


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 29 August 2012
Sec. Alzheimer's Disease and Related Dementias
Volume 4 - 2012 | https://doi.org/10.3389/fnagi.2012.00024
This article is part of the Research TopicSpatial memory – a unique window into healthy and pathological ageingView all 14 articles
The hippocampus is a prime target for glucocorticoids (GCs) and a brain structure particularly vulnerable to aging. Prolonged exposure to excess GCs compromises hippocampal electrophysiology, structure, and function. Blood GC levels tend to increase with aging and correlate with impaired spatial memory in aging rodents and humans. The magnitude of GC action within tissues depends not only on levels of steroid hormone that enter the cells from the periphery and the density of intracellular receptors but also on the local metabolism of GCs by 11β-hydroxysteroid dehydrogenases (11β-HSD). The predominant isozyme in the adult brain, 11β-HSD1, locally regenerates active GCs from inert 11-keto forms thus amplifying GC levels within specific target cells including in the hippocampus and cortex. Aging associates with elevated hippocampal and neocortical 11β-HSD1 and impaired spatial learning while deficiency of 11β-HSD1 in knockout (KO) mice prevents the emergence of cognitive decline with age. Furthermore, short-term pharmacological inhibition of 11β-HSD1 in already aged mice reverses spatial memory impairments. Here, we review research findings that support a key role for GCs with special emphasis on their intracellular regulation by 11β-HSD1 in the emergence of spatial memory deficits with aging, and discuss the use of 11β-HSD1 inhibitors as a promising novel treatment in ameliorating/improving age-related memory impairments.
Cognitive decline is a key feature of aging but significant impairments of learning and memory are not inevitable or strictly linked to chronological age. Marked inter-individual variability exists, ranging from almost no decline through mild impairments to frank dementia. This phenomenon has been described in several species including rodents and humans but the mechanisms underlying the individual differences remain poorly understood. One important mechanistic hypothesis is that variations in hypothalamic-pituitary-adrenal (HPA) activity and consequent exposure to glucocorticoids (GCs; cortisol in humans, corticosterone in rodents) during life may contribute to the inter-individual differences in cognitive decline in animals and humans.
The adrenal cortex synthesizes GCs and these steroid hormones are released directly into the peripheral circulation following stimulation of the HPA axis in response to external (stress) and internal (circadian) cues. Collectively, GCs released in coordination with the rapidly acting sympathetic-adrenomedullary system, help an organism respond to “stressors” or threats to homeostasis by mobilizing energy stores, suppressing nonessential physiological processes (e.g., reproduction, digestion) and initiating behavioral responses. Circulating GC levels are normally tightly regulated by negative feedback inhibition upon the HPA axis where GCs act back on the hypothalamus and pituitary glucocorticoid receptors (GRs) (to suppress CRH and ACTH production) to terminate its own release. GC feedback also occurs in higher centers such as the hippocampus and cingulate cortex.
GCs readily enter the brain, a major target for GC action (McEwen et al., 1968). Here GCs bind to classical nuclear (hormone) receptors to regulate the transcription of specific genes, either by direct binding of receptor homodimers to DNA (Datson et al., 2001) or via protein-protein interactions with other transcription factors such as fos and Jun-1 (Heck et al., 1994; Hayashi et al., 2004). Through activation of their intracellular receptors, GCs affect a wide range of processes including altering neurotransmission, electrophysiological activity, cellular metabolism, and structure, as well as neuronal division, maturation, and death.
Two types of corticosteroid receptors exist, the type I high affinity mineralocorticoid receptors (MR) and type II lower affinity GRs (Reul and de Kloet, 1985). Although sharing almost identical DNA-binding domains, MR and GR can exert distinct cellular functions as the genes they bind to show little overlap (Datson et al., 2001). GRs are widely distributed throughout the brain in most neurons and glia. GRs have a lower affinity for physiological GCs and only become substantially activated as hormone levels rise following stress. MR is expressed in neurons only and has a more restricted distribution with high expression confined particularly to the hippocampus (Figure 1), and septum. MRs having a 10-fold higher affinity for physiological corticosteroids (GCs and the mineralocorticoid aldosterone) are extensively occupied under basal conditions when hormone levels are low (Reul and de Kloet, 1985; McEwen et al., 1986).
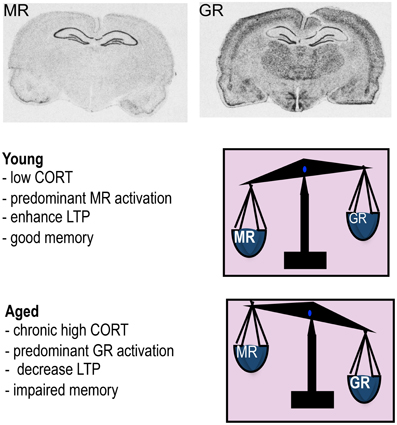
Figure 1. Mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) mRNA expression in rat brain. The high affinity MR is predominantly occupied under low corticosterone (CORT) levels typically observed in young animals under basal conditions and this leads to enhanced LTP and good memory. When CORT levels rises such as in aging, MR is fully occupied and the excess GCs now occupy more GR so the balance of receptors activated is now predominantly GR which decreases LTP and impairs memory.
In addition to the delayed genomic effects via intracellular MR and GR, it has become evident from recent work that GCs also affect brain function through rapid non-genomic membrane-associated mechanisms (Groeneweg et al., 2011) (Figure 2). The latter mode of action explains the rapid (minutes) effects of GCs on the excitability and activation of neurons in several brain regions (e.g., hypothalamus, hippocampus, amygdala, and prefrontal cortex) and provides a physiological basis for rapid effects on behavior (de Kloet et al., 2008). A membrane-localized form of MR appears to mediate the rapid GC signaling in the hippocampus (Karst et al., 2005). However, not all rapid GC effects occur via MRs with some [e.g., increase in spine density of hippocampal neurons (Komatsuzaki et al., 2005)] depending on membrane-located GRs rather than MRs and others [e.g., long-term potentiation (LTP) induction (Wiegert et al., 2006), NMDA-dependent neurotoxicity (Xiao et al., 2010)] occurring independent of MR or GR but mediated possibly through as yet unidentified membrane-localized receptors.
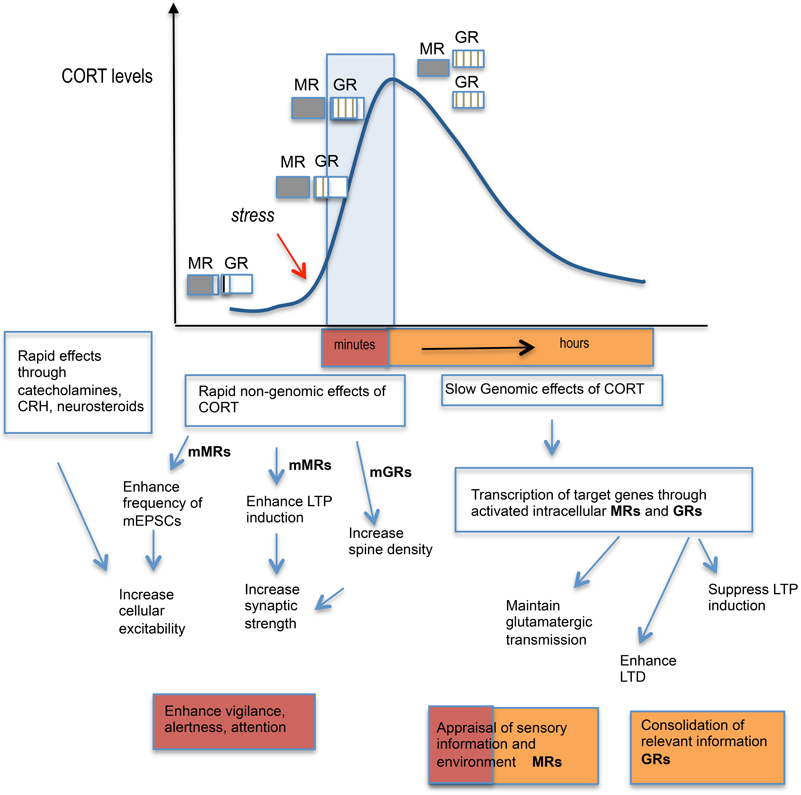
Figure 2. Schematic diagram showing stress hormone effects: from cellular responses to behavior. Activation of the HPA axis by stress leads to a rapid rise in circulating corticosterone (CORT) levels which starts to fully occupy MR and increasingly occupy GRs. The varying level of occupancy of MRs and GRs are represented by extent of shading or lines within the boxes. The CORT levels normally return to baseline levels by 2 h through negative feedback regulation upon the HPA axis. The initial rapid effects of CORT via activation of membrane-located MRs enhance LTP induction and enhance miniature excitatory postsynaptic currents in the hippocampus while membrane-located GRs appear to increase neuronal spine density. Together with other stress hormones released such as the catecholamines (noradrenaline and adrenaline) and corticotropin-releasing hormone (CRH), which also strengthen synaptic transmission in the hippocampus, these rapid events contribute to enhanced vigilance, alertness, and attention. The slower genomic effects of CORT through activation of intracellular MRs and GRs and the transcription of target genes, leads to the maintenance of glutamatergic transmission via MRs and suppression of LTP induction but enhanced LTD via GRs in the hippocampus. MRs and GRs contributes to different aspects of cognitive processing with MR activation responsible for the appraisal of sensory information and environmental cues, while GRs are important for the consolidation of events/information in memory.
Short term increases in GC levels are normally adaptive and beneficial but prolonged exposure to elevated GC levels such as during chronic stress or as a consequence of failure or impairment of negative feedback control of GC secretion (e.g., Cushing's syndrome, major depression) will lead to excessive GC responses and to pathology in the periphery (e.g., diabetes, hypertension, osteoporosis, central obesity) and CNS (e.g., depression, impaired learning, and memory). Cushing's syndrome patients with hyper-secretion of cortisol show reduced hippocampal volume and impaired performance on hippocampal learning tasks (Starkman et al., 1992). Notably, successful treatment to correct the excessive secretion of GCs reverses the pathology including hippocampal structural recovery and the restoration, to some extent, of mood, learning, and memory (Starkman et al., 1999). Conversely, severely reduced GC levels, as in Addison's disease, also result in pathology and cognitive impairments (Tytherleigh et al., 2004) and are treated with lifelong corticosteroid replacement. Thus both too little and too much GCs can have detrimental effects on memory, emphasizing the crucial need to maintain optimal levels of GCs for health and survival.
GCs via binding to abundant MRs and GRs in the hippocampus control the excitability of neuronal networks that underlie learning and memory processes. While not the focus of this review, it is worth noting that other brain areas rich in GRs such as the basolateral amygdala (BLA) and prefrontal cortex, which controls emotional and working memory respectively, interacts with the hippocampus to modulate cognitive function (Roozendaal and McGaugh, 1997, 2011). An efficient interplay between activation of MR and GR appears essential for maximal learning. The two receptors have different roles in learning as evident from rodent studies, especially in the watermaze using antagonists of MR and GR at the various phases of spatial learning (Oitzl and de Kloet, 1992); MRs have a major role in behavioral reactivity toward stimuli while GRs are involved in consolidating learned information. Furthermore, the ratio of occupation and presumably activation of MR/GR appears to determine whether GCs improve or impair memory (de Kloet et al., 1999). Thus, optimal enhanced memory occurs when GC levels are mildly elevated such that most MRs and only some GRs are activated (i.e., increased MR/GR ratio) but impaired memory results when circulating GC levels are greatly increased such as found in some aging individuals (i.e., low MR/GR ratio) (Kim and Diamond, 2002; Tytherleigh et al., 2004; Herbert et al., 2006) (Figure 1). Indeed, circulating basal (nadir) GC levels do not always increase as a function of chronological age with only a proportion of individuals showing increasingly high GC levels with advancing age while some show levels within the young normal range (Lupien et al., 1994).
Whether the consequence of the receptor activation is positive or negative for memory depend largely on the context of the situation/event to be remembered, the timing and the magnitude of the increased GC levels (de Kloet et al., 1999; Joels et al., 2006). Acute stress or increased GC levels occurring around the time of learning and within the context of the event to be remembered enhances memory consolidation. In contrast, they impair memory if occurring either before or a considerable time after the learning task (Quervain et al., 1998; Cazakoff et al., 2010). Evidence, mainly from animal studies, suggests that GCs preferentially enhances memory consolidation of emotionally arousing experiences (Roozendaal et al., 2006). Animal learning tasks, including the Morris watermaze and radial arm maze for spatial memory training, are designed to be generally affectively arousing because they require motivation to elicit changes in behavior. Even learning tasks that include no rewarding or aversive stimulation, such as the object recognition task, induces modest novelty-induced stress or arousal during training (Okuda et al., 2004). GCs released during learning appear essential for establishing enduring memories (De Kloet et al., 1998). Thus, small increases in GCs enhance hippocampus-mediated learning and memory while larger, prolonged elevations impair memory (Lupien and McEwen, 1997; Kim and Diamond, 2002). This follows the inverted U-shaped dose-response relationship between GC levels and effects on hippocampal LTP, an electrophysiological phenomenon associated with synaptic strengthening which is one of the major cellular mechanisms underlying learning and memory. Low moderate GC levels occupy predominantly high affinity MR which increases LTP and memory, while high GC levels occupy the lower affinity GR (in addition to MR) and impair LTP and memory (Pavlides et al., 1995, 1996; Kim and Diamond, 2002; Kim et al., 2007). One recent study supports the inverted-U-shaped relationship between intrinsic stress intensity (i.e., increased endogenous GCs induced by factors associated with the learning task, in this case water temperature) and spatial memory in the radial arm watermaze. Thus, rats trained at 19°C made fewer errors than rats trained at either more (16°C) or less (25°C) stressful conditions (Salehi et al., 2010).
The hippocampus not only plays a central role in the processing of spatial and contextual information (Morris et al., 1982; Moser et al., 1995) but also exerts an inhibitory influence over HPA function (Jacobson and Sapolsky, 1991). With its high density of MRs and GRs, the hippocampus is also particularly sensitive to the deleterious actions of chronic GC excess, potentiating neurotoxicity, dendritic atrophy, and perhaps even neuronal loss (Sapolsky, 1987). The idea that excess GCs could promote aging of the hippocampus was first established over 30 years ago following a study that showed a positive correlation between hippocampal aging (astrocyte reactivity as a marker of neuronal damage) and plasma levels of corticosterone in aging rats (Landfield et al., 1978). While only a few studies have shown high GC levels or stress actually cause hippocampal neuron loss (Uno et al., 1989; Sousa et al., 1998), much evidence support chronic stress (or high GCs) causing hippocampal atrophy (Watanabe et al., 1992; Magarinos and McEwen, 1995).
In humans, including those with Cushing's disease, Alzheimer's disease, depression, and normal aging, higher cortisol levels have been associated with poorer memory and hippocampal shrinkage/neuronal loss (De Leon et al., 1988; Wolkowitz et al., 1990; Newcomer et al., 1994; Mitchell and Dening, 1996; Lupien et al., 1998; Karlamangla et al., 2005; MacLullich et al., 2005). Moreover, increased HPA activity, as a consequence of impaired HPA axis negative feedback control, has been hypothesized to contribute to the decline in cognitive function, including deficits on spatial tasks with aging (Ohta, 1981; Lupien et al., 1998; McEwen et al., 1999). In support, the extent of age-related cognitive impairments in rodents and humans correlates with increased HPA activity (Issa et al., 1990; Meaney et al., 1995; Yau et al., 1995; Lupien et al., 1996, 1998). Aging rodents also show difficulty with hippocampus-dependent tasks that require a spatial mapping strategy; acquisition deficits have been reported in spatial information processing tests including Barnes hole-board task, radial arm mazes, and Morris watermaze (Ingram et al., 1981; Barnes and McNaughton, 1985; Gage et al., 1989; Issa et al., 1990; Gallagher et al., 1993; Yau et al., 1994, 1995).
Although GCs act on the hippocampus to modulate the formation of new memories, it also acts at other sites, notably the amygdala and prefrontal cortex, which functionally interact. Thus, GC effects on hippocampal LTP and memory can be blocked by lesions to the BLA (Roozendaal and McGaugh, 1997; Kim et al., 2001) and much evidence supports a role of the BLA via emotional arousal-induced noradrenergic activation and GR actions in the modulation of memory consolidation and working memory (Roozendaal and McGaugh, 2011). Given that amygdala function is affected by aging (Iidaka et al., 2002; Charles et al., 2003), it might be expected that fear conditioning would also be disrupted with aging and hence influence hippocampal memory deficits. However, age-related impairments in spatial memory appear not to be influenced by emotional and contextual memories, which tend to be preserved with aging (Comblain et al., 2004; Gould and Feiro, 2005; May et al., 2005; Bergado et al., 2011; Broster et al., 2012).
As a group, aged rats show impaired spatial learning. However, there are substantial inter-individual differences in performance (Yau et al., 1995). This increased variation in aged rats allows subdivision into categories (cognitively-unimpaired, cognitively-impaired) in the watermaze according to their latency to find the submerged platform on the last days of training relative to the mean latency of young controls (Issa et al., 1990; Tombaugh et al., 2002) or to a learning index score computed from probe trials (during retraction of the platform to the bottom of the pool during 30 s of 90 s trials) (Gallagher et al., 1993; Wilson et al., 2004; Robitsek et al., 2008). Aged rats have also been subdivided into impaired and unimpaired categories according to their spatial memory performance in a Y-maze two-trial spatial recognition task where the mean value of the percentage of visits in the novel arm of impaired rats were not different from chance values (Vallee et al., 1997). We typically find approximately ~20–25% of aged Lister hooded rat cohorts differ significantly (>2.5 SD) from young controls in the watermaze (escape latency and probe times) and another ~20–25% not significantly different (<0.5 SD) from young controls (Figure 3). It is the cognitively impaired groups that selectively show elevated plasma corticosterone levels (Issa et al., 1990; Yau et al., 1995), and reduced hippocampal corticosteroid receptor density (Issa et al., 1990; Yau et al., 1995) compared to both young and similarly aged cognitively unimpaired rats (Issa et al., 1990). While aged rats with watermaze performances not significantly different from young rats have been sub-classed as “cognitively unimpaired,” it is important to note that they are not cognitively the same as young rats. Indeed, we found that aged Lister hooded rats, categorized as unimpaired by their performance in the conventional spatial memory watermaze task, were impaired in a more cognitively demanding delayed matching-to-place paradigm spatial memory watermaze task (Steele and Morris, 1999) when compared to young controls (Yau and Seckl, unpublished).
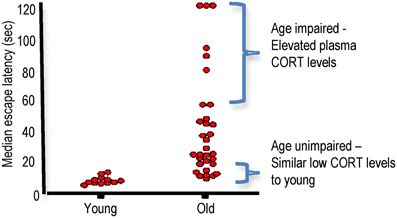
Figure 3. Aged Lister hooded rats show inter-individual differences in spatial learning in the watermaze. 24 months old male Lister hooded rats show impaired spatial learning and memory in the watermaze as a group compared to 6 months old controls. However, individual performances taking the median of the escape latencies (4 trials/day) for the last 2 days of training shows a large spread of learning abilities for aged rats. Plasma corticosterone (CORT) levels in the aged rats correlated negatively with spatial memory with impaired aged rats showing increased CORT while unimpaired aged rats had lower CORT similar to young controls (Based on Yau et al., 1995, with permission from Elsevier).
Aged mice also show impaired spatial memory in the watermaze (Verbitsky et al., 2004; Pawlowski et al., 2009) and increased plasma CORT levels (Yau et al., 2001, 2007; Holmes et al., 2010) such that higher CORT levels correlates with impaired learning in the watermaze (Yau et al., 2001). However, in contrast to aged rats, subdividing aged mice cohorts into cognitively aged-impaired and aged-unimpaired groups is less clear-cut with large variations in performance found even in young controls (Pawlowski et al., 2009). Mice in general take longer to train in the watermaze than rats and show less consistent performance with some aged mice tending to float rather than swim. The Y-maze spatial recognition task, in contrast to the watermaze, is an ethologically relevant test based on the rodent's innate curiosity to explore novel areas and presents no negative or positive reinforcers and little stress for the rodents. When spatial memory was tested in the Y-maze using a 2 h inter-trial interval, young C57BL/6J mice can still remember the novel arm (exploring it more than the other two arms) while aged mice were overall impaired as a group (not distinguishing the novel arm between the three arms). As in the watermaze, inter-individual differences in Y-maze spatial memory performances exist in aged mice, but with a smaller percentage of aged mice “unimpaired” (<20%) while the majority were “impaired” (Yau and Seckl, unpublished).
In rats, manipulations which keep GC levels low throughout life, such as postnatal handling during the first two weeks of life (which permanently increases hippocampal expression of GR, thus improving HPA axis negative feedback to reduce circulating GC levels), denser maternal care (licking and arched back nursing of her offspring), or adrenalectomy at middle age with low dose corticosterone replacement, prevent later hippocampal morphological changes and spatial memory deficits with aging (Landfield et al., 1981; Meaney et al., 1988). Although such manipulations are probably not clinically utilizable, they suggest that pharmacological treatments to increase GR density in the adult hippocampus may reduce GC levels long-term and ameliorate or prevent the emergence of spatial memory impairments with aging. One potent long-term regulator of MR and GR in the hippocampus is serotonin; this neurotransmitter directly increases GR in primary neuronal cultures and in vivo following postnatal handling of rat pups (Mitchell et al., 1990; Meaney et al., 1994; Lai et al., 2003) while lesions of the serotonergic pathway reduces hippocampal MR and GR (Yau et al., 1997). Antidepressants, which amongst other effects, increases serotonin levels, increase hippocampal GR density, improve HPA feedback regulation and thus reduce GC levels in adult rats and mice (Reul et al., 1993; Montkowski et al., 1995; Barden, 1996). Chronic (2 months) treatment of aged Lister hooded rats with amitriptyline, however, did not prevent spatial memory impairments but treatment of young (8 months) animals improved spatial memory, reduced plasma corticosterone levels, and increased hippocampal MR mRNA expression (Yau et al., 1995). Since hippocampal MR enhances LTP (Pavlides et al., 1994) and has a positive influence on memory while central MR blockade impairs spatial memory in adult rats (Yau et al., 1999), the antidepressant induced increase in hippocampal MR may, in part, underlie the better spatial memory in the young rats. Aged rats may lack the plasticity for antidepressants to be effective at enhancing memory later in life. In support, earlier treatment with antidepressants from middle age (for 6 months) improved HPA negative feedback (Rowe et al., 1997) and reduced the emergence of spatial memory impairments in a cohort of aged rats (Yau et al., 2002).
While many studies have measured blood GC levels and correlated this to GC actions within tissues of interest, the principal determinant of GC action is the level of hormone inside the cell. The magnitude of intracellular GC action has long been thought to be determined by the concentration of active hormone in the circulation [modulated by hormone binding to plasma proteins, mainly corticosteroid binding globulin (CBG)] and the density of intracellular receptors in target tissues. However, during the past two decades, enzymic pre-receptor metabolism of GCs by 11β-hydroxysteroid dehydrogenases (11β-HSDs) has emerged as a key mechanism for tissue specific control of active GC levels (Seckl, 1997). 11β-HSDs are microsomal (endoplasmic reticulum) enzymes which catalyse the interconversion of active GCs (corticosterone in rodents, cortisol in humans) and inert 11-keto forms [11-dehydrocorticosterone (11-DHC), cortisone]. They thus, potently regulate steroid access to receptors within specific tissues (Seckl, 1997).
11β-HSD1 is the predominant isoform in the adult rodent and human brain, where it is widely distributed with particularly high expression in the hippocampus, cerebellum, and cortex in both neurons and glia cells (Moisan et al., 1990; Sandeep et al., 2004). 11β-HSD2 whilst highly expressed in the fetal CNS until mid-gestation, in the adult brain is restricted to the nucleus of the solitary tract (NTS) in mice and this plus a few other scattered brain stem and diencephalic nuclei in rats. 11β-HSD2 acts as a dehydrogenase to inactivate GCs before they can bind to receptors. It is best noted for its role to exclude GCs from otherwise non-selective MRs in the distal nephron, thus allowing aldosterone selectivity. 11β-HSD1 in contrast functions as a 11β-reductase (regenerating active GCs from inert 11-DHC) in intact cells, thus locally “amplifying” GC levels within brain cells as well as in liver, adipose tissue, immune system cells etc (Figure 4). This direction of action, far from protecting neurons against GC excess, would be anticipated to increase local intraneuronal GC levels, potentiating their effects including toxicity. Consistent with this hypothesis, in vitro otherwise inert 11-DHC potentiates kainate neurotoxicity in hippocampal cells in culture, as a consequence of its conversion to active corticosterone by 11β-HSD1 expressed in the neurons, an effect lost in the presence of an 11β-HSD inhibitor (Rajan et al., 1996). The importance of such regeneration of GCs within cells in vivo was shown in mice homozygous for targeted disruption of the 11β-HSD1 gene (Kotelevtsev et al., 1997). 11β-HSD1 appears to be the only 11β-reductase, at least in mice, since knockout (KO) animals cannot convert 11-DHC to active corticosterone and showed evidence of reduced tissue GC actions (e.g., resist hyperglycaemia induced by obesity or stress) (Kotelevtsev et al., 1997). So are there brain effects of 11β-HSD1 deficiency?
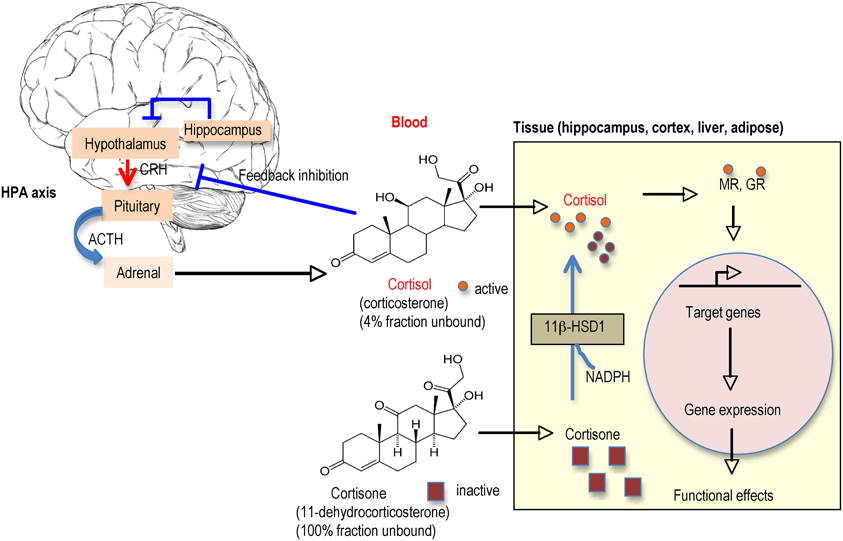
Figure 4. Regulation of glucocorticoid (GC) levels in blood and tissues. Circulating GC levels are tightly regulated by negative feedback inhibition at the hypothalamic-pituitary-adrenal (HPA) axis. Active GCs (cortisol in humans, corticosterone in rodents) circulate mostly bound to CBG (corticosteroid binding globulin) such that only ~4% are free to enter tissues. The inactive GCs (cortisone, 11-dehydrocorticosterone) the products of 11β-HSD2 metabolism mainly in kidney, circulates unbound to plasma proteins so all are available to enter tissues. Once inside the cell, there is another level of control by the presence of 11β-HSD1 (found in specific cells in the adult brain such as in hippocampus and cortex and other tissues such as liver and adipose). 11β-HSD1 regenerates active GCs from their inactive forms thereby effectively amplifying intracellular GC levels before they bind to MR and/or GR (depending on brain region). The activated receptors then translocate into the nucleus to activate the transcription of target genes.
Expression of 11β-HSD1 in brain sites (prefrontal cortex, hippocampus, hypothalamus) and pituitary that underpin negative feedback actions of GCs suggests that this enzyme may influence HPA axis activity. In order to maintain tissue GC levels in feedback sites that normally express 11β-HSD1, higher levels of plasma GCs would be predicted to result as a consequence of loss of local production of active GCs at these sites. Indeed mice lacking 11β-HSD1 on the 129/MF1 strain showed evidence of reduced HPA axis feedback sensitivity (elevated nadir levels of plasma corticosterone, enlarged adrenal glands, and exaggerated GC response to an acute stressor) (Harris et al., 2001). However, the effects of 11ß-HSD1 deficiency on HPA axis activity were lost when the mice were bred onto another genetic background strain. Thus, 11β-HSD1 KO mice congenic on a C57BL/6J background show normal nadir plasma corticosterone levels and efficient negative feedback regulation thought to be due to compensatory increased GR expression in the hippocampus and paraventricular nucleus of the hypothalamus (Carter et al., 2009). Activation of the HPA axis is therefore not an inevitable consequence of 11β-HSD1 deficiency or inhibition. The genetic background appears crucial in governing the HPA axis response to 11β-HSD1 deficiency or inhibition.
If 11β-HSD1 locally regenerates active GCs thus amplifying GC action within specific cells in vivo, this direction of enzyme activity would be anticipated to impair spatial memory in the aging brain when there is the additional contribution of increased “free” unbound GCs from the periphery in individuals with elevated blood GC levels. Indeed, a lack of the enzyme in the aged brain appears beneficial to memory processes as shown in 11β-HSD1 KO mice (Yau et al., 2001). Thus, aged 11β-HSD1 KO mice congenic on the 129 strain, despite modestly elevated plasma corticosterone levels throughout life, show ameliorated GC-related learning impairments in the watermaze (Yau et al., 2001). This is the opposite of what would be anticipated if only circulating corticosterone levels were taken into consideration. Hippocampal tissue corticosterone levels measured ex-vivo were significantly lower in aged 11β-HSD1 KO mice than aged controls (Yau et al., 2001, 2011), supporting the important role the enzyme plays in determining the levels of GCs within cells and thus their action.
Different genetic background mouse strains can exhibit marked differences in learning and memory (Owen et al., 1997). The original 11β-HSD1 KO mice generated on the 129 Ola background learnt a cued version of a watermaze task (Yau et al., 2001), but had difficulty learning the classical task. Subsequently, aged 11β-HSD1 KO mice congenic on the C57BL/6J background, the strain of choice in many behavioral tests, also showed an improved cognitive phenotype resisting the spatial memory impairments observed in many aged control mice, this time observed in the standard reference memory watermaze and also Y-maze spatial recognition tasks (Yau et al., 2007). Even a 50% reduction in 11β-HSD1 in heterozygous 11β-HSD1 KOs is enough to prevent age-related spatial memory impairments (Sooy et al., 2010).
Aged C57BL/6J mice showed increased 11β-HSD1 expression in the cortex (layer V) and CA3 cells of the hippocampus compared to young controls. Moreover, hippocampal and cortical 11β-HSD1 mRNA levels correlate with impaired spatial learning in the watermaze (Holmes et al., 2010). The selective increase of 11β-HSD1 in the CA3 subregion of the memory-impaired hippocampus may be of functional significance since it is the CA3 cells that undergo dendritic atrophy following chronic restraint stress or corticosterone injections (Magarinos and McEwen, 1995), both of which impair spatial memory (Luine et al., 1994; Conrad et al., 1996; Wright and Conrad, 2005; Hoffman et al., 2011). CA3 appears crucial for memory acquisition and consolidation in the watermaze (Florian and Roullet, 2004). Furthermore, CA3 cells selectively fail to rapidly encode new spatial information in memory-impaired aged rats (Wilson et al., 2005). This suggests that the implied increase of GC levels driven by variable overexpression of 11β-HSD1 in CA3 of aged mice may be a major contributor to the spatial memory deficits with aging. The mechanisms underlying the upregulation of 11β-HSD1 in CA3 and cortex of aged mice are unknown. GCs and stress elevate hippocampal 11β-HSD1 in young animals affording a possible feed-forward system to amplify GC action (Low et al., 1994). Consistent with increased 11β-HSD1 levels in the hippocampus impairing memory with aging, transgenic mice overexpressing 11β-HSD1 in the forebrain under the CAMIIK promoter resulting in 50% increase in the hippocampus, developed premature memory impairments with deficits in hippocampus-dependent learning tasks (watermaze spatial reference memory and passive avoidance memory) at 18 months (Holmes et al., 2010).
In the absence of hippocampal neuron loss, age-related spatial learning deficits in rats may be the result of more subtle changes in synaptic structure or function (Rapp and Gallagher, 1996; Smith et al., 2000). Consistent with this idea, impaired hippocampal synaptic plasticity, specifically LTP in aged rats relates to individual differences in spatial learning ability (Deupree et al., 1991; Bach et al., 1999). Indeed, LTP induced in the CA1 region using theta-frequency stimulation (5 Hz) was selectively impaired in hippocampal slices from a subpopulation of aged rats that had previously shown poor spatial learning in the water maze (Tombaugh et al., 2002). Elevated corticosterone levels (presumably occupying both MR and GR) also impairs Primed Burst Potentiation (PBP), a low threshold form of LTP, in the hippocampal CA1 in the rat, while low levels of corticosterone (that would occupy mostly MRs) facilitates PBP (Diamond et al., 1992). Although the involvement of other hormones and/or brain regions may also play a role, high levels of corticosterone directly impairs hippocampal synaptic potentiation (Alfarez et al., 2002). In addition, induction of long-term depression (LTD), which in contrast to LTP weakens rather than strengthens synaptic contacts by repeated stimulation (Bear and Malenka, 1994), is enhanced by high corticosterone levels (Coussens et al., 1997) and during aging (Norris et al., 1996, 1998).
Primed burst stimulation induces lower potentiation in the hippocampus of young animals compared to the LTP induction protocol (Diamond et al., 1994, 1996; Alfarez et al., 2002), but few reports have shown this reliably in aged animals (Moore et al., 1993). We found very little PBP (a single 200 Hz stimulus followed 180 ms later by a burst of four stimuli at 200 Hz) in hippocampal slices from either 25 months old 11β-HSD1 KO or age matched controls. However, LTP induced with a single 100 Hz tetanus, was increased in hippocampal slices from aged 11β-HSD1 KO mice compared to aged-matched C57BL/6J controls (Yau et al., 2007). Thus, an increase in LTP in the hippocampus of aged 11β-HSD1 KO mice may, in part, underlie their retention of spatial memory with age.
Adult neurogenesis, the generation of new neurons via mitotic cell division, occurs in the dendate gyrus of the hippocampus throughout life. Although the full functional significance of these new neurons is not fully understood, there is increasing evidence to support the notion that these newborn neurons can mature, form synapses, integrate with the local circuitry, and are involved in hippocampus-dependent learning (Lemaire et al., 2012; Marin-Burgin and Schinder, 2012). Of relevance to aging, high levels of GCs or stress reduce neurogenesis (Czeh et al., 2002; Wong and Herbert, 2006). Indeed, neurogenesis is substantially reduced with aging in rodents (Seki and Arai, 1995; Kuhn et al., 1996) and this may be in part related to increased GC levels since within a cohort of aged rats, those with the highest GC levels had the lowest levels of neurogenesis (Montaron et al., 2006). Furthermore, when aged rats were subdivided into aged unimpaired and aged impaired according to their spatial learning abilities (the top and bottom 30% of the population), cell proliferation in the granule cell layer of the dentate gyrus correlated with spatial memory performances (Drapeau et al., 2003). However, when neurogenesis was examined in the aged 11β-HSD1 KO, there was no significant difference compared to aged C57BL/6J controls, although an increase in neurogenesis was observed in young 11β-HSD1 KO mice (Yau et al., 2007). This suggests that the maintained spatial memory in the aged 11β-HSD1 KO mice is not a consequence of increased neurogenesis and that reduced intrahippocampal GCs is insufficient to overcome the other factors linked with aging (e.g., decreased serotonin) that regulate neurogenesis.
Since aged mice with complete or partial 11β-HSD1 deficiency throughout life are protected from spatial memory impairments (Yau et al., 2007; Sooy et al., 2010), inhibiting 11β-HSD1 activity might benefit cognitive function in the aging brain. The big question is can short-term pharmacological inhibition of 11β-HSD1 have memory-enhancing effects in aged rodents and humans? In vivo studies in humans are hampered by the non-selectivity of the originally available liquorice-based inhibitors. Carbenoxolone, an old drug formerly used clinically to treat peptic ulcers, inhibits both 11β-HSD1 and 11β-HSD2. This may not matter in the adult brain as the predominant isozyme is 11β-HSD1. Initial small exploratory studies, albeit randomized, double-blind, and placebo-controlled in healthy elderly men and middle-aged patients with type 2 diabetes (52–75 years) showed that carbenoxolone improved aspects of cognitive function (verbal fluency and verbal memory) after 4–6 weeks treatment (Sandeep et al., 2004). Note that amilioride was also given to prevent renal mineralocorticoid excess and hence hypertension. This gave the first indication that inhibition of 11β-HSD1 (assuming the effects were centrally mediated) may be a promising new approach to prevent/ameliorate cognitive decline in humans.
Selective 11β-HSD1 inhibitors that can cross the blood brain barrier have recently been developed (Webster et al., 2007). Two weeks peripheral treatment with a CNS active selective 11β-HSD1 inhibitor (UE1961) in aged C57BL/6J mice improved spatial memory in the Y-maze compared to vehicle treated age-matched controls (Sooy et al., 2010). Moreover, intracerebroventricular administration of another selective 11β-HSD1 inhibitor (UE2316) for 2 weeks also reversed spatial memory impairments in aged C57BL/6J mice confirming mediation by brain 11β-HSD1 inhibition (Yau et al., unpublished). Thus, spatial memory impairments in aged mice are not always a consequence of irreversible brain structural changes and the effects of the inhibitor are most likely the consequence of reduced intracellular GC levels during spatial learning and recall. Previous studies have shown that it is the consequence of increased GCs as a result of hippocampal structural changes (e.g., chronic stress-induced dendritic atrophy) impairing HPA axis feedback that has the more important influence on spatial memory performance since such impairments can be prevented on the day of testing by blocking GC synthesis (Roozendaal et al., 2001; Wright et al., 2006).
Depending on the duration of treatment, selective 11β-HSD1 inhibitors may result in resistance of target tissues to GCs, including HPA axis regulatory centers of the brain and pituitary. This could lead to compensatory activation of the HPA axis in an attempt to compensate for the GC deficiency in negative feedback loci expressing 11β-HSD1 in a similar manner to that observed in 11β-HSD1 KO mice on the 129 genetic background (Harris et al., 2001). A hyperactive HPA axis would lead not only to excess plasma cortisol, but perhaps also to excess mineralocorticoid precursors (corticosterone and deoxycorticosterone) and androgens, all of which may manifest as unwanted side effects, such as hypertension and hirsutism/hyperandrogenization in women. The degree of HPA axis activation may depend on the genetic background of the individual, as in mice, as well as the levels of 11β-HSD1 in these tissues, which may differ under various environmental conditions (e.g., with stress, diet, aging). There is also the question of whether inhibition of 11β-HSD1 will affect other types of memory other than spatial memory in cognitively impaired aged individuals. In particular, emotional and fear associated memories, involving the basolateral amygdala, are known to be enhanced with increased GCs (Roozendaal and McGaugh, 2011). Hence, 11β-HSD1 inhibitors may compromise the strength of such memories but whether this is the case remains to be determined.
Impaired spatial memory in aged C57BL/6J mice correlates with higher plasma GCs which are thought to activate the lower affinity GR. Indeed central blockade of GR (but not MR) for 2 weeks reversed the impaired spatial memory in aged C57BL/6J mice in the Y-maze (Yau et al., 2011). This again suggests that the impaired memory in aged C57BL/6J mice is not a direct consequence of irreversible structural changes. The improved spatial memory in aged 11β-HSD1 KO mice has been proposed to occur via reduced intracellular GC levels altering the balance of receptor activation in favor of MR activation. In support, central blockade of MR but not GR in aged 11β-HSD1 KO mice reversed their improved spatial memory phenotype such that they were now impaired in the Y-maze (Yau et al., 2011). Therefore, decreasing GR/increasing MR activation appears effective at preventing spatial memory impairments even in already aged mice. Reducing GC action by altering the receptor balance from predominant GR activation to predominant MR activation may be achieved by the use of selective 11β-HSD1 inhibitors or GR antagonists. Long-term treatment with GR antagonists in humans, however, may potentially cause compensatory activation of the HPA axis to overcome the blockade thus producing generalized GC resistance (Bamberger and Chrousos, 1995). While the use of selective 11β-HSD1 inhibitors causes compensatory activation of the HPA axis simply to accommodate the reduced regeneration of GCs, elevation of cortisol levels was not observed with carbenoxolone in elderly humans (Sandeep et al., 2004) or with selective 11β-HSD1 inhibitors in clinical trials (Rosenstock et al., 2010). Similarly, corticosterone levels were not increased in aged C57BL/6J mice treated short-term with a selective 11β-HSD1 inhibitor (Sooy et al., 2010).
GCs play a role in the regulation of peripheral glucose mobilization and metabolism. Much less is known about the effects of GCs on glucose metabolism in brain tissues. GCs inhibit glucose utilization in the hippocampus (Sapolsky, 1986). Adrenalectomy decreases serum glucose levels and increases hippocampal glucose utilization (Kadekaro et al., 1988). While exogenously administered GCs adversely affect human memory consolidation and recall (Newcomer et al., 1994), glucose improves memory performance in normal elderly individuals (Hall et al., 1989) and in subjects with probable Alzheimer's disease (Craft et al., 1993). Studies in rodents suggest that hyperglycemia and high-fat diets adversely affect hippocampal function (Kamal et al., 2000; Molteni et al., 2002). In humans, hyperglycemia associate with decline in cognitive function with aging (Convit et al., 2003). Since 11β-HSD1 KO mice lack the enzyme in all tissues, it is possible that the cognitive protection in aged 11β-HSD1 KO mice may reflect, in part, their improved metabolic profiles (Kotelevtsev et al., 1997; Morton et al., 2001), rather than solely the direct effects of enzyme deficiency in the brain. However, aged 11β-HSD1 KO mice show no significant difference in glucose tolerance compared to age-matched controls (Yau et al., 2007). Moreover, the 11β-HSD inhibitor carbenoxolone improved cognitive function in aged humans without altering plasma glucose levels (Sandeep et al., 2004). Thus, the implication is that direct CNS effects of 11β-HSD1 deficiency or inhibition are more likely responsible for the observed improved cognitive function.
To further our understanding of how GC action affects age-related memory performance, it will be important to determine the downstream genes/pathways activated by hippocampal MR and GRs that leads to the impaired and improved spatial memory of aged mice with and without 11β-HSD1. Several studies have examined hippocampal gene expression changes associated with age-related cognitive decline in rats and mice (Burger et al., 2007; Rowe et al., 2007; Pawlowski et al., 2009). Using microarrays, many hippocampal genes were found to be altered with aging regardless of cognitive status. Down-regulated genes in rat hippocampus are involved in mitochrondrial function, cell signaling, neural plasticity, and synaptic function whereas up-regulated genes underpin inflammation, glial structure, cholesterol transport, and lipid/protein degradation. Genes selectively downregulated in spatial memory impaired aged rats characterized in the watermaze and culled soon after training include a number of immediate early genes (IEGs) such as Arc and Zif268 (also called Egr-1 and NGFI-A) and glucose ultilization/insulin signaling genes such as Irs1, GcK, and Insr (Rowe et al., 2007). Zif268, is thought to activate the transcription of genes essential for hippocampus-dependent long-term memory (Jones et al., 2001). Reduced resting levels of Zif268 mRNA in the CA1 hippocampal area of aged rats with impaired spatial learning have been observed by microarray analysis and in situ hybridization histochemistry (Yau et al., 1996; Blalock et al., 2003). Whether the selective alterations in gene transcription in cognitively impaired but not unimpaired aged animals is related to their HPA activity and hence circulating GC levels is not known. Hippocampal gene expression microarrays and proteomic approaches in behaviorally defined aged mice including aged 11β-HSD1 KO mice and animals given selective 11β-HSD1 inhibitors may indicate new drug targets for the enhancement of memory in the aged individual.
Recent studies have revealed additional substrates for 11β-HSD1. These include additional functions in the metabolism of neurosteroids such as dehydroepiandrosterone (DHEA) and pregnenolone, metabolism of 7-oxysterols, as well as in the detoxification of various xenobiotics (Odermatt and Nashev, 2010). Neurosteroids have long been implicated to play an important role in the modulation of spatial learning and memory processes (Vallee et al., 2001). Circulating levels of DHEA derive from the adrenal cortex and decline significantly with aging in humans and this parallels cognitive decline (Orentreich et al., 1992). In rodents there is very little DHEA in brain because they lack adrenal expression of the enzyme cyp17α that converts pregnenolone to DHEA (Le Goascogne et al., 1991) though there may be limited local CNS synthesis (Liu et al., 2009). However, high levels of its precursor pregnenolone have been measured in the hippocampus of young rats and this declines with aging correlating with spatial memory impairments (Vallee et al., 1997). Pregnenolone, DHEA, and related 3β-hydroxysteroids are 7α-hydroxylated in brain by the cytochrome P450-7B1 (CYP7B1) enzyme into their bioactive steroid metabolites (Rose et al., 1997). High levels of CYP7B1 is expressed in the hippocampus and enzyme activity is selectively decreased in aged rats with impaired spatial memory but not in aged cognitively unimpaired rats (Yau et al., 2006). In the memory-impaired aged rats, central administration of an active product, 7α-hydroxypregnenolone, reversed the spatial memory deficits and improved memory in a radial arm watermaze memory task thus effectively overcoming reduced CYP7B1 activity in the aged brain (Yau et al., 2006). It has recently been proposed that the products of CYP7B1, 7α-hydroxypregnenolone, and 7α-hydroxyDHEA, may exert anti-GC effects in target tissues by competing with 11-keto GCs for access to 11β-HSD1, thus attenuating regeneration of active GCs (Muller et al., 2006a). Thus, 11β-HSD1, in addition to its known role in reactivating GCs within target cells, has also been shown to carry out the inter-conversion of 7α-hydroxyDHEA into 7β-hydroxyDHEA, at least in vitro (Muller et al., 2006b) (Figure 5). Enzyme kinetic data from yeast-expressed human 11β-HSD1 implies that the 7-hydroxysteroid substrates are preferred to cortisone. Hence in tissues, 7-hydroxysteroid substrates may act like endogenous “inhibitors” of 11β-HSD1, reducing their regeneration of active GCs. This adds another level of potential fine tuning of GC action within specific target CNS cells but whether this occurs in vivo under normal or pathological conditions is not known.
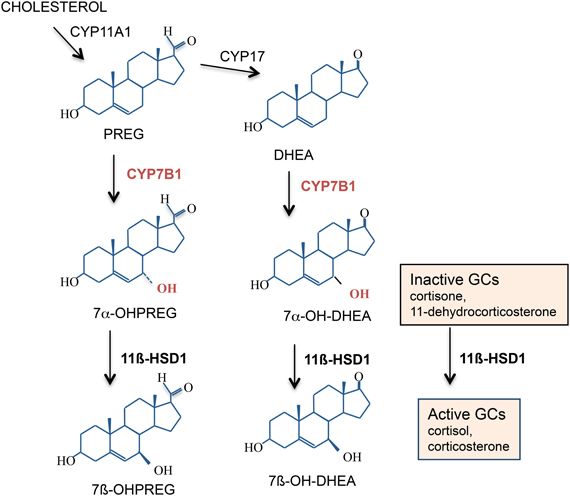
Figure 5. 7α-Hydroxylated steroids are also substrates for 11β-HSD1. Both 11β-HSD1 and CYP7B1 are expressed in the rodent and human hippocampus. CYP7B1 7α-hydroxylates pregnenolone (PREG) and dehydroepiandrosterone (DHEA) into their more active 7α-hydroxylated metabolites which are also substrates for 11β-HSD1. If levels of these 7α-hydroxylated metabolites within specific cells become high enough, they may compete with the inactive GCs for 11β-HSD1 and effectively act as endogenous “inhibitors” of 11β-HSD1 amplification of intracellular GC action.
The local amplification of GC action in the brain by 11β-HSD1 plays a pivotal role in the emergence of spatial learning impairments with aging. Evidence over the past decade has confirmed that while elevated plasma GCs correlate with impaired spatial learning with aging, it is the level of active GCs within specific brain cells regulated by 11β-HSD1 that appears essential for the control of spatial memory in the aged animal. Importantly, whereas treatments to lower blood GC levels need to be maintained long-term to ameliorate spatial memory deficits in aged rats, lowering GC levels inside cells expressing the enzyme by selective 11β-HSD1 inhibitors requires only short-term treatment and can reverse spatial memory deficits in already aged mice. Whether this is the case in humans remains to be tested. The data also show that spatial memory deficits in aged rodents are not necessarily irreversible due to structural changes but appear to be regulated in the short-term by intracellular GC actions activating MR and GR to regulate the transcription of target genes that influence memory formation. Future investigations to determine the mechanisms whereby 11β-HSD1 in the brain is upregulated by aging and the downstream pathways following predominant brain MR activation in aged 11β-HSD1 KO mice will be crucial in the understanding of how GCs maintain or impair hippocampus-dependent spatial learning and memory with aging. Whether or not this approach is of utility in pathological brain aging, such as Alzheimer's disease and other dementias, remains to be determined, but of substantial interest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors' work was supported by project grants from The Wellcome Trust, BBSRC, MRC, and fellowships (Joyce L. W. Yau) from Alzheimer's Research UK and RCUK. The authors are members of The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, part of the cross council Lifelong Health and Wellbeing Initiative. Funding from the BBSRC, EPSRC, ESRC, and MRC is gratefully acknowledged.
Alfarez, D. N., Wiegert, O., Joels, M., and Krugers, H. J. (2002). Corticosterone and stress reduce synaptic potentiation in mouse hippocampal slices with mild stimulation. Neuroscience 115, 1119–1126.
Bach, M. E., Barad, M., Son, H., Zhuo, M., Lu, Y. F., Shih, R., Mansuy, I., Hawkins, R. D., and Kandel, E. R. (1999). Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5280–5285.
Bamberger, C. M., and Chrousos, G. P. (1995). The glucocorticoid receptor and RU 486 in man. Ann. N.Y. Acad. Sci. 761, 296–310.
Barden, N. (1996). Modulation of glucocorticoid receptor gene expression by antidepressant drugs. Pharmacopsychiatry 29, 12–22.
Barnes, C. A., and McNaughton, B. L. (1985). An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behav. Neurosci. 99, 1040–1048.
Bear, M. F., and Malenka, R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 4, 389–399.
Bergado, J. A., Almaguer, W., Rojas, Y., Capdevila, V., and Frey, J. U. (2011). Spatial and emotional memory in aged rats: a behavioral-statistical analysis. Neuroscience 172, 256–269.
Blalock, E. M., Chen, K. C., Sharrow, K., Herman, J. P., Porter, N. M., Foster, T. C., and Landfield, P. W. (2003). Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci. 23, 3807–3819.
Broster, L. S., Blonder, L. X., and Jiang, Y. (2012). Does emotional memory enhancement assist the memory-impaired? Front. Aging Neurosci. 4:2. doi: 10.3389/fnagi.2012.00002
Burger, C., Lopez, M. C., Feller, J. A., Baker, H. V., Muzyczka, N., and Mandel, R. J. (2007). Changes in transcription within the CA1 field of the hippocampus are associated with age-related spatial learning impairments. Neurobiol. Learn. Mem. 87, 21–41.
Carter, R. N., Paterson, J. M., Tworowska, U., Stenvers, D. J., Mullins, J. J., Seckl, J. R., and Holmes, M. C. (2009). Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11beta-HSD1 is strain-dependent. J. Neuroendocrinol. 21, 879–887.
Cazakoff, B. N., Johnson, K. J., and Howland, J. G. (2010). Converging effects of acute stress on spatial and recognition memory in rodents: a review of recent behavioural and pharmacological findings. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 733–741.
Charles, S. T., Mather, M., and Carstensen, L. L. (2003). Aging and emotional memory: the forgettable nature of negative images for older adults. J. Exp. Psychol. Gen. 132, 310–324.
Comblain, C., D'Argembeau, A., Van der Linden, M., and Aldenhoff, L. (2004). The effect of ageing on the recollection of emotional and neutral pictures. Memory 12, 673–684.
Conrad, C. D., Galea, L. A., Kuroda, Y., and McEwen, B. S. (1996). Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav. Neurosci. 110, 1321–1334.
Convit, A., Wolf, O. T., Tarshish, C., and de Leon, M. J. (2003). Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. Proc. Natl. Acad. Sci. U.S.A. 100, 2019–2022.
Coussens, C. M., Kerr, D. S., and Abraham, W. C. (1997). Glucocorticoid receptor activation lowers the threshold for NMDA-receptor-dependent homosynaptic long-term depression in the hippocampus through activation of voltage-dependent calcium channels. J. Neurophysiol. 78, 1–9.
Craft, S., Dagogo-Jack, S. E., Wiethop, B. V., Murphy, C., Nevins, R. T., Fleischman, S., Rice, V., Newcomer, J. W., and Cryer, P. E. (1993). Effects of hyperglycaemia on memory and hormone levels in dementia of the Alzheimer type: a longitudinal study. Behav. Neurosci. 107, 926–940.
Czeh, B., Welt, T., Fischer, A. K., Erhardt, A., Schmitt, W., Muller, M. B., Toschi, N., Fuchs, E., and Keck, M. E. (2002). Chronic psychosocial stress and concomitant repetitive transcranial magnetic stimulation: effects on stress hormone levels and adult hippocampal neurogenesis. Biol. Psychiatry 52, 1057–1065.
Datson, N. A., van der Perk, J., de Kloet, E. R., and Vreugdenhil, E. (2001). Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur. J. Neurosci. 14, 675–689.
de Kloet, E. R., Karst, H., and Joels, M. (2008). Corticosteroid hormones in the central stress response: quick-and-slow. Front. Neuroendocrinol. 29, 268–272.
de Kloet, E. R., Oitzl, M. S., and Joels, M. (1999). Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 22, 422–426.
De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S., and Joels, M. (1998). Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 19, 269–301.
De Leon, M. J., McRae, T., Tsai, J. R., George, A. E., Marcus, D. L., Freedman, M., Wolf, A. P., and McEwen, B. S. (1988). Abnormal cortisol response in Alzheimer's disease linked to hippocampal atrophy. Lancet 2, 391–392.
Deupree, D. L., Turner, D. A., and Watters, C. L. (1991). Spatial performance correlates with in vitro potentiation in young and aged Fischer 344 rats. Brain Res. 554, 1–9.
Diamond, D. M., Bennett, M. C., Fleshner, M., and Rose, G. M. (1992). Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2, 421–430.
Diamond, D. M., Fleshner, M., Ingersoll, N., and Rose, G. M. (1996). Psychological stress impairs spatial working memory: relevance to electrophysiological studies of hippocampal function. Behav. Neurosci. 110, 661–672.
Diamond, D. M., Fleshner, M., and Rose, G. M. (1994). Psychological stress repeatedly blocks hippocampal primed burst potentiation in behaving rats. Behav. Brain Res. 62, 1–9.
Drapeau, E., Mayo, W., Aurousseau, C., Le Moal, M., Piazza, P. V., and Abrous, D. N. (2003). Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 14385–14390.
Florian, C., and Roullet, P. (2004). Hippocampal CA3-region is crucial for acquisition and memory consolidation in Morris water maze task in mice. Behav. Brain Res. 154, 365–374.
Gage, F. H., Dunnett, S. B., and Bjorklund, A. (1989). Age-related impairments in spatial memory are independent of those in sensorimotor skills. Neurobiol. Aging 10, 347–352.
Gallagher, M., Burwell, R., and Burchinal, M. (1993). Severity of spatial learning impairment in aging: development of a learning index for performance in the Morris water maze. Behav. Neurosci. 107, 618–626.
Gould, T. J., and Feiro, O. R. (2005). Age-related deficits in the retention of memories for cued fear conditioning are reversed by galantamine treatment. Behav. Brain Res. 165, 160–171.
Groeneweg, F. L., Karst, H., de Kloet, E. R., and Joels, M. (2011). Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 209, 153–167.
Hall, J. L., Gonder-Frederick, L. A., Chewning, W. W., Silveira, J., and Gold, P. E. (1989). Glucose enhancement of performance on memory tests in young and aged humans. Neuropsychologia 27, 1129–1138.
Harris, H. J., Kotelevtsev, Y., Mullins, J. J., Seckl, J. R., and Holmes, M. C. (2001). Intracellular regeneration of glucocorticoids by 11ß-HSD-1 plays a key role in regulation of the HPA axis: analysis of 11ß-HSD-1 deficient mice. Endocrinology 142, 114–120.
Hayashi, R., Wada, H., Ito, K., and Adcock, I. M. (2004). Effects of glucocorticoids on gene transcription. Eur. J. Pharmacol. 500, 51–62.
Heck, S., Kullmann, M., Gast, A., Ponta, H., Rahmsdorf, H. J., Herrlich, P., and Cato, A. C. (1994). A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 13, 4087–4095.
Herbert, J., Goodyer, I. M., Grossman, A. B., Hastings, M. H., de Kloet, E. R., Lightman, S. L., Lupien, S. J., Roozendaal, B., and Seckl, J. R. (2006). Do corticosteroids damage the brain? J. Neuroendocrinol. 18, 393–411.
Hoffman, A. N., Krigbaum, A., Ortiz, J. B., Mika, A., Hutchinson, K. M., Bimonte-Nelson, H. A., and Conrad, C. D. (2011). Recovery after chronic stress within spatial reference and working memory domains: correspondence with hippocampal morphology. Eur. J. Neurosci. 34, 1023–1030.
Holmes, M. C., Carter, R. N., Noble, J., Chitnis, S., Dutia, A., Paterson, J. M., Mullins, J. J., Seckl, J. R., and Yau, J. L. (2010). 11beta-hydroxysteroid dehydrogenase type 1 expression is increased in the aged mouse hippocampus and parietal cortex and causes memory impairments. J. Neurosci. 30, 6916–6920.
Iidaka, T., Okada, T., Murata, T., Omori, M., Kosaka, H., Sadato, N., and Yonekura, Y. (2002). Age-related differences in the medial temporal lobe responses to emotional faces as revealed by fMRI. Hippocampus 12, 352–362.
Ingram, D. K., London, E. D., and Goodrick, C. L. (1981). Age and neurochemical correlates of radial maze performance in rats. Neurobiol. Aging 2, 41–47.
Issa, A. M., Rowe, W., Gauthier, S., and Meaney, M. J. (1990). Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J. Neurosci. 10, 3247–3254.
Jacobson, L., and Sapolsky, R. (1991). The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenal axis. Endocr. Rev. 12, 118–134.
Joels, M., Pu, Z., Wiegert, O., Oitzl, M. S., and Krugers, H. J. (2006). Learning under stress: how does it work? Trends Cogn. Sci. 10, 152–158.
Jones, M. W., Errington, M. L., French, P. J., Fine, A., Bliss, T. V., Garel, S., Charnay, P., Bozon, B., Laroche, S., and Davis, S. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296.
Kadekaro, M., Ito, M., and Gross, P. M. (1988). Local cerebral glucose utilization is increased in acutely adrenalectomized rats. Neuroendocrinology 47, 329–334.
Kamal, A., Biessels, G. J., Duis, S. E., and Gispen, W. H. (2000). Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia 43, 500–506.
Karlamangla, A. S., Singer, B. H., Chodosh, J., McEwen, B. S., and Seeman, T. E. (2005). Urinary cortisol excretion as a predictor of incident cognitive impairment. Neurobiol. Aging 26(Suppl. 1), 80–84.
Karst, H., Berger, S., Turiault, M., Tronche, F., Schutz, G., and Joels, M. (2005). Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Nat. Acad. Sci. U.S.A. 102, 19204–19207.
Kim, J. J., and Diamond, D. M. (2002). The stressed hippocampus, synaptic plasticity and lost memories. Nat. Rev. 3, 453–462.
Kim, J. J., Lee, H. J., Han, J. S., and Packard, M. G. (2001). Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J. Neurosci. 21, 5222–5228.
Kim, J. J., Lee, H. J., Welday, A. C., Song, E., Cho, J., Sharp, P. E., Jung, M. W., and Blair, H. T. (2007). Stress-induced alterations in hippocampal plasticity, place cells, and spatial memory. Proc. Nat. Acad. Sci. U.S.A. 104, 18297–18302.
Komatsuzaki, Y., Murakami, G., Tsurugizawa, T., Mukai, H., Tanabe, N., Mitsuhashi, K., Kawata, M., Kimoto, T., Ooishi, Y., and Kawato, S. (2005). Rapid spinogenesis of pyramidal neurons induced by activation of glucocorticoid receptors in adult male rat hippocampus. Biochem. Biophys. Res. Commun. 335, 1002–1007.
Kotelevtsev, Y., Holmes, M. C., Burchell, A., Houston, P. M., Schmoll, D., Jamieson, P., Best, R., Brown, R., Edwards, C. R. W., Seckl, J. R., and Mullins, J. J. (1997). 11ß-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 94, 14924–14929.
Kuhn, H. G., Dickinson-Anson, H., and Gage, F. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033.
Lai, M., McCormick, J. A., Chapman, K. E., Kelly, P. A., Seckl, J. R., and Yau, J. L. (2003). Differential regulation of corticosteroid receptors by monoamine neurotransmitters and antidepressant drugs in primary hippocampal culture. Neuroscience 118, 975–984.
Landfield, P., Waymire, J., and Lynch, G. (1978). Hippocampal aging and adrenocorticoids: a quantitative correlation. Science 202, 1098–1102.
Landfield, P. W., Baskin, R. K., and Pitler, T. A. (1981). Brain aging correlates: retardation by hormonal-pharmacological treatments. Science 214, 581–584.
Le Goascogne, C., Sananes, N., Gouezou, M., Takemori, S., Kominami, S., Baulieu, E. E., and Robel, P. (1991). Immunoreactive cytochrome P-450(17 alpha) in rat and guinea-pig gonads, adrenal glands and brain. J. Reprod. Fertil. 93, 609–622.
Lemaire, V., Tronel, S., Montaron, M. F., Fabre, A., Dugast, E., and Abrous, D. N. (2012). Long-lasting plasticity of hippocampal adult-born neurons. J. Neurosci. 32, 3101–3108.
Liu, Y., Pocivavsek, A., and Papadopoulos, V. (2009). Dehydroepiandrosterone formation is independent of cytochrome P450 17alpha-hydroxylase/17, 20 lyase activity in the mouse brain. J. Steroid Biochem. Mol. Biol. 115, 86–90.
Low, S. C., Moisan, M.-P., Edwards, C. R. W., and Seckl, J. R. (1994). Glucocorticoids and chronic stress up-regulate 11ß-hydroxysteroid dehydrogenase activity and gene expression in the hippocampus. J. Neuroendocrinol. 6, 285–290.
Luine, V., Villegas, M., Martinez, C., and McEwen, B. S. (1994). Repeated stress causes reversible impairments of spatial memory performance. Brain Res. 639, 167–170.
Lupien, S., Lecours, A. R., Lussier, I., Schwartz, G., Nair, N., and Meaney, M. J. (1994). Basal cortisol-levels and cognitive deficits in human aging. J. Neurosci. 14, 2893–2903.
Lupien, S., Lecours, A. R., Schwartz, G., Sharma, S., Hauger, R. L., Meaney, M. J., and Nair, N. (1996). Longitudinal-study of basal cortisol-levels in healthy elderly subjects - evidence for subgroups. Neurobiol. Aging 17, 95–105.
Lupien, S. J., de Leon, M., de Santi, S., Convit, A., Tarshish, C., Nair, N. P. V., Thakur, M., McEwen, B. S., Hauger, R. L., and Meaney, M. J. (1998). Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1, 69–73.
Lupien, S. J., and McEwen, B. S. (1997). The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res. Rev. 24, 1–27.
MacLullich, A. M., Deary, I. J., Starr, J. M., Ferguson, K. J., Wardlaw, J. M., and Seckl, J. R. (2005). Plasma cortisol levels, brain volumes and cognition in healthy elderly men. Psychoneuroendocrinology 30, 505–515.
Magarinos, A. M., and McEwen, B. S. (1995). Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69, 89–98.
Marin-Burgin, A., and Schinder, A. F. (2012). Requirement of adult-born neurons for hippocampus-dependent learning. Behav. Brain Res. 227, 391–399.
May, C. P., Rahhal, T., Berry, E. M., and Leighton, E. A. (2005). Aging, source memory, and emotion. Psychol. Aging 20, 571–578.
McEwen, B. S., DeKloet, E. R., and Rostene, W. (1986). Adrenal steroid receptors and action in the nervous system. Physiol. Rev. 66, 1121–1188.
McEwen, B. S., de Leon, M. J., Lupien, S. J., and Meaney, M. J. (1999). Corticosteroids, the aging brain and cognition. Trends Endocrinol. Metab. 10, 92–96.
McEwen, B., Weiss, J., and Schwartz, L. (1968). Selective retention of corticosterone by limbic structures in the rat brain. Nature 220, 911–912.
Meaney, M. J., Aitken, D. H., van Berkel, C., Bhatnagar, S., and Sapolsky, R. M. (1988). Effect of neonatal handling on age-related impairments associated with the hippocampus. Science 239, 766–768.
Meaney, M. J., Diorio, J., Francis, D., Larocque, S., Odonnell, D., Smythe, J. W., Sharma, S., and Tannenbaum, B. (1994). Environmental-regulation of the development of glucocorticoid receptor systems in the rat forebrain - the role of serotonin. Ann. N.Y. Acad. Sci. 746, 260–274.
Meaney, M. J., Odonnell, D., Rowe, W., Tannenbaum, B., Steverman, A., Walker, M., Nair, N., and Lupien, S. (1995). Individual-differences in hypothalamic-pituitary-adrenal activity in later life and hippocampal aging. Exp. Gerontol. 30, 229–251.
Mitchell, A. J., and Dening, T. R. (1996). Depression-related cognitive impairment: possibilities for its pharmacological treatment. J. Affect. Disord. 36, 79–87.
Mitchell, J. B., Rowe, W., Boksa, P., and Meaney, M. (1990). Serotonin regulates type II corticosteroid receptor binding in hippocampal cell cultures. J. Neurosci. 10, 1745–1752.
Moisan, M.-P., Seckl, J. R., and Edwards, C. R. W. (1990). 11ß-hydroxysteroid dehydrogenase bioactivity and messenger RNA expression in rat forebrain: localization in hypothalamus, hippocampus and cortex. Endocrinology 127, 1450–1455.
Molteni, R., Barnard, R. J., Ying, Z., Roberts, C. K., and Gomez-Pinilla, F. (2002). A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112, 803–814.
Montaron, M. F., Drapeau, E., Dupret, D., Kitchener, P., Aurousseau, C., Le Moal, M., Piazza, P. V., and Abrous, D. N. (2006). Lifelong corticosterone level determines age-related decline in neurogenesis and memory. Neurobiol. Aging 27, 645–654.
Montkowski, A., Barden, N., Wotjak, C., Stec, I., Ganster, J., Meaney, M., Engelmann, M., Reul, J., Landgraf, R., and Holsboer, F. (1995). Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J. Neuroendocrinol. 7, 841–845.
Moore, C. I., Browning, M. D., and Rose, G. M. (1993). Hippocampal plasticity induced by primed burst, but not long-term potentiation, stimulation is impaired in area CA1 of aged Fischer 344 rats. Hippocampus 3, 57–66.
Morris, R. G. M., Garrud, P., Rawline, J. N. P., and O'Keefe, J. (1982). Place navigation is impaired in rats with hippocampal lesions. Nature 297, 681–683.
Morton, N. M., Holmes, M. C., Fievet, C., Staels, B., Tailleux, A., Mullins, J. J., and Seckl, J. R. (2001). Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 276, 41293–41300.
Moser, M. B., Moser, E. I., Forrest, E., Andersen, P., and Morris, R. (1995). Spatial learning with a minislab in the dorsal hippocampus. Proc. Nat. Acad. Sci. U.S.A. 92, 9697–9701.
Muller, C., Hennebert, O., and Morfin, R. (2006a). The native anti-glucocorticoid paradigm. J. Steroid Biochem. Mol. Biol. 100, 95–105.
Muller, C., Pompon, D., Urban, P., and Morfin, R. (2006b). Inter-conversion of 7alpha- and 7beta-hydroxy-dehydroepiandrosterone by the human 11beta-hydroxysteroid dehydrogenase type 1. J. Steroid Biochem. Mol. Biol. 99, 215–222.
Newcomer, J. W., Craft, S., Hershey, T., Askins, K., and Bardgett, M. E. (1994). Glucocorticoid-induced impairment in declarative memory performance in adult humans. J. Neurosci. 14, 2047–2053.
Norris, C. M., Korol, D. L., and Foster, T. C. (1996). Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J. Neurosci. 16, 5382–5392.
Norris, C. M., Halpain, S., and Foster, T. C. (1998). Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J. Neurosci. 18, 3171–3179.
Odermatt, A., and Nashev, L. G. (2010). The glucocorticoid-activating enzyme 11beta-hydroxysteroid dehydrogenase type 1 has broad substrate specificity: physiological and toxicological considerations. J. Steroid Biochem. Mol. Biol. 119, 1–13.
Ohta, R. J. (1981). Spatial problem solving: the response selection tendencies of young and elderly adults. Exp. Aging Res. 7, 81–84.
Oitzl, M. S., and de Kloet, E. R. (1992). Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav. Neurosci. 106, 62–71.
Okuda, S., Roozendaal, B., and McGaugh, J. L. (2004). Glucocorticoid effects on object recognition memory require training-associated emotional arousal. Proc. Nat. Acad. Sci. U.S.A. 101, 853–858.
Orentreich, N., Brind, J. L., Vogelman, J. H., Andres, R., and Baldwin, H. (1992). Long-term longitudinal measurements of plasma dehydroepiandrosterone sulphate in normal men. J. Clin. Endocrinol. Metab. 75, 1002–1004.
Owen, E. H., Logue, S. F., Rasmussen, D. L., and Wehner, J. M. (1997). Assessment of learning by the Morris water task and fear conditioning in inbred mouse strains and F1 hybrids: implications of genetic background for single gene mutations and quantitative trait loci analyses. Neuroscience 80, 1087–1099.
Pavlides, C., Kimura, A., Magarinos, A.-M., and McEwen, B. S. (1994). Type I adrenal steroid receptors prolong hippocampal long-term potentiation. Neuroreport 5, 2673–2677.
Pavlides, C., Ogawa, S., Kimura, A., and McEwen, B. S. (1996). Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res. 738, 229–235.
Pavlides, C., Watanabe, Y., Magarinos, A. M., and McEwen, B. S. (1995). Opposing roles of type I and type II adrenal steroid receptors in hippocampal long-term potentiation. Neuroscience 68, 387–394.
Pawlowski, T. L., Bellush, L. L., Wright, A. W., Walker, J. P., Colvin, R. A., and Huentelman, M. J. (2009). Hippocampal gene expression changes during age-related cognitive decline. Brain Res. 1256, 101–110.
Quervain, D. J.-F., Roozendaal, B., and McGaugh, J. L. (1998). Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394, 787–790.
Rajan, V., Edwards, C. R. W., and Seckl, J. R. (1996). 11ß-hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J. Neurosci. 16, 65–70.
Rapp, P. R., and Gallagher, M. (1996). Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. U.S.A. 93, 9926–9930.
Reul, J. M. H. M., and de Kloet, E. R. (1985). Two receptor systems for corticosterone in rat brain: microdissection and differential occupation. Endocrinology 117, 2505–2511.
Reul, J. M. H. M., Stec, I., Söder, M., and Holsboer, F. (1993). Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology 133, 312–320.
Robitsek, R. J., Fortin, N. J., Koh, M. T., Gallagher, M., and Eichenbaum, H. (2008). Cognitive aging: a common decline of episodic recollection and spatial memory in rats. J. Neurosci. 28, 8945–8954.
Roozendaal, B., and McGaugh, J. L. (1997). Basolateral amygdala lesions block the memory-enhancing effect of glucocorticoid administration in the dorsal hippocampus of rats. Eur. J. Neurosci. 9, 76–83.
Roozendaal, B., Okuda, S., Van der Zee, E. A., and McGaugh, J. L. (2006). Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 103, 6741–6746.
Roozendaal, B., Phillips, R. G., Power, A. E., Brooke, S. M., Sapolsky, R. M., and McGaugh, J. L. (2001). Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat. Neurosci. 4, 1169–1171.
Rose, K. A., Stapleton, G., Dott, K., Kieny, M. P., Best, R., Schwarz, M., Russell, D. W., Björkhem, I., Seckl, J., and Lathe, R. (1997). Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7a-hydroxy dehydroepiandrosterone and 7a-hydroxy pregnenolone. Proc. Natl. Acad. Sci. U.S.A. 94, 4925–4930.
Rosenstock, J., Banarer, S., Fonseca, V. A., Inzucchi, S. E., Sun, W., Yao, W., Hollis, G., Flores, R., Levy, R., Williams, W. V., Seckl, J. R., and Huber, R. (2010). The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33, 1516–1522.
Rowe, W. B., Blalock, E. M., Chen, K. C., Kadish, I., Wang, D., Barrett, J. E., Thibault, O., Porter, N. M., Rose, G. M., and Landfield, P. W. (2007). Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J. Neurosci. 27, 3098–3110.
Rowe, W., Steverman, A., Walker, M., Sharma, S., Barden, N., Seckl, J. R., and Meaney, M. J. (1997). Antidepressants restore hypothalamic-pituitary-adrenal feedback function in aged, cognitively-impaired rats. Neurobiol. Aging 18, 527–533.
Salehi, B., Cordero, M. I., and Sandi, C. (2010). Learning under stress: the inverted-U-shape function revisited. Learn. Mem. 17, 522–530.
Sandeep, T. C., Yau, J. L., MacLullich, A. M., Noble, J., Deary, I. J., Walker, B. R., and Seckl, J. R. (2004). 11Beta-hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc. Natl. Acad. Sci. U.S.A. 101, 6734–6739.
Sapolsky, R. M. (1986). Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J. Neurosci. 2, 2240–2247.
Seckl, J. R. (1997). 11ß-hydroxysteroid dehydrogenase in the brain: a novel regulator of glucocorticoid action? Front. Neuroendocrinol. 18, 49–99.
Seki, T., and Arai, Y. (1995). Age-related production of new granule cells in the adult dentate gyrus. Neuroreport 6, 2479–2482.
Smith, T. D., Adams, M. M., Gallagher, M., Morrison, J. H., and Rapp, P. R. (2000). Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J. Neurosci. 20, 6587–6593.
Sooy, K., Webster, S. P., Noble, J., Binnie, M., Walker, B. R., Secki, J. R., and Yau, J. L. W. (2010). Partial deficiency or short-term inhibition of 11β-hydroxysteroid dehydrogenase type 1 improves cognitive function in ageing mice. J. Neurosci. 30, 13867–13872.
Sousa, N., Almeida, O. F., Holsboer, F., Paula-Barbosa, M. M., and Madeira, M. D. (1998). Maintenance of hippocampal cell numbers in young and aged rats submitted to chronic unpredictable stress. Comparison with the effects of corticosterone treatment. Stress 2, 237–249.
Starkman, M. N., Gebarski, S. S., Berent, S., and Schteingart, D. E. (1992). Hippocampal formation volume, memory dysfunction and cortisol levels in patients with Cushing's syndrome. Biol. Psychiatry 32, 756–765.
Starkman, M. N., Giordani, B., Gebarski, S. S., Berent, S., Schork, M. A., and Schteingart, D. E. (1999). Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing's disease. Biol. Psychiatry 46, 1595–1602.
Steele, R. J., and Morris, R. G. (1999). Delay-dependent impairment of a matching-to-place task with chronic and intrahippocampal infusion of the NMDA-antagonist D-AP5. Hippocampus 9, 118–136.
Tombaugh, G. C., Rowe, W. B., Chow, A. R., Michael, T. H., and Rose, G. M. (2002). Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats. J. Neurosci. 22, 9932–9940.
Tytherleigh, M. Y., Vedhara, K., and Lightman, S. L. (2004). Mineralocorticoid and glucocorticoid receptors and their differential effects on memory performance in people with Addison's disease. Psychoneuroendocrinology 29, 712–723.
Uno, H., Tarara, R., Else, J. G., Suleman, M. A., and Sapolsky, R. M. (1989). Hippocampal damage associated with prolonged and fatal stress in primates. J. Neurosci. 9, 1705–1711.
Vallee, M., Mayo, W., Darnaudery, M., Corpechot, C., Young, J., Koehl, M., Le Moal, M., Baulieu, E. E., Robel, P., and Simon, H. (1997). Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc. Nat. Acad. Sci. U.S.A. 94, 14865–14870.
Vallee, M., Mayo, W., and Le Moal, M. (2001). Role of pregnenolone, dehydroepiandrosterone and their sulfate esters on learning and memory in cognitive aging. Brain Res. Rev. 37, 301–312.
Verbitsky, M., Yonan, A. L., Malleret, G., Kandel, E. R., Gilliam, T. C., and Pavlidis, P. (2004). Altered hippocampal transcript profile accompanies an age-related spatial memory deficit in mice. Learn. Mem. 11, 253–260.
Watanabe, Y., Gould, E., and McEwen, B. (1992). Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 588, 341–346.
Webster, S. P., Ward, P., Binnie, M., Craigie, E., McConnell, K. M., Sooy, K., Vinter, A., Seckl, J. R., and Walker, B. R. (2007). Discovery and biological evaluation of adamantyl amide 11beta-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 17, 2838–2843.
Wiegert, O., Joels, M., and Krugers, H. (2006). Timing is essential for rapid effects of corticosterone on synaptic potentiation in the mouse hippocampus. Learn. Mem. 13, 110–113.
Wilson, I. A., Ikonen, S., Gallagher, M., Eichenbaum, H., and Tanila, H. (2005). Age-associated alterations of hippocampal place cells are subregion specific. J. Neurosci. 25, 6877–6886.
Wilson, I. A., Ikonen, S., Gureviciene, I., McMahan, R. W., Gallagher, M., Eichenbaum, H., and Tanila, H. (2004). Cognitive aging and the hippocampus: how old rats represent new environments. J. Neurosci. 24, 3870–3878.
Wolkowitz, O. M., Reus, V. I., Weingartner, H., Thompson, K., Breier, A., Doran, A., Rubinow, D., and Pickar, D. (1990). Cognitive effects of corticosteroids. Am. J. Psychiatry 147, 1297–1303.
Wong, E. Y., and Herbert, J. (2006). Raised circulating corticosterone inhibits neuronal differentiation of progenitor cells in the adult hippocampus. Neuroscience 137, 83–92.
Wright, R. L., and Conrad, C. D. (2005). Chronic stress leaves novelty-seeking behavior intact while impairing spatial recognition memory in the Y-maze. Stress 8, 151–154.
Wright, R. L., Lightner, E. N., Harman, J. S., Meijer, O. C., and Conrad, C. D. (2006). Attenuating corticosterone levels on the day of memory assessment prevents chronic stress-induced impairments in spatial memory. Eur. J. Neurosci. 24, 595–605.
Xiao, L., Feng, C., and Chen, Y. (2010). Glucocorticoid rapidly enhances NMDA-evoked neurotoxicity by attenuating the NR2A-containing NMDA receptor-mediated ERK1/2 activation. Mol. Endocrinol. 24, 497–510.
Yau, J., Noble, J., and Seckl, J. R. (1997). Site-specific regulation of corticosteroid and serotonin receptor subtype gene expression in the rat hippocampus following 3, 4- methylenedioxymethamphetamine: role of corticosterone and serotonin. Neuroscience 78, 111–121.
Yau, J., Olsson, T., Morris, R., Noble, J., and Seckl, J. R. (1996). Decreased NGFI-A gene expression in the hippocampus of cognitively impaired aged rats. Mol. Brain Res. 42, 354–357.
Yau, J. L., McNair, K. M., Noble, J., Brownstein, D., Hibberd, C., Morton, N., Mullins, J. J., Morris, R. G., Cobb, S., and Seckl, J. R. (2007). Enhanced hippocampal long-term potentiation and spatial learning in aged 11beta-hydroxysteroid dehydrogenase type 1 knock-out mice. J. Neurosci. 27, 10487–10496.
Yau, J. L., Morris, R. G., and Seckl, J. R. (1994). Hippocampal corticosteroid receptor mRNA expression and spatial learning in the aged Wistar rat. Brain Res. 657, 59–64.
Yau, J. L., Noble, J., Graham, M., and Seckl, J. R. (2006). Central administration of a cytochrome P450-7B product 7 alpha-hydroxypregnenolone improves spatial memory retention in cognitively impaired aged rats. J. Neurosci. 26, 11034–11040.
Yau, J. L., Noble, J., and Seckl, J. R. (1999). Continuous blockade of brain mineralocorticoid receptors impairs spatial learning in rats. Neurosci. Lett. 277, 45–48.
Yau, J. L., Noble, J., and Seckl, J. R. (2011). 11beta-hydroxysteroid dehydrogenase type 1 deficiency prevents memory deficits with aging by switching from glucocorticoid receptor to mineralocorticoid receptor-mediated cognitive control. J. Neurosci. 31, 4188–4193.
Yau, J. L., Noble, J., Kenyon, C. J., Hibberd, C., Kotelevtsev, Y., Mullins, J. J., and Seckl, J. R. (2001). Lack of tissue glucocorticoid reactivation in 11beta-hydroxysteroid dehydrogenase type 1 knockout mice ameliorates age-related learning impairments. Proc. Natl. Acad. Sci. U.S.A. 98, 4716–4721.
Yau, J. L., Noble, J., Hibberd, C., Rowe, W. B., Meaney, M. J., Morris, R. G., and Seckl, J. R. (2002). Chronic treatment with the antidepressant amitriptyline prevents impairments in water maze learning in aging rats. J. Neurosci. 22, 1436–1442.
Keywords: corticosterone, cortisol, watermaze, hippocampus, 11β-HSD1, neurosteroids, CYP7B1
Citation: Yau JLW and Seckl JR (2012) Local amplification of glucocorticoids in the aging brain and impaired spatial memory. Front. Ag. Neurosci. 4:24. doi: 10.3389/fnagi.2012.00024
Received: 25 May 2012; Paper pending published: 18 June 2012;
Accepted: 02 August 2012; Published online: 29 August 2012.
Edited by:
Emma R. Wood, University of Edinburgh, UKReviewed by:
Jacques Micheau, University of Bordeaux 1, FranceCopyright © 2012 Yau and Seckl. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Joyce L. W. Yau, Queen's Medical Research Institute, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK. e-mail:am95Y2UueWF1QGVkLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.