 Russell T. Hepple
Russell T. Hepple
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 10 September 2014
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 6 - 2014 | https://doi.org/10.3389/fnagi.2014.00211
This article is part of the Research Topic Pathophysiological Mechanisms of Sarcopenia in Aging and in Muscular Dystrophy: a Translational Approach View all 28 articles
Atrophy is a defining feature of aging skeletal muscle that contributes to progressive weakness and an increased risk of mobility impairment, falls, and physical frailty in very advanced age. Amongst the most frequently implicated mechanisms of aging muscle atrophy is mitochondrial dysfunction. Recent studies employing methods that are well-suited to interrogating intrinsic mitochondrial function find that mitochondrial respiration and reactive oxygen species emission changes are inconsistent between aging rat muscles undergoing atrophy and appear normal in human skeletal muscle from septuagenarian physically active subjects. On the other hand, a sensitization to permeability transition seems to be a general property of atrophying muscle with aging and this effect is even seen in atrophying muscle from physically active septuagenarian subjects. In addition to this intrinsic alteration in mitochondrial function, factors extrinsic to the mitochondria may also modulate mitochondrial function in aging muscle. In particular, recent evidence implicates oxidative stress in the aging milieu as a factor that depresses respiratory function in vivo (an effect that is not present ex vivo). Furthermore, in very advanced age, not only does muscle atrophy become more severe and clinically relevant in terms of its impact, but also there is evidence that this is driven by an accumulation of severely atrophied denervated myofibers. As denervation can itself modulate mitochondrial function and recruit mitochondrial-mediated atrophy pathways, future investigations need to address the degree to which skeletal muscle mitochondrial alterations in very advanced age are a consequence of denervation, rather than a primary organelle defect, to refine our understanding of the relevance of mitochondria as a therapeutic target at this more advanced age.
Progressive atrophy is a defining feature of aging skeletal muscle and when it becomes severe in very advanced age (≥80 years of age), it can lead to weakness that precipitates mobility impairment, an increased risk of falls, and physically frailty (Cruz-Jentoft et al., 2010). For this reason, understanding the mechanisms underlying aging muscle atrophy so that suitable therapeutic targets can be identified is key to promoting health and mobility in the elderly. Whilst many possible targets have been suggested, the most effective therapeutic targets will be those that serve as a nexus point for modulating a wide range of cellular functions that are affected with aging; a concept that brings to mind the role of the mitochondrion.
Mitochondria serve a central role as an integrator of a variety of signals within the cell, and accordingly vary their function to modulate energy supply, reactive oxygen species (ROS) signaling, and intrinsic pathways of apoptosis (Figure 1). For this reason, mitochondria have been frequently studied as a target for combating cellular aging. This is also true of muscle cells where one of the first studies to suggest impaired mitochondrial function may be involved in muscle aging was performed in flies and poignantly found that mitochondrial dysfunction was associated with loss of the wings (Rockstein and Brandt, 1963)! A great many studies have followed and although a substantial number have found evidence for impaired mitochondrial function in aging muscle (Rumsey et al., 1987; Trounce et al., 1989; Cooper et al., 1992; Conley et al., 2000; Gouspillou et al., 2010, 2014a,b; Picard et al., 2011a), others have not (Kent-Braun and Ng, 2000; Rasmussen et al., 2003; Lanza et al., 2005), underscoring the complexity of understanding in this area. The purpose of this review, therefore, is to address the basis for these complexities, identify relevant mechanisms therein, and finally to provide some suggestions for future investigation.
Figure 1. Mitochondria respond to a wide array of intracellular signals and accordingly modulate their function [ATP production, reactive oxygen species (ROS) production, and sensitivity to permeability transition] across a large dynamic range to meet the cellular needs. It is thus important to understand whether alterations observed in aging muscle represent a primary organelle defect versus a secondary (potentially adaptive) response to a changing cellular environment.
There are numerous issues that need to be considered when evaluating the involvement of mitochondria in aging of skeletal muscle. The first issue to consider is the means by which the mitochondrial function is interrogated. For example, direct interrogation of mitochondrial function frequently employs mechanical isolation of mitochondria, a method that itself can induce alterations in the intrinsic function of the organelle (Picard et al., 2011c). Indeed, mechanical isolation of mitochondria not only exaggerates the magnitude of age-related impairment in various aspects of mitochondrial function but also induces changes not seen in a preparation where mitochondrial structure is preserved (Picard et al., 2010).
A second issue to consider is which functional outputs are quantified as it relates to the different roles that mitochondria serve in skeletal muscle, including energy provision (respiration), signaling (ROS emission), and regulation of apoptosis (e.g., through permeability transition and release of mitochondrial-derived apoptotic factors) (Picard et al., 2011a). Further to this point, it is also essential to account for the fact that some mitochondrial functional properties vary between fast twitch and slow twitch muscles (Picard et al., 2012). For example, slow twitch muscles generally exhibit lower mitochondrial ROS emission (Anderson and Neufer, 2006; Picard et al., 2008) and a lower capacity for calcium retention before permeability transition occurs (Picard et al., 2008) when compared to fast twitch muscles. Thus, in situations where muscle exhibits a shift in fiber type with aging, the impact of this shift on the mitochondrial function properties needs to be considered.
The third issue to consider is the age studied and in this respect, it is important to address mitochondrial function across the aging continuum, including study of ages that are most affected by the consequences of aging muscle atrophy. As noted in the section “Introduction,” this is typically ≥80 years of age (Cruz-Jentoft et al., 2010) and thus far there is very little known about muscle mitochondrial alterations in this age group from human studies, although several studies in the animal literature exist (Chabi et al., 2008; Ljubicic et al., 2009; Joseph et al., 2010; Picard et al., 2011a). Related to this point, when using an animal model system the age studied should represent points along the trajectory of muscle aging that are relevant for human muscle.
A fourth issue to consider is that of physical activity because it is well-established that skeletal muscle mitochondrial content is highly adaptable and not only increases in response to elevated metabolic demand but also decreases in response to reduced metabolic demand, with the latter being the typical situation in aging (Martin et al., 2014). As such, understanding the degree to which mitochondrial content and functional alterations are an obligatory consequence of aging versus being wholly or partially avoidable through a physically active lifestyle, is of critical importance.
Finally, since mitochondria serve as central integrators of a wide variety of cellular signals and thus exhibit a wide range of what should be considered physiological (rather than pathological) function (Picard et al., 2011b), it is essential to consider the involvement of cellular alterations that are extrinsic to the mitochondria as modulating factors in their function before concluding that a primary mitochondrial defect exists. For example, aging in muscle has long been known to induce neurological alterations such as neuromuscular junction instability leading to denervation (Tomonaga, 1977; Oda, 1984; Edstrom et al., 2007; Rudolf et al., 2014), and these changes may themselves exert an impact on the cellular environment that modulates mitochondrial function (Csukly et al., 2006; Muller et al., 2007; Bhattacharya et al., 2009). The suitability of the mitochondrion as a primary therapeutic target in this scenario needs to be carefully considered as reversing mitochondrial alterations that are secondary to denervation [e.g., those that result in activation of atrophy pathways (Romanello et al., 2010; Rowan et al., 2012)] could make the situation worse rather than better by preventing atrophy of denervated fibers and thereby increasing the burden on the remaining muscle fibers. These issues will be addressed in detail in the sections that follow to inform our current understanding of the role played by mitochondrial dysfunction in aging muscle atrophy and the resulting implications for intervening at this level.
Many different methodological approaches have been taken to evaluate mitochondrial function in aging muscle, including direct measures (e.g., isolated mitochondria, permeabilized myofibers) and indirect measures [e.g., phosphocreatine (PCr) recovery kinetics following muscle contractions, enzyme assays, protein content, etc.]. Similarly, a variety of functional outputs of mitochondria have been examined, with indices related to mitochondrial respiration being by far the most frequent and measures of ROS emission second most frequent, whereas to date there are relatively few studies examining mitochondrial sensitivity to an apoptotic challenge, particularly in the human literature. Importantly, the impact of aging on the different aspects of mitochondrial function is often quite variable from one function to the next and this has implications for understanding the involvement of mitochondria in aging muscle. For example, a reduced respiratory capacity could impact muscle fatigue by limiting energy provision to the myocyte (Stary et al., 2004; Chabi et al., 2008), an increase in ROS emission could induce cellular and organelle oxidative stress and thereby increase the requirement for removal of resulting damage (Fulle et al., 2004; Mansouri et al., 2006), and an increased susceptibility to permeability transition could increase the release of mitochondrial-localized apoptotic factors thereby contributing to nuclear loss and myocyte atrophy (Marzetti et al., 2010; Gouspillou et al., 2014b).
By far the most common approach to gaining insights about mitochondrial function in aging muscle is the use of enzymatic assays of representative mitochondrial enzymes, such as citrate synthase and electron transport chain complexes, and these measures provide indirect insights into the energy producing (respiratory) capacity of the mitochondria. Most of the studies performed to date indicate a marked decline in mitochondrial enzyme activities from aging muscle (Bass et al., 1975; Lezza et al., 1994; Desai et al., 1996; Hagen et al., 2004), although some studies find this to be maintained (Orlander et al., 1978; Barrientos et al., 1996) and others report it to be highly variable from one muscle to the next (Houmard et al., 1998; Lyons et al., 2006). Importantly, individual enzyme activities do not provide unambiguous insights about the respiratory function of the intact mitochondrion because individual enzyme activities can become very disparately related to whole organelle function when the mitochondrion becomes defective. Thus, more direct and integrated measures of function are required to interpret aging impact.
Amongst the most frequently used methodological approaches to directly interrogate mitochondrial function involves the mechanical isolation of mitochondria (Chance and Williams, 1956). Although this method has been very widely used in the aging literature (Trounce et al., 1989; Boffoli et al., 1994; Capel et al., 2005; Chabi et al., 2008; Gouspillou et al., 2010), as we have recently reviewed (Picard et al., 2011b), not only does mechanical isolation markedly disrupt the normal architecture of skeletal muscle mitochondria from an irregular tubular network into smaller, relatively homogenous, spherical structures, but it also profoundly potentiates both mitochondrial ROS emission and sensitivity to permeability transition (Figure 2). The importance of this to studies in aging is illustrated by the fact that when we compared the apparent effect of aging on mitochondrial function in mitochondria isolated from skeletal muscle versus saponin-permeabilized myofibers (where mitochondrial structure is preserved) in very old rat muscle (35% survival rate, which is a similar relative age to ≥80 years old humans), we found the isolated mitochondria profoundly exaggerated the impact of aging (Picard et al., 2010). Specifically, with aging the decline in maximal mitochondrial respiratory capacity was fourfold greater, the increase in ROS emission was twofold greater and the reduction in time to permeability transition was twofold greater when examined in isolated mitochondria versus saponin-permeabilized myofibers (Figure 3). Not only this, but there were significant alterations in mitochondrial enzyme activity stoichiometry (reduced ratio of cytochrome oxidase to citrate synthase activity) and stoichiometry of respiratory states (suggestive of a defect in complex I) in isolated mitochondria with aging that were not seen in saponin-permeabilized myofibers. We have suggested that these latter observations may be indicative of a lesser ability of aged mitochondria to reseal during isolation procedures, since isolation induces transient disruption of tubular mitochondrial structures and subsequent reconstitution into the spherical organelles typical of isolates, resulting in greater contamination and/or loss of mitochondrial matrix constituents in isolates prepared from aged muscles (Picard et al., 2010) (Figure 4). Collectively, therefore, our data show that the method used to interrogate mitochondrial function can have a profound influence on the degree to which mitochondrial function appears to be altered in aging muscle and that generally speaking, the degree to which mitochondrial function is altered in aging muscle is considerably less severe than has often been considered. This notion is consistent with other studies that have used the saponin-permeabilized myofibers method, which have found relatively mild (Joseph et al., 2012) or in other cases no impairments in mitochondrial respiratory capacity in aging muscle (Tonkonogi et al., 2000; Hutter et al., 2007; Gouspillou et al., 2014b).
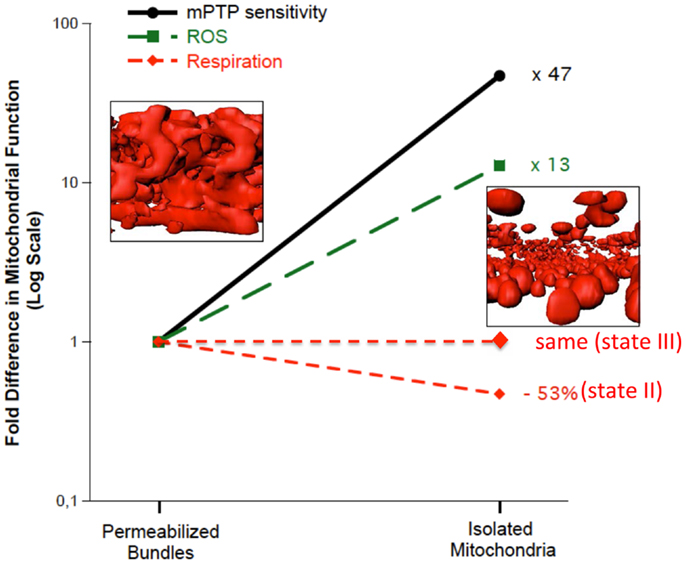
Figure 2. Multiple features of mitochondrial structure and function are altered in mechanically isolated organelles compared to saponin-permeabilized myofibers (representing a preparation where all mitochondria are represented and their structure remains intact). In particular, the irregular tubular structure is lost following isolation, producing more homogenous spherical structures. Accompanying these structural changes is a marked potentiation of reactive oxygen species (ROS) emission and sensitization to mitochondrial permeability transition pore (mPTP) opening. Data are re-plotted from Picard et al. (2011c).
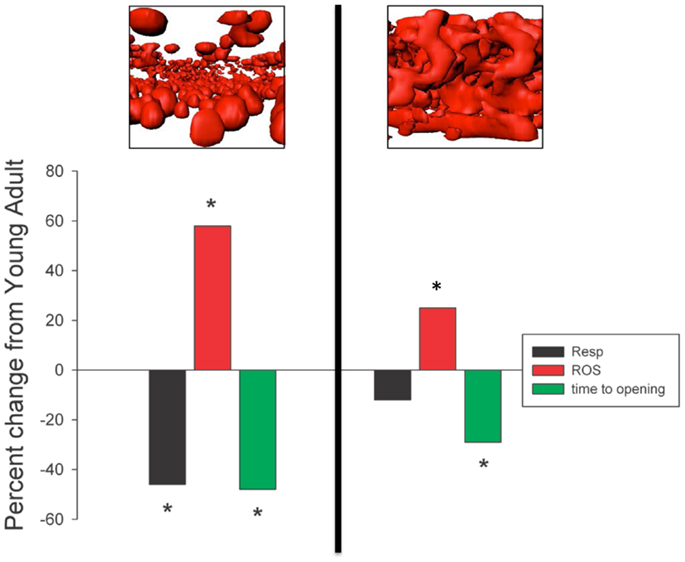
Figure 3. Mechanical isolation (to the left of the vertical line) markedly exaggerates the apparent impact of aging on skeletal muscle mitochondria respiratory function, reactive oxygen species (ROS) emission, and sensitivity to mitochondrial permeability transition pore (mPTP) opening (permeability transition) compared to saponin-permeabilized myofibers (to the right of the vertical line). *P < 0.05 versus SEN. Data are re-plotted from Picard et al. (2010).
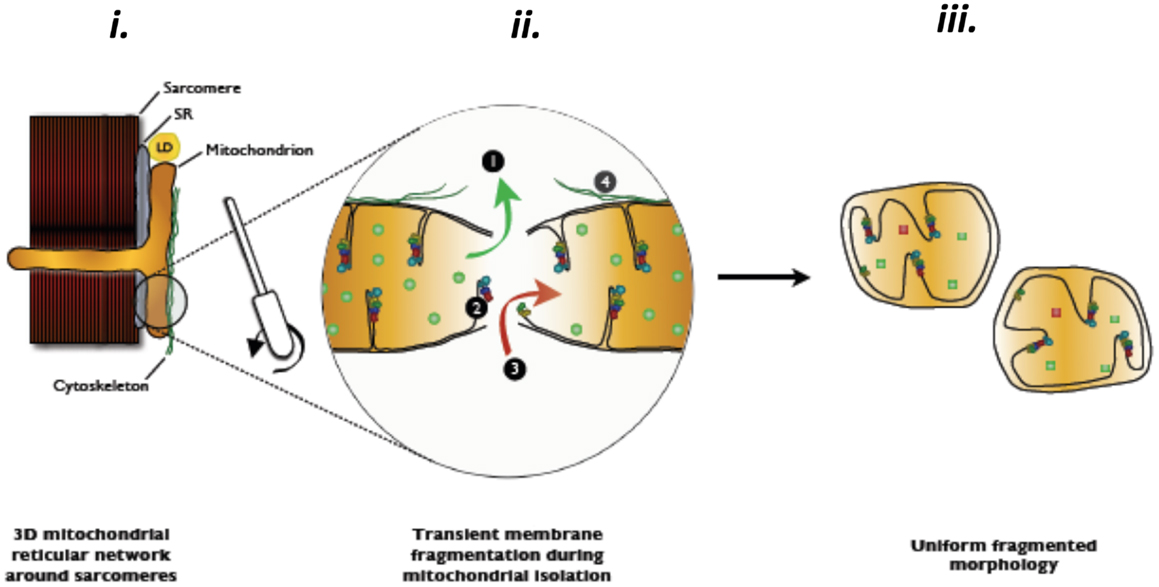
Figure 4. Hypothesized contamination and/or matrix dilution occurring with the transient rupture of mitochondrial membranes during mitochondrial isolation procedures. Reproduced with permission from Picard et al. (2011b).
Notwithstanding the point about mitochondrial respiratory alterations in aging muscle being less severe than often considered, in a follow-up study where we compared indices of mitochondrial function in saponin-permeabilized myofibers prepared from four rat muscles that exhibited contrasting fiber type and contrasting atrophy susceptibility with aging, we did find mitochondrial alterations that we believe bear consideration as a contributing factor in atrophy of aging muscle (Picard et al., 2011a). Our comparison involved two fast twitch muscles [extensor digitorum longus (EDL), gastrocnemius (Gas)] and two slow twitch muscles [soleus (Sol), adductor longus (AL)], where the Gas exhibited the most severe atrophy with aging, the EDL and Sol were similarly affected, and most strikingly, the AL actually hypertrophied by nearly 50% and the mitochondrial function was largely preserved in this muscle. Although the factors accounting for this profoundly better adjustment to aging in the rat AL remain under study, the other muscles exhibited varying degrees of mitochondrial functional alteration with aging. In particular, the Sol was the only muscle to exhibit a reduction in muscle respiratory capacity (Figure 5A) and in this case it was secondary to reduced mitochondrial content. In contrast, although the muscle respiratory capacity was largely preserved in both of the fast muscles, these two muscles had higher levels of representative subunits of the mitochondrial electron transport complexes, suggesting that the intrinsic respiratory capacity of the mitochondrial electron transport system was impaired in these fast muscles with aging. Muscle ROS emission normalized for respiration (an index of the proportion of O2 consumption lost to ROS leak) demonstrated a general trend to be elevated in all four muscles under state II and state III conditions, although after normalizing for mitochondrial content only the Sol demonstrated an increase, suggesting an increase in intrinsic mitochondrial ROS emission in aging muscle is not a general feature of aging muscle (Figure 5A). On the other hand, the sensitivity to permeability transition was increased in the fast twitch muscles, suggesting greater apoptotic potential. Although we did not observe a change in this property in the Sol muscle, despite its marked atrophy with aging, as noted in Section “Addressing the Question of Mitochondrial Dysfunction in Aging Muscle,” the time to mitochondrial permeability transition pore (mPTP) opening (permeability transition) in response to a calcium challenge is typically lower in slow twitch muscle (and higher in fast twitch muscle). As the aged Sol is characterized by a striking increase in the fraction of fibers that express fast myosin heavy chain isoforms (Edstrom and Ulfhake, 2005; Snow et al., 2005; Carter et al., 2010), after taking this shift into account, the aged Sol should have a longer time to mPTP opening than observed, meaning it too exhibits a sensitization to permeability transition with aging. This is depicted in Figure 5B as a time to pore (mPTP) opening that falls below the line predicted by the relationship between time to mPTP opening and percentage of fibers positive for fast myosin heavy chain. Collectively, therefore, since all three muscles, which atrophied with aging (EDL, Gas, Sol) exhibited a reduced time to permeability transition, whereas the only muscle that did not atrophy (AL) was unaffected, our results reveal that a general property of aging muscles undergoing atrophy is an increased mitochondrial sensitivity to an apoptotic challenge. Taken in context with the abundance of evidence for elevated recruitment of mitochondrial-mediated pathways of apoptosis in aging muscle (Dirks and Leeuwenburgh, 2002; Leeuwenburgh et al., 2005; Siu et al., 2005; Chabi et al., 2008; Marzetti et al., 2008; Gouspillou et al., 2014b), this change in mitochondrial function in aging muscle may be a key to induction of myofiber atrophy.
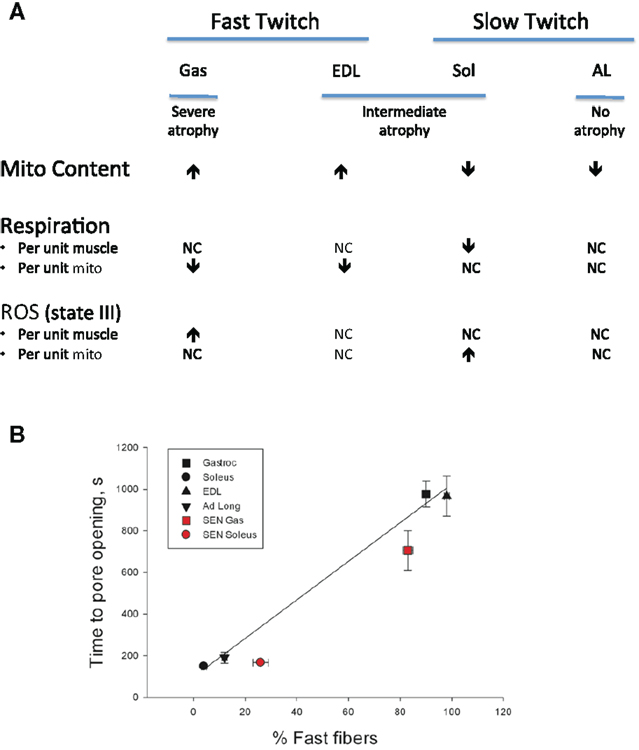
Figure 5. Impact of aging on mitochondrial function in saponin- permeabilized myofibers prepared from rat muscles of contrasting fiber type and atrophy susceptibility is shown. Gas = gastrocnemius muscle, EDL = extensor digitorum longus muscle, Sol = soleus muscle, AL = adductor longus muscle. (A) Impact of aging on mitochondrial content, respiration, and reactive oxygen species (ROS) emission responses. (B) Sensitivity of mitochondria to permeability transition following a calcium challenge and how this varies as a function of fast twitch fiber abundance. Solid black symbols represent the responses of young adult muscles, whereas the solid red symbols represent responses of senescent muscles. After taking into account the shift in proportion of fibers positive for fast myosin heavy chain with aging in each muscle, both the soleus and gastrocnemius muscles exhibit a sensitization to permeability transition. Data for mitochondrial content, respiration, ROS, and time to pore opening are taken from Picard et al. (2011a). Data for proportion of fibers positive for fast myosin heavy chain are taken from Carter et al. (2010).
The final methodological approach to be discussed in this review is the use of non-invasive spectroscopic techniques to interrogate mitochondrial function in aging muscle. Although the value of these measures is that they provide insights into the function of the organelle in its native environment, as will be discussed here, under some circumstances this can pose challenges in deciphering whether a primary mitochondrial defect exists.
The most common non-invasive spectroscopic approach for monitoring mitochondrial function in vivo involves 31Phosphorous spectroscopy to determine PCr recovery time following muscle contractions, as the rate of PCr recovery is proportional to mitochondrial respiration and content (Mahler, 1985; Meyer, 1988; Prompers et al., 2014). While this is the only way currently available to gain insights into mitochondrial energy production in vivo; it is, however, important to note that the rate of PCr resynthesis following muscle contractions also depends upon oxygen supply (Haseler et al., 1999). Thus, if muscle blood flow is at all reduced in aging muscle during the recovery from contractions, noting that some studies have observed lower blood flow during dynamic exercise in the elderly (Lawrenson et al., 2003; Poole et al., 2003), the resulting lower muscle oxygen delivery with aging is a confounding factor in the interpretation of reduced mitochondrial energetic capacity in the elderly based upon slower PCr recovery kinetics alone.
Keeping this point in mind, although several studies report a reduced rate of PCr recovery following muscle contractions in aging human gastrocnemius (McCully et al., 1993) and vastus lateralis muscles (Conley et al., 2000), this was not seen in aging human tibialis anterior muscle of subjects matched for physical activity levels to the young comparison group (Kent-Braun and Ng, 2000; Lanza et al., 2005). Although differences in physical activity status may be part of the explanation for the different results between studies (Kent-Braun and Ng, 2000), it may also relate to the aforementioned impact of oxygen delivery and/or reflect differences in the impact of aging on mitochondrial function in different muscles, as we have shown that occurs in aged rodents (Picard et al., 2011a) (see Mechanically Isolated Mitochondria versus Saponin-Permeabilized Myocytes).
Non-invasive spectroscopic methods have also been used to evaluate mitochondrial coupling in skeletal muscle in vivo. In particular, several studies have now combined measures of PCr resynthesis (to derive what has been termed “ATPmax”) with optical spectroscopic assessment of myoglobin and hemoglobin oxygen saturations (to derive intramuscular oxygen consumption) to yield a measure of coupling efficiency based upon the quotient of ATP turnover (derived from the PCr spectroscopy measures) and oxygen consumption (Amara et al., 2007). These studies have identified mild uncoupling of mitochondria in both mouse (Marcinek et al., 2005) and human (Amara et al., 2007) skeletal muscle. As discussed in the following section, mild uncoupling has also been reported in saponin-permeabilized myofibers from physically active septuagenarian humans (Gouspillou et al., 2014b). Since uncoupling of mitochondria has been argued as one strategy for reducing mitochondrial ROS emission and promoting healthy longevity (Speakman et al., 2004), the mild uncoupling observed in aged muscle may represent an adaptive response to keep ROS within physiological levels, rather than a defect per se (Amara et al., 2007).
Amongst the most controversial issues about mitochondrial impact in aging muscle is that of mitochondrial content. Whilst some studies have concluded that mitochondrial content is reduced in aging muscle (Kerner et al., 2001; Chabi et al., 2008), some find no change (Mathieu-Costello et al., 2005; Callahan et al., 2014; Gouspillou et al., 2014b; Konopka et al., 2014), and others find it to vary between muscles (Lyons et al., 2006; Picard et al., 2011a). As was the case with measuring mitochondrial function, many different approaches have been taken to characterize mitochondrial content in aging muscle, including mitochondrial marker enzymes (e.g., citrate synthase or cytochrome oxidase activities), select mitochondrial proteins (e.g., porin, electron transport system complex subunits), mitochondrial DNA (mtDNA) copy number, and the gold standard, electron microscopic quantitation of mitochondrial volume density. Although the wide variety of approaches to inferring mitochondrial content changes in aging muscle has no doubt contributed to the variability in interpretation herein, even in studies using the gold standard of electron microscopy significant variation has been observed. For example, in two studies examining aged rodent muscle (Mathieu-Costello et al., 2005) and aged human muscle (Callahan et al., 2014), there was no change in skeletal muscle mitochondrial content assessed by electron microscopy with aging, whereas another study in aged humans did observe a decrease with aging using the same method and examining the same muscle (vastus lateralis muscle) (Conley et al., 2000), underscoring the discrepancy on the question of how aging impacts muscle mitochondrial content. Amongst the factors that contribute to this variability between studies, as noted in Section “Addressing the Question of Mitochondrial Dysfunction in Aging Muscle,” physical activity levels can play an important modulating role, where activity-matched subjects consistently show no decline in indices of mitochondrial content in aging muscle (Lanza et al., 2005; Gouspillou et al., 2014b). However, whether differences in physical activity status alone can explain the discordant results remains to be clarified.
Regardless of the controversy about the impact of aging on mitochondrial content, the capacity for generation of new mitochondria declines with aging at ages where the clinical consequences of aging in muscle are most relevant. Specifically, the ability of aged muscle to augment mitochondrial biogenesis in response to both endurance exercise training (Betik et al., 2009) and more severe muscle activity induced by chronic electrical stimulation (Ljubicic and Hood, 2009; Ljubicic et al., 2009) is severely compromised in rat muscle from animals at a similar relative age as humans ≥80 years of age. This impairment appears to be secondary to a failure to upregulate the machinery regulating mitochondrial biogenesis, including peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC-1α) (Betik et al., 2009), and an accelerated degradation of precursor proteins destined for import into mitochondria (Huang et al., 2010). To date, no therapeutic advances have been developed to restore the mitochondrial biogenesis response in very old muscle, designating this as an issue of high priority for future investigations.
Coming back to ages where muscle mitochondrial content remains adaptable (≤80 years of age), not only does physical activity level modulate mitochondrial content, but it can also modulate indices of mitochondrial function in aging muscle. In particular, previous studies have shown that when younger and older subjects are matched for physical activity levels, there is no reduction in muscle mitochondrial respiratory capacity with aging (Kent-Braun and Ng, 2000; Lanza et al., 2005; Safdar et al., 2010). We recently extended these results to show that, like respiratory capacity (Figure 6A), mitochondrial ROS emission is also not elevated with aging (Figure 6B) when comparing young adult and septuagenarian subjects with relatively high levels of recreational physical activity, although as seen in very old rodent muscle (see Mechanically Isolated Mitochondria versus Saponin-Permeabilized Myocytes; Picard et al., 2011a), we did observe a pronounced sensitization of the mitochondria to permeability transition (Gouspillou et al., 2014b) (Figure 6C). Indeed, the increased apoptotic sensitivity of aged mitochondria was also accompanied by a marked translocation of a mitochondrial-derived pro-apoptotic molecule, endonuclease G, to myonuclei in atrophied septuagenarian human muscle of recreationally active subjects, suggesting that this may be a key mechanism by which aged mitochondria contribute to aging muscle atrophy and one which cannot be circumvented through an active lifestyle (Gouspillou et al., 2014b). Potential mechanisms for this will be discussed in Section “Mechanisms of Primary Defects in Mitochondrial Function in Aging Muscle.”
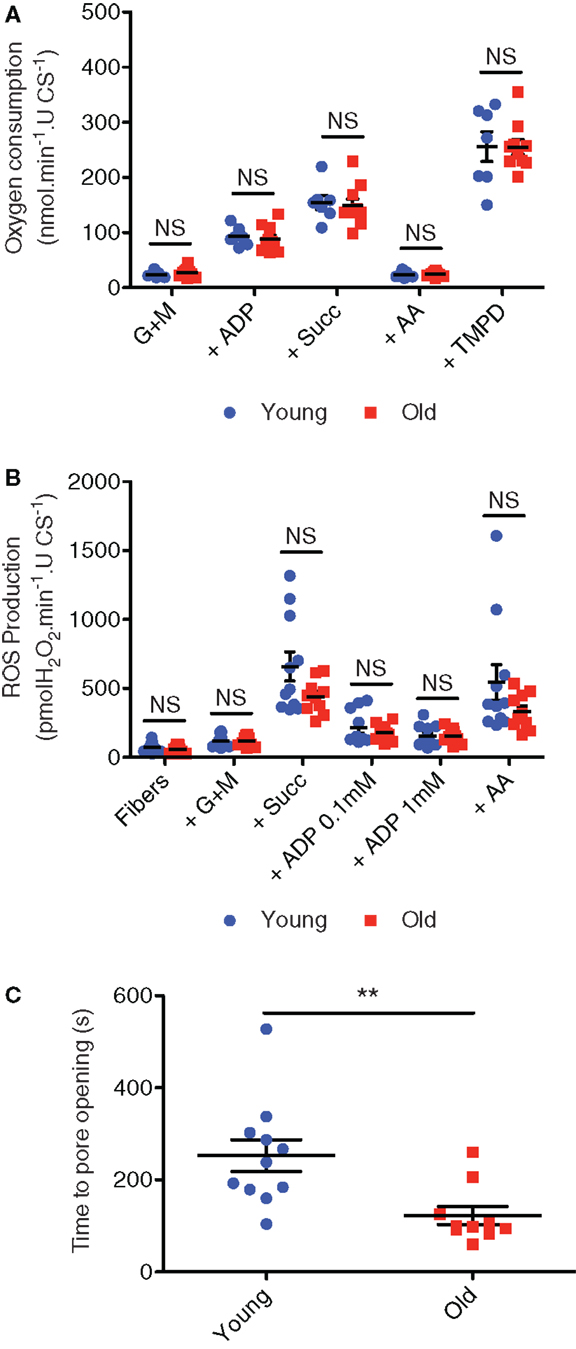
Figure 6. Mitochondrial respiration (A), hydrogen peroxide emission (B), and sensitivity to permeability transition following a calcium challenge (C) in saponin-permeabilized myofibers prepared from vastus lateralis muscle biopsies obtained from recreationally active healthy young (24 ± 1 years; mean ± SE) and older (71 ± 2 years) men. Figures are reproduced with permission from Gouspillou et al. (2014b). G + M = glutamate + malate; Succ = succinate; AA = antimycin A, TMPD = artificial electron donor. **P < 0.05 versus old.
As noted above, a sensitization of mitochondria to permeability transition (Picard et al., 2011a) and release of mitochondrial-derived pro-apoptotic factors (Chabi et al., 2008; Marzetti et al., 2008) is evident in aged muscle, even in those who remain physically active (Gouspillou et al., 2014b). In accounting for the reasons for mitochondrial dysfunction in aging muscle, two primary hypotheses have been put forth to explain declining mitochondrial function with aging in general. The first hypothesis relates to the fact that mitochondria have their own genome, which, although only encoding for a small fraction of all mitochondrial proteins, is essential for production of normally functioning mitochondria. Since this genome accumulates damage progressively with aging (Richter et al., 1988; Katayama et al., 1991; Simonetti et al., 1992; Melov et al., 1995), it is posited that this leads to impaired synthesis of mitochondria and/or synthesis of mitochondria with aberrant function (Hiona and Leeuwenburgh, 2008). The second hypothesis relates to the fact that mitochondria must be regularly removed and replaced to preserve their fidelity (Schiavi and Ventura, 2014); it is posited that this process becomes impaired with aging, resulting in accumulation of damaged mitochondria with aberrant function (Terman et al., 2010; Joseph et al., 2013). The available evidence relating to these two hypotheses will be discussed in detail below.
The mtDNA molecule is a small circular genome (approximately 16 k base pairs in length) encoding 37 genes, of which 22 are for transfer RNAs, 2 are for ribosomal RNAs, and 13 are for polypeptide subunits of the respiratory chain and ATP synthase. In this last respect, the mtDNA encodes for 7 subunits of complex I, 1 subunit of complex III, 3 subunits of complex IV, and 2 subunits of the ATP synthase, and these components are essential to normal respiratory function. As evidence of the importance of mtDNA, there is marked mitochondrial dysfunction in patients with mtDNA mutations and when these mutations occur in skeletal muscle they can result in profound exercise intolerance (Taivassalo et al., 2003). As noted above, mtDNA mutations increase progressively with increasing age in multiple tissues, including skeletal muscle (Katayama et al., 1991; Melov et al., 1995; Bua et al., 2006). Furthermore, the focal accumulation of mtDNA mutations to high levels in skeletal myocytes with aging has been implicated in fiber atrophy, breakage, and loss (Lee et al., 1998; Wanagat et al., 2001; Bua et al., 2002). However, this latter aspect seems to be losing favor for several reasons. In particular, the proportion of fibers with severe mitochondrial dysfunction arising from mtDNA mutations is very low and these fibers do not seem to be consistently smaller than fibers with healthy mitochondria (Rowan et al., 2011). Further to this point, the proportion of fibers with focal atrophy at regions coinciding with high levels of mutant mtDNA and severe electron transport system dysfunction in human skeletal myocytes is also too low (in two subjects >65 years of age, only 5% of fibers with electron transport chain dysfunction exhibited focal atrophy; Bua et al., 2006) to be biologically meaningful compared to other causes of atrophy in aging muscle, particularly denervation, which is the primary cause of myofiber atrophy in very old rat muscle (Rowan et al., 2012).
Although focal accumulation of mtDNA mutations does not appear to be a primary cause of skeletal myocyte atrophy with aging, a mouse engineered with a faulty mtDNA proof-reading enzyme, the PolG mutant mouse, exhibits a dramatic increase in rate of mtDNA mutation, a markedly shortened lifespan, and numerous features that resemble premature aging (Trifunovic et al., 2004, 2005; Kujoth et al., 2005), including muscle atrophy (Hiona et al., 2010). Interestingly, the mitochondrial phenotypes observed in this so-called mtDNA mutator mouse differ in important ways from what is seen in normally aging muscle. In particular, whereas the PolG mutant mouse exhibits reduced levels of mitochondrial electron transport system complex subunits in skeletal muscle, as discussed in Sections “Mechanically Isolated Mitochondria versus Saponin-Permeabilized Myocytes” and “Mitochondrial Content and Function in Relation to Age and Physical Activity” this is not a consistent finding in aging rat (Picard et al., 2011a) or human muscle (Gouspillou et al., 2014b) where levels of mitochondrial electron transport system complex subunits are often higher in aged muscle. Similarly, whereas markers of mitochondrial fission and autophagy are higher in the PolG mouse muscle, normally aged mouse muscle exhibits higher levels of markers of mitochondrial fusion and lower levels of markers of autophagy (Joseph et al., 2013). On this basis, mtDNA mutations per se are unlikely to be the root cause of mitochondrial dysfunction in aging muscle.
As mentioned above, mitochondria normally undergo degradation and replacement to ensure the fidelity of mitochondrial function. Indeed, impairment in mitochondrial autophagy (mitophagy) is implicated in a wide variety of neudegenerative disorders (Chu, 2010; Scheibye-Knudsen et al., 2014), in addition to normal aging (Schiavi and Ventura, 2014). Although in skeletal muscle the rate of mitochondrial turnover is unknown (nor it is understood if it differs between species), mitochondria have a half-life of approximately 2 days in mouse liver (Miwa et al., 2008), which when scaled up to the lifespan of an organism means that mitochondria undergo hundreds to thousands of cycles of turnover throughout the aging process. Clearly, therefore, if this rate of renewal were to decline, it seems a likely basis leading to accumulation of mitochondria with aberrant function in aging muscle.
Mitophagy is a tightly regulated process whereby dysfunctional/damaged mitochondria are targeted for removal by incorporation into autophagosomes for subsequent degradation by lysosomes. This targeting of dysfunctional mitochondria for removal depends upon several mechanisms that are likely relevant for aging. For example, mitophagy is elevated in response to mitochondrial fragmentation, reduced mitochondrial membrane potential (e.g., as occurs in mitochondria undergoing permeability transition), and increased mitochondrial ROS emission (Romanello et al., 2010; Twig and Shirihai, 2011; Schiavi and Ventura, 2014). Evidence that mitophagy is impaired in aging muscle is only now beginning to accumulate. Firstly, the fact that mitochondria with sensitization to permeability transition accumulate in aging muscle (Gouspillou et al., 2014b) is evidence that mitophagy of damaged mitochondria is impaired since permeability transition causes loss of mitochondrial membrane potential, a potent stimulus for mitophagy (Twig et al., 2008). In addition, two studies have now shown that Parkin, a mitochondrial-targeted ubiquitin ligase, which interacts with the autophagy protein LC3 to induce formation of autophagosomes around dysfunctional mitochondria, is reduced in skeletal muscle of both physically active septuagenarian men (Gouspillou et al., 2014b) and in physically inactive frail older women (Drummond et al., 2014). Furthermore, impairments in the signaling pathway that regulates mitochondrial quality control have also been reported in aging mouse muscle (Joseph et al., 2013) and aging human muscle (Koltai et al., 2012). Thus, whereas the significance of mtDNA mutations in mitochondrial dysfunction in aging muscle is less clear, impaired mitophagy appears a likely contributor to accumulation of dysfunctional mitochondria in aging muscle. Importantly, that this appears to also occur in physically active subjects (Gouspillou et al., 2014b) underscores that new therapeutic approaches will be required to address this problem.
Although their particular role in aging remains to be established, two promising new targets involved in regulating degradation of damaged mitochondria are the micro RNA miR137 and the AAA ATPase p97. miR137 has been identified to inhibit mitophagy occurring in response to hypoxia, through reducing the interaction of LC3 with two mitophagy receptors, FUNDC1 and NIX (Li et al., 2014). p97 is specifically involved in targeting and removal of carbonylated mitochondrial proteins for subsequent degradation by the ubiquitin proteasome (Hemion et al., 2014). If these pathways are disrupted in aging muscle, they are likely targets for improving removal of damaged mitochondria and thus, ameliorating aging impact.
Although mitochondrial changes in aging muscle are most often considered as primary dysfunction suitable for therapeutic targeting, this view does not consider the influence of factors extrinsic to the mitochondrion that constitute the aging intracellular milieu. For example, age-related changes in the intracellular milieu could depress certain aspects of mitochondrial function independent of the intrinsic mitochondrial functional capacity per se and recent data support this idea. In addition, other changes impacting aging muscle, such as impaired neuromuscular junction stability and myofiber denervation, could also modulate mitochondrial function as a secondary consequence.
In addressing the potential involvement of the aging milieu as an inhibitory influence on mitochondrial function in aging muscle, Siegel et al. (2013) recently characterized mitochondrial function in vivo in distal hindlimb muscles of young (5 months old) and aged (27 months old) mice using optical and 31Phosphorous magnetic resonance spectroscopy, finding a decrease in mitochondrial respiratory function (reduced maximal ATP production rate, reduced coupling, and reduced rate of PCr resynthesis). Strikingly, these age-related impairments in mitochondrial respiratory function were restored to youthful levels 1 h following the administration of a mitochondria-specific antioxidant peptide, SS-31 (Figures 7A–E), and this was also accompanied by an increased muscle fatigue resistance and running time to exhaustion in the aged mice but not younger mice. In stark contrast to these acute in vivo results, when mitochondrial respiratory function was examined in saponin-permeabilized myofibers there was no detectable impairment in mitochondrial respiratory function with aging and administration of SS-31 had no effect (Figures 7F,G), despite inducing a reduction in mitochondrial ROS emission. The implications of these results is that, independent of the intrinsic function of the organelle, mitochondrial function can be impeded in vivo by oxidative stress in the intracellular environment of aging muscle. Thus, these recent findings underscore the importance of considering aspects of the aging milieu as contributing factors to impairing mitochondrial function in vivo, and demand further study of how this may affect other aspects of mitochondrial function (e.g., ROS emission, sensitivity to permeability transition) with aging. Furthermore, these results also show that manipulation of the aging milieu, rather than the mitochondrion directly, can be an effective strategy for improving mitochondrial function in aging muscle.
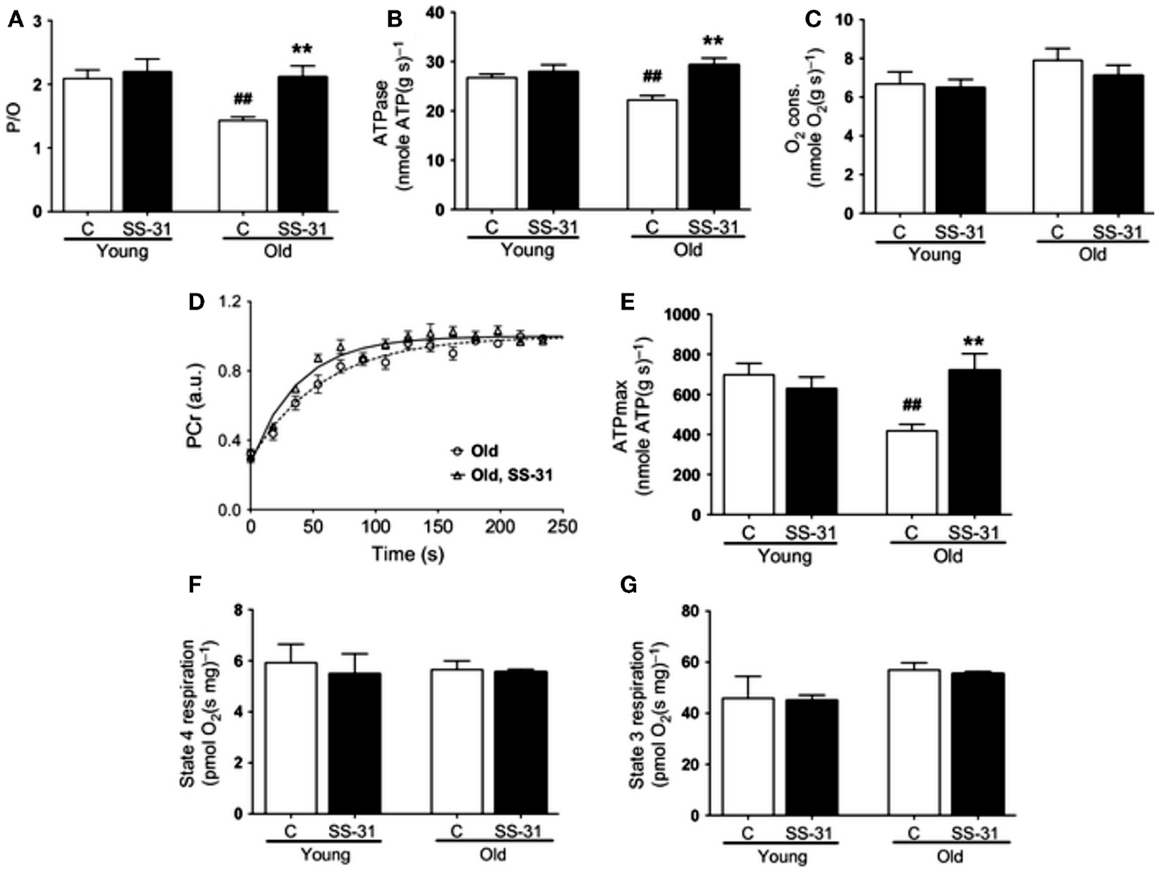
Figure 7. Influence of the aging intracellular milieu on mitochondrial function in vivo in mouse skeletal muscle. (A) P/O ratio, (B) in vivo ATPase rate; (C) in vivo rate of oxygen consumption; (D) in vivo rate of phosphocreatine (PCr) resynthesis following muscle contractions; (E) in vivo maximal rate of ATP synthesis (ATPmax); (F) state 4 respiration in saponin-permeabilized myofibers; (G) state 3 respiration in saponin-permeabilized myofibers. Figures are reproduced with permission from Siegel et al. (2013).
As noted in the section “Introduction,” mitochondria serve as cellular rheostats that integrate a wide variety of intracellular signals and modify aspects of their function as appropriate to those cellular conditions. Although intrinsic impairment in mitochondrial function involving an increased sensitivity to permeability transition in mitochondria from aged muscle has been demonstrated in both a sedentary rat model (Picard et al., 2011a) and in physically active aging humans (Gouspillou et al., 2014b), whether factors upstream of the mitochondrion can also modulate mitochondrial function is unclear. This question may be particularly relevant when aging muscle atrophy becomes severe and more likely to yield clinical impact because it is likely that additional mechanisms come into play in driving an acceleration of muscle atrophy. In this respect, as noted from the outset of this review, the age at which aging muscle atrophy is most likely to cause clinical consequences such as an increased risk of falls and physical frailty, is in individuals ≥80 years of age (Cruz-Jentoft et al., 2010). Significantly, in an aging rat model this corresponds to a period where muscle atrophy accelerates through the accumulation of small angular fibers characteristic of long-term denervation (Rowan et al., 2011).
Although evidence of an accumulation of denervated myofibers in skeletal muscle at very advanced age in humans thus far is based only upon the morphological appearance of small angular fibers (Scelsi et al., 1980) that are typical of neuropathologies such as amyotrophic lateral sclerosis and polyneurophathy (Baloh et al., 2007), the presence of denervated myofibers in very old mouse (Wang et al., 2005) and rat muscle (Rowan et al., 2012) has been confirmed by probing in situ for a sodium channel isoform typical of denervation in adult muscle, NAV1.5 (Kallen et al., 1990; Yang et al., 1991). Importantly, these denervated myofibers also demonstrate a marked up-regulation of the ubiquitin ligases muscle atrophy F-box (MAFbx) and muscle ring finger 1 (MuRF1) (Rowan et al., 2012), and the up-regulation of these proteins in response to denervation is dependent upon release of mitochondrial lipid hydroperoxides that can be detected in some ROS assays (Bhattacharya et al., 2009). Thus, denervation can also recruit mitochondrial-mediated proteolytic pathways, raising the likelihood that at least some of the alterations in mitochondria seen in skeletal muscle at very advanced age are a secondary event to denervation rather than a primary organelle defect. A schematic representation of how we hypothesize that denervation may modulate mitochondrial pathways of atrophy in aging muscle is provided in Figure 8. Since understanding this issue is key to advancing to new therapeutic targets, future study of how sporadic denervation may modulate mitochondrial function across the continuum of aging muscle atrophy is clearly warranted to better define when intervening at the level of the mitochondrion is most appropriate.
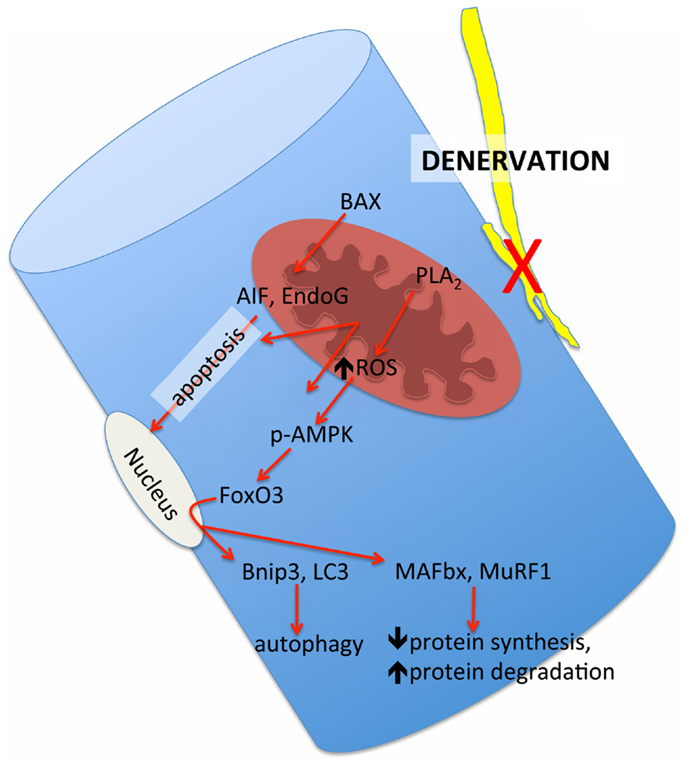
Figure 8. Schematic representation of the mitochondrial-mediated atrophy pathways that have been observed to be up-regulated in aging muscle and their hypothesized link to the upstream influence of sporadic denervation in very advanced age where denervation becomes pronounced. BAX = Bcl2-associated X protein, PLA2 = cytoplasmic phospholipase A2, AIF = apoptosis inducing factor; EndoG = endonuclease G, p-AMPK = phosphorylated adenosine monophosphate kinase, FoxO3 = forkhead box O3, BNIP3 = BCl2/adenovirus E1B 19 kDa protein-interacting protein 3, LC3 = microtubule associated protein light chain 3, MAFbx = muscle atrophy F-box, MuRF1 = muscle ring finger 1.
Our understanding of the impact of aging on mitochondrial function in aging skeletal muscle continues to evolve and has recently undergone significant revision. In particular, it now appears that mitochondrial functional alterations are more subtle than initially indicated, owing to an exaggeration of mitochondrial impact with aging when using isolated organelles (a common approach in early studies). Furthermore, whereas mitochondrial respiratory function and ROS emission changes are highly variable between muscles and are also largely attenuated by maintaining physical activity, a sensitization of the mitochondria to permeability transition seems to be a general property of aging muscles undergoing atrophy regardless of physical activity status. The mechanisms contributing to this mitochondrial functional alteration in aging muscle remains an area of intensive study. Presently, it seems that mtDNA-driven mechanisms are incompatible with the available evidence and instead impairment in mitophagy is appearing the more likely culprit based upon data from recent studies showing alterations in mitophagy signaling. In this respect, there is also emerging evidence that the aging milieu can itself depress mitochondrial respiratory function in vivo, even in the absence of an intrinsic organelle defect in mitochondria studied ex vivo. Furthermore, the likelihood that other extrinsic factors, such as denervation, may also be modulating mitochondrial function at very advanced and clinically relevant ages requires careful consideration of the most appropriate ages at which to target the mitochondrion in seeking more effective treatments for aging muscle. Based on current evidence, it is suggested that an increased susceptibility to permeability transition at ages preceding the most severe clinical impact of aging muscle atrophy (≤75 years) is an appropriate therapeutic target and should be pursued. However, at clinically relevant ages where an increased falls risk, mobility impairment, and physical frailty are more likely to result from aging muscle impact, it remains to be determined whether mitochondrial alterations are now largely secondary to denervation, rendering the mitochondrion a less attractive therapeutic target for the ≥ 80 years age group.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Amara, C. E., Shankland, E. G., Jubrias, S. A., Marcinek, D. J., Kushmerick, M. J., and Conley, K. E. (2007). Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 1057–1062. doi: 10.1073/pnas.0610131104
Anderson, E. J., and Neufer, P. D. (2006). Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am. J. Physiol. Cell Physiol. 290, C844–C851. doi:10.1152/ajpcell.00402.2005
Baloh, R. H., Rakowicz, W., Gardner, R., and Pestronk, A. (2007). Frequent atrophic groups with mixed-type myofibers is distinctive to motor neuron syndromes. Muscle Nerve 36, 107–110. doi:10.1002/mus.20755
Barrientos, A., Casademont, J., Rotig, A., Miro, O., Urbano-Marquez, A., Rustin, P., et al. (1996). Absence of relationship between the level of electron transport chain activities and aging in human skeletal muscle. Biochem. Biophys. Res. Commun. 229, 536–539. doi:10.1006/bbrc.1996.1839
Bass, A., Gutmann, E., and Hanzlikova, V. (1975). Biochemical and histochemical changes in energy supply enzyme pattern of muscles of the rat during old age. Gerontologia 21, 31–45. doi:10.1159/000212028
Betik, A. C., Thomas, M. M., Wright, K. J., Riel, C. D., and Hepple, R. T. (2009). Exercise training from late middle age until senescence does not attenuate the declines in skeletal muscle aerobic function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R744–R755. doi:10.1152/ajpregu.90959.2008
Bhattacharya, A., Muller, F. L., Liu, Y., Sabia, M., Liang, H., Song, W., et al. (2009). Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J. Biol. Chem. 284, 46–55. doi:10.1074/jbc.M806311200
Boffoli, D., Scacco, S. C., Vergari, R., Solarino, G., Santacroce, G., and Papa, S. (1994). Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1226, 73–82. doi:10.1016/0925-4439(94)90061-2
Bua, E., Johnson, J., Herbst, A., Delong, B., McKenzie, D., Salamat, S., et al. (2006). Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480. doi:10.1086/507132
Bua, E. A., McKiernan, S. H., Wanagat, J., McKenzie, D., and Aiken, J. M. (2002). Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J. Appl. Physiol. 92, 2617–2624. doi:10.1152/japplphysiol.01102.2001
Callahan, D. M., Bedrin, N. G., Subramanian, M., Berking, J., Ades, P. A., Toth, M. J., et al. (2014). Age-related structural alterations in human skeletal muscle fibers and mitochondria are sex specific: relationship to single-fiber function. J. Appl. Physiol. 116, 1582–1592. doi:10.1152/japplphysiol.01362.2013
Capel, F., Rimbert, V., Lioger, D., Diot, A., Rousset, P., Mirand, P. P., et al. (2005). Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech. Ageing Dev. 126, 505–511. doi:10.1016/j.mad.2004.11.001
Carter, E. E., Thomas, M. M., Murynka, T., Rowan, S. L., Wright, K. J., Huba, E., et al. (2010). Slow twitch soleus muscle is not protected from sarcopenia in senescent rats. Exp. Gerontol. 45, 662–670. doi:10.1016/j.exger.2010.04.001
Chabi, B., Ljubicic, V., Menzies, K. J., Huang, J. H., Saleem, A., and Hood, D. A. (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. doi:10.1111/j.1474-9726.2007.00347.x
Chance, B., and Williams, G. R. (1956). Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J. Biol. Chem. 221, 477–489.
Chu, C. T. (2010). A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum. Mol. Genet. 19, R28–R37. doi:10.1093/hmg/ddq143
Conley, K. E., Jubrias, S. A., and Esselman, P. C. (2000). Oxidative capacity and aging in human muscle. J. Physiol. 526, 203–210. doi:10.1111/j.1469-7793.2000.t01-1-00203.x
Cooper, J. M., Mann, V. M., and Schapira, A. H. (1992). Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J. Neurol. Sci. 113, 91–98. doi:10.1016/0022-510X(92)90270-U
Cruz-Jentoft, A. J., Landi, F., Topinkova, E., and Michel, J. P. (2010). Understanding sarcopenia as a geriatric syndrome. Curr. Opin. Clin. Nutr. Metab. Care 13, 1–7. doi:10.1097/MCO.0b013e328333c1c1
Csukly, K., Ascah, A., Matas, J., Gardiner, P. F., Fontaine, E., and Burelle, Y. (2006). Muscle denervation promotes opening of the permeability transition pore and increases the expression of cyclophilin D. J. Physiol. 574, 319–327. doi:10.1113/jphysiol.2006.109702
Desai, V. G., Weindruch, R., Hart, R. W., and Feuers, R. J. (1996). Influences of age and dietary restriction on gastrocnemius electron transport system activities in mice. Arch. Biochem. Biophys. 333, 145–151. doi:10.1006/abbi.1996.0375
Dirks, A., and Leeuwenburgh, C. (2002). Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R519–R527. doi:10.1152/ajpregu.00458.2001
Drummond, M. J., Addison, O., Brunker, L., Hopkins, P. N., McClain, D. A., LaStayo, P. C., et al. (2014). Downregulation of e3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 69, 1040–1048. doi:10.1093/gerona/glu004
Edstrom, E., Altun, M., Bergman, E., Johnson, H., Kullberg, S., Ramirez-Leon, V., et al. (2007). Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol. Behav. 92, 129–135. doi:10.1016/j.physbeh.2007.05.040
Edstrom, E., and Ulfhake, B. (2005). Sarcopenia is not due to lack of regenerative drive in senescent skeletal muscle. Aging Cell 4, 65–77. doi:10.1111/j.1474-9728.2005.00145.x
Fulle, S., Protasi, F., Di Tano, G., Pietrangelo, T., Beltramin, A., Boncompagni, S., et al. (2004). The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp. Gerontol. 39, 17–24. doi:10.1016/j.exger.2003.09.012
Gouspillou, G., Bourdel-Marchasson, I., Rouland, R., Calmettes, G., Biran, M., Deschodt-Arsac, V., et al. (2014a). Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 13, 39–48. doi:10.1111/acel.12147
Gouspillou, G., Sgarioto, N., Kapchinsky, S., Purves-Smith, F., Norris, B., Pion, C. H., et al. (2014b). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 28, 1621–1633. doi:10.1096/fj.13-242750
Gouspillou, G., Bourdel-Marchasson, I., Rouland, R., Calmettes, G., Franconi, J. M., Deschodt-Arsac, V., et al. (2010). Alteration of mitochondrial oxidative phosphorylation in aged skeletal muscle involves modification of adenine nucleotide translocator. Biochim. Biophys. Acta 1797, 143–151. doi:10.1016/j.bbabio.2009.09.004
Hagen, J. L., Krause, D. J., Baker, D. J., Fu, M., Tarnopolsky, M. A., and Hepple, R. T. (2004). Skeletal muscle aging in F344BN F1-hybrid rats: I. Mitochondrial dysfunction contributes to the age-associated reduction in VO2max. J. Gerontol. A Biol. Sci. Med. Sci. 59A, 1099–1110. doi:10.1093/gerona/59.11.1099
Haseler, L. J., Hogan, M. C., and Richardson, R. S. (1999). Skeletal muscle phosphocreatine recovery in exercise-trained humans is dependent on O 2 availability. J. Appl. Physiol. 86, 2013–2018.
Hemion, C., Flammer, J., and Neutzner, A. (2014). Quality control of oxidatively-damaged mitochondrial proteins is mediated by p97 and the proteasome. Free Radic. Biol. Med. doi:10.1016/j.freeradbiomed.2014.07.016
Hiona, A., and Leeuwenburgh, C. (2008). The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 43, 24–33. doi:10.1016/j.exger.2007.10.001
Hiona, A., Sanz, A., Kujoth, G. C., Pamplona, R., Seo, A. Y., Hofer, T., et al. (2010). Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 5:e11468. doi:10.1371/journal.pone.0011468
Houmard, J. A., Weidner, M. L., Gavigan, K. E., Tyndall, G. L., Hickey, M. S., and Alshami, A. (1998). Fiber type and citrate synthase activity in the human gastrocnemius and vastus lateralis with aging. J. Appl. Physiol. 85, 1337–1341.
Huang, J. H., Joseph, A. M., Ljubicic, V., Iqbal, S., and Hood, D. A. (2010). Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 65, 138–146. doi:10.1093/gerona/glp201
Hutter, E., Skovbro, M., Lener, B., Prats, C., Rabol, R., Dela, F., et al. (2007). Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 6, 245–256. doi:10.1111/j.1474-9726.2007.00282.x
Joseph, A. M., Adhihetty, P. J., Buford, T. W., Wohlgemuth, S. E., Lees, H. A., Nguyen, L. M., et al. (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 11, 801–809. doi:10.1111/j.1474-9726.2012.00844.x
Joseph, A. M., Adhihetty, P. J., Wawrzyniak, N. R., Wohlgemuth, S. E., Picca, A., Kujoth, G. C., et al. (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 8:e69327. doi:10.1371/journal.pone.0069327
Joseph, A. M., Ljubicic, V., Adhihetty, P. J., and Hood, D. A. (2010). Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am. J. Physiol. Cell Physiol. 298, C1308–C1314. doi:10.1152/ajpcell.00644.2008
Kallen, R. G., Sheng, Z. H., Yang, J., Chen, L. Q., Rogart, R. B., and Barchi, R. L. (1990). Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron 4, 233–242. doi:10.1016/0896-6273(90)90098-Z
Katayama, M., Tanaka, M., Yamamoto, H., Ohbayashi, T., Nimura, Y., and Ozawa, T. (1991). Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem. Int. 25, 47–56.
Kent-Braun, J. A., and Ng, A. V. (2000). Skeletal muscle oxidative capacity in young and older women and men. J. Appl. Physiol. 89, 1072–1078.
Kerner, J., Turkaly, P. J., Minkler, P. E., and Hoppel, C. L. (2001). Aging skeletal muscle mitochondria in the rat: decreased uncoupling protein-3 content. Am. J. Physiol. Endocrinol. Metab. 281, E1054–E1062.
Koltai, E., Hart, N., Taylor, A. W., Goto, S., Ngo, J. K., Davies, K. J., et al. (2012). Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 303, R127–R134. doi:10.1152/ajpregu.00337.2011
Konopka, A. R., Suer, M. K., Wolff, C. A., and Harber, M. P. (2014). Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J. Gerontol. A Biol. Sci. Med. Sci. 69, 371–378. doi:10.1093/gerona/glt107
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. doi:10.1126/science.1112125
Lanza, I. R., Befroy, D. E., and Kent-Braun, J. A. (2005). Age-related changes in ATP-producing pathways in human skeletal muscle in vivo. J. Appl. Physiol. 99, 1736–1744. doi:10.1152/japplphysiol.00566.2005
Lawrenson, L., Poole, J. G., Kim, J., Brown, C., Patel, P., and Richardson, R. S. (2003). Vascular and metabolic response to isolated small muscle mass exercise: effect of age. Am. J. Physiol. Heart Circ. Physiol. 285, H1023–H1031. doi:10.1152/ajpheart.00135.2003
Lee, C. M., Lopez, M. E., Weindruch, R., and Aiken, J. M. (1998). Association of age-related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic. Biol. Med. 25, 964–972. doi:10.1016/S0891-5849(98)00185-3
Leeuwenburgh, C., Gurley, C. M., Strotman, B. A., and Dupont-Versteegden, E. E. (2005). Age-related differences in apoptosis with disuse atrophy in soleus muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1288–R1296. doi:10.1152/ajpregu.00576.2004
Lezza, A. M., Boffoli, D., Scacco, S., Cantatore, P., and Gadaleta, M. N. (1994). Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem. Biophys. Res. Commun. 205, 772–779. doi:10.1006/bbrc.1994.2732
Li, W., Zhang, X., Zhuang, H., Chen, H. G., Chen, Y., Tian, W., et al. (2014). MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 289, 10691–10701. doi:10.1074/jbc.M113.537050
Ljubicic, V., and Hood, D. A. (2009). Diminished contraction-induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell 8, 394–404. doi:10.1111/j.1474-9726.2009.00483.x
Ljubicic, V., Joseph, A. M., Adhihetty, P. J., Huang, J. H., Saleem, A., Uguccioni, G., et al. (2009). Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany NY) 1, 818–830.
Lyons, C. N., Mathieu-Costello, O., and Moyes, C. D. (2006). Regulation of skeletal muscle mitochondrial content during aging. J. Gerontol. A Biol. Sci. Med. Sci. 61, 3–13. doi:10.1093/gerona/61.1.3
Mahler, M. (1985). First-order kinetics of muscle oxygen consumption, and an equivalent proportionality between QO 2 and phosphoryl-creatine level. J. Gen. Physiol. 86, 165.
Mansouri, A., Muller, F. L., Liu, Y., Ng, R., Faulkner, J., Hamilton, M., et al. (2006). Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech. Ageing Dev. 127, 298–306. doi:10.1016/j.mad.2005.11.004
Marcinek, D. J., Schenkman, K. A., Ciesielski, W. A., Lee, D., and Conley, K. E. (2005). Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 569, 467–473. doi:10.1113/jphysiol.2005.097782
Martin, K. R., Koster, A., Murphy, R. A., Van Domelen, D. R., Hung, M. Y., Brychta, R. J., et al. (2014). Changes in daily activity patterns with age in U.S. men and women: National Health and Nutrition Examination Survey 2003-04 and 2005-06. J. Am. Geriatr. Soc. 62, 1263–1271. doi:10.1111/jgs.12893
Marzetti, E., Hwang, J. C., Lees, H. A., Wohlgemuth, S. E., Dupont-Versteegden, E. E., Carter, C. S., et al. (2010). Mitochondrial death effectors: relevance to sarcopenia and disuse muscle atrophy. Biochim. Biophys. Acta 1800, 235–244. doi:10.1016/j.bbagen.2009.05.007
Marzetti, E., Wohlgemuth, S. E., Lees, H. A., Chung, H. Y., Giovannini, S., and Leeuwenburgh, C. (2008). Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech. Ageing Dev. 129, 542–549. doi:10.1016/j.mad.2008.05.005
Mathieu-Costello, O., Ju, Y., Trejo-Morales, M., and Cui, L. (2005). Greater capillary-fiber interface per fiber mitochondrial volume in skeletal muscles of old rats. J. Appl. Physiol. 99, 281–289. doi:10.1152/japplphysiol.00750.2004
McCully, K. K., Fielding, R. A., Evans, W. J., Leigh, J. S. Jr., and Posner, J. D. (1993). Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. J. Appl. Physiol. 75, 813–819.
Melov, S., Shoffner, J. M., Kaufman, A., and Wallace, D. C. (1995). Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 23, 4122–4126. doi:10.1093/nar/23.23.4938-a
Meyer, R. A. (1988). A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am. J. Physiol. 254, C548–C553.
Miwa, S., Lawless, C., and von Zglinicki, T. (2008). Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell 7, 920–923. doi:10.1111/j.1474-9726.2008.00426.x
Muller, F. L., Song, W., Jang, Y. C., Liu, Y., Sabia, M., Richardson, A., et al. (2007). Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1159–R1168. doi:10.1152/ajpregu.00767.2006
Oda, K. (1984). Age changes of motor innervation and acetylcholine receptor distribution on human skeletal muscle fibres. J. Neurol. Sci. 66, 327–338. doi:10.1016/0022-510X(84)90021-2
Orlander, J., Kiessling, K. H., Larsson, L., Karlsson, J., and Aniansson, A. (1978). Skeletal muscle metabolism and ultrastructure in relation to age in sedentary men. Acta Physiol. Scand. 104, 249–261. doi:10.1111/j.1748-1716.1978.tb06277.x
Picard, M., Csukly, K., Robillard, M. E., Godin, R., Ascah, A., Bourcier-Lucas, C., et al. (2008). Resistance to Ca2+-induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R659–R668. doi:10.1152/ajpregu.90357.2008
Picard, M., Hepple, R. T., and Burelle, Y. (2012). Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am. J. Physiol. Cell Physiol. 302, C629–C641. doi:10.1152/ajpcell.00368.2011
Picard, M., Ritchie, D., Thomas, M. M., Wright, K. J., and Hepple, R. T. (2011a). Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 10, 1047–1055. doi:10.1111/j.1474-9726.2011.00745.x
Picard, M., Taivassalo, T., Gouspillou, G., and Hepple, R. T. (2011b). Mitochondria: isolation, structure and function. J. Physiol. 589, 4413–4421. doi:10.1113/jphysiol.2011.212712
Picard, M., Taivassalo, T., Ritchie, D., Wright, K. J., Thomas, M. M., Romestaing, C., et al. (2011c). Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 6:e18317. doi:10.1371/journal.pone.0018317
Picard, M., Ritchie, D., Wright, K. J., Romestaing, C., Thomas, M. M., Rowan, S. L., et al. (2010). Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 9, 1032–1046. doi:10.1111/j.1474-9726.2010.00628.x
Poole, J. G., Lawrenson, L., Kim, J., Brown, C., and Richardson, R. S. (2003). Vascular and metabolic response to cycle exercise in sedentary humans: effect of age. Am. J. Physiol. Heart Circ. Physiol. 284, H1251–H1259. doi:10.1152/ajpheart.00790.2002
Prompers, J. J., Wessels, B., Kemp, G. J., and Nicolay, K. (2014). MITOCHONDRIA: investigation of in vivo muscle mitochondrial function by 31P magnetic resonance spectroscopy. Int. J. Biochem. Cell Biol. 50, 67–72. doi:10.1016/j.biocel.2014.02.014
Rasmussen, U. F., Krustrup, P., Kjaer, M., and Rasmussen, H. N. (2003). Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes of ATP formation and mitochondrial capacity. Pflugers Arch. 446, 270–278. doi:10.1007/s00424-003-1022-2
Richter, C., Park, J. W., and Ames, B. N. (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. U.S.A. 85, 6465–6467. doi:10.1073/pnas.85.17.6465
Rockstein, M., and Brandt, K. F. (1963). Enzyme changes in flight muscle correlated with aging and flight ability in the male housefly. Science 139, 1049–1051. doi:10.1126/science.139.3559.1049
Romanello, V., Guadagnin, E., Gomes, L., Roder, I., Sandri, C., Petersen, Y., et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785. doi:10.1038/emboj.2010.60
Rowan, S. L., Purves-Smith, F. M., Solbak, N. M., and Hepple, R. T. (2011). Accumulation of severely atrophic myofibers marks the acceleration of sarcopenia in slow and fast twitch muscles. Exp. Gerontol. 46, 660–669. doi:10.1016/j.exger.2011.03.005
Rowan, S. L., Rygiel, K., Purves-Smith, F. M., Solbak, N. M., Turnbull, D. M., and Hepple, R. T. (2012). Denervation causes fiber atrophy and Myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 7:e29082. doi:10.1371/journal.pone.0029082
Rudolf, R., Khan, M. M., Labeit, S., and Deschenes, M. R. (2014). Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 6:99. doi:10.3389/fnagi.2014.00099
Rumsey, W. L., Kendrick, Z. V., and Starnes, J. W. (1987). Bioenergetics in the aging Fischer 344 rat: effects of exercise and food restriction. Exp. Gerontol. 22, 271–287. doi:10.1016/0531-5565(87)90006-4
Safdar, A., Hamadeh, M. J., Kaczor, J. J., Raha, S., Debeer, J., and Tarnopolsky, M. A. (2010). Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 5:e10778. doi:10.1371/journal.pone.0010778
Scelsi, R., Marchetti, C., and Poggi, P. (1980). Histochemical and ultrastructural aspects of m. vastus lateralis in sedentary old people (aged 65-89 years). Acta Neuropathol. 51, 99–105. doi:10.1007/BF00690450
Scheibye-Knudsen, M., Fang, E. F., Croteau, D. L., and Bohr, V. A. (2014). Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 10, doi:10.4161/auto.29321
Schiavi, A., and Ventura, N. (2014). The interplay between mitochondria and autophagy and its role in the aging process. Exp. Gerontol. 56, 147–153. doi:10.1016/j.exger.2014.02.015
Siegel, M. P., Kruse, S. E., Percival, J. M., Goh, J., White, C. C., Hopkins, H. C., et al. (2013). Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763–771. doi:10.1111/acel.12102
Simonetti, S., Chen, X., DiMauro, S., and Schon, E. A. (1992). Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim. Biophys. Acta 1180, 113–122. doi:10.1016/0925-4439(92)90059-V
Siu, P. M., Pistilli, E. E., and Alway, S. E. (2005). Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1015–R1026. doi:10.1152/ajpregu.00198.2005
Snow, L. M., McLoon, L. K., and Thompson, L. V. (2005). Adult and developmental myosin heavy chain isoforms in soleus muscle of aging Fischer Brown Norway rat. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 286, 866–873. doi:10.1002/ar.a.20218
Speakman, J. R., Talbot, D. A., Selman, C., Snart, S., McLaren, J. S., Redman, P., et al. (2004). Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell 3, 87–95. doi:10.1111/j.1474-9728.2004.00097.x
Stary, C. M., Mathieu-Costello, O., and Hogan, M. C. (2004). Resistance to fatigue of individual Xenopus single skeletal muscle fibres is correlated with mitochondrial volume density. Exp. Physiol. 89, 617–621. doi:10.1113/expphysiol.2004.027763
Taivassalo, T., Jensen, T. D., Kennaway, N., DiMauro, S., Vissing, J., and Haller, R. G. (2003). The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain 126, 413–423. doi:10.1093/brain/awg028
Terman, A., Kurz, T., Navratil, M., Arriaga, E. A., and Brunk, U. T. (2010). Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 12, 503–535. doi:10.1089/ars.2009.2598
Tomonaga, M. (1977). Histochemical and ultrastructural changes in senile human skeletal muscle. J. Am. Geriatr. Soc. 25, 125–131.
Tonkonogi, M., Walsh, B., Svensson, M., and Sahlin, K. (2000). Mitochondrial function and antioxidative defence in human muscle: effects of endurance training and oxidative stress. J. Physiol. 528, 379–388. doi:10.1111/j.1469-7793.2000.00379.x
Trifunovic, A., Hansson, A., Wredenberg, A., Rovio, A. T., Dufour, E., Khvorostov, I., et al. (2005). From the cover: somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 102, 17993–17998. doi:10.1073/pnas.0508886102
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi:10.1038/nature02517
Trounce, I., Byrne, E., and Marzuki, S. (1989). Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet 1, 637–639. doi:10.1016/S0140-6736(89)92143-0
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi:10.1038/sj.emboj.7601963
Twig, G., and Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 14, 1939–1951. doi:10.1089/ars.2010.3779
Wanagat, J., Cao, Z., Pathare, P., and Aiken, J. M. (2001). Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332. doi:10.1096/fj.00-0320com
Wang, Z. M., Zheng, Z., Messi, M. L., and Delbono, O. (2005). Extension and magnitude of denervation in skeletal muscle from ageing mice. J. Physiol. (Lond.) 565, 757–764. doi:10.1113/jphysiol.2005.087601
Keywords: sarcopenia, bioenergetics, mitochondria, skeletal muscle, reactive oxygen species, permeability transition, oxidative stress, muscle atrophy
Citation: Hepple RT (2014) Mitochondrial involvement and impact in aging skeletal muscle. Front. Aging Neurosci. 6:211. doi: 10.3389/fnagi.2014.00211
Received: 15 July 2014; Accepted: 30 July 2014;
Published online: 10 September 2014.
Edited by:
Emanuele Marzetti, Catholic University of the Sacred Heart, ItalyReviewed by:
Riccardo Calvani, Institute of Crystallography – Italian National Research Council, ItalyCopyright: © 2014 Hepple. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Russell T. Hepple, Department of Critical Care Medicine, Royal Victoria Hospital, 675 Pine Avenue West, Montreal, QC H3A1A1, Canada e-mail:cnVzc2VsbC5oZXBwbGVAbWNnaWxsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.