 Akanksha Jarwal
Akanksha Jarwal Anjali Dhall
Anjali Dhall Akanksha Arora
Akanksha Arora Sumeet Patiyal
Sumeet Patiyal Gajendra P. S. Raghava
Gajendra P. S. Raghava
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 30 May 2024
Sec. Molecular Diagnostics and Therapeutics
Volume 11 - 2024 | https://doi.org/10.3389/fmolb.2024.1395721
This article is part of the Research TopicNovel Biomarkers and Big Data-Based Biomedical Studies in Cancer Diagnosis and ManagementView all 21 articles
Background: Head and Neck Squamous Cell Carcinoma (HNSCC) is the seventh most highly prevalent cancer type worldwide. Early detection of HNSCC is one of the important challenges in managing the treatment of the cancer patients. Existing techniques for detecting HNSCC are costly, expensive, and invasive in nature.
Methods: In this study, we aimed to address this issue by developing classification models using machine learning and deep learning techniques, focusing on single-cell transcriptomics to distinguish between HNSCC and normal samples. Furthermore, we built models to classify HNSCC samples into HPV-positive (HPV+) and HPV-negative (HPV−) categories. In this study, we have used GSE181919 dataset, we have extracted 20 primary cancer (HNSCC) samples, and 9 normal tissues samples. The primary cancer samples contained 13 HPV− and 7 HPV+ samples. The models developed in this study have been trained on 80% of the dataset and validated on the remaining 20%. To develop an efficient model, we performed feature selection using mRMR method to shortlist a small number of genes from a plethora of genes. We also performed Gene Ontology (GO) enrichment analysis on the 100 shortlisted genes.
Results: Artificial Neural Network based model trained on 100 genes outperformed the other classifiers with an AUROC of 0.91 for HNSCC classification for the validation set. The same algorithm achieved an AUROC of 0.83 for the classification of HPV+ and HPV− patients on the validation set. In GO enrichment analysis, it was found that most genes were involved in binding and catalytic activities.
Conclusion: A software package has been developed in Python which allows users to identify HNSCC in patients along with their HPV status. It is available at https://webs.iiitd.edu.in/raghava/hnscpred/.
Head and neck cancer, encompasses a variety of malignancies that affect the respiratory tract and upper digestive tract. Head and Neck Squamous Cell Carcinoma (HNSCC) is the most typical kind among the head and neck cancer (Mody et al., 2021). In 2020, 562,328 people were diagnosed with head and neck cancer (HNC) worldwide, with a total count of 277,587 deaths due to the disease (Broutian et al., 2020). These carcinomas often develop in the salivary glands, larynx, oral cavity, throat, and sino-nasal tract epithelium. A number of head and neck malignancies are linked to the human papillomavirus (HPV) infection, notably HPV-16. However, some malignancies are also related to the other carcinogens like smoking, excessive alcohol, and other factors depending on the country or area. Hence, we can classify this cancer into two major categories—HPV-negative and HPV-positive. The median age of diagnosis for HPV associated HNSCC is about 66 years, whereas for HPV-associated oropharyngeal cancer the median age is ∼53 years (Johnson et al., 2020).
Distinguishing between HPV-positive and HPV-negative Head and Neck Squamous Cell Carcinoma (HNSCC) samples holds profound significance in clinical practice as it unveils distinct molecular mechanisms underlying tumorigenesis and guides tailored therapeutic interventions. HPV-positive HNSCCs, primarily driven by high-risk human papillomavirus (HPV) infection, often manifest with activated cell cycle pathways, particularly the retinoblastoma protein (pRB) pathway, leading to enhanced cell proliferation (Leemans et al., 2018). Conversely, HPV-negative tumors frequently arise from genomic instability induced by environmental factors such as tobacco and alcohol exposure, resulting in diverse genetic alterations, such as mutations in tumor suppressor genes and oncogenes. Consequently, HPV-positive tumors exhibit heightened sensitivity to radiotherapy and chemotherapy due to their intact DNA repair mechanisms and increased expression of apoptosis-regulating proteins (Fakhry et al., 2008). Conversely, HPV-negative tumors, characterized by aberrant DNA repair pathways and resistance to apoptosis, necessitate more aggressive therapeutic strategies. Understanding the HPV status in HNSCC thus facilitates personalized treatment approaches, optimizing patient outcomes by targeting specific molecular vulnerabilities (Dok and Nuyts, 2016). Better understanding the HPV status of HNSCC tumors enables clinicians to tailor treatment strategies and provide accurate prognostic information, ultimately improving patient management and outcomes (Ang et al., 2010; Chaturvedi et al., 2011; Gillison et al., 2012). The mechanisms of HPV+ and HPV- associated HNSCC are explained in Figure 1.
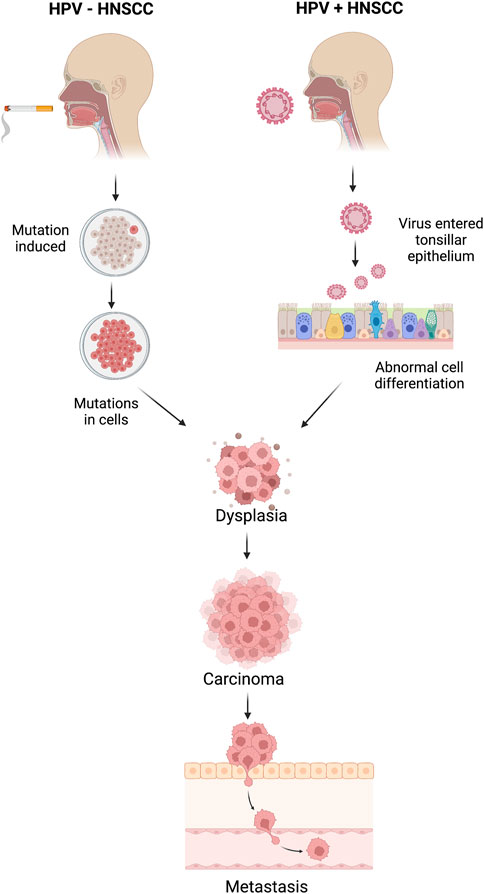
Figure 1. Mechanisms of head and neck squamous cell carcinoma (HNSCC) for HPV-positive and HPV-negative HNSCC patients.
Despite thorough and targeted treatment efforts, the chances of survival are reduced due to the majority of head and neck cancer cases being diagnosed at advanced stages. The traditional diagnosis of HNSCC is based on the physical examination, radiological investigation, and histological analysis of the tissue sections obtained from biopsies or surgical resections. These procedures can take a lot of time and are susceptible to mistakes in observation or interpretation, which can lead to discrepancies in cancer grading and prognostication (Mahmood et al., 2021). In addition to this, most of the HNSCC cancers are detected at a later stage. The reasons range from limited symptomatology in early-stage patients, swift progression from early to advanced stage, indistinctive diagnostic characteristics, and imprecise history information (Basheeth and Patil, 2019).
Identification of molecular biomarkers of HNSCC can lead to early diagnosis of this cancer and can also help in preventive management of HNSCC. The cancer biomarkers not only influence diagnosis but they also have the potential to improve the treatment outcomes using targeted therapy. The currently known biomarker of HNSCC is PD-L1 which is commonly used in treatment decision making in advanced stage of HNSCC. It has a moderate predictive value and has many limitations due to the lack of standardization and highly dynamic nature of PD-L1 expression. Currently, there are no any other FDA approved molecular biomarkers for HNSCC diagnosis or prognosis (Basheeth and Patil, 2019).
In this study, we made an attempt to identify biomarkers for HNSCC using single-cell sequencing data. On the basis of the 100 biomarkers identified in this study, we have developed a method that can predict the HNSCC cancer along with HPV+ or HPV− status. Single-cell data collected from individual cells using next-generation sequencing methods provides a better knowledge of the activity of a single cell in relation to its microenvironment (Eberwine et al., 2014). Cell-to-cell variation can be revealed by single-cell sequencing of RNA or epigenetic alterations, which may aid the populations in quickly adapting to new circumstances (Saliba et al., 2014). The significance of gene mosaicism, as well as intra-tumor genetic heterogeneity in the genesis of cancer or response to therapy, can be uncovered by single-cell precision (Gawad et al., 2016). Single-cell technology makes it possible to detect molecular alterations in individual cancer cells. This can increase the research of more specialized biomarkers with excellent resolution, leading to the development of a complete landscape of distinct cell types within tumors (Radpour and Forouharkhou, 2018). The full workflow of this study is described in Figure 2.
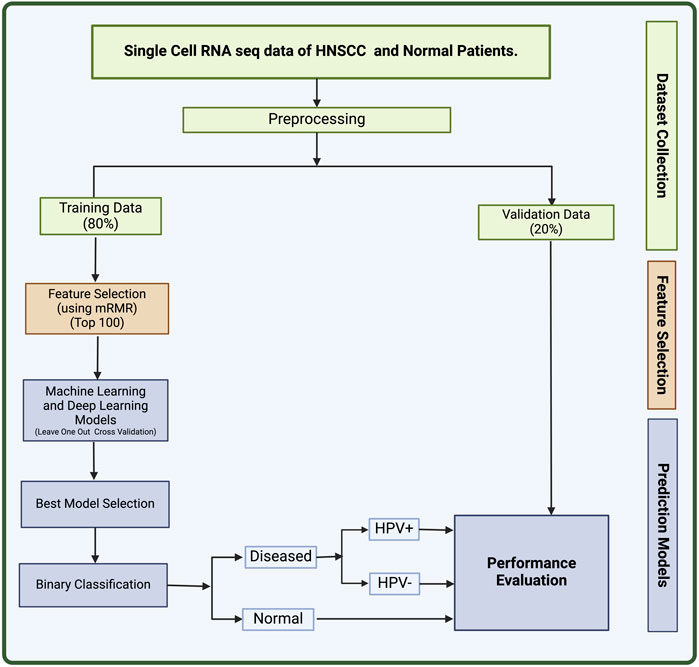
Figure 2. The full workflow of the study.
We retrieved the dataset used in this study (GSE181919) from National Centre for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) (Clough and Barrett, 2016; Choi et al., 2023). The GSE181919 dataset comprise of 37 tissue specimens from 23 patients with Head and Neck Squamous Cell Carcinoma (HNSCC), covering a range of tissues, including normal tissues (n = 9), precancerous leukoplakia (n = 4), primary HNSCC (n = 20), and metastasized tumors (n = 4). Choi et al. methodology involved aligning sequencing data to the human reference genome (GRCh38) and processing it using CellRanger 2.1.1 by 10X Genomics. Subsequently, cell-level transcripts were clustered using the “Seurat”package’s shared nearest neighbor method. To ensure the clarity in the dataset, we chose two distinct groups: normal tissue (n = 9) and primary HNSCC tissues (n = 20). Therefore, in this study, we have only taken 29 total samples comprising 20 primary cancer samples, and 9 normal samples. In addition, these cancer samples are divided into 13 HPV− and 7 HPV+ samples. The information on whether the samples were derived from HPV+ or HPV− patients was derived from the metadata provided on GEO. This dataset used Illumina HiSeq 4000 as the platform for scRNA sequencing. The 80% of this dataset was used to train machine learning (ML) and deep learning (DL) models and 20% was used as validation set.
After the retrieval of data from GEO, we processed the data using in-house python scripts. Firstly, we converted the sparse data into a matrix and removed insignificant columns from our training data. The genes that had no mapped readings to more than 80% of the cells were eliminated, and cells containing zeroes were filtered leading to 2,604 genes. The sequencing depth affects the range of values for the features, which necessitates normalizing the count data before doing any sort of analysis. Hence, we performed counts per million (CPM) normalization and log transformation on the data using scanpy package in python (Wolf et al., 2018).
We applied feature selection to the set of 2,604 genes obtained after pre-processing to obtain a set of biomarkers for HNSCC. This was achieved using mRMR (Minimum Redundancy and Maximum Relevance) feature selection algorithm (Radovic et al., 2017). mRMR selects a subset of features that have the least correlation amongst themselves but high correlation with the output class. The advantage of using this method is that it provides with a small set of features with high predictive potential. The redundancy between genes is taken into account in this technique in addition to the relationship between samples and genes. The most relevant feature will be considered out of the numerous identical features. We used the value K = 100 for mRMR to extract 100 most relevant genes for the prediction of HNSCC (Zhao et al., 2019a). This strategy has been previously demonstrated to be useful and often utilized in single-cell RNA sequencing analysis (Ding and Peng, 2005; Radovic et al., 2017).
After extracting the top 100 genes from feature selection, we performed a T-test analysis for the mean expression of genes in the cells of the groups Normal vs. Cancer and Cancer HPV+ vs. Cancer HPV−. We also wished to identify the top most dysregulated genes in both the comparisons. To achieve this, we found mean difference between the two classes in both comparisons (Normal vs. Cancer and Cancer HPV+ vs. Cancer HPV−), and reported the 10 most dysregulated (5 upregulated and 5 downregulated) genes with the highest difference in means for each comparison.
We have developed various machine learning (ML) models to classify between normal subjects and HNSCC patients. In addition, we have also classified HNSCC patients into HPV positive and HPV negative. These machine learning models include Extreme Gradient Boosting (XGB), Decision Tree (DT), K-Nearest Neighbors (KNN), Extra Trees (ET), Logistic Regression (LR), and Random Forest (RF) algorithms. Hyperparameter tuning was also used to optimise the parameters of these algorithms. The DT classifier is a supervised machine learning model that classifies the output by learning decision rules from input, the KNN classifier predicts on the basis of the maximum number of votes cast in support of the class that is closest to the nearest neighbouring data point, LR classifier uses a logistic function to calculate the likelihood of an event, XGB Classifier is a distributed gradient-boosted decision tree machine learning package that offers simultaneous tree boosting, and RF classifier trains a number of decision trees to produce a single tree. A technique for ensemble supervised machine learning that makes use of decision trees is called extra trees. (Breiman, 2001; Wu et al., 2002; Geurts et al., 2006; Stoltzfus, 2011; Bulac and Bulac, 2016; Chen and Guestrin, 2016). These methods have previously been used in many studies (Aggarwal et al., 2023; Arora et al., 2023; Kaur et al., 2023; Srivastava et al., 2023).
Along with the ML models, we have also applied deep learning classification technique—Artificial Neural Network (ANN) to classify the data (Wang, 2003). In this method, networks are composed of multiple layers, and each layer has a number of nodes (or neurons) that support decision making. The model architecture of ANN used in this study includes three hidden layers and an output layer. A dropout of 0.5 is implemented at each step to lessen the overfitting by neural network. Biological neuron networks served as the basis for this strategy. Artificial neurons, which are constructed from a network of connected units or nodes and are conceptually similar to the neurons in the human brain, are used to build ANNs. They consist of several layers, and inside each layer there are multiple nodes (or neurons) that support decision-making. The anticipated label (Diseased or Normal) of the sample is the final result. The final result classifies the samples into HNSCC positive or negative, and if found HNSCC positive then whether the patient is HPV positive or negative is identified.
The dataset was primarily composed of training data, which made up 80% of it and validation set, which made up the remaining 20%. In the LOOCV (Leave One out Cross Validation) approach, the whole training set is separated into N equivalent folds using the LOOCV technique, with (N-1) being utilized for training and the single fold being used for testing. Each fold serves as testing data for the technique’s N iterations. The overall performance was calculated as the mean of N iterations. This is a common practice in many types of studies (Peng et al., 2015; Vabalas et al., 2019).
To evaluate the efficacy of various prediction models, we employed a number of evaluation indicators. In this study, we used both threshold-independent and threshold-dependent parameters. To calculate threshold-dependent characteristics like sensitivity (Sens), specificity (Spec), precision, F1-Score, and accuracy (Acc), we utilised the following formulae. We also used the conventional threshold-independent parameter Area Under the Curve (AUC) to assess the performance of the models. The metrics calculated in this study are mentioned in Eqs 1–5.
Where, Pt is true positive, Nt is true negative, Pf is false positive, and Nf is false negative.
We applied a feature selection technique called mRMR to obtain a list of highly relevant features (genes) for the detection of HNSCC samples from a set of 2,604 genes that were obtained after data pre-processing (Zhao et al., 2019b). We obtained a subset of 100 genes that were able to classify HNSCC and non-HNSCC samples as well as HPV+ and HPV− samples correctly.
After performing T-test on selected 100 genes for Normal vs. Cancer and Cancer HPV+ vs. Cancer HPV− groups. It was found that all the genes were significantly differentially expressed with p-values<0.05 in Normal vs. Cancer comparison whereas 94 genes out of 100 were significantly differentially expressed with p-values<0.05 in Cancer HPV+ vs. Cancer HPV− comparison. The list of selected 100 genes along with their p-values, mean gene expressions, mean expression difference, and t-statistics for Normal vs. Cancer and Cancer HPV+ vs. Cancer HPV− are given in Supplementary Tables S1, S2, respectively. We also identified the top 10 dysregulated genes (5 upregulated and 5 downregulated) on the basis of mean difference between two classes in both the comparisons. The top 10 dysregulated genes for Normal vs. Cancer and Cancer HPV+ vs. Cancer HPV− are given in Tables 1, 2, respectively.
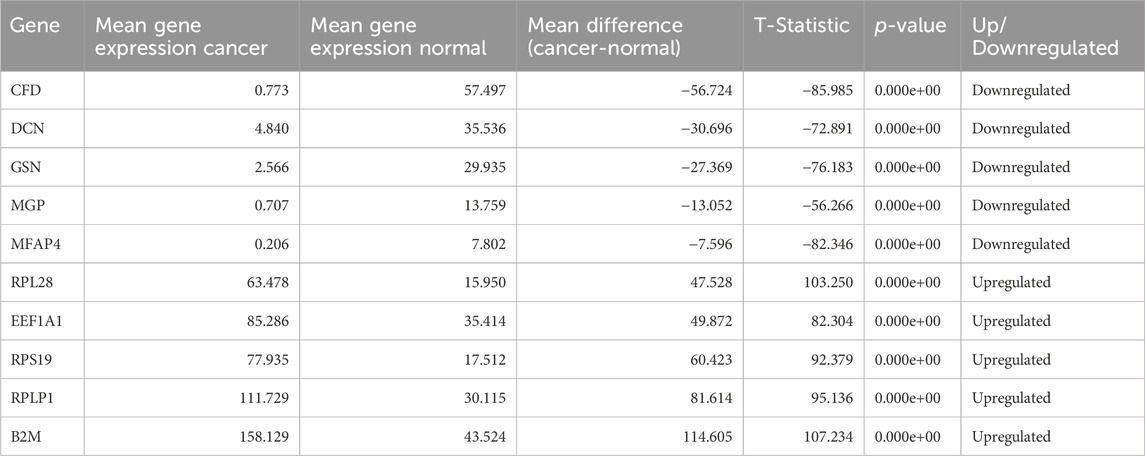
Table 1. Top 10 dysregulated genes for Normal vs. Cancer.
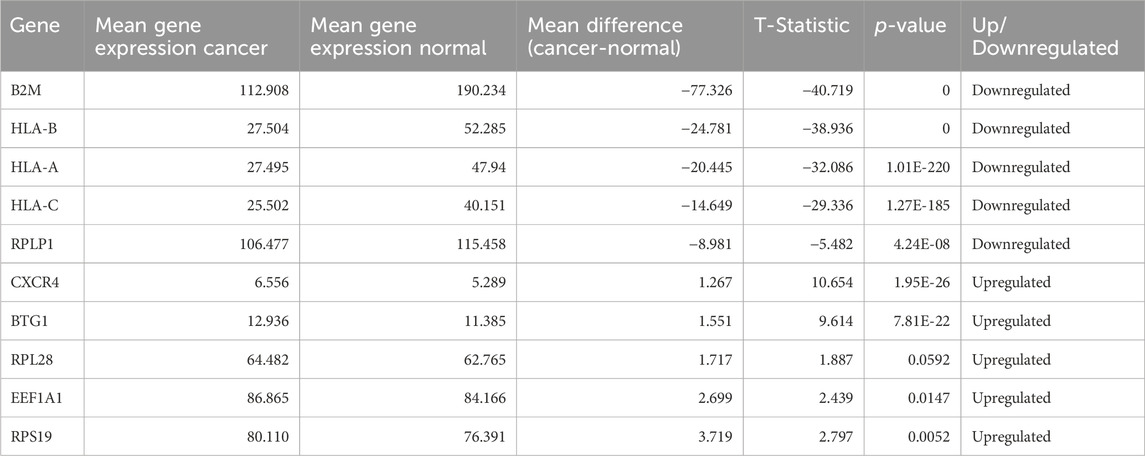
Table 2. Top 10 dysregulated genes for Cancer HPV- vs. Cancer HPV+.
We applied various ML models like DT, RF, ET, XGB, and KNN on our dataset, where we used 80% of dataset GSE181919 for training, 20% of dataset GSE181919 as validation set, and. It was observed that machine learning models were able to achieve an AUROC of 0.85 (XGB, ET) on the validation set. In order to increase the AUROC, we applied DL algorithm—ANN on the dataset and observed that the AUROCs increased to 0.91 for the validation set. The complete results for the ML and DL performances are given in Table 3.
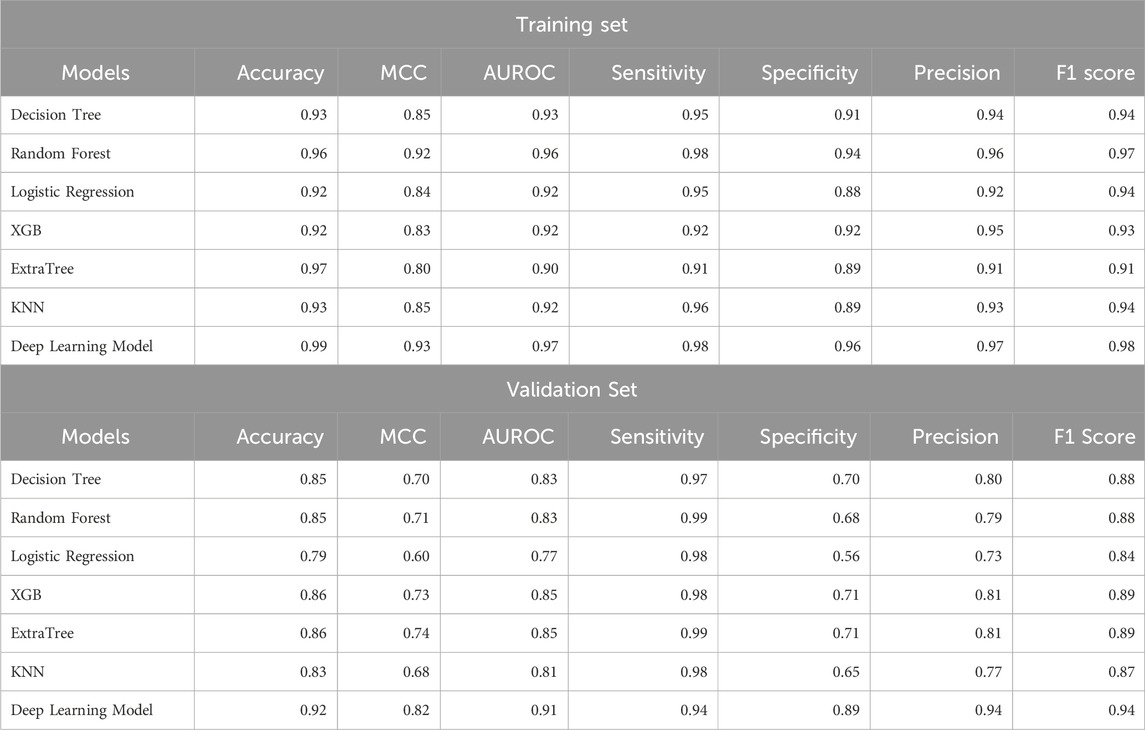
Table 3. Performance of ML and DL models for the classification of HNSCC patients and normal subjects.
After classification of samples as HNSCC or non-HNSCC, we attempted to classify whether an HNSCC sample belonged to an HPV+ or HPV− class. Hence, we developed ML and DL models to further classify the HNSCC samples as HPV+ and HPV−. The maximum AUROC achieved by ML models was 0.81 (XGB) for the validation set. After employing ANN classifier to the data, it was observed that the AUROC increased to 0.84 for the validation set. The results for HPV+ and HPV− classification from HNSCC patients are summarized in Table 4.
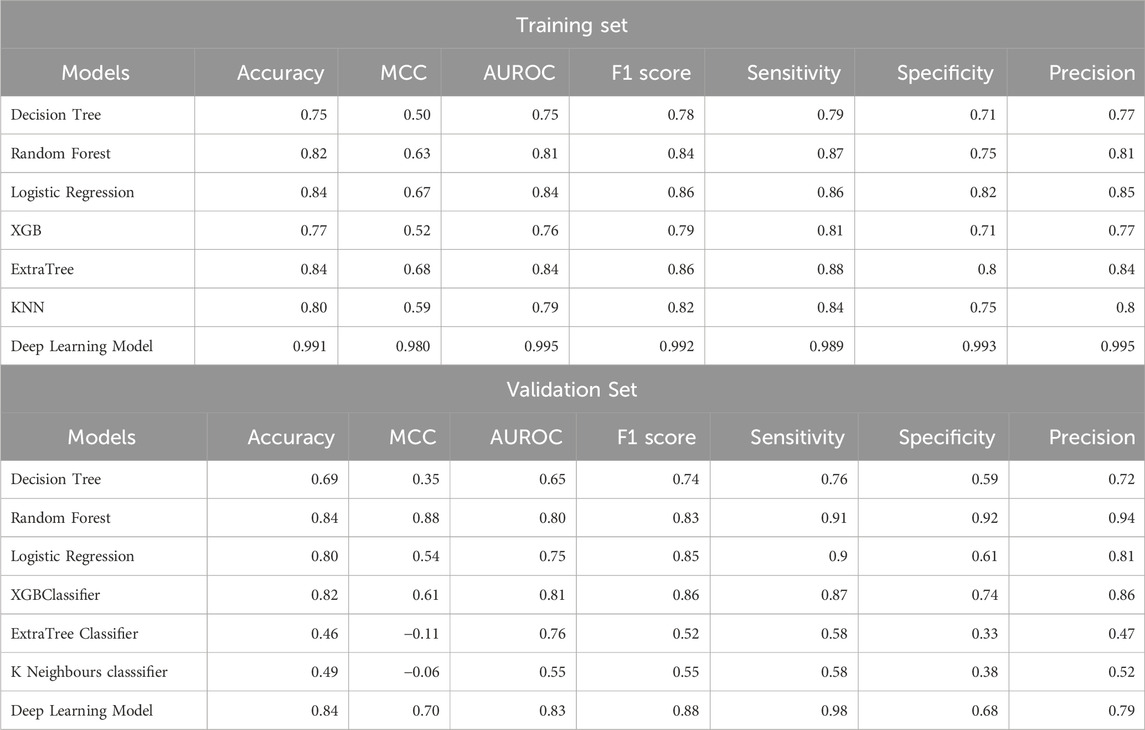
Table 4. Performance of ML and DL models for the classification of HPV+ and HPV− patients from HNSCC patients.
The Gene Ontology (GO) encapsulates our understanding of the biological world in three ways: molecular function, cellular component, and biological process (Ashburner et al., 2000; Gene Ontology Consortium, 2021). 100 genes that may serve as potential biomarkers of HNSCC were retrieved once mRMR analysis was complete. On these 100 retrieved genes, we next ran Gene Ontology (GO) enrichment analysis using PantherDB to map the biological processes, cellular components, and molecular functions of the chosen genes (Mi et al., 2013). The findings of the GO enrichment analysis for all 100 selected genes are displayed in Supplementary Tables S3–S5, respectively. The results of Gene Ontology for Biological Processes and Cellular Component are shown in Figure 3A, B respectively. We see that the majority of genes have a role in the binding activities of many metabolic processes as shown in Figure 3C. The genes and their roles are described in Figure 3.
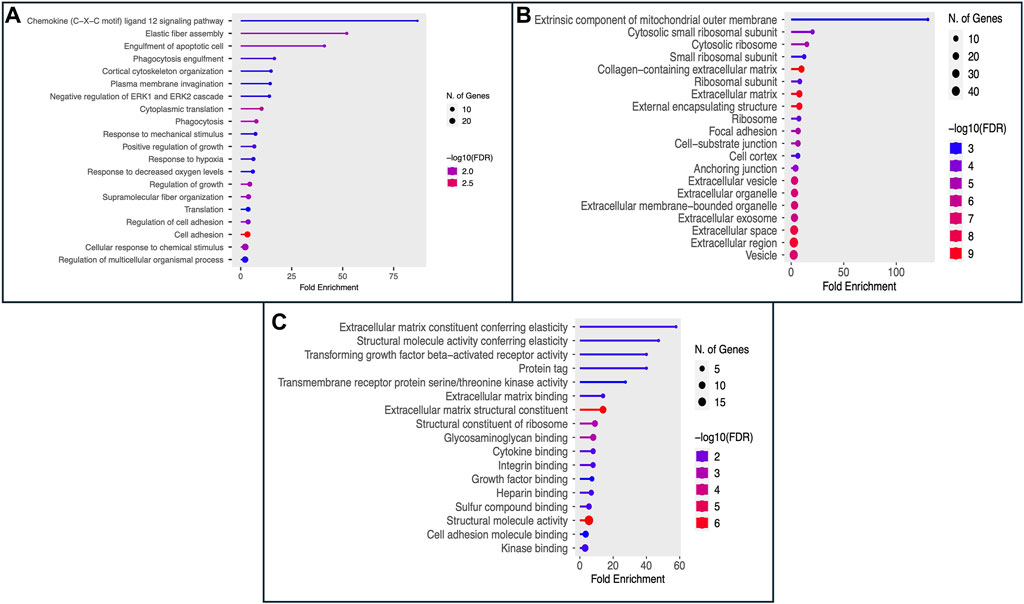
Figure 3. The figure displays the Gene Ontology (GO) enrichment analysis results as (A) Biological Process, (B) Cellular Component (CC), and (C) Molecular Function (MF) for the top 100 selected genes.
One of the heterogeneous diseases, HNSCC affects the head and neck region, namely the oral cavity, paranasal sinuses, larynx, nasal cavity, hypopharynx, and oropharynx. It is described by malignant and uncontrollable cell proliferation in these locations (Hsieh et al., 2019). Advancement in the sequence technology allows the researchers to find various biomarkers such as diagnostic, predictive, and prognostic biomarkers. These biomarkers help in better understanding of the disease as well as may aids in designing novel and effective diagnosis and treatment. A biomarker is described as a biological molecule present in the blood, other body fluids, as well as in tissues, that serves as a sign of a normal or aberrant process, a condition, or a disease by the National Cancer Institute (NCI). To determine how effectively the body will react to an illness or condition medication, a biomarker could well be utilized (Hsieh et al., 2019). This study aims to find out a set of potential biomarkers from single-cell transcriptomic data of head and neck cancer patients that can classify HNSCC patients and normal individuals with reliable accuracy. In addition, we have also attempted to classify HNSCC patients as HPV+ or HPV−. The biomarkers identified in this study could aid in the early diagnosis and screening of HNSCC.
To categorize non-cancer and HNSCC disease cells from their single-cell RNA seq data, we employed a variety of machine learning models, including an ANN deep learning model. We also further tried to categorize the diseased patients into HPV+ and HPV−. We trained the model with 80% of the dataset GSE181919, 20% of the dataset GSE181919 as validation set. The datasets were originally quite extensive and had a significant number of features. During the preprocessing step, the feature count was decreased to a shallow level of 2,604 genes (features). One of the feature selection techniques known as mRMR was used to obtain the limited set of features which could be helpful in categorizing the samples because many characteristics were correlated. The top 100 genes with the least amount of redundancy and the most relevance were extracted from these 2,604 genes (features) using mRMR. Furthermore, 100 genes (features) separated the HNSCC patients from non-cancer with an accuracy of around 92%, an AUROC of 0.91 in the validation set. Whereas in the case of HPV classification, the metrics obtained were, AUROC 0.83% and 98% accuracy on the validation set. For the detection and categorization of biomarkers, ANN has proven to be an effective technique among all machine learning models.
After obtaining the top 100 most relevant genes for the classification of HNSCC, we performed Gene Ontology (GO) enrichment analysis using PantherDB and most of the genes were observed to be related to catalytic and binding activities (Mi et al., 2013). Some of them also had a role in other essential processes like ATP-dependent activity, molecular function regulator, molecular transducer, structural molecule activity, translation regulator activity, transcription regulator, and transporter activity. Many of the genes identified in this study have been previously linked to HNSCC in earlier studies. The gene PLAC9’s overexpression has been reported in connection with the inhibition of cell growth regulation and has also been reported in connection with cancers such as ovarian cancer and breast cancers as prognostic biomarkers (Ouyang et al., 2018). Gene “ACKR1”, along with other 3 genes in a study, was reported to be downregulated in HNSCC patients, which was correlated with poor prognosis (p < 0.05) (Liu et al., 2022). Also, gene “AQP7,” which is involved in physiologically functional cell migration, was upregulated in MSR of patients with ten tumors (Zivicova et al., 2018). Whereas, gene FXYD1 was reported to be downregulated in the cancer samples, while FXYD4 and FXYD5 were overexpressed (p < 0.05, fold change>1.5) (Jin et al., 2021). In a study on cancer cells, it was observed that BTG1 gene overexpression was linked to tumor growth or lung metastasis, inhibited proliferation, and induced differentiation in different types of cancer cells (Zhao et al., 2020). Also, mutations occurring in different genes, including B2M, CDKN2A, is found to be related with the occurrence and development of tumors in Head and neck cancer patients (Wang et al., 2020). As shown in the study Sun et al., 2020, genes such as MFAP4, CD37, CXCL12, ADH1B, SOD3, SCARA5, ANGPTL1, FHL1, F10, CXCR4, MEG3, TXNIP, GDF10, and ABI3BP are downregulated in head and neck squamous cell carcinoma as they operate as potential tumor suppressor genes, inhibiting tumor cell proliferation, invasion, and migration while also promoting apoptosis (Sun et al., 2020). By controlling the expression of miR-421 and E-cadherin, MEG3 long-encoding RNA inhibits the development of head and neck squamous cell carcinoma. However, additional research into MEG3’s downstream mechanism in controlling the molecular process of epithelial-mesenchymal transformation (EMT) in head and neck squamous cell carcinoma (HNSCC) development is required (Ji et al., 2020). Growth differentiation factor-10 (GDF10), also known as BMP3b, is a tumor suppressor that belongs to the transforming growth factor-b (TGF-b) superfamily (Cheng et al., 2016). CIB1, PIM3, SLC16A3, VOPP1, BMP4, TIGIT, ADAR, and LRRN4CL are studied as upregulated genes in various cancer types such as squamous carcinoma cells, breast cancer, head and neck squamous cell carcinoma, and pancreatic cancer (Baras et al., 2011; Alarmo et al., 2013; Zheng and Tian, 2014; de Jong et al., 2018; Notarangelo, 2018; Broutian et al., 2020; Yu et al., 2020; Huo et al., 2021; Wen et al., 2021; Yang et al., 2022). A complex that is important in the keratinocyte-intrinsic immune response to human papillomaviruses (-HPVs) is formed when CIB1 interacts with the EVER1, and EVER2 proteins (de Jong et al., 2018; Notarangelo, 2018). It has been observed that nearly all primary HNSCCs express at least one PIM kinase member at high levels (Broutian et al., 2020). Immunological checkpoint T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) is essential for immune suppression. However, it has a connection to genetics and epigenetics, and a role in tumor immunity (Wen et al., 2021). The transforming growth factor (TGF) superfamily includes extracellular signaling molecules known as bone morphogenetic proteins (BMPs), which are known to control cell proliferation, differentiation, and motility, particularly during development. Functional research shows that, particularly in HNSCC cancer, has connected BMP4 to the encouragement of cell migration and the suppression of cell proliferation (Alarmo et al., 2013).
Overall, most of the genes which were obtained from our study have been reported as promising candidate for biomarkers in various studies (Zivicova et al., 2018; Broutian et al., 2020; Sun et al., 2020; Wang et al., 2020; Zhao et al., 2020; Jin et al., 2021; Liu et al., 2022). However, some genes have not yet been reported in connection with Head and Neck cancer. These genes may require further investigation and study. These genes may act as novel findings which could help in diagnose patients with Head and neck cancer. In order to help the scientific community, we created a Python package called “HNSCPred” based on the aforementioned work (https://webs.iiitd.edu.in/raghava/hnscpred/). To fully understand how the discovered genes impact and contribute to the progression of HNSCC disease, further clinical investigations on these genes are necessary.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://webs.iiitd.edu.in/raghava/hnscpred/.
AJ: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing–review and editing. AD: Data curation, Formal Analysis, Investigation, Methodology, Software, Supervision, Visualization, Writing–review and editing. AA: Methodology, Supervision, Visualization, Writing–original draft, Writing–review and editing. SP: Formal Analysis, Methodology, Project administration, Software, Supervision, Writing–review and editing. AS: Methodology, Software, Writing–review and editing. GR: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors are thankful to the Council of Scientific and Industrial Research (CSIR), Department of Biotechnology (DBT), and Department of Science and Technology (DST) for providing fellowships and financial support. We are thankful to the Department of Biotechnology (DBT) for providing an infrastructure grant to the institute (grant number - BT/PR40158/BTIS/137/24/2021).
The authors are thankful to the Department of Computational Biology, IIITD, New Delhi for its infrastructure and facilities. We would like to acknowledge that the figures were created using BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2024.1395721/full#supplementary-material
Aggarwal, S., Dhall, A., Patiyal, S., Choudhury, S., Arora, A., and Raghava, G. P. S (2023). An ensemble method for prediction of phage-based therapy against bacterial infections. Front. Microbiol. 14, 1148579. doi:10.3389/fmicb.2023.1148579
Alarmo, E.-L., Huhtala, H., Korhonen, T., Pylkkänen, L., Holli, K., Kuukasjärvi, T., et al. (2013). Bone morphogenetic protein 4 expression in multiple normal and tumor tissues reveals its importance beyond development. Mod. Pathol. 26, 10–21. doi:10.1038/modpathol.2012.128
Ang, K. K., Harris, J., Wheeler, R., Weber, R., Rosenthal, D. I., Nguyen-Tân, P. F., et al. (2010). Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 363 (1), 24–35. doi:10.1056/NEJMoa0912217
Arora, A., Patiyal, S., Sharma, N., Devi, N. L., Kaur, D., and Raghava, G. P. S. (2023). A random forest model for predicting exosomal proteins using evolutionary information and motifs. Proteomics 24, e2300231. doi:10.1002/pmic.202300231
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi:10.1038/75556
Baras, A. S., Solomon, A., Davidson, R., and Moskaluk, C. A. (2011). Loss of VOPP1 overexpression in squamous carcinoma cells induces apoptosis through oxidative cellular injury. Lab. Investig. 91, 1170–1180. doi:10.1038/labinvest.2011.70
Basheeth, N., and Patil, N. (2019). Biomarkers in head and neck cancer an update. Indian J. Otolaryngol. Head. Neck Surg. 71, 1002–1011. doi:10.1007/s12070-019-01683-1
Broutian, T. R., Jiang, B., Li, J., Akagi, K., Gui, S., Zhou, Z., et al. (2020). Human papillomavirus insertions identify the PIM family of serine/threonine kinases as targetable driver genes in head and neck squamous cell carcinoma. Cancer Lett. 476, 23–33. doi:10.1016/j.canlet.2020.01.012
Bulac, C., and Bulac, A. (2016). “Decision trees,” in Advanced solutions in power systems: HVDC, FACTS, and AI techniques. doi:10.1002/9781119175391.ch18
Chaturvedi, A. K., Engels, E. A., Pfeiffer, R. M., Hernandez, B. Y., Xiao, W., Kim, E., et al. (2011). Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 29 (32), 4294–4301. doi:10.1200/JCO.2011.36.4596
Chen, T., and Guestrin, C. (2016). “XGBoost: a scalable tree boosting system,” in Proceedings of the ACM SIGKDD international conference on knowledge discovery and data mining. doi:10.1145/2939672.2939785
Cheng, C.-W., Hsiao, J.-R., Fan, C.-C., Lo, Y.-K., Tzen, C.-Y., Wu, L.-W., et al. (2016). Loss of GDF10/BMP3b as a prognostic marker collaborates with TGFBR3 to enhance chemotherapy resistance and epithelial-mesenchymal transition in oral squamous cell carcinoma. Mol. Carcinog. 55, 499–513. doi:10.1002/mc.22297
Choi, J.-H., Lee, B.-S., Jang, J. Y., Lee, Y. S., Kim, H. J., Roh, J., et al. (2023). Single-cell transcriptome profiling of the stepwise progression of head and neck cancer. Nat. Commun. 14, 1055. doi:10.1038/s41467-023-36691-x
Clough, E., and Barrett, T. (2016). The gene expression Omnibus database. Methods Mol. Biol. 1418, 93–110. doi:10.1007/978-1-4939-3578-9_5
de Jong, S. J., Créquer, A., Matos, I., Hum, D., Gunasekharan, V., Lorenzo, L., et al. (2018). The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. J. Exp. Med. 215, 2289–2310. doi:10.1084/jem.20170308
Ding, C., and Peng, H. (2005). Minimum redundancy feature selection from microarray gene expression data. J. Bioinform Comput. Biol. 3, 185–205. doi:10.1142/s0219720005001004
Dok, R., and Nuyts, S. (2016). HPV positive head and neck cancers: molecular pathogenesis and evolving treatment strategies. Cancers (Basel) 8 (4), 41. doi:10.3390/cancers8040041
Eberwine, J., Sul, J.-Y., Bartfai, T., and Kim, J. (2014). The promise of single-cell sequencing. Nat. Methods 11, 25–27. doi:10.1038/nmeth.2769
Fakhry, C., Westra, W. H., Li, S., Cmelak, A., Ridge, J. A., Pinto, H., et al. (2008). Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 100 (4), 261–269. doi:10.1093/jnci/djn011
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188. doi:10.1038/nrg.2015.16
Gene Ontology Consortium (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. doi:10.1093/nar/gkaa1113
Geurts, P., Ernst, D., and Wehenkel, L. (2006). Extremely randomized trees. Mach. Learn 63, 3–42. doi:10.1007/s10994-006-6226-1
Gillison, M. L., Zhang, Q., Jordan, R., Xiao, W., Westra, W. H., Trotti, A., et al. (2012). Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol. 30 (17), 2102–2111. doi:10.1200/JCO.2011.38.4099
Hsieh, J. C.-H., Wang, H.-M., Wu, M.-H., Chang, K.-P., Chang, P.-H., Liao, C.-T., et al. (2019). Review of emerging biomarkers in head and neck squamous cell carcinoma in the era of immunotherapy and targeted therapy. Head. Neck 41 (Suppl. 1), 19–45. doi:10.1002/hed.25932
Huo, X.-X., Wang, S.-J., Song, H., Li, M., Yu, H., Wang, M., et al. (2021). Roles of major RNA adenosine modifications in head and neck squamous cell carcinoma. Front. Pharmacol. 12, 779779. doi:10.3389/fphar.2021.779779
Ji, Y., Feng, G., Hou, Y., Yu, Y., Wang, R., and Yuan, H. (2020). Long noncoding RNA MEG3 decreases the growth of head and neck squamous cell carcinoma by regulating the expression of miR-421 and E-cadherin. Cancer Med. 9, 3954–3963. doi:10.1002/cam4.3002
Jin, M., Zhang, H., Yang, J., Zheng, Z., and Liu, K. (2021). Expression mode and prognostic value of FXYD family members in colon cancer. Aging 13, 18404–18422. doi:10.18632/aging.203290
Johnson, D. E., Burtness, B., Leemans, C. R., Lui, V. W. Y., Bauman, J. E., and Grandis, J. R. (2020). Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 6, 92. doi:10.1038/s41572-020-00224-3
Kaur, D., Arora, A., Vigneshwar, P., and Raghava, G. P. S. (2023). Prediction of peptide hormones using an ensemble of machine learning and similarity-based methods. bioRxiv 2023 (15), 540764. doi:10.1101/2023.05.15.540764
Leemans, C. R., Snijders, P. J. F., and Brakenhoff, R. H. (2018). The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18 (5), 269–282. doi:10.1038/nrc.2018.11
Liu, H., Hei, G., Zhang, L., Jiang, Y., and Lu, H. (2022). Identification of a novel ceRNA network related to prognosis and immunity in HNSCC based on integrated bioinformatic investigation. Sci. Rep. 12, 17560. doi:10.1038/s41598-022-21473-0
Mahmood, H., Shaban, M., Rajpoot, N., and Khurram, S. A. (2021). Artificial Intelligence-based methods in head and neck cancer diagnosis: an overview. Br. J. Cancer 124, 1934–1940. doi:10.1038/s41416-021-01386-x
Mi, H., Muruganujan, A., and Thomas, P. D. (2013). PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386. doi:10.1093/nar/gks1118
Mody, M. D., Rocco, J. W., Yom, S. S., Haddad, R. I., and Saba, N. F. (2021). Head and neck cancer. Lancet 398, 2289–2299. doi:10.1016/S0140-6736(21)01550-6
Notarangelo, L. D. (2018). HPV: CIB1 is for EVER and EVER. J. Exp. Med. 215, 2229–2231. doi:10.1084/jem.20181207
Ouyang, C., Pu, Y.-Z., Qin, X.-H., Shen, J., Liu, Q.-H., Ma, L., et al. (2018). Placenta-specific 9, a putative secretory protein, induces G2/M arrest and inhibits the proliferation of human embryonic hepatic cells. Biosci. Rep. 38. doi:10.1042/BSR20180820
Peng, L., Bian, X. W., Li, D. K., Xu, C., Wang, G. M., Xia, Q. Y., et al. (2015). Large-scale RNA-seq transcriptome analysis of 4043 cancers and 548 normal tissue controls across 12 TCGA cancer types. Sci. Rep. 5, 13413. doi:10.1038/srep13413
Radovic, M., Ghalwash, M., Filipovic, N., and Obradovic, Z. (2017). Minimum redundancy maximum relevance feature selection approach for temporal gene expression data. BMC Bioinforma. 18, 9. doi:10.1186/s12859-016-1423-9
Radpour, R., and Forouharkhou, F. (2018). Single-cell analysis of tumors: creating new value for molecular biomarker discovery of cancer stem cells and tumor-infiltrating immune cells. World J. Stem Cells 10, 160–171. doi:10.4252/wjsc.v10.i11.160
Saliba, A.-E., Westermann, A. J., Gorski, S. A., and Vogel, J. (2014). Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 42, 8845–8860. doi:10.1093/nar/gku555
Srivastava, A., Dhall, A., Patiyal, S., Arora, A., Jarwal, A., and Raghava, G. P. S. (2023) Prediction of alzheimer’s disease from single cell transcriptomics using deep learning. doi:10.1101/2023.07.07.548171
Stoltzfus, J. C. (2011). Logistic regression: a brief primer. Acad. Emerg. Med. 18, 1099–1104. doi:10.1111/j.1553-2712.2011.01185.x
Sun, Y., Zhang, Q., Yao, L., Wang, S., and Zhang, Z. (2020). Comprehensive analysis reveals novel gene signature in head and neck squamous cell carcinoma: predicting is associated with poor prognosis in patients. Transl. Cancer Res. 9, 5882–5892. doi:10.21037/tcr-20-805
Vabalas, A., Gowen, E., Poliakoff, E., and Casson, A. J. (2019). Machine learning algorithm validation with a limited sample size. PLoS One 14, e0224365. doi:10.1371/journal.pone.0224365
Wang, J., Chen, X., Tian, Y., Zhu, G., Qin, Y., Chen, X., et al. (2020). Six-gene signature for predicting survival in patients with head and neck squamous cell carcinoma. Aging 12, 767–783. doi:10.18632/aging.102655
Wang, S.-C. (2003). “Artificial neural network,” in Interdisciplinary computing in java programming (Boston, MA: Springer US), 81–100. doi:10.1007/978-1-4615-0377-4_5
Wen, J., Mao, X., Cheng, Q., Liu, Z., and Liu, F. (2021). A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci. Rep. 11, 22502. doi:10.1038/s41598-021-01933-9
Wolf, F. A., Angerer, P., and Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. doi:10.1186/s13059-017-1382-0
Wu, Y., Ianakiev, K., and Govindaraju, V. (2002). Improved k-nearest neighbor classification. Pattern Recognit. 35, 2311–2318. doi:10.1016/S0031-3203(01)00132-7
Yang, F., Zhou, L.-Q., Yang, H.-W., and Wang, Y.-J. (2022). Nine-gene signature and nomogram for predicting survival in patients with head and neck squamous cell carcinoma. Front. Genet. 13, 927614. doi:10.3389/fgene.2022.927614
Yu, S., Wu, Y., Li, C., Qu, Z., Lou, G., Guo, X., et al. (2020). Comprehensive analysis of the SLC16A gene family in pancreatic cancer via integrated bioinformatics. Sci. Rep. 10, 7315. doi:10.1038/s41598-020-64356-y
Zhao, S., Xue, H., Hao, C.-L., Jiang, H.-M., and Zheng, H.-C. (2020). BTG1 overexpression might promote invasion and metastasis of colorectal cancer via decreasing adhesion and inducing epithelial-mesenchymal transition. Front. Oncol. 10, 598192. doi:10.3389/fonc.2020.598192
Zhao, Z., Anand, R., and Wang, M. (2019a) Maximum relevance and minimum redundancy feature selection methods for a marketing machine learning platform.
Zhao, Z., Anand, R., and Wang, M. (2019b) Maximum relevance and minimum redundancy feature selection methods for a marketing machine learning platform.
Zheng, D., and Tian, B. (2014). RNA-binding proteins in regulation of alternative cleavage and polyadenylation. Adv. Exp. Med. Biol. 825, 97–127. doi:10.1007/978-1-4939-1221-6_3
Zivicova, V., Gal, P., Mifkova, A., Novak, S., Kaltner, H., Kolar, M., et al. (2018). Detection of distinct changes in gene-expression profiles in specimens of tumors and transition zones of tenascin-positive/-negative head and neck squamous cell carcinoma. Anticancer Res. 38, 1279–1290. doi:10.21873/anticanres.12350
Keywords: HNSCC, gene biomarkers, single cell transcriptomics, machine learning, deep learning, classification models
Citation: Jarwal A, Dhall A, Arora A, Patiyal S, Srivastava A and Raghava GPS (2024) A deep learning method for classification of HNSCC and HPV patients using single-cell transcriptomics. Front. Mol. Biosci. 11:1395721. doi: 10.3389/fmolb.2024.1395721
Received: 04 March 2024; Accepted: 13 May 2024;
Published: 30 May 2024.
Edited by:
Qingyu Luo, Dana–Farber Cancer Institute, United StatesReviewed by:
Lin Han, Dana–Farber Cancer Institute, United StatesCopyright © 2024 Jarwal, Dhall, Arora, Patiyal, Srivastava and Raghava. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gajendra P. S. Raghava, cmFnaGF2YUBpaWl0ZC5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.