with various common pathological features, including impaired insulin resistance, endoplasmic reticulum stress, oxidative stress, protein aggregation, inflammation and altered gene expression (Vannuvel et al., 2013; Hokama et al., 2014; Singhal et al., 2014; Rosales-Corral et al., 2015). Furthermore, it was described that vascular dysfunction caused by T2DM might be relevant to AD (Wang et al., 2014). These results, however, were pathological observations, and do not necessarily explain the rationale for the emergence of these two comorbid aging-related disorders in evolution.
Comorbidity of Alzheimer’s Disease and Type 2 Diabetes Mellitus From the Viewpoint of Evolvability
In this regard, evolvability might then underlie these biological phenomena. Provided that both T2DM and AD might be antagonistic phenomena derived from evolvability, it follows that amylin and amyloid-β (Aβ) and/or tau might interact cooperatively rather than function independently. Given the prevailing concept of cross-seeding (CS) of APs (Morales et al., 2013), perhaps the CS of amylin and Aβ and/or tau may be more potent than either monomer to stimulate various aspects of evolvability, such as hormesis and transgenerational transmission. Yet CS of these APs may manifest later in life as comorbid T2DM and AD through antagonistic pleiotropy in parental aging. Indeed, amylin interacts with Aβ and tau in both the pancreas and hippocampus in the brain of diabetic patients with AD (Figure 2A; Jackson et al., 2013).
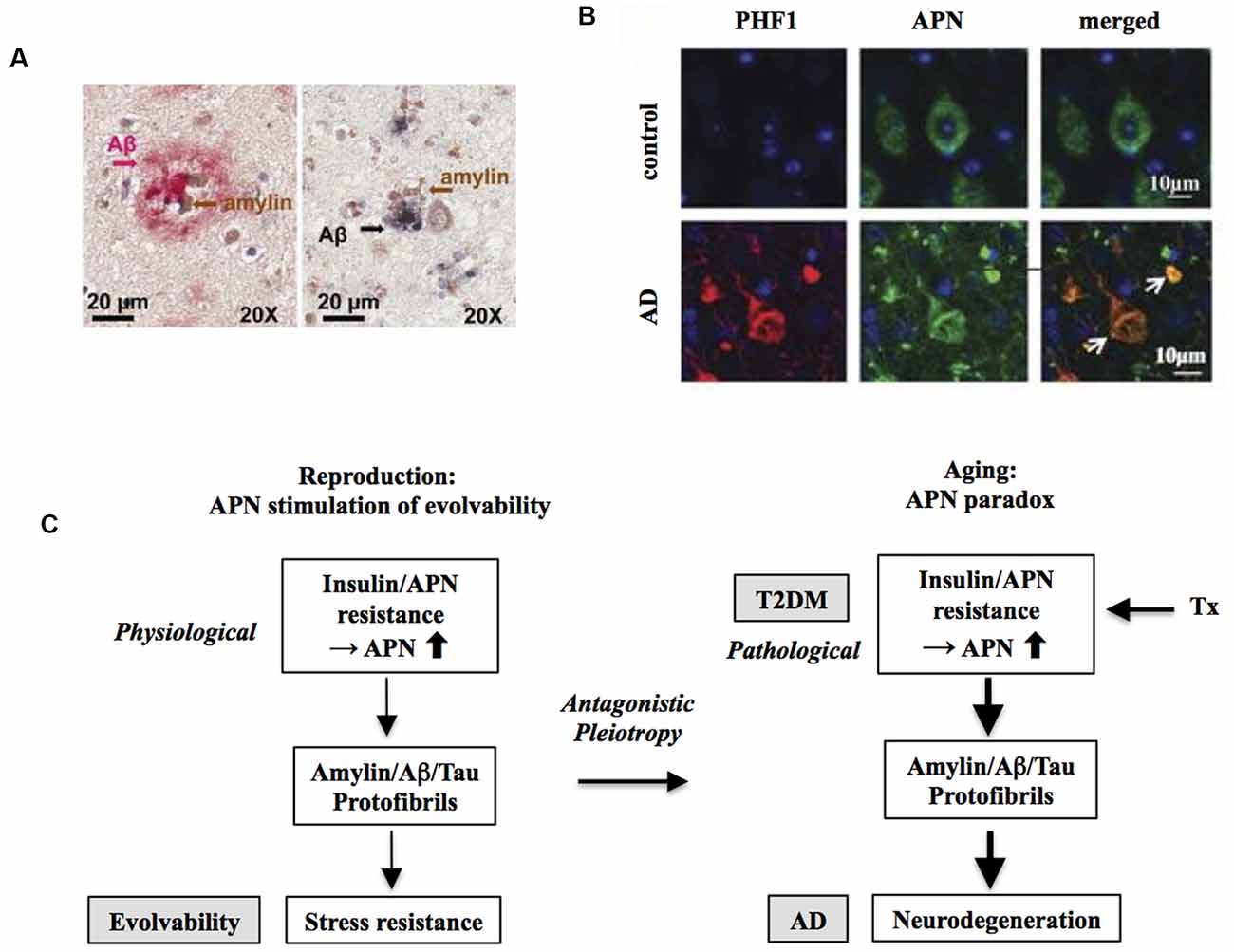
Figure 2. Increased risk of Alzheimer’s disease (AD) by T2DM as an antagonistic pleiotropy of evolvability. (A) Co-localization of amylin and amyloid-β (Aβ) is shown in brain parenchyma of AD patients with anti-amylin and anti-Aβ antibodies. The left panel shows a brain section from an AD patient without clinically demonstrated T2DM, while the right panel is derived from a diabetic patient with AD. Clusters of small amylin plaques adjacent to, or surrounded by larger Aβ deposits, are present. Modified from Jackson et al. (2013) with permission. (B) Adiponectin (APN)-positive neurofibrillary tangles (NFTs) in AD brain. Confocal laser scanning microscopy of double immunofluorescence of control and AD brains using anti-tau antibody PHF (lower) and anti-APN antibody showed that APN co-localized with tau in AD. White arrows indicate NFTs identified by the presence of PHF-1 immunoreactive abnormal fibrous inclusions, which were within the perikaryal cytoplasm of neurons. Scale bar = 10 μm. Modified from Waragai et al. (2016) with permission. (C) Schematic diagram of the mechanism underlying the APN paradox in T2DM-mediated increased AD risk. Presumably, amylin protofibrillar formation caused by APN resistance could up-regulate AP protofibrils in evolvability, which might manifest as T2DM stimulation of AD as the antagonistic pleiotropy in aging.
Importance of Common Modifiers of Alzheimer’s Disease and Type 2 Diabetes Mellitus From the Viewpoint of Evolvability
Notably, abnormally high levels of D-ribose have been observed in both T1DM (Yu et al., 2019) and T2DM (Su and He, 2014). Since administration of D-ribose induces the yield of Aβ and hyper-phosphorylated tau in the brain (Wu et al., 2015, 2019; Li et al., 2020), D-ribose might be also important for induction of AD. Thus, D-ribose might affect evolvability of amylin and Aβ, the dysmetabolism of which might lead to manifestation as comorbidity of AD and T2DM in aging. Indeed, it is possible that there might be many factors/stressors that may be attributed to the comorbidity of AD and T2DM. These include formaldehyde and amylin glycation.
Comorbidity of Alzheimer’s Disease With Other Metabolic Disorders
Considering that AD is frequently comorbid with many other types of aging-associated diseases, similar mechanisms might be applied to the comorbidity of AD with these other conditions. Indeed, an increasing prevalence of associated metabolic disorders, including obesity, hypertension, hyperlipidemia, and atherosclerosis, is associated with an expanding adult and elderly population worldwide (Takamatsu et al., 2017). One may argue, however, that metabolic disorders other than T2DM are not always associated with amyloid fibril formation yet frequently promote neurodegeneration (Pugazhenthi, 2017). In this regard, evolvability of APs, including Aβ and tau, might be increased through mechanisms other than aggregative protein–protein interactions. For instance, cholesterol, which may play a central role in dyslipidemia, might stimulate misfolding and aggregation of APs in evolvability as well as in neurodegeneration and T2DM (Singh et al., 2015). Also, catecholamines possibly related to essential hypertension might be relevant (Goldstein, 1983). Aβ was shown to undergo regulated co-secretion with neuropeptide and catecholamine neurotransmitters (Toneff et al., 2013), and catecholamines were also shown to stimulate protein deposition in AD and PD (Bharath and Andersen, 2004). Thus, catecholamines might be involved in evolvability, which may manifest as essential hypertension as an antagonistic pleiotropy relationship in aging. Collectively, it is possible that the comorbidity of AD with metabolic disorders might represent antagonistic pleiotropy attributed to increased evolvability due to the CS of APs and other factors.
Role of Adiponectin Paradox Linking Type 2 Diabetes Mellitus and Alzheimer’s Disease
The concept of the CS of APs suggests that the pathogenesis of T2DM might be equivalent to that in AD but fails to explain how T2DM could be upstream of AD. Given that AD risk might be increased by T2DM, perhaps the mechanistic action of amylin in evolvability may be situated upstream to APs, including Aβ and tau.
In this regard, we hypothesize that APN might be involved. APN is a multifunctional adipokine that is important in insulin receptor signaling sensitization and an anti-inflammatory role (Waragai et al., 2017). Although APN is generally protective, it has been well characterized that APN might be critically involved in promoting aging-associated chronic diseases, such as chronic heart failure and chronic kidney disease, the so-called APN paradox (Menon et al., 2006; Kizer, 2014). Notably, a recent study suggests that the APN paradox is also observed in the nervous system (Waragai et al., 2020). APN is neuroprotective and mitigates neurodegeneration in both cellular and animal experimental systems. Nonetheless, in cohort studies, it was shown that hyperadiponectinemia correlates well with neuropathological features, such as amyloidosis and cognitive deficits, suggesting that APN paradox might be involved in AD pathogenesis (Wennberg et al., 2016; Waragai et al., 2020). Consistent with this notion, it was shown that APN associated with phospho-tau in the AD brain, suggesting that APN might be involved in the tangle formation (Figure 2B).
Given that the APN paradox might be critical in promoting AD (Waragai et al., 2020), it is possible that the APN paradox in aging might be an antagonistic pleiotropy phenomenon derived from evolvability during reproduction (Waragai et al., 2020). In this context, APN resistance/insulin resistance might be a physiologically caused by fibrillar amylin in evolvability, which might later be manifest as T2DM stimulation of AD (Figure 2C). Consistently, insulin resistance was previously shown in cavefish as an adaptation to a nutrient-limited environment (Riddle et al., 2018), indicating that insulin resistance can be beneficially activated under the physiological conditions. Notably, it was recently described that large-scale proteomic analysis of AD brain and cerebrospinal fluid revealed early changes in energy metabolism associated with glial activation (Johnson et al., 2020), which is consistent with involvement of the APN paradox in the pathogenesis of AD (Waragai et al., 2020).
Similar to AD, a recent prospective cohort study showed that higher serum APN concentrations were observed in incident cancers and are associated with cancer-related deaths in T2DM, suggesting that the APN paradox might be significant in cancer related to T2DM (Lee et al., 2020).
Implications for Therapeutic Interventions
In view of the possible involvement of the APN paradox in linking neurodegeneration to metabolic disease, this may account for the pathological positioning of T2DM upstream of AD in aging, suggesting that therapeutic targeting of APN might be a viable strategy.
Targeting Adiponectin Expression
For this purpose, suppressing APN expression by various methods, such as antisense oligonucleotides against APN mRNA (Rinaldi and Wood, 2018) and immunotherapy against the APN protein (Lannfelt et al., 2014), might effectively decrease formation of APs protofibrils, leading to suppression of APN paradox and AD (Figure 2C). However, the majority of the previous studies describe that the potential of APN activity for therapeutic purposes based on the presumption that APN is protective. Therefore, it is necessary to bear in mind the possibility of a therapeutic “trade-off” by suppression of APN expression.
Notably, administration of D-ribose leads to an increase of visceral triglycerides in mice (Chen et al., 2019b); and since high levels of D-ribose (Su et al., 2013; Chen et al., 2019a) and high HbA1c (Chen et al., 2017) are associated with diabetes, this raises a possibility that D-ribose could be utilized as a biomarker to assess the therapeutic efficiency.
Anti-Diabetes Mellitus and Metabolic Syndrome
It is expected that combining APN with treatment against T2DM and other related metabolic disorders might enhance the therapeutic efficiency of targeting of APN. Because of an increased risk of neurodegenerative disorders related to T2DM, previous studies have strongly suggested that treatment of T2DM might be beneficial for and repurposed to therapy of neurodegenerative disorders. Indeed, a recent phase II clinical trial of glucagon-like peptide-1 (GLP-1)/incretin for PD appeared promising (Hölscher, 2012; Athauda et al., 2017), prompting initiation of a subsequent clinical trial of GLP-1/incretin in AD (Gejl et al., 2016). Although metformin has long been used to treat T2DM, there is growing evidence for the benefits of metformin to counteract age-related diseases such as cancer, cardiovascular disease, and neurodegenerative diseases (Rotermund et al., 2018). Moreover, studies are in progress to assess the therapeutic potential of other anti-T2DM molecules, such as DPP-4 inhibitors (Kosaraju et al., 2017) and APN (Sekiyama et al., 2014; Waragai et al., 2018). In relation to the current state of neurodegenerative therapy in relation to T2DM, our current view regarding evolvability may offer several novel insights.
Recently, chronic hypertension has been suggested as one of the largest modifiable risk factors for developing AD (Marfany et al., 2018). Indeed, several epidemiological studies reveal that β-blocker treatment reduces the prevalence of AD in patients suffering from hypertension (Yu et al., 2011). Since it is possible that Aβ evolvability might be promoted not only by amylin protofibrils but also by other molecules, such as cholesterol and catecholamines, it follows that disease-modifying therapies targeted against other metabolic syndrome disorders in addition to T2DM, especially anti-hypertension agents, might increase overall treatment efficacy.
Concluding Remarks
In the present discussion, the concept of amylin evolvability was shown to provide novel insights into the T2DM and related diseases, which are currently elusive. First, we argue that T1DM and T2DM, etiologically distinct types of DM, might be linked through amylin evolvability. Furthermore, the comorbidity of AD and T2DM in aging might be an antagonistic pleiotropy phenomenon derived from evolvability regulated by AD-relevant APs, including Aβ, tau, and amylin. Importantly, it was suggested that APN resistance might stimulate amylin protofibrils, leading to Aβ protofibrillar formation in evolvability, which might manifest as an increased AD risk driven by T2DM through an antagonistic pleiotropy mechanism in aging. Taken together, a better understanding of the mechanism of evolvability may shed light on novel evolvability-based therapeutic strategies.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
MH conceived the study. MH and GH wrote the article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer NK declared a past co-authorship with one of the authors MW to the handling editor.
Acknowledgments
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago).
References
Abedini, A., and Schmidt, A. M. (2013). Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 587, 1119–1127. doi: 10.1016/j.febslet.2013.01.017
Akter, R., Cao, P., Noor, H., Ridgway, Z., Tu, L. H., Wang, H., et al. (2016). Islet amyloid polypeptide: structure, function, and pathophysiology. J. Diabetes Res. 2016:2798269. doi: 10.2210/pdb5e61/pdb
Athauda, D., Maclagan, K., Skene, S. S., Bajwa-Joseph, M., Letchford, D., Chowdhury, K., et al. (2017). Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. doi: 10.1016/S0140-6736(17)31585-4
Atlantis, E., Fahey, P., and Foster, J. (2014). Collaborative care for comorbid depression and diabetes: a systematic review and meta-analysis. BMJ Open 4:e004706. doi: 10.1136/bmjopen-2013-004706
Aviles-Olmos, I., Limousin, P., Lees, A., and Foltynie, T. (2013). Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 136, 374–384. doi: 10.1093/brain/aws009
Berveiller, P., Marty, O., Vialard, F., and Mir, O. (2016). Use of anticancer agents in gynecological oncology during pregnancy: a systematic review of maternal pharmacokinetics and transplacental transfer. Expert Opin. Drug Metab. Toxicol. 12, 523–531. doi: 10.1517/17425255.2016.1167187
Bharadwaj, P., Wijesekara, N., Liyanapathirana, M., Newsholme, P., Ittner, L., Fraser, P., et al. (2017). The link between type 2 diabetes and neurodegeneration: roles for amyloid-β, amylin and tau proteins. J. Alzheimers Dis. 59, 421–432. doi: 10.3233/jad-161192
Bharath, S., and Andersen, J. K. (2004). Catecholamines and protein deposition in Parkinson’s and Alzheimer’s disease: old medicine, new targets. Rejuvenation Res. 7, 92–94. doi: 10.1089/1549168041553071
Brange, J., and Langkjoer, L. (1993). Insulin structure and stability. Pharm. Biotechnol. 5, 315–350. doi: 10.1007/978-1-4899-1236-7_11
Carpenter, M. W. (2007). Gestational diabetes, pregnancy hypertension, and late vascular disease. Diabetes Care 30, S246–S250. doi: 10.2337/dc07-s224
Catalano, P. M. (2014). Trying to understand gestational diabetes. Diabetic Med. 31, 273–281. doi: 10.1111/dme.12381
Chen, X., Su, T., Chen, Y., He, Y., Liu, Y., Xu, Y., et al. (2017). d-Ribose as a contributor to glycated haemoglobin. EBioMedicine 25, 143–153. doi: 10.1016/j.ebiom.2017.10.001
Chen, Y., Yu, L., Wang, Y., Wei, Y., Xu, Y., He, T., et al. (2019a). d-Ribose contributes to the glycation of serum protein. Biochim. Biophys. Acta 1865, 2285–2292. doi: 10.1016/j.bbadis.2019.05.005
Chen, Y., Yu, L., Wei, Y., Long, Y., Xu, Y., He, T., et al. (2019b). d-Ribose increases triglyceride via upregulation of DGAT in the liver. Sci. China Life Sci. 62, 858–861. doi: 10.1007/s11427-019-9542-2
Dadon, D., Tornovsky-Babaey, S., Furth-Lavi, J., Ben-Zvi, D., Ziv, O., Schyr-Ben, R.-H., et al. (2012). Glucose metabolism: key endogenous regulator of beta-cell replication and survival. Diabetes Obes. Metab. 14, 101–108. doi: 10.1111/j.1463-1326.2012.01646.x
Deshpande, A. D., Harris-Hayes, M., and Schootman, M. (2008). Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 88, 1254–1264. doi: 10.2522/ptj.20080020
Egan, A. M., Vellinga, A., Harreiter, J., Simmons, D., Desoye, G., Corcoy, R., et al. (2017). Epidemiology of gestational diabetes mellitus according to IADPSG/WHO 2013 criteria among obese pregnant women in Europe. Diabetologia 60, 1913–1921. doi: 10.1007/s00125-017-4353-9
Gejl, M., Gjedde, A., Egefjord, L., Moller, A., Hansen, S. B., Vang, K., et al. (2016). In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 8:108. doi: 10.3389/fnagi.2016.00108
Goldstein, D. S. (1983). Plasma catecholamines and essential hypertension. An analytical review. Hypertension 5, 86–99. doi: 10.1161/01.hyp.5.1.86
Guo, S. W., and Tuomilehto, J. (2002). Preferential transmission of type 1 diabetes from parents to offspring: fact or artifact? Genet. Epidemiol. 23, 323–334. doi: 10.1002/gepi.10183
Hashimoto, M., Ho, G., Sugama, S., Takamatsu, Y., Shimizu, Y., Takenouchi, T., et al. (2018a). Evolvability of amyloidogenic proteins in human brain. J. Alzheimers Dis. 62, 73–83. doi: 10.3233/jad-170894
Hashimoto, M., Ho, G., Takamatsu, Y., Shimizu, Y., Sugama, S., Takenouchi, T., et al. (2018b). Evolvability and neurodegenerative disease: antagonistic pleiotropy phenomena derived from amyloid aggregates. J. Parkinsons Dis. 8, 405–408. doi: 10.3233/jpd-181365
Hashimoto, M., Ho, G., Takamatsu, Y., Wada, R., Sugama, S., Takenouchi, T., et al. (2018c). Possible role of the polyglutamine elongation in evolution of amyloid-related evolvability. J. Huntingtons Dis. 7, 297–307. doi: 10.3233/jhd-180309
Hokama, M., Oka, S., Leon, J., Ninomiya, T., Honda, H., Sasaki, K., et al. (2014). Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488. doi: 10.1093/cercor/bht101
Hölscher, C. (2012). Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs 26, 871–882. doi: 10.2165/11635890-000000000-00000
Jackson, K., Barisone, G. A., Diaz, E., Jin, L. W., DeCarli, C., and Despa, F. (2013). Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann. Neurol. 74, 517–526. doi: 10.1002/ana.23956
Jaikaran, E. T., and Clark, A. (2001). Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 1537, 179–203. doi: 10.1016/s0925-4439(01)00078-3
John, C. M., Mohamed Yusof, N. I. S., and Abdul Aziz, S. H. F. (2018). Mohd fauzi, maternal cognitive impairment associated with gestational diabetes mellitus—a review of potential contributing mechanisms. Int. J. Mol. Sci. 19:3894. doi: 10.3390/ijms19123894
Johnson, E. C. B., Dammer, E. B., Duong, D. M., Ping, L., Zhou, M., Yin, L., et al. (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780. doi: 10.1038/s41591-020-0815-6
Kampmann, U., Madsen, L. R., Skajaa, G. O., Iversen, D. S., Moeller, N., and Ovesen, P. (2015). Gestational diabetes: a clinical update. World J. Diabetes 6, 1065–1072. doi: 10.4239/wjd.v6.i8.1065
Kawasaki, M., Arata, N., Miyazaki, C., Mori, R., Kikuchi, T., Ogawa, Y., et al. (2018). Obesity and abnormal glucose tolerance in offspring of diabetic mothers: a systematic review and meta-analysis. PLoS One 13:e0190676. doi: 10.1371/journal.pone.0190676
Kirschner, M., and Gerhart, J. (1998). Evolvability. Proc. Natl. Acad. Sci. U S A 95, 8420–8427. doi: 10.1073/pnas.95.15.8420
Kizer, J. R. (2014). Adiponectin, cardiovascular disease and mortality: parsing the dual prognostic implications of a complex adipokine. Metabolism 63, 1079–1083. doi: 10.1016/j.metabol.2014.06.011
Kosaraju, J., Holsinger, R. M. D., Guo, L., and Tam, K. Y. (2017). Linagliptin, a dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3×Tg-AD mouse model of Alzheimer’s disease. Mol. Neurobiol. 54, 6074–6084. doi: 10.1007/s12035-016-0125-7
Lannfelt, L., Relkin, N. R., and Siemers, E. R. (2014). Amyloid-ss-directed immunotherapy for Alzheimer’s disease. J. Intern. Med. 275, 284–295. doi: 10.1111/joim.12168
Lee, C. H., Lui, D. T. W., Cheung, C. Y. Y., Fong, C. H. Y., Yuen, M. M. A., Chow, W. S., et al. (2020). Higher circulating adiponectin concentrations predict incident cancer in type 2 diabetes—the adiponectin paradox. J. Clin. Endocrinol. Metab. 105:dgaa075. doi: 10.1210/clinem/dgaa385
Li, Z.-H., He, X. P., Li, H., He, R. Q., and Hu, X. T. (2020). Age-associated changes in amyloid-β and formaldehyde concentrations in cerebrospinal fluid of rhesus monkeys. Zool. Res. 41, 444–448. doi: 10.24272/j.issn.2095-8137.2020.088
Luca, S., Yau, W. M., Leapman, R., and Tycko, R. (2007). Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry 46, 13505–13522. doi: 10.1021/bi701427q
Marfany, A., Sierra, C., Camafort, M., Domenech, M., and Coca, A. (2018). High blood pressure, Alzheimer disease and antihypertensive treatment. Panminerva Med. 60, 8–16. doi: 10.23736/S0031-0808.18.03360-8
Menke, A., Orchard, T. J., Imperatore, G., Bullard, K. M., Mayer-Davis, E., and Cowie, C. C. (2013). The prevalence of type 1 diabetes in the United States. Epidemiology 24, 773–774. doi: 10.1097/EDE.0b013e31829ef01a
Menon, V., Li, L., Wang, X., Greene, T., Balakrishnan, V., Madero, M., et al. (2006). Adiponectin and mortality in patients with chronic kidney disease. J. Am. Soc. Nephrol. 17, 2599–2606. doi: 10.1681/ASN.2006040331
Mietlicki-Baase, E. G. (2016). Amylin-mediated control of glycemia, energy balance and cognition. Physiol. Behav. 162, 130–140. doi: 10.1016/j.physbeh.2016.02.034
Montojo, M. T., Aganzo, M., and Gonzalez, N. (2017). Huntington’s disease and diabetes: chronological sequence of its association. J. Huntingtons Dis. 6, 179–188. doi: 10.3233/jhd-170253
Moore, S. J., Sonar, K., Bharadwaj, P., Deplazes, E., and Mancera, R. L. (2018). Characterisation of the structure and oligomerisation of islet amyloid polypeptides (IAPP): a review of molecular dynamics simulation studies. Molecules 23:2142. doi: 10.3390/molecules23092142
Morales, R., Moreno-Gonzalez, I., and Soto, C. (2013). Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 9:e1003537. doi: 10.1371/journal.ppat.1003537
Mulder, H., Gebre-Medhin, S., Betsholtz, C., Sundler, F., and Ahren, B. (2000). Islet amyloid polypeptide (amylin)-deficient mice develop a more severe form of alloxan-induced diabetes. Am. J. Physiol. Endocrinol. Metab. 278, E684–E691. doi: 10.1152/ajprenal.2000.278.4.F684
Ng, T. P., Feng, L., Nyunt, M. S., Feng, L., Gao, Q., Lim, M. L., et al. (2016). Metabolic syndrome and the risk of mild cognitive impairment and progression to dementia: follow-up of the singapore longitudinal ageing study cohort. JAMA Neurol. 73, 456–463. doi: 10.1001/jamaneurol.2015.4899
Pugazhenthi, S. (2017). Metabolic syndrome and the cellular phase of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 146, 243–258. doi: 10.1016/bs.pmbts.2016.12.016
Riddle, M. R., Aspiras, A. C., Gaudenz, K., Peuss, R., Sung, J. Y., Martineau, B., et al. (2018). Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 555, 647–651. doi: 10.1038/nature26136
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Roan, N. R., Sandi-Monroy, N., Kohgadai, N., Usmani, S. M., Hamil, K. G., Neidleman, J., et al. (2017). Semen amyloids participate in spermatozoa selection and clearance. eLife 6:e24888. doi: 10.7554/eLife.24888
Rosales-Corral, S., Tan, D. X., Manchester, L., and Reiter, R. J. (2015). Diabetes and Alzheimer disease, two overlapping pathologies with the same background: oxidative stress. Oxid. Med. Cell. Longev. 2015:985845. doi: 10.1155/2015/985845
Rotermund, C., Machetanz, G., and Fitzgerald, J. C. (2018). The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 9:400. doi: 10.3389/fendo.2018.00400
Sekiyama, K., Waragai, M., Akatsu, H., Sugama, S., Takenouchi, T., Takamatsu, Y., et al. (2014). Disease-modifying effect of adiponectin in model of alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 1, 479–489. doi: 10.1002/acn3.77
Singh, S., Trikha, S., Bhowmick, D. C., Sarkar, A. A., and Jeremic, A. M. (2015). Role of cholesterol and phospholipids in amylin misfolding, aggregation and etiology of islet amyloidosis. Adv. Exp. Med. Biol. 855, 95–116. doi: 10.1007/978-3-319-17344-3_4
Singhal, G., Jaehne, E. J., Corrigan, F., Toben, C., and Baune, B. T. (2014). Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front. Neurosci. 8:315. doi: 10.3389/fnins.2014.00315
Steck, A. K., and Rewers, M. J. (2011). Genetics of type 1 diabetes. Clin. Chem. 57, 176–285. doi: 10.1373/clinchem.2010.148221
Stewart, C. E., and Rotwein, P. (1996). Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol. Rev. 76, 1005–1026. doi: 10.1152/physrev.1996.76.4.1005
Su, T., and He, R. (2014). d-Ribose, an overlooked player in type 2 diabetes mellitus? Sci. China Life Sci. 57:361. doi: 10.1007/s11427-014-4614-5
Su, T., Xin, L., He, Y., Song, Y., Li, W., Wei, Y., et al. (2013). Abnormally high level of uric D-ribose for type 2 diabetics. Prog. Biochem. Biophys. 40, 816–825. doi: 10.1111/j.1558-5646.1957.tb02911.x
Takamatsu, Y., Fujita, M., Ho, G. J., Wada, R., Sugama, S., Takenouchi, T., et al. (2018). Motor and nonmotor symptoms of Parkinson’s disease: antagonistic pleiotropy phenomena derived from α-synuclein evolvability? Parkinsons Dis. 2018:5789424. doi: 10.1155/2018/5789424
Takamatsu, Y., Ho, G., Koike, W., Sugama, S., Takenouchi, T., Waragai, M., et al. (2017). Combined immunotherapy with “anti-insulin resistance” therapy as a novel therapeutic strategy against neurodegenerative diseases. NPJ Parkinsons Dis. 3:4. doi: 10.1038/s41531-016-0001-1
Takamatsu, Y., Ho, G., Waragai, M., Wada, R., Sugama, S., Takenouchi, T., et al. (2019). Transgenerational interaction of Alzheimer’s disease with schizophrenia through amyloid evolvability. J. Alzheimers Dis. 68, 473–481. doi: 10.3233/jad-180986
Thorn, L. M., Forsblom, C., Waden, J., Soderlund, J., Rosengard-Barlund, M., Saraheimo, M., et al. (2009). Effect of parental type 2 diabetes on offspring with type 1 diabetes. Diabetes Care 32, 63–68. doi: 10.2337/dc08-0472
Toneff, T., Funkelstein, L., Mosier, C., Abagyan, A., Ziegler, M., and Hook, V. (2013). Beta-amyloid peptides undergo regulated co-secretion with neuropeptide and catecholamine neurotransmitters. Peptides 46, 126–135. doi: 10.1016/j.peptides.2013.04.020
Vannuvel, K., Renard, P., Raes, M., and Arnould, T. (2013). Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 228, 1802–1818. doi: 10.1002/jcp.24360
Wang, F., Guo, X., Shen, X., Kream, R. M., Mantione, K. J., and Stefano, G. B. (2014). Vascular dysfunction associated with type 2 diabetes and Alzheimer’s disease: a potential etiological linkage. Med. Sci. Monit. Basic Res. 20, 118–129. doi: 10.12659/msmbr.891278
Waragai, M., Adame, A., Trinh, I., Sekiyama, K., Takamatsu, Y., Une, K., et al. (2016). Possible involvement of adiponectin, the anti-diabetes molecule, in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 52, 1453–1459. doi: 10.3233/jad-151116
Waragai, M., Ho, G., Takamatsu, Y., Sekiyama, K., Sugama, S., Takenouchi, T., et al. (2017). Importance of adiponectin activity in the pathogenesis of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 4, 591–600. doi: 10.1002/acn3.436
Waragai, M., Ho, G., Takamatsu, Y., Shimizu, Y., Sugino, H., Sugama, S., et al. (2018). Dual-therapy strategy for modification of adiponectin receptor signaling in aging-associated chronic diseases. Drug Discov. Today 23, 1305–1311. doi: 10.1016/j.drudis.2018.05.009
Waragai, M., Ho, G., Takamatsu, Y., Wada, R., Sugama, S., Takenouchi, T., et al. (2020). Adiponectin paradox in Alzheimer’s disease; relevance to amyloidogenic evolvability? Front. Endocrinol. 11:108. doi: 10.3389/fendo.2020.00108
Watts, A. S., Loskutova, N., Burns, J. M., and Johnson, D. K. (2013). Metabolic syndrome and cognitive decline in early Alzheimer’s disease and healthy older adults. J. Alzheimers Dis. 35, 253–265. doi: 10.3233/jad-121168
Wennberg, A. M., Gustafson, D., Hagen, C. E., Roberts, R. O., Knopman, D., Jack, C., et al. (2016). Serum adiponectin levels, neuroimaging and cognition in the mayo clinic study of aging. J. Alzheimers Dis. 53, 573–581. doi: 10.3233/jad-151201
Williams, G. C. (1957). Pleiotropy, natural selection and the evolution of senescence. Evolution 11, 398–411. doi: 10.1111/j.1558-5646.1957.tb02911.x
Wu, B., Wang, Y., Shi, C., Chen, Y., Yu, L., Li, J., et al. (2019). Ribosylation-derived advanced glycation end products induce tau hyperphosphorylation through brain-derived neurotrophic factor reduction. J. Alzheimers Dis. 71, 291–305. doi: 10.3233/jad-190158
Wu, B., Wei, Y., Wang, Y., Su, T., Zhou, L., Liu, Y., et al. (2015). Gavage of D-Ribose induces Abeta-like deposits, Tau hyperphosphorylation as well as memory loss and anxiety-like behavior in mice. Oncotarget 6, 34128–34142. doi: 10.18632/oncotarget.6021
Yu, L., Chen, Y., Xu, Y., He, T., Wei, Y., and He, R. (2019). d-Ribose is elevated in T1DM patients and can be involved in the onset of encephalopathy. Aging 11, 4943–4969. doi: 10.18632/aging.102089
Keywords: Alzheimer’s disease, diabetes mellitus, evolvability, antagonistic pleiotropy, adiponectin paradox
Citation: Ho G, Takamatsu Y, Wada R, Sugama S, Waragai M, Takenouchi T, Masliah E and Hashimoto M (2020) Connecting Alzheimer’s Disease With Diabetes Mellitus Through Amyloidogenic Evolvability. Front. Aging Neurosci. 12:576192. doi: 10.3389/fnagi.2020.576192
Received: 25 June 2020; Accepted: 28 August 2020;
Published: 28 October 2020.
Edited by:
Jiehui Jiang, Shanghai University, ChinaReviewed by:
Nobuyuki Kimura, National Center for Geriatrics and Gerontology (NCGG), JapanRongqiao He, Institute of Biophysics (CAS), China
Copyright © 2020 Ho, Takamatsu, Wada, Sugama, Waragai, Takenouchi, Masliah and Hashimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Makoto Hashimoto, aGFzaGltb3RvLW1rQGlnYWt1a2VuLm9yLmpw