 Isaac Micallef
Isaac Micallef Kimberly Fenech
Kimberly Fenech Byron Baron
Byron Baron- 1Centre for Molecular Medicine and Biobanking, University of Malta, Msida, Malta
- 2Department of Tumor Genetics and Biology, Graduate School of Medical Sciences, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan
Cancer treatments have continued to improve tremendously over the past decade, but therapy resistance is still a common, major factor encountered by patients diagnosed with cancer. Chemoresistance arises due to various circumstances and among these causes, increasing evidence has shown that enzymes referred to as protein methyltransferases (PMTs) play a significant role in the development of chemoresistance in various cancers. These enzymes are responsible for the methylation of different amino acids, particularly lysine and arginine, via protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs), respectively. Various PMTs have been identified to be dysregulated in the development of cancer and chemoresistance. Nonetheless, the functional role of these PMTs in the development of chemoresistance is poorly characterised. This advocates the need for innovative approaches and technologies suitable for better characterisation of these PMTs and their potential clinical inhibitors. In the case of a handful of PMTs, inhibitory small molecules which can function as anticancer drugs have been developed and have also entered clinical trials. Considering all this, PMTs have become a promising and valuable target in cancer chemoresistance related research. This review will give a small introduction on the different PKMTs and PRMTs families which are dysregulated in different cancers and the known proteins targeted by the respective enzymes. The focus will then shift towards PMTs known to be involved in chemoresistance development and the inhibitors developed against these, together with their mode of action. Lastly, the current obstacles and future perspectives of PMT inhibitors in cancer chemoresistance will be discussed.
1 Introduction
Chemotherapy is one of the main modes of cancer treatments, however its efficacy is limited by chemoresistance. Chemoresistance is the ability of cancer cells to survive in the presence of commonly used chemotherapeutics, ultimately increasing the morbidity and mortality of the cancer (Yeldag et al., 2018). For this reason, drug resistance is a key issue that cancer research seeks to understand and address (Yeldag et al., 2018).
Recently, several studies have demonstrated that post translational modifications (PTMs), particularly protein methylation can play a critical role in chemoresistance. PTMs are chemical modifications on certain amino acid residues, employed by cells to alter the stability, interactions, activity, distribution and conformation of various proteins (Zhu et al., 2022). Over 200 different types of PTMs have been identified, which alter cellular metabolism, signalling cascades, gene expression, DNA repair and cell cycle control (Duan and Walther, 2015). Protein methylation long recognised for its role in transcription regulation through its addition on histones, has also been identified on non-histone proteins, such as ribosomal proteins, transcription factors, structural complexes and enzymes (Dai et al., 2021; Levy, 2019). Protein methylation is added by ‘writers’ known as histone or protein methyltransferases (HMTs or PMTs), while ‘erasers’ referred to as histone or protein demethylases (HDMs or PDMs) remove methyl groups from amino acid residues (Kaniskan et al., 2018).
The addition and removal of methyl groups from proteins, together with the enzymes responsible for these processes, are known to be dysregulated in cancer. However, the relationship between protein methylation, the responsible PMTs and cancer chemoresistance remains largely unexplored. Furthermore, numerous groups have been investigating the potential of inhibiting methylation using small molecules as a therapeutic approach to counter chemoresistance.
This review will introduce the main PMT families known to be dysregulated in cancer and implicated in chemoresistance. The focus will then shift onto the available small molecule inhibitors, their mode of action and enzyme specificity. Finally, the challenges of applying these molecules in a clinical setting as well as the future prospects of PMTs in cancer chemoresistance will be discussed.
2 Protein methylation
Protein methylation is the transfer of a CH3 group from the methyl donor S-adenosyl-l-methionine (AdoMet or SAM) to the amino acid residues of a protein. Most of the methylation substrates are histones, particularly Histone 3 and 4, however various non-histone methylated proteins such as tumour protein 53 (p53), heat shock proteins 70 or 90 (HSP70 or HSP90), Nuclear factor kappa B (NF-κB), Retinoblastoma 1 (RB1), among other have been identified over the past few years (Tables 1, 2) (Hamamoto et al., 2015; Wei et al., 2014).

Table 1. Histone Methylation markers contributing to chemoresistance development.
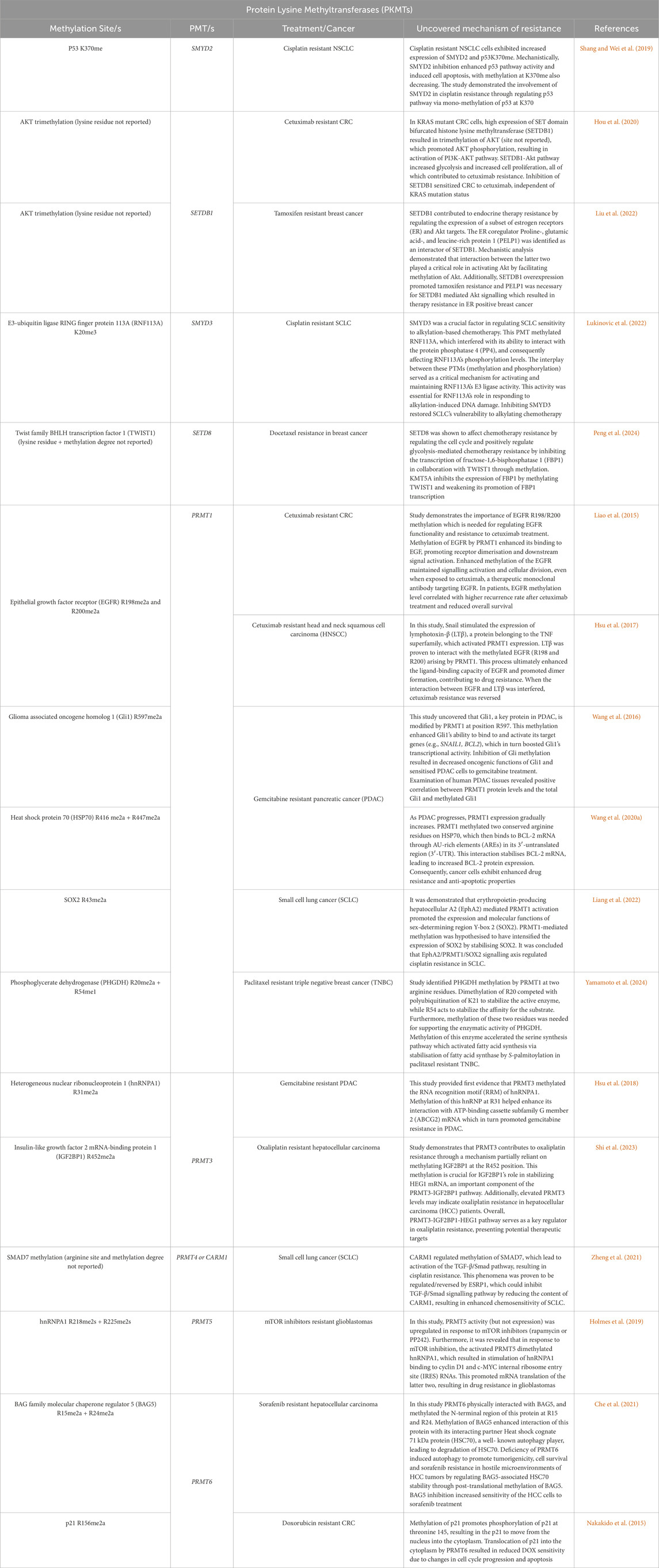
Table 2. Non-histone Methylation markers contributing to chemoresistance development.
One or more CH3 groups are predominantly added to lysine (K) and arginine (R) residues giving rise to N-methylation. However, several other amino acids can undergo N-, S- or O- methylation, with methionine (M) and cysteine (C) undergoing S-methylation, aspartate (D) and glutamate (E) undergoing O-methylation and lastly, asparagine (N), histidine (H), proline (P) and alanine (A) undergoing N-methylation (Deng et al., 2016; Cloutier et al., 2013; Clarke, 2013). Methylation does not alter the overall charged state of the residue; however, it increases the hydrophobicity and bulkiness of the protein. This can affect protein-protein interactions and recognitions, which in turn alters gene regulation (Kaniskan H. U. et al., 2015).
2.1 Protein methyltransferases (PMTs)
PMTs are responsible for catalysing the transfer of a CH3 group to methylated or unmethylated lysine or arginine residues from the methyl donor SAM (Levy, 2019). Each PMT within its respective family is structurally distinct, with the main differences being dependent on the amino acid sequences they recognise for catalysis, the residues (or side chains) they methylate and their 3D structure (Richon et al., 2011). However, a common feature is the overall structure of their catalytic active site composed of a SAM-binding pocket and a residue acceptor channel (Copeland et al., 2009). These two sites are linked together by a hydrophobic channel, which permits the transfer of a CH3 group from the SAM onto the amino acid through a state known as the SN2 transition. This gives rise to the methylated product and the release of S-5′-adenosyl-L-homocysteine (SAH), the cofactor product (Kaniskan et al., 2018). The vast majority of the currently identified PMTs are categorised into three superfamilies, according to their overall structure and sequence similarities, namely, the Seven-β-strand (7βS) domain family, the SpoU-TrmD (SPOUT) domain family and the Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) (SET) domain family (Clarke, 2013).
The 7βS family is the most diverse family among proteomes, with around 125 enzymes harbouring this domain that features a Rossmann-like structural core and can methylate amino acids, RNA and DNA (Petrossian and Clarke, 2010; Carlson and Gozani, 2016). The SPOUT methyltransferase family is the second largest SAM-dependent enzyme family within all proteomes containing a distinctive α/β knot-like structure and methylating RNA substrates (Petrossian and Clarke, 2010; Tkaczuk et al., 2007; Clarke, 2013). The SET domain family is the largest family in humans with around 56 SET domain-containing methyltransferases being identified to date (Clarke, 2013). It contains folds of multiple tiny β-sheets, which encircle into a knot-like conformation, wrapped with a pre- and post-SET domain. An i-SET domain is found within the SET domain itself and all domains are essential for enzymatic activity (Cheng et al., 2005; Kaniskan et al., 2018). Additionally, the SET proteins also contain four conserved sequence motifs, SET motif I (GxG), SET motif II (YxG), SET motif III (RFINHxCxPN), and SET motif IV (ELxFDY), where each motif is involved in the methylation reactions (Cheng et al., 2005).
Some PMTs can perform one round of catalysis, while some can perform multiple rounds. A processive mechanism is when multiple CH3 groups are added to the protein before the protein dissociates from the enzyme. On the other hand, if the protein is dissociated after the first CH3 group is added, this is referred to as a distributive mechanism. With the latter mechanism, the methylated protein can rebind to the same or different PMT for additional methyl modifications (Copeland et al., 2009; Al-Hamashi et al., 2020; Fulton et al., 2018). Both PKMTs and PRMTs make use of these two mechanisms.
For the scope of this review the focus will be on the two major groups of PMTs, based on which residue they modify, i.e., protein lysine methyltransferases (PKMTs), which are responsible for methylating lysine residues, and protein arginine methyltransferases (PRMTs), which methylate arginine residues (Tables 1, 2) (Al-Hamashi et al., 2020; Fulton et al., 2018).
2.1.1 Protein lysine methyltransferases (PKMTs)
Lysine (K) residues can be mono-, di- or tri-methylated on their epsilon (Ԑ)-amine group (Greer and Yang, 2012). Lysine methylation partakes in regulating gene expression via histone methylation, which can activate or repress transcription depending on which lysine residues are methylated and the degree of methylation. For example, mono-methylation of Histone 3 Lysine 4 (H3K4me1), mono-methylation of Histone 3 Lysine 27 (H3K27me1), tri-methylation of Histone 3 Lysine 36 (H3K36me3) and tri-methylation of histone 3 lysine 79 (H3K79me3) are linked to transcription activation, while di- or tri-methylation of Histone 3 Lysine 9 (H3K9me2 and H3K9me3) and tri-methylation of Histone 3 Lysine 27 (H3K27me3) are linked to transcription repression (Barski et al., 2007). Overall, lysine methylation is responsible for PTM crosstalk, subcellular localisation, protein stability, DNA promoter binding affinity alteration and protein-protein interactions (Cornett et al., 2019; Hamamoto et al., 2015).
The PKMTs are generally subdivided into two groups, the SET-domain containing and the non-SET domain containing enzymes (Kaniskan et al., 2018). The disruptor of telomeric silencing-1-like (DOT1L) enzyme is the only member of the latter group (Kaniskan et al., 2018; Richon et al., 2011). DOT1L is similar in structure to the PRMT enzymes (Type I) (as will be explained in Section 2.1.2. It is composed of an N-terminal helical domain and a 7βS harbouring the methyl donor binding site and an active site pocket covered with conserved hydrophobic residues (Falnes et al., 2016). The core of the DOT1L also contains seven conserved sequence motifs (I, II, III, IV, VI, VIII and X), which are scattered across the conserved core (Cheng et al., 2005). There are other PKMTs bearing the 7βS domain, known as the methyltransferase-like (METTL) proteins, however this review will not tackle this group (Falnes et al., 2016; Wong and Eirin-Lopez, 2021).
The SET domain PKMTs are further distributed among eight groups (KMT1-KMT8) based on the sequence similarities surrounding the SET domain, their enzymatic activity and their methylation targets (Allis et al., 2007; Kaniskan et al., 2018). Most of the SET domain enzymes primarily methylate histones, some can methylate both histones and non-histone proteins, while very few primarily methylate non-histones. However, there is still quite a large number of SET-domain enzymes that have no known substrates and their enzyme activity is still uncertain (Carlson and Gozani, 2016). As a result of this, it is difficult to precisely sort SET domain enzymes based on their substrates and activity on protein substrates, despite these two factors being considered when the eight KMT sub-groups were created.
2.1.2 Protein arginine methyltransferases (PRMTs)
Arginine (R) residues can be monomethylated (MMA) or di-methylated either symmetrically (me2s or SDMA) or asymmetrically (me2a or ADMA) on the guanidyl group (Greer and Yang, 2012). Arginine methylation has been observed mostly on H3 (H3R2me2a, H3R2me2s, H3R8me2s, H3R8me2a, H3R17me2a and H3R26me2a) and H4 (H4R3me2a, H4R3me2s and H4R17me), which in turn regulates transcription. Histone arginine methylation can activate or repress the expression of tumour suppressor proteins, depending on which histones are methylated. For example, methylation of H3R8 (H3R8me2s) and H4R3 (H4R3me2s) by PRMT5 results in silencing of the tumour suppressor Rb1 (Wang et al., 2008). In general, transcription activation in linked to ADMA residues while SDMA residues are associated with transcriptional repression. Having said that, there are exceptions where this is not always the resulting outcome (Bedford and Clarke, 2009; Yang and Bedford, 2013). Moreover, arginine methylation is involved in RNA processing and transcriptional elongation, cell cycle and DNA damage repair, apoptosis and signal transduction (Blanc and Richard, 2017; Clarke, 2013; Wei et al., 2014).
A total of nine human PRMTs have been identified and studied to date, with these being divided into three sub-types: Type I, Type II and Type III. All three sub-types can give rise to MMA, however Type I and Type II PRMTs can also give rise to ADMAs and SDMAs, respectively. PRMTs are found in the nucleus or cytoplasm, except for PRMT8, which is predominantly found on the plasma membrane. Among the known PRMTs, PRMT1 was the first one to be identified and thus is the most extensively studied, mediating the majority of arginine methylation events taking place inside cells (Obianyo et al., 2011).
The Type I PRMTs include PRMT 1, 2, 3, 6, 8 and Coactivator-Associated Arginine Methyltransferase 1 (CARM1; PRMT4), the Type II enzymes include PRMT5 and 9, while PRMT7 falls under Type III (Al-Hamashi et al., 2020; Fulton et al., 2018). Normally, Type II PRMTs (PRMT5) and some Type I PRMTs (except PRMT8) can methylate both histones and non-histones, with the majority methylating glycine and arginine rich (GAR/RGG/RG) motifs (Al-Hamashi et al., 2020). However, CARM1 exclusively methylates proline-, glycine-, and methionine-rich (PGM) motifs. Some PRMTs can also read their own unique substrate motifs. For instance, PRMT5 can recognise PGM motifs, while PRMT7 can read RXR sequences (X is equivalent to any amino acid) (Kaniskan et al., 2018; Al-Hamashi et al., 2020).
The PRMT family shares one methyltransferase (MTase) domain (except PRMT7 which has two), a 7-β-barrel Rossmann fold domain (interrelates with SAM) and a dimerization arm (Kaniskan et al., 2018; Wei et al., 2014; Al-Hamashi et al., 2020). The MTase domain is highly conserved and is composed of a THW loop and three motifs; motif I (VLD/EVGXGXG), post I (V/IXG/AXD/E), motif II (F/I/VDI/L/K) and motif III (LR/KXXG) (Wei et al., 2014). Further, some PRMTs have additional domains, such as PRMT2 having a SRC homology 3 (SH3) domain, PRMT3 and 9 having a zinc finger domain, PRMT9 having an F-box domain, PRMT9 having a tetratricopeptide repeat 2 (TPR2) domain and PRMT8 having an N-terminal myristoylation (myr) motif (Wei et al., 2014).
2.2 PMTs in cancer
PMTs are implicated in important cellular processes such as gene expression, cell proliferation, growth and differentiation. Dysregulation in PMT expression and activity interferes with such signalling pathways and cellular functions, giving rise to certain diseases like cancer. PMT activity in carcinogenesis and metastasis is affected by point mutations, gene rearrangements and gene amplifications (Richon et al., 2011; Au et al., 2013; Morin et al., 2010). Moreover, the involvement of PMTs in cancer development has become a hot topic in the methyl proteome field in recent years, with various reviews already published in this aspect (Bennett et al., 2017; Huang et al.,2017; Hamamoto and Nakamura, 2016; Husmann and Gozani, 2019; Hwang et al., 2021; Wu et al., 2021). More recently, research has also shifted towards investigating how PMTs are contributing to cancer chemoresistance development, an area which is still underexplored. Certain methylation histone and non-histone markers arising due to PMTs dysregulation have already been reported to be contributing to cancer chemoresistance development (Yang et al., 2020; Zhang L. et al., 2024; Zhu et al., 2023), as summarised in Tables 1, 2 below. However, how PMTs are contributing to chemoresistance development and how this phenomenon can be reversed will be further discussed in Section 3.1.
3 Chemoresistance
The development of chemoresistant tumours remains one of the main challenges of cancer treatment. Resistance to therapeutic agents in cancer patients can be classified as inherent or acquired (Mansoori et al., 2017). Inherent resistance refers to genetic characteristics within the cancer cell that provide a protective mechanism for survival, making the therapy ineffective (Mansoori et al., 2017). In contrast, acquired resistance develops after drug exposure in tumours that were initially sensitive (Mansoori et al., 2017). Further, tumours contain a high degree of molecular heterogeneity, thus even a small number of resistant cancer cells or the presence of cancer stem cells (CSC) can give rise to a chemoresistant phenotype (Mansoori et al., 2017; Phi et al., 2018).
There are several mechanisms of drug resistance. For instance, chemotherapeutic agents can induce tumour cells to express more of the therapeutic target or activate other compensatory signalling pathways such as the phosphoinositide 3-kinase (PI3K) pathway or mitogen-activated protein kinase (MAPK) signalling (Yeldag et al., 2018). Additionally, cancer cells can actively remove the drug by expressing ATP binding cassette (ABC) transporters on the cell membrane (Sun et al., 2012). The Epithelial-to-Mesenchymal transition (EMT) programme enables epithelial cells to lose cell polarity and cell-to-cell adhesion to gain a mesenchymal, stem cell-like phenotype. These mesenchymal cells show higher resistance to chemotherapy, in various cancers (Creighton et al., 2009; El Amrani et al., 2019).
When it comes to the role of PMTs in relation to altered gene expression in cancer cells, alternative splicing is one of the mechanisms tumours employ to gain drug resistance and this is achieved either through mutations in intragenic regions or altered expression of splicing factors and splicing patterns (Bonner and Lee, 2023). Dysregulated splicing factors in solid tumours include multiple serine and arginine-rich (SR) family proteins (particularly Serine/arginine-rich splicing factor 1; SRSF1), heterogeneous nuclear ribonucleoproteins (hnRNPs), and members of other RNA binding proteins (including RBM5, RBM10, and RBFOX2) (Bradley and Anczuków, 2023). Arginine methylation, particularly by Type 1 PRMTs and PRMT5 on such proteins has been shown to play a key role and its inhibition has shown promise in pre-clinical therapeutic testing (Fong et al., 2019).
3.1 PMTs in cancer chemoresistance
Upon development of acquired chemoresistance, the therapeutic drugs lose their effectiveness or do not kill all the cancer cells, leading to proliferation of the remaining cells. Dysregulation of protein methylation and the PMTs that bring it about, is one of the mechanisms involved in drug resistance. This section together with Tables 3, 4 focus on recent studies concerning the main PKMTs and PRMTs that are involved in chemoresistance or the mechanisms underlying chemoresistance. However, whether any methylation substrate is targeted by said PMTs in these studies is unknown. Moreover, Table 5 summarises and focuses on the completed/ongoing studies on the use of PMT inhibitors in reversing cancer chemoresistance.
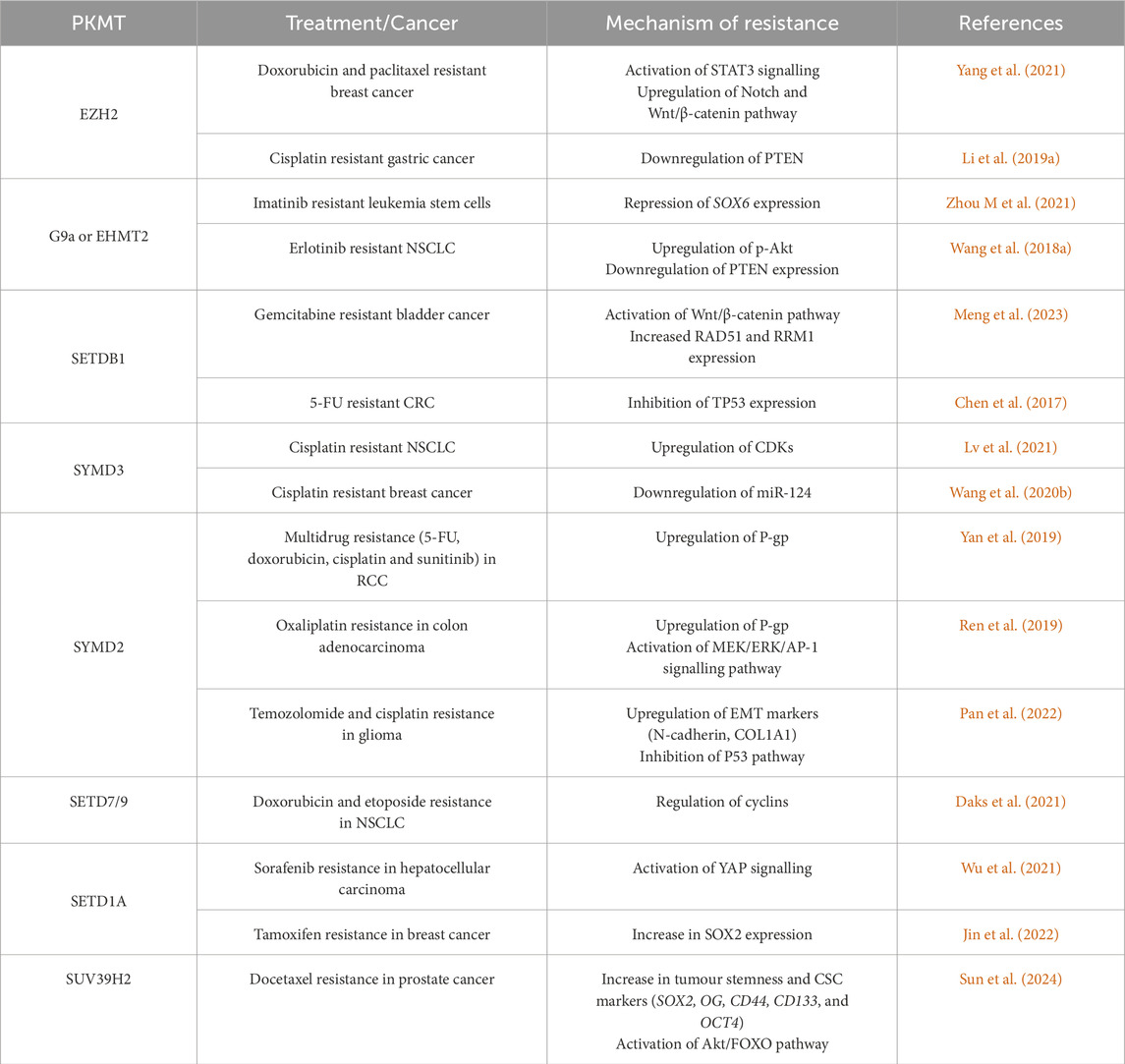
Table 3. The role of PKMTs in cancer drug resistance.
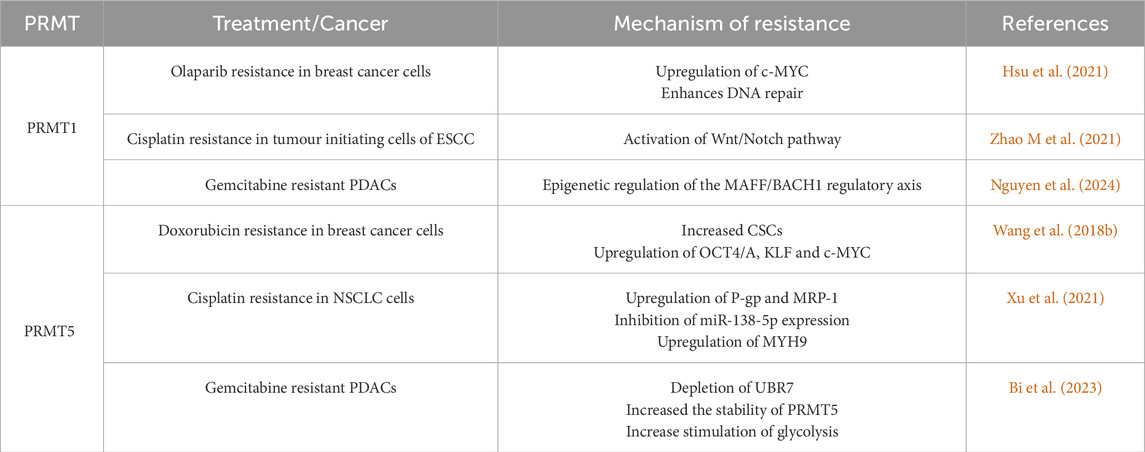
Table 4. The role of PRMTs in cancer drug resistance.
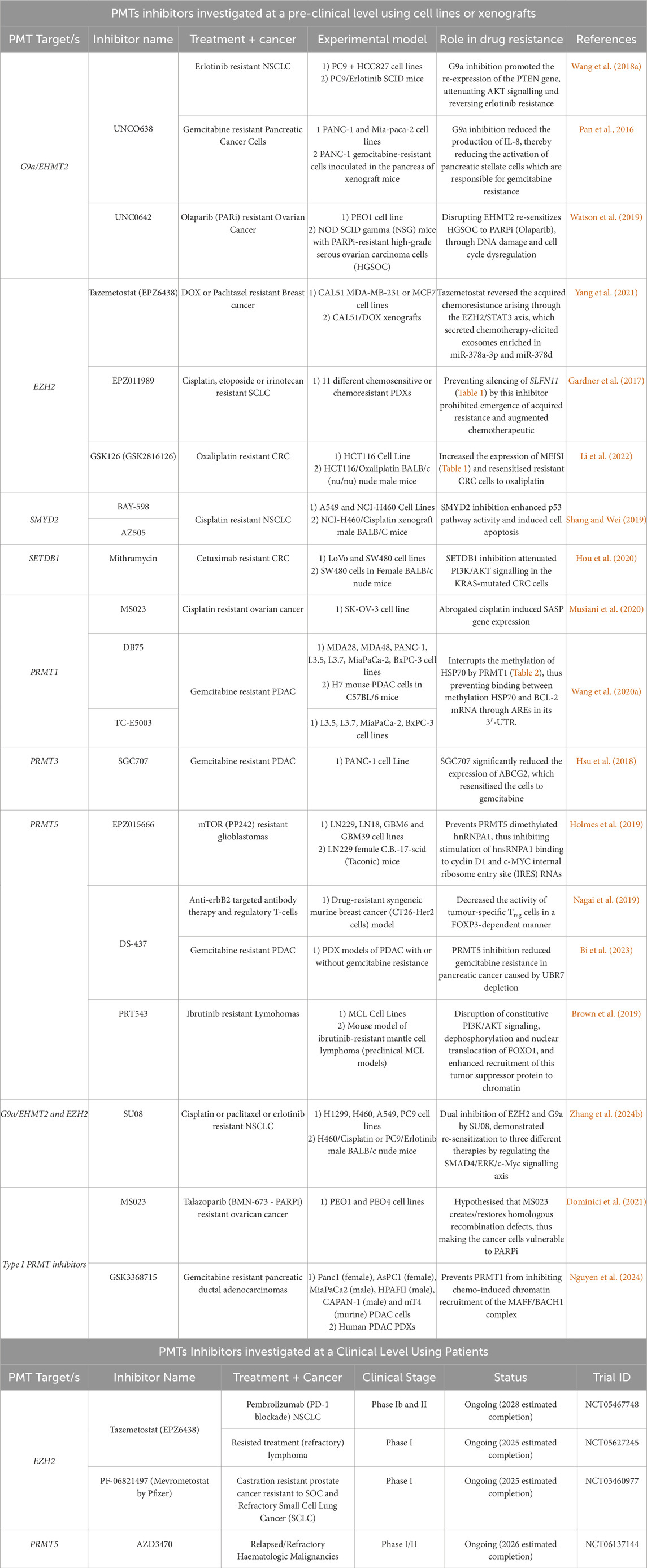
Table 5. Current inhibitors developed against PMTs which have shown potential in reversing chemoresistance at a pre-clinical or clinical level.
3.1.1 PKMTs and drug resistance
3.1.1.1 EZH2
The Enhancer of zest homolog 2 (EZH2) is responsible for catalysing the mono-, di- and tri-methylation of H3K27 that are associated with transcriptional repression. EZH2 has been known to be significantly overexpressed in drug-resistant cancer cells of multiple myeloma, leukemia, breast, colorectal (CRC), prostate and ovarian tumours (Li et al., 2017; Li Q. et al., 2019; Li X. et al., 2021; Rastgoo et al., 2018; Wen et al., 2021; Yang et al., 2021). For instance, Zhou M et al., 2021 showed that oxaliplatin resistance in CRC arises from high EZH2 protein expression. Here, the tripartite motif containing 25 (TRIM 25) prevented the ubiquitination and degradation of EZH2 (Zhou S et al., 2021).
Although the exact mechanism behind the involvement of EZH2 in drug resistance is unclear, many studies reported the role of EZH2 in maintaining CSCs through the modulation of proteins such as Signal transducer and activator of transcription 3 (STAT3) and key pathways like the Wingless-related integration site (Wnt)/β-catenin pathway. Indeed Yang et al. (2021) revealed that the increased expression of EZH2 in both doxorubicin- and paclitaxel-resistant breast cancer cells activated STAT3 signalling, which promoted the expression of miR-378a-3p and miR-378d and induced their exosomal secretion. The miRNAs targeted the WNT antagonist Dickkopf WNT signaling pathway inhibitor 3 (DKK3) and the Notch suppressor Endocytic Adaptor Protein (NUMB) in the cancer cells that remained viable following chemotherapy. In this way, the miRNAs modulated the WNT/β-catenin and Notch signalling pathways, both of which are linked with CSC maintenance and drug resistance.
In osteosarcoma, circular PR domain zinc finger protein 2 (circPRDM2) positively modulated EZH2 expression in doxorubicin-resistant cells by targeting miR-760, inhibiting its tumour suppressive roles (Yuan et al., 2021). However, the relationship between circPRDM2, EZH2 and miR-760 in chemoresistance is unclear and requires further investigation. Furthermore, the long non-coding RNA (lncRNA) prostate cancer-associated transcript 1 (PCAT-1) was found to be upregulated in cisplatin resistant gastric cancer cells (Li H. et al., 2019). Li et al. (2019a) found that PCAT-1 recruited EZH2, which epigenetically supressed the expression of the tumour suppressor Phosphatase and tensin homolog (PTEN) by increasing H3K27me3 levels.
In another study, EZH2 interacted with DNMT3a and was proven to contribute to oxaliplatin resistance in CRC by epigenetically silencing and inhibiting the expression of MEIS1 (Table 1) (Li et al., 2022). Additionally, EZH2 was also found to bind to the Euchromatic histone-lysine N-methyltransferase 2 (EHMT2) driving cisplatin and paclitaxel resistance in NSCLC cells (Zhang Q. et al., 2024). EZH2 was found to work with EHMT2 to silence the expression of the tumour suppressor of SMAD family member (SMAD) 4 which in turn activates the extracellular signal-regulated kinase (ERK)/cellular myelocytomatosis oncogene (c-MYC) pathway, controlling drug resistance (Table 1) (Zhang Q. et al., 2024).
3.1.1.2 Euchromatic histone-lysine N-methyltransferase 2 (EHMT2)/G9a
Euchromatic histone-lysine N-methyltransferase 2 (EHMT2), also known as G9a, modifies H3K9 together with SETDB1, Suv39H1 and Suv39H2. Specifically, G9a catalyses the formation of H3K9me1 and H3K9me2 which results in gene expression repression, apart from also methylating other lysine residues on H3).
Several studies report the role of G9a in cancer drug resistance. For instance, G9a was shown to be significantly upregulated in patients with resistant acute myeloid leukemia (AML) in comparison with the complete remission group (Gouda et al., 2022). In chronic myeloid leukemia (CML) stem cells (LSCs) resistant to imatinib, G9a was also found to be overexpressed and responsible for repressing the transcription of the tumour suppressor SRY-Box Transcription Factor 6 (SOX6) (Zhou M. et al., 2021).
Moreover, G9a was found to be overexpressed in erlotinib resistant (i.e., Epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor (TKI) resistant) non-small cell lung cancer (NSCLC) cells (Table 1) (Wang L. et al., 2018). In these cells, G9a epigenetically inhibited PTEN transcription by depositing H3K9me2 at its promoter region (Wang L. et al., 2018). This resulted in the activation of PI3K/AKT signalling which promoted the migration and self-renewal capacity of the NSCLC cells (Wang L. et al., 2018). In contrast, in another study on NSCLC, G9a was found to have an opposing effect. In this case, G9a prevented paclitaxel resistance by transcriptionally repressing the levels of aldehyde dehydrogenase 2 (ALDH2) and G9a inhibition upregulated ALDH2 levels (Wang W. et al., 2022). ALDH2 is a CSC marker associated with tumour cell proliferation and paclitaxel resistance in NSCLC cells (Wang W. et al., 2022). Hence, the role of G9a in NSCLC chemoresistance depended on which gene it transcriptionally repressed.
3.1.1.3 SETDB1
SETDB1 is responsible for the di–and tri-methylation of H3K9, which is associated with gene silencing. It is involved in the methylation of many key proteins and signalling cascades implicated in oncogenesis and drug resistance such as p53, alpha serine/threonine-protein kinase or protein kinase B (AKT), Wnt/β-catenin, Janus tyrosine kinase (JAK)/STAT3 and SMAD2-3 (Chen et al., 2017; Hou et al., 2020; Liu et al., 2022; Meng et al., 2023).
Increased expression of SETDB1 is found in several drug-resistant cancer cells. For instance, the circular non-coding RNA generated from the SETDB1 mRNA, serum circSETDB1, was found to be upregulated in ovarian cancer patients that displayed lymph node metastasis and resistance to platinum-taxane combined chemotherapy (Wang et al., 2019). Additionally, Meng et al. (2023) demonstrated how the protein tyrosine kinase 2 (PTK2)- Polyadenylate-binding protein 1 (PABPC1) complex stabilised the SETDB1 mRNA, preventing its degradation and hence promoting its protein expression in gemcitabine resistant bladder cancer cells. The increased SETDB1 expression was then responsible for facilitating EMT by activating the Wnt/β-catenin pathway, contributing to cancer progression (Meng et al., 2023). Gene set enrichment analysis revealed that SETDB1 is strongly linked to DNA repair and drug resistance. Further analysis revealed that SETDB1 increases the expression of RAD51 recombinase (RAD51), a protein involved in homologous recombination and ribonucleotide reductase catalytic subunit M1 (RRM1) which contributes to gemcitabine resistance (Meng et al., 2023).
SETDB1 was also found to be overexpressed in cetuximab resistant CRC cells, where it promoted AKT activation (Table 2) (Hou et al., 2020). Inhibition of SETDB1 then limited AKT hyperactivation and reversed cetuximab resistance, even in Kirsten rat sarcoma virus (KRAS)-mutated CRC cells (Hou et al., 2020). In agreement, Chen et al. (2017) showed that SETDB1 expression levels were correlated to poor prognosis in CRC patients and its overexpression suppressed apoptosis induced by 5-FU. SETDB1 inhibited the expression of TP53 by methylating its promoter, promoting cell survival in the presence of 5-FU (Chen et al., 2017). Similarly, in breast cancer cells, the interaction of SETDB1 and PELP1 was necessary to promote AKT methylation and activation, which in turn led to tamoxifen therapy resistance (Table 2) (Liu et al., 2022).
3.1.1.4 SYMD3
A notable number of research studies identified the role of SYMD3 in drug resistance. For example, SYMD3 was found to control the alkylation damage response in small cell lung cancer (SCLC) (Lukinovic et al., 2022). SYMD3 methylated and activated the E3-ubiquitin ligase RING finger protein 113A (RNF113A), which has been linked to alkylation damage repair, thus inducing resistance against alkylating chemotherapy (Table 2) (Lukinovic et al., 2022). Additionally, it was correlated with cisplatin treatment resistance in NSCLC and breast cancer (Lv et al., 2021; Wang et al., 2020b). In NSCLC, the interaction between SMAD3 and Ankyrin Repeat and KH Domain Containing 1 (ANKHD1) was shown to be essential for the upregulation of cyclin dependant kinases (CDKs) and in particular CDK2, which was found to confer cisplatin resistance (Lv et al., 2021). In breast cancer, SYMD3 negatively regulated miR-124, which is involved in cancer cell sensitivity to cisplatin (Wang et al., 2020b).
3.1.1.5 SYMD2
SYMD2 is involved in cancer cell proliferation, migration, apoptosis and drug resistance (Pan et al., 2022). SYMD2 was found to promote the expression of P-glycoprotein (P-gp) also known as multidrug resistance protein 1 (MDR1) or ATP binding cassette B1 (ABCB1) in renal cell carcinoma (RCC) and CRC cells. (Yan et al., 2019). In addition, in RCC, SYMD2 was found to promote the expression of miR-125b, in turn reducing the levels of DKK3. Inhibition of SYMD2 in RCC cells and in xenograft tumour models through the inhibitor AZ505 sensitised the cancer cells to cisplatin, doxorubicin, 5-FU and sunitinib confirming its role in chemoresistance (Yan et al., 2019). Then in CRC, SYMD2 regulated P-gp expression by activating the Mitogen-activated protein kinase (MEK)/Extracellular signal-regulated kinase (ERK)/activator protein 1 (AP-1) signalling pathway, promoting oxaliplatin resistance (Ren et al., 2019).
Pan et al. (2022) showed that inhibiting SMYD2 by AZ505 (a SYMD2 inhibitor) in glioma cells leads to a reduction in EMT markers such as N-cadherin and Collagen 1A1 (COL1A1). It also activated the expression of genes and proteins involved in the p53 signalling pathway, as evidenced by the upregulation of p21, Growth arrest and DNA damage-inducible protein (GADD45) and Bcl-2-associated protein x (Bax), along with an increase in H2A histone family member X (γH2AX), a marker of DNA damage, and a higher Bax/B-cell leukemia/lymphoma 2 protein (Bcl-2) ratio. In contrast, cyclin D1 was downregulated. Additionally, genes related to steroid metabolism, including LDL receptors (LDLR) were also downregulated. These molecular changes collectively enhanced the sensitivity of glioma cells to temozolomide and cisplatin (Pan et al., 2022).
3.1.1.6 Other PKMTs
In addition to the PKMTs discussed above, others have also been slightly investigated in the context of chemoresistance. In particular, SETD7/9 was found to be implicated in resistance to doxorubicin and etoposide in NSCLC cells (Daks et al., 2021). The loss of SETD7/9 in the cells, increased the levels of cyclins, which may have increased the sensitivity to genotoxic stress by the chemotherapy (Daks et al., 2021). Moreover, in multiple myeloma, SETD8 was associated with melphalan resistance and its expression was found to be significantly higher in relapsed patients than in newly diagnosed patients (Herviou et al., 2021). Then, inhibition of SETD8 in multiple myeloma cells, overcame melphalan resistance and thus improved the treatment for multiple myeloma patients (Herviou et al., 2021). Wu et al. (2020) uncovered the role of SETD1A in sorafenib resistance in hepatocellular carcinoma by activating Yes-associates protein (YAP) signalling. Similarly, SETD1A was also linked to tamoxifen resistance in breast cancer by promoting the expression of sex-determining region Y-box 2 SOX2 (Jin et al., 2022).
The suppressor of variegation 3–9 homolog 2 (SUV39H2) was identified as a critical enhancer of CSC populations in prostate cancer (Sun et al., 2024). Specifically, the knockdown of SUV39H2 in prostate cancer cells led to a marked reduction in the expression of key stemness markers, including SOX2, NANOG, CD44, CD133, and octamer-binding transcription factor 4 (OCT4) (Sun et al., 2024). Additionally, SUV39H2 knockdown significantly impaired cell viability in response to docetaxel treatment compared to control cells, indicating that SUV39H2 contributes to docetaxel resistance in prostate cancer (Sun et al., 2024). Furthermore, the knockdown of SUV39H2 resulted in decreased levels of AKT and forkhead box O3a (FOXO3a), highlighting its role in modulating these markers to confer drug resistance (Sun et al., 2024).
3.1.2 PRMTs and drug resistance
3.1.2.1 PRMT1
PRMT1, the major PRMT found in mammals catalyses the methylation of H4R3 together with other non-histone substrates. In cancer, PRMT1 was found to be associated with lower efficacy of chemotherapy (Matsubara et al., 2021; Shimomura et al., 2021). Cervical and ovarian cancer patients with lower PRMT1 levels were likely to benefit more from cisplatin-based chemotherapy and had a longer overall survival period when compared to the patients with high PRMT1 expression (Matsubara et al., 2021; Shimomura et al., 2021). Indeed, the downregulation of PRMT1 in both cervical cancer and ovarian cancer cells improved their sensitivity to cisplatin treatment (Matsubara et al., 2021; Shimomura et al., 2021). Likewise, PRMT1 was found to be overexpressed in triple negative breast cancer (TNBC) and oesophageal squamous cell carcinoma (ESCC) (Suresh et al., 2022; Zhao et al., 2019).
In TNBC, PRMT1 positively regulated c-MYC protein stability, which controls the expression of homologous recombination gene expression, enhancing DNA repair (Hsu et al., 2021). This resulted in the resistance to the poly (ADP-ribose) polymerase (PARP) inhibitor Olaparib, which works by inhibiting DNA repair mechanisms in the TNBC cells (Hsu et al., 2021). PRMT1 was also responsible for promoting the Wnt and Notch pathways in ESCC, leading to increased tumour initiating cells with enhanced self-renewal capabilities and resistance to cisplatin treatment (Zhao et al., 2019). Additionally, in SCLC, SOX2 methylation by PRMT1, promoted its expression and molecular functions in cancer stemness, self-renewal and chemoresistance (Table 2) (Liang et al., 2022). The increased H4R3me2a mark deposited by PRMT1 upon cisplatin treatment induced the expression of senescence-associated secretory phenotype (SASP) genes such as interleukin (IL)1Α, IL-1Β, IL-6, and tumour necrosis factor-alpha (TNFα) (Table 1) (Musiani et al., 2020). These genes protected ovarian cancer cells from DNA damage and apoptosis, promoting cisplatin resistance (Musiani et al., 2020). Lastly, a recent study demonstrated the involvement of PRMT1 in acquired gemcitabine resistance of pancreatic ductal adenocarcinoma (PDAC) cells (Nguyen et al., 2024). Mechanistically, gemcitabine treatment induced translocation of PRMT1 into the nucleus, where its enzymatic activity limited the assembly of chromatin-bound MAF Basic Leucine Zipper Transcription Factor F/BTB and CNC homology 1 (MAFF/BACH1) transcriptional complexes (Nguyen et al., 2024). In this study, no direct ADMA modifications of MAFF and BACH1 were detected, thus it was hypothesised that PRMT1 indirectly regulates the formation of MAFF/BACH1 complex and its chromatin recruitment. It remains unknown what additional factors together with PRMT1 modulate this complex in response to gemcitabine (Nguyen et al., 2024).
3.1.2.2 PRMT5
PRMT5 is the main type II PRMT, which promotes gene silencing through the methylation of H2AR3, H3R8 and H4R3 (Zhu and Rui, 2019). Like the other PMTs, PRMT5 can also methylate a number of non-histone proteins (Zhu and Rui, 2019).
In glioblastoma, PRMT5 activity conferred therapeutic resistance to mammalian target of rapamycin (mTOR) inhibitors by mediating the methylation of heterogeneous nuclear ribonucleoprotein 1 (hnRNP A1) to promote the translation of cyclin D1 and c-MYC, leading to drug resistance (Table 2) (Holmes et al., 2019). In fact, subsequent inhibition of PRMT5 sensitised the glioblastoma cells to mTOR inhibition (Holmes et al., 2019). In agreement, Wang Z. et al. (2018) found that PRMT5 mediated doxorubicin resistance in breast cancer by inducing the expression of the stem cell markers octamer-binding transcription factor 4 (OCT4/A), Krupple-like factor 4 (KLF4) and c-MYC (Wang Z. et al., 2018). Further, circ-PRMT5 was found to be increased in cisplatin-resistant NSCLC cells (Xu et al., 2021). Its knockdown reduced the expression of the drug transporter P-gp and multidrug resistance-associated protein 1 (MRP-1)and induced an immune response by increasing the expression of IL-2, TNF-a and decreasing the levels of transforming growth factor beta (TGF-β) (Xu et al., 2021). Further analysis revealed that circ-PRMT5 controls these effects by sponging miR-158-5p, supporting the expression of myosin heavy chain 9 (MYH9) which promotes cancer progression (Xu et al., 2021). Lastly, gemcitabine resistance in PDAC patient derived xenografts (PDXs) was demonstrated to mechanistically arise due to depleted UBR7 which resulted in increased stability of PRMT5 and increased glycolysis promotion (Bi et al., 2023).
3.1.2.3 Other PRMTs
Overexpression of other PRMTs has also been implicated in chemoresistance. For instance, the overexpression of PRMT3 led to gemcitabine resistance in pancreatic cancer cells by upregulating the expression of the drug transporter ABCG2 (Hsu et al., 2018) (Table 2). Then, in SCLC, CARM1 was found to regulate SMAD7 methylation to activate TGF-β/SMAD signalling, promoting EMT and chemoresistance (Zheng et al., 2021) (Table 2). In contrast, knockout of CARM1 induced drug resistance in breast cancer cell lines whereas high CARM1 expression in breast cancer patients increased their overall survival after adjuvant chemotherapy (Wang et al., 2015). Finally, PRMT6 has been shown to methylate p21, increasing its cytoplasmic localisation and inhibiting its growth suppressive roles (Nakakido et al., 2015) (Table 2).
4 Potential targeting of PMTs to reverse chemoresistance
PMT inhibitors have different modes of action, as well reviewed by Wang and his colleagues (2022). They either compete with SAM, which is the methylation cofactor, occupying the SAM-binding pocket or compete with the substrate and occupy the substrate-binding site of the methyltransferase (Ferreira de Freitas et al., 2019; Wang M. Y. et al., 2022). Cofactor inhibitors are more challenging for drug discovery than substrate competitors due to the hydrophilicity of the SAM-binding pocket (Schapira, 2016). Developing compounds that are polar enough for the SAM-binding site but also hydrophobic enough to cross the cell membrane is very difficult (Schapira, 2016). Moreover, the inhibitors have to compete with the high cellular levels of SAM. However, cofactor competitors were the first to reach the market. For instance, the PRMT5 inhibitors LLY-283 and JNJ-64619178 have been the starting point for SAM mimetics with good potency and cellular activity (Wang M. Y. et al., 2022). Others like the SYMD2 inhibitor PFI-5 retain the adenine ring of the cofactor or are chemically unrelated to SAM but still sufficiently inhibit its function (Ferreira de Freitas et al., 2019).
In contrast, the substrate-binding site is less polar than the SAM-binding pocket and is more structurally-druggable, hence the majority of PMT inhibitors are substrate competitors (Schapira., 2016). However, SAM or SAH may be required for the binding of substrate inhibitors to their target enzyme (Ferreira de Freitas et al., 2019). The substrate competitor may either interact directly with the cofactor or the cofactor may allosterically stabilise the substrate-binding pocket (Ferreira de Freitas et al., 2019). For instance, some PKMTs like SETD7 require the binding of SAM to generate the substrate-binding groove (Ferreira de Freitas et al., 2019).
Other PMT inhibitors focused on targeting allosteric sites to modulate PMT function. For example, MAK683 is an EZH2 inhibitor that binds to the embryonic ectoderm development (EED) domain. EZH2 is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2) that is only active in complexes with EED and SUZ12 polycomb repressive complex 2 subunit (SUZ12). Thus, by targeting the EED domain, MAK683 inhibits the catalytic activity of EZH2 (Schapira., 2016).
Targeting key PMTs in cancer can prevent metastatic growth and reverse drug resistance. Most of the currently developed inhibitors against PMTs have been investigated in there use to prevent cancer growth, with a number of reviews covering the different inhibitors synthesised to date, together with their mode of action (Feoli et al., 2022; Hu et al., 2016; Kaniskan H. U. et al., 2015; Kaniskan et al., 2018; Song et al., 2016; Wang M. Y. et al., 2022; Wu et al., 2021). However, whether such inhibitors could also help in reversing cancer chemoresistance is still being investigated. Moreover, combining PMT inhibitors with other anti-cancer therapeutics can prolong cellular effectiveness, reverse drug resistance and improve the response and overall survival rate of cancer patients. This section will focus on the major PKMT and PRMT inhibitors that have shown outstanding pre-clinical potential or are currently being studied in clinical trials for reversing cancer chemoresistance.
4.1 Targeting EZH2
Tazemetostat (EPZ6438) is a SAM-competitive EZH2 inhibitor that has been approved by the FDA for the treatment of epithelial sarcoma characterised by loss of integrase interactor 1/SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (INI1/SMARCB1) and follicular lymphoma (FDA, 2020a; FDA, 2020b). The anti-tumourigenic properties have been studied in several malignancies. For example, in breast cancer xenograft models, Tazemetostat reversed doxorubicin resistance by reducing the number of chemotherapy-induced exosomal miR-378a-3p and miR-378d (Yang et al., 2021). In biliary tract cancer cell lines, Tazemetostat has been demonstrated to effectively reduce levels of H3K27me3 and upregulate the gene expression of fructose-bisphosphatase 1 (FBP1) (Bekric et al., 2023). FBP1 is recognised for its role as a tumour suppressor in some cancers such as cholangiocarcinoma (Zhao et al., 2018). Moreover, Tazemetostat is being investigated in more than 50 clinical trials (ClinicalTrials.gov accessed August 2024). These include nerve sheath tumours (NCT04917042 - ClinicalTrials.gov, 2024L), AT-rich interacting domain-containing protein 1A (ARID1A) mutated malignancies (NCT05023655 - ClinicalTrials.gov, 2024e), patients with relapsed or refractory tumours harbouring EZH2, SWI/SNF related matrix associated actin dependent regulator of chromatin subfamily a, member 4 (SMARCA4) or SMARCB1 mutations (NCT03213665 - ClinicalTrials.gov, 2024m), T-cell lymphoma (NCT05983965 - ClinicalTrials.gov, 2024i), NSCLC in combination with pembrolizumab (NCT05467748 - ClinicalTrials.gov, 2024b), metastatic castration-resistant prostate cancer in combination with Talazoparib (NCT04846478 - ClinicalTrials.gov, 2024d) and combination therapy with belinostat (Histome deacetylases inhibitor) in treating patients with lymphomas that have returned (relapsed) or resisted treatment (refractory) (NCT05627245 - ClinicalTrials.gov, 2024n). Additionally, the success of Tazemetostat in treating some cancers has led to an ongoing study evaluating its long-term safety in patients who have previously benefited from the therapy (NCT02875548 - ClinicalTrials.gov, 2024j). In a completed clinical trial (NCT02860286 - ClinicalTrials.gov, 2021) involving 74 patients with relapsed or refractory BRCA1-Associated Protein 1 (BAP1)-inactivated pleural mesothelioma, Tazemetostat showed limited efficacy: only two patients had partial responses, and none achieved a complete response (Zauderer et al., 2022). This suggests that tumours with specific biomarkers might benefit more from this therapy (Zauderer et al., 2022).
GSK126 (GSK2816126) is another selective, SAM-competitive small molecule inhibitor of EZH2, which inhibits both wildtype and mutant EZH2 (Gulati et al., 2018). In a phase I clinical trial (NCT02082977 - ClinicalTrials.gov, 2020) which took place between 2014 and 2017, a study was performed to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK2816126 in 41 subjects having either relapsed/refractory diffuse large B cell lymphoma, transformed follicular lymphoma, other Non-Hodgkin’s lymphomas, solid tumors and multiple myeloma (Gulati et al., 2018). Due to different adverse events (e.g., fatigue, nausea, anemia and vomiting), as well as insufficient evidence of clinical activity, further clinical investigation of this inhibitor were terminated in this study, and since then, no additional clinical studies were performed. However, in recent years, this inhibitor has shown to also resensitize different resistant cancers to therapy (Li et al., 2022; Zhang Q. et al., 2024). For instance, this inhibitor increased the expression of MEISI (Table 1) and made resistant CRC cells sensitive to oxaliplatin again (Li et al., 2022). Similarly, Zhang and his colleagues (2024) also demonstrated that GSK126 increased the therapy sensitivity to cisplatin or paclitaxel resistant NSCLC. Moreover, in the same study performed (Zhang Q. et al., 2024), a novel inhibitor which targets not only EZH2, but also G9a, was developed to reverse to cisplatin, paclitaxel or erlotinib resistance in NSCLC, named SU08. Molecular docking analysis demonstrated that SU08 accommodates in the substrate-binding cavities of G9a and EZH2, thus acting as a substrate competitor. SU08 could inhibit the enzyme activity of EZH2 and G9a in a concentration-dependent manner, with IC50 values close to those of single agents GSK126 (EZH2) and UNC0638 (G9a/EHMT2). Despite demonstrating significant potential under both in vitro and in vivo conditions (Table 5), it was stated that SU08 also requires extensive further work to improve its drug activity and intensive preclinical studies, as well as its use in other resistant cancers (Zhang Q. et al., 2024).
EPZ011989 is another potent, orally-available EZH2 inhibitor that increased the sensitivity of cisplatin, etoposide and irinotecan in both chemosensitive and chemoresistant models of SCLC (Gardner et al., 2017). Ramakrishnan et al. (2019) also reported that EPZ011989 increased the number of natural killer cells (NKCs) in cisplatin-treated bladder cancer cells. These NKCs have the potential to enhance cisplatin sensitivity by increasing the cytotoxicity of stem-like cells or promoting the differentiation of tumour cells.
PF-06821497, CPI-205, MAK683 and DS-3201 are other EZH2 inhibitors that are currently being studied in clinical trials on different cancer sub-types as a monotherapy or in combination with other therapeutics. For example, PF-06821497 (Mevrometostat by Pfizer) is currently being investigated in patients with advanced prostate cancer in combination with enzalutamide (NCT06551324 - ClinicalTrials.gov, 2024k). Ongoing clinical trials for Mevrometostat are also being carried out for relapsed and refractory SCLC, castration resistant prostate cancer (including those resistant to standard or care (SOC)) and follicular lymphoma (NCT03460977 - ClinicalTrials.gov, 2024c). Recently, a phase I study has been completed for the evaluation of CPI-1205 in 32 patients with B-cell lymphoma (NCT02395601 - ClinicalTrials.gov, 2022d). CPI-1205 was well-tolerated with manageable adverse effects and showed evidence of antitumor activity (Harb et al., 2018). CPI-1205 was also studied in combination with ipilimumab in patients with advanced solid tumours (NCT03525795 - ClinicalTrials.gov, 2022b) and advanced prostate cancer in combination with enzalutamide or abiraterone/prednisone (NCT03480646), however, despite both studies having concluded, the results have not yet been published or made publicly available. Furthermore, MAK683 is currently being investigated in adults with advanced malignancies (NCT02900651 - ClinicalTrials.gov, 2024f). Similarly, DS-3201 (Valemetostat) is currently being investigated together with ipilimumab for the treatment of patients with metastatic prostate, urothelial and renal cell cancers (NCT04388852 - ClinicalTrials.gov, 2024a) and in patients with T-cell lymphoma (NCT04703192 - ClinicalTrials.gov, 2024o).
From all the known PKMTs, the inhibitors developed against EZH2 are arguably the most investigated at a clinical level (Chen et al., 2024; Duan et al., 2020). However, most inhibitors tested at clinical level focusing mostly on inhibiting tumour growth, with combination of said inhibitors with other therapeutics to inhibit tumour growth also gaining attention at a clinical level. However, whether such inhibitors also have potential in reversing chemoresistance has been given less importance at a clinical level. Thus, apart from the emphasis given to test the significance of these drugs at a preclinical level as evident from the information provided above or summarised in Table 5, further work is needed at a clinical level.
4.2 Targeting EHMT2/G9a
Several small-molecule G9a inhibitors have been investigated for their anti-cancer effects and have been used to enhance the sensitivity of cancer cells to traditional chemotherapy treatments. For instance, UNC0638 is a substrate-competitive, quinazoline-derivative G9A inhibitor that has been shown to reduce cancer cell viability, suppress migration and invasion and trigger apoptosis in neuroblastoma, breast and kidney cancer cells (Bellamy et al., 2020; Li R. et al., 2021; Liu et al., 2018). Furthermore, UNCO638 was found to induce apoptosis in EGFR-TKI resistant cells and its combination with erlotinib reduced tumour growth in NSCLC xenograft models (Wang L. et al., 2018). G9a inhibition promoted the re-expression of the PTEN gene, attenuating AKT signalling and reversing TKI resistance (Wang L. et al., 2018). Comparably, UNCO638 was found to enhance the sensitivity of resistant pancreatic cancer cells to gemcitabine by reducing the production of IL-8, which is implicated in stem-like properties and drug resistance (Pan et al., 2016). Indeed, the combination of UNC0638 and gemcitabine decreased cancer growth and metastatic properties of the gemcitabine-resistant cancer cells (Pan et al., 2016). In breast cancer, the combination of UNC0638 and the HDAC inhibitor CI-994 also induced apoptosis, decreased cancer stemness and reversed drug resistance (Lin et al., 2022).
UNC0642 is another substrate-competitive, quinazoline-derivate G9a small molecule inhibitor with better in vivo pharmacokinetic properties than UNC0638 and minimal cytotoxicity (Casciello et al., 2015). Its anticancer effects were shown in melanoma, bladder, breast and lung cancers (Cao et al., 2019; Dang et al., 2020; Pangeni et al., 2020; Yaqub et al., 2022). Moreover, G9a disruption through UNC0642 sensitised ovarian cancer cells to the PARP inhibitor Olaparib by disrupting DNA repair pathways and promoting DNA damage (Watson et al., 2019). Pangeni et al. (2020) also demonstrated that UNC0642 repressed the expression of CSC markers in lung cancer. Similarly, Zhou M et al., 2021 showed that UNC0642 reduced the survival and self-renewal capacity of leukemia stem cells, which are responsible for TKI resistance in CML.
Other G9a small molecule inhibitors like CM-272 and EML-741 that have demonstrated anti-tumoural and anti-neoplastic effects in different cancer sub-types, however more studies need to be carried out to investigate their effects on chemoresistance (Feoli et al., 2022).
4.3 Targeting other PKMTs
Although the majority of PKMT inhibitors are directed against EZH2 and G9a, other small molecules have been designed to target SYMD2, SYMD3, SETD7, SETD8 and SETDB1, with some found to aid in chemosensitivity.
Some SYMD2 inhibitors include BAY-598, AZ505, A893, EPZ033294 and LLY-507 (Fabini et al., 2019). In one study, BAY-598 reversed cisplatin resistance in NSCLC as it significantly decreased cell migration, tumour sphere number and induced apoptosis of cisplatin-treated lung cancer cells through the upregulation of p53 target genes including cyclin-dependent kinase inhibitor 1 (p21), Bcl-2-associated protein x (Bax) and Growth arrest and DNA damage-inducible protein GADD45 (GADD45) (Shang and Wei, 2019). The combination of AZ505,another a substrate-competitive SYMD2 inhibitor, together with cisplatin significantly inhibited tumour growth in NSCLC xenograft models when compared to AZ505 and cisplatin monotherapy groups (Shang and Wei, 2019).
SYMD3 is also a druggable target in cancer. For instance, BCI-121 is a substrate-competitive SYMD3 inhibitor that was found to impair the cell viability of medulloblastoma, CRC and ovarian cancer cells (Asuthkar et al., 2022; Jiang et al., 2019; Peserico et al., 2015). BCI-121 also downregulated cyclin D1/D3 and upregulated retinoblastoma (Rb) expression in medulloblastoma patient-derived xenografts, however at a high concentration BCI-121 was found to be significantly cytotoxic in the primary tumour samples (Asuthkar et al., 2022). Additionally, BCI-121 produced a dose-dependent inhibition of adhesion and invasion in ovarian cancer spheroids (Lyu et al., 2020). Nonetheless, more studies are required to investigate its effect with other chemotherapeutics and its benefit in reversing drug resistance. EPZ031686, diperodon and MS2177 are different SYMD3 inhibitors that also require further investigation as anti-cancer therapeutics.
(R)-PFI-2 is a SETD7 inhibitor that has been shown to increase the sensitivity of NSCLC cells to doxorubicin (Daks et al., 2021). Cyproheptadine is an antagonist of the histamine and serotonin receptor but also a SETD7 inhibitor. It was found to induce apoptosis in multiple myeloma cells by inhibiting PI3K/AKT signalling, a key signalling cascade involved in chemoresistance (Li et al., 2013). Hence, cyproheptadine could potentially aid in the reversal of SETD7-induced chemoresistance, however further investigations are required.
No specific small molecules directed towards SETDB1 have yet been characterised. However, studies have shown that this PKMT can be targeted through non-specific compounds such as chaetocin, paclitaxel, mithramycin and 3-deazaneplanocin A (DZNep) (Karanth et al., 2017; Liao et al., 2023). All have shown positive therapeutic effects in cancer cell lines, as reviewed by Karanth et al. (2017). For example, Hou et al. (2020) showed that mithramycin enhanced cetuximab sensitivity in KRAS-mutated CRC cells. The combination of cetuximab and mithramycin significantly reduced the size of CRC tumour xenografts as compared to either of the therapies administered individually. Further, treatment with this combination also attenuated PI3K/AKT signalling in the KRAS-mutated CRC cells (Hou et al., 2020).
UNC0379 is a substrate-competitive SETD8 inhibitor that has shown synergistic effects with the alkylating agent, melphalan in multiple myeloma (Herviou et al., 2021). In addition, UNC0379 suppressed the invasion and migration abilities of pancreatic cancer cells and decreased the proliferation of ovarian cancer cells through the induction of apoptosis (Liu M. et al., 2021; Wada et al., 2020).
Lastly, Zhang and his colleagues (2024a) have recently reviewed other PKMT inhibitors and their contribution to reversing chemoresistance in lung cancer. The inhibitors discussed in relation to resensitising lung cancer cells target either EZH2 (GSK126, GSK343, DZNep, GSK343, EPZ011989), G9a/EHMT2 (UNC0642, UNC0638), DOT1L (SGC0946), SMYD3 (EPZ031686) or SMYD2 (BAY-598), some of which were also discussed in this review.
4.4 Targeting type I PRMTs
Type I PRMT inhibitors are typically not designed to be selective towards one particular PRMT. For instance, MS023 is a potent substrate binding-site inhibitor of type I PRMTs (Eram et al., 2016). It has shown anti-tumour effects in breast cancer cells by activating an antiviral response characterised by increased interferon expression and double-stranded RNA (dsRNA) accumulation, which induces cell death (Wu et al., 2022). Treatment with MS023 in HT29 colon cancer cells significantly downregulated pathways involved in migration and invasion and increased the expression of adhesion proteins (Plotnikov et al., 2020). MS023 also reduced the clonogenic growth of ovarian cancer cells exposed to cisplatin, sensitising them to apoptosis (Musiani et al., 2020). MS023 was also found to upregulate genes involved in mitosis and cell-cycle regulation inducing apoptosis in Srsf2P95H mutant AML preferentially to wild-type cells (Fong et al., 2019). As for the use/role of MS023 in chemoresistance, its potential use is still poorly understood, however when used in with combination Talazoparib (PARP inhibitor - PARPi), it helped resensitise PARPi resistant ovarian cells to treatment, thus suggesting that type I PRMT inhibitors could mitigate resistance to PARPi inhibitors (Dominici et al., 2021).
GSK3368715 is a SAM uncompetitive type I PRMT inhibitor that entered the clinical trials (NCT03666988 - ClinicalTrials.gov, 2022a) for the treatment of solid tumours and diffuse large B-cell lymphoma. Unfortunately, it was terminated as the benefit/risk profile did not favour continuation of the study. Nonetheless, recently this inhibitor has shown to increase gemcitabine sensitivity in human and murine PDAC and PDAC PDXs, as well as delay the development of gemcitabine resistance in human PDAC xenograft models (Nguyen et al., 2024). Thus, there is still potential that this inhibitory may be of benefit in future clinical studies, particularly for reversing or preventing chemoresistance.
DB75 and TC-E5003 are two selective PRTM1 inhibitors which have been reported to reverse gemcitabine resistance in PDAC at pre-clinical level, by inhibiting the HSP70–BCL2 pathway (Wang et al., 2020a). However, their potential at a clinical level has not been tested yet, and their exact mode of inhibition is also not fully understood.
SGC707 is an allosteric PRMT3 inhibitor (Kaniskan H.Ü et al., 2015). Treatment of pancreatic cancer cells with SGC707 significantly reduced the expression of ABCG2, which sensitised the cells to gemcitabine (Hsu et al., 2018). In addition, SGC707 has been shown to inhibit glycolysis and tumour growth in hepatocellular carcinoma and glioblastoma cells (Lei et al., 2022; Liao et al., 2022). In CRC cells, SGC707 inhibited Vascular Endothelial Growth Factor A (VEGFA) expression and significantly reduced their migration and invasion abilities (Zhang et al., 2021).
4.5 Targeting PRMT5
Many PRMT5 inhibitors have shown promising pre-clinical efficacy, with some now being investigated in clinical trials. The review by Feustel and Falchook (2022) discusses the recent PRMT5 inhibitors that have made it to oncology clinical trials. One is JNJ-64619178, an inhibitor of the PRMT5-methylosome protein 50 (MEP50) complex that binds the SAM- and substrate-binding pockets (Brehmer et al., 2021). It is currently being studied in phase I clinical trials (NCT03573310 - ClinicalTrials.gov, 2024h) as a monotherapy in patients with advanced solid tumours and B-cell non-Hodgkin lymphoma (Vieito et al., 2023). Additionally, JNJ-64619178 was investigated as a synergistic tool to enhance the efficacy of Trametinib (MEK1/2 kinase inhibitor) in glioblastoma. The combination of both drugs showed an increase in the number of apoptotic cancer cells, caspase 3/7 activity and the number of cells in G1 cell cycle arrest than either individual therapy alone (Banasavadi-Siddegowda et al., 2022). Likewise, the combination of the PRMT5 inhibitor and LB100 (a protein phosphatase 2A (PP2A) inhibitor) enhanced G1 cell cycle arrest in glioblastoma cells (Otani et al., 2021).
GSK3326595 is a non-SAM competitive and substrate-competitive PRMT5 inhibitor. In mantle cell lymphoma cell lines, treatment with GSK3326595 resulted in a loss of viability and increased DNA damage characterised by downregulation of defects in DNA-damage response (DDR) genes, increased reactive oxygen species (ROS) and gamma-H2AX foci (Che et al., 2023). GSK3326595 has been studied in patients with solid tumors and non-Hodgkin’s lymphoma (NCT02783300 - ClinicalTrials.gov, 2023a), where it demonstrated positive clinical activity and manageable side effects (Siu et al., 2019). It has also been investigated in early-stage breast cancer (NCT04676516 - ClinicalTrials.gov, 2022c), but no results have been published yet for this study.
The inhibitor EPZ015666 targets the peptide-binding site of PRMT5. It has shown significant anti-tumour activities in retinoblastoma, multiple myeloma, breast, bladder and cervical cancers (Liu X. et al., 2021; Gao et al., 2021; Gullà et al., 2018; Hu et al., 2018; Vinet et al., 2019). In glioblastoma, EPZ015666 overcame therapeutic resistance to the mTOR inhibitor PP242 via synergistic tumour inhibitory effects in vitro and xenograft mouse models in combination with the mTOR inhibitor PP242 (Holmes et al., 2019). Glioblastoma cells treated with a combination of EPZ015666 and PP242 resulted in a significant loss of cell viability, an increase in the number of apoptotic cells and a reduction in the expression of c-MYC and cyclin D1 (Holmes et al., 2019). Similarly, in xenograft models, the combinatorial treatment was markedly more effective in terms of tumour growth inhibition than either monotherapy (Holmes et al., 2019).
GSK3203591 (a.k.a GSK591) is another inhibitor of PRMT5-MEP50 activity. It has been shown to induce cancer apoptosis and promote chemosensitivity to resveratrol in lung cancer cells by downregulating the AKT/GSK3β/cyclin D and E signalling pathway (Li Y. et al., 2019). In agreement, Zhang et al. (2019) showed that GSK591 suppressed lung cancer cell proliferation by downregulating phosphorylated AKT together with cyclins D and E, preventing cell cycle progression. Moreover, GSK591 significantly reduced the proliferation and self-renewal potential of breast CSCs, an effect that was enhanced in combination with 4-hydroxytamoxifen (Chiang et al., 2017). It was also shown to be 10-fold more effective at killing Srsf2P95H mutant AML compared to the WT counterpart (Fong et al., 2019).
PRT543 is a potent, selective, oral PRMT5 inhibitor that binds to the substrate recognition site of PRMT5 and inhibits its methyltransferase activity. It was shown to have pre-clinical antitumour activity in Splicing Mutant Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML) (Schwartz et al., 2021). Also at pre-clinical stage, PRT543 on Splicing factor 3b subunit 1 (SF3B1) uveal melanoma MEL202 (SF3B1R625G active mutant) and MEL270 (SF3B1WT) cell lines showed downregulation of SF3B1 target genes associated with increased intron retention, predominantly in the MEL202 mutant. In combination with DNA-alkylating agents or PARP inhibitors, PRT543 produced a synergistic reduction in cell viability through the regulation of cancer-associated RNA splicing machinery and the DNA damage response (Ito et al., 2021). This led to the open-label phase 1 study NCT03886831 (ClinicalTrials.gov, 2023b), which assessed the safety and efficacy of PRT543 in 23 unselected patients with refractory disease to established therapies, 12 with myelofibrosis and 11 with myelodysplastic syndrome. The results showed that PRT543 was safe and tolerated, presenting a dose-dependent inhibition of PRMT5. It is thus being evaluated either as monotherapy in patients with at least one spliceosome mutation or in combination with ruxolitinib in myelofibrosis patients demonstrating a sub-optimal response to ruxolitinib (Patel et al., 2021). The safety, tolerability and preliminary efficacy of PRT543 was also analysed in another cohort consisting of 56 patients with recurrent and/or metastatic adenoid cystic carcinoma (ACC), since there is currently no approved systemic treatment. The results showed that PRT543 was tolerable and 57% of the patients benefited from PRT543 administration, with 70% of patients presenting stable disease, however the efficacy was limited (Ferrarotto et al., 2024). At pre-clinical level, PRT543 has shown that when used in combination with venetoclax (a BCL-2 inhibitor), it sensitised a mouse model of ibrutinib-resistant mantle cell lymphoma (MCL). Combination treatment with well-tolerated doses of venetoclax and PRT543 in MCL in vivo models showed synergistic anti-tumour activity without evidence of toxicity, thus justifying that this combination strategy could be advanced to clinical setting (Brown et al., 2019).
The recent inhibitor, AZD3470, which inhibits PRMT5 activity by binding to the PRMT in the presence of methylthioadenosine (MTA), in methylthioadenosine phosphorylase (MTAP)-deficient tumor cells (Spira et al., 2024), has recently been developed by AstraZeneca and is currently being tested in two different clinical trials (NCT06137144 - ClinicalTrials.gov, 2024p and NCT06130553 - ClinicalTrials.gov, 2024g). In one of the clinical trials undergoing (NCT06137144), this inhibitor is being tested as a monotherapy in combination with other anticancer agents (not provided) in patients with relapsed/refractory haematologic malignancies. Among the participants anticipated in this study, patients who have previously received at least three prior lines of therapy for the treatment of Classical Hodgkin lymphoma (cHL), and must have exhausted all available therapies with demonstrated clinical benefit are expected to take part. Through this study, the safety, tolerability, pharmacokinetics and preliminary efficacy following oral administration of AZD3470 as monotherapy or in combination will be investigated. Thus, this novel PRMT5 inhibitor can serve as potential therapy to sensitise patient to therapy used for cHL.
The inhibitor DS-47, which targets not only PRMT5, but also PRMT7, is a SAM-competitive inhibitor which has been tested against PRMT5 inhibition. Pharmacological inhibition of PRMT5 by DS-437 resulted in reduced human regulatory T cells functions and inhibited the methylation of FOXP3 at R51. Furthermore, this inhibition resulted in enhanced anti-tumour effects of anti-erbB2/neu monoclonal antibody targeted therapy in Balb/c mice having CT26Her2 tumours by inhibiting regulatory T cells function and induction of tumour immunity (Nagai et al., 2019). Moreover, the same inhibitor also demonstrated that inhibition of PRMT5 significantly reduced gemcitabine resistance in pancreatic cancer caused by UBR7 depletion (Table 4) (Bi et al., 2023). Despite presenting potential for reversing resistance, this inhibitor is yet to be investigated at a clinical level. Lastly, there are other PRMT5 inhibitors which have been tested at pre-clinical level, including some for reversing chemoresistance (PRT382, YQ36286), as well reviewed by others (Feustel and Falchook, 2022).
5 Conclusion and future directions
Drug resistance is one of the major challenges of cancer treatment since it greatly limits the therapeutic options for the patient and worsens the prognosis. With the current push towards more personalised medicine, combining current chemotherapeutic regimens with specific PMT inhibitors could in the future be a strategy to ensure that a sub-set of patients can successfully benefit in the long-term from the treatment being administered. However, in order to achieve this there is a need for better understanding of the role of PKMTs and PRMTs in chemoresistance, through the collection of more comprehensive data from well-characterised patient cohorts. This is because most of the current published work on PMT inhibitors has focused on inhibiting enzymes which are dysregulated in relation to tumorigenesis and not chemoresistance, such that a more focused investigation is required in relation to whether chemoresistance can be reversed when administering such inhibitors and the identification of chemoresistance-specific targets.
Despite being discovered years ago (Ambler and Rees, 1959), protein methylation has not been as widely studied as other PTMs due to the lack of confident identification, efficient enrichment strategies and experimental techniques (Deng et al., 2016; Levy, 2019; Micallef and Baron, 2023; Wang et al., 2017). In particular there needs to be a thorough understanding of the biological significance and role of non-histone proteins as substrates of PMTs in chemoresistance, which is still far from properly characterised. Technological advances in mass spectrometry for PTM identification as well as various methodological developments in the analysis of methyltransferase expression and activity will be required to obtain crucial information from clinical samples.
Another important aspect to investigate is how PMTs are involved in the mechanism of the different chemotherapeutic drugs and lead to cancer cell evasion once dysregulated, which also requires the collection of much more evidence from different sources. The dysregulation of both PKMTs (such as EZH2, G9A, SETD7, SETD8, SYMD2 and SYMD3) and PRMTs (such as PRMT1, PRMT3, PRMT5 and PRMT6) in tumours has been shown to promote drug resistance in various ways such as by CSC renewal, activation of key signalling pathways like the Wnt/Notch pathway and through cell cycle progression. This would provide key mechanistic information and insight into how to design future inhibitors which would be specifically synergistic with selected chemotherapeutic drugs. Targeting PMTs strategically using small inhibitory molecules would aid in the reversal of drug resistance, thus promoting cell death within the evading cancer cells.
Currently inhibitors are only available against a very small number of the known PMTs and the specificity and sensitivity of the available molecules are not always of the desired quality. In order to improve their efficacy, apart from developing more iterations of these inhibitory molecules, it would be ideal to design new inhibitory mechanisms. However, this option is somewhat limited considering that all PMTs use the same donor and mechanism to add methylation, and they fall within 2 families making their active sites particularly similar. Recently, the use of proteolysis-targeting chimera (PROTAC) or enzyme-substrate adaptor interaction inhibitors to inhibit methylation events mediate by PMTs have started to be implement against some PKMTs (Velez et al., 2023; Velez et al., 2024) and PRMTs (Martin et al., 2024; McKinney et al., 2021; Shen et al., 2020). These two approaches could be implemented to the development of future inhibitors against PMTs, offering additional novel inhibitory mechanisms to the ones already implemented (Wang M. Y. et al., 2022). Nevertheless, irrespective of the route chosen, therapeutic targeting of PKMTs and PRMTs is expected to continue expanding in the near future.
However, to do this effectively there needs to be a clear understanding of why a number of these inhibitors have failed in either pre-clinical or clinical trials. PMT inhibitors have failed to demonstrate sufficient therapeutic efficacy at non-toxic doses in clinical trials. Their broad range of targets results in unacceptable side effects that outweigh their potential benefits. This effectively translates into a very narrow therapeutic range for an effective dose that has acceptable toxicity in humans, making achieving therapeutic effects safely extremely challenging. Building on this, more clinically relevant inhibitor testing and better biochemical understanding will also allow for an improvement in silico prediction and assist in shortlisting further potential PMT inhibitors. Moreover, we still lack many specifics on the mode of action of each inhibitor, hindering the design of better inhibitors. The similarity between the active sites of enzymes within the same family make PMT inhibitor specificity probably the greatest challenge to overcome in PMT inhibitor targeting for safe clinical application. Considering that each enzyme has hundreds of substrate proteins fulfilling very diverse cellular roles it is expected that perturbing the enzyme concentration will have implications on numerous biochemical pathways, some of which may be essential and non-redundant for non-cancerous cells.
A final point to consider is that the ability of cancer cells to alter the activity of specific proteins through PTMs including methylation is only one of the mechanisms by which tumours develop chemoresistance. Thus it might be necessary to use multiple inhibitory molecules in combination in order to reverse chemoresistance. Further investigating the significance of combinatory targeting with other, already approved, drugs such as immunomodulatory (particularly immune checkpoint inhibitors) or signalling pathway modulatory molecules (PI3K, MAPK or Wnt pathway inhibitors) along with PMT inhibitors might be one way of going about it. Finding the right balance between the different inhibitors will be extremely challenging, given the key role such pathways play, but a potential avenue nonetheless. Moreover, a similar issue which will have to be faced has to do with the development of resistance against the PMT inhibitors themselves, something which has already started being observed for some PMTs, such as the EZH2 inhibitors (Chen et al., 2024; Duan et al., 2020; Feoli et al., 2022). Thus, solving one issue can result in another problem arising and so forth.
Author contributions
IM: Conceptualization, Writing–original draft, Writing–review and editing. KF: Writing–original draft, Writing–review and editing. BB: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abumustafa, W., Castven, D., Becker, D., Salih, S. S., Manzoor, S., Zamer, B. A., et al. (2024). Inhibition of PRMT5-mediated regulation of DKK1 sensitizes colorectal cancer cells to chemotherapy. Cell. Signal. 119, 111166. doi:10.1016/j.cellsig.2024.111166
Al-Hamashi, A. A., Diaz, K., and Huang, R. (2020). Non-histone arginine methylation by protein arginine methyltransferases. Curr. Protein Peptide Sci. 21 (7), 699–712. doi:10.2174/1389203721666200507091952
Allis, C. D., Berger, S. L., Cote, J., Dent, S., Jenuwien, T., Kouzarides, T., et al. (2007). New Nomenclature for chromatin-Modifying enzymes. Cell 131 (4), 633–636. doi:10.1016/j.cell.2007.10.039
Ambler, R. P., and Rees, M. W. (1959). Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature 184 (4679), 56–57. doi:10.1038/184056b0
Asuthkar, S., Venkataraman, S., Avilala, J., Shishido, K., Vibhakar, R., Veo, B., et al. (2022). SMYD3 promotes cell cycle progression by inducing cyclin D3 transcription and stabilizing the cyclin D1 protein in medulloblastoma. Cancers 14 (7), 1673. doi:10.3390/cancers14071673
Au, S. L. K., Wong, C. C. L., Lee, J. M. F., Wong, C. M., and Ng, I. O. L. (2013). EZH2-mediated H3K27me3 is involved in epigenetic repression of deleted in liver cancer 1 in human cancers. PloS one 8 (6), 68226. doi:10.1371/journal.pone.0068226
Banasavadi-Siddegowda, Y. K., Namagiri, S., Otani, Y., Sur, H., Rivas, S., Bryant, J., et al. (2022). Targeting protein arginine methyltransferase 5 sensitizes glioblastoma to trametinib. Neuro-Oncology Adv. 4 (1), 95. doi:10.1093/noajnl/vdac095
Barski, A., Cuddapah, S., Cui, K., Roh, T.-Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129 (4), 823–837. doi:10.1016/j.cell.2007.05.009
Bedford, M. T., and Clarke, S. G. (2009). Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33 (1), 1–13. doi:10.1016/j.molcel.2008.12.013
Bekric, D., Neureiter, D., Ablinger, C., Dobias, H., Beyreis, M., Ritter, M., et al. (2023). Evaluation of tazemetostat as a therapeutically relevant substance in biliary tract cancer. Cancers 15 (5), 1569. doi:10.3390/cancers15051569
Bellamy, J., Szemes, M., Melegh, Z., Dallosso, A., Kollareddy, M., Catchpoole, D., et al. (2020). Increased efficacy of histone methyltransferase G9a inhibitors against MYCN-amplified neuroblastoma. Front. Oncol. 10, 818. doi:10.3389/fonc.2020.00818
Bennett, R. L., Swaroop, A., Troche, C., and Licht, J. D. (2017). The role of nuclear receptor–binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med. 7 (6), a026708. doi:10.1101/cshperspect.a026708
Bi, L., Zhao, M., Ru, K., Tang, B., and Wang, Y. (2023). Effect of UBR7 deficiency on glycolysis and PDAC resistance to gemcitabine by promoting PRMT5 stability. J. Clin. Oncol. 14 (16), e16265. doi:10.1200/JCO.2023.41.16_suppl.e16265
Blanc, R. S., and Richard, S. (2017). Arginine methylation: the coming of Age. Mol. Cell 65 (1), 8–24. doi:10.1016/j.molcel.2016.11.003
Bonner, E. A., and Lee, S. C. (2023). Therapeutic targeting of RNA splicing in cancer. Genes 14 (7), 1378. doi:10.3390/genes14071378
Bradley, R. K., and Anczuków, O. (2023). RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer 23 (3), 135–155. doi:10.1038/s41568-022-00541-7
Brehmer, D., Beke, L., Wu, T., Millar, H. J., Moy, C., Sun, W., et al. (2021). Discovery and pharmacological characterization of JNJ-64619178, a novel small-molecule inhibitor of PRMT5 with potent antitumor activity. Mol. Cancer Ther. 20 (12), 2317–2328. doi:10.1158/1535-7163.MCT-21-0367
Brown, F., Zhang, Y., Hinterschied, C., Prouty, A., Sloan, S., Helmig-Mason, J., et al. (2019). PRMT5 inhibition drives therapeutic vulnerability to BCL-2 inhibition with Venetoclax and provides rationale for combination therapy in mantle cell lymphoma. Blood 134, 302. doi:10.1182/blood-2019-130797
Cao, Y., Sun, J., Li, M., Dong, Y., Zhang, Y., Yan, J., et al. (2019). Inhibition of G9a by a small molecule inhibitor, UNC0642, induces apoptosis of human bladder cancer cells. Acta Pharmacol. Sin. 40 (8), 1076–1084. doi:10.1038/s41401-018-0205-5
Carlson, S. M., and Gozani, O. (2016). Nonhistone lysine methylation in the regulation of cancer pathways. Cold Spring Harb. Perspect. Med. 6 (11), a026435. doi:10.1101/cshperspect.a026435
Casciello, F., Windloch, K., Gannon, F., and Lee, J. S. (2015). Functional role of G9a histone methyltransferase in cancer. Front. Immunol. 6, 487. doi:10.3389/fimmu.2015.00487
Che, N., Ng, K., Wong, T., Tong, M., Kau, P. W., Chan, L., et al. (2021). PRMT6 deficiency induces autophagy in hostile microenvironments of hepatocellular carcinoma tumors by regulating BAG5-associated HSC70 stability. Cancer Lett. 501, 247–262. doi:10.1016/j.canlet.2020.11.002
Che, Y., Liu, Y., Yao, Y., Hill, H. A., Li, Y., Cai, Q., et al. (2023). Exploiting PRMT5 as a target for combination therapy in mantle cell lymphoma characterized by frequent ATM and TP53 mutations. Blood Cancer J. (New York) 13 (1), 27. doi:10.1038/s41408-023-00799-6
Chen, K., Zhang, F., Ding, J., Liang, Y., Zhan, Z., Zhan, Y., et al. (2017). Histone methyltransferase SETDB1 promotes the progression of colorectal cancer by inhibiting the expression of TP53. J. Cancer 8 (16), 3318–3330. doi:10.7150/jca.20482
Chen, Y., Zhu, H., Luo, Y., Tong, S., and Liu, Y. (2024). EZH2: the roles in targeted therapy and mechanisms of resistance in breast cancer. Biomed. and Pharmacother. 175, 116624. doi:10.1016/j.biopha.2024.116624
Cheng, X., Collins, R. E., and Zhang, X. (2005). Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophysics Biomol. Struct. 34 (1), 267–294. doi:10.1146/annurev.biophys.34.040204.144452
Chiang, K., Zielinska, A. E., Shaaban, A. M., Sanchez-Bailon, M. P., Jarrold, J., Clarke, T. L., et al. (2017). PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep. 21 (12), 3498–3513. doi:10.1016/j.celrep.2017.11.096
Clarke, S. G. (2013). Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem. Sci. 38 (5), 243–252. doi:10.1016/j.tibs.2013.02.004
ClinicalTrials.gov (2020). A study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK2816126 in subjects with relapsed/Refractory diffuse large B cell lymphoma, transformed follicular lymphoma, other non-hodgkin's lymphomas, solid tumors and multiple myeloma [NCT02082977]. Available at: https://clinicaltrials.gov/study/NCT02082977?term=NCT02082977&limit=10&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2021). Study of the EZH2 inhibitor tazemetostat in malignant mesothelioma. (NCT02860286). Available at: https://clinicaltrials.gov/study/NCT02860286?cond=Cancer&intr=Tazemetostat&page=2&rank=19#publications(Accessed August, 2024).
ClinicalTrials.gov (2022a). First time in humans (FTIH) study of GSK3368715 in participants with solid tumors and diffuse large B-cell lymphoma (DLBCL) [NCT03666988]. Available at: https://clinicaltrials.gov/study/NCT03666988?term=NCT03666988&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2022b). ORIOn-E: a study evaluating CPI-1205 in patients with advanced solid tumors. (NCT03525795). Available at: https://clinicaltrials.gov/study/NCT03525795#more-information (Accessed August, 2024).
ClinicalTrials.gov (2022c). A phase II window of opportunity trial of PRMT5 inhibitor, GSK3326595, in early stage breast cancer (OTT-19-06). (NCT04676516). Available at: https://clinicaltrials.gov/study/NCT04676516?intr=GSK3326595&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2022d). A study evaluating CPI-1205 in patients with B-cell lymphomas. (NCT02395601). Available at: https://clinicaltrials.gov/study/NCT02395601?intr=CPI-1205&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2023a). An open-label, dose escalation study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK3326595 in participants with solid tumors and non-hodgkin's lymphoma (meteor 1). (NCT02783300). Available at: https://clinicaltrials.gov/study/NCT02783300?intr=GSK3326595&rank=3&tab=results [Accessed August 2024].
ClinicalTrials.gov (2023b). A study of PRT543 in participants with advanced solid tumors and hematologic malignancies. (NCT03886831). Available at: https://clinicaltrials.gov/study/NCT03886831?intr=PRT543&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2024a). DS3201 and ipilimumab for the treatment of metastatic prostate, urothelial and renal cell cancers. (NCT04388852). Available at: https://clinicaltrials.gov/study/NCT04388852?intr=DS-3201%20&rank=2 (Accessed August, 2024).
ClinicalTrials.gov (2024b). EZH2 inhibitor, tazemetostat, and PD-1 blockade for treatment of advanced non-small cell lung cancer. (NCT05467748). Available at: https://clinicaltrials.gov/study/NCT05467748?cond=Cancer&intr=Tazemetostat&page=2&rank=11 (Accessed August, 2024).
ClinicalTrials.gov (2024c). Mevrometostat treatment of relapsed/Refractory SCLC, castration resistant prostate cancer, and follicular lymphoma. (NCT03460977). Available at: https://clinicaltrials.gov/study/NCT03460977?cond=PF-06821497&rank=4 (Accessed August, 2024).
ClinicalTrials.gov (2024d). Phase ia/Ib talazoparib + tazemetostat for mCRPC. (NCT04846478). Available at: https://clinicaltrials.gov/study/NCT04846478?cond=Cancer&intr=Tazemetostat&page=2&rank=16 (Accessed August, 2024).
ClinicalTrials.gov (2024e). Phase II study of tazemetostat in solid tumors harboring an ARID1A mutation. (NCT05023655). Available at: https://clinicaltrials.gov/study/NCT05023655?cond=Cancer&intr=Tazemetostat&rank=3 (Accessed August, 2024).
ClinicalTrials.gov (2024f). Safety and efficacy of MAK683 in adult patients with advanced malignancies. (NCT02900651). Available at: https://clinicaltrials.gov/study/NCT02900651?intr=MAK683&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2024g). A study of AZD3470, a PRMT5 inhibitor, in patients with MTAP deficient advanced/Metastatic solid tumours (PRIMROSE). Available at: https://clinicaltrials.gov/study/NCT06130553?term=NCT06130553&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2024h). A study of JNJ-64619178, an inhibitor of PRMT5 in participants with advanced solid tumors, NHL, and lower risk MDS. Available at: https://clinicaltrials.gov/study/NCT03573310?intr=JNJ-64619178&rank=1 (Accessed August, 2024).NCT03573310
ClinicalTrials.gov (2024i). Study of tazemetostat in lymphoid malignancies. (NCT05983965). Available at: https://clinicaltrials.gov/study/NCT05983965?cond=Cancer&intr=Tazemetostat&rank=6 (Accessed August, 2024).
ClinicalTrials.gov (2024j). A study to assess the long-term safety of tazemetostat (TRuST). (NCT02875548). Available at: https://clinicaltrials.gov/study/NCT02875548?cond=Cancer&intr=Tazemetostat&page=2&rank=12 (Accessed August, 2024).
ClinicalTrials.gov (2024k). A study to learn about the investigational medicine called PF-06821497 mevrometostat in men with mCRPC who were previously treated with abiraterone acetate for prostate cancer (MEVPRO-1). (NCT06551324). Available at: https://clinicaltrials.gov/study/NCT06551324?cond=PF-06821497&rank=3 (Accessed August, 2024).
ClinicalTrials.gov (2024l). Tazemetostat in malignant peripheral nerve sheath tumors. (NCT04917042). Available at: https://clinicaltrials.gov/study/NCT04917042?cond=Cancer&intr=Tazemetostat&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2024m). Tazemetostat in treating patients with relapsed or refractory advanced solid tumors, non-hodgkin lymphoma, or histiocytic disorders with EZH2, SMARCB1, or SMARCA4 gene mutations (A pediatric MATCH treatment trial). (NCT03213665). Available at: https://clinicaltrials.gov/study/NCT03213665?cond=Cancer&intr=Tazemetostat&page=2&rank=13 (Accessed August, 2024).
ClinicalTrials.gov (2024n). Testing the safety of the anti-cancer drugs tazemetostat and belinostat in patients with lymphomas that have resisted treatment [NCT05627245]. Available at: https://clinicaltrials.gov/study/NCT05627245?term=NCT05627245&rank=1 (Accessed August, 2024).
ClinicalTrials.gov (2024o). Valemetostat tosylate (DS-3201b), an enhancer of zeste homolog (EZH) 1/2 dual inhibitor, for relapsed/Refractory peripheral T-cell lymphoma (VALENTINE-PTCL01). (NCT04703192). Available at: https://clinicaltrials.gov/study/NCT04703192?intr=%5D&term=NCT04703192&rank=1&tab=results (Accessed August, 2024).
ClinicalTrials.gov (2024p). AZD3470 as monotherapy and in combination with anticancer agents in participants with relapsed/Refractory haematologic malignancies. (PRIMAVERA) [NCT06137144]. Available at: https://clinicaltrials.gov/study/NCT06137144?term=NCT06137144&rank=1 (Accessed August, 2024).
Cloutier, P., Lavallée-Adam, M., Faubert, D., Blanchette, M., and Coulombe, B. (2013). A newly uncovered group of Distantly related lysine methyltransferases preferentially interact with molecular Chaperones to regulate their activity. PLoS Genet. 9 (1), 1003210. doi:10.1371/journal.pgen.1003210
Copeland, R. A., Solomon, M. E., and Richon, V. M. (2009). Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 8 (9), 724–732. doi:10.1038/nrd2974
Cornett, E. M., Ferry, L., Defossez, P.-A., and Rothbart, S. B. (2019). Lysine methylation regulators Moonlighting outside the Epigenome. Mol. Cell 75 (6), 1092–1101. doi:10.1016/j.molcel.2019.08.026
Creighton, C. J., Li, X., Landis, M., Dixon, J. M., Neumeister, V. M., Sjolund, A., et al. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. 106 (33), 13820–13825. doi:10.1073/pnas.0905718106
Dai, X., Ren, T., Zhang, Y., and Nan, N. (2021). Methylation multiplicity and its clinical values in cancer. Expert Rev. Mol. Med. 23, 2. doi:10.1017/erm.2021.4
Daks, A., Mamontova, V., Fedorova, O., Petukhov, A., Shuvalov, O., Parfenyev, S., et al. (2021). Set7/9 controls proliferation and genotoxic drug resistance of NSCLC cells. Biochem. Biophysical Res. Commun. 572, 41–48. doi:10.1016/j.bbrc.2021.07.086
Dang, N., Jiao, J., Meng, X., An, Y., Han, C., and Huang, S. (2020). Abnormal overexpression of G9a in melanoma cells promotes cancer progression via upregulation of the Notch1 signaling pathway. Aging (Albany NY) 12 (3), 2393–2407. doi:10.18632/aging.102750
Deng, W., Wang, Y., Ma, L., Zhang, Y., Ullah, S., and Xue, Y. (2016). Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Briefings Bioinforma. 18 (4), 647–658. doi:10.1093/bib/bbw041
Deng, X., Kong, F., Li, S., Jiang, H., Dong, L., Xu, X., et al. (2021). A KLF4/PiHL/EZH2/HMGA2 regulatory axis and its function in promoting oxaliplatin-resistance of colorectal cancer. Cell death and Dis. 12 (5), 485. doi:10.1038/s41419-021-03753-1
Dominici, C., Sgarioto, N., Yu, Z., Sesma-Sanz, L., Masson, J. Y., Richard, S., et al. (2021). Synergistic effects of type I PRMT and PARP inhibitors against non-small cell lung cancer cells. Clin. Epigenetics 13, 54. doi:10.1186/s13148-021-01037-1
Duan, G., and Walther, D. (2015). The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 11 (2), e1004049. doi:10.1371/journal.pcbi.1004049
Duan, R., Du, W., and Guo, W. (2020). EZH2: a novel target for cancer treatment. J. Hematol. and Oncol. 13, 104. doi:10.1186/s13045-020-00937-8
El Amrani, M., Corfiotti, F., Corvaisier, M., Vasseur, R., Fulbert, M., Skrzypczyk, C., et al. (2019). Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol. Carcinog. 58 (11), 1985–1997. doi:10.1002/mc.23090
Eram, M. S., Shen, Y., Szewczyk, M. M., Wu, H., Senisterra, G., Li, F., et al. (2016). A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 11 (3), 772–781. doi:10.1021/acschembio.5b00839
Fabini, E., Manoni, E., Ferroni, C., Rio, A. D., and Bartolini, M. (2019). Small-molecule inhibitors of lysine methyltransferases SMYD2 and SMYD3: current trends. Future Med. Chem. 11 (8), 901–921. doi:10.4155/fmc-2018-0380
Falnes, P. O., Jakobsson, M. E., Davydova, E., Ho, A., and Ma ecki, J. (2016). Protein lysine methylation by seven-β-strand methyltransferases. Biochem. J. 473 (14), 1995–2009. doi:10.1042/bcj20160117
FDA (2020a). FDA approves tazemetostat for advanced epithelioid sarcoma. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tazemetostat-advanced-epithelioid-sarcoma (Accessed August, 2024).doi:10.31525/cmr-279c348
FDA (2020b). FDA granted accelerated approval to tazemetostat for follicular lymphoma. Available at: https://www.fda.gov/drugs/fda-granted-accelerated-approval-tazemetostat-follicular-lymphoma#:∼:text=On%20June%2018%2C%202020%2C%20the,approved%20test%20and%20who%20have (Accessed August, 2024).
Feoli, A., Viviano, M., Cipriano, A., Milite, C., Castellano, S., and Sbardella, G. (2022). Lysine methyltransferase inhibitors: where we are now. RSC Chem. Biol. 3 (4), 359–406. doi:10.1039/d1cb00196e
Ferrarotto, R., Swiecicki, P. L., Zandberg, D. P., Baiocchi, R. A., Wesolowski, R., Rodriguez, C. P., et al. (2024). PRT543, a protein arginine methyltransferase 5 inhibitor, in patients with advanced adenoid cystic carcinoma: an open-label, phase I dose-expansion study. Oral Oncol. 149, 106634. doi:10.1016/j.oraloncology.2023.106634
Ferreira de Freitas, R., Ivanochko, D., and Schapira, M. (2019). Methyltransferase inhibitors: Competing with, or exploiting the bound cofactor. Molecules 24 (24), 4492. doi:10.3390/molecules24244492
Feustel, K., and Falchook, G. S. (2022). Protein arginine methyltransferase 5 (PRMT5) inhibitors in oncology clinical trials: a review. J. Immunother. Precis. Oncol. 5 (3), 58–67. doi:10.36401/JIPO-22-1
Fong, J. Y., Pignata, L., Goy, P. A., Kawabata, K. C., Lee, S. C., Koh, C. M., et al. (2019). Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36 (2), 194–209. doi:10.1016/j.ccell.2019.07.003
Fulton, M. D., Brown, T., and Zheng, Y. G. (2018). Mechanisms and inhibitors of histone arginine methylation. Chem. Rec. 18 (12), 1792–1807. doi:10.1002/tcr.201800082
Gao, J., Liu, R., Feng, D., Huang, W., Huo, M., Zhang, J., et al. (2021). Snail/PRMT5/NuRD complex contributes to DNA hypermethylation in cervical cancer by TET1 inhibition. Cell Death Differ. 28 (9), 2818–2836. doi:10.1038/s41418-021-00786-z
Gardner, E. E., Lok, B. H., Schneeberger, V. E., Desmeules, P., Miles, L. A., Arnold, P. K., et al. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31 (2), 286–299. doi:10.1016/j.ccell.2017.01.006
Gouda, M. B. Y., Zidane, M. A., Abdelhady, A. S., and Hassan, N. M. (2022). Expression and prognostic significance of chromatin modulators EHMT2/G9a and KDM2b in acute myeloid leukemia. J. Cell. Biochem. 123 (8), 1340–1355. doi:10.1002/jcb.30297
Greer, E. L., and Yang, S. (2012). Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13 (5), 343–357. doi:10.1038/nrg3173
Gulati, N., Béguelin, W., and Giulino-Roth, L. (2018). Enhancer of zeste homolog 2 (EZH2) inhibitors. Leukemia and lymphoma 59 (7), 1574–1585. doi:10.1080/10428194.2018.1430795
Gullà, A., Hideshima, T., Bianchi, G., Fulciniti, M., Kemal Samur, M., Qi, J., et al. (2018). Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 32 (4), 996–1002. doi:10.1038/leu.2017.334
Hamamoto, R., and Nakamura, Y. (2016). Dysregulation of protein methyltransferases in human cancer: an emerging target class for anticancer therapy. Cancer Sci. 107 (4), 377–384. doi:10.1111/cas.12884
Hamamoto, R., Saloura, V., and Nakamura, Y. (2015). Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 15 (2), 110–124. doi:10.1038/nrc3884
Harb, W., Abramson, J., Lunning, M., Goy, A., Maddocks, K., Lebedinsky, C., et al. (2018). A phase 1 study of CPI-1205, a small molecule inhibitor of EZH2, preliminary safety in patients with B-cell lymphomas. Ann. Oncol. 29 (29), 7. doi:10.1093/annonc/mdy048.001
He, C., Liu, C., Wang, L., Sun, Y., Jiang, Y., and Hao, Y. (2019). Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death and Dis. 10 (2), 65. doi:10.1038/s41419-019-1347-1
Herviou, L., Ovejero, S., Izard, F., Karmous-Gadacha, O., Gourzones, C., Bellanger, C., et al. (2021). Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma. Clin. Epigenetics 13 (1), 174. doi:10.1186/s13148-021-01160-z
Holmes, B., Benavides-Serrato, A., Saunders, J. T., Landon, K. A., Schreck, A. J., Nishimura, R. N., et al. (2019). The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J. Neuro-Oncology 145 (1), 11–22. doi:10.1007/s11060-019-03274-0
Hou, Z., Sun, L., Xu, F., Hu, F., Lan, J., Song, D., et al. (2020). Blocking histone methyltransferase SETDB1 inhibits tumorigenesis and enhances cetuximab sensitivity in colorectal cancer. Cancer Lett. 487, 63–73. doi:10.1016/j.canlet.2020.05.029
Hsu, D. S. S., Hwang, W. L., Yuh, C. H., Chu, C. H., Ho, Y. H., Chen, P. B., et al. (2017). Lymphotoxin-β interacts with methylated EGFR to mediate acquired resistance to cetuximab in head and neck cancer. Clin. Cancer Res. 23 (15), 4388–4401. doi:10.1158/1078-0432.CCR-16-1955
Hsu, M. C., Pan, M. R., Chu, P. Y., Tsai, Y. L., Tsai, C. H., Shan, Y. S., et al. (2018). Protein arginine methyltransferase 3 enhances chemoresistance in pancreatic cancer by methylating hnRNPA1 to increase ABCG2 expression. Cancers 11 (1), 8. doi:10.3390/cancers11010008
Hsu, W., Chen, C., Chang, Y., Cheng, C., TsaI, Y., and Lin, C. (2021). PRMT1 confers resistance to olaparib via modulating MYC signaling in triple-negative breast cancer. J. Personalized Med. 11 (10), 1009. doi:10.3390/jpm11101009
Hu, G., Wang, X., Han, Y., and Wang, P. (2018). Protein arginine methyltransferase 5 promotes bladder cancer growth through inhibiting NF-kB dependent apoptosis. EXCLI J. 17, 1157–1166. doi:10.17179/excli2018-1719
Hu, H., Qian, K., Ho, M. C., and Zheng, Y. G. (2016). Small molecule inhibitors of protein arginine methyltransferases. Expert Opin. investigational drugs 25 (3), 335–358. doi:10.1517/13543784.2016.1144747
Huang, T., Lin, C., Zhong, L. L., Zhao, L., Zhang, G., Lu, A., et al. (2017). Targeting histone methylation for colorectal cancer. Ther. Adv. gastroenterology 10 (1), 114–131. doi:10.1177/1756283X16671287
Husmann, D., and Gozani, O. (2019). Histone lysine methyltransferases in biology and disease. Nat. Struct. and Mol. Biol. 26 (10), 880–889. doi:10.1038/s41594-019-0298-7
Hwang, J. W., Cho, Y., Bae, G. U., Kim, S. N., and Kim, Y. K. (2021). Protein arginine methyltransferases: promising targets for cancer therapy. Exp. and Mol. Med. 53 (5), 788–808. doi:10.1038/s12276-021-00613-y
Ito, K., Thodima, V., Carter, J., Bhagwat, N., Sivakumar, M., Grego, A., et al. (2021). Abstract 1137: PRMT5 inhibition regulates alternative splicing and DNA damage repair pathways in SF3B1 R625G expressing uveal melanoma cells. Cancer Res. 81 (13), 1137. doi:10.1158/1538-7445.AM2021-1137
Jiang, Y., Lyu, T., Che, X., Jia, N., Li, Q., and Feng, W. (2019). Overexpression of SMYD3 in ovarian cancer is associated with ovarian cancer proliferation and apoptosis via methylating H3K4 and H4K20. J. Cancer 10 (17), 4072–4084. doi:10.7150/jca.29861
Jin, M. L., Yang, L., and Jeong, K. W. (2022). SETD1A-SOX2 axis is involved in tamoxifen resistance in estrogen receptor α-positive breast cancer cells. Theranostics 12 (13), 5761–5775. doi:10.7150/thno.72599
Kaniskan, H. U., Konze, K. D., and Jin, J. (2015a). Selective inhibitors of protein methyltransferases. J. Med. Chem. 58 (4), 1596–1629. doi:10.1021/jm501234a
Kaniskan, H. U., Martini, M. L., and Jin, J. (2018). Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 118 (3), 989–1068. doi:10.1021/acs.chemrev.6b00801
Kaniskan, H. Ü., Szewczyk, M. M., Yu, Z., Eram, M. S., Yang, X., Schmidt, K., et al. (2015b). A potent, selective and cell-active allosteric inhibitor of protein arginine Methyltransferase 3 (PRMT3). Angew. Chem. Int. Ed. 54 (17), 5166–5170. doi:10.1002/anie.201412154
Karanth, A. V., Maniswami, R. R., Prashanth, S., Govindaraj, H., Padmavathy, R., Jegatheesan, S. K., et al. (2017). Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets 21 (3), 319–331. doi:10.1080/14728222.2017.1279604
Lei, Y., Han, P., Chen, Y., Wang, H., Wang, S., Wang, M., et al. (2022). Protein arginine methyltransferase 3 promotes glycolysis and hepatocellular carcinoma growth by enhancing arginine methylation of lactate dehydrogenase A. Clin. Transl. Med. 12 (1), 686. doi:10.1002/ctm2.686
Levy, D. (2019). Lysine methylation signaling of non-histone proteins in the nucleus. Cellular and Molecular Life Sciences. Cell. Mol. Life Sci. 76 (15), 2873–2883. doi:10.1007/s00018-019-03142-0
Li, H., Ma, X., Yang, D., Suo, Z., Dai, R., and Liu, C. (2019a). PCAT-1 contributes to cisplatin resistance in gastric cancer through epigenetically silencing PTEN via recruiting EZH2. J. Cell. Biochem. 121 (2), 1353–1361. doi:10.1002/jcb.29370
Li, J., Cao, B., Zhou, S., Zhu, J., Zhang, Z., Hou, T., et al. (2013). Cyproheptadine-induced myeloma cell apoptosis is associated with inhibition of the PI3K/AKT signaling. Eur. J. Haematol. 91 (6), 514–521. doi:10.1111/ejh.12193
Li, P., Zhang, X., Wang, H., Wang, L., Liu, T., Du, L., et al. (2017). MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol. Cancer Ther. 16 (4), 739–751. doi:10.1158/1535-7163.MCT-16-0591
Li, Q., Song, W., and Wang, J. (2019b). TUG1 confers adriamycin resistance in acute myeloid leukemia by epigenetically suppressing miR-34a expression via EZH2. Biomed. and Pharmacother. 109, 1793–1801. doi:10.1016/j.biopha.2018.11.003
Li, R., Deng, H., Liu, X., Chen, Z., Wan, S., and Wang, L. (2021a). Histone methyltransferase G9a promotes the development of renal cancer through epigenetic silencing of tumor suppressor gene SPINK5. Oxidative Med. Cell. Longev. 2021, 6650781–6650816. doi:10.1155/2021/6650781
Li, X., Gera, L., Zhang, S., Chen, Y., Lou, L., Wilson, L. M., et al. (2021b). Pharmacological inhibition of noncanonical EED-EZH2 signaling overcomes chemoresistance in prostate cancer. Theranostics 11 (14), 6873–6890. doi:10.7150/thno.49235
Li, Y., Gan, Y., Liu, J., Li, J., Zhou, Z., Tian, R., et al. (2022). Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct. Target. Ther. 7 (1), 87. doi:10.1038/s41392-022-00902-6
Li, Y., Yang, Y., Liu, X., Long, Y., and Zheng, Y. (2019c). PRMT5 promotes human lung cancer cell apoptosis via akt/Gsk3β signaling induced by resveratrol. Cell Transplant. 28 (12), 1664–1673. doi:10.1177/0963689719885083
Liang, S., Wang, Q., Wen, Y., Wang, Y., Li, M., Wang, Q., et al. (2022). Ligand-independent EphA2 contributes to chemoresistance in small-cell lung cancer by enhancing PRMT1-mediated SOX2 methylation. Cancer Sci. 114 (3), 921–936. doi:10.1111/cas.15653
Liao, H. W., Hsu, J. M., Xia, W., Wang, H. L., Wang, Y. N., Chang, W. C., et al. (2015). PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. investigation 125 (12), 4529–4543. doi:10.1172/jci82826
Liao, Q., Yang, J., Ge, S., Chai, P., Fan, J., and Jia, R. (2023). Novel insights into histone lysine methyltransferases in cancer therapy: from epigenetic regulation to selective drugs. J. Pharm. Analysis 13 (2), 127–141. doi:10.1016/j.jpha.2022.11.009
Liao, Y., Luo, Z., Lin, Y., Chen, H., Chen, T., Xu, L., et al. (2022). PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death and Dis. 13 (11), 943. doi:10.1038/s41419-022-05389-1
Lin, H., Wu, H., Chen, S., Hou, M., Lin, C., and Chu, P. (2022). Epigenetic therapy combination of UNC0638 and CI-994 suppresses breast cancer via epigenetic remodeling of BIRC5 and GADD45A. Biomed. and Pharmacother. 145, 112431. doi:10.1016/j.biopha.2021.112431
Liu, C. W., Hua, K. T., Li, K. C., Kao, H. F., Hong, R. L., Ko, J. Y., et al. (2017). Histone methyltransferase G9a drives chemotherapy resistance by regulating the glutamate–cysteine ligase catalytic subunit in head and neck squamous cell carcinoma. Mol. cancer Ther. 16 (7), 1421–1434. doi:10.1158/1535-7163.MCT-16-0567-T
Liu, M., Shi, Y., Hu, Q., Qin, Y., Ji, S., Liu, W., et al. (2021a). SETD8 induces stemness and epithelial–mesenchymal transition of pancreatic cancer cells by regulating ROR1 expression. Acta Biochimica Biophysica Sinica 53 (12), 1614–1624. doi:10.1093/abbs/gmab140
Liu, X., He, J., Mao, L., Zhang, Y., Cui, W., Duan, S., et al. (2021b). EPZ015666, a selective protein arginine methyltransferase 5 (PRMT5) inhibitor with an antitumour effect in retinoblastoma. Exp. Eye Res. 202, 108286. doi:10.1016/j.exer.2020.108286
Liu, X., Zhou, L., Hu, J., Liu, L., Wan, H., and Zhang, X. (2018). UNC0638, a G9a inhibitor, suppresses epithelial-mesenchymal transition-mediated cellular migration and invasion in triple negative breast cancer. Mol. Med. Rep. 17 (2), 2239–2244. doi:10.3892/mmr.2017.8190
Liu, Z., Liu, J., Ebrahimi, B., Pratap, U. P., He, Y., Altwegg, K. A., et al. (2022). SETDB1 interactions with PELP1 contributes to breast cancer endocrine therapy resistance. Breast Cancer Res. BCR 24 (1), 26. doi:10.1186/s13058-022-01520-4
Lukinovic, V., Hausmann, S., Roth, G. S., Oyeniran, C., Ahmad, T., Tsao, N., et al. (2022). SMYD3 impedes small cell lung cancer sensitivity to alkylation damage through RNF113A methylation-phosphorylation cross-talk. Cancer Discov. 12 (9), 2158–2179. doi:10.1158/2159-8290.CD-21-0205
Lv, H., Xing, W., Ba, Y., Li, H., Wang, H., and Li, Y. (2021). SMYD3 confers cisplatin chemoresistance of NSCLC cells in an ANKHD1-dependent manner. Transl. Oncol. 14 (6), 101075. doi:10.1016/j.tranon.2021.101075
Lyu, T., Jiang, Y., Jia, N., Che, X., Li, Q., Yu, Y., et al. (2020). SMYD3 promotes implant metastasis of ovarian cancer via H3K4 trimethylation of integrin promoters. Int. J. Cancer 146 (6), 1553–1567. doi:10.1002/ijc.32673
Mansoori, B., Mohammadi, A., Davudian, S., Shirjang, S., and Baradaran, B. (2017). The different mechanisms of cancer drug resistance: a brief review. Adv. Pharm. Bull. 7 (3), 339–348. doi:10.15171/apb.2017.041
Martin, P. L., Pérez-Areales, F. J., Rao, S. V., Walsh, S. J., Carroll, J. S., and Spring, D. R. (2024). Towards the targeted protein degradation of PRMT1. ChemMedChem 19 (16), 202400269. doi:10.1002/cmdc.202400269
Matsubara, H., Fukuda, T., Awazu, Y., Nanno, S., Shimomura, M., Inoue, Y., et al. (2021). PRMT1 expression predicts sensitivity to platinum-based chemotherapy in patients with ovarian serous carcinoma. Oncol. Lett. 21 (2), 162. doi:10.3892/ol.2020.12423
McKinney, D. C., McMillan, B. J., Ranaghan, M. J., Moroco, J. A., Brousseau, M., Mullin-Bernstein, Z., et al. (2021). Discovery of a first-in-class inhibitor of the PRMT5–substrate adaptor interaction. J. Med. Chem. 64 (15), 11148–11168. doi:10.1021/acs.jmedchem.1c00507
Meng, X., Xiao, W., Sun, J., Li, W., Yuan, H., Yu, T., et al. (2023). CircPTK2/PABPC1/SETDB1 axis promotes EMT-mediated tumor metastasis and gemcitabine resistance in bladder cancer. Cancer Lett. 554, 216023. doi:10.1016/j.canlet.2022.216023
Micallef, I., and Baron, B. (2023). Proteomic strategies for methylation analysis in colorectal cancer chemoresistance. J. Proteome Data Methods 5, 16. doi:10.14889/jpdm.2023.0016
Morin, R. D., Johnson, N. A., Severson, T. M., Mungall, A. J., An, J., Goya, R., et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42 (2), 181–185. doi:10.1038/ng.518
Musiani, D., Giambruno, R., Massignani, E., Ippolito, M. R., Maniaci, M., Jammula, S., et al. (2020). PRMT1 is recruited via DNA-PK to chromatin where it sustains the senescence-associated secretory phenotype in response to cisplatin. Cell Rep. 30 (4), 1208–1222. doi:10.1016/j.celrep.2019.12.061
Nagai, Y., Ji, M. Q., Zhu, F., Xiao, Y., Tanaka, Y., Kambayashi, T., et al. (2019). PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185erbB2/neu tumor immunotherapy. Front. Immunol. 10, 174. doi:10.3389/fimmu.2019.00174
Nakakido, M., Deng, Z., Suzuki, T., Dohmae, N., Nakamura, Y., and Hamamoto, R. (2015). PRMT6 increases cytoplasmic localization of p21CDKN1A in cancer cells through arginine methylation and makes more resistant to cytotoxic agents. Oncotarget 6 (31), 30957–30967. doi:10.18632/oncotarget.5143
Nguyen, C. D., Colón-Emeric, B. A., Murakami, S., Shujath, M. N., and Yi, C. (2024). PRMT1 promotes epigenetic reprogramming associated with acquired chemoresistance in pancreatic cancer. Cell Rep. 43 (5), 114176. doi:10.1016/j.celrep.2024.114176
Obianyo, O., Causey, C. P., Jones, J. E., and Thompson, P. R. (2011). Activity-based protein profiling of protein arginine methyltransferase 1. ACS Chem. Biol. 6 (10), 1127–1135. doi:10.1021/cb2001473
Otani, Y., Sur, H. P., Rachaiah, G., Namagiri, S., Chowdhury, A., Lewis, C. T., et al. (2021). Inhibiting protein phosphatase 2A increases the antitumor effect of protein arginine methyltransferase 5 inhibition in models of glioblastoma. Neuro-Oncology Charlottesv. Va. 23 (9), 1481–1493. doi:10.1093/neuonc/noab014
Pan, K., Hu, B., Wang, L., Yuan, J., and Xu, W. (2022). STUB1-SMYD2 axis regulates drug resistance in glioma cells. J. Mol. Neurosci. 72 (9), 2030–2044. doi:10.1007/s12031-022-02051-5
Pan, M., Hsu, M., Luo, C., Chen, L., Shan, Y., and Hung, W. (2016). The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget 7 (38), 61136–61151. doi:10.18632/oncotarget.11256
Pangeni, R. P., Yang, L., Zhang, K., Wang, J., Li, W., Guo, C., et al. (2020). G9a regulates tumorigenicity and stemness through genome-wide DNA methylation reprogramming in non-small cell lung cancer. Clin. Epigenetics 12 (1), 88. doi:10.1186/s13148-020-00879-5
Paschall, A. V., Yang, D., Lu, C., Choi, J. H., Li, X., Liu, F., et al. (2015). H3K9 trimethylation silences Fas expression to confer colon carcinoma immune escape and 5-fluorouracil chemoresistance. J. Immunol. 195 (4), 1868–1882. doi:10.4049/jimmunol.1402243
Patel, M. R., Monga, V., Jauhari, S., Stevens, D., Masarova, L., McKean, M., et al. (2021). A phase 1 dose escalation study of protein arginine methyltransferase 5 (PRMT5) inhibitor PRT543 in patients with myeloid malignancies. Blood 138, 2609. doi:10.1182/blood-2021-150938
Peng, X., Ma, L., Chen, X., Tang, F., and Zong, X. (2024). Inhibition of FBP1 expression by KMT5A through TWIST1 methylation is one of the mechanisms leading to chemoresistance in breast cancer. Oncol. Rep. 52 (2), 110. doi:10.3892/or.2024.8769
Peserico, A., Germani, A., Sanese, P., Barbosa, A. J., Di Virgilio, V., Fittipaldi, R., et al. (2015). A SMYD3 small-molecule inhibitor impairing cancer cell growth. J. Cell. Physiology 230 (10), 2447–2460. doi:10.1002/jcp.24975
Petrossian, T. C., and Clarke, S. G. (2010). Uncovering the human methyltransferasome. Mol. and Cell. Proteomics 10 (1), M110.000976. doi:10.1074/mcp.m110.000976
Phi, L. T. H., Sari, I. N., Yang, Y., Lee, S., Jun, N., Kim, K. S., et al. (2018). Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 5416923–5417016. doi:10.1155/2018/5416923
Plotnikov, A., Kozer, N., Cohen, G., Carvalho, S., Duberstein, S., Almog, O., et al. (2020). PRMT1 inhibition induces differentiation of colon cancer cells. Sci. Rep. 10 (1), 20030. doi:10.1038/s41598-020-77028-8
Ramakrishnan, S., Granger, V., Rak, M., Hu, Q., Attwood, K., Aquila, L., et al. (2019). Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ. 26 (10), 2100–2114. doi:10.1038/s41418-019-0278-9
Rastgoo, N., Pourabdollah, M., Abdi, J., Reece, D., and Chang, H. (2018). Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 32 (11), 2471–2482. doi:10.1038/s41375-018-0140-y
Ren, H., Wang, Z., Chen, Y., Liu, Y., Zhang, S., Zhang, T., et al. (2019). SMYD2-OE promotes oxaliplatin resistance in colon cancer through MDR1/P-glycoprotein via MEK/ERK/AP1 pathway. OncoTargets Ther. 12, 2585–2594. doi:10.2147/OTT.S186806
Richon, V. M., Johnston, D., Sneeringer, C. J., Jin, L., Majer, C. R., Elliston, K., et al. (2011). Chemogenetic analysis of human protein methyltransferases. Chem. Biol. and Drug Des. 78 (2), 199–210. doi:10.1111/j.1747-0285.2011.01135.x
Schapira, M. (2016). Chemical inhibition of protein methyltransferases. Cell Chem. Biol. 23 (9), 1067–1076. doi:10.1016/j.chembiol.2016.07.014
Schwartz, L. Y., Choudhary, G. S., Ramachandra, N., Sahu, S., Gordon, S., Ruggeri, B., et al. (2021). Preclinical activity of the clinical stage protein arginine methyltransferase 5 (PRMT5) inhibitor PRT543 in splicing mutant myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Blood 138, 2597. doi:10.1182/blood-2021-148494
Shang, L., and Wei, M. (2019). Inhibition of SMYD2 sensitized cisplatin to resistant cells in NSCLC through activating p53 pathway. Front. Oncol. 9, 306. doi:10.3389/fonc.2019.00306
Shen, Y., Gao, G., Yu, X., Kim, H., Wang, L., Xie, L., et al. (2020). Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem. 63 (17), 9977–9989. doi:10.1021/acs.jmedchem.0c01111
Shen, Y., Zhao, P., Dong, K., Wang, J., Li, H., Li, M., et al. (2022). Tadalafil increases the antitumor activity of 5-FU through inhibiting PRMT5-mediated glycolysis and cell proliferation in colorectal cancer. Cancer and Metabolism 10 (1), 22. doi:10.1186/s40170-022-00299-4
Shi, Y., Niu, Y., Yuan, Y., Li, K., Zhong, C., Qiu, Z., et al. (2023). PRMT3-mediated arginine methylation of IGF2BP1 promotes oxaliplatin resistance in liver cancer. Nat. Commun. 14 (1), 1932. doi:10.1038/s41467-023-37542-5
Shimomura, M., Fukuda, T., Awazu, Y., Nanno, S., Inoue, Y., Matsubara, H., et al. (2021). PRMT1 expression predicts response to neoadjuvant chemotherapy for locally advanced uterine cervical cancer. Oncol. Lett. 21 (2), 150. doi:10.3892/ol.2020.12411
Siu, L. L., Rasco, D. W., Vinay, S. P., Romano, P. M., Menis, J., Opdam, F. L., et al. (2019). METEOR-1: A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumours. Ann. Oncol. 30, 159. doi:10.1093/annonc/mdz244
Song, Y., Wu, F., and Wu, J. (2016). Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J. Hematol. and Oncol. 9, 49. doi:10.1186/s13045-016-0279-9
Spira, A. I., Lau, J., Hattersley, M. M., Aronson, B. E., Peters, J., Soo-Hoo, Y., et al. (2024). PRIMROSE: a modular phase 1/2a study of AZD3470, an MTA-cooperative PRMT5 inhibitor, in patients with MTAP deficient advanced solid tumors. J. Clin. Oncol. 42 (16), TPS3179. doi:10.1200/JCO.2024.42.16_suppl.TPS3179
Sun, D., Guo, J., Liang, W., Chen, Y., Wei, S., Li, A., et al. (2024). Histone methyltransferase SUV39H2 regulates apoptosis and chemosensitivity in prostate cancer through AKT/FOXO signaling pathway. Med. Oncol. N. Lond. Engl. 41 (2), 44. doi:10.1007/s12032-023-02252-x
Sun, Y., Patel, A., Kumar, P., and Chen, Z. (2012). Role of ABC transporters in cancer chemotherapy. Ai Zheng 31 (2), 51–57. doi:10.5732/cjc.011.10466
Suresh, S., Huard, S., Brisson, A., Némati, F., Dakroub, R., Poulard, C., et al. (2022). PRMT1 regulates EGFR and wnt signaling pathways and is a promising target for combinatorial treatment of breast cancer. Cancers 14 (2), 306. doi:10.3390/cancers14020306
Tkaczuk, K. L., Dunin-Horkawicz, S., Purta, E., and Bujnicki, J. M. (2007). Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinforma. 8, 73. doi:10.1186/1471-2105-8-73
Velez, J., Han, Y., Yim, H., Yang, P., Deng, Z., Park, K. S., et al. (2024). Discovery of the first-in-class G9a/GLP PROTAC degrader. J. Med. Chem. 67 (8), 2024.02.26.582210–6409. doi:10.1101/2024.02.26.582210
Velez, J., Kaniskan, H. Ü., and Jin, J. (2023). Recent advances in developing degraders and inhibitors of lysine methyltransferases. Curr. Opin. Chem. Biol. 76, 102356. doi:10.1016/j.cbpa.2023.102356
Vieito, M., Moreno, V., Spreafico, A., Brana, I., Wang, J. S., Preis, M., et al. (2023). Phase 1 study of JNJ-64619178, a protein arginine methyltransferase 5 inhibitor, in advanced solid tumors. Clin. Cancer Res. 29 (18), 3592–3602. doi:10.1158/1078-0432.CCR-23-0092
Vinet, M., Suresh, S., Maire, V., Monchecourt, C., Némati, F., Lesage, L., et al. (2019). Protein arginine methyltransferase 5: a novel therapeutic target for triple-negative breast cancers. Cancer Med. 8 (5), 2414–2428. doi:10.1002/cam4.2114
Wada, M., Kukita, A., Sone, K., Hamamoto, R., Kaneko, S., Komatsu, M., et al. (2020). Epigenetic modifier SETD8 as a therapeutic target for high-grade serous ovarian cancer. Biomol. Basel, Switz. 10 (12), 1686. doi:10.3390/biom10121686
Wang, J., Yang, B., Wang, Y., Liu, S., Ma, C., Piao, J., et al. (2024). CBX2 enhances the progression and TMZ chemoresistance of glioma via EZH2-mediated epigenetic silencing of PTEN expression. Front. Pharmacol. 15, 1430891. doi:10.3389/fphar.2024.1430891
Wang, L., Dong, X., Ren, Y., Luo, J., Liu, P., Su, D., et al. (2018a). Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 9 (2), 129–213. doi:10.1038/s41419-017-0120-6
Wang, L., Jia, Z., Xie, D., Zhao, T., Tan, Z., Zhang, S., et al. (2020a). Methylation of HSP70 orchestrates its binding to and stabilization of BCL2 mRNA and renders pancreatic cancer cells resistant to therapeutics. Cancer Res. 80 (20), 4500–4513. doi:10.1158/0008-5472.CAN-19-1738
Wang, L., Pal, S., and Sif, S. (2008). Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 28 (20), 6262–6277. doi:10.1128/MCB.00923-08
Wang, L., Xu, M., Wang, C., Dong, Q., Miao, Z., Chen, X., et al. (2020b). SET and MYND domain-containing protein 3 inhibits tumor cell sensitivity to cisplatin. Oncol. Lett. 19 (5), 3469–3476. doi:10.3892/ol.2020.11465
Wang, L., Zeng, H., Wang, Q., Zhao, Z., Boyer, T. G., Bian, X., et al. (2015). MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci. Adv. 1 (9), 1500463. doi:10.1126/sciadv.1500463
Wang, M. Y., Liow, P., Guzman, M. I. T., and Qi, J. (2022a). Exploring methods of targeting histone methyltransferases and their applications in cancer therapeutics. ACS Chem. Biol. 17 (4), 744–755. doi:10.1021/acschembio.2c00062
Wang, Q., Wang, K., and Ye, M. (2017). Strategies for large-scale analysis of non-histone protein methylation by LC-MS/MS. Analyst 142 (19), 3536–3548. doi:10.1039/c7an00954b
Wang, W., Wang, J., Liu, S., Ren, Y., Wang, J., Liu, S., et al. (2022b). An EHMT2/NFYA-ALDH2 signaling axis modulates the RAF pathway to regulate paclitaxel resistance in lung cancer. Mol. Cancer 21 (1), 106. doi:10.1186/s12943-022-01579-9
Wang, W., Wang, J., Zhang, X., and Liu, G. (2019). Serum circSETDB1 is a promising biomarker for predicting response to platinum-taxane-combined chemotherapy and relapse in high-grade serous ovarian cancer. OncoTargets Ther. 12, 7451–7457. doi:10.2147/OTT.S220700
Wang, Y., Hsu, J. M., Kang, Y., Wei, Y., Lee, P. C., Chang, S. J., et al. (2016). Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer Res. 76 (23), 7049–7058. doi:10.1158/0008-5472.CAN-16-0715
Wang, Z., Kong, J., Wu, Y., Zhang, J., Wang, T., Li, N., et al. (2018b). PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res. Treat. 168 (2), 531–542. doi:10.1007/s10549-017-4597-6
Watson, Z. L., Yamamoto, T. M., McMellen, A., Kim, H., Hughes, C. J., Wheeler, L. J., et al. (2019). Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenetics 11 (1), 165. doi:10.1186/s13148-019-0758-2
Wei, H., Mundade, R., Lange, K., and Lu, T. (2014). Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 13 (1), 32–41. doi:10.4161/cc.27353
Wen, Y., Hou, Y., Yi, X., Sun, S., Guo, J., He, X., et al. (2021). EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics 11 (4), 1795–1813. doi:10.7150/thno.48101
Wong, J. M., and Eirin-Lopez, J. M. (2021). Evolution of methyltransferase-like (METTL) proteins in Metazoa: a complex gene family involved in Epitranscriptomic regulation and other epigenetic processes. Mol. Biol. Evol. 38 (12), 5309–5327. doi:10.1093/molbev/msab267
Wu, J., Chai, H., Li, F., Ren, Q., and Gu, Y. (2020). SETD1A augments sorafenib primary resistance via activating YAP in hepatocellular carcinoma. Life Sci. 260, 118406–118410. doi:10.1016/j.lfs.2020.118406
Wu, Q., Nie, D. Y., Ba-Alawi, W., Ji, Y., Zhang, Z., Cruickshank, J., et al. (2022). PRMT inhibition induces a viral mimicry response in triple-negative breast cancer. Nat. Chem. Biol. 18 (8), 821–830. doi:10.1038/s41589-022-01024-4
Wu, Q., Schapira, M., Arrowsmith, C. H., and Barsyte-Lovejoy, D. (2021). Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 20 (7), 509–530. doi:10.1038/s41573-021-00159-8
Xu, Y., Zhao, R., Wang, H., Jiang, J., Wang, Z., Wang, J., et al. (2021). Circular RNA PRMT5 knockdown enhances cisplatin sensitivity and immune response in non-small cell lung cancer by regulating miR-138-5p/MYH9 axis. J. B.U.ON:Official J. Balkan Union Oncol. 26 (5), 1850–1861. Available at: https://pubmed.ncbi.nlm.nih.gov/34761592/.
Yamamoto, T., Hayashida, T., Masugi, Y., Oshikawa, K., Hayakawa, N., Itoh, M., et al. (2024). PRMT1 sustains de novo fatty acid synthesis by methylating PHGDH to drive chemoresistance in triplenegative breast cancer. Cancer Res. 84 (7), 1065–1083. doi:10.1158/0008-5472.CAN-23-2266
Yan, L., Ding, B., Liu, H., Zhang, Y., Zeng, J., Hu, J., et al. (2019). Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics 9 (26), 8377–8391. doi:10.7150/thno.37628
Yang, C., Zhang, J., Ma, Y., Wu, C., Cui, W., and Wang, L. (2020). Histone methyltransferase and drug resistance in cancers. J. Exp. and Clin. Cancer Res. 39, 173. doi:10.1186/s13046-020-01682-z
Yang, Q., Zhao, S., Shi, Z., Cao, L., Liu, J., Pan, T., et al. (2021). Chemotherapy-elicited exosomal miR-378a-3p and miR-378d promote breast cancer stemness and chemoresistance via the activation of EZH2/STAT3 signaling. J. Exp. and Clin. Cancer Res. 40 (1), 120. doi:10.1186/s13046-021-01901-1
Yang, Y., and Bedford, M. T. (2013). Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13 (1), 37–50. doi:10.1038/nrc3409
Yaqub, A., Amin, M., Fatima, Q., Toor, R. H., Aftab, S., and Shakoori, A. R. (2022). Effect of inhibitor UNC0642 on expression of G9a and its proliferative and apoptotic markers in breast cancer cell line MCF-7. Pak. J. Zoology 54 (6), 2745. doi:10.17582/journal.pjz/20220512100555
Yeldag, G., Rice, A., and Del Río Hernández, A. (2018). Chemoresistance and the self-maintaining tumor microenvironment. Cancers 10 (12), 471. doi:10.3390/cancers10120471
Yuan, J., Liu, Y., Zhang, Q., Ren, Z., Li, G., and Tian, R. (2021). CircPRDM2 contributes to doxorubicin resistance of osteosarcoma by elevating EZH2 via sponging miR-760. Cancer Manag. Res. 13, 4433–4445. doi:10.2147/CMAR.S295147
Zauderer, M. G., Szlosarek, P. W., Le Moulec, S., Popat, S., Taylor, P., Planchard, D., et al. (2022). EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma: a multicentre, open-label, phase 2 study. Lancet Oncol. 23 (6), 758–767. doi:10.1016/S1470-2045(22)00277-7
Zhang, L., Zhang, X., Shi, Y., Ni, Y., Fei, J., Jin, Z., et al. (2024a). Role and potential therapeutic value of histone methyltransferases in drug resistance mechanisms in lung cancer. Front. Oncol. 14, 1376916. doi:10.3389/fonc.2024.1376916
Zhang, Q., Shi, Y., Liu, S., Yang, W., Chen, H., Guo, N., et al. (2024b). EZH2/G9a interact to mediate drug resistance in non-small-cell lung cancer by regulating the SMAD4/ERK/c-Myc signaling axis. Cell Rep. 43 (2), 113714. doi:10.1016/j.celrep.2024.113714
Zhang, S., Ma, Y., Hu, X., Zheng, Y., and Chen, X. (2019). Targeting PRMT5/akt signalling axis prevents human lung cancer cell growth. J. Cell. Mol. Med. 23 (2), 1333–1342. doi:10.1111/jcmm.14036
Zhang, X., Wang, K., Feng, X., Wang, J., Chu, Y., Jia, C., et al. (2021). PRMT3 promotes tumorigenesis by methylating and stabilizing HIF1α in colorectal cancer. Cell Death and Dis. 12 (11), 1066. doi:10.1038/s41419-021-04352-w
Zhao, W., Yang, S., Chen, J., Zhao, J., and Dong, J. (2018). Forced overexpression of FBP1 inhibits proliferation and metastasis in cholangiocarcinoma cells via Wnt/β-catenin pathway. Life Sci. 210, 224–234. doi:10.1016/j.lfs.2018.09.009
Zhao, Y., Lu, Q., Li, C., Wang, X., Jiang, L., Huang, L., et al. (2019). PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death and Dis. 10 (5), 359. doi:10.1038/s41419-019-1595-0
Zheng, M., Niu, Y., Bu, J., Liang, S., Zhang, Z., Liu, J., et al. (2021). ESRP1 regulates alternative splicing of CARM1 to sensitize small cell lung cancer cells to chemotherapy by inhibiting TGF-β/smad signaling. Aging (Albany, NY.) 13 (3), 3554–3572. doi:10.18632/aging.202295
Zhou M., M., Zhang, X., Liu, C., Nie, D., Li, S., Lai, P., et al. (2021). Targeting protein lysine methyltransferase G9A impairs self-renewal of chronic myelogenous leukemia stem cells via upregulation of SOX6. Oncogene 40 (20), 3564–3577. doi:10.1038/s41388-021-01799-1
Zhou S, S., Peng, J., Xiao, L., Zhou, C., Fang, Y., Ou, Q., et al. (2021). TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death and Dis. 12 (5), 463. doi:10.1038/s41419-021-03734-4
Zhu, F., and Rui, L. (2019). PRMT5 in gene regulation and hematologic malignancies. Genes and Dis. 6 (3), 247–257. doi:10.1016/j.gendis.2019.06.002
Zhu, G., Jin, L., Sun, W., Wang, S., and Liu, N. (2022). Proteomics of post-translational modifications in colorectal cancer: discovery of new biomarkers. Biochimica Biophysica Acta (BBA)-Reviews Cancer 1877 (4), 188735. doi:10.1016/j.bbcan.2022.188735
Keywords: chemoresistance, methylproteomics, protein methylation, protein methyltransferases (PMTs), PMT inhibitors
Citation: Micallef I, Fenech K and Baron B (2024) Therapeutic targeting potential of the protein lysine and arginine methyltransferases to reverse cancer chemoresistance. Front. Mol. Biosci. 11:1455415. doi: 10.3389/fmolb.2024.1455415
Received: 26 June 2024; Accepted: 10 October 2024;
Published: 05 December 2024.
Edited by:
Han Peng, University of California, Davis, United StatesReviewed by:
Enrico Massignani, Flemish Institute for Biotechnology, BelgiumKrishna Ghosh, University of Texas MD Anderson Cancer Center, United States
Copyright © 2024 Micallef, Fenech and Baron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byron Baron, Ynlyb24uYmFyb25AdW0uZWR1Lm10