 Maria Tsekrekou
Maria Tsekrekou Maria Giannakou
Maria Giannakou Katerina Papanikolopoulou
Katerina Papanikolopoulou Georgios Skretas
Georgios Skretas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 24 May 2024
Sec. Cellular Biochemistry
Volume 11 - 2024 | https://doi.org/10.3389/fmolb.2024.1383453
This article is part of the Research Topic Cellular Contributors and Consequences of Protein Misfolding and Aggregation View all 5 articles
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with severe socio-economic impact. A hallmark of ALS pathology is the presence of aberrant cytoplasmic inclusions composed of misfolded and aggregated proteins, including both wild-type and mutant forms. This review highlights the critical role of misfolded protein species in ALS pathogenesis, particularly focusing on Cu/Zn superoxide dismutase (SOD1) and TAR DNA-binding protein 43 (TDP-43), and emphasizes the urgent need for innovative therapeutic strategies targeting these misfolded proteins directly. Despite significant advancements in understanding ALS mechanisms, the disease remains incurable, with current treatments offering limited clinical benefits. Through a comprehensive analysis, the review focuses on the direct modulation of the misfolded proteins and presents recent discoveries in small molecules and peptides that inhibit SOD1 and TDP-43 aggregation, underscoring their potential as effective treatments to modify disease progression and improve clinical outcomes.
Precise activity of cellular protein networks is essential for the proper function and fitness of cells and organisms. Protein function highly depends on its three-dimensional structure, i.e., its folding state (Yadav et al., 2019). To ensure functionality, biological systems have evolved protein quality control mechanisms comprising molecular chaperones, proteases and regulatory factors, which supervise protein folding and direct protein species which fail to meet cellular quality control criteria for degradation. Despite the existence of highly specialized protein folding surveillance mechanisms, protein misfolding and aggregation are hallmarks of numerous human diseases (Chiti and Dobson, 2006; Chiti and Dobson, 2017). These diseases range from systemic amyloidoses, where amyloid fibrils accumulate throughout the human body, to neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and ALS, where protein aggregates are mainly deposited either extracellularly (AD) or intracellularly (PD, ALS) within the central nervous system (CNS).
ALS is a neurodegenerative disease (Hardiman et al., 2017) characterized by the selective degeneration of both upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord (Tandan and Bradley, 1985; Hardiman et al., 2017; van Es et al., 2017). It is the most common form of motor neuron disease with adult onset and the third most common neurodegenerative disease (Hou et al., 2019). The survival rate is highly variable, with a median rate of about 3–5 years after symptom onset; however, up to 10% of ALS patients survive for more than 10 years (Chio et al., 2009; van Es et al., 2017; Longinetti and Fang, 2019). ALS affects approximately two to four people per 100,000 individuals per year in Caucasian populations and about one person or less per 100,000 individuals annually in Asian and Hispanic populations (Cronin et al., 2007; Al-Chalabi and Hardiman, 2013; Hardiman et al., 2017; Longinetti and Fang, 2019), with the exception of Japan where the incidence rate is closer to the that of Western countries (Doi et al., 2014). Recent studies indicate a trend of rising incidence rates for ALS (Collaborators, 2019; Longinetti and Fang, 2019; Ryan et al., 2019).
ALS has traditionally been classified as either sporadic (sALS), representing approximately 90%–95% of cases, or familial (fALS), typically characterized by Mendelian autosomal dominant inheritance (van Es et al., 2017; Ranganathan et al., 2020; Kirola et al., 2022). The term “sporadic” is applied to cases which are unrelated to family incidence, even when gene mutations associated with or causing ALS are present (Mejzini et al., 2019). Comprehensive analyses from large-scale genome-wide association studies (GWAS) on sALS patients have revealed that the genetic landscape of ALS predominantly comprises rare variants (van Rheenen et al., 2016; van Rheenen et al., 2021). Consequently, the field is progressing toward a more precise genetic classification, emphasizing the identification of risk genes (Cooper-Knock et al., 2021; Brenner and Freischmidt, 2022). Regardless of the subclassification criteria, both forms of ALS are clinically indistinguishable (Couratier et al., 2021), differing only in the age of onset, with fALS occurring about a decade earlier than sALS (Wijesekera and Leigh, 2009).
Familial cases of ALS have facilitated the identification of the involvement of the genetic background in disease pathogenesis (Gros-louis et al., 2006). Currently more than 40 genes have been associated with fALS (van Rheenen et al., 2021; Brenner and Freischmidt, 2022; Suzuki et al., 2023), enabling the generation of animal models carrying ALS-related gene mutations (Bonifacino et al., 2021; Zhu et al., 2023). In vivo modelling of ALS has provided a valuable tool to elucidate the pathogenic mechanisms contributing to disease onset and progression, and allowing for targeted drug development (Hardiman et al., 2017; Bonifacino et al., 2021; Zhu et al., 2023). Importantly, research of ALS animal models has revealed an intertwining network of molecular (e.g., aberrant RNA metabolism, impaired protein homeostasis, oxidative stress, mitochondrial dysfunction, impaired DNA repair and dysregulated vesicle transport) and cellular disruptions (such as hyperexcitability, glial dysfunction and axonopathy) that build up to systemic aberrations, ultimately leading to the disease (Hardiman et al., 2017).
ALS is a complex disease, characterized by significant clinical heterogeneity. This heterogeneity is evident in the variability of the site and age of disease onset, the rate of progression, and the degree of cognitive impairment (Chio et al., 2011; Ferrari et al., 2011; Statland et al., 2015). Despite this clinical variability, a cellular hallmark of ALS is the presence of ubiquitinated skein-like or dense and round cytoplasmic inclusions of certain proteins, such as SOD1, TDP-43, and fused in sarcoma (FUS) in motor neurons (Leigh et al., 1988; Lowe et al., 1988). Approximately 97% of ALS cases exhibit TDP-43-positive inclusions (Mackenzie et al., 2007). Notably, TDP-43 cytoplasmic inclusions are also found in other neurodegenerative disorders including Frontotemporal lobar degeneration (FTLD), combined ALS-FTLD, AD and atypical Parkinsonism (de Boer et al., 2020). TDP-43 inclusions are absent in ALS cases caused by mutations in SOD1 and FUS; in these instances, protein deposits consist of the respective mutated gene products (Neumann et al., 2009; Vance et al., 2009).
Considering that aberrant cytoplasmic inclusions are a ubiquitous finding in ALS patients, numerous research efforts have been directed toward inhibiting the aggregation of ALS-related proteins. In this review, we focus on two extensively studied ALS-related proteins associated with protein misfolding and aggregation, SOD1 and TDP-43. We examine the contribution of their accumulation to disease phenotypes and review the recent progress in identifying inhibitors of their aggregation, which could serve as lead molecules for the development of effective anti-ALS treatments.
SOD1 plays a major role in regulating redox potential by catalyzing the conversion of the superoxide anion O2− into hydrogen peroxide, which is further processed by other enzymes (Bunton-Stasyshyn et al., 2015; Huai and Zhang, 2019). In its functional form, SOD1 forms a homodimer, with each subunit containing a copper and a zinc ion at its [Cu/Zn] site and an intra-subunit C57-C146 disulfide bond (Valentine et al., 2005; Huai and Zhang, 2019). The discovery of SOD1 mutations causing fALS in 1993 was a milestone for ALS research (Rosen et al., 1993). To date, more than 200 mutations in human SOD1 have been associated with ALS (van der Spek et al., 2019). Mutations in SOD1, lead to classical dominantly inherited ALS, accounting for about 10%–15% of fALS cases and approximately 1.5% of sALS cases (Gros-louis et al., 2006). Although SOD1 misfolding and aggregation are considered hallmarks of SOD1-fALS (Saccon et al., 2013), SOD1-immunoreactive inclusions have also been detected in a significant percentage of sALS cases (Bosco et al., 2010; Forsberg et al., 2019). As the longest-studied ALS-related protein, SOD1 has been associated with a large number of pathophysiological mechanisms causing neurotoxicity, including protein misfolding, excitotoxicity, oxidative stress, impaired axonal transport, inflammation and mitochondrial dysfunction (Ferraiuolo et al., 2011).
Wild-type SOD1 is an unusually stable protein, which remains active even under denaturing conditions (Arnesano et al., 2004). It is able to maintain its disulfide status inside the reducing cytosolic environment and is highly resistant to proteolysis (Arnesano et al., 2004; Valentine et al., 2005). This remarkable stability is regulated by the disulfide bond and the metalation status, which mutually affect each other (Arnesano et al., 2004; Tiwari et al., 2005; Nordlund et al., 2009). Disruption of the disulfide bond or loss of the metal co-factors can lead to pathogenic misfolding. Specific post-translational modifications (PTMs), including phosphorylation, acetylation, and succinylation, play crucial roles in regulating the structure and functions of SOD1, such as ROS scavenging, cytoskeletal organization, and transcriptional activity (Banks and Andersen, 2019), while others like sumoylation and oxidation can lead to misfolding and aggregation (Fei et al., 2006; Xu et al., 2018; Trist et al., 2022).
Interestingly, mutations associated with ALS have been identified across all five exons of human SOD1. These mutations impact the structure of SOD1 in different ways, often leading to varying degrees of misfolding, aggregation and consequent variable toxicities (Brasil et al., 2018). Initially, it was proposed that ALS-causing mutations in SOD1 were linked to a loss of enzymatic function (Rosen et al., 1993; Saccon et al., 2013); however, subsequent studies in SOD1 mice contradicted this notion (Gurney et al., 1994; Reaume et al., 1996). Mutant SOD1 retaining partial or complete enzymic function was found to induce ALS-like phenotypes (Gurney et al., 1994), and Sod1 knockout mice did not develop ALS (Reaume et al., 1996). In contrast, ALS-related SOD1 variants are associated with decreased metal binding, reduced formation of a stabilizing intramolecular disulfide bond, diminished structural stability and an increased tendency to monomerize and aggregate (Ray et al., 2004). Importantly, the aggregation propensity of several SOD1 variants has been linked to life expectancy after the onset of ALS symptoms in both humans and transgenic mouse models (Wang et al., 2008; Prudencio et al., 2009; Pratt et al., 2014; Lang et al., 2015; McAlary et al., 2020).
TDP-43, encoded by the TARDBP gene, was identified in 2006 as a major component of cytoplasmic protein inclusions accompanied by nuclear clearance of the protein, as observed in motor neurons of ALS cases (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2007). TDP-43 pathology is present in nearly all ALS cases except for those caused by mutations in SOD1 or FUS. TARDBP mutations account for approximately 5% of familial and almost 1% of sporadic ALS cases. It is an RNA-binding protein (RBP) involved in the regulation of gene expression at multiple levels by playing an active role in alternative splicing of pre-mRNAs (Bhardwaj et al., 2013; Lukavsky et al., 2013; Kuo et al., 2014), splicing of non-coding RNAs (Tollervey et al., 2011) and mRNA stability (Fukushima et al., 2019), thus affecting diverse cell processes, including mitochondrial homeostasis (Izumikawa et al., 2017), DNA damage response (Konopka et al., 2020; Riancho et al., 2020; Wood et al., 2020) and axonal transport (Fallini et al., 2012; Tripathi et al., 2014). While TDP-43 primarily localizes in the nucleus, upon cellular stress, it translocates to the cytoplasm to form stress granules together with other RBPs and stalled ribosomes (Colombrita et al., 2009). Interestingly, single cell proteomic analysis of somatic motor neurons (MNs) derived from postmortem spinal cords of ALS donors with TDP-43 pathology, not only facilitated the differentiation of disease states in individual MNs but also uncovered significant reduction in the abundance of proteins with critical roles in cell energetics, protein translation, proteostasis, and trafficking mechanisms such as Golgi-lysosome trafficking (Guise et al., 2024). Furthermore, C9orf72 mutations lead to the formation of RNA foci which sequester a range of RBPs including TDP-43, resulting in its aberrant accumulation in the cytoplasm (Lee et al., 2013; Chew et al., 2019).
TDP-43 consists of four domains that mediate distinct activities: (a) a globular N-terminal domain (NTD) with a nuclear localization signal (NLS). The NTD is crucial for the formation of the TDP-43 functional dimer/oligomer (Qin et al., 2014; Mompean et al., 2016; Afroz et al., 2017; Jiang et al., 2017; Mompean et al., 2017) and recruits RNA for splicing (Jiang et al., 2017); (b) two tandem RNA recognition motifs (RRM1 and RRM2) (Lukavsky et al., 2013) with a nuclear export signal (NES) in RRM2 (Winton et al., 2008), although the NES functionality has been questioned (Archbold et al., 2018; Pinarbasi et al., 2018), and (c) a prion-like domain (PrLD) at the C-terminus encompassing two subdomains, a glutamine/asparagine-rich and a glycine-rich region, which is disordered and essential for protein-protein interactions (Buratti, 2020; Jiang et al., 2013). Six cysteine residues are present in TDP-43, with four located in the two RRM domains (Cys173, Cys175, Cys198, and Cys244), while the other two (Cys39 and Cys50) in the N-terminal domain (Valle and Carri, 2017). The oxidation of cysteines within the two RRMs decreases protein solubility and induces the formation of intra- and inter-molecular disulfide linkages (Cohen et al., 2012; Chang et al., 2013). The PrLD mediates TDP-43 intrinsic aggregation propensity (Johnson et al., 2009) and its incorporation into stress granules via its ability to undergo liquid-liquid phase separation (LLPS) (Conicella et al., 2016; Wang et al., 2018). Interestingly, the majority (∼95%) of the ALS-linked TARDBP mutations are localized at the C-terminal domain (Prasad et al., 2019); these TDP-43 variants show increased stress granule formation upon oxidative stress (Liu-Yesucevitz et al., 2010) and higher aggregation propensity (Conicella et al., 2016). Moreover, certain TARDBP-linked mutations enhance cytoplasmic mislocalization of TDP-43 (Barmada et al., 2010; Mutihac et al., 2015; Mitsuzawa et al., 2018). Finally, in addition to cysteine oxidation, also other PTMs such as phosphorylation, acetylation, ubiquitination and the proteolytic processing of TDP-43 that leads to the formation of C-terminal fragments are closely associated with the misfolding and aggregation of the protein (Buratti, 2018; Farina et al., 2021).
Cell culture and animal models have provided compelling evidence indicating a significant involvement of TDP-43 in the initiation and progression of motor neuron degeneration (Bonifacino et al., 2021). Transgenic mice overexpressing TDP-43 with familial fALS mutations, develop aggregates and manifest a full spectrum of ALS-like phenotypes at the molecular, cellular and behavioral levels (Ke et al., 2015; Huang et al., 2020). Similarly, the overexpression of a TDP-43 variant lacking the nuclear localization signal (dNLS), leads to the accumulation of insoluble, phosphorylated cytoplasmic TDP-43 in the brain and spinal cord. This is accompanied by brain atrophy, muscle denervation, significant motor neuron loss, and the development of progressive motor impairment (Walker et al., 2015). Notably, suppression of TDP-43 overexpression leads to a rapid clearance of TDP-43 pathology and rescues motor deficits in inducible mouse models (Ke et al., 2015; Walker et al., 2015). These collective findings strongly suggest that TDP-43 is a key protein implicated in neurodegenerative processes in motor neurons.
The term protein aggregation describes the transition of a protein from its native biologically functional state to the formation of oligomers and medium- or higher-order aggregates through self-association. Protein aggregation is usually caused by the presence of unfolded or misfolded species of the protein. Misfolded conformations typically expose hydrophobic segments within a hydrophilic- either intracellular or extracellular- environment, which subsequently tend to self-associate into soluble oligomers and eventually to larger insoluble aggregates. Most often, the building block of oligomeric and aggregated species is the protein’s unfolded/misfolded/partially folded monomer. The structure of such aggregates highly depends on the conformation of the monomeric state: disordered amorphous aggregates are usually formed by unfolded or native monomers while well-defined fibrils with cross-β structure (amyloid fibrils) can originate from partially folded monomers (Almeida and Brito, 2020). Amyloid deposits have been extensively implicated in disease pathogenesis, while there are few disorders associated with amorphous aggregates, such as cataracts caused by γD-crystallin disordered deposition (Moreau and King, 2012).
The amyloidogenic pathway has been extensively studied and two distinct mechanisms, the downhill polymerization and the nucleation-growth model, have been described. Both are characterized by sigmoidal kinetics and differ in the factor that determines the rate-limiting step. In each case, the mechanism of aggregation employed depends on the protein. Downhill polymerization (Figure 1A) is typically observed in proteins with oligomeric native conformation, such as transthyretin (TTR) (Hammarstrom et al., 2003; Eisele et al., 2015). The rate-limiting step is the disassembly of the native oligomer into unstable monomers, which then rapidly form aggregates (Lai et al., 1996; Hurshman et al., 2004; Eisele et al., 2015). Importantly, the lag phase is governed by the slow dissociation of native oligomeric state and seeding does not accelerate the aggregation process. The nucleation-growth model (Figure 1B), characteristic of the aggregation of amyloid-β peptide (Knowles et al., 2014), resembles the crystallization process (Teplow, 1998; Scherzinger et al., 1999; Wood et al., 1999; Knowles et al., 2014). The rate-limiting step for amyloid fibril development is the formation of protein oligomers (nuclei or seeds), which subsequently drive the rapid elongation of the fibrils by further monomeric or oligomeric species binding to the nuclei. The slow lag phase can be eliminated by adding pre-formed nuclei.
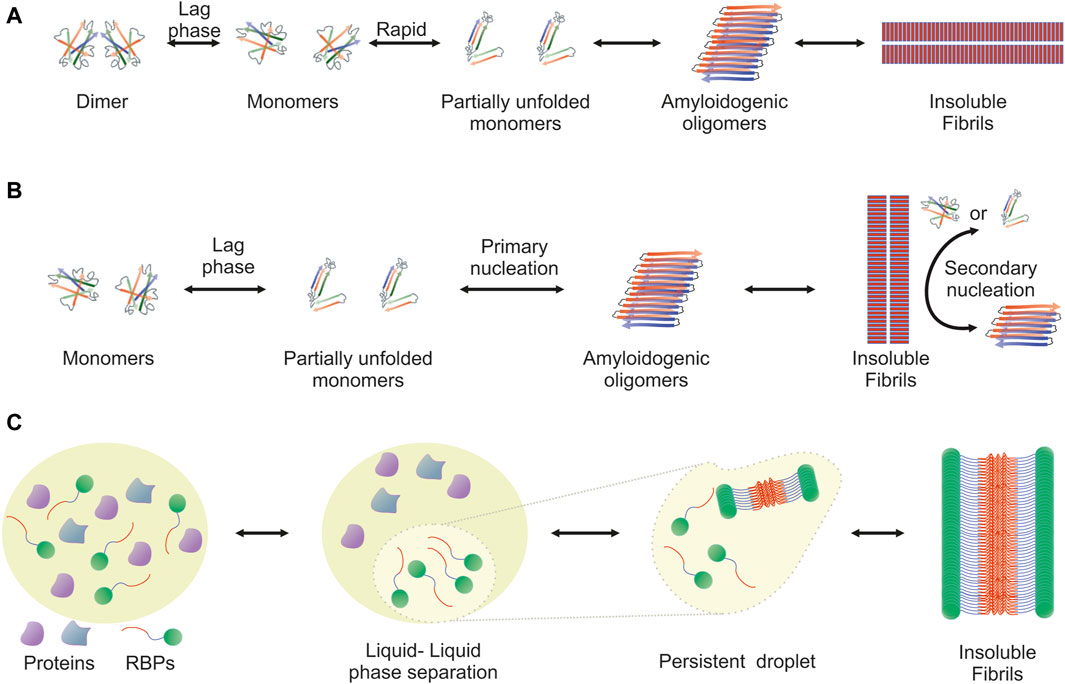
Figure 1. Visualization of the amyloidogenic pathway featuring three distinct models. Detailed overview of the diverse mechanisms through which amyloid fibrils can form, each highlighting different aspects and stages of the amyloidogenic process. (A) Downhill Polymerization Model: The protein first dissociates into native monomers, which is the rate-limiting step in the formation of fibrils. Subsequently, the monomeric species partially unfold to form the aggregation intermediates. Once such intermediates are formed, the following self-assembly process is a downhill polymerization. (B) Nucleation-Growth Model: Amyloid aggregation is kinetically dependent on nucleation events, which include primary and secondary nucleation. Primary nucleation refers to initial events where protein monomers associate into nucleation “seeds.” This is a rate-limiting, kinetically unfavorable process, but once the nuclei are formed, addition of further monomers is more favorable through an elongation process. Primary nucleation causes elongation into fibrils, which eventually leads to the formation of a critical mass of fibrils capable of catalyzing a secondary nucleation phase in which nucleus formation from monomers is catalyzed by existing aggregates. This creates a positive feedback loop of aggregation, where small and large oligomers are constantly formed. (C) Liquid-Liquid Phase Separation (LLPS) Model: It proposes that amyloidogenic proteins first undergo liquid-liquid phase separation, forming protein-rich droplets. Within these droplets, the concentration of proteins reaches a threshold that facilitates the transition from a disordered liquid state to a more ordered solid state, leading to amyloid fibril formation. This model provides insight into the spatial and temporal regulation of amyloidogenesis within cellular environments.
Another mechanism that leads to the formation of ordered aggregates is mediated by LLPS, typically observed in RBPs (Figure 1C). Approximately 30% of the genes associated with ALS encode RBPs, such as TDP-43, FUS, TATA-box binding protein associated factor 15 (TAF15) and proteins from the heterogeneous nuclear ribonucleoprotein (hnRNP) family, including hnRNPA1 and hnRNPA2B1 (Wroe et al., 2008). These proteins form cytoplasmic inclusions in motor neurons of ALS patients and their aggregation propensity is linked to the PrLDs they harbor (Molliex et al., 2015; Patel et al., 2015). PrLDs are a subset of intrinsic disordered regions (IDRs), characterized by low sequence complexity, comprising only a subset of the 20 amino acids; in particular, PrLDs are enriched in uncharged polar amino acids and glycine (Alberti et al., 2009). Intriguingly, about 30% of the 240 human proteins containing predicted PrLDs are involved in RNA processing (March et al., 2016; Verdile et al., 2019). LLPS describes liquid-liquid de-mixing: a high local concentration of IDRs promotes LLPS through transient and weak interactions among the IDRs of RBPs. Importantly, association of RBPs into “droplets” which are separated from the nucleoplasm or the cytoplasm is dynamic and transient. However, persistent association of RBPs with the separated liquid state results in the formation of stable hydrogels and eventually fibrils (Kato et al., 2012; Lin Y. et al., 2015; Guo and Shorter, 2015; Molliex et al., 2015; Murakami et al., 2015; Patel et al., 2015; Kato and McKnight, 2017; Shin and Brangwynne, 2017).
Until recently, there was intense debate over whether the protein aggregates detected in ALS patients are typical amyloids, primarily due to controversial findings. Initially, reports indicated that the TDP-43 positive filamentous aggregates found in ALS-affected motor neurons (Hasegawa et al., 2008; Lin and Dickson, 2008) did not stain with the amyloid-specific dyes Congo red and Thioflavin S (ThS) (Kerman et al., 2010). This finding was further supported by in vitro experiments, where recombinant TDP-43 formed Thioflavin T (ThT)-negative granulo-filamentous aggregates similar to those observed in ALS patients (Johnson et al., 2009) or amorphous aggregates (Capitini et al., 2014). On the other hand, it has been shown that peptide fragments from the C-terminal domain and certain TDP-43 variants form ThT-positive aggregates in vitro, acquiring a β-sheet-enriched structure (Chen et al., 2010; Guo et al., 2011; Zhu et al., 2014; Jiang et al., 2016). These conflicting findings can now be explained based on the structure of TDP-43 aggregates from ALS with FTLD (Arseni et al., 2022), revealing the formation of filaments which are structurally distinct from cross-β amyloid fibrils and a novel fold that bears no similarity to TDP-43 filaments formed in vitro (Cao et al., 2019; Li et al., 2021).
The Falcon group employed cryogenic electron microscopy to determine the structures of aggregated TDP-43 in the frontal and motor cortices of an individual who succumbed to ALS with FTLD, as well as in the frontal cortex of another individual with the same diagnosis (Arseni et al., 2022). Remarkably, they identified an identical amyloid-like filament structure consisting of a single protofilament in both brain regions and individuals. The ordered filament core, spanning residues 282 to 360 in the TDP-43 low-complexity domain, adopts a unique double-spiral-shaped fold. The filament structure lacks the typical β-sheet stacking associated with cross-β amyloid formation due to an abundance of glycine and neutral polar residues, facilitating numerous turns and restricting β-strand length. The uneven distribution of residues results in structurally and chemically distinct surfaces, suggesting potential ligand binding sites on external densities.
This study also revealed that the brain-derived TDP-43 filaments are structurally distinct compared to filaments assembled in vitro from its LCD under mildly acidic conditions (pH 4), or related fragments (Li et al., 2021). These differences include opposite chirality and variations in both protein fold and secondary structure. For the LCD, the structure revealed single protofilament fibrils featuring a large core comprised of 139 residues out of the 148 present in the LCD, tightly packed together. The C-terminal segment of this core exhibits mainly planar characteristics and is marked by a small proportion of hydrophobic amino acids. In contrast, the N-terminal region includes numerous hydrophobic residues and adopts a non-planar backbone conformation, leading to the rugged surfaces observed at the ends of the fibrils.
Cryo-electron microscopy was also employed to elucidate the structures of two segments identified as the pathogenic cores in human TDP-43 aggregation (Cao et al., 2019). The first segment, SegA (residues 311–360), exhibits three polymorphs, each characterized by a dagger-shaped fold, spanning residues 312–346. Tight hydrophobic interactions are formed by hydrophobic residues ranging from Phe313 to Ala341 whereas the dagger tip is created by a sharp 160° kink at Gln327. The variations among the three SegA polymorphs primarily arise from differences in protofilament numbers and symmetry. In contrast to the polymorphic nature of SegA, the second segment, SegB A315E (residues 286–331, containing the ALS hereditary mutation A315E), forms fibrils with a consistent morphology. These fibrils adopt an R-shaped fold spanning residues 288–319, which, overall, displays a more pronounced kink compared to the dagger-shaped fold, likely attributable to the higher prevalence of glycine residues. Each fibril is composed of four protofilaments, and notably, all four R-folds are characterized by the presence of a salt-bridge between Arg293 and Glu315, facilitated by the pathogenic A315E mutation. This salt-bridge may impact the kinetics of fibril growth and nucleation, providing a mechanistic explanation for A315E’s propensity to promote TDP-43 aggregation through electrostatic attraction with Arg293.
Along the same lines, SOD1-positive inclusions from ALS cases display a fibrillary morphology (Kato et al., 2000) but do not react with ThS (Kerman et al., 2010). In contrast, SOD1 cytoplasmic aggregates from a transgenic SOD1-fALS mouse model could be stained with ThS (Furukawa et al., 2008). Importantly, the aggregation kinetics of SOD1 are typically monitored by ThT staining (Rakhit et al., 2002; Furukawa et al., 2008; Iwakawa et al., 2017; Shvil et al., 2018) and pre-formed SOD1 aggregates exhibit seeding activity both in vitro and intracellularly (Furukawa et al., 2008; Furukawa et al., 2013). Moreover, ALS-related SOD1 mutant proteins crystallize in forms comprising higher-order assemblies of aligned β-sheets (Elam et al., 2003). On the other hand, in vitro monitoring of SOD1 aggregation has demonstrated that a competition between amorphous and amyloid aggregation occurs (Abdolvahabi et al., 2016), proposing a possible factor for the wide variability in kinetic results among publications (Crown et al., 2019).
In a recent study, cytotoxic amyloid fibrils were generated from full-length human apo-SOD1 under reducing conditions, and their atomic structure was elucidated using cryo-EM (Wang LQ. et al., 2022). The SOD1 fibril is composed of a singular protofilament featuring a left-handed helix. Within the fibril core, a serpentine fold is formed, incorporating six β-strands of the N-terminal segment (residues 3–55) and seven β-strands of the C-terminal segment (residues 86–153), with an unstructured region in between. This novel amyloid fibril structure displays a very compact fold, and the connection of the two segments is stabilized by three pairs of salt bridges, effectively “zipping up” the structure.
A multitude of pathogenic mechanisms have been described for ALS, including protein misfolding and aggregation, glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, declined autophagy, neuroinflammation and DNA damage. However, the drive behind ALS onset and progression, as well as the degree to which each mechanism contributes to disease phenotypes and heterogeneity, remains unclear. Current evidence supports that protein misfolding and aggregation exert toxic effects on cellular fitness, eventually compromising organismal health. Regarding the toxic agent in ALS and other conformational neurodegenerative diseases, the debate continuous on whether soluble misfolded species (monomeric or oligomeric) or insoluble aggregates confer toxicity (Gelpi and Colom-Cadena, 2019).
Recent work has highlighted SOD1 oligomers, particularly the trimeric variants, as the neurotoxic entities (Proctor et al., 2016). The study revealed that SOD1 mutants promoting trimerization led to increased neuronal cell death and established a direct association between misfolded oligomers and neuron death by linking cytotoxicity to trimer stability. Additional studies showed that large aggregates are non-toxic and actually play a neuroprotective role (Zhu et al., 2018; Gill et al., 2019). Interestingly, SOD1 mutants designed to promote the formation of large aggregates and destabilize toxic trimers did not impact cell viability (Zhu et al., 2018). The mechanism leading to the formation of non-native trimers remains unclear, however, in order to occur, SOD1 must undergo dissociation into monomers (Khare et al., 2004). External factors, such as oxidative stress (Wilcox et al., 2009) or exposure to the toxin β-methylamino-L-alanine (BMMA) (Proctor et al., 2019), may play a role in the transition of SOD1 dimers into monomers. At present, there are two hypotheses regarding trimer formation (Choi and Dokholyan, 2021). The first suggests that trimers occur on the pathway from monomers to native dimers (dissociating into monomers), then to trimers and finally to larger aggregates. The second posits that trimer formation is an off-pathway phenomenon, meaning it does not lead to the generation of large aggregates, and current experimental evidence favors this scenario (Hnath and Dokholyan, 2022). Indicatively, the abundance of soluble misfolded SOD1 subfractions, in the range from monomeric to trimeric in size, has been associated with a reduced lifespan in SOD1-ALS mouse models (Zetterstrom et al., 2007).
Despite the association of TDP-43 aggregation with disease (Neumann et al., 2006; Hasegawa et al., 2008), several studies have highlighted the functional oligomerization of TDP-43 in physiological contexts. The NTD residues 1–105 and 1–265 induce the formation of homo-oligomeric species in a concentration-dependent manner, initiated by the formation of intermolecular disulfide bonds ultimately leading to its tetramerization (Chang et al., 2012; Jiang et al., 2017). Oligomerization of TDP-43 is crucial for its nuclear RNA splicing activity and, interestingly, serves to prevent the aggregation of the C-terminal TDP-43 within the nucleus of neuronal cells (Afroz et al., 2017; Jiang et al., 2017). The crystal structure of a purified recombinant human TDP-43 fragment (residues 1–80) was determined at a resolution of 2.1 Å, revealing a unique pattern of head-to-tail interactions between monomers, resulting in the formation of solenoid-like polymers (Afroz et al., 2017). In contrast to physiological oligomers, pathological TDP-43 oligomers exhibit distinct characteristics. Detectable in the early stages of aggregation, their formation is accelerated in the presence of disease-associated TDP-43 mutations (Johnson et al., 2009; French et al., 2019). Using a polyclonal TDP-43 oligomer-specific antibody (Fang et al., 2014), pathological TDP-43 oligomers were identified in FTLD patients (Kao et al., 2015).
The TDP-43 oligomers manifest as a diverse spectrum of molecules, ranging from low-molecular-weight species like dimers, trimers, and tetramers, to a variety of high-molecular-weight species (Johnson et al., 2009; Guo et al., 2011; Choksi et al., 2014; Fang et al., 2014; French et al., 2019). Described as heterogeneous structures at the intermediate stage of aggregate formation, the oligomerization process may involve cysteine oxidation, particularly in the initial stages (Bozzo et al., 2016). Extracellular exposure to TDP-43 oligomers demonstrated cytotoxicity in neuroblastome cells (Choksi et al., 2014), and intrahippocampal injection of these oligomers caused damage to hippocampal neurons in wild-type mice (Fang et al., 2014), further substantiating their neurotoxicity. Interestingly, in an animal model expressing the ALS-related TDP-43 (M377V) variant at nearly endogenous levels, there was complete absence of mature insoluble aggregates (Gordon et al., 2019). However, these mice progressively developed motor function deficits concomitant with the loss of neuromuscular junction integrity, thus recapitulating ALS phenotypes (Gordon et al., 2019). Taken together, current evidence is not sufficient to argue towards one or the other direction, though it seems possible that both oligomeric soluble species and insoluble aggregates contribute differentially to ALS pathogenesis (Arnold et al., 2013; Gordon et al., 2019).
The ability to isolate, cultivate, and reprogram cells obtained from patients, such as fibroblasts, into central nervous system (CNS) cells, has introduced a novel approach that complements traditional preclinical in vitro and in vivo models (Ferraiuolo and Maragakis, 2021; Du et al., 2023). There are two primary approaches to this method. Several laboratories have investigated the use of induced pluripotent stem cells (iPSCs), focusing on generating motor neurons from patients with mutations in TARDBP, FUS, SOD1, and C9orf72, or those with sporadic disease (Ferraiuolo and Maragakis, 2021). For instance, motor neurons derived from ALS patients with mutated TDP-43, developed cytosolic aggregates resembling those observed in postmortem tissues and displayed shorter neurites (Egawa et al., 2012). Importantly, the accumulation of insoluble protein inclusions has been observed in numerous studies using iPSC-derived motor neurons (Dimos et al., 2008; Burkhardt et al., 2013; Chen et al., 2014; Seminary et al., 2018), although neuronal cell death is not observed, supporting the non-cell-autonomous hypothesis of ALS neurodegeneration (Ilieva et al., 2009; Wachter et al., 2015).
Limiting the screening approach to monocultured motor neurons restricts the influence of non-neuronal cells on observed phenotypes. In ALS, neighboring cells like astrocytes and microglia significantly impact neurodegenerative phenotypes and neuronal survival (Boillee et al., 2006). Additionally, iPSC technology often results in the loss of the cellular aging signature (Lapasset et al., 2011; Patterson et al., 2012). An alternative method involves trans-differentiating patient fibroblasts into neural progenitors, which can then be further differentiated into neurons or astrocytes (Meyer et al., 2014; Mertens et al., 2016; Tang et al., 2017). These astrocytes exhibit expected toxicity toward co-cultured motor neurons and seem to maintain the associated aging phenotype (Gatto et al., 2021). Interestingly, a study using iPSC-derived motor neurons and astrocytes revealed that recombinant TDP-43 oligomers, can induce neuronal toxicity, leading to increased caspase-3 activity and to apoptosis of neurons but not astrocytes (Smethurst et al., 2020).
Concerning the mechanisms underlying aggregation-mediated toxicity in ALS and other protein misfolding diseases, two major models have been proposed: the loss-of-function hypothesis and the gain-of-toxic activity hypothesis. While both terms are borrowed from genetics, mutations are not always relevant within the context of misfolding and aggregation. In the majority of ALS cases, misfolded species originate from a wild-type protein devoid of inherited mutations. Thus, in this section we focus on how the aggregation process itself, rather than the genetic background (as reviewed in Kim et al., 2020), can lead to phenotypes resembling loss and gain of activity. Importantly, these mechanisms are not mutually exclusive; in fact, some alterations observed in ALS-related cellular physiology can be better explained by a combination of loss and gain of activity.
In genetics, the term “loss-of-function” is used to describe a mutation that partially or completely disrupts the activity of a gene product. Accordingly, during the aggregation process, sequestration and often mislocalization of a protein within aggregates, reduce its abundance, thereby leading to impaired activity. Indicatively, most ALS-affected motor neurons exhibit wild-type TDP-43 cytoplasmic inclusions concomitant with TDP-43 clearance from the nucleus, indicating that a loss of TDP-43 splicing activity could contribute to disease phenotypes (Polymenidou et al., 2011; Tollervey et al., 2011). In support of this, Tardbp knockout mice display early embryonic lethality (Kraemer et al., 2010); moreover, TDP-43 is essential for normal motor neuron function in flies (Feiguin et al., 2009) and mice (Yang et al., 2014), as evidenced by motor dysfunction and deficits in the neuromuscular junction after TDP-43 depletion. Conditional Tardbp knockout mice, when crossed with various tissue-specific Cre lines, exhibit diverse cellular and behavioral phenotypes, ranging from electrophysiological abnormalities to deficits in motor movement (Iguchi et al., 2013; Wu et al., 2019). TDP-43 has been demonstrated to interact with RNA transcripts of over 6,000 genes in mice. Accordingly, the reduction of TDP-43 levels in adult mouse brains using antisense oligonucleotides (ASOs) led to the differential regulation of approximately 600 genes, mostly involved in synaptic activity and neuronal development, and to the alteration of 1,000 splicing events (Polymenidou et al., 2011).
Loss of function, at least partially, can also be attributed to SOD1-linked ALS. Although Sod1 knockout mice (Sod1−/−) develop normally and do not display evident ALS-like phenotypes (Reaume et al., 1996), they do exhibit- together with other pathologies-progressive motor neuropathy characterized by peripheral motor axon degeneration (Flood et al., 1999; Fischer et al., 2011; Larkin et al., 2011; Fischer et al., 2012; Shi et al., 2014; Ivannikov and Van Remmen, 2015). Furthermore, recent human genetics studies indicate that SOD1 homozygous loss-of-function can lead to debilitating phenotypes, including progressive loss of motor abilities, tetraspasticity, and hyperreflexia, but not ALS. In contrast, heterozygous carriers remained unaffected (Andersen et al., 2019; Park et al., 2019).
The accumulation of insoluble proteins is a prominent feature in ALS pathology, suggesting an interconnection between protein aggregation and disease development. The hypothesis posits that protein aggregates may initiate the disease process by acquiring toxic properties, which are mainly exerted through the sequestration of crucial components of physiological processes, such as RNA metabolism, DNA damage response, synaptic signaling and axonal trafficking, thereby disrupting these cellular functions and contributing to disease progression (Ilieva et al., 2009). For instance, TDP-43 aggregates have been found to contain proteins like the RNA-processing factor Matrin 3 (MATR3) (Tada et al., 2018), the ubiquitin-proteasome system factor Ubiquilin 2 (UBQLN2) (Deng et al., 2011; Williams et al., 2012), the Golgi complex and membrane trafficking factor Optineurin (OPN) (Hortobagyi et al., 2011) and the Rho guanine nucleotide exchange factor 28 (RGNEF) (Keller et al., 2012) involved in synapse formation and dendritic morphogenesis. Other potential mechanisms for the induction of cell toxicity by protein aggregation in ALS include disruption of the proteostasis network, leading to impairment of crucial protein degradation pathways like the ubiquitin proteasome system and autophagy (Medinas et al., 2017; Ramesh and Pandey, 2017; Montibeller et al., 2020). Compelling evidence also connects aggregated SOD1 and TDP-43 to mitochondrial dysfunction and degeneration (Jankovic et al., 2021). TDP-43 aggregates sequester specific microRNAs and mitochondrial proteins encoded by the nuclear genome, with dysregulation of their expression levels leading to mitochondrial dysfunction and oxidative stress (Zuo et al., 2021). Aggregates of mutant SOD1 in the intermembrane space of mitochondria diminish the activity of the electron transport chain (ETC) complexes in rats (Pickles et al., 2013) and affect mitophagy by disabling recruitment of autophagy receptors on damaged mitochondria in N2a cells (Tak et al., 2020).
Alternatively, aberrant protein aggregates can induce a chronic inflammatory response in the brain contributing to disease progression (Appel et al., 2021). In a recent study, Yu et al. demonstrated that TDP-43 induces inflammation in ALS by initiating the release of mitochondrial DNA into the cytoplasm. This, in turn, activates the cytoplasmic DNA-sensing cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway (Yu et al., 2020). Separately, the nuclear factor-kappa β (NF-κB) protein has been reported as a master regulator of inflammation in ALS (Haidet-Phillips et al., 2011); in mutant SOD1 mice, NF-κB signaling becomes activated within glia as the disease progresses (Frakes et al., 2014). In support of these findings, extensive astrocytosis (Kushner et al., 1991; Nagy et al., 1994; Reischauer et al., 2018; Schiffer et al., 1996), microglial activation (Henkel et al., 2004; Turner et al., 2004; Zurcher et al., 2015), as well as increased levels of inflammatory cytokines (Moreau et al., 2005; Liu et al., 2015; Lu et al., 2016) and elevated levels of chitotriosidase (Chit-1) and chitinase-3-like protein 1 (CHI3L1) in cerebrospinal fluid (CSF) correlated to disease progression (Vu et al., 2020), have been detected in ALS patients.
Among neurodegenerative diseases, ALS stands out as one of the few for which disease-modifying therapies have gained approval. The current standard of care for ALS patients involves multidisciplinary symptom management, both pharmacological and non-pharmacological, such as nutritional and respiratory support (Hardiman et al., 2017; Mejzini et al., 2019). Clinical trials in ALS have assessed over 60 compounds (Petrov et al., 2017), each with distinct mechanisms of action; however, only four- riluzole, edaravone, AMX0035 and tofersen- have been granted regulatory clearance for clinical use. Riluzole, the first FDA-approved ALS therapy in 1995 (Lacomblez et al., 1996a; Lacomblez et al., 1996b), is not a cure for ALS. Instead, it is a neuroprotective drug that decreases glutamate release into the synaptic cleft by blocking voltage-gated sodium channels on presynaptic neurons, thereby mitigating excitotoxicity (Wang et al., 2004; Bissaro and Moro, 2019). While Riluzole does not modify the course of the disease (Fang et al., 2018), it prolongs the patient’s survival from 6 to 19 months (Andrews et al., 2020). Edaravone is a potent anti-oxidant and a free-radical scavenger, preventing oxidative stress-induced motor neuron death (Jami et al., 2015; Soejima-Kusunoki et al., 2022). A randomized controlled trial conducted in Japan demonstrated the effectiveness of Edaravone in slowing the rate of motor function deterioration, particularly in selected patients with early disease onset and rapid progression (Writing Group and Edaravone MCI-186 ALS 19 Study Group, 2017). Its impact on survival is yet to be determined. Apart from Japan and other Asian countries, its use has been approved by the FDA and Health Canada, but not yet from the EMA. A recently developed oral formulation in the United States is expected to replace the intravenous version (Pattee et al., 2023). AMX0035 was approved in 2022 for the treatment of ALS in the United States and Canada, and it is currently being evaluated in the PHOENIX phase III clinical trial (Nikitin et al., 2023). It is a coformulation of sodium phenylbutyrate, a chemical chaperone that improves endoplasmic reticulum (ER) folding capacity by upregulating heat-shock proteins (Suaud et al., 2011), and taurursodiol (tauroursodeoxycholic acid), an inhibitor of mitochondrial-associated apoptosis (Rodrigues et al., 2003). This therapeutic approach is designed to address various ALS-associated pathophysiological mechanisms such as mitochondrial dysfunction and ER stress that ultimately lead to motor neuron injury and cell death.
Tofersen is an antisense oligonucleotide (ASO) that obtained its initial approval in the United States on April 25, 2023, and in Europe on February 22, 2024, for the treatment of ALS in patients with a confirmed SOD1 mutation. It has been designed to mediate RNase H–dependent degradation of SOD1 mRNA to reduce the synthesis of SOD1 protein (McCampbell et al., 2018; Rinaldi and Wood, 2018). The preclinical rationale supporting the ASO knockdown of SOD1 was very robust, stemming from several years of systematic analysis showing increased survival and enhanced motor performance in SOD1 (G93A) rats and mice and decreased SOD1 protein levels in nonhuman primates (Iannitti et al., 2018; McCampbell et al., 2018). Its intrathecal administration in clinical trials demonstrated a deceleration in the decline of participants with rapidly progressing disease and apparent clinical stabilization in those with slower progressing disease (Miller et al., 2022). During the intervention period, concentrations of phosphorylated neurofilament heavy and neurofilament light (NFL) in plasma and CSF, biomarkers indicative of axonal injury and neurodegeneration, were found to be decreased (Miller et al., 2022). Tofersen is currently being evaluated in the phase III ATLAS study for its ability to delay the clinical onset of ALS in pre-symptomatic individuals with a confirmed SOD1 mutation (Benatar et al., 2022).
The complex nature of ALS genetics, coupled with the disease’s high heterogeneity, presents a great challenge in developing treatment strategies that universally benefit every patient. Consequently, there has been a shift in focus towards identifying converging paths and common pathologies that contribute to motor neuron vulnerability and degeneration in ALS. Among these factors, protein aggregation emerges as a prevalent underlying cause not only in ALS but also in various other neurodegenerative diseases. Despite the diversity of mutations in different genes, leading to potential variations in the aggregated proteins, the shared problem of protein aggregation spans a wide spectrum of patients, encompassing familial ALS (fALS), sporadic ALS (sALS), and ALS with frontotemporal dementia (ALS/FTLD) (Blokhuis et al., 2013). As a result, the pursuit of therapies targeting protein aggregation holds significant promise and represents a crucial step forward in addressing ALS and related disorders (Elliott et al., 2020).
Several approaches have been applied to reduce the levels of toxic variants; these include proteolysis targeting chimeras (PROTACs) inducing protein degradation (Tseng et al., 2023), antibodies (Maier et al., 2018; Pozzi et al., 2019; Afroz et al., 2023; Audrain et al., 2023; Bakavayev et al., 2023), vaccines (Zhao et al., 2019), antisense oligonucleotides (Iannitti et al., 2018; Mead et al., 2023), RNA interference (RNAi) with small RNAs (shRNA and miRNA) (Bravo-Hernandez et al., 2020; Mueller et al., 2020), and CRISPR/Cas9 gene editing (Duan et al., 2020). Another way of reducing the accumulation of toxic aggregates is to restore dysfunctional proteostasis by upregulating chaperones (Kalmar and Greensmith, 2017; Kinger et al., 2023), inducing autophagy (Chen et al., 2020) and activating the proteasome (Webster et al., 2017). Currently, many compounds targeting proteostasis, such as trehalose (Seelos Therapeutics) and Trametinib (GENUV) are being explored in clinical trials. Moreover, PMN-267 (ProMIS Neurosciences), an antibody that targets the formation of misfolded TDP-43, is under preclinical development (Mead et al., 2023). Despite the success of Tofersen, a similar strategy utilizing intrathecally delivered ASOs for C9orf72-associated ALS did not show clinical benefits, leading to the termination of the clinical trial in 2022 (Mead et al., 2023).
In this review, we focus on molecules that directly target the misfolded species (monomers, oligomers or mature aggregates) to (a) stabilize the native protein conformation, (b) restore the native conformation of misfolded species and (c) inhibit protein oligomerization. Such molecules fall into two categories depending on the specificity to bind to their target, namely, chemical and pharmacological chaperones.
The term “chaperone” is borrowed from the name of a class of cellular proteins that assist polypeptide chains in acquiring their native, functionally active, conformation. They participate in (a) the folding of nascent polypeptides (de novo folding) or the refolding of stress-mediated misfolding, (b) the disaggregation of protein aggregates and (c) proteostatic pathways that direct denatured and misfolded/unfolded proteins for degradation (Wentink and Rosenzweig, 2023). However, pharmacological chaperones, unlike proteins, are low molecular weight chemical molecules that exert their action by specifically binding to their target proteins. They typically stabilize an already folded or partially folded protein, protect it from thermal denaturation, prevent proteolytic degradation and also inhibit aggregation, restoring proper steady-state levels (Convertino et al., 2016; Bose and Cho, 2017). Pharmacological chaperones have successfully been used experimentally in vitro and in vivo (Leidenheimer and Ryder, 2014; Hou et al., 2018), and they have entered clinical trials, to restore the function of specific misfolded proteins (Liguori et al., 2020; Tran et al., 2020). For example, Tafamidis has been approved for the treatment of transthyretin amyloidosis (ATTR) (Bulawa et al., 2012; Maurer et al., 2018) and Migalastat has been licensed for the treatment of Fabry disease (FD) in patients carrying specific lysosomal enzyme α-galactosidase A (GLA) variants (Germain et al., 2016).
Chemical chaperones are also low-molecular-weight compounds that lack specificity since they can bind to and stabilize virtually any protein without having a designated binding site (Cortez and Sim, 2014). They can be divided into two groups: osmolytes and hydrophobic compounds. Osmolytes, such as glycerol, produce a hydrophobic environment around proteins by removing water molecules, thereby increasing the free energy of the unfolded state and eventually shifting the equilibrium towards the folded state (Wang and Bolen, 1997; Street et al., 2006). Hydrophobic chaperones facilitate proper folding by directly interacting with the exposed hydrophobic regions of unfolded proteins, resembling the action of molecular chaperones (Kuzuhara et al., 2008). For example, the green tea catechin, (−)-epigallocatechin gallate (EGCG), seems to exert its general anti-amyloidogenic properties by directly binding to unfolded/misfolded proteins (Debnath et al., 2016). As mentioned above, the best-known example of chemical chaperone for ALS treatment is sodium phenylbutyrate (Suaud et al., 2011).
Developing a new drug is a complex and challenging process that yields significant benefits for both society and the scientific community. High-throughput screening (HTS) is crucial in the early stages of small-molecule drug development, particularly when insufficient information limits structure-based approaches (Blay et al., 2020). For targets such as oligomeric species and aggregates, which lack well-defined structures and exist in dynamically varying mixtures, HTS requires innovative strategies to discover effective treatments. The utilized methods must be biologically relevant, sensitive, robust, and cost-effective, targeting diseases of significant relevance. HTS integrates biochemical and cell-based assays with computational strategies to identify potential drug candidates. In silico screening employs computational methods to predict interactions between molecules and targets, a critical step given the extensive chemical space. Unlike traditional HTS, which assesses thousands of compounds, virtual screening has the capacity to evaluate billions (Bohacek et al., 1996; Gorgulla et al., 2022), showcasing the expansive scope of modern drug discovery efforts.
In 2005, Ray et al. employed an in silico screening strategy of approximately 1.5 million compounds from commercial libraries (Ray et al., 2005). The objective was to identify molecules capable of stabilizing the SOD1 (A4V) dimer. The screening process yielded 100 hits, and subsequent mutagenesis studies using various in vitro aggregation assay protocols helped narrow down the selection to the 15 most effective compounds. Notably, three of the top five compounds shared pyrimidine-like structures. However, subsequent co-crystal structures of SOD1 with various small molecules and pyrimidine-like compounds challenged the initial assumption that the observed protection was solely due to binding to the hydrophobic cavity formed at the dimer interface. Instead, these structures suggested that the ligands interacted with Trp32, offering new insights into the mechanism of action (Antonyuk et al., 2010; Wright et al., 2013).
Nowak et al. developed (Nowak et al., 2011) an algorithm for the in silico screening of an extensive library comprising 2.2 million small molecules sourced from 11 commercially available databases. The primary objective was to discern compounds exhibiting selective binding affinity for mutant SOD1 over plasma proteins. Their investigation revealed a substantial subset of compounds displaying robust binding capabilities to SOD1, particularly within a hydrophobic cavity encompassing Val7–Gly147–Val148 at the dimerization interface. Notably, in vitro experimentation demonstrated the pronounced inhibitory effects of both isoproterenol and 5-fluorouridine on the aggregation process of mutant SOD1.
In another computational screen involving 4,400 drugs and compounds, three flavonoids, quercitrin, quercetin-3-β-D-glucoside (Q3BDG) and EGCG, were predicted to bind around the dimer interface and stabilize SOD1, potentially inhibiting its aggregation, predictions which were confirmed experimentally (Ip et al., 2017). Interestingly, quercitrin and Q3BDG outperformed EGCG both in silico and in vitro. The tripeptide CGH (Srinivasan and Rajasekaran, 2019), hesperidin and 2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside retrieved from the traditional Chinese medicine database (Huang et al., 2014), and the natural polyphenols, kaempferol, and kaempferide (Srinivasan and Rajasekaran, 2018) have all been predicted to bind SOD1 and inhibit its aggregation, necessitating experimental verification.
This type of screens involves the use of a purified target protein of interest to assess the aggregation-inhibitory activity of test compounds. To quantify the outcomes of these assays, various optical methods such as absorbance, fluorescence, or luminescence are employed as readouts (Fang, 2012). For example, a screening assay of 640 FDA-approved drugs, aiming to evaluate their impact on the abnormal oligomerization of SOD1 (G37R) has been conducted in vitro. The primary objective was to identify molecules capable of inhibiting the formation of insoluble disulfide-linked oligomers. Among the extensive drug repertoire, six compounds - simvastatin, lovastatin, mevastatin, miltefosine, alfacalcidol and calcitriol-exhibited remarkable efficacy in almost completely suppressing the increase in solution turbidity (Anzai et al., 2016).
Cell-based assays play a crucial role at every stage of the drug discovery process, serving various purposes from target identification and validation to lead optimization and safety screening (Macarron and Hertzberg, 2009; McGown and Stopford, 2018). For ALS, cell models employed in these assays include the NSC-34 cell line (Barber et al., 2009), created by fusing MN from the spinal cords of mouse embryos with mouse neuroblastoma cells, human astrocyte-derived H4 cells (Murakami et al., 2011), and PC12 cells from the rat adrenal gland (Benmohamed et al., 2011; Xia et al., 2011; Zhang W. et al., 2012; Wright et al., 2012; Boyd et al., 2014). Emphasis has also been given to the use of primary rat MN cultures (Vincent et al., 2005) or mouse embryonic stem cell-derived MNs in small molecule screening studies (Yang et al., 2013; Thams et al., 2019). Finally, the use of iPSC technology has significantly contributed to the development of current ALS drug-screening platforms (Burkhardt et al., 2013; Imamura et al., 2017).
In the following section, we describe molecules that have emerged from drug-screens based on the above platforms and target misfolded species of SOD1 and TDP-43 (Figure 2).
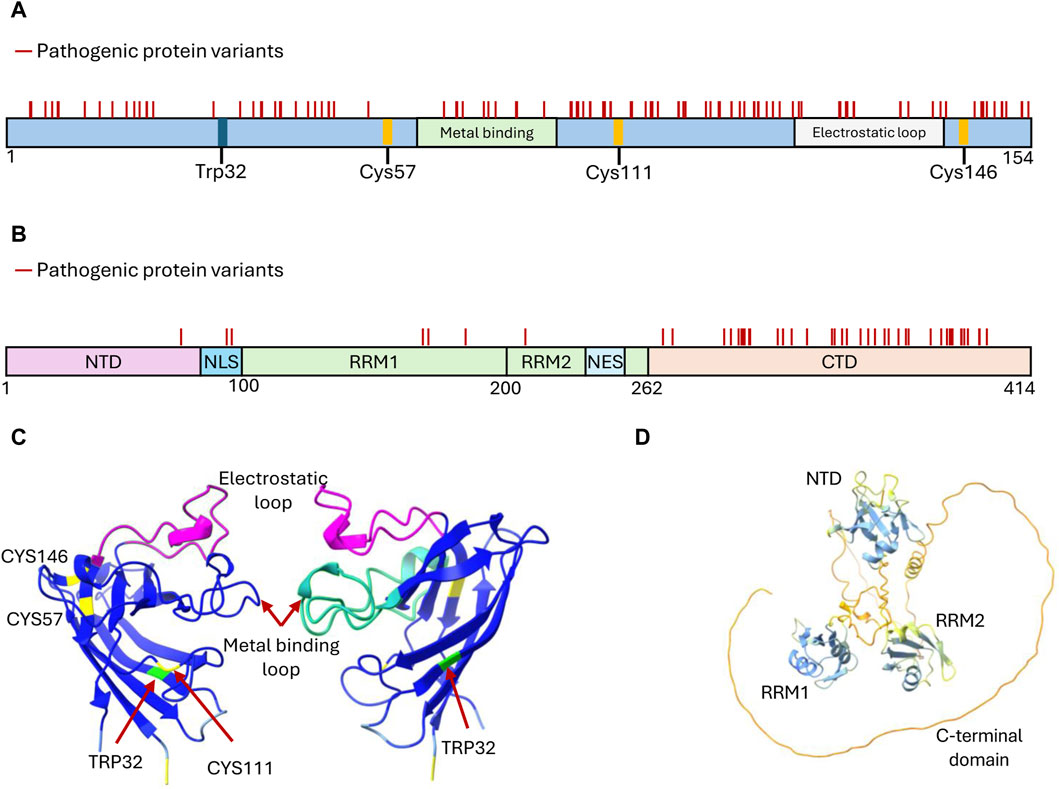
Figure 2. Structural features of SOD1 and TDP-43 proteins targeted by developed aggregation inhibitors. (A) Domain organization of SOD1 depicting structural features critical for its function, i.e., the cysteine residues participating in disulfide bond formation (Cys57 and Cys146), the metal-binding domain, and the electrostatic loop. These sites, along with Trp32 and Cys111, have been explored for drug discovery targeting SOD1 inhibition of aggregate formation. Mutations that generate amino acid substitutions in SOD1 are depicted with solid lines. (B) Domain organization of TDP-43 depicting the features that mediate functional oligomerization (N terminal domain-NTD), RNA binding (RNA Recognition motifs RRM1 and RRM2), protein interactions and liquid-liquid phase separation (C terminal domain-CTD), and nucleo-cytoplasmic shuffling (Nuclear Localization Signal-NLS and Nuclear Export Signal-NES). Mutations leading to amino acid substitutions in TDP-43 are represented by solid lines. (C) Structure of two SOD1 subunits rotated 180° to showcase the arrangement of key residues and domains (Uniprot P00441). Reconstituted using Chimera X (AlphaFold). (D) Structure of TDP-43 (Uniprot Q13148) highlighting the arrangement of its domains. Predicted AlphaFold structure reconstituted using Chimera X (Pettersen et al., 2021).
Upon translation, SOD1 undergoes spontaneous folding, primarily initiated around hydrophobic residues, culminating in the formation of a monomeric Greek-key β-barrel structure. For SOD1 to achieve activity and stability, three essential PTMs are required. The binding of zinc and the formation of an intra-subunit disulphide bond establish the SOD1 homodimer interface in conjunction with the N- and C-termini and four inter-subunit hydrogen bonds are formed. The introduction of copper to the SOD1 active site enhances thermal stability and activates enzymatic functionality (Hornberg et al., 2007). In the absence of these PTMs and the subsequent homodimer formation, SOD1 exhibits diminished thermal stability, populates unfolded conformations, and is susceptible to self-association (Culik et al., 2018). Interestingly, the engineering of an inter-subunit disulfide bond in the SOD1 (A4V) variant alleviated aggregation and restored enzymic activity (Ray et al., 2004), suggesting that stabilizing the dimer is a promising therapeutic strategy (McAlary and Yerbury, 2019; Yerbury and Cashman, 2020).
The covalent attachment of small molecules to Cys111 has been a frequently employed strategy in the discovery of pharmacological chaperones for SOD1. This is attributed to their accessibility, reactivity, and potential for linking or stabilizing the SOD1 homodimer, as opposing Cys111 residues are separated by a short distance across the dimer interface and possess a highly reactive sulfhydryl group. Tethering Cys111 residues with maleimide linkers to bridge the 9 Å gap across the SOD1 dimer interface substantially increases the thermal stability of the SOD1 (G93A) and SOD1 (G85R) ALS-linked variants, restoring the enzymatic activity and metal-binding ability of the otherwise inactive G85R variant (Auclair et al., 2010). However, maleimide compounds do not exhibit favorable drug-like properties (Gavrin et al., 2012), while maleimide crosslinking inhibits SOD1 complex formation with the human copper chaperone for SOD1 (CCS), an interaction essential for SOD1 activation (Wright et al., 2016). Similarly, cyclic disulfides have demonstrated the ability to link opposing Cys111 residues and exhibited cell penetrability (Wright, 2020). For example, mono-S-oxo derivatives were able to bind and tether 95% of SOD1 monomers as dimers within cells (Donnelly et al., 2018). Taking a different approach, cisplatin enhances the thermal stability of metal-free SOD1 and can effectively prevent or reverse SOD1 aggregation within cells without the need for subunit tethering (Banci et al., 2012) but hinders complex formation between SOD1 and hCCS (Wright et al., 2016).
Synthetic organo-selenium pharmaceutical compounds, such as ebselen and ebsulphur, function as bifunctional small molecule chaperones for various mutant forms of SOD1 by forming covalent bonds with Cys111 (Capper et al., 2018). The SOD1 dimer interface groove can accommodate two ebselen molecules, binding to opposing Cys111 residues and expanding the hydrophobic SOD1 homodimer interface through aromatic π–π stacking of adjacent ligands. As a result, the affinity between monomers significantly increases, particularly for the SOD1 (A4V) mutant, shifting the monomer-dimer equilibrium towards the dimer (Capper et al., 2018). Ebsulphur exhibits similar binding properties. Importantly, ebselen does not interfere with the SOD1-CCS interaction (Capper et al., 2018), giving it a significant advantage over other Cys111-binding compounds, such as cisplatin. Ebselen is also known for its anti-inflammatory, antioxidant, and cytoprotective properties (Schewe, 1995; Saito et al., 1998; Yamaguchi et al., 1998; Wood-Allum et al., 2006). However, achieving target engagement and specificity remains challenging due to the abundance of free thiols and the large number of ebselen-binding proteins within cells (Chen et al., 2018). Thus, a medium-throughput screen using a differential scanning fluorescence assay to investigate the stabilization of SOD1 (A4V) mutants by ebselen derivatives was developed, aiming for improved SOD1 dimer stabilization and drug-like properties (Amporndanai et al., 2020; Chantadul et al., 2020). Potent candidates exhibited remarkable neuroprotective activities without observable toxicity in cells, while in vivo experiments with a selected compound demonstrated a significant delay in ALS disease onset in SOD1(G93A) mice, showcasing its potential therapeutic effect (Amporndanai et al., 2020).
The sole tryptophan residue within SOD1 has been identified as a key factor in the misfolding of both mutant and wild-type proteins, through oxidative and prion-like aggregation mechanisms (Coelho et al., 2014; Pokrishevsky et al., 2018; Duval et al., 2019; Gill et al., 2019; Manjula et al., 2019; Crown et al., 2020). Trp32, residing on the solvent-exposed external layer of the protein, distanced from the Zn/Cu active site and the homodimer interface, is subject to oxidative post-translational modifications in vivo (Medinas et al., 2010; Figueroa et al., 2020). The bicarbonate-dependent peroxidase activity of SOD1 further enhances the oxidation rate of Trp32 in vitro (Coelho et al., 2014), while upon chronic ER stress conditions Trp32 oxidation is increased in cytosolic and mitochondrial extracts from transgenic WT SOD1 mice (Medinas et al., 2018). Site-directed mutagenesis studies have emphasized the pivotal role of Trp32 in aggregate formation as substitution of this residue in SOD1 (G93A) cultured motor neurons (Taylor et al., 2007) and in HEK cells expressing SOD1 (G127X) and SOD1 (G85R) (Grad et al., 2011) considerably reduced mutant cytotoxicity and aggregation propensity.
Prion-like propagation and in vivo conversion of WT SOD1 to a misfolded state linked to Trp32, has been identified following exposure to misfolded SOD1 mutant seed species. SOD1 (G127X) can convert WT SOD1 to the misfolded state in various cell lines (Grad et al., 2011) while SOD1 (G93A) induces WT SOD1 aggregation accelerating disease progression in mice (Bruijn et al., 1998; Deng et al., 2006; Wang et al., 2009; Ayers et al., 2016). Notably, human SOD1 mutants did not induce misfolding of murine SOD1 protein both in vivo (Wang et al., 2009; Audet et al., 2010) and in cells (Grad et al., 2011), due to a different codon for the residue at position 32 between humans and mice. This differentiation, from a tryptophan to a serine, has been associated with the observed aggregation resistance in mice. Intriguingly, the SOD1(W32F) variant significantly reduced the formation of high-molecular weight and disulfide-dependent SOD1 aggregates in NSC-34 cells (Medinas et al., 2018). Moreover, zebrafish embryos injected with SOD1(W32S) showed mitigated motor deficiency and motor neuron axonopathy compared to embryos injected with WT or ALS-related SOD1 variants (Duval et al., 2019), supporting the notion that reducing SOD1 self-interactions at Trp32 might be of therapeutic interest.
Recognizing the role of Trp32 oxidation in potentiating protein aggregation and cytotoxicity has spurred the development of small molecules targeting Trp32 in SOD1 (Ray et al., 2005; Antonyuk et al., 2010; Wright et al., 2013; Manjula et al., 2019). Pyrimidine-like molecules, such as 5-Fluorouridine (5-FUrd), 5-Fluorouracil (5-FU) and Telbivudine (a synthetic thymine analogue), have all demonstrated the ability to decrease SOD1 aggregation in cellular and animal models of ALS (Pokrishevsky et al., 2018; Duval et al., 2019; Rando et al., 2019). In particular, 5-FU administration in SOD1 (G93A) transgenic mice delayed disease onset, increased survival and ameliorated motor performance deficits (Rando et al., 2019). Additionally, telbivudine improved motor performance and reduced SOD1 toxicity in a SOD1-ALS zebrafish model (Duval et al., 2019). Both 5-FU and telbivudine are licensed in the United States and EU as chemotherapeutic and antiviral agents, respectively, and importantly, both compounds can also penetrate the BBB (Formica et al., 2006; Ahrens and Manno, 2014).
The structure and activity of SOD1 depend on two important loops, the Zn-binding (residues 49-82) and the electrostatic loop (E-loop or loop VII, residues 121-142). A highly conserved cluster of charged residues situated on the surface of the electrostatic loop forms an active site channel, facilitating effective interaction with superoxide (Getzoff et al., 1992; Polticelli et al., 1995). NMR analysis of SOD1 folding and misfolding pathways identified thermal fluctuations within the electrostatic loop that mediate the formation of aberrant intermolecular interactions and the development of abnormal oligomers (Sekhar et al., 2015). Furthermore, structural and dynamic changes affecting this loop have been previously identified as a shared property of 13 familial ALS-related SOD1 variants (Molnar et al., 2009), while mutations dispersed throughout the SOD1 sequence that destabilize the electrostatic loop aberrantly expose the metal site of the protein to water (Souza et al., 2019). Thus, the binding of small molecules to the loop could alleviate the impact of mutations on the structure. Interestingly, the cyclic peptide HGG4FFQ (4FF = 4-fluorophenylalanine), selected by an epitope-specific high-throughput screen for binding to the peptide sequence of the electrostatic loop, can bind to both mature and metal-free SOD1 at the electrostatic loop, stabilize the native state and accelerate the WT and SOD1 (G93A) folding rates (Bunck et al., 2018).
Short segments of SOD1 have a tendency to aggregate into oligomers and fibrils through the establishment of intramolecular hydrogen bonds and hydrophobic interactions (Ivanova et al., 2014; Sangwan et al., 2017). Additionally, SOD1 can create non-native oligomers in which monomers are connected through intermolecular disulfide bonds (Furukawa et al., 2006; Toichi et al., 2013). Several symptomatic transgenic ALS mouse models exhibit disulfide cross-linked SOD1 multimers detected in insoluble fractions derived from the animals’ spinal cords (Furukawa et al., 2006; Anzai et al., 2017). Importantly, such multimers are absent from transgenic mice expressing a C-terminally truncated SOD1 lacking Cys146, suggesting that this residue has a significant role in disulfide-linked multimerization in vivo (Furukawa et al., 2006). A screening of 640 FDA-approved drugs was conducted to evaluate their impact on abnormally cross-linked SOD1 oligomers derived from metal deficient apo-SOD1 (G37R)S−S in an in vitro assay. The goal was to identify compounds that could inhibit the formation of insoluble S-S oligomers. Six compounds, namely, simvastatin, lovastatin, mevastatin, miltefosine, alfacalcidol and calcitriol were discovered to significantly suppress solution turbidity (Anzai et al., 2016). Despite this finding, treatment of an ALS mouse model expressing human SOD1 (G93A) with simvastatin resulted in accelerated disease progression and shortened survival (Su et al., 2016).
Apart from stabilizing the SOD1 dimer and promoting proper folding to prevent SOD1 aggregation, molecules which can interfere with the self-assembly, the nucleation, or the fibrillation process are of high therapeutic interest. A prime example is Methylene Blue (MB), a phenothiazine dye, which has shown considerable potential in modulating the aggregation of various amyloidogenic proteins (Gureev et al., 2022). The ability of MB to readily cross the blood-brain barrier has significantly increased interest in its use as a therapeutic agent for central nervous system disorders (Rojas et al., 2012). In vitro assays have demonstrated that MB inhibits both the spontaneous amyloid aggregation of SOD1 and the elongation of pre-existing fibrils (Malik et al., 2020; Musteikyte et al., 2020). However, MB treatment of SOD1 (G93A) mice had no effect on motor neuron loss, or on the aggregation or misfolding of SOD1 (Audet et al., 2012).
Flavonoids are the most common phytochemicals, exerting pharmacological effects and possessing antioxidant properties (Nday et al., 2015), metal chelator abilities (Nday et al., 2015), anti-inflammatory effects (Lin L. et al., 2015) and neuroprotective properties (Prakash and Sudhandiran, 2015). Importantly, some flavonoids can cross the BBB, rendering them attractive drug candidates for CNS diseases, including AD, PD and ALS (de Andrade Teles et al., 2018; Maher, 2019). Certain flavonoids such as myricetin, lacmoid, EGCG and curcumin (Zhao et al., 2018) have demonstrated the ability to reduce apo-SOD1 aggregation in an in vitro assay by restraining the nucleation step and inhibiting fibril elongation (Malik et al., 2020). Despite evidence of a direct inhibitory role in SOD1 aggregation (Malik et al., 2020), myricetin may exert its beneficial effects through additional cellular mechanisms. For example, the observed decrease in intracellular accumulation of ubiquitin-positive SOD1 inclusions in myricetin-treated cells expressing SOD1 (G37R) may also be mediated by modulation of the endogenous levels of Hsp70 and the quality control E3 ubiquitin ligase E6-AP (Joshi et al., 2019). EGCG is another flavonoid that has shown beneficial effects on onset delay, motor function, neuroprotection and extension of the lifespan of SOD1 (G93A) mice (Koh et al., 2006; Xu et al., 2006), combined with the suppression of microglial activation and inflammation (Xu et al., 2006). Finally, curcumin in an in vitro assay, reduced the fibril formation propensity of DTT-treated SOD1, favoring the formation of small amorphous aggregates which are not cytotoxic (Bhatia et al., 2015).
Molecular tweezers are small, horseshoe-shaped artificial receptors that can form complexes with guest molecules, such as a nucleic acids, carbohydrates, amino acids or proteins, via non-covalent interactions (Mbarek et al., 2019). The molecular tweezer CLR01 selectively binds to positively charged lysine and arginine residues, interfering with the self-assembly process by disrupting hydrophobic and electrostatic interactions, thereby preventing the aggregation process in various amyloidogenic proteins (Sinha et al., 2011; Attar et al., 2012; Acharya et al., 2014; Ferreira et al., 2014; Herzog et al., 2015; Lopes et al., 2015; Zheng et al., 2015; Vopel et al., 2017; Mittal et al., 2018; Malishev et al., 2021). CLR01 also inhibits mutant SOD1 aggregation, as measured by ThT fluorescence assays, but leads to a marginal reduction in disease duration in the SOD1 (G93A) ALS mouse model despite reduced levels of misfolded SOD1 in the spinal cord (Malik et al., 2019). Recently, Lys61 and Lys92 have been identified as the preferential binding sites of CLR01 to SOD1, with the authors providing further evidence of CLR01’s ability to stabilize the monomer (Samanta et al., 2022).
Peptides constitute a promising class of therapeutic molecules (Wang L. et al., 2022). Synthetic peptides from structure-based designs possess the ability to function as beta-sheet breakers of amyloid forming proteins (Sievers et al., 2011; Young et al., 2017), and their use in preventing aggregate formation has demonstrated promising outcomes in various disorders, including Alzheimer’s, prion, and polyglutamic diseases (Truex and Nowick, 2016; Alam et al., 2017; Goyal et al., 2017; Mukundan et al., 2017; Ryan et al., 2018). Peptide inhibitors, as potential drug candidates, combine several advantages such as high specificity, low toxicity and good tolerance (Hard and Lendel, 2012; Goyal et al., 2017). The peptide SE-12 (LSGDHCIIGRTLVVHEKADD) has been shown to bind SOD1 variants and to direct the aggregation of ALS-related SOD1 mutants from the typical amyloid to an amorphous aggregation pathway, which is presumably less toxic (Banerjee et al., 2016). Further studies were based on this peptide for the in silico design of tripeptides with inhibitory activity against SOD1 (A4V) aggregation, identifying CGH as the one with the greatest binding affinity among the tested sequences (Srinivasan and Rajasekaran, 2019). However, none of these peptides have been evaluated in cells or in vivo. In another approach, a peptide library comprising engineered variants of the hyperthermophilic variant of protein G (HTB1) was constructed and subsequently evaluated for specificity and affinity by a yeast display platform, identifying HTB1M as a selective inhibitor of SOD1 (G93A) and SOD1 (G85R) aggregation (Banerjee et al., 2017). Moreover, HTB1M prevented the accumulation of misfolded proteins in living cells and reduced the cytotoxicity of SOD1 (G93A) expressed in NSC-34 motor neuron–like cells. Importantly, the peptide library was designed using a computational algorithm for mapping protein surfaces predisposed to HTB1 intermolecular interactions. This conversion of an IgG-binding domain into a targeted SOD1 inhibitor showcases the potential usefulness of rational design for discovering peptide drug candidates. Finally, a bacterial platform was used to screen a combinatorial library of cyclic tetra-, penta- and hexapeptides for discovering rescuers of the misfolding and aggregation of the ALS-associated variant SOD1 (A4V). Among other hits, this high-throughput screen revealed that cyclic pentapeptides with the consensus cyclo-T (Φ1, S)SΦ2W motif, where Φ1 is one of the hydrophobic (Φ) amino acids A, W or F, while Φ2 is V, W or F, are efficient rescuers of SOD1 (A4V) aggregation (Matis et al., 2017).
Despite its central role in cellular health and disease, most strategies for removing pathological TDP-43 primarily focus on cellular clearance pathways (i.e., ubiquitin/proteasome system, autophagy) (Babazadeh et al., 2023) rather than directly targeting TDP-43 to inhibit its aggregation (Buratti et al., 2005), possibly due to the lack of typical druggable pockets in its structure (Francois-Moutal et al., 2019). Additionally, TDP-43 demonstrates a significant tendency to aggregate either during or shortly after the purification process (Wright et al., 2020; Yang et al., 2023), constraining the development of high-throughput assays for screening molecules inhibiting TDP-43 misfolding and aggregation. Notably, the design of stable monomeric TDP-43 variants (i.e., double mutant S333D/S342D) with the ability to be purified as stable monomers enabled the identification of small molecules capable of stabilizing the protein’s native state and preventing aggregation (Yang et al., 2023). Compounds with the ability to modulate the aggregation pathways and toxicity of amyloidogenic proteins such as MB and curcumin (Lo Cascio et al., 2021; Gureev et al., 2022) also prevent TDP-43 aggregation. MB reduces cytoplasmic TDP-43 inclusions in cells (Yamashita et al., 2009) and suppresses mutant TDP-43-mediated toxicity in C. elegans and zebrafish ALS (Vaccaro et al., 2012; Vaccaro et al., 2013) but was ineffective in reducing disease symptoms and cytoplasmic accumulation of TDP-43 in transgenic TDP-43 (G348C) mice (Audet et al., 2012). Similarly, the improved curcumin analogue monocarbonyl dimethoxy-curcumin C has been shown to inhibit the aggregation of mutant TDP-43 and to diminish oxidative stress in cells (Duan et al., 2014).
The extended C-terminal disordered region includes a hydrophobic α-helix (residues 320–340) and dispersed aromatic residues that facilitate the LLPS of TDP-43, allowing its incorporation to stress granules (Li et al., 2018). A substantial body of evidence indicates the Q/N-rich segment (residues 341–366) as the pivotal region for amyloid formation (Johnson et al., 2009; Budini et al., 2012; Kametani et al., 2016), further supported by the recently determined cryo-EM structure of aggregated TDP-43 (Arseni et al., 2022). Inhibitors that block the aggregation of TDP-43 mediated by the Q/N-rich segment without interfering with the hydrophobic segment’s ability to drive association with stress granules could be an effective therapeutic approach. Polyglutamine binding peptide 1 (QBP1) is an octapeptide, designed to prevent polyglutamine amyloid formation (Nagai et al., 2000; Nagai et al., 2003; Takeuchi and Nagai, 2017). QBP1 exhibits a relatively broad specificity, capable of hindering amyloid formation by various amyloid-prone polypeptides, including huntingtin, the yeast Sup35 prion domain, α-synuclein, and the functional amyloid CPEB3. However, it does not affect certain amyloids like Aβ (Bauer et al., 2010; Hervas et al., 2012; Popiel et al., 2013; Hervas et al., 2016). Biochemical in vitro assays showed that QBP1 binds to the Q/N rich region of TDP-43 C-terminal domain and inhibits its aggregation (Mompean et al., 2019).
The small planar molecule AIM4 [4,5-bis acridine] is an acridine derivative, which has been predicted to bind amino acids 288-319 in the C-terminal domain of TDP-43 (Girdhar et al., 2020). AIM4 exhibited pronounced inhibitory effects on TDP-43 aggregation in both a yeast model expressing TDP-43-YFP and in vitro, using a recombinantly purified C-terminal fragment spanning amino acids 193 to 414 (Prasad et al., 2016). Recently, the amyloid fiber-detecting molecules, 4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS) and Congo Red, have been shown to modulate LLPS of TDP-43 (Babinchak et al., 2020). Importantly, these molecules allow TDP-43 LLPS at low concentrations while decondensing the liquid droplets at higher concentrations, indicating that they do not interfere with physiological phase transitions. Although bis-ANS and Congo red are toxic and therefore not suitable for drugs, their use as lead compounds could assist in the development of optimized molecules.
In an effort to design aggregation blockers, Liu and coworkers developed peptides based on sequence motifs of TDP-43. Through this approach, they discovered that two synthetic peptides from the C-terminal region effectively hindered the formation of TDP-43 protein aggregates in a concentration-dependent manner within HeLa cells overexpressing a mutant TDP-43. However, it's noteworthy that despite their ability to reduce aggregation, these peptides did not mitigate or prevent cell death (Liu et al., 2013). In another attempt, TDP-43-derived peptides based on two core aggregation-prone motifs of TDP-43 (Saini and Chauhan, 2011), 246-EDLIIKGISV-255 and 311-MNFGAFSINP-320, have been functionalized to enhance desired properties (Gao et al., 2019). These multi-functional peptides comprise three moieties: (a) a hydrophobic motif consisting of adamantane, (b) a TDP-43-derived peptide and (c) a Tat (RRRQRRKKRG) or Poly-D-Arginine (D-Arg) tag, each one conferring to the functionality of the compound. Adamantane mimics the unfolded state of proteins, and consequently, adamantane-tagged proteins are directed to the degradation machineries of the cell (Coll-Martinez et al., 2020). Moreover, adamantane increases the BBB permeability of conjugated molecules (Tsuzuki et al., 1994). Tat or (D-Arg)8 tags are used to increase cell-permeability of conjugated molecules (Szabo et al., 2022). While the peptide M311/P320 was not further examined due to low solubility, the peptide E246/V255 exhibited a potent capacity to trigger TDP-43 degradation within cells and to diminish TDP-43-induced cytotoxicity. Additionally, it was effective in lowering TDP-43 levels in a transgenic Drosophila model. Importantly, peptides lacking the adamantane moiety had no effect.
TDP-43 localization to stress granules (SGs) is mediated by both its RRM1 and the C-terminal domain (Colombrita et al., 2009; Dewey et al., 2011). To discover small-molecule modulators influencing SG formation and decrease TDP-43 accumulation in induced pluripotent stem cell-derived motor neurons (iPSC-MN) from ALS patients, a cellular screen was conducted using a 50,000-member small molecule library (Fang et al., 2019). The screen involved HEK293T and neural precursor cells treated with NaAsO2 to induce SG formation. Approximately 100 hits were identified, and among them, mitoxantrone, quinacrine and pyrvinium planar compounds which act as nucleic acid intercalating molecules, showed reduced recruitment of TDP-43 to SGs. More recently, it has been reported that small molecules inhibiting the binding of TDP-43 to RNA could reduce neuronal toxicity (Francois-Moutal et al., 2019). Using an amplified luminescent proximity assay, the investigators observed that compound rTRD01 displaced (G4C2)4 RNA of the disease-linked c9orf72 gene from TDP-43 and improved larval neuromuscular coordination of Drosophila overexpressing mutant TDP-43 (Francois-Moutal et al., 2019). Despite being targeted to an aggregation-mediating domain of the protein, it has not been examined whether rTRD01 reduces TDP-43 aggregation propensity.
In another study, through in silico docking of 50,000 compounds to the N-terminal domain of TDP-43, a small molecule (nTRD22) that binds specifically to the NTD domain has been identified. Notably, nTRD22 induced allosteric modulation of the RRM of TDP-43, leading to reduced RNA binding in vitro (Mollasalehi et al., 2020). Furthermore, exposure of primary motor neurons to nTRD22 resulted in a decrease in TDP-43 protein levels, akin to the effect observed with TDP-43 RNA binding-deficient mutants (Zacco et al., 2018; Flores et al., 2019), supporting the idea that nTRD22 disrupts the binding of TDP-43 to RNA. Interestingly, in a Drosophila ALS model, nTRD22 alleviated motor impairment (Mollasalehi et al., 2020). Additionally, several studies have demonstrated that nucleotides rich in UG/TG sequences can impede the aggregation of TDP-43 using in vitro biochemical assays (Huang et al., 2013; Sun et al., 2014; Rengifo-Gonzalez et al., 2021) but also in cells (Yang et al., 2023).
Auranofin [gold (1+)-3,4,5- triacetyloxy- 6- (acetyloxymethyl) oxane-2 - thiolate-triethyl-phosphanium] is an organic gold thiol compound currently used to treat rheumatoid arthritis due to its anti-inflammatory properties (Madeira et al., 2013; Madeira et al., 2014; Madeira et al., 2015). Auranofin has been shown to inhibit TDP-43 self-interaction in mouse N2a neuroblastoma cells without affecting endogenous levels of the protein and without conferring cellular toxicity (Oberstadt et al., 2018). This effect of auranofin was discovered in a cell-based screen of 1280 pharmacologically active compounds where TDP-43 self-interaction was monitored by establishing a NanoBit luciferase complementation assay in which the presence of auranofin enhances the protein’s partitioning in the soluble fraction (Oberstadt et al., 2018). Although its mechanism of action is not known, it has been proposed that auranofin modulates the cysteine residues of TDP-43 located in the RRMs, thus affecting its self-interaction and eventually aggregation (Oberstadt et al., 2018). Importantly, auranofin holds desirable pharmacokinetic properties as it is able to reach the CNS of mice and rats after oral administration (Madeira et al., 2013), rendering it an attractive drug candidate.
The role of protein aggregation in the pathogenesis of ALS is critical, owing to its adverse effects on neuronal function and survival. Proteins such as SOD1 and TDP-43, when aggregated, can disrupt cellular processes, impair protein quality control mechanisms, and trigger toxicity, ultimately resulting in the degeneration of motor neurons, a hallmark of ALS. Present therapeutic strategies aim to mitigate these effects through various approaches, including the degradation of mRNA or the inhibition of its translation, the clearance of protein aggregates via antibodies, the disruption of protein-protein interactions that facilitate aggregation, the prevention of aggregate formation through vaccines, and the activation of protein clearance pathways. Nonetheless, an approach focused on directly targeting misfolded proteins might offer a more precise method for altering the course of the disease. Such specific interventions could provide greater accuracy in mitigating the detrimental impacts of certain protein aggregates. A better understanding of the aggregation mechanisms could lead to the development of molecules or screening techniques aimed at particular stages of the fibrillation process. For instance, targeting the dimerization of SOD1 at late folding stages may not yield effective in vivo results. Instead, compounds that facilitate metal binding or disulfide bond formation at earlier stages of SOD1 maturation could prove more beneficial therapeutically. Information on how misfolded and aggregated species confer toxicity via gains and losses of activity may also guide candidate drug decisions.
Despite the authorization of Tofersen, an antisense oligonucleotide targeting SOD1, there remains a critical need for additional therapeutic interventions to manage SOD1 aggregation. The specificity of Tofersen for SOD1-linked familial ALS, which accounts for only a fraction of ALS cases, underscores the necessity for a broader range of therapeutic strategies that can effectively reduce SOD1 aggregation. In this context, a strategy aiming at restoring SOD1’s conformation and eventually its function could be invaluable, serving as either a standalone or complementary therapeutic intervention. Furthermore, the ubiquity of TDP-43 in ALS pathology across the majority of cases positions it as a primary focus of ALS research. Efforts to mitigate TDP-43 aggregation are promising and could benefit a broad spectrum of ALS patients. However, challenges such as differentiating between the protein’s physiological and pathological states within cells, and the obstacles in crossing the blood-brain barrier with therapeutic agents, must be addressed. Significantly, evidence from cellular and animal studies has shown that identifying compounds capable of inhibiting misfolding and/or aggregation or promoting disaggregation may lead to disease-modifying effects (both in vitro and in vivo, as summarized in Table 1), yet none has proceeded to clinical trials. This situation highlights the urgent need for continued research and development in this area.
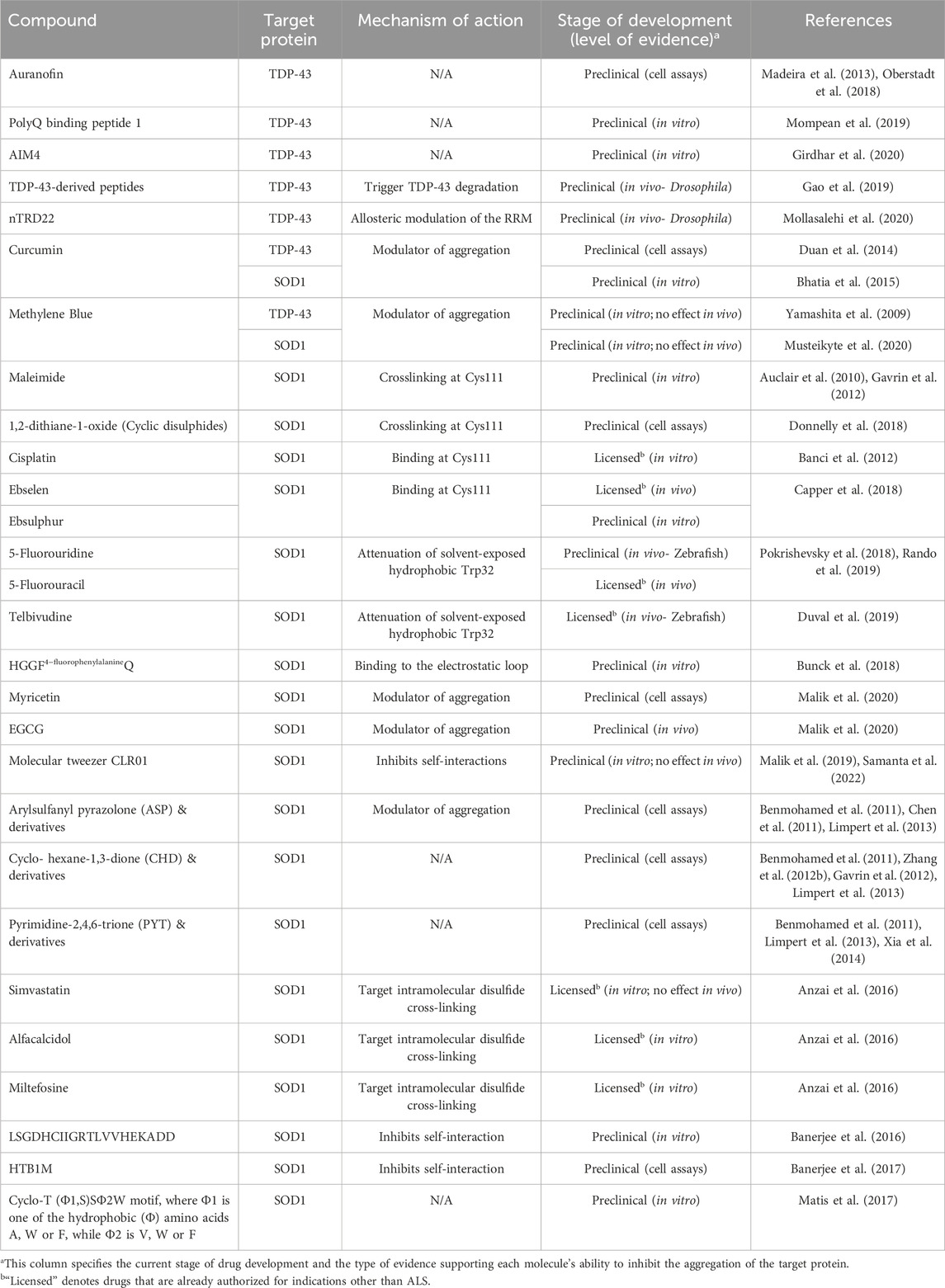
Table 1. A summary of compounds identified by their ability to inhibit aggregation of TDP-43 or SOD1.
The high rate of failure in ALS clinical trials can be attributed to multiple pivotal factors. Historically, ALS has been treated as a uniform condition, overlooking its inherent heterogeneity. Future research must categorize ALS patients by specific genetic determinants and disease progression rates. Enhancing the use of biomarkers, refining preclinical models, improving trial design, and facilitating early intervention are crucial strategies to advance the development of ALS therapies. A holistic approach that integrates target-specific in vitro screening, validation using patient-derived iPSC neurons and glia, and mechanistic studies in model organisms such as zebrafish, fruit flies, and mice, can enhance the prediction of therapeutic effectiveness and its translation to clinical settings.
MT: Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. MG: Data curation, Investigation, Methodology, Writing–original draft. KP: Data curation, Investigation, Writing–original draft, Writing–review and editing. GS: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was financed for research and authorship by: (i) the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Project “ProMiDis”; grant agreement no. 819934; support for GS and MT); (ii) Greece and the European Union (European Social Fund- ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research–2nd Cycle” (MIS 5000432), implemented by the State Scholarships Foundation (IKY) (support for MG); (iii) the Horizon Europe Programme under the “Widening Participation and Spreading Excellence” component (call Twinning “HORIZON-WIDERA-2021-ACCESS- 03-1”); Project “Twin4Promis”; Grant Agreement No. 101079363 (support for GS); (v) the Horizon Europe Programme under the “Widening Participation & Spreading Excellence” component (call Twinning “HORIZON-WIDERA-2022- TALENTS-01-01–ERA Chairs”); Project “Boost4Bio”; Grant Agreement No. 101087471 (support for GS and KP). Financial support for the publication of this article was provided by the Horizon Europe Programme under the “Widening Participation and Spreading Excellence” component (call Twinning “HORIZON-WIDERA-2021-ACCESS- 03-1”); Project “Twin4Promis”; Grant Agreement No. 101079363.
Author GS is co-founder and shareholder of the company ResQ Biotech and author KP was employed by the company ResQ Biotech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdolvahabi, A., Shi, Y., Chuprin, A., Rasouli, S., and Shaw, B. F. (2016). Stochastic Formation of fibrillar and amorphous superoxide dismutase oligomers linked to amyotrophic lateral sclerosis. ACS Chem. Neurosci. 7 (6), 799–810. doi:10.1021/acschemneuro.6b00048
Acharya, S., Safaie, B. M., Wongkongkathep, P., Ivanova, M. I., Attar, A., Klarner, F. G., et al. (2014). Molecular basis for preventing α-synuclein aggregation by a molecular tweezer. J. Biol. Chem. 289 (15), 10727–10737. doi:10.1074/jbc.M113.524520
Afroz, T., Chevalier, E., Audrain, M., Dumayne, C., Ziehm, T., Moser, R., et al. (2023). Immunotherapy targeting the C-terminal domain of TDP-43 decreases neuropathology and confers neuroprotection in mouse models of ALS/FTD. Neurobiol. Dis. 179, 106050. doi:10.1016/j.nbd.2023.106050
Afroz, T., Hock, E. M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L. A. B., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8 (1), 45. doi:10.1038/s41467-017-00062-0
Ahrens, C. L., and Manno, E. M. (2014). Neurotoxicity of commonly used hepatic drugs. Handb. Clin. Neurol. 120, 675–682. doi:10.1016/B978-0-7020-4087-0.00046-2
Alam, P., Siddiqi, K., Chturvedi, S. K., and Khan, R. H. (2017). Protein aggregation: from background to inhibition strategies. Int. J. Biol. Macromol. 103, 208–219. doi:10.1016/j.ijbiomac.2017.05.048
Alberti, S., Halfmann, R., King, O., Kapila, A., and Lindquist, S. (2009). A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137 (1), 146–158. doi:10.1016/j.cell.2009.02.044
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9 (11), 617–628. doi:10.1038/nrneurol.2013.203
Almeida, Z. L., and Brito, R. M. M. (2020). Structure and aggregation mechanisms in amyloids. Molecules 25 (5), 1195. doi:10.3390/molecules25051195
Amporndanai, K., Rogers, M., Watanabe, S., Yamanaka, K., Neill, P. M. O., and Hasnain, S. S. (2020). Novel Selenium-based compounds with therapeutic potential for SOD1-linked amyotrophic lateral sclerosis. EBioMedicine 59, 102980. doi:10.1016/j.ebiom.2020.102980
Andersen, P. M., Nordstrom, U., Tsiakas, K., Johannsen, J., Volk, A. E., Bierhals, T., et al. (2019). Phenotype in an infant with SOD1 homozygous truncating mutation. N. Engl. J. Med. 381 (5), 486–488. doi:10.1056/NEJMc1905039
Andrews, J. A., Jackson, C. E., Heiman-Patterson, T. D., Bettica, P., Brooks, B. R., and Pioro, E. P. (2020). Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 21 (7-8), 509–518. doi:10.1080/21678421.2020.1771734
Antonyuk, S., Strange, R. W., and Hasnain, S. S. (2010). Structural discovery of small molecule binding sites in Cu-Zn human superoxide dismutase familial amyotrophic lateral sclerosis mutants provides insights for lead optimization. J. Med. Chem. 53 (3), 1402–1406. doi:10.1021/jm9017948
Anzai, I., Toic, Hi K., Tokuda, E., Mukaiyama, A., Akiyama, S., and Furukawa, Y. (2016). Screening of drugs inhibiting in vitro oligomerization of Cu/Zn-superoxide dismutase with a mutation causing amyotrophic lateral sclerosis. Front. Mol. Biosci. 3, 1–11. doi:10.3389/fmolb.2016.00040
Anzai, I., Tokuda, E., Mukaiyama, A., Akiyama, S., Endo, F., Yamanaka, K., et al. (2017). A misfolded dimer of Cu/Zn-superoxide dismutase leading to pathological oligomerization in amyotrophic lateral sclerosis. Protein Sci. a Publ. Protein Soc. 26 (3), 484–496. doi:10.1002/pro.3094
Appel, S. H., Beers, D. R., and Zhao, W. (2021). Amyotrophic lateral sclerosis is a systemic disease: peripheral contributions to inflammation-mediated neurodegeneration. Curr. Opin. Neurol. 34 (5), 765–772. doi:10.1097/WCO.0000000000000983
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351 (3), 602–611. doi:10.1016/j.bbrc.2006.10.093
Archbold, H. C., Jackson, K. L., Arora, A., Weskamp, K., Tank, E. M., Li, X., et al. (2018). TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 8 (1), 4606. doi:10.1038/s41598-018-22858-w
Arnesano, F., Banci, L., Bertini, I., Martinelli, M., Furukawa, Y., and O'Halloran, T. V. (2004). The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 279 (46), 47998–48003. doi:10.1074/jbc.M406021200
Arnold, E. S., Ling, S. C., Huelga, S. C., Lagier-Tourenne, C., Polymenidou, M., Ditsworth, D., et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. U. S. A. 110 (8), E736–E745. doi:10.1073/pnas.1222809110
Arseni, D., Hasegawa, M., Murzin, A. G., Kametani, F., Arai, M., Yoshida, M., et al. (2022). Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601 (7891), 139–143. doi:10.1038/s41586-021-04199-3
Attar, A., Ripoli, C., Riccardi, E., Maiti, P., Li Puma, D. D., Liu, T., et al. (2012). Protection of primary neurons and mouse brain from Alzheimer's pathology by molecular tweezers. Brain 135 (Pt 12), 3735–3748. doi:10.1093/brain/aws289
Auclair, J. R., Boggio, K. J., Petsko, G. A., Ringe, D., and Agar, J. N. (2010). Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 107, 21394–21399. doi:10.1073/pnas.1015463107
Audet, J. N., Gowing, G., and Julien, J. P. (2010). Wild-type human SOD1 overexpression does not accelerate motor neuron disease in mice expressing murine Sod1 G86R. Neurobiol. Dis. 40 (1), 245–250. doi:10.1016/j.nbd.2010.05.031
Audet, J. N., Soucy, G., and Julien, J. P. (2012). Methylene blue administration fails to confer neuroprotection in two amyotrophic lateral sclerosis mouse models. Neuroscience 209, 136–143. doi:10.1016/j.neuroscience.2011.12.047
Audrain, M., Egesipe, A. L., Tentillier, N., Font, L., Ratnam, M., Mottier, L., et al. (2023). Targeting amyotrophic lateral sclerosis by neutralizing seeding-competent TDP-43 in CSF. Brain Commun. 5 (6), fcad306. doi:10.1093/braincomms/fcad306
Ayers, J. I., Fromholt, S. E., O'Neal, V. M., Diamond, J. H., and Borchelt, D. R. (2016). Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 131 (1), 103–114. doi:10.1007/s00401-015-1514-0
Babazadeh, A., Rayner, S. L., Lee, A., and Chung, R. S. (2023). TDP-43 as a therapeutic target in neurodegenerative diseases: focusing on motor neuron disease and frontotemporal dementia. Ageing Res. Rev. 92, 102085. doi:10.1016/j.arr.2023.102085
Babinchak, W. M., Dumm, B. K., Venus, S., Boyko, S., Putnam, A. A., Jankowsky, E., et al. (2020). Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 11 (1), 5574. doi:10.1038/s41467-020-19211-z
Bakavayev, S., Stavsky, A., Argueti-Ostrovsky, S., Yehezkel, G., Fridmann-Sirkis, Y., Barak, Z., et al. (2023). Blocking an epitope of misfolded SOD1 ameliorates disease phenotype in a model of amyotrophic lateral sclerosis. Brain 146 (11), 4594–4607. doi:10.1093/brain/awad222
Banci, L., Bertini, I., Blaz, O., Calderone, V., Cantini, F., Mao, J., et al. (2012). Interaction of cisplatin with human superoxide dismutase. JACS 134, 7009–7014. doi:10.1021/ja211591n
Banerjee, V., Oren, O., Ben-Zeev, E., Taube, R., Engel, S., and Papo, N. (2017). A computational combinatorial approach identifies a protein inhibitor of superoxide dismutase 1 misfolding, aggregation, and cytotoxicity. J. Biol. Chem. 292 (38), 15777–15788. doi:10.1074/jbc.M117.789610
Banerjee, V., Shani, T., Katzman, B., Vyazmensky, M., Papo, N., Israelson, A., et al. (2016). Superoxide dismutase 1 (SOD1)-Derived peptide inhibits amyloid aggregation of familial amyotrophic lateral sclerosis SOD1 mutants. ACS Chem. Neurosci. 7, 1595–1606. doi:10.1021/acschemneuro.6b00227
Banks, C. J., and Andersen, J. L. (2019). Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 26, 101270. doi:10.1016/j.redox.2019.101270
Barber, S. C., Higginbottom, A., Mead, R. J., Barber, S., and Shaw, P. J. (2009). An in vitro screening cascade to identify neuroprotective antioxidants in ALS. Free Radic. Biol. Med. 46 (8), 1127–1138. doi:10.1016/j.freeradbiomed.2009.01.019
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30 (2), 639–649. doi:10.1523/JNEUROSCI.4988-09.2010
Bauer, P. O., Goswami, A., Wong, H. K., Okuno, M., Kurosawa, M., Yamada, M., et al. (2010). Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 28 (3), 256–263. doi:10.1038/nbt.1608
Benatar, M., Wuu, J., Andersen, P. M., Bucelli, R. C., Andrews, J. A., Otto, M., et al. (2022). Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics 19 (4), 1248–1258. doi:10.1007/s13311-022-01237-4
Benmohamed, R., Arvanites, A. C., Kim, J., Ferrante, R. J., Silverman, R. B., Morimoto, R. I., et al. (2011). Identification of compounds protective against G93A-SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12 (2), 87–96. doi:10.3109/17482968.2010.522586
Bhardwaj, A., Myers, M. P., Buratti, E., and Baralle, F. E. (2013). Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 41 (9), 5062–5074. doi:10.1093/nar/gkt189
Bhatia, N. K., Srivastava, A., Katyal, N., Jain, N., Khan, M. A., Kundu, B., et al. (2015). Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta 1854 (5), 426–436. doi:10.1016/j.bbapap.2015.01.014
Bissaro, M., and Moro, S. (2019). Rethinking to riluzole mechanism of action: the molecular link among protein kinase CK1δ activity, TDP-43 phosphorylation, and amyotrophic lateral sclerosis pharmacological treatment. Neural Regen. Res. 14 (12), 2083–2085. doi:10.4103/1673-5374.262578
Blay, V., Tolani, B., Ho, S. P., and Arkin, M. R. (2020). High-Throughput Screening: today's biochemical and cell-based approaches. Drug Discov. Today 25 (10), 1807–1821. doi:10.1016/j.drudis.2020.07.024
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125 (6), 777–794. doi:10.1007/s00401-013-1125-6
Bohacek, R. S., McMartin, C., and Guida, W. C. (1996). The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 16 (1), 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6
Boillee, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52 (1), 39–59. doi:10.1016/j.neuron.2006.09.018
Bonifacino, T., Zerbo, R. A., Balbi, M., Torazza, C., Frumento, G., Fedele, E., et al. (2021). Nearly 30 Years of animal models to study amyotrophic lateral sclerosis: a historical overview and future perspectives. Int. J. Mol. Sci. 22 (22), 12236. doi:10.3390/ijms222212236
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13 (11), 1396–1403. doi:10.1038/nn.2660
Bose, S., and Cho, J. (2017). Targeting chaperones, heat shock factor-1, and unfolded protein response: promising therapeutic approaches for neurodegenerative disorders. Ageing Res. Rev. 35, 155–175. doi:10.1016/j.arr.2016.09.004
Boyd, J. D., Lee, P., Feiler, M. S., Zauur, N., Liu, M., Concannon, J., et al. (2014). A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J. Biomol. Screen 19 (1), 44–56. doi:10.1177/1087057113501553
Bozzo, F., Salvatori, I., Iacovelli, F., Mirra, A., Rossi, S., Cozzolino, M., et al. (2016). Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol. Dis. 94, 63–72. doi:10.1016/j.nbd.2016.06.006
Brasil, A. A., Magalhaes, R. S. S., De Carvalho, M. D. C., Paiva, I., Gerhardt, E., Pereira, M. D., et al. (2018). Implications of fALS mutations on Sod1 function and oligomerization in cell models. Mol. Neurobiol. 55 (6), 5269–5281. doi:10.1007/s12035-017-0755-4
Bravo-Hernandez, M., Tadokoro, T., Navarro, M. R., Platoshyn, O., Kobayashi, Y., Marsala, S., et al. (2020). Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med. 26 (1), 118–130. doi:10.1038/s41591-019-0674-1
Brenner, D., and Freischmidt, A. (2022). Update on genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 35 (5), 672–677. doi:10.1097/WCO.0000000000001093
Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., et al. (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281 (5384), 1851–1854. doi:10.1126/science.281.5384.1851
Budini, M., Buratti, E., Stuani, C., Guarnaccia, C., Romano, V., De Conti, L., et al. (2012). Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287 (10), 7512–7525. doi:10.1074/jbc.M111.288720
Bulawa, C. E., Connelly, S., Devit, M., Wang, L., Weigel, C., Fleming, J. A., et al. (2012). Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U. S. A. 109 (24), 9629–9634. doi:10.1073/pnas.1121005109
Bunck, D., Atsavapranee, B., Museth, A., VanderVelde, D., and Heath, J. (2018). Modulating the folding landscape of superoxide dismutase 1 with targeted molecular binders. Angew. Chem. Int. Ed. 57, 6212–6215. doi:10.1002/anie.201802269
Bunton-Stasyshyn, R. K., Saccon, R. A., Fratta, P., and Fisher, E. M. (2015). SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist 21 (5), 519–529. doi:10.1177/1073858414561795
Buratti, E. (2018). TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. targets 22 (3), 279–293. doi:10.1080/14728222.2018.1439923
Buratti, E. (2020). Targeting TDP-43 proteinopathy with drugs and drug-like small molecules. Br. J. Pharmacol. 178, 1298–1315. doi:10.1111/bph.15148
Buratti, E., Brindisi, A., Giombi, M., Tisminetzky, S., Ayala, Y. M., and Baralle, F. E. (2005). TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280 (45), 37572–84. doi:10.1074/jbc.M505557200
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell Neurosci. 56, 355–364. doi:10.1016/j.mcn.2013.07.007
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2019). Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 26 (7), 619–627. doi:10.1038/s41594-019-0248-4
Capitini, C., Conti, S., Perni, M., Guidi, F., Cascella, R., De Poli, A., et al. (2014). TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One 9 (1), e86720. doi:10.1371/journal.pone.0086720
Capper, M. J., Wright, G. S. A., Barbieri, L., Luchinat, E., Mercatelli, E., Mcalary, L., et al. (2018). The cysteine-reactive small molecule ebselen facilitates effective SOD1 maturation. Nat. Commun. 9, 1693–1699. doi:10.1038/s41467-018-04114-x
Chang, C. K., Chiang, M. H., Toh, E. K., Chang, C. F., and Huang, T. H. (2013). Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 587 (6), 575–582. doi:10.1016/j.febslet.2013.01.038
Chang, C. K., Wu, T. H., Wu, C. Y., Chiang, M. H., Toh, E. K., Hsu, Y. C., et al. (2012). The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 425 (2), 219–224. doi:10.1016/j.bbrc.2012.07.071
Chantadul, V., Wright, G. S. A., Amporndanai, K., Shahid, M., Antonyuk, S. V., Washbourn, G., et al. (2020). Ebselen as template for stabilization of A4V mutant dimer for motor neuron disease therapy. Commun. Biol. 3 (1), 97. doi:10.1038/s42003-020-0826-3
Chen, A. K., Lin, R. Y., Hsieh, E. Z., Tu, P. H., Chen, R. P., Liao, T. Y., et al. (2010). Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132 (4), 1186–1187. doi:10.1021/ja9066207
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14 (6), 796–809. doi:10.1016/j.stem.2014.02.004
Chen, T., Benmohamed, R., Arvanites, A. C., Ralay, H., Morimoto, R. I., Ferrante, R. J., et al. (2011). Arylsulfanyl pyrazolones block mutant SOD1-G93A aggregation. Potential application for the treatment of amyotrophic lateral sclerosis. Bioorg. Med. Chem. 19, 613–622. doi:10.1016/j.bmc.2010.10.052
Chen, Y., Wang, H., Ying, Z., and Gao, Q. (2020). Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: the new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem. Biophys. Res. Commun. 526 (1), 231–238. doi:10.1016/j.bbrc.2020.03.051
Chen, Z., Jiang, Z., Chen, N., Shi, Q., Tong, L., Kong, F., et al. (2018). Target discovery of ebselen with a biotinylated probe. Chem. Commun. 54 (68), 9506–9509. doi:10.1039/c8cc04258f
Chew, J., Cook, C., Gendron, T. F., Jansen-West, K., Del Rosso, G., Daughrity, L. M., et al. (2019). Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 14 (1), 9. doi:10.1186/s13024-019-0310-z
Chio, A., Calvo, A., Moglia, C., Mazzini, L., Mora, G., and group, P. (2011). Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J. Neurol. Neurosurg. Psychiatry 82 (7), 740–746. doi:10.1136/jnnp.2010.235952
Chio, A., Logroscino, G., Hardiman, O., Swingler, R., Mitchell, D., Beghi, E., et al. (2009). Prognostic factors in ALS: a critical review. Amyotroph. Lateral Scler. 10 (5-6), 310–323. doi:10.3109/17482960802566824
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi:10.1146/annurev.biochem.75.101304.123901
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi:10.1146/annurev-biochem-061516-045115
Choi, E. S., and Dokholyan, N. V. (2021). SOD1 oligomers in amyotrophic lateral sclerosis. Curr. Opin. Struct. Biol. 66, 225–230. doi:10.1016/j.sbi.2020.12.002
Choksi, D. K., Roy, B., Chatterjee, S., Yusuff, T., Bakhoum, M. F., Sengupta, U., et al. (2014). TDP-43 Phosphorylation by casein kinase Iε promotes oligomerization and enhances toxicity in vivo. Hum. Mol. Genet. 23 (4), 1025–1035. doi:10.1093/hmg/ddt498
Coelho, F. R., Iqbal, A., Linares, E., Silva, D. F., Lima, F. S., Cuccovia, I. M., et al. (2014). Oxidation of the tryptophan 32 residue of human superoxide dismutase 1 caused by its bicarbonate-dependent peroxidase activity triggers the non-amyloid aggregation of the enzyme. J. Biol. Chem. 289 (44), 30690–30701. doi:10.1074/jbc.M114.586370
Cohen, T. J., Hwang, A. W., Unger, T., Trojanowski, J. Q., and Lee, V. M. (2012). Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 31 (5), 1241–1252. doi:10.1038/emboj.2011.471
Collaborators, GBDN (2019). Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurology 18 (5), 459–480. doi:10.1016/S1474-4422(18)30499-X
Coll-Martinez, B., Delgado, A., and Crosas, B. (2020). The potential of proteolytic chimeras as pharmacological tools and therapeutic agents. Molecules 25 (24), 5956. doi:10.3390/molecules25245956
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111 (4), 1051–1061. doi:10.1111/j.1471-4159.2009.06383.x
Conicella, A. E., Zerze, G. H., Mittal, J., and Fawzi, N. L. (2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24 (9), 1537–1549. doi:10.1016/j.str.2016.07.007
Convertino, M., Das, J., and Dokholyan, N. V. (2016). Pharmacological chaperones: design and development of new therapeutic strategies for the treatment of conformational diseases. ACS Chem. Biol. 11 (6), 1471–1489. doi:10.1021/acschembio.6b00195
Cooper-Knock, J., Harvey, C., Zhang, S., Moll, T., Timpanaro, I. S., Kenna, K. P., et al. (2021). Advances in the genetic classification of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 34 (5), 756–764. doi:10.1097/WCO.0000000000000986
Cortez, L., and Sim, V. (2014). The therapeutic potential of chemical chaperones in protein folding diseases. Prion 8 (2), 197–202. doi:10.4161/pri.28938
Couratier, P., Lautrette, G., Luna, J. A., and Corcia, P. (2021). Phenotypic variability in amyotrophic lateral sclerosis. Rev. Neurol. 177 (5), 536–543. doi:10.1016/j.neurol.2021.03.001
Cronin, S., Hardiman, O., and Traynor, B. J. (2007). Ethnic variation in the incidence of ALS: a systematic review. Neurology 68 (13), 1002–1007. doi:10.1212/01.wnl.0000258551.96893.6f
Crown, A., Mcalary, L., Fagerli, E., Brown, H., Yerbury, J. J., Galaleldeen, A., et al. (2020). Tryptophan residue 32 in human Cu-Zn superoxide dismutase modulates prion-like propagation and strain selection. PLoS ONE 15, e0227655. doi:10.1371/journal.pone.0227655
Crown, A. M., Roberts, B. L., Crosby, K., Brown, H., Ayers, J. I., Hart, P. J., et al. (2019). Experimental mutations in superoxide dismutase 1 provide insight into potential mechanisms involved in aberrant aggregation in familial amyotrophic lateral sclerosis. G3 (Bethesda) 9 (3), 719–728. doi:10.1534/g3.118.200787
Culik, R. M., Sekhar, A., Nagesh, J., Deol, H., Rumfeldt, J. A. O., Meiering, E. M., et al. (2018). Effects of maturation on the conformational free-energy landscape of SOD1. Proc. Natl. Acad. Sci. U. S. A. 115 (11), E2546–E55. doi:10.1073/pnas.1721022115
de Andrade Teles, R. B., Diniz, T. C., Costa Pinto, T. C., de Oliveira Junior, R. G., Gama, E. S. M., de Lavor, E. M., et al. (2018). Flavonoids as therapeutic agents in Alzheimer's and Parkinson's diseases: a systematic review of preclinical evidences. Oxid. Med. Cell Longev. 2018, 7043213. doi:10.1155/2018/7043213
Debnath, K., Shekhar, S., Kumar, V., Jana, N. R., and Jana, N. R. (2016). Efficient inhibition of protein aggregation, disintegration of aggregates, and lowering of cytotoxicity by green tea polyphenol-based self-assembled polymer nanoparticles. ACS Appl. Mater Interfaces 8 (31), 20309–20318. doi:10.1021/acsami.6b06853
de Boer, E. M. J., Orie, V. K., Williams, T., Baker, M. R., De Oliveira, H. M., Polvikoski, T., et al. (2020). TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 92, 86–95. doi:10.1136/jnnp-2020-322983
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477 (7363), 211–215. doi:10.1038/nature10353
Deng, H. X., Shi, Y., Furukawa, Y., Zhai, H., Fu, R., Liu, E., et al. (2006). Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 103 (18), 7142–7147. doi:10.1073/pnas.0602046103
Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P., Good, S. K., et al. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31 (5), 1098–1108. doi:10.1128/MCB.01279-10
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321 (5893), 1218–1221. doi:10.1126/science.1158799
Doi, Y., Atsuta, N., Sobue, G., Morita, M., and Nakano, I. (2014). Prevalence and incidence of amyotrophic lateral sclerosis in Japan. J. Epidemiol. 24 (6), 494–499. doi:10.2188/jea.JE20140059
Donnelly, D. P., Dowgiallo, M. G., Salisbury, J. P., Krishna, C., Iyengar, S., Chaudhari, M., et al. (2018). Cyclic thiosulfinates and cyclic disulfides selectively crosslink thiols while avoiding modification of lone thiols. JACS 140, 7377–7380. doi:10.1021/jacs.8b01136
Du, H., Huo, Z., Chen, Y., Zhao, Z., Meng, F., Wang, X., et al. (2023). Induced pluripotent stem cells and their applications in amyotrophic lateral sclerosis. Cells 12 (6), 971. doi:10.3390/cells12060971
Duan, W., Guo, M., Yi, L., Liu, Y., Li, Z., Ma, Y., et al. (2020). The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 27 (3-4), 157–169. doi:10.1038/s41434-019-0116-1
Duan, W., Guo, Y., Xiao, J., Chen, X., Li, Z., Han, H., et al. (2014). Neuroprotection by monocarbonyl dimethoxycurcumin C: ameliorating the toxicity of mutant TDP-43 via HO-1. Mol. Neurobiol. 49 (1), 368–379. doi:10.1007/s12035-013-8525-4
Duval, M. G., Hinge, V. K., Snyder, N., Kanyo, R., Bratvold, J., Pokrishevsky, E., et al. (2019). Tryptophan 32 mediates SOD1 toxicity in a in vivo motor neuron model of ALS and is a promising target for small molecule therapeutics. Neurobiol. Dis. 124, 297–310. doi:10.1016/j.nbd.2018.11.025
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4 (145), 145ra104. doi:10.1126/scitranslmed.3004052
Eisele, Y. S., Monteiro, C., Fearns, C., Encalada, S. E., Wiseman, R. L., Powers, E. T., et al. (2015). Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14 (11), 759–780. doi:10.1038/nrd4593
Elam, J. S., Taylor, A. B., Strange, R., Antonyuk, S., Doucette, P. A., Rodriguez, J. A., et al. (2003). Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 10 (6), 461–467. doi:10.1038/nsb935
Elliott, E., Bailey, O., Waldron, F. M., Hardingham, G. E., Chandran, S., and Gregory, J. M. (2020). Therapeutic targeting of proteostasis in amyotrophic lateral sclerosis-a systematic review and meta-analysis of preclinical research. Front. Neurosci. 14, 511. doi:10.3389/fnins.2020.00511
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21 (16), 3703–3718. doi:10.1093/hmg/dds205
Fang, M. Y., Markmiller, S., Vu, A. Q., Javaherian, A., Dowdle, W. E., Jolivet, P., et al. (2019). Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103 (5), 802–819. doi:10.1016/j.neuron.2019.05.048
Fang, T., Al Khleifat, A., Meurgey, J. H., Jones, A., Leigh, P. N., Bensimon, G., et al. (2018). Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurology 17 (5), 416–422. doi:10.1016/S1474-4422(18)30054-1
Fang, Y. (2012). Ligand-receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 7 (10), 969–988. doi:10.1517/17460441.2012.715631
Fang, Y. S., Tsai, K. J., Chang, Y. J., Kao, P., Woods, R., Kuo, P. H., et al. (2014). Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 5, 4824. doi:10.1038/ncomms5824
Farina, S., Esposito, F., Battistoni, M., Biamonti, G., and Francia, S. (2021). Post-translational modifications modulate proteinopathies of TDP-43, FUS and hnRNP-A/B in amyotrophic lateral sclerosis. Front. Mol. Biosci. 8, 693325. doi:10.3389/fmolb.2021.693325
Fei, E., Jia, N., Yan, M., Ying, Z., Sun, Q., Wang, H., et al. (2006). SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 347 (2), 406–412. doi:10.1016/j.bbrc.2006.06.092
Feiguin, F., Godena, V. K., Romano, G., D'Ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583 (10), 1586–1592. doi:10.1016/j.febslet.2009.04.019
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7 (11), 616–630. doi:10.1038/nrneurol.2011.152
Ferraiuolo, L., and Maragakis, N. J. (2021). Mini-Review: induced pluripotent stem cells and the search for new cell-specific ALS therapeutic targets. Neurosci. Lett. 755, 135911. doi:10.1016/j.neulet.2021.135911
Ferrari, R., Kapogiannis, D., Huey, E. D., and Momeni, P. (2011). FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 8 (3), 273–294. doi:10.2174/156720511795563700
Ferreira, N., Pereira-Henriques, A., Attar, A., Klarner, F. G., Schrader, T., Bitan, G., et al. (2014). Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics 11 (2), 450–461. doi:10.1007/s13311-013-0256-8
Figueroa, J. D., Zarate, A. M., Fuentes-Lemus, E., Davies, M. J., and Lopez-Alarcon, C. (2020). Formation and characterization of crosslinks, including Tyr-Trp species, on one electron oxidation of free Tyr and Trp residues by carbonate radical anion. RSC Adv. 10 (43), 25786–25800. doi:10.1039/d0ra04051g
Fischer, L. R., Igoudjil, A., Magrane, J., Li, Y., Hansen, J. M., Manfredi, G., et al. (2011). SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 134 (Pt 1), 196–209. doi:10.1093/brain/awq314
Fischer, L. R., Li, Y., Asress, S. A., Jones, D. P., and Glass, J. D. (2012). Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 233 (1), 163–171. doi:10.1016/j.expneurol.2011.09.020
Flood, D. G., Reaume, A. G., Gruner, J. A., Hoffman, E. K., Hirsch, J. D., Lin, Y. G., et al. (1999). Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am. J. Pathol. 155 (2), 663–672. doi:10.1016/S0002-9440(10)65162-0
Flores, B. N., Li, X., Malik, A. M., Martinez, J., Beg, A. A., and Barmada, S. J. (2019). An intramolecular salt bridge linking TDP43 RNA binding, protein stability, and TDP43-dependent neurodegeneration. Cell Rep. 27 (4), 1133–1150. doi:10.1016/j.celrep.2019.03.093
Formica, V., Leary, A., Cunningham, D., and Chua, Y. J. (2006). 5-Fluorouracil can cross brain-blood barrier and cause encephalopathy: should we expect the same from capecitabine? A case report on capecitabine-induced central neurotoxicity progressing to coma. Cancer Chemother. Pharmacol. 58 (2), 276–278. doi:10.1007/s00280-005-0159-4
Forsberg, K., Graffmo, K., Pakkenberg, B., Weber, M., Nielsen, M., Marklund, S., et al. (2019). Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J. Neurol. Neurosurg. Psychiatry 0, 861–869. doi:10.1136/jnnp-2018-319386
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81 (5), 1009–1023. doi:10.1016/j.neuron.2014.01.013
Francois-Moutal, L., Felemban, R., Scott, D. D., Sayegh, M. R., Miranda, V. G., Perez-Miller, S., et al. (2019). Small molecule targeting TDP-43's RNA recognition motifs reduces locomotor defects in a Drosophila model of amyotrophic lateral sclerosis (ALS). ACS Chem. Biol. 14 (9), 2006–2013. doi:10.1021/acschembio.9b00481
French, R. L., Grese, Z. R., Aligireddy, H., Dhavale, D. D., Reeb, A. N., Kedia, N., et al. (2019). Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 294 (17), 6696–6709. doi:10.1074/jbc.RA118.005889
Fukushima, M., Hosoda, N., Chifu, K., and Hoshino, S. I. (2019). TDP-43 accelerates deadenylation of target mRNAs by recruiting Caf1 deadenylase. FEBS Lett. 593 (3), 277–287. doi:10.1002/1873-3468.13310
Furukawa, Y., Fu, R., Deng, H. X., Siddique, T., and O'Halloran, T. V. (2006). Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U. S. A. 103 (18), 7148–7153. doi:10.1073/pnas.0602048103
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2013). Intracellular seeded aggregation of mutant Cu,Zn-superoxide dismutase associated with amyotrophic lateral sclerosis. FEBS Lett. 587 (16), 2500–2505. doi:10.1016/j.febslet.2013.06.046
Furukawa, Y., Kaneko, K., Yamanaka, K., O'Halloran, T. V., and Nukina, N. (2008). Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 283 (35), 24167–24176. doi:10.1074/jbc.M802083200
Gao, N., Huang, Y. P., Chu, T. T., Li, Q. Q., Zhou, B., Chen, Y. X., et al. (2019). TDP-43 specific reduction induced by Di-hydrophobic tags conjugated peptides. Bioorg. Chem. 84, 254–259. doi:10.1016/j.bioorg.2018.11.042
Gatto, N., Dos Santos Souza, C., Shaw, A. C., Bell, S. M., Myszczynska, M. A., Powers, S., et al. (2021). Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell 20 (1), e13281. doi:10.1111/acel.13281
Gavrin, L. K., Denny, R. A., and Saiah, E. (2012). Small molecules that target protein misfolding. J. Med. Chem. 55, 10823–10843. doi:10.1021/jm301182j
Gelpi, E., and Colom-Cadena, M. (2019). Oligomers: a hot topic for neurodegeneration and a note of caution for experimental models. Brain 142 (2), 228–230. doi:10.1093/brain/awy342
Germain, D. P., Hughes, D. A., Nicholls, K., Bichet, D. G., Giugliani, R., Wilcox, W. R., et al. (2016). Treatment of fabry's disease with the pharmacologic chaperone Migalastat. N. Engl. J. Med. 375 (6), 545–555. doi:10.1056/NEJMoa1510198
Getzoff, E. D., Cabelli, D. E., Fisher, C. L., Parge, H. E., Viezzoli, M. S., Banci, L., et al. (1992). Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 358 (6384), 347–351. doi:10.1038/358347a0
Gill, C., Phelan, J. P., Hatzipetros, T., Kidd, J. D., Tassinari, V. R., Levine, B., et al. (2019). SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 9 (1), 6724. doi:10.1038/s41598-019-43164-z
Girdhar, A., Bharathi, V., Tiwari, V. R., Abhishek, S., Deeksha, W., Mahawar, U. S., et al. (2020). Computational insights into mechanism of AIM4-mediated inhibition of aggregation of TDP-43 protein implicated in ALS and evidence for in vitro inhibition of liquid-liquid phase separation (LLPS) of TDP-43(2C)-A315T by AIM4. Int. J. Biol. Macromol. 147, 117–130. doi:10.1016/j.ijbiomac.2020.01.032
Gordon, D., Dafinca, R., Scaber, J., Alegre-Abarrategui, J., Farrimond, L., Scott, C., et al. (2019). Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 121, 148–162. doi:10.1016/j.nbd.2018.09.024
Gorgulla, C., Jayaraj, A., Fackeldey, K., and Arthanari, H. (2022). Emerging frontiers in virtual drug discovery: from quantum mechanical methods to deep learning approaches. Curr. Opin. Chem. Biol. 69, 102156. doi:10.1016/j.cbpa.2022.102156
Goyal, D., Shuaib, S., Mann, S., and Goyal, B. (2017). Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: potential therapeutics of Alzheimer's disease. ACS Comb. Sci. 19 (2), 55–80. doi:10.1021/acscombsci.6b00116
Grad, L. I., Guest, W. C., Yanai, A., Pokrishevsky, E., O'Neill, M. A., Gibbs, E., et al. (2011). Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. U. S. A. 108 (39), 16398–16403. doi:10.1073/pnas.1102645108
Gros-louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972. doi:10.1016/j.bbadis.2006.01.004
Guise, A. J., Misal, S. A., Carson, R., Chu, J. H., Boekweg, H., Van Der Watt, D., et al. (2024). TDP-43-stratified single-cell proteomics of postmortem human spinal motor neurons reveals protein dynamics in amyotrophic lateral sclerosis. Cell Rep. 43 (1), 113636. doi:10.1016/j.celrep.2023.113636
Guo, L., and Shorter, J. (2015). It's raining liquids: RNA tunes viscoelasticity and dynamics of membraneless organelles. Mol. Cell 60 (2), 189–192. doi:10.1016/j.molcel.2015.10.006
Guo, W., Chen, Y., Zhou, X., Kar, A., Ray, P., Chen, X., et al. (2011). An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18 (7), 822–830. doi:10.1038/nsmb.2053
Gureev, A. P., Sadovnikova, I. S., and Popov, V. N. (2022). Molecular mechanisms of the neuroprotective effect of methylene blue. Biochem. Biokhimiia 87 (9), 940–956. doi:10.1134/S0006297922090073
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264 (5166), 1772–1775. doi:10.1126/science.8209258
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29 (9), 824–828. doi:10.1038/nbt.1957
Hammarstrom, P., Wiseman, R. L., Powers, E. T., and Kelly, J. W. (2003). Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 299 (5607), 713–716. doi:10.1126/science.1079589
Hard, T., and Lendel, C. (2012). Inhibition of amyloid formation. J. Mol. Biol. 421 (4-5), 441–465. doi:10.1016/j.jmb.2011.12.062
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3, 17071. doi:10.1038/nrdp.2017.71
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64 (1), 60–70. doi:10.1002/ana.21425
Henkel, J. S., Engelhardt, J. I., Siklos, L., Simpson, E. P., Kim, S. H., Pan, T., et al. (2004). Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 55 (2), 221–235. doi:10.1002/ana.10805
Hervas, R., Li, L., Majumdar, A., Fernandez-Ramirez Mdel, C., Unruh, J. R., Slaughter, B. D., et al. (2016). Molecular basis of Orb2 amyloidogenesis and blockade of memory consolidation. PLoS Biol. 14 (1), e1002361. doi:10.1371/journal.pbio.1002361
Hervas, R., Oroz, J., Galera-Prat, A., Goni, O., Valbuena, A., Vera, A. M., et al. (2012). Common features at the start of the neurodegeneration cascade. PLoS Biol. 10 (5), e1001335. doi:10.1371/journal.pbio.1001335
Herzog, G., Shmueli, M. D., Levy, L., Engel, L., Gazit, E., Klarner, F. G., et al. (2015). The lys-specific molecular tweezer, CLR01, modulates aggregation of the mutant p53 DNA binding domain and inhibits its toxicity. Biochemistry 54 (24), 3729–3738. doi:10.1021/bi501092p
Hnath, B., and Dokholyan, N. V. (2022). Toxic SOD1 trimers are off-pathway in the formation of amyloid-like fibrils in ALS. Biophysical J. 121 (11), 2084–2095. doi:10.1016/j.bpj.2022.04.037
Hornberg, A., Logan, D. T., Marklund, S. L., and Oliveberg, M. (2007). The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J. Mol. Biol. 365 (2), 333–342. doi:10.1016/j.jmb.2006.09.048
Hortobagyi, T., Troakes, C., Nishimura, A. L., Vance, C., van Swieten, J. C., Seelaar, H., et al. (2011). Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 121 (4), 519–527. doi:10.1007/s00401-011-0813-3
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15 (10), 565–581. doi:10.1038/s41582-019-0244-7
Hou, Z. S., Ulloa-Aguirre, A., and Tao, Y. X. (2018). Pharmacoperone drugs: targeting misfolded proteins causing lysosomal storage-ion channels-and G protein-coupled receptors-associated conformational disorders. Expert Rev. Clin. Pharmacol. 11 (6), 611–624. doi:10.1080/17512433.2018.1480367
Huai, J., and Zhang, Z. (2019). Structural properties and interaction partners of familial ALS-associated SOD1 mutants. Front. Neurol. 10, 527. doi:10.3389/fneur.2019.00527
Huang, H. J., Chang, T. T., Chen, H. Y., and Chen, C. Y. (2014). Finding inhibitors of mutant superoxide dismutase-1 for amyotrophic lateral sclerosis therapy from traditional Chinese medicine. Evidence-based complementary Altern. Med. eCAM. 2014, 156276. doi:10.1155/2014/156276
Huang, S. L., Wu, L. S., Lee, M., Chang, C. W., Cheng, W. C., Fang, Y. S., et al. (2020). A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 8 (1), 3. doi:10.1186/s40478-020-0881-5
Huang, Y. C., Lin, K. F., He, R. Y., Tu, P. H., Koubek, J., Hsu, Y. C., et al. (2013). Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One 8 (5), e64002. doi:10.1371/journal.pone.0064002
Hurshman, A. R., White, J. T., Powers, E. T., and Kelly, J. W. (2004). Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43 (23), 7365–7381. doi:10.1021/bi049621l
Iannitti, T., Scarrott, J. M., Likhite, S., Coldicott, I. R. P., Lewis, K. E., Heath, P. R., et al. (2018). Translating SOD1 gene silencing toward the clinic: a highly efficacious, off-target-free, and biomarker-supported strategy for fALS. Mol. Ther. Nucleic acids 12, 75–88. doi:10.1016/j.omtn.2018.04.015
Iguchi, Y., Katsuno, M., Niwa, J., Takagi, S., Ishigaki, S., Ikenaka, K., et al. (2013). Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136 (Pt 5), 1371–1382. doi:10.1093/brain/awt029
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187 (6), 761–772. doi:10.1083/jcb.200908164
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9 (391), eaaf3962. doi:10.1126/scitranslmed.aaf3962
Ip, P., Sharda, P. R., Cunningham, A., Chakrabartty, S., Pande, V., and Chakrabartty, A. (2017). Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 30, 431–440. doi:10.1093/protein/gzx025
Ivannikov, M. V., and Van Remmen, H. (2015). Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free Radic. Biol. Med. 84, 254–262. doi:10.1016/j.freeradbiomed.2015.03.021
Ivanova, M. I., Sievers, S. A., Guenther, E. L., Johnson, L. M., Winkler, D. D., Galaleldeen, A., et al. (2014). Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. PNAS 111, 197–201. doi:10.1073/pnas.1320786110
Iwakawa, N., Morimoto, D., Walinda, E., Kawata, Y., Shirakawa, M., and Sugase, K. (2017). Real-Time observation of the interaction between Thioflavin T and an amyloid protein by using high-sensitivity rheo-NMR. Int. J. Mol. Sci. 18 (11), 2271. doi:10.3390/ijms18112271
Izumikawa, K., Nobe, Y., Yoshikawa, H., Ishikawa, H., Miura, Y., Nakayama, H., et al. (2017). TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 7 (1), 7709. doi:10.1038/s41598-017-06953-y
Jami, M. S., Salehi-Najafabadi, Z., Ahmadinejad, F., Hoedt, E., Chaleshtori, M. H., Ghatrehsamani, M., et al. (2015). Edaravone leads to proteome changes indicative of neuronal cell protection in response to oxidative stress. Neurochem. Int. 90, 134–141. doi:10.1016/j.neuint.2015.07.024
Jankovic, M., Novakovic, I., Gamil, A. D. P., Gamil, A. D. A., Drinic, A., Abdel, M. F. I., et al. (2021). Current concepts on genetic aspects of mitochondrial dysfunction in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22 (18), 9832. doi:10.3390/ijms22189832
Jiang, L. L., Che, M. X., Zhao, J., Zhou, C. J., Xie, M. Y., Li, H. Y., et al. (2013). Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288 (27), 19614–19624. doi:10.1074/jbc.M113.463828
Jiang, L. L., Xue, W., Hong, J. Y., Zhang, J. T., Li, M. J., Yu, S. N., et al. (2017). The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 7 (1), 6196. doi:10.1038/s41598-017-06263-3
Jiang, L. L., Zhao, J., Yin, X. F., He, W. T., Yang, H., Che, M. X., et al. (2016). Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 6, 23928. doi:10.1038/srep23928
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284 (30), 20329–20339. doi:10.1074/jbc.M109.010264
Joshi, V., Poluri, K. M., Upadhyay, A., Amanullah, A., Singh, S., Kumar, A., et al. (2019). Polyphenolic flavonoid (Myricetin) upregulated proteasomal degradation mechanisms: eliminates neurodegenerative proteins aggregation. J. Cell. Physiology 234 (2), 20900–20914. doi:10.1002/jcp.28695
Kalmar, B., and Greensmith, L. (2017). Cellular chaperones as therapeutic targets in ALS to restore protein homeostasis and improve cellular function. Front. Mol. Neurosci. 10, 251. doi:10.3389/fnmol.2017.00251
Kametani, F., Obi, T., Shishido, T., Akatsu, H., Murayama, S., Saito, Y., et al. (2016). Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 6, 23281. doi:10.1038/srep23281
Kao, P. F., Chen, Y. R., Liu, X. B., DeCarli, C., Seeley, W. W., and Jin, L. W. (2015). Detection of TDP-43 oligomers in frontotemporal lobar degeneration-TDP. Ann. Neurol. 78 (2), 211–221. doi:10.1002/ana.24431
Kato, M., Han, T. W., Xie, S., Shi, K., Du, X., Wu, L. C., et al. (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149 (4), 753–767. doi:10.1016/j.cell.2012.04.017
Kato, M., and McKnight, S. L. (2017). Cross-β polymerization of low complexity sequence domains. Cold Spring Harb. Perspect. Biol. 9 (3), a023598. doi:10.1101/cshperspect.a023598
Kato, S., Takikawa, M., Nakashima, K., Hirano, A., Cleveland, D. W., Kusaka, H., et al. (2000). New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 1 (3), 163–184. doi:10.1080/14660820050515160
Ke, Y. D., van Hummel, A., Stevens, C. H., Gladbach, A., Ippati, S., Bi, M., et al. (2015). Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol. 130 (5), 661–678. doi:10.1007/s00401-015-1486-0
Keller, B. A., Volkening, K., Droppelmann, C. A., Ang, L. C., Rademakers, R., and Strong, M. J. (2012). Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. 124 (5), 733–747. doi:10.1007/s00401-012-1035-z
Kerman, A., Liu, H. N., Croul, S., Bilbao, J., Rogaeva, E., Zinman, L., et al. (2010). Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 119, 335–344. doi:10.1007/s00401-010-0646-5
Khare, S. D., Caplow, M., Dokholyan, N. V., and O, M. (2004). The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. PNAS 101, 15094–15099. doi:10.1073/pnas.0406650101
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020). ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842. doi:10.1016/j.neuron.2020.08.022
Kinger, S., Dubey, A. R., Kumar, P., Jagtap, Y. A., Choudhary, A., Kumar, A., et al. (2023). Molecular chaperones' potential against defective proteostasis of amyotrophic lateral sclerosis. Cells 12 (9), 1302. doi:10.3390/cells12091302
Kirola, L., Mukherjee, A., and Mutsuddi, M. (2022). Recent updates on the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Mol. Neurobiol. 59 (9), 5673–5694. doi:10.1007/s12035-022-02934-z
Knowles, T. P., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15 (6), 384–396. doi:10.1038/nrm3810
Koh, S. H., Lee, S. M., Kim, H. Y., Lee, K. Y., Lee, Y. J., Kim, H. T., et al. (2006). The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci. Lett. 395 (2), 103–107. doi:10.1016/j.neulet.2005.10.056
Konopka, A., Whelan, D. R., Jamali, M. S., Perri, E., Shahheydari, H., Toth, R. P., et al. (2020). Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 15 (1), 51. doi:10.1186/s13024-020-00386-4
Kraemer, B. C., Schuck, T., Wheeler, J. M., Robinson, L. C., Trojanowski, J. Q., Lee, V. M., et al. (2010). Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 119 (4), 409–419. doi:10.1007/s00401-010-0659-0
Kuo, P. H., Chiang, C. H., Wang, Y. T., Doudeva, L. G., and Yuan, H. S. (2014). The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 42 (7), 4712–4722. doi:10.1093/nar/gkt1407
Kushner, P. D., Stephenson, D. T., and Wright, S. (1991). Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. neuropathology Exp. neurology 50 (3), 263–277. doi:10.1097/00005072-199105000-00008
Kuzuhara, T., Suganuma, M., and Fujiki, H. (2008). Green tea catechin as a chemical chaperone in cancer prevention. Cancer Lett. 261 (1), 12–20. doi:10.1016/j.canlet.2007.10.037
Lacomblez, L., Bensimon, G., Leigh, P. N., Guillet, P., and Meininger, V. (1996a). Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 347 (9013), 1425–1431. doi:10.1016/s0140-6736(96)91680-3
Lacomblez, L., Bensimon, G., Leigh, P. N., Guillet, P., Powe, L., Durrleman, S., et al. (1996b). A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology 47 (6 Suppl. 4), S242–S250. doi:10.1212/wnl.47.6_suppl_4.242s
Lai, Z., Colon, W., and Kelly, J. W. (1996). The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35 (20), 6470–6482. doi:10.1021/bi952501g
Lang, L., Zetterstrom, P., Brannstrom, T., Marklund, S. L., Danielsson, J., and Oliveberg, M. (2015). SOD1 aggregation in ALS mice shows simplistic test tube behavior. Proc. Natl. Acad. Sci. U. S. A. 112 (32), 9878–9883. doi:10.1073/pnas.1503328112
Lapasset, L., Milhavet, O., Prieur, A., Besnard, E., Babled, A., Ait-Hamou, N., et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25 (21), 2248–2253. doi:10.1101/gad.173922.111
Larkin, L. M., Davis, C. S., Sims-Robinson, C., Kostrominova, T. Y., Van Remmen, H., Richardson, A., et al. (2011). Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301 (5), R1400–R1407. doi:10.1152/ajpregu.00093.2011
Lee, Y. B., Chen, H. J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 5 (5), 1178–1186. doi:10.1016/j.celrep.2013.10.049
Leidenheimer, N. J., and Ryder, K. G. (2014). Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol. Res. 83, 10–19. doi:10.1016/j.phrs.2014.01.005
Leigh, P. N., Anderton, B. H., Dodson, A., Gallo, J. M., Swash, M., and Power, D. M. (1988). Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 93 (2-3), 197–203. doi:10.1016/0304-3940(88)90081-x
Li, H. R., Chiang, W. C., Chou, P. C., Wang, W. J., and Huang, J. R. (2018). TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293 (16), 6090–6098. doi:10.1074/jbc.AC117.001037
Li, Q., Babinchak, W. M., and Surewicz, W. K. (2021). Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nat. Commun. 12 (1), 1620. doi:10.1038/s41467-021-21912-y
Liguori, L., Monticelli, M., Allocca, M., Hay Mele, B., Lukas, J., Cubellis, M. V., et al. (2020). Pharmacological chaperones: a therapeutic approach for diseases caused by destabilizing missense mutations. Int. J. Mol. Sci. 21 (2), 489. doi:10.3390/ijms21020489
Limpert, A. S., Mattmann, M. E., and Cosford, N. D. P. (2013). Recent progress in the discovery of small molecules for the treatment of amyotrophic lateral sclerosis (ALS). Beilstein J. Org. Chem. 9, 717–732. doi:10.3762/bjoc.9.82
Lin, L., Ni, B., Lin, H., Zhang, M., Li, X., Yin, X., et al. (2015b). Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum Thunb.: a review. J. Ethnopharmacol. 159, 158–183. doi:10.1016/j.jep.2014.11.009
Lin, W. L., and Dickson, D. W. (2008). Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 116 (2), 205–213. doi:10.1007/s00401-008-0408-9
Lin, Y., Protter, D. S., Rosen, M. K., and Parker, R. (2015a). Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60 (2), 208–219. doi:10.1016/j.molcel.2015.08.018
Liu, J., Gao, L., and Zang, D. (2015). Elevated levels of IFN-gamma in CSF and serum of patients with amyotrophic lateral sclerosis. PLoS One 10 (9), e0136937. doi:10.1371/journal.pone.0136937
Liu, R., Yang, G., Nonaka, T., Arai, T., Jia, W., and Cynader, M. S. (2013). Reducing TDP-43 aggregation does not prevent its cytotoxicity. Acta Neuropathol. Commun. 1, 49. doi:10.1186/2051-5960-1-49
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5 (10), e13250. doi:10.1371/journal.pone.0013250
Lo Cascio, F., Marzullo, P., Kayed, R., and Palumbo Piccionello, A. (2021). Curcumin as scaffold for drug discovery against neurodegenerative diseases. Biomedicines 9 (2), 173. doi:10.3390/biomedicines9020173
Longinetti, E., and Fang, F. (2019). Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr. Opin. Neurol. 32 (5), 771–776. doi:10.1097/WCO.0000000000000730
Lopes, D. H., Attar, A., Nair, G., Hayden, E. Y., Du, Z., McDaniel, K., et al. (2015). Molecular tweezers inhibit islet amyloid polypeptide assembly and toxicity by a new mechanism. ACS Chem. Biol. 10 (6), 1555–1569. doi:10.1021/acschembio.5b00146
Lowe, J., Lennox, G., Jefferson, D., Morrell, K., McQuire, D., Gray, T., et al. (1988). A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci. Lett. 94 (1-2), 203–210. doi:10.1016/0304-3940(88)90296-0
Lu, C. H., Allen, K., Oei, F., Leoni, E., Kuhle, J., Tree, T., et al. (2016). Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurology(R) Neuroimmunol. neuroinflammation 3 (4), e244. doi:10.1212/NXI.0000000000000244
Lukavsky, P. J., Daujotyte, D., Tollervey, J. R., Ule, J., Stuani, C., Buratti, E., et al. (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20 (12), 1443–1449. doi:10.1038/nsmb.2698
Macarron, R., and Hertzberg, R. P. (2009). Design and implementation of high-throughput screening assays. Methods Mol. Biol. 565, 1–29. doi:10.1385/1-59259-180-9:001
Mackenzie, I. R., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61 (5), 427–434. doi:10.1002/ana.21147
Madeira, J. M., Bajwa, E., Stuart, M. J., Hashioka, S., and Klegeris, A. (2014). Gold drug auranofin could reduce neuroinflammation by inhibiting microglia cytotoxic secretions and primed respiratory burst. J. Neuroimmunol. 276 (1-2), 71–79. doi:10.1016/j.jneuroim.2014.08.615
Madeira, J. M., Renschler, C. J., Mueller, B., Hashioka, S., Gibson, D. L., and Klegeris, A. (2013). Novel protective properties of auranofin: inhibition of human astrocyte cytotoxic secretions and direct neuroprotection. Life Sci. 92 (22), 1072–1080. doi:10.1016/j.lfs.2013.04.005
Madeira, J. M., Schindler, S. M., and Klegeris, A. (2015). A new look at auranofin, dextromethorphan and rosiglitazone for reduction of glia-mediated inflammation in neurodegenerative diseases. Neural Regen. Res. 10 (3), 391–393. doi:10.4103/1673-5374.153686
Maher, P. (2019). The potential of flavonoids for the treatment of neurodegenerative diseases. Int. J. Mol. Sci. 20, 3056. doi:10.3390/ijms20123056
Maier, M., Welt, T., Wirth, F., Montrasio, F., Preisig, D., McAfoose, J., et al. (2018). A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci. Transl. Med. 10 (470), eaah3924. doi:10.1126/scitranslmed.aah3924
Malik, R., Corrales, C., Sepanj, N., and Bitan, G. (2020). Examination of SOD1 aggregation modulators and their effect on SOD1 enzymatic activity as a proxy for potential toxicity. FASEB J. 34 (9), 1–12. doi:10.1096/fj.202000948R
Malik, R., Meng, H., Wongkongkathep, P., Corrales, C. I., Sepanj, N., Atlasi, R. S., et al. (2019). The molecular tweezer CLR01 inhibits aberrant superoxide dismutase 1 (SOD1) self-assembly in vitro and in the G93A-SOD1 mouse model of ALS. J. Biol. Chem. 294, 3501–3513. doi:10.1074/jbc.RA118.005940
Malishev, R., Salinas, N., Gibson, J., Eden, A. B., Mieres-Perez, J., Ruiz-Blanco, Y. B., et al. (2021). Inhibition of Staphylococcus aureus biofilm-forming functional amyloid by molecular tweezers. Cell Chem. Biol. 28 (9), 1310–1320.e5. doi:10.1016/j.chembiol.2021.03.013
Manjula, R., Unni, S., Wright, G. S. A., Bharath, M. M. S., and Padmanabhan, B. (2019). Rational discovery of a SOD1 tryptophan oxidation inhibitor with therapeutic potential for amyotrophic lateral sclerosis. J. Biomol. Struct. Dyn. 37 (15), 3936–3946. doi:10.1080/07391102.2018.1531787
March, Z. M., King, O. D., and Shorter, J. (2016). Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 1647, 9–18. doi:10.1016/j.brainres.2016.02.037
Matis, I., Delivoria, D. C., Mavroidi, B., Papaevgeniou, N., Panoutsou, S., Bellou, S., et al. (2017). An integrated bacterial system for the discovery of chemical rescuers of disease-associated protein misfolding. Nat. Biomed. Eng. 1 (10), 838–852. doi:10.1038/s41551-017-0144-3
Maurer, M. S., Schwartz, J. H., Gundapaneni, B., Elliott, P. M., Merlini, G., Waddington-Cruz, M., et al. (2018). Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379 (11), 1007–1016. doi:10.1056/NEJMoa1805689
Mbarek, A., Moussa, G., and Chain, J. L. (2019). Pharmaceutical applications of molecular tweezers, clefts and clips. Molecules 24 (9), 1803. doi:10.3390/molecules24091803
McAlary, L., Chew, Y. L., Lum, J. S., Geraghty, N. J., Yerbury, J. J., and Cashman, N. R. (2020). Amyotrophic lateral sclerosis: proteins, proteostasis, prions, and promises. Front. Cell. Neurosci. 14, 581907. doi:10.3389/fncel.2020.581907
McAlary, L., and Yerbury, J. J. (2019). Strategies to promote the maturation of ALS-associated SOD1 mutants: small molecules return to the fold. Neural Regen Res 9, 1511–1512. doi:10.4103/1673-5374.255962
McCampbell, A., Cole, T., Wegener, A. J., Tomassy, G. S., Setnicka, A., Farley, B. J., et al. (2018). Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. investigation 128 (8), 3558–3567. doi:10.1172/JCI99081
McGown, A., and Stopford, M. J. (2018). High-throughput drug screens for amyotrophic lateral sclerosis drug discovery. Expert Opin. Drug Discov. 13 (11), 1015–1025. doi:10.1080/17460441.2018.1533953
Mead, R. J., Shan, N., Reiser, H. J., Marshall, F., and Shaw, P. J. (2023). Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 22 (3), 185–212. doi:10.1038/s41573-022-00612-2
Medinas, D. B., Gozzo, F. C., Santos, L. F., Iglesias, A. H., and Augusto, O. (2010). A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic. Biol. Med. 49 (6), 1046–1053. doi:10.1016/j.freeradbiomed.2010.06.018
Medinas, D. B., Rozas, P., Martínez, F., Woehlbier, U., Brown, R. H., Bosco, D. A., et al. (2018). Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. PNAS 115, 8209–8214. doi:10.1073/pnas.1801109115
Medinas, D. B., Valenzuela, V., and Hetz, C. (2017). Proteostasis disturbance in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26 (R2), R91–R104. doi:10.1093/hmg/ddx274
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now?. Front. Neurosci. 13, 1–27. doi:10.3389/fnins.2019.01310
Mertens, J., Marchetto, M. C., Bardy, C., and Gage, F. H. (2016). Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17 (7), 424–437. doi:10.1038/nrn.2016.46
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U. S. A. 111 (2), 829–832. doi:10.1073/pnas.1314085111
Miller, T. M., Cudkowicz, M. E., Genge, A., Shaw, P. J., Sobue, G., Bucelli, R. C., et al. (2022). Trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 387 (12), 1099–1110. doi:10.1056/NEJMoa2204705
Mitsuzawa, S., Akiyama, T., Nishiyama, A., Suzuki, N., Kato, M., Warita, H., et al. (2018). TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci 11, 20–22. doi:10.1016/j.ensci.2018.04.001
Mittal, S., Bravo-Rodriguez, K., and Sanchez-Garcia, E. (2018). Mechanism of inhibition of beta amyloid toxicity by supramolecular tweezers. J. Phys. Chem. B 122 (15), 4196–4205. doi:10.1021/acs.jpcb.7b10530
Mollasalehi, N., Francois-Moutal, L., Scott, D. D., Tello, J. A., Williams, H., Mahoney, B., et al. (2020). An allosteric modulator of RNA binding targeting the N-terminal domain of TDP-43 yields neuroprotective properties. ACS Chem. Biol. 15 (11), 2854–2859. doi:10.1021/acschembio.0c00494
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163 (1), 123–133. doi:10.1016/j.cell.2015.09.015
Molnar, K. S., Karabacak, N. M., Johnson, J. L., Wang, Q., Tiwari, A., Hayward, L. J., et al. (2009). A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the copper/zinc superoxide dismutase electrostatic loop. J. Biol. Chem. 284 (45), 30965–30973. doi:10.1074/jbc.M109.023945
Mompean, M., Ramirez de Mingo, D., Hervas, R., Fernandez-Ramirez, M. D. C., Carrion-Vazquez, M., and Laurents, D. V. (2019). Molecular mechanism of the inhibition of TDP-43 amyloidogenesis by QBP1. Arch. Biochem. Biophys. 675, 108113. doi:10.1016/j.abb.2019.108113
Mompean, M., Romano, V., Pantoja-Uceda, D., Stuani, C., Baralle, F. E., Buratti, E., et al. (2016). The TDP-43 N-terminal domain structure at high resolution. FEBS J. 283 (7), 1242–1260. doi:10.1111/febs.13651
Mompean, M., Romano, V., Pantoja-Uceda, D., Stuani, C., Baralle, F. E., Buratti, E., et al. (2017). Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem. 292 (28), 11992–12006. doi:10.1074/jbc.M117.775965
Montibeller, L., Tan, L. Y., Kim, J. K., Paul, P., and de Belleroche, J. (2020). Tissue-selective regulation of protein homeostasis and unfolded protein response signalling in sporadic ALS. J. Cell. Mol. Med. 24 (11), 6055–6069. doi:10.1111/jcmm.15170
Moreau, C., Devos, D., Brunaud-Danel, V., Defebvre, L., Perez, T., Destee, A., et al. (2005). Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology 65 (12), 1958–1960. doi:10.1212/01.wnl.0000188907.97339.76
Moreau, K. L., and King, J. A. (2012). Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol. Med. 18 (5), 273–282. doi:10.1016/j.molmed.2012.03.005
Mueller, C., Berry, J. D., McKenna-Yasek, D. M., Gernoux, G., Owegi, M. A., Pothier, L. M., et al. (2020). SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. N. Engl. J. Med. 383 (2), 151–158. doi:10.1056/NEJMoa2005056
Mukundan, V., Maksoudian, C., Vogel, M. C., Chehade, I., Katsiotis, M. S., Alhassan, S. M., et al. (2017). Cytotoxicity of prion protein-derived cell-penetrating peptides is modulated by pH but independent of amyloid formation. Arch. Biochem. Biophys. 613, 31–42. doi:10.1016/j.abb.2016.11.001
Murakami, G., Inoue, H., Tsukita, K., Asai, Y., Amagai, Y., Aiba, K., et al. (2011). Chemical library screening identifies a small molecule that downregulates SOD1 transcription for drugs to treat amyotrophic lateral sclerosis. J. Biomol. Screen. 16, 405–414. doi:10.1177/1087057110397888
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88 (4), 678–690. doi:10.1016/j.neuron.2015.10.030
Musteikyte, G., Ziaunys, M., and Smirnovas, V. (2020). Methylene blue inhibits nucleation and elongation of SOD1 amyloid fibrils. PeerJ 8, e9719. doi:10.7717/peerj.9719
Mutihac, R., Alegre-Abarrategui, J., Gordon, D., Farrimond, L., Yamasaki-Mann, M., Talbot, K., et al. (2015). TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca²⁺ signaling in motor neurons. Neurobiol. Dis. 75, 64–77. doi:10.1016/j.nbd.2014.12.010
Nagai, Y., Fujikake, N., Ohno, K., Higashiyama, H., Popiel, H. A., Rahadian, J., et al. (2003). Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum. Mol. Genet. 12 (11), 1253–1259. doi:10.1093/hmg/ddg144
Nagai, Y., Tucker, T., Ren, H., Kenan, D. J., Henderson, B. S., Keene, J. D., et al. (2000). Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J. Biol. Chem. 275 (14), 10437–10442. doi:10.1074/jbc.275.14.10437
Nagy, D., Kato, T., and Kushner, P. D. (1994). Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 38 (3), 336–347. doi:10.1002/jnr.490380312
Nday, C. M., Halevas, E., Jackson, G. E., and Salifoglou, A. (2015). Quercetin encapsulation in modified silica nanoparticles: potential use against Cu(II)-induced oxidative stress in neurodegeneration. J. Inorg. Biochem. 145, 51–64. doi:10.1016/j.jinorgbio.2015.01.001
Neumann, M., Roeber, S., Kretzschmar, H. A., Rademakers, R., Baker, M., and Mackenzie, I. R. (2009). Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 118 (5), 605–616. doi:10.1007/s00401-009-0581-5
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 (5796), 130–133. doi:10.1126/science.1134108
Nikitin, D., Makam, A. N., Suh, K., McKenna, A., Carlson, J. J., Richardson, M., et al. (2023). The effectiveness and value of AMX0035 and oral edaravone for amyotrophic lateral sclerosis: a summary from the Institute for clinical and economic review's midwest comparative effectiveness public advisory Council. J. Manag. care & specialty Pharm. 29 (2), 216–221. doi:10.18553/jmcp.2023.29.2.216
Nordlund, A., Leinartaite, L., Saraboji, K., Aisenbrey, C., Grobner, G., Zetterstrom, P., et al. (2009). Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc. Natl. Acad. Sci. U. S. A. 106 (24), 9667–9672. doi:10.1073/pnas.0812046106
Nowak, R. J., Cuny, G. D., Choi, S., Lansbury, P. T., and Ray, S. S. (2011). Improving binding specificity of pharmacological chaperones that target mutant superoxide dismutase-1 linked to familial amyotrophic lateral sclerosis using computational methods. J. Med. Chem. 53, 2709–2718. doi:10.1021/jm901062p
Oberstadt, M., Stieler, J., Simpong, D. L., Romuss, U., Urban, N., Schaefer, M., et al. (2018). TDP-43 self-interaction is modulated by redox-active compounds Auranofin, Chelerythrine and Riluzole. Sci. Rep. 8 (1), 2248. doi:10.1038/s41598-018-20565-0
Park, J. H., Elpers, C., Reunert, J., McCormick, M. L., Mohr, J., Biskup, S., et al. (2019). SOD1 deficiency: a novel syndrome distinct from amyotrophic lateral sclerosis. Brain 142, 2230–2237. doi:10.1093/brain/awz182
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162 (5), 1066–1077. doi:10.1016/j.cell.2015.07.047
Pattee, G. L., Genge, A., Couratier, P., Lunetta, C., Sobue, G., Aoki, M., et al. (2023). Oral edaravone - introducing a flexible treatment option for amyotrophic lateral sclerosis. Expert Rev. Neurother. 23 (10), 859–866. doi:10.1080/14737175.2023.2251687
Patterson, M., Chan, D. N., Ha, I., Case, D., Cui, Y., Van Handel, B., et al. (2012). Defining the nature of human pluripotent stem cell progeny. Cell Res. 22 (1), 178–193. doi:10.1038/cr.2011.133
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS clinical trials review: 20 Years of failure. Are We Any Closer Registering a New Treat. ? 9, 1–11. doi:10.3389/fnagi.2017.00068
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein science a publication of the Protein Society 30 (1), 70–82. doi:10.1002/pro.3943
Pickles, S., Destroismaisons, L., Peyrard, S. L., Cadot, S., Rouleau, G. A., Brown, R. H., et al. (2013). Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Human molecular genetics 22 (19), 3947–3959. doi:10.1093/hmg/ddt249
Pinarbasi, E. S., Cagatay, T., Fung, H. Y. J., Li, Y. C., Chook, Y. M., and Thomas, P. J. (2018). Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep 8 (1), 7083. doi:10.1038/s41598-018-25008-4
Pokrishevsky, E., Mcalary, L., Farrawell, N. E., Zhao, B., Sher, M., Yerbury, J. J., et al. (2018). Tryptophan 32-mediated SOD1 aggregation is attenuated by pyrimidine-like compounds in living cells. Scientific Reports 8, 15590. doi:10.1038/s41598-018-32835-y
Polticelli, F., Bottaro, G., Battistoni, A., Carri, M. T., Djinovic-Carugo, K., Bolognesi, M., et al. (1995). Modulation of the catalytic rate of Cu,Zn superoxide dismutase in single and double mutants of conserved positively and negatively charged residues. Biochemistry 34 (18), 6043–6049. doi:10.1021/bi00018a006
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14 (4), 459–468. doi:10.1038/nn.2779
Popiel, H. A., Takeuchi, T., Burke, J. R., Strittmatter, W. J., Toda, T., Wada, K., et al. (2013). Inhibition of protein misfolding/aggregation using polyglutamine binding peptide QBP1 as a therapy for the polyglutamine diseases. Neurotherapeutics 10 (3), 440–446. doi:10.1007/s13311-013-0184-7
Pozzi, S., Thammisetty, S. S., Codron, P., Rahimian, R., Plourde, K. V., Soucy, G., et al. (2019). Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. The Journal of clinical investigation 129 (4), 1581–1595. doi:10.1172/JCI123931
Prakash, D., and Sudhandiran, G. (2015). Dietary flavonoid fisetin regulates aluminium chloride-induced neuronal apoptosis in cortex and hippocampus of mice brain. J Nutr Biochem. 26 (12), 1527–1539. doi:10.1016/j.jnutbio.2015.07.017
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12, 25. doi:10.3389/fnmol.2019.00025
Prasad, A., Raju, G., Sivalingam, V., Girdhar, A., Verma, M., Vats, A., et al. (2016). An acridine derivative, [4,5-bis{(N-carboxy methyl imidazolium)methyl}acridine] dibromide, shows anti-TDP-43 aggregation effect in ALS disease models. Sci Rep 6, 39490. doi:10.1038/srep39490
Pratt, A. J., Shin, D. S., Merz, G. E., Rambo, R. P., Lancaster, W. A., Dyer, K. N., et al. (2014). Aggregation propensities of superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes. Proceedings of the National Academy of Sciences of the U. S. A. 111 (43), E4568–E4576. doi:10.1073/pnas.1308531111
Proctor, E. A., Fee, L., Tao, Y., Redler, R. L., Fay, J. M., Zhang, Y., et al. (2016). Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the U. S. A. 113 (3), 614–619. doi:10.1073/pnas.1516725113
Proctor, E. A., Mowrey, D. D., and Dokholyan, N. V. (2019). β-Methylamino-L-alanine substitution of serine in SOD1 suggests a direct role in ALS etiology. PLoS computational biology 15 (7), e1007225. doi:10.1371/journal.pcbi.1007225
Prudencio, M., Hart, P. J., Borchelt, D. R., and Andersen, P. M. (2009). Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Human molecular genetics 18 (17), 3217–3226. doi:10.1093/hmg/ddp260
Qin, H., Lim, L. Z., Wei, Y., and Song, J. (2014). TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proceedings of the National Academy of Sciences of the U. S. A. 111 (52), 18619–18624. doi:10.1073/pnas.1413994112
Rakhit, R., Cunningham, P., Furtos-Matei, A., Dahan, S., Qi, X. F., Crow, J. P., et al. (2002). Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 277 (49), 47551–47556. doi:10.1074/jbc.M207356200
Ramesh, N., and Pandey, U. B. (2017). Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci 10, 263. doi:10.3389/fnmol.2017.00263
Rando, A., de la Torre, M., Martinez-Muriana, A., Zaragoza, P., Musaro, A., Hernandez, S., et al. (2019). Chemotherapeutic agent 5-fluorouracil increases survival of SOD1 mouse model of ALS. PLoS One 14 (1), e0210752. doi:10.1371/journal.pone.0210752
Ranganathan, R., Haque, S., Coley, K., Shepheard, S., Cooper-Knock, J., and Kirby, J. (2020). Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Frontiers in neuroscience 14, 684. doi:10.3389/fnins.2020.00684
Ray, S. S., Nowak, R. J., Brown, R. H., and Lansbury, P. T. (2005). Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. PNAS 102, 3639–3644. doi:10.1073/pnas.0408277102
Ray, S. S., Nowak, R. J., Strokovich, K., Brown, R. H., Walz, T., and Lansbury, P. T. (2004). An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry 43 (17), 4899–4905. doi:10.1021/bi030246r
Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., et al. (1996). Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13 (1), 43–47. doi:10.1038/ng0596-43
Reischauer, C., Gutzeit, A., Neuwirth, C., Fuchs, A., Sartoretti-Schefer, S., Weber, M., et al. (2018). In-vivo evaluation of neuronal and glial changes in amyotrophic lateral sclerosis with diffusion tensor spectroscopy. Neuroimage Clin. 20, 993–1000.
Rengifo-Gonzalez, J. C., El Hage, K., Clement, M. J., Steiner, E., Joshi, V., Craveur, P., et al. (2021). The cooperative binding of TDP-43 to GU-rich RNA repeats antagonizes TDP-43 aggregation. Elife 10, e67605. doi:10.7554/eLife.67605
Riancho, J., Castanedo-Vazquez, D., Gil-Bea, F., Tapia, O., Arozamena, J., Duran-Vian, C., et al. (2020). ALS-derived fibroblasts exhibit reduced proliferation rate, cytoplasmic TDP-43 aggregation and a higher susceptibility to DNA damage. J Neurol 267 (5), 1291–1299. doi:10.1007/s00415-020-09704-8
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 14 (1), 9–21. doi:10.1038/nrneurol.2017.148
Rodrigues, C. M., Sola, S., Sharpe, J. C., Moura, J. J., and Steer, C. J. (2003). Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry 42 (10), 3070–3080. doi:10.1021/bi026979d
Rojas, J. C., Bruchey, A. K., and Gonzalez-Lima, F. (2012). Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog Neurobiol 96 (1), 32–45. doi:10.1016/j.pneurobio.2011.10.007
Rosen, D. R., Siddiquet, T., Pattersont, D., Figlewicz, D. A., Ii, P. S., Hentatit, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi:10.1038/362059a0
Ryan, M., Heverin, M., McLaughlin, R. L., and Hardiman, O. (2019). Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA neurology 76 (11), 1367–1374. doi:10.1001/jamaneurol.2019.2044
Ryan, P., Patel, B., Makwana, V., Jadhav, H. R., Kiefel, M., Davey, A., et al. (2018). Peptides, peptidomimetics, and carbohydrate-peptide conjugates as amyloidogenic aggregation inhibitors for Alzheimer's disease. ACS Chem Neurosci 9 (7), 1530–1551. doi:10.1021/acschemneuro.8b00185
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136 (Pt 8), 2342–2358. doi:10.1093/brain/awt097
Saini, A., and Chauhan, V. S. (2011). Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. Chembiochem a European journal of chemical biology 12 (16), 2495–2501. doi:10.1002/cbic.201100427
Saito, I., Asano, T., Sano, K., Takakura, K., Abe, H., Yoshimoto, T., et al. (1998). Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery 42 (2), 269–277. doi:10.1097/00006123-199802000-00038
Samanta, N., Ruiz-Blanco, Y. B., Fetahaj, Z., Gnutt, D., Lantz, C., Loo, J. A., et al. (2022). Superoxide dismutase 1 folding stability as a target for molecular tweezers in SOD1-related amyotrophic lateral sclerosis. Chembiochem a European journal of chemical biology 23 (21), e202200396. doi:10.1002/cbic.202200396
Sangwan, S., Zhao, A., Adams, K. L., Jayson, C. K., Sawaya, M. R., Guenther, E. L., et al. (2017). Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proceedings of the National Academy of Sciences of the U. S. A. 114 (33), 8770–8775. doi:10.1073/pnas.1705091114
Scherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R., Hasenbank, R., et al. (1999). Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proceedings of the National Academy of Sciences of the U. S. A. 96 (8), 4604–4609. doi:10.1073/pnas.96.8.4604
Schewe, T. (1995). Molecular actions of ebselen--an antiinflammatory antioxidant. Gen Pharmacol 26 (6), 1153–1169. doi:10.1016/0306-3623(95)00003-j
Schiffer, D., Cordera, S., Cavalla, P., and Migheli, A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139 (Suppl. l), 27–33. doi:10.1016/0022-510x(96)00073-1
Sekhar, A., Rumfeldt, J. A., Broom, H. R., Doyle, C. M., Bouvignies, G., Meiering, E. M., et al. (2015). Thermal fluctuations of immature SOD1 lead to separate folding and misfolding pathways. Elife 4, e07296. doi:10.7554/eLife.07296
Seminary, E. R., Sison, S. L., and Ebert, A. D. (2018). Modeling protein aggregation and the heat shock response in ALS iPSC-derived motor neurons. Frontiers in neuroscience 12, 86. doi:10.3389/fnins.2018.00086
Shi, Y., Ivannikov, M. V., Walsh, M. E., Liu, Y., Zhang, Y., Jaramillo, C. A., et al. (2014). The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS One 9 (6), e100834. doi:10.1371/journal.pone.0100834
Shin, Y., and Brangwynne, C. P. (2017). Liquid phase condensation in cell physiology and disease. Science. 357 (6357), eaaf4382. doi:10.1126/science.aaf4382
Shvil, N., Banerjee, V., Zoltsman, G., Shani, T., Kahn, J., Abu-hamad, S., et al. (2018). MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: implications for familial ALS. Cell Death and Disease 9, 107. doi:10.1038/s41419-017-0130-4
Sievers, S. A., Karanicolas, J., Chang, H. W., Zhao, A., Jiang, L., Zirafi, O., et al. (2011). Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475 (7354), 96–100. doi:10.1038/nature10154
Sinha, S., Lopes, D. H., Du, Z., Pang, E. S., Shanmugam, A., Lomakin, A., et al. (2011). Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc 133 (42), 16958–16969. doi:10.1021/ja206279b
Smethurst, P., Risse, E., Tyzack, G. E., Mitchell, J. S., Taha, D. M., Chen, Y. R., et al. (2020). Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 143 (2), 430–440. doi:10.1093/brain/awz419
Soejima-Kusunoki, A., Okada, K., Saito, R., and Watabe, K. (2022). The protective effect of edaravone on TDP-43 Plus oxidative stress-induced neurotoxicity in neuronal cells: analysis of its neuroprotective mechanisms using RNA sequencing. Pharmaceuticals 15 (7), 842. doi:10.3390/ph15070842
Souza, P. C. T., Thallmair, S., Marrink, S. J., and Mera-Adasme, R. (2019). An allosteric pathway in copper, zinc superoxide dismutase unravels the molecular mechanism of the G93A amyotrophic lateral sclerosis-linked mutation. J Phys Chem Lett 10 (24), 7740–7744. doi:10.1021/acs.jpclett.9b02868
Srinivasan, E., and Rajasekaran, R. (2018). Comparative binding of kaempferol and kaempferide on inhibiting the aggregate formation of mutant (G85R) SOD1 protein in familial amyotrophic lateral sclerosis: a quantum chemical and molecular mechanics study. BioFactors 44, 431–442. doi:10.1002/biof.1441
Srinivasan, E., and Rajasekaran, R. (2019). Rational design of linear tripeptides against the aggregation of human mutant SOD1 protein causing amyotrophic lateral sclerosis. J Neurol Sci 405, 116425. doi:10.1016/j.jns.2019.116425
Statland, J. M., Barohn, R. J., McVey, A. L., Katz, J. S., and Dimachkie, M. M. (2015). Patterns of weakness, classification of motor neuron disease, and clinical diagnosis of sporadic amyotrophic lateral sclerosis. Neurol Clin 33 (4), 735–748. doi:10.1016/j.ncl.2015.07.006
Street, T. O., Bolen, D. W., and Rose, G. D. (2006). A molecular mechanism for osmolyte-induced protein stability. Proceedings of the National Academy of Sciences of the U. S. A. 103 (38), 13997–14002. doi:10.1073/pnas.0606236103
Su, X. W., Nandar, W., Neely, E. B., Simmons, Z., and Connor, J. R. (2016). Statins accelerate disease progression and shorten survival in SOD1(G93A) mice. Muscle & nerve 54 (2), 284–291. doi:10.1002/mus.25048
Suaud, L., Miller, K., Panichelli, A. E., Randell, R. L., Marando, C. M., and Rubenstein, R. C. (2011). 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J Biol Chem 286 (52), 45083–45092. doi:10.1074/jbc.M111.293282
Sun, Y., Arslan, P. E., Won, A., Yip, C. M., and Chakrabartty, A. (2014). Binding of TDP-43 to the 3'UTR of its cognate mRNA enhances its solubility. Biochemistry 53 (37), 5885–5894. doi:10.1021/bi500617x
Suzuki, N., Nishiyama, A., Warita, H., and Aoki, M. (2023). Genetics of amyotrophic lateral sclerosis: seeking therapeutic targets in the era of gene therapy. Journal of human genetics 68 (3), 131–152. doi:10.1038/s10038-022-01055-8
Szabo, I., Yousef, M., Soltesz, D., Bato, C., Mezo, G., and Banoczi, Z. (2022). Redesigning of cell-penetrating peptides to improve their efficacy as a drug delivery system. Pharmaceutics 14 (5), 907. doi:10.3390/pharmaceutics14050907
Tada, M., Doi, H., Koyano, S., Kubota, S., Fukai, R., Hashiguchi, S., et al. (2018). Matrin 3 is a component of neuronal cytoplasmic inclusions of motor neurons in sporadic amyotrophic lateral sclerosis. Am J Pathol 188 (2), 507–514. doi:10.1016/j.ajpath.2017.10.007
Tak, Y. J., Park, J. H., Rhim, H., and Kang, S. (2020). ALS-related mutant SOD1 aggregates interfere with mitophagy by sequestering the autophagy receptor Optineurin. Int J Mol Sci 21 (20), 7525. doi:10.3390/ijms21207525
Takeuchi, T., and Nagai, Y. (2017). Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci 7 (10), 128. doi:10.3390/brainsci7100128
Tandan, R., and Bradley, W. G. (1985). Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann Neurol 18 (3), 271–280. doi:10.1002/ana.410180302
Tang, Y., Liu, M. L., Zang, T., and Zhang, C. L. (2017). Direct reprogramming rather than iPSC-based reprogramming maintains aging hallmarks in human motor neurons. Front Mol Neurosci 10, 359. doi:10.3389/fnmol.2017.00359
Taylor, D. M., Gibbs, B. F., Kabashi, E., Minotti, S., Durham, H. D., and Agar, J. N. (2007). Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J Biol Chem 282 (22), 16329–16335. doi:10.1074/jbc.M610119200
Teplow, D. B. (1998). Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid 5 (2), 121–142. doi:10.3109/13506129808995290
Thams, S., Lowry, E. R., Larraufie, M. H., Spiller, K. J., Li, H., Williams, D. J., et al. (2019). A stem cell-based screening platform identifies compounds that desensitize motor neurons to endoplasmic reticulum stress. Molecular therapy the journal of the American Society of Gene Therapy 27 (1), 87–101. doi:10.1016/j.ymthe.2018.10.010
Tiwari, A., Xu, Z., and Hayward, L. J. (2005). Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem 280 (33), 29771–29779. doi:10.1074/jbc.M504039200
Toichi, K., Yamanaka, K., and Furukawa, Y. (2013). Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J Biol Chem 288, 4970–4980. doi:10.1074/jbc.M112.414235
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14 (4), 452–458. doi:10.1038/nn.2778
Tran, M. L., Genisson, Y., Ballereau, S., and Dehoux, C. (2020). Second-generation pharmacological chaperones: beyond inhibitors. Molecules 25 (14), 3145. doi:10.3390/molecules25143145
Tripathi, V. B., Baskaran, P., Shaw, C. E., and Guthrie, S. (2014). Tar DNA-binding protein-43 (TDP-43) regulates axon growth in vitro and in vivo. Neurobiol Dis 65, 25–34. doi:10.1016/j.nbd.2014.01.004
Trist, B. G., Genoud, S., Roudeau, S., Rookyard, A., Abdeen, A., Cottam, V., et al. (2022). Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain 145 (9), 3108–3130. doi:10.1093/brain/awac165
Truex, N. L., and Nowick, J. S. (2016). Coassembly of peptides derived from β-sheet regions of β-amyloid. J Am Chem Soc 138 (42), 13891–13900. doi:10.1021/jacs.6b06001
Tseng, Y. L., Lu, P. C., Lee, C. C., He, R. Y., Huang, Y. A., Tseng, Y. C., et al. (2023). Degradation of neurodegenerative disease-associated TDP-43 aggregates and oligomers via a proteolysis-targeting chimera. Journal of biomedical science 30 (1), 27. doi:10.1186/s12929-023-00921-7
Tsuzuki, N., Hama, T., Kawada, M., Hasui, A., Konishi, R., Shiwa, S., et al. (1994). Adamantane as a brain-directed drug carrier for poorly absorbed drug. 2. AZT derivatives conjugated with the 1-adamantane moiety. Journal of pharmaceutical sciences 83 (4), 481–484. doi:10.1002/jps.2600830407
Turner, M. R., Cagnin, A., Turkheimer, F. E., Miller, C. C., Shaw, C. E., Brooks, D. J., et al. (2004). Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15 (3), 601–609. doi:10.1016/j.nbd.2003.12.012
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol Dis 55, 64–75. doi:10.1016/j.nbd.2013.03.015
Vaccaro, A., Patten, S. A., Ciura, S., Maios, C., Therrien, M., Drapeau, P., et al. (2012). Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One 7 (7), e42117. doi:10.1371/journal.pone.0042117
Valentine, J. S., Doucette, P. A., and Potter, S. Z. (2005). Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev Biochem. 74, 563–593. doi:10.1146/annurev.biochem.72.121801.161647
Valle, C., and Carri, M. T. (2017). Cysteine modifications in the pathogenesis of ALS. Front Mol Neurosci 10, 5. doi:10.3389/fnmol.2017.00005
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 (5918), 1208–1211. doi:10.1126/science.1165942
van der Spek, R. A. A., van Rheenen, W., Pulit, S. L., Kenna, K. P., van den Berg, L. H., Veldink, J. H., et al. (2019). The project MinE databrowser: bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph Lateral Scler Frontotemporal Degener 20 (5-6), 432–440. doi:10.1080/21678421.2019.1606244
van Es, M. A., Hardiman, O., Chio, A., Al-Chalabi, A., Pasterkamp, R. J., Veldink, J. H., et al. (2017). Amyotrophic lateral sclerosis. The Lancet 390, 2084–2098. doi:10.1016/S0140-6736(17)31287-4
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48 (9), 1043–1048. doi:10.1038/ng.3622
van Rheenen, W., van der Spek, R. A. A., Bakker, M. K., van Vugt, J., Hop, P. J., Zwamborn, R. A. J., et al. (2021). Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 53 (12), 1636–1648. doi:10.1038/s41588-021-00973-1
Verdile, V., De Paola, E., and Paronetto, M. P. (2019). Aberrant phase transitions: side effects and novel therapeutic strategies in human disease. Front Genet 10, 173. doi:10.3389/fgene.2019.00173
Vincent, A. M., Backus, C., Taubman, A. A., and Feldman, E. L. (2005). Identification of candidate drugs for the treatment of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 6 (1), 29–36. doi:10.1080/14660820510026171
Vopel, T., Bravo-Rodriguez, K., Mittal, S., Vachharajani, S., Gnutt, D., Sharma, A., et al. (2017). Inhibition of huntingtin exon-1 aggregation by the molecular tweezer CLR01. J Am Chem Soc. 139 (16), 5640–5643. doi:10.1021/jacs.6b11039
Vu, L., An, J., Kovalik, T., Gendron, T., Petrucelli, L., and Bowser, R. (2020). Cross-sectional and longitudinal measures of chitinase proteins in amyotrophic lateral sclerosis and expression of CHI3L1 in activated astrocytes. J Neurol Neurosurg Psychiatry 91 (4), 350–358. doi:10.1136/jnnp-2019-321916
Wachter, N., Storch, A., and Hermann, A. (2015). Human TDP-43 and FUS selectively affect motor neuron maturation and survival in a murine cell model of ALS by non-cell-autonomous mechanisms. Amyotroph Lateral Scler Frontotemporal Degener 16 (7-8), 431–441. doi:10.3109/21678421.2015.1055275
Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130 (5), 643–660. doi:10.1007/s00401-015-1460-x
Wang, A., and Bolen, D. W. (1997). A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 36 (30), 9101–9108. doi:10.1021/bi970247h
Wang, A., Conicella, A. E., Schmidt, H. B., Martin, E. W., Rhoads, S. N., Reeb, A. N., et al. (2018). A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 37 (5). doi:10.15252/embj.201797452
Wang, L., Deng, H. X., Grisotti, G., Zhai, H., Siddique, T., and Roos, R. P. (2009). Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Human molecular genetics 18 (9), 1642–1651. doi:10.1093/hmg/ddp085
Wang, L., Wang, N., Zhang, W., Cheng, X., Yan, Z., Shao, G., et al. (2022b). Therapeutic peptides: current applications and future directions. Signal transduction and targeted therapy 7 (1), 48. doi:10.1038/s41392-022-00904-4
Wang, Q., Johnson, J. L., Agar, N. Y., and Agar, J. N. (2008). Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol. 6 (7), e170. doi:10.1371/journal.pbio.0060170
Wang, L. Q., Ma, Y., Yuan, H. Y., Zhao, K., Zhang, M. Y., Wang, Q., et al. (2022a). Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion. Nature communications 13 (1), 3491. doi:10.1038/s41467-022-31240-4
Wang, S. J., Wang, K. Y., and Wang, W. C. (2004). Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience 125 (1), 191–201. doi:10.1016/j.neuroscience.2004.01.019
Webster, C. P., Smith, E. F., Shaw, P. J., and De Vos, K. J. (2017). Protein homeostasis in amyotrophic lateral sclerosis: therapeutic opportunities? Front Mol Neurosci 10, 123. doi:10.3389/fnmol.2017.00123
Wentink, A., and Rosenzweig, R. (2023). Protein disaggregation machineries in the human cytosol. Current opinion in structural biology 83, 102735. doi:10.1016/j.sbi.2023.102735
Wijesekera, L. C., and Leigh, P. N. (2009). Amyotrophic lateral sclerosis. Orphanet Journal of Rare Diseases 4, 3–22. doi:10.1186/1750-1172-4-3
Wilcox, K. C., Zhou, L., Jordon, J. K., Huang, Y., Yu, Y., Redler, R. L., et al. (2009). Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem 284, 13940–13947. doi:10.1074/jbc.M809687200
Williams, K. L., Warraich, S. T., Yang, S., Solski, J. A., Fernando, R., Rouleau, G. A., et al. (2012). UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 33 (10), 2527 e3–e10. doi:10.1016/j.neurobiolaging.2012.05.008
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283 (19), 13302–13309. doi:10.1074/jbc.M800342200
Wood, M., Quinet, A., Lin, Y. L., Davis, A. A., Pasero, P., Ayala, Y. M., et al. (2020). TDP-43 dysfunction results in R-loop accumulation and DNA replication defects. J Cell Sci 133 (20), jcs244129. doi:10.1242/jcs.244129
Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M., and Biere, A. L. (1999). alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem 274 (28), 19509–19512. doi:10.1074/jbc.274.28.19509
Wood-Allum, C. A., Barber, S. C., Kirby, J., Heath, P., Holden, H., Mead, R., et al. (2006). Impairment of mitochondrial anti-oxidant defence in SOD1-related motor neuron injury and amelioration by ebselen. Brain 129 (Pt 7), 1693–1709. doi:10.1093/brain/awl118
Wright, G. S. A. (2020). Molecular and pharmacological chaperones for SOD1. Biochem. Soc Trans. 48, 1795–1806. doi:10.1042/BST20200318
Wright, G. S. A., Antonyuk, S. V., and Hasnain, S. S. (2016). A faulty interaction between SOD1 and hCCS in neurodegenerative disease. Sci Rep 6, 27691–27699. doi:10.1038/srep27691
Wright, G. S. A., Antonyuk, S. V., Kershaw, N. M., Strange, R. W., and Samar Hasnain, S. (2013). Ligand binding and aggregation of pathogenic SOD1. Nat Commun 4, 1758–58. doi:10.1038/ncomms2750
Wright, G. S. A., Watanabe, T. F., Amporndanai, K., Plotkin, S. S., Cashman, N. R., Antonyuk, S. V., et al. (2020). Purification and structural characterization of aggregation-prone human TDP-43 involved in neurodegenerative diseases. iScience 23 (6), 101159. doi:10.1016/j.isci.2020.101159
Wright, P. D., Wightman, N., Huang, M., Weiss, A., Sapp, P. C., Cuny, G. D., et al. (2012). A high-throughput screen to identify inhibitors of SOD1 transcription. Frontiers in bioscience 4 (8), 2701–2708. doi:10.2741/e584
Writing Group and Edaravone (MCI-186) ALS 19 Study Group (2017). Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. The Lancet Neurology 16 (7), 505–512. doi:10.1016/S1474-4422(17)30115-1
Wroe, R., Wai-Ling, B. A., Andersen, P. M., Powell, J. F., and Al-Chalabi, A. (2008). ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Lateral Scler 9 (4), 249–250. doi:10.1080/17482960802146106
Wu, L. S., Cheng, W. C., Chen, C. Y., Wu, M. C., Wang, Y. C., Tseng, Y. H., et al. (2019). Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol Commun 7 (1), 50. doi:10.1186/s40478-019-0674-x
Xia, G., Benmohamed, R., Kim, J., Arvanites, A. C., Morimoto, R. I., Ferrante, R. J., et al. (2011). Pyrimidine-2, 4, 6-trione derivatives and their inhibition of mutant SOD1-dependent protein aggregation. Toward a treatment for amyotrophic lateral sclerosis. J Med Chem 54, 2409–2421. doi:10.1021/jm101549k
Xia, G., Benmohamed, R., Morimoto, R. I., Kirsch, D. R., and Silverman, R. B. (2014). Deuteration and fluorination of 1,3-bis(2-phenylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione to improve its pharmacokinetic properties. Bioorganic & Medicinal Chemistry Letters 24, 5098–5101. doi:10.1016/j.bmcl.2014.08.066
Xu, W. C., Liang, J. Z., Li, C., He, Z. X., Yuan, H. Y., Huang, B. Y., et al. (2018). Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell death & disease 9 (2), 67. doi:10.1038/s41419-017-0106-4
Xu, Z., Chen, S., Li, X., Luo, G., Li, L., and Le, W. (2006). Neuroprotective effects of (-)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem. Res 31 (10), 1263–1269. doi:10.1007/s11064-006-9166-z
Yadav, K., Yadav, A., Vashistha, P., Pandey, V. P., and Dwivedi, U. N. (2019). Protein misfolding diseases and therapeutic approaches. Curr Protein Pept Sci 20 (12), 1226–1245. doi:10.2174/1389203720666190610092840
Yamaguchi, T., Sano, K., Takakura, K., Saito, I., Shinohara, Y., Asano, T., et al. (1998). Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke 29 (1), 12–17. doi:10.1161/01.str.29.1.12
Yamashita, M., Nonaka, T., Arai, T., Kametani, F., Buchman, V. L., Ninkina, N., et al. (2009). Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett 583 (14), 2419–2424. doi:10.1016/j.febslet.2009.06.042
Yang, C., Wang, H., Qiao, T., Yang, B., Aliaga, L., Qiu, L., et al. (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the U. S. A. 111 (12), E1121–E1129. doi:10.1073/pnas.1322641111
Yang, L., Jasiqi, Y., Zettor, A., Vadas, O., Chiaravalli, J., Agou, F., et al. (2023). Effective inhibition of TDP-43 aggregation by native state stabilization. Angewandte Chemie 63, e202314587. doi:10.1002/anie.202314587
Yang, Y. M., Gupta, S. K., Kim, K. J., Powers, B. E., Cerqueira, A., Wainger, B. J., et al. (2013). A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12 (6), 713–726. doi:10.1016/j.stem.2013.04.003
Yerbury, J. J., and Cashman, N. R. (2020). Selenium-based compounds: emerging players in the ever-unfolding story of SOD1 in amyotrophic lateral sclerosis. EBioMedicine 59, 102997. doi:10.1016/j.ebiom.2020.102997
Young, L. M., Ashcroft, A. E., and Radford, S. E. (2017). Small molecule probes of protein aggregation. Current opinion in chemical biology 39, 90–99. doi:10.1016/j.cbpa.2017.06.008
Yu, C. H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., Laohamonthonkul, P., et al. (2020). TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183 (3), 636–649. doi:10.1016/j.cell.2020.09.020
Zacco, E., Martin, S. R., Thorogate, R., and Pastore, A. (2018). The RNA-recognition motifs of TAR DNA-binding protein 43 may play a role in the aberrant self-assembly of the protein. Front Mol Neurosci 11, 372. doi:10.3389/fnmol.2018.00372
Zetterstrom, P., Stewart, H. G., Bergemalm, D., Jonsson, P. A., Graffmo, K. S., Andersen, P. M., et al. (2007). Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proceedings of the National Academy of Sciences of the U. S. A. 104 (35), 14157–14162. doi:10.1073/pnas.0700477104
Zhang, W., Benmohamed, R., Arvanites, A. C., Morimoto, R. I., Ferrante, R. J., Kirsch, D. R., et al. (2012a). Cyclohexane 1, 3-diones and their inhibition of mutant SOD1-dependent protein aggregation and toxicity in PC12 cells. Bioorg Med Chem 20, 1029–1045. doi:10.1016/j.bmc.2011.11.039
Zhang, Y., Benmohamed, R., Zhang, W., Kim, J., Edgerly, C. K., Zhu, Y., et al. (2012b). Chiral cyclohexane 1,3-diones as inhibitors of mutant SOD1- dependent protein aggregation for the treatment of ALS. ACS Med Chem Lett 3, 584–587. doi:10.1021/ml3000963
Zhao, B., Marciniuk, K., Gibbs, E., Yousefi, M., Napper, S., and Cashman, N. R. (2019). Therapeutic vaccines for amyotrophic lateral sclerosis directed against disease specific epitopes of superoxide dismutase 1. Vaccine 37 (35), 4920–4927. doi:10.1016/j.vaccine.2019.07.044
Zhao, B., Zhuang, X., Pi, Z., Liu, S., Liu, Z., and Song, F. (2018). Determining the effect of catechins on SOD1 conformation and aggregation by ion mobility mass spectrometry combined with optical spectroscopy. J Am Soc Mass Spectrom 29, 734–741. doi:10.1007/s13361-017-1864-z
Zheng, X., Liu, D., Klarner, F. G., Schrader, T., Bitan, G., and Bowers, M. T. (2015). Amyloid β-protein assembly: the effect of molecular tweezers CLR01 and CLR03. The journal of physical chemistry B 119 (14), 4831–4841. doi:10.1021/acs.jpcb.5b00692
Zhu, C., Beck, M. V., Griffith, J. D., Deshmukh, M., and Dokholyan, N. V. (2018). Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the U. S. A. 115 (18), 4661–4665. doi:10.1073/pnas.1800187115
Zhu, L., Li, S., Li, X. J., and Yin, P. (2023). Pathological insights from amyotrophic lateral sclerosis animal models: comparisons, limitations, and challenges. Translational neurodegeneration 12 (1), 46. doi:10.1186/s40035-023-00377-7
Zhu, L., Xu, M., Yang, M., Yang, Y., Li, Y., Deng, J., et al. (2014). An ALS-mutant TDP-43 neurotoxic peptide adopts an anti-parallel β-structure and induces TDP-43 redistribution. Human molecular genetics 23 (25), 6863–6877. doi:10.1093/hmg/ddu409
Zuo, X., Zhou, J., Li, Y., Wu, K., Chen, Z., Luo, Z., et al. (2021). TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nature structural & molecular biology 28 (2), 132–142. doi:10.1038/s41594-020-00537-7
Keywords: amyotrophic lateral sclerosis, SOD1, TDP-43, protein misfolding, protein aggregation, neurotoxicity, therapeutics
Citation: Tsekrekou M, Giannakou M, Papanikolopoulou K and Skretas G (2024) Protein aggregation and therapeutic strategies in SOD1- and TDP-43- linked ALS. Front. Mol. Biosci. 11:1383453. doi: 10.3389/fmolb.2024.1383453
Received: 07 February 2024; Accepted: 02 May 2024;
Published: 24 May 2024.
Edited by:
Emily Sontag, Marquette University, United StatesReviewed by:
Valentina Bonetto, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyCopyright © 2024 Tsekrekou, Giannakou, Papanikolopoulou and Skretas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgios Skretas, c2tyZXRhc0BmbGVtaW5nLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.