 Vladimir N. Uversky1,2,3,4*
Vladimir N. Uversky1,2,3,4*- 1Department of Molecular Medicine, USF Health Byrd Alzheimer's Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, USA
- 2Biology Department, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
- 3Institute for Biological Instrumentation, Russian Academy of Sciences, Pushchino, Russia
- 4Laboratory of Structural Dynamics, Stability and Folding of Proteins, Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia
The recent years have witnessed a rise in the number of intrinsically disordered proteins (IDPs), also known as hybrid proteins, which possess both structured domains and biologically important intrinsically disordered protein regions (IDPRs). These proteins challenge the “one sequence—one structure—one function” concept by demonstrating that the lack of stable tertiary and/or secondary structure does not preclude proteins from being biologically active (Wright and Dyson, 1999; Uversky et al., 2000; Dunker et al., 2001; Tompa, 2002; Uversky and Dunker, 2010). Both ordered and disordered/hybrid proteins tend to misfold under certain conditions, and the aggregation that typically accompanies protein misfolding is associated with the pathogenesis of several human diseases, particularly those that originate from the deposition of protein aggregates in a variety of organs and tissues (Kelly, 1998; Bellotti et al., 1999; Dobson, 1999; Uversky et al., 1999a,b; Rochet and Lansbury, 2000; Uversky and Fink, 2004; Gasperini et al., 2012; Moreau and King, 2012; Safar, 2012; Walker and LeVine, 2012; Cuanalo-Contreras et al., 2013; Mulligan and Chakrabartty, 2013; Hipp et al., 2014). Misfolding and aggregation of IDPs/IDPRs are especially common in neurodegeneration (Uversky, 2010, 2014a; Breydo and Uversky, 2011). An incomplete list of human neurodegenerative diseases associated with IDPs/IDPRs is provided below. This list shows that some IDPs are involved in several diseases and that some neurodegenerative diseases are associated with misbehavior of several IDPs/IDPRs. The list includes Alzheimer's disease [AD, where the deposition of amyloid-β, tau-protein, and α-synuclein fragment NAC (Glenner and Wong, 1984; Ueda et al., 1993) takes place]; Niemann-Pick disease type C, subacute sclerosing panencephalitis, argyrophilic grain disease, myotonic dystrophy, and motor neuron disease with neurofibrillary tangles (NFTs) (accumulation of tau-protein in the form of NFTs, Lee et al., 1991); Down's syndrome (nonfilamentous amyloid-β deposits, Wisniewski et al., 1985); Parkinson's disease (PD), dementia with Lewy body (LB), diffuse LB disease, LB variant of AD, multiple system atrophy (MSA), and Hallervorden-Spatz disease [all characterized by the deposition of α-synuclein in a form of LB, or Lewy neurites (LNs), Dev et al., 2003]; amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTD) [both characterized by the presence of the cytoplasmic inclusions rich in transactive response element DNA-binding protein of 43 kDa (TDP43) (Nass et al., 2012; Barmada et al., 2014)]; aberrant accumulation of the wild type and mutated fused in sarcoma/translocated in liposarcoma (FUS/TLS) in the cytosol of voluntary motor neurons in sporadic and familial ALS (Pokrishevsky et al., 2012; Sreedharan and Brown, 2013; Ajroud-Driss and Siddique, 2015); prion diseases (deposition of PrPSC, Prusiner, 2001); and a family of polyQ diseases, a group of neurodegenerative disorders caused by the expansion of GAC trinucleotide repeats that code for polyQ in the gene products (Zoghbi and Orr, 1999).
There are several reasons for why IDPs/IDPRs are so common in neurodegenerative diseases. Firstly, these proteins/regions, with their unique structural plasticity, conformational adaptability, ability to react quickly in response to changes in their environment, and their binding promiscuity, are abundantly involved in various signaling, regulation, and recognition processes, and play diverse roles in the modulation and control of the functions of their binding partners (Dyson and Wright, 2005; Oldfield et al., 2008; Uversky and Dunker, 2010; Cozzetto and Jones, 2013; Ferreon et al., 2013). Secondly, the biological activities of IDPs/IDPRs are under tight control and are regulated by means of extensive posttranslational modifications (PTMs), such as phosphorylation, acetylation, glycosylation (Collins et al., 2008; Uversky and Dunker, 2010; Kurotani et al., 2014; Pejaver et al., 2014), and by alternative splicing (Romero et al., 2006; Buljan et al., 2012, 2013; Uversky, 2014b). Thirdly, many IDPs and hybrid proteins are able to interact with a large number of unrelated partners, thereby serving as hubs in cellular protein-protein interaction networks (Dunker et al., 2005; Uversky et al., 2005; Dosztanyi et al., 2006; Ekman et al., 2006; Haynes et al., 2006; Patil and Nakamura, 2006; Singh et al., 2007; Singh and Dash, 2007). Lastly, IDPs/IDPRs are often able to fold differently while interacting with different binding partners (Dyson and Wright, 2005; Oldfield et al., 2008; Hsu et al., 2013).
Since IDPs/IDPRs play a number of crucial roles in numerous biological processes, it is not surprising that some of these proteins are related to the pathogenesis of human disease, and to neurodegenerative processes in particular. In fact, dysregulation and misfolding of the otherwise tightly controlled IDPs/IDPRs can result in their dysfunction, ultimately leading to the development of life-threatening pathological conditions. Mutations and/or changes in the environment may reduce the capability of a protein to recognize proper binding partners, leading to the formation of nonfunctional complexes and aggregates (Uversky et al., 2005). The topic of the involvement of IDPs/IDPRs in neurodegeneration was covered in recent reviews (Uversky, 2010, 2014a; Breydo and Uversky, 2011). Table 1 clearly shows that some individual proteins involved in the pathogenesis of human neurodegenerative diseases are either completely disordered or contain long disordered regions. Table 1 also illustrates that neurodegeneration-related IDPs are characterized by astonishing binding promiscuity, as they are able to interact with a large number of unrelated partners. It is worth noting that the numbers shown in Table 1 correspond to the minimal interactomes, or “first interaction shells”; i.e., they correspond to the numbers of proteins directly interacting with a given neurodegeneration-related protein. Many of the proteins in such “first interaction shells” interact with other proteins, thereby generating a broadened “second interaction shell.”
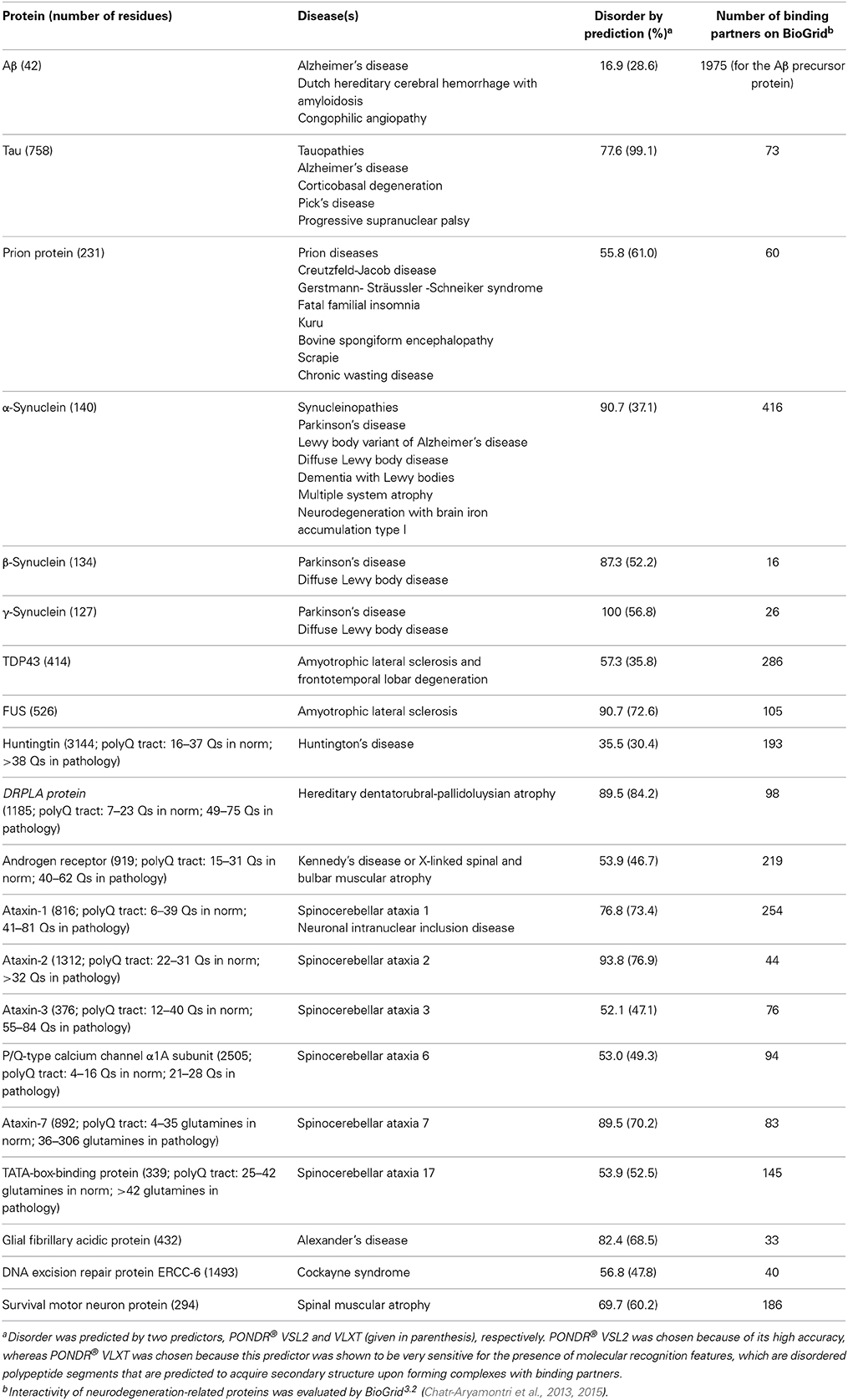
Table 1. IDPs and associated neurodegenerative diseases.
Neurodegeneration-related proteins are intrinsically disordered hubs with highly extended proteomes. Due to the overall abundance of IDPs, at least some of these binding partners are disordered proteins themselves. This gives an interesting twist to the involvement of IDPs in neurodegeneration, since the neurodegeneration-related IDPs listed in Table 1 are not only abundantly found in neurodegenerative diseases, but are also regulated and controlled by other IDPs. A few related examples are provided below to illustrate the ensuing “chaos to control chaos” concept.
The first example deals with the proteins directly involved in the control of normal cellular proteostasis: protein chaperones. Protein chaperones form a specific network that constitutes the major component of the cellular quality control (McClellan et al., 2005; Bukau et al., 2006; Leidhold and Voos, 2007; Witt, 2010). Computational studies revealed that ~40% of chaperones' residues are located within the disordered regions, with ~15% of the residues falling within long IDPRs exceeding 30 consecutive residues (Tompa and Csermely, 2004). Studies have shown that many neuroprotective chaperones/co-chaperones are either completely disordered or possess long IDPRs (Uversky, 2011). The illustrative examples of these disordered/hybrid chaperones include Hsp70 (C-terminal part of substrate binding domain and lid domain); members of the human DnaJ homolog subfamilies A, B, and C; various Hsp70 co-chaperones [Hip, Hsp100, BAG family molecular chaperone regulator 1 L, CHIP/STIP1, Hop (Hsp70/Hsp90-organization protein)]; linker regions in human Hsp90α and Hsp90β; Hsp90 co-chaperones p23 and FKBP52; small heat shock proteins Hsp27/HspB1, HspB2, HspB3, αA-crystallin/HspB4, αB-crystallin/HspB5, Hsp20/HspB6, cvHsp/HspB7, H11/HspB8, HspB9, and outer dense fiber protein 1 (ODF1); human α-, β-, and γ-synucleins; prefoldin subunits. (Uversky, 2011). It was demonstrated that the presence of disorder determines the promiscuity of chaperones, allowing them to act as pliable molecular recognition elements, to wrap misfolded chains, and to participate in disaggregation and local unfolding of the aggregated and misfolded species (Uversky, 2011). Furthermore, disorder plays a number of important roles in the precise orchestration of coordinated actions of chaperones, co-chaperones, and their auxiliary and regulatory proteins, which intricately communicate with each other to form a sophisticated and highly flexible network of malleable guardians (Uversky, 2011).
The second example is related to sirtuins that constitute an important family of regulatory proteins involved in several physiological functions, including control of gene expression, metabolism, and aging (Paraiso et al., 2013). In mammals, there are seven sirtuins (Sirt1 to Sirt7), with members of this protein family being highly expressed in various regions of the brain. Here, sirtuins are involved in cognitive functions and regulate cellular protection against oxidative stress in many neurodegenerative diseases (Gan and Mucke, 2008). For example, in animal PD models, Sirt1 regulates autophagy (Lee et al., 2008), which is responsible for the aggregated α-synuclein clearance (Paraiso et al., 2013), and therefore Sirt1 can reduce α-synuclein aggregation (Zhang et al., 2012). In AD models, the overexpression of Sirt1 modulates the processing of APP by increasing the activity of α-secretase (Bonda et al., 2011), which shifts APP processing toward the non-amyloidogenic Aβ forms (Bonda et al., 2011). Furthermore, Sirt1 deactivation has been associated with increased levels of acetylated and pathogenic phosphorylated forms of tau protein (Min et al., 2010). In HD mouse models, some neuroprotection was reported for Sirt1 overexpression (Jeong et al., 2012). These data show that sirtuins can act as important regulators of several neurodegenerative diseases caused by the misbehavior of several neurodegeneration-promoting IDPs (α-synuclein, tau, Aβ, and huntingtin). Recent in silico analysis of the sirtuin family members showed that all these proteins have long disordered arms that play crucial biological roles such as recognition and interaction with other protein molecules (Costantini et al., 2013).
The third example emphasizes the important role of intrinsic disorder in the maintenance of stress granules (SGs) that are potentially related to the pathology of some neurodegenerative diseases (Wolozin, 2012; Bentmann et al., 2013). These SGs are formed as a cellular stress response and are cytoplasmic membrane-less organelles that contain several RNA-binding proteins and RNA molecules that are stalled at the pre-initiation stage. These RNA molecules and binding proteins possess defined cytoprotective function (Bentmann et al., 2013). Importantly, all the major players responsible for the nucleation and maturation of SGs are either IDPs or hybrid proteins containing long, functionally important IDPRs (Uversky et al., 2015). Furthermore, SGs were shown to co-localize with insoluble protein aggregates in many neurodegenerative diseases (Wolozin, 2012). They commonly include RNA-binding proteins related to the pathogenesis of various neurodegenerative diseases, such as TDP-43 and FUS (related to the pathology of ALS and FTD), SMN (related to SMA pathology), ataxin-2 (related to SCA2), optineurin (related to primary open angle glaucoma and ALS12), and angiogenin (involved in ALS9 pathology). They may also contain, and be regulated by, proteins such as tau (Wolozin, 2012), PrPSC (Goggin et al., 2008), huntingtin (Waelter et al., 2001), and some other Q/N-rich proteins (Furukawa et al., 2009).
The last example emphasizes the role of the tubulin polymerization promoting protein (TPPP)/p25 in regulating α-synuclein aggregation in multiple system atrophy (MSA). The α-synuclein-containing glial cytoplasmic inclusions are the neuropathological hallmark of MSA. Recent studies revealed that the pathological aggregation of α-synuclein in oligodendroglia is dramatically accelerated by TPPP/p25 (Hasegawa et al., 2010), which is a highly disordered and widely expressed protein that possesses multiple PTM sites, and is involved in a multitude of interactions with unrelated partners. Structural analysis revealed that TPPP/p25 is a typical IDP that partially folds as a result of Zn2+ binding (Zotter et al., 2011).
Conclusions
Human neurodegenerative diseases are commonly associated with the misbehavior of IDPs and hybrid proteins containing ordered domains and IDPRs. This link between intrinsic disorder and neurodegeneration is determined by the specific structural and functional features of IDPs/IDPRs, which are some of the major cellular regulators, recognizers, and signal transducers. The normal functionality and pathological misbehavior of IDPs/IDPRs are both modulated by various PTMs and alternative splicing, with dysregulation of these regulatory mechanisms being a crucial contributing factor to dysfunction of related proteins and, consequently, to neurodegeneration. IDPs/IDPRs are promiscuous binders that can fold differently upon interaction with different binding partners. Furthermore, proteomes of such neurodegeneration-related IDPs/IDPRs are vast and contain numerous regulatory IDPs. Therefore, pathogenesis of neurodegenerative diseases is commonly driven by the dysfunction of corresponding IDPs, and the normal and pathogenic behavior of such disease-related IDPs are controlled by other IDPs.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am thankful to Alexey Uversky for careful reading and editing this manuscript. This work was supported in part by a grant from Russian Science Foundation RSCF No. 14-24-00131.
References
Ajroud-Driss, S., and Siddique, T. (2015). Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta 1852, 679–684. doi: 10.1016/j.bbadis.2014.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barmada, S. J., Serio, A., Arjun, A., Bilican, B., Daub, A., Ando, D. M., et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685. doi: 10.1038/nchembio.1563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellotti, V., Mangione, P., and Stoppini, M. (1999). Biological activity and pathological implications of misfolded proteins. Cell. Mol. Life Sci. 55, 977–991. doi: 10.1007/s000180050348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bentmann, E., Haass, C., and Dormann, D. (2013). Stress granules in neurodegeneration – lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 280, 4348–4370. doi: 10.1111/febs.12287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonda, D. J., Lee, H. G., Camins, A., Pallas, M., Casadesus, G., Smith, M. A., et al. (2011). The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 10, 275–279. doi: 10.1016/S1474-4422(11)70013-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breydo, L., and Uversky, V. N. (2011). Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 3, 1163–1180. doi: 10.1039/c1mt00106j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451. doi: 10.1016/j.cell.2006.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buljan, M., Chalancon, G., Dunker, A. K., Bateman, A., Balaji, S., Fuxreiter, M., et al. (2013). Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol. 23, 443–450. doi: 10.1016/j.sbi.2013.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buljan, M., Chalancon, G., Eustermann, S., Wagner, G. P., Fuxreiter, M., Bateman, A., et al. (2012). Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883. doi: 10.1016/j.molcel.2012.05.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatr-Aryamontri, A., Breitkreutz, B. J., Heinicke, S., Boucher, L., Winter, A., Stark, C., et al. (2013). The BioGRID interaction database: 2013 update. Nucleic Acids Res. 41, D816–D823. doi: 10.1093/nar/gks1158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatr-Aryamontri, A., Breitkreutz, B. J., Oughtred, R., Boucher, L., Heinicke, S., Chen, D., et al. (2015). The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–D478. doi: 10.1093/nar/gku1204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collins, M. O., Yu, L., Campuzano, I., Grant, S. G., and Choudhary, J. S. (2008). Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol. Cell. Proteomics 7, 1331–1348. doi: 10.1074/mcp.M700564-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Costantini, S., Sharma, A., Raucci, R., Costantini, M., Autiero, I., and Colonna, G. (2013). Genealogy of an ancient protein family: the Sirtuins, a family of disordered members. BMC Evol. Biol. 13:60. doi: 10.1186/1471-2148-13-60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cozzetto, D., and Jones, D. T. (2013). The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr. Opin. Struct. Biol. 23, 467–472. doi: 10.1016/j.sbi.2013.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cuanalo-Contreras, K., Mukherjee, A., and Soto, C. (2013). Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013:638083. doi: 10.1155/2013/638083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dev, K. K., Hofele, K., Barbieri, S., Buchman, V. L., and van der Putten, H. (2003). Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 45, 14–44. doi: 10.1016/S0028-3908(03)00140-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobson, C. M. (1999). Protein misfolding, evolution and disease. Trends Biochem. Sci. 24, 329–332. doi: 10.1016/S0968-0004(99)01445-0
Dosztanyi, Z., Chen, J., Dunker, A. K., Simon, I., and Tompa, P. (2006). Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 5, 2985–2995. doi: 10.1021/pr060171o
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005). Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. (2001). Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59. doi: 10.1016/S1093-3263(00)00138-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyson, H. J., and Wright, P. E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. doi: 10.1038/nrm1589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ekman, D., Light, S., Bjorklund, A. K., and Elofsson, A. (2006). What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 7:R45. doi: 10.1186/gb-2006-7-6-r45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferreon, A. C., Ferreon, J. C., Wright, P. E., and Deniz, A. A. (2013). Modulation of allostery by protein intrinsic disorder. Nature 498, 390–394. doi: 10.1038/nature12294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Furukawa, Y., Kaneko, K., Matsumoto, G., Kurosawa, M., and Nukina, N. (2009). Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J. Neurosci. 29, 5153–5162. doi: 10.1523/JNEUROSCI.0783-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gan, L., and Mucke, L. (2008). Paths of convergence: sirtuins in aging and neurodegeneration. Neuron 58, 10–14. doi: 10.1016/j.neuron.2008.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gasperini, R. J., Klaver, D. W., Hou, X., Aguilar, M. I., and Small, D. H. (2012). Mechanisms of transthyretin aggregation and toxicity. Subcell. Biochem. 65, 211–224. doi: 10.1007/978-94-007-5416-4_9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/S0006-291X(84)80190-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goggin, K., Beaudoin, S., Grenier, C., Brown, A. A., and Roucou, X. (2008). Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim. Biophys. Acta 1783, 479–491. doi: 10.1016/j.bbamcr.2007.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasegawa, T., Baba, T., Kobayashi, M., Konno, M., Sugeno, N., Kikuchi, A., et al. (2010). Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem. Int. 57, 857–866. doi: 10.1016/j.neuint.2010.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haynes, C., Oldfield, C. J., Ji, F., Klitgord, N., Cusick, M. E., Radivojac, P., et al. (2006). Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2:e100. doi: 10.1371/journal.pcbi.0020100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hipp, M. S., Park, S. H., and Hartl, F. U. (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514. doi: 10.1016/j.tcb.2014.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, W. L., Oldfield, C. J., Xue, B., Meng, J., Huang, F., Romero, P., et al. (2013). Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 22, 258–273. doi: 10.1002/pro.2207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeong, H., Cohen, D. E., Cui, L., Supinski, A., Savas, J. N., Mazzulli, J. R., et al. (2012). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165. doi: 10.1038/nm.2559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelly, J. W. (1998). The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8, 101–106. doi: 10.1016/S0959-440X(98)80016-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurotani, A., Tokmakov, A. A., Kuroda, Y., Fukami, Y., Shinozaki, K., and Sakurai, T. (2014). Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics 30, 1095–1103. doi: 10.1093/bioinformatics/btt762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379. doi: 10.1073/pnas.0712145105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, V. M., Balin, B. J., Otvos, L. Jr., and Trojanowski, J. Q. (1991). A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678. doi: 10.1126/science.1899488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leidhold, C., and Voos, W. (2007). Chaperones and proteases–guardians of protein integrity in eukaryotic organelles. Ann. N.Y. Acad. Sci. 1113, 72–86. doi: 10.1196/annals.1391.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McClellan, A. J., Tam, S., Kaganovich, D., and Frydman, J. (2005). Protein quality control: chaperones culling corrupt conformations. Nat. Cell Biol. 7, 736–741. doi: 10.1038/ncb0805-736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreau, K. L., and King, J. A. (2012). Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol. Med. 18, 273–282. doi: 10.1016/j.molmed.2012.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mulligan, V. K., and Chakrabartty, A. (2013). Protein misfolding in the late-onset neurodegenerative diseases: common themes and the unique case of amyotrophic lateral sclerosis. Proteins 81, 1285–1303. doi: 10.1002/prot.24285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nass, R. D., Meister, I. G., Haupt, W. F., and Fink, G. R. (2012). [ALS and frontotemporal dementia - case report and review of the literature]. Fortschr. Neurol. Psychiatr. 80, 711–719. doi: 10.1055/s-0032-1325604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oldfield, C. J., Meng, J., Yang, J. Y., Yang, M. Q., Uversky, V. N., and Dunker, A. K. (2008). Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9(Suppl. 1):S1. doi: 10.1186/1471-2164-9-S1-S1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paraiso, A. F., Mendes, K. L., and Santos, S. H. (2013). Brain activation of SIRT1: role in neuropathology. Mol. Neurobiol. 48, 681–689. doi: 10.1007/s12035-013-8459-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patil, A., and Nakamura, H. (2006). Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 580, 2041–2045. doi: 10.1016/j.febslet.2006.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pejaver, V., Hsu, W. L., Xin, F., Dunker, A. K., Uversky, V. N., and Radivojac, P. (2014). The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 23, 1077–1093. doi: 10.1002/pro.2494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pokrishevsky, E., Grad, L. I., Yousefi, M., Wang, J., Mackenzie, I. R., and Cashman, N. R. (2012). Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 7:e35050. doi: 10.1371/journal.pone.0035050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prusiner, S. B. (2001). Shattuck lecture–neurodegenerative diseases and prions. N. Engl. J. Med. 344, 1516–1526. doi: 10.1056/NEJM200105173442006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rochet, J. C., and Lansbury, P. T. Jr. (2000). Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 10, 60–68. doi: 10.1016/S0959-440X(99)00049-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Romero, P. R., Zaidi, S., Fang, Y. Y., Uversky, V. N., Radivojac, P., Oldfield, C. J., et al. (2006). Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. U.S.A. 103, 8390–8395. doi: 10.1073/pnas.0507916103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Safar, J. G. (2012). Molecular pathogenesis of sporadic prion diseases in man. Prion 6, 108–115. doi: 10.4161/pri.18666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, G. P., and Dash, D. (2007). Intrinsic disorder in yeast transcriptional regulatory network. Proteins 68, 602–605. doi: 10.1002/prot.21497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, G. P., Ganapathi, M., and Dash, D. (2007). Role of intrinsic disorder in transient interactions of hub proteins. Proteins 66, 761–765. doi: 10.1002/prot.21281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sreedharan, J., and Brown, R. H. Jr. (2013). Amyotrophic lateral sclerosis: Problems and prospects. Ann. Neurol. 74, 309–316. doi: 10.1002/ana.24012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tompa, P. (2002). Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533. doi: 10.1016/S0968-0004(02)02169-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tompa, P., and Csermely, P. (2004). The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18, 1169–1175. doi: 10.1096/fj.04-1584rev
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N. (2010). Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept. Expert Rev. Proteomics 7, 543–564. doi: 10.1586/epr.10.36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N. (2011). Flexible nets of malleable guardians: intrinsically disordered chaperones in neurodegenerative diseases. Chem. Rev. 111, 1134–1166. doi: 10.1021/cr100186d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N. (2014a). The triple power of D(3): protein intrinsic disorder in degenerative diseases. Front. Biosci. (Landmark Ed.) 19, 181–258. doi: 10.2741/4204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N. (2014b). Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front. Mol. Biosci. 1:6. doi: 10.3389/fmolb.2014.00006
Uversky, V. N., and Dunker, A. K. (2010). Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264. doi: 10.1016/j.bbapap.2010.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., and Fink, A. L. (2004). Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta 1698, 131–153. doi: 10.1016/j.bbapap.2003.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., Gillespie, J. R., and Fink, A. L. (2000). Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41: 415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., Kuznetsova, I. M., Turoverov, K. K., and Zaslavsky, B. (2015). Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 589, 15–22. doi: 10.1016/j.febslet.2014.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2005). Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384. doi: 10.1002/jmr.747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N., Talapatra, A., Gillespie, J. R., and Fink, A. L. (1999a). Protein deposits as the molecular basis of amyloidosis. I. Systemic amyloidoses. Med. Sci. Monit. 5, 1001–1012.
Uversky, V. N., Talapatra, A., Gillespie, J. R., and Fink, A. L. (1999b). Protein deposits as the molecular basis of amyloidosis. II. Localized amyloidosis and neurodegenerative disordres. Med. Sci. Monit. 5, 1238–1254.
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H., et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407. doi: 10.1091/mbc.12.5.1393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walker, L. C., and LeVine, H. III. (2012). Corruption and spread of pathogenic proteins in neurodegenerative diseases. J. Biol. Chem. 287, 33109–33115. doi: 10.1074/jbc.R112.399378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wisniewski, K. E., Dalton, A. J., McLachlan, C., Wen, G. Y., and Wisniewski, H. M. (1985). Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology 35, 957–961. doi: 10.1212/WNL.35.7.957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Witt, S. N. (2010). Hsp70 molecular chaperones and Parkinson's disease. Biopolymers 93, 218–228. doi: 10.1002/bip.21302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolozin, B. (2012). Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegener. 7, 56. doi: 10.1186/1750-1326-7-56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, P. E., and Dyson, H. J. (1999). Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331. doi: 10.1006/jmbi.1999.3110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, A., Wang, H., Qin, X., Pang, S., and Yan, B. (2012). Genetic analysis of SIRT1 gene promoter in sporadic Parkinson's disease. Biochem. Biophys. Res. Commun. 422, 693–696. doi: 10.1016/j.bbrc.2012.05.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zoghbi, H. Y., and Orr, H. T. (1999). Polyglutamine diseases: protein cleavage and aggregation. Curr. Opin. Neurobiol. 9, 566–570. doi: 10.1016/S0959-4388(99)00013-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zotter, A., Olah, J., Hlavanda, E., Bodor, A., Perczel, A., Szigeti, K., et al. (2011). Zn(2)+-induced rearrangement of the disordered TPPP/p25 affects its microtubule assembly and GTPase activity. Biochemistry 50, 9568–9578. doi: 10.1021/bi201447w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: intrinsically disordered proteins, protein-protein interaction, posttranslational modification, neurodegenerative diseases, proteome
Citation: Uversky VN (2015) Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7:18. doi: 10.3389/fnagi.2015.00018
Received: 29 December 2014; Accepted: 13 February 2015;
Published online: 02 March 2015.
Edited by:
Vinood B. Patel, University of Westminster, UKReviewed by:
Jonathon Hull, University of the West of England, UKCopyright © 2015 Uversky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:dnV2ZXJza3lAaGVhbHRoLnVzZi5lZHU=