 Holly M. Brothers
Holly M. Brothers Maya L. Gosztyla
Maya L. Gosztyla Stephen R. Robinson
Stephen R. Robinson- 1Department of Psychology, The Ohio State University Columbus, Columbus, OH, United States
- 2Department of Neuroscience, The Ohio State University Columbus, Columbus, OH, United States
- 3Discipline of Psychology, School of Health and Biomedical Sciences, RMIT University, Melbourne, VIC, Australia
Amyloid-ß (Aß) is best known as the misfolded peptide that is involved in the pathogenesis of Alzheimer's disease (AD), and it is currently the primary therapeutic target in attempts to arrest the course of this disease. This notoriety has overshadowed evidence that Aß serves several important physiological functions. Aß is present throughout the lifespan, it has been found in all vertebrates examined thus far, and its molecular sequence shows a high degree of conservation. These features are typical of a factor that contributes significantly to biological fitness, and this suggestion has been supported by evidence of functions that are beneficial for the brain. The putative roles of Aß include protecting the body from infections, repairing leaks in the blood-brain barrier, promoting recovery from injury, and regulating synaptic function. Evidence for these beneficial roles comes from in vitro and in vivo studies, which have shown that the cellular production of Aß rapidly increases in response to a physiological challenge and often diminishes upon recovery. These roles are further supported by the adverse outcomes of clinical trials that have attempted to deplete Aß in order to treat AD. We suggest that anti-Aß therapies will produce fewer adverse effects if the known triggers of Aß deposition (e.g., pathogens, hypertension, and diabetes) are addressed first.
Introduction
The presence of large numbers of “senile plaques” in the hippocampus and overlying cortical regions is one of the definitive features of Alzheimer's disease (AD). These spherical proteinaceous deposits consist primarily of a 38–42 amino acid long peptide known as amyloid-ß (Aß). The Aß peptide is derived from a transmembrane protein called amyloid-β precursor protein (APP). β-site APP cleaving enzyme 1 (BACE1) cleaves APP to release the C99 fragment of APP. This fragment gives rise to various species of Aβ peptide during subsequent cleavage by γ-secretase. In the brain, Aß is produced by astrocytes and neurons; however, non-neural tissues such as skin, skeletal muscle and intestinal epithelium also secrete Aß (Puig and Combs, 2013). Normally present in a soluble form, Aß is secreted into the extracellular space of the brain and then cleared by the cerebrospinal fluid (CSF) and vascular system. In the CSF of cognitively normal humans, the most abundant isoform is 40 amino acids long (Aß40; 2–3 ng/mL), while the second most common isoform (Aß42) is present at approximately 0.75 ng/mL (Ida et al., 1996; Mo et al., 2015). Experiments with transgenic mice that overexpress Aß have revealed that the turnover of soluble Aß is rapid, and it is cleared from the extracellular space and CSF with a half-life of just 0.7–2.0 h (Savage et al., 1998; Abramowski et al., 2008). Radioactive tracer studies have shown that Aß is removed from the circulation by the capillary beds of the kidneys, liver, gastrointestinal tract, and skin (Xiang et al., 2015).
Soluble Aß can bind to other molecules of Aß to form oligomers that are cleared more slowly from the brain, or which can accrete to form insoluble Aß plaques. Numerous in vivo and in vitro experiments demonstrated that the oligomeric and insoluble forms of Aß are toxic to brain cells. These findings have led to the prevailing view that Aß exhibits a “toxic gain-of-function” when it forms oligomers and aggregates into plaques, thereby directly contributing to the pathogenesis of AD, and making it the logical target for therapeutic intervention (Masters and Selkoe, 2012). However, of more than 200 clinical trials that specifically targeted Aß between 1984 and 2013, none improved clinical outcomes in AD patients (Schneider et al., 2014). Indeed, some of these trials were associated with adverse outcomes. This situation has continued through to the present day, with not a single clinical trial between 2012 and 2017 producing a significant cognitive benefit. This frustrating lack of progress has led to suggestions that Aß needs to be targeted at an earlier stage of the disease, prior to the onset of dementia or even before any cognitive changes are detectable (Tarawneh and Holtzman, 2009).
The term “amyloidogenic” is applied to any soluble peptide or protein that has the capacity to interact with similar molecules to self-assemble into insoluble fibrils, which then bond with other fibrils to form a regular β-pleated sheet. The molecular conformation of these amyloid sheets makes them strongly resistant to degradation by proteolytic enzymes. Functional amyloids and amyloidogenic peptides are common in biological systems. For instance, colonial bacteria utilize amyloids to aggregate, attach to a substrate, and improve the strength of their protective biofilms (Dueholm et al., 2013). Plants produce amyloids with strong antifungal and antimicrobial properties (Villar-Piqué et al., 2010; Garvey et al., 2013).
A meta-analysis of APP-like and Aß-like sequences in living species has found that these sequences are present in hydra and sea anemones, indicating that the sequences must have evolved prior to the evolution of arthropods, around 500 million years ago (Tharp and Sarkar, 2013). All vertebrates produce APP, ß-secretase, and Aß; Aß in birds, reptiles and amphibians has a >90% sequence homology with human Aß, while in mammals the sequence homology exceeds 95% (Tharp and Sarkar, 2013). The conservation of the Aß molecular sequence throughout vertebrate evolution implies that it must confer a selective advantage for species survival. This notion is further supported by evidence that depletion of endogenous Aß has adverse consequences in a variety of species and animal models (summarized in Table 1). Although this concept runs counter to research that has focused on Aß's neurotoxic potential in AD, enough evidence has accumulated to suggest that Aß serves a beneficial role in human physiology, where it may contribute to:
• Antimicrobial activity: Aß has antibacterial, antifungal, and antiviral properties that are effective against at least eleven species of microbes.
• Tumor suppression: Aß may intercept oncogenic viruses and suppress tumor growth.
• Sealing leaks in the blood-brain barrier (BBB): Aß binds blood-borne solutes together to form a plug that prevents the spread of neuroactive and toxic components into the brain.
• Promoting recovery from brain injury: The presence of Aß results in better outcomes in animal models of controlled cortical impact, spinal cord injury, hypoxia, and autoimmune disease.
• Regulating synaptic function: Aß regulates the responsiveness of glutamatergic and cholinergic synapses in the hippocampus, thereby contributing to memory consolidation.
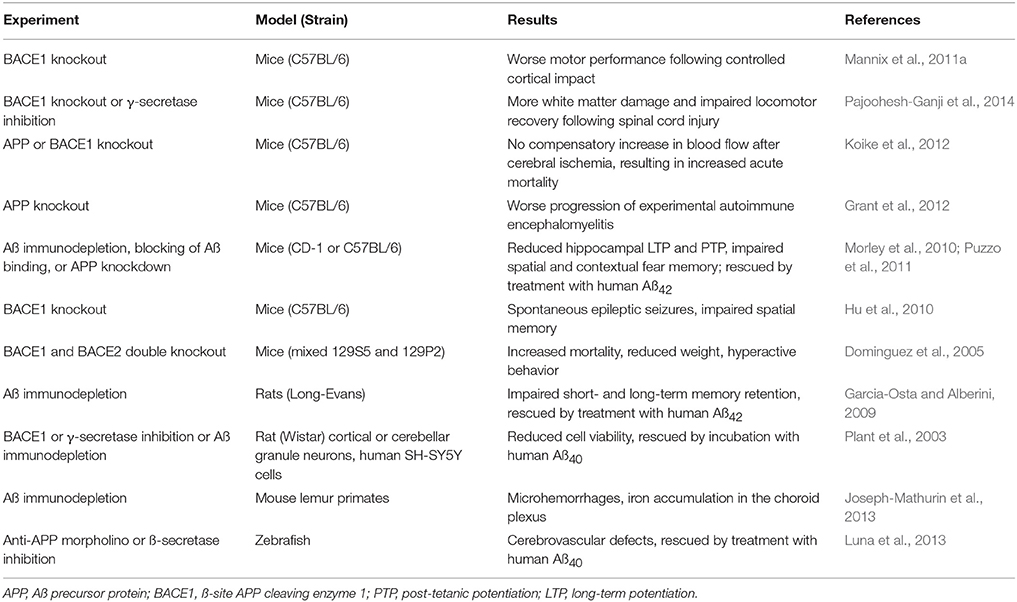
Table 1. Adverse consequences of endogenous Aß depletion.
Such beneficial properties may explain the persistence of Aß throughout the vertebrate series. The following sections consider the evidence that supports each of these putative functions.
Aß Has Antimicrobial Properties
Among the first physiological functions of Aß to be proposed was the “Bioflocculant hypothesis” (Bishop and Robinson, 2002; Robinson and Bishop, 2002), where we noted that the widespread occurrence of Aß in healthy individuals suggests that Aß plays a natural physiological role, one that is most probably protective. We suggested that “Aß may have a broader role as a general chelator and flocculant of potentially toxic agents that are dissolved in the extracellular fluid. In addition to metal ions, this would include bacteria and viruses, proteins, and neuroactive molecules that have been inadvertently released into the extracellular fluid.” Once bound and taken out of solution, we envisaged that these pathogens could be phagocytosed and cleared by microglia and macrophages. In our review of the recent evidence for Aß's role as an antimicrobial peptide (AMP), a class of innate immune molecules with broad-spectrum antimicrobial properties, we noted that Aß not only binds and intercepts microbial pathogens, as suggested by the Bioflocculant hypothesis, but also possesses microbicidal activity that enables it to directly kill bacteria and viruses (Gosztyla et al., 2018).
The notion that Aß is an AMP is consistent with reports that several other amyloid peptides have antimicrobial properties, including serum amyloid A, microcin E492, temporins, and protegrin-1 (for reviews see Bishop and Robinson, 2004a; Kagan et al., 2012). Their antimicrobial activity may be partly due to the capacity of these peptides to form fibrils that insert into cell membranes and create pores that permit the unregulated passage of solutes into and out of microbes, leading to the death of these cells (Kagan et al., 2012). Similarly, Aß may capture and perforate microbes with its hairpin loop, while aggregates of Aß may immobilize microbes, akin to neutrophil extracellular traps, and the destruction of microbes may be accelerated by increased oxidation in the presence of iron from ferritin-rich cells like microglia (Batton et al., 1997; Robinson et al., 2003; Bishop and Robinson, 2004b; Wang et al., 2012).
The antimicrobial activity of human Aß was confirmed by Soscia et al. who demonstrated that the addition of 25 μg/mL of synthetic Aß42 slowed the proliferation of seven different bacteria and one fungal species in culture as effectively or better than the innate defensin LL-37. Aß42 was found to be slightly more potent than Aß40 when delivered at the same concentrations. It may be argued that this antimicrobial effect is not representative of the in vivo situation, because the concentrations of Aß used by Soscia et al. exceeded the physiological range by 4–5 orders of magnitude. However, this limitation was addressed by demonstrating that homogenates of temporal cortex from AD brain are more effective at inhibiting the growth of Candida albicans cultures than homogenates from cognitively normal subjects. This inhibitive effect was neutralized by pre-incubating the homogenates with anti-Aß antisera, indicating that the antimicrobial activity was probably due to the higher Aß burden in the AD brains (Soscia et al., 2010). Additionally, if Aß responds to pathogens, it is likely that the concentration of Aß in the immediate vicinity would far exceed the concentration of Aß averaged across a given brain region. These findings were confirmed by Spitzer et al. who found that 25–50 μg/mL of Aß42 agglutinated and reduced the viability of four bacterial species and the yeast C. albicans (Spitzer et al., 2016). Direct evidence for Aß's antimicrobial activity in vivo was reported in a recent study by Kumar et al. Here, mice and nematodes that overexpressed human Aß demonstrated enhanced resistance to bacterial or yeast infections. Electron microscopy images revealed that Aß fibrils entrapped bacterial and yeast cells in vitro and in vivo (Kumar et al., 2016).
In addition to bactericidal and fungicidal activity, Aß has virucidal properties (reviewed by Bourgade et al., 2016a). Lukiw et al. demonstrated that high concentrations of Aß42 inhibit the infection of human neuron-glia co-cultures by herpes simplex virus 1 (HSV1) as effectively as the antiviral agent acyclovir (Lukiw et al., 2010). An in vitro study by Bourgade et al. solidified this finding by demonstrating that Aß42 and Aß40 prevent HSV1 infection as effectively as LL-37, by binding to the virus and preventing its uptake into cells (Bourgade et al., 2015). This team further demonstrated that human H4 neuroglioma cells produce Aß upon exposure to HSV1 and that transfer of cell media containing Aß to naive H4 cells prevented HSV1 infection (Bourgade et al., 2016b). Aß was ineffective against the non-enveloped human adenovirus, leading Bourgade et al. to conclude that the antiviral activity of Aß is associated with a capacity to interact with viral coat proteins.
This conclusion is consistent with the findings of White et al. who showed that Aß42, and to a lesser extent Aß40, are effective at preventing infection of cultured cells by the pandemic strains H1N1 and H3N2 of the influenza virus (an enveloped virus) (White et al., 2014). The primary antiviral mechanism involves Aß binding viral particles into extracellular aggregates that were then precipitated from the supernatant and unable to infect cultured fibroblasts. Furthermore, pre-incubation of the virus with soluble Aß stimulated the subsequent uptake of virus by phagocytes but the virus did not replicate within these cells, indicating that the aggregated viral particles had been neutralized by the Aß. The strength of these effects increased as a function of Aß concentration. It appears that the antimicrobial property of Aß is largely due to its capacities to permeablize cells and to bind and aggregate pathogens. A recent study by the same group found that the Aß42 C-terminal amino acids 41 and 42 are critical for this function, as fragments lacking these amino acids show an impaired ability to flocculate and promote neutrophil uptake of viruses and bacteria (White et al., 2018).
Indirect evidence from animal studies has shown that the production of Aß waxes and wanes in response to immune challenge and healthy resolution. For example, Aß deposits have been noted in wild-type mice infected with Chlamydia pneumoniae (Little et al., 2004, 2014; Boelen et al., 2007), HSV1 (Wozniak et al., 2007), pseudorabies virus (Tanaka and Nagashima, 2017), or Toxoplasma gondii (Torres et al., 2018), while transgenic-AD mice infected with Porphyromonas gingivalis showed increased Aß deposition (Ishida et al., 2017). Notably, Aß returned to normal levels after the C. pneumoniae infection had resolved. In addition, wild-type mice infected with persistent cerebral toxocariasis acquire insoluble Aß deposits in the hippocampus at concentrations 10–20-fold higher than in uninfected mice (Chou et al., 2017). The presence of Aß deposits in non-transgenic mice is significant because it may provide insight into the presence of Aß in the 95% or more AD patients who have non-hereditary “sporadic” AD. This interpretation is further supported by evidence from human studies. CSF levels of soluble APP or Aß decrease during CNS infection (Sjögren et al., 2001; Gisslén et al., 2009; Jesse et al., 2010; Mattsson et al., 2010; Angel et al., 2012; Krut et al., 2013), indicating that APP and Aß42 are sequestered within the brain during this time. After resolution of Lyme neuroborreliosis and bacterial meningitis, CSF levels of APP, or Aß42 return to normal (Sjögren et al., 2001; Angel et al., 2012).
Several researchers have championed the “Pathogen hypothesis” of AD (for review see Robinson et al., 2004b; Itzhaki et al., 2016), which postulates that AD may be caused by a cerebral infection of HSV1 (Ball, 1982; Ball et al., 2013; Itzhaki, 2014), Borrelia (Miklossy, 2015), or C. pneumoniae (Balin et al., 2008). While a discussion of that literature is beyond the scope of the present review, it is pertinent to note a variety of microbial species have been observed in Aß plaques or AD brains. Specifically, HSV1 DNA (Wozniak et al., 2009), and Borrelia antigen and DNA (Miklossy, 2016) have been found in plaque cores, both Borrelia and C. pneumoniae have been cultured from AD brain tissue (Balin et al., 1998; Gérard et al., 2006; Dreses-Werringloer et al., 2009), and extracellular and intracellular C. pneumoniae and various intraneuronal fungal infections have been reported in AD brain tissue (Hammond et al., 2010; Alonso et al., 2014, 2015; Pisa et al., 2015a,b). These observations can be viewed within the context of work by Michael D'Andrea and others, which have demonstrated that intracellular accumulations of Aß can burst out following cell death to produce extracellular dense-core plaques (reviewed by D'Andrea, 2014, 2016). A viral infection could contribute to intraneuronal deposition of Aß, resulting in cell lysis, and the release of dense-core plaques containing viral DNA into the extracellular space. In contrast, extracellular Aß accumulation could be attributed to the interception of bacterial or fungal pathogens. Taken together, these data suggest that Aß responds to and limits various types of infections in cells, animals, and humans (Figure 1).
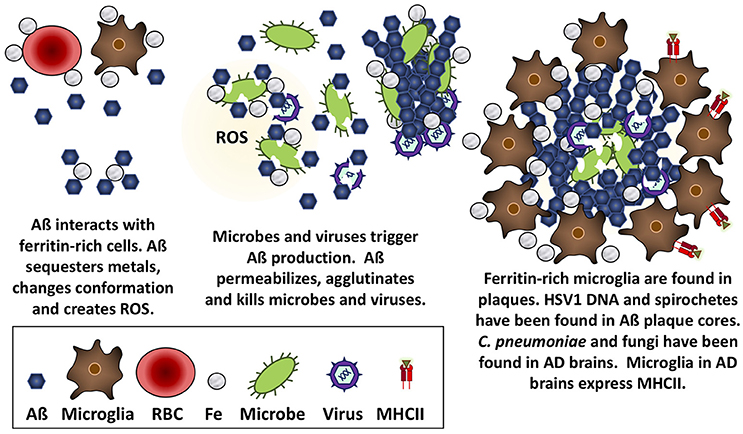
Figure 1. Aß is antimicrobial against bacteria, fungi, and viruses. Aß has the mechanical properties to trap microbes, insert into and permeabilize their membranes, and create a toxic oxidative response that is likely accelerated in the presence of iron obtained from nearby ferritin-rich cells.
Since Aß acts as an AMP, depletion of Aß would be expected to increase infection rates or infection severity. Indeed, clinical trials that have targeted Aß in AD patients have provided support for this notion (Table 2). For instance, approximately 6% of trial participants that received AN-1792, an active anti-Aß immunization, developed meningoencephalitis (Orgogozo et al., 2003; Robinson et al., 2004a; Gilman et al., 2005; Patton et al., 2006). Increased infection rates, including orolabial herpes relapse, and upper respiratory infections, have been reported in clinical trials of ß- or γ-secretase inhibitors [Green et al., 2009; Doody et al., 2013; AlzForum (n.d.-d, h, m)] and the Aß-binding compound ELND005 (Salloway et al., 2011; Alonso et al., 2014). A recent meta-analysis of ten clinical trials concluded that γ-secretase inhibitors are associated with an increased risk of infections (Penninkilampi et al., 2016). While these observations are not direct evidence of an antimicrobial role for Aß, they are certainly consistent with this possibility and indicate that future clinical trials should be alert to the potential for such outcomes.
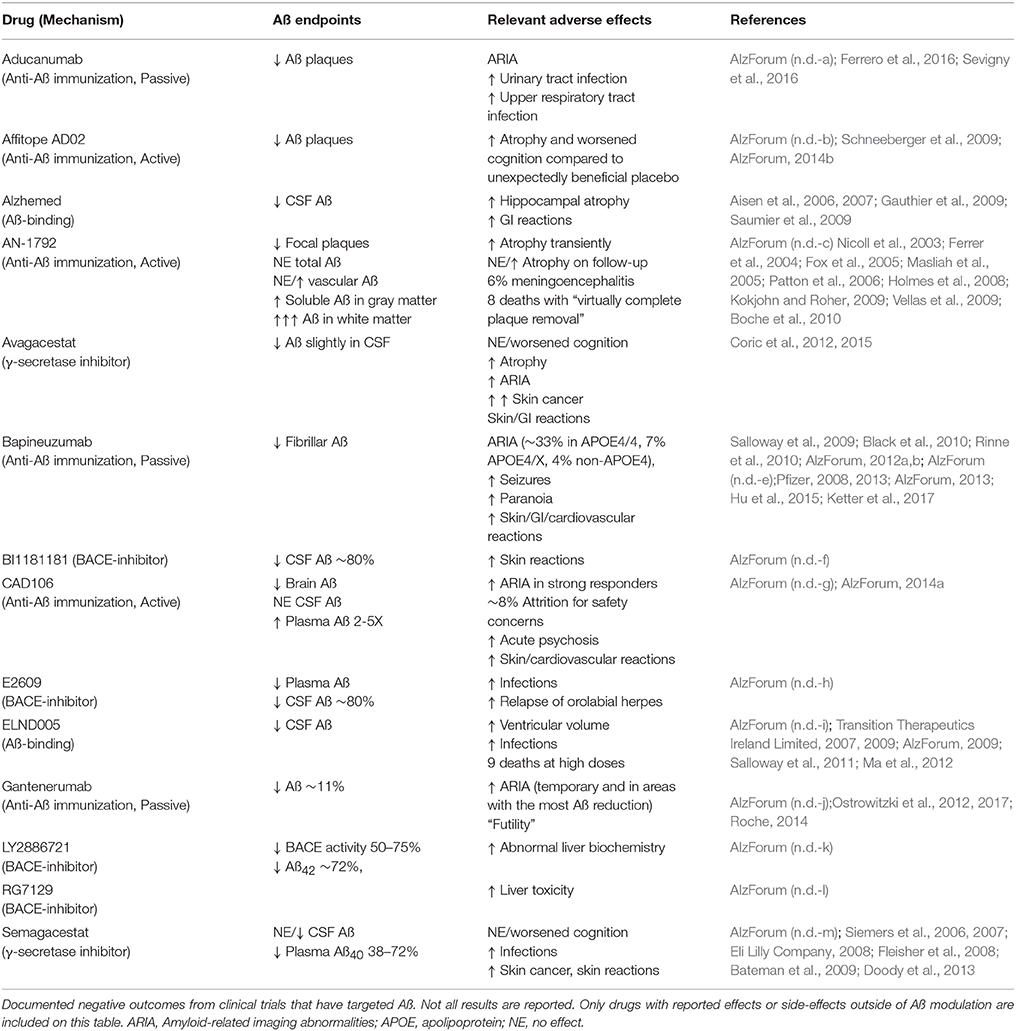
Table 2. Adverse events in Aß-targeted clinical trials.
Aß May Protect Against Some Forms of Cancer
There is an impressive inverse relationship between AD and cancer. One interesting example is the naked mole rate, a notoriously cancer-resistant rodent that accumulates Aß at levels similar to AD-mice bearing at least three human transgenes without developing memory impairment (Edrey et al., 2013; Deweerdt, 2014). Multiple studies have demonstrated that cognitively normal elderly patients who are diagnosed with cancer are less likely to subsequently develop AD, whereas patients who have been diagnosed with probable AD are half as likely to have had cancer or to develop cancer compared to age-matched, cognitively-normal peers (Driver et al., 2012; White et al., 2013; Catalá-López et al., 2014; Ma et al., 2014; Shi et al., 2015; Yarchoan et al., 2017). While some reports have suggested that this relationship may be due to ascertainment bias (Freedman et al., 2016; Bowles et al., 2017; Hanson et al., 2017), a recent examination of nearly 3.5 million veterans found that the risk of several types of cancer is lower in AD patients, even after accounting for bias (Frain et al., 2017).
AD patients have significantly lower incidences of non-melanoma skin cancer, head and neck cancer, colorectal cancer, lung cancer, breast cancer, bladder cancer, and hematologic malignancies (reviewed by Shi et al., 2015). A recent analysis of 4,357 subjects observed a reduced risk of AD following a diagnosis with incident cancer, though no difference in AD risk was found for prevalent cancers. However, when the analysis was restricted to late-stage prevalent cancers, diagnosis was associated with a 50% reduction in AD or dementia risk (Bowles et al., 2017). The fact that patients with vascular dementia have incidences of cancer that are comparable to those in the general elderly population (Roe et al., 2010) implies that there is something specific about AD that confers protection against cancer. This conclusion is supported by a meta-analysis of over 50 clinical studies involving more than half a million participants, in which it was found that AD is associated with a greatly reduced risk of cancer (Catalá-López et al., 2014).
While the basis of the protection against cancer is unknown, the outcomes of the AD clinical trials do not support a direct role for Aß in the repression of cancer. While some of the trials reported increased rates of cancer, such trials involved γ-secretase inhibitors, rather than inhibitors of BACE1 or immunotherapy against Aß (Table 2). For instance, clinical trials of the γ-secretase inhibitor Avagacestat were halted early, in part because seven patients developed squamous-cell or basal-cell carcinomas of the skin, compared to none in the placebo group (Coric et al., 2012, 2015). Similarly, a phase III trial of the γ-secretase inhibitor Semagacestat was discontinued after 5–6% of patients developed squamous-cell carcinomas of the skin and 15–16% developed “neoplasms,” compared to the placebo group who had rates of 1 and 5%, respectively (Doody et al., 2013). A meta-analysis found that γ-secretase inhibitors are associated with a nearly five-fold increase in skin cancer risk (Penninkilampi et al., 2016). It should be noted that increased rates of cancer have not been reported for clinical trials that have specifically targeted Aß, so it is likely that the adverse effects were related to functions of γ-secretase that are separate from its cleavage of Aß, such as a loss of the APP cleavage product, Notch (Roperch et al., 1998; Paris et al., 2005).
Although the evidence from clinical trials does not support an active role for Aß in the suppression of cancer, it is possible that circulating Aß plays an indirect role by intercepting oncogenic viruses. Up to 18% of cancers are thought to be induced by oncogenic viruses (Parkin, 2006). For example, most non-melanoma skin cancers, including squamous-cell and basal cell carcinomas, contain human papillomavirus (Arroyo Mühr et al., 2015). It is notable that these forms of cancer are underrepresented in AD. The finding that the oncogenic virus Epstein-Barr stimulates the production of anti-Aß antibodies, raises the possibility that some viruses may benefit from the elimination of Aß (Xu and Gaskin, 1997). Since high titers of anti-Epstein-Barr virus antibodies in patients with mild cognitive impairment are predictive of future cognitive decline (Shim et al., 2017), it is tempting to speculate that AD may involve attempts by oncogenic viruses to neutralize the defenses provided by Aß, which are then countered by increased production of antibodies against the virus. Such counter-responses could account for the lower rate of some cancers in AD.
Experimental evidence indicates that Aß is capable of inhibiting tumor cell growth. For instance, the treatment of cultured cancer cell lines with conditioned media containing Aß significantly reduced the rate of proliferation of human glioblastoma, human breast adenocarcinoma, and mouse melanoma cells (Zhao et al., 2009). The extent of the reduction was associated with the concentration of Aß present in the medium and was not linked to the presence of APP. In mice, Aß suppresses tumor growth when injected directly into human glioblastoma and human lung adenocarcinoma xenografts (Paris et al., 2004). Similarly, Aß delivered into the peritoneal cavity reduces the growth of lung adenocarcinoma xenografts (Paris et al., 2004). In transgenic mouse lines that overexpress Aß, the rates of growth of implanted glioma tumor masses are suppressed by 40–50% at 8 months of age compared to tumor masses in wild-type mice (Paris et al., 2010).
Paris et al. demonstrated that high concentrations of Aß inhibit capillary growth both in vivo and in vitro, and when present at very high concentrations it causes capillaries to degenerate. They concluded that Aß may slow tumor growth by retarding neovascularization (Paris et al., 2004, 2010). An alternative possibility, suggested by the present authors, relates to the exceptionally high binding affinity of Aß for iron, copper, and zinc (Bishop and Robinson, 2002; Robinson and Bishop, 2002). By scavenging free metal ions, Aß may limit the availability of these essential micronutrients and slow the proliferation of tumor cells. Evidently there are several potential mechanisms that could account for the inverse relationship between AD and some forms of cancer, and until further research has been conducted, the basis of this relationship will remain a matter for speculation.
Aß Seals Leaks in the Blood-Brain Barrier
In contrast to cancer, the link between Aß and the integrity of the BBB is firmly established. Aß plaques in AD brain tissue contain many different blood proteins and peptides, including serum albumin, fibrinogen, thrombin, IgG, von Willebrand factor, collagen IV, and hemin (Cullen et al., 2006), which are normally foreign to the brain. Hemoglobin also binds to Aß in an iron-dependent manner and colocalizes with Aß plaques and vascular deposits in post-mortem AD brains (Oyama et al., 2000; Wu et al., 2004; Chuang et al., 2012). In 2002, one of us proposed that if the BBB becomes leaky, allowing pro-inflammatory and neuroactive compounds to enter the brain, soluble Aß will bind these compounds into an insoluble mass to prevent their spread through the neuropil (Bishop and Robinson, 2002). Other researchers have built on this idea. Stone demonstrated that practically all plaques in AD are closely associated with a capillary, thereby supporting a causal link between a leaky BBB and Aß deposition (Stone, 2008), while Atwood et al. (2003) proposed that Aß may serve as a vascular “scab” that seals breaches of the BBB.
Aß may slow bleeding with filamentous aggregates that pull the walls of the capillary endothelial cells back together (Atwood et al., 2003). In fact, incorporation of Aß into the surface of either red blood cells or endothelial cells increases the probability that the cells will adhere to the microvasculature (Ravi et al., 2004; Figure 2). Viewed from this perspective, the heavier plaque burden in AD may be because the BBB is more porous than in non-demented elders. A recent imaging study reported that the BBB becomes more permeable in the human hippocampus with age, and that permeability is more pronounced in individuals with mild cognitive impairment than in age-matched controls or those with multiple sclerosis (Montagne et al., 2015). In support of this hypothesis, cortical superficial siderosis is seven times more common in AD than in age-matched controls (Dubessy et al., 2012; Wollenweber et al., 2014); this condition is due to the accumulation of iron from extravasated hemoglobin, and its presence is indicative of a history of micro-hemorrhages (Charidimou et al., 2015). It follows that removal of Aß in AD is likely to lead to increased BBB permeability and an increase in micro-hemorrhages and subsequent brain edema.
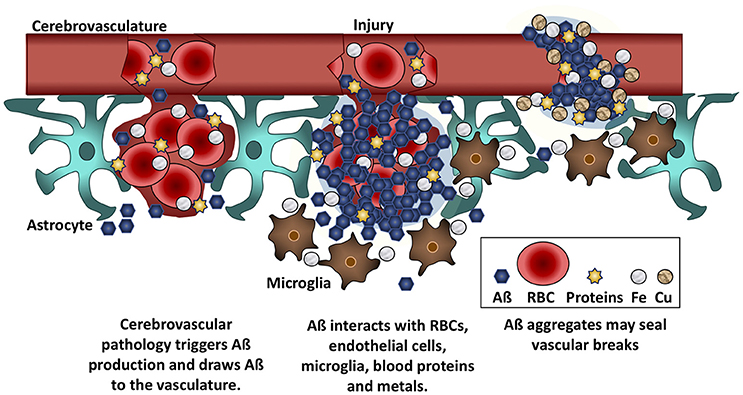
Figure 2. Aß seals leaky vessels. Traumatic brain injury and cerebrovascular insults stimulate Aß production and draw Aß to the vasculature. Aß binds to red blood cells (RBCs), blood proteins, and to iron (Fe) and copper (Cu) ions. These interactions cause Aß to aggregate at the site of hemorrhage or breaches of the BBB. Aß anchors into cell membranes and increases adherence between RBCs and vascular endothelial cells, helping to seal leaks in the vasculature.
Clinical trials targeting Aß have provided dramatic confirmation of this hypothesis. The most common adverse side-effect of these trials has been MRI evidence of brain edema and/or micro-hemorrhages, sometimes accompanied by increased confusion and disorientation. This pattern of pathological change, so characteristic of amyloid depletion, has been termed “Amyloid-Related Imaging Abnormalities” (ARIA). The prevalence of ARIA has been very high in clinical trials of passive immunotherapies (e.g., Bapineuzumab, Solenezumab, Aducanumab, and Gantenerumab) (reviewed by Lannfelt et al., 2014; DiFrancesco et al., 2015). In the clinical trials with Bapineuzumab for instance, the incidence of ARIA with edema increased from 7.1% of patients on the lowest dose of the drug to 30.8% of patients on the highest dose; 47.2% of these patients also exhibited evidence of micro-hemorrhages, while a further 4% had micro-hemorrhages without evidence of increased edema (Sperling et al., 2012). In the Aducanumab trials, the incidence of ARIA was even higher, at 47–55% of participants in the highest dosage group (AlzForum (n.d.-a); DiFrancesco et al., 2015; Sevigny et al., 2016). A meta-analysis of fourteen clinical trials found that anti-Aß immunotherapies are associated with a nearly five-fold increase in ARIA (Penninkilampi et al., 2017). Unlike trials targeting secretases, these immunotherapies targeted Aß without altering APP processing, suggesting that ARIA is directly related to the loss of Aß. ARIA resulting from the targeting of Aß is more likely to occur in carriers of the apolipoprotein ε4 (APOE ε4) allele AlzForum (n.d.-e), and imaging confirms that the edema occurs at focal sites that exhibit the greatest reductions in Aß (Ostrowitzki et al., 2012). The edema results from the entry of blood solutes into the neuropil, followed by an influx of water into the brain along the osmotic gradient.
Although ARIA was not observed in the early experiments with transgenic mice, subsequent experiments have confirmed that the link exists. For instance, anti-Aß immunotherapy of PDAPP mice, which overexpress APP, leads to an increase in BBB permeability in a subset of mice that is accompanied by cerebral microbleeds, siderosis, and localized edema, which are the hallmarks of ARIA (Blockx et al., 2016). Similarly, aged mouse lemur primates, which exhibit an age-associated accumulation of endogenous Aß with a peptide sequence that is similar to that in humans, also develop ARIA following anti-Aß immunotherapy (Joseph-Mathurin et al., 2013).
Several lines of experimental evidence from mice indicate that BBB breakdown leads to increased Aß deposition. Mice that overexpress endothelin-1, resulting in a weakened BBB, show increased astrocytic secretion of Aß following an ischemic stroke (Hung et al., 2015). Micro-hemorrhages created with Rose Bengal dye in transgenic-AD mice drove transient increases in Aß plaques in infarcted and adjacent areas (Garcia-Alloza et al., 2011). Similarly, micro-hemorrhages created with diet-induced hyperhomocysteinemia in transgenic-AD mice shifted the distribution of Aß deposits from the parenchyma to the vasculature (Sudduth et al., 2014). Furthermore, hemorrhages induced by needle stick lesions in wild-type rats led to a transient up-regulation of APP, Aß, and phosphorylated tau near the lesion site and a longer-lasting deposition of Aß along the needle tract (Purushothuman et al., 2013). Mice that have been chronically subjected to high blood pressure develop Aß deposits around their cerebral blood vessels and display learning impairments (Carnevale et al., 2016).
Collectively, the preceding observations provide powerful support for the view that Aß seals leaks in the BBB, a role that probably becomes increasingly important as the aging BBB gradually loses its integrity. Viewed from this perspective, comorbid conditions that are likely to enhance the permeability of the BBB, such as diabetes and vascular hypertension, should be correlated with a heavier plaque burden. Indeed, a diabetic phenotype does increase the expression of Aß in AD-Tg mice (Ho et al., 2004), is sufficient to do the same in wild-type rabbits (Bitel et al., 2012), and is associated with greater plaque pathology in the subset of diabetic AD patients with an APOE ε4 genotype (Malek-Ahmadi et al., 2013). Similarly, vascular hypertension is associated with higher Aß burdens in wild-type animal models (Gentile et al., 2009; Schreiber et al., 2014; reviewed by Bueche et al., 2014), increased Aß aggregates in the human placenta (Kalkunte et al., 2013; Buhimschi et al., 2014), and increased Aß deposits in the AD brain (Ashby et al., 2016).
Aß May Improve Recovery From Brain Injury
The presence of Aß plaques in the hippocampus and cerebral cortex has become synonymous with AD, to the point where cognitively normal persons with significant Aß burdens are assumed to have incipient AD. However, as was seen in the preceding sections, the presence of Aß deposits does not necessarily indicate AD; they may indicate sites where pathogens have been intercepted and neutralized or where a leaky BBB has been repaired. Another well-documented role of Aß is in assisting the brain to recover from traumatic and ischemic injuries.
In humans, a traumatic brain injury (TBI) elevates APP levels in the brain within 2 h (McKenzie et al., 1996) and Aß plaques become evident within 4 h (Roberts et al., 1994; Graham et al., 1995; Johnson et al., 2010). Two microdialysis studies of brain extracellular fluid from TBI patients found that those with the higher titers of Aß experienced better outcomes (Brody et al., 2008; Magnoni et al., 2012). Aß plaques form routinely after a head injury, even in children as young as 10 years, who presumably would not otherwise have plaques (Roberts et al., 1994; Graham et al., 1995), and they are more likely to form in APOE ε4 carriers (Nicoll et al., 1995; Zunarelli et al., 1996; Mauri et al., 2006; Abu Hamdeh et al., 2017). A PET study of TBI patients found that while Aß deposits colocalized with areas of white matter damage, the Aß burden was not significantly correlated with the extent of neuropsychological impairment (Scott et al., 2016). Though Aß accumulation occurs immediately after a traumatic injury and can persist in damaged axons for years (Johnson et al., 2012; Scott et al., 2016; Bagnato et al., 2017), the brains of long-term survivors of head injury do not have greater plaque numbers or increased APP expression when compared to age- and APOE-matched controls (Macfarlane et al., 1999; Chen et al., 2009). This suggests that Aß accumulates transiently in response to injury.
Evidence from animal models also shows that Aß expression responds rapidly to injury, resolves over time, and may be necessary for a good recovery. A recent meta-analysis of 19 animal studies reported that Aß expression consistently increases within 24 h of TBI, including in models that lack AD-related transgenes (Bird et al., 2016). For example, controlled cortical impact leads to an upregulation of APP and BACE1 expression in wild-type rats (Blasko et al., 2004; Acosta et al., 2017) and accelerates Aß deposition in transgenic-AD mice (Tajiri et al., 2013; Washington et al., 2014; Shishido et al., 2016). Controlled cortical impact increases the expression of Aß40 and Aß42 in transgenic-AD mice within one day, with levels returning to baseline within one week (Washington et al., 2014). During this early period, post-injury macrophage activation is suppressed (Kokiko-Cochran et al., 2016), which may provide injured neurons with time to repair and recover, instead of being phagocytosed.
It is notable that controlled cortical impact results in worse motor performance in mice that are deficient in BACE1 compared to wild-type mice (Mannix et al., 2011a), and when the mice are treated with intra-ventricular injections of Aß40 after the injury, motor performance improves in the knock-out mice but worsens in the wild-type mice (Mannix et al., 2013). These results suggest that the level of Aß production in wild-type mice after controlled cortical impact was tuned and appropriate. Recovery after controlled cortical impact is also modulated by age and APOE genotype; in immature and adult mice expressing human APOE ε4, only adults showed worse spatial memory performance compared to wild-type, as well as high Aß40 levels 1 month after injury (Mannix et al., 2011b). Similarly, Aß expression increases during the first 3 days after spinal cord injury, and if Aß production is prevented by BACE1 knock-out or by treatment with a γ-secretase inhibitor, the mice develop more white matter damage, and display impaired recovery from locomotor deficits (Pajoohesh-Ganji et al., 2014).
Steinman et al. noted that Aß42 is prominent in cerebral lesions and in the damaged axons of patients with multiple sclerosis, and postulated that this might represent a protective response to injury (Ferguson et al., 1997; Trapp et al., 1998; Han et al., 2008). Steinman's group delivered intraperitoneal injections of Aß40 or Aß42 into four different animal models of multiple sclerosis. Stunningly, they found that the treatment attenuated motor paralysis, reduced the extent of demyelinated lesions, suppressed lymphocyte activation, and lowered pro-inflammatory cytokine expression in blood. In contrast, APP knockout mice fared much worse than wild-type mice. It is important to note that protection in this model was associated with a hexameric form of Aß that reduces T-cell activation (Grant et al., 2012).
Another example of the protection that Aß affords from brain injury comes from experimental models of stroke. In wild-type mice, occlusion of the common carotid artery drives a compensatory increase in blood flow in the cerebral arteries, but this compensatory increase is attenuated in APP knock-out mice (Koike et al., 2012). This reduction of compensatory blood flow is lethal, and consequently APP knock-out mice die shortly after bilateral occlusion of the common carotid artery, whereas wild-type mice survive; since BACE1 knockout mice suffer the same fate, this loss of viability is likely due to the absence of Aß rather than other products of APP cleavage (Koike et al., 2012). Further evidence that Aß is protective during a stroke comes from a rat model of middle cerebral artery occlusion, in which the mean infarct volume is significantly reduced in transgenic-AD rats compared to wild-type rats (Clarke et al., 2007).
The preceding observations show that the presence of Aß improves outcomes after injury to the central nervous system. Consequently, pre-emptive anti-Aß treatments for AD are likely to increase the probability that the treated individuals will display poorer prognoses if they have the misfortune of sustaining a TBI (e.g., from a fall) or a stroke.
Aß May Regulate Activity at Hippocampal Synapses
A growing body of evidence demonstrates that soluble Aß is necessary for synaptic plasticity and memory (reviewed by Puzzo and Arancio, 2013; Morley and Farr, 2014). During periods of neuronal activity, APP is transported anterogradely to synapses, where Aß is cleaved and released along with neurotransmitter into the synaptic cleft (Kamenetz et al., 2003; Cirrito et al., 2005; Tampellini et al., 2009). Aß then acts on presynaptic neurons to increase the probability of further neurotransmitter release (Fedele et al., 2015; Puzzo et al., 2015). Depletion of endogenous Aß in rodents greatly reduces LTP and short- and long-term memory; this can be rescued by the addition of human Aß42 (Garcia-Osta and Alberini, 2009; Morley et al., 2010; Puzzo et al., 2011). Additionally, rodents treated with picomolar concentrations of human Aß42 show enhanced memory compared to a scrambled control peptide or vehicle (Garcia-Osta and Alberini, 2009; Morley et al., 2010). The duration of Aß exposure seems to be important for this process. Mouse hippocampal neurons exposed to physiological concentrations of oligomeric Aß42 show enhanced plasticity within minutes, but reduced plasticity with prolonged exposure (Koppensteiner et al., 2016). This was confirmed in vivo, as brief hippocampal infusions of Aß42 enhanced contextual memory in mice, while longer infusions impaired memory (Koppensteiner et al., 2016).
Aß may enhance long-term potentiation (LTP) by increasing the amount of acetylcholine released into the synaptic cleft and increasing the probability that the postsynaptic neuron will depolarize. Mice injected with low concentrations of Aß into the hippocampus showed enhanced memory retention in two memory tasks and increased acetylecholine production in the hippocampus (Morley et al., 2010). Picomolar concentrations of Aß directly activate α7-nicotinic acetylcholine receptors, whereas nanomolar concentrations of Aß block and inactivate the receptors. Similarly, picomolar concentrations of Aß42 enhance LTP, and memory consolidation in mice, while nanomolar concentrations impair memory (Fedele et al., 2015; Puzzo et al., 2015; Ricciarelli and Fedele, 2017). Furthermore, Aß-mediated enhancement of memory is ineffective in the absence of α7-nicotinic acetylcholine receptors (Fedele et al., 2015; Puzzo et al., 2015; Ricciarelli and Fedele, 2017).
In addition to interacting with acetylcholine signaling, Aß can also stimulate glutamatergic receptors. Nanomolar concentrations of Aß facilitate NMDA (N-methyl-D-aspartate) receptor-mediated LTP, while picomolar concentrations enhance contextual fear memories. When Aß is present at high picomolar concentrations (which are pathological), it can disrupt the clearance of glutamate from the extracellular space by astrocytes, leading to a build-up of extracellular glutamate that then causes aberrant activation of NMDA receptors and eventual synaptic dysfunction (Tu et al., 2014).
Some clinical trials that have depleted patients' brains of Aß have reported increased rates of adverse events that might be attributable to synaptic dysfunction. For instance, increased seizure activity was reported in clinical trials of the anti-Aß immunotherapy Bapineuzamab (AlzForum (n.d.-e)). In other clinical trials the removal of Aß has been accompanied by worse cognitive outcomes. For example, the γ-secretase inhibitor Semagacestat caused significantly lower scores on tests of cognitive status, functional status, and dementia in a dose-dependent manner, which did not resolve until 32 weeks after termination of drug dosing (Doody et al., 2013). Avagacestat, another γ-secretase inhibitor, was also associated with a decrease in performance on these same cognitive tests, though it did not reach statistical significance (Coric et al., 2012, 2015). In trials of active anti-Aß immunotherapies, Affitope AD02 counteracted the surprisingly beneficial effect of the placebo on cognitive function (AlzForum (n.d.-b); Schneeberger et al., 2009; AlzForum, 2014b), while CAD106 caused an increase in acute psychosis (AlzForum (n.d.-g); AlzForum, 2014a). The negative cognitive effects of anti-Aß immunotherapies support a direct role of Aß depletion in contributing to synaptic dysfunction in these trials.
While most animal studies have not reported adverse cognitive or behavioral effects after immunization with Aß, two anti-Aß immunotherapies (one was the rodent equivalent of Bapineuzumab) were recently tested in two transgenic mouse models of AD and all four conditions resulted in neuronal hyperactivity and dysfunction, independent of the effects of these antibodies on the clearance of Aß plaques (Busche et al., 2015). Notably, however, others have reported that anti-APP/Aß immunotherapy successfully reduced neuronal hyperexcitability and epileptiform discharges in triple transgenic-AD mice (Kazim et al., 2017). This may be explained by the use of older mice in the former study, compared to younger pre-plaque mice in the latter. Additionally, the inability of the antibody used in the latter study to differentiate between APP and Aß could also be a contributing factor to the discrepancy. Other studies have reported that the absence of APP and Aß, due to the knockout of APP or BACE1, increases spontaneous seizure activity and potentiates elicited seizures (Steinbach et al., 1998; Hu et al., 2010). Finally, the treatment of healthy wild-type mice with Aß immunotherapy or with antisense directed at APP significantly impaired their learning on a T-maze foot-shock avoidance task (Morley et al., 2010). Collectively, the evidence reviewed in this section provide strong evidence for Aß serving a physiological role in hippocampal LTP and memory retention.
Conclusion
The research reviewed in the current paper reveals that the Aß peptide is involved in the protection and repair of the central nervous system. Aß regulates synaptic function and contributes to memory consolidation; it may also protect from some forms of cancer and aid recovery from TBI. There is solid evidence that pathogens or a breach in the BBB trigger soluble Aß to aggregate into insoluble deposits in order to intercept the pathogen or seal the leak. Some of the adverse outcomes associated with clinical trials can be understood from this perspective: a reduction in the capacity to intercept pathogens leads to a higher incidence of infections, while a loss of capacity to seal leaks in the BBB leads to increased numbers of micro-bleeds and brain edema (ARIA). This being the case, targeting the production or removal of Aß earlier in the course of AD is likely to be associated with the same adverse events, except that the longer duration between start of treatment and patient death will increase the likelihood of these adverse events occurring during the lifetime of the patient and may reduce the patients' capacity to recover.
More favorable outcomes might be achieved by treating the known triggers of Aß deposition before targeting Aß production. This would involve screening patients for potential causes of BBB leakage (such as diabetes or vascular hypertension) and/or for evidence of latent microbial infections, and then treating accordingly. Such treatments should slow the rate of Aß deposition, with corresponding benefits for cognition. Once these known sources of Aß deposition have been addressed, we would expect subsequent anti-Aß therapies to be associated with fewer instances of ARIA or brain infection. However, it remains possible that anti-Aß therapies will adversely affect learning and memory, recovery from TBI or the incidence of some forms of cancer.
Author Contributions
HB: Wrote the first draft of the manuscript; MG and SR: Substantially revised subsequent drafts. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michael D'Andrea and Keith Crutcher for their valuable feedback and commentary. This work was supported by The Ohio State University.
Abbreviations
Aß, Amyloid-ß; AD, Alzheimer's disease; APP, amyloid-ß precursor protein; AMP, antimicrobial protein; BACE1, β-site APP cleaving enzyme 1; HSV1, herpes simplex virus-1; APOE, apolipoprotein; CSF, cerebrospinal fluid; ARIA, amyloid-related imaging abnormalities; BBB, blood-brain barrier; TBI, traumatic brain injury; LTP, long-term potentiation.
References
Abramowski, D., Wiederhold, K.-H., Furrer, U., Jaton, A.-L., Neuenschwander, A., Runser, M.-J., et al. (2008). Dynamics of Abeta turnover and deposition in different beta-amyloid precursor protein transgenic mouse models following gamma-secretase inhibition. J. Pharmacol. Exp. Ther. 327, 411–424. doi: 10.1124/jpet.108.140327
Abu Hamdeh, S., Waara, E. R., Möller, C., Söderberg, L., Basun, H., Alafuzoff, I., et al. (2017). Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol. doi: 10.1111/bpa.12532. [Epub ahead of print].
Acosta, S. A., Tajiri, N., Sanberg, P. R., Kaneko, Y., and Borlongan, C. V. (2017). Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J. Cell. Physiol. 232, 665–677. doi: 10.1002/jcp.25629
Aisen, P. S., Gauthier, S., Vellas, B., Briand, R., Saumier, D., Laurin, J., et al. (2007). Alzhemed: a potential treatment for Alzheimer's disease. Curr. Alzheimer Res. 4, 473–478. doi: 10.2174/156720507781788882
Aisen, P. S., Saumier, D., Briand, R., Laurin, J., Gervais, F., Tremblay, P., et al. (2006). A phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 67, 1757–1763. doi: 10.1212/01.wnl.0000244346.08950.64
Alonso, R., Pisa, D., Marina, A. I., Morato, E., Rábano, A., and Carrasco, L. (2014). Fungal infection in patients with Alzheimer's disease. J. Alzheimers Dis. 41, 301–311. doi: 10.3233/JAD-132681.
Alonso, R., Pisa, D., Rábano, A., Rodal, I., and Carrasco, L. (2015). Cerebrospinal fluid from Alzheimer's disease patients contains fungal proteins and DNA. J. Alzheimers Dis. 47, 873–876. doi: 10.3233/JAD-150382
AlzForum (2009). Drug Brief—Adverse Events Prompt Dose Drop in Elan Trial. Available online at: http://www.alzforum.org/news/research-news/drug-brief-adverse-events-prompt-dose-drop-elan-trial
AlzForum (2012a). Bapineuzumab Phase 3: Target Engagement, But No Benefit. Available online at: http://www.alzforum.org/news/conference-coverage/bapineuzumab-phase-3-target-engagement-no-benefit
AlzForum (2012b). Clinical Trials of Intravenous Bapineuzumab Halted. Available online at: http://www.alzforum.org/news/research-news/clinical-trials-intravenous-bapineuzumab-halted
AlzForum (2013). Anti-Amyloid Results Show Modest Benefits, Mild Side Effects. Available online at: http://www.alzforum.org/news/conference-coverage/anti-amyloid-results-show-modest-benefits-mild-side-effects
AlzForum (2014a). Denied Breakthroughs, Researchers Keep Hunting New Therapies. Available online at: http://www.alzforum.org/news/conference-coverage/denied-breakthroughs-researchers-keep-hunting-new-therapies
AlzForum (2014b). In Surprise, Placebo, not Aß Vaccine, Said to Slow Alzheimer's. Available online at: http://www.alzforum.org/news/research-news/surprise-placebo-not-av-vaccine-said-slow-alzheimers
AlzForum (n.d.-a). Aducanumab. Available online at: http://www.alzforum.org/therapeutics/aducanumab
AlzForum (n.d.-b). Affitope AD02. Available online at: http://www.alzforum.org/therapeutics/affitope-ad02
AlzForum (n.d.-c). AN-1792. Available online at: http://www.alzforum.org/therapeutics/1792
AlzForum (n.d.-d). Avagacestat. Available online at: http://www.alzforum.org/therapeutics/avagacestat
AlzForum (n.d.-e). Bapineuzumab. Available online at: http://www.alzforum.org/therapeutics/bapineuzumab
AlzForum (n.d.-f). BI 1181181. Available online at: http://www.alzforum.org/therapeutics/bi-1181181
AlzForum (n.d.-g). CAD106. Available online at: http://www.alzforum.org/therapeutics/cad106
AlzForum (n.d.-h). E2609. Available online at: http://www.alzforum.org/therapeutics/e2609
AlzForum (n.d.-i). ELND005. Available online at: http://www.alzforum.org/therapeutics/elnd005
AlzForum (n.d.-j). Gantenerumab. Available online at: http://www.alzforum.org/therapeutics/gantenerumab
AlzForum (n.d.-k). LY2886721. Available online at: http://www.alzforum.org/therapeutics/ly2886721
AlzForum (n.d.-l). RG7129. Available online at: http://www.alzforum.org/therapeutics/rg7129
AlzForum (n.d.-m). Semagacestat. Available online at: http://www.alzforum.org/therapeutics/semagacestat
Angel, T. E., Jacobs, J. M., Smith, R. P., Pasternack, M. S., Elias, S., Gritsenko, M. A., et al. (2012). Cerebrospinal fluid proteome of patients with acute Lyme disease. J. Proteome Res. 11, 4814–4822. doi: 10.1021/pr300577p
Arroyo Mühr, L. S., Hultin, E., Bzhalava, D., Eklund, C., Lagheden, C., Ekström, J., et al. (2015). Human papillomavirus type 197 is commonly present in skin tumors. Int. J. Cancer. 136, 2546–2555. doi: 10.1002/ijc.29325
Ashby, E. L., Miners, J. S., Kehoe, P. G., and Love, S. (2016). Effects of hypertension and anti-hypertensive treatment on amyloid-β (Aβ) plaque load and Aβ-synthesizing and Aβ-degrading enzymes in frontal cortex. J. Alzheimers Dis. 50, 1191–1203. doi: 10.3233/JAD-150831
Atwood, C. S., Bowen, R. L., Smith, M. A., and Perry, G. (2003). Cerebrovascular requirement for sealant, anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply. Brain Res. Brain Res. Rev. 43, 164–178. doi: 10.1016/S0165-0173(03)00206-6
Bagnato, S., Andriolo, M., Boccagni, C., and Galardi, G. (2017). Prolonged changes in amyloid-β metabolism after a severe traumatic brain injury. Neuroreport 28, 250–252. doi: 10.1097/WNR.0000000000000748
Balin, B. J., Gérard, H. C., Arking, E. J., Appelt, D. M., Branigan, P. J., Abrams, J. T., et al. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med. Microbiol. Immunol. 187, 23–42. doi: 10.1007/s004300050071
Balin, B. J., Little, C. S., Hammond, C. J., Appelt, D. M., Whittum-Hudson, J. A., Gérard, H. C., et al. (2008). Chlamydophila pneumoniae and the etiology of late-onset Alzheimer's disease. J. Alzheimers Dis. 13, 371–380. doi: 10.3233/JAD-2008-13403
Ball, M. J. (1982). Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can. J. Neurol. Sci. 9, 303–306.
Ball, M. J., Lukiw, W. J., Kammerman, E. M., and Hill, J. M. (2013). Intracerebral propagation of Alzheimer's disease: strengthening evidence of a herpes simplex virus etiology. Alzheimers Dement. 9, 169–175. doi: 10.1016/j.jalz.2012.07.005
Bateman, R. J., Siemers, E. R., Mawuenyega, K. G., Wen, G., Browning, K. R., Sigurdson, W. C., et al. (2009). A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann. Neurol. 66, 48–54. doi: 10.1002/ana.21623
Batton, C. I., O'Dowd, B. S., Noone, D. F., Kril, J., and Robinson, S. R. (1997). Ferritin-rich microglia are concentrated within ß-amyloid plaques. Alzheimers Res. 3, 23–28.
Bird, S. M., Sohrabi, H. R., Sutton, T. A., Weinborn, M., Rainey-Smith, S. R., Brown, B., et al. (2016). Cerebral amyloid-β accumulation and deposition following traumatic brain injury–A narrative review and meta-analysis of animal studies. Neurosci. Biobehav. Rev. 64, 215–228. doi: 10.1016/j.neubiorev.2016.01.004
Bishop, G. M., and Robinson, S. R. (2002). The amyloid hypothesis: let sleeping dogmas lie? Neurobiol. Aging 23, 1101–1105. doi: 10.1016/S0197-4580(02)00050-7
Bishop, G. M., and Robinson, S. R. (2004a). Physiological roles of amyloid-beta and implications for its removal in Alzheimer's disease. Drugs Aging 21, 621–630. doi: 10.2165/00002512-200421100-00001
Bishop, G. M., and Robinson, S. R. (2004b). The amyloid paradox: amyloid-beta-metal complexes can be neurotoxic and neuroprotective. Brain Pathol. 14, 448–452. doi: 10.1111/j.1750-3639.2004.tb00089.x
Bitel, C. L., Kasinathan, C., Kaswala, R. H., Klein, W. L., and Frederikse, P. H. (2012). Amyloid-β and tau pathology of Alzheimer's disease induced by diabetes in a rabbit animal model. J. Alzheimers Dis. 32, 291–305. doi: 10.3233/JAD-2012-120571
Black, R. S., Sperling, R. A., Safirstein, B., Motter, R. N., Pallay, A., Nichols, A., et al. (2010). A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 24, 198–203. doi: 10.1097/WAD.0b013e3181c53b00
Blasko, I., Beer, R., Bigl, M., Apelt, J., Franz, G., Rudzki, D., et al. (2004). Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1). J. Neural Trans. 111, 523–536. doi: 10.1007/s00702-003-0095-6
Blockx, I., Einstein, S., Guns, P. J., Van Audekerke, J., Guglielmetti, C., Zago, W., et al. (2016). Monitoring blood-brain barrier integrity following amyloid-beta immunotherapy using gadolinium-enhanced MRI in a PDAPP mouse model. J. Alzheimers Dis. 54, 723–735. doi: 10.3233/JAD-160023
Boche, D., Donald, J., Love, S., Harris, S., Neal, J. W., Holmes, C., et al. (2010). Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol. 120, 13–20. doi: 10.1007/s00401-010-0705-y
Boelen, E., Stassen, F. R., van der Ven, A. J., Lemmens, M. A., Steinbusch, H. P., Bruggeman, C. A., et al. (2007). Detection of amyloid beta aggregates in the brain of BALB/c mice after Chlamydia pneumoniae infection. Acta Neuropathol. 114, 255–261. doi: 10.1007/s00401-007-0252-3
Bourgade, K., Dupuis, G., Frost, E. H., and Fülöp, T. S. (2016a). Anti-viral properties of amyloid-β peptides. J. Alzheimers Dis. 54, 859–878. doi: 10.3233/JAD-160517
Bourgade, K., Garneau, H., Giroux, G., Le Page, A. Y., Bocti, C., Dupuis, G., et al. (2015). β-Amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology 16, 85–98. doi: 10.1007/s10522-014-9538-8
Bourgade, K., Le Page, A., Bocti, C., Witkowski, J. M., Dupuis, G., Frost, E. H., et al. (2016b). Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimers Dis. 50, 1227–1241. doi: 10.3233/JAD-150652
Bowles, E. J. A., Walker, R. L., Anderson, M. L., Dublin, S., Crane, P. K., and Larson, E. B. (2017). Risk of Alzheimer's disease or dementia following a cancer diagnosis. PLoS ONE 12:e0179857. doi: 10.1371/journal.pone.0179857
Brody, D. L., Magnoni, S., Schwetye, K. E., Spinner, M. L., Esparza, T. J., Stocchetti, N., et al. (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224. doi: 10.1126/science.1161591
Bueche, C. Z., Hawkes, C., Garz, C., Vielhaber, S., Attems, J., Knight, R. T., et al. (2014). Hypertension drives parenchymal β-amyloid accumulation in the brain parenchyma. Ann. Clin. Transl. Neurol. 1, 124–129. doi: 10.1002/acn3.27
Buhimschi, I. A., Nayeri, U. A., Zhao, G., Shook, L. L., Pensalfini, A., Funai, E. F., et al. (2014). Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 6:245ra292. doi: 10.1126/scitranslmed.3008808
Busche, M. A., Grienberger, C., Keskin, A. D., Song, B., Neumann, U., Staufenbiel, M., et al. (2015). Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer's models. Nat. Neurosci. 18, 1725–1727. doi: 10.1038/nn.4163
Carnevale, D., Perrotta, M., Lembo, G., and Trimarco, B. (2016). Pathophysiological links among hypertension and Alzheimer's disease. High Blood Press. Cardiovasc. Prev. 23, 3–7. doi: 10.1007/s40292-015-0108-1
Catalá-López, F., Suárez-Pinilla, M., Suárez-Pinilla, P., Valderas, J. M., Gómez-Beneyto, M., Martinez, S., et al. (2014). Inverse and direct cancer comorbidity in people with central nervous system disorders: a meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother. Psychosom. 83, 89–105. doi: 10.1159/000356498
Charidimou, A., Martinez-Ramirez, S., Shoamanesh, A., Oliveira-Filho, J., Frosch, M., Vashkevich, A., et al. (2015). Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology 84, 1206–1212. doi: 10.1212/WNL.0000000000001398
Chen, X. H., Johnson, V. E., Uryu, K., Trojanowski, J. Q., and Smith, D. H. (2009). A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 19, 214–223. doi: 10.1111/j.1750-3639.2008.00176.x
Chou, C. H., Lee, J. T., Lin, C. C., Sung, Y. F., Muo, C. H., Yang, F. C., et al. (2017). Septicemia is associated with increased risk for dementia: a population-based longitudinal study. Oncotarget 8, 84300–84308. doi: 10.18632/oncotarget.20899
Chuang, J.-Y., Lee, C.-W., Shih, Y.-H., Yang, T., Yu, L., and Kuo, Y.-M. (2012). Interactions between amyloid-β and hemoglobin: implications for amyloid plaque formation in Alzheimer's disease. PLoS ONE 7:e33120. doi: 10.1371/journal.pone.0033120
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clarke, J., Thornell, A., Corbett, D., Soininen, H., Hiltunen, M., and Jolkkonen, J. (2007). Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur. J. Neurosci. 26, 1845–1852. doi: 10.1111/j.1460-9568.2007.05807.x
Coric, V., Salloway, S., van Dyck, C. H., Dubois, B., Andreasen, N., Brody, M., et al. (2015). Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 72, 1324–1333. doi: 10.1001/jamaneurol.2015.0607
Coric, V., van Dyck, C. H., Salloway, S., Andreasen, N., Brody, M., Richter, R. W., et al. (2012). Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 69, 1430–1440. doi: 10.1001/archneurol.2012.2194
Cullen, K. M., Kócsi, Z., and Stone, J. (2006). Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol. Aging 27, 1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016
D'Andrea, M. R. (2014). Bursting Neurons and Fading Memories: An Alternative Hypothesis of the Pathogenesis of Alzheimer's Disease, 1st Edn. New York, NY: Academic Press.
D'Andrea, M. R. (2016). Intracellular Consequences of Amyloid in Alzheimer's Disease. Amsterdam: Academic Press.
DiFrancesco, J. C., Longoni, M., and Piazza, F. (2015). Anti-Aβ autoantibodies in amyloid related imaging abnormalities (ARIA): candidate biomarker for immunotherapy in Alzheimer's disease and cerebral amyloid angiopathy. Front. Neurol. 6:207. doi: 10.3389/fneur.2015.00207
Dominguez, D., Tournoy, J., Hartmann, D., Huth, T., Cryns, K., Deforce, S., et al. (2005). Phenotypic and biochemical analysis of BACE1- and BACE2-deficient mice. J. Biol. Chem. 280, 30797–30806. doi: 10.1074/jbc.M505249200
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N. Engl. J. Med. 369, 341–350. doi: 10.1056/NEJMoa1210951
Dreses-Werringloer, U., Bhuiyan, M., Zhao, Y., Gérard, H. C., Whittum-Hudson, J. A., and Hudson, A. P. (2009). Initial characterization of Chlamydophila (Chlamydia) pneumoniae cultured from the late-onset Alzheimer brain. Int. J. Med. Microbiol. 299, 187–201. doi: 10.1016/j.ijmm.2008.07.002
Driver, J. A., Beiser, A., Au, R., Kreger, B. E., Splansky, G. L., Kurth, T., et al. (2012). Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. BMJ 344:e1442. doi: 10.1136/bmj.e1442
Dubessy, A.-L., Ursu, R., Maillet, D., Augier, A., Le Guilloux, J., Carpentier, A. F., et al. (2012). Superficial siderosis of the central nervous system: a rare cause of dementia with therapeutic consequences. Age Ageing 41, 275–277. doi: 10.1093/ageing/afr177
Dueholm, M. S., Otzen, D., and Nielsen, P. H. (2013). Evolutionary insight into the functional amyloids of the pseudomonads. PLoS ONE 8:e76630. doi: 10.1371/journal.pone.0076630
Edrey, Y. H., Medina, D. X., Gaczynska, M., Osmulski, P. A., Oddo, S., Caccamo, A., et al. (2013). Amyloid β and the longest-lived rodent: the naked mole-rat as a model for natural protection from Alzheimer's disease. Neurobiol. Aging 34, 2352–2360. doi: 10.1016/j.neurobiolaging.2013.03.032
Eli Lilly and Company (2008). Effect of LY450139 on the Long Term Progression of Alzheimer's Disease.
Fedele, E., Rivera, D., Marengo, B., Pronzato, M. A., and Ricciarelli, R. (2015). Amyloid β: walking on the dark side of the moon. Mech. Ageing Dev. 152, 1–4. doi: 10.1016/j.mad.2015.09.001
Ferguson, B., Matyszak, M. K., Esiri, M. M., and Perry, V. H. (1997). Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3), 393–399. doi: 10.1093/brain/120.3.393
Ferrer, I., Boada Rovira, M., Sánchez Guerra, M. L., Rey, M. J., and Costa-Jussá, F. (2004). Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 14, 11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x
Ferrero, J., Williams, L., Stella, H., Leitermann, K., Mikulskis, A., O'Gorman, J., et al. (2016). First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer's disease. Alzheimers Dement. 2, 169–176. doi: 10.1016/j.trci.2016.06.002
Fleisher, A. S., Raman, R., Siemers, E. R., Becerra, L., Clark, C. M., Dean, R. A., et al. (2008). Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch. Neurol. 65, 1031–1038. doi: 10.1001/archneur.65.8.1031
Fox, N. C., Black, R. S., Gilman, S., Rossor, M. N., Griffith, S. G., Jenkins, L., et al. (2005). Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64, 1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99
Frain, L., Swanson, D., Cho, K., Gagnon, D., Lu, K. P., Betensky, R. A., et al. (2017). Association of cancer and Alzheimer's disease risk in a national cohort of veterans. Alzheimers Dement. 13, 1364–1370. doi: 10.1016/j.jalz.2017.04.012
Freedman, D. M., Wu, J., Chen, H., Kuncl, R. W., Enewold, L. R., Engels, E. A., et al. (2016). Associations between cancer and Alzheimer's disease in a U.S. Medicare population. Cancer Med. 5, 2965–2976. doi: 10.1002/cam4.850
Garcia-Alloza, M., Gregory, J., Kuchibhotla, K. V., Fine, S., Wei, Y., Ayata, C., et al. (2011). Cerebrovascular lesions induce transient β-amyloid deposition. Brain 134(Pt 12), 3697–3707. doi: 10.1093/brain/awr300
Garcia-Osta, A., and Alberini, C. M. (2009). Amyloid beta mediates memory formation. Learn. Mem. 16, 267–272. doi: 10.1101/lm.1310209
Garvey, M., Meehan, S., Gras, S. L., Schirra, H. J., Craik, D. J., Van der Weerden, N. L., et al. (2013). A radish seed antifungal peptide with a high amyloid fibril-forming propensity. Biochim. Biophys. Acta 1834, 1615–1623. doi: 10.1016/j.bbapap.2013.04.030
Gauthier, S., Aisen, P. S., Ferris, S. H., Saumier, D., Duong, A., Haine, D., et al. (2009). Effect of tramiprosate in patients with mild-to-moderate Alzheimer's disease: exploratory analyses of the MRI sub-group of the Alphase study. J. Nutr. Health Aging 13, 550–557. doi: 10.1007/s12603-009-0106-x
Gentile, M. T., Poulet, R., Di Pardo, A., Cifelli, G., Maffei, A., Vecchione, C., et al. (2009). Beta-amyloid deposition in brain is enhanced in mouse models of arterial hypertension. Neurobiol. Aging 30, 222–228. doi: 10.1016/j.neurobiolaging.2007.06.005
Gérard, H. C., Dreses-Werringloer, U., Wildt, K. S., Deka, S., Oszust, C., Balin, B. J., et al. (2006). Chlamydophila (Chlamydia) pneumoniae in the Alzheimer's brain. FEMS Immunol. Med. Microbiol. 48, 355–366. doi: 10.1111/j.1574-695X.2006.00154.x
Gilman, S., Koller, M., Black, R. S., Jenkins, L., Griffith, S. G., Fox, N. C., et al. (2005). Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C
Gisslén, M., Krut, J., Andreasson, U., Blennow, K., Cinque, P., Brew, B. J., et al. (2009). Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol. 9:63. doi: 10.1186/1471-2377-9-63
Gosztyla, M. L., Brothers, H. M., and Robinson, S. R. (2018). Alzheimer's amyloid-β is an antimicrobial peptide: a review of the evidence. J. Alzheimers Dis. 62, 1495–1506. doi: 10.3233/JAD-171133
Graham, D. I., Gentleman, S. M., Lynch, A., and Roberts, G. W. (1995). Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol. Appl. Neurobiol. 21, 27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x
Grant, J. L., Ghosn, E. E. B., Axtell, R. C., Herges, K., Kuipers, H. F., Woodling, N. S., et al. (2012). Reversal of paralysis and reduced inflammation from peripheral administration of ß -amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med. 4:145ra105. doi: 10.1126/scitranslmed.3004145
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., et al. (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564. doi: 10.1001/jama.2009.1866
Hammond, C. J., Hallock, L. R., Howanski, R. J., Appelt, D. M., Little, C. S., and Balin, B. J. (2010). Immunohistological detection of Chlamydia pneumoniae in the Alzheimer's disease brain. BMC Neurosci. 11:121. doi: 10.1186/1471-2202-11-121
Han, M. H., Hwang, S.-I., Roy, D. B., Lundgren, D. H., Price, J. V., Ousman, S. S., et al. (2008). Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081. doi: 10.1038/nature06559
Hanson, H. A., Horn, K. P., Rasmussen, K. M., Hoffman, J. M., and Smith, K. R. (2017). Is cancer protective for subsequent Alzheimer's disease risk? Evidence from the Utah population database. J. Gerontol. B Psychol. Sci. Soc. Sci. 72, 1032–1043. doi: 10.1093/geronb/gbw040
Ho, L., Qin, W., Pompl, P. N., Xiang, Z., Wang, J., Zhao, Z., et al. (2004). Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 18, 902–904. doi: 10.1096/fj.03-0978fje
Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., et al. (2008). Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223. doi: 10.1016/S0140-6736(08)61075-2
Hu, C., Adedokun, O., Ito, K., Raje, S., and Lu, M. (2015). Confirmatory population pharmacokinetic analysis for bapineuzumab phase 3 studies in patients with mild to moderate Alzheimer's disease. J. Clin. Pharmacol. 55, 221–229. doi: 10.1002/jcph.393
Hu, X., Zhou, X., He, W., Yang, J., Xiong, W., Wong, P., et al. (2010). BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010
Hung, V. K., Yeung, P. K., Lai, A. K., Ho, M. C., Lo, A. C., Chan, K. C., et al. (2015). Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. J. Cereb. Blood Flow Metab. 35, 1687–1696 doi: 10.1038/jcbfm.2015.109
Ida, N., Hartmann, T., Pantel, J., Schröder, J., Zerfass, R., Förstl, H., et al. (1996). Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J. Biol. Chem. 271, 22908–22914. doi: 10.1074/jbc.271.37.22908
Ishida, N., Ishihara, Y., Ishida, K., Tada, H., Funaki-Kato, Y., Hagiwara, M., et al. (2017). Periodontitis induced by bacterial infection exacerbates features of Alzheimer's disease in transgenic mice. NPJ Aging Mech. Dis. 3, 15. doi: 10.1038/s41514-017-0015-x
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer's disease: increasing evidence for a major role of the virus. Front. Aging Neurosci. 6, 202–202. doi: 10.3389/fnagi.2014.00202
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer's Disease. J. Alzheimers Dis. 51, 979–984. doi: 10.3233/JAD-160152
Jesse, S., Steinacker, P., Lehnert, S., Sdzuj, M., Cepek, L., Tumani, H., et al. (2010). A proteomic approach for the diagnosis of bacterial meningitis. PLoS ONE 5:e10079. doi: 10.1371/journal.pone.0010079
Johnson, V. E., Stewart, W., and Smith, D. H. (2010). Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370. doi: 10.1038/nrn2808
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Joseph-Mathurin, N., Dorieux, O., Trouche, S. G., Boutajangout, A., Kraska, A., Fontès, P., et al. (2013). Amyloid beta immunization worsens iron deposits in the choroid plexus and cerebral microbleeds. Neurobiol. Aging 34, 2613–2622. doi: 10.1016/j.neurobiolaging.2013.05.013
Kagan, B. L., Jang, H., Capone, R., Teran Arce, F., Ramachandran, S., Lal, R., et al. (2012). Antimicrobial properties of amyloid peptides. Mol. Pharm. 9, 708–717. doi: 10.1021/mp200419b
Kalkunte, S. S., Neubeck, S., Norris, W. E., Cheng, S.-B., Kostadinov, S., Vu Hoang, D., et al. (2013). Transthyretin is dysregulated in preeclampsia, and its native form prevents the onset of disease in a preclinical mouse model. Am. J. Pathol. 183, 1425–1436. doi: 10.1016/j.ajpath.2013.07.022
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kazim, S. F., Chuang, S. C., Zhao, W., Wong, R. K., Bianchi, R., and Iqbal, K. (2017). Early-onset network hyperexcitability in presymptomatic Alzheimer's disease transgenic mice is suppressed by passive immunization with anti-human APP/Abeta antibody and by mGluR5 blockade. Front. Aging Neurosci. 9:71. doi: 10.3389/fnagi.2017.00071
Ketter, N., Brashear, H. R., Bogert, J., Di, J., Miaux, Y., Gass, A., et al. (2017). Central review of amyloid-related imaging abnormalities in two phase III clinical trials of bapineuzumab in mild-to-moderate Alzheimer's Disease patients. J. Alzheimers Dis. 57, 557–573. doi: 10.3233/JAD-160216
Koike, M. A., Lin, A. J., Pham, J., Nguyen, E., Yeh, J. J., Rahimian, R., et al. (2012). APP knockout mice experience acute mortality as the result of ischemia. PLoS ONE 7:e42665. doi: 10.1371/journal.pone.0042665
Kokiko-Cochran, O. N., Ransohoff, L., Veenstra, M., Lee, S., Saber, M., Sikora, M., et al. (2016). Altered neuroinflammation and behavior following traumatic brain injury in a mouse model of Alzheimer's disease. J. Neurotrauma 33, 625–640. doi: 10.1089/neu.2015.3970
Kokjohn, T. A., and Roher, A. E. (2009). Antibody responses, amyloid-beta peptide remnants and clinical effects of AN-1792 immunization in patients with AD in an interrupted trial. CNS Neurol. Disord. Drug Targets 8, 88–97. doi: 10.2174/187152709787847315
Koppensteiner, P., Trinchese, F., Fà, M., Puzzo, D., Gulisano, W., Yan, S., et al. (2016). Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Abeta42: an early index of Alzheimer's disease. Sci. Rep. 6:32553. doi: 10.1038/srep32553
Krut, J. J., Zetterberg, H., Blennow, K., Cinque, P., Hagberg, L., Price, R. W., et al. (2013). Cerebrospinal fluid Alzheimer's biomarker profiles in CNS infections. J. Neurol. 260, 620–626. doi: 10.1007/s00415-012-6688-y
Kumar, D. K. V., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 8:340ra372. doi: 10.1126/scitranslmed.aaf1059
Lannfelt, L., Relkin, N. R., and Siemers, E. R. (2014). Amyloid-ß-directed immunotherapy for Alzheimer's disease. J. Intern. Med. 275, 284–295. doi: 10.1111/joim.12168
Little, C. S., Hammond, C. J., MacIntyre, A., Balin, B. J., and Appelt, D. M. (2004). Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol. Aging 25, 419–429. doi: 10.1016/S0197-4580(03)00127-1
Little, C. S., Joyce, T. A., Hammond, C. J., Matta, H., Cahn, D., Appelt, D. M., et al. (2014). Detection of bacterial antigens and Alzheimer's disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 6, 304–304. doi: 10.3389/fnagi.2014.00304
Lukiw, W. J., Cui, J. G., Yuan, L. Y., Bhattacharjee, P. S., Corkern, M., Clement, C., et al. (2010). Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport 21, 922–927. doi: 10.1097/WNR.0b013e32833da51a
Luna, S., Cameron, D. J., and Ethell, D. W. (2013). Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS ONE 8:e75052. doi: 10.1371/journal.pone.0075052
Ma, K., Thomason, L. A., and McLaurin, J. (2012). scyllo-Inositol, preclinical, and clinical data for Alzheimer's disease. Adv. Pharmacol. 64, 177–212. doi: 10.1016/B978-0-12-394816-8.00006-4
Ma, L.-L., Yu, J.-T., Wang, H.-F., Meng, X.-F., Tan, C.-C., Wang, C., et al. (2014). Association between cancer and Alzheimer's disease: systematic review and meta-analysis. J. Alzheimer's Dis. 42, 565–573. doi: 10.3233/JAD-140168
Macfarlane, D. P., Nicoll, J. A., Smith, C., and Graham, D. I. (1999). APOE epsilon4 allele and amyloid beta-protein deposition in long term survivors of head injury. Neuroreport 10, 3945–3948. doi: 10.1097/00001756-199912160-00040
Magnoni, S., Esparza, T. J., Conte, V., Carbonara, M., Carrabba, G., Holtzman, D. M., et al. (2012). Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 135(Pt 4), 1268–1280. doi: 10.1093/brain/awr286
Malek-Ahmadi, M., Beach, T., Obradov, A., Sue, L., Belden, C., Davis, K., et al. (2013). Increased Alzheimer's disease neuropathology is associated with type 2 diabetes and ApoE ε.4 carrier status. Curr. Alzheimer Res. 10, 654–659. doi: 10.2174/15672050113109990006
Mannix, R. C., Zhang, J., Berglass, J., Qui, J., and Whalen, M. J. (2013). Beneficial effect of amyloid beta after controlled cortical impact. Brain Injury 27, 743–748. doi: 10.3109/02699052.2013.771797
Mannix, R. C., Zhang, J., Park, J., Lee, C., and Whalen, M. J. (2011a). Detrimental effect of genetic inhibition of B-site APP-cleaving enzyme 1 on functional outcome after controlled cortical impact in young adult mice. J. Neurotrauma 28, 1855–1861. doi: 10.1089/neu.2011.1759
Mannix, R. C., Zhang, J., Park, J., Zhang, X., Bilal, K., Walker, K., et al. (2011b). Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 31, 351–361. doi: 10.1038/jcbfm.2010.99
Masliah, E., Hansen, L., Adame, A., Crews, L., Bard, F., Lee, C., et al. (2005). Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64, 129–131. doi: 10.1212/01.WNL.0000148590.39911.DF
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006262
Mattsson, N., Bremell, D., Anckarsäter, R., Blennow, K., Anckarsäter, H., Zetterberg, H., et al. (2010). Neuroinflammation in Lyme neuroborreliosis affects amyloid metabolism. BMC Neurol. 10:51. doi: 10.1186/1471-2377-10-51
Mauri, M., Sinforiani, E., Bono, G., Cittadella, R., Quattrone, A., Boller, F., et al. (2006). Interaction between apolipoprotein epsilon 4 and traumatic brain injury in patients with Alzheimer's disease and mild cognitive impairment. Funct. Neurol. 21, 223–228.
McKenzie, K. J., McLellan, D. R., Gentleman, S. M., Maxwell, W. L., Gennarelli, T. A., and Graham, D. I. (1996). Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol. 92, 608–613. doi: 10.1007/s004010050568
Miklossy, J. (2015). Historic evidence to support a causal relationship between spirochetal infections and Alzheimer's disease. Front. Aging Neurosci. 7, 46–46. doi: 10.3389/fnagi.2015.00046
Miklossy, J. (2016). Bacterial Amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimer's Dis. 53, 1459–1473. doi: 10.3233/JAD-160451
Mo, J.-A., Lim, J.-H., Sul, A.-R., Lee, M., Youn, Y. C., and Kim, H.-J. (2015). Cerebrospinal fluid β-amyloid1-42 levels in the differential diagnosis of Alzheimer's disease–systematic review and meta-analysis. PLoS ONE 10:e0116802. doi: 10.1371/journal.pone.0116802
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Morley, J. E., and Farr, S. A. (2014). The role of amyloid-beta in the regulation of memory. Biochem. Pharmacol. 88, 479–485. doi: 10.1016/j.bcp.2013.12.018
Morley, J. E., Farr, S. A., Banks, W. A., Johnson, S. N., Yamada, K. A., and Xu, L. (2010). A physiological role for amyloid-beta protein:enhancement of learning and memory. J. Alzheimers. Dis. 19, 441–449. doi: 10.3233/JAD-2010-1230
Nicoll, J. A., Wilkinson, D., Holmes, C., Steart, P., Markham, H., and Weller, R. O. (2003). Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat. Med. 9, 448–452. doi: 10.1038/nm840
Nicoll, J. A., Roberts, G. W., and Graham, D. I. (1995). Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat. Med. 1, 135–137. doi: 10.1038/nm0295-135
Orgogozo, J. M., Gilman, S., Dartigues, J. F., Laurent, B., Puel, M., Kirby, L. C., et al. (2003). Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61, 46–54. doi: 10.1212/01.WNL.0000073623.84147.A8
Ostrowitzki, S., Deptula, D., Thurfjell, L., Barkhof, F., Bohrmann, B., Brooks, D. J., et al. (2012). Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 69, 198–207. doi: 10.1001/archneurol.2011.1538
Ostrowitzki, S., Lasser, R. A., Dorflinger, E., Scheltens, P., Barkhof, F., Nikolcheva, T., et al. (2017). A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers. Res. Ther. 9:95. doi: 10.1186/s13195-017-0318-y
Oyama, R., Yamamoto, H., and Titani, K. (2000). Glutamine synthetase, hemoglobin alpha-chain, and macrophage migration inhibitory factor binding to amyloid β-protein: their identification in rat brain by a novel affinity chromatography and in Alzheimer's disease brain by immunoprecipitation. Biochim. Biophys. Acta 1479, 91–102. doi: 10.1016/S0167-4838(00)00057-1
Pajoohesh-Ganji, A., Burns, M. P., Pal-Ghosh, S., Tadvalkar, G., Hokenbury, N. G., Stepp, M. A., et al. (2014). Inhibition of amyloid precursor protein secretases reduces recovery after spinal cord injury. Brain Res. 1560, 73–82. doi: 10.1016/j.brainres.2014.02.049
Paris, D., Ganey, N., Banasiak, M., Laporte, V., Patel, N., Mullan, M., et al. (2010). Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer's disease. J. Neurosci. 30, 11251–11258. doi: 10.1523/JNEUROSCI.2586-10.2010
Paris, D., Quadros, A., Patel, N., DelleDonne, A., Humphrey, J., and Mullan, M. (2005). Inhibition of angiogenesis and tumor growth by beta and gamma-secretase inhibitors. Eur. J. Pharmacol. 514, 1–15. doi: 10.1016/j.ejphar.2005.02.050
Paris, D., Townsend, K., Quadros, A., Humphrey, J., Sun, J., Brem, S., et al. (2004). Inhibition of angiogenesis by Abeta peptides. Angiogenesis 7, 75–85. doi: 10.1023/B:AGEN.0000037335.17717.bf
Parkin, D. M. (2006). The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118, 3030–3044. doi: 10.1002/ijc.21731
Patton, R. L., Kalback, W. M., Esh, C. L., Kokjohn, T. A., Van Vickle, G. D., Luehrs, D. C., et al. (2006). Amyloid-β peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am. J. Pathol. 169, 1048–1063. doi: 10.2353/ajpath.2006.060269
Penninkilampi, R., Brothers, H. M., and Eslick, G. D. (2016). Pharmacological agents targeting γ-secretase increase risk of cancer and cognitive decline in Alzheimer's disease patients: a systematic review and meta-analysis. J. Alzheimers. Dis. 53, 1395–1404. doi: 10.3233/JAD-160275
Penninkilampi, R., Brothers, H. M., and Eslick, G. D. (2017). Safety and efficacy of anti-amyloid-β Immunotherapy in Alzheimer's disease: a systematic review and meta-analysis. J. Neuroimmune Pharmacol. 12, 194–203. doi: 10.1007/s11481-016-9722-5
Pfizer (2013). A Long-Term Safety and Tolerability Study of Bapineuzumab in Alzheimer Disease Patients.
Pisa, D., Alonso, R., Juarranz, A., Rábano, A., and Carrasco, L. (2015a). Direct visualization of fungal infection in brains from patients with Alzheimer's disease. J. Alzheimer's Dis. 43, 613–624. doi: 10.3233/JAD-141386
Pisa, D., Alonso, R., Rábano, A., Rodal, I., and Carrasco, L. (2015b). Different brain regions are infected with fungi in Alzheimer's disease. Sci. Rep. 5:15015. doi: 10.1038/srep15015
Plant, L. D., Boyle, J. P., Smith, I. F., Peers, C., and Pearson, H. A. (2003). The production of amyloid β peptide is a critical requirement for the viability of central neurons. J. Neurosci. 23, 5531–5535. doi: 10.1523/JNEUROSCI.23-13-05531.2003
Puig, K. L., and Combs, C. K. (2013). Expression and function of APP and its metabolites outside the central nervous system. Exp. Gerontol. 48, 608–611. doi: 10.1016/j.exger.2012.07.009
Purushothuman, S., Marotte, L., Stowe, S., Johnstone, D. M., and Stone, J. (2013). The response of cerebral cortex to haemorrhagic damage: experimental evidence from a penetrating injury model. PLoS ONE 8:e59740. doi: 10.1371/journal.pone.0059740
Puzzo, D., and Arancio, O. (2013). Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimer's Dis. 33 (Suppl. 1), S111–S120. doi: 10.3233/JAD-2012-129033
Puzzo, D., Gulisano, W., Arancio, O., and Palmeri, A. (2015). The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience 307, 26–36. doi: 10.1016/j.neuroscience.2015.08.039
Puzzo, D., Privitera, L., Fa, M., Staniszewski, A., Hashimoto, G., Aziz, F., et al. (2011). Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830. doi: 10.1002/ana.22313
Ravi, L. B., Mohanty, J. G., Chrest, F. J., Jayakumar, R., Nagababu, E., Usatyuk, P. V., et al. (2004). Influence of beta-amyloid fibrils on the interactions between red blood cells and endothelial cells. Neurol. Res. 26, 579–585. doi: 10.1179/016164104225016227
Ricciarelli, R., and Fedele, E. (2017). The amyloid cascade hypothesis in alzheimer's disease: it's time to change our mind. Curr. Neuropharmacol. 15, 926–935. doi: 10.2174/1570159X15666170116143743
Rinne, J. O., Brooks, D. J., Rossor, M. N., Fox, N. C., Bullock, R., Klunk, W. E., et al. (2010). 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet. Neurol. 9, 363–372. doi: 10.1016/S1474-4422(10)70043-0
Roberts, G. W., Gentleman, S. M., Lynch, A., Murray, L., Landon, M., and Graham, D. I. (1994). Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatr. 57, 419–425. doi: 10.1136/jnnp.57.4.419
Robinson, S. R., and Bishop, G. M. (2002). Aβ as a bioflocculant: implications for the amyloid hypothesis of Alzheimer's disease. Neurobiol. Aging 23, 1051–1072. doi: 10.1016/S0197-4580(01)00342-6
Robinson, S. R., Bishop, G. M., and Münch, G. (2003). Alzheimer vaccine: amyloid-beta on trial. BioEssays 25, 283–288. doi: 10.1002/bies.10236
Robinson, S. R., Bishop, G. M., Lee, H. G., and Münch, G. (2004a). Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol. Aging 25, 609–615. doi: 10.1016/j.neurobiolaging.2003.12.020
Robinson, S. R., Dobson, C., and Lyons, J. (2004b). Challenges and directions for the pathogen hypothesis of Alzheimer's disease. Neurobiol. Aging 25, 629–637. doi: 10.1016/j.neurobiolaging.2003.12.022
Roe, C. M., Fitzpatrick, A. L., Xiong, C., Sieh, W., Kuller, L., Miller, J. P., et al. (2010). Cancer linked to Alzheimer disease but not vascular dementia. Neurology 74, 106–112. doi: 10.1212/WNL.0b013e3181c91873
Roperch, J. P., Alvaro, V., Prieur, S., Tuynder, M., Nemani, M., Lethrosne, F., et al. (1998). Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat. Med. 4, 835–838. doi: 10.1038/nm0798-835
Salloway, S., Sperling, R., Gilman, S., Fox, N. C., Blennow, K., Raskind, M., et al. (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070. doi: 10.1212/WNL.0b013e3181c67808
Salloway, S., Sperling, R., Keren, R., Porsteinsson, A. P., van Dyck, C. H., Tariot, P. N., et al. (2011). A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 77, 1253–1262. doi: 10.1212/WNL.0b013e3182309fa5
Saumier, D., Duong, A., Haine, D., Garceau, D., and Sampalis, J. (2009). Domain-specific cognitive effects of tramiprosate in patients with mild to moderate Alzheimer's disease: ADAS-cog subscale results from the alphase study. J. Nutr. Health Aging 13, 808–812. doi: 10.1007/s12603-009-0217-4
Savage, M. J., Trusko, S. P., Howland, D. S., Pinsker, L. R., Mistretta, S., Reaume, A. G., et al. (1998). Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 18, 1743–1752. doi: 10.1523/JNEUROSCI.18-05-01743.1998
Schneeberger, A., Mandler, M., Otawa, O., Zauner, W., Mattner, F., and Schmidt, W. (2009). Development of AFFITOPE vaccines for Alzheimer's disease (AD)–from concept to clinical testing. J. Nutr. Health Aging 13, 264–267. doi: 10.1007/s12603-009-0070-5
Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., et al. (2014). Clinical trials and late-stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283. doi: 10.1111/joim.12191
Schreiber, S., Drukarch, B., Garz, C., Niklass, S., Stanaszek, L., Kropf, S., et al. (2014). Interplay between age, cerebral small vessel disease, parenchymal amyloid-β, and tau pathology: longitudinal studies in hypertensive stroke-prone rats. J. Alzheimer's Dis. 42(Suppl. 3), S205–S215. doi: 10.3233/JAD-132618.
Scott, G., Ramlackhansingh, A. F., Edison, P., Hellyer, P., Cole, J., Veronese, M., et al. (2016). Amyloid pathology and axonal injury after brain trauma. Neurology 86, 821–828. doi: 10.1212/WNL.0000000000002413
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature 537, 50–56. doi: 10.1038/nature19323
Shi, H.-B., Tang, B., Liu, Y.-W., Wang, X.-F., and Chen, G.-J. (2015). Alzheimer disease and cancer risk: a meta-analysis. J. Cancer Res. Clin. Oncol. 141, 485–494. doi: 10.1007/s00432-014-1773-5
Shim, S. M., Cheon, H. S., Jo, C., Koh, Y. H., Song, J., and Jeon, J. P. (2017). Elevated epstein-barr virus antibody level is associated with cognitive decline in the korean elderly. J. Alzheimers. Dis. 55, 293–301. doi: 10.3233/JAD-160563
Shishido, H., Kishimoto, Y., Kawai, N., Toyota, Y., Ueno, M., Kubota, T., et al. (2016). Traumatic brain injury accelerates amyloid-β deposition and impairs spatial learning in the triple-transgenic mouse model of Alzheimer's disease. Neurosci. Lett. 629, 62–67. doi: 10.1016/j.neulet.2016.06.066
Siemers, E. R., Dean, R. A., Friedrich, S., Ferguson-Sells, L., Gonzales, C., Farlow, M. R., et al. (2007). Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin. Neuropharmacol. 30, 317–325. doi: 10.1097/WNF.0b013e31805b7660
Siemers, E. R., Quinn, J. F., Kaye, J., Farlow, M. R., Porsteinsson, A., Tariot, P., et al. (2006). Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology 66, 602–604. doi: 10.1212/01.WNL.0000198762.41312.E1
Sjögren, M., Gisslén, M., Vanmechelen, E., and Blennow, K. (2001). Low cerebrospinal fluid beta-amyloid 42 in patients with acute bacterial meningitis and normalization after treatment. Neurosci. Lett. 314, 33–36. doi: 10.1016/S0304-3940(01)02285-6
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 5:e9505. doi: 10.1371/journal.pone.0009505
Sperling, R., Salloway, S., Brooks, D. J., Tampieri, D., Barakos, J., Fox, N. C., et al. (2012). Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet. Neurol. 11, 241–249. doi: 10.1016/S1474-4422(12)70015-7
Spitzer, P., Condic, M., Herrmann, M., Oberstein, T. J., Scharin-Mehlmann, M., Gilbert, D. F., et al. (2016). Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 6:32228. doi: 10.1038/srep32228
Steinbach, J. P., Müller, U., Leist, M., Li, Z. W., Nicotera, P., and Aguzzi, A. (1998). Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ. 5, 858–866. doi: 10.1038/sj.cdd.4400391
Stone, J. (2008). What initiates the formation of senile plaques? The origin of Alzheimer-like dementias in capillary haemorrhages. Med Hypotheses 71, 347–359. doi: 10.1016/j.mehy.2008.04.007
Sudduth, T. L., Weekman, E. M., Brothers, H. M., Braun, K., and Wilcock, D. M. (2014). β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimer's Res. Ther. 6, 32–32. doi: 10.1186/alzrt262
Tajiri, N., Kellogg, S. L., Shimizu, T., Arendash, G. W., and Borlongan, C. V. (2013). Traumatic brain injury precipitates cognitive impairment and extracellular Aβ aggregation in Alzheimer's disease transgenic mice. PLoS ONE 8:e78851. doi: 10.1371/journal.pone.0078851
Tampellini, D., Rahman, N., Gallo, E. F., Huang, Z., Dumont, M., Capetillo-Zarate, E., et al. (2009). Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J. Neurosci. 29, 9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009
Tanaka, S., and Nagashima, H. (2017). Establishment of an Alzheimer's disease model with latent herpesvirus infection using PS2 and Tg2576 double transgenic mice. Exp. Anim. doi: 10.1538/expanim.17-0066. [Epub ahead of print].
Tarawneh, R., and Holtzman, D. M. (2009). Critical issues for successful immunotherapy in Alzheimer's disease: development of biomarkers and methods for early detection and intervention. CNS Neurol. Disord. Drug Targets 8, 144–159. doi: 10.2174/187152709787847324
Tharp, W. G., and Sarkar, I. N. (2013). Origins of amyloid-β. BMC Genomics 14:290. doi: 10.1186/1471-2164-14-290
Torres, L., Robinson, S. A., Kim, D. G., Yan, A., Cleland, T. A., and Bynoe, M. S. (2018). Toxoplasma gondii alters NMDAR signaling and induces signs of Alzheimer's disease in wild-type, C57BL/6 mice. J. Neuroinflammation 15:57. doi: 10.1186/s12974-018-1086-8
Transition Therapeutics Ireland Limited (2007). ELND005 in Patients With Mild to Moderate Alzheimer's Disease.
Transition Therapeutics Ireland Limited (2009). [ELND005 Long-Term Follow-up Study in Subjects With Alzheimer's Disease.
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., and Mörk, S., and, B.ö, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Tu, S., Okamoto, S.-I., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Vellas, B., Black, R., Thal, L. J., Fox, N. C., Daniels, M., McLennan, G., et al. (2009). Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr. Alzheimer Res. 6, 144–151. doi: 10.2174/156720509787602852
Villar-Piqué, A., Sabaté, R., Lopera, O., Gibert, J., Torne, J. M., Santos, M., et al. (2010). Amyloid-like protein inclusions in tobacco transgenic plants. PLoS ONE 5:e13625. doi: 10.1371/journal.pone.0013625
Wang, L., Xi, G., Keep, R. F., and Hua, Y. (2012). Iron enhances the neurotoxicity of amyloid β. Transl. Stroke Res. 3, 107–113. doi: 10.1007/s12975-011-0099-8
Washington, P. M., Morffy, N., Parsadanian, M., Zapple, D. N., and Burns, M. P. (2014). Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-β in an Alzheimer's disease mouse model. J. Neurotrauma 31, 125–134. doi: 10.1089/neu.2013.3017
White, M. R., Kandel, R., Hsieh, I. N., De Luna, X., and Hartshorn, K. L. (2018). Critical role of the C-terminal residues of the Alzheimer's associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS ONE 13(3):e0194001. doi: 10.1371/journal.pone.0194001
White, M. R., Kandel, R., Tripathi, S., Condon, D., Qi, L., Taubenberger, J., et al. (2014). Alzheimer's associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS ONE 9:e101364. doi: 10.1371/journal.pone.0101364
White, R. S., Lipton, R. B., Hall, C. B., and Steinerman, J. R. (2013). Nonmelanoma skin cancer is associated with reduced Alzheimer disease risk. Neurology 80, 1966–1972. doi: 10.1212/WNL.0b013e3182941990
Wollenweber, F. A., Buerger, K., Mueller, C., Ertl-Wagner, B., Malik, R., Dichgans, M., et al. (2014). Prevalence of cortical superficial siderosis in patients with cognitive impairment. J. Neurol. 261, 277–282. doi: 10.1007/s00415-013-7181-y
Wozniak, M. A., Itzhaki, R. F., Shipley, S. J., and Dobson, C. B. (2007). Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci. Lett. 429, 95–100. doi: 10.1016/j.neulet.2007.09.077
Wozniak, M. A., Mee, A. P., and Itzhaki, R. F. (2009). Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J. Pathol. 217, 131–138. doi: 10.1002/path.2449
Wu, C.-W., Liao, P.-C., Yu, L., Wang, S.-T., Chen, S.-T., Wu, C.-M., et al. (2004). Hemoglobin promotes Abeta oligomer formation and localizes in neurons and amyloid deposits. Neurobiol. Dis. 17, 367–377. doi: 10.1016/j.nbd.2004.08.014
Xiang, Y., Bu, X. L., Liu, Y. H., Zhu, C., Shen, L. L., Jiao, S. S., et al. (2015). Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 130, 487–499. doi: 10.1007/s00401-015-1477-1
Xu, S., and Gaskin, F. (1997). Increased incidence of anti-beta-amyloid autoantibodies secreted by Epstein-Barr virus transformed B cell lines from patients with Alzheimer's disease. Mech. Ageing Dev. 94, 213–222. doi: 10.1016/S0047-6374(96)01861-1
Yarchoan, M., James, B. D., Shah, R. C., Arvanitakis, Z., Wilson, R. S., Schneider, J., et al. (2017). Association of cancer history with Alzheimer's disease dementia and neuropathology. J. Alzheimers. Dis. 56, 699–706. doi: 10.3233/JAD-160977
Zhao, H., Zhu, J., Cui, K., Xu, X., O'Brien, M., Wong, K. K., et al. (2009). Bioluminescence imaging reveals inhibition of tumor cell proliferation by Alzheimer's amyloid beta protein. Cancer Cell Int. 9:15. doi: 10.1186/1475-2867-9-15
Keywords: infection, antimicrobial, cancer, traumatic injury, cerebrovascular, immune system, seizure, ARIA
Citation: Brothers HM, Gosztyla ML and Robinson SR (2018) The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer's Disease. Front. Aging Neurosci. 10:118. doi: 10.3389/fnagi.2018.00118
Received: 09 February 2018; Accepted: 06 April 2018;
Published: 25 April 2018.
Edited by:
Rommy Von Bernhardi, Pontificia Universidad Católica de Chile, ChileReviewed by:
Walter E. Müller, Goethe University Frankfurt, GermanyRichard Lathe, University of Edinburgh, United Kingdom
Copyright © 2018 Brothers, Gosztyla and Robinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen R. Robinson, c3RlcGhlbi5yb2JpbnNvbkBybWl0LmVkdS5hdQ==