 Micol Avenali
Micol Avenali Fabio Blandini
Fabio Blandini Silvia Cerri
Silvia Cerri
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 21 April 2020
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 12 - 2020 | https://doi.org/10.3389/fnagi.2020.00097
This article is part of the Research TopicProgress in Risk Factors and Development of Parkinson’s DiseaseView all 28 articles
Heterozygous mutations of the GBA1 gene, encoding for lysosomal enzyme glucocerebrosidase (GCase), occur in a considerable percentage of all patients with sporadic Parkinson’s disease (PD), varying between 8% and 12% across the world. Genome wide association studies have confirmed the strong correlation between PD and GBA1 mutations, pointing to this element as a major risk factor for PD, possibly the most important one after age. The pathobiological mechanisms underlying the link between a defective function of GCase and the development of PD are still unknown and are currently the focus of intense investigation in the community of pre-clinical and clinical researchers in the PD field. A major controversy regards the fact that, despite the unequivocal correlation between the presence of GBA1 mutations and the risk of developing PD, only a minority of asymptomatic carriers with GBA1 mutations convert to PD in their lifetime. GBA1 mutations reduce the enzymatic function of GCase, impairing lysosomal efficiency and the cellular ability to dispose of pathological alpha-synuclein. Changes in the cellular lipidic content resulting from the accumulation of glycosphingolipids, triggered by lysosomal dysfunction, may contribute to the pathological modification of alpha-synuclein, due to its ability to interact with cell membrane lipids. Mutant GCase can impair mitochondrial function and cause endoplasmic reticulum stress, thereby impacting on cellular energy production and proteostasis. Importantly, reduced GCase activity is associated with clear activation of microglia, a major mediator of neuroinflammatory response within the brain parenchyma, which points to neuroinflammation as a major consequence of GCase dysfunction. In this present review article, we summarize the current knowledge on the role of GBA1 mutations in PD development and their phenotypic correlations. We also discuss the potential role of the GCase pathway in the search for PD biomarkers that may enable the development of disease modifying therapies. Answering these questions will aid clinicians in offering more appropriate counseling to the patients and their caregivers and provide future directions for PD preclinical research.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease, with a 0.3% prevalence in the general population, increasing to 1% in people older than 60 years and to 3% in the over 80s (Lee and Gilbert, 2016).
PD is characterized by a wide spectrum of motor and non-motor symptoms (Lee and Gilbert, 2016). According to clinical and imaging findings, the neurodegenerative process of PD may begin 7–10 years before the appearance of the classic motor symptoms bradykinesia, rigidity, and tremor (Hawkes, 2008; Schapira and Tolosa, 2010). Non-motor symptoms, a crucial component of the PD clinical picture, include olfactory loss, rapid eye movement sleep behavior disorders (RBD), constipation, dysautonomia, and depression, and can precede PD motor symptoms by many years (Schapira and Tolosa, 2010; Schapira et al., 2017; Heinzel et al., 2019).
The majority of PD cases are idiopathic, with a 10–15% fraction being associated with gene mutations (Sidransky et al., 2009; Westbroek et al., 2011; Schapira, 2015). In the context of PD genetics, the attention of many groups have converged, in recent years, onto the GBA1 gene, which encodes glucocerebrosidase (GCase), a lysosomal enzyme that breaks down glucocerebroside into glucose and ceramide (Beutler et al., 2005; Grabowski, 2008).
Homozygous mutations in the GBA1 gene cause Gaucher’s disease (GD), which may present with either neurological involvement (neuronopathic) or not (non-neuronopathic). Interest in GBA1, as a causative factor for PD, was initially sparkled by the observation, in the 1990s, that among GD patients and GBA1 mutation carriers, a higher proportion developed parkinsonian symptoms (Neudorfer et al., 1996; Machaczka et al., 1999). Moreover, the association between GBA1 mutation and PD development was confirmed by a large multicenter study demonstrating a significantly higher prevalence of GBA1 mutations among the PD population (Sidransky et al., 2009). Although the proportion of PD patients with GBA1 mutations varies by ethnicity and sequencing methods used, recent studies suggested that the heterozygous status confers a cumulative risk of developing PD of 5% at age 60, rising to 15–30% at age 80 (Schapira, 2015; Balestrino and Schapira, 2018). The GBA1 mutation prevalence is estimated between 2.35% and 9.4% in the PD population, climbing to 31.3% in the PD Ashkenazi Jewish (AJ) population (Schapira, 2015) indicating these mutations as the most common genetic risk factor for PD.
GD patients and asymptomatic heterozygous GBA1 mutation carriers share the same risk of developing PD (Alcalay et al., 2012), but not all GBA1 mutant carriers will develop the disease. Interestingly, PD risk seems to be affected by GBA1 mutations with varying degrees of severity (Sidransky and Lopez, 2012; Migdalska-Richards and Schapira, 2016).
GBA1 mutations reduce the enzymatic function of GCase, which may favor toxic accumulation of alpha-synuclein fibrils in the typical intra-neuronal inclusions (Lewy bodies) found throughout the central nervous system of PD patients (Balestrino and Schapira, 2018). Alternatively, or in addition to, changes in the cellular lipidic content, resulting from the accumulation of glycosphingolipids associated with lysosomal dysfunction, may contribute to the pathological modification of alpha-synuclein. Various classes of lipids can strongly interact with alpha-synuclein, causing modifications of its native structure, which can favor the formation of fibrils and subsequent aggregation of the protein (Morabito et al., 2017; Fecchio et al., 2018).
GCase defects may also contribute to neuroinflammation, a phenomenon that is increasingly implicated in PD pathogenesis, as suggested by various studies demonstrating that a reduction in GCase activity is associated with clear activation of microglia in the brain parenchyma (Ginns et al., 2014; Rocha et al., 2015c; Soria et al., 2017; Mus et al., 2019).
Moreover, GCase deficiency has been observed in idiopathic PD (iPD) patients without GBA1 mutation, suggesting a central, more widespread involvement of this enzyme in PD pathogenesis (Chiasserini et al., 2015; Parnetti et al., 2017).
From a clinical point of view, GBA1-associated PD (GBA-PD) is very similar to iPD, except for an earlier onset and higher prevalence of cognitive deterioration and non-motor features, manifesting before the development of the PD motor features (Siebert et al., 2014). The risk of cognitive impairment in GBA-PD patients is 2.4-fold higher than in non-carriers (Nalls et al., 2013). GBA1 heterozygous carriers have an increased chance of developing either PD or dementia with Lewy bodies at an earlier age of onset, a higher risk for progression to dementia, visual hallucinations, autonomic dysfunction and faster progression of motor symptoms than non-carriers, and an overall decreased survival rate (Winder-Rhodes et al., 2013; Cilia et al., 2016).
GBA1 mutations causing GD have been categorized as “severe” (L444P, for example) or “mild” (N370S, for example), with a residual GCase activity of 13–24% and 32–38%, respectively (Beutler et al., 2005). N370S and L444P mutations are the two most relevant GBA1 mutations in the PD population although, recently, the E236K mutation, which is absent in the GD population, has been identified as the most prevalent PD-associated GBA1 mutation (Duran et al., 2013). Mutation severity can influence PD phenotype profoundly. A meta-analysis revealed that the risk for dementia in PD patients carrying “severe” mutations is 2- to 3-fold higher than in those carrying “mild” mutations (Cilia et al., 2016). A more recent study extended this notion demonstrating that psychiatric symptoms, cognitive impairment, and olfactory deficiency are more pronounced in PD patients carrying severe GBA1 mutations (Thaler et al., 2018a).
In this review article, we summarize the current knowledge on the multiple roles that GBA1 mutations, and resulting GCase/lysosomal impairment, may play in the pathogenesis of PD and their phenotypic correlations. We also discuss the potential role of the GCase pathway in the search for PD biomarkers and molecular targets that may help the development of disease modifying therapies.
The mechanism by which GBA1 mutations are linked to PD is still poorly understood. However, as in iPD, several mechanisms may be involved in both the development and progression of GBA-PD, such as alpha-synuclein accumulation, neuroinflammation, mitochondrial deficiency, autophagic dysfunction, and oxidative stress (Schapira and Tolosa, 2010; Blandini et al., 2019).
The relationship between alpha-synuclein accumulation and GCase is still a matter of study. Experimental findings and clinical observations led to three main hypotheses that may explain this relationship. These hypotheses may not be mutually exclusive and multiple mechanisms might be in place.
The gain-of-function hypothesis suggests that misfolded GCase directly interacts with alpha-synuclein, leading to an increase of alpha-synuclein accumulation and aggregation (Sidransky and Lopez, 2012). Indeed, GCase was observed in abnormal protein aggregates, such as Lewy bodies and neurites, from GBA-PD brains, confirming a co-localization of mutant GCase and alpha-synuclein in vivo (Goker-Alpan et al., 2008). Cullen et al. (2011) reported increased alpha-synuclein levels in GBA mutant neural cells. This finding was confirmed by Xu et al. (2011) in the forebrain and cerebellum of mice carrying the homozygous V394L GBA1 mutation. However, this hypothesis is at odds with the general observation that congenital diseases involving enzymes almost exclusively cause a loss of function and that null GBA1 mutations are associated with PD risk (Neumann et al., 2009; Gan-Or et al., 2015; Franco et al., 2018).
According to the loss-of-function hypothesis, GBA1 mutations may affect GCase protein structure, leading to decreased enzymatic activity via multiple mechanisms (e.g., the failure of the GCase protein to exit the endoplasmic reticulum or to link with its transporter). The reduction in GCase activity causes an accumulation of its substrates with subsequent perturbation of lipid homeostasis. This affects trafficking, processing, and clearance of alpha-synuclein, resulting in accumulation and aggregation of the protein (Mazzulli et al., 2011; Westbroek et al., 2011; Sidransky and Lopez, 2012; Suzuki et al., 2007; PMID: 26362253). In line with this, Rocha et al. (2015a) observed that increased levels of glucosylsphingosine are associated with reduced GCase activity in the substantia nigra and hippocampus of iPD patients. Daily systemic treatment with selective GCase inhibitor conduritol-β-epoxide (CBE) in mice promoted the accumulation of lipid substrates and insoluble α-synuclein aggregates in the substantia nigra (Rocha et al., 2015a). Huebecker et al. (2019) recently reported a significant increase in glucosylceramide levels in the substantia nigra of iPD patients in association with deficiencies in multiple lysosomal hydrolases, including GCase, α-galactosidase, and β-hexosaminidase. These findings partially contradict previous studies that did not report GCase substrate accumulation in the brain of PD patients with heterozygote GBA1 mutations or iPD patients with reduced GCase activity (Murphy et al., 2014; Gegg et al., 2015). These discrepancies are not easy to reconcile, but might be due, at least in part, to differences in the brain regions examined and/or to the different methodologies used to measure lipid substrates. Still at odds with the loss-of-function hypothesis, pharmacological inhibition of GCase activity by CBE in cells overexpressing alpha-synuclein and primary neurons did not alter alpha-synuclein levels or solubility (Zurbruegg et al., 2019; Henderson et al., 2020). On the contrary, siRNA knockdown of the GBA1 gene significantly increased alpha-synuclein levels, suggesting that alpha-synuclein accumulation is unlikely to be solely due to decreased GCase activity (Zurbruegg et al., 2019).
A third hypothesis proposes a bidirectional loop in which GCase deficiency facilitates alpha-synuclein oligomer formation that, in turn, impairs GCase activity, ultimately promoting further formation of alpha-synuclein oligomers (Mazzulli et al., 2011). Accordingly, overexpression of exogenous alpha-synuclein in in vitro models causes reduction of GCase activity and protein levels (Gegg et al., 2012). In addition, Wong and Krainc (2016) have demonstrated that loss of GCase activity in human midbrain neuron cultures promotes alpha-synuclein accumulation and toxicity, which in turn disrupts trafficking to lysosomes of GCase and additional lysosomal hydrolases, further contributing to GCase deficits and—more in general—to lysosomal dysfunction. This hypothesis is not completely supported by in vivo studies, since no changes in GCase protein levels and activity were observed in the brain of different mouse models overexpressing human wild-type or A53T (M83KO) alpha-synuclein (Richter et al., 2014; Henderson et al., 2020). In addition, it should be noted that build-up of alpha-synuclein has been documented in many lysosomal disorders (LSD; Saito et al., 2004; Suzuki et al., 2007; Nelson et al., 2014; Smith et al., 2014). In Krabbe disease, a demyelinating LSD caused by deficiency in galactosylceramidase, the accumulation of substrate psychosine would promote the disruption of lipid raft architecture, which in turn may affect alpha-synuclein localization to synapses and increase its aggregation in the neuronal cytoplasm (Smith et al., 2014). Phosphorylated alpha-synuclein accumulation was found in the brain of patients with Niemann-Pick type C1 disease as a consequence of defects in intracellular cholesterol trafficking (Saito et al., 2004). Together, these findings indicate that, as in LSD, lipid accumulation could trigger a cascade of events driving PD pathology.
Interestingly, a recent study highlighted the importance of the lysosomal protein cathepsin D in mediating the increase of monomeric alpha-synuclein levels associated with GBA1 mutations, indicating a new player in the relationship between GCase and alpha-synuclein (Yang et al., 2020).
Despite the large body of evidence in favor of the link between GCase deficiency and alpha-synuclein accumulation, the question of why only a small proportion of individuals with GBA1 mutations develops PD remains unanswered, suggesting that other factors must be playing a role.
Mitochondria dysfunctions have been classically involved in the pathogenesis of iPD (Schapira et al., 1989, 1990; Schapira, 2008; Mounsey and Teismann, 2011; Schapira and Gegg, 2011) and may also be a feature of GBA PD pathobiology. Cleeter et al. (2013) demonstrated that GCase loss-of-function triggers oxidative stress and mitochondrial dysfunction in human dopaminergic cell lines, together with an accumulation of alpha-synuclein. Similar findings were observed in in vivo models of GCase deficiency. Osellame et al. (2013) found an accumulation of dysfunctional mitochondria with subsequent reduction in respiratory chain complex activities and oxygen consumption in a mouse model carrying homozygous knock-out of the GBA1 gene.
Similarly, a reduction of oxygen consumption and mitochondrial adenosine triphosphate production was demonstrated in neuronopathic GD mouse models generated from backcrossing of the homozygous point-mutated GBA1 mice with hypomorphic prosaposin mice (Xu et al., 2014).
Recently, Li et al. (2019) investigated the link between heterozygous GBA1 mutation and mitochondrial dysfunction showing that in GBA1 L444P/WT knock-in mice, the GBA1 mutation triggers mitochondrial dysfunction by blocking autophagy and mitochondrial priming. Moreover, Schöndorf et al. (2018) demonstrated that neurons differentiated from GBA-PD patients induced pluripotent stem cells showed alterations in morphology, energy metabolism, and mitochondrial function.
Endoplasmic reticulum-associated degradation (ERAD) promotes balanced cell proteostasis, degrading altered proteins through ubiquitination and proteosomal degradation (Migdalska-Richards and Schapira, 2016). ERAD impairment leads to the accumulation of misfolded proteins, promoting ER stress and apoptosis.
Emerging evidence shows that ER stress has a pivotal role in the pathobiology of PD (Mercado et al., 2013; Sardi et al., 2015), but few studies have analyzed, so far, the link between ER stress and the pathogenesis of GBA-PD. Mutant misfolded GCase triggers ER stress and evokes proteasomal degradation (Ron et al., 2010; Maor et al., 2013). Indeed, a study on human GD fibroblasts showed misfolded GCase retained in the ER (Bendikov-Bar et al., 2011). Moreover, the degree of GCase retention in ER and proteasomal degradation seems to be correlated with GD severity (Ron and Horowitz, 2005). Gegg et al. (2012) found that ERAD was increased in the GBA-PD brains. Furthermore, a study using induced pluripotent stem cells from GBA1 carriers confirmed increased ER stress and ERAD (Fernandes et al., 2016).
Interestingly, blocking GCase activity with CBE also stimulated ER stress in neuroblastoma cells, suggesting that GCase activity reduction, also in the absence of a GBA1 mutation, may play a role in this context (Gegg and Schapira, 2018).
Neuroinflammation—mostly expressed as microglia activation—is clearly implicated in PD progression and accompanies dopaminergic cell death since the very early phase of the disease (Fuzzati-Armentero et al., 2019). GCase defects may substantially contribute to this phenomenon. Marked increases in the level of inflammatory mediators have been reported in the brain of a neuronopathic GD mouse model (Vitner et al., 2012). Subsequent studies have further detailed how a reduction in GCase activity is associated with clear glial activation. Ginns et al. (2014) reported astrocytosis and microglia activation in the nigrostriatal tract of GBA1-mutant mice, as well as in mice treated sub-chronically with GCase inhibitor CBE, which also induced alpha-synuclein accumulation. These results were confirmed by the studies from Rocha et al. (2015b) and Mus et al. (2019), both showing that a CBE-induced drop in GCase enzymatic activity activates microglia throughout the brain. Interestingly, using a DAT-GBA1-KO mouse displaying selective homozygous GBA1 disruption in midbrain dopamine neurons, Soria et al. (2017) demonstrated strong microglial activation in the substantia nigra of these mice, without any evidence of dopaminergic cell loss, alpha-synuclein accumulation, or motor abnormalities.
More than 300 mutations in the GBA1 gene, such as insertions, deletions, and point mutations, have been discovered so far (O’Regan et al., 2017; Zhang et al., 2018a). The N370S (c.1226A > G) and the L444P (c.1448T > C) mutations are the most common mutations worldwide (Zhang et al., 2018b). A recent meta-analysis described other GBA1 variants, such as R120W, IVS2 + 1G > A, H255Q, D409H, RecNciI, E326K, and T369M related to PD risk. Ethnic heterogeneity of GBA1 mutations in PD was also reported. R496H and 84insGG increase PD risks exclusively in AJ populations, while L444P, E326K, T369M, R120W, IVS2 + 1G > A, H255Q, D409H, and RecNciI were found more frequently in non-AJ subjects. N370S is correlated to an increased PD risk in all populations (Zhang et al., 2018b).
A substantial body of evidence suggests the existence of a prodromal phase of iPD, of variable length, preceding the onset of the classical signs of the disease (Schapira and Tolosa, 2010; Schapira et al., 2017; Heinzel et al., 2019). The existence of such a premotor period has been proposed based on imaging, pathology, clinical, and epidemiological studies, suggesting that the nigrostriatal lesion might evolve in 7–10 years before becoming clinically manifest (Hawkes, 2008; Schapira and Tolosa, 2010). Although stiffness, tremors, or imbalance may be present as prodromal features (de Lau et al., 2006), the vast majority of early symptoms and signs in PD are of the non-motor type, including hyposmia, REM behavior disorder (RBD), constipation, depression, and color discrimination (Hawkes, 2008; Postuma and Berg, 2016; Schapira et al., 2017).
A prodromal phase was also described in both GD and heterozygous GBA1 carriers without a clinical diagnosis of PD (McNeill et al., 2012a,b). Cognitive and olfactory functions were significantly impaired and motor testing was abnormal in GD patients and GBA1 mutation carriers without PD, compared to healthy subjects. No differences were found in terms of sleep abnormalities or autonomic function (Beavan et al., 2015; Avenali et al., 2019).
In a web-based evaluation of prodromal markers designed to estimate PD risk, Noyce et al. (2014, 2015) observed that the odds of having a GBA1 variant was 9.5 times higher in the “high risk group” compared to other groups.
Compared to iPD, GBA-PD patients are characterized by a slightly younger age at onset, higher incidence of neuropsychiatric features (such as hallucination, depression, and anxiety), greater incidence of sleep disturbances, and earlier development of cognitive deficits (Neumann et al., 2009; Sidransky et al., 2009; Brockmann et al., 2011; McNeill et al., 2012b; Winder-Rhodes et al., 2013). In general, the disease course tends to be more aggressive in GBA-PD patients (Winder-Rhodes et al., 2013; Zokaei et al., 2014; Brockmann et al., 2015b; Cilia et al., 2016; Jesús et al., 2016; Figure 1).
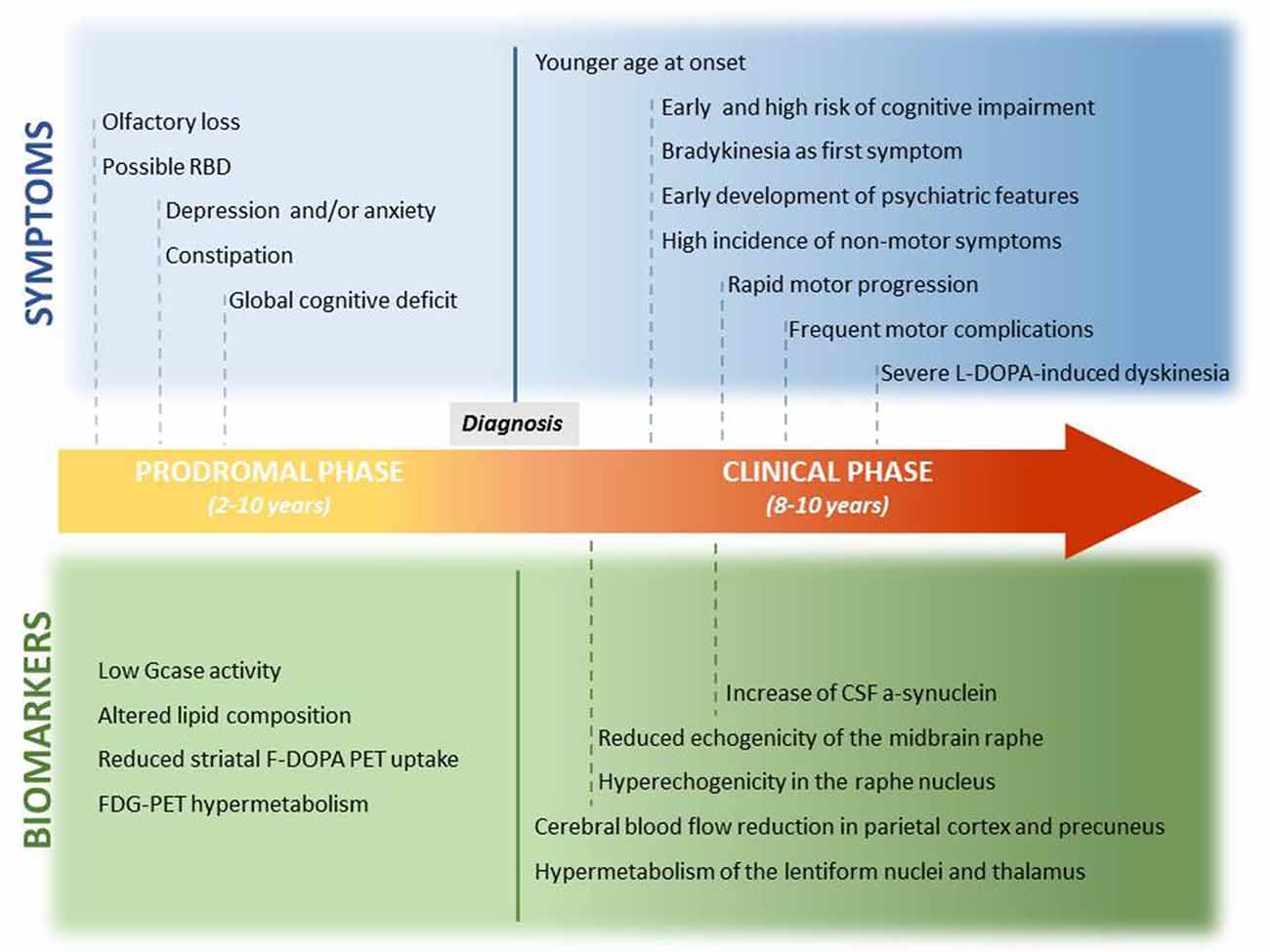
Figure 1. Prodromal and clinical features of GBA-PD and related biomarkers. This is a graphic representation of the potential timeline by which the preclinical symptoms and motor and non-motor features of GBA-PD may manifest. Potential biomarkers are also reported, and they are correlated with the onset of clinical features.
GBA-PD patients exhibit an asymmetric onset with the classic triad of motor symptoms, bradykinesia, rigidity, and tremors (Goker-Alpan et al., 2008), although bradykinesia seems to be more common, as an initial symptom, than in iPD (Ziegler et al., 2007; Gan-Or et al., 2010; Lesage et al., 2011).
Longitudinal cohort studies using the Unified Parkinson’s Disease Rating Scale, motor subscale, and Hoehn and Yahr staging showed a more rapid motor progression in GBA-PD than iPD patients (Winder-Rhodes et al., 2013; Brockmann et al., 2015b). One study in a large European series (Lesage et al., 2011) reported that L-Dopa induced dyskinesias were more severe in GBA-PD patients, but no difference was observed in another study (Zhang et al., 2015).
Motor complications, such as swallowing disorders, dysarthria, and freezing of gait, are observed more frequently in GBA-PD patients (Jesús et al., 2016). The risk of developing motor fluctuations and dyskinesia seems to be higher in GBA-PD subjects carrying a severe mutation (Cilia et al., 2016; Jesús et al., 2016; Lythe et al., 2017).
Non-motor symptoms are the earliest presenting symptoms in GBA-PD, as well as in iPD, but some of them may be more common in GBA-PD patients.
Compared with iPD, GBA-PD patients show a higher risk for cognitive deterioration (Gan-Or et al., 2010). Several studies investigated the difference in cognitive performance between iPD and GBA-PD patients (Goker-Alpan et al., 2008; Alcalay et al., 2012; McNeill et al., 2012b; Nalls et al., 2013; Malec-Litwinowicz et al., 2014; Cilia et al., 2016). In a meta-analysis conducted by Ziegler et al. (2007), the risk of cognitive impairment was three times higher for patients with GBA1 mutations. Cognitive impairment seems to affect memory and visuo-spatial domains (Alcalay et al., 2012), working memory, executive and visuospatial abilities (Mata et al., 2016), and visual short-term memory (Zokaei et al., 2014) in particular. GBA-PD patients may undergo cognitive decline sooner than iPD patients (Winder-Rhodes et al., 2013; Brockmann et al., 2015b; Oeda et al., 2015; Liu et al., 2016), although this evidence was not reported by all studies (Aharon-Peretz et al., 2005; Clark et al., 2007; Nichols et al., 2009).
GBA-PD shows increased frequency of psychiatric symptoms at an earlier stage (Oeda et al., 2015; Cilia et al., 2016), as well as a higher risk for delirium and hallucinations when compared with iPD (Aharon-Peretz et al., 2005; Wang et al., 2014), without differences linked to mutation severity (Barrett et al., 2014).
Olfactory functions are affected in GBA1 mutation carriers, as they are in early iPD patients (McNeill et al., 2012b).
A substantial impact of depression was initially reported in GBA-PD patients (Machaczka et al., 1999), but was not confirmed by subsequent studies (Winder-Rhodes et al., 2013; Zhang et al., 2015). Analogously, conflicting results have been reported for anxiety and apathy, whose frequency in GBA-PD patients was found to be increased (Brockmann et al., 2011; Wang et al., 2014) compared to iPD patients.
Dysautonomic symptoms such as bowel alteration, sexual impairment, orthostatic symptoms, and urinary dysfunction are other non-motor symptoms frequently reported in GBA-PD individuals (Brockmann et al., 2011; Wang et al., 2014).
As for pain, contradictory findings have been reported, without a clear indication that pain may afflict GBA-PD more than iPD patients (Gan-Or et al., 2010; Kresojević et al., 2015).
GBA1 mutations may have differential effects on PD risks, depending on the specific variant, as well as on the clinical profile and disease progression rate. For mild GBA1 mutation carriers, the odds ratios for developing PD ranged between 2.84 and 4.94, while for severe GBA1 mutation carriers the odds ratios were between 9.92 and 21.29 (Gan-Or et al., 2015). The risk for dementia in GBA-PD subjects bearing severe mutations (L444P, splicing mutation IVS10 + 1G > T) is 5.6 times greater than in iPD patients and 2.9 times greater than in patients carrying mild GBA1 mutations (N370S; Cilia et al., 2016). The risk of hallucination development is not affected by the severity of GBA1 mutations (Barrett et al., 2014).
In a more recent study, motor and some non-motor features (depression, RBD, and olfactory loss) were significantly worse in GBA-PD patients with severe mutations than in those bearing mild mutations (Thaler et al., 2018a).
E326K is the most prevalent PD-associated GBA1 mutation and has not been described in GD (Duran et al., 2013). PD patients bearing E236K mutations show a faster progression of motor symptoms, with gait disorders and postural instability. They also have a higher risk of cognitive decline, but a lesser risk for motor complications (Winder-Rhodes et al., 2013; Davis et al., 2016; Mata et al., 2016).
Data on genotype-phenotype correlations between other GBA1 polymorphisms, clinical features, and risk rate of progression are lacking. Further research is therefore mandatory, in order to better clarify the role of GBA1 variants in specific clinical manifestations and disease progression.
No reliable biomarker that may inform on the ongoing neuronal loss, before the onset of PD clinical features, is yet available. The knowledge that has been accumulating on GBA1 defects in PD may help us fill this gap (Figure 1).
Positron emission tomography (PET) employing several different radioisotopes [fluorodeoxyglucose (FDG), F-Dopa, and C-CFT, 2β-carbomethoxy-3β-(4-fluorophenyl-tropane)] has been used to assess brain metabolism in GBA-PD. Kono et al. (2010) first described a significant FDG decrease in the supplementary motor area of the frontal cortex in GBA1 mutants (one GD-PD patient, two GBA-PD patients, and three GBA1 heterozygous carriers). GBA-PD patients also showed hypometabolism in the parieto-occipital cortices, while GBA heterozygous carriers without PD showed normal metabolism in the putamen and increased metabolism in the caudate nucleus (Kono et al., 2010). Using a different tracer (11C-CFT, binding to the dopamine transporter) the same group also demonstrated a significant presynaptic dysfunction in GBA-PD, with a severe reduction of striatal uptake in both putamen and caudate nucleus, whereas asymptomatic GBA carriers had increased caudate uptake with normal putamen uptake (Kono et al., 2010).
Fluorodopa PET or single-photon emission computerized tomography (SPECT) with dopamine-sensitive ligands in GD-PD patients showed asymmetric striatal dopaminergic neuronal loss with the highest reduction in the caudate and posterior putamen nuclei; this reduction was also observed in iPD brains, although less represented (Goker-Alpan et al., 2012). By analyzing the resting regional cerebral blood flow, the same authors reported bilateral hypoperfusion in the lateral parieto-occipital association cortex and precuneus in GD-PD, but not in iPD patients. They also demonstrated that in GD subjects without PD-like manifestations, the F-Dopa uptake was decreased in the striatum, with a marked neuronal loss in the tail of the putamen, whereas this reduction was not found in GBA1 heterozygous asymptomatic carriers (Goker-Alpan et al., 2012). More recently, a large cohort study conducted within the Parkinson’s Progression Markers Initiative of the Michael J. Fox Foundation reported increased DAT striatal binding ratios in GBA asymptomatic carriers, compared with healthy controls (Simuni et al., 2020).
A magnetic resonance spectroscopy study conducted by Brockmann et al. (2012) showed significant reduction of N-acetylaspartate and choline levels in the putamen and in the midbrain of GBA-PD patients, compared to healthy subjects, along with increased levels of glycerophosphoethalonamine, pointing to a reduction in neuronal integrity and altered membrane phospholipid metabolism.
As for MRI structural data, Agosta et al. (2013) reported a diffused pattern of white matter alterations involving the parahippocampal tract and the parietal and occipital lobes, including interhemispheric and frontal cortical connections, in GBA-PD patients compared to iPD patients. On the other hand, when cortical thickness and subcortical volumes were more recently analyzed, no differences between GBA-PD and iPD patients were observed (Thaler et al., 2018b).
Transcranial sonography is able to detect substantia nigra hyperechogenicity in 90% of iPD patients and has been proposed as a potential screening tool for the early detection of PD (Beavan and Schapira, 2013). The few studies conducted on GBA1 positive individuals (Saunders-Pullman et al., 2010; Brockmann et al., 2011; Barrett et al., 2013) reported a greater substantia nigra echogenicity when compared to healthy controls, but without any differences with iPD. More recently, Arkadir et al. (2019) reported an enlarged area of hyperechogenicity also in the substantia nigra of asymptomatic GBA1 carriers and GD patients without PD, with respect to healthy controls.
Finally, myocardial 123I-metaiodobenzylguanidine scintigraphy showed a marked reduction of tracer accumulation in PD patients carrying GBA1 mutation compared to iPD patients (Li et al., 2014), suggesting that neurodegeneration of myocardial nerve fibers had begun before clinical presentation and/or that disease progression is more rapid in this population. Reduced myocardial uptake of the tracer was also demonstrated by Oeda et al. (2015) in GBA-PD patients, but in this case without differences with respect to iPD.
Although several imaging biomarkers have been proposed to discriminate PD subjects with and without GBA1 mutations, no conclusive evidence has yet been produced. Further imaging studies on wider populations will be needed to better clarify the neuroimaging correlates of GBA-PD.
Several studies investigated the possible value of GCase enzymatic activity as a potential biomarker for PD. Decreased GCase activity has been observed in fibroblasts and in the cerebrospinal fluid (CSF) from GD, GBA-PD, and also iPD patients (Parnetti et al., 2009, 2014; Gegg et al., 2012; Ambrosi et al., 2015). Moreover, Parnetti et al. (2014) observed that PD patients at an early stage of the disease showed lower GCase activity levels than patients at later stages, suggesting that GCase could have a potential role as a marker of early PD phases.
Data on GCase activity in iPD subjects are controversial. Indeed, a study on a Dutch cohort of de novo iPD patients (van Dijk et al., 2013) reported only a trend toward a decrease in GCase activity. These mixed results may be attributed to different collection and storage procedures for CSF, but also to the intrinsic instability of GCase in CSF (Persichetti et al., 2014).
GCase enzymatic activity is also detectable in leucocytes isolated from peripheral blood, which can be obtained with a far simpler and less invasive procedure than CSF. Raghavan et al. (1980) first developed a reliable and reproducible assay technique to measure GCase enzymatic activity in human leukocytes, by using the artificial substrate 4MU-/3-glucoside. Kim et al. (2016) used this technique to explore if GCase activity was altered in the leucocytes of iPD and subjects with PD associated to a genetic mutations. They did not find differences in GCase activity between groups of patients, but GCase activity was positively associated with disease duration in iPD. A reduction of GCase activity has also been observed in blood samples from both iPD and GBA-PD patients (Alcalay et al., 2015). In these cohorts, the authors tested the association between GCase enzymatic activity and PD disease severity. They reported that in iPD patients, higher GCase enzymatic activity was correlated with a longer disease duration and a milder disease progression.
Recently, in a pilot study, Ortega et al. (2016) measured GCase activity from leucocytes isolated from drawn heparinized blood in GBA1 heterozygous and homozygous mutation carriers, with and without PD, and iPD. They observed that GCase activity was significantly lower in GBA-PD than iPD patients and even lower in GD-PD patients, suggesting that GCase activity could be a possible marker of the GBA1 mutated condition.
Other metabolites linked to GCase deficiency have been analyzed in GBA-PD patients. Indeed, GCase deficiency in the brain of PD subjects may also be correlated to pathological alterations in the lipid metabolism (Schapira et al., 2016). In a gas chromatography analysis, free fatty acids in the CSF of GBA-PD patients showed a different pattern compared to iPD and control subjects (Schmid et al., 2012). Alterations of fatty acid metabolites have been found also in the plasma of iPD patients, with an increase of the glucosylcerebroside substrate and different ceramide species (Mielke et al., 2013). These results were confirmed by Pchelina et al. (2018) in a recent study where GBA-PD patients showed increased hexosylsphingosine levels in plasma when compared with iPD and controls.
Alterations of other lysosomal enzymes have been described in iPD subjects. Huebecker et al. (2019) reported that reduction in GCase activity in human post-mortem substantia nigra of iPD were correlated with alterations in lysosomal sphingolipid hydrolases and concomitant glycosphingolipid substrates accumulation. In addition, in this study iPD showed a distinctly significant reduction in levels of complex gangliosides (e.g., GM1a) in substantia nigra, as well as in CSF and serum. Same findings, albeit less marked, were also observed in elderly controls as well as in subjects with RBD (Huebecker et al., 2019).
Although cognitive deficits are frequently reported in GBA-PD, CSF levels of tau and β-amyloid—which are altered in patients with Alzheimer’s disease—were unchanged in GBA-PD patients (Beavan and Schapira, 2013). However, higher baseline CSF levels of p-Tau in GBA-PD patients seem to be related with increased deterioration of cognitive functions over time (24 months; Brockmann et al., 2015a). Moreover, by assessing CSF profiles longitudinally, GBA-PD showed lower levels of Aβ1–42, t-Tau, and p-Tau than control subjects and LRRK2-PD.
Finally, alpha-synuclein was also measured in plasma and in CSF in subjects carrying GBA1 mutations. Parnetti et al. (2014) evaluated alpha-synuclein levels in CSF of iPD subjects, showing decreased levels of total alpha-synuclein in iPD compared with healthy controls in contrast to an increased level of oligomeric form of alpha-synuclein and a higher oligomeric/total alpha-syn ratio in iPD than controls. In the same study they also revealed that in PD patients carrying GBA1 mutations, CSF levels of total and oligomeric alpha-synuclein were higher than iPD (Parnetti et al., 2014).
Moreover, by combining GCase activity, oligomeric/total alpha-synuclein ratio in CSF, and age they discriminated PD patients from controls with a high sensitivity (82%) and specificity (71%; Parnetti et al., 2014).
Nuzhnyi et al. (2015) showed a negative association between plasma oligomeric alpha-synuclein and GCase activity in leucocytes from GD patients, speculating that the reduced GCase activity may be correlated to the peripheral aggregation of toxic alpha-synuclein species.
Recently, Lerche et al. (2020) showed reduced CSF levels of total alpha-synuclein in GBA-PD compared to iPD and controls. Moreover, in this study, GBA-PD subjects bearing a severe mutation showed lower CSF levels of total alpha-synuclein than GBA-PD with mild or risk variant mutations or iPD patients.
Overall, reduced CSF alpha-synuclein levels might reflect its accumulation in the brain, but further studies are warranted to better understand the meaning of alpha-synuclein changes in the CSF of PD patients carrying different pathogenic GBA1 mutations.
GBA-PD individuals show, especially in the initial phases of the disease, a good response to L-Dopa, although motor symptoms seem to progress more rapidly than in iPD. Therefore, a specific therapeutic management should be considered in these patients.
A recent article investigating the outcomes of treatment with deep brain stimulation in a cohort of GBA-PD patients after a 7.5-year follow-up demonstrated similar outcomes compared to iPD in terms of motor symptoms, whereas cognitive deterioration and non-motor features were more represented among GBA-PD patients (Lythe et al., 2017). However, because of the positive effects on motor impairment, deep brain stimulation should also be considered as a suitable therapeutic option for these patients.
Despite the availability of successful treatments for systemic manifestations in GD patients, these approaches do not affect glycosphingolipid accumulation in the central nervous system, because they are not able to cross the blood-brain barrier. Current experimental approaches available in clinical trials (Table 1) are founded on the main hypothesis that the loss of function determined by GBA1 mutations causes abnormal glycosphingolipid accumulation, leading to neuronal dysfunction and associated proteinopathy. Increasing GCase activity via small chaperones showed good efficacy in modulating pathological alterations in models of disease. In order to address this pathogenetic mechanism, two different chaperones that increase GCase activity in the brain were investigated. The mucolytic ambroxol was described to improve GCase enzymatic activity and reduce total and phosphorylated alpha-synuclein levels in models of PD (McNeill et al., 2014; Ambrosi et al., 2015). Currently, two clinical trials assessing the safety, tolerability and efficacy of ambroxol are ongoing. In 2016, a phase II, single-center (Canada), randomized placebo-controlled, double-blind trial started to assess the safety, tolerability, and efficacy of ambroxol on cognitive and motor symptoms of PD Dementia (PDD; ClinicalTrials.gov NCT02914366). Preliminary results showed that higher dose ambroxol are effective and safe in PDD (Silveira et al., 2019). A similar approach is being tested by Schapira’s group in a cohort of GBA-PD patients (ClinicalTrials.gov: NCT02941822).
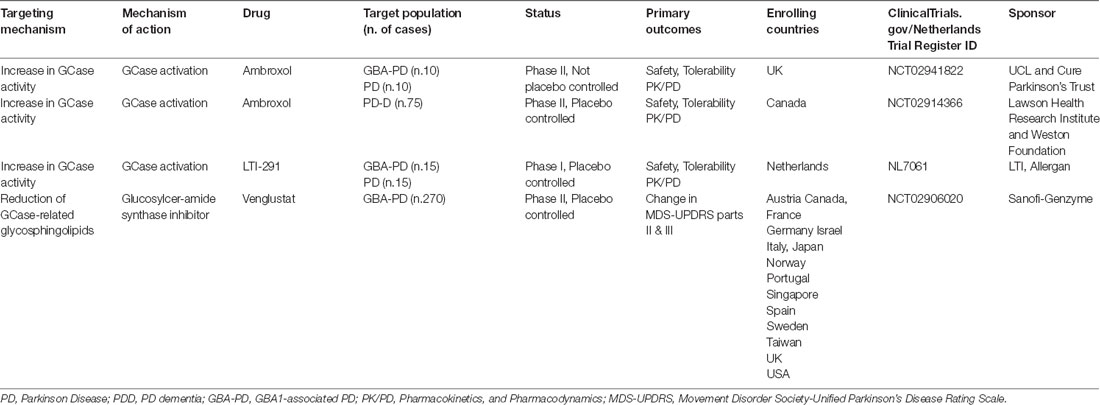
Table 1. Disease-modifying therapies in active GBA-PD trials.
Another approach is being tested in the Netherlands by Allergan in a phase I clinical trial with LTI-291, a non-inhibitory chaperone of GCase able to both increase its catalytic activity and extend its half-life (Nederlands Trial Register: NL7061).
An alternative therapeutic strategy that is being explored is to reduce the accumulation of glucosylceramide (the substrate degraded by the normal GCase) by targeting the enzyme glucosylceramide synthetase. The substrate reduction restored glycosphingolipid balance and reduced alpha-synuclein accumulation in neuronal cells of PD with and without GBA1 mutations (Kim et al., 2016; Zunke et al., 2018). In mouse models of GBA-related synucleinopathy, the use of a glucosylceramide synthetase inhibitor improved alpha-synuclein degradation and the behavioral alterations (Sardi et al., 2017). Based on these findings, a multicenter phase II, randomized, double-blind, placebo-controlled study supported by Sanofi-Genzyme, is investigating the safety, pharmacokinetics, and pharmacodynamics of Venglustat (GZ/SAR402671), an oral inhibitor of glucosylceramide synthase in GBA1 carriers with early-stage PD (MOVES-PD study: NCT02906020).
Gene therapy approaches are also being developed as new therapeutic strategies for PD. The most common mechanism used to cross the blood-brain barrier is the delivery of genes via viral vectors to the affected areas of the brain. The most common vector used in preclinical studies on animal models in relation to GBA1 is the adeno-associated virus (AAV). Based on the assumption that a decrease in GCase activity might favor alpha-synuclein accumulation and synucleinopathy development, AAV- mediated GBA1 delivery has been tested in mouse models of GD-related PD to assess its effects on GCase and alpha-synuclein modulation (Sardi et al., 2011, 2013; Rocha et al., 2015b). Sardi et al. (2011) first demonstrated that treatment with AAV-GBA1 reduced alpha-synuclein levels, increased GCase activity, reduced glucosylsphingosine and glucoceramide levels, and improved the cognitive deficits in a pre-symptomatic mouse model of GD. The same author replicated the experiment in a symptomatic mouse model of GD-associated PD (Sardi et al., 2013). In this case, the increased GCase was able to modulate the aberrant lipids accumulations, reduce alpha-synuclein and tau aggregation, and ameliorate cognitive functions. When tested in an A53T alpha-synuclein mouse model, the increase in GCase levels reduced alpha-synuclein levels, demonstrating the relationship between alpha-synuclein and GCase activity and suggesting that this approach may also be beneficial for PD without GBA1 mutations (Sardi et al., 2013).
Rocha et al. (2015b) also demonstrated the neuroprotective effects of GBA1 gene transfer in rodent models of PD. They observed that in a transgenic mouse model overexpressing wild-type alpha-synuclein, the increased GCase activity was able to reduce alpha-synuclein accumulation in the substantia nigra and striatum by improving insoluble alpha-synuclein clearance. Moreover, in a rat model of selective dopamine neuron degeneration, they showed that the overexpression of GBA1 preserved dopaminergic neurons from neurodegeneration (Rocha et al., 2015b).
Recently, Morabito et al. (2017) developed a non-invasive approach for the AAV delivery, using intravenous injection. This method has the advantage to treat non-selective brain areas and prolong gene overexpression. Transduction of A53T- SNCA transgenic mice with AAV-PHP.B-GBA1 restored GCase activity and improved alpha-synuclein pathology (Morabito et al., 2017).
Interestingly, Massaro et al. (2019) tested a gene therapy approach on a neuronopathic Gaucher disease mouse model. Authors demonstrated that fetal intracranial injections of AAV-GBA1 increased GCase activity, reduced neuroinflammation, blocked neurodegeneration, and improved motor and systemic symptoms (Massaro et al., 2019). Finally, the same authors assessed the effects of a systemic AAV-GBA1 injection, showing prolongation of the life span of treated mice, reduction of visceral and motor symptoms, and protection from fatal neurodegeneration (Massaro et al., 2020).
Gene therapy may therefore hold fascinating promises for PD patients with and without GBA1 mutations, but clinical trials to assess this type of approach will be needed.
Although genetic information could potentially help in the diagnosis, prognosis, and treatment of PD, clinical practice guidelines for PD genetic testing are not available yet, and genetic analyses are still limited in PD patients. In the past, the majority of the medical community was reluctant to recommend genetic testing for PD, which was mostly restricted to familial PD. However, in the last few years, the recent development of clinical trials targeting patients with GBA-PD and the launch of personalized medicine has prompted interest in receiving genotype information and increased the demand for genetic counselling regarding GBA1 mutations. In this view, the European Federation of Neurological Societies re-evaluated the recommendations for genetic testing, proposing that it should be considered for PD firstly when the following risk factors are present:
(1) Family history of PD; (2) early onset of PD (before 50 years old); or (3) individual among “population at risk,” such as AJ individuals.
Genetic screening should be also performed to: (1) stratify patients into subtypes to better understand the disease process; and (2) provide patients with the opportunity to participate in clinical trials with experimental therapies designed for patients with specific genotypes (Payne et al., 2019). For all these reasons, genetic counselling should be warranted and implemented.
GBA1 mutations play a pivotal role in the molecular pathogenesis of PD and are considered one of the most frequent risk factors for the disease. Increasing evidence suggests that GCase deficiency might also have a role in iPD.
The pathobiological mechanisms underlying the link between a defective function of GCase and the development of PD are still poorly understood, but interactions with alpha-synuclein, lysosomal dysfunction, ER stress, and neuroinflammation may all play a crucial role.
Further studies on large GBA1-positive populations will be needed to precisely identify the biological factors that link GBA1 mutations to PD. This will improve the diagnostic and caring process, possibly leading to the discovery of new therapeutic targets to engage in the frame of an innovative, personalized approach to PD therapy.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was funded by the Italian Ministry of Health (Ricerca Corrente) in the frame of the JPND project GBAPARK. This is an EU Joint Programme–Neurodegenerative Disease Research (JPND) project (GBAPARK). The project is supported through the following funding organizations under the aegis of JPND (www.jpnd.eu): Canada, Canadian Institutes of Health Research-University of Ottawa; Germany, DZNE; Italy, Ministry of Health; United Kingdom, Medical Research Council-University College London.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agosta, F., Kostic, V. S., Davidovic, K., Kresojević, N., Sarro, L., Svetel, M., et al. (2013). White matter abnormalities in Parkinson’s disease patients with glucocerebrosidase gene mutations. Mov. Disord. 28, 772–778. doi: 10.1002/mds.25397
Aharon-Peretz, J., Badarny, S., Rosenbaum, H., and Gershoni-Baruch, R. (2005). Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype-genotype correlation. Neurology 65, 1460–1461. doi: 10.1212/01.wnl.0000176987.47875.28
Alcalay, R. N., Caccappolo, E., Mejia-Santana, H., Tang, M., Rosado, L., Orbe Reilly, M., et al. (2012). Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78, 1434–1440. doi: 10.1212/WNL.0b013e318253d54b
Alcalay, R. N., Levy, O. A., Waters, C. C., Fahn, S., Ford, B., Kuo, S. H., et al. (2015). Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658. doi: 10.1093/brain/awv179
Ambrosi, G., Ghezzi, C., Zangaglia, R., Levandis, G., Pacchetti, C., and Blandini, F. (2015). Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 82, 235–242. doi: 10.1016/j.nbd.2015.06.008
Arkadir, D., Dinur, T., Becker Cohen, M., Revel-Vilk, S., Tiomkin, M., Brüggemann, N., et al. (2019). Prodromal substantia nigra sonography undermines suggested association between substrate accumulation and the risk for GBA -related Parkinson’s disease. Eur. J. Neurol. 26, 1013–1018. doi: 10.1111/ene.13927
Avenali, M., Toffoli, M., Mullin, S., McNeil, A., Hughes, D. A., Mehta, A., et al. (2019). Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J. Neurol. Neurosurg. Psychiatry 90, 1091–1097. doi: 10.1136/jnnp-2019-320394
Balestrino, R., and Schapira, A. H. V. (2018). Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist 24, 540–559. doi: 10.1177/1073858417748875
Barrett, M. J., Hagenah, J., Dhawan, V., Peng, S., Stanley, K., Raymond, D., et al. (2013). Transcranial sonography and functional imaging in glucocerebrosidase mutation Parkinson disease. Parkinsonism Relat. Disord. 19, 186–191. doi: 10.1016/j.parkreldis.2012.09.007
Barrett, M. J., Shanker, V. L., Severt, W. L., Raymond, D., Gross, S. J., Schreiber-Agus, N., et al. (2014). Cognitive and antipsychotic medication use in monoallelic GBA-related Parkinson disease. JIMD Rep. 16, 31–38. doi: 10.1007/8904_2014_315
Beavan, M., McNeill, A., Proukakis, C., Hughes, D. A., Mehta, A., and Schapira, A. H. V. (2015). Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 72, 201–208. doi: 10.1001/jamaneurol.2014.2950
Beavan, M. S., and Schapira, A. H. V. (2013). Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann. Med. 45, 511–521. doi: 10.3109/07853890.2013.849003
Bendikov-Bar, I., Ron, I., Filocamo, M., and Horowitz, M. (2011). Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 46, 4–10. doi: 10.1016/j.bcmd.2010.10.012
Beutler, E., Gelbart, T., and Scott, C. R. (2005). Hematologically important mutations: gaucher disease. Blood Cells Mol. Dis. 35, 355–364. doi: 10.1016/j.bcmd.2005.07.005
Blandini, F., Cilia, R., Cerri, S., Pezzoli, G., Schapira, A. H. V., Mullin, S., et al. (2019). Glucocerebrosidase mutations and synucleinopathies: toward a model of precision medicine. Mov. Disord. 34, 9–21. doi: 10.1002/mds.27583
Brockmann, K., Hilker, R., Pilatus, U., Baudrexel, S., Srulijes, K., Magerkurth, J., et al. (2012). GBA-associated PD: neurodegeneration, altered membrane metabolism, and lack of energy failure. Neurology 79, 213–220. doi: 10.1212/wnl.0b013e31825dd369
Brockmann, K., Schulte, C., Deuschle, C., Hauser, A.-K., Heger, T., Gasser, T., et al. (2015a). Neurodegenerative CSF markers in genetic and sporadic PD: classification and prediction in a longitudinal study. Parkinsonism Relat. Disord. 21, 1427–1434. doi: 10.1016/j.parkreldis.2015.10.008
Brockmann, K., Srulijes, K., Pflederer, S., Hauser, A. K., Schulte, C., Maetzler, W., et al. (2015b). GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 30, 407–411. doi: 10.1002/mds.26071
Brockmann, K., Srulijes, K., Hauser, A.-K., Schulte, C., Csoti, I., Gasser, T., et al. (2011). GBA-associated PD presents with nonmotor characteristics. Neurology 77, 276–280. doi: 10.1212/wnl.0b013e318225ab77
Chiasserini, D., Paciotti, S., Eusebi, P., Persichetti, E., Tasegian, A., Kurzawa-Akanbi, M., et al. (2015). Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 10:15. doi: 10.1186/s13024-015-0010-2
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Clark, L. N., Ross, B. M., Wang, Y., Mejia-Santana, H., Harris, J., Louis, E. D., et al. (2007). Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69, 1270–1277. doi: 10.1212/01.wnl.0000276989.17578.02
Cleeter, M. W. J., Chau, K. Y., Gluck, C., Mehta, A., Hughes, D. A., Duchen, M., et al. (2013). Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. doi: 10.1016/j.neuint.2012.10.010
Cullen, V., Sardi, S. P., Ng, J., Xu, Y. H., Sun, Y., Tomlinson, J. J., et al. (2011). Acid β-glucosidase mutants linked to gaucher disease, parkinson disease and lewy body dementia alter α-synuclein processing. Ann. Neurol. 69, 940–953. doi: 10.1002/ana.22400
Davis, M. Y., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., et al. (2016). Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in parkinson disease. JAMA Neurol. 73, 1217–1224. doi: 10.1001/jamaneurol.2016.2245
de Lau, L. M. L., Koudstaal, P. J., Hofman, A., and Breteler, M. M. B. (2006). Subjective complaints precede Parkinson disease: the Rotterdam study. Arch. Neurol. 63, 362–365. doi: 10.1001/archneur.63.3.noc50312
Duran, R., Mencacci, N. E., Angeli, A. V., Shoai, M., Deas, E., Houlden, H., et al. (2013). The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 28, 232–236. doi: 10.1002/mds.25248
Fecchio, C., Palazzi, L., and de Laureto, P. P. (2018). α-synuclein and polyunsaturated fatty acids: molecular basis of the interaction and implication in neurodegeneration. Molecules 23:E1531. doi: 10.3390/molecules23071531
Fernandes, H. J. R., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Franco, R., Sánchez-Arias, J. A., Navarro, G., and Lanciego, J. L. (2018). Glucocerebrosidase mutations and synucleinopathies. Potential role of sterylglucosides and relevance of studying Both GBA1 and GBA2 genes. Front. Neuroanat. 12:52. doi: 10.3389/fnana.2018.00052
Fuzzati-Armentero, M. T., Cerri, S., and Blandini, F. (2019). Peripheral-central neuroimmune crosstalk in Parkinson’s disease: what do patients and animal models tell us? Front. Neurol. 10:232. doi: 10.3389/fneur.2019.00232
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., Mirelman, A., et al. (2015). Differential effects of severe vs. mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/wnl.0000000000001315
Gan-Or, Z., Bar-Shira, A., Mirelman, A., Gurevich, T., Kedmi, M., Giladi, N., et al. (2010). LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 11, 121–125. doi: 10.1007/s10048-009-0198-9
Gegg, M. E., Burke, D., Heales, S. J. R., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Gegg, M. E., and Schapira, A. H. V. (2018). The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 285, 3591–3603. doi: 10.1111/febs.14393
Gegg, M. E., Sweet, L., Wang, B. H., Shihabuddin, L. S., Sardi, S. P., and Schapira, A. H. V. (2015). No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 30, 1085–1089. doi: 10.1002/mds.26278
Ginns, E. I., Mak, S. K. K., Ko, N., Karlgren, J., Akbarian, S., Chou, V. P., et al. (2014). Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 111, 152–162. doi: 10.1016/j.ymgme.2013.12.003
Goker-Alpan, O., Lopez, G., Vithayathil, J., Davis, J., Hallett, M., and Sidransky, E. (2008). The spectrum of Parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 65, 1353–1357. doi: 10.1001/archneur.65.10.1353
Goker-Alpan, O., Masdeu, J. C., Kohn, P. D., Ianni, A., Lopez, G., Groden, C., et al. (2012). The neurobiology of glucocerebrosidase-associated parkinsonism: a positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain 135, 2440–2448. doi: 10.1093/brain/aws174
Grabowski, G. A. (2008). Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 372, 1263–1271. doi: 10.1016/S0140-6736(08)61522-6
Hawkes, C. H. (2008). The prodromal phase of sporadic Parkinson’s disease: does it exist and if so how long is it? Mov. Disord. 23, 1799–1807. doi: 10.1002/mds.22242
Heinzel, S., Berg, D., Gasser, T., Chen, H., Yao, C., and Postuma, R. B. (2019). Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 34, 1464–1470. doi: 10.1002/mds.27802
Henderson, M. X., Sedor, S., McGeary, I., Cornblath, E. J., Peng, C., Riddle, D. M., et al. (2020). Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron 105, 822.e7–836.e7. doi: 10.1016/j.neuron.2019.12.004
Huebecker, M., Moloney, E. B., Van Der Spoel, A. C., Priestman, D. A., Isacson, O., Hallett, P. J., et al. (2019). Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 14:40. doi: 10.1186/s13024-019-0339-z
Jesús, S., Huertas, I., Bernal-Bernal, I., Bonilla-Toribio, M., Cáceres-Redondo, M. T., Vargas-González, L., et al. (2016). GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS One 11:e0167749. doi: 10.1371/journal.pone.0167749
Kim, H.-J., Jeon, B., Song, J., Lee, W.-W., Park, H., and Shin, C.-W. (2016). Leukocyte glucocerebrosidase and β-hexosaminidase activity in sporadic and genetic Parkinson disease. Parkinsonism Relat. Disord. 23, 99–101. doi: 10.1016/j.parkreldis.2015.12.002
Kono, S., Ouchi, Y., Terada, T., Ida, H., Suzuki, M., and Miyajima, H. (2010). Functional brain imaging in glucocerebrosidase mutation carriers with and without parkinsonism. Mov. Disord. 25, 1823–1829. doi: 10.1002/mds.23213
Kresojević, N., Janković, M., Petrović, I., Kumar, K. R., Dragašević, N., Dobričić, V., et al. (2015). Presenting symptoms of GBA-related Parkinson’s disease. Parkinsonism Relat. Disord. 21, 804–807. doi: 10.1016/j.parkreldis.2015.04.028
Lee, A., and Gilbert, R. M. (2016). Epidemiology of Parkinson disease. Neurol. Clin. 34, 955–965. doi: 10.1016/j.ncl.2016.06.012
Lerche, S., Wurster, I., Roeben, B., Zimmermann, M., Riebenbauer, B., Deuschle, C., et al. (2020). Parkinson’s disease: glucocerebrosidase 1 mutation severity is associated with CSF α-synuclein profiles. Mov. Disord. 35, 495–499. doi: 10.1002/mds.27884
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., et al. (2011). Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210. doi: 10.1093/hmg/ddq454
Li, H., Ham, A., Ma, T. C., Kuo, S. H., Kanter, E., Kim, D., et al. (2019). Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130. doi: 10.1080/15548627.2018.1509818
Li, Y., Sekine, T., Funayama, M., Li, L., Yoshino, H., Nishioka, K., et al. (2014). Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol. Aging 35:935.e3-8. doi: 10.1016/j.neurobiolaging.2013.09.019
Liu, G., Boot, B., Locascio, J. J., Jansen, I. E., Winder-Rhodes, S., Eberly, S., et al. (2016). Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 80, 674–685. doi: 10.1002/ana.24781
Lythe, V., Athauda, D., Foley, J., Mencacci, N. E., Jahanshahi, M., Cipolotti, L., et al. (2017). GBA-Associated Parkinson’s disease: progression in a deep brain stimulation cohort. J. Parkinsons Dis. 7, 635–644. doi: 10.3233/JPD-171172
Machaczka, M., Rucinska, M., Skotnicki, A. B., and Jurczak, W. (1999). Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease. Am. J. Hematol. 61, 216–217. doi: 10.1002/(sici)1096-8652(199907)61:3<216::aid-ajh12>3.0.co;2-b
Malec-Litwinowicz, M., Rudzińska, M., Szubiga, M., Michalski, M., Tomaszewski, T., and Szczudlik, A. (2014). Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson disease patients. Neurol. Neurochir. Pol. 48, 258–261. doi: 10.1016/j.pjnns.2014.07.005
Maor, G., Rencus-Lazar, S., Filocamo, M., Steller, H., Segal, D., and Horowitz, M. (2013). Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J. Rare Dis. 8:140. doi: 10.1186/1750-1172-8-140
Massaro, G., Hughes, M. P., Whaler, S. M., Wallom, K.-L., Priestman, D. A., Platt, F. M., et al. (2020). Systemic AAV9 gene therapy using the synapsin I promoter rescues a mouse model of neuronopathic Gaucher disease but with limited cross-correction potential to astrocytes. Hum. Mol. Genet. doi: 10.1093/hmg/ddz317 [Epub ahead of print].
Massaro, G., Mattar, C. N. Z., Wong, A. M. S., Sirka, E., Suzanne, M. K., Herbert, B. R., et al. (2019). Europe PMC Funders Group Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 24, 1317–1323. doi: 10.1038/s41591-018-0106-7
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., et al. (2016). GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 31, 95–102. doi: 10.1002/mds.26359
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
McNeill, A., Duran, R., Hughes, D. A., Mehta, A., and Schapira, A. H. V. (2012a). A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 83, 853–854. doi: 10.1136/jnnp-2012-302402
McNeill, A., Duran, R., Proukakis, C., Bras, J., Hughes, D., Mehta, A., et al. (2012b). Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov. Disord. 27, 526–532. doi: 10.1002/mds.24945
McNeill, A., Magalhaes, J., Shen, C., Chau, K. Y., Hughes, D., Mehta, A., et al. (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495. doi: 10.1093/brain/awu020
Mercado, G., Valdés, P., and Hetz, C. (2013). An ERcentric view of Parkinson’s disease. Trends Mol. Med. 19, 165–175. doi: 10.1016/j.molmed.2012.12.005
Mielke, M. M., Maetzler, W., Haughey, N. J., Bandaru, V. V. R., Savica, R., Deuschle, C., et al. (2013). Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One 8:e73094. doi: 10.1371/journal.pone.0073094
Migdalska-Richards, A., and Schapira, A. H. V. (2016). The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 139, 77–90. doi: 10.1111/jnc.13385
Morabito, G., Giannelli, S. G., Ordazzo, G., Bido, S., Castoldi, V., Indrigo, M., et al. (2017). AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol. Ther. 25, 2727–2742. doi: 10.1016/j.ymthe.2017.08.004
Mounsey, R. B., and Teismann, P. (2011). Mitochondrial dysfunction in Parkinson’s disease: pathogenesis and neuroprotection. Parkinsons Dis. 2011:617472. doi: 10.4061/2011/617472
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased δ-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848. doi: 10.1093/brain/awt367
Mus, L., Siani, F., Giuliano, C., Ghezzi, C., Cerri, S., and Blandini, F. (2019). Development and biochemical characterization of a mouse model of Parkinson’s disease bearing defective glucocerebrosidase activity. Neurobiol. Dis. 124, 289–296. doi: 10.1016/j.nbd.2018.12.001
Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., McKeith, I. G., Chinnery, P. F., et al. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735. doi: 10.1001/jamaneurol.2013.1925
Nelson, M. P., Tse, T. E., O’Quinn, D. B., Percival, S. M., Jaimes, E. A., Warnock, D. G., et al. (2014). Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of α-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2:20. doi: 10.1186/2051-5960-2-20
Neudorfer, O., Giladi, N., Elstein, D., Abrahamov, A., Turezkite, T., Aghai, E., et al. (1996). Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89, 691–694. doi: 10.1093/qjmed/89.9.691
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Nichols, W. C., Pankratz, N., Marek, D. K., Pauciulo, M. W., Elsaesser, V. E., Halter, C. A., et al. (2009). Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316. doi: 10.1212/01.wnl.0000327823.81237.d1
Noyce, A. J., Bestwick, J. P., Silveira-Moriyama, L., Hawkes, C. H., Knowles, C. H., Hardy, J., et al. (2014). PREDICT-PD: identifying risk of Parkinson’s disease in the community: methods and baseline results. J. Neurol. Neurosurg. Psychiatry 85, 31–37. doi: 10.1136/jnnp-2013-305420
Noyce, A. J., Mencacci, N. E., Schrag, A., Bestwick, J. P., Giovannoni, G., Lees, A. J., et al. (2015). Web-based assessment of Parkinson’s prodromal markers identifies GBA variants. Mov. Disord. 30, 1002–1003. doi: 10.1002/mds.26249
Nuzhnyi, E., Emelyanov, A., Boukina, T., Usenko, T., Yakimovskii, A., Zakharova, E., et al. (2015). Plasma oligomeric α-synuclein is associated with glucocerebrosidase activity in gaucher disease. Mov. Disord. 30, 989–991. doi: 10.1002/mds.26200
O’Regan, G., Desouza, R. M., Balestrino, R., and Schapira, A. H. (2017). Glucocerebrosidase mutations in Parkinson disease. J. Parkinsons Dis. 7, 411–422. doi: 10.3233/JPD-171092
Oeda, T., Umemura, A., Mori, Y., Tomita, S., Kohsaka, M., Park, K., et al. (2015). Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol. Aging 36, 3306–3313. doi: 10.1016/j.neurobiolaging.2015.08.027
Ortega, R. A., Torres, P. A., Swan, M., Nichols, W., Boschung, S., Raymond, D., et al. (2016). Glucocerebrosidase enzyme activity in GBA mutation Parkinson disease. J. Clin. Neurosci. 28, 185–186. doi: 10.1016/j.jocn.2015.12.004
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of gaucher disease—links to Parkinson’s disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Parnetti, L., Balducci, C., Pierguidi, L., De Carlo, C., Peducci, M., D’Amore, C., et al. (2009). Cerebrospinal fluid β-glucocerebrosidase activity is reduced in dementia with lewy bodies. Neurobiol. Dis. 34, 484–486. doi: 10.1016/j.nbd.2009.03.002
Parnetti, L., Chiasserini, D., Persichetti, E., Eusebi, P., Varghese, S., Qureshi, M. M., et al. (2014). Cerebrospinal fluid lysosomal enzymes and α-synuclein in Parkinson’s disease. Mov. Disord. 29, 1019–1027. doi: 10.1002/mds.25772
Parnetti, L., Paciotti, S., Eusebi, P., Dardis, A., Zampieri, S., Chiasserini, D., et al. (2017). Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 32, 1423–1431. doi: 10.1002/mds.27136
Payne, K., Walls, B., and Wojcieszek, J. (2019). Approach to assessment of Parkinson disease with emphasis on genetic testing. Med. Clin. North Am. 103, 1055–1075. doi: 10.1016/j.mcna.2019.08.003
Pchelina, S., Baydakova, G., Nikolaev, M., Senkevich, K., Emelyanov, A., Kopytova, A., et al. (2018). Blood lysosphingolipids accumulation in patients with parkinson’s disease with glucocerebrosidase 1 mutations. Mov. Disord. 33, 1325–1330. doi: 10.1002/mds.27393
Persichetti, E., Chiasserini, D., Parnetti, L., Eusebi, P., Paciotti, S., De Carlo, C., et al. (2014). Factors influencing the measurement of lysosomal enzymes activity in human cerebrospinal fluid. PLoS One 9:e101453. doi: 10.1371/journal.pone.0101453
Postuma, R. B., and Berg, D. (2016). Advances in markers of prodromal Parkinson disease. Nat. Rev. Neurol. 12, 622–634. doi: 10.1038/nrneurol.2016.152
Raghavan, S. S., Topol, J., and Kolodny, E. H. (1980). Leukocyte β-glucosidase in homozygotes and heterozygotes for Gaucher disease. Am. J. Hum. Genet. 32, 158–173.
Richter, F., Fleming, S. M., Watson, M., Lemesre, V., Pellegrino, L., Ranes, B., et al. (2014). A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 11, 840–856. doi: 10.1007/s13311-014-0294-x
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hallett, P., et al. (2015a). Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2, 433–438. doi: 10.1002/acn3.177
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hayes, M. A., et al. (2015b). Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 82, 495–503. doi: 10.1016/j.nbd.2015.09.009
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Graham, A.-R., Brown, E., et al. (2015c). Sustained systemic glucocerebrosidase inhibition induces brain α-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid. Redox Signal. 23, 550–564. doi: 10.1089/ars.2015.6307
Ron, I., and Horowitz, M. (2005). ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 14, 2387–2398. doi: 10.1093/hmg/ddi240
Ron, I., Rapaport, D., and Horowitz, M. (2010). Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 19, 3771–3781. doi: 10.1093/hmg/ddq292
Saito, Y., Suzuki, K., Hulette, C. M., and Murayama, S. (2004). Aberrant phosphorylation of α-synuclein in human Niemann-Pick type C1 disease. J. Neuropathol. Exp. Neurol. 63, 323–328. doi: 10.1093/jnen/63.4.323
Sardi, S. P., Cheng, S. H., and Shihabuddin, L. S. (2015). Gaucher-related synucleinopathies: the examination of sporadic neurodegeneration from a rare (disease) angle. Prog. Neurobiol. 125, 47–62. doi: 10.1016/j.pneurobio.2014.12.001
Sardi, S. P., Clarke, J., Kinnecom, C., Tamsett, T. J., Li, L., Stanek, L. M., et al. (2011). CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U S A 108, 12101–12106. doi: 10.1073/pnas.1108197108
Sardi, S. P., Clarke, J., Viel, C., Chan, M., Tamsett, T. J., Treleaven, C. M., et al. (2013). Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. U S A 110, 3537–3542. doi: 10.1073/pnas.1220464110
Sardi, S. P., Viel, C., Clarke, J., Treleaven, C. M., Richards, A. M., Park, H., et al. (2017). Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. U S A 114, 2699–2704. doi: 10.1073/pnas.1616152114
Saunders-Pullman, R., Hagenah, J., Dhawan, V., Stanley, K., Pastores, G., Sathe, S., et al. (2010). Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov. Disord. 25, 1364–1372. doi: 10.1002/mds.23046
Schapira, A. H. V. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109. doi: 10.1016/S1474-4422(07)70327-7
Schapira, A. H. V. (2015). Glucocerebrosidase and Parkinson disease: recent advances europe PMC funders group. Mol. Cell. Neurosci. 66, 37–42. doi: 10.1016/j.mcn.2015.03.013
Schapira, A. H. V., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1:1269. doi: 10.1016/s0140-6736(89)92366-0
Schapira, A. H. V., Chaudhuri, K. R., and Jenner, P. (2017). Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 435–450. doi: 10.1038/nrn.2017.62
Schapira, A. H. V., Chiasserini, D., Beccari, T., and Parnetti, L. (2016). Glucocerebrosidase in Parkinson’s disease: insights into pathogenesis and prospects for treatment. Mov. Disord. 31, 830–835. doi: 10.1002/mds.26616
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x
Schapira, A. H. V., and Gegg, M. (2011). Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis. 2011:159160. doi: 10.4061/2011/159160
Schapira, A. H. V., and Tolosa, E. (2010). Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 6, 309–317. doi: 10.1038/nrneurol.2010.52
Schmid, S. P., Schleicher, E. D., Cegan, A., Deuschle, C., Baur, S., Hauser, A. K., et al. (2012). Cerebrospinal fluid fatty acids in glucocerebrosidase-associated Parkinson’s disease. Mov. Disord. 27, 288–293. doi: 10.1002/mds.23984
Schöndorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in ipsc and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988. doi: 10.1016/j.celrep.2018.05.009
Sidransky, E., and Lopez, G. (2012). The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. doi: 10.1016/s1474-4422(12)70190-4
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Siebert, M., Sidransky, E., and Westbroek, W. (2014). Glucocerebrosidase is shaking up the synucleinopathies. Brain 137, 1304–1322. doi: 10.1093/brain/awu002
Silveira, C. R. A., MacKinley, J., Coleman, K., Li, Z., Finger, E., Bartha, R., et al. (2019). Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 19:20. doi: 10.1186/s12883-019-1252-3
Simuni, T., Uribe, L., Cho, H. R., Caspell-Garcia, C., Coffey, C. S., Siderowf, A., et al. (2020). Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol. 19, 71–80. doi: 10.1016/S1474-4422(19)30319-9
Smith, B. R., Santos, M. B., Marshall, M. S., Cantuti-Castelvetri, L., Lopez-Rosas, A., Li, G., et al. (2014). Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 232, 509–521. doi: 10.1002/path.4328
Soria, F. N., Engeln, M., Martinez-Vicente, M., Glangetas, C., López-González, M. J., Dovero, S., et al. (2017). Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum. Mol. Genet. 26, 2603–2615. doi: 10.1093/hmg/ddx120
Suzuki, K., Iseki, E., Togo, T., Yamaguchi, A., Katsuse, O., Katsuyama, K., et al. (2007). Neuronal and glial accumulation of α- and β-synucleins in human lipidoses. Acta Neuropathol. 114, 481–489. doi: 10.1007/s00401-007-0264-z
Thaler, A., Bregman, N., Gurevich, T., Shiner, T., Dror, Y., Zmira, O., et al. (2018a). Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat. Disord. 55, 45–49. doi: 10.1016/j.parkreldis.2018.05.009
Thaler, A., Kliper, E., Maidan, I., Herman, T., Rosenberg-Katz, K., Bregman, N., et al. (2018b). Cerebral imaging markers of GBA and LRRK2 related Parkinson’s disease and their first-degree unaffected relatives. Brain Topogr. 31, 1029–1036. doi: 10.1007/s10548-018-0653-8
van Dijk, K. D., Persichetti, E., Chiasserini, D., Eusebi, P., Beccari, T., Calabresi, P., et al. (2013). Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 28, 747–754. doi: 10.1002/mds.25495
Vitner, E. B., Farfel-Becker, T., Eilam, R., Biton, I., and Futerman, A. H. (2012). Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 135, 1724–1735. doi: 10.1093/brain/aws095
Wang, C., Cai, Y., Gu, Z., Ma, J., Zheng, Z., Tang, B. S., et al. (2014). Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol. Aging 35, 725.e1–725.e6. doi: 10.1016/j.neurobiolaging.2013.08.012
Westbroek, W., Gustafson, A. M., and Sidransky, E. (2011). Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 17, 485–493. doi: 10.1016/j.molmed.2011.05.003
Winder-Rhodes, S. E., Evans, J. R., Ban, M., Mason, S. L., Williams-Gray, C. H., Foltynie, T., et al. (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136, 392–399. doi: 10.1093/brain/aws318
Wong, Y. C., and Krainc, D. (2016). Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov. Disord. 31, 1610–1618. doi: 10.1002/mds.26802
Xu, Y. H., Sun, Y., Ran, H., Quinn, B., Witte, D., and Grabowski, G. A. (2011). Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 102, 436–447. doi: 10.1016/j.ymgme.2010.12.014
Xu, Y. H., Xu, K., Sun, Y., Liou, B., Quinn, B., Li, R. H., et al. (2014). Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 23, 3943–3957. doi: 10.1093/hmg/ddu105
Yang, S.-Y., Gegg, M., Chau, D., and Schapira, A. (2020). Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 134:104620. doi: 10.1016/j.nbd.2019.104620
Zhang, Y., Shu, L., Sun, Q., Zhou, X., Pan, H., Guo, J., et al. (2018a). Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front. Mol. Neurosci. 11:43. doi: 10.3389/fnmol.2018.00043
Zhang, Y., Shu, L., Zhou, X., Pan, H., Xu, Q., Guo, J., et al. (2018b). A meta-analysis of GBA-related clinical symptoms in Parkinson’s disease. Parkinsons Dis. 2018:3136415. doi: 10.1155/2018/3136415
Zhang, Y., Sun, Q. Y., Zhao, Y. W., Shu, L., Guo, J. F., Xu, Q., et al. (2015). Effect of GBA mutations on phenotype of Parkinson’s disease: a study on chinese population and a meta-analysis. Parkinsons Dis. 2015:916971. doi: 10.1155/2015/916971
Ziegler, S. G., Eblan, M. J., Gutti, U., Hruska, K. S., Stubblefield, B. K., Goker-Alpan, O., et al. (2007). Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol. Genet. Metab. 91, 195–200. doi: 10.1016/j.ymgme.2007.03.004
Zokaei, N., McNeill, A., Proukakis, C., Beavan, M., Jarman, P., Korlipara, P., et al. (2014). Visual short-term memory deficits associated with GBA mutation and Parkinson’s disease. Brain 137, 2303–2311. doi: 10.1093/brain/awu143
Zunke, F., Moise, A. C., Belur, N. R., Gelyana, E., Stojkovska, I., Dzaferbegovic, H., et al. (2018). Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron 97, 92–107.e10. doi: 10.1016/j.neuron.2017.12.012
Keywords: Parkinson’s disease, glucocerebrosidase, alpha-synuclein, GBA1, risk factor, synucleinopathy
Citation: Avenali M, Blandini F and Cerri S (2020) Glucocerebrosidase Defects as a Major Risk Factor for Parkinson’s Disease. Front. Aging Neurosci. 12:97. doi: 10.3389/fnagi.2020.00097
Received: 21 November 2019; Accepted: 23 March 2020;
Published: 21 April 2020.
Edited by:
Annalisa Pinna, Italian National Research Council, ItalyReviewed by:
Lamya Shihabuddin, Sanofi Genzyme, United StatesCopyright © 2020 Avenali, Blandini and Cerri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Blandini, ZmFiaW8uYmxhbmRpbmlAbW9uZGluby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.