 Alquiandra Stefani Ferreira Mançano
Alquiandra Stefani Ferreira Mançano Juliana Guanaes Pina
Juliana Guanaes Pina Juliana Mozer Sciani
Juliana Mozer Sciani- Laboratório de Produtos Naturais, Universidade São Francisco, Bragança Paulista, São Paulo, Brazil
Alzheimer’s disease (AD) is characterized by neuronal loss, attributed to amyloid-beta (Aβ) aggregation and accumulation. The autophagy-lysosomal pathway, including cathepsins B and D, is crucial for protein degradation and clearance, but it is impaired in some diseases. This review summarizes current knowledge on the dysregulation of this pathway in AD. Accumulating evidence suggests that Aβ overload impairs autophagy-lysosomal function and cathepsin activity, exacerbating Aβ accumulation and neurodegeneration. However, the precise mechanisms underlying these interactions remain elusive. Despite these challenges, targeting the lysosomal pathway emerges as a promising therapeutic strategy, and a comprehensive understanding of the autophagy-lysosomal system is essential to develop effective interventions for AD.
1 Introduction
The aging population has led to a significant increase in Alzheimer’s disease (AD) prevalence, making it a major public health concern, with projections estimating a doubling of cases every 5 years (D’Cruz and Banerjee, 2021). By 2050, this number is expected to soar to around 152 million (Janoutová et al., 2021).
AD is associated with the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles in the brain. Aβ peptides, mainly Aβ40 and Aβ42 fragments (Meng et al., 2024), are generated from the amyloid precursor protein (APP) through sequential cleavage by β- and γ-secretase, and are primarily cleared through the autophagy-lysosomal pathway (Breijyeh and Karaman, 2020). These peptides aggregate and accumulate to form plaques in various areas of brain tissue, and also contribute to the alteration of Tau protein, another cause of AD (Guan et al., 2021; Ge et al., 2022).
These Aβ subunits are formed in intra and extracellular environment, and targeted to lysosomes and mitochondria (HAASS et al., 1993). Once Aβ is intracellular environment, it is internalized into autophagic vacuoles within lysosomes for subsequent degradation and elimination of these toxic compounds for the cells (Perez et al., 2014). Meanwhile, within mitochondria, the accumulation of these plaques on the inner membrane destabilizes it and disrupts the function of electron transport chain components, increasing reactive oxygen species (ROS) production (Milane et al., 2021; Song et al., 2023). However, it is observed that this autophagy-lysosomal system is not functional in AD, and amyloid plaques are not degraded, causing neuron’s death. Figure 1 depicted the action of amyloid peptides in autophagy-lysosomal system, including cathepsins and the consequences for mitochondria and neuron death.
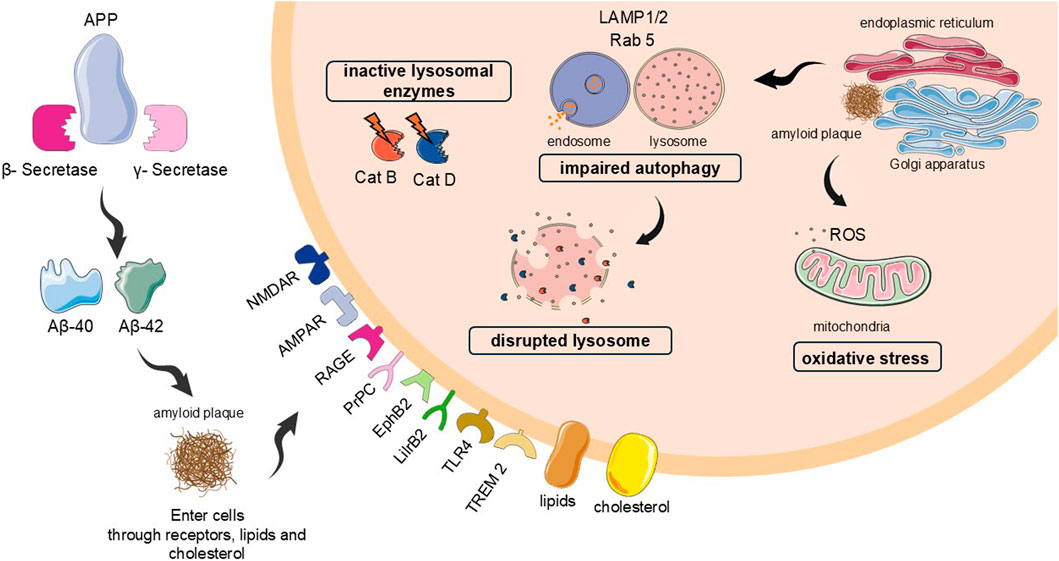
Figure 1. Implications of amyloid plaques in autophagy and lysosomal system, including cathepsins: the fusion of vesicles is impaired, reducing the autophagy, and the membrane of lysosome is disrupted, leaking the cathepsins D and B, the most abundant, to the cytosol, with inadequate pH for their better activity. These mechanisms contributed even more to the protein accumulation, and consequently neuron death. The disrupted lysosomes also cause release of reactive oxygen species (ROS), causing oxidative stress. Cat B and D = cathepsins B and D, respectively. ROS = reactive oxygen species.
The precise mechanisms underlying this lysosomal dysfunction are not fully understood, but it is believed to involve factors such as lysosomal membrane permeabilization related to ApoE4, impaired acidification, and reduced cathepsin activity, compromising this aspect of the proteostase system (Ji et al., 2002). The topic of lysosomal dysfunction in Alzheimer’s disease is controversial, as animal models may not fully recapitulate all aspects of the human disease, and cellular studies are often performed post-mortem. This review compiles these studies and results, as well as discussing medications that target autophagy currently in clinical trials for AD.
Due to its controversial nature, many studies investigating the role of the autophagy-lysosomal system in AD were discontinued or discouraged. However, recent studies increasingly highlight the importance of this topic in the disease and its potential as a key therapeutic target for novel treatments, including lysosomal cathepsins.
Given the critical role of the autophagy-lysosomal pathway in AD pathogenesis, a comprehensive understanding of this system is essential for developing effective therapeutic strategies. This review offers a comprehensive overview of AD, delving into its physiopathology, proteostasis, autophagy, lysosomal system, and the crucial function of cathepsins as lysosomal proteolytic enzymes. We examine how these systems contribute to the development and progression of AD.
2 Alzheimer’s disease - definition and physiopathology
Alzheimer’s disease (AD) was first described by Alois Alzheimer in 1906, who observed amyloid deposits and severe neuronal degeneration in the brain of a deceased patient. This pioneering work laid the groundwork for understanding the pathological hallmarks of the disease (Breijyeh and Karaman, 2020; Zhang et al., 2021).
AD is characterized by progressive neuronal loss in the hippocampus and cortex, leading to brain atrophy and cognitive decline. As the disease progresses, these changes become more pronounced, resulting in severe cognitive impairment. Neuropathological studies have identified a characteristic pattern of amyloid plaque accumulation, beginning in the pre-olfactory cortex and spreading to the entorhinal cortex, hippocampus, frontal, parietal, and temporal lobes (Tangaro et al., 2014; Nagata et al., 2016; Pini et al., 2016).
The onset of the patient’s symptoms is characterized by recent memory loss, and this condition worsens as the disease progresses. Over time, the patient begins to experience other symptoms such as behavioral and psychological changes, slurred speech, confusion, impaired judgment, and other abnormalities in brain function (Tarawneh and Holtzman, 2012).
The most accepted theory for the cause of AD is the synthesis and accumulation of Aβ. It is a peptide generated from the amyloid precursor protein (APP) through sequential cleavage by β- and γ-secretase enzymes. This process, termed the amyloidogenic pathway, produces primarily Aβ40 and Aβ42 variants. The acidic environment of endosomes and lysosomes is conducive to β-secretase activity. In contrast, the non-amyloidogenic pathway involves cleavage of APP by α-secretase, resulting in non-toxic fragments that inhibit Aβ formation and protect against AD (Meng et al., 2024).
β-amyloid fragments, especially the Aβ42 isoform, exhibit cytotoxic properties, particularly for neurons, also promoting the formation of ROS and induction of the release of inflammatory mediators in microglia (Forloni and Balducci, 2018; Jankovska et al., 2020).
Another component of AD is the Tau protein, responsible for promoting the specific assembly of the tubulin protein. In the pathogenesis of AD, the neurofibrillary tangles formed are the result of hyperphosphorylation of the Tau protein mediated by ROS, which can stimulate Tau protein hyperphosphorylation and disrupt calcium homeostasis in neuronal cell membranes, inducing the process of apoptosis (Guan et al., 2021; Ge et al., 2022).
While mitochondria possess intrinsic antioxidant systems, such as cytochrome c oxidase, their activity is significantly reduced in the AD hippocampus. This compromised antioxidant capacity renders mitochondria vulnerable to oxidative stress, leading to a vicious cycle of ROS generation and further mitochondrial damage. Such mitochondrial dysfunction is associated with the impairment of mechanisms that protect mitochondria against the action of ROS (Giorgi et al., 2018; Zhang et al., 2018).
Mitochondria possess antioxidant defenses, such as cytochrome c oxidase, and their activity is significantly reduced in the AD hippocampus (Gowda et al., 2022). Mitochondrial enzymes and complexes I, IV and V, besides pyruvate desydrogenase and α-ketoglutarate dehydrogenase complexes area altered in AD (Johri, 2021). This vulnerability, coupled with the generation of ROS, exacerbates mitochondrial damage, compromising the general metabolism and energy generation Furthermore, Aβ oligomers can directly infiltrate mitochondria via the ER-mitochondria interface or through the outer mitochondrial membrane (Huang and Liu, 2020). Their accumulation disrupts electron transport chain function, leading to a vicious cycle of increased ROS production and further mitochondrial impairment (Milane et al., 2021; Song et al., 2023).
The combined effects of Aβ accumulation and mitochondrial dysfunction contribute to impaired proteostasis and ultimately neuronal death.
3 Proteostasis
Proteostasis is the intricate process of maintaining protein homeostasis, and plays a pivotal role in safeguarding cellular health. This multifaceted mechanism encompasses three key systems: chaperones, the ubiquitin-proteasome system (UPS), and autophagy. These systems work in concert to ensure that proteins with faulty primary, secondary, or tertiary structures, as well as aggregated proteins, are either removed from the cell or properly refolded (Labbadia and Morimoto, 2015).
The endosome-lysosome system, a remarkable intracellular machinery, plays a pivotal role in maintaining cellular homeostasis by orchestrating the uptake of extracellular material and the intricate trafficking of macromolecules and organelles for recycling purposes. This intricate process, known as autophagy, serves as a cellular self-cleaning mechanism, ensuring the proper functioning and survival of cells (Vilchez et al., 2014).
3.1 Autophagy
Autophagy is a cellular degradation process essential for proteostasis, and it is initiated by nutrient deprivation, cellular stress or the presence of misfolding/mutated/aggregated proteins. There are four primary types:
(1) Macroautophagy involves the formation of double-membrane structures called autophagosomes, which engulf cellular components and fuse with lysosomes for degradation (Feng et al., 2014).
(2) Microautophagy is a more selective process where cellular components are directly engulfed by lysosomes. This can occur through various mechanisms, including chaperone-mediated autophagy and peroxisome degradation (peroxophagy) (Kunz et al., 2004).
(3) Chaperone-mediated autophagy, for degradation of cytosolic proteins, targeting them to the lysosomes (Kaushik and Cuervo, 2012).
(4) Mitophagy is a specialized form of autophagy targeting damaged mitochondria, facilitating their clearance and preventing cellular dysfunction (Gómez-Sintes et al., 2016).
Thus, lysosomes, the central components of this degradation machinery, are acidic organelles (pH 4.5–5.5) containing hydrolytic enzymes, primarily cathepsins (Saftig and Klumperman, 2009). They fuse with endosomes and autophagosomes, delivering their contents for degradation. This intricate network, encompassing endocytosis, autophagy, and lysosomal activity, is vital for recycling cellular components, generating energy, and maintaining cellular health (Yorimitsu and Klionsky, 2005).
Endocytosis brings extracellular substances into the cell within vesicles that mature into early and late endosomes before fusing with lysosomes. Autophagy delivers intracellular components for degradation, and the intricate interplay between these pathways, often referred to as the “lysosomal network,” is essential for cellular homeostasis. This network not only recycles cellular components and generates energy but also provides critical information about the cell’s internal and external environment (Nixon, 2013).
Macromolecules or ligands enter in the cell through present in clathrin regions, composed by plasma membrane, to then be delivered to the lysosomes. This process regulates the interaction of cells and provides energy by capturing substrates important for molecule synthesis (Doherty and McMahon, 2009).
The autophagy pathways rely on important proteins for vesicle fusion and transport. Rab5a regulates the fusion of endocytic vesicles with early endosomes. Its function, as proposed by de Hoop et al. (1994), is primarily related to the regulation and fusion of early endocytosis, playing a crucial role in the formation and transport of endosomes, cellular structures responsible for the internalization of cell membrane and surface proteins. Additionally, Rab5a may also be involved in the recycling of membrane components from synaptic vesicles in neurons, through its association with early endosomes both in dendrites and axons (de Hoop et al., 1994).
Another important protein for autophagy is LAMP, a lysosome-associated membrane protein. These membrane proteins of types 1 and 2 (LAMP-1 and LAMP-2) associate and stabilize polypeptide translocation systems, such as the transporter associated with antigen processing (TAPL). Dysfunction in the autophagy-lysosomal pathway of neurons, as seen in diseases like Alzheimer’s, leads to exosomal uptake of lysosomal membrane proteins and lysosomal granule enzyme cathepsin D. Consequently, levels of LAMP-1 are elevated in neural-derived plasma exosomes from patients with AD compared to those without the disease, as will be commented below (Goetzl et al., 2015). Moreover, LAMP-2A also is a receptor for intracellular protein transport in the chaperone-mediated autophagy (Qiao et al., 2023).
Lysosomes contain several hydrolases for a wide range of substrates, such as DNA, RNA, proteins, and lipids. However, the main ones being cathepsins. Its environment is acidic due to the action of the proton pump vacuolar-type ATPase, localized into the lumen of lysosomes, which ensuring the maintenance of pH, and consequently the proper functioning of the organelle (Turk et al., 2012; Futai et al., 2019). In mice, G subunit isoforms (G1 and G2) were found in neurons, strongly expressed in cortical and hippocampal areas, important for the acidification of organelles and synaptic vesicles (Murata et al., 2002). In general, neurons are rich in lysosomes, as this cell type needs to remove proteins through autophagy, maintaining proteostasis. Studies show that the reduced autophagy is related to neurodegeneration (Song et al., 2020).
The correct acidification of lysosomes and good functioning of the organelle reflects on mitochondrial function. The control of mitochondrial processes is essential for protecting the cell from aging and related diseases, such as dementia. Abnormal mitochondria are removed by autophagy and mitophagy defects are linked to mitochondrial dysfunction (Mindell, 2012).
4 Cathepsins
Cathepsins are peptidases belonging to the families of the serine peptidases (cathepsins A and G), aspartic peptidases (cathepsins D and E) or cysteine peptidases (cathepsins B, C, L, F, H, K, O, S, V, X and W), which are different in their structures, cellular and tissue localization and expression (Saftig et al., 1998). They are related to several biological processes, including lysosomal activities (Brömme, 2000).
Cathepsins are initially inactive and require activation through proteolytic removal of the N-terminal propeptide (Neurath, 1999). This activation process is crucial for triggering their biological functions, allowing them to play important roles in various physiological and pathological pathways, including protein digestion, antigen processing, and tissue remodeling (Sonkar et al., 2022).
Once activated, they play crucial roles in protein turnover. Beyond their degradative functions, cathepsins are involved in diverse cellular processes, such as angiogenesis and apoptosis (Premzl et al., 2006). Cathepsin B, for instance, participates in mammary gland involution and is implicated in lysosome-mediated cell death (Kreuzaler et al., 2011). Overexpressed cathepsins B and L, regulated by the transcription factor Stat3, can be released into the cytosol, where they act as potent proteases, triggering apoptosis (Lockshin and Zakeri, 2004). While distinct from classical apoptosis, this process involves the activation of pro-apoptotic proteins such as caspases (Boya and Kroemer, 2008).
The multifaceted functions of cathepsins highlight their significance in both physiological and pathological processes, underscoring their potential as therapeutic targets for various diseases (Cunha et al., 2022).
Interestingly, cathepsin D also influences cellular processes beyond its proteolytic function. Berchem and colleagues reported its mitogenic and angiogenic effects in cancer cells. While the exact mechanisms remain unclear, both catalytically active and inactive forms of cathepsin D contribute to tumor growth and progression (Berchem et al., 2002).
5 The endosomal-lysosomal pathway and AD
Neurons heavily rely on this autophagy system, given that the brain is often the organ most severely affected in primary lysosomal disorders. Consequently, mutations in genes involved in the global lysosomal network are causally linked to neurodegenerative disorders frequently (Komatsu et al., 2006).
Neurons possess extensive dendritic and axonal cytoplasmic extensions, which is why they face specific challenges in preventing dysfunctional organelles and cellular debris that may accumulate throughout life without the assistance of cellular division. Young neurons efficiently perform this task by eliminating autophagic substrates (Boland et al., 2008). However, the long distances that the many autophagic vacuoles generated in axons must travel to reach lysosomes, which are primarily concentrated near the cell body, it is not surprising that neurons are particularly vulnerable to slowdowns in the proteolytic clearance of autolysosomal substrates (Lee et al., 2011). In the absence of competent autophagy, neurons accumulate protein aggregates and degenerate (Hara et al., 2006; Komatsu et al., 2006). In neurodegenerative diseases, autophagy goes awry at various points along the way, giving rise to distinct pathologies (Nixon, 2013).
Therefore, lysosomal alterations and LCD (lysosomal cell death) have been identified as key pathological events in diseases that develop with age and neurodegenerative diseases (Serrano-Puebla and Boya, 2016). In AD, several alterations in proteostasis, the ubiquitin-proteasome system, autophagy, and chaperones have been observed. One of the compartments where Aβ is generated through sequential cleavages of APP is the endoplasmic reticulum (ER), in addition to the medial and trans-Golgi complex, where it is compartmentalized and exported to other cellular locations. Aβ40 is exclusively generated in the trans-Golgi portion, while Aβ42 is generated within the ER before reaching the Golgi via secretory vesicles. The amyloid peptide, whether generated intra- or extracellularly, is recognized as abnormal, and in the cytosol, it undergoes ubiquitination to be targeted either to the proteasome for degradation, to macroautophagy or to chaperone-mediated autophagy (CMA) (Choy et al., 2012; Mputhia et al., 2019).
Extracellular Aβ oligomers can enter in the cells by interacting to lipids and cholesterol from neuron’s membrane. However, it has been well described that the monomers, oligomers or fibrils bind to cellular receptors, such as NMDAR and AMPAR (Zhang et al., 2023). Other receptors also are important for Aβ binding, such as RAGE receptor (receptor for advanced glycosylation end-products), responsible for capturing circulating Aβ to the brain (Deane et al., 2003; Jarosz-Griffiths et al., 2016).
Cellular prion protein (PrPC) and Ephrin receptor B2 (EphB2) have been identified as Aβ receptors, and downstream signaling of both alters N-methyl-D-aspartate (NMDA) receptor function (Laurén et al., 2009; Cissé et al., 2011; Um et al., 2012) Furthermore, LilrB2, a neuron surface receptor, associated with growth cones and synapses, has been shown to capture oligomeric forms of Aβ42 (Cao et al., 2018). Toll-like receptor 4 (TLR4) are the main receptor in microglia that interacts to aggregated Aβ to activate the cell, activating the complement system, NLRP3 inflammasome and release of cytokines (Yang et al., 2020).
Another receptor, called TREM2, is a single-pass transmembrane structure expressed in microglia and myeloid cells 2, binds to a variety of ligands, for example, lipoproteins, phospholipids and oligomeric Aβ (oAβ) (Wang et al., 2022). A study conducted by Zhao et al. (2018) showed that TREM2 binds directly to β-amyloid (Aβ) oligomers with nanomolar affinity, and its deficiency impairs Aβ degradation in primary microglial culture and in the mouse brain.
Marshall et al. (2020) showed that oligomeric Aβ binds to extracellular membrane and also is internalized in rats’ primary neurons through plasma membrane to reach cytoplasm, in a time-dependent manner. Moreover, the oligomeric peptide is endocyted by dynamin-dependent mechanism, being addressed to lysosomes. Interestingly, Aβ42, when reach lysosomes, impair its function, influencing the traffic of other proteins inside the cell.
The uptake of oligomeric Aβ was observed using 10 μM of the peptide in the Marshall’s study, but was also demonstrated with 1 μM in SH-SY5Y neurons. Hu et al. (2009) showed that 1 nM of extracellular Aβ could be uptaken and accumulated in lysosomes in concentration higher than 2.5 μM, enough to impair the organelle activity.
The ubiquitin-proteasome system activity is compromised in aging, a common condition in AD patients, and therefore not a feasible pathway for Aβ removal. The main consequences of Aβ and Tau oligomer presence in the cells that impair the proteasome activity is its localization and accumulation of polyubiquitinylated substrates (Tai et al., 2012). Continuous proteasome inhibition leads to excessive activation of the lysosomal system, resulting in increased autophagy (Ihara et al., 2012). Thus, intracellular Aβ is internalized into autophagic vacuoles containing apolipoprotein E (ApoE) for subsequent degradation, while misfolded Tau protein is delivered to the same organelle by chaperone-mediated autophagy (Fuentealba et al., 2010; Caballero et al., 2018). Many studies describe the secretory pathway from the endoplasmic reticulum (ER), Golgi, and endosomal pathway in APP processing for Aβ generation and transport, including the processing of both Aβ40 and Aβ42. Early endosomes are identified as the primary targeting site for Aβ (Perez et al., 2014).
Lysosomes, in turn, are responsible for intracellular degradation of this Aβ, ensuring cellular homeostasis maintenance, as previously discussed. However, in the context of AD, there is dysfunction in autophagy, which can lead to even greater accumulation of Aβ in neurons, further contributing to neuronal death (Reddy and Oliver, 2019).
There is substantial evidence indicating that one mechanism for autophagy disfunction in AD is the lysosomal rupture. One piece of evidence is related to ApoE4, which is the strongest genetic risk factor for late-onset AD and functions as a lipid transport protein. ApoE4 has been shown to destabilize lysosomal membranes. It is proposed that in the presence of amyloid peptide, ApoE4 forms a reactive molecular intermediate that binds to phospholipids and integrates into the lysosomal membrane. This integration destabilizes the membrane, leading to lysosomal leakage and ultimately apoptosis (Perez et al., 2014).
Besides, when the Aβ is present in early endosomes, they become enlarged, reinforcing the importance of the endocytic pathway in AD. This pathway is related to the internalization of both Aβ and ApoE (Vidoni et al., 2016). Moreover, the accumulation of Aβ42 in the lysosomes of AD neurons leads to lysosomal leakage and the release of enzymes into the cytoplasm, both of which correlate with morphological evidence of cellular toxicity (Yang et al., 1998).
In aging, senescence or dementia, there is dysfunction of the v-ATPase or disruption of the lysosome membrane, leading to alkalinization of the organelle, compromising the catalytic activity of the enzymes present. In the case of lysosome disruption, the extravasated protons decrease the pH of the cytosol, altering cellular metabolism. Without hydrolysis, there is no elimination of undesirable proteins and lipofuscin stores, and consequently the accumulation of metabolites and cellular debris. Some compounds and elements, such as the amino acid cysteine, reduce the availability of iron ions, compromising mitochondrial respiration, besides the lysosome (Mindell, 2012).
Early onset AD can be caused by presenilin 1 mutations, which alter lysosomal proteolysis and autophagy (Lee et al., 2010) leading to calcium dyshomeostasis due to impaired V-ATPase-mediated lysosomal acidification (Lee et al., 2015). Increases in calcium levels, calpain activation, and LCD are observed in many models of neuronal cell death including excitotoxicity and stroke (Yamashima and Oikawa, 2009).
Kim et al. (2023) showed a destruction of endolysosomes by neurons rich in Aβ and brains from AD patients, related to the impairment of the V-ATPase activity. Interestingly, a gene suppressor reverted the lysosomal dysfunction and this intervention could increase the memory of transgenic mice for AD.
In addition to this mechanism of lysosomal disruption, there are studies showing alterations in autophagy and proteostasis pathways in AD. Decreased levels of Hsp70 and increased levels of cathepsin D, LAMP-1, and ubiquitinated proteins are found in neurally-derived exosomes taken from AD patients prior to the onset of clinical signs (Goetzl et al., 2015). Furthermore, alterations in mTOR signaling and reduced autophagy in postmortem tissue from AD patients correlate with decreased expression of the autophagy regulator Beclin1 (Tramutola et al., 2015). Collectively, these observations indicate alterations occurring at multiple levels within the endolysosomal system in AD patients.
The alteration in proteins from autophagy pathway was also observed in a study that analyzed the brains of pre-AD patients, who had the presence of Aβ but no clinical manifestations of the disease, compared to age-matched controls without the comorbidity or presence of the peptide. In control patients, endosomes appeared normal in size, as visualized by an early endosome marker, anti-rab5. In pre-AD patients, however, endosomes in hippocampal and prefrontal cortex neurons were enlarged. Another marker, rabaptin 5, which regulates endosomal anchoring and fusion, was observed in the cytosol of control cells but was found within endosomes in the pre-AD group. Additionally, an increase in Rab4, a protein involved in directing the recycling of early endosomes to the cell surface, was observed in these patients (Tramutola et al., 2015). These endosomal abnormalities were more pronounced in patients with the ε4 allele and even more so in patients with late-onset FAD-APOE ε4 and Down Syndrome, who develop AD in aging due to Aβ production, as well as in patients with PS mutations causing early-onset (Cataldo et al., 1997).
The results from analyses of plasma-derived neuronal exosomal proteins, derived from lysosomes and related cellular organelles, enhance the understanding that lysosomal dysfunction in AD may provide useful biomarkers for the recognition of preclinical AD (Goetzl et al., 2015).
6 Cathepsins and Alzheimer’s
Cathepsins have been demonstrated to have a strong relation in the physiopathology of diseases, including Alzheimer’s (Jung et al., 1999; Lynch and Bi, 2003).
Levels of cathepsin D are increased in AD and we found in extra lysosomal localization in brains from old rats (Jung et al., 1999; Lynch and Bi, 2003). Also, an increase in cathepsin B and E activities have been found with age, whereas a decrease in cathepsin L is observed (Nakanishi et al., 1994). Moreover, these changes in cathepsin levels and localization are common to neurons and glial cells in the brain and are restricted to areas in the brain with increased vulnerability to age related diseases (Nakanishi et al., 1994; Jung et al., 1999; Lynch and Bi, 2003).
Interestingly, extracellular accumulations of cathepsins B, L, and D associated with Aβ deposits have been described in the brains of patients with AD, but not in other neurodegenerative diseases such as Parkinson’s or Huntington’s disease. Specific sections from AD patients display cathepsin antigens intracellularly within lysosomes and related structures, as well as extracellularly in senile plaques, within degenerated neurons (non-neuronal cells with plaques do not show labeling). Cathepsins B and D are associated with amyloid deposits within extracellular lipofuscins. Controlled patients (without the disease) showed no reactivity (Cataldo and Nixon, 1990).
Cathepsins are known to activate and/or degrade various important neuronal proteins, thus playing significant roles in neurodegenerative disorders (Stoka et al., 2016; Vidoni et al., 2016).
6.1 Cathepsin D
Lysosomal cathepsin D is a fundamental aspartic endopeptidase for cellular processes, meticulously regulated by a series of intracellular mechanisms. These include enzyme synthesis regulation, post-translational modifications, activation of the pro-enzyme form, and maintenance of lysosomal pH. The enzyme is synthesized in an inactive form, and a further processing for an N-terminal propeptide removal results in its active form and an enzyme of 48 kDa (Benes et al., 2008).
Cathepsin D plays a crucial role in neuronal homeostasis, and its dysfunction leads to impaired proteolysis of target proteins such as huntingtin, α-synuclein, Tau, lipofuscin, apoE, which can result in neurological diseases including Parkinson’s and Alzheimer’s, among others (Vidoni et al., 2016).
In general, proteolysis mediated by cathepsin D is essential for neuronal cell homeostasis through the degradation of unfolded or oxidized protein aggregates delivered to lysosomes via autophagy or endocytosis. Specifically, many altered neuronal proteins characteristic of neurodegenerative diseases (such as APP, α-synuclein, huntingtin) are physiological substrates of cathepsin D and would accumulate abnormally if not degraded by this enzyme. Furthermore, experimental evidence indicates that cathepsin D activity is linked to cholesterol and glycosaminoglycan metabolism, which explains its involvement in plasticity (Vidoni et al., 2016).
Nakamura et al. (1991) reported an abnormal distribution of both cathepsin D and cathepsin B in brains of early-onset AD patients, indicating that the lysosomal proteases could be altered in the disease.
Cataldo et al. (1994) showed that the lysosomal system is altered in AD patients, being the disturbance related to the progression of the disease. In this study, authors showed that the cathepsin D immunoreactivity was increased according to the quantity of Aβ.
Lysosomal cathepsin D plays a crucial role in degrading unfolded protein aggregates delivered through endocytosis or autophagy, relevant for diseases that are caused by protein accumulation, such as AD (Vidoni et al., 2016).
Besides this overlapping between amyloid plaque and cathepsin D, and the similarity to peptidases that produce Aβ, an important question emerged: has the enzyme a role in the APP processing, and consequently formation of Aβ or did the enzyme could process and eliminate the β-amyloid peptide and amyloid plaque?
The APP from the endolysosomal system is cleaved by acidic peptidases, generating Aβ, secreted to the extracellular media, and also addressed to lysosomes. Such enzymes are β and γ-secretases, with share common features of cathepsin D (Haass et al., 1993).
Schechter and Ziv (2008) demonstrated that the kinetic parameters of cathepsin D and BACE-1 are similar, using the APP as substrate (β-sites of the wild type or mutated Swedish families), but both with slow cleavage, with (kcat/Km: approx. 50 m−1 s−1). On the other hand, in the human brain, cathepsin D was 280-fold more abundant compared to BACE-1 and was inhibited by pepstatin A, in contrast to BACE-1, which was not inhibited, showing important differences between them.
Higaki et al. (1996) demonstrated that cathepsin D and γ-secretase do not have the same cleavage site of two recombinants APPs (containing 156 and 100 amino acids of its carboxyl terminus). Interestingly, they showed that the cleavage of APP by the cathepsin D changes according to the tridimensional structure of the substrate. Processing of beta-amyloid precursor protein by cathepsin D was demonstrated by Higaki.
Previous studies had shown similar results Dreyer et al. (1994), Gallwitz et al. (2022) showed that cathepsin D could cleave APP, but under some parameters: the enzyme hydrolyzed APP between Glu593 and Val594 residues. Interestingly, they showed that the velocity of the reaction increased according to the mutations of APP, such as Asn instead Lys595 and Leu instead Met.
Therefore, cathepsin D can generate Aβ species in some circumstances, but not in the amyloidogenic pathway, being not relevant for the disease cause or development. Moreover, considering that there are reported several variants for APP, cathepsins and secretases, further studies have to be performed to confirm the activity of cathepsin D as a APP processing (Schechter and Ziv, 2008).
On the other hand, it is well described that the cathepsin D is able to cleave Aβ40 and 42. Gallwitz et al. (2022) demonstrated that cathepsin D cleaved Aβ42, without affecting the intracellular APP processing and consequently Aβ formation by in vitro and in vivo models.
This data has been earlier demonstrated by McDermott and Gibson (1996), Hamazaki (1996) and by Kenessey et al. (1997), which showed the ability of cathepsin D in the processing of Tau protein as well, using in vitro technique. Moreover, the deletion of the cathepsin D gene caused an increase (x4) of intracerebral Aβ Suire et al. (2020) confirming the correlation between the enzyme activity and the amyloid peptide processing.
Studies show the upregulation of cathepsin D in the brain from AD patients. Elevated levels of cathepsin D have also been observed in amyloid plaques in the brain and cerebrospinal fluid (CSF) of AD patients. However, studies on cathepsin D levels in the plasma of AD patients have shown inconsistent results. While studies show increased levels of cathepsin D in plasma samples, others report negative regulation in fibroblasts and monocytes derived from AD patients (Kim et al., 2021).
In the study conducted by Kim et al. (2021), using immunoblotting and ELISA techniques, a reduction in plasma levels of cathepsin D was observed in patients with amyloid defects (AD group). Furthermore, a negative correlation was found between plasma levels of cathepsin D and the CDR-SB score, one of the clinical cognitive standards commonly used in AD diagnosis. On the other hand, cathepsin D was found in high levels in plasma from AD patients, presenting cognitive decline and brain atrophy. These findings suggest that plasma levels of cathepsin D may serve as a novel biomarker for the diagnosis of AD (Drobny et al., 2022; Chai et al., 2023).
This overexpression of cathepsin D is probably due to the impaired activity of the enzyme. Cathepsin D was found oxidized in the AD brain, compared to healthy people. Moreover, the oxidized V-ATPase was also found, which indicates that the impairment of cathepsin D enzymatic activity is related to the incorrect lysosome acidification (Di Domenico et al., 2013). This is an important aspect, considering that cathepsin D is the primary soluble protease that degrades Aβ at acidic pH (Benes et al., 2008).
Loss-of-function mutations and variations of cathepsin D lead to multiple forms of neurodegeneration in humans (Steinfeld et al., 2006) and is related to the risk of late-onset AD (Davidson et al., 2006).
Suire et al. (2020) demonstrated a critical role for cathepsin D in maintaining cerebral proteostasis. CatD-knockout (CatD-KO) mice exhibited increased levels of insoluble Aβ42 and Aβ40, correlating with a reduction in soluble Aβ species. This suggests that cathepsin D is the primary protease responsible for degrading soluble Aβ within the brain. Notably, the absence of cathepsin D did not affect the levels of APP, its C-terminal fragments, or other major Aβ-degrading enzymes like IDE and NEP, indicating a specific role for de enzyme in Aβ clearance. The accumulation of insoluble Aβ within lysosomes of CatD-KO mice further supports the hypothesis that CatD is crucial for Aβ degradation in this organelle. Interestingly, the inverse relationship between soluble and insoluble Aβ levels suggests that Aβ42 may inhibit cathepsin D activity.
Thus, cathepsin D has an important function of eliminating Aβ42, as a protective (Suire et al., 2020) rule in the AD, but the enzyme activity may be compromised, increasing the risk of the disease.
A large number of genetic association studies have investigated a non-synonymous single nucleotide polymorphism (SNP) located in exon 2 (C to T substitution leading to the amino acid change from alanine to valine) of the cathepsin D gene (CTSD), which has previously been associated with AD, where the T allele increases the risk of developing AD. This finding is consistent with previous in vitro studies that linked the CTSD*T allele to increased pro-cathepsin D secretion and alteration of the enzyme’s intracellular maturation (Touitou et al., 1994).
The association of the CTSD polymorphism with AD was replicated in a subsequent study involving the Irish population (Touitou et al., 1994).
Crawford et al. (2000) reported an ethnicity-dependent association of the CTSD polymorphism with AD, where the CTSD∗C allele was significantly overrepresented in Hispanic patients with AD.
Menzer et al. (2001) did not detect a significant association in the overall population of German patients with AD and control individuals. However, they reported significant results in interactions between APOE4 and CTSD∗T alleles in men. Additionally, they found that the CTSD∗T allele exerts a significant influence on dementia-free survival.
However, two studies in the United States failed to confirm the association between the DA genotype and CTSD (Bhojak et al., 2000; Bertram and Tanzi, 2001). In order to clarify these findings, Papassotiropoulos et al. (2002) measured the concentration of Aβ42 and Aβ40 peptides in patients and control individuals. As a result, the CTSD genotype T allele was associated with a 50% reduction in Aβ42 levels in cerebrospinal fluid.
Discrepancies among genetic association studies are common and can be attributed to various causes: initial findings may be false positives due to multiple testing or population heterogeneity, negative studies may result from low statistical power or population admixture, or initial findings may be true only in certain populations (Suire et al., 2020). As stated by Roses, (1998) genetic associations should be validated by assessing their biological relevance to the disease.
Thus, when considered individually, these studies yielded conflicting results. However, utilizing data from AlzGene Bertram et al. (2007) a meta-analysis of all 18 reports exclusively from Caucasian populations published to date, excluding those with Hardy-Weinberg and balance violations, yielded a statistically significant result (Suire et al., 2020).
6.2 Cathepsin B
Cathepsin B is a cysteine protease enzyme able to degrade peptides and proteins that achieve the lysosomal pathway by endocytosis or phagocytosis (Chapman et al., 1997), playing a crucial role in the degradation of intracellular proteins. Its presence is widely observed in a variety of human cells (Cunha et al., 2022) and is primarily located in the lysosomal and endosomal compartments (Sun et al., 2008). However, it has the ability to act both intracellularly and extracellularly, in various types of cells. The enzyme plays an important role in AD, as it can degrade oligomeric Aβ42 and is subsequently secreted by exocytosis (Mueller-Steiner et al., 2006). Thus, it could help to remove Aβ from the extracellular environment or attenuate the intracellular toxicity of Aβ, which has been associated with the accumulation of Aβ1-42 aggregates in endosomal vesicles and other cellular organelles (Zhang et al., 2002; Esposito et al., 2004; Takahashi et al., 2004; Oddo et al., 2006).
Therefore, cathepsin B emerges as a promising target for therapeutic interventions in AD. Substantial research implicates significant involvement of category B in the cognitive and behavioral deficits associated with AD. They also highlight a comprehensive approach to clinical studies and animal models of AD, including the use of cathepsin B gene knockout and chemical inhibitors, highlighting the importance of this enzyme for new therapeutic approaches that could be used in AD (Hook et al., 2020).
Cathepsin B possesses the unique ability to act both as a dipeptidyl carboxypeptidase and as an endopeptidase. This is due to having two histidine residues (His110 and His111) in a 20-residue occlusion loop on the primer side of its catalytic site (Khouri et al., 1991; Illy et al., 1997; Nägler et al., 1997). This segment called the “occlusion loop,” plays a crucial role in regulating enzymatic activity, triggered as an autoinhibition mechanism. When not involved in proteolytic manipulation processes, the occlusion loop blocks the active site of cathepsin B, preventing interaction with its substrates. This fine-tuned adjustment helps prevent indiscriminate manipulation of cellular proteins and contributes to protein homeostasis within the cell (Cunha et al., 2022).
Furthermore, optimal activity of cathepsin B occurs in an acidic environment, with pH between 4.5 and 5.0, consistent with its primary activity site in lysosomes, where pH is low (Cunha et al., 2022).
However, cathepsin B is also found in the extracellular environment (Cataldo and Nixon, 1990; Hook et al., 2020). Several studies have shown elevated levels of cathepsin B protein or its activity in plasma, cerebrospinal fluid, and amyloid plaques in individuals with AD. It is highlighted that increased plasma concentration of cathepsin B is associated with cognitive dysfunction in AD, as well as severe brain lesion outcomes (Sun et al., 2015).
In patients with AD, serum levels of cathepsin B were increased compared to healthy controls of the same age group. Importantly, the rise in cathepsin B has been correlated with dementia scores and cognitive impairment in the Mini-Mental State Examination (MMSE) in AD patients. Elevated plasma cathepsin B has been observed in both mild and severe AD compared to healthy controls. In patients with mild cognitive impairment (MCI), plasma levels of cathepsin B were similar to those of controls. These results indicate that increases in plasma cathepsin B are associated with the cognitive status of AD and occur in both early and advanced stages of the disease (Morena et al., 2017).
The results of these investigations support the hypothesis that the release of cathepsin B from lysosomes to the cytosol contributes to neurodegenerative processes and behavioral deficits in AD. These findings underscore the importance of exploring cathepsin B as a potential therapeutic target for the development of innovative treatment strategies targeting AD (Hook et al., 2020).
The study conducted by Mueller-Steiner where Cathepsin B was inhibited through genetic inactivation of the enzyme in mice expressing human familial Alzheimer’s disease mutant APP, resulted in increased levels of intracellular Aβ1-42, worsening plaque deposition and other Alzheimer’s disease-related pathologies. In contrast, when Cathepsin B was present, it reduced levels of Aβ42 peptides through proteolytic cleavage and decreased preexisting amyloid deposits. Furthermore, Cathepsin B cleaved both fibrillar and non-fibrillar assemblies of Aβ42 into shorter Aβ peptides (Mueller-Steiner et al., 2006).
It was also found that Cathepsin B is capable of cleaving the C-terminal portion of Aβ42, generating truncated Aβ peptides that are less amyloidogenic and more easily degraded by other proteases. Therefore, Cathepsin B likely plays anti-amyloidogenic and neuroprotective roles. Insufficient Cathepsin B activity may promote AD, whereas increasing Cathepsin B activity could potentially neutralize the neuropathology of this disease (Mueller-Steiner et al., 2006).
Cermak et al. (2016) confirmed these findings and demonstrated that cathepsin B regulates lysosomal function and intracellular cholesterol trafficking, which is crucial for the autophagy-lysosomal pathway. In in vitro experiments, enzyme inhibition with 100 μM PADK compromised this pathway, leading to alterations in LC3II, NPC1, and NPC2 proteins. Additionally, cathepsin B degraded Aβ as well as the C-terminal fragment of APP, facilitating subsequent degradation by BACE-1.
It is important to highlight that Cathepsin B contributes to the global degradation of Aβ in the brain, targeting different forms of Aβ due to its distinct proteolytic properties and subcellular localizations (Mueller-Steiner et al., 2006). These findings suggest that increasing endogenous Cathepsin B activity could potentially reduce Aβ levels, particularly Aβ1-42, and protect against Alzheimer’s disease-related deficits (Sun et al., 2008).
Genetic linkage studies have shown an association between a CST3 polymorphism and increased risk of late-onset sporadic Alzheimer’s disease (AD), supported by subsequent systematic meta-analyses (Crawford et al., 2000; Finckh et al., 2000; Beyer et al., 2001; Bertram et al., 2007). In the study by Sun et al. (2008), it was demonstrated that CysC is a key inhibitor of Cathepsin B-dependent Aβ degradation in vivo. The relative abundance of Aβ42 is modulated by the CysC-CatB axis, and Cathepsin B-induced truncation of Aβ42 is typically suppressed by endogenous CysC in vivo (Sun et al., 2008).
The authors observed that the significant reduction in plaque burden induced by CysC ablation depends on Cathepsin B. These intriguing effects could reflect the different mechanisms by which CysC regulates soluble and insoluble Aβ—whereby it inhibits Cathepsin B for the former and binds to Aβ for the latter (Kaeser et al., 2007; Mi et al., 2007). Therefore, in mice with reduced CysC levels, there is an increase in Cathepsin B activity, resulting in a marked decrease in overall Aβ deposits (Sun et al., 2008).
The increase in intracellular CysC may impair Cathepsin B-dependent degradation of Aβ in the endosomal/lysosomal pathways and contribute to Aβ accumulation. The finding that CysC regulates soluble Aβ by inhibiting Cathepsin B could lead to new, highly specific strategies for reducing Aβ (Sun et al., 2008).
In addition to its role in proteostasis, cathepsin B plays an important function in the brain’s immune system. Microglia express higher levels of cathepsin B compared to other cell types, and cathepsin B can promote the elimination of Aβ through microglia-mediated phagocytosis, cleaving internalized Aβ aggregates (Mueller-Steiner et al., 2006).
Besides, the cathepsin B activity is frequently elevated in inflammatory neurological diseases and it has relation with the alterations caused by inflammatory activity and it could result in organ failure (Hook et al., 2020).
The excessive concentration of Cathepsin B disrupts the microglial clearance functions, mainly in the hippocampal area, and these factors stimulate the IL-1β liberation, which intensifies the inflammation activity. Moreover, a cathepsin B inhibitor, CA-074, reduced the inflammatory response and neuronal death mediated by Aβ, which shows the importance of the enzyme in reducing neurodegeneration (Sun et al., 2008).
Table 1 summarizes the key alterations in proteostasis, autophagy, and lysosomes in AD, as discussed in this review.
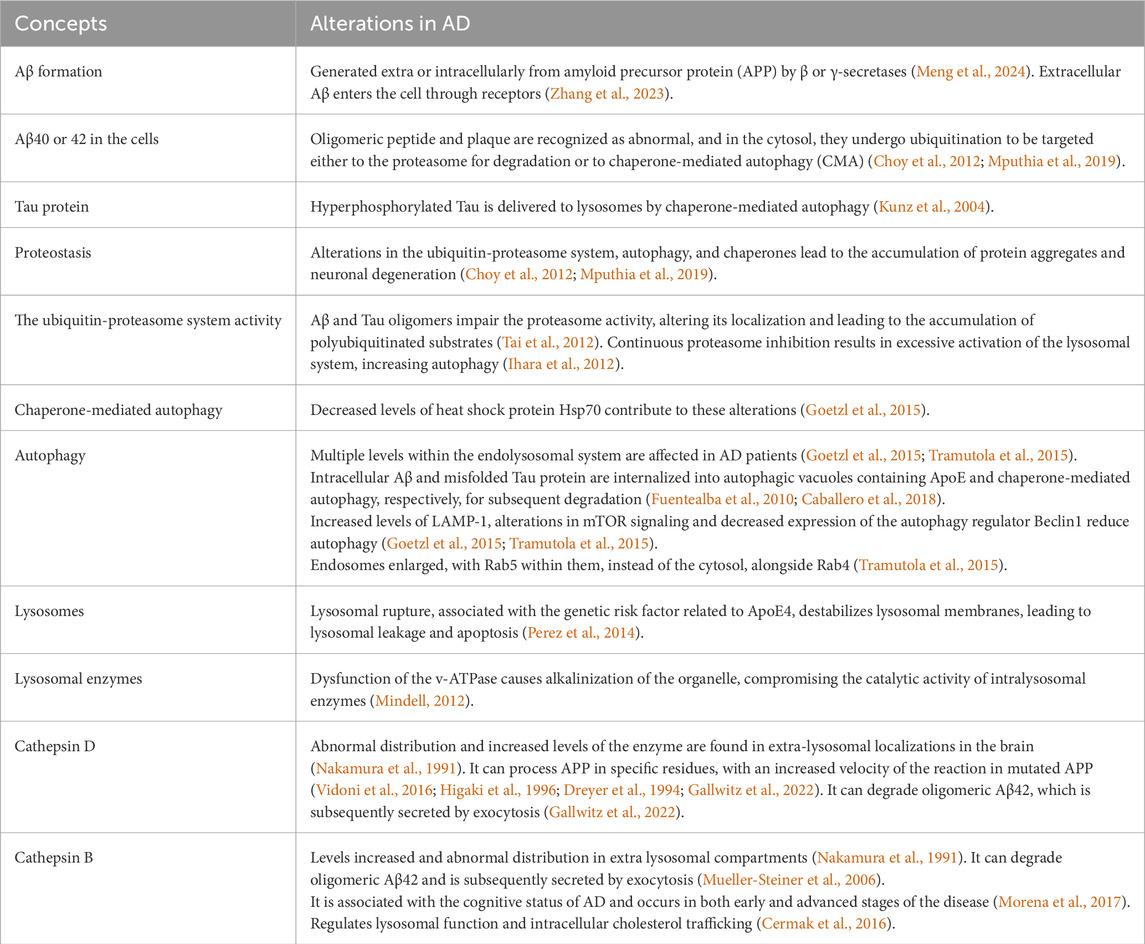
Table 1. Summary of the key alterations in proteostasis, autophagy, and lysosomes in AD, as discussed in this review.
7 Novel drugs for autophagy modulation in Alzheimer’s
Few clinical studies are currently underway to restore the autophagy-lysosomal system. One such study involves hydralazine, which is in phase 3 clinical trials for mild-to-moderate AD and, among various mechanisms, activates autophagy (Huang et al., 2023). Nilotinib, an FDA-approved drug for chronic myeloid leukemia, is in phase III clinical trials for AD due to its effect on increasing autophagy. Animal studies have shown that this increase in autophagy led to a decrease in amyloid plaques and an increase in cognition (Nobili et al., 2023).
In phase II clinical trials, GSK4527226, a monoclonal antibody against the sortilin protein (SORT1), increased lysosomal function. Rapamycin, also in phase II clinical trials, increases autophagy and inhibits mTOR (NCT04629495).
However, many compounds show, in clinical studies, the disaggregation of amyloid plaques, fibrils, or oligomers, leading to improved memory and cognition, but with mechanisms not yet fully elucidated. Restoration of this system, along with de-oligomerization, may be the mechanism underlying this improvement (Cummings et al., 2024).
While monoclonal antibodies have shown efficacy in reducing amyloid burden in Alzheimer’s disease, the underlying mechanisms of action may involve the autophagy-lysosomal system. Pre-clinical and clinical studies support the notion that compounds targeting this system can enhance clearance of toxic protein aggregates, suggesting a potential synergistic effect when combined with amyloid-directed therapies.
8 Conclusion
Lysosomal dysfunction, characterized by impaired clearance and biogenesis, plays a pivotal role in the pathogenesis of AD by exacerbating protein aggregation, characteristic of the disease. The intricate interplay between autophagy and the endosome-lysosomal system is essential for maintaining cellular homeostasis and is compromised in AD. Strategies that enhance lysosomal clearance or enhance lysosomal biogenesis are thus emerging as potential therapies (Nixon, 2013).
It is well established that the increased levels of Aβ and protein-Tau can impair autophagic and mitochondrial function, contributing to disease pathogenesis (Xu et al., 2024). While there is compelling evidence supporting the involvement of the lysosomal pathway in AD, the precise mechanisms, particularly the role of cathepsins, remain to be fully elucidated. Further research is necessary to clarify the contributions of lysosomal enzymes to disease progression and to identify novel therapeutic targets. Enhancing lysosomal function and promoting proteostase represent promising avenues for the development of effective treatments for AD.
Author contributions
AM: Investigation, Writing–original draft. JP: Investigation, Writing–original draft. BF: Investigation, Writing–original draft. JS: Conceptualization, Data curation, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. JMS is funded by the grants 19/19929-6 (FAPESP) and 313402/2023-0 (CNPq).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Benes, P., Vetvicka, V., and Fusek, M. (2008). Cathepsin D—many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68 (1), 12–28. doi:10.1016/j.critrevonc.2008.02.008
Berchem, G., Glondu, M., Gleizes, M., Brouillet, J.-P., Vignon, F., Garcia, M., et al. (2002). Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene 21 (38), 5951–5955. doi:10.1038/sj.onc.1205745
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39 (1), 17–23. doi:10.1038/ng1934
Bertram, L., and Tanzi, R. E. (2001). Of replications and refutations: the status of Alzheimer’s disease genetic research. Curr. Neurol. Neurosci. Rep. 1 (5), 442–450. doi:10.1007/s11910-001-0104-9
Beyer, K., Lao, J. I., Gómez, M., Riutort, N., Latorre, P., Mate, J. L., et al. (2001). Alzheimer’s disease and the cystatin C gene polymorphism: an association study. Neurosci. Lett. 315 (1-2), 17–20. doi:10.1016/S0304-3940(01)02307-2
Bhojak, T. J., DeKosky, S. T., Ganguli, M., and Kamboh, M. I. (2000). Genetic polymorphisms in the cathespin D and interleukin-6 genes and the risk of Alzheimer’s disease. Neurosci. Lett. 288 (1), 21–24. doi:10.1016/S0304-3940(00)01185-X
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28 (27), 6926–6937. doi:10.1523/JNEUROSCI.0800-08.2008
Boya, P., and Kroemer, G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene 27 (50), 6434–6451. doi:10.1038/onc.2008.310
Breijyeh, Z., and Karaman, R. (2020). Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25 (24), 5789. doi:10.3390/molecules25245789
Brömme, D. (2000). Papain-like cysteine proteases. Curr. Protoc. Protein. Sci. 21, Unit 21.2. doi:10.1002/0471140864.ps2102s21
Caballero, B., Wang, Y., Diaz, A., Tasset, I., Juste, Y. R., Stiller, B., et al. (2018). Interplay of pathogenic forms of human tau with different autophagic pathways. Aging. Cell 17 (1), e12692. doi:10.1111/acel.12692
Cao, Q., Shin, W. S., Chan, H., Vuong, C. K., Dubois, B., Li, B., et al. (2018). Inhibiting amyloid-β cytotoxicity through its interaction with the cell surface receptor LilrB2 by structure-based design. Nat. Chem. 10 (12), 1213–1221. doi:10.1038/s41557-018-0147-z
Cataldo, A. M., Barnett, J. L., Pieroni, C., and Nixon, R. A. (1997). Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J. Neurosci. 17 (16), 6142–6151. doi:10.1523/JNEUROSCI.17-16-06142.1997
Cataldo, A. M., Hamilton, D. J., and Nixon, R. A. (1994). Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain. Res. 640 (1-2), 68–80. doi:10.1016/0006-8993(94)91858-9
Cataldo, A. M., and Nixon, R. A. (1990). Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. U. S. A. 87 (10), 3861–3865. doi:10.1073/pnas.87.10.3861
Cermak, S., Kosicek, M., Mladenovic-Djordjevic, A., Smiljanic, K., Kanazir, S., and Hecimovic, S. (2016). Loss of cathepsin B and L leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS. One. 11 (11), e0167428. doi:10.1371/journal.pone.0167428
Chai, Y. L., Liang, N. H. P., Chong, J. R., Venketasubramanian, N., Tan, B. Y., Hilal, S., et al. (2023). Serum cathepsin D is a potential biomarker for Alzheimer’s disease dementia and cognitive decline. J.Alzheimer’s Dis. 91 (3), 989–998. doi:10.3233/JAD-220852
Chapman, H. A., Riese, R. J., and Shi, G.-P. (1997). Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 59, 63–88. doi:10.1146/annurev.physiol.59.1.63
Choy, R. W.-Y., Cheng, Z., and Schekman, R. (2012). Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U. S. A. 109 (30), E2077–E2082. doi:10.1073/pnas.1208635109
Cissé, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. doi:10.1038/nature09635
Crawford, F. C., Freeman, M. J., Schinka, J., Abdullah, L. I., Richards, D., Sevush, S., et al. (2000). The genetic association between Cathepsin D and Alzheimer’s disease. Neurosci. Lett. 289 (1), 61–65. doi:10.1016/S0304-3940(00)01260-X
Cummings, J., Zhou, Y., Lee, G., Zhong, K., Fonseca, J., and Cheng, F. (2024). Alzheimer’s disease drug development pipeline: 2024. Alzheimer’s Dement. (NY) 10 (2), e12465. doi:10.1002/trc2.12465
Cunha, M., Fernandes, P., and Ramos, M. (2022). Estudo das proteases Cat B e L. Rev. Ciência Elem. 10 (1), 008. doi:10.24927/rce2022.008
Davidson, Y., Gibbons, L., Pritchard, A., Hardicre, J., Wren, J., Tian, J., et al. (2006). Genetic associations between cathepsin D exon 2 C->T polymorphism and Alzheimer’s disease, and pathological correlations with genotype. J. Neurol. Neurosurg. Psychiatry. 77 (4), 515–517. doi:10.1136/jnnp.2005.063917
D’Cruz, M., and Banerjee, D. (2021). The person is not the disease – revisiting Alzheimer’s dementia after 120 years. J. Geriatr. Ment. Health 8 (2), 136–137. doi:10.4103/jgmh.jgmh_39_21
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi:10.1038/nm890
de Hoop, M. J., Huber, L. A., Stenmark, H., Williamson, E., Zerial, M., Parton, R. G., et al. (1994). The involvement of the small GTP-binding protein Rab5a in neuronal endocytosis. Neuron 13 (1), 11–22. doi:10.1016/0896-6273(94)90456-1
Di Domenico, F., Coccia, R., Cocciolo, A., Murphy, M. P., Cenini, G., Head, E., et al. (2013). Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim. Biophys. Acta. - Mol. Basis Dis. 1832 (8), 1249–1259. doi:10.1016/j.bbadis.2013.04.013
Doherty, G. J., and McMahon, H. T. (2009). Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902. doi:10.1146/annurev.biochem.78.081307.110540
Dreyer, R. N., Bausch, K. M., Fracasso, P., Hammond, L. J., Wunderlich, D., Wirak, D. O., et al. (1994). Processing of the pre-beta-amyloid protein by cathepsin D is enhanced by a familial Alzheimer's disease mutation. Eur. J. Biochem. 224 (2), 265–271. doi:10.1111/j.1432-1033.1994.00265.x
Drobny, A., Prieto Huarcaya, S., Dobert, J., Kluge, A., Bunk, J., Schlothauer, T., et al. (2022). The role of lysosomal cathepsins in neurodegeneration: mechanistic insights, diagnostic potential and therapeutic approaches. Biochim. Biophys. Acta. Mol. Cell. Res. 1869 (7), 119243. doi:10.1016/j.bbamcr.2022.119243
Esposito, L., Gan, L., Yu, G., Essrich, C., and Mucke, L. (2004). Intracellularly generated amyloid-beta peptide counteracts the antiapoptotic function of its precursor protein and primes proapoptotic pathways for activation by other insults in neuroblastoma cells. J. Neurochem. 91 (6), 1260–1274. doi:10.1111/j.1471-4159.2004.02816.x
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell. Res. 24 (1), 24–41. doi:10.1038/cr.2013.168
Finckh, U., von der Kammer, H., Velden, J., Michel, T., Andresen, B., Deng, A., et al. (2000). Genetic association of a cystatin C gene polymorphism with late-onset alzheimer disease. Arch. Neurol. 57 (11), 1579–1583. doi:10.1001/archneur.57.11.1579
Forloni, G., and Balducci, C. (2018). Alzheimer’s disease, oligomers, and inflammation. J. Alzheimers. Dis. 62 (3), 1261–1276. doi:10.3233/JAD-170819
Fuentealba, R. A., Liu, Q., Zhang, J., Kanekiyo, T., Hu, X., Lee, J.-M., et al. (2010). Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS. One. 5 (7), e11884. doi:10.1371/journal.pone.0011884
Futai, M., Sun-Wada, G.-H., Wada, Y., Matsumoto, N., and Nakanishi-Matsui, M. (2019). Vacuolar-type ATPase: a proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 95 (6), 261–277. doi:10.2183/pjab.95.018
Gallwitz, L., Schmidt, L., Marques, A. R. A., Tholey, A., Cassidy, L., Ulku, I., et al. (2022). Cathepsin D: analysis of its potential role as an amyloid beta degrading protease. Neurobiol. Dis. 175, 105919. doi:10.1016/j.nbd.2022.105919
Ge, M., Zhang, J., Chen, S., Huang, Y., Chen, W., He, L., et al. (2022). Role of calcium homeostasis in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 18, 487–498. doi:10.2147/NDT.S350939
Giorgi, C., Marchi, S., Simoes, I. C. M., Ren, Z., Morciano, G., Perrone, M., et al. (2018). Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell. Mol. Biol. 340, 209–344. doi:10.1016/bs.ircmb.2018.05.006
Goetzl, E. J., Boxer, A., Schwartz, J. B., Abner, E. L., Petersen, R. C., Miller, B. L., et al. (2015). Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85 (1), 40–47. doi:10.1212/WNL.0000000000001702
Gómez-Sintes, R., Ledesma, M. D., and Boya, P. (2016). Lysosomal cell death mechanisms in aging. Ageing. Res. Rev. 32, 150–168. doi:10.1016/j.arr.2016.02.009
Gowda, P., Reddy, P. H., and Kumar, S. (2022). Deregulated mitochondrial microRNAs in Alzheimer’s disease: focus on synapse and mitochondria. Ageing. Res. Rev. 73, 101529. doi:10.1016/j.arr.2021.101529
Guan, P.-P., Cao, L.-L., and Wang, P. (2021). Elevating the levels of calcium ions exacerbate Alzheimer’s disease via inducing the production and aggregation of β-amyloid protein and phosphorylated tau. Int. J. Mol. Sci. 22 (11), 5900. doi:10.3390/ijms22115900
Haass, C., Hung, A. Y., Schlossmacher, M. G., Oltersdorf, T., Teplow, D. B., and Selkoe, D. J. (1993). Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann. N. Y. Acad. Sci. 695, 109–116. doi:10.1111/j.1749-6632.1993.tb23037.x
Hamazaki, H. (1996). Cathepsin D is involved in the clearance of Alzheimer's beta-amyloid protein. Febs. Lett. 396 (2-3), 139–142. doi:10.1016/0014-5793(96)01087-3
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441 (7095), 885–889. doi:10.1038/nature04724
Higaki, J., Catalano, R., Guzzetta, A. W., Quon, D., Navé, J.-F., Tarnus, C., et al. (1996). Processing of beta-amyloid precursor protein by cathepsin D. J. Biol. Chem. 271 (50), 31885–31893. doi:10.1074/jbc.271.50.31885
Hook, V., Yoon, M., Mosier, C., Ito, G., Podvin, S., Head, B. P., et al. (2020). Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta. Proteins. Proteom. 1868 (8), 140428. doi:10.1016/j.bbapap.2020.140428
Hu, X., Crick, S. L., Bu, G., Frieden, C., Pappu, R. V., and Lee, J.-M. (2009). Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. U. S. A. 106 (48), 20324–20329. doi:10.1073/pnas.0911281106
Huang, L.-K., Kuan, Y.-C., Lin, H.-W., and Hu, C.-J. (2023). Clinical trials of new drugs for Alzheimer disease: a 2020–2023 update. J. Biomed. Sci. 30 (1), 83. doi:10.1186/s12929-023-00976-6
Huang, Y., and Liu, R. (2020). The toxicity and polymorphism of β-amyloid oligomers. Int. J. Mol. Sci. 21 (12), 4477. doi:10.3390/ijms21124477
Ihara, Y., Morishima-Kawashima, M., and Nixon, R. (2012). The ubiquitin-proteasome system and the autophagic-lysosomal system in alzheimer disease. Cold. Spring. Harb. Perspect. Med. 2 (8), a006361. doi:10.1101/cshperspect.a006361
Illy, C., Quraishi, O., Wang, J., Purisima, E., Vernet, T., and Mort, J. S. (1997). Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 272 (2), 1197–1202. doi:10.1074/jbc.272.2.1197
Jankovska, N., Olejar, T., and Matej, R. (2020). Extracellular amyloid deposits in Alzheimer’s and creutzfeldt–jakob disease: similar behavior of different proteins? Int. J. Mol. Sci. 22 (1), 7. doi:10.3390/ijms22010007
Janoutová, J., Kovalová, M., Machaczka, O., Ambroz, P., Zatloukalová, A., Němček, K., et al. (2021). Risk factors for Alzheimer’s disease: an epidemiological study. Curr. Alzheimer. Res. 18 (5), 372–379. doi:10.2174/1567205018666210820124135
Jarosz-Griffiths, H. H., Noble, E., Rushworth, J. V., and Hooper, N. M. (2016). Amyloid-β receptors: the good, the bad, and the prion protein. J. Biol. Chem. 291 (7), 3174–3183. doi:10.1074/jbc.R115.702704
Ji, Z.-S., Miranda, R. D., Newhouse, Y. M., Weisgraber, K. H., Huang, Y., and Mahley, R. W. (2002). Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J. Biol. Chem. 277 (24), 21821–21828. doi:10.1074/jbc.M112109200
Johri, A. (2021). Disentangling mitochondria in Alzheimer’s disease. Int. J. Mol. Sci. 22 (21), 11520. doi:10.3390/ijms222111520
Jung, H., Lee, E. Y., and Lee, S. I. (1999). Age-related changes in ultrastructural features of cathepsin B- and D-containing neurons in rat cerebral cortex. Brain. Res. 844 (1-2), 43–54. doi:10.1016/S0006-8993(99)01888-0
Kaeser, S. A., Herzig, M. C., Coomaraswamy, J., Kilger, E., Selenica, M.-L., Winkler, D. T., et al. (2007). Cystatin C modulates cerebral beta-amyloidosis. Nat. Genet. 39 (12), 1437–1439. doi:10.1038/ng.2007.23
Kaushik, S., and Cuervo, A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends. Cell. Biol. 22 (8), 407–417. doi:10.1016/j.tcb.2012.05.006
Kenessey, A., Nacharaju, P., Ko, L., and Yen, S. (1997). Degradation of tau by lysosomal enzyme cathepsin D: implication for alzheimer neurofibrillary degeneration. J. Neurochem. 69 (5), 2026–2038. doi:10.1046/j.1471-4159.1997.69052026.x
Khouri, H. E., Plouffe, C., Hasnain, S., Hirama, T., Storer, A. C., and Ménard, R. (1991). A model to explain the pH-dependent specificity of cathepsin B-catalysed hydrolyses. Biochem. J. 275 (3), 751–757. doi:10.1042/bj2750751
Kim, J.-W., Jung, S.-Y., Kim, Y., Heo, H., Hong, C.-H., Seo, S.-W., et al. (2021). Identification of cathepsin D as a plasma biomarker for Alzheimer’s disease. Cells 10 (1), 138. doi:10.3390/cells10010138
Kim, S.-H., Cho, Y.-S., Kim, Y., Park, J., Yoo, S.-M., Gwak, J., et al. (2023). Endolysosomal impairment by binding of amyloid beta or MAPT/Tau to V-ATPase and rescue via the HYAL-CD44 axis in Alzheimer disease. Autophagy 19 (8), 2318–2337. doi:10.1080/15548627.2023.2181614
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441 (7095), 880–884. doi:10.1038/nature04723
Kreuzaler, P. A., Staniszewska, A. D., Li, W., Omidvar, N., Kedjouar, B., Turkson, J., et al. (2011). Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell. Biol. 13 (3), 303–309. doi:10.1038/ncb2171
Kunz, J. B., Schwarz, H., and Mayer, A. (2004). Determination of four sequential stages during Microautophagy in vitro. J. Biol. Chem. 279 (11), 9987–9996. doi:10.1074/jbc.M307905200
Labbadia, J., and Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi:10.1146/annurev-biochem-060614-033955
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457 (7233), 1128–1132. doi:10.1038/nature07761
Lee, J.-H., McBrayer, M. K., Wolfe, D. M., Haslett, L. J., Kumar, A., Sato, Y., et al. (2015). Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell. Rep. 12 (9), 1430–1444. doi:10.1016/j.celrep.2015.07.050
Lee, J.-H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by alzheimer-related PS1 mutations. Cell 141 (7), 1146–1158. doi:10.1016/j.cell.2010.05.008
Lee, S., Sato, Y., and Nixon, R. A. (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an alzheimer’s-like axonal dystrophy. J. Neurosci. 31 (21), 7817–7830. doi:10.1523/JNEUROSCI.6412-10.2011
Lockshin, R. A., and Zakeri, Z. (2004). Caspase-independent cell death? Oncogene 23 (16), 2766–2773. doi:10.1038/sj.onc.1207514
Lynch, G., and Bi, X. (2003). Lysosomes and brain aging in mammals. Neurochem. Res. 28 (11), 1725–1734. doi:10.1023/A:1026069223763
Marshall, K. E., Vadukul, D. M., Staras, K., and Serpell, L. C. (2020). Misfolded amyloid-β-42 impairs the endosomal-lysosomal pathway. Cell. Mol. Life. Sci. 77 (23), 5031–5043. doi:10.1007/s00018-020-03464-4
McDermott, J. R., and Gibson, A. M. (1996). Degradation of Alzheimer's beta-amyloid protein by human cathepsin D. Neuroreport. 7 (13), 2163–2166. doi:10.1097/00001756-199609020-00021
Meng, X., Song, Q., Liu, Z., Liu, X., Wang, Y., and Liu, J. (2024). Neurotoxic β-amyloid oligomers cause mitochondrial dysfunction—the trigger for PANoptosis in neurons. Front. Aging. Neurosci. 16, 1400544. doi:10.3389/fnagi.2024.1400544
Menzer, G., Müller-Thomsen, T., Meins, W., Alberici, A., Binetti, G., Hock, C., et al. (2001). Non-replication of association between cathepsin D genotype and late onset alzheimer disease. Am. J. Med. Genet. 105 (2), 179–182. doi:10.1002/ajmg.1204
Mi, W., Pawlik, M., Sastre, M., Jung, S. S., Radvinsky, D. S., Klein, A. M., et al. (2007). Cystatin C inhibits amyloid-beta deposition in Alzheimer's disease mouse models. Nat. Genet. 39 (12), 1440–1442. doi:10.1038/ng.2007.29
Milane, L., Dolare, S., Jahan, T., and Amiji, M. (2021). Mitochondrial nanomedicine: subcellular organelle-specific delivery of molecular medicines. Nanomedicine 37, 102422. doi:10.1016/j.nano.2021.102422
Mindell, J. A. (2012). Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86. doi:10.1146/annurev-physiol-012110-142317
Morena, F., Argentati, C., Trotta, R., Crispoltoni, L., Stabile, A., Pistilli, A., et al. (2017). A comparison of lysosomal enzymes expression levels in peripheral blood of mild- and severe-alzheimer’s disease and MCI patients: implications for regenerative medicine approaches. Int. J. Mol. Sci. 18 (8), 1806. doi:10.3390/ijms18081806
Mputhia, Z., Hone, E., Tripathi, T., Sargeant, T., Martins, R., and Bharadwaj, P. (2019). Autophagy modulation as a treatment of amyloid diseases. Molecules 24 (18), 3372. doi:10.3390/molecules24183372
Mueller-Steiner, S., Zhou, Y., Arai, H., Roberson, E. D., Sun, B., Chen, J., et al. (2006). Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714. doi:10.1016/j.neuron.2006.07.027
Murata, Y., Sun-Wada, G.-H., Yoshimizu, T., Yamamoto, A., Wada, Y., and Futai, M. (2002). Differential localization of the vacuolar H+ pump with G subunit isoforms (G1 and G2) in mouse neurons. J. Biol. Chem. 277 (39), 36296–36303. doi:10.1074/jbc.M200586200
Nagata, K., Yamazaki, T., Takano, D., Maeda, T., Fujimaki, Y., Nakase, T., et al. (2016). Cerebral circulation in aging. Ageing. Res. Rev. 30, 49–60. doi:10.1016/j.arr.2016.06.001
Nägler, D. K., Storer, A. C., Portaro, F. C., Carmona, E., Juliano, L., and Ménard, R. (1997). Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry 36 (41), 12608–12615. doi:10.1021/bi971264+
Nakamura, Y., Takeda, M., Suzuki, H., Hattori, H., Tada, K., Hariguchi, S., et al. (1991). Abnormal distribution of cathepsins in the brain of patients with Alzheimer’s disease. Neurosci. Lett. 130 (2), 195–198. doi:10.1016/0304-3940(91)90395-A
Nakanishi, H., Tominaga, K., Amano, T., Hirotsu, I., Inoue, T., and Yamamoto, K. (1994). Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp. Neurol. 126 (1), 119–128. doi:10.1006/exnr.1994.1048
Neurath, H. (1999). Proteolytic enzymes, past and future. Proc. Natl. Acad. Sci. U. S. A. 96 (20), 10962–10963. doi:10.1073/pnas.96.20.10962
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19 (8), 983–997. doi:10.1038/nm.3232
Nobili, A., D’Amelio, M., and Viscomi, M. (2023). Nilotinib: from animal-based studies to clinical investigation in Alzheimer’s disease patients. Neural. Regen. Res. 18 (4), 803–804. doi:10.4103/1673-5374.350700
Oddo, S., Caccamo, A., Smith, I. F., Green, K. N., and LaFerla, F. M. (2006). A dynamic relationship between intracellular and extracellular pools of Abeta. Am. J. Pathol. 168 (1), 184–194. doi:10.2353/ajpath.2006.050593
Papassotiropoulos, A., Lewis, H. D., Bagli, M., Jessen, F., Ptok, U., Schulte, A., et al. (2002). Cerebrospinal fluid levels of β-amyloid(42) in patients with Alzheimer's disease are related to the exon 2 polymorphism of the cathepsin D gene. Neuroreport 13 (10), 1291–1294. doi:10.1097/00001756-200207190-00015
Perez, F. P., Bose, D., Maloney, B., Nho, K., Shah, K., and Lahiri, D. K. (2014). Late-onset alzheimer’s disease, heating up and foxed by several proteins: pathomolecular effects of the aging process. J. Alzheimer’s Dis. 40 (1), 1–17. doi:10.3233/JAD-131544
Pini, L., Pievani, M., Bocchetta, M., Altomare, D., Bosco, P., Cavedo, E., et al. (2016). Brain atrophy in Alzheimer’s Disease and aging. Ageing. Res. Rev. 30, 25–48. doi:10.1016/j.arr.2016.01.002
Premzl, A., Turk, V., and Kos, J. (2006). Intracellular proteolytic activity of cathepsin B is associated with capillary-like tube formation by endothelial cells in vitro. J. Cell. Biochem. 97 (6), 1230–1240. doi:10.1002/jcb.20720
Qiao, L., Hu, J., Qiu, X., Wang, C., Peng, J., Zhang, C., et al. (2023). LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy 19 (11), 2837–2852. doi:10.1080/15548627.2023.2235196
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8 (5), 488. doi:10.3390/cells8050488
Roses, A. D. (1998). Genetic associations. Lancet 351 (9106), 916. doi:10.1016/S0140-6736(05)70341-X
Saftig, P., Hunziker, E., Wehmeyer, O., Jones, S., Boyde, A., Rommerskirch, W., et al. (1998). Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 95 (23), 13453–13458. doi:10.1073/pnas.95.23.13453
Saftig, P., and Klumperman, J. (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell. Biol. 10 (9), 623–635. doi:10.1038/nrm2745
Schechter, I., and Ziv, E. (2008). Kinetic properties of cathepsin D and BACE 1 indicate the need to search for additional beta-secretase candidate(s). Biol. Chem. 389 (3), 313–320. doi:10.1515/BC.2008.025
Serrano-Puebla, A., and Boya, P. (2016). Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 1371 (1), 30–44. doi:10.1111/nyas.12966
Song, L. L., Qu, Y. Q., Tang, Y. P., Chen, X., Lo, H. H., Qu, L. Q., et al. (2023). Hyperoside alleviates toxicity of β-amyloid via endoplasmic reticulum-mitochondrial calcium signal transduction cascade in APP/PS1 double transgenic Alzheimer’s disease mice. Redox. Biol. 61, 102637. doi:10.1016/j.redox.2023.102637
Song, Q., Meng, B., Xu, H., and Mao, Z. (2020). The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl. Neurodegener. 9 (1), 17. doi:10.1186/s40035-020-00196-0
Sonkar, S. K., Singh, P. K., Chandra, S., Sonkar, G. K., Bhosale, V., and Sharma, S. (2022). Cathepsin S as an early biomarker for cardiovascular disease in chronic kidney disease patients. J. Bras. Nephrol. 44 (3), 329–335. doi:10.1590/2175-8239-jbn-2021-0135
Steinfeld, R., Reinhardt, K., Schreiber, K., Hillebrand, M., Kraetzner, R., Brück, W., et al. (2006). Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 78 (6), 988–998. doi:10.1086/504159
Stoka, V., Turk, V., and Turk, B. (2016). Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing. Res. Rev. 32, 22–37. doi:10.1016/j.arr.2016.04.010
Suire, C. N., Abdul-Hay, S. O., Sahara, T., Kang, D., Brizuela, M. K., Saftig, P., et al. (2020). Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40. Alzheimers. Res. Ther. 12 (1), 80. doi:10.1186/s13195-020-00649-8
Sun, B., Zhou, Y., Halabisky, B., Lo, I., Cho, S.-H., Mueller-Steiner, S., et al. (2008). Cystatin C-cathepsin B Axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 60 (2), 247–257. doi:10.1016/j.neuron.2008.10.001
Sun, Y., Rong, X., Lu, W., Peng, Y., Li, J., Xu, S., et al. (2015). Translational study of Alzheimer's disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J. Alzheimer’s. Dis. 45 (1), 269–282. doi:10.3233/JAD-142805
Tai, H.-C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181 (4), 1426–1435. doi:10.1016/j.ajpath.2012.06.033
Takahashi, R. H., Almeida, C. G., Kearney, P. F., Yu, F., Lin, M. T., Milner, T. A., et al. (2004). Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24 (14), 3592–3599. doi:10.1523/JNEUROSCI.5167-03.2004
Tangaro, S., Amoroso, N., Boccardi, M., Bruno, S., Chincarini, A., Ferraro, G., et al. (2014). Automated voxel-by-voxel tissue classification for hippocampal segmentation: methods and validation. Phys. Med. 30 (8), 878–887. doi:10.1016/j.ejmp.2014.06.044
Tarawneh, R., and Holtzman, D. M. (2012). The clinical problem of symptomatic alzheimer disease and mild cognitive impairment. Cold. Spring. Harb. Perspect. Med. 2 (5), a006148. doi:10.1101/cshperspect.a006148
Touitou, I., Capony, F., Brouillet, J.-P., and Rochefort, H. (1994). Missense polymorphism (C/T224) in the human cathepsin D pro-fragment determined by polymerase chain reaction—single strand conformational polymorphism analysis and possible consequences in cancer cells. Eur. J. Cancer. 30 (3), 390–394. doi:10.1016/0959-8049(94)90261-5
Tramutola, A., Triplett, J. C., Di Domenico, F., Niedowicz, D. M., Murphy, M. P., Coccia, R., et al. (2015). Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 133 (5), 739–749. doi:10.1111/jnc.13037
Turk, V., Stoka, V., Vasiljeva, O., Renko, M., Sun, T., Turk, B., et al. (2012). Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta. 1824 (1), 68–88. doi:10.1016/j.bbapap.2011.10.002
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15 (9), 1227–1235. doi:10.1038/nn.3178
Vidoni, C., Follo, C., Savino, M., Melone, M. A. B., and Isidoro, C. (2016). The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 36 (5), 845–870. doi:10.1002/med.21394
Vilchez, D., Saez, I., and Dillin, A. (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659. doi:10.1038/ncomms6659
Wang, S., Sudan, R., Peng, V., Zhou, Y., Du, S., Yuede, C. M., et al. (2022). TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185 (22), 4153–4169.e19. doi:10.1016/j.cell.2022.09.033
Xu, Z., Hu, J., Wei, Z., Lei, Y., Afewerky, H. K., Gao, Y., et al. (2024). Dynamic changes in lysosome-related pathways in APP/PS1 mice with aging. MedComm 5 (4), e540. doi:10.1002/mco2.540
Yamashima, T., and Oikawa, S. (2009). The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 89 (4), 343–358. doi:10.1016/j.pneurobio.2009.09.003
Yang, A. J., Chandswangbhuvana, D., Margol, L., and Glabe, C. G. (1998). Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 52 (6), 691–698. doi:10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3
Yang, J., Wise, L., and Fukuchi, K. (2020). TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 11, 724. doi:10.3389/fimmu.2020.00724
Yorimitsu, T., and Klionsky, D. J. (2005). Autophagy: molecular machinery for self-eating. Cell. death. Differ. 12 (Suppl. 2), 1542–1552. doi:10.1038/sj.cdd.4401765
Zhang, D., Li, Y., Heims-Waldron, D., Bezzerides, V., Guatimosim, S., Guo, Y., et al. (2018). Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 122 (1), 74–87. doi:10.1161/CIRCRESAHA.117.311349
Zhang, X.-X., Tian, Y., Wang, Z.-T., Ma, Y.-H., Tan, L., and Yu, J.-T. (2021). The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J. Prev. Alzheimers. Dis. 8 (3), 313–321. doi:10.14283/jpad.2021.15
Zhang, Y., Chen, H., Li, R., Sterling, K., and Song, W. (2023). Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future. Signal. Transduct. Target. Ther. 8 (1), 248. doi:10.1038/s41392-023-01484-7
Zhang, Y., McLaughlin, R., Goodyer, C., and LeBlanc, A. (2002). Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J. Cell. Biol. 156 (3), 519–529. doi:10.1083/jcb.200110119
Keywords: Alzheimer’s disease, autophagy, lysosome, β-amyloid, cathepsin B, cathepsin D
Citation: Mançano ASF, Pina JG, Froes BR and Sciani JM (2024) Autophagy-lysosomal pathway impairment and cathepsin dysregulation in Alzheimer’s disease. Front. Mol. Biosci. 11:1490275. doi: 10.3389/fmolb.2024.1490275
Received: 02 September 2024; Accepted: 15 October 2024;
Published: 31 October 2024.
Edited by:
Alice S. Pereira, New University of Lisboa, PortugalReviewed by:
Virginia Actis Dato, University of California San Diego, United StatesJyoti Bala Kaushal, University of Nebraska Medical Center, United States
Copyright © 2024 Mançano, Pina, Froes and Sciani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juliana Mozer Sciani, anVsaWFuYS5zY2lhbmlAdXNmLmVkdS5icg==, am1zY2lhbmlAZ21haWwuY29t