 Paola Gamba
Paola Gamba Gabriella Testa
Gabriella Testa Simona Gargiulo
Simona Gargiulo Erica Staurenghi
Erica Staurenghi Giuseppe Poli
Giuseppe Poli Gabriella Leonarduzzi
Gabriella Leonarduzzi- Department of Clinical and Biological Sciences, School of Medicine, University of Turin, Orbassano, Torino, Italy
Alzheimer’s disease (AD), the most common neurodegenerative disorder associated with dementia, is typified by the pathological accumulation of amyloid Aβ peptides and neurofibrillary tangles (NFT) within the brain. Considerable evidence indicates that many events contribute to AD progression, including oxidative stress, inflammation, and altered cholesterol metabolism. The brain’s high lipid content makes it particularly vulnerable to oxidative species, with the consequent enhancement of lipid peroxidation and cholesterol oxidation, and the subsequent formation of end products, mainly 4-hydroxynonenal and oxysterols, respectively from the two processes. The chronic inflammatory events observed in the AD brain include activation of microglia and astrocytes, together with enhancement of inflammatory molecule and free radical release. Along with glial cells, neurons themselves have been found to contribute to neuroinflammation in the AD brain, by serving as sources of inflammatory mediators. Oxidative stress is intimately associated with neuroinflammation, and a vicious circle has been found to connect oxidative stress and inflammation in AD. Alongside oxidative stress and inflammation, altered cholesterol metabolism and hypercholesterolemia also significantly contribute to neuronal damage and to progression of AD. Increasing evidence is now consolidating the hypothesis that oxidized cholesterol is the driving force behind the development of AD, and that oxysterols are the link connecting the disease to altered cholesterol metabolism in the brain and hypercholesterolemia; this is because of the ability of oxysterols, unlike cholesterol, to cross the blood brain barrier (BBB). The key role of oxysterols in AD pathogenesis has been strongly supported by research pointing to their involvement in modulating neuroinflammation, Aβ accumulation, and cell death. This review highlights the key role played by cholesterol and oxysterols in the brain in AD pathogenesis.
Introduction
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder associated with dementia, and the progressive deterioration of mental capacities. It is a complex disease characterized by progressive memory impairment, cognitive deficit, and personality changes; these symptoms are due to substantial synaptic and neuronal loss occuring in specific brain areas, especially in the neocortex, hippocampus, and other subcortical regions. Brains from AD patients show distinct neuropathological features, which we now know are the hallmarks of the disease: extracellular deposits of amyloid β (Aβ) peptides in the form of senile plaques, Aβ deposits in the cerebral blood vessels, and intracellular inclusion of neurofibrillary tangles (NFT) composed of hyperphosphorylated tau protein (Querfurth and LaFerla, 2010; Chopra et al., 2011).
One of the events that promotes AD pathogenesis is the abnormal processing of amyloid precursor protein (APP), which leads to excess production of Aβ peptides through the sequential enzymatic actions of beta-site APP cleaving enzyme 1 (BACE1), a β-secretase, and γ-secretase, both enzymes of the amiloidogenic pathway. An imbalance between the production and clearance of Aβ peptides in the brain, and their aggregation, cause Aβ to accumulate, with subsequent formation of senile plaques. Depending on the site of γ-secretase cleavage, two major forms of Aβ are generated: a peptide of 40 amino acids (Aβ1–40), and one of 42 amino acids (Aβ1–42). Aβ1–42 is the predominant species of Aβ in senile plaques, and the insoluble oligomers and intermediate amyloids are its most neurotoxic forms (Walsh and Selkoe, 2007). Aβ oligomers are hypothesized to cause neuronal damage and cognitive failure by generating free radicals, as well as mitochondrial oxidative damage, synaptic failure, and inflammatory changes in AD brains (Oddo et al., 2003; Mattson, 2004; Reddy and Beal, 2005; Castellani et al., 2010). Different assembly states of Aβ, and its accumulation in different cellular compartments, can then affect critical pathways, thereby facilitating the development of tau pathology. Experimental evidence confirms that Aβ accumulation precedes and drives NFT formation (Götz et al., 2001; Lewis et al., 2001).
Besides aging, which is the most obvious risk factor for the disease, a number of theories point to other risk factors, such as culture, lifestyle, head injury, and genetics. Other risk factors are associated with vascular disease, including hypercholesterolemia, hypertension, atherosclerosis, coronary heart disease, smoking, obesity, and diabetes (Mayeux, 2003). Some evidence suggests that the dietary intake of homocysteine-related vitamins (vitamin B12 and folate), antioxidants (vitamins C and E), unsaturated fatty acids, and also a moderate alcohol intake, especially in the form of wine, may reduce the risk of AD (Luchsinger and Mayeux, 2004).
Although environmental factors increase the risk of AD, this disease can also be caused by various gene mutations (Gatz et al., 2006). In this contest, mutation in the genes APP, presenilin 1 (PSEN1), and presenilin 2 (PSEN2) accounts for most cases of the familial (or early-onset) form of AD, by increasing the production and aggregation of Aβ and amyloid plaque formation (Tanzi and Bertram, 2005). Conversely, the apolipoprotein E (ApoE) gene is an important genetic risk factor for the sporadic (or late-onset) form of AD (Raber et al., 2004). The contribution of other candidate genes is probably less important, and none has been verified. A possible explanation for this difficulty in clarifying the genetic background might be that the sporadic form of AD is not a uniform disease entity, and that several susceptibility-enhancing genes may act in concert, each conferring only a small increase in risk, in a complex interaction with environmental factors.
However, although the pathophysiology of AD is still not clearly understood, considerable evidence indicates that many events participate in the development and progression of the disease, including oxidative stress, inflammation, glial cell activation, dysregulation of metal ions and calcium, presence of ApoEε4, altered cholesterol metabolism, and dysregulation of intercellular communication among brain cells (Quintanilla et al., 2012).
Oxidative Stress in AD Pathogenesis
It has been extensively reported that free radicals are pathologically important in neurodegenerative diseases, and that the brain tissue is exposed to oxidative damage during the development of AD, already from its early onset (Smith et al., 2000, 2010; Mariani et al., 2005; Zhu et al., 2005; Reynolds et al., 2007). Because age is a significant risk factor for AD, it is also widely accepted that oxidative stress increases with age leading to the accumulation of oxidative damage in biomolecules (Butterfield and Kanski, 2001; Martin and Grotewiel, 2006; Jacob et al., 2013).
The brain is particularly vulnerable to oxidative damage for numerous reasons, but chiefly because it utilizes about 25% of the respired oxygen, with a consequent increase of free radicals; it also contains high concentrations of catalytic iron and lipids, which are easily oxidized by free radicals. Further, the brain contains relatively low levels of antioxidants and antioxidant defense enzymes, and is thus not very efficient at removing free radicals (Ansari and Scheff, 2010; Mazzetti et al., 2015). Because the brain has a high lipid content, it is extremely vulnerable to oxidative species, with the consequent enhancement of lipid peroxidation and cholesterol oxidation, and the subsequent formation of end products, mainly 4-hydroxynonenal and oxysterols, respectively from the two processes (Sottero et al., 2009; Reed, 2011).
Within the brain, neurons are the cells most vulnerable to excess reactive oxygen species (ROS) and reactive nitrose species (RNS), and their survival depends on the antioxidant action of astrocytes. Astrocytes are very important for normal brain function, because of their ability to actively promote neuroprotection, in particular by releasing glutathione, which protects neurons from oxidative stress (Shih et al., 2003). However, neurons can also defend themselves through an intrinsic mechanism of antioxidant defense involving the glucose metabolism (Fernandez-Fernandez et al., 2012).
AD brains display high levels of oxidative stress, and a direct association between free radical generation and the presence of Aβ plaques has been shown both in living AD mouse models and in human AD tissue (McLellan et al., 2003). However, it is still not clear whether oxidative stress is a cause or a consequence of the neuropathology associated with AD (Zhu et al., 2007; Bonda et al., 2010; Smith et al., 2010; Luque-Contreras et al., 2014). In support of its being a cause, it has been proposed that oxidative stress precedes the onset of clinical and pathological AD symptoms, including Aβ deposition, NFT formation, vascular malfunction, and cognitive decline (Nunomura et al., 2001; Praticò et al., 2001). In this connection, in a triple-transgenic mouse model of AD, mimicking AD progression in humans, the levels of antioxidants (glutathione and vitamin E) were found to be decreased, and the extent of lipid peroxidation increased, before the appearance of senile plaques and NFT (Resende et al., 2008). Moreover, as a prominent early event, oxidative stress is believed to contribute to tau hyperphosphorylation in neurons (Su et al., 2010) and, of note, it has been observed that neuritic and cored amyloid plaques show evidence of oxidatively modified Aβ (Head et al., 2002). Conversely, Aβ is a potent promoter of oxidative stress, since it is a potent generator of both ROS (Ding et al., 2007) and RNS (Combs et al., 2001). Within the Aβ sequence, it has been suggested that methionine 35 plays an important role in promoting oxidative activity. When this amino acid is replaced by cysteine, the oxidative stress induced by Aβ is greatly attenuated (Butterfield and Boyd-Kimball, 2005; Butterfield et al., 2013). In this connection, it has also been proposed that the amyloid oligomers may insert themselves into the lipid bilayer, causing lipid peroxidation and, consequently, oxidative damage to proteins and other biomolecules (Butterfield et al., 2001). Aβ oligomers are, indeed, the strongest inducers of oxidative stress among all Aβ species (Tamagno et al., 2006; Naylor et al., 2008). Generation of free radicals, altered membrane properties, as well as disturbed calcium homeostasis, may also underlie the apoptotic effect of Aβ oligomers (Malaplate-Armand et al., 2006). It has also been observed, not only in cultured neurons but also in vivo using the double transgenic model of AD (APP/PS1), that Aβ causes an increase in oxidative stress that leads to phosphorylation of p38, which in turn phosphorylates tau at its T231 residue (Giraldo et al., 2014).
Mitochondrial dysfunction is another feature of AD pathogenesis (Castellani et al., 2002). Defects in the mitochondria are typically defects of the electron transport chain; these contribute both to the hyperproduction of a variety of ROS, and to the deficiency of several key enzymes responsible for oxidative metabolism that, in turn, cause cell damage and eventual death (Cottrell et al., 2001). Moreover, it has been shown that oxidative species, through mitochondrial impairment, cause tau hyperphosphorylation leading to neuron and synapse loss (Melov et al., 2007). Mitochondrial dysfunction and oxidative damage have been investigated in triple-transgenic mice that develop both Aβ and tau disorders. These mice exhibited increased oxidative stress, manifested by increased hydrogen peroxide production and lipid peroxidation (Yao et al., 2009; Reddy, 2011).
APP and Aβ have also been associated with dysfunctional consequences for mitochondrial homeostasis and cell death (Manczak et al., 2006). APP impairs mitochondrial energy metabolism, thus causing mitochondrial abnormalities leading to ROS production (Anandatheerthavarada et al., 2003); accumulation of APP in the mitochondrial import channel then potentially inhibits mitochondrial import (Reddy et al., 2004). Aβ is also considered a potent mitochondrial poison, especially affecting the synaptic pool. It appears that Aβ enters the mitochondria, induces free radical generation, disrupts the electron transport chain, and ultimately causes mitochondrial dysfunction (Mungarro-Menchaca et al., 2002). Aβ, then, inhibits key mitochondrial enzymes in the brain and in isolated mitochondria (Caspersen et al., 2005; Reddy and Beal, 2008), and cytochrome c oxidase is also specifically attacked (Crouch et al., 2005). A recent study on the transgenic mouse brain confirmed that Aβ accumulates in neuronal mitochondria, thus affecting mitochondrial function, as shown by increased mitochondrial permeability, the decline of both respiratory function and cytochrome c oxidase activity, and increased mitochondrial oxidative stress (Du et al., 2010).
An association between ApoEε4 and oxidative stress-mediated damage in AD has been also suggested. Despite playing a beneficial role, by maintaining lipid homeostasis and redox balance, ApoE can also contribute to oxidative damage in an isoform-dependent manner, the ApoEε4 isoform being the most harmful in AD (Luque-Contreras et al., 2014). The ApoEε4 genotype is also involved in mitochondrial dysfunction (Chang et al., 2005), and might be a risk for potential antioxidant system loss in AD (Shea et al., 2002).
Alterations in cerebrovascular regulation have recently been ascribed to the early stages of AD, and the vascular endothelium is also a target for oxidative stress leading to endothelium dysfunction (Iadecola, 2004; Park et al., 2008). Chronic hypoperfusion may thus play an important role in the pathophysiology of AD, because it induces oxidative stress, and over time this damage could initiate mitochondrial failure (Sochocka et al., 2013). A recent study has shown that inhibition of NADPH oxidase activity can mitigate cognitive impairment in rodent models of hypoperfusion (Kim et al., 2012). Oxidative stress is intimately associated with neuroinflammation, and there has been found to be a vicious circle connecting oxidative stress and inflammation in AD (Rosales-Corral et al., 2010; Quintanilla et al., 2012; Joshi and Praticò, 2015). It has been observed that the redox status modulates inflammatory factors involvement in signaling processes, which are critical mediators of oxidative stress and inflammation, causing neurodegeneration (Mrak and Griffin, 2005; Kierdorf et al., 2010). Activation of glial cells and increased cytokine production is also induced by oxidative stress and, in turn, glial activation leads to the release of other neurotoxic factors such as ROS and nitric oxide (NO), which further exacerbate neuronal damage (Town et al., 2005; Block et al., 2007; Michelucci et al., 2009; von Bernhardi et al., 2010). Consequently, the resultant cellular damage amplifies the inflammatory response, with glial activation and leukocyte recruitment, leading to further inflammation in the AD brain (Figure 1).
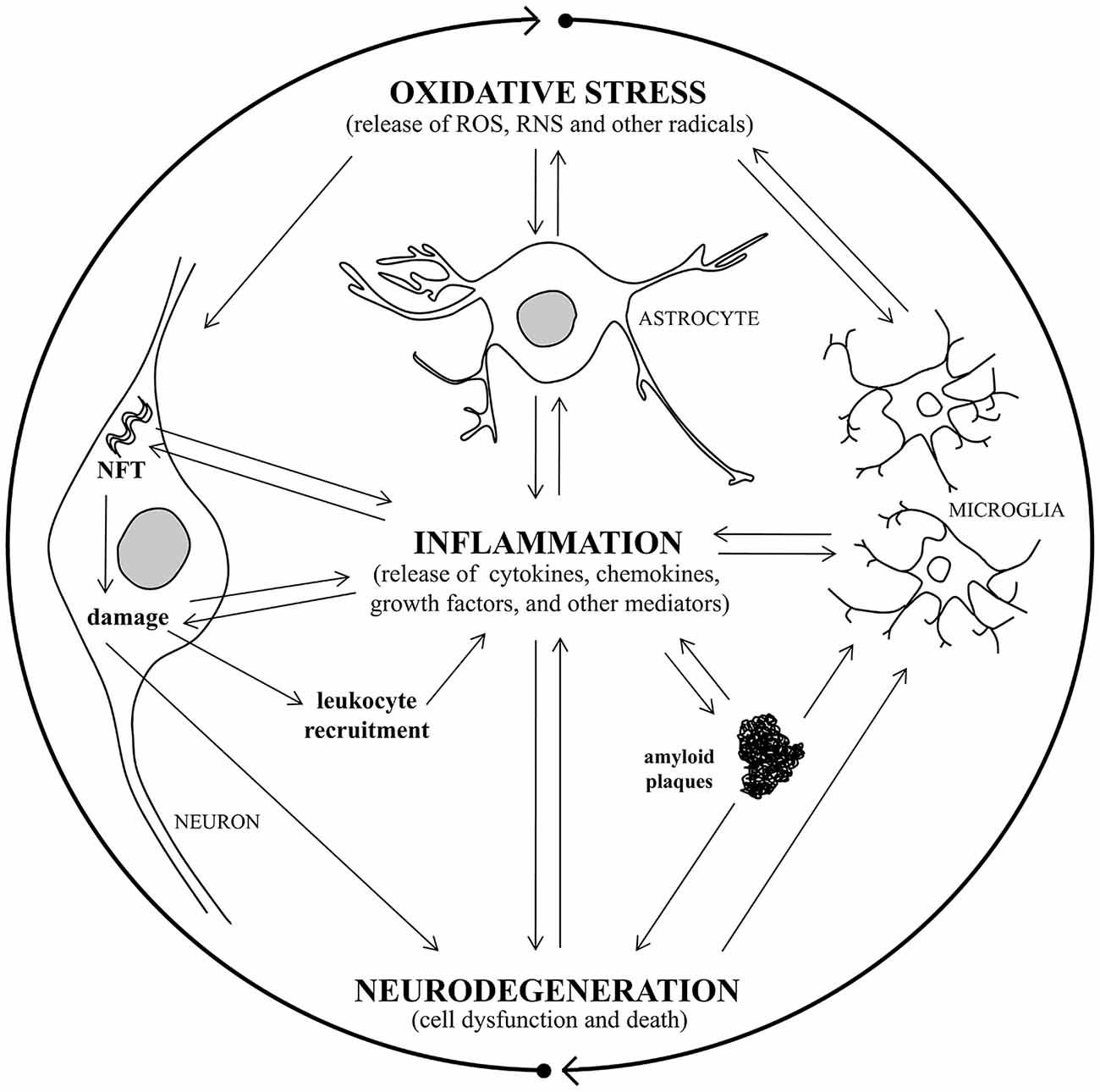
Figure 1. A vicious circle connects oxidative stress, inflammation, and neurodegeneration in Alzheimer’s disease (AD). Oxidative stress damage and inflammatory response are closely associated with AD, causing neurodegeneration. Oxidative stress induces activation of microglia and astrocytes with a consequent increase of pro-inflammatory mediator production and, in turn, glial activation leads to toxic radical release, exacerbating neuronal damage. Consequently, the resultant cellular damage amplifies the inflammatory response, with glial activation and leukocyte recruitment, leading to further inflammation in the AD brain. The release of inflammatory cytokines leads to amyloid plaque and neurofibrillary tangle (NFT) formation, that triggers inflammatory molecule release and causes neuronal damage, with consequent neurodegeneration.
Inflammation in AD Pathogenesis
Besides oxidative stress damage, inflammatory responses are also closely associated with AD pathology. Inflammation occurs in pathologically vulnerable regions of the AD brain, with increased expression of acute-phase proteins and pro-inflammatory molecules, which create a chronic and self-sustaining inflammatory process, involving activated glia cells, and stressed neurons (Mrak and Griffin, 2005; Perry et al., 2010; Morales et al., 2014). Thus chronic inflammation plays a basic role in the progression of neuropathological changes in AD, resulting in neuronal dysfunction and death. The importance of neuroinflammation has emerged from several studies of AD brains, which have evidenced the activation and proliferation of glial cells, together with enhanced release of inflammatory mediators (cytokines, chemokines, growth factors, and other mediators) and free-radical-mediated oxidative stress (ROS, NO, and other radicals) (Glass et al., 2010; Heneka et al., 2010; McGeer and McGeer, 2010; Holmes and Butchart, 2011; Azizi and Mirshafiey, 2012; Rubio-Perez and Morillas-Ruiz, 2012; Lyman et al., 2014; Figueiredo-Pereira et al., 2015). Microglia are key players in the disease process, and once activated they present cell-surface antigens commonly present on monocytes and macrophages (Latta et al., 2014). Microglial activation thus leads to the initiation of an innate immune response, dominated by the release of pro-inflammatory cytokines (Town et al., 2005; Mrak, 2012; Weitz and Town, 2012). Incidental to this is also the phagocytosis of fibrils and large aggregates of Aβ, suggesting an initial neuroprotective defense mechanism (D’Andrea et al., 2004; Colton and Wilcock, 2010). Further, microglia can also secrete a number of soluble factors, such as glia-derived neurotrophic factor, which are potentially beneficial to the survival of neurons (Liu and Hong, 2003). Although the initial purpose of microglial activation is to counteract the detrimental effects induced by the pathological features, it subsequently leads to the release of high concentrations of neurotoxic factors, such as inflammatory molecules, NO, ROS, proteolitic enzymes, complementary factors, and excitatory amino acids, which further exacerbate cell damage (Michelucci et al., 2009). Moreover, in later stages of the disease, the overproduction of inflammatory cytokines makes the microglia phagocytically inactive, especially against insoluble oligomers and high concentrations of Aβ (Hickman et al., 2008; Krabbe et al., 2013).
A growing body of evidence also suggests that the central nervous system (CNS) and systemic inflammation cannot be viewed in isolation. Systemic inflammation might exarcerbate behavioral symptoms and accelerate AD progression by increasing the production of local pro-inflammatory cytokines and chemokines, as well as of ROS and NO (Holmes, 2013). The detrimental effects of peripheral inflammatory molecules in the brain of AD patients chiefly occur because these mediators can easily enter the brain, together with infiltrating leukocytes, thanks to increased permeability of the blood-brain barrier (BBB) as the disease progresses (Leoni et al., 2003; Popescu et al., 2009; Takeda et al., 2014). Attention is also now being paid to the participation of Toll-like receptors (TLRs) in inflammation and neurodegeneration. The recruitment of TLRs contributes to inflammation by amplifying the release of inflammatory molecules, thus playing an important role in the impact of inflammation on neuronal function and death (Drouin-Ouellet and Cicchetti, 2012).
However, it also remains unclear in the case of inflammation whether this process is a cause or a consequence of AD. Clinical and experimental studies support the appearance of neuroinflammatory changes already at the early stages of AD, even before the formation of extracellular Aβ deposits and intracellular NFT accumulation (Sheng et al., 2000). The release of pro-inflammatory cytokines and enzymes could affect the normal behavior of neuronal cells, leading to cell dysfunction and abnormalities such as Aβ peptides and NFT accumulation, events in the pathway leading to neuronal degeneration. Inflammatory molecules, and a number of stress conditions, enhance APP levels and the amyloidogenic processing of APP to induce Aβ1–42 peptide production. This, in turn, inhibits the formation of the soluble APP fraction that seems to have a neuronal protective effect (Fassbender et al., 2000; Misonou et al., 2000). Conversely, it has been demonstrated that intraneuronal Aβ and soluble Aβ oligomers activate microglia in the earliest stages of AD, even before senile plaque and NFT formation, in particular when cells are stressed (Ferretti and Cuello, 2011; Khandelwal et al., 2011). Moreover, Aβ and NFT have also been shown to trigger the release by activated glia cells of pro-inflammatory mediators, free radicals, and other neurotoxic substances, implying that the development of AD leads to the initiation of several self-propagating cycles (Vukic et al., 2009; Morales et al., 2013). Fibrillar Aβ can also activate microglia by binding to cell membranes via specific receptors, including a multi-receptor complex involving CD36, α6β1-integrin, and CD47 (Verdier et al., 2004; Yu and Ye, 2015), while the disruption of the APP gene and of its proteolytic products delays and decreases microglial activation (DeGiorgio et al., 2002). Once activated, microglia may also recruit and activate astrocytes, which actively enhance the inflammatory response to extracellular Aβ deposits (Jo et al., 2014). Aggregated amyloid fibrils and neurotoxic inflammatory molecules, secreted by glial cells, intensify neuronal dysfunction and death, either alone or in concert (Brown and Bal-Price, 2003; Findeis, 2007). In this context, activated glia cells and the released inflammatory molecules, together with other components of the immune response, are often present in proximity to neurons and areas of amyloid plaque (Abbas et al., 2002; Serrano-Pozo et al., 2013). Of note, microglia have also been suggested to be preferentially associated with certain types of amyloid deposits (D’Andrea et al., 2004). Furthermore, astrocytes are known to play a critical role in Aβ clearance, in providing trophic support to neurons, and in forming a protective barrier between Aβ deposits and neurons. The presence of large numbers of astrocytes associated with Aβ plaques suggests that these lesions induce the release of chemotactic molecules that mediate astrocyte recruitment. However, it has been suggested that astrocytes could also be a source for Aβ peptides, because they overexpress BACE1 in response to chronic stress (Roßner et al., 2005; Wang et al., 2011). While Aβ has been widely demonstrated to be pro-inflammatory, the association between microglial activation and tau pathology development is still not supported by direct evidence, since neurofibrillary disorder occurs both in the presence and absence of neuroinflammation (Streit et al., 2014), and intraneuronal NFT lesions usually precede the formation of Aβ aggregates (Braak and Del Tredici, 2004).
Although it has been reported that microglia and astrocytes actively promote disease development, and play pivotal roles in amyloid deposition (Wegiel et al., 2004), conversely it has also been reported that microglia are protective and may remove amyloid deposits, thus having no effect on AD development (Simard et al., 2006; Grathwohl et al., 2009). Moreover, and interestingly, it has been suggested that infiltrating macrophages from the circulation, rather than microglia, play a central role in clearing Aβ deposits in cerebral amyloid angiopathy (Hawkes and McLaurin, 2009), but that these cells can also play a determinant role in AD development (Gate et al., 2010; Rezai-Zadeh et al., 2011).
Of note, while neurons were traditionally believed to be passive bystanders in neuroinflammation, some recent evidence suggests that not only astrocytes and microglia, but also neurons themselves, contribute to the chronic inflammation in AD, by serving as a source of inflammatory molecules (Tchelingerian et al., 1994; Yan et al., 1995; Murphy et al., 1999; Suzuki et al., 1999; Acarin et al., 2000; Heneka and O’Banion, 2007; Rubio-Perez and Morillas-Ruiz, 2012; Ramesh et al., 2013). An increase in pro-inflammatory molecule expression has also been observed in human neuroblastoma SH-SY5Y cells after incubation with some cholesterol oxidation products (known as oxysterols) potentially implicated in AD pathogenesis (Testa et al., 2014).
Brain Cholesterol Metabolism and AD
The human brain contains approximately 25% of the body’s cholesterol; this is essential for its normal functioning, being a major component of neuronal cell membranes and an essential factor in membrane fluidity. In the adult brain, cholesterol is mostly present in its non-esterified form; however, small amounts of desmosterol and cholesteryl ester are also present. The brain is separated from the peripheral circulation by the BBB, which prevents the dietary intake of cholesterol being transported from the circulation to the brain, since lipoproteins do not cross the BBB. This means that nearly all the brain cholesterol is synthesized de novo within the CNS, from 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, through a complex series of reactions involving more than 20 enzymes; cholesterol metabolism is, thus, regulated independently of that in the peripheral tissues (Vance et al., 2005; Dietschy, 2009). Because neurons do not efficiently synthesize cholesterol, they rely on astrocytes as an external source. Astrocytes meet neuronal cholesterol demands by secreting ApoE-cholesterol complexes, which are transported to the neurons for their development and function. On note, ApoE transcription is regulated by 24-hydroxycholesterol (24-OH) released by the neurons via the liver X receptor (LXR), this cholesterol oxidation product being one of its natural ligands (Pfrieger, 2003; Nieweg et al., 2009). LXR is a nuclear receptor that regulates the expression of specific genes involved in cholesterol efflux and metabolism, such as ATP-binding cassette transporters A1 and G1 (ABCA1 and ABCG1), and ApoE (Sodhi and Singh, 2013).
ApoE is the brain’s principal cholesterol-carrier protein; through its receptors it regulates the redistribution and homeostasis of cholesterol within the brain. Humans express three naturally-occurring alleles of the ApoE gene: ε2, ε3, and ε4. Murine studies have shown that astrocytes and microglia are the primary ApoE secreting cells in the brain, but neurons can express ApoE under conditions of excitotoxic injury (Xu et al., 2006).
The association between ApoE polymorphism and AD is presumably related to disorders in cholesterol transport. In this connection, it has been found that receptors recognizing ApoE are widely expressed in the brain of AD patients. Of note, AD is accelerated in ApoEε4 individuals: those who are homozygous for the ε4 allelic variant of ApoE have a 50–90% higher chance of developing AD by age 85 years than those carrying ε2 and ε3, showing that this ApoE isoform is one of the major risk factors for AD (Puglielli et al., 2003; Evans et al., 2004; Bu, 2009; Kim et al., 2009a; Martins et al., 2009; Schipper, 2011). However, the mechanisms whereby ApoE isoforms affect the risk of AD are still obscure. One hypothesis is that ApoEε4 protein accelerates the uptake of cholesterol-rich particles by the vasculature, leading to more rapid disease progression: compared to ApoEε4, ApoEε2 and ApoEε3 proteins show reduced receptor binding.
Although ApoE facilitates intracellular Aβ degradation by microglia, reducing intracellular cholesterol levels (Lee et al., 2011) by accelerating its clearance by binding Aβ and forming a stable complex (Sagare et al., 2007; Jiang et al., 2008), ApoE is, conversely, essential for Aβ aggregation and deposition, promoting Aβ fibrillization and plaque formation, as well as tau hyperphosphorylation and NFT formation (Cam and Bu, 2006; Bu, 2009; Marzolo and Bu, 2009). Of note, in the AD-affected brain, ApoE colocalizes with cholesterol and fibrillar Aβ in senile plaques (Burns et al., 2003b). It has also been shown that the ApoEε4 genotype enhances Aβ production and Aβ fibril formation in vitro, as well as in transgenic mice with mutated APP, more than in mice expressing ApoEε3 (Holtzman et al., 2000; Carter et al., 2001; Ye et al., 2005). In addition, ApoEε4 synergizes with Aβ toxicity (Ji et al., 2002). Conversely, a striking reduction in amyloid deposits has been observed in all brain regions of ApoE-null mice (Bales et al., 1999). It has also been suggested that, in AD patients with ApoEε4, a decreased ability to clear Aβ contributes to increasing Aβ accumulation and amyloid plaque formation (Deane et al., 2008). In addition, ApoE may mediate Aβ cell internalization, by binding to the low density lipoprotein receptor-related protein (LRP; Herz and Beffert, 2000). Further, like APP, ApoE also undergoes cleavage, and ApoEε4 is more susceptible to cleavage than is ApoEε3. Fragments of ApoE, such as Aβ, can be toxic, causing AD-like neurotoxicity in mouse models (Harris et al., 2003; Brecht et al., 2004); the lipid-binding region of ApoE is required for this toxicity (Chang et al., 2005).
Accumulating evidence also supports a key role for human ApoE in modulating neuroinflammation (Maezawa et al., 2006c; Keene et al., 2011; Tai et al., 2015). Many studies have reported that ApoEε4 induces a detrimental neuroinflammatory phenotype, both in peripheral and CNS inflammation. In vitro data, using microglia isolated from ApoE transgenic mice, has demonstrated ApoE genotype-specific modulation of TLR4/lipopolisaccaride (LPS) induced inflammation. In particular, LPS-induced pro-inflammatory cytokines are more strongly expressed in ApoEε4 and ApoEε3 (Maezawa et al., 2006b). Moreover, in microglia treated with LPS/interferon γ, pro-inflammatory cytokine levels were higher, and anti-inflammatory cytokine levels were lower, in ApoEε4 compared to ApoEε3. Unlike what occurs in microglia, in astrocytes TLR4/LPS-induced secretion of pro-inflammatory cytokines follows the pattern: ApoEε2 > ApoEε3 > ApoEε4 via differential nuclear factor-κB (NF-κB) activation (Maezawa et al., 2006a). With regard to ApoE modulation of Aβ-induced neuroinflammation, the available results are scarse. It has been demonstrated that impaired ApoEε4 function modulates the effects induced by Aβ on inflammatory receptor signaling, by amplifying the detrimental pathway TLR4-p38α and suppressing the beneficial pathway interleukin-4R-nuclear receptor (Tai et al., 2015). Importantly, ApoE also modulates BBB function, by a process that may be considered neuroinflammatory. It has been observed that a lack of murine ApoE combined with the expression of ApoEε4, but not of ApoEε2 nor ApoEε3, leads to BBB breakdown by activating the pro-inflammatory pathway cyclophilin A-NF-κB-matrix metalloproteinase 9 in pericytes. Consequently, blood-derived neurotoxic proteins are taken up by neurons, and microvascular and cerebral blood flows are reduced. It has been shown that the vascular defects in ApoE-deficient and ApoEε4-expressing mice precede neuronal dysfunction and can initiate neurodegenerative changes (Bell et al., 2012).
In addition to ApoE, other LXR-responsive genes involved in cholesterol efflux are the ABCA1 and ABCG1 (Voloshyna and Reiss, 2011). ABCA1 is reported to be involved in ApoE metabolism and Aβ production, as well as in the modulation of amyloid plaque formation in the CNS (Koldamova et al., 2010; Kim et al., 2011). A close correlation between Aβ and ABCA1 levels has been demonstrated, since, in cultured astrocytes, Aβ inhibits ABCA1 expression (Canepa et al., 2011). In addition, it has also been shown that AD transgenic mice lacking ABCA1 develop increased Aβ levels and senile plaques, in the absence of changes in APP processing (Wahrle et al., 2004). By contrast, in transgenic mice overexpressing ABCA1, the increased ABCA1 function significantly decreases Aβ deposition (Wahrle et al., 2008). Another ABC transporter, ABCA7, has been found to stimulate cellular cholesterol efflux to ApoE-containing particles in the same way as ABCA1 (Chan et al., 2008).
Cholesterol transport and homeostasis are thus closely linked to multiple aspects of Aβ biology, since cholesterol levels influence the production and deposition of the pathogenic Aβ peptides (Burns et al., 2003a; Hughes et al., 2014; Lane-Donovan et al., 2014). Cholesterol has indeed been shown to directly modulate the processing of APP in neuronal cell cultures (Ehehalt et al., 2003), probably by promoting β-secretase activity (Xiong et al., 2008). However, the mechanisms whereby cholesterol affects Aβ production and deposition are still not fully understood. A change in membrane properties and distribution of cholesterol has been suggested as a possible mechanism (Shobab et al., 2005). Cholesterol is mainly concentrated in membrane microdomains termed lipid rafts, which are considered to be the site of the amyloidogenic pathway (Cordy et al., 2006; Vetrivel and Thinakaran, 2010). Further, it has been shown that cellular cholesterol, especially when levels in the membrane are elevated, binds directly to APP at its C terminal transmembrane domain (Harris, 2008; Beel et al., 2010); as a consequence of the binding APP is inserted into the phospholipid monolayers of the lipid rafts and other organelles, where β- and γ-secretases reside (Wahrle et al., 2002; Beel et al., 2010). The amyloidogenic pathway of APP processing is thus linked to cholesterol levels in these microdomains: β- and γ-secretase activities are stimulated by high, and inhibited by low levels of cholesterol (Grimm et al., 2008; Xiong et al., 2008). Conversely, in the non-amyloidogenic pathway, APP is processed by α-secretase in non-raft domains, and this event is promoted by a decreased cellular cholesterol level (Reid et al., 2007). Moreover, α-secretase can be forced to associate with lipid rafts thus inactivating the amyloidogenic pathway (Harris et al., 2009). Of note, it has also been suggested that high levels of non-esterified cholesterol alone might not affect APP processing, while the conversion of free cholesterol to esterified cholesterol might up-regulate APP processing and Aβ generation (Puglielli et al., 2001). Conversely, inhibition of the enzyme acyl-coenzyme A:cholesterol acyl-transferase 1, which esterifies cholesterol, leads to the formation of smaller amounts of cholesteryl esters and Aβ (Bhattacharyya and Kovacs, 2010). These observations suggest that the balance between non-esterified and esterified cholesterol is a fundamental point controlling the amyloidogenic pathway.
It has also been shown that increased cholesterol levels in the lipid bilayers favor Aβ binding to cell membranes (Kakio et al., 2001). Additionally, cholesterol interacts with the peptide as soon as it inserts into the lipid bilayer, and accelerates its recruitment and oligomerization (Fantini and Yahi, 2010). Indeed, cholesterol enhances Aβ to form neurotoxic aggregates (Yanagisawa, 2005), and binds avidly to Aβ protofibrils at level of lipid rafts where fibrillogenesis of these peptides has been proposed to take place (Kakio et al., 2002; Harris, 2008). Conversely, other research has demonstrated that cholesterol decreases the Aβ-induced changes in structure and morphology of lipid rafts, hindering β-sheet formation in membranes, and thereby reducing peptide insertion, aggregation, and cytotoxicity (Arispe and Doh, 2002; Curtain et al., 2003; Qiu et al., 2011).
The effect of Aβ on cholesterol metabolism has also been investigated: Aβ, especially the oligomeric rather than the monomeric form, may alter the intracellular trafficking and homeostasis of cholesterol, by promoting the release from the cells of cholesterol and other lipids, in the form of Aβ-lipid complexes (Michikawa et al., 2001). It has also been reported that Aβ fibrils down-regulate cholesterol metabolism in cultured neurons (Gong et al., 2002). Additionally, the intracellular domain of APP, which is released upon γ-secretase cleavage of APP, may act as a transcriptional suppressor of LRP1, leading to the down-regulation of cellular cholesterol uptake (Liu et al., 2007) and synaptic failure (Koudinov and Koudinova, 2005), as well as enhancement of tau phosphorylation (Fan et al., 2001). It has also been shown that extracellular cholesterol accumulates in the senile plaques of AD patients, and in transgenic mice expressing the Swedish Alzheimer mutation APP751, by binding to aggregated Aβ (Mori et al., 2001).
Given the above considerations, it appears that cholesterol distribution and trafficking within brain cells, rather than the total amount of cholesterol in the neurons, play key roles in APP processing and in the amyloid cascade during AD progression. Conversely, Aβ may affect cellular cholesterol dynamics, such as transport, distribution, and binding, which in turn have a variety of effects on AD-related pathologic changes leading to neurodegeneration.
A number of epidemiological studies also suggest a positive correlation between hypercholesterolemia and susceptibility to AD, in particular in individuals with the ApoEε4 genotype, which influences cholesterol metabolism, although this relationship is still the subject of considerable controversy (Shobab et al., 2005; Panza et al., 2006; Jenner et al., 2010; Wood et al., 2014; Luckhoff et al., 2015).
Elevated dietary cholesterol has been reported to increase senile plaque formation in numerous animal models. In double transgenic (APP/PS) mice, consumption of a high-cholesterol diet (7 weeks) elevated Aβ deposition in the CNS (Refolo et al., 2000). In addition, in transgenic mice, a typical Western diet (1% cholesterol), increased Aβ accumulation and plaque burden, particularly in the dentate gyrus of the hippocampus (Hooijmans et al., 2007). APP23 mice fed a high-fat/high-cholesterol diet for 4 months showed the AD phenotype. This resulted in significantly worse memory deficits than in the same mice fed with a normal diet (Fitz et al., 2010). Furthermore, studies using New Zealand white rabbits have demonstrated that a diet inducing hypercholesterolemia doubles the Aβ concentration in the hippocampal cortex (Sparks et al., 2000). In addition to increasing Aβ, cholesterol-enriched diets increased tau phosphorylation and oxidative stress in rabbit brains (Ghribi et al., 2006; Jaya Prasanthi et al., 2008). It has also been shown that cholesterol colocalizes with fibrillar Aβ in the amyloid plaques of transgenic mice (Burns et al., 2003b). Conversely, a study on guinea pigs showed that lowering total cholesterol profile by administering statins decreased cerebral Aβ production and accumulation (Fassbender et al., 2001).
Although systemic lipoprotein carrying cholesterol cannot cross the BBB, oxysterols, formed during oxidative stress, can cross the BBB, partly because they may have damaging effects on the BBB’s integrity and function (Dias et al., 2014). This consideration supports the idea that oxysterols are the link between hypercholesterolemia and AD.
The Involvement of Oxysterols in AD Pathogenesis
To maintain cholesterol homeostasis in the brain, and because the brain cannot degrade cholesterol, there must be a mechanism to eliminate excess cholesterol, transporting it through the BBB into the systemic circulation, thus preventing its accumulation. The most important such mechanism operates via the conversion of cholesterol to oxysterols. Cholesterol is converted into the relatively polar oxysterol 24-OH, which is produced in the brain almost exclusively by cholesterol 24-hydroxylase (CYP46A1) expressed in neurons, and which, unlike cholesterol itself, diffuses across the BBB into the systemic circulation. To a lesser extent, cholesterol is also converted into 27-OH in the brain by cholesterol 27-hydroxylase (CYP27A1), and then into 7α-hydroxy-3-oxo-4-cholestenoic acid (7-OH-4-C) by the enzyme CYP7B; crossing the BBB, 7-OH-4-C reaches the liver where it is eliminated (Björkhem, 2006; Meaney et al., 2007; Björkhem et al., 2009). However, most 27-OH has been found to flow from the systemic circulation into the brain, since it can cross the BBB; here it acts as an important link between extracerebral and intracerebral pools of cholesterol, and may contribute to the negative effects of hypercholesterolemia in the brain (Heverin et al., 2005; Björkhem, 2006; Sharma et al., 2008). A further oxysterol, 7β-hydroxycholesterol (7β-OH), derives from cholesterol oxidation in the brain, following its interaction with APP and Aβ (Nelson and Alkon, 2005). In addition to those oxysterols, others including 7α-hydroxycholesterol (7α-OH), 4β-hydroxycholestrerol (4β-OH), 5α, 6α- and 5β, 6β-epoxicholesterol (α- and β-EPOX) and 7-ketocholesterol (7-K), have recently been identified post-mortem in human AD brains, and their concentrations compared across the disease states (Hascalovici et al., 2009). Of note, the study authors observed that the levels of potentially amyloidogenic sterol species derived from the auto-oxidation of cholesterol, like β-EPOX, were higher at the mild cognitive impairment disease stage. Oxysterols have also been identified in mouse brains and, in addition to the above cited cholesterol derivatives, consistent levels of 25-hydroxycholesterol (25-OH) were found (Ahonen et al., 2014). Increased levels of 24-OH, 7-K, and β-EPOX have also been detected in areas of the rat hippocampus undergoing gliosis and neuroinflammation, after excitotoxic injury (He et al., 2006; Kim et al., 2010). Further, because oxysterols can cross the BBB, the flux of more than 20 cholesterol metabolites between brain and circulation has very recently been verified in 20 patients. Differences in concentrations, between jugular and forearm veins, of 18 oxysterols, 5 cholestenoic acids and 3 cholenoic acids were measured. The study reported that 24-OH and 7-OH-4-C, of enzymatic origin, but also 6-oxo-5α-hydroxycholesterol, 7β-OH, 7-K, which are formed from cholesterol by ROS, are exported from the brain, while 27-OH is imported into brain (Iuliano et al., 2015). Of these cholesterol metabolites, that exported in the largest quantities is 24-OH. Two other cholesterol metabolites, 7α, 25-dihydroxycholest-4-en-3-one and 7α, (25R)26-hydroxycholest-4-en-3-one, were reported to be exported from the brain (Crick et al., 2015b). Oxysterols and cholestenoic acids have also been identified and quantified in mouse cerebrospinal fluid (CSF), and the findings compared with concentrations of the same metabolites found in the plasma, in order to clarify cholesterol metabolism. Concentrations of oxysterols were lower in the CSF than in the plasma, but 7α, 24-dihydroxycholesterol and 7α, 24-dihydroxycholest-4-en-3-one, both of enzymatic origin, were only identified in the CSF (Crick et al., 2015a). These data clearly demonstrate that there are several routes by which cholesterol metabolites may be exported or imported from the brain (Figure 2).
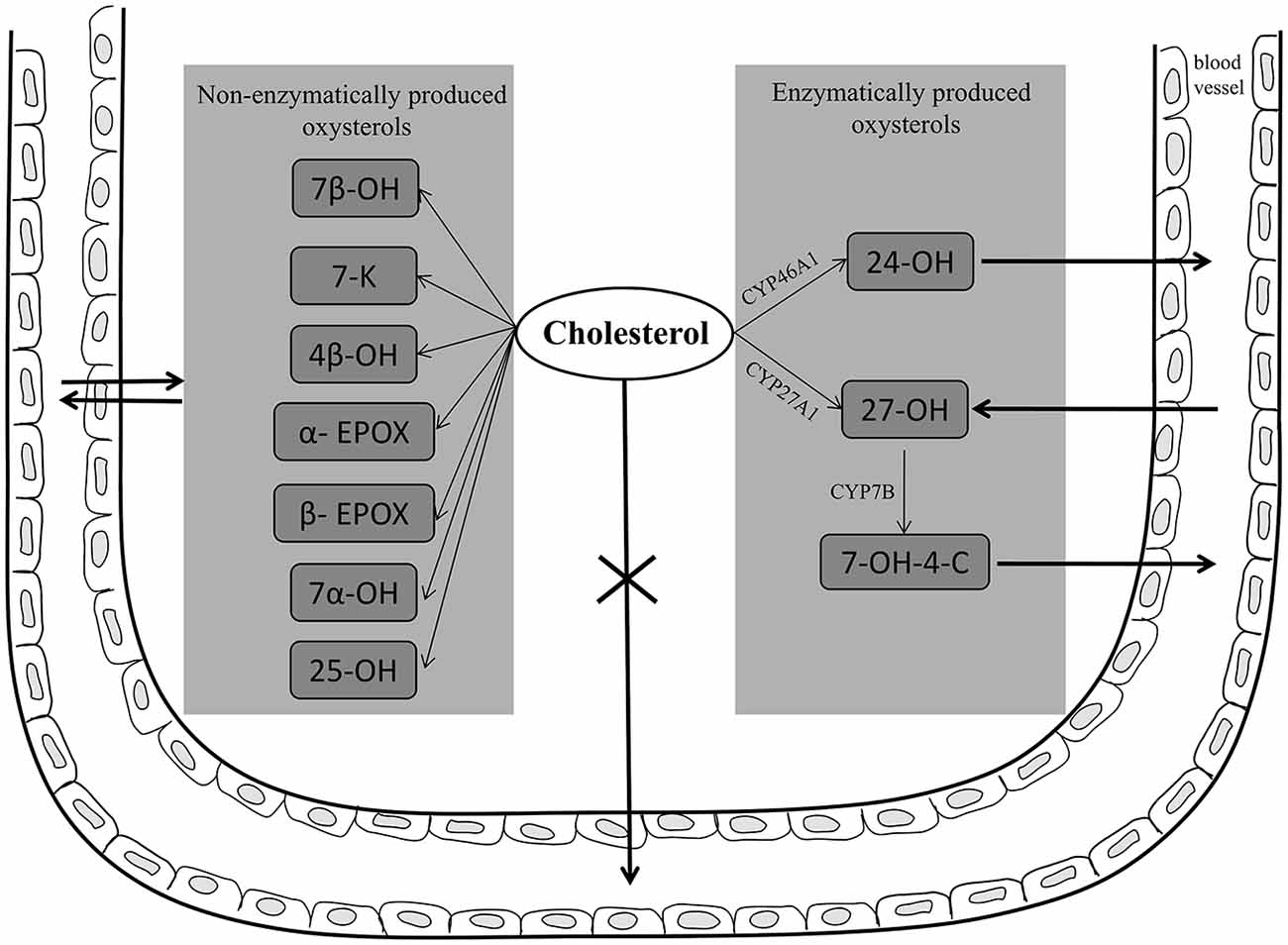
Figure 2. Enzymatically and non-enzymatically produced oxysterols in AD brain and their fluxes across the blood brain barrier (BBB). In neuronal cells, cholesterol is converted into 24-hydroxycholesterol (24-OH) by the enzyme CYP46A1; 24-OH, unlike cholesterol, diffuses across the BBB into the systemic circulation. To a lesser extent, cholesterol is also converted into 27-hydroxycholesterol (27-OH) by the enzyme CYP27A1, and then into 7α-hydroxy-3-oxo-4-cholestenoic acid (7-OH-4-C) by the enzyme CYP7B; crossing the BBB, 7-OH-4-C reaches the liver where it is eliminated. However, most 27-OH flows from the circulation into the brain, since it can cross the BBB. In addition, other oxysterols, such as 7β-hydroxycholesterol (7β-OH), 7-ketocholesterol (7-K), 7α-hydroxycholesterol (7α-OH), 4β-hydroxycholestrerol (4β-OH), 5α, 6α- and 5β, 6β-epoxicholesterol (α- and β-EPOX), and 25-hydroxycholesterol (25-OH), have been found in AD brain deriving from brain cholesterol autoxidation. Potentially these oxysterols, as well as other cholesterol metabolites, can cross the BBB.
The idea has therefore gained ground that, owing to their ability, unlike cholesterol, to cross the BBB, oxysterols might be the missing link between altered brain cholesterol metabolism and AD pathogenesis, as well as between hypercholesterolemia and AD (Gamba et al., 2012). Although this means that the brain can eliminate excess amounts of oxysterols, it could conversely allow toxic amounts of these compounds, present in the bloodstream, to accumulate in the brain, as in the case of 27-OH. The key role of oxysterols in AD pathogenesis has been strongly supported by the last decade’s research, pointing to the involvement of these oxysterols in the amyloid cascade.
To date, although contradictory results could obviously arise from future findings on the other cholesterol metabolites found in the brain, the oxysterols most widely considered to be potentially implicated in the pathogenesis of AD are 24-OH and 27-OH, both of enzymatic origin (Iuliano, 2011; Jeitner et al., 2011; Leoni and Caccia, 2011; Gamba et al., 2012; Hughes et al., 2013; Marwarha and Ghribi, 2014; Noguchi et al., 2014; Table 1).
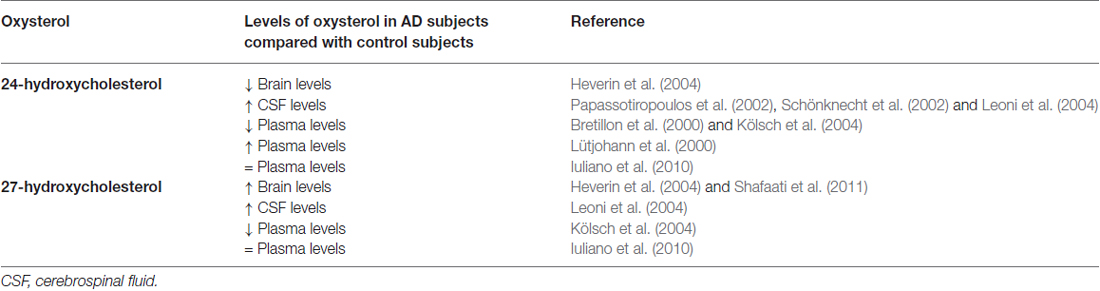
Table 1. 24-hydroxycholesterol and 27-hydroxycholesterol levels in Alzheimer’s disease patients compared with healthy controls.
Higher levels of 24-OH than in unaffected individuals have been found in the peripheral circulation and CSF of early-stage AD patients, suggesting that cholesterol turnover in the brain increases during the neurodegenerative changes of AD (Lütjohann et al., 2000; Papassotiropoulos et al., 2002; Kölsch et al., 2004). Conversely, plasma levels of 24-OH were lower in patients with later stages of AD than in unaffected individuals, suggesting that the rate of cholesterol transport slows as the disease progresses (Bretillon et al., 2000; Kölsch et al., 2004). These apparently contradictory results may be rationalized by considering that increased plasma levels of 24-OH reflect ongoing neurodegeneration and/or demyelinization, whereas decreased plasma levels in later stages reflect a selective loss of neuronal cells expressing the enzyme CYP46A1 (Björkhem and Meaney, 2004). The decrease of 24-OH in the AD brain may also be the result of an increase in total free cholesterol (likely derived from cell membrane collapse and widespread myelin release) that exceeds the brain’s capacity to convert it to 24-OH (Vaya and Schipper, 2007). However, it has been observed that, in glial cells, and especially around senile plaques, there is an ectopic induction of CYP46A1, leading to some 24-OH production, but without compensating for the decrease of that oxysterol (Bogdanovic et al., 2001; Brown et al., 2004). Another study, however, has found that plasma levels of 24-OH and 27-OH in AD patients are not significantly different from control values (Iuliano et al., 2010). A small fraction of total 24-OH excretion occurs via the CSF, and the 24-OH concentration is increased in the CSF of AD patients, probably reflecting neuronal damage and loss rather then metabolically active neuronal cells (Schönknecht et al., 2002; Leoni et al., 2004).
It has also been reported that, in critical areas of post-mortem AD brains, as well as in aged mice expressing the Swedish Alzheimer mutation APP751, 24-OH levels are decreased and those of 27-OH increased (Heverin et al., 2004). Increased levels of 27-OH were also found in the brains of patients carrying the Swedish APP670/671 mutation (Shafaati et al., 2011). As a consequence of neuron loss, expression of CYP27A1 may also be reduced; however, 27-OH levels remain elevated because CYP27A1 is also expressed in astrocytes and oligodendrocytes, leading to in situ generation of 27-OH (Brown et al., 2004). Accumulation of 27-OH in the brain is also due to the increased flux of this oxysterol across the BBB, because of either hypercholesterolemia associated to oxidative stress (Björkhem, 2006), or damaged BBB integrity (Leoni et al., 2003). There is a positive correlation between levels of cholesterol and those of 27-OH in the circulation, and the high flux of 27-OH from the peripheral circulation into the brain suggests that this oxysterol may be the link between hypercholesterolemia and AD. An alternative explanation for the accumulation of 27-OH is reduced activity of CYP7B, the neuronal enzyme responsible for 27-OH metabolism; this reduction arises from the reduced CYP7B expression in the brain of AD patients, because of neuron loss (Yau et al., 2003). Moreover, high CSF levels of 27-OH were found in mild cognitive impairment and AD patients (Leoni et al., 2004).
From these considerations, it is clear that the balance between 24-OH and 27-OH levels is important. Oxysterol homeostasis in the brain is tightly regulated, specific levels being maintained in various brain regions. For example, the 27-OH:24-OH ratio is ~1:8 in the frontal cortex, 1:5 in the occipital cortex, and 1:10 in the basal ganglia, and the increased ratio of 27-OH to 24-OH in AD brains is consistent with AD pathogenesis. Thus it is likely that reduced levels of 24-OH may accelerate disease progression, and that the increased levels of 27-OH may be insufficient to compensate for this: the shift in balance between the two oxysterols might lead to increased generation and accumulation of Aβ with consequent neurodegeneration (Heverin et al., 2004; Björkhem, 2006; Björkhem et al., 2009). In a retrospective study on cardiovascular patients with evidence of cerebrovascular disease, higher plasma levels of 24-OH and a higher 24-OH/27-OH ratio were found to be associated with the development of incidental cognitive impairment over 8 years of follow-up (Hughes et al., 2012). However, opinions still differ about the involvement of 24-OH and 27-OH in APP processing and Aβ production.
To date, the conversion of cholesterol into 24-OH, by inducing CYP46A1 activity, has been considered to exert a protective action on the brain, mainly by regulating cholesterol homeostasis and favoring the efflux of its excess from the brain to the blood, but also by preventing Aβ generation (Björkhem et al., 2009). Astrocytes are sensitive to levels of 24-OH, which regulates the expression of LXR-responsive genes involved in cholesterol homeostasis (i.e., ABCA1, ABCG1 and ApoE) (Abildayeva et al., 2006). Indeed, 24-OH acts as an endogenous ligand of LXR. Conversely, it has been reported that brain accumulation of 27-OH antagonizes the preventive effect of 24-OH on generation of Aβ and that it is potentially toxic (Shafaati et al., 2011). Since the flux of 27-OH across the BBB increases under conditions of hypercholesterolemia (Björkhem, 2006), or in the case of increased BBB permeability (Leoni et al., 2003), the inhibitory effect of 24-OH on Aβ generation would consequently be reduced.
Studies on human neuroblastoma cells and on brain tissues have somewhat clarified the different effects of 24-OH and 27-OH on APP levels and processing: 24-OH may favor the non-amyloidogenic pathway, with consequent inhibition of Aβ formation, whereas 27-OH is thought to stimulate the amyloidogenic pathway, with production of Aβ as well as tau hyperphosphorylation (Bu, 2009; Prasanthi et al., 2009; Marwarha et al., 2010). However, the mechanisms underlying their different effects are still unclear.
In human SH-SY5Y neuroblastoma cells, 24-OH appears to exert a unique modulatory effect on APP processing: it directly increases α-secretase activity, as well as elevating the α/β-secretase activity ratio; conversely, 27-OH enhances the generation of Aβ (Famer et al., 2007). In vitro experiments suggest that 24-OH reduces Aβ production, by down-regulating APP trafficking via enhancement of the complex formation of APP, also up-regulating glucose-regulated protein 78 in the endoplasmic reticulum. The inhibitory effect of 24-OH was reduced in glucose-regulated protein 78 knockdowned cells (Urano et al., 2013). In rat primary cortical neurons, both 24-OH and 27-OH were found to be inhibitors of Aβ secretion, 24-OH being approximately 1000 times more potent than 27-OH (Brown et al., 2004). SH-SY5Y cells incubated with 27-OH release higher levels of Aβ1–42 and APP as well as of BACE1. Conversely, cells incubated with 24-OH do not release increased Aβ1–42 levels, and are associated with increased levels of sAPPα, suggesting that 24-OH favors APP processing via the non-amyloidogenic pathway (Prasanthi et al., 2009). Another study, on hippocampal slices from adult rabbits, found that 27-OH increases Aβ accumulation by reducing levels of insulin-like growth factor 1, a neurotrophic factor that promotes neurogenesis and has a neuroprotective effect (Sharma et al., 2008). This oxysterol has also been found to increase both BACE1 and Aβ levels in retinal pigment epithelial cells (Dasari et al., 2010). In neuronal SK-N-BE cells, 24-OH and 27-OH have both been shown to enhance expression and activity of the β-secretase of the amyloidogenic pathway of APP processing, leading to increased Aβ generation and accumulation in those cells (Gamba et al., 2014; Table 2).
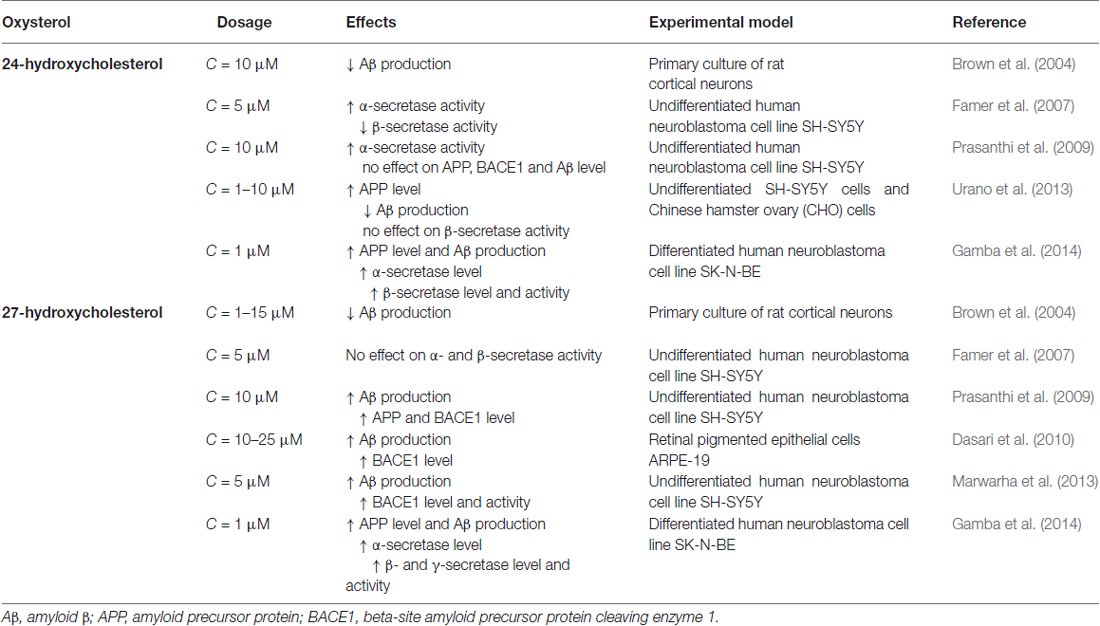
Table 2. Effects of 24-hydroxycholesterol and 27-hydroxycholesterol on the amyloidogenic pathway.
24-OH has also been reported to be neurotoxic, but this effect may depend on its concentration. It has been demonstrated that high concentrations of 24-OH (25–50 μM) caused cell death when added to undifferentiated or differentiated SH-SY5Y cells: the effect was mediated by increased generation of free radicals (Kölsch et al., 2001). High concentrations of 24-OH (50 μM) also induce necroptosis, a form of programmed necrosis in neuronal SH-SY5Y cells (Yamanaka et al., 2011). In contrast, pretreatment of human neuroblastoma SH-SY5Y cells with sub-lethal concentrations of 24-OH induces adaptive responses, and protects the cells against subsequent cytotoxic stress induced by 7-K treatment, via transcriptional activation of the LXR signaling pathway. The cytoprotective effects of 24-OH disappeared in LXRβ-knockdowned cells, suggesting that this nuclear receptor may play a key role in the 24-OH-induced adaptive response. Adaptive responses are also induced by other oxysterols, such as 25-OH and 27-OH, both ligands of LXR, similarly to 24-OH (Okabe et al., 2013). However, in our recent studies, a very low concentration (1 μM) of 24-OH was found to markedly potentiate both the apoptotic and the necrogenic effects exerted by the Aβ1–42 peptide, on two human differentiated neuronal cell lines (SK-N-BE and NT-2) (Gamba et al., 2011), but also on human dental pulp progenitor cells differentiated into neuron-like cells (Testa et al., 2012). 24-OH appeared to interact with Aβ1–42 by strongly increasing intracellular ROS steady-state levels, an action not exerted by either 27-OH or 7β-OH (Gamba et al., 2011). This evidence supports the opinion that Aβ must form oligomers in order to induce neurotoxicity, and that the latter process is probably enhanced by redox imbalance. Additionally, 50 μM 24-OH has been shown to enhance the neurotoxic effect of the Aβ1–42 peptide in the human differentiated neuroblastoma cell line MSN, as well as augmenting ROS generation (Ferrera et al., 2008). Although in many of the various in vitro tests performed in our laboratory, 27-OH (1 μM) did not display neurotoxicity, in terms of necrosis and apoptosis (Gamba et al., 2011), conversely, the toxicity of 27-OH (5–20 μM) has been demonstrated in astrocyte cells (C6 cells). This oxysterol increased ROS levels and decreased antioxidant defense system levels, with consequent decrease of cell viability. In addition, 27-OH down-regulated the expression of the nuclear factor E2-related factor 2 signaling pathway (Ma et al., 2015).
It is known that hypertension is a risk factor for AD, and that angiotensin converting enzyme activity is increased in AD brains. In this connection, it has been suggested that 27-OH might up-regulate the renin-angiotensin system in AD brains. A positive correlation between angiotensin converting enzyme activity in the CSF, and both plasma and CSF levels of 27-OH, has been shown, as well as an increased production of angiotensinogen in rat primary neurons, astrocytes, and human neuroblastoma cells treated with 27-OH (Mateos et al., 2011).
Moreover, 7β-OH has been found to be neurotoxic at very low concentrations on cultured rat hippocampal neuronal cells, and may therefore contribute to neurodegeneration in AD brains. In the same study, it has also been shown that Aβ can oxidize cholesterol to form 7β-OH in a highly efficient mechanism and more actively than APP. Oxidation of cholesterol was accompanied by hydrogen peroxide production, suggesting that Aβ could contribute to the oxidative damage observed in AD (Nelson and Alkon, 2005). 7β-OH causes re-arrangement of the liquid-ordered phase which results in the formation of lipid rafts (Wang et al., 2008; Mitomo et al., 2009); it is also a potent inhibitor of α protein kinase C, an enzyme critical for memory consolidation and synaptic plasticity that is implicated in AD (Nelson and Alkon, 2005). Another oxysterol that might derive from the autoxidation of cellular cholesterol released during neurodegeneration is 7α-OH, which appeared to be responsible for SH-SY5Y cell death (Kölsch et al., 2000); a further possibility is 7-K. 7-K has been shown to enhance mitochondrial dysfunction in the neuronal PC12 cell line, leading to cell death (Kim et al., 2006; Kim and Lee, 2010; Jang and Lee, 2011). It has also been reported that incorporation of 7-K to lipid raft domains of plasma membranes triggers apoptotic signaling; α-tochopherol (vitamin E) reduces the cytotoxicity of 7-K by inhibiting its distribution to the lipid raft domains (Berthier et al., 2004; Royer et al., 2009). It has recently been suggested that 25-OH is an important regulator of cholesterol metabolism, as well as of humoral immunity (Diczfalusy, 2013; Waltl et al., 2013). Of note, it has also been hypothesized that Aβ deposition is not a central event in AD, but rather is subservient to 25-OH. Confirming this hypothesis, it has been observed that, in a large cohort of AD patients, specific 25-hydroxylase haplotypes were associated with a complete absence of Aβ deposits in the brain, despite all other aspects of AD pathology being present (Papassotiropoulos et al., 2005). This suggests that neuroinflammation and 25-hydroxylase activation precede Aβ generation in the sequence of events leading to the disease. An interesting study on different immortalized, tumoral and normal cells of the CNS has found that oxysterols oxidized at C4, such as 4α-OH and 4β-OH, have no effect on cell viability and almost no effect on cell growth; conversely, oxysterols oxidized at C7, such as 7-K, 7α-OH, and 7β-OH, inhibit cell growth and decrease viability through their cytotoxic activity. These data suggest that 4α-OH and 4β-OH, the only oxysterols identified as having cytostatic properties, may be of some interest for attempts to counteract cell proliferation (Nury et al., 2013).
Oxysterols have also been shown to modify specific sites of the Aβ peptide thus enhancing Aβ aggregation and its neurotoxicity. Following Aβ modification at Lys-16, peptide aggregates were formed faster than in the case of modification at Lys-28 or at Asp-1 (Usui et al., 2009).
Further, evidence has emerged that Aβ has predominant cholesterol oxidase activity, particularly in the presence of divalent cations such as Cu2+. Significantly elevated levels of 4-cholesten-3-one were reported in brains of Aβ transgenic mice and in brain tissue of AD patients (Puglielli et al., 2005; Yoshimoto et al., 2005).
The interaction of Aβ with cell membranes is the crucial event in AD pathogenesis. Of note, there is less evidence to date on the negative effects of oxysterols on Aβ binding to the cell membranes. Because the orientation of oxysterols in the cell membrane differs from that of cholesterol, they are less able to condense lipids, thus modifying some physical properties of membrane, including raft domains; it has thus been suggested that oxysterols may facilitate Aβ interaction with cell membranes. The effects of 7-K and 7β-OH on enhancing Aβ insertion into the lipid bilayer, by decreasing intermolecular cohesive interaction, have been demonstrated (Kim and Frangos, 2008). Using a model membrane, it was shown that 25-OH and 7-K render the membrane more sensitive to Aβ, in contrast to the role played by cholesterol, which inhibits Aβ’s interaction with membranes. 7-K facilitated Aβ’s localization in the membrane, while 25-OH stimulated the peptide’s insertion but lead to membrane modification. In addition, the higher potential of Aβ1–42, compared to Aβ1–40, to interact with the membrane has also been demonstrated (Phan et al., 2013). Further, it is hypothesized that increased oxysterol concentrations, mainly of 7-K, but also of 24-OH and β-EPOX, may enhance exocytosis and neurotransmitter release in damaged areas of the brain, thereby aggravating neuronal excitotoxicity (Ma et al., 2010). Further, in our study we observed that 24-OH, 27-OH, and 7β-OH markedly enhanced the binding of Aβ1–42 on membranes of human differentiated neuronal cell lines (SK-N-BE and NT-2), by up-regulating CD36 and β1-integrin receptors (Gamba et al., 2011), two components of the multireceptor complex CD36/β1-integrin/CD47, through which Aβ peptide binds to cell membranes (Verdier et al., 2004; Yu and Ye, 2015). This event might favor the accumulation of the toxic Aβ1–42 peptide into neurons.
Although oxysterols have been analyzed for their involvement in neurotoxicity and Aβ production during AD progression, their role as natural ligands for LXR is now emerging (e.g., 24-OH, 27-OH, 22-OH, 25-OH, 4β-OH, and 7α-OH) (Vaya and Schipper, 2007). Indeed, astrocytes are sensitive to 24-OH-mediated up-regulation of ApoE, a LXR-target gene involved in cholesterol efflux (Abildayeva et al., 2006). Moreover, it has been reported that 27-OH prevents Aβ generation from primary human neurons, not by modulating α-, β-, or γ-secretase, but rather by overexpressing LXR-responsive genes (ABCA1, ABCG1 and ApoE) (Kim et al., 2009b). Moreover, incubation of primary brain cells with 22-OH significantly reduced Aβ secretion in a dose-dependent manner, while ABCA1 expression and cholesterol efflux were induced (Koldamova et al., 2003).
Recent in vitro evidence also suggests that 24-OH and 27-OH might contribute to decreasing the influx of Aβ peptide into the brain across the BBB, increasing expression of the ABCB1 transporter in brain capillary endothelial cells, resulting in protection from peripheral Aβ entry (Saint-Pol et al., 2013). Of note, ABCB1 has never been described as an LXR target gene, and other nuclear receptors might control its transcription. Conversely, treatment of brain pericytes with 24-OH up-regulated ABCA1 expression that was correlated with an increase of cholesterol efflux, whereas 24-OH treatment did not reduce the pericytes’ ability to accumulate Aβ in the cells (Saint-Pol et al., 2012). The clearance of Aβ might also be mediated through microglia-induced phagocytosis of Aβ, which depends on LXR activation (Terwel et al., 2011).
Of note, LXR activation not only regulates cholesterol homeostasis, and Aβ peptide transport and clearance, but also neuroinflammation. Studies have shown that LXR activation inhibits inflammatory gene expression, pointing to the ability of LXRs to inactivate promoters of pro-inflammatory genes (Wang et al., 2002; Cao et al., 2007; Zelcer et al., 2007; Sodhi and Singh, 2013; Steffensen et al., 2013). Moreover, LXR activation may prevent neuroinflammation, by indirectly down-regulating TLR target genes. However, although LXR-activating oxysterols might reduce membrane cholesterol content and inflammation, they may also activate opposing pathways, and induce inflammation independently of LXRs. In our very recent study, we observed that 27-OH, 24-OH, and 7β-OH enhanced inflammatory molecule expression in human neuroblastoma SH-SY5Y cells via TLR4/cyclooxygenase-2/membrane bound prostaglandin E synthase; this clearly indicates that oxysterols may promote neuroinflammation in AD (Testa et al., 2014).
Although it can be assumed that oxysterols may increase the activation of microglia promoting their phagocytosis, there is less evidence to date on their effects on microglial phagocytosis during neuroninflammation. The phagocytosis of fibrils and large aggregates of Aβ by microglia is an important neuroprotective mechanism for Aβ peptide clearance (D’Andrea et al., 2004; Colton and Wilcock, 2010) but, in later stages of AD, the increased inflammatory molecule release makes the microglia phagocytically inactive leading to neuronal death (Hickman et al., 2008; Krabbe et al., 2013). Among the receptors promoting Aβ phagocytosis and clearance by microglia, the CD36 scavenger receptor appears to be involved and its increased expression may be crucial in preventing AD (Verdier et al., 2004; Yu and Ye, 2015): CD36 initiates a signaling cascade that promotes microglial activation and recruitment to β-amyloid deposits in the brain (Stuart et al., 2007). Concernig sterols, it has been shown that cholesterol (20 μM) and α-EPOX (20 μM) do not interfere with CD36 membrane distribution but both compounds were found to up-regulate the total CD36 levels in the mouse microglial cell line BV-2 potentiating phagocytosis in LPS-stimulated cells (Račková, 2013). Moreover, treatment with methyl-β-cyclodextrin, a reagent able to remove cholesterol from cell membranes, inhibited phagocytosis in LPS-activated microglia, indirectly supporting the potential role of sterols in phagocytosis (Churchward and Todd, 2014).
Conclusion
This review has pointed up the vicious circle connecting oxidative stress and inflammation in AD. Alongside oxidative stress and neuroinflammation, altered cholesterol metabolism in the brain and hypercholesterolemia also significantly contribute to AD pathogenesis. Thanks to consistent research evidence, it is now believed that oxidized cholesterol is the driving force behind the development of AD and that oxysterols are the link connecting altered cholesterol metabolism and hypercholesterolemia to this neurodegenerative disease. Oxysterols play a fundamental role, by enhancing inflammation, Aβ generation and accumulation, and neuron death.
The involvement of oxysterols in AD pathogenesis, and the analysis of such products in the plasma and CSF, may contribute to clarifying the role of cholesterol metabolism in AD; ultimately, it may be helpful in developing therapeutic strategies to prevent or slow AD pathogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the CRT Foundation (Turin) and the University of Turin (Italy).
Abbreviations
α-EPOX, 5α, 6α-epoxicholesterol; β-EPOX, 5β, 6β-epoxicholesterol; 24-OH, 24-hydroxycholesterol; 25-OH, 25-hydroxycholesterol; 4β-OH, 4β-hydroxycholestrerol; 7β-OH, 7β-hydroxycholesterol; 7α-OH, 7α-hydroxycholesterol; 7-OH-4-C, 7α-hydroxy-3-oxo-4-cholestenoic acid; 7-K, 7-ketocholesterol; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; AD, Alzheimer’s disease; ApoE, apolipoprotein E; APP, amyloid precursor protein; Aβ, amyloid β; BACE1, beta-site amyloid precursor protein cleaving enzyme 1; BBB, blood-brain barrier; CNS, central nervous system; CSF, cerebrospinal fluid; CYP27A1, 27-hydroxylase; CYP46A1, 24-hydroxylase; LPS, lipopolisaccaride; LXR, liver X receptor; NF-κB, nuclear factor-κB; NFT, neurofibrillary tangles; NO, nitric oxide; PSEN1, presenilin 1; PSEN2, presenilin 2; RNS, reactive nitrose species; ROS, reactive oxygen species; TLR, Toll-like receptor.
References
Abbas, N., Bednar, I., Mix, E., Marie, S., Paterson, D., Ljungberg, A., et al. (2002). Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. 126, 50–57. doi: 10.1016/s0165-5728(02)00050-4
Abildayeva, K., Jansen, P. J., Hirsch-Reinshagen, V., Bloks, V. W., Bakker, A. H., Ramaekers, F. C., et al. (2006). 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 281, 12799–12808. doi: 10.1074/jbc.m601019200
Acarin, L., González, B., and Castellano, B. (2000). Neuronal, astroglial and microglial cytokine expression after an excitotoxic lesion in the immature rat brain. Eur. J. Neurosci. 12, 3505–3520. doi: 10.1046/j.1460-9568.2000.00226.x
Ahonen, L., Maire, F. B., Savolainen, M., Kopra, J., Vreeken, R. J., Hankemeier, T., et al. (2014). Analysis of oxysterols and vitamin D metabolites in mouse brain and cell line samples by ultra-high-performance liquid chromatography-atmospheric pressure photoionization-mass spectrometry. J. Chromatogr. A. 1364, 214–222. doi: 10.1016/j.chroma.2014.08.088
Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell. Biol. 161, 41–54. doi: 10.1083/jcb.200207030
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
Arispe, N., and Doh, M. (2002). Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AbetaP (1-40) and (1-42) peptides. FASEB J. 16, 1526–1536. doi: 10.1096/fj.02-0829com
Azizi, G., and Mirshafiey, A. (2012). The potential role of proinflammatory and antiinflammatory cytokines in Alzheimer disease pathogenesis. Immunopharmacol. Immunotoxicol. 34, 881–895. doi: 10.3109/08923973.2012.705292
Bales, K. R., Verina, T., Cummins, D. J., Du, Y., Dodel, R. C., Saura, J., et al. (1999). Apolipoprotein E is essential for amyloid deposition in the APP (V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 96, 15233–15238. doi: 10.1073/pnas.96.26.15233
Beel, A. J., Sakakura, M., Barrett, P. J., and Sanders, C. R. (2010). Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim Biophys. Acta 1801, 975–982. doi: 10.1016/j.bbalip.2010.03.008
Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. doi: 10.1038/nature11087
Berthier, A., Lemaire-Ewing, S., Prunet, C., Monier, S., Athias, A., Bessède, G., et al. (2004). Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ. 11, 897–905. doi: 10.1038/sj.cdd.4401434
Bhattacharyya, R., and Kovacs, D. M. (2010). ACAT inhibition and amyloid beta reduction. Biochim. Biophys. Acta 1801, 960–965. doi: 10.1016/j.bbalip.2010.04.003
Björkhem, I. (2006). Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Intern. Med. 260, 493–508. doi: 10.1111/j.1365-2796.2006.01725.x
Björkhem, I., and Meaney, S. (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815. doi: 10.1161/01.atv.0000120374.59826.1b
Björkhem, I., Cedazo-Minguez, A., Leoni, V., and Meaney, S. (2009). Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 30, 171–179. doi: 10.1016/j.mam.2009.02.001
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Bogdanovic, N., Bretillon, L., Lund, E. G., Diczfalusy, U., Lannfelt, L., Winblad, B., et al. (2001). On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci. Lett. 314, 45–48. doi: 10.1016/s0304-3940(01)02277-7
Bonda, D. J., Wang, X., Perry, G., Nunomura, A., Tabaton, M., Zhu, X., et al. (2010). Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59, 290–294. doi: 10.1016/j.neuropharm.2010.04.005
Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G. Q., Xu, Q., et al. (2004). Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534. doi: 10.1523/jneurosci.4315-03.2004
Bretillon, L., Sidén, A., Wahlund, L. O., Lütjohann, D., Minthon, L., Crisby, M., et al. (2000). Plasma levels of 24S-hydroxycholesterol in patients with neurological diseases. Neurosci. Lett. 293, 87–90. doi: 10.1016/s0304-3940(00)01466-x
Brown, G. C., and Bal-Price, A. (2003). Inflammatory neurodegeneration mediated by nitric oxide, glutamate and mitochondria. Mol. Neurobiol. 27, 325–355. doi: 10.1385/mn:27:3:325
Brown, J. 3rd, Theisler, C., Silberman, S., Magnuson, D., Gottardi-Littell, N., Lee, J. M., et al. (2004). Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J. Biol. Chem. 279, 34674–34681. doi: 10.1074/jbc.m402324200
Braak, H., and Del Tredici, K. (2004). Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-beta formation. Neurobiol. Aging 25, 713–718. doi: 10.1016/j.neurobiolaging.2003.12.015
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burns, M. P., Noble, W. J., Olm, V., Gaynor, K., Casey, E., LaFrancois, J., et al. (2003b). Co-localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques. Brain Res. Mol. Brain Res. 110, 119–125. doi: 10.1016/s0169-328x(02)00647-2
Burns, M., Gaynor, K., Olm, V., Mercken, M., LaFrancois, J., Wang, L., et al. (2003a). Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 23, 5645–5649.
Butterfield, D. A., and Boyd-Kimball, D. (2005). The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 1703, 149–156. doi: 10.1016/j.bbapap.2004.10.014
Butterfield, D. A., and Kanski, J. (2001). Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 122, 945–962. doi: 10.1016/s0047-6374(01)00249-4
Butterfield, D. A., Drake, J., Pocernich, C., and Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol. Med. 7, 548–554. doi: 10.1016/s1471-4914(01)02173-6
Butterfield, D. A., Swomley, A. M., and Sultana, R. (2013). Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 19, 823–835. doi: 10.1089/ars.2012.5027
Cam, J. A., and Bu, G. (2006). Modulation of beta-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol. Neurodegener. 18, 1–8. doi: 10.1186/1750-1326-1-8
Canepa, E., Borghi, R., Viña, J., Traverso, N., Gambini, J., Domenicotti, C., et al. (2011). Cholesterol and amyloid-A: evidence for a cross-talk between astrocytes and neuronal cells. J. Alzheimers Dis. 25, 645–653. doi: 10.3233/JAD-2011-110053
Cao, G., Bales, K. R., DeMattos, R. B., and Paul, S. M. (2007). Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: relevance to Alzheimer’s disease therapeutics. Curr. Alzheimer Res. 4, 179–184. doi: 10.2174/156720507780362173
Carter, D. B., Dunn, E., McKinley, D. D., Stratman, N. C., Boyle, T. P., Kuiper, S. L., et al. (2001). Human apolipoprotein E4 accelerates beta-amyloid deposition in APPsw transgenic mouse brain. Ann. Neurol. 50, 468–475. doi: 10.1002/ana.1134
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Castellani, R. J., Rolston, R. K., and Smith, M. A. (2010). Alzheimer disease. Dis. Mon. 56, 484–546. doi: 10.1016/j.disamonth.2010.06.001
Castellani, R., Hirai, K., Aliev, G., Drew, K. L., Nunomura, A., Takeda, A., et al. (2002). Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 70, 357–360. doi: 10.1002/jnr.10389
Chan, S. L., Kim, W. S., Kwok, J. B., Hill, A. F., Cappai, R., Rye, K. A., et al. (2008). ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 106, 793–804. doi: 10.1111/j.1471-4159.2008.05433.x
Chang, S., ran Ma, T., Miranda, R. D., Balestra, M. E., Mahley, R. W., and Huang, Y. (2005). Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U S A 102, 18694–18699. doi: 10.1073/pnas.0508254102
Chopra, K., Misra, S., and Kuhad, A. (2011). Current perspectives on pharmacotherapy of Alzheimer’s disease. Expert Opin. Pharmacother. 12, 335–350. doi: 10.1517/14656566.2011.520702
Churchward, M. A., and Todd, K. G. (2014). Statin treatment affects cytokine release and phagocytic activity in primary cultured microglia through two separable mechanisms. Mol. Brain 7:85. doi: 10.1186/s13041-014-0085-7
Colton, C., and Wilcock, D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. doi: 10.2174/187152710791012053
Combs, C. K., Karlo, J. C., Kao, S. C., and Landreth, G. E. (2001). β-amyloid stimulation of microglia anti monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 21, 1179–1188.
Cordy, J. M., Hooper, N. M., and Turner, A. J. (2006). The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol. 23, 111–122. doi: 10.1080/09687860500496417
Cottrell, D. A., Blakely, E. L., Johnson, M. A., Ince, P. G., and Turnbull, D. M. (2001). Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 57, 260–264. doi: 10.1212/wnl.57.2.260
Crick, P. J., Beckers, L., Baes, M., Van Veldhoven, P. P., Wang, Y., and Griffiths, W. J. (2015a). The oxysterol and cholestenoic acid profile of mouse cerebrospinal fluid. Steroids doi: 10.1016/j.steroids.2015.02.021 [Epub ahead of print]
Crick, P. J., William Bentley, T., Abdel-Khalik, J., Matthews, I., Clayton, P. T., Morris, A. A., et al. (2015b). Quantitative charge-tags for sterol and oxysterol analysis. Clin. Chem. 61, 400–411. doi: 10.1373/clinchem.2014.231332
Crouch, P. J., Blake, R., Duce, J. A., Ciccotosto, G. D., Li, Q. X., Barnham, K. J., et al. (2005). Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J. Neurosci. 25, 672–679. doi: 10.1523/jneurosci.4276-04.2005
Curtain, C. C., Ali, F. E., Smith, D. G., Bush, A. I., Masters, C. L., and Barnham, K. J. (2003). Metal ions, pH and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J. Biol. Chem. 278, 2977–2982. doi: 10.1074/jbc.m205455200
D’Andrea, M. R., Cole, G. M., and Ard, M. D. (2004). The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol. Aging 25, 675–683. doi: 10.1016/j.neurobiolaging.2003.12.026
Dasari, B., Prasanthi, J. R., Marwarha, G., Singh, B. B., and Ghribi, O. (2010). The oxysterol 27-hydroxycholesterol increases β-amyloid and oxidative stress in retinal pigment epithelial cells. BMC Ophthalmol. 10:22. doi: 10.1186/1471-2415-10-22
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
DeGiorgio, L. A., Shimizu, Y., Chun, H. S., Kim, Y. S., Sugama, S., Son, J. H., et al. (2002). Amyloid precursor protein gene disruption attenuates degeneration of substantia nigra compacta neurons following axotomy. Brain Res. 938, 38–44. doi: 10.1016/s0006-8993(02)02483-6
Dias, I. H., Polidori, M. C., and Griffiths, H. R. (2014). Hypercholesterolaemia-induced oxidative stress at the blood-brain barrier. Biochem. Soc. Trans. 42, 1001–1005. doi: 10.1042/BST20140164
Diczfalusy, U. (2013). On the formation and possible biological role of 25-hydroxycholesterol. Biochimie 95, 455–460. doi: 10.1016/j.biochi.2012.06.016
Dietschy, J. M. (2009). Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 390, 287–293. doi: 10.1515/BC.2009.035
Ding, Q., Dimayuga, E., and Keller, J. N. (2007). Oxidative damage, protein synthesis and protein degradation in Alzheimer’s disease. Curr. Alzheimer Res. 4, 73–79. doi: 10.2174/156720507779939788
Drouin-Ouellet, J., and Cicchetti, F. (2012). Inflammation and neurodegeneration: the story ‘retolled’. Trends Pharmacol. Sci. 33, 542–551. doi: 10.1016/j.tips.2012.07.002
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U S A 107, 18670–18675. doi: 10.1073/pnas.1006586107
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell. Biol. 160, 113–123. doi: 10.1083/jcb.200207113
Evans, R. M., Hui, S., Perkins, A., Lahiri, D. K., Poirier, J., and Farlow, M. R. (2004). Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology 62, 1869–1871. doi: 10.1212/01.wnl.0000125323.15458.3f
Famer, D., Meaney, S., Mousavi, M., Nordberg, A., Björkhem, I., and Crisby, M. (2007). Regulation of alpha- and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem. Biophys. Res. Commun. 359, 46–50. doi: 10.1016/j.bbrc.2007.05.033
Fan, Q. W., Yu, W., Senda, T., Yanagisawa, K., and Michikawa, M. (2001). Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J. Neurochem. 76, 391–400. doi: 10.1046/j.1471-4159.2001.00063.x
Fantini, J., and Yahi, N. (2010). Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 12:e27. doi: 10.1017/s1462399410001602
Fassbender, K., Masters, C., and Beyreuther, K. (2000). Alzheimer’s disease: an inflammatory disease? Neurobiol. Aging 21, 433–436. doi: 10.1016/S0197-4580(00)00147-0
Fassbender, K., Simons, M., Bergmann, C., Stroick, M., Lütjohann, D., Keller, P., et al. (2001). Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. U S A 98, 5856–5861. doi: 10.1073/pnas.081620098
Fernandez-Fernandez, S., Almeida, A., and Bolaños, J. P. (2012). Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 443, 3–11. doi: 10.1042/bj20111943
Ferrera, P., Mercado-Gómez, O., Silva-Aguilar, M., Valverde, M., and Arias, C. (2008). Cholesterol potentiates beta-amyloid-induced toxicity in human neuroblastoma cells: involvement of oxidative stress. Neurochem. Res. 33, 1509–1517. doi: 10.1007/s11064-008-9623-y
Ferretti, M. T., and Cuello, A. C. (2011). Does a pro-inflammatory process precede Alzheimer’s disease and mild cognitive impairment? Curr. Alzheimer Res. 8, 164–174. doi: 10.2174/1567211213451702050
Figueiredo-Pereira, M. E., Rockwell, P., Schmidt-Glenewinkel, T., and Serrano, P. (2015). Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 7:104. doi: 10.3389/fnmol.2014.00104
Findeis, M. A. (2007). The role of amyloid β peptide 42 in Alzheimer’s disease. Pharmacol. Ther. 116, 266–286. doi: 10.1016/j.pharmthera.2007.06.006
Fitz, N. F., Cronican, A., Pham, T., Fogg, A., Fauq, A. H., Chapman, R., et al. (2010). LXR agonist treatment ameliorates amyloid pathology and memory deficits caused by high fat diet in APP23 mice. J. Neurosci. 30, 6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010
Gamba, P., Guglielmotto, M., Testa, G., Monteleone, D., Zerbinati, C., Gargiulo, S., et al. (2014). Up-regulation of β-amyloidogenesis in neuron-like human cells by both 24- and 27-hydroxycholesterol: protective effect of N-acetyl-cysteine. Aging Cell. 13, 561–572. doi: 10.1111/acel.12206
Gamba, P., Leonarduzzi, G., Tamagno, E., Guglielmotto, M., Testa, G., Sottero, B., et al. (2011). Interaction between 24-hydroxycholesterol, oxidative stress and amyloid-β in amplifying neuronal damage in Alzheimer’s disease: three partners in crime. Aging Cell 10, 403–417. doi: 10.1111/j.1474-9726.2011.00681.x
Gamba, P., Testa, G., Sottero, B., Gargiulo, S., Poli, G., and Leonarduzzi, G. (2012). The link between altered cholesterol metabolism and Alzheimer’s disease. Ann. N Y Acad. Sci. 1259, 54–64. doi: 10.1111/j.1749-6632.2012.06513.x
Gate, D., Rezai-Zadeh, K., Jodry, D., Rentsendorj, A., and Town, T. (2010). Macrophages in Alzheimer’s disease: the blood-borne identity. J. Neural Transm. 117, 961–970. doi: 10.1007/s00702-010-0422-7
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Ghribi, O., Larsen, B., Schrag, M., and Herman, M. M. (2006). High cholesterol content in neurons increases BACE, A-amyloid and phosphorylated tau levels in rabbit hippocampus. Exp. Neurol. 200, 460–467. doi: 10.1016/j.expneurol.2006.03.019
Giraldo, E., Lloret, A., Fuchsberger, T., and Viña, J. (2014). Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2, 873–877. doi: 10.1016/j.redox.2014.03.002
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Gong, J. S., Sawamura, N., Zou, K., Sakai, J., Yanagisawa, K., and Michikawa, M. (2002). Amyloid beta-protein affects cholesterol metabolism in cultured neurons: implications for pivotal role of cholesterol in the amyloid cascade. J. Neurosci. Res. 70, 438–446. doi: 10.1002/jnr.10347
Götz, J., Chen, F., van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Grimm, M. O., Grimm, H. S., Tomic, I., Beyreuther, K., Hartmann, T., and Bergmann, C. (2008). Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 283, 11302–11311. doi: 10.1074/JBC.M801520200
Harris, B., Pereira, I., and Parkin, E. (2009). Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 1296, 203–215. doi: 10.1016/j.brainres.2009.07.105
Harris, F. M., Brecht, W. J., Xu, Q., Tesseur, I., Kekonius, L., Wyss-Coray, T., et al. (2003). Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. U S A 100, 10966–10971. doi: 10.1073/pnas.1434398100
Harris, J. R. (2008). Cholesterol binding to amyloid-beta fibrils: a TEM study. Micron 39, 1192–1196. doi: 10.1016/j.micron.2008.05.001
Hascalovici, J. R., Vaya, J., Khatib, S., Holcroft, C. A., Zukor, H., Song, W., et al. (2009). Brain sterol dysregulation in sporadic AD and MCI: relationship to heme oxygenase-1. J. Neurochem. 110, 1241–1253. doi: 10.1111/j.1471-4159.2009.06213.x
Hawkes, C. A., and McLaurin, J. (2009). Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U S A 106, 1261–1266. doi: 10.1073/pnas.0805453106
He, X., Jenner, A. M., Ong, W. Y., Farooqui, A. A., and Patel, S. C. (2006). Lovastatin modulates increased cholesterol and oxysterol levels and has a neuroprotective effect on rat hippocampal neurons after kainate injury. J. Neuropathol. Exp. Neurol. 65, 652–663. doi: 10.1097/01.jnen.0000225906.82428.69
Head, E., Lott, I. T., Cribbs, D. H., Cotman, C. W., and Rohn, T. T. (2002). Beta-amyloid deposition and neurofibrillary tangle association with caspase activation in Down syndrome. Neurosci. Lett. 330, 99–103. doi: 10.1016/s0304-3940(02)00705-x
Heneka, M. T., and O’Banion, M. K. (2007). Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 184, 69–91. doi: 10.1016/j.jneuroim.2006.11.017
Heneka, M. T., O’Banion, M. K., Terwel, D., and Kummer, M. P. (2010). Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm. 117, 919–947. doi: 10.1007/s00702-010-0438-z
Herz, J., and Beffert, U. (2000). Apolipoprotein E receptors: linking brain development and Alzheimer’s disease. Nat. Rev. Neurosci. 1, 51–58. doi: 10.1038/35036221
Heverin, M., Bogdanovic, N., Lütjohann, D., Bayer, T., Pikuleva, I., Bretillon, L., et al. (2004). Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 45, 186–193. doi: 10.1194/jlr.m300320-jlr200
Heverin, M., Meaney, S., Lütjohann, D., Diczfalusy, U., Wahren, J., and Björkhem, I. (2005). Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J. Lipid Res. 46, 1047–1052. doi: 10.1194/jlr.m500024-jlr200
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Holmes, C. (2013). Review: systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 39, 51–68. doi: 10.1111/j.1365-2990.2012.01307.x
Holmes, C., and Butchart, J. (2011). Systemic inflammation and Alzheimer’s disease. Biochem. Soc. Trans. 39, 898–901. doi: 10.1042/BST0390898
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., et al. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 2892–2897. doi: 10.1073/pnas.050004797
Hooijmans, C. R., Rutters, F., Dederen, P. J., Gambarota, G., Veltien, A., van Groen, T., et al. (2007). Changes in cerebral blood volume and amyloid pathology in aged Alzheimer APP/PS1 mice on a docosahexaenoic acid (DHA) diet or cholesterol enriched Typical Western Diet (TWD). Neurobiol. Dis. 28, 16–29. doi: 10.1016/j.nbd.2007.06.007
Hughes, T. M., Kuller, L. H., Lopez, O. L., Becker, J. T., Evans, R. W., Sutton-Tyrrell, K., et al. (2012). Markers of cholesterol metabolism in the brain show stronger association with cerebrovascular disease than Alzheimer’s disease. J. Alzheimers Dis. 30, 53–61. doi: 10.3233/JAD-2012-111460
Hughes, T. M., Lopez, O. L., Evans, R. W., Kamboh, M. I., Williamson, J. D., Klunk, W. E., et al. (2014). Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol. Aging 35, 802–807. doi: 10.1016/j.neurobiolaging.2013.09.040
Hughes, T. M., Rosano, C., Evans, R. W., and Kuller, L. H. (2013). Brain cholesterol metabolism, oxysterols and dementia. J. Alzheimers Dis. 33, 891–911. doi: 10.3233/JAD-2012-121585
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iuliano, L. (2011). Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 164, 457–458. doi: 10.1016/j.chemphyslip.2011.06.006
Iuliano, L., Crick, P. J., Zerbinati, C., Tritapepe, L., Abdel-Khalik, J., Poirot, M., et al. (2015). Cholesterol metabolites exported from human brai. Steroids doi: 10.1016/j.steroids.2015.01.026 [Epub ahead of print]
Iuliano, L., Monticolo, R., Straface, G., Spoletini, I., Gianni, W., Caltagirone, C., et al. (2010). Vitamin E and enzymatic/oxidative stress-driven oxysterols in amnestic mild cognitive impairment subtypes and Alzheimer’s disease. J. Alzheimers Dis. 21, 1383–1392. doi: 10.3233/JAD-2010-100780
Jacob, K. D., Noren Hooten, N., Trzeciak, A. R., and Evans, M. K. (2013). Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech. Ageing Dev. 134, 139–157. doi: 10.1016/j.mad.2013.02.008
Jang, E. R., and Lee, C. S. (2011). 7-ketocholesterol induces apoptosis in differentiated PC12 cells via reactive oxygen species-dependent activation of NF-κB and Akt pathways. Neurochem. Int. 58, 52–59. doi: 10.1016/j.neuint.2010.10.012
Jaya Prasanthi, R. P., Schommer, E., Thomasson, S., Thompson, A., Feist, G., and Ghribi, O. (2008). Regulation of beta-amyloid levels in the brain of cholesterol-fed rabbit, a model system for sporadic Alzheimer’s disease. Mech. Ageing Dev. 129, 649–655. doi: 10.1016/j.mad.2008.09.002
Jeitner, T. M., Voloshyna, I., and Reiss, A. B. (2011). Oxysterol derivatives of cholesterol in neurodegenerative disorders. Curr. Med. Chem. 18, 1515–1525. doi: 10.2174/092986711795328445
Jenner, A. M., Lim, W. L., Ng, M. P., Wenk, M. R., Shui, G., Sharman, M. J., et al. (2010). The effect of APOE genotype on brain levels of oxysterols in young and old human APOE epsilon2, epsilon3 and epsilon4 knock-in mice. Neuroscience 169, 109–115. doi: 10.1016/j.neuroscience.2010.04.026
Ji, Z. S., Miranda, R. D., Newhouse, Y. M., Weisgraber, K. H., Huang, Y., and Mahley, R. W. (2002). Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J. Biol. Chem. 277, 21821–21828. doi: 10.1074/jbc.m112109200
Jiang, Q., Lee, C. Y., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., et al. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. doi: 10.1016/j.neuron.2008.04.010
Jo, W. K., Law, A. C., and Chung, S. K. (2014). The neglected co-star in the dementia drama: the putative roles of astrocytes in the pathogeneses of major neurocognitive disorders. Mol. Psychiatry 19, 159–167. doi: 10.1038/mp.2013.171
Joshi, Y. B., and Praticò, D. (2015). The 5-lipoxygenase pathway: oxidative and inflammatory contributions to the Alzheimer’s disease phenotype. Front. Cell Neurosci. 8:436. doi: 10.3389/fncel.2014.00436
Kakio, A., Nishimoto, S. I., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2001). Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276, 24985–24990. doi: 10.1074/jbc.m100252200
Kakio, A., Nishimoto, S., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2002). Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41, 7385–7390. doi: 10.1021/bi0255874
Keene, C. D., Cudaback, E., Li, X., Montine, K. S., and Montine, T. J. (2011). Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 21, 920–928. doi: 10.1016/j.conb.2011.08.002
Khandelwal, P. J., Herman, A. M., and Moussa, C. E. (2011). Inflammation in the early stages of neurodegenerative pathology. J. Neuroimmunol. 238, 1–11. doi: 10.1016/j.jneuroim.2011.07.002
Kierdorf, K., Wang, Y., and Neumann, H. (2010). . Immune-mediated CNS damage. Results Probl. Cell Differ. 51, 173–196. doi: 10.1007/400_2008_15
Kim, D. H., and Frangos, J. A. (2008). Effects of amyloid beta-peptides on the lysis tension of lipid bilayer vesicles containing oxysterols. Biophys. J. 95, 620–628. doi: 10.1529/biophysj.107.114983
Kim, H. A., Miller, A. A., Drummond, G. R., Thrift, A. G., Arumugam, T. V., Phan, T. G., et al. (2012). Vascular cognitive impairment and Alzheimer’s disease: role of cerebral hypoperfusion and oxidative stress. Naunyn Schmiedebergs Arch. Pharmacol. 385, 953–959. doi: 10.1007/s00210-012-0790-7
Kim, J. H., Jittiwat, J., Ong, W. Y., Farooqui, A. A., and Jenner, A. M. (2010). Changes in cholesterol biosynthetic and transport pathways after excitotoxicity. J. Neurochem. 112, 34–41. doi: 10.1111/j.1471-4159.2009.06449.x
Kim, J., Basak, J. M., and Holtzman, D. M. (2009a). The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. doi: 10.1016/j.neuron.2009.06.026
Kim, W. S., Chan, S. L., Hill, A. F., Guillemin, G. J., and Garner, B. (2009b). Impact of 27-hydroxycholesterol on amyloid-beta peptide production and ATP-binding cassette transporter expression in primary human neurons. J. Alzheimers Dis. 16, 121–131. doi: 10.3233/JAD-2009-0944
Kim, W. S., Hill, A. F., Fitzgerald, M. L., Freeman, M. W., Evin, G., and Garner, B. (2011). Wild type and tangier disease ABCA1 mutants modulate cellular amyloid-A production independent of cholesterol efflux activity. J. Alzheimers Dis. 27, 441–452. doi: 10.3233/JAD-2011-110521
Kim, Y. J., and Lee, C. S. (2010). Tyrosine kinase inhibitor AG126 reduces 7-ketocholesterol-induced cell death by suppressing mitochondria-mediated apoptotic process. Neurochem. Res. 35, 603–612. doi: 10.1007/s11064-009-0105-7
Kim, Y. J., Han, J. H., Han, E. S., and Lee, C. S. (2006). 7-Ketocholesterol enhances 1-methyl-4-phenylpyridinium-induced mitochondrial dysfunction and cell death in PC12 cells. J. Neural Transm. 113, 1877–1885. doi: 10.1007/s00702-006-0486-6
Koldamova, R. P., Lefterov, I. M., Ikonomovic, M. D., Skoko, J., Lefterov, P. I., Isanski, B. A., et al. (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J. Biol. Chem. 278, 13244–13256. doi: 10.1074/jbc.m300044200
Koldamova, R., Fitz, N. F., and Lefterov, I. (2010). The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 1801, 824–830. doi: 10.1016/j.bbalip.2010.02.010
Koudinov, A. R., and Koudinova, N. V. (2005). Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci. 229–230, 233–240. doi: 10.1016/j.jns.2004.11.036
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8:e60921. doi: 10.1371/journal.pone.0060921
Kölsch, H., Heun, R., Kerksiek, A., Bergmann, K. V., Maier, W., and Lütjohann, D. (2004). Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci. Lett. 368, 303–308. doi: 10.1016/j.neulet.2004.07.031
Kölsch, H., Ludwig, M., Lütjohann, D., Prange, W., and Rao, M. L. (2000). 7alpha-Hydroperoxycholesterol causes CNS neuronal cell death. Neurochem. Int. 36, 507–512. doi: 10.1016/s0197-0186(99)00157-6
Kölsch, H., Ludwig, M., Lütjohann, D., and Rao, M. L. (2001). Neurotoxicity of 24-hydroxycholesterol, an important cholesterol elimination product of the brain, may be prevented by vitamin E and estradiol-17beta. J. Neural. Transm. 108, 475–488. doi: 10.1007/s007020170068
Lane-Donovan, C., Philips, G. T., and Herz, J. (2014). More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83, 771–787. doi: 10.1016/j.neuron.2014.08.005
Latta, C. H., Brothers, H. M., and Wilcock, D. M. (2014). Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience doi: 10.1007/978-1-4615-5405-9_3 [Epub ahead of print]
Lee, C. Y., Tse, W., Smith, J. D., and Landreth, G. E. (2011). ApoE promotes AA trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044. doi: 10.1074/jbc.M111.295451
Leoni, V., and Caccia, C. (2011). Oxysterols as biomarkers in neurodegenerative diseases. Chem .Phys. Lipids 164, 515–524. doi: 10.1016/j.chemphyslip.2011.04.002
Leoni, V., Masterman, T., Mousavi, F. S., Wretlind, B., Wahlund, L. O., Diczfalusy, U., et al. (2004). Diagnostic use of cerebral and extracerebral oxysterols. Clin. Chem. Lab. Med. 42, 186–191. doi: 10.1515/cclm.2004.034
Leoni, V., Masterman, T., Patel, P., Meaney, S., Diczfalusy, U., and Björkhem, I. (2003). Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J. Lipid Res. 44, 793–799. doi: 10.1194/jlr.m200434-jlr200
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. doi: 10.1126/science.1058189
Liu, B., and Hong, J. S. (2003). Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7. doi: 10.1124/jpet.102.035048
Liu, Q., Zerbinatti, C. V., Zhang, J., Hoe, H. S., Wang, B., Cole, S. L., et al. (2007). Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78. doi: 10.1016/j.neuron.2007.08.008
Luchsinger, J. A., and Mayeux, R. (2004). Dietary factors and Alzheimer’s disease. Lancet. Neurol. 3, 579–587. doi: 10.1016/S1474-4422(04)00878-6
Luckhoff, H. K., Brand, T., van Velden, D. P., Kidd, M., Fisher, L. R., van Rensburg, S. J., et al. (2015). Clinical relevance of apolipoprotein e genotyping based on a family history of Alzheimer’s disease. Curr. Alzheimer Res. 12, 210–217. doi: 10.2174/1567205012666150302154354
Luque-Contreras, D., Carvajal, K., Toral-Rios, D., Franco-Bocanegra, D., and Campos-Peña, V. (2014). Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer’s disease? Oxid. Med. Cell. Longev. 2014:497802. doi: 10.1155/2014/497802
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: the role and consequences. Neurosci. Res. 79, 1–12. doi: 10.1016/j.neures.2013.10.004
Lütjohann, D., Papassotiropoulos, A., Björkhem, I., Locatelli, S., Bagli, M., Oehring, R. D., et al. (2000). Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 41, 195–198.
Ma, M. T., Zhang, J., Farooqui, A. A., Chen, P., and Ong, W. Y. (2010). Effects of cholesterol oxidation products on exocytosis. Neurosci. Lett. 476, 36–41. doi: 10.1016/j.neulet.2010.03.078
Ma, W. W., Li, C. Q., Yu, H. L., Zhang, D. D., Xi, Y. D., Han, J., et al. (2015). The oxysterol 27-hydroxycholesterol increases oxidative stress and regulate nrf2 signaling pathway in astrocyte cells. Neurochem. Res. 40, 758–766. doi: 10.1007/s11064-015-1524-2
Maezawa, I., Maeda, N., Montine, T. J., and Montine, K. S. (2006a). Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J. Neuroinflammation 7, 3–10. doi: 10.1186/1742-2094-3-10
Maezawa, I., Nivison, M., Montine, K. S., Maeda, N., and Montine, T. J. (2006b). Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 20, 797–799. doi: 10.1096/fj.05-5423fje
Maezawa, I., Zaja-Milatovic, S., Milatovic, D., Stephen, C., Sokal, I., Maeda, N., et al. (2006c). Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J. Neuroinflammation 25, 3–21. doi: 10.1186/1742-2094-3-21
Malaplate-Armand, C., Florent-Béchard, S., Youssef, I., Koziel, V., Sponne, I., Kriem, B., et al. (2006). Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 23, 178–189. doi: 10.1016/j.nbd.2006.02.010
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Mariani, E., Polidori, M. C., Cherubini, A., and Mecocci, P. (2005). Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 827, 65–75. doi: 10.1016/j.jchromb.2005.04.023
Martin, I., and Grotewiel, M. S. (2006). Oxidative damage and age-related functional declines. Mech. Ageing Dev. 127, 411–423. doi: 10.1016/j.mad.2006.01.008
Martins, I. J., Berger, T., Sharman, M. J., Verdile, G., Fuller, S. J., and Martins, R. N. (2009). Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J. Neurochem. 111, 1275–1308. doi: 10.1111/j.1471-4159.2009.06408.x
Marwarha, G., Dasari, B., Prasanthi, J. R., Schommer, J., and Ghribi, O. (2010). Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J. Alzheimers Dis. 19, 1007–1019. doi: 10.3233/JAD-2010-1298
Marwarha, G., and Ghribi, O. (2014). Does the oxysterol 27-hydroxycholesterol underlie Alzheimer’s disease-Parkinson’s disease overlap? Exp. Gerontol. doi: 10.1016/j.exger.2014.09.013 [Epub ahead of print]
Marwarha, G., Raza, S., Prasanthi, J. R., and Ghribi, O. (2013). Gadd153 and NF-κB crosstalk regulates 27-hydroxycholesterol-induced increase in BACE1 and β-amyloid production in human neuroblastoma SH-SY5Y cells. PLoS One 8:e70773. doi: 10.1371/journal.pone.0070773
Marzolo, M. P., and Bu, G. (2009). Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin. Cell Dev. Biol. 20, 191–200. doi: 10.1016/j.semcdb.2008.10.005
Mateos, L., Ismail, M. A., Gil-Bea, F. J., Leoni, V., Winblad, B., Björkhem, I., et al. (2011). Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer’s disease. J. Alzheimers Dis. 24, 669–679. doi: 10.3233/JAD-2011-101512
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
Mayeux, R. (2003). Epidemiology of neurodegeneration. Annu. Rev. Neurosci. 26, 81–104. doi: 10.1146/annurev.neuro.26.043002.094919
Mazzetti, A. P., Fiorile, M. C., Primavera, A., and Lo Bello, M. (2015). Glutathione transferases and neurodegenerative diseases. Neurochem. Int. 82, 10–18. doi: 10.1016/j.neuint.2015.01.008
McGeer, E. G., and McGeer, P. L. (2010). Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J. Alzheimers Dis. 19, 355–361. doi: 10.3233/JAD-2010-1219
McLellan, M. E., Kajdasz, S. T., Hyman, B. T., and Bacskai, B. J. (2003). In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J. Neurosci. 23, 2212–2217.
Meaney, S., Heverin, M., Panzenboeck, U., Ekström, L., Axelsson, M., Andersson, U., et al. (2007). Novel route for elimination of brain oxysterols across the blood-brain barrier: conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J. Lipid Res. 48, 944–951. doi: 10.1194/jlr.m600529-jlr200
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2:e536. doi: 10.1371/journal.pone.0000536
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E., and Heuschling, P. (2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 210, 3–12. doi: 10.1016/j.jneuroim.2009.02.003
Michikawa, M., Gong, J. S., Fan, Q. W., Sawamura, N., and Yanagisawa, K. (2001). A novel action of alzheimer’s amyloid beta-protein (Abeta): oligomeric Abeta promotes lipid release. J. Neurosci. 21, 7226–7235.
Misonou, H., Morishima-Kawashima, M., and Ihara, Y. (2000). Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 39, 6951–6959. doi: 10.1021/bi000169p
Mitomo, H., Chen, W. H., and Regen, S. L. (2009). Oxysterol-induced rearrangement of the liquid-ordered phase: a possible link to Alzheimer’s disease? J. Am. Chem. Soc. 131, 12354–12357. doi: 10.1021/ja904308y
Morales, I., Guzmán-Martínez, L., Cerda-Troncoso, C., Farías, G. A., and Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell Neurosci. 8:112. doi: 10.3389/fncel.2014.00112
Morales, I., Jiménez, J. M., Mancilla, M., and Maccioni, R. B. (2013). Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 37, 849–856. doi: 10.3233/JAD-131843
Mori, T., Paris, D., Town, T., Rojiani, A. M., Sparks, D. L., Delledonne, A., et al. (2001). Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J. Neuropathol. Exp. Neurol. 60, 778–785.
Mrak, R. E. (2012). Microglia in Alzheimer brain: a neuropathological perspective. Int. J. Alzheimers Dis. 2012:165021. doi: 10.1155/2012/165021
Mrak, R. E., and Griffin, W. S. (2005). Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 26, 349–354. doi: 10.1016/j.neurobiolaging.2004.05.010
Mungarro-Menchaca, X., Ferrera, P., Morán, J., and Arias, C. (2002). beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J. Neurosci. Res. 68, 89–96. doi: 10.1002/jnr.10193
Murphy, P. G., Borthwick, L. S., Johnston, R. S., Kuchel, G., and Richardson, P. M. (1999). Nature of the retrograde signal from injured nerves that induces interleukin-6 mRNA in neurons. J. Neurosci. 19, 3791–3800.
Naylor, R., Hill, A. F., and Barnham, K. J. (2008). Neurotoxicity in Alzheimer’s disease: is covalently crosslinked A beta responsible? Eur. Biophys. J. 37, 265–268. doi: 10.1007/s00249-007-0243-2
Nelson, T. J., and Alkon, D. L. (2005). Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J. Biol. Chem. 280, 7377–7387. doi: 10.1074/jbc.m409071200
Nieweg, K., Schaller, H., and Pfrieger, F. W. (2009). Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 109, 125–134. doi: 10.1111/j.1471-4159.2009.05917.x
Noguchi, N., Saito, Y., and Urano, Y. (2014). Diverse functions of 24(S)-hydroxycholesterol in the brain. Biochem. Biophys. Res. Commun. 446, 692–696. doi: 10.1016/j.bbrc.2014.02.010
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
Nury, T., Samadi, M., Zarrouk, A., Riedinger, J. M., and Lizard, G. (2013). Improved synthesis and in vitro evaluation of the cytotoxic profile of oxysterols oxidized at C4 (4α- and 4β-hydroxycholesterol) and C7 (7-ketocholesterol, 7α- and 7β-hydroxycholesterol) on cells of the central nervous system. Eur. J. Med. Chem. 70, 558–567. doi: 10.1016/j.ejmech.2013.09.028
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/s0896-6273(03)00434-3
Okabe, A., Urano, Y., Itoh, S., Suda, N., Kotani, R., Nishimura, Y., et al. (2013). Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2, 28–35. doi: 10.1016/j.redox.2013.11.007
Panza, F., D’Introno, A., Colacicco, A. M., Capurso, C., Pichichero, G., Capurso, S. A., et al. (2006). Lipid metabolism in cognitive decline and dementia. Brain Res. Rev. 51, 275–292. doi: 10.1016/j.brainresrev.2005.11.007
Papassotiropoulos, A., Lambert, J. C., Wavrant-De Vrièze, F., Wollmer, M. A., von der Kammer, H., Streffer, J. R., et al. (2005). Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neurodegener. Dis. 2, 233–241. doi: 10.1159/000090362
Papassotiropoulos, A., Lütjohann, D., Bagli, M., Locatelli, S., Jessen, F., Buschfort, R., et al. (2002). 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J. Psychiatr. Res. 36, 27–32. doi: 10.1016/s0022-3956(01)00050-4
Park, L., Zhou, P., Pitstick, R., Capone, C., Anrather, J., Norris, E. H., et al. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U S A 105, 1347–1352. doi: 10.1073/pnas.0711568105
Perry, V. H., Nicoll, J. A., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Pfrieger, F. W. (2003). Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays 25, 72–78. doi: 10.1002/bies.10195
Phan, H. T., Hata, T., Morita, M., Yoda, T., Hamada, T., Vestergaard, M. C., et al. (2013). The effect of oxysterols on the interaction of Alzheimer’s amyloid beta with model membranes. Biochim. Biophys. Acta 1828, 2487–2495. doi: 10.1016/j.bbamem.2013.06.021
Popescu, B. O., Toescu, E. C., Popescu, L. M., Bajenaru, O., Muresanu, D. F., Schultzberg, M., et al. (2009). Blood-brain barrier alterations in ageing and dementia. J. Neurol. Sci. 283, 99–106. doi: 10.1016/j.jns.2009.02.321
Prasanthi, J. R., Huls, A., Thomasson, S., Thompson, A., Schommer, E., and Ghribi, O. (2009). Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 4:1. doi: 10.1186/1750-1326-4-1
Praticò, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 21, 4183–4187.
Puglielli, L., Friedlich, A. L., Setchell, K. D., Nagano, S., Opazo, C., Cherny, R. A., et al. (2005). Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J. Clin. Invest. 115, 2556–2563. doi: 10.1172/JCI23610
Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A., Berezovska, O., Hyman, B. T., et al. (2001). Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 3, 905–912. doi: 10.1038/ncb1001-905
Puglielli, L., Tanzi, R. E., and Kovacs, D. M. (2003). Alzheimer’s disease: the cholesterol connection. Nat. Neurosci. 6, 345–351. doi: 10.1038/nn0403-345
Qiu, L., Buie, C., Reay, A., Vaughn, M. W., and Cheng, K. H. (2011). Molecular dynamics simulations reveal the protective role of cholesterol in β-amyloid protein-induced membrane disruptions in neuronal membrane mimics. J. Phys. Chem. B. 115, 9795–9812. doi: 10.1021/jp2012842
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Quintanilla, R. A., Orellana, J. A., and von Bernhardi, R. (2012). Understanding risk factors for Alzheimer’s disease: interplay of neuroinflammation, connexin-based communication and oxidative stress. Arch. Med. Res. 43, 632–644. doi: 10.1016/j.arcmed.2012.10.016
Raber, J., Huang, Y., and Ashfordm, J. W. (2004). ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 25, 641–650. doi: 10.1016/j.neurobiolaging.2003.12.023
Račková, L. (2013). Cholesterol load of microglia: contribution of membrane architecture changes to neurotoxic power? Arch Biochem Biophys. 537, 91–103. doi: 10.1016/j.abb.2013.06.015
Ramesh, G., MacLean, A. G., and Philipp, M. T. (2013). Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration and neuropathic pain. Mediators Inflamm. 2013:480739. doi: 10.1155/2013/480739
Reddy, P. H. (2011). Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria and synaptic deprivation in Alzheimer’s disease. Brain Res. 1415, 136–148. doi: 10.1016/j.brainres.2011.07.052
Reddy, P. H., and Beal, M. F. (2005). Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev. 49, 618–632. doi: 10.1016/j.brainresrev.2005.03.004
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Reddy, P. H., McWeeney, S., Park, B. S., Manczak, M., Gutala, R. V., Partovi, D., et al. (2004). Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 13, 1225–1240. doi: 10.1093/hmg/ddh140
Reed, T. T. (2011). Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 51, 1302–1319. doi: 10.1016/j.freeradbiomed.2011.06.027
Refolo, L. M., Malester, B., LaFrancois, J., Bryant-Thomas, T., Wang, R., Tint, G. S., et al. (2000). Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 4, 321–331. doi: 10.1006/nbdi.2000.0304
Reid, P. C., Urano, Y., Kodama, T., and Hamakubo, T. (2007). Alzheimer’s disease: cholesterol, membrane rafts, isoprenoids and statins. J. Cell Mol. Med. 11, 383–392. doi: 10.1111/j.1582-4934.2007.00054.x
Resende, R., Moreira, P. I., Proença, T., Deshpande, A., Busciglio, J., Pereira, C., et al. (2008). Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 44, 2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012
Reynolds, A., Laurie, C., Mosley, R. L., and Gendelman, H. E. (2007). Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 82, 297–325. doi: 10.1016/s0074-7742(07)82016-2
Rezai-Zadeh, K., Gate, D., Gowing, G., and Town, T. (2011). How to get from here to there: macrophage recruitment in Alzheimer’s disease. Curr. Alzheimer Res. 8, 156–163. doi: 10.2174/1567211213451662050
Rosales-Corral, S., Reiter, R. J., Tan, D. X., Ortiz, G. G., and Lopez-Armas, G. (2010). Functional aspects of redox control during neuroinflammation. Antioxid Redox Signal. 13, 193–247. doi: 10.1089/ars.2009.2629
Roßner, S., Lange-Dohna, C., Zeitschel, U., and Perez-Polo, J. R. (2005). Alzheimer’s disease β-secretase BACE1 is not a neuron-specific enzyme. J. Neurochem. 92, 226–234. doi: 10.1111/j.1471-4159.2004.02857.x
Royer, M. C., Lemaire-Ewing, S., Desrumaux, C., Monier, S., Pais de Barros, J. P., Athias, A., et al. (2009). 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched (lipid raft) domains is impaired by vitamin E: a specific role for alpha-tocopherol with consequences on cell death. J. Biol. Chem. 284, 15826–15834. doi: 10.1074/jbc.m808641200
Rubio-Perez, J. M., and Morillas-Ruiz, J. M. (2012). A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012:756357. doi: 10.1100/2012/756357
Sagare, A., Deane, R., Bell, R. D., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Saint-Pol, J., Candela, P., Boucau, M. C., Fenart, L., and Gosselet, F. (2013). Oxysterols decrease apical-to-basolateral transport of Aß peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 1517, 1–15. doi: 10.1016/j.brainres.2013.04.008
Saint-Pol, J., Vandenhaute, E., Boucau, M. C., Candela, P., Dehouck, L., Cecchelli, R., et al. (2012). Brain pericytes ABCA1 expression mediates cholesterol efflux but not cellular amyloid-β peptide accumulation. J. Alzheimers Dis. 30, 489–503. doi: 10.3233/JAD-2012-112090
Schipper, H. M. (2011). Apolipoprotein E: implications for AD neurobiology, epidemiology and risk assessment. Neurobiol. Aging 32, 778–790. doi: 10.1016/j.neurobiolaging.2009.04.021
Schönknecht, P., Lütjohann, D., Pantel, J., Bardenheuer, H., Hartmann, T., von Bergmann, K., et al. (2002). Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controls. Neurosci. Lett. 324, 83–85. doi: 10.1016/s0304-3940(02)00164-7
Serrano-Pozo, A., Muzikansky, A., Gómez-Isla, T., Growdon, J. H., Betensky, R. A., Frosch, M. P., et al. (2013). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol Exp Neurol. 72, 462–471. doi: 10.1097/NEN.0b013e3182933788
Shafaati, M., Marutle, A., Pettersson, H., Lövgren-Sandblom, A., Olin, M., Pikuleva, I., et al. (2011). Marked accumulation of 27-hydroxycholesterol in the brains of Alzheimer’s patients with the Swedish APP 670/671 mutation. J. Lipid Res. 52, 1004–1010. doi: 10.1194/jlr.m014548
Sharma, S., Prasanthi, R. P. J., Schommer, E., Feist, G., and Ghribi, O. (2008). Hypercholesterolemia-induced Abeta accumulation in rabbit brain is associated with alteration in IGF-1 signaling. Neurobiol. Dis. 32, 426–432. doi: 10.1016/j.nbd.2008.08.002
Shea, T. B., Rogers, E., Ashline, D., Ortiz, D., and Sheu, M. S. (2002). Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic. Biol. Med. 33, 1115–1120. doi: 10.1016/s0891-5849(02)01001-8
Sheng, J. G., Mrak, R. E., Bales, K. R., Cordell, B., Paul, S. M., and Jones, R. A. (2000). Overexpression of the neuritotrophic cytokine S100beta precedes the appearance of neuritic beta-amyloid plaques in APPV717F mice. J. Neurochem. 74, 295–301. doi: 10.1046/j.1471-4159.2000.0740295.x
Shih, A. Y., Johnson, D. A., Wong, G., Kraft, A. D., Jiang, L., Erb, H., et al. (2003). Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 23, 3394–3406.
Shobab, L. A., Hsiung, G. Y., and Feldman, H. H. (2005). Cholesterol in Alzheimer’s disease. Lancet Neurol. 4, 841–852. doi: 10.1016/S1474-4422(05)70248-9
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502. doi: 10.1016/j.neuron.2006.01.022
Smith, M. A., Rottkamp, C. A., Nunomura, A., Raina, A. K., and Perry, G. (2000). Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta 1502, 139–144. doi: 10.1016/S0925-4439(00)00040-5
Smith, M. A., Zhu, X., Tabaton, M., Liu, G., McKeel, D. W. Jr., Cohen, M. L., et al. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 363–372. doi: 10.3233/JAD-2010-1239
Sochocka, M., Koutsouraki, E. S., Gasiorowski, K., and Leszek, J. (2013). Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: a new approach to therapy. CNS Neurol. Disord. Drug Targets 12, 870–881. doi: 10.2174/18715273113129990072
Sodhi, R. K., and Singh, N. (2013). Liver X receptors: emerging therapeutic targets for Alzheimer’s disease. Pharmacol. Res. 72, 45–51. doi: 10.1016/j.phrs.2013.03.008
Sottero, B., Gamba, P., Gargiulo, S., Leonarduzzi, G., and Poli, G. (2009). Cholesterol oxidation products and disease: an emerging topic of interest in medicinal chemistry. Curr. Med. Chem. 16, 685–705. doi: 10.2174/092986709787458353
Sparks, D. L., Kuo, Y. M., Roher, A., Martin, T., and Lukas, R. J. (2000). Alterations of Alzheimer’s disease in the cholesterol-fed rabbit, including vascular inflammation. Preliminary observations. Ann. N Y Acad. Sci. 903, 335–344. doi: 10.1111/j.1749-6632.2000.tb06384.x
Steffensen, K. R., Jakobsson, T., and Gustafsson, J. Å. (2013). Targeting liver X receptors in inflammation. Expert Opin. Ther. Targets 17, 977–990. doi: 10.1517/14728222.2013.806490
Streit, W. J., Xue, Q. S., Braak, H., and del Tredici, K. (2014). Presence of severe neuroinflammation does not intensify neurofibrillary degeneration in human brain. Glia 62, 96–105. doi: 10.1002/glia.22589
Stuart, L. M., Bell, S. A., Stewart, C. R., Silver, J. M., Richard, J., Goss, J. L., et al. (2007). CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J. Biol. Chem. 282, 27392–27401. doi: 10.1074/jbc.m702887200
Su, B., Wang, X., Lee, H. G., Tabaton, M., Perry, G., Smith, M. A., et al. (2010). Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 468, 267–271. doi: 10.1016/j.neulet.2009.11.010
Suzuki, S., Tanaka, K., Nagata, E., Ito, D., Dembo, T., and Fukuuchi, Y. (1999). Cerebral neurons express interleukin-6 after transient forebrain ischemia in gerbils. Neurosci. Lett. 262, 117–120. doi: 10.1016/s0304-3940(99)00051-8
Tai, L. M., Ghura, S., Koster, K. P., Liakaite, V., Maienschein-Cline, M., Kanabar, P., et al. (2015). APOE-modulated Aβ-induced neuroinflammation in Alzheimer’s disease: current landscape, novel data and future perspective. J. Neurochem. 133, 465–488. doi: 10.1111/jnc.13072
Takeda, S., Sato, N., and Morishita, R. (2014). Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front. Aging Neurosci. 6:171. doi: 10.3389/fnagi.2014.00171
Tamagno, E., Bardini, P., Guglielmotto, M., Danni, O., and Tabaton, M. (2006). The various aggregation states of beta-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration and BACE-1 expression. Free Radic Biol. Med. 41, 202–212. doi: 10.1016/j.freeradbiomed.2006.01.021
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Tchelingerian, J. L., Vignais, L., and Jacque, C. (1994). TNF alpha gene expression is induced in neurones after a hippocampal lesion. Neuroreport 5, 585–588. doi: 10.1097/00001756-199401000-00013
Terwel, D., Steffensen, K. R., Verghese, P. B., Kummer, M. P., Gustafsson, J. Å., Holtzman, D. M., et al. (2011). Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J. Neurosci. 31, 7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011
Testa, G., Gamba, P., Badilli, U., Gargiulo, S., Maina, M., Guina, T., et al. (2014). Loading into nanoparticles improves quercetin’s efficacy in preventing neuroinflammation induced by oxysterols. PLoS One 9:e96795. doi: 10.1371/journal.pone.0096795
Testa, G., Gamba, P., Di Scipio, F., Sprio, A. E., Salamone, P., Gargiulo, S., et al. (2012). Potentiation of amyloid-β peptide neurotoxicity in human dental neuron-like cells by the membrane lipid peroxidation product 4-hydroxynonenal. Free Radic. Biol. Med. 53, 1708–1717. doi: 10.1016/j.freeradbiomed.2012.08.581
Town, T., Nikolic, V., and Tan, J. (2005). The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation 2:24. doi: 10.1186/1742-2094-2-24
Urano, Y., Ochiai, S., and Noguchi, N. (2013). Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 27, 4305–4315. doi: 10.1096/fj.13-231456
Usui, K., Hulleman, J. D., Paulsson, J. F., Siegel, S. J., Powers, E. T., and Kelly, J. W. (2009). Site-specific modification of Alzheimer’s peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc. Natl. Acad. Sci. U S A 106, 18563–18568. doi: 10.1073/pnas.0804758106
Vance, J. E., Hayashi, H., and Karten, B. (2005). Cholesterol homeostasis in neurons and glial cells. Semin. Cell Dev. Biol. 16, 193–212. doi: 10.1016/j.semcdb.2005.01.005
Vaya, J., and Schipper, H. M. (2007). Oxysterols, cholesterol homeostasis and Alzheimer disease. J. Neurochem. 102, 1727–1737. doi: 10.1111/j.1471-4159.2007.04689.x
Verdier, Y., Zarándi, M., and Penke, B. (2004). Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 10, 229–248. doi: 10.1002/psc.573
Vetrivel, K. S., and Thinakaran, G. (2010). Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta. 1801, 860–867. doi: 10.1016/j.bbalip.2010.03.007
Voloshyna, I., and Reiss, A. B. (2011). The ABC transporters in lipid flux and atherosclerosis. Prog. Lipid Res. 50, 213–224. doi: 10.1016/j.plipres.2011.02.001
von Bernhardi, R., Tichauer, J. E., and Eugenín, J. (2010). Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 112, 1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x
Vukic, V., Callaghan, D., Walker, D., Lue, L. F., Liu, Q. Y., Couraud, P. O., et al. (2009). Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 34, 95–106. doi: 10.1016/j.nbd.2008.12.007
Wahrle, S., Das, P., Nyborg, A. C., McLendon, C., Shoji, M., Kawarabayashi, T., et al. (2002). Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23. doi: 10.1006/nbdi.2001.0470
Wahrle, S. E., Jiang, H., Parsadanian, M., Kim, J., Li, A., Knoten, A., et al. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682. doi: 10.1172/jci33622
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., et al. (2004). ABCA1 is required for normal central nervous system APOE levels and for lipidation of astrocyte-secreted APOE. J. Biol. Chem. 279, 40987–40993. doi: 10.1074/jbc.m407963200
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Waltl, S., Patankar, J. V., Fauler, G., Nusshold, C., Ullen, A., Eibinger, G., et al. (2013). 25-Hydroxycholesterol regulates cholesterol homeostasis in the murine CATH.a neuronal cell line. Neurosci. Lett. 539, 16–21. doi: 10.1016/j.neulet.2013.01.014
Wang, L., Schuster, G. U., Hultenby, K., Zhang, Q., Andersson, S., and Gustafsson, J. A. (2002). Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc. Natl. Acad. Sci. U S A 99, 13878–13883. doi: 10.1073/pnas.172510899
Wang, N., Yvan-Charvet, L., Lütjohann, D., Mulder, M., Vanmierlo, T., Kim, T. W., et al. (2008). ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J. 22, 1073–1082. doi: 10.1096/fj.07-9944com
Wang, Y., Li, M., Tang, J., Song, M., Xu, X., Xiong, J., et al. (2011). Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 152, 2704–2715. doi: 10.1210/en.2011-0145
Wegiel, J., Imaki, H., Wang, K. C., Wegiel, J., and Rubenstein, R. (2004). Cells of monocyte/microglial lineage are involved in both microvessel amyloidosis and fibrillar plaque formation in APPsw tg mice. Brain Res. 1022, 19–29. doi: 10.1016/j.brainres.2004.06.058
Weitz, T. M., and Town, T. (2012). Microglia in Alzheimer’s Disease: It’s All About Context. Int. J. Alzheimers Dis. 2012:314185. doi: 10.1155/2012/314185
Wood, W. G., Li, L., Müller, W. E., and Eckert, G. P. (2014). Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J. Neurochem. 129, 559–572. doi: 10.1111/jnc.12637
Xiong, H., Callaghan, D., Jones, A., Walker, D. G., Lue, L. F., Beach, T. G., et al. (2008). Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 29, 422–437. doi: 10.1016/j.nbd.2007.10.005
Xu, Q., Bernardo, A., Walker, D., Kanegawa, T., Mahley, R. W., and Huang, Y. (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. doi: 10.1523/jneurosci.5476-05.2006
Yamanaka, K., Saito, Y., Yamamori, T., Urano, Y., and Noguchi, N. (2011). 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J. Biol. Chem. 286, 24666–24673. doi: 10.1074/jbc.m111.236273
Yan, S. D., Yan, S. F., Chen, X., Fu, J., Chen, M., Kuppusamy, P., et al. (1995). Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat. Med. 1, 693–699. doi: 10.1038/nm0795-693
Yanagisawa, K. (2005). Cholesterol and amyloid beta fibrillogenesis. Subcell. Biochem. 38, 179–202. doi: 10.1007/0-387-23226-5_9
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yau, J. L., Rasmuson, S., Andrew, R., Graham, M., Noble, J., Olsson, T., et al. (2003). Dehydroepiandrosterone 7-hydroxylase CYP7B: predominant expression in primate hippocampus and reduced expression in Alzheimer’s disease. Neuroscience 121, 307–314. doi: 10.1016/s0306-4522(03)00438-x
Ye, S., Huang, Y., Müllendorff, K., Dong, L., Giedt, G., Meng, E. C., et al. (2005). Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. U S A 102, 18700–18705. doi: 10.1073/pnas.0508693102
Yoshimoto, N., Tasaki, M., Shimanouchi, T., Umakoshi, H., and Kuboi, R. (2005). Oxidation of cholesterol catalyzed by amyloid beta-peptide (Abeta)-Cu complex on lipid membrane. J. Biosci. Bioeng. 100, 455–459. doi: 10.1263/jbb.100.455
Yu, Y., and Ye, R. D. (2015). Microglial Aβ receptors in Alzheimer’s disease. Cell Mol. Neurobiol. 35, 71–83. doi: 10.1007/s10571-014-0101-6
Zelcer, N., Khanlou, N., Clare, R., Jiang, Q., Reed-Geaghan, E. G., Landreth, G. E., et al. (2007). Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U S A 104, 10601–10606. doi: 10.1073/pnas.0701096104
Zhu, X., Lee, H. G., Casadesus, G., Avila, J., Drew, K., Perry, G., et al. (2005). Oxidative imbalance in Alzheimer’s disease. Mol. Neurobiol. 31, 205–217. doi: 10.1385/mn:31:1-3:205
Keywords: Alzheimer’s disease, oxidative stress, inflammation, oxidized cholesterol, oxysterols
Citation: Gamba P, Testa G, Gargiulo S, Staurenghi E, Poli G and Leonarduzzi G (2015) Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front. Aging Neurosci. 7:119. doi: 10.3389/fnagi.2015.00119
Received: 30 April 2015; Accepted: 03 June 2015;
Published online: 19 June 2015.
Edited by:
Enrique Cadenas, University of Southern California, USAReviewed by:
Giuseppe Valacchi, University of Ferrara, ItalyJoão Laranjinha, University of Coimbra, Portugal
Copyright © 2015 Gamba, Testa, Gargiulo, Staurenghi, Poli and Leonarduzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Poli, Department of Clinical and Biological Sciences, School of Medicine, University of Turin, San Luigi Hospital, 10043 Orbassano, Torino, Italy,Z2l1c2VwcGUucG9saUB1bml0by5pdA==
† These authors have contributed equally to this work.