
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 07 August 2018
Sec. Neurocognitive Aging and Behavior
Volume 10 - 2018 | https://doi.org/10.3389/fnagi.2018.00241
 Amal Bouzid1
Amal Bouzid1 Ibtihel Smeti1
Ibtihel Smeti1 Amine Chakroun2,3Salma Loukil1
Amine Chakroun2,3Salma Loukil1 Abdullah Ahmed Gibriel4
Abdullah Ahmed Gibriel4 Mhamed Grati5†Abdelmonem Ghorbel2,3
Mhamed Grati5†Abdelmonem Ghorbel2,3 Saber Masmoudi1*
Saber Masmoudi1*Introduction: Presbycusis, an age-related hearing impairment (ARHI) disease, is the most common cause for HI in adults worldwide. One of the best candidate genes for ARHI susceptibility is Cadherin 23 (CDH23) which encodes stereocilia tip-links of the inner ear sensory hair cell. Although alterations in the methylation status of CpG dinucleotides across various genes were reported to be associated with HI, methylation changes in CDH23 gene have not been reported previously.
Objectives: This study aimed at investigating whether DNA methylation level of CDH23 gene at intragenic CpG island overlapping an exonic-intronic region at position chr10:73565570-73565827 (GRCh37/hg19) could be risk factor associated with ARHI.
Materials and Methods: We screened for methylation changes in this particular position for CDH23 gene in 50 blood samples of elderly women affected with presbycusis and healthy control cohort. Methylation of CpG sites were assessed using Quantitative methylation-specific PCR (qMSP) following sodium bisulfite DNA conversion chemistry. Methylation levels were normalized against TSH2B reference gene.
Results: DNA methylation analysis for the common CpG islands in CDH23 gene revealed 3.27-folds significant increase (p < 0.0001) in methylation profile for ARHI women as compared to healthy controls with an elevated risk odds ratio (OR) of 2.219 [95% CI 1.071–4.597].
Conclusion: Our study is the first of its kind to prove that higher CpG site methylation levels in CDH23 gene are likely to be associated with ARHI.
Age-related hearing impairment (ARHI), named also presbycusis, is the most common form of sensory disability in elderly individuals. The World Health Organization (WHO) estimated that 466 million people worldwide are disabled with HI1. Presbycusis is characterized by bilateral, symmetrical and non-conductive type of hearing defects. It is associated with negative consequences in communication and social life quality. The causative molecular pathogenesis for ARHI is still unknown (Tu and Friedman, 2018). Therefore, investigating molecular pathogenic mechanisms underlying presbycusis is very crucial prior to development of cost-effective therapeutic strategies.
Cadherin 23 (CDH23), also known as otocadherin, is part of the cadherin super-family of calcium-dependent cell-surface adhesion proteins. CDH23 encompasses 69 exons (NM_022124) and encodes a 3354 amino acid protein (NP_071407.4) with 27 extracellular cadherin domains, a single trans-membrane domain and a short cytoplasmic domain. CDH23 was reported to play a key role in lateral (Lagziel et al., 2005) and stereociliary tip-links (Siemens et al., 2004) of the inner ear sensory hair cells that control hearing process. Tip-links are extracellular filaments that are the “gate cables” for opening mechano-transduction channels which transduces mechanical forces arising from sound waves and head movement, allowing hearing and balance to occur (Pickles et al., 1984). Thus, mutations in CDH23 gene are associated with distinct sensory impairments (Siemens et al., 2004). Missense mutations in CDH23 gene (NM_022124) have been associated with severe to profound non-syndromic pre-lingual hearing loss DFNB12 (McHugh and Friedman, 2006). Meanwhile, deletions, nonsense or frame-shift mutations were reported to cause Usher syndrome (USH1D). This clearly indicates that mutations classes are closely related to the phenotype (Petit et al., 2001).
Qualitative and quantitative linkage studies on mice linked ARHI genetic susceptibility to Ahl locus on chromosome 10. The Ahl allele, also known as Cdh23753A, is a recessive synonymous single nucleotide polymorphism (SNP; c.753G>A) in exon 7 of Cdh23 gene that was significantly associated with ARHI in common inbred mouse strains (Johnson et al., 2000). The hypomorphic Cdh23 Ahl allele was reported to be common in at least C57BL/6, BALB, and 129S6 ARHI models and to cause in-frame skipping of exon 7 (Noben-Trauth et al., 2003). Although the Ahl allele of Cdh23 gene was proven to be linked with ARHI in mice, there is no reported evidence for the implication of CDH23 intron-7 SNP in ARHI in humans (Hwang et al., 2012).
Epigenetic hearing modifications in elderly individuals were responsible for impaired hearing in both ARHI and syndromic cases (Provenzano and Domann, 2007). Interestingly, recent investigations demonstrated that aberrant methylation of HI genes in children could lead to the development of ARHI. Hypermethylation of GJB2 promoter region in the mimetic aging rat cochlea was associated with down regulation of connexin 26 that resulted in the development of presbycusis (Wu et al., 2014). Moreover, hypermethylation of CpG sites in the promoter region of SLC26A4 (DFNB4) and P2RX2 (DFNA41) genes resulted in increased risk for presbycusis in men (Xu et al., 2017), and a down-expression in elderly women with presbycusis (Bouzid et al., 2018), respectively. The aim of the present study was to correlate CDH23 methylation profiles with ARHI phenotype characteristics to investigate possible use of CDH23 gene as a novel epigenetic marker in ARHI. This could pave the way towards understanding complex pathogenic mechanisms underlying ARHI.
The present study was approved by ethics committee of the University Hospital of Sfax (Tunisia). Informed and written consent was obtained from all individual participated in this study. Clinical, biological and pure tone audiometric data were collected from 50 unrelated age-matched subjects (25 patients and 25 controls) ranging from 50 years to 75 years.
Selected women were classified into controls and affected groups based on mean bone conduction threshold as a function of age as indicated in our previously published study (Bouzid et al., 2018). Wherein, it was reported that hearing loss thresholds were assessed through 95% confidence interval (CI) for means of bone conduction values across increasing ages (p < 0.05). Subjects with hearing loss below the lower limit of the 95% CI were considered as control group (with hearing loss thresholds <20 dB) while subjects with hearing loss above the upper limit of the 95% CI were classified in ARHI group (with hearing loss thresholds ≥20 dB) showed typically presbycusis audiograms with a downgrade into higher frequencies. Individuals having ARHI risk factors such as occupational noise, medication induced ototoxicity, otitis media infection history, hypertension, diabetes, dyslipidemia and family history for hereditary HI were excluded from this study.
CpG island sequence conservation for CDH23 gene was evaluated using tools provided by the UCSC Genome Browser interface. Evolutionary conservation in 100 vertebrates is generated using multiz alignment and scored by the phastCons and phyloP programs (Siepel et al., 2005). Scores of evolutionarily constrained elements were estimated using the GERP++ method (Davydov et al., 2010). Classification of CDH23 CpG-rich regions according to their evolutionary dynamics was examined using parameter-rich evolutionary model and clustering analysis (Cohen et al., 2011). For the CDH23 CpG island sites, chromatin non-condensed DNaseI Hypersensitivity regions in 125 cell types tracks were assessed from ENCODE project (V3).
The bisulfite conversion method was implemented using innuconvert bisulfite basic kit (AnalytikJena, Jena, Germany) according to the manufacturer’s recommendations. Bisulfite-modified DNA was then used as template for the real-time PCR. The CDH23 primers, F: 5’-CAAAGTCACAGGAAGTGTGC-3’ and R: 5’-CATTCACATCCAGGACAGTG-3’, were designed to amplify an amplicon size of 137 bp in the CpG island site of the junction exon 54–intron 54 (NM_022124). For endogenous normalization purposes, TSH2B promoter primers were used to amplify a highly methylated CG rich region in all somatic cells (Diagenode, Liège, Belgium). Quantitative methylation-specific PCR (qMSP) was performed for both CDH23 and TSH2B genes.
To ensure selectivity of those primers in CDH23 gene, a positive control genomic methylated bisulfite converted DNA and a negative control genomic unmethylated bisulfite converted DNA (Diagenode, Liège, Belgium) were used. The former control contains methylated cytosine residues to ensure amplification of methylated products. The positive control would also evaluate methylation levels for both ARHI and control samples. The latter contains uracil residues to ensure no amplification for un-methylated DNA.
The PCR reaction mix contained Syber Premix of the Episcope MSP kit (TakaRa, Otsu, Japan), 10 μM of each primer and 100 ng of bisulfite-modified genomic DNA template in a final volume of 10 μL. PCR amplification was performed using CFX96 Real-Time PCR detection system (Bio Rad, Redmond, WA, USA). All samples were run in duplicates for both target and reference genes. The real-time PCR conditions were as follows; begun with an initial denaturation step at 95°C for 30 s, followed by 40 repetitive cycles at 98°C for 5 s, 60°C for 40 s and extension at 72°C for 30 s. Melt curve analysis was performed for all samples following RT-PCR run. The Bio-Rad CFX Manager software (Bio Rad, Redmond, WA, USA) was used to interpret data.
The quantification cycle was defined for each reaction and the ratio of methylated to total amplifiable bisulfite-treated DNA was determined (Bustin et al., 2009). The relative level of methylated DNA for each sample group of patients with presbycusis and controls was carried out using comparative threshold cycle. To evaluate different CpG methylation levels between ARHI patients and controls, the ΔΔCq method was applied using the TSH2B as a reference gene (Schmittgen and Livak, 2008).
Statistical analyses were performed with GraphPad Prism software version 5.0 (La Jolla, CA, USA). Differences in methylation levels between presbycusis patients and controls were compared with Student’s t-test. P-values ≤0.05 were considered statistically significant. To assess correlation between CDH23 gene methylation levels and hearing impairment, the two tailed Fisher’s exact test was used. Odds ratio (OR) was explored as a part of association with a 95% CI as an appraisal of precision for this evaluation.
Total RNA extraction from peripheral blood of three individuals, with different ages 60, 64 and 70 years old, was performed using PAXgene Blood RNA Kit (PreAnalytiX, Hombrechtikon, Switzerland) according to manufacturer’s instructions. Extraction yield was evaluated with Nanodrop2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Complementary DNA was synthesized using High Capacity RNA-to-cDNA Kit based on manufacturer’s protocol (Foster City, CA, USA). The expression of exon 54 (NM_022124) was checked on CFX96 Real Time PCR detection system (Bio-Rad, Hercules, CA, USA) using Syber Premix Ex Taq at 2× (Tli RNaseH Plus, Japan). Primers sequences (listed in Table 1) were carefully designed to amplify relevant transcripts without genomic DNA contamination. Validation of amplification specificity and accuracy was confirmed through the presence of a single peak on melt curve at expected Tm and also through the generation of the correct amplicon size on agarose gel electrophoresis.
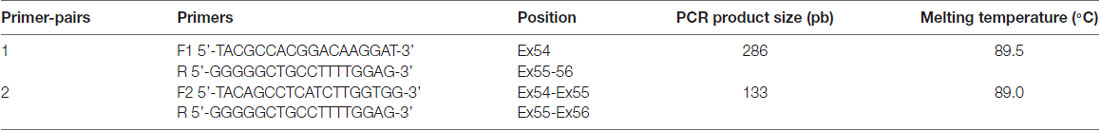
Table 1. CDH23 primers for RT-PCR analysis.
Presbycusis and healthy groups were carefully evaluated in terms of age match. Pure tone audiometry for air and bone conduction thresholds show no air-bone gap. The distribution of mean bone conduction threshold for each participant as function of age is plotted in Figure 1. The distribution of mean bone conduction threshold for healthy controls do not change significantly as function of the age ranging from 50 to 75 years. Whereas, for ARHI cases, the mean bone conduction threshold increases gradually with age as indicated from the linear trend line y = 0.609x − 5.584.
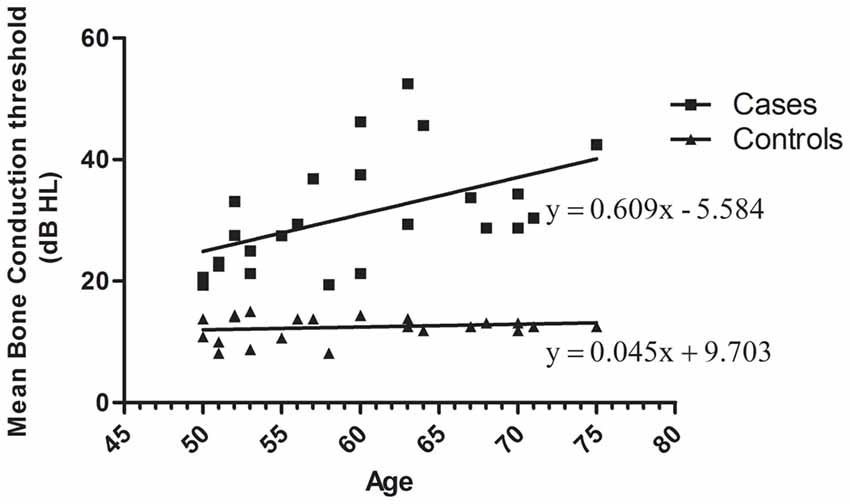
Figure 1. Distribution of mean bone conduction threshold as a function of age in age-related hearing impairment (ARHI) cases and healthy controls, generated using GraphPad Prism 5.0. Means of the right and left ear hearing thresholds at 500, 1000, 2000 and 4000 Hz for bone conduction in patients were plotted as a function against subjects’ age ranging from 50 to 75 years. Hearing loss thresholds are expressed in decibels (dB). Linear regression equation and line were added for each data series of cases and controls.
To investigate CDH23 epigenetic modifications in ARHI, we screened for CpG-rich region within this gene in both ARHI cases and healthy controls. An intragenic CpG island at genomic position chr10:73, 565, 570-73, 565, 827 (GRCh37/hg19) overlapping with the junction exon 54—intron 54 (NM 022124) was selected. This region was chosen based on the high conservation score with vertebrate multiz alignment and conservation for 100 species in addition to genomic evolutionary rate profiling for mammalian alignments (Table 2). This CpG island was found to be conserved between human and mouse species genomes with 89.7% of identity. The conserved region of mouse Cdh23 gene is localized on chr10:60,312,545-60,312,660 (GRCm38/mm10) at exon 55 for both isoforms (NM_023370) and (NM_001252635). Mouse Cdh23 gene contains 69 exons encoding two Cdh23 transcripts differing only by exon 68 splicing (Bork et al., 2001; Di Palma et al., 2001). Notably, these two isoforms including exon 55 are expressed in the inner ear tissues (Siemens et al., 2004; Lagziel et al., 2005; Xu et al., 2008). Moreover, RT-PCR melt curve results for peripheral blood human samples proved the amplification of the correct products of the exon 54 (NM 022124) while agarose gel electrophoresis confirmed the amplification of the expected amplicons sizes (Figure 2).
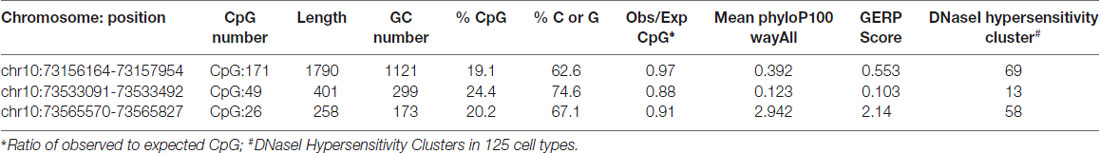
Table 2. Characteristic and evidence for conservation of intragenic Cadherin 23 (CDH23) CpG island in the human (GRCh37/hg19) and other species genomes, generated by UCSC genome Browser.
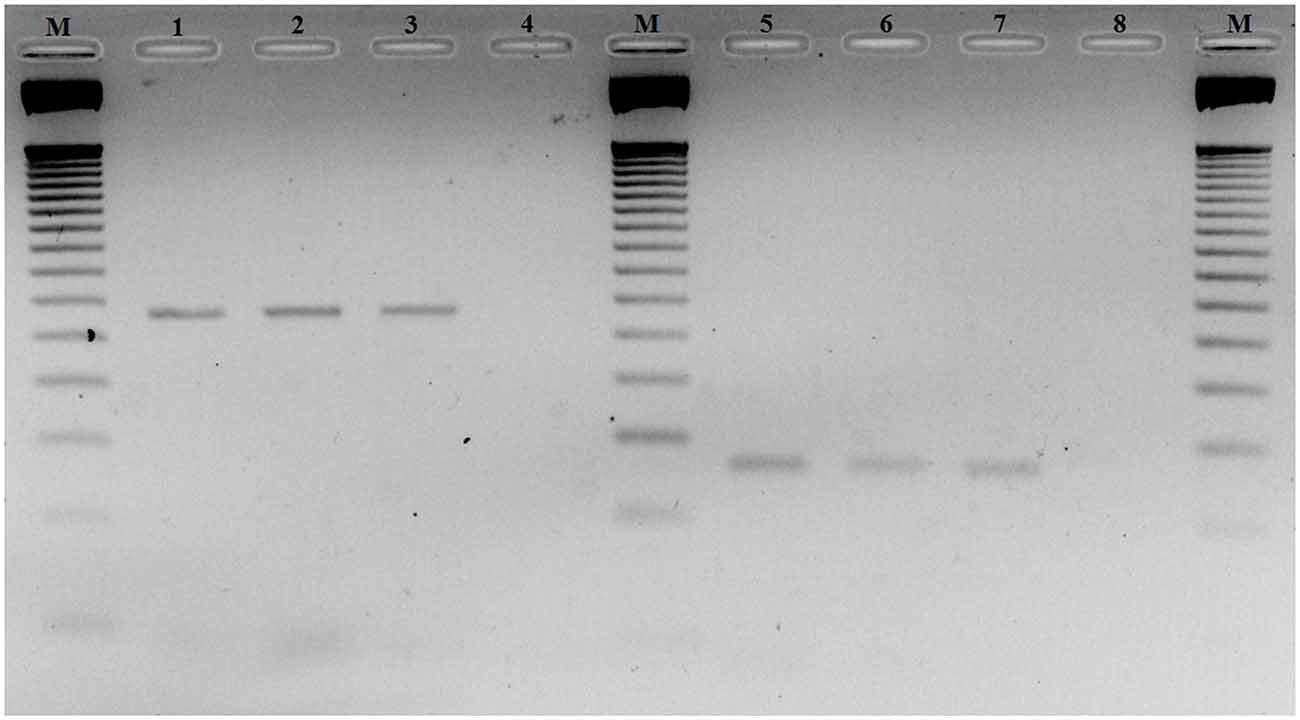
Figure 2. Agarose gel electrophoresis of RT-PCR products of exon 54 Cadherin 23 (CDH23) gene (NM_022124). Lanes M: 50-pb DNA molecular weight markers. Lanes 1, 2 and 3: RT-PCR products using primer-pairs 1 with different ages 60, 64 and 70 years old, respectively. Lanes 5, 6 and 7: RT-PCR products using primer-pairs 2 with different ages 60, 64 and 70 years old, respectively. Lanes 4 and 8: negative controls primer-pairs 1 and 2, respectively.
To determine methylation patterns for human CDH23 CpG island, we carried out qMSP in a cohort of ARHI patients and healthy subjects of matched age. After bisulfite conversion of selected DNA samples, we performed amplification in the CpG island region as explained in Subjects and methods section. Different CpG methylation levels between ARHI patients and controls were evaluated applying the ΔΔCq method using the TSH2B as a reference gene. Methylated DNA quantification revealed 3.27-fold increase in methylation levels at common CpG island of CDH23 in blood samples from women with presbycusis as compared to controls.
Relative level of DNA methylation was about 71.52 ± 0.45 for ARHI group, and 67.40 ± 0.61 for control group (Figure 3). Using unpaired t-test, this relative level of methylation was found to be significantly different (p < 0.0001) between the two groups. The difference in the relative level of DNA methylation between ARHI and control groups resulted in hypermethylation changes of 4.12 ± 0.76 value in ARHI patients as compared to healthy controls with a 95% CI range of 2.59–5.64.
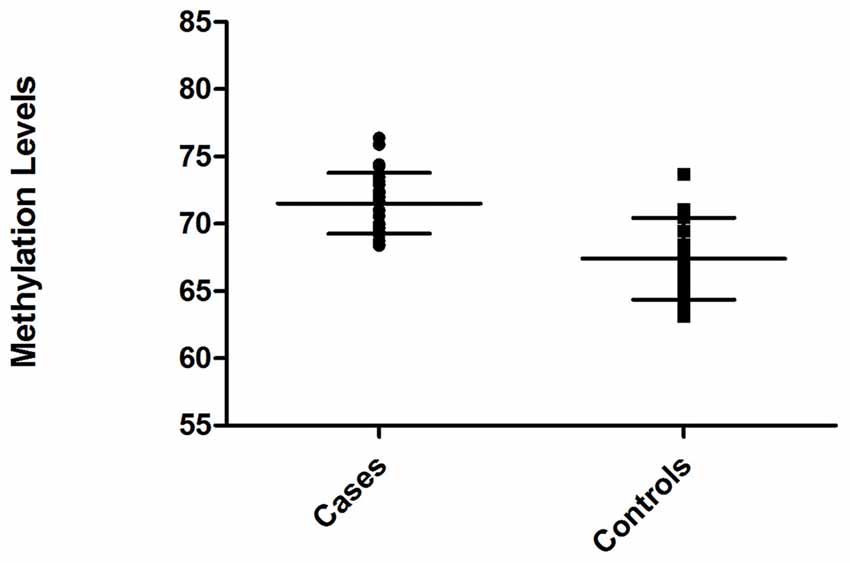
Figure 3. Distribution of methylation levels of intragenic CDH23 CpG island for ARHI patients (cases) and healthy controls (Controls), generated using GraphPad Prism 5.0.
Moreover, correlation analysis was assessed between the DNA methylation levels of CDH23 gene for ARHI and control groups. For DNA methylation levels and hearing impairment values of ARHI and control groups, Fisher’s exact test resulted in statistically significant correlation with p-value of 0.036. The strength of correlation was determined with a relative risk of about 1.85 through the 95% CI [1.04–3.30]. The DNA methylation level of CDH23 was significantly related to an increase of ARHI risk with OR of 2.22 and a 95% CI range of [1.07–4.60].
Presbycusis is one of the major health problems affecting elderly people, with an expanding incidence as the population age increases. It is estimated that the number of people suffering from hearing impairment will increase considerably to reach over 900 million people by 2050 (WHO, March 2018). Although several chromosomal loci and mutations have been determined in non-syndromic hearing impairment patients, many cases remain undiagnosed especially those affected with presbycusis. It develops in a multistep manner which is most likely a multifactorial process. The difficulty to identify ARHI genes could suggest the fact that it is a complex genetic disorder resulting from accumulation of several mutations in different interacting genes with lifelong cumulative effects. Environmental factors such as cumulative noise or ototoxic drugs cause sensorineural cell damage (Yang et al., 2015). Hypertension and diabetes are also reported to be major risk factors for ARHI disorder (Oron et al., 2014). Furthermore, genetic predisposition factors represent a significant scope for hearing impairment with age increase (Kvestad et al., 2012). On the other hand, progressive phenotype implies that epigenetic mechanisms could be involved in this process. Epigenetic regulation has been involved in various types of age-related human diseases (García-Giménez et al., 2012). Given the fact that they play an important role in cell development and differentiation, epigenetics are expected to significantly contribute to HI sensory disorders (Friedman and Avraham, 2009). DNA methylation, as a crucial epigenetic regulatory mechanism, is sensitive to the interaction between genetic and environmental factors. It may provide a link between the environment (Fraga et al., 2005) and genetic background (Bell et al., 2011).
In the present study, we performed a case-control comparison of intragenic CDH23 methylation levels in peripheral blood samples. Our study suggests that there is an increased risk for ARHI through DNA methylation changes in CDH23 gene. Obviously, DNA methylation profiles are likely to be dependent from specific conditions and to be variable from one cell-type to another. However, it was reported for instance that age effects on DNA methylation levels are well maintained between human brain and blood tissue (Horvath et al., 2012). Due to difficulty in accessing human cochlear tissue, we opted to use peripheral blood cells as propitious surrogate as reported in other disorders in which tissues are inaccessible (Mohr and Liew, 2007). Nevertheless, it is still an interesting approach with more practical convenience and significance for the discovery of potential and systemic biomarkers for presbycusis.
Empirically, DNA methylation occurring at the cytosine nucleotides in the CpG islands that are mainly located at promoter region controls gene expression (Larsen et al., 1992; Cheng et al., 2014). In fact, it was well proven that hyper-methylation of promoters correlates with a decrease or even silencing of gene expression (Suzuki and Bird, 2008). Interestingly, methylome studies showed a high density of intragenic DNA methylation compared to promoter regions (Cokus et al., 2008). Moreover, DNA methylation within body genes was found to interfere with transcript elongation in Arabidopsis thaliana instead of causing gene expression variation or silencing (Zilberman et al., 2007). These findings in turn raise more interests in investigating intragenic DNA methylation implications in human diseases. Here, we report for the first time DNA hypermethylation changes occurring in exon-intron junction of CDH23 gene. It has been reported that exons were more highly sensitive to methylation than introns, and changeover in the methylation levels occurring in exon-intron junctions were considered to be important for splicing regulation (Laurent et al., 2010; Gelfman et al., 2013). CDH23 encodes nine different isoforms (GRCh37/hg19). Transcript variant NM_022124 (isoform 1) represents the longest transcript with 68 total exons of which 67 are coding (GRCh37/hg19). Isoforms 2, 3, 4 and 5 differ in the 3’-untranslated and 3’-coding regions compared to variant 1. They are shorter than variant 1 with different C-termini. Compared to variant 1, isoforms 6 (NM_001171933.1) and 7 (NM_001171934.1) differ in the 5’-untranslated region as they lack a section of the 5’-coding region and begin translation at a downstream start codon. Also, isoform 7 lacks an in-frame exon in the 3’-coding region. Finally, isoforms 8 (NM_001171935) and 9 (NM_001171936) include only 5 and 4 coding exons, respectively. The evaluated CpG region was only common to transcript variants corresponding to isoforms 1, 6 and 7. It occurs at an exon-intron junction, which corresponds to exon-intron 54 for isoform 1 and exon-intron 9 for both isoforms 6 and 7. The comprehensive transcriptomic study (Schrauwen et al., 2016) reported CDH23 expression levels for cochlear isoforms in elder controls. Isoform 1 (NM_022124) was the most abundant in cochlear tissues and this correlates with our findings in blood cells.
DNA methylation changes at this CpG region may cause in frame skipping of 192 bp exonic region that encodes a peptide of 64 amino acids (Glu2625-Ser2688) located at the extracellular region. DNA methylation could inhibit binding of post-transcription regulatory factors that interact with pre-mRNA to regulate exons splicing (Lev Maor et al., 2015). One in-frame and four missense variations in exon 54 (NM_022124) of CDH23 gene are predicted as pathogenic and likely pathogenic, respectively, to cause hearing loss according to Deafness Variation Database2. Thus, the possible alternative skipping of exon 54 will affect CDH23 protein function which plays a key role in hearing process. Likewise, mouse Cdh23753A allele, underlying Ahl phenotype, results in-frame exon-skipping (Johnson et al., 2017). The skipped exon 7 encodes a peptide of 43 amino acids that is also located at the extracellular amino-terminus, which is in the homodimerization site and part of the 2nd and 3rd ectodomains of Cadherin 23 (Noben-Trauth et al., 2003). In-frame exon-skipping of the Cdh23 is likely to cause partial function defect that could progressively lead to hair cell degeneration.
Considering the relevance of CDH23 biomarker as a congenital hearing impairment gene and a good candidate gene for ARHI, risk associated studies for this particular biomarker should be replicated in large and different elderly populations. Moreover, similar studies on DNA methylation changes in other known candidate genes of ARHI should be undertaken.
This study provides more evidence in respect to the identification of new epigenetic ARHI biomarkers with a guidance value to better understand the pathogenicity of ARHI complex disease. We used a non-invasive approach to compare CDH23 methylation levels in ARHI patients against age-matched healthy subjects. Our study demonstrates the involvement of CDH23 hypermethylation in ARHI disorder. Further investigations of DNA methylation in CDH23 and other candidate genes would certainly assist in understanding complex pathogenic mechanisms involved in ARHI.
SM conceived and designed this study and had contributions to all its stages. AB designed and performed experiments, analyzed data and wrote the article. IS and SL collected and evaluated DNA samples. AC and AAG ascertained patients and obtained clinical data. AAG and MG critically revised the manuscript.
This work was financially supported by the Ministry of Higher Education and Research of Tunisia (LR15CBS07).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully thank all the subjects in this study for their collaboration.
Bell, J. T., Pai, A. A., Pickrell, J. K., Gaffney, D. J., Pique-Regi, R., Degner, J. F., et al. (2011). DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 12:R10. doi: 10.1186/gb-2011-12-1-r10
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. doi: 10.1086/316954
Bouzid, A., Smeti, I., Dhouib, L., Roche, M., Achour, I., Khalfallah, A., et al. (2018). Down-expression of P2RX2, KCNQ5, ERBB3 and SOCS3 through DNA hypermethylation in elderly women with presbycusis. Biomarkers 23, 347–356. doi: 10.1080/1354750x.2018.1427795
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Cheng, J., Tang, L., Hong, Q., Ye, H., Xu, X., Xu, L., et al. (2014). Investigation into the promoter DNA methylation of three genes (CAMK1D, CRY2 and CALM2) in the peripheral blood of patients with type 2 diabetes. Exp. Ther. Med. 8, 579–584. doi: 10.3892/etm.2014.1766
Cohen, N. M., Kenigsberg, E., and Tanay, A. (2011). Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell 145, 773–786. doi: 10.1016/j.cell.2011.04.024
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., Batzoglou, S., et al. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
Di Palma, F., Pellegrino, R., and Noben-Trauth, K. (2001). Genomic structure, alternative splice forms and normal and mutant alleles of cadherin 23 (Cdh23). Gene 281, 31–41. doi: 10.1016/s0378-1119(01)00761-2
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U S A 102, 10604–10609. doi: 10.1073/pnas.0500398102
Friedman, L. M., and Avraham, K. B. (2009). MicroRNAs and epigenetic regulation in the mammalian inner ear: implications for deafness. Mamm. Genome 20, 581–603. doi: 10.1007/s00335-009-9230-5
García-Giménez, J. L., Sanchis-Gomar, F., Lippi, G., Mena, S., Ivars, D., Gomez-Cabrera, M. C., et al. (2012). Epigenetic biomarkers: a new perspective in laboratory diagnostics. Clin. Chim. Acta 413, 1576–1582. doi: 10.1016/j.cca.2012.05.021
Gelfman, S., Cohen, N., Yearim, A., and Ast, G. (2013). DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 23, 789–799. doi: 10.1101/gr.143503.112
Horvath, S., Zhang, Y., Langfelder, P., Kahn, R. S., Boks, M. P. M., van Eijk, K., et al. (2012). Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 13:R97. doi: 10.1186/gb-2012-13-10-r97
Hwang, J.-H., Liu, K. S., Wu, C.-C., and Liu, T.-C. (2012). Association of Cadherin23 single nucleotide polymorphism with age-related hearing impairment in Han Chinese. Otolaryngol. Head Neck Surg. 147, 531–534. doi: 10.1177/0194599812446904
Johnson, K. R., Tian, C., Gagnon, L. H., Jiang, H., Ding, D., Salvi, R., et al. (2017). Effects of Cdh23 single nucleotide substitutions on age-related hearing loss in C57BL/6 and 129S1/Sv mice and comparisons with congenic strains. Sci. Rep. 7:44450. doi: 10.1038/srep44450
Johnson, K. R., Zheng, Q. Y., and Erway, L. C. (2000). A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics 70, 171–180. doi: 10.1006/geno.2000.6377
Kvestad, E., Czajkowski, N., Krog, N. H., Engdahl, B., and Tambs, K. (2012). Heritability of hearing loss. Epidemiology 23, 328–331. doi: 10.1097/ede.0b013e318245996e
Lagziel, A., Ahmed, Z. M., Schultz, J. M., Morell, R. J., Belyantseva, I. A., Friedman, T. B., et al. (2005). Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev. Biol. 280, 295–306. doi: 10.1016/j.ydbio.2005.01.015
Larsen, F., Gundersen, G., Lopez, R., and Prydz, H. (1992). CpG islands as gene markers in the human genome. Genomics 13, 1095–1107. doi: 10.1016/0888-7543(92)90024-m
Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C. T., et al. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331. doi: 10.1101/gr.101907.109
Lev Maor, G., Yearim, A., and Ast, G. (2015). The alternative role of DNA methylation in splicing regulation. Trends Genet. 31, 274–280. doi: 10.1016/j.tig.2015.03.002
McHugh, R. K., and Friedman, R. A. (2006). Genetics of hearing loss: Allelism and modifier genes produce a phenotypic continuum. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288, 370–381. doi: 10.1002/ar.a.20297
Mohr, S., and Liew, C.-C. (2007). The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol. Med. 13, 422–432. doi: 10.1016/j.molmed.2007.08.003
Noben-Trauth, K., Zheng, Q. Y., and Johnson, K. R. (2003). Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat. Genet. 35, 21–23. doi: 10.1038/ng1226
Oron, Y., Elgart, K., Marom, T., and Roth, Y. (2014). Cardiovascular risk factors as causes for hearing impairment. Audiol. Neurootol. 19, 256–260. doi: 10.1159/000363215
Petit, C., Levilliers, J., and Hardelin, J.-P. (2001). Molecular genetics of hearing loss. Annu. Rev. Genet. 35, 589–645. doi: 10.1146/annurev.genet.35.102401.091224
Pickles, J. O., Comis, S. D., and Osborne, M. P. (1984). Cross-links between stereocilia in the guinea pig organ of Corti and their possible relation to sensory transduction. Hear. Res. 15, 103–112. doi: 10.1016/0378-5955(84)90041-8
Provenzano, M. J., and Domann, F. E. (2007). A role for epigenetics in hearing: establishment and maintenance of auditory specific gene expression patterns. Hear. Res. 233, 1–13. doi: 10.1016/j.heares.2007.07.002
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Schrauwen, I., Hasin-Brumshtein, Y., Corneveaux, J. J., Ohmen, J., White, C., Allen, A. N., et al. (2016). A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear. Res. 333, 266–274. doi: 10.1016/j.heares.2015.08.013
Siemens, J., Lillo, C., Dumont, R. A., Reynolds, A., Williams, D. S., Gillespie, P. G., et al. (2004). Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 428, 950–955. doi: 10.1038/nature02483
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. doi: 10.1101/gr.3715005
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. doi: 10.1038/nrg2341
Tu, N. C., and Friedman, R. A. (2018). Age-related hearing loss: unraveling the pieces. Laryngoscope Investig. Otolaryngol. 21, 68–72. doi: 10.1002/lio2.134
Wu, X., Wang, Y., Sun, Y., Chen, S., Zhang, S., Shen, L., et al. (2014). Reduced expression of Connexin26 and its DNA promoter hypermethylation in the inner ear of mimetic aging rats induced by d-galactose. Biochem. Biophys. Res. Commun. 452, 340–346. doi: 10.1016/j.bbrc.2014.08.063
Xu, Z., Peng, A. W., Oshima, K., and Heller, S. (2008). MAGI-1, A candidate stereociliary scaffolding protein, associates with the tip-link component cadherin (23). J. Neurosci. 28, 11269–11276. doi: 10.1523/JNEUROSCI.3833-08.2008
Xu, J., Zheng, J., Shen, W., Ma, L., Zhao, M., Wang, X., et al. (2017). Elevated SLC26A4 gene promoter methylation is associated with the risk of presbycusis in men. Mol. Med. Rep. 16, 347–352. doi: 10.3892/mmr.2017.6565
Yang, C.-H., Schrepfer, T., and Schacht, J. (2015). Age-related hearing impairment and the triad of acquired hearing loss. Front. Cell. Neurosci. 9:276. doi: 10.3389/fncel.2015.00276
Keywords: presbycusis, CDH23, DNA methylation, CpG sites, quantitative methylation-specific PCR
Citation: Bouzid A, Smeti I, Chakroun A, Loukil S, Gibriel AA, Grati M, Ghorbel A and Masmoudi S (2018) CDH23 Methylation Status and Presbycusis Risk in Elderly Women. Front. Aging Neurosci. 10:241. doi: 10.3389/fnagi.2018.00241
Received: 07 April 2018; Accepted: 23 July 2018;
Published: 07 August 2018.
Edited by:
P. Hemachandra Reddy, Texas Tech University Health Sciences Center, United StatesReviewed by:
Hela Azaiez, University of Iowa, United StatesCopyright © 2018 Bouzid, Smeti, Chakroun, Loukil, Gibriel, Grati, Ghorbel and Masmoudi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saber Masmoudi, c2FiZXIubWFzbW91ZGlAY2JzLnJucnQudG4=
† Present address: Mhamed Grati, Laboratory of Cell Structure and Dynamics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Maryland, MD, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.