
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci., 03 November 2017
Sec. Neuroinflammation and Neuropathy
Volume 9 - 2017 | https://doi.org/10.3389/fnagi.2017.00365
This article is part of the Research TopicHormones and Neural Aging: Lessons from Experimental ModelsView all 12 articles
 Jose L. Labandeira-Garcia1,2*
Jose L. Labandeira-Garcia1,2* Maria A. Costa-Besada1,2
Maria A. Costa-Besada1,2 Carmen M. Labandeira3
Carmen M. Labandeira3 Begoña Villar-Cheda1,2
Begoña Villar-Cheda1,2 Ana I. Rodríguez-Perez1,2
Ana I. Rodríguez-Perez1,2Insulin-like growth factor-1 (IGF-1) effects on aging and neurodegeneration is still controversial. However, it is widely admitted that IGF-1 is involved in the neuroinflammatory response. In peripheral tissues, several studies showed that IGF-1 inhibited the expression of inflammatory markers, although other studies concluded that IGF-1 has proinflammatory functions. Furthermore, proinflammatory cytokines such as TNF-α impaired IGF-1 signaling. In the brain, there are controversial results on effects of IGF-1 in neuroinflammation. In addition to direct protective effects on neurons, several studies revealed anti-inflammatory effects of IGF-1 acting on astrocytes and microglia, and that IGF-1 may also inhibit blood brain barrier permeability. Altogether suggests that the aging-related decrease in IGF-1 levels may contribute to the aging-related pro-inflammatory state. IGF-1 inhibits the astrocytic response to inflammatory stimuli, and modulates microglial phenotype (IGF-1 promotes the microglial M2 and inhibits of M1 phenotype). Furthermore, IGF-1 is mitogenic for microglia. IGF-1 and estrogen interact to modulate the neuroinflammatory response and microglial and astrocytic phenotypes. Brain renin-angiotensin and IGF-1 systems also interact to modulate neuroinflammation. Induction of microglial IGF-1 by angiotensin, and possibly by other pro-inflammatory inducers, plays a major role in the repression of the M1 microglial neurotoxic phenotype and the enhancement of the transition to an M2 microglial repair/regenerative phenotype. This mechanism is impaired in aged brains. Aging-related decrease in IGF-1 may contribute to the loss of capacity of microglia to undergo M2 activation. Fine tuning of IGF-1 levels may be critical for regulating the neuroinflammatory response, and IGF-1 may be involved in inflammation in a context-dependent mode.
Insulin-like growth factor-1 (IGF-1) is a protein produced in several organs, such as gonads, muscle, bones, liver, gut and brain and is also present in plasma. IGF-1 signals primarily via IGF-1 receptors (IGF-1R), but IGF-1 can act also through the insulin receptor. IGF-1 is actively transported to the central nervous system (CNS) from plasma through the choroid plexus (Carro et al., 2000; Santi et al., 2017), and it is also locally produced in the brain by neurons and glial cells (Quesada et al., 2007; Suh et al., 2013; Rodriguez-Perez et al., 2016). IGF-1 has multiple effects in the CNS, regulating early brain development, myelination, synapse formation, adult neurogenesis, production of neurotransmitters and cognition (Nieto-Estévez et al., 2016; Wrigley et al., 2017). Furthermore, it is usually considered that IGF-1 is a potent neuroprotective compound (Carro et al., 2003; Tien et al., 2017), and that this is, at least partially, due to inhibition of neuroinflammation (Sukhanov et al., 2007; Park et al., 2011). Consistent with this, a decrease in IGF-1 signaling has been related with neurodegeneration, depressive disorders and other brain diseases, in which IGF-1 has been suggested as a possible therapy (Torres Aleman, 2012; Guan et al., 2013). Several decades ago, circulating GH and IGF-1 have been shown to decrease with aging. However, the possible relationship between IGF-1 effects and aging is still controversial, and opposite concepts can be found in the literature. Both the increase in IGF-1 levels and the inhibition of the IG-1R signal appear to induce beneficial effects in the CNS, and exert either aging or anti-aging effects (Cohen and Dillin, 2008; Fernandez and Torres-Alemán, 2012; Sonntag et al., 2012).
This apparent contradiction has been related to different explanations. Mild decrease in IGF-1 signal may lead to mild metabolic changes that induce protective defenses against more intense and deleterious conditions associated to aging, finally leading to an increase in lifespan (Fernandez and Torres-Alemán, 2012; Sonntag et al., 2012). IGF-1 decrease may lead to increased resistance to oxidative stress (OS), mild mitochondrial dysfunction leading to hormesis (Troulinaki and Bano, 2012), or increased resistance to proteotoxicity (Cohen et al., 2009; George et al., 2017). It has also been proposed that deletion of IGF-1R may counteract possible deleterious IGF-1R signaling independently of IGF-1 (Torres Aleman, 2012). Inhibition of IGF-1-related tumor development may also result in increased longevity (Novosyadlyy and Leroith, 2012). We suggest that development of compensatory mechanisms against mild dysregulation of the neuroinflammatory response may also be involved. Actions of IGF-1 may be context-dependent (Fernandez and Torres-Alemán, 2012), and IGF-1 may be involved in inflammation in a context-dependent mode, which may explain controversial results on the role of IGF-1 in neuroinflammation.
In summary, it is known that IGF-1 plays a major role in regulation of brain cells in health and disease conditions, although this role is controversial and has not been totally clarified. However, it is widely admitted that IGF-1 levels increase in response to brain injury, and IGF-1 is involved in the neuroinflammatory response to injury. Since neuroinflammation plays a major role in brain aging and neurodegeneration, clarification of the role of IGF-1 in the neuroinflammatory process may shed light on the above mentioned controversy.
In peripheral tissues, several studies have shown that IGF-1 regulates macrophagic functions, inhibits expression of pro-inflammatory cytokines and decreases disease progression (Sukhanov et al., 2007; Hijikawa et al., 2008). However, other studies concluded that IGF-1 has proinflammatory functions, as it was observed that IGF-1 increased chemotactic migration and TNF-α expression in macrophages (Renier et al., 1996). Conversely, proinflammatory cytokines such as TNF-α impaired IGF-1 signaling (Hotamisligil et al., 1993). Controversial results on effects of IGF-1 in neuroinflammation have also been reported. IGF-1 (Park et al., 2011) and IGF-1 gene transfer (Hung et al., 2007; Dodge et al., 2008) inhibited neuroinflammatory responses. However, other studies observed that inhibition of IGF-1R signaling decreased neuroinflammation and neuronal death in Alzheimer’s disease (AD) mice models: IGF1R-deficient mice were more resistant to amyloid-β oligomer-induced proteotoxicity, showing notably less activated astrocytes and less microgliosis (Cohen et al., 2009; George et al., 2017). As previously observed in peripheral tissues, TNF-α inhibited IGF-1 signaling in neurons (Venters et al., 1999).
The mechanisms responsible for the above mentioned effects are unclear. The anti-inflammatory properties of IGF-1 may be related to regulation of infiltration of inflammatory cells into tissues (Motani et al., 1996), including CNS, rather than a direct effect on the inflammatory cells (i.e., IGF-1 may act on the blood brain barrier; BBB). Consistent with this, IGF-1 ameliorated the breakdown of the BBB in a traumatic lesion of the spinal cord (Sharma, 2005), and in a model of multiple sclerosis (Liu et al., 1995). However, other studies showed that, IGF-1 enhanced BBB permeability and leukocyte infiltration (Pang et al., 2010): co-administration of IGF-1 with LPS further enhanced BBB permeability induced by LPS alone, while no change of BBB integrity was found in rats treated with IGF-1 alone. It was suggested that the effects of IGF-1 may depend on pathological conditions, and that in an acute inflammation, IGF-1 may have detrimental effects. However, several in vitro experiments revealed direct anti-inflammatory effects of IGF-1 on astrocytes and microglia (Bluthé et al., 2006; Palin et al., 2007). A simultaneous effect on the BBB and glial cells was also suggested (Bake et al., 2014).
On the basis of the anti-inflammatory effects of IGF-1, it has been suggested that development of resistance to IGF-1 may contribute to neuroinflammation and progression of major brain diseases. Furthermore, neuroinflammation may contribute to disease progression by decreasing levels of neuroprotective molecules such as IGF-1. Therefore, aging-related decrease in IGF-1 levels may contribute to the aging-related pro-inflammatory state and vice versa. However, the molecular mechanisms involved in the above mentioned controversial effects of IGF-1 have not been clarified. Investigation of the specific role of IGF-1 in neurons, astrocytes and microglia in different experimental contexts may shed light on the controversy (Figure 1).
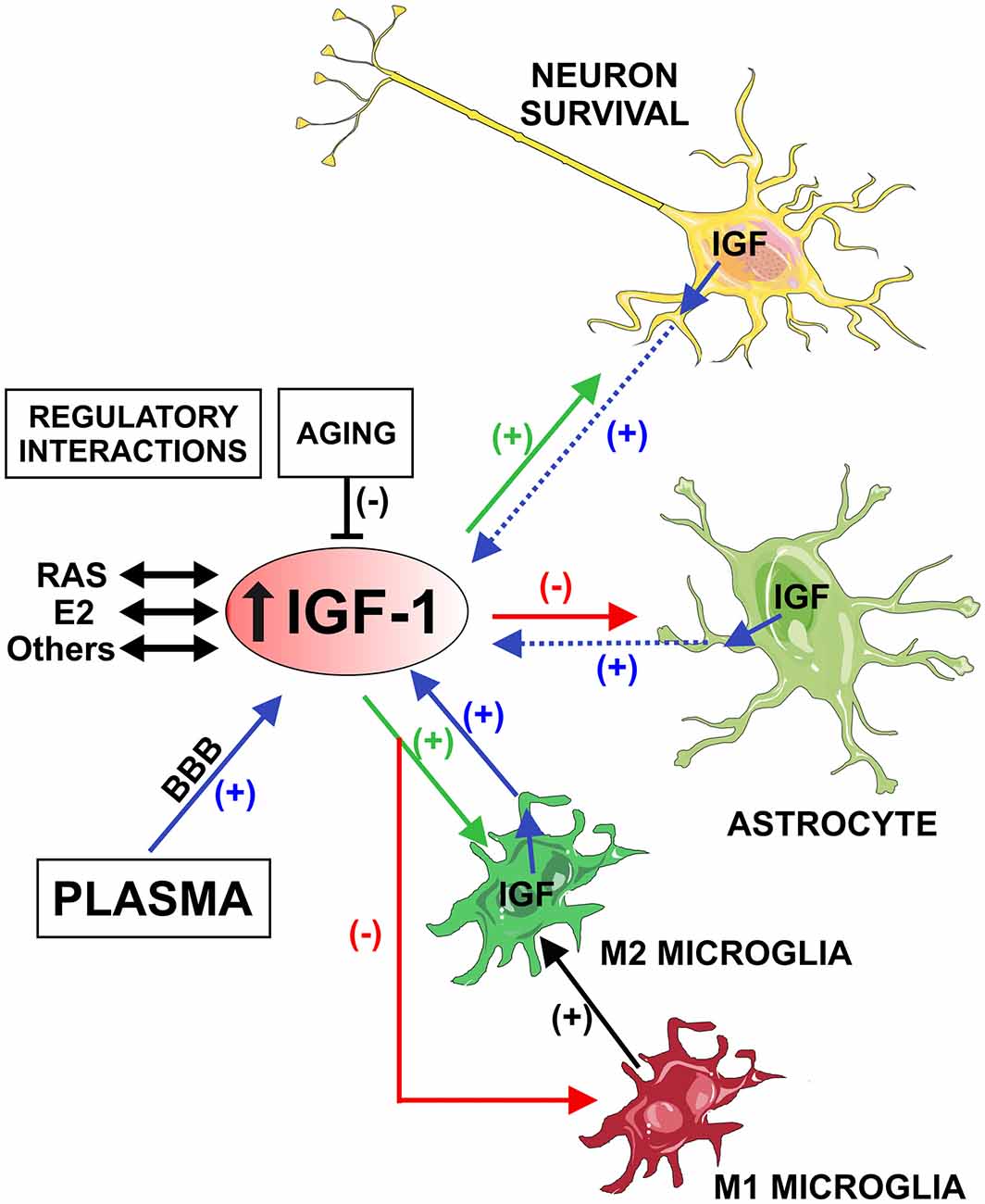
Figure 1. Schematic model showing the possible role of Insulin-like growth factor-1 (IGF-1) in neuroinflammation. IGF-1 is actively transported from plasma and locally produced in the brain by neurons and glial cells (blue arrows). Microglial cells are a major source of IGF-1(blue arrow) in comparison with astrocytes and neurons (dashed blue arrows). IGF-1 receptors are predominantly expressed in neurons and astrocytes, which appear to be targeted by IGF-1 in lesioned regions. IGF-1 promotes neuronal survival and the M2 microglial repair/regenerative phenotype (green arrows), and inhibits the astrocytic response to inflammatory stimuli and the M1 microglial phenotype (red arrows). Therefore, IGF-1 induces repression of the M1 microglial neurotoxic phenotype and enhancement of the transition to M2 (black arrow). Aging-related decrease in IGF-1 may contribute to the loss of capacity of microglia to undergo M2 activation, leading to an aging-related pro-inflammatory state. Brain IGF-1, estrogen and angiotensin interact to modulate the neuroinflammatory response. However, these regulatory mechanisms are impaired in aged brains. Abbreviations: BBB, blood-brain barrier; E2, estrogen; RAS, renin-angiotensin system. Figure was produced using Servier Medical Art (http://www.servier.com).
Our recent studies and others (Zhou et al., 1999; Rodriguez-Perez et al., 2016) have shown the presence of IGF-1 and IGF-1R in neurons, astrocytes and microglia. IGF-1 is synthesized by both neurons and glial cells, although its role is different in each cell type. IGF-1 protects neurons from neurotoxins in the presence of glia, which may be related to direct IGF-1 regulation of the glial inflammatory response (Nadjar et al., 2009). However, IGF-1 may also modulate neuroinflammation indirectly through effects on neurons, which modulate the neuroinflammatory response. IGF-1 directly protects neurons in pure neuronal cultures (Offen et al., 2001; Rodriguez-Perez et al., 2016). The mechanisms responsible for the direct neuronal protection of IGF-1 have not been clarified. However, effects on mitochondrial function (Puche et al., 2008), inhibition of OS, Sirtuin-1 activation (Vinciguerra et al., 2009; Tran et al., 2014) and other possible mechanisms have been suggested (Fernandez and Torres-Alemán, 2012; Torres Aleman, 2012; Werner and Leroith, 2014).
IGF-1 is also synthesized by astrocytes, and several studies indicate that IGF-1 plays a major role in modulating the astrocytic activity. IGF-1 regulates astrocytic glucose control and CNS glucose metabolism (Hernandez-Garzón et al., 2016). Furthermore, astrocytes contribute to regulation of IGF-1R expression in neurons (Costantini et al., 2010). Astrocytic IGF-1 protects neurons against oxidative damage (Genis et al., 2014) and traumatic brain injury (Madathil et al., 2013). IGF-1 administration or IGF-1 gene therapy inhibited expression of toll-like receptor 4, and reduced the astrocytic response to inflammatory stimuli (Bellini et al., 2011) and the expression of inflammatory mediators such as TNF-α, IL-1β and iNOS (Park et al., 2011). However, it was also found that IGF1R-deficient mice showed less amyloid-β oligomer-induced activation of astrocytes and less microgliosis (Cohen et al., 2009; George et al., 2017). Furthermore, astrocytes modulate the inflammatory response both directly and indirectly by releasing mediators that control the microglial response (Dominguez-Meijide et al., 2017).
Microglia act as resident macrophages in the CNS and major mediators of neuroinflammatory responses (Prinz and Priller, 2014). Classically, microglia present two functional states (i.e., resting and activated microglia). In a healthy state, neurons release immunosuppressive signals, which induce the classical inactivated state in the surrounding microglial cells (Harrison et al., 1998). However, it is now considered that the classical microglial activation comprises a group of “activated” states, and that microglia, in response to their environment, can adopt multiple phenotypes and functions to control CNS homeostasis (Ransohoff, 2016; Labandeira-Garcia et al., 2017; Nissen, 2017). Appropriate progression of the microglial response from the so-called proinflammatory/M1 to the so-called immunoregulatory/M2 phenotype is required for an efficient repair of brain injuries. A dysregulation of this process, leading to continued release reactive nitrogen species (RNS), reactive oxygen species (ROS) and inflammatory cytokines, results in progression neuronal death and brain diseases (Kettenmann et al., 2011; Labandeira-Garcia et al., 2017).
Microglial cells are a major source of IGF-1, which is upregulated during the inflammatory process. IGF-1R are predominantly expressed in neurons and astrocytes, which appear to be targeted by IGF-1 in lesioned regions (Butovsky et al., 2006; Suh et al., 2013) to promote neuronal survival (Arroba et al., 2011; Ueno et al., 2013; Figure 1). The immunoregulatory/M2 phenotype is typically promoted by cytokines such as IL-4 or IL-13. However, a number of data suggest that IGF-1 may also modulate the microglial phenotype: an increase in IGF-1 levels promotes the M2 phenotype (Lee et al., 2013), and treatment with IL-4 increases IGF-1 release by microglial cells (Ferger et al., 2010). IGF-1 also inhibits microglial ROS and markers of M1 phenotype such as TNF-α (Grinberg et al., 2013). Consistent with this, microglia showed a marked upregulation of IGF-1 levels and down-regulation of IL-6 in a transgenic mouse model of amyotrophic lateral sclerosis, which was related to modulation of a beneficial inflammatory response to neuronal damage (Chiu et al., 2008). Similarly, in rat models of ischemic lesion, IGF-1 mRNA was overexpressed in astrocytes and microglia surrounding surviving neurons (Beilharz et al., 1998), and elimination of proliferating microglia enhanced infarct volume and reduced levels of IGF-1 induced by the ischemic lesion (Lalancette-Hébert et al., 2007). Furthermore, microglia may be responsible for the neuroprotective effects of peripheral IGF-1 transported into the CNS, since treatment of microglia with IGF-1 was mitogenic (O’Donnell et al., 2002), and proliferation of microglia is usually considered a major regulator of the neuroinflammatory response, and a source of neuroprotective molecules such as IGF-1 (Lalancette-Hébert et al., 2007). Apparently controversial results were also observed: activation of human microglia with LPS reduced IGF-1 levels, which suggested that chronic neuroinflammation and increased inflammatory cytokines may lead to neurodegeneration by inhibiting the release of microglia-derived neuronal trophic factors, such as IGF-1 (Suh et al., 2013). However, a sudden increase in IGF levels may induce feedback signals decreasing responsiveness of the IGF-1R and IGF signaling, which may be different to the aging and neurodegenerative context (Suh et al., 2008).
Interestingly, aging (Lee et al., 2013) and mitochondrial dysfunctions (Ferger et al., 2010) inhibited IL-4-induced M2 phenotype and IL-4-induced increase in IGF-1 levels. This is consistent with recent studies suggesting that mitochondrial metabolism may modulate microglial polarization (Orihuela et al., 2016).
Sex hormones induce trophic effects on neurons and glia, enhance neuron survival and modulate several CNS functions (Rettberg et al., 2014). As in the case of IGF-1, estrogens are transported to the brain through the BBB and, in addition, the CNS can synthesize some endogenous estrogens (Azcoitia et al., 2011). Several studies have shown the presence of estrogen receptors (ERs) in neurons, astrocytes and microglia (Rettberg et al., 2014; Rodriguez-Perez et al., 2015). The mechanism responsible for estrogen-induced neuroprotection is still unclear. Direct anti-apoptotic (Brendel et al., 2013) and trophic (Campos et al., 2012) effects on neurons have been observed. However, a number of recent studies have revealed that regulation of the neuroinflammatory response constitutes a major mechanism involved in estrogen neuroprotective effects (Morale et al., 2006; Vegeto et al., 2008).
Several studies have shown that neurons and glial cells co-express ERs and IGF-1R (Cardona-Gómez et al., 2000; Quesada et al., 2007), and that estrogen and IGF-1 interact in the CNS for regulation of developmental and synaptic plasticity events and adult neurogenesis (Garcia-Segura et al., 2010). Furthermore, IGF-1 may mediate neuroprotection induced by estrogen. In aged ovariectomized rats, JB-1 (IGF-1R inhibitor) inhibited the beneficial effects of estrogen administration, suggesting that the IGF-1R signaling may mediate the effects of estrogen (Witty et al., 2013). Similarly, in a rat model of stroke, JB-1 blocked the estrogen neuroprotective effects (Selvamani and Sohrabji, 2010; Sohrabji, 2015). Several possible molecular mechanisms have been involved in the IGF-1/estrogen interaction, such as cross-regulation of ER and IGF-1R expression, IGF-1 regulation of ER-induced transcription, or regulation of IGF-1R signaling by estrogen (Garcia-Segura et al., 2010). Estrogen and IGF-1 may also stimulate common signaling pathways such as MAP kinases, PI3kinase/AKT ERK1 and ERK2 (Singh et al., 1999; Sohrabji, 2015).
Administration of either IGF-1 or estrogen can modify the inflammatory response and microglial and astrocytic phenotypes. However, they may also induce synergistic effects on neuroinflammation by still unclear mechanisms. Brain lesions stimulate production of IGF-1 and estrogen by reactive astrocytes, and increase ER and IGF-1R expression in activated glial cells (Garcia-Estrada et al., 1992; Blurton-Jones and Tuszynski, 2001; García-Ovejero et al., 2002; Hwang et al., 2004). Similar to IGF-1, estrogen has complex effects on the inflammatory response (Bellini et al., 2011; Rodriguez-Perez et al., 2015, 2016). Both estrogen and IGF-1 produced by activated glial cells may act directly on the same glial cells and on the surrounding neurons, modulating both the neuroinflammatory response and neuronal survival. Furthermore, as indicated above for IGF-1, effects of estrogen on neuroinflammation may be in part related to regulation of the BBB, since estrogen-deficient conditions such as menopause or reproductive senescence increase permeability of BBB (Bake and Sohrabji, 2004).
Consistent with the important interactions between estrogen and brain IGF-1 system, gender differences have been observed in several responses mediated by IGF-1 such as effects of exercise (Munive et al., 2016). Furthermore, in a series of studies using cell cultures, young rodents and menopausal rats (Rodriguez-Perez et al., 2010, 2011, 2012, 2015, 2016; Labandeira-Garcia et al., 2016), we have revealed important interactions and mutual regulation between estrogen and brain renin-angiotensin system (RAS). Interestingly, we also observed mutual regulation between RAS and IGF-1 (see below).
As in the case of IGF-1, the brain RAS has been associated with longevity, neuroinflammation and aging-related neuronal vulnerability to degeneration (Labandeira-Garcia et al., 2011, 2013, 2017; Labandeira-García et al., 2014). Angiotensin II (AII) is classically considered the main effector peptide of the RAS. AII signaling is mediated via AII type 1 and 2 receptors (AT1R and AT2R). AT2R induce effects that counteract those induced by AT1R stimulation (McCarthy et al., 2013). Overstimulation of the local/paracrine/tissue RAS, through AT1R, leads to OS due to NADPH oxidase overactivation, and triggers inflammatory responses. Consistent with this, RAS overactivation reduces longevity and induces aging-related degeneration in several tissues (Benigni et al., 2009, 2013; de Cavanagh et al., 2015).
Mutual regulation between RAS and IGF-1 was observed in peripheral cells, particularly in vascular smooth muscle cells (Ma et al., 2006; Jia et al., 2011) and cardiomyocytes (Leri et al., 1999; Kajstura et al., 2001). In a recent study, we have investigated possible interactions between both systems in the brain and in the neuroinflammatory process in particular (Rodriguez-Perez et al., 2016), and the results may shed light on the role of IGF-1 in the neuroinflammatory response. We observed reciprocal regulation between RAS and IGF-1: IGF-1 administration decreased RAS activity in neurons and glial cells (i.e., IGF-1 decreased AT1R, increased AT2R and reduced angiotensinogen/angiotensin levels). Inversely, AT1R activation increased IGF-1 and IGF-1R levels in microglia, while AT2R stimulation reduced IGF-1 and IGF-1R expression. AII administration promoted the microglial M1 phenotype via AT1R, which was blocked by activation of AT2R. Consistent with the above-mentioned interactions, the AII-induced enhancement of M1 phenotype markers was inhibited by administration of IGF-1. This suggests that induction of microglial IGF-1 production by AII, and possibly by other OS and pro-inflammatory inducers, plays a major role in the repression the M1 microglial neurotoxic phenotype and the enhancement of the transition to an M2 microglial repair/regenerative phenotype.
According to previous observations in plasma, and other brain regions and tissues (Bartke et al., 2003; Brown-Borg, 2015), we observed a reduction in IGF-1 levels in the substantia nigra of aged rats in comparison with young controls. In young animals and cultures, AT1R stimulation induced an increase in IGF-1 levels (see above). Since AII/AT1R activity is enhanced in the nigra of aged rats (Villar-Cheda et al., 2012, 2014), a counterregulatory increase rather than a decrease in IGF-1 levels may be expected. This suggests an aging-related loss of the IGF-1-mediated counterregulatory mechanism, which may lead to the pro-oxidative and pro-inflammatory state observed aged brains (Villar-Cheda et al., 2012; Lee et al., 2013). Low levels of IGF-1 may contribute to the loss of capacity of microglia to undergo M2 activation in the aged brain.
In addition to IGF-1-induced neuronal protection by modulation of the microglial inflammatory response, IGF-1 mediate a direct effect on neurons (see above), which may lead to indirect (i.e., neuron-mediated) modulation of the glial inflammatory response. This is consistent with our recent studies on the intraneuronal or intracrine RAS, in which we observed that activation of nuclear AT1R induces several intraneuronal protective mechanisms that may counteract the deleterious effects of activation of plasmatic membrane pro-oxidative AT1R (Valenzuela et al., 2016; Villar-Cheda et al., 2017). This protective response includes an increase in transcription of IGF-1. Interestingly, this intracrine protective response was impaired in nuclei isolated from aged brains (Villar-Cheda et al., 2017).
Experimental manipulation of IGF-1 and IGF-1R led to some controversial findings, possibly because a fine tuning of IGF-1 levels is necessary for each specific situation. This may be critical for regulating the neuroinflammatory response, as well as other IGF-1 functions and brain health. Furthermore, IGF-1 may be involved in inflammation in a context-dependent mode. Views of IGF-1 as beneficial or detrimental appear over-simplistic. Future studies taking into account different experimental contexts and progression in the knowledge of the microglial responses and phenotypes will help to solve current controversies.
All authors have contributed to this work and approved its final version for submission. JLL-G developed the idea for this review and wrote the manuscript. AIR-P prepared the figure and was involved in literature review and revision of the manuscript. MAC-B, CML and BV-C were involved in literature review and preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work reported here was supported by Spanish Ministry of Science and Innovation (BFU2015-70523), Spanish Ministry of Health, Instituto de Salud Carlos III (RD12/0019/0020, RD16/0011/0016 and CIBERNED), Galician Government, Consellería de Cultura, Educación e Ordenación Universitaria, Xunta de Galicia (XUGA GRC2014/002; CIMUS accreditation 2016-2019, ED431G/05) and European Regional Development Fund (FEDER).
Arroba, A. I., Alvarez-Lindo, N., Van Rooijen, N., and de la Rosa, E. J. (2011). Microglia-mediated IGF-I neuroprotection in the rd10 mouse model of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 52, 9124–9130. doi: 10.1167/iovs.11-7736
Azcoitia, I., Yague, J. G., and Garcia-Segura, L. M. (2011). Estradiol synthesis within the human brain. Neuroscience 191, 139–147. doi: 10.1016/j.neuroscience.2011.02.012
Bake, S., Selvamani, A., Cherry, J., and Sohrabji, F. (2014). Blood brain barrier and neuroinflammation are critical targets of IGF-1-mediated neuroprotection in stroke for middle-aged female rats. PLoS One 9:e91427. doi: 10.1371/journal.pone.0091427
Bake, S., and Sohrabji, F. (2004). 17β-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology 145, 5471–5475. doi: 10.1210/en.2004-0984
Bartke, A., Chandrashekar, V., Dominici, F., Turyn, D., Kinney, B., Steger, R., et al. (2003). Insulin-like growth factor 1 (IGF-1) and aging: controversies and new insights. Biogerontology 4, 1–8. doi: 10.1023/A:1022448532248
Beilharz, E. J., Russo, V. C., Butler, G., Baker, N. L., Connor, B., Sirimanne, E. S., et al. (1998). Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Mol. Brain Res. 59, 119–134. doi: 10.1016/s0169-328x(98)00122-3
Bellini, M. J., Hereñú, C. B., Goya, R. G., and Garcia-Segura, L. M. (2011). Insulin-like growth factor-I gene delivery to astrocytes reduces their inflammatory response to lipopolysaccharide. J. Neuroinflammation 8:21. doi: 10.1186/1742-2094-8-21
Benigni, A., Corna, D., Zoja, C., Sonzogni, A., Latini, R., Salio, M., et al. (2009). Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530. doi: 10.1172/JCI36703
Benigni, A., Orisio, S., Noris, M., Iatropoulos, P., Castaldi, D., Kamide, K., et al. (2013). Variations of the angiotensin II type 1 receptor gene are associated with extreme human longevity. Age 35, 993–1005. doi: 10.1007/s11357-012-9408-8
Blurton-Jones, M., and Tuszynski, M. H. (2001). Reactive astrocytes express estrogen receptors in the injured primate brain. J. Comp. Neurol. 433, 115–123. doi: 10.1002/cne.1129
Bluthé, R. M., Kelley, K. W., and Dantzer, R. (2006). Effects of insulin-like growth factor-I on cytokine-induced sickness behavior in mice. Brain Behav. Immun. 20, 57–63. doi: 10.1016/j.bbi.2005.02.003
Brendel, A., Felzen, V., Morawe, T., Manthey, D., and Behl, C. (2013). Differential regulation of apoptosis-associated genes by estrogen receptor α in human neuroblastoma cells. Restor. Neurol. Neurosci. 31, 199–211. doi: 10.3233/RNN-120272
Brown-Borg, H. M. (2015). The somatotropic axis and longevity in mice. Am. J. Physiol. Endocrinol. Metab. 309, E503–E510. doi: 10.1152/ajpendo.00262.2015
Butovsky, O., Ziv, Y., Schwartz, A., Landa, G., Talpalar, A. E., Pluchino, S., et al. (2006). Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 31, 149–160. doi: 10.1016/j.mcn.2005.10.006
Campos, F. L., Cristovão, A. C., Rocha, S. M., Fonseca, C. P., and Baltazar, G. (2012). GDNF contributes to oestrogen-mediated protection of midbrain dopaminergic neurones. J. Neuroendocrinol. 24, 1386–1397. doi: 10.1111/j.1365-2826.2012.02348.x
Cardona-Gómez, G. P., Doncarlos, L., and Garcia-Segura, L. M. (2000). Insulin-like growth factor I receptors and estrogen receptors colocalize in female rat brain. Neuroscience 99, 751–760. doi: 10.1016/s0306-4522(00)00228-1
Carro, E., Nuñez, A., Busiguina, S., and Torres-Aleman, I. (2000). Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 20, 2926–2933.
Carro, E., Trejo, J. L., Núñez, A., and Torres-Aleman, I. (2003). Brain repair and neuroprotection by serum insulin-like growth factor I. Mol. Neurobiol. 27, 153–162. doi: 10.1385/mn:27:2:153
Chiu, I. M., Chen, A., Zheng, Y., Kosaras, B., Tsiftsoglou, S. A., Vartanian, T. K., et al. (2008). T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. U S A 105, 17913–17918. doi: 10.1073/pnas.0804610105
Cohen, E., and Dillin, A. (2008). The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 9, 759–767. doi: 10.1038/nrn2474
Cohen, E., Paulsson, J. F., Blinder, P., Burstyn-Cohen, T., Du, D., Estepa, G., et al. (2009). Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169. doi: 10.1016/j.cell.2009.11.014
Costantini, C., Lorenzetto, E., Cellini, B., Buffelli, M., Rossi, F., and Della-Bianca, V. (2010). Astrocytes regulate the expression of insulin-like growth factor 1 receptor (IGF1-R) in primary cortical neurons during in vitro senescence. J. Mol. Neurosci. 40, 342–352. doi: 10.1007/s12031-009-9305-5
de Cavanagh, E. M., Inserra, F., and Ferder, L. (2015). Angiotensin II blockade: how its molecular targets may signal to mitochondria and slow aging. Coincidences with calorie restriction and mTOR inhibition. Am. J. Physiol. Heart Circ. Physiol. 309, H15–H44. doi: 10.1152/ajpheart.00459.2014
Dodge, J. C., Haidet, A. M., Yang, W., Passini, M. A., Hester, M., Clarke, J., et al. (2008). Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 16, 1056–1064. doi: 10.1038/mt.2008.60
Dominguez-Meijide, A., Rodriguez-Perez, A. I., Diaz-Ruiz, C., Guerra, M. J., and Labandeira-García, J. L. (2017). Dopamine modulates astroglial and microglial activity via glial renin-angiotensin system in cultures. Brain Behav. Immun. 62, 277–290. doi: 10.1016/j.bbi.2017.02.013
Ferger, A. I., Campanelli, L., Reimer, V., Muth, K. N., Merdian, I., Ludolph, A. C., et al. (2010). Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflammation 7:45. doi: 10.1186/1742-2094-7-45
Fernandez, A. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Garcia-Estrada, J., Garcia-Segura, L. M., and Torres-Aleman, I. (1992). Expression of insulin-like growth factor I by astrocytes in response to injury. Brain Res. 592, 343–347. doi: 10.1016/0006-8993(92)91695-b
García-Ovejero, D., Veiga, S., García-Segura, L. M., and Doncarlos, L. L. (2002). Glial expression of estrogen androgen receptors after rat brain injury. J. Comp. Neurol. 450, 256–271. doi: 10.1002/cne.10325
Garcia-Segura, L. M., Arévalo, M. A., and Azcoitia, I. (2010). Interactions of estradiol and insulin-like growth factor-I signalling in the nervous system: new advances. Prog. Brain Res. 181, 251–272. doi: 10.1016/s0079-6123(08)81014-x
Genis, L., Dávila, D., Fernandez, S., Pozo-Rodrigálvarez, A., Martinez-Murillo, R., and Torres-Aleman, I. (2014). Astrocytes require insulin-like growth factor I to protect neurons against oxidative injury. F1000Res. 3:28. doi: 10.12688/f1000research.3-28.v2
George, C., Gontier, G., Lacube, P., François, J. C., Holzenberger, M., and Aid, S. (2017). The Alzheimer’s disease transcriptome mimics the neuroprotective signature of IGF-1 receptor-deficient neurons. Brain 140, 2012–2027. doi: 10.1093/brain/awx132
Grinberg, Y. Y., Dibbern, M. E., Levasseur, V. A., and Kraig, R. P. (2013). Insulin-like growth factor-1 abrogates microglial oxidative stress and TNF-α responses to spreading depression. J. Neurochem. 126, 662–672. doi: 10.1111/jnc.12267
Guan, J., Mathai, S., Liang, H. P., and Gunn, A. J. (2013). Insulin-like growth factor-1 and its derivatives: potential pharmaceutical application for treating neurological conditions. Recent Pat. CNS Drug Discov. 8, 142–160. doi: 10.2174/1574889811308020004
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U S A 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hernandez-Garzón, E., Fernandez, A. M., Perez-Alvarez, A., Genis, L., Bascuñana, P., Fernandez de la Rosa, R., et al. (2016). The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia 64, 1962–1971. doi: 10.1002/glia.23035
Hijikawa, T., Kaibori, M., Uchida, Y., Yamada, M., Matsui, K., Ozaki, T., et al. (2008). Insulin-like growth factor 1 prevents liver injury through the inhibition of TNF-α and iNOS induction in D-galactosamine and LPS-treated rats. Shock 29, 740–747. doi: 10.1097/shk.0b013e31815d0780
Hotamisligil, G. S., Shargill, N. S., and Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259, 87–91. doi: 10.1126/science.7678183
Hung, K. S., Tsai, S. H., Lee, T. C., Lin, J. W., Chang, C. K., and Chiu, W. T. (2007). Gene transfer of insulin-like growth factor-I providing neuroprotection after spinal cord injury in rats. J. Neurosurg. Spine 6, 35–46. doi: 10.3171/spi.2007.6.1.35
Hwang, I. K., Yoo, K. Y., Park, S. K., An, S. J., Lee, J. Y., Choi, S. Y., et al. (2004). Expression and changes of endogenous insulin-like growth factor-1 in neurons and glia in the gerbil hippocampus and dentate gyrus after ischemic insult. Neurochem. Int. 45, 149–156. doi: 10.1016/j.neuint.2003.10.006
Jia, G., Aggarwal, A., Yohannes, A., Gangahar, D. M., and Agrawal, D. K. (2011). Cross-talk between angiotensin II and IGF-1-induced connexin 43 expression in human saphenous vein smooth muscle cells. J. Cell. Mol. Med. 15, 1695–1702. doi: 10.1111/j.1582-4934.2010.01161.x
Kajstura, J., Fiordaliso, F., Andreoli, A. M., Li, B., Chimenti, S., Medow, M. S., et al. (2001). IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 50, 1414–1424. doi: 10.2337/diabetes.50.6.1414
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Labandeira-García, J. L., Garrido-Gil, P., Rodriguez-Pallares, J., Valenzuela, R., Borrajo, A., and Rodriguez-Perez, A. I. (2014). Brain renin-angiotensin system and dopaminergic cell vulnerability. Front. Neuroanat. 8:67. doi: 10.3389/fnana.2014.00067
Labandeira-Garcia, J. L., Rodriguez-Pallares, J., Dominguez-Meijide, A., Valenzuela, R., Villar-Cheda, B., and Rodriguez-Perez, A. I. (2013). Dopamine-angiotensin interactions in the basal ganglia and their relevance for Parkinson’s disease. Mov. Disord. 28, 1337–1342. doi: 10.1002/mds.25614
Labandeira-Garcia, J. L., Rodriguez-Pallares, J., Villar-Cheda, B., Rodríguez-Perez, A. I., Garrido-Gil, P., and Guerra, M. J. (2011). Aging, Angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2, 257–274.
Labandeira-Garcia, J. L., Rodriguez-Perez, A. I., Garrido-Gil, P., Rodriguez-Pallares, J., Lanciego, J. L., and Guerra, M. J. (2017). Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front. Aging Neurosci. 9:129. doi: 10.3389/fnagi.2017.00129
Labandeira-Garcia, J. L., Rodriguez-Perez, A. I., Valenzuela, R., Costa-Besada, M. A., and Guerra, M. J. (2016). Menopause and Parkinson’s disease. Interaction between estrogens and brain renin-angiotensin system in dopaminergic degeneration. Front. Neuroendocrinol. 43, 44–59. doi: 10.1016/j.yfrne.2016.09.003
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/jneurosci.5360-06.2007
Lee, D. C., Ruiz, C. R., Lebson, L., Selenica, M. L., Rizer, J., Hunt, J. B. Jr., et al. (2013). Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 34, 1610–1620. doi: 10.1016/j.neurobiolaging.2012.12.014
Leri, A., Liu, Y., Wang, X., Kajstura, J., Malhotra, A., Meggs, L. G., et al. (1999). Overexpression of insulin-like growth factor-1 attenuates the myocyte renin-angiotensin system in transgenic mice. Circ. Res. 84, 752–762. doi: 10.1161/01.res.84.7.752
Liu, X., Yao, D. L., and Webster, H. (1995). Insulin-like growth factor I treatment reduces clinical deficits and lesion severity in acute demyelinating experimental autoimmune encephalomyelitis. Mult. Scler. 1, 2–9. doi: 10.1177/135245859500100102
Ma, Y., Zhang, L., Peng, T., Cheng, J., Taneja, S., Zhang, J., et al. (2006). Angiotensin II stimulates transcription of insulin-like growth factor I receptor in vascular smooth muscle cells: role of nuclear factor-κB. Endocrinology 147, 1256–1263. doi: 10.1210/en.2005-0888
Madathil, S. K., Carlson, S. W., Brelsfoard, J. M., Ye, P., D’Ercole, A. J., and Saatman, K. E. (2013). Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One 8:e67204. doi: 10.1371/journal.pone.0067204
McCarthy, C. A., Widdop, R. E., Denton, K. M., and Jones, E. S. (2013). Update on the angiotensin AT2 receptor. Curr. Hypertens. Rep. 15, 25–30. doi: 10.1007/s11906-012-0321-4
Morale, M. C., Serra, P. A., L’episcopo, F., Tirolo, C., Caniglia, S., Testa, N., et al. (2006). Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138, 869–878. doi: 10.1016/j.neuroscience.2005.07.060
Motani, A., Forster, L., Tull, S., Anggard, E. E., and Ferns, G. A. (1996). Insulin-like growth factor-I modulates monocyte adhesion to EAhy 926 endothelial cells. Int. J. Exp. Pathol. 77, 31–35. doi: 10.1046/j.1365-2613.1996.960098.x
Munive, V., Santi, A., and Torres-Aleman, I. (2016). A concerted action of estradiol and insulin like growth factor I underlies sex differences in mood regulation by exercise. Sci. Rep. 6:25969. doi: 10.1038/srep25969
Nadjar, A., Berton, O., Guo, S., Leneuve, P., Dovero, S., Diguet, E., et al. (2009). IGF-1 signaling reduces neuro-inflammatory response and sensitivity of neurons to MPTP. Neurobiol. Aging 30, 2021–2030. doi: 10.1016/j.neurobiolaging.2008.02.009
Nieto-Estévez, V., Defterali, Ç., and Vicario-Abejón, C. (2016). IGF-I: a key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front. Neurosci. 10:52. doi: 10.3389/fnins.2016.00052
Nissen, J. C. (2017). Microglial function across the spectrum of age and gender. Int. J. Mol. Sci 18:E561. doi: 10.3390/ijms18030561
Novosyadlyy, R., and Leroith, D. (2012). Insulin-like growth factors and insulin: at the crossroad between tumor development and longevity. J. Gerontol. A Biol. Sci. Med. Sci. 67, 640–651. doi: 10.1093/gerona/gls065
O’Donnell, S. L., Frederick, T. J., Krady, J. K., Vannucci, S. J., and Wood, T. L. (2002). IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39, 85–97. doi: 10.1002/glia.10081
Offen, D., Shtaif, B., Hadad, D., Weizman, A., Melamed, E., and Gil-Ad, I. (2001). Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: possible implications for Parkinson’s disease. Neurosci. Lett. 316, 129–132. doi: 10.1016/s0304-3940(01)02344-8
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Palin, K., Bluthé, R. M., McCusker, R. H., Moos, F., Dantzer, R., and Kelley, K. W. (2007). TNFα-induced sickness behavior in mice with functional 55 kD TNF receptors is blocked by central IGF-I. J. Neuroimmunol. 187, 55–60. doi: 10.1016/j.jneuroim.2007.04.011
Pang, Y., Zheng, B., Campbell, L. R., Fan, L. W., Cai, Z., and Rhodes, P. G. (2010). IGF-1 can either protect against or increase LPS-induced damage in the developing rat brain. Pediatr. Res. 67, 579–584. doi: 10.1203/PDR.0b013e3181dc240f
Park, S. E., Dantzer, R., Kelley, K. W., and McCusker, R. H. (2011). Central administration of insulin-like growth factor-I decreases depressive-like behavior and brain cytokine expression in mice. J. Neuroinflammation 8:12. doi: 10.1186/1742-2094-8-12
Prinz, M., and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. doi: 10.1038/nrn3722
Puche, J. E., García-Fernández, M., Muntané, J., Rioja, J., González-Barán, S., and Castilla Cortazar, I. (2008). Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 149, 2620–2627. doi: 10.1210/en.2007-1563
Quesada, A., Romeo, H. E., and Micevych, P. (2007). Distribution and localization patterns of estrogen receptor-β and insulin-like growth factor-1 receptors in neurons and glial cells of the female rat substantia nigra: localization of ERβ and IGF-1R in substantia nigra. J. Comp. Neurol. 503, 198–208. doi: 10.1002/cne.21358
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Renier, G., Clement, I., Desfaits, A. C., and Lambert, A. (1996). Direct stimulatory effect of insulin-like growth factor-I on monocyte and macrophage tumor necrosis factor-α production. Endocrinology 137, 4611–4618. doi: 10.1210/en.137.11.4611
Rettberg, J. R., Yao, J., and Brinton, R. D. (2014). Estrogen: a master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 35, 8–30. doi: 10.1016/j.yfrne.2013.08.001
Rodriguez-Perez, A. I., Borrajo, A., Diaz-Ruiz, C., Garrido-Gil, P., and Labandeira-Garcia, J. L. (2016). Crosstalk between insulin-like growth factor-1 and angiotensin-II in dopaminergic neurons and glial cells: role in neuroinflammation and aging. Oncotarget 7, 30049–30067. doi: 10.18632/oncotarget.9174
Rodriguez-Perez, A. I., Borrajo, A., Valenzuela, R., Lanciego, J. L., and Labandeira-Garcia, J. L. (2015). Critical period for dopaminergic neuroprotection by hormonal replacement in menopausal rats. Neurobiol. Aging 36, 1194–1208. doi: 10.1016/j.neurobiolaging.2014.10.028
Rodriguez-Perez, A. I., Valenzuela, R., Joglar, B., Garrido-Gil, P., Guerra, M. J., and Labandeira-Garcia, J. L. (2011). Renin angiotensin system and gender differences in dopaminergic degeneration. Mol. Neurodegener. 6:58. doi: 10.1186/1750-1326-6-58
Rodriguez-Perez, A. I., Valenzuela, R., Villar-Cheda, B., Guerra, M. J., and Labandeira-Garcia, J. L. (2012). Dopaminergic neuroprotection of hormonal replacement therapy in young and aged menopausal rats: role of the brain angiotensin system. Brain 135, 124–138. doi: 10.1093/brain/awr320
Rodriguez-Perez, A. I., Valenzuela, R., Villar-Cheda, B., Guerra, M. J., Lanciego, J. L., and Labandeira-Garcia, J. L. (2010). Estrogen and angiotensin interaction in the substantia nigra. Relevance to postmenopausal Parkinson’s disease. Exp. Neurol. 224, 517–526. doi: 10.1016/j.expneurol.2010.05.015
Santi, A., Genis, L., and Torres Aleman, I. (2017). A coordinated action of blood-borne and brain insulin-like growth factor I in the response to traumatic brain injury. Cereb. Cortex doi: 10.1093/cercor/bhx106 [Epub ahead of print].
Selvamani, A., and Sohrabji, F. (2010). The neurotoxic effects of estrogen on ischemic stroke in older female rats is associated with age-dependent loss of insulin-like growth factor-1. J. Neurosci. 30, 6852–6861. doi: 10.1523/JNEUROSCI.0761-10.2010
Sharma, H. S. (2005). Neuroprotective effects of neurotrophins and melanocortins in spinal cord injury: an experimental study in the rat using pharmacological and morphological approaches. Ann. N Y Acad. Sci. 1053, 407–421. doi: 10.1196/annals.1344.036
Singh, M., Sétáló, G. Jr., Guan, X., Warren, M., and Toran-Allerand, C. D. (1999). Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J. Neurosci. 19, 1179–1188.
Sohrabji, F. (2015). Estrogen-IGF-1 interactions in neuroprotection: ischemic stroke as a case study. Front. Neuroendocrinol. 36, 1–14. doi: 10.1016/j.yfrne.2014.05.003
Sonntag, W. E., Csiszar, A., deCabo, R., Ferrucci, L., and Ungvari, Z. (2012). Diverse roles of growth hormone and insulin-like growth factor-1 in mammalian aging: progress and controversies. J. Gerontol. A Biol. Sci. Med. Sci. 67, 587–598. doi: 10.1093/gerona/gls115
Suh, Y., Atzmon, G., Cho, M. O., Hwang, D., Liu, B., Leahy, D. J., et al. (2008). Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. U S A 105, 3438–3442. doi: 10.1073/pnas.0705467105
Suh, H. S., Zhao, M. L., Derico, L., Choi, N., and Lee, S. C. (2013). Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J. Neuroinflammation 10:37. doi: 10.1186/1742-2094-10-37
Sukhanov, S., Higashi, Y., Shai, S. Y., Vaughn, C., Mohler, J., Li, Y., et al. (2007). IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 2684–2690. doi: 10.1161/atvbaha.107.156257
Tien, L. T., Lee, Y. J., Pang, Y., Lu, S., Lee, J. W., Tseng, C. H., et al. (2017). Neuroprotective effects of intranasal IGF-1 against neonatal lipopolysaccharide-induced neurobehavioral deficits and neuronal inflammation in the substantia nigra and locus coeruleus of juvenile rats. Dev. Neurosci. doi: 10.1159/000477898 [Epub ahead of print].
Torres Aleman, I. (2012). Insulin-like growth factor-1 and central neurodegenerative diseases. Endocrinol. Metab. Clin. North. Am. 41, 395–408, vii. doi: 10.1016/j.ecl.2012.04.016
Tran, D., Bergholz, J., Zhang, H., He, H., Wang, Y., Zhang, Y., et al. (2014). Insulin-like growth factor-1 regulates the SIRT1–p53 pathway in cellular senescence. Aging Cell 13, 669–678. doi: 10.1111/acel.12219
Troulinaki, K., and Bano, D. (2012). Mitochondrial deficiency: a double-edged sword for aging and neurodegeneration. Front. Genet. 3:244. doi: 10.3389/fgene.2012.00244
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M., et al. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551. doi: 10.1038/nn.3358
Valenzuela, R., Costa-Besada, M. A., Iglesias-Gonzalez, J., Perez-Costas, E., Villar-Cheda, B., Garrido-Gil, P., et al. (2016). Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 7:e2427. doi: 10.1038/cddis.2016.327
Vegeto, E., Benedusi, V., and Maggi, A. (2008). Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol. 29, 507–519. doi: 10.1016/j.yfrne.2008.04.001
Venters, H. D., Tang, Q., Liu, Q., VanHoy, R. W., Dantzer, R., and Kelley, K. W. (1999). A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc. Natl. Acad. Sci. U S A 96, 9879–9884. doi: 10.1073/pnas.96.17.9879
Villar-Cheda, B., Costa-Besada, M. A., Valenzuela, R., Perez-Costas, E., Melendez-Ferro, M., and Labandeira-Garcia, J. L. (2017). The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 8:e3044. doi: 10.1038/cddis.2017.439
Villar-Cheda, B., Dominguez-Meijide, A., Valenzuela, R., Granado, N., Moratalla, R., and Labandeira-Garcia, J. L. (2014). Aging-related dysregulation of dopamine and angiotensin receptor interaction. Neurobiol. Aging 35, 1726–1738. doi: 10.1016/j.neurobiolaging.2014.01.017
Villar-Cheda, B., Valenzuela, R., Rodriguez-Perez, A. I., Guerra, M. J., and Labandeira-Garcia, J. L. (2012). Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol. Aging 33, 204.e1–204.e11. doi: 10.1016/j.neurobiolaging.2010.08.006
Vinciguerra, M., Santini, M. P., Claycomb, W. C., Ladurner, A. G., and Rosenthal, N. (2009). Local IGF-1 isoform protects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging (Albany NY) 2, 43–62. doi: 10.18632/aging.100107
Werner, H., and Leroith, D. (2014). Insulin and insulin-like growth factor receptors in the brain: physiological and pathological aspects. Eur. Neuropsychopharmacol. 24, 1947–1953. doi: 10.1016/j.euroneuro.2014.01.020
Witty, C. F., Gardella, L. P., Perez, M. C., and Daniel, J. M. (2013). Short-term estradiol administration in aging ovariectomized rats provides lasting benefits for memory and the hippocampus: a role for insulin-like growth factor-I. Endocrinology 154, 842–852. doi: 10.1210/en.2012-1698
Wrigley, S., Arafa, D., and Tropea, D. (2017). Insulin-like growth factor 1: at the crossroads of brain development and aging. Front. Cell Neurosci. 11:14. doi: 10.3389/fncel.2017.00014
Keywords: aging, angiotensin, astrocytes, estrogen, IGF-1, insulin, microglia, neurodegeneration
Citation: Labandeira-Garcia JL, Costa-Besada MA, Labandeira CM, Villar-Cheda B and Rodríguez-Perez AI (2017) Insulin-Like Growth Factor-1 and Neuroinflammation. Front. Aging Neurosci. 9:365. doi: 10.3389/fnagi.2017.00365
Received: 09 September 2017; Accepted: 23 October 2017;
Published: 03 November 2017.
Edited by:
Luis Miguel Garcia-Segura, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Maria Jose Bellini, Argentinian National Council of Research, ArgentiniaCopyright © 2017 Labandeira-Garcia, Costa-Besada, Labandeira, Villar-Cheda and Rodríguez-Perez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose L. Labandeira-Garcia, am9zZWx1aXMubGFiYW5kZWlyYUB1c2MuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.