 Gabriela Mercado1,2*
Gabriela Mercado1,2* Claudio Hetz
Claudio Hetz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Aging Neurosci. , 27 March 2015
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 7 - 2015 | https://doi.org/10.3389/fnagi.2015.00039
This article is part of the Research Topic Dysregulation of Protein Folding in Neurodegenerative Disorders View all 12 articles
Parkinson's disease (PD) is characterized by the selective loss of dopaminergic neurons of the substantia nigra pars compacta (SNpc). Proteostasis impairment at the level of the endoplasmic reticulum (ER) is emerging as a driving factor of dopaminergic neuron loss in PD. ER stress engages the activation of an adaptive reaction known as the unfolded protein response (UPR) to recover proteostasis or trigger apoptosis of damaged cells. The therapeutic potential of the UPR as a target has been recently validated using pharmacological and gene therapy approaches. A complex view is emerging where ER stress may have a dual role in PD, both in maintaining cell survival during initial stages of the diseases and trigger neuronal degeneration when the stress levels are sustained. Here we overview recent advances in determining the impact of ER stress to PD.
PD is a progressive neurodegenerative disease that affects movement control, characterized by the loss of dopaminergic neurons in the SNpc. In most PD cases the presence of intracellular inclusions, termed Lewy bodies (LBs) is observed, where fibrillar aggregates of αSynuclein constitute a major component. Many cellular processes are altered in PD, including redox control, mitochondrial function, autophagy/lysosomal function, protein quality control mechanisms, and vesicle trafficking, among other processes. Accumulating evidence supports disruption in the secretory pathway as a triggering factor of proteostasis dysfunction in PD, mediating in part the selective degeneration of dopaminergic neurons (Chua and Tang, 2013; Mercado et al., 2013). Importantly, in addition to PD, ER stress is emerging as a relevant driver of most common neurodegenerative diseases (Hetz and Mollereau, 2014).
ER stress activates the UPR, a complex signaling transduction pathway that mediates cellular adaptation to restore ER function (reviewed in Ron and Walter, 2007; Hetz, 2012). In this article we discuss recent insights on the significance of ER stress as a driver of dopaminergic neuron loss in PD and the potential of targeting UPR components to augment the homeostatic capacity of the ER and reduce pro-apoptotic signals.
The UPR is a signaling network mediated by the activation of three stress sensors located at the ER membrane, including inositol requiring kinase 1α (IRE1α), activating transcription factor 6 (ATF6), and protein kinase RNA-like ER kinase (PERK) (Figure 1A). These UPR transducers control the expression of a variety of genes involved in almost every aspect of the secretory pathway, resulting in a reduction in the load of misfolded proteins at the ER. Activation of the UPR improves the efficiency of protein folding and quality control mechanisms, in addition to enhance ER and Golgi biogenesis, protein secretion and the clearance of abnormally folded proteins through the autophagy and ER-associated degradation (ERAD) pathways. However, under chronic ER stress UPR sensors shifts their signaling toward induction of cell death by apoptosis (Urra et al., 2013).
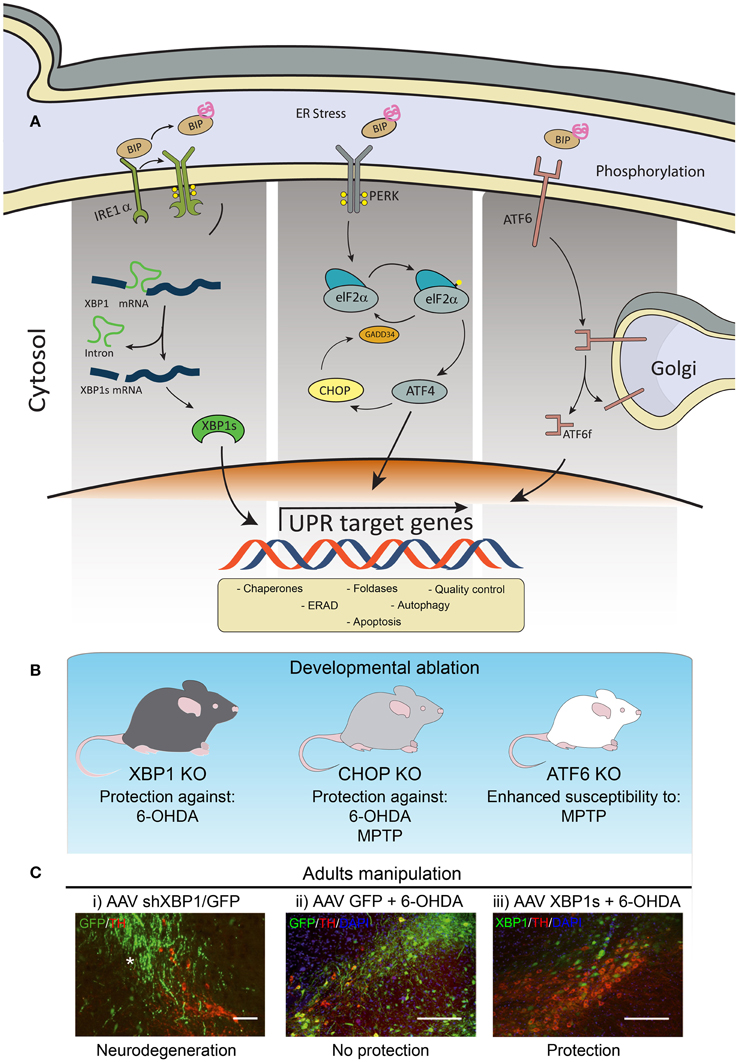
Figure 1. Involvement of ER stress in PD. (A) Schematic representation of the three branches of the UPR. (B) Knockout (KO) animals for XBP1, CHOP or ATF6 have been tested to manipulate the UPR in PD models. (C) Images modified from Valdes et al. (2014): Wild-type mice were injected into the with (i) AAV expressing an shRNA against XBP1 (shXBP1/GFP), (ii) EGFP alone, or (iii) a vector to overexpress XBP1s. One month after injection, experimental PD was induced using the 6-OHDA model to monitor dopaminergic neuron loss at the SNpc. Green: AAV transduced cells expressing GFP. Red: dopaminergic neurons stained with anti-tyrosine hydroxylase (TH). Scale bar: 200 μm.
IRE1α is an endoribonuclease that processes the mRNA encoding the transcription factor X-Box binding protein-1 (XBP1) which results in the expression of a more stable and active transcription factor, termed XBP1s (Ron and Walter, 2007). Upon activation, ATF6 traffics to the Golgi and undergoes subsequent proteolytic processing to release ATF6f, an active transcriptional factor (Ron and Walter, 2007). PERK is an ER-located kinase that upon activation phosphorylates the eukaryotic initiation factor 2α (eIF2α), attenuating general protein translation. In turn, eIF2α phosphorylation leads to the specific translation of activating transcription factor 4 (ATF4), which up-regulates many important genes functioning in redox control, amino acid metabolism and protein folding (Harding et al., 2003). Under chronic stress, ATF4 regulates the expression of pro-apoptotic genes such as CHOP.
The mechanisms leading to ER stress in PD and the actual impact of the UPR on the degeneration cascade are just starting to be uncovered. A genetic screening in yeast revealed that one of the major physical targets of αSynuclein is Rab1, an essential component of the ER-to-Golgi trafficking machinery (Cooper et al., 2006; Gitler et al., 2008). Over-expression of Rab1 in animal models of PD reduced stress levels and protected dopaminergic neurons against degeneration (Coune et al., 2011). Importantly, the generation of neuronal cultures from induced pluripotent stem cells (iPSC)-derived from PD patients revealed major proteostasis alterations (Chung et al., 2013). The authors provided evidence indicating that ER stress is a salient molecular signature of human PD neurons. There are many other studies linking other PD genes with alteration of the secretory pathway, including LRRK2, Parkin, Pael-R, DJ-1, ATP13A2 (reviewed in Mercado et al., 2013), and VPS35 (Zimprich et al., 2011). These reports suggest that secretory pathway dysfunction is a common hallmark of PD, which may result in pathological levels of ER stress contributing to the etiology of the disease.
Genetic manipulation of essential UPR components in the context of PD had been performed only in a few studies (Figure 1B). For example, ATF6α knockout animals showed increased accumulation of ubiquitin-positive inclusions and enhanced loss of dopaminergic neurons induced by a PD-triggering neurotoxin (Egawa et al., 2011). Although ATF6 is not essential for development and survival of dopaminergic neurons in mice, this stress sensor controls the levels of the chaperone BiP and ERAD components under resting conditions in these neurons (Egawa et al., 2011). A recent study determined that ATF6 is a direct target of αSynuclein. Expression of αSynuclein was shown to inhibit the processing of ATF6 through a physical association, leading to an impaired up-regulation of ERAD genes, which sensitized cells to apoptosis (Credle et al., 2015).
We recently reported a set of in vivo studies uncovering the significance of the UPR transcription factor XBP1 in controlling the survival of dopaminergic neurons (Valdes et al., 2014). We found that the developmental ablation of Xbp1 in the nervous system preconditioned dopaminergic neurons and rendered them resistant to the PD-triggering neurotoxin 6-hydroxydopamine (6-OHDA) (Figure 1B). This neuroprotective effect was accompanied by the up-regulation of several UPR effectors in the SNpc of animals in the absence of pro-apoptotic markers such as Chop. This phenotype correlated with the presence of poly-ubiquitinated proteins and large inclusion bodies in dopaminergic neurons of XBP1 deficient animals, resembling the classical alterations observed in PD. Remarkably, dopaminergic neurons were prompt to undergo proteostasis alterations in the absence of XBP1, a phenomenon not observed in other brain areas including cortex, striatum, or spinal cord (Hetz et al., 2009; Valenzuela et al., 2012; Vidal et al., 2012; Valdes et al., 2014). We proposed that developmental targeting of XBP1 provides neuroprotection through an “ER-hormesis” mechanism where the occurrence of mild non-lethal ER stress engages an adaptive response that sustains neuronal function in the absence of XBP1, which also renders dopaminergic neurons more resistance to a PD-inducing stimulus. In agreement with this concept, establishment of an ER-hormesis condition (Matus et al., 2012) by the administration of low doses of the ER stress agent tunicamicyn on a rodent and fly model of PD selectively engaged adaptive UPR signaling events involving the expression of XBP1s (Fouillet et al., 2012).
Since genetic manipulations during development can lead to compensatory mechanisms that mask the direct biological effects of a certain gene, we then targeted XBP1 in adult animals locally at the SNpc (Valdes et al., 2014). Knocking down XBP1 resulted in chronic ER stress involving the up-regulation of Chop, causing spontaneous neurodegeneration of dopaminergic neurons (Figure 1C). These results highlight the importance of XBP1 in sustaining dopaminergic neuron function and viability, reinforcing the concept that ER stress is a factor underling their differential neuronal vulnerability. Therapeutic strategy to artificially engage a UPR adaptive program has been developed to pre-adapt dopaminergic neurons to a PD-inducing event. Using a gene therapy approach, we delivered active XBP1s into the SNpc of adult mice using adeno-asociated viral (AAVs) vectors (Valdes et al., 2014). This strategy conferred a dramatic protection against 6-OHDA (Figure 1C), in addition to reduce striatal denervation. Similarly, a previous report also indicated that XBP1s gene transfer also protects dopaminergic neurons against the PD-inducing neurotoxin MPTP (Sado et al., 2009).
XBP1 has a conserved role in sustaining dopaminergic neuron survival. Recently, the over-expression of XBP1 was shown to protect against αSynuclein-induced dopaminergic neuron degeneration in C. elegans, whereas neuron-specific RNAi knockdown of xbp1 exacerbates the neurodegeneration process (Ray et al., 2014). The unconventional splicing of XBP1 mRNA, in addition to require the endoribonuclease IRE1α, it involves the RNA ligase RTCB-1. This ligase also confers protection to dopaminergic neurons against αSynuclein overexpression in C. elegans, uncovering for the first time a functional relationship between XBP1 and its ligase in the regulation of proteostasis in neurons (Ray et al., 2014). XBP1 expression has been shown to be neuroprotective also when it is delivered into neural stem cells that are then transfer into the brain. This strategy, increased the survival of the graft and improved the motor performance in a rotenone-induced rat model of PD (Lihui et al., 2012). Finally, an AAV-based gene therapy strategy to enhance the folding capacity of the ER was also evaluated on a genetic model of PD (Gorbatyuk et al., 2012). Thus, increasing evidence indicates that the local modulation of the UPR in the nigrostriatal circuit may have important therapeutic potential in PD.
The UPR is a double-edged sword, cytoprotective when activated to a moderate extent, but degenerative when it is sustained over time. Markers of PERK/eIF2α activation have been found in PD post-mortem brain tissue, where nigral dopaminergic neurons displaying αSynuclein inclusion are also positive for phosphorylated PERK and eIF2α (Hoozemans et al., 2007). Deletion of the pro-apoptotic factor CHOP protects dopaminergic neurons against 6-OHDA and MPTP (Silva et al., 2005) (Figure 1B). Several strategies are now available to modulate PERK signaling in different disease contexts, including inhibitors of PERK activity, eIF2α phosphatases, and ATF4 expression (reviewed in Hetz et al., 2013). Salubrinal, a small compound that enhances eIF2α (Boyce et al., 2005), was shown to delay disease onset and attenuate motor deficits induced by αSynuclein over-expression (Colla et al., 2012). Unexpectedly, although salubrinal treatment attenuated disease symptoms, its administration did not protect dopaminergic neurons from degeneration (Colla et al., 2012). In the last 2 years, new exciting findings implicate the PERK/ATF4 signaling branch of the UPR as an interesting target to treat neurodegenerative diseases (Halliday et al., 2014). In this scenario, additional tools are available to systematically test the consequences of inhibiting the PERK pathway in PD models at the level of PERK, eIF2α, or ATF4, respectively.
Many important questions remain to be solved in this growing field. Since distinct UPR signaling branches could have specific and even opposite consequences on neuronal survival depending on the disease input (Hetz and Mollereau, 2014), a systematic approach is needed to determine what are the optimal components of the UPR pathway as possible targets to develop future therapeutic interventions. Gene therapy strategies are currently been developed in PD patients and the first results of phase I and II clinical trials are available showing excellent safety profiles (Coune et al., 2012). In this context, the possible therapeutic potential and side effects of delivering active UPR components into the SNpc in the long term remains to be determined in non-human primates since most of the available studies only used rapid-evolving PD rodent models. Another interesting aspect to explore in the future is the cell-non-autonomous control of the UPR in PD, which may propagate protective responses to other brain areas and tissues (Mardones et al., 2015). Overall all these novel insights have placed ER proteostasis in the center of the etiology of PD, which may translate in the near future into the development of prototypic strategies to alleviate dopaminergic neuron loss.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funded by FONDECYT-11140738 (GM), FONDECYT-1140549 (CH) Michael J. Fox Foundation for Parkinson Research, The Frick Foundation, COPEC-UC Foundation, ACT1109, FONDEF D11I1007, Millennium Institute P09-015-F. RV is supported by FONDECYT-1150608, VC is supported by CONICYT fellowship.
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chua, C. E., and Tang, B. L. (2013). Linking membrane dynamics and trafficking to autophagy and the unfolded protein response. J. Cell. Physiol. 228, 1638–1640. doi: 10.1002/jcp.24341
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, C. Y., Khurana, V., Auluck, P. K., Tardiff, D. F., Mazzulli, J. R., Soldner, F., et al. (2013). Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. doi: 10.1126/science.1245296
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328. doi: 10.1126/science.1129462
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coune, P. G., Bensadoun, J. C., Aebischer, P., and Schneider, B. L. (2011). Rab1A over-expression prevents Golgi apparatus fragmentation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson's disease. J. Parkinsons Dis. 1, 373–387. doi: 10.3233/JPD-2011-11058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coune, P. G., Schneider, B. L., and Aebischer, P. (2012). Parkinson's disease: gene therapies. Cold Spring Harb. Perspect. Med. 2:a009431. doi: 10.1101/cshperspect.a009431
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Credle, J. J., Forcelli, P. A., Delannoy, M., Oaks, A. W., Permaul, E., Berry, D. L., et al. (2015). Alpha-synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson's disease. Neurobiol. Dis. 76, 112–125. doi: 10.1016/j.nbd.2015.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Egawa, N., Yamamoto, K., Inoue, H., Hikawa, R., Nishi, K., Mori, K., et al. (2011). The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 286, 7947–7957. doi: 10.1074/jbc.M110.156430
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi: 10.4161/auto.19716
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150. doi: 10.1073/pnas.0710685105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., et al. (2012). Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 20, 1327–1337. doi: 10.1038/mt.2012.28
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halliday, M., Radford, H., and Mallucci, G. R. (2014). Prions: generation and spread versus neurotoxicity. J. Biol. Chem. 289, 9862–19868. doi: 10.1074/jbc.R114.568477
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/S1097-2765(03)00105-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoozemans, J. J., van Haastert, E. S., Eikelenboom, P., de Vos, R. A., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson's disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lihui, S., Tianmin, X., Fengzhang, W., Qun, L., and Manhua, C. (2012). X-box-binding protein 1-modified neural stem cells for treatment of Parkinson's disease. Neural Regen. Res. 7, 736–740. doi: 10.3969/j.issn.1673-5374.2012.10.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mardones, P., Martinez, G., and Hetz, C. (2015). Control of systemic proteostasis by the nervous system. Trends Cell Biol. 25, 1–10. doi: 10.1016/j.tcb.2014.08.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matus, S., Castillo, K., and Hetz, C. (2012). Hormesis: protecting neurons against cellular stress in Parkinson disease. Autophagy 8, 997–1001. doi: 10.4161/auto.20748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mercado, G., Valdes, P., and Hetz, C. (2013). An ERcentric view of Parkinson's disease. Trends Mol. Med. 19, 165–175. doi: 10.1016/j.molmed.2012.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ray, A., Zhang, S., Rentas, C., Caldwell, K. A., and Caldwell, G. A. (2014). RTCB-1 mediates neuroprotection via XBP-1 mRNA splicing in the unfolded protein response pathway. J. Neurosci. 34, 16076–16085. doi: 10.1523/JNEUROSCI.1945-14.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sado, M., Yamasaki, Y., Iwanaga, T., Onaka, Y., Ibuki, T., Nishihara, S., et al. (2009). Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 1257, 16–24. doi: 10.1016/j.brainres.2008.11.104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Silva, R. M., Ries, V., Oo, T. F., Yarygina, O., Jackson-Lewis, V., Ryu, E. J., et al. (2005). CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 95, 974–986. doi: 10.1111/j.1471-4159.2005.03428.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Urra, H., Dufey, E., Lisbona, F., Rojas-Rivera, D., and Hetz, C. (2013). When ER stress reaches a dead end. Biochim. Biophys. Acta 1833, 3507–3517. doi: 10.1016/j.bbamcr.2013.07.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Valdes, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U.S.A. 111, 6804–6809. doi: 10.1073/pnas.1321845111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Valenzuela, V., Collyer, E., Armentano, D., Parsons, G. B., Court, F. A., and Hetz, C. (2012). Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 3:e272. doi: 10.1038/cddis.2012.8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C., Wirth, C., et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. doi: 10.1093/hmg/dds040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zimprich, A., Benet-Pages, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. doi: 10.1016/j.ajhg.2011.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: UPR signaling pathways, Parkinson disease, gene therapy, preclinical models, dopaminergic neurons, ER stress
Citation: Mercado G, Castillo V, Vidal R and Hetz C (2015) ER proteostasis disturbances in Parkinson's disease: novel insights. Front. Aging Neurosci. 7:39. doi: 10.3389/fnagi.2015.00039
Received: 27 February 2015; Accepted: 10 March 2015;
Published: 27 March 2015.
Edited by:
Vinood B. Patel, University of Westminster, UKReviewed by:
Rafael Linden, Federal University of Rio de Janeiro, BrazilCopyright © 2015 Mercado, Castillo, Vidal and Hetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Mercado and Claudio Hetz,bWFyaWFtZXJjYWRvQG1lZC51Y2hpbGUuY2w=;Y2hldHpAbWVkLnVjaGlsZS5jbA==;Y2hldHpAaHNwaC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.