
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 03 September 2020
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 12 - 2020 | https://doi.org/10.3389/fnagi.2020.571185
This article is part of the Research TopicProtein Misfolding and Proteostasis Impairment in Aging and Neurodegeneration: From Spreading Studies to Therapeutic ApproachesView all 14 articles
 Paulina Troncoso-Escudero1,2,3,4
Paulina Troncoso-Escudero1,2,3,4 Denisse Sepulveda1,2,4
Denisse Sepulveda1,2,4 Rodrigo Pérez-Arancibia1,2,4
Rodrigo Pérez-Arancibia1,2,4 Alejandra V. Parra1,2,3,4
Alejandra V. Parra1,2,3,4 Javiera Arcos1,2,4
Javiera Arcos1,2,4 Felipe Grunenwald1,2,4
Felipe Grunenwald1,2,4 Rene L. Vidal1,2,4*
Rene L. Vidal1,2,4*Movement disorders are neurological conditions in which patients manifest a diverse range of movement impairments. Distinct structures within the basal ganglia of the brain, an area involved in movement regulation, are differentially affected for every disease. Among the most studied movement disorder conditions are Parkinson’s (PD) and Huntington’s disease (HD), in which the deregulation of the movement circuitry due to the loss of specific neuronal populations in basal ganglia is the underlying cause of motor symptoms. These symptoms are due to the loss principally of dopaminergic neurons of the substantia nigra (SN) par compacta and the GABAergic neurons of the striatum in PD and HD, respectively. Although these diseases were described in the 19th century, no effective treatment can slow down, reverse, or stop disease progression. Available pharmacological therapies have been focused on preventing or alleviating motor symptoms to improve the quality of life of patients, but these drugs are not able to mitigate the progressive neurodegeneration. Currently, considerable therapeutic advances have been achieved seeking a more efficacious and durable therapeutic effect. Here, we will focus on the new advances of several therapeutic approaches for PD and HD, starting with the available pharmacological treatments to alleviate the motor symptoms in both diseases. Then, we describe therapeutic strategies that aim to restore specific neuronal populations or their activity. Among the discussed strategies, the use of Neurotrophic factors (NTFs) and genetic approaches to prevent the neuronal loss in these diseases will be described. We will highlight strategies that have been evaluated in both Parkinson’s and Huntington’s patients, and also the ones with strong preclinical evidence. These current therapeutic techniques represent the most promising tools for the safe treatment of both diseases, specifically those aimed to avoid neuronal loss during disease progression.
Movement disorders are characterized by disabilities in speed, fluency, quality, and ease of motor execution, impairments that could be due to an excess or lack of voluntary movements (Shipton, 2012). Movement is produced by the coordinated action of several cortical and subcortical brain structures such as the spinal cord, brainstem, cerebral cortex, cerebellum, and basal ganglia, which collectively fine-tune voluntary and involuntary movements (Ferreira-Pinto et al., 2018). Particularly, the basal ganglia structure comprises a group of subcortical nuclei including the striatum (subdivided in most mammals in caudate and putamen), internal globus pallidus (GPi) and external globus pallidus (GPe), substantia nigra (SN) pars reticulata (SNpr) and compacta (SNpc), and subthalamic nucleus (STN), that together with the primary motor cortex and the thalamus, comprise the motor circuit involved in the control of voluntary movement (Albin et al., 1989; Obeso et al., 2008; Calabresi et al., 2014). The striatum is composed of medium spiny neurons (MSNs), a type of GABAergic neurons representing 90–95% of striatal neurons (Dubé et al., 1988), cholinergic and GABAergic interneurons (Lapper and Bolam, 1992). MSNs are innervated by glutamatergic (excitatory) inputs from the cortex and thalamus, together with dopaminergic inputs from the SN (Pickel et al., 1992). In turn, MSNs differentially express dopamine (DA) D1 or D2 receptors, which determine their participation in two motor circuits: the direct or indirect pathways. The direct pathway is composed of the striatum, GPi, SNpr, thalamus, and motor cortex, promoting the activation of movement through the inhibition of the GPi, in addition to the consequent disinhibition of the thalamus. The indirect pathway, formed by the striatum, GPe, STN, SNpr, thalamus, and motor cortex favors the inhibition of movement by activating the STN with the consequent inhibition of the thalamus (Figure 1A; Calabresi et al., 2014). Alterations in these brain regions are associated with a spectrum of abnormal movement disorders. Among the most studied movement disorders are Parkinson’s (PD) and Huntington’s disease (HD; Figures 1B,C), which are the main focus of this review.
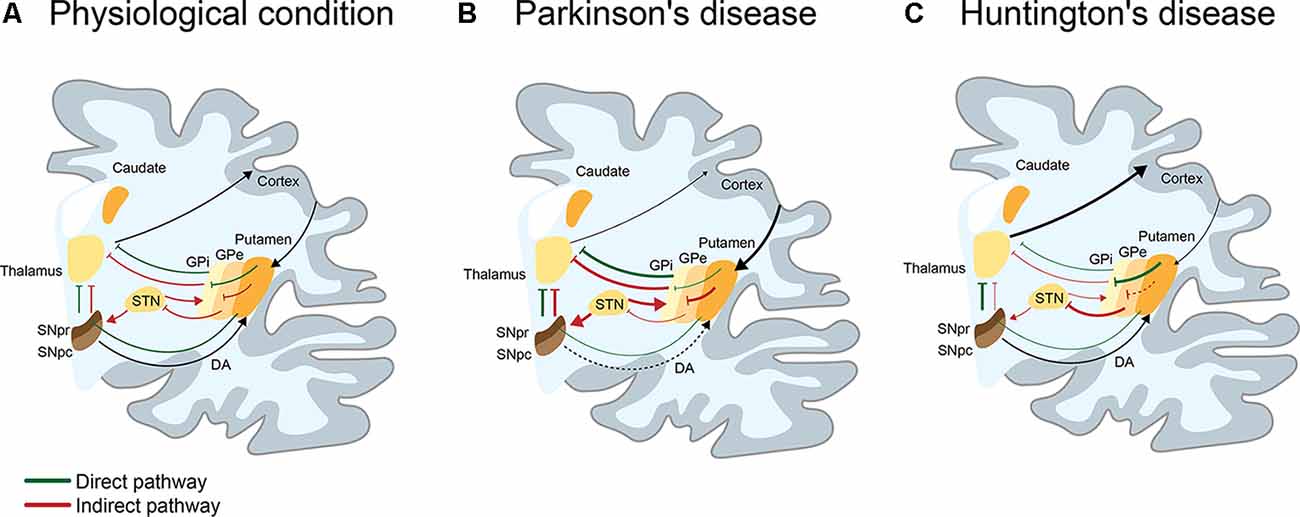
Figure 1. I Schematic representation of the direct and indirect pathways in basal ganglia in physiological, Parkinson’s (PD), and Huntington’s disease (HD) conditions. (A) Physiologically, the direct (green line) pathway participates in the activation of movement. This pathway is engaged when the activation of the cortex produces a release of glutamate into the striatum, activating GABAergic medium spiny neurons (MSNs) of the direct pathway. By releasing GABA to the substantia nigra pars reticulata (SNpr) and the internal globus pallidus (GPi), MSNs inhibit neurons of the SNpr/GPi that are also GABAergic. This causes activation of the glutamatergic neurons present in the thalamus, which projects to the cortex, resulting in the activation of movements. On the contrary, the indirect pathway (red line) participates in the inhibition of movement. When GABAergic MSNs that indirectly project to the SNpr through the external globus pallidus (GPe) and the subthalamic nucleus (STN), release GABA into the GPe, inhibits GABAergic neurons present in the GPe. This leads to the disinhibition of the glutamatergic neurons of the STN, which activates GABAergic neurons of the SNpr/GPi. These neurons inhibit neurons present in the thalamus, resulting in a reduction of movement. The selection and execution of movement reflect a dynamic balance between both pathways. (B) In PD, the loss of dopaminergic neurons of the SNpc, induces an overactivation of the indirect pathway and decrease of movement. Consequently, there is an increase of GABAergic activity of the GPi/SNpr over thalamic neurons that project to the cortex, leading to loss of movement (hypokinetic disorder). (C) In HD (early stage), MSNs of the indirect pathway appear to be affected before the MSNs of the direct pathway. This induces an increase of GABAergic (or inhibitor) activity of the GPe over the STN, which causes the loss of inhibitory activity of the GPi/SNpr over thalamic neurons that project to cortex, leading to the appearance of choreic movements (hyperkinetic disorder).
PD is the second most common chronic and progressive neurological disorder with no effective cure (Emamzadeh and Surguchov, 2018) PD incidence is currently around 200 in 100,000 persons (Ball et al., 2019) and the number of PD patients is expected to reach 10 million by 2030 (Dorsey et al., 2007; Ball et al., 2019). PD is mainly an idiopathic disorder with multifactorial etiology, with aging being one of its main risk factors (Hou et al., 2019). Nevertheless, genetic factors have been associated with 5–10% of PD cases (Kim and Alcalay, 2017). Clinical features of PD are a combination of motor symptoms including muscle rigidity, gait difficulty, postural instability, bradykinesia, and tremor at rest (Rees et al., 2018). Also, non-motor symptoms include sensory and sleep alterations, constipation, cognitive impairment, dementia, anxiety, depression, and mood disorders at an early stage of the disease (Munhoz et al., 2015).
One of the two neuropathological criteria required for the diagnosis of PD is the progressive loss of dopaminergic neurons within the SNpc of the basal ganglia (Figure 1B; Agid, 1991; Hirsch et al., 1999). However, not all dopaminergic neurons are equally vulnerable: those that project their axons to the putamen are more vulnerable than those that project their axons to cognitive areas. The second pathological criteria is the presence of α-synuclein (α-syn)-positive inclusions accumulated in Lewy bodies in neurons (Double, 2012). Although the main hallmarks of PD are well described, diagnosis before classic clinical features occur is not currently achievable. Motor symptoms are observed when 70% of the striatal dopaminergic neurons terminals are lost and half of the dopaminergic neurons in the whole brain have degenerated (Double, 2012; Surmeier et al., 2017; Fu et al., 2018).
The current PD treatment is a pharmacological therapy that increases DA levels to provide symptomatic relief. Unfortunately, this therapeutic strategy has limited efficacy. However, new therapeutic alternatives under development attempt to modify the pathology of PD by increasing DA production or improving neuronal health. These approaches efficiently delay neurodegeneration as well as PD symptoms in preclinical models and patients. Currently, finding new treatments that modify pathological PD progression, prevent the dopaminergic neuronal loss, counteract aberrant neuronal activity, and delay the appearance of motor symptoms is the principal goal of many investigations.
HD is the world’s most common monogenic neurological disorder (Huntington’s Disease Collaborative Research Group, 1993), characterized by its autosomal dominant inheritance, midlife onset and progressive course with a combination of motor, cognitive and behavioral features. HD is caused by a mutation in the gene that encodes for the protein huntingtin (HTT), which leads to an expanded CAG trinucleotide, causing an abnormally long polyglutamine (polyQ) tract in HTT. This repetition ranges between 6–35 glutamine units in the normal population. When this tract is ≥40 glutamines long, the mutation is highly penetrant, triggering a disease process that leads to the onset of motor symptoms. Mutant HTT (mHTT) exhibits gain-of-toxic properties, causing dysfunction and death of GABAergic MSNs of the striatum, which is particularly vulnerable to mHTT toxicity (Figure 1C). Once signs and symptoms begin, they progress inexorably throughout the illness, which is inevitably fatal, with a median survival from motor onset of 18 years (Ross et al., 2014).
We describe current pharmacological therapies for both diseases, cellular replacement strategies to restore lost neuronal populations, administration of Neurotrophic factors (NTFs) to increase neuronal viability and health, electrical neuromodulation to restore movement circuitry lost, and genetic approaches to decrease mHTT and α-syn levels. We focused on studies that have reached clinical testing, highlighting preclinical evidence that supports those clinical trials. The clinical trials mentioned throughout this review are summarized in Tables 1, 2. For those strategies in which clinical trials have not been performed, we present the current state of investigations and remark the important drawbacks that must be solved before these therapeutic approaches jump into clinical trials.
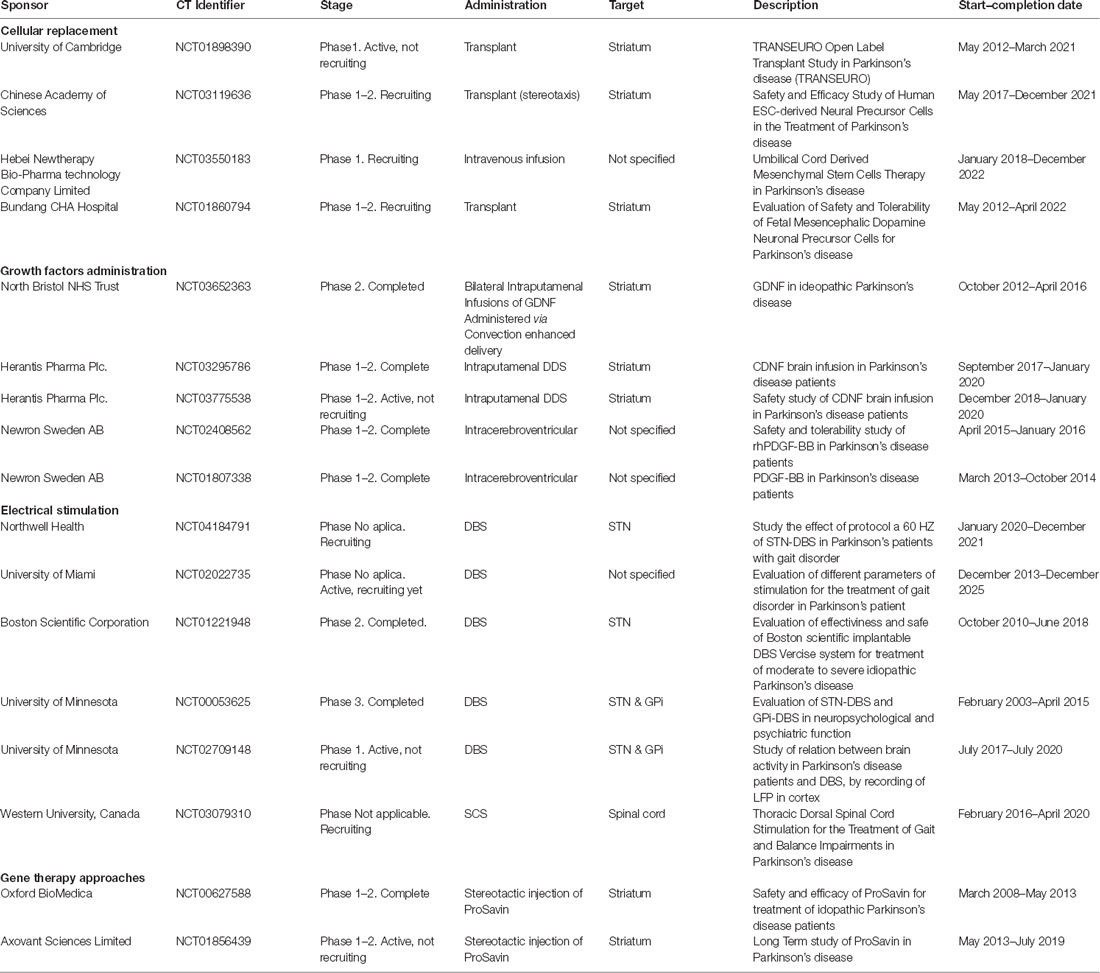
Table 1. Parkinson’s disease (PD) therapies under clinical trials.
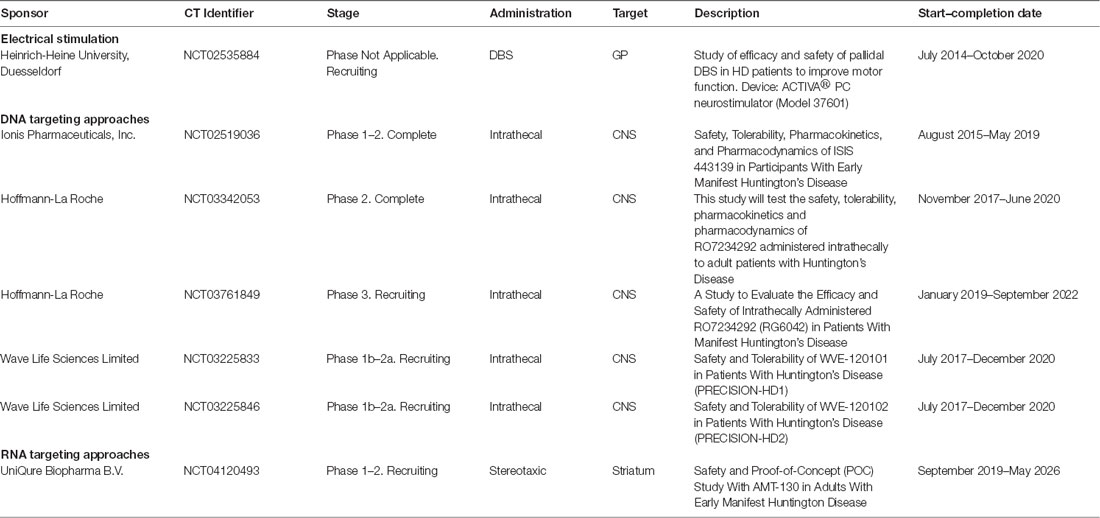
Table 2. Huntington’s disease therapies under clinical trials.
Currently, no approved drugs can modify the progression of either PD or HD. Available pharmacological therapies only aim to treat motor symptoms to improve the quality of life of patients but, ultimately, these drugs do not mitigate the progressive neurodegeneration.
For PD, pharmacological approaches are designed to reestablish DA levels through: (i) increasing DA availability using DA precursors like levodopa (L-dopa) or dopaminergic agonists like pramipexole, and (ii) inhibiting DA degradation by monoamine oxidase B inhibitors [MAO-BI, like selegiline (Eldepryl®) and rasagiline (Azilect®)] or catechol-O-methyl transferase inhibitors [COMTI, like entacapone (Comtan®)] and tolcapone (Tasmar®; Van de Schyf, 2015; Teijido and Cacabelos, 2018; Carrera and Cacabelos, 2019). A caveat in the chronic administration of anti-parkinsonian drugs is the “wearing-off” phenomenon, which produces additional psychomotor and autonomic complications, like levodopa-induced dyskinesia (LID; Fahn et al., 2004; Stacy, 2009; Ammal Kaidery et al., 2013; Cacabelos, 2017). Moreover, L-dopa pharmacokinetics is unpredictable and commonly leads to administration increase, complex regimens, and poor patient compliance. Nevertheless, L-dopa remains as the gold standard pharmacological intervention for motor symptoms in PD patients (Oertel and Schulz, 2016; Fox et al., 2018). Although the pathophysiology of wearing-off and dyskinesia is complex and not completely understood, it appears to be linked to the short plasma half-life of L-dopa, as short-acting dopaminergic drugs can induce alterations in brain DA concentrations which lead to motor dysfunction (Olanow et al., 2006; Rajan et al., 2017). To assess this problem, strategies that prolong L-dopa plasma half-life have been developed, including the administration of COMTI. A significant quantity of orally administered L-dopa is metabolized to 3-O-methyldopa (a useless metabolite) by COMT in the gastrointestinal tract. By inhibiting COMT, more L-dopa will be absorbed, increasing its bioavailability and extending its half-life (Oertel and Schulz, 2016). Some of these drugs are tolcapone, entacapone, and opicapone (Parkinson Study Group, 1997; Rinne et al., 1998; Poewe et al., 2002; Cacabelos, 2017). However, the FDA has restricted the use of tolcapone due to hepatic necrosis leading to death (Haasio et al., 2001). Common combinations include L-dopa/carbidopa (an L-dopa decarboxylation inhibitor; Sinemet®) and L-dopa/benserazide (Madopar®), looking to prevent systemic adverse effects related to the peripheral metabolism of L-dopa to DA, including nausea, dyskinesia, motor fluctuations, hypotension, psychiatric symptoms (especially hallucinations) and diaphoresis. Duodopa, an intestinal gel form of L-dopa/carbidopa, provides a more stable response to L-dopa. Stalevo® is a triple-drug, containing carbidopa, L-dopa, and entacapone (Rezak, 2007). Another strategy for prolonging L-dopa half-life is the inhibition of MAO-B, the breakdown-enzyme of DA in the brain, increasing DA levels at synapses. Selegiline, through its metabolite desmethylselegiline, has also shown anti-apoptotic effects (Tatton et al., 1996; Parkinson Study Group, 2005; Cacabelos, 2017). Rasagiline is a significantly more potent MAO-BI that reduces the motor symptoms in PD patients and has disease-modifying potentials (Parkinson Study Group, 2005; Rascol et al., 2005; Olanow et al., 2009). IPX066 (Rytary) is an improved oral formulation of L-dopa approved in 2015 by the FDA containing both an immediate-release and a sustained-release L-dopa. Published phase II and III studies show that IPX066 improved Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores compared to placebo in early PD patients and reduced wearing-off in advanced PD patients (Kestenbaum and Fahn, 2015). Before L-dopa, anticholinergics were the treatment of choice for PD. However, MAO-BI and modern DA agonists have largely replaced these drugs. Trihexyphenidyl (Artane®), benztropine (Cogentin®), and procyclidine (Kemadrin®) is usually reserved for tremor resistant to dopaminergic agents. The adverse effects of anticholinergics include blurred vision, dry mucus membranes, urinary retention, and cognitive changes (Rezak, 2007).
Dopamine receptor (DR) agonists mimic DA actions in the brain by directly stimulating DRs. Two common DR agonists are pramipexole (Mirapex®) and ropinirole (Requip®). These directly stimulate post-synaptic D2 and D3 receptors in the striatum and are prescribed as monotherapy or combined with L-dopa. Potential side effects of pramipexole and ropinirole include hypotension, sleep, cognitive/psychiatric alterations, dyskinesias, and compulsive behavior. The latter is believed to be a result of DA dysregulation in the limbic and frontal circuits that are connected to the basal ganglia (Evans and Lees, 2004). Polyoxazoline (POZ) polymer conjugation for continuous dopaminergic drug delivery may improve motor symptoms while avoiding side effects. The in vitro and in vivo pharmacokinetics of POZ-conjugated rotigotine (DA agonist) was characterized, demonstrating that the sustained dopaminergic stimulation profile achieved by POZ-conjugated rotigotine formulations, could represent a significant advance in the treatment of PD. POZ polymer administration can improve motor symptoms in a rat model of PD (Eskow Jaunarajs et al., 2013; Fox et al., 2018). For HD, the only approved pharmacological therapy for the treatment of motor symptoms is tetrabenazine, a vesicular monoamine transporter 2 (VMAT-2) inhibitor that reduces DA neurotransmission via its depletion from presynaptic vesicles, resulting in a reduction of chorea manifested by HD patients (Wyant et al., 2017). However, its side effects, which include sedation, anxiety, depression, and suicidality have limited its use (Wyant et al., 2017; Dean and Sung, 2018). In 2017, the FDA approved a deuterated derivative of tetrabenazine, deutetrabenazine, with improved pharmacokinetic profile, allowing a less frequent daily dosage with comparable systemic exposure of the drug, resulting in less adverse events (Dean and Sung, 2018). No other small molecules are currently used for HD treatment, but several therapeutic approaches are in clinical trials, which will be discussed later.
In 1967, in an important breakthrough, Cotzias et al. (1967) demonstrated that the administration of a precursor of DA, L-dopa, improved motor function in PD patients, leading to the thought that the cure for PD was discovered. Also in the 1960s, tetrabenazine was introduced as an antipsychotic but also showed beneficial effects for the treatment of hyperkinetic motor symptoms, like chorea in HD patients (Dalby, 1969; Huntington Study Group, 2006). To date, it is known that these drugs do not reverse disease progression and in many cases do not have the desired effects. This has brought the idea that local production of DA and GABA, and therefore the replacement of the neurons that produce it, would be the ideal treatment for these diseases. The fact that the major symptoms present in PD and HD patients are due to the loss of dopaminergic and GABAergic neurons in specific brain regions, respectively, means that replacing these specific cell types could help relieve some of the symptoms present in patients. This has given rise to different branches of investigations seeking cellular replacement-based therapies, which have shown promising results in animal models for these diseases as well as in affected patients (Figure 2).
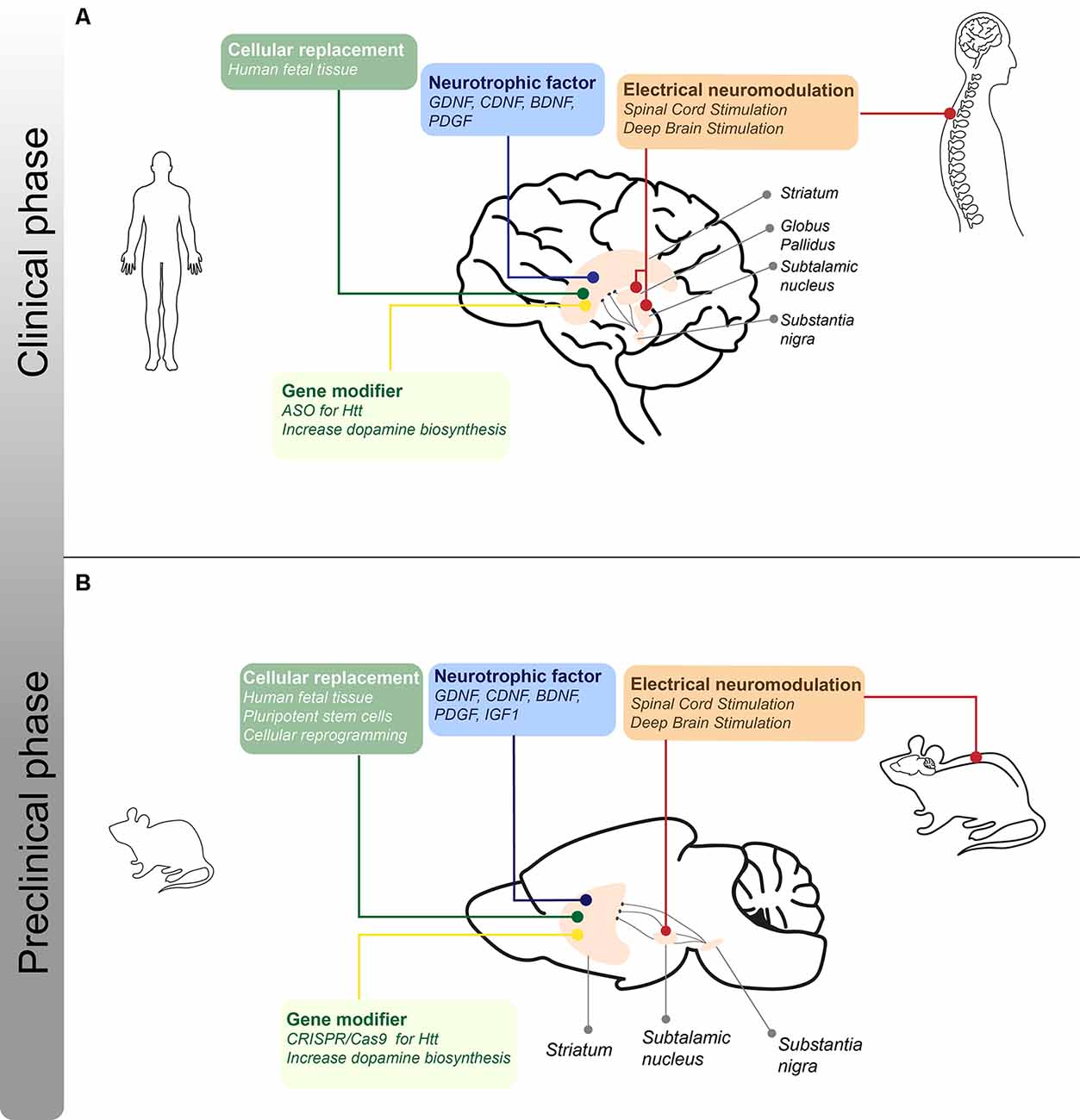
Figure 2. I Schematic representation of the different treatments for motor symptoms for PD and HD under clinical and preclinical phases. (A) Approaches regarding cellular, neurotrophic, electrical, and gene-modifying therapies that are in clinical trials or stage of optimization for PD and HD patients. The lines indicate the different brain structures that are the target of the therapeutic strategies mentioned. (B) Approaches regarding cellular, neurotrophic, electrical, and disease-modifying therapies in the preclinical phase using rodent models of PD and/or HD. The lines indicate the different brain structures that are the target of the therapeutic strategies mentioned.
The first study demonstrating that dopaminergic neurons could be replaced using fetal tissue was performed using 6-hydroxydopamine (6-OHDA)-lesioned rats that were implanted with DA-rich ventral mesencephalic tissue from rat fetuses (Björklund and Stenevi, 1979; Perlow et al., 1979). These studies were followed by the generation of the first non-human primates PD model: monkeys lesioned with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; Burns et al., 1983). This model manifested several of the patient’s symptoms, and transplanting primate fetal mesencephalic tissue into their striatum showed to alleviate these symptoms (Bakay et al., 1985; Sladek et al., 1987; Taylor et al., 1991). These studies set foot for the first PD cell replacement therapy in humans. These clinical trials were performed using dopaminergic neuron precursors from human fetal tissue, which were transplanted into the striatum of PD patients (Lindvall et al., 1989, 1992; Freed et al., 1990, 1992). Transplanted tissue presented no negative effects at the transplantation site, was functional and survived in the transplanted brain region, but clinical benefits were variable (Freed et al., 1992; Kordower et al., 1998; Hagell et al., 1999; Li et al., 2016).
It is important to understand that dopaminergic neurons engrafted in the striatum are deprived of their SNpc afferents. Instead, when dopaminergic neurons are transplanted in the striatum, they may form connections with cortical, intrastriatal, and thalamic neurons, which are not normally connected with nigral dopaminergic neurons. Moreover, considering the nature of fetal tissue, possibly other types of neurons and glial cells differentiate in the grafted brain area. Additionally, only a portion of the grafted fetal tissue corresponds to cells that needed restoration in the specific brain region. Another important concern is the presence of α-syn aggregates and Lewy body inclusions in the grafted cells in PD patients (Kordower et al., 2008; Li et al., 2008). This highlights the necessity of using a complementary strategy that allows donor cells to be resistant to the spreading of α-syn.
On the other hand, by the mid-1980s, the first studies using fetal tissue were performed in rat models of HD (Table 4). These studies demonstrated that the grafted tissue survived, was functional and recovered some of the behavioral alterations present in HD rats (Deckel et al., 1983; Isacson et al., 1984, 1985; Sanberg et al., 1986). The in vivo functionality of the grafted striatal fetal tissue was also assessed, showing GABA release upon dopaminergic and glutamatergic inputs (Campbell et al., 1993). Diverse studies further demonstrated the effect of striatal fetal tissue transplantation in diverse pharmacological models of HD (Hantraye et al., 1992; Nakao et al., 1999) and by late 1990s, the first clinical trials in HD patients were performed. HD patients were injected unilaterally or bilaterally with fetal tissue, which was originated from various donated embryos. Given the amount of tissue needed and their origin from different embryos, this strategy causes immune rejection by the patient’s immune system. Therefore, complementary immunosuppression therapy is needed, which has shown no adverse effects on patients (Freeman et al., 2000). After fetal cell transplantation, patients presented improved cognitive function and stabilization of motor functions, which worsen a few years after the surgery (Kopyov et al., 1998; Gallina et al., 2008; also see Table 1).
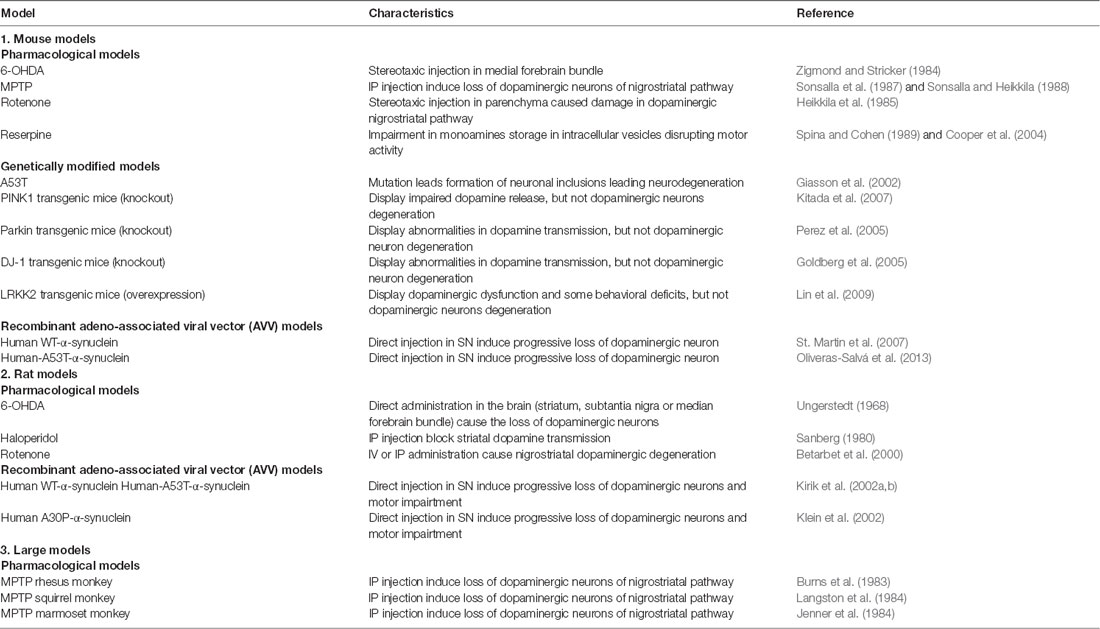
Table 3. Common animal models of Parkinson’s Disease.
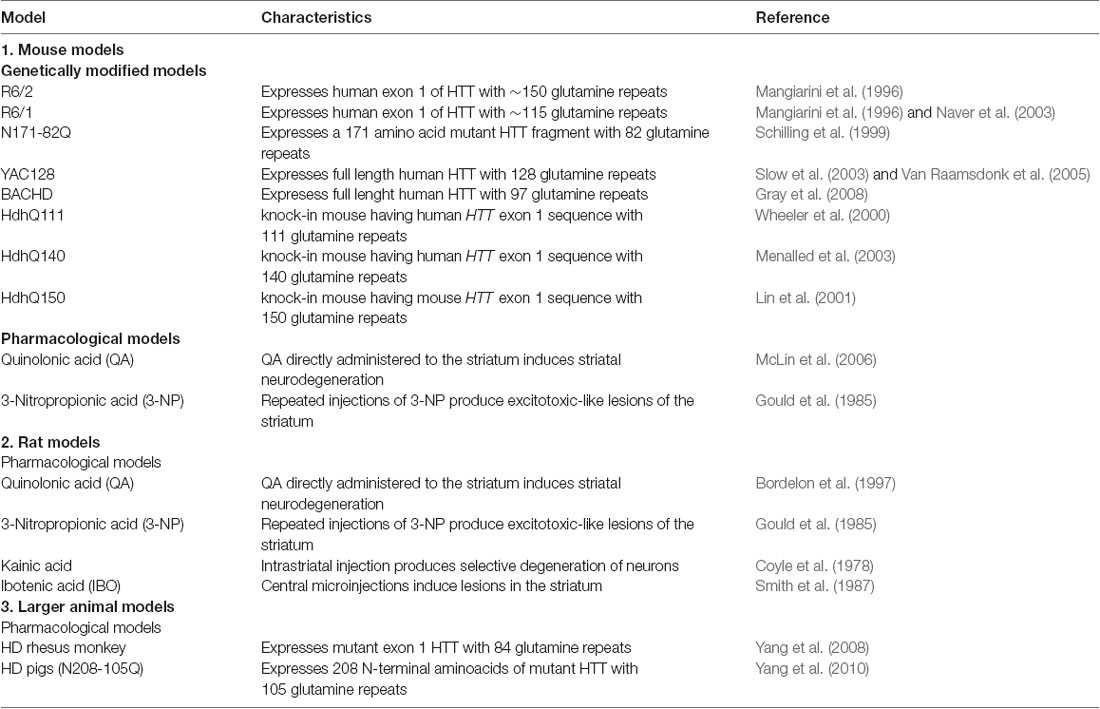
Table 4. Common animal models of Huntington’s disease.
Although several clinical trials have been performed using fetal tissue transplants to treat PD and HD patients, this technique has a few but important limitations. First, is important to consider that when using human fetal tissue, tumors can develop, which could be explained by the presence of actively dividing immature neuroepithelial cells (Keene et al., 2009). Second, not only neuronal loss must be corrected, but also the loss of glial cells. Astrocytes are the most abundant cell type found in the brain (Miller, 2018) and play important roles in maintaining brain homeostasis, supporting a neuronal activity, and metabolism. In PD and HD, there is a deregulation of astrocyte activity, including electrophysiological changes, calcium homeostasis, glutamate reuptake, and metabolism, among others (Booth et al., 2017; Garcia et al., 2019; Gray, 2019). Therefore, the replacement of the lost neurons should be accompanied by a replacement or modification of the glial cells that support them.
Pluripotent stem cells (PSCs) are an unlimited source of cells with the potential to give rise to any type of cell of the body. Cells differentiated from embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs; Takahashi and Yamanaka, 2006) are widely used as in vitro models for many diseases, including neurodegenerative diseases, and also as a source of cell-replacement therapies. Initial studies demonstrated that, when midbrain-derived dopaminergic neurons where grafted in the striatum of rodent models of PD (Table 3), long-term survival of these cells was observed, which were tyrosine hydroxylase (TH)-positive neurons, completely reversed amphetamine-induced rotational behavior and lacked neuronal overgrowth (Kriks et al., 2011). Importantly, midbrain human dopaminergic neurons grafted in MPTP-lesioned non-human primates survived in the grafted area, expressed TH, extended fibers to the surrounding striatum, and did not present neuronal overgrowth (Kriks et al., 2011).
However, one limitation regarding the use of PSCs-derived neurons is the greater immune reaction observed with allogeneic grafts compared with isogenic cells (Duan et al., 1995; Morizane et al., 2013). Using both approaches, Hallett et al. (2015) demonstrated that in a non-human primate PD model autologous iPSCs-derived midbrain-like dopaminergic neurons could successfully engraft and survive for as long as 2 years. This led to improving motor function and complete re-innervation in the striatum with extensive axonal outgrowth, and no graft overgrowth, tumor formation or inflammation was observed (Hallett et al., 2015). Also, as clinical trials are underway, establishing the optimal and safest protocol for dopaminergic neurons differentiation is necessary to obtain the adequate number of neurons that permit improvements in patient’s motor symptoms. Also, the characterization of the graft is relevant, as the differentiation protocol could give rise to other cell types that may alter the physiological conditions in the grafted site. One important concern about the use of autologous transplantation using iPSC-derived dopaminergic neurons of PD patients is that these cells will carry any intracellular dysfunction related to disease pathogenesis. It is important to remember that dopaminergic neuron replacement is primarily focused on the treatment of motor symptoms. Patients in which symptoms like dementia or other cognitive impairment are also present may not be benefited completely with this type of therapy.
One key issue for cellular transplantation into human brains is the necessity of an important amount of cells. Fetal tissue cells not only provide a limited number of cells but also come with ethical and religious concerns. Therefore, the use of iPSCs obtained from somatic cells of patients is excellent for personalized cell-based therapy and to model HD in vitro. The first research group to obtain striatal neurons from HD iPSCs-derived neuronal stem cells (NSCs) was Zhang et al. (2010), who used iPSCs derived from an HD patient (Park et al., 2008). These cells not only express MSNs markers but also could be used as an excellent model for drug screening in HD research (Zhang et al., 2010). Using the same HD iPSCs, Zhang et al. (2010) demonstrated that these cells can be corrected for the CAG mutation by replacing the expanded 72 CAG repeat with a normal 20–21 CAG repeat (An et al., 2012). Finally, the corrected iPSCs-derived NSCs could be successfully differentiated into MSNs in vitro, and when transplanted into the striatum of R6/2 mice (Table 4) they differentiated into MSNs neurons (An et al., 2012). The genetic modification of human iPSCs not only brings us closer to the proper modeling of diseases but also provides a potential therapy. It has been demonstrated that iPSCs-derived neuronal cells from an HD preclinical model develop cellular features of HD cells, which could be rescued by genetic suppression of HTT and pharmacological treatment (Carter et al., 2014). Using human ESCs (hESCs) or human iPSCs (hiPSCs) differentiated into MSNs progenitors, it has been demonstrated that the transplantation of these cells into the striatum of rodent HD models can form functional connections with other cells, and project their axons to other structures involved in the movement circuitry, like the SN (Faedo et al., 2017; Adil et al., 2018).
As highlighted previously, since HD is caused by a genetic mutation, and differentiated MSNs progenitors come from HD patients, it is imperative to correct the mutation present in these cells, along with the replacement of the target neurons and other cell types, like interneurons and glial cells, as they may provide a healthy and functional environment for the new neurons to integrate to the local circuitry and survive. Currently, no clinical trials are assessing the use of PSCs in HD patients.
In the adult brain, NSCs are present in the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus. These NSCs are capable of generating neuroblasts, which differentiate into mature neurons (Zhao et al., 2008; Ma et al., 2009). Despite the presence of a niche for the generation of new neurons, these cells have limited migration to remote regions, like the SN and striatum. Hence, the idea to generate new local neurons from preexisting cells has been studied for the last 10 years. Initial studies have demonstrated that fibroblasts can be reprogrammed to dopaminergic neurons through the ectopic expression of transcription factors (Caiazzo et al., 2011; Kim et al., 2011; Pfisterer et al., 2011). Considering the reprogramming of cells for the treatment of neurodegenerative diseases, astrocytes were initially considered as an attractive alternative and their reprogramming to neurons forming functional synapses was demonstrated (Berninger et al., 2007; Heinrich et al., 2010).
The first studies in which dopaminergic neurons were generated by reprogramming human and mouse fibroblasts using lineage-specific factors for the conversion to dopaminergic neurons (Caiazzo et al., 2011; Kim et al., 2011). The converted neurons were positive for several dopaminergic markers and expressed genes related to the dopaminergic lineage rather than with the fibroblast of origin (Caiazzo et al., 2011; Kim et al., 2011). Converted dopaminergic neurons formed synapses, had synaptic activity in culture, and showed electrophysiological properties similar to dopaminergic neurons (Caiazzo et al., 2011). The transplantation of these cells into the brain of wild type (Caiazzo et al., 2011) and PD mice (Kim et al., 2011) showed that converted neurons integrated with the host tissue expressed dopaminergic markers and had electrophysiological responses, which led to an improvement in mice behavior (Kim et al., 2011).
Also, Addis et al. (2011) were the first to demonstrate that astrocytes could be reprogrammed into dopaminergic neurons in vitro. The obtained neurons displayed an up-regulation of genes expressed by dopaminergic neurons, along with electrophysiological properties, including the spontaneous firing of action potentials observed in dopaminergic neurons (Addis et al., 2011). A few years later, the first human astrocytes were directly converted into functionally competent dopaminergic neurons in vitro (Rivetti di Val Cervo et al., 2017) and it was described for the first time the in vivo conversion of astrocytes into dopaminergic neurons in the 6-OHDA mouse model of PD (Rivetti di Val Cervo et al., 2017). These converted dopaminergic neurons were excitable and expressed dopaminergic neuron markers, which helped relieve the cycling behavior observed in these animals. These and other reports have shown that the cellular reprogramming relies on the expression of lineage-specific transcription factors. However, it has been shown that downregulation of PTB in mouse and human fibroblasts, an RNA-binding protein negatively controlling neuronal induction and maturation, induces the conversion of these cells into functional neurons (Xue et al., 2013, 2016). Considering these finding and that downregulation of PTB occurs during neurogenesis (Hu et al., 2018), Qian et al. (2020) recently demonstrated using the 6-OHDA mouse model of PD that adeno-associated virus (AAV)-mediated downregulation of PTB using an shRNA convert nigral astrocytes into functional dopaminergic neurons. These converted neurons integrate into the nigrostriatal pathway, extending their axons into the striatum and other brain regions. These neurons were electrophysiologically functional and restored the striatal dopamine lost due to the 6-OHDA treatment, leading to a reversal of the motor deficits observed in these mice (Qian et al., 2020). Importantly, these results were also observed using an antisense oligonucleotide against PTB, giving these findings a potentially clinical approach for the treatment of PD in patients (Qian et al., 2020).
Not only glial cells can originate dopaminergic neurons. In an elegant work performed by Niu et al. (2018), authors demonstrated that striatal neurons could be reprogramed to dopaminergic-like neurons in the adult mouse striatum. These neurons, although expressing both dopaminergic and GABAergic markers, have electrophysiological properties like endogenous dopaminergic but no MSNs neurons. These dopaminergic-like neurons were also functionally connected with surrounding neurons, confirmed by the presence of spontaneous postsynaptic currents (Niu et al., 2018). Hence, these results seem to be promising for converting MSNs into dopaminergic neurons under pathological conditions.
Initial studies have shown that striatal astrocytes can be reprogramed into proliferative neuroblasts in young, adult and aged mice brains (Niu et al., 2013), which are interesting especially for HD, a disease of adult-onset. Furthermore, when these neuroblasts were treated with NTFs or histone deacetylase inhibitor, they differentiated into mature neurons with electrophysiological properties (Niu et al., 2013). Using (AAV)-based conversion, striatal GABAergic, and glutamatergic neurons could be originated after reprogramming NG2 glial cells (Torper et al., 2015). Newly generated neurons presented electrophysiological properties of functional neurons, remained stable for a long period, and even integrated into local neuronal circuitry (Torper et al., 2015). Consequently, the use of endogenous glial cells for the regeneration of neuronal population lost under neurodegenerative conditions has emerged as an interesting source, avoiding the use of differentiated external cells and therefore minimizing the possible immunorejection of foreign cells (Li and Chen, 2016; Srivastava and DeWitt, 2016; Barker et al., 2018).
Recently, using AAV-based reprogramming of striatal astrocytes, Wu et al. (2020) demonstrated that astrocytes could be converted to MSNs in the striatum of R6/2 and YAC128 mice. Converted neurons expressed specific MSNs markers, showed electrophysiological properties, and projected their axonal terminals to the GP and SNpr. All these findings were accompanied by a reduction in striatal atrophy, attenuation of the phenotypic deficit, and an extended life span of R6/2 mice with converted MSNs (Wu et al., 2020).
Nevertheless, it is important to complement these reprogram therapies with a therapy that targets the mutation in the HTT gene. Converted neurons will sooner or later express and accumulate mHTT, which will eventually lead to neurodegeneration. Therefore, in vivo reprogramming of glial cells into healthy MSNs has an important clinical potential, which must also be combined with gene therapy strategies to reduce or ablate mHTT expression in these new neurons. Also, the application of this approach in the clinic is challenging by the lack of standardized protocols for cellular reprogramming, as well as the efficiency of converted cells. This depends on the donor cell, the type of cell that is needed, the characteristics of patients that must be considered, i.e., age, the severity of the disease, and treatment with other drugs, among others. Although challenging, in vivo cell reprogramming appears as the most promising therapy candidate for cellular replacement for PD and HD patients.
NTFs are molecules that promote the differentiation, myelination, and survival of neurons, which are also involved in the neuroinflammatory response (Fernandez and Torres-Aleman, 2012; Labandeira-Garcia et al., 2017). A reduction in the bioavailability of NTFs in the peripheral and central system during aging suggests a role of these factors during neurodegenerative disorders such as PD and HD (Zuccato and Cattaneo, 2007; Gasperi and Castellano, 2010; Procaccini et al., 2016; Salem et al., 2016). NTFs, like Glial cell-line Derived Neurotrophic Factor (GDNF), Brain-Derived Neurotrophic Factor (BDNF), Cerebral Dopamine Neurotrophic Factor (CDNF), Mesencephalic astrocyte-derived Neurotrophic Factor (MANF), Platelet-Derived Growth Factor (PDGF), Insulin-like Growth Factors (IGFs), and others have been through preclinical and clinical trials for PD and HD (Figure 2). Here, we describe the recently therapeutic approaches based on the restoration of NTFs levels in the brain to prevent and/or stop the neurodegenerative process describe in PD and HD.
GDNF is considered as a neuro-restorative therapeutic protein that induces the regeneration of dopaminergic neurons given that enhances dopaminergic cell survival and differentiation in vitro (Lin et al., 1993; Zurn et al., 2001). Besides, GDNF has shown a protective effect on the survival of noradrenergic neurons in the locus coeruleus (Arenas et al., 1995), an affected region in neurodegenerative diseases such as PD and HD (Zweig et al., 1992; Oertel and Schulz, 2016). The neuroprotective effects of GDNF have prompted preclinical and clinical studies. Chronic infusion of GDNF into the lateral ventricle or the striatum promoted the restoration of the nigrostriatal dopaminergic system and significantly improved motor functions in a rhesus monkey PD model (Grondin et al., 2002). Moreover, GDNF protects nigral dopaminergic neurons from degeneration and improves motor behavior in 6-OHDA rat models of PD (Tereshchenko et al., 2014). However, GDNF has limited use due to its inability to cross the blood-brain barrier (BBB), therefore new administration methods have been explored, including: (1) the delivery of GDNF in biodegradable microspheres (Garbayo et al., 2009); (2) gene therapy using DNA nanoparticle (DNP) technology for the expression of human GDNF (hGDNF) in the striatum (Fletcher et al., 2011); (3) the use of intra-cerebroventricular (ICV) catheters implanted into the basal ganglia (Gill et al., 2003; Nutt et al., 2003); (4) the use of viral vectors (Kordower et al., 2000); and (5) GDNF-producing fibroblasts (Grandoso et al., 2007). ICV administration of recombinant hGDNF to non-human primates showed to significantly improve locomotor activity after 4 months of treatment (Zhang et al., 1997). Also, intraputamenal (Ipu) delivery of GDNF in MPTP-lesioned non-human primates significantly increased DA release (Grondin et al., 2003). Despite the positive results in the survival of dopaminergic neurons and improvements in motor behavior (Gill et al., 2003; Patel et al., 2005; Lang et al., 2006), the invasiveness of the delivery of GDNF to the brain represents a limitation for its use.
The first attempt to probe the benefits of GDNF in PD patients consisted of the ICV administration through catheter implantation in 50 PD patients for 8 months. Patients presented side effects after drug administration, mainly weight loss, nausea, and vomiting. At the end of treatment, patients did not present improvements in the UPDRS motor scores (Nutt et al., 2003). Similarly, delivery through Ipu infusion of recombinant hGDNF in 34 PD patients did not observe significant improvements in UPDRS motor scores (Lang et al., 2006). Furthermore, in a completed clinical study with 42 PD patients, bilateral Ipu GDNF infusions every 4 weeks for 9 months showed that 18F-DOPA analyzed through PET scan imaging had a significant increase in the putamen, but no significant changes in UPDRS scores were registered. However, the extended treatment for 18 months showed significant improvements in the UPDRS motor scores (Whone A. et al., 2019; Whone A. L. et al., 2019). Additionally, a phase I study showed an important improvement in UPDRS motor scores after 1 year of GDNF Ipu therapy (Slevin et al., 2005, 2007). Completed clinical studies have demonstrated the safety and potential efficacy of Ipu GDNF infusion, with no evidence of GDNF-induced toxicity (Slevin et al., 2005). However, antibodies were detected in some patients and device-related problems were reported (Lang et al., 2006; Slevin et al., 2007). It has been shown that the effect of GDNF in vitro and in vivo requires TGF-β (Peterziel et al., 2002). The combined effect of GDNF-TGFβ showed a strong neuroprotective effect in rodent PD models (Peterziel et al., 2002) and future therapies may include the simultaneous use of both molecules. Finding a non-invasive and safe way to deliver GDNF is key to evaluate this NTF as an effective treatment for PD.
Preclinical studies in rat models of HD have demonstrated the benefits of ICV injection of GDNF in restoring the excitotoxic-induced damage in the striatum, amelioration of amphetamine-induced rotational behavior (Araujo and Hilt, 1997) and locomotor activity improvement (Araujo and Hilt, 1998). However, no registered clinical trials are testing the efficacy of GDNF in HD patients.
In 2003, a protein called mesencephalic astrocyte-derived neurotrophic factor (MANF) was characterized and demonstrated to promote survival of embryonic dopaminergic neurons in vitro (Petrova et al., 2003). Then, a homologous protein called CDNF was discovered with a protective role for dopaminergic neurons. Several studies evidence the protective role of CDNF and MANF in dopaminergic neurons against the injury caused by α-syn oligomers (Latge et al., 2015). The intrastriatal injection of CDNF prevents the loss of TH-positive neurons in a 6-OHDA-lesioned rat model of PD (Lindholm et al., 2007), and protected dopaminergic neurons in 6-OHDA and MPTP mouse models of PD (Lindholm et al., 2007; Voutilainen et al., 2009). MANF has been tested in the 6-OHDA-lesioned rat model showing beneficial effects (Voutilainen et al., 2009). CDNF and MANF diffuse to the brain significantly better than GDNF, and CDNF was more efficient in reducing amphetamine-induced ipsilateral rotations in the 6-OHDA rat PD model in comparison with GDNF treatment (Voutilainen et al., 2011). In 6-OHDA-lesioned monkeys, PET imaging showed a significant increase of DA transporter (DAT) ligand-binding activity in lesioned animals treated with CDNF (Garea-Rodríguez et al., 2016).
The first phase I–II clinical trial using CDNF in PD patients is being conducted since 2017. In this study, an implanted drug delivery system (DDS) for Ipu of recombinant human CDNF is used in patients with idiopathic mild-advanced PD (Table 1, NCT03295786). Additionally, another phase I–II clinical trial to evaluate the beneficial effects of CDNF in PD patients is still on course (Table 1, NCT03775538). Currently, the delivery of CDNF for HD treatment has not been described.
BDNF is the most abundant NTFs in the brain (Barde et al., 1982), mostly involved in physiological processes including morphological and functional synaptic plasticity, long-term potentiation, learning and memory (Bramham and Messaoudi, 2005; Lu et al., 2014). In the CNS, BDNF binds specifically to tropomyosin-related kinase receptors B (TrkB) receptors and its signaling cascade is involved in neuronal survival (Kaplan and Miller, 2000). Interestingly, MAOI (i.e., rasagiline and selegiline) prevent dopaminergic neuron loss by increasing BDNF expression and other NTFs (Weinreb et al., 2007; Maruyama and Naoi, 2013).
Studies have demonstrated that a decrease of BDNF is implicated in neurological disorders (Siegel and Chauhan, 2000; Takahashi et al., 2012; Lu et al., 2013). Thus, strategies for developing quantification and modulation of BDNF levels represent a viable approach for biomarker and treatment development, respectively, being useful for a variety of neurodegenerative diseases (Lu et al., 2013; Song et al., 2015). Post-mortem studies reveal that BDNF is significantly reduced in nigrostriatal dopaminergic neurons from PD patients (Mogi et al., 1999; Parain et al., 1999; Howells et al., 2000). A decrease of BDNF in serum from PD patients has also been observed (Wang et al., 2016). Also, BDNF offers neuroprotection of striatal neurons, and supporting studies have shown that BDNF levels are decreased in the brains of HD rodent models (Conforti et al., 2008) and patients (Ferrer et al., 2000; Zuccato and Cattaneo, 2007).
A study with 42 HD patients revealed that BDNF serum concentrations were significantly lower in patients compared to healthy controls (Ciammola et al., 2007). However, a later study analyzed 398 blood samples, indicating that mRNA and protein levels of BDNF between HD and healthy controls were not significantly different, questioning its potential as a biomarker for early diagnosis of HD (Zuccato et al., 2011).
Although the contribution of BDNF on PD and HD pathology is robust, no clinical trials are currently testing its safety and efficacy for the treatment of these diseases.
Classic studies indicate that PDGF has diverse functions in organs, including the stimulation of cell proliferation (Heldin and Westermark, 1999). Different isoforms of PDGF can be found in tissues, in which the PDGF-BB isoform has shown a protective effect in cultured dopaminergic neurons (Pietz et al., 1996). After the treatment of rats with 6-OHDA, PDGF-BB was increased, suggesting a compensatory response (Funa et al., 1996) and PDGF-BB injections induced functional recovery and provided neuroprotection of the nigrostriatal system in a PD mouse model (Zachrisson et al., 2011). PDGF-BB might be acting on neural progenitors and stem cells in the subventricular zone, promoting neurogenesis (Zachrisson et al., 2011). A study with 12 PD patients demonstrated that the administration of PDGF-BB into the brain ventricles for 2 weeks was well tolerated with no evident or aggressive side effects, and an increase in DAT binding was noted in the putamen of PDGF-BB-treated patients (Paul et al., 2015). Considering that PDGF-BB can stimulate neurogenesis, it may be possible to evaluate the co-treatment with PDGF-BB and other NTFs or drugs to restore the nigrostriatal pathway and promote neuroprotection in PD.
Both in vitro (Nakao et al., 1994) and in vivo reports (Sjöborg et al., 1998) have related the effect of PDGF in HD. PDGF-BB exerts trophic effects in developing rat DARPP32-positive striatal neurons in culture, suggesting the possibility that PDGF-BB might participate in the development and maintenance of striatal neurons in vivo, and could be used to modulate the neurodegeneration in HD models (Nakao et al., 1994). Then, the same group published the expression profile of PDGF in a rat model of HD, generated by unilateral intrastriatal ibotenic acid injections (Table 4). The evidence showed the accumulation of PDGF in astrocytes, suggesting a role of PDGF in a repair process in neurodegeneration (Sjöborg et al., 1998). Currently, no PDGF-based clinical trials are in course for HD treatment.
The IGF system is composed of insulin, IGF1, and IGF2, and its receptors: IR, IGF1R, and IGF2R, respectively (Cohen et al., 1991). The use of IGFs as therapy might represent a novel tool for the treatment of neurodegenerative disorders (Ebert et al., 2008). Studies in vitro and in preclinical models have demonstrated the neuroprotective effects of IGFs (Jarvis et al., 2007). Administration of IGF1 after injury reduced neuronal loss against several stressors such as oxidative stress, excitotoxicity, hypoxia, hypoglycemia, among others (Suh et al., 2013). Several studies using in vivo models of PD demonstrated beneficial effects of IGF1 treatment by preventing dopaminergic neuronal loss in the SN (Ebert et al., 2008), improving motor performance in a rat model of PD (Guan et al., 2000; Krishnamurthi et al., 2004).
IGFs have received interest due to its role in preventing and rescuing striatal neuronal damage which is observed in HD (Lewitt and Boyd, 2019). Increased IGF1 plasma levels were observed in the YAC128 mouse model of HD (Pouladi et al., 2010). Similarly, high IGF1 plasma levels were observed in HD patients and this was associated with cognitive impairment characteristic in this disorder (Saleh et al., 2010), however, the correlation between elevated IGF1 plasma levels and the motor and cognitive impairment in HD remains to be elucidated. Despite the high peripheral levels of IGF1, only small amounts of IGF1 cross the BBB into the brain. In this context, Lopes et al. (2014) showed that intranasal administration of IGF1 significantly improve motor function and restores metabolic changes in YAC128 mice model of HD, demonstrating that intranasal administration allows for the direct delivery of IGF1 into the CNS through the olfactory pathway.
In a recently published study, it has been demonstrated that the AAV administration of IGF2 into the striatum of YAC128 and R6/2 mice decreased the levels of mHTT and increased the levels of DARPP-32, a marker used to assess striatal neurons survival (García-Huerta et al., 2020). Interestingly, neuroprotective effects of IGF2 treatment have been described in preclinical models of others neurodegenerative diseases such as amyotrophic lateral sclerosis (Allodi et al., 2016), spinal muscular atrophy (Brown et al., 2009) and Alzheimer’s disease (Pascual-Lucas et al., 2014). However, today no clinical trials are studying the safety and efficacy of IGF2 as a possible treatment for PD or HD.
The cardinal motor symptoms of PD and chorea in HD are caused by the progressive degeneration of dopaminergic neurons in the SNpc (Figure 1B) and the loss of MSNs in the striatum (Figure 1C), respectively. In both cases, the motor impairment is attributed to alteration of functional connectivity of the striatum, a principal input of the basal ganglia (Albin et al., 1989).
Hyperkinetic movement disorders are characterized by uncontrollable and excessive motor activity, as chorea in HD. Reports published between 1987 and 1989 showed that blocking the activity of the STN produces hyperkinetic motor symptoms. Similar results are observed when the GABAergic inputs from the striatum are blocked, favoring the inhibitory (GABAergic) modulation of the GPe over the STN (Crossman, 1987; Crossman et al., 1988; Robertson et al., 1989). On the other hand, hypokinetic disorders like akinesia and bradykinesia have been described in PD. In this case, the decrease in striatal DA levels, as a result of the decrease in its synthesis, release, and reuptake (Lundblad et al., 2012) alter the corticostriatal balance causing an increase in the activity of the indirect pathway and reducing the activity of the direct pathway, that leads to a breakdown of the internal balance of the basal ganglia, and consequently the loss of movement control (Obeso et al., 2008; Galvan et al., 2015). These symptoms, unlike hyperkinetic movements, are treated with DA agonists as L-dopa (Cotzias et al., 1967, 1969). However, as stated in previous sections, the chronic use of this pharmacological therapy has a limited effect, which in the case of PD can induce a motor complication known as LID and on-off phenomenon (Fahn et al., 2004; Stacy, 2009; Ammal Kaidery et al., 2013; Cacabelos, 2017).
Among the therapeutic alternatives proposed in recent decades, electrical neuromodulation therapies, i.e., Deep Brain Stimulation (DBS) and Spinal Cord Stimulation (SCS) have emerged as interesting options for treatment of neurodegenerative pathologies associated with movement disorders such as PD and HD (Figure 2). The effect of stimulation on different basal ganglia nuclei in patients is described below.
DBS is a neurosurgical strategy based on the implantation of electrodes on subcortical nuclei that, through electrical signals, can modulate the neuronal activity of different regions of the brain. The use of DBS for the treatment of motor disorders was first proposed in 1971 by Natalia Bekhtereva (Bekhtereva et al., 1972; Hariz et al., 2010). But it was until 1996 when the FDA approved the stimulation of the ventral intermediate nucleus (VIM) of the thalamus for the treatment of essential tremor and severe tremor in PD. Later in 2002, the stimulation of the STN and GPi for the treatment of bradykinesia and rigidity in advanced cases of PD was included (Strotzer et al., 2019). It has been suggested that the stimulation of the STN induce the suppression of aberrant oscillatory synchronization at low frequency (13–35 Hz, beta band) and this contributes to motor amelioration, mainly bradykinesia and rigidity. However, more studies are necessary to understand the mechanisms involved. Currently, a group at the University of Minnesota is performing a clinical trial to investigate how the brain activity in PD patients is related to DBS and pharmacological management, by the chronic recording of local field potential (LFP) in the cortex (Table 1, NCT02709148).
The underlying molecular and cellular mechanisms of DBS are still not sufficiently identified and different assumptions about functional principles have been proposed (Jakobs et al., 2019). Studies in neurotoxin pre-clinical models of PD (Table 3) have suggested that STN-DBS can induce neuroprotective effects by reducing the loss of dopaminergic neurons (Maesawa et al., 2004; Temel et al., 2006; Wallace et al., 2007; Harnack et al., 2008; Spieles-Engemann et al., 2010; Wu et al., 2012). Additionally, it has been described that STN-DBS induces an increase in the striatal expression of BDNF (Spieles-Engemann et al., 2011), which is described as a powerful anti-apoptotic factor that favors the maintenance and survival of the nigral dopaminergic population (Zhao et al., 2017). As stated before, BDNF would exert its effect by signaling downstream of the TrkB receptor (Fischer et al., 2017). Nevertheless, reports in a genetic model of PD have revealed contradictory results. Musacchio et al. (2017) have reported that DBS reduces the loss of dopaminergic neurons, but it did not rescue the DA deficit. On the other side, Fischer et al. (2017) revealed that STN-DBS cannot counteract the axonopathy and dopaminergic loss progression induced by α-syn overexpression, but the differences in the starting time of application protocol must be considered. Moreover, the application of STN-DBS in pre-clinical models of PD has shown both metabolic and physiological effects on the nigrostriatal DA system, inducing an increase in the levels of striatal DA and its metabolites (Bruet et al., 2001; Meissner et al., 2003; He et al., 2014). Genetic screening has revealed that high frequency-DBS can induce a differential expression of genes involved in apoptosis, growth, and neuroplasticity (Lortet et al., 2013). Studies also revealed that DBS increased the activity of the enzyme TH (Meissner et al., 2003) and the expression of the D1 receptor (Carcenac et al., 2015).
For this reason, different ongoing clinical trials are assessing the long-term neurological, neurophysiological, and neuropsychological effects of DBS treatment (Table 1, NCT00053625, NCT03021031), as well as improving the administration of the treatment (Table 1, NCT03021031). The technical difficulty of STN-DBS in PD patients is that a case-by-case parameter adjustment is necessary. Therefore, different studies are looking for electrophysiology biomarkers that can predict and improve the effectiveness of DBS therapy, as well as new stimulation methodologies (directional DBS system) by invasive and non-invasive recording (Table 1, NCT03353688). Another limitation of DBS is the incapacity to alleviate the freezing of gait (FoG), a symptom observed in more than half of PD patients. Using electroencephalography (EEG) it is crucial to determine electrophysiological biomarkers present in FoG episodes that can be modulated by GPi-DBS or pedunculopontine nucleus stimulation (PPN-DBS, Table 1, NCT02548897). Furthermore, studies are evaluating different parameters of STN-DBS protocols to improve gait disorders (Table 1, NCT04184791, NCT02022735).
DBS of the GP is a promising alternative target for the treatment of chorea, motor symptoms classic in the early stages of HD (Albin et al., 1990). DBS has been considered an alternative to pallidotomy, a neurosurgical strategy for the treatment of choreic movement published in 1952, showing good effects in four HD patients (di Cianni et al., 1992). In 2004, Moro and colleagues (Moro et al., 2004) reported the first case of a 43-year-old patient with an 8-year history of HD submitted to bilateral GP stimulation. They found that stimulation at 40 Hz and 130 Hz improved motor symptoms, reducing chorea and dystonia, although the high frequency worsened bradykinesia. The improvement in the patient’s performance of a motor task was associated with an increase of the activation of the sensorimotor cortex, premotor cortex, supplementary motor area, and anterior cingulate cortex, although high frequency did not modulate the activity in premotor cortex (Moro et al., 2004). Moreover, the simultaneous stimulation of the STN and GPi has shown beneficial effects (Gruber et al., 2014), however, if the simultaneous stimulation has an additive effect is not clear. To date, two prospective, randomized, and double blind studies have been posted. One published in 2015 (Wojtecki et al., 2015), and others still on course (Table 2, NCT02535884). In the first study, six patients, four with chorea-dominant and two with Westphal-variant (rigid-hypokinetic syndrome, often associated with juvenile-onset of HD), underwent DBS treatment for 6 months and showed a reduction of chorea (Wojtecki et al., 2015). Nevertheless, the Westphal-variant patients did not show any improvements, suggesting that GP-DBS cannot ameliorate bradykinesia.
There is no doubt that optimizing DBS systems is a great challenge. Furthermore, understanding the correlation between abnormal brain activity and motor symptoms is necessary to obtain major beneficial results. For a more detailed review of this topic, we suggest some reviews published elsewhere (Anderson and Lenz, 2006; Miocinovic et al., 2013; Fasano and Lozano, 2015).
Generally used for the treatment of chronic pain since 1967 (Shealy et al., 1967; Dones and Levi, 2018), SCS consists of the application of electrical pulses in the dorsal columns of the spinal cord (directly in the epidural space). It has been suggested that the mechanism of action is based on the antidromic activation of the dorsal column fibers, which activate the inhibitory interneurons within the dorsal horn (Yampolsky et al., 2012). However, the exact mechanisms are not fully elucidated.
Currently, the use of this therapeutic strategy has gone beyond nociceptive control. In 2009, Fuentes and colleagues (Fuentes et al., 2009) proposed the use of SCS for the treatment of motor symptoms in PD. Like this report, in the last decade several studies have evaluated SCS effects in advanced cases of PD, showing interesting effects in axial symptoms (gait and postural dysfunction) both in preclinical (Santana et al., 2014) and clinical studies. These benefits are not observed with other treatments like DBS or L-dopa. The mechanism involved in the effects of SCS in motor symptoms of PD is not fully understood. It has been proposed that SCS, by releasing biphasic electrical pulses of high frequencies (300 Hz), would increase locomotor activity mainly through modulation of activity in the cortex and basal ganglia (Fuentes et al., 2009; Santana et al., 2014). This modulation would occur by activating the path of the dorsal columns, which in turn would modulate the activity of the thalamus, and from there to the cortex and striatum, causing the breakdown of aberrant low-frequency oscillations [beta (10–30 Hz)] observed in preclinical models (Fuentes et al., 2009; Santana et al., 2014) and PD patients (Kuhn et al., 2006), similarly as described for DBS treatment. Nevertheless, this does not explain the effect of SCS on gait and postural dysfunction. The participation of the brainstem, specifically the pedunculopontine nucleus (PPN), has been suggested, given its connection with different structures involved in the motor system, summarized by Chambers et al. (2019).
SCS treatment application methodologies differ among reports. The position of the electrodes varies between patients depending on the study. Electrodes can be placed at the cervical or thoracic level (Cai et al., 2020), showing that in general, both strategies ameliorate motor impairments. The first studies assessing SCS as a treatment for motor symptoms involved PD patients with chronic pain as a primary indication. In most cases, SCS reduced the pain and improved motor function, which could reflect a synergistic effect (Thevathasan et al., 2010; Fénelon et al., 2012; Hassan et al., 2013; Nishioka and Nakajima, 2015; Kobayashi et al., 2018; Mazzone et al., 2019). Recently, Samotus et al. (2018) showed the effectiveness of SCS (300–400 μs/30–130 Hz) in the treatment of FoG and gait in five patients without pain for 6 months. This evidence is encouraging for the search of treatments for symptoms that, to date, have not been satisfactorily controlled. This is the case for gait dysfunctions that affect nearly 40–60% of PD patients and are not improved by dopaminergic therapy (Giladi et al., 2001). Additionally, Pinto de Souza et al. (2017) reported the use and effectiveness of SCS as a complementary strategy to rescue the loss of efficacy of DBS and DA medication. But other studies are currently in a course to evaluate the efficacy of SCS in gait disorders (Table 1, NCT03079310).
To date, three studies in preclinical models have suggested that SCS could counteract the progression of PD through a neuroprotective effect. Shinko et al. (2014) showed in rats that SCS applied regularly for 2 weeks (one session of 1 h daily) starting 2 days before 6-OHDA injection, decreases dopaminergic neuronal loss by 20–25%, and increase the expression of VEGF, which could favor neuroprotection given its ability to reduce dopaminergic neuronal death by suppressing apoptosis (Yasuhara et al., 2004). Similarly, Yadav et al. (2014) demonstrated that the application of an SCS protocol for 6 weeks (two sessions of 30 min per week) that started a week post-6-OHDA injection reduced the loss of dopaminergic neurons by 10%. Furthermore, both studies showed a positive effect in preserving the dopaminergic innervation of the striatum, reducing its loss by 30–35%. Recently, an investigation showed that continuous application of SCS (24 h ON SCS), starting immediately after injection of 6-OHDA, reduced the loss of dopaminergic neurons and their striatal projections by ~35% and ~32% respectively, while a regular SCS protocol (8 h SCS ON/16 h SVS OFF) counteracted the degeneration of the nigrostriatal dopaminergic pathway only by ~23% (Kuwahara et al., 2020). Thus, the periodicity of the treatment application reflects on different degrees of neuroprotection (Kuwahara et al., 2020). These data suggest that SCS could have a neuroprotective effect that might contribute to the relief of the observed motor symptoms in PD, given the relevance of dopaminergic projections in modulating the functioning of the circuit of the nuclei of the base through the regulation of cortical and subcortical neuronal activity during movement (Gerfen and Surmeier, 2011; Canessa et al., 2016). Nevertheless, additional studies are necessary to understand the mechanism involved in the beneficial effects associated with SCS in the long-term. More detailed reviews on this topic can be found elsewhere (de Andrade et al., 2016; Cai et al., 2020).
For HD, SCS treatment has not been suggested, probably because SCS has better effects in alleviating gait and posture problems, while DBS is still the best option for the treatment of involuntary movements, such as chorea.
For the development of new therapies for PD and HD, it is important to include, especially for HD and genetic forms of PD, genetic correction/editing of the mutated gene(s). Nowadays, there are several gene silencing/editing technologies, including RNA interference (RNAi), antisense oligonucleotides (ASO), and clustered interspaced short palindromic repeats (CRISPR/cas9), which can be used as therapies for the treatment of PD and HD. For a more in-depth knowledge of gene therapy delivery systems and other cellular targets, reviews are published elsewhere (Sudhakar and Richardson, 2019; Chen et al., 2020).
As previously stated, PD is characterized by the selective degeneration of dopaminergic neurons in the SN, thus approaches aiming to revert this loss based on the delivery of genes encoding for enzymes required for DA synthesis could be useful. The first enzyme for DA synthesis is TH, which requires the enzyme GTP-cyclohydrolase-1 (GCH-1) to synthesize a cofactor for DA biosynthesis (Daubner et al., 2011). TH converts tyrosine into L-dopa, which finally is converted into DA by the aromatic L-amino acid decarboxylase (AADC; Hadjiconstantinou and Neff, 2008). Therapies to deliver enzymes involved in DA synthesis have been proved in preclinical and clinical studies showing its benefits.
Initially, gene therapy was based on the delivery of separate AAV vectors to transfer two or three enzymes critical for DA biosynthesis. These strategies showed behavioral benefits in rat and non-human primate PD models (Kirik et al., 2002a; Muramatsu et al., 2002). Furthermore, a clinical study in patients with moderate to advanced PD demonstrated the safety and tolerability of a 6 months treatment with a bilateral Ipu of AAV vector encoding for the human AADC gene (AAV-hAADC; Eberling et al., 2008). Importantly, PET scans using an AADC tracer demonstrated an increase in gene expression throughout the study. Similarly, administrating the AAV-hAADC vector in the putamen of PD patients showed to be safe and tolerable (Muramatsu et al., 2010). This study demonstrated the efficacies of AAV vectors for gene delivery, which persisted up to 2 years with a 46% improvement of the UPDRS motor scores (Muramatsu et al., 2010). Another completed study showed that bilateral Ipu infusion of the AAV-hAADC vector improves UPDRS mean scores by 30% in PD patients after 6 months (Table 1, NCT00229736). However, this study reported 3 cases in which surgical procedures caused intracranial hemorrhage, showing an important limitation of the surgical procedure, but not necessarily of the therapeutic strategy of gene delivery (Christine et al., 2009). Consequently, a long-term evaluation study of AAV-hAADC demonstrated stable transgene expression over 4 years after vector delivery in PD patients (Mittermeyer et al., 2012).
To potentiate the effects and benefits of the enzyme therapy for DA synthesis, researchers have developed a strategy using a simple vector, which carries the genes that encode for the three key enzymes for DA biosynthesis. One of these strategies uses a lentiviral-based vector derived from the equine infectious anemia virus (EIAV; Azzouz et al., 2002). This tricistronic lentivirus vector encodes TH, AADC and CH1 (Lenti-TH-AADC-CH1) in a single vector (ProSavin). Delivery of Lenti-TH-AADC-CH1 vector into the striatum of a non-human primate PD model restored extracellular concentrations of DA and improved motor function for up to 12 months (Jarraya et al., 2009). Furthermore, ProSavin was administered into the striatum of PD patients, demonstrating a significant improvement in mean UPDRS motor scores at 6 months post-treatment, with no adverse effects detected (Table 1, NCT00627588). Additionally, a long-term study of ProSavin showed that the treatment was well-tolerated and safe, but its clinical benefits are still under observation after 4 years (Palfi et al., 2014).
Other alternatives using gene delivery for PD treatment are focused on lowering α-syn levels in dopaminergic neurons. One study used a ribozyme combined with an AAV vector (rAAV-SynRz). Nigrostriatal injection of rAAV-SynRz in MPP+-treated adult rats, which has increased expression of α-syn (Kühn et al., 2003), resulted in down-regulation of α-syn, preventing its accumulation in the SN, and significantly protected TH-positive neurons (Hayashita-Kinoh et al., 2006). Despite these positive results, no clinical trials using gene therapy for silencing α-syn to treat PD are being conducted.
One important branch of investigation regarding HD therapies is the reduction of HTT DNA, RNA, and/or protein levels. Using a conditional transgenic mouse model of HD, it has been demonstrated that the reduction of mHTT ameliorates motor and psychiatric-like deficits, forebrain weight loss, cortical and striatal volume decrease, presynaptic and postsynaptic markers changes, and electrophysiological changes in these mice (Wang et al., 2014). One possible alternative to reduce mHTT levels is the use of DNA-targeting strategies (Ambrose et al., 1994). Thus, permanent and selective deletion of mHTT gene could be an interesting therapeutic approach for HD with no negative effect on patient health. The CRISPR/Cas9 system has been investigated for its utility for HD therapy but is still in the preclinical stage. Using fibroblasts (Shin et al., 2016; Monteys et al., 2017; Dabrowska et al., 2018) and iPSCs-derived NPCs (Shin et al., 2016) from an HD patient, it has been demonstrated that the selective deletion of a fragment of the mHTT gene using the CRISPR/Cas9 strategy is possible. This approach led to a near-complete reduction of mHTT protein and left intact the wild type HTT gene (Shin et al., 2016). Furthermore, using CRISPR/Cas9 and a transposon-based approach, the precise correction of the CAG expansion in HD iPSCs has been achieved (Xu et al., 2017). These cells retain pluripotency, have a normal karyotype, and could be differentiated into NPCs and MSNs-like neurons, which presented some electrical properties of neurons. Interestingly, the corrected cells were deprived of some phenotypic abnormalities observed in the HD iPSC-derived neurons, including increased susceptibility to growth factor withdrawal, impaired neural rosette formation, and mitochondrial dysfunction (Xu et al., 2017).
Using BACHD mice (Table 4) it has been shown that mHTT expression is reduced when Cas9 and a single guide RNA (sgRNA) are delivered using AAVs in the striatum, causing the deletion of a fragment around exon 1 (Monteys et al., 2017). Likewise, AAV-mediated injection of Cas9 and 2 sgRNA into the striatum of HD140Q-KI mice (Table 4) achieved a significant reduction in mHTT levels, accompanied by improved motor performance, decrease in reactive astrocytes and attenuation of bodyweight reduction (Yang et al., 2017). Similar results have been found using the R6/2 mice model, in which AAV-mediated delivery of Cas9 and sgRNA into the striatum caused a near 40% reduction in mHTT inclusions and around a 50% decrease in mHTT protein levels (Ekman et al., 2019). These mice presented increased mean survival, better motor performance, and decreased hindlimb clasping, an established indicator of dystonia (Ekman et al., 2019).
Despite the promising results, there is still a need for validation of this approach for human research. Given that many studies use CRISPR/Cas9 technology based in the recognition of short repeat sequences in the DNA, intensive studies must be performed to find these short sequences in the mHTT gene that is not present in another codifying gene sequence, so off-target genomic removal could be avoided. Moreover, single-nucleotide changes can occur in the mHTT gene in every patient affected, therefore sequencing of the mutant gene would be necessary for each patient. Another important issue of using CRISPR/Cas9 technology in humans is that the deletion of the mutated gene is permanent and irreversible. Also, the transduction of Cas9, a protein of bacterial origin, could activate the patients’ immune system and edited cells would be effectively eliminated (Wignakumar and Fairchild, 2019). Despite this, CRISPR/Cas9-mediated deletion of the mHTT gene could be perfectly coupled with cell replacement strategies, with a correction of the mutation.
A second strategy for lowering mHTT levels is based on RNA-targeting strategies. One of the most studied methods to achieve this is through RNAi. These include the use of microRNAs (miRNAs) and short hairpin RNAs (shRNAs), all using the Dicer-RISC machinery for the targeted degradation of mRNAs (Ghosh and Tabrizi, 2017). Initially, using shRNAs and HD-N171-82Q HD mice (Table 4), it was shown a partial reduction of mHTT mRNA by 50% and decreased mHTT protein accumulation in the striatum, accompanied by a significant improvement in motor performance (Harper et al., 2005). Similarly, using R6/1 HD mice (Table 4), it has been demonstrated that AAV-mediated delivery of shRNAs against mHTT significantly reduces mHTT mRNA and protein levels (Rodriguez-Lebron et al., 2005). shRNA-injected striatum had smaller and less intense intranuclear mHTT inclusions, which was accompanied by increased MSNs mRNA markers. However, this group showed just a mild effect on the clasping phenotype observed in these mice (Rodriguez-Lebron et al., 2005).
These and other studies have demonstrated that the reduction of mHTT using RNAi in the brain of HD mice decreases both mHTT mRNA and protein levels, which is accompanied by improved motor behavior. However, in these studies the RNAi was designed to target human mHTT, leaving endogenous mouse HTT unaltered. This represents a problem when we think about patients’ treatment, given that both alleles differ in the expanded polyQ in the mutated gene and different single nucleotide polymorphisms (SNPs) present. Even though several SNPs have been identified to be differentially present in the mutant and wild type allele, this accounts just for 80% of the population with HD, leaving an important number of patients without a therapeutic alternative (Pfister et al., 2009). Therefore, RNAi has been tested for the reduction of both mHTT and wild type HTT mRNA. Using an HD mouse model that expresses both mouse wild type and human mutant HTT, non-allele specific miRNA-mediated knockdown of both mRNAs in the striatum can significantly improve motor coordination (Boudreau et al., 2009). Notably, these mice survived and had no phenotypic changes after a 75% reduction of wild type HTT for 4 months (Boudreau et al., 2009). Using AAV-mediated miRNA injection into the putamen of rhesus monkeys to target both HTT and mHTT mRNAs, it has been demonstrated that a 45% reduction of HTT mRNA did not induce motor deficits in these animals, nor neurodegeneration, astrogliosis, microglial activation or neuroinflammation (McBride et al., 2011). These phenotypes were also maintained in rhesus monkeys treated with AAV-shRNA for HTT mRNA for 6 months (Grondin et al., 2012).
One important limitation of double-stranded RNAs (shRNAs and miRNAs) is that they are not capable of crossing the BBB and do not easily cross the plasma membrane of cells. Given that they demonstrated positive effects when targeted to the brain, methods to deliver these molecules are under study (Chernikov et al., 2019). Despite this limitation, in 2019 the first clinical trial using AAV5-mediated delivery of a miRNA against HTT was initiated (Table 2, NCT04120493). Early manifest HD patients will be assessed for motor and cognitive functions, with miRNA and exploratory biomarkers measured in the CSF. The same AAV5-miRNA was proven to lower mHTT mRNA specifically in rat and minipig HD model. Suppression of mHTT mRNA levels was associated with lower mHTT protein levels, inclusion formation, and neuronal dysfunction (Miniarikova et al., 2017; Evers et al., 2018).
Another strategy used for RNA targeting is through ASOs. These are short, synthetic single-stranded DNA molecules that are complementary to a pre-mRNA target sequence in the nucleus, and have been widely studied for the treatment of neurological disorders (Rinaldi and Wood, 2018). Using IONIS-HTTRx against HTT mRNA, Tabrizi et al. (2019) reported the first results obtained from a phase I–II multicenter clinical trial in which this ASO was intrathecally administered in early-manifest HD patients. IONIS-HTTRx demonstrated to be safe and well-tolerated, and importantly, mHTT levels in the cerebrospinal fluid (CSF) of patients treated with IONIS-HTTRx decreased dose-dependently. In this trial, no changes in motor and cognitive functions were observed between the placebo and the treated group, which can be explained by the slow progression of the disease and the narrow window of time in which these changes were measured (Tabrizi et al., 2019). Some of these patients participated in a 15-month extension study, which ended by October 2019 (Table 2, NCT03342053). The original clinical trial is now under phase III, with more than 900 patients in 101 locations around the world, which is expected to end by September 2022 (Table 2, NCT03761849).
A second and third phase Ib/IIa clinical trials are underway to evaluate single and multiple doses of two ASOs against two specific SNPs found in the mHTT gene. PRECISION-HD1 clinical trial will evaluate the ASO WVE-120101 against the SNP rs362307 (Table 2, NCT03225833) and PRECISION-HD2 clinical trial will use the ASO WVE-120102 against the SNP rs362331 (Table 2, NCT03225846). These multicenter clinical trials, in which 60 patients with early-manifest HD are participating, started in 2017 and are expected to finish by the end of this year.
The results of the IONIS-HTTRx clinical trial are encouraging and bring us closer to therapy for HD. Given that ASOs bind directly to the DNA sequence, it is less likely that off-target suppression occurs. Moreover, as in the PRECISION clinical trials, unique SNPs found specifically in the mHTT DNA sequence allows for the design of specific ASOs, avoiding off-target suppression. However, the use of ASOs has disadvantages. In the PRECISION clinical trials, the ASOs evaluated target specific SNPs found in the mHTT gene, but they are not present in all HD patients. On the other hand, the IONIS-HTTRx clinical trial targets both wild type and mHTT genes, so it can be used in all HD patients. However, to date, there is no data available on the possible effects of wild type HTT suppression in patients. As mentioned before, non-human primates with sustained suppression of wild type HTT did not show adverse effects in motor and cognitive skills (McBride et al., 2011; Grondin et al., 2012), results that could be escalated to humans. Nevertheless, evaluation of long-term suppression of wild type HTT is key in HD patients. Also, repeated administration of ASOs could be necessary to maintain therapeutic benefits.
PD and HD are movement disorders characterized by the presence of aberrant and unwanted involuntary movements. The main cause for the variety of motor symptoms observed in patients is the selective neuronal loss in brain areas implicated in movement fine-tuning. Despite this knowledge, current treatments are not able to stop, reverse, or slow down PD and HD. Current treatments are directed to keep to line the motor symptoms, but with limited efficacy. Thus, continuous strong efforts are being made for the development of new therapeutic strategies for both diseases. Moreover, the development of small molecules for the treatment of these neurodegenerative diseases has a particularly high failure rate. Thus, strong efforts are being made for the development of new therapeutic strategies for both diseases.
Currently, no approved drugs can modify the progression of either PD or HD. Nevertheless, pharmacology provided therapies to reestablish DA levels in PD patients and to reduce DA neurotransmission in HD patients. Unfortunately, different and aggressive side effects appear during these treatments. New formulations have been developed to control the additional psychomotor and autonomic complications produced by some anti-parkinsonians drugs, however, it is important to reduce all the events that can affect or deteriorate the quality life of patients, and maybe the answers are out of the classical pharmacology approach.
Among the first strategies used for the treatment of PD and HD were cellular replacement therapies, in which fetal tissue was transplanted into the patient’s brain. Even though some functional recovery was observed, this strategy had several limitations. The number of fetuses used generates important religious and ethical concerns. Besides, in some cases, tumor formation was observed in the site of transplantation, and most of the studies required immunosuppressive therapy. Although new strategies have been developed for obtaining dopaminergic and GABAergic neurons from ESCs and iPSCs, these have not yet escalated into clinical trials.
Deregulation of NTFs’ levels has been reported in PD and HD patients, and the administration of NTFs has shown beneficial effects in patients with these diseases. The reported neuroprotective effect can be explained by the prevention of neurodegeneration, restoring, and delaying the occurrence of motor symptoms. Moreover, therapy with NTFs has proven to be effective not only in preclinical stages but has also shown a positive effect in clinical trials. However, one main limitation of NTFs therapy is its poor brain availability after oral administration, which can only be solved by direct administration to the brain. Clinical studies directly administrating NTFs to the brain have reported side effects such as nausea, vomiting, headache, as well as infection when catheter implantation is used. Despite side effects, these studies have observed promising effects with this therapy, however, improvement and development of new delivery methods are key for successful NTF-base treatments.
Without a doubt, the development of electrical neuromodulatory therapies to treat motor symptoms in PD and HD has brought encouraging news for patients, doctors, and their families. Clinical reports carried out in recent decades have validated the use of DBS or SCS as a complement to classical pharmacological therapies that, due to their chronic use, decrease their effectiveness or induce more complex motor symptoms. Additionally, studies in PD patients have reported that the simultaneous use of DBS and SCS would bring important benefits, since DBS is effective in relieving motor symptoms such as tremor, bradykinesia, and stiffness, while SCS has to relieve symptoms associated with walking and posture. Although this has not been validated in the clinic, it opens the discussion about the possibility of using both strategies for the synergistic treatment of cardinal and axial symptoms of PD. When it comes to HD treatment, DBS has emerged as a promising alternative after showing positive effects similar to pallidotomy, reducing choreic movements and dystonia, but with the advantage of being a flexible option that adjusts to the patient’s requirements. Since the mechanisms by which both strategies exert their effect have not been fully elucidated, their use requires personalized patient-to-patient care throughout the treatment, complicating its application in countries with weak health systems. Moreover, given their surgical complexity and high monetary cost, these therapies continue to be considered as a last option, even when reports in pre-clinical models, particularly in the case of PD, have reported encouraging results regarding their effect in counteracting neurodegeneration processes.
Worldwide excitement among HD patients and their families arose after the report of a promising potential therapy (IONIS-HTTRx) that was able to decrease mHTT levels in the CSF of patients. Now, this treatment is under phase III clinical trial, in which patients of more than 100 centers around the world have been recruited. This is by far the closest that we have got to a definite treatment for HD. Nevertheless, many questions need to be answered during this clinical trial to ensure safety for patients. Given that IONIS-HTTRx targets both HTT and mHTT, one important question is whether long-term suppression of HTT is deleterious for neurons and if patients present improvements in motor and cognitive function.
Along with the development of new therapeutic approaches, like the ones described in this review, comes the important need to assess therapy response and disease progression. A key line of investigation is focused on finding new and reliable biomarkers that allow assessing changes in the disease course, helping in the evaluation of therapeutic response. Several biomarkers have been described for both PD and HD patients, but global multicenter studies are necessary to validate those biomarkers worldwide. Another important aspect to be considered in the development of new therapies is the heterogeneity of symptoms observed in both PD and HD patients, which suggest that not all treatments are the best option for all cases, as well as not all treatments are the best options for the same symptoms, suggesting that future clinical approaches will personalize the treatment or combine the strategies presented in this article to increase their efficacy and/or reduce its adverse effects.
Altogether, the presented data highlights the idea that we are on the right track for the development of new therapeutic strategies for PD and HD, but there is still a long road ahead to find a definitive treatment that may stop the progression or even cure the disease.
PT-E, DS, RP-A, AP, JA, and RV wrote and edited the manuscript. RV and FG critically analyzed the data. PT-E, AP, JA, and RV prepared the figures and tables. RV planned the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by Fondo Nacional de Desarrollo Científico, Tecnológico y de Innovación Tecnológica (FONDECYT) 1191003, Millennium Institute No. P09-015-F, and FONDAP 15150012 (RV), CONICYT PhD fellowship 21160843 (PT-E), and CONICYT PhD fellowship 21181114 (AP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Addis, R. C., Hsu, F. C., Wright, R. L., Dichter, M. A., Coulter, D. A., and Gearhart, J. D. (2011). Efficient conversion of astrocytes to functional midbrain dopaminergic neurons using a single polycistronic vector. PLoS One 6:e28719. doi: 10.1371/journal.pone.0028719
Adil, M. M., Gaj, T., Rao, A. T., Kulkarni, R. U., Fuentes, C. M., Ramadoss, G. N., et al. (2018). hPSC-derived striatal cells generated using a scalable 3D hydrogel promote recovery in a huntington disease mouse model. Stem Cell Reports 10, 1481–1491. doi: 10.1016/j.stemcr.2018.03.007
Agid, Y. (1991). Parkinson’s disease: pathophysiology. Lancet 337, 1321–1324. doi: 10.1016/0140-6736(91)92989-f
Albin, R. L., Reiner, A., Anderson, K. D., Penney, J. B., and Young, A. B. (1990). Striatal and nigral neuron subpopulations in rigid Huntington’s disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann. Neurol. 27, 357–365. doi: 10.1002/ana.410270403
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. doi: 10.1016/0166-2236(89)90074-x
Allodi, I., Comley, L., Nichterwitz, S., Nizzardo, M., Simone, C., Benitez, J. A., et al. (2016). Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 6:25960. doi: 10.1038/srep25960
Ambrose, C. M., Duyao, M. P., Barnes, G., Bates, G. P., Lin, C. S., Srinidhi, J., et al. (1994). Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet. 20, 27–38. doi: 10.1007/BF02257483
Ammal Kaidery, N., Tarannum, S., and Thomas, B. (2013). Epigenetic landscape of Parkinson’s disease: emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 10, 698–708. doi: 10.1007/s13311-013-0211-8
An, M. C., Zhang, N., Scott, G., Montoro, D., Wittkop, T., Mooney, S., et al. (2012). Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11, 253–263. doi: 10.1016/j.stem.2012.04.026
Anderson, W. S., and Lenz, F. A. (2006). Surgery insight: deep brain stimulation for movement disorders. Nat. Clin. Pract. Neurol. 2, 310–320. doi: 10.1038/ncpneuro0193
Araujo, D. M., and Hilt, D. C. (1997). Glial cell line-derived neurotrophic factor attenuates the excitotoxin-induced behavioral and neurochemical deficits in a rodent model of Huntington’s disease. Neuroscience 81, 1099–1110. doi: 10.1016/s0306-4522(97)00079-1
Araujo, D. M., and Hilt, D. C. (1998). Glial cell line-derived neurotrophic factor attenuates the locomotor hypofunction and striatonigral neurochemical deficits induced by chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid. Neuroscience 82, 117–127. doi: 10.1016/s0306-4522(97)00266-2
Arenas, E., Trupp, M., Akerud, P., and Ibáñez, C. F. (1995). GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 15, 1465–1473. doi: 10.1016/0896-6273(95)90024-1
Azzouz, M., Martin-Rendon, E., Barber, R. D., Mitrophanous, K. A., Carter, E. E., Rohll, J. B., et al. (2002). Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson’s disease. J. Neurosci. 22, 10302–10312. doi: 10.1523/JNEUROSCI.22-23-10302.2002
Bakay, R. A., Fiandaca, M. S., Barrow, D. L., Schiff, A., and Collins, D. C. (1985). Preliminary report on the use of fetal tissue transplantation to correct MPTP-induced Parkinson-like syndrome in primates. Appl. Neurophysiol. 48, 358–361. doi: 10.1159/000101157
Ball, N., Teo, W. P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Barde, Y. A., Edgar, D., and Thoenen, H. (1982). Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x
Barker, R. A., Götz, M., and Parmar, M. (2018). New approaches for brain repair-from rescue to reprogramming. Nature 557, 329–334. doi: 10.1038/s41586-018-0087-1
Bekhtereva, N. P., Bondarchuk, A. N., Smirnov, V. M., and Meliucheva, L. A. (1972). [Therapeutic electric stimulation of deep brain structures]. Vopr. Neirokhir. 36, 7–12.
Berninger, B., Costa, M. R., Koch, U., Schroeder, T., Sutor, B., Grothe, B., et al. (2007). Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J. Neurosci. 27, 8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Björklund, A., and Stenevi, U. (1979). Reconstruction of the nigrostriatal dopamine pathway by intracerebral nigral transplants. Brain Res. 177, 555–560. doi: 10.1016/0006-8993(79)90472-4
Booth, H. D. E., Hirst, W. D., and Wade-Martins, R. (2017). The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 40, 358–370. doi: 10.1016/j.tins.2017.04.001
Bordelon, Y. M., Chesselet, M. F., Nelson, D., Welsh, F., and Erecinska, M. (1997). Energetic dysfunction in quinolinic acid-lesioned rat striatum. J. Neurochem. 69, 1629–1639. doi: 10.1046/j.1471-4159.1997.69041629.x
Boudreau, R. L., McBride, J. L., Martins, I., Shen, S., Xing, Y., Carter, B. J., et al. (2009). Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 17, 1053–1063. doi: 10.1038/mt.2009.17
Bramham, C. R., and Messaoudi, E. (2005). BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog. Neurobiol. 76, 99–125. doi: 10.1016/j.pneurobio.2005.06.003
Brown, J., Jones, E. Y., and Forbes, B. E. (2009). Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends Biochem. Sci. 34, 612–619. doi: 10.1016/j.tibs.2009.07.003
Bruet, N., Windels, F., Bertrand, A., Feuerstein, C., Poupard, A., and Savasta, M. (2001). High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J. Neuropathol. Exp. Neurol. 60, 15–24. doi: 10.1093/jnen/60.1.15
Burns, R. S., Chiueh, C. C., Markey, S. P., Ebert, M. H., Jacobowitz, D. M., and Kopin, I. J. (1983). A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. U S A 80, 4546–4550. doi: 10.1073/pnas.80.14.4546
Cacabelos, R. (2017). Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 18:551. doi: 10.3390/ijms18030551
Cai, Y., Reddy, R. D., Varshney, V., and Chakravarthy, K. V. (2020). Spinal cord stimulation in Parkinson’s disease: a review of the preclinical and clinical data and future prospects. Bioelectron. Med. 6:5. doi: 10.1186/s42234-020-00041-9
Caiazzo, M., Dell’Anno, M. T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D., et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. doi: 10.1038/nature10284
Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V., and Di Filippo, M. (2014). Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030. doi: 10.1038/nn.3743
Campbell, K., Kalén, P., Wictorin, K., Lundberg, C., Mandel, R. J., and Björklund, A. (1993). Characterization of GABA release from intrastriatal striatal transplants: dependence on host-derived afferents. Neuroscience 53, 403–415. doi: 10.1016/0306-4522(93)90204-s
Canessa, A., Pozzi, N. G., Arnulfo, G., Brumberg, J., Reich, M. M., Pezzoli, G., et al. (2016). Striatal dopaminergic innervation regulates subthalamic beta-oscillations and cortical-subcortical coupling during movements: preliminary evidence in subjects with Parkinson’s disease. Front. Hum. Neurosci. 10:611. doi: 10.3389/fnhum.2016.00611
Carcenac, C., Favier, M., Vachez, Y., Lacombe, E., Carnicella, S., Savasta, M., et al. (2015). Subthalamic deep brain stimulation differently alters striatal dopaminergic receptor levels in rats. Mov. Disord. 30, 1739–1749. doi: 10.1002/mds.26146
Carrera, I., and Cacabelos, R. (2019). Current drugs and potential future neuroprotective compounds for Parkinson’s disease. Curr. Neuropharmacol. 17, 295–306. doi: 10.2174/1570159x17666181127125704
Carter, R. L., Chen, Y., Kunkanjanawan, T., Xu, Y., Moran, S. P., Putkhao, K., et al. (2014). Reversal of cellular phenotypes in neural cells derived from Huntington’s disease monkey-induced pluripotent stem cells. Stem Cell Reports 3, 585–593. doi: 10.1016/j.stemcr.2014.07.011
Chambers, N. E., Lanza, K., and Bishop, C. (2019). Pedunculopontine nucleus degeneration contributes to both motor and non-motor symptoms of Parkinson’s disease. Front. Pharmacol. 10:1494. doi: 10.3389/fphar.2019.01494
Chen, W., Hu, Y., and Ju, D. (2020). Gene therapy for neurodegenerative disorders: advances, insights and prospects. Acta Pharm. Sin. B doi: 10.1016/j.apsb.2020.01.015 [Epub ahead of print].
Chernikov, I. V., Vlassov, V. V., and Chernolovskaya, E. L. (2019). Current development of siRNA bioconjugates: from research to the clinic. Front. Pharmacol. 10:444. doi: 10.3389/fphar.2019.00444
Christine, C. W., Starr, P. A., Larson, P. S., Eberling, J. L., Jagust, W. J., Hawkins, R. A., et al. (2009). Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669. doi: 10.1212/WNL.0b013e3181c29356
Ciammola, A., Sassone, J., Cannella, M., Calza, S., Poletti, B., Frati, L., et al. (2007). Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 574–577. doi: 10.1002/ajmg.b.30501
Cohen, P., Peehl, D. M., Lamson, G., and Rosenfeld, R. G. (1991). Insulin-like growth factors (IGFs), IGF receptors and IGF-binding proteins in primary cultures of prostate epithelial cells. J. Clin. Endocrinol. Metab. 73, 401–407. doi: 10.1210/jcem-73-2-401
Conforti, P., Ramos, C., Apostol, B. L., Simmons, D. A., Nguyen, H. P., Riess, O., et al. (2008). Blood level of brain-derived neurotrophic factor mRNA is progressively reduced in rodent models of Huntington’s disease: restoration by the neuroprotective compound CEP-1347. Mol. Cell. Neurosci. 39, 1–7. doi: 10.1016/j.mcn.2008.04.012
Cooper, J. R., Bloom, F. E., and Roth, R. H. (2004). The Biochemical Basis of Neuropharmacology. New York, NY: Oxford University Press.
Cotzias, G. C., Papavasiliou, P. S., and Gellene, R. (1969). Modification of Parkinsonism—chronic treatment with L-dopa. N. Engl. J. Med. 280, 337–345. doi: 10.1056/NEJM196902132800701
Cotzias, G. C., Van Woert, M. H., and Schiffer, L. M. (1967). Aromatic amino acids and modification of Parkinsonism. N. Engl. J. Med. 276, 374–379. doi: 10.1056/nejm196702162760703
Coyle, J. T., Molliver, M. E., and Kuhar, M. J. (1978). In situ injection of kainic acid: a new method for selectively lesioning neural cell bodies while sparing axons of passage. J. Comp. Neurol. 180, 301–323. doi: 10.1002/cne.901800208
Crossman, A. R. (1987). Primate models of dyskinesia: the experimental approach to the study of basal ganglia-related involuntary movement disorders. Neuroscience 21, 1–40. doi: 10.1016/0306-4522(87)90322-8
Crossman, A. R., Mitchell, I. J., Sambrook, M. A., and Jackson, A. (1988). Chorea and myoclonus in the monkey induced by γ-aminobutyric acid antagonism in the lentiform complex. The site of drug action and a hypothesis for the neural mechanisms of chorea. Brain 111, 1211–1233. doi: 10.1093/brain/111.5.1211
Dabrowska, M., Juzwa, W., Krzyzosiak, W. J., and Olejniczak, M. (2018). Precise excision of the CAG tract from the huntingtin gene by Cas9 nickases. Front. Neurosci. 12:75. doi: 10.3389/fnins.2018.00075
Dalby, M. A. (1969). Effect of tetrabenazine on extrapyramidal movement disorders. Br. Med. J. 2, 422–423. doi: 10.1136/bmj.2.5654.422
Daubner, S. C., Le, T., and Wang, S. (2011). Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 508, 1–12. doi: 10.1016/j.abb.2010.12.017
Dean, M., and Sung, V. W. (2018). Review of deutetrabenazine: a novel treatment for chorea associated with Huntington’s disease. Drug Des. Devel. Ther. 12, 313–319. doi: 10.2147/dddt.s138828
de Andrade, E. M., Ghilardi, M. G., Cury, R. G., Barbosa, E. R., Fuentes, R., Teixeira, M. J., et al. (2016). Spinal cord stimulation for Parkinson’s disease: a systematic review. Neurosurg. Rev. 39, 27–35; discussion 35. doi: 10.1007/s10143-015-0651-1
Deckel, A. W., Robinson, R. G., Coyle, J. T., and Sanberg, P. R. (1983). Reversal of long-term locomotor abnormalities in the kainic acid model of Huntington’s disease by day 18 fetal striatal implants. Eur. J. Pharmacol. 93, 287–288. doi: 10.1016/0014-2999(83)90150-4
di Cianni, G., Benzi, L., Ciccarone, A. M., Cecchetti, P., and Navalesi, R. (1992). Bed time NPH-insulin plus combined sulfonylurea-biguanide oral therapy for treating refractory non insulin dependent diabetic patients. Diabete Metab. 18, 468–469.
Dones, I., and Levi, V. (2018). Spinal cord stimulation for neuropathic pain: current trends and future applications. Brain Sci. 8:138. doi: 10.3390/brainsci8080138
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., et al. (2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68, 384–386. doi: 10.1212/01.wnl.0000247740.47667.03
Double, K. L. (2012). Neuronal vulnerability in Parkinson’s disease. Parkinsonism Relat. Disord. 18, S52–S54. doi: 10.1016/S1353-8020(11)70018-9
Duan, W. M., Widner, H., and Brundin, P. (1995). Temporal pattern of host responses against intrastriatal grafts of syngeneic, allogeneic or xenogeneic embryonic neuronal tissue in rats. Exp. Brain Res. 104, 227–242. doi: 10.1007/bf00242009
Dubé, L., Smith, A. D., and Bolam, J. P. (1988). Identification of synaptic terminals of thalamic or cortical origin in contact with distinct medium-size spiny neurons in the rat neostriatum. J. Comp. Neurol. 267, 455–471. doi: 10.1002/cne.902670402
Eberling, J. L., Jagust, W. J., Christine, C. W., Starr, P., Larson, P., Bankiewicz, K. S., et al. (2008). Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 70, 1980–1983. doi: 10.1212/01.wnl.0000312381.29287.ff
Ebert, A. D., Beres, A. J., Barber, A. E., and Svendsen, C. N. (2008). Human neural progenitor cells over-expressing IGF-1 protect dopamine neurons and restore function in a rat model of Parkinson’s disease. Exp. Neurol. 209, 213–223. doi: 10.1016/j.expneurol.2007.09.022
Ekman, F. K., Ojala, D. S., Adil, M. M., Lopez, P. A., Schaffer, D. V., and Gaj, T. (2019). CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol. Ther. Nucleic Acids 17, 829–839. doi: 10.1016/j.omtn.2019.07.009
Emamzadeh, F. N., and Surguchov, A. (2018). Parkinson’s disease: biomarkers, treatment, and risk factors. Front. Neurosci. 12:612. doi: 10.3389/fnins.2018.00612
Eskow Jaunarajs, K. L., Standaert, D. G., Viegas, T. X., Bentley, M. D., Fang, Z., Dizman, B., et al. (2013). Rotigotine polyoxazoline conjugate SER-214 provides robust and sustained antiparkinsonian benefit. Mov. Disord. 28, 1675–1682. doi: 10.1002/mds.25625
Evans, A. H., and Lees, A. J. (2004). Dopamine dysregulation syndrome in Parkinson’s disease. Curr. Opin. Neurol. 17, 393–398. doi: 10.1097/01.wco.0000137528.23126.41
Evers, M. M., Miniarikova, J., Juhas, S., Valles, A., Bohuslavova, B., Juhasova, J., et al. (2018). AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol. Ther. 26, 2163–2177. doi: 10.1016/j.ymthe.2018.06.021
Faedo, A., Laporta, A., Segnali, A., Galimberti, M., Besusso, D., Cesana, E., et al. (2017). Differentiation of human telencephalic progenitor cells into MSNs by inducible expression of Gsx2 and Ebf1. Proc. Natl. Acad. Sci. U S A 114, E1234–E1242. doi: 10.1073/pnas.1611473114
Fahn, S., Oakes, D., Shoulson, I., Kieburtz, K., Rudolph, A., Lang, A., et al. (2004). Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 351, 2498–2508. doi: 10.1056/NEJMoa033447
Fasano, A., and Lozano, A. M. (2015). Deep brain stimulation for movement disorders: 2015 and beyond. Curr. Opin. Neurol. 28, 423–436. doi: 10.1097/WCO.0000000000000226
Fénelon, G., Goujon, C., Gurruchaga, J. M., Cesaro, P., Jarraya, B., Palfi, S., et al. (2012). Spinal cord stimulation for chronic pain improved motor function in a patient with Parkinson’s disease. Parkinsonism Relat. Disord. 18, 213–214. doi: 10.1016/j.parkreldis.2011.07.015
Fernandez, A. M., and Torres-Aleman, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Ferreira-Pinto, M. J., Ruder, L., Capelli, P., and Arber, S. (2018). Connecting circuits for supraspinal control of locomotion. Neuron 100, 361–374. doi: 10.1016/j.neuron.2018.09.015
Ferrer, I., Goutan, E., Marin, C., Rey, M. J., and Ribalta, T. (2000). Brain-derived neurotrophic factor in Huntington disease. Brain Res. 866, 257–261. doi: 10.1016/s0006-8993(00)02237-x
Fischer, D. L., Kemp, C. J., Cole-Strauss, A., Polinski, N. K., Paumier, K. L., Lipton, J. W., et al. (2017). Subthalamic nucleus deep brain stimulation employs trkB signaling for neuroprotection and functional restoration. J. Neurosci. 37, 6786–6796. doi: 10.1523/JNEUROSCI.2060-16.2017
Fletcher, A. M., Kowalczyk, T. H., Padegimas, L., Cooper, M. J., and Yurek, D. M. (2011). Transgene expression in the striatum following intracerebral injections of DNA nanoparticles encoding for human glial cell line-derived neurotrophic factor. Neuroscience 194, 220–226. doi: 10.1016/j.neuroscience.2011.07.072
Fox, S. H., Katzenschlager, R., Lim, S. Y., Barton, B., de Bie, R. M. A., Seppi, K., et al. (2018). International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 33, 1248–1266. doi: 10.1002/mds.27372
Freed, C. R., Breeze, R. E., Rosenberg, N. L., Schneck, S. A., Kriek, E., Qi, J. X., et al. (1992). Survival of implanted fetal dopamine cells and neurologic improvement 12 to 46 months after transplantation for Parkinson’s disease. N. Engl. J. Med. 327, 1549–1555. doi: 10.1056/nejm199211263272202
Freed, C. R., Breeze, R. E., Rosenberg, N. L., Schneck, S. A., Wells, T. H., Barrett, J. N., et al. (1990). Transplantation of human fetal dopamine cells for Parkinson’s disease. Results at 1 year. Arch. Neurol. 47, 505–512. doi: 10.1001/archneur.1990.00530050021007
Freeman, T. B., Cicchetti, F., Hauser, R. A., Deacon, T. W., Li, X. J., Hersch, S. M., et al. (2000). Transplanted fetal striatum in Huntington’s disease: phenotypic development and lack of pathology. Proc. Natl. Acad. Sci. U S A 97, 13877–13882. doi: 10.1073/pnas.97.25.13877
Fu, H., Hardy, J., and Duff, K. E. (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21, 1350–1358. doi: 10.1038/s41593-018-0221-2
Fuentes, R., Petersson, P., Siesser, W. B., Caron, M. G., and Nicolelis, M. A. (2009). Spinal cord stimulation restores locomotion in animal models of Parkinson’s disease. Science 323, 1578–1582. doi: 10.1126/science.1164901
Funa, K., Yamada, N., Brodin, G., Pietz, K., Ahgren, A., Wictorin, K., et al. (1996). Enhanced synthesis of platelet-derived growth factor following injury induced by 6-hydroxydopamine in rat brain. Neuroscience 74, 825–833. doi: 10.1016/0306-4522(96)00152-2
Gallina, P., Paganini, M., Lombardini, L., Saccardi, R., Marini, M., De Cristofaro, M. T., et al. (2008). Development of human striatal anlagen after transplantation in a patient with Huntington’s disease. Exp. Neurol. 213, 241–244. doi: 10.1016/j.expneurol.2008.06.003
Galvan, A., Devergnas, A., and Wichmann, T. (2015). Alterations in neuronal activity in basal ganglia-thalamocortical circuits in the parkinsonian state. Front. Neuroanat. 9:5. doi: 10.3389/fnana.2015.00005
Garbayo, E., Montero-Menei, C. N., Ansorena, E., Lanciego, J. L., Aymerich, M. S., and Blanco-Prieto, M. J. (2009). Effective GDNF brain delivery using microspheres—a promising strategy for Parkinson’s disease. J. Control Release 135, 119–126. doi: 10.1016/j.jconrel.2008.12.010
Garcia, V. J., Rushton, D. J., Tom, C. M., Allen, N. D., Kemp, P. J., Svendsen, C. N., et al. (2019). Huntington’s disease patient-derived astrocytes display electrophysiological impairments and reduced neuronal support. Front. Neurosci. 13:669. doi: 10.3389/fnins.2019.00669
García-Huerta, P., Troncoso-Escudero, P., Wu, D., Thiruvalluvan, A., Cisternas-Olmedo, M., Henriquez, D. R., et al. (2020). Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. doi: 10.1007/s00401-020-02183-1 [Epub ahead of print].
Garea-Rodríguez, E., Eesmaa, A., Lindholm, P., Schlumbohm, C., Konig, J., Meller, B., et al. (2016). Comparative analysis of the effects of neurotrophic factors CDNF and GDNF in a nonhuman primate model of Parkinson’s disease. PLoS One 11:e0149776. doi: 10.1371/journal.pone.0149776
Gasperi, M., and Castellano, A. E. (2010). Growth hormone/insulin-like growth factor I axis in neurodegenerative diseases. J. Endocrinol. Invest. 33, 587–591. doi: 10.1007/BF03346653
Gerfen, C. R., and Surmeier, D. J. (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. doi: 10.1146/annurev-neuro-061010-113641
Ghosh, R., and Tabrizi, S. J. (2017). Gene suppression approaches to neurodegeneration. Alzheimers Res. Ther. 9:82. doi: 10.1186/s13195-017-0307-1
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2002). Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34, 521–533. doi: 10.1016/s0896-6273(02)00682-7
Giladi, N., Treves, T. A., Simon, E. S., Shabtai, H., Orlov, Y., Kandinov, B., et al. (2001). Freezing of gait in patients with advanced Parkinson’s disease. J. Neural Transm. 108, 53–61. doi: 10.1007/s007020170096
Gill, S. S., Patel, N. K., Hotton, G. R., O’Sullivan, K., McCarter, R., Bunnage, M., et al. (2003). Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 9, 589–595. doi: 10.1038/nm850
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Gould, D. H., Wilson, M. P., and Hamar, D. W. (1985). Brain enzyme and clinical alterations induced in rats and mice by nitroaliphatic toxicants. Toxicol. Lett. 27, 83–89. doi: 10.1016/0378-4274(85)90123-7
Grandoso, L., Ponce, S., Manuel, I., Arrue, A., Ruiz-Ortega, J. A., Ulibarri, I., et al. (2007). Long-term survival of encapsulated GDNF secreting cells implanted within the striatum of parkinsonized rats. Int. J. Pharm. 343, 69–78. doi: 10.1016/j.ijpharm.2007.05.027
Gray, M. (2019). Astrocytes in Huntington’s disease. Adv. Exp. Med. Biol. 1175, 355–381. doi: 10.1007/978-981-13-9913-8_14
Gray, M., Shirasaki, D. I., Cepeda, C., Andre, V. M., Wilburn, B., Lu, X. H., et al. (2008). Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 28, 6182–6195. doi: 10.1523/jneurosci.0857-08.2008
Grondin, R., Cass, W. A., Zhang, Z., Stanford, J. A., Gash, D. M., and Gerhardt, G. A. (2003). Glial cell line-derived neurotrophic factor increases stimulus-evoked dopamine release and motor speed in aged rhesus monkeys. J. Neurosci. 23, 1974–1980. doi: 10.1523/jneurosci.23-05-01974.2003
Grondin, R., Kaytor, M. D., Ai, Y., Nelson, P. T., Thakker, D. R., Heisel, J., et al. (2012). Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 135, 1197–1209. doi: 10.1093/brain/awr333
Grondin, R., Zhang, Z., Yi, A., Cass, W. A., Maswood, N., Andersen, A. H., et al. (2002). Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain 125, 2191–2201. doi: 10.1093/brain/awf234
Gruber, D., Kuhn, A. A., Schoenecker, T., Kopp, U. A., Kivi, A., Huebl, J., et al. (2014). Quadruple deep brain stimulation in Huntington’s disease, targeting pallidum and subthalamic nucleus: case report and review of the literature. J. Neural Transm. 121, 1303–1312. doi: 10.1007/s00702-014-1201-7
Guan, J., Krishnamurthi, R., Waldvogel, H. J., Faull, R. L., Clark, R., and Gluckman, P. (2000). N-terminal tripeptide of IGF-1 (GPE) prevents the loss of TH positive neurons after 6-OHDA induced nigral lesion in rats. Brain Res. 859, 286–292. doi: 10.1016/s0006-8993(00)01988-0
Haasio, K., Sopanen, L., Vaalavirta, L., Linden, I. B., and Heinonen, E. H. (2001). Comparative toxicological study on the hepatic safety of entacapone and tolcapone in the rat. J. Neural Transm. 108, 79–91. doi: 10.1007/s007020170099
Hadjiconstantinou, M., and Neff, N. H. (2008). Enhancing aromatic L-amino acid decarboxylase activity: implications for L-DOPA treatment in Parkinson’s disease. CNS Neurosci. Ther. 14, 340–351. doi: 10.1111/j.1755-5949.2008.00058.x
Hagell, P., Schrag, A., Piccini, P., Jahanshahi, M., Brown, R., Rehncrona, S., et al. (1999). Sequential bilateral transplantation in Parkinson’s disease: effects of the second graft. Brain 122, 1121–1132. doi: 10.1093/brain/122.6.1121
Hallett, P. J., Deleidi, M., Astradsson, A., Smith, G. A., Cooper, O., Osborn, T. M., et al. (2015). Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 16, 269–274. doi: 10.1016/j.stem.2015.01.018
Hantraye, P., Riche, D., Maziere, M., and Isacson, O. (1992). Intrastriatal transplantation of cross-species fetal striatal cells reduces abnormal movements in a primate model of Huntington disease. Proc. Natl. Acad. Sci. U S A 89, 4187–4191. doi: 10.1073/pnas.89.9.4187
Hariz, M. I., Blomstedt, P., and Zrinzo, L. (2010). Deep brain stimulation between 1947 and 1987: the untold story. Neurosurg. Focus 29:E1. doi: 10.3171/2010.4.focus10106
Harnack, D., Meissner, W., Jira, J. A., Winter, C., Morgenstern, R., and Kupsch, A. (2008). Placebo-controlled chronic high-frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive Parkinsonism. Exp. Neurol. 210, 257–260. doi: 10.1016/j.expneurol.2007.10.002
Harper, S. Q., Staber, P. D., He, X., Eliason, S. L., Martins, I. H., Mao, Q., et al. (2005). RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. U S A 102, 5820–5825. doi: 10.1073/pnas.0501507102
Hassan, S., Amer, S., Alwaki, A., and Elborno, A. (2013). A patient with Parkinson’s disease benefits from spinal cord stimulation. J. Clin. Neurosci. 20, 1155–1156. doi: 10.1016/j.jocn.2012.08.018
Hayashita-Kinoh, H., Yamada, M., Yokota, T., Mizuno, Y., and Mochizuki, H. (2006). Down-regulation of α-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 341, 1088–1095. doi: 10.1016/j.bbrc.2006.01.057
He, Z., Jiang, Y., Xu, H., Jiang, H., Jia, W., Sun, P., et al. (2014). High frequency stimulation of subthalamic nucleus results in behavioral recovery by increasing striatal dopamine release in 6-hydroxydopamine lesioned rat. Behav. Brain Res. 263, 108–114. doi: 10.1016/j.bbr.2014.01.014
Heikkila, R. E., Nicklas, W. J., Vyas, I., and Duvoisin, R. C. (1985). Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci. Lett. 62, 389–394. doi: 10.1016/0304-3940(85)90580-4
Heinrich, C., Blum, R., Gascon, S., Masserdotti, G., Tripathi, P., Sanchez, R., et al. (2010). Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 8:e1000373. doi: 10.1371/journal.pbio.1000373
Heldin, C. H., and Westermark, B. (1999). Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316. doi: 10.1152/physrev.1999.79.4.1283
Hirsch, E. C., Hunot, S., Faucheux, B., Agid, Y., Mizuno, Y., Mochizuki, H., et al. (1999). Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov. Disord. 14, 383–385. doi: 10.1002/1531-8257(199903)14:2<383::aid-mds1037>3.0.co;2-f
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Howells, D. W., Porritt, M. J., Wong, J. Y., Batchelor, P. E., Kalnins, R., Hughes, A. J., et al. (2000). Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 166, 127–135. doi: 10.1006/exnr.2000.7483
Hu, J., Qian, H., Xue, Y., and Fu, X. D. (2018). PTB/nPTB: master regulators of neuronal fate in mammals. Biophys. Rep. 4, 204–214. doi: 10.1007/s41048-018-0066-y
Huntington Study Group. (2006). Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66, 366–372. doi: 10.1212/01.wnl.0000198586.85250.13
Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-e
Isacson, O., Brundin, P., Gage, F. H., and Bjorklund, A. (1985). Neural grafting in a rat model of Huntington’s disease: progressive neurochemical changes after neostriatal ibotenate lesions and striatal tissue grafting. Neuroscience 16, 799–817. doi: 10.1016/0306-4522(85)90095-8
Isacson, O., Brundin, P., Kelly, P. A., Gage, F. H., and Bjorklund, A. (1984). Functional neuronal replacement by grafted striatal neurones in the ibotenic acid-lesioned rat striatum. Nature 311, 458–460. doi: 10.1038/311458a0
Jakobs, M., Fomenko, A., Lozano, A. M., and Kiening, K. L. (2019). Cellular, molecular and clinical mechanisms of action of deep brain stimulation-a systematic review on established indications and outlook on future developments. EMBO Mol. Med. 11:e9575. doi: 10.15252/emmm.201809575
Jarraya, B., Boulet, S., Ralph, G. S., Jan, C., Bonvento, G., Azzouz, M., et al. (2009). Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci. Transl. Med. 1:2ra4. doi: 10.1126/scitranslmed.3000130
Jarvis, K., Assis-Nascimento, P., Mudd, L. M., and Montague, J. R. (2007). Beta-amyloid toxicity and reversal in embryonic rat septal neurons. Neurosci. Lett. 423, 184–188. doi: 10.1016/j.neulet.2007.06.058
Jenner, P., Rupniak, N. M., Rose, S., Kelly, E., Kilpatrick, G., Lees, A., et al. (1984). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in the common marmoset. Neurosci. Lett. 50, 85–90. doi: 10.1016/0304-3940(84)90467-1
Kaplan, D. R., and Miller, F. D. (2000). Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 10, 381–391. doi: 10.1016/s0959-4388(00)00092-1
Keene, C. D., Chang, R. C., Leverenz, J. B., Kopyov, O., Perlman, S., Hevner, R. F., et al. (2009). A patient with Huntington’s disease and long-surviving fetal neural transplants that developed mass lesions. Acta Neuropathol. 117, 329–338. doi: 10.1007/s00401-008-0465-0
Kestenbaum, M., and Fahn, S. (2015). Safety of IPX066, an extended release carbidopa-levodopa formulation, for the treatment of Parkinson’s disease. Expert Opin. Drug Saf. 14, 761–767. doi: 10.1517/14740338.2015.1015986
Kim, C. Y., and Alcalay, R. N. (2017). Genetic forms of Parkinson’s disease. Semin. Neurol. 37, 135–146. doi: 10.1055/s-0037-1601567
Kim, J., Su, S. C., Wang, H., Cheng, A. W., Cassady, J. P., Lodato, M. A., et al. (2011). Functional integration of dopaminergic neurons directly converted from mouse fibroblasts. Cell Stem Cell 9, 413–419. doi: 10.1016/j.stem.2011.09.011
Kirik, D., Georgievska, B., Burger, C., Winkler, C., Muzyczka, N., Mandel, R. J., et al. (2002a). Reversal of motor impairments in parkinsonian rats by continuous intrastriatal delivery of L-dopa using rAAV-mediated gene transfer. Proc. Natl. Acad. Sci. U S A 99, 4708–4713. doi: 10.1016/s1525-0016(16)43116-3
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., et al. (2002b). Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791. doi: 10.1523/jneurosci.22-07-02780.2002
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U S A 104, 11441–11446. doi: 10.1073/pnas.0702717104
Klein, R. L., King, M. A., Hamby, M. E., and Meyer, E. M. (2002). Dopaminergic cell loss induced by human A30P α-synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 13, 605–612. doi: 10.1089/10430340252837206
Kobayashi, R., Kenji, S., Taketomi, A., Murakami, H., Ono, K., and Otake, H. (2018). New mode of burst spinal cord stimulation improved mental status as well as motor function in a patient with Parkinson’s disease. Parkinsonism Relat. Disord. 57, 82–83. doi: 10.1016/j.parkreldis.2018.07.002
Kopyov, O. V., Jacques, S., Lieberman, A., Duma, C. M., and Eagle, K. S. (1998). Safety of intrastriatal neurotransplantation for Huntington’s disease patients. Exp. Neurol. 149, 97–108. doi: 10.1006/exnr.1997.6685
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 14, 504–506. doi: 10.1038/nm1747
Kordower, J. H., Emborg, M. E., Bloch, J., Ma, S. Y., Chu, Y., Leventhal, L., et al. (2000). Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290, 767–773. doi: 10.1126/science.290.5492.767
Kordower, J. H., Freeman, T. B., Chen, E. Y., Mufson, E. J., Sanberg, P. R., Hauser, R. A., et al. (1998). Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson’s disease. Mov. Disord. 13, 383–393. doi: 10.1002/mds.870130303
Kriks, S., Shim, J. W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. doi: 10.1038/nature10648
Krishnamurthi, R., Stott, S., Maingay, M., Faull, R. L., McCarthy, D., Gluckman, P., et al. (2004). N-terminal tripeptide of IGF-1 improves functional deficits after 6-OHDA lesion in rats. Neuroreport 15, 1601–1604. doi: 10.1097/01.wnr.0000127461.15985.07
Kuhn, A. A., Kupsch, A., Schneider, G. H., and Brown, P. (2006). Reduction in subthalamic 8–35 Hz oscillatory activity correlates with clinical improvement in Parkinson’s disease. Eur. J. Neurosci. 23, 1956–1960. doi: 10.1111/j.1460-9568.2006.04717.x
Kühn, K., Wellen, J., Link, N., Maskri, L., Lubbert, H., and Stichel, C. C. (2003). The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur. J. Neurosci. 17, 1–12. doi: 10.1046/j.1460-9568.2003.02408.x
Kuwahara, K., Sasaki, T., Yasuhara, T., Kameda, M., Okazaki, Y., Hosomoto, K., et al. (2020). Long-term continuous cervical spinal cord stimulation exerts neuroprotective effects in experimental Parkinson’s disease. Front. Aging Neurosci. 12:164. doi: 10.3389/fnagi.2020.00164
Labandeira-Garcia, J. L., Costa-Besada, M. A., Labandeira, C. M., Villar-Cheda, B., and Rodriguez-Perez, A. I. (2017). Insulin-like growth factor-1 and neuroinflammation. Front. Aging Neurosci. 9:365. doi: 10.3389/fnagi.2017.00365
Lang, A. E., Gill, S., Patel, N. K., Lozano, A., Nutt, J. G., Penn, R., et al. (2006). Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 59, 459–466. doi: 10.1002/ana.20737
Langston, J. W., Forno, L. S., Rebert, C. S., and Irwin, I. (1984). Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 292, 390–394. doi: 10.1016/0006-8993(84)90777-7
Lapper, S. R., and Bolam, J. P. (1992). Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience 51, 533–545. doi: 10.1016/0306-4522(92)90293-b
Latge, C., Cabral, K. M., de Oliveira, G. A., Raymundo, D. P., Freitas, J. A., Johanson, L., et al. (2015). The solution structure and dynamics of full-length human cerebral dopamine neurotrophic factor and its neuroprotective role against α-synuclein oligomers. J. Biol. Chem. 290, 20527–20540. doi: 10.1074/jbc.m115.662254
Lewitt, M. S., and Boyd, G. W. (2019). The role of insulin-like growth factors and insulin-like growth factor-binding proteins in the nervous system. Biochem. Insights 12:1178626419842176. doi: 10.1177/1178626419842176
Li, H., and Chen, G. (2016). In vivo reprogramming for CNS repair: regenerating neurons from endogenous glial cells. Neuron 91, 728–738. doi: 10.1016/j.neuron.2016.08.004
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Li, W., Englund, E., Widner, H., Mattsson, B., van Westen, D., Latt, J., et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. U S A 113, 6544–6549. doi: 10.1073/pnas.1605245113
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S., and Collins, F. (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260, 1130–1132. doi: 10.1126/science.8493557
Lin, X., Parisiadou, L., Gu, X. L., Wang, L., Shim, H., Sun, L., et al. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant α-synuclein. Neuron 64, 807–827. doi: 10.1016/j.neuron.2009.11.006
Lin, C. H., Tallaksen-Greene, S., Chien, W. M., Cearley, J. A., Jackson, W. S., Crouse, A. B., et al. (2001). Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 10, 137–144. doi: 10.1093/hmg/10.2.137
Lindholm, P., Voutilainen, M. H., Lauren, J., Peranen, J., Leppanen, V. M., Andressoo, J. O., et al. (2007). Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448, 73–77. doi: 10.1038/nature05957
Lindvall, O., Rehncrona, S., Brundin, P., Gustavii, B., Astedt, B., Widner, H., et al. (1989). Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 46, 615–631. doi: 10.1001/archneur.1989.00520420033021
Lindvall, O., Widner, H., Rehncrona, S., Brundin, P., Odin, P., Gustavii, B., et al. (1992). Transplantation of fetal dopamine neurons in Parkinson’s disease: one-year clinical and neurophysiological observations in two patients with putaminal implants. Ann. Neurol. 31, 155–165. doi: 10.1002/ana.410310206
Lopes, C., Ribeiro, M., Duarte, A. I., Humbert, S., Saudou, F., Pereira de Almeida, L., et al. (2014). IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 49, 1126–1142. doi: 10.1007/s12035-013-8585-5
Lortet, S., Lacombe, E., Boulanger, N., Rihet, P., Nguyen, C., Kerkerian-Le Goff, L., et al. (2013). Striatal molecular signature of subchronic subthalamic nucleus high frequency stimulation in parkinsonian rat. PLoS One 8:e60447. doi: 10.1371/journal.pone.0060447
Lu, B., Nagappan, G., Guan, X., Nathan, P. J., and Wren, P. (2013). BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 14, 401–416. doi: 10.1038/nrn3505
Lu, B., Nagappan, G., and Lu, Y. (2014). BDNF and synaptic plasticity, cognitive function and dysfunction. Handb. Exp. Pharmacol. 220, 223–250. doi: 10.1007/978-3-642-45106-5_9
Lundblad, M., Decressac, M., Mattsson, B., and Bjorklund, A. (2012). Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U S A 109, 3213–3219. doi: 10.1073/pnas.1200575109
Ma, D. K., Bonaguidi, M. A., Ming, G. L., and Song, H. (2009). Adult neural stem cells in the mammalian central nervous system. Cell Res. 19, 672–682. doi: 10.1038/cr.2009.56
Maesawa, S., Kaneoke, Y., Kajita, Y., Usui, N., Misawa, N., Nakayama, A., et al. (2004). Long-term stimulation of the subthalamic nucleus in hemiparkinsonian rats: neuroprotection of dopaminergic neurons. J. Neurosurg. 100, 679–687. doi: 10.3171/jns.2004.100.4.0679
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/s0092-8674(00)81369-0
Maruyama, W., and Naoi, M. (2013). “70th Birthday Professor Riederer” induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (−)deprenyl: a way to a disease-modifying therapy? J. Neural Transm. 120, 83–89. doi: 10.1007/s00702-012-0876-x
Mazzone, P., Viselli, F., Ferraina, S., Giamundo, M., Marano, M., Paoloni, M., et al. (2019). High cervical spinal cord stimulation: a one year follow-up study on motor and non-motor functions in Parkinson’s disease. Brain Sci. 9:78. doi: 10.3390/brainsci9040078
McBride, J. L., Pitzer, M. R., Boudreau, R. L., Dufour, B., Hobbs, T., Ojeda, S. R., et al. (2011). Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 19, 2152–2162. doi: 10.1038/mt.2011.219
McLin, J. P., Thompson, L. M., and Steward, O. (2006). Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur. J. Neurosci. 24, 3134–3140. doi: 10.1111/j.1460-9568.2006.05198.x
Meissner, W., Harnack, D., Reese, R., Paul, G., Reum, T., Ansorge, M., et al. (2003). High-frequency stimulation of the subthalamic nucleus enhances striatal dopamine release and metabolism in rats. J. Neurochem. 85, 601–609. doi: 10.1046/j.1471-4159.2003.01665.x
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 465, 11–26. doi: 10.1002/cne.10776
Miller, S. J. (2018). Astrocyte heterogeneity in the adult central nervous system. Front. Cell. Neurosci. 12:401. doi: 10.3389/fncel.2018.00401
Miniarikova, J., Zimmer, V., Martier, R., Brouwers, C. C., Pythoud, C., Richetin, K., et al. (2017). AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 24, 630–639. doi: 10.1038/gt.2017.71
Miocinovic, S., Somayajula, S., Chitnis, S., and Vitek, J. L. (2013). History, applications, and mechanisms of deep brain stimulation. JAMA Neurol. 70, 163–171. doi: 10.1001/2013.jamaneurol.45
Mittermeyer, G., Christine, C. W., Rosenbluth, K. H., Baker, S. L., Starr, P., Larson, P., et al. (2012). Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum. Gene Ther. 23, 377–381. doi: 10.1089/hum.2011.220
Mogi, M., Togari, A., Kondo, T., Mizuno, Y., Komure, O., Kuno, S., et al. (1999). Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 270, 45–48. doi: 10.1016/s0304-3940(99)00463-2
Monteys, A. M., Ebanks, S. A., Keiser, M. S., and Davidson, B. L. (2017). CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 25, 12–23. doi: 10.1016/j.ymthe.2016.11.010
Morizane, A., Doi, D., Kikuchi, T., Okita, K., Hotta, A., Kawasaki, T., et al. (2013). Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a non-human primate. Stem Cell Reports 1, 283–292. doi: 10.1016/j.stemcr.2013.08.007
Moro, E., Lang, A. E., Strafella, A. P., Poon, Y. Y., Arango, P. M., Dagher, A., et al. (2004). Bilateral globus pallidus stimulation for Huntington’s disease. Ann. Neurol. 56, 290–294. doi: 10.1002/ana.20183
Munhoz, R. P., Moro, A., Silveira-Moriyama, L., and Teive, H. A. (2015). Non-motor signs in Parkinson’s disease: a review. Arq. Neuropsiquiatr. 73, 454–462. doi: 10.1590/0004-282X20150029
Muramatsu, S., Fujimoto, K., Ikeguchi, K., Shizuma, N., Kawasaki, K., Ono, F., et al. (2002). Behavioral recovery in a primate model of Parkinson’s disease by triple transduction of striatal cells with adeno-associated viral vectors expressing dopamine-synthesizing enzymes. Hum. Gene Ther. 13, 345–354. doi: 10.1089/10430340252792486
Muramatsu, S., Fujimoto, K., Kato, S., Mizukami, H., Asari, S., Ikeguchi, K., et al. (2010). A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol. Ther. 18, 1731–1735. doi: 10.1038/mt.2010.135
Musacchio, T., Rebenstorff, M., Fluri, F., Brotchie, J. M., Volkmann, J., Koprich, J. B., et al. (2017). Subthalamic nucleus deep brain stimulation is neuroprotective in the A53T α-synuclein Parkinson’s disease rat model. Ann. Neurol. 81, 825–836. doi: 10.1002/ana.24947
Nakao, N., Brundin, P., Funa, K., Lindvall, O., and Odin, P. (1994). Platelet-derived growth factor exerts trophic effects on rat striatal DARPP-32-containing neurons in culture. Exp. Brain Res. 101, 291–296. doi: 10.1007/bf00228749
Nakao, N., Ogura, M., Nakai, K., and Itakura, T. (1999). Embryonic striatal grafts restore neuronal activity of the globus pallidus in a rodent model of Huntington’s disease. Neuroscience 88, 469–477. doi: 10.1016/s0306-4522(98)00197-3
Naver, B., Stub, C., Moller, M., Fenger, K., Hansen, A. K., Hasholt, L., et al. (2003). Molecular and behavioral analysis of the R6/1 Huntington’s disease transgenic mouse. Neuroscience 122, 1049–1057. doi: 10.1016/j.neuroscience.2003.08.053
Nishioka, K., and Nakajima, M. (2015). Beneficial therapeutic effects of spinal cord stimulation in advanced cases of Parkinson’s disease with intractable chronic pain: a case series. Neuromodulation 18, 751–753. doi: 10.1111/ner.12315
Niu, W., Zang, T., Wang, L. L., Zou, Y., and Zhang, C. L. (2018). Phenotypic reprogramming of striatal neurons into dopaminergic neuron-like cells in the adult mouse brain. Stem Cell Reports 11, 1156–1170. doi: 10.1016/j.stemcr.2018.09.004
Niu, W., Zang, T., Zou, Y., Fang, S., Smith, D. K., Bachoo, R., et al. (2013). In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 15, 1164–1175. doi: 10.1038/ncb2843
Nutt, J. G., Burchiel, K. J., Comella, C. L., Jankovic, J., Lang, A. E., Laws, E. R. Jr., et al. (2003). Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60, 69–73. doi: 10.1212/wnl.60.1.69
Obeso, J. A., Rodriguez-Oroz, M. C., Benitez-Temino, B., Blesa, F. J., Guridi, J., Marin, C., et al. (2008). Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 23, S548–S559. doi: 10.1002/mds.22062
Oertel, W., and Schulz, J. B. (2016). Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J. Neurochem. 139, 325–337. doi: 10.1111/jnc.13750
Olanow, C. W., Obeso, J. A., and Stocchi, F. (2006). Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 5, 677–687. doi: 10.1016/s1474-4422(06)70521-x
Olanow, C. W., Rascol, O., Hauser, R., Feigin, P. D., Jankovic, J., Lang, A., et al. (2009). A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 361, 1268–1278. doi: 10.1056/NEJMoa0809335
Oliveras-Salvá, M., Van der Perren, A., Casadei, N., Stroobants, S., Nuber, S., D’Hooge, R., et al. (2013). rAAV2/7 vector-mediated overexpression of α-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol. Neurodegener. 8:44. doi: 10.1186/1750-1326-8-44
Palfi, S., Gurruchaga, J. M., Ralph, G. S., Lepetit, H., Lavisse, S., Buttery, P. C., et al. (2014). Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146. doi: 10.1016/S0140-6736(13)61939-X
Parain, K., Murer, M. G., Yan, Q., Faucheux, B., Agid, Y., Hirsch, E., et al. (1999). Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 10, 557–561. doi: 10.1097/00001756-199902250-00021
Park, I. H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A., et al. (2008). Disease-specific induced pluripotent stem cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Parkinson Study Group. (1997). Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann. Neurol. 42, 747–755. doi: 10.1002/ana.410420511
Parkinson Study Group. (2005). A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch. Neurol. 62, 241–248. doi: 10.1001/archneur.62.2.241
Pascual-Lucas, M., Viana da Silva, S., Di Scala, M., Garcia-Barroso, C., Gonzalez-Aseguinolaza, G., Mulle, C., et al. (2014). Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med. 6, 1246–1262. doi: 10.15252/emmm.201404228
Patel, N. K., Bunnage, M., Plaha, P., Svendsen, C. N., Heywood, P., and Gill, S. S. (2005). Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Ann. Neurol. 57, 298–302. doi: 10.1002/ana.20374
Paul, G., Zachrisson, O., Varrone, A., Almqvist, P., Jerling, M., Lind, G., et al. (2015). Safety and tolerability of intracerebroventricular PDGF-BB in Parkinson’s disease patients. J. Clin. Invest. 125, 1339–1346. doi: 10.1172/JCI79635
Perez, F. A., Curtis, W. R., and Palmiter, R. D. (2005). Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 6:71. doi: 10.1186/1471-2202-6-71
Perlow, M. J., Freed, W. J., Hoffer, B. J., Seiger, A., Olson, L., and Wyatt, R. J. (1979). Brain grafts reduce motor abnormalities produced by destruction of nigrostriatal dopamine system. Science 204, 643–647. doi: 10.1126/science.571147
Peterziel, H., Unsicker, K., and Krieglstein, K. (2002). TGFβ induces GDNF responsiveness in neurons by recruitment of GFRα1 to the plasma membrane. J. Cell Biol. 159, 157–167. doi: 10.1083/jcb.200203115
Petrova, P., Raibekas, A., Pevsner, J., Vigo, N., Anafi, M., Moore, M. K., et al. (2003). MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J. Mol. Neurosci. 20, 173–188. doi: 10.1385/jmn:20:2:173
Pfister, E. L., Kennington, L., Straubhaar, J., Wagh, S., Liu, W., DiFiglia, M., et al. (2009). Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 19, 774–778. doi: 10.1016/j.cub.2009.03.030
Pfisterer, U., Kirkeby, A., Torper, O., Wood, J., Nelander, J., Dufour, A., et al. (2011). Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. U S A 108, 10343–10348. doi: 10.1073/pnas.1105135108
Pickel, V. M., Chan, J., and Sesack, S. R. (1992). Cellular basis for interactions between catecholaminergic afferents and neurons containing Leu-enkephalin-like immunoreactivity in rat caudate-putamen nuclei. J. Neurosci. Res. 31, 212–230. doi: 10.1002/jnr.490310203
Pietz, K., Odin, P., Funa, K., and Lindvall, O. (1996). Protective effect of platelet-derived growth factor against 6-hydroxydopamine-induced lesion of rat dopaminergic neurons in culture. Neurosci. Lett. 204, 101–104. doi: 10.1016/0304-3940(96)12326-0
Pinto de Souza, C., Hamani, C., Oliveira Souza, C., Lopez Contreras, W. O., Dos Santos Ghilardi, M. G., Cury, R. G., et al. (2017). Spinal cord stimulation improves gait in patients with Parkinson’s disease previously treated with deep brain stimulation. Mov. Disord. 32, 278–282. doi: 10.1002/mds.26850
Poewe, W. H., Deuschl, G., Gordin, A., Kultalahti, E. R., Leinonen, M., and Celomen Study. (2002). Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol. Scand. 105, 245–255. doi: 10.1034/j.1600-0404.2002.1o174.x
Pouladi, M. A., Xie, Y., Skotte, N. H., Ehrnhoefer, D. E., Graham, R. K., Kim, J. E., et al. (2010). Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum. Mol. Genet. 19, 1528–1538. doi: 10.1093/hmg/ddq026
Procaccini, C., Santopaolo, M., Faicchia, D., Colamatteo, A., Formisano, L., de Candia, P., et al. (2016). Role of metabolism in neurodegenerative disorders. Metabolism 65, 1376–1390. doi: 10.1016/j.metabol.2016.05.018
Qian, H., Kang, X., Hu, J., Zhang, D., Liang, Z., Meng, F., et al. (2020). Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556. doi: 10.1038/s41586-020-2388-4
Rajan, R., Popa, T., Quartarone, A., Ghilardi, M. F., and Kishore, A. (2017). Cortical plasticity and levodopa-induced dyskinesias in Parkinson’s disease: connecting the dots in a multicomponent network. Clin. Neurophysiol. 128, 992–999. doi: 10.1016/j.clinph.2017.03.043
Rascol, O., Brooks, D. J., Melamed, E., Oertel, W., Poewe, W., Stocchi, F., et al. (2005). Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 365, 947–954. doi: 10.1016/s0140-6736(05)71083-7
Rees, R. N., Acharya, A. P., Schrag, A., and Noyce, A. J. (2018). An early diagnosis is not the same as a timely diagnosis of Parkinson’s disease. F1000Res. 7:F1000 Faculty Rev-1106. doi: 10.12688/f1000research.14528.1
Rezak, M. (2007). Current pharmacotherapeutic treatment options in Parkinson’s disease. Dis. Mon. 53, 214–222. doi: 10.1016/j.disamonth.2007.05.002
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Rinne, U. K., Larsen, J. P., Siden, A., and Worm-Petersen, J. (1998). Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 51, 1309–1314. doi: 10.1212/wnl.51.5.1309
Rivetti di Val Cervo, P., Romanov, R. A., Spigolon, G., Masini, D., Martin-Montanez, E., Toledo, E. M., et al. (2017). Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 35, 444–452. doi: 10.1038/nbt.3835
Robertson, R. G., Farmery, S. M., Sambrook, M. A., and Crossman, A. R. (1989). Dyskinesia in the primate following injection of an excitatory amino acid antagonist into the medial segment of the globus pallidus. Brain Res. 476, 317–322. doi: 10.1016/0006-8993(89)91252-3
Rodriguez-Lebron, E., Denovan-Wright, E. M., Nash, K., Lewin, A. S., and Mandel, R. J. (2005). Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. 12, 618–633. doi: 10.1016/j.ymthe.2005.05.006
Ross, C. A., Aylward, E. H., Wild, E. J., Langbehn, D. R., Long, J. D., Warner, J. H., et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216. doi: 10.1038/nrneurol.2014.24
Saleh, N., Moutereau, S., Azulay, J. P., Verny, C., Simonin, C., Tranchant, C., et al. (2010). High insulinlike growth factor I is associated with cognitive decline in Huntington disease. Neurology 75, 57–63. doi: 10.1212/WNL.0b013e3181e62076
Salem, L., Saleh, N., Desamericq, G., Youssov, K., Dolbeau, G., Cleret, L., et al. (2016). Insulin-like growth factor-1 but not insulin predicts cognitive decline in Huntington’s disease. PLoS One 11:e0162890. doi: 10.1371/journal.pone.0162890
Samotus, O., Parrent, A., and Jog, M. (2018). Spinal cord stimulation therapy for gait dysfunction in advanced Parkinson’s disease patients. Mov. Disord. 33, 783–792. doi: 10.1002/mds.27299
Sanberg, P. R. (1980). Haloperidol-induced catalepsy is mediated by postsynaptic dopamine receptors. Nature 284, 472–473. doi: 10.1038/284472a0
Sanberg, P. R., Henault, M. A., and Deckel, A. W. (1986). Locomotor hyperactivity: effects of multiple striatal transplants in an animal model of Huntington’s disease. Pharmacol. Biochem. Behav. 25, 297–300. doi: 10.1016/0091-3057(86)90269-8
Santana, M. B., Halje, P., Simplicio, H., Richter, U., Freire, M. A. M., Petersson, P., et al. (2014). Spinal cord stimulation alleviates motor deficits in a primate model of Parkinson disease. Neuron 84, 716–722. doi: 10.1016/j.neuron.2014.08.061
Schilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K., Kotzuk, J. A., et al. (1999). Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8, 397–407. doi: 10.1093/hmg/8.3.397
Shealy, C. N., Mortimer, J. T., and Reswick, J. B. (1967). Electrical inhibition of pain by stimulation of the dorsal columns: preliminary clinical report. Anesth. Analg. 46, 489–491. doi: 10.1213/00000539-196707000-00025
Shin, J. W., Kim, K. H., Chao, M. J., Atwal, R. S., Gillis, T., MacDonald, M. E., et al. (2016). Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 25, 4566–4576. doi: 10.1093/hmg/ddw286
Shinko, A., Agari, T., Kameda, M., Yasuhara, T., Kondo, A., Tayra, J. T., et al. (2014). Spinal cord stimulation exerts neuroprotective effects against experimental Parkinson’s disease. PLoS One 9:e101468. doi: 10.1371/journal.pone.0101468
Shipton, E. A. (2012). Movement disorders and neuromodulation. Neurol. Res. Int. 2012:309431. doi: 10.1155/2012/309431
Siegel, G. J., and Chauhan, N. B. (2000). Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res. Rev. 33, 199–227. doi: 10.1016/s0165-0173(00)00030-8
Sjöborg, M., Pietz, K., Ahgren, A., Yamada, N., Lindvall, O., Funa, K., et al. (1998). Expression of platelet-derived growth factor after intrastriatal ibotenic acid injury. Exp. Brain Res. 119, 245–250. doi: 10.1007/s002210050338
Sladek, J. R. Jr., Collier, T. J., Haber, S. N., Deutch, A. Y., Elsworth, J. D., Roth, R. H., et al. (1987). Reversal of parkinsonism by fetal nerve cell transplants in primate brain. Ann. N Y Acad. Sci. 495, 641–657. doi: 10.1111/j.1749-6632.1987.tb23706.x
Slevin, J. T., Gash, D. M., Smith, C. D., Gerhardt, G. A., Kryscio, R., Chebrolu, H., et al. (2007). Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: response to 1 year of treatment and 1 year of withdrawal. J. Neurosurg. 106, 614–620. doi: 10.3171/jns.2007.106.4.614
Slevin, J. T., Gerhardt, G. A., Smith, C. D., Gash, D. M., Kryscio, R., and Young, B. (2005). Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg. 102, 216–222. doi: 10.3171/jns.2005.102.2.0216
Slow, E. J., van Raamsdonk, J., Rogers, D., Coleman, S. H., Graham, R. K., Deng, Y., et al. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567. doi: 10.1093/hmg/ddg169
Smith, D. E., Saji, M., Joh, T. H., Reis, D. J., and Pickel, V. M. (1987). Ibotenic acid-induced lesions of striatal target and projection neurons: ultrastructural manifestations in dopaminergic and non-dopaminergic neurons and in glia. Histol. Histopathol. 2, 251–263.
Song, J. H., Yu, J. T., and Tan, L. (2015). Brain-derived neurotrophic factor in Alzheimer’s disease: risk, mechanisms and therapy. Mol. Neurobiol. 52, 1477–1493. doi: 10.1007/s12035-014-8958-4
Sonsalla, P. K., and Heikkila, R. E. (1988). Neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and methamphetamine in several strains of mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 12, 345–354. doi: 10.1016/0278-5846(88)90054-1
Sonsalla, P. K., Youngster, S. K., Kindt, M. V., and Heikkila, R. E. (1987). Characteristics of 1-methyl-4–(2′-methylphenyl)-1,2,3,6-tetrahydropyridine-induced neurotoxicity in the mouse. J. Pharmacol. Exp. Ther. 242, 850–857.
Spieles-Engemann, A. L., Behbehani, M. M., Collier, T. J., Wohlgenant, S. L., Steece-Collier, K., Paumier, K., et al. (2010). Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol. Dis. 39, 105–115. doi: 10.1016/j.nbd.2010.03.009
Spieles-Engemann, A. L., Steece-Collier, K., Behbehani, M. M., Collier, T. J., Wohlgenant, S. L., Kemp, C. J., et al. (2011). Subthalamic nucleus stimulation increases brain derived neurotrophic factor in the nigrostriatal system and primary motor cortex. J. Parkinsons Dis. 1, 123–136. doi: 10.3233/jpd-2011-11008
Spina, M. B., and Cohen, G. (1989). Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc. Natl. Acad. Sci. U S A 86, 1398–1400. doi: 10.1073/pnas.86.4.1398
Srivastava, D., and DeWitt, N. (2016). In vivo cellular reprogramming: the next generation. Cell 166, 1386–1396. doi: 10.1016/j.cell.2016.08.055
Stacy, M. (2009). Medical treatment of Parkinson disease. Neurol. Clin. 27, 605–631. doi: 10.1016/j.ncl.2009.04.009
St. Martin, J. L., Klucken, J., Outeiro, T. F., Nguyen, P., Keller-McGandy, C., Cantuti-Castelvetri, I., et al. (2007). Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of α-synuclein in mouse substantia nigra. J. Neurochem. 100, 1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x
Strotzer, Q. D., Anthofer, J. M., Faltermeier, R., Brawanski, A. T., Torka, E., Waldthaler, J. A., et al. (2019). Deep brain stimulation: connectivity profile for bradykinesia alleviation. Ann. Neurol. 85, 852–864. doi: 10.1002/ana.25475
Sudhakar, V., and Richardson, R. M. (2019). Gene therapy for neurodegenerative diseases. Neurotherapeutics 16, 166–175. doi: 10.1007/s13311-018-00694-0
Suh, H. S., Zhao, M. L., Derico, L., Choi, N., and Lee, S. C. (2013). Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J. Neuroinflammation 10:37. doi: 10.1186/1742-2094-10-37
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Tabrizi, S. J., Leavitt, B. R., Landwehrmeyer, G. B., Wild, E. J., Saft, C., Barker, R. A., et al. (2019). Targeting Huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 380, 2307–2316. doi: 10.1056/NEJMoa1900907
Takahashi, M., Ishikawa, K., Sato, N., Obayashi, M., Niimi, Y., Ishiguro, T., et al. (2012). Reduced brain-derived neurotrophic factor (BDNF) mRNA expression and presence of BDNF-immunoreactive granules in the spinocerebellar ataxia type 6 (SCA6) cerebellum. Neuropathology 32, 595–603. doi: 10.1111/j.1440-1789.2012.01302.x
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tatton, W. G., Wadia, J. S., Ju, W. Y., Chalmers-Redman, R. M., and Tatton, N. A. (1996). (−)-Deprenyl reduces neuronal apoptosis and facilitates neuronal outgrowth by altering protein synthesis without inhibiting monoamine oxidase. J. Neural Transm. Suppl. 48, 45–59. doi: 10.1007/978-3-7091-7494-4_5
Taylor, J. R., Elsworth, J. D., Roth, R. H., Sladek, J. R. Jr., Collier, T. J., and Redmond, D. E. Jr. (1991). Grafting of fetal substantia nigra to striatum reverses behavioral deficits induced by MPTP in primates: a comparison with other types of grafts as controls. Exp. Brain Res. 85, 335–348. doi: 10.1007/bf00229411
Teijido, O., and Cacabelos, R. (2018). Pharmacoepigenomic interventions as novel potential treatments for Alzheimer’s and Parkinson’s diseases. Int. J. Mol. Sci. 19:3199. doi: 10.3390/ijms19103199
Temel, Y., Visser-Vandewalle, V., Kaplan, S., Kozan, R., Daemen, M. A., Blokland, A., et al. (2006). Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res. 1120, 100–105. doi: 10.1016/j.brainres.2006.08.082
Tereshchenko, J., Maddalena, A., Bahr, M., and Kugler, S. (2014). Pharmacologically controlled, discontinuous GDNF gene therapy restores motor function in a rat model of Parkinson’s disease. Neurobiol. Dis. 65, 35–42. doi: 10.1016/j.nbd.2014.01.009
Thevathasan, W., Mazzone, P., Jha, A., Djamshidian, A., Dileone, M., Di Lazzaro, V., et al. (2010). Spinal cord stimulation failed to relieve akinesia or restore locomotion in Parkinson disease. Neurology 74, 1325–1327. doi: 10.1212/WNL.0b013e3181d9ed58
Torper, O., Ottosson, D. R., Pereira, M., Lau, S., Cardoso, T., Grealish, S., et al. (2015). In vivo reprogramming of striatal NG2 glia into functional neurons that integrate into local host circuitry. Cell Rep. 12, 474–481. doi: 10.1016/j.celrep.2015.06.040
Ungerstedt, U. (1968). 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–110. doi: 10.1016/0014-2999(68)90164-7
Van de Schyf, C. J. (2015). Rational durg discovery design approaches for treating Parkinson’s disease. Expert Opin. Drug Discov. 10, 713–741. doi: 10.1517/17460441.2015.1041495
Van Raamsdonk, J. M., Pearson, J., Slow, E. J., Hossain, S. M., Leavitt, B. R., and Hayden, M. R. (2005). Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 25, 4169–4180. doi: 10.1523/jneurosci.0590-05.2005
Voutilainen, M. H., Back, S., Peranen, J., Lindholm, P., Raasmaja, A., Mannisto, P. T., et al. (2011). Chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson’s disease. Exp. Neurol. 228, 99–108. doi: 10.1016/j.expneurol.2010.12.013
Voutilainen, M. H., Back, S., Porsti, E., Toppinen, L., Lindgren, L., Lindholm, P., et al. (2009). Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J. Neurosci. 29, 9651–9659. doi: 10.1523/JNEUROSCI.0833-09.2009
Wallace, B. A., Ashkan, K., Heise, C. E., Foote, K. D., Torres, N., Mitrofanis, J., et al. (2007). Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain 130, 2129–2145. doi: 10.1093/brain/awm137
Wang, N., Gray, M., Lu, X. H., Cantle, J. P., Holley, S. M., Greiner, E., et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat. Med. 20, 536–541. doi: 10.1038/nm.3514
Wang, Y., Liu, H., Zhang, B. S., Soares, J. C., and Zhang, X. Y. (2016). Low BDNF is associated with cognitive impairments in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 29, 66–71. doi: 10.1016/j.parkreldis.2016.05.023
Weinreb, O., Amit, T., Bar-Am, O., and Youdim, M. B. (2007). Induction of neurotrophic factors GDNF and BDNF associated with the mechanism of neurorescue action of rasagiline and ladostigil: new insights and implications for therapy. Ann. N Y Acad. Sci. 1122, 155–168. doi: 10.1196/annals.1403.011
Wheeler, V. C., White, J. K., Gutekunst, C. A., Vrbanac, V., Weaver, M., Li, X. J., et al. (2000). Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 9, 503–513. doi: 10.1093/hmg/9.4.503
Whone, A. L., Boca, M., Luz, M., Woolley, M., Mooney, L., Dharia, S., et al. (2019). Extended treatment with glial cell line-derived neurotrophic factor in Parkinson’s disease. J. Parkinsons Dis. 9, 301–313. doi: 10.3233/JPD-191576
Whone, A., Luz, M., Boca, M., Woolley, M., Mooney, L., Dharia, S., et al. (2019). Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 142, 512–525. doi: 10.1093/brain/awz023
Wignakumar, T., and Fairchild, P. J. (2019). Evasion of pre-existing immunity to Cas9: a prerequisite for successful genome editing in vivo? Curr. Transplant. Rep. 6, 127–133. doi: 10.1007/s40472-019-00237-2
Wojtecki, L., Groiss, S. J., Ferrea, S., Elben, S., Hartmann, C. J., Dunnett, S. B., et al. (2015). A prospective pilot trial for pallidal deep brain stimulation in Huntington’s disease. Front. Neurol. 6:177. doi: 10.3389/fneur.2015.00177
Wu, S. T., Ma, Y., Zhang, K., and Zhang, J. G. (2012). Effect of deep brain stimulation on substantia nigra neurons in a rat model of Parkinson’s disease. Chin. Med. J. 125, 4072–4075. doi: 10.3760/cma.j.issn0366-6999.2012.22.029
Wu, Z., Parry, M., Hou, X. Y., Liu, M. H., Wang, H., Cain, R., et al. (2020). Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat. Commun. 11:1105. doi: 10.1038/s41467-020-14855-3
Wyant, K. J., Ridder, A. J., and Dayalu, P. (2017). Huntington’s disease-update on treatments. Curr. Neurol. Neurosci. Rep. 17:33. doi: 10.1007/s11910-017-0739-9
Xu, X., Tay, Y., Sim, B., Yoon, S. I., Huang, Y., Ooi, J., et al. (2017). Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports 8, 619–633. doi: 10.1016/j.stemcr.2017.01.022
Xue, Y., Ouyang, K., Huang, J., Zhou, Y., Ouyang, H., Li, H., et al. (2013). Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96. doi: 10.1016/j.cell.2012.11.045
Xue, Y., Qian, H., Hu, J., Zhou, B., Zhou, Y., Hu, X., et al. (2016). Sequential regulatory loops as key gatekeepers for neuronal reprogramming in human cells. Nat. Neurosci. 19, 807–815. doi: 10.3410/f.726310460.793517647
Yadav, A. P., Fuentes, R., Zhang, H., Vinholo, T., Wang, C. H., Freire, M. A., et al. (2014). Chronic spinal cord electrical stimulation protects against 6-hydroxydopamine lesions. Sci. Rep. 4:3839. doi: 10.1038/srep03839
Yampolsky, C., Hem, S., and Bendersky, D. (2012). Dorsal column stimulator applications. Surg. Neurol. Int. 3, S275–289. doi: 10.4103/2152-7806.103019
Yang, S., Chang, R., Yang, H., Zhao, T., Hong, Y., Kong, H. E., et al. (2017). CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Invest. 127, 2719–2724. doi: 10.1172/jci92087
Yang, S. H., Cheng, P. H., Banta, H., Piotrowska-Nitsche, K., Yang, J. J., Cheng, E. C., et al. (2008). Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 453, 921–924. doi: 10.1038/nature06975
Yang, D., Wang, C. E., Zhao, B., Li, W., Ouyang, Z., Liu, Z., et al. (2010). Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet. 19, 3983–3994. doi: 10.1093/hmg/ddq313
Yasuhara, T., Shingo, T., Kobayashi, K., Takeuchi, A., Yano, A., Muraoka, K., et al. (2004). Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur. J. Neurosci. 19, 1494–1504. doi: 10.1111/j.1460-9568.2004.03254.x
Zachrisson, O., Zhao, M., Andersson, A., Dannaeus, K., Haggblad, J., Isacson, R., et al. (2011). Restorative effects of platelet derived growth factor-BB in rodent models of Parkinson’s disease. J. Parkinsons Dis 1, 49–63. doi: 10.3233/JPD-2011-0003
Zhang, N., An, M. C., Montoro, D., and Ellerby, L. M. (2010). Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2:RRN1193. doi: 10.1371/currents.RRN1193
Zhang, Z., Miyoshi, Y., Lapchak, P. A., Collins, F., Hilt, D., Lebel, C., et al. (1997). Dose response to intraventricular glial cell line-derived neurotrophic factor administration in parkinsonian monkeys. J. Pharmacol. Exp. Ther. 282, 1396–1401.
Zhao, H., Alam, A., San, C. Y., Eguchi, S., Chen, Q., Lian, Q., et al. (2017). Molecular mechanisms of brain-derived neurotrophic factor in neuro-protection: recent developments. Brain Res. 1665, 1–21. doi: 10.1016/j.brainres.2017.03.029
Zhao, C., Deng, W., and Gage, F. H. (2008). Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660. doi: 10.1016/j.cell.2008.01.033
Zigmond, M. J., and Stricker, E. M. (1984). Parkinson’s disease: studies with an animal model. Life Sci. 35, 5–18. doi: 10.1016/0024-3205(84)90147-4
Zuccato, C., and Cattaneo, E. (2007). Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 81, 294–330. doi: 10.1016/j.pneurobio.2007.01.003
Zuccato, C., Marullo, M., Vitali, B., Tarditi, A., Mariotti, C., Valenza, M., et al. (2011). Brain-derived neurotrophic factor in patients with Huntington’s disease. PLoS One 6:e22966. doi: 10.14711/thesis-991012662566803412
Zurn, A. D., Widmer, H. R., and Aebischer, P. (2001). Sustained delivery of GDNF: towards a treatment for Parkinson’s disease. Brain Res. Rev. 36, 222–229. doi: 10.1016/s0165-0173(01)00098-4
Keywords: Parkinson’s disease, Huntington’s disease, neurotrophic factors, pharmacological therapy, gene modifiers, cellular replacement
Citation: Troncoso-Escudero P, Sepulveda D, Pérez-Arancibia R, Parra AV, Arcos J, Grunenwald F and Vidal RL (2020) On the Right Track to Treat Movement Disorders: Promising Therapeutic Approaches for Parkinson’s and Huntington’s Disease. Front. Aging Neurosci. 12:571185. doi: 10.3389/fnagi.2020.571185
Received: 10 June 2020; Accepted: 17 August 2020;
Published: 03 September 2020.
Edited by:
Ines Moreno-Gonzalez, University of Malaga, SpainReviewed by:
Gabriela Mercado, Van Andel Institute, United StatesCopyright © 2020 Troncoso-Escudero, Sepulveda, Pérez-Arancibia, Parra, Arcos, Grunenwald and Vidal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rene L. Vidal, cmVuZS52aWRhbEB1bWF5b3IuY2w=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.