 Alejandra María Rivera Nieves
Alejandra María Rivera Nieves Brian Michael Wauford
Brian Michael Wauford Accalia Fu
Accalia Fu- 1Diabetes Center of Excellence, University of Massachusetts Chan Medical School, Worcester, MA, United States
- 2Program in Molecular Medicine, University of Massachusetts Chan Medical School, Worcester, MA, United States
In Type 1 and Type 2 diabetes, pancreatic β-cell survival and function are impaired. Additional etiologies of diabetes include dysfunction in insulin-sensing hepatic, muscle, and adipose tissues as well as immune cells. An important determinant of metabolic health across these various tissues is mitochondria function and structure. This review focuses on the role of mitochondria in diabetes pathogenesis, with a specific emphasis on pancreatic β-cells. These dynamic organelles are obligate for β-cell survival, function, replication, insulin production, and control over insulin release. Therefore, it is not surprising that mitochondria are severely defective in diabetic contexts. Mitochondrial dysfunction poses challenges to assess in cause-effect studies, prompting us to assemble and deliberate the evidence for mitochondria dysfunction as a cause or consequence of diabetes. Understanding the precise molecular mechanisms underlying mitochondrial dysfunction in diabetes and identifying therapeutic strategies to restore mitochondrial homeostasis and enhance β-cell function are active and expanding areas of research. In summary, this review examines the multidimensional role of mitochondria in diabetes, focusing on pancreatic β-cells and highlighting the significance of mitochondrial metabolism, bioenergetics, calcium, dynamics, and mitophagy in the pathophysiology of diabetes. We describe the effects of diabetes-related gluco/lipotoxic, oxidative and inflammation stress on β-cell mitochondria, as well as the role played by mitochondria on the pathologic outcomes of these stress paradigms. By examining these aspects, we provide updated insights and highlight areas where further research is required for a deeper molecular understanding of the role of mitochondria in β-cells and diabetes.
1 Introduction
Mitochondria are integral to pancreatic β-cell function and survival. The multifaceted role of mitochondria in β-cells encompasses pivotal processes and pathways, including mitochondrial bioenergetics and metabolism, proton leak, calcium (Ca2+), structural integrity, dynamics, as well as turnover/mitophagy. In this review, we delve into the nuanced roles played by β-cell mitochondria. Remarkably, an individual pancreatic β-cell is estimated to have hundreds to thousands of mitochondria due to its high reliance on mitochondrial activity. Understanding the interplay between β-cell mitochondrial structure and function is imperative in unraveling the intricate role of mitochondria in the pathogenesis of Type 1 and Type 2 diabetes (T1 and T2D). In this comprehensive review, we compare mitochondria in healthy and diabetic β-cells and highlight the evidence that mitochondria play a causal role in β-cell demise and diabetes development.
2 The good: mitochondrial roles in healthy β-cells
Healthy pancreatic β-cells are metabolic machines with adapted fuel preferences and metabolism required to perform their functions as glucose sensors. They contribute to systemic metabolic homeostasis by dosing the body with insulin, released via an orchestrated program of cellular excitation and metabolism. Mitochondrial activity is required for insulin secretion, β-cell expansion in response to increased functional demand, and balance of turnover and renewal/replication for maintaining β-cell mass. In the next sections, we detail the involvement of mitochondria as a homeostat in healthy β-cell function and survival.
The events required for insulin secretion have been extensively studied in β-cell lines and rodent primary whole islets and have been comprehensively reviewed (Prentki et al., 2013; Campbell and Newgard, 2021; Merrins et al., 2022; Rutter et al., 2023). In both mouse and human β-cells, glucokinase (HKIV; GCK) plays the dominant rate-limiting role in insulin secretion as it initiates phosphorylation and breakdown of glucose (De Vos et al., 1995; Matschinsky and Wilson, 2019), and contributes to a biphasic pattern of insulin release (Henquin et al., 2015). Although, the kinetics and amplitude of response may differ slightly between mouse and human β-cells due to differential expression of glucose transporter isoforms (GLUT 1 and 3 in humans, GLUT2 in rodents) (De Vos et al., 1995; McCulloch et al., 2011; Berger and Zdzieblo, 2020). Upon glucose uptake and subsequent rises in ATP levels, KATP-channels close and voltage-gated calcium channels (VGCC) open to allow for an influx of Ca2+ critical for the rapid first phase of insulin release. This is the electrical excitation-dependent phase of insulin secretion (Ashcroft and Rorsman, 2013; Rorsman and Braun, 2013). Glycolysis rates increase immediately and stabilize around 5 min after glucose stimulation and within another 10–15 min, generate notable levels of pyruvate apportioned for mitochondrial metabolism. This enhances production of suggested ‘coupling factors’ that potentiate the second phase of insulin release. These include ATP, TCA intermediates, mitochondrial GTP/phosphoenolpyruvate (Stark et al., 2009; Lewandowski et al., 2020), isocitrate/glutamine decarboxylation/SENP1 (Zhang et al., 2021), monoacyl-glycerol (Zhao et al., 2014; Attane et al., 2016), and acetyl- and malonyl-coA (Prentki et al., 2020) (Figure 1).
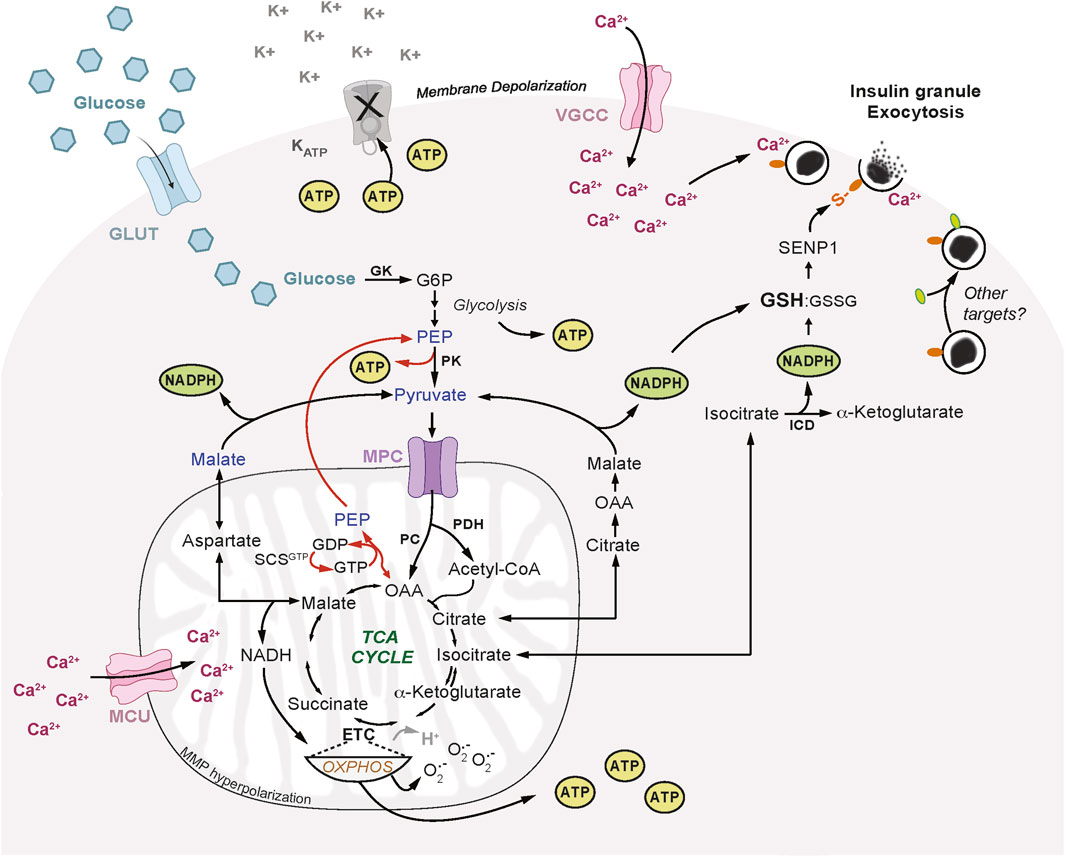
FIGURE 1. Mitochondrial Pathways Required for Insulin Secretion. Glucose enters pancreatic β-cells through glucose transporters (GLUT) and is phosphorylated by glucokinase (GK) to form glucose-6-phosphate (G6P) which is further metabolized in glycolysis. Pyruvate generated from glycolysis enters mitochondria via the mitochondrial pyruvate carrier (MPC), to participate in several metabolic cycles that are required for insulin secretion. Pyruvate is hydrolyzed by pyruvate dehydrogenase (PDH) to form acetyl-coA and contribute to the production of tricarboxylic acid (TCA) cycle intermediates. TCA intermediate reactions supply the electron transport chain (ETC) with reducing equivalents and contribute to ATP synthesis driven by the proton motive force generated by mitochondrial membrane hyperpolarization. Pyruvate can also enter the TCA cycle via pyruvate carboxylase (PC) to produce oxaloacetic acid (OAA) and regenerate pyruvate via the GTP-specific succinyl-CoA synthetase (SCS-GTP) and GTP-phosphoenolpyruvate (PEP) cycle. Mitochondrial-cytosolic isocitrate exchange can generate nicotinamide adenine dinucleotide phosphate (NADPH) via isocitrate dehydrogenase (ICD) which increases reduced glutathione (GSH) and lowers oxidized GSH (GSSG). The anapleurotic malate-aspartate and citrate-malate shuttles can support additional NAPDH synthesis. GSH can activate sentrin/SUMO-specific protease-1 (SENP1) that deSUMOylates proteins on insulin granules and enhances granule docking and exocytosis at the plasma membrane. There may be targets and modifications beyond deSUMOylation that contribute to insulin granule docking and exocytosis. Mitochondrial membrane potential (MMP) decreases or hyperpolarizes. Oxidative phosphorylation (OXPHOS) increases in coupling to ATP synthesis, proton leak (H+) is reduced, and superoxide (O2•-) production is increased. ATP generated from glycolysis and mitochondrial OXPHOS causes KATP-channel (KATP) closure and plasma membrane depolarization which leads to opening of voltage-gated calcium channels (VGCC) and calcium (Ca2+) influx. As Ca2+ increases in the cytosol this enhances mitochondrial Ca2+ uptake through the mitochondrial calcium uniporter (MCU) which may also be influenced by mitochondrial membrane hyperpolarization. Insulin granule exocytosis requires this influx in both cytosolic and mitochondrial Ca2+.
2.1 Mitochondrial metabolism
The impact of mitochondrial metabolism on insulin secretion is the most well-studied area of mitochondrial β-cell research and has been reviewed extensively (Maechler and Wollheim, 2000; Maechler et al., 2006; Jitrapakdee et al., 2010; Kaufman et al., 2015; Campbell and Newgard, 2021; Rutter et al., 2023). Glucose carbons are preferentially supplied to mitochondria as pyruvate, this results from downregulation of alternate or anaerobic routes of glycolysis synthesizing products other than pyruvate. For example, only ∼3% of glucose carbons enter the pentose phosphate pathway (PPP) (MacDonald, 1993), and minimal amounts are used for lactate production (Sekine et al., 1994; Schuit et al., 1997) due to low lactate dehydrogenase activity and expression in β-cells (Sekine et al., 1994; Pullen et al., 2010). Pyruvate enters the mitochondria through mitochondrial pyruvate carriers 1 and 2 (MPC1 and 2), and their genetic depletion or pharmacologic inhibition reduces glucose-stimulated insulin secretion (GSIS) in rodent and human islets (Patterson et al., 2014; Vigueira et al., 2014), underscoring the importance of mitochondrial pyruvate entry for the β-cell functional response to glucose. Pyruvate is then broken down to form acetyl-coA by pyruvate dehydrogenase (PDH). Pyruvate can also enter the tricarboxylic acid (TCA) cycle via a carboxylation reaction to form oxaloacetate by pyruvate carboxylase (PC). PDH and PC may have near equal rates in β-cells, however, PC activity positively correlates with insulin secretion and increasing glucose concentrations, unlike PDH (Khan et al., 1996; Schuit et al., 1997; Sugden and Holness, 2011) (Figure 1).
The relatively high activities of β-cell PC and the mitochondrial phosphoenolpyruvate (PEP) cycle further confine glucose to pyruvate metabolism in the mitochondria and are beneficial for insulin release, glucose homeostasis, and high fat diet (HFD)-induced insulin resistance (Abulizi et al., 2020). High PC activity is also linked to other metabolic intermediates including those involved in arginine metabolism, the urea cycle, and glutathione de novo synthesis which improve β-cell resilience against diabetes-related inflammation and oxidative stress (Fu et al., 2020; Fu et al., 2021). Furthermore, PC activity provides another means by which β-cells can impede lactate production, and interestingly, lactate accumulates in humans with PC deficiency (Habarou et al., 2015). Mitochondrial metabolism also impacts insulin secretion via the electron transport chain (ETC) and the TCA cycle, which generate ATP and other coupling factors essential for insulin release (Figure 1). Few studies have made direct biochemical links from mitochondrial metabolism to insulin granule exocytosis. One example is that mitochondrially-derived isocitrate, through its metabolism by cytosolic isocitrate dehydrogenase (ICD), enhances reductive potential that then activates sentrin/SUMO-specific protease-1 (SENP1), a deSUMOylating protein (Ferdaoussi et al., 2015). SENP1KO β-cells have reduced insulin exocytosis but no defects in upstream insulin events, thus the authors conclude this was due to a direct effect on exocytosis, and while the precise biochemical mechanism remains unknown, it presumably involves deSUMOylation of multiple granule-regulating proteins (Ferdaoussi et al., 2015). TCA activity can also support the malate–aspartate shuttle that converts cytosolic NADH to mitochondrial FADH and NADH, enhancing ATP production which leads to KATP-channel closure (Eto et al., 1999). The malate-aspartate shuttle and glutamate dehydrogenase (GDH) may also directly impact insulin secretion via influencing glutamate levels (Vetterli et al., 2012; Gheni et al., 2014). Direct uptake of glutamate potentiated by incretins underlies the amplification of insulin secretion, since glutamate transporter knockdown within insulin granules reduces GSIS (Gheni et al., 2014). TCA anapleurotic activity could also support insulin secretion via a mitochondrial GTP-PEP cycle that operates independently of OXPHOS to enhance insulin release (Jesinkey et al., 2019). This pathway of GSIS involves mitochondrial GTP and PEP cycling (Jesinkey et al., 2019), and likely implicates cytosolic pyruvate kinase (PK) (Lewandowski et al., 2020), since PKm1/2 β-cell KO mice have no change in in vivo glucose tolerance and homeostasis (Foster et al., 2022). PK influence over GSIS is thought to involve ATP-dependent K+ATP-channel closure, but how and whether this activity depends on mitochondrial GTP cycling is not fully understood. The increase in ATP independent of OXPHOS presents a new model that stipulates a “silent phase” and avoids ADP deprivation via over-activity of OXPHOS, thus allowing for subsequent rounds of OXPHOS. How this impacts ADP within mitochondria while being driven by cytosolic PK which is presumed to be at the plasma membrane (PM) is uncertain. It would be interesting to see if mitochondrial heterogeneity between those located near the PM versus those further away might be involved. It will also be important to know whether this is part of the natural GSIS pathway for human β-cells.
At their most proximal steps, most of the pathways of mitochondrial metabolism impacting GSIS in rodent studies are conserved in human β-cells. Of note, human β-cells have lower rates of mitochondrial metabolism across various studies, although this may be explained by the lower relative abundance of β-cells in human islets or experimental caveats such as poorer islet culture quality compared to freshly isolated rodent islets. How these many arms of mitochondrial metabolism integrate in an epistatic fashion or act independently to regulate insulin secretion remains to be determined. Nonetheless, the importance of mitochondrial activity in general is evident in a study that depleted mitochondrial DNA with ethidium bromide which reduced glucose-stimulated ETC activity, ATP production, Ca2+ influx, and insulin secretion (Noda et al., 2002). It would be interesting to see how newer models of mitochondrial depletion, including the use of genetic tools, impact β-cell biology and insulin secretion (Stewart, 2021).
2.2 Mitochondrial oxidative phosphorylation and proton leak
Oxidative metabolism is favored by β-cells, this encompasses the oxidative arm of glycolysis and mitochondrial respiration, which is the consumption of oxygen molecules to produce metabolic intermediates of the TCA and ETC, and synthesize ATP. The energy in mitochondrial substrates, such as TCA intermediates, is transferred through the ETC by reducing NAD+ at Complex I (NADH dehydrogenase) and FAD+ at Complex II (Succinate dehydrogenase; SDH). This energy is ultimately used by Complex IV (cytochrome c oxidase) to reduce oxygen to water and remove electrons so that the carriers can be re-oxidized to continue the transport of energy through the ETC (Martinez-Reyes and Chandel, 2020). The proton gradient created by the ETC drives ATP synthesis via ATP synthase, and this coupling is known as OXPHOS. Mitochondrial respiration in β-cells increases to an exceptionally high level in response to increasing glucose concentrations, surpassing even muscle cells, despite an unusually high basal level of proton leak (Affourtit and Brand, 2006; Wikstrom et al., 2012). However, coupling to ATP synthesis increases upon glucose stimulation (Affourtit and Brand, 2006; Wikstrom et al., 2012) resulting in a relative reduction of leak compared to basal glucose levels (Figure 1).
It is important to consider caveats in studies on mitochondrial bioenergetics, electron, and proton leak including the penetrance of mitochondrial inhibitors and the challenge of uniformly delivering drugs to all cells within islets. Recent advances in approaches for measuring oxygen consumption in individual large-sized islets have revealed considerable heterogeneity between islets of the same donor or mouse (Taddeo et al., 2018). In addition, intact islets, as opposed to re-aggregated ones, demonstrate a higher capacity for respiration (Taddeo et al., 2018). Conducting bioenergetics analyses on single islets can improve our overall understanding of islet bioenergetics (Taddeo et al., 2018). However, even with these advancements current methods are unable to distinguish islet cell type-specific contributions to mitochondrial respiration.
Mitochondria can generate reactive oxygen species (ROS) in the form of superoxide primarily due to electron leak at Complex I and III (Zhao et al., 2019). This phenomenon is often associated with proton leak (Zhao et al., 2019). Acute mitochondrial reactive oxygen species (mtROS) can amplify GSIS, but prolonged elevated exposure impairs insulin release and β-cell survival (Graciano et al., 2011). In low glucose, limiting mtROS may prevent inappropriate insulin release (Mailloux et al., 2012). Conversely, high levels of matrix ROS can activate a supraphysiologic uncoupling protein 2 (UCP2)-dependent increase in proton leak, impairing GSIS (Mailloux et al., 2012). The impact of different sources of ROS on insulin secretion is highly context-dependent. Some studies observe amplification or positive correlation (Pi et al., 2007; Graciano et al., 2011; Rebelato et al., 2011; Mailloux et al., 2012; Lagundzin et al., 2019), while others report impairment or inverse correlation with GSIS (Sakai et al., 2003; Munhoz et al., 2016; Mukai et al., 2022). While signaling mtROS may be beneficial, excess ROS accumulation from any source can uncouple OXPHOS, decrease ATP synthesis, and elevate mitochondrial membrane potential (MMP), all of which are associated with reduced insulin secretion (Newsholme et al., 2019).
High proton leak could increase superoxide/mtROS if it exceeds OXPHOS coupling to ATP synthesis. One study found that in INS1 β-cells, high glucose decreased superoxide/mtROS production, which was attributed to pyruvate cycling activity from the malate-aspartate and citrate-isocitrate shuttle substrates (Plecita-Hlavata et al., 2020). However, it should be noted that this study used oxidation of mitoSOX dye to measure mtROS, which is an indirect measure, and its oxidation may depend on redox metal ions and/or an unknown peroxidation mechanism which may be reduced by glucose independently of OXPHOS (Kalyanaraman, 2020). Furthermore, the coupling of OXPHOS relative to oxygen consumption is elevated beyond the increase in proton leak compared to low glucose (Leloup et al., 2009; Taddeo et al., 2020). The enhanced coupling contributes to the production of signaling mtROS required to amplify insulin secretion (Leloup et al., 2009; Taddeo et al., 2020). At low glucose proton leak was shown to enhance insulin secretion independent of OXPHOS and required cyclophilin D activity, but this effect was not observed in high glucose-dependent proton leak (Taddeo et al., 2020). Proton leak may be less pronounced in human β-cells (Wikstrom et al., 2012), or high heterogeneity between islets within a human donor may impede large batch islet measurements (Taddeo et al., 2020). UCP2 was an early candidate for producing proton leak, however, UCP2KO β-cells have increased OXPHOS and ATP levels, with no change in proton leak (Zhang et al., 2001; Robson-Doucette et al., 2011). Thus, the source of leak during GSIS remains unknown but may involve cyclophilin D, a regulator of the mitochondrial permeability transition pore (Taddeo et al., 2020). Mitochondrial membrane potential (MMP) also hyperpolarizes in response to glucose (Figure 1), contributing to significant ATP production in human islets (Gerencser et al., 2016; Gerencser, 2018). The role of MMP is thought to be dependent on OXPHOS, and their contributions to insulin secretion are usually associated (Gerencser, 2018), however the precise connections linking these processes in β-cells, and whether they are bi-directional, are not known. Despite these unknowns, mitochondrial respiration is essential for the dynamic glucose-stimulated increase in ATP:ADP and GSIS in the INS1 β-cell line and mouse and human β-cells (De Marchi et al., 2014).
2.3 Mitochondrial calcium
Cytosolic Ca2+ is integral for insulin secretion and a growing body of evidence reveals that mitochondrial Ca2+ regulation, specifically by the mitochondrial calcium uniporter (MCU) and its associated proteins, mitochondrial calcium uptake 1 and 2 (MICU1 and MICU2), are also critical for maintaining insulin secretion (Alam et al., 2012; Georgiadou et al., 2020; Vishnu et al., 2021). The MCU transports Ca2+ into mitochondria in a membrane potential-driven manner that does not require ATP (Vais et al., 2020). Ca2+ influx into the mitochondrial matrix via MCU and MICU1 is required for oxidative metabolism of glucose and ATP generation in INS1 cells and rat islets (Wiederkehr et al., 2011; Alam et al., 2012; Tarasov et al., 2012). MCU and MICU1 are necessary for proper insulin release in INS1 cells, as knockdown of MCU or MICU1 decreases GSIS (Alam et al., 2012). Specific deletion of MCU in β-cells in mice led to significantly higher blood glucose levels upon glucose challenge due to impaired in vivo insulin release (Georgiadou et al., 2020). In line with these results, MICU2KO β-cells have decreased GSIS and ATP/ADP, absent mitochondrial membrane hyperpolarization, and diminished influx of cytosolic Ca2+ in response to elevated glucose (Vishnu et al., 2021).
The kinetics of mitochondrial matrix Ca2+ accumulation is delayed by seconds compared to the rapid increases in cytosolic Ca2+ in response to glucose (Waldeck-Weiermair et al., 2012). At rest, cytosolic Ca2+ is ∼50–70 nM which increases to ∼300–400 nM after glucose stimulation (Zhang et al., 2003; Wiederkehr et al., 2011), and there are large oscillations thought to be essential for GSIS and may involve mitochondrial and ER exchange. INS1 β-cell mitochondrial matrix Ca2+ increases from ∼100 to 200 nM to ∼600–800 nM, which is slightly higher compared to the increase in cytosolic Ca2+, and gradually increases without the oscillatory nature (Wiederkehr et al., 2011). Glucose-stimulated mitochondrial uptake of Ca2+ occurs at a constant rate and has been proposed to serve as a sink for cytosolic Ca2+ (Rutter et al., 2023). Such a sink would further potentiate the demand for ATP synthesis after initial increased ATP production, and increased activation of ETC and dehydrogenase by Ca2+ (Jouaville et al., 1999). Efflux of Ca2+ from mitochondria may be low in β-cells, which could further contribute to its accumulation upon glucose stimulation. For example, the efflux Na+/Ca2+ exchanger restricts mitochondrial Ca2+ accumulation, promotes cytosolic oscillations in Ca2+ and contributes to insulin release in MIN6 β-cells to a minor degree (Nita et al., 2012). Tarasov et al. proposed that slow mitochondrial Ca2+ accumulation transmits a constant, averaged signal from oscillations in cytosolic Ca2+ to allow for ATP synthesis, presumably via TCA cycle enzyme activation (Tarasov et al., 2013). Interestingly, when Ca2+ is depleted in human β-cells, glucose-stimulated ATP-coupled mitochondrial respiration is completely blocked (De Marchi et al., 2014). On the other hand, mitochondrial metabolism may also be required for mitochondrial Ca2+ influx, as revealed in studies that inhibited the ETC in INS1 cells and found lower glucose-induced ATP synthesis and mitochondrial Ca2+ influx (Jeyarajan et al., 2023). Thus, the impact of mitochondrial Ca2+ on insulin secretion via plasma membrane depolarization and/or mitochondrial bioenergetics and metabolism is complex and multidirectional (Zhang et al., 2003). Mitochondria and ER also exchange Ca2+ which may further exert control over insulin secretion (Alam et al., 2012; Klec et al., 2019; Jeyarajan et al., 2023), although these aspects are less well-defined and merit further testing in primary islets and in vivo models.
2.4 Mitochondrial dynamics and mitophagy
Mitochondrial dynamics, fission, fusion, and mitophagy/turnover are also important regulators of β-cell function, mass, and survival (Molina et al., 2009; Rutter et al., 2023). Dynamin-related protein 1 (DRP1), a regulator of fission, is required for the metabolic amplifying phase of insulin secretion and led to disrupted hyperfused mitochondrial networks yet did not have any effect on oxygen consumption nor Ca2+ influx when depleted in β-cells in vivo (Reinhardt et al., 2016; Hennings et al., 2018). Impaired insulin secretion in DRP1-deficient MIN6 β-cells was fully restored with pyruvate supplementation, indicating that the major defect is one of substrate generation as opposed to mitochondrial function (Kabra et al., 2022). This implies that control of insulin by mitochondrial pyruvate cycles is dependent on structural dynamics. Inducing mitochondrial fragmentation with mitofusin1 and 2 (MFN1/2) knockout in β-cells results in hyperglycemia, and reduced circulating insulin, mitochondrial length, MMP hyperpolarization, ATP synthesis, and Ca2+ influx in response to glucose (Georgiadou et al., 2022). In complementary studies, depletion of MFN1/2 found similar hyperglycemia, reductions in insulin secretion, and mitochondrial network fusion and respiration, however, these effects correlate more with effects on mitochondrial DNA (mtDNA) content rather than changes in fission/fusion (Sidarala et al., 2022). MFN1/2 knockout had minimal effects on β-cell mass, underscoring a stronger impact of mitochondrial dynamics on β-cell function (Georgiadou et al., 2022; Sidarala et al., 2022). On the other hand, depletion of optic atrophy 1 (OPA1), the mitochondrial dynamin-like GTPase controlling membrane fusion and cristae formation, decreases complex IV activity and function, impaired insulin release and ATP synthesis, and impaired β-cell proliferation and mass (Zhang et al., 2011). These studies reveal a complex role for mitochondrial dynamics in insulin secretion and β-cell mass.
Mitochondrial turnover, or mitophagy, is a homeostat for population control over mitochondria and is also a key determinant of β-cell health. Studying mitophagy has been challenging in β-cells, and its relevance was brought into question when loss of function studies on Parkin, a key regulator of mitophagy through ubiquitin-dependent autophagosome degradation, in β-cells revealed no overt phenotype (Corsa et al., 2019). However, depletion of the Parkin activator PTEN-induced putative kinase 1 (PINK1) impaired glucose uptake and yet increased basal insulin release with an overall improvement in glucose tolerance in vivo, thus suggesting that PINK1 mediates glucose-sensing coupling to insulin release (Deas et al., 2014). Most studies on mitophagy have been conducted in pathogenic and diabetic contexts and are briefly described in The Bad sections below and are reviewed elsewhere in greater detail (Lee et al., 2019; Blagov et al., 2023). Our knowledge of bona fide mitophagy in healthy β-cells may improve with the development of new tools to study mitophagy directly or using orthogonal indirect tools (Aoyagi et al., 2023; Levi-D'Ancona et al., 2023). In general, we know very little about the requirements for mitophagy at the subcellular and biochemical level in β-cells but there is compelling evidence for its importance in diabetes, and it is an expanding area we look forward to learning more about in future studies.
3 The Bad: mitochondrial dysfunction in diabetes
Mitochondria have a powerful impact on insulin secretion and β-cell function and survival. In the context of diabetes, countless lines of evidence reveal that mitochondria are dysfunctional and may exacerbate disease progression (Figure 2). However, it is unclear if mitochondria play a direct, causal role in the development of diabetes. In the next sections, we detail the mitochondrial dysfunction in response to diabetogenic stress in terms of cellular respiration, Ca2+ levels, ER stress, and mitophagy.
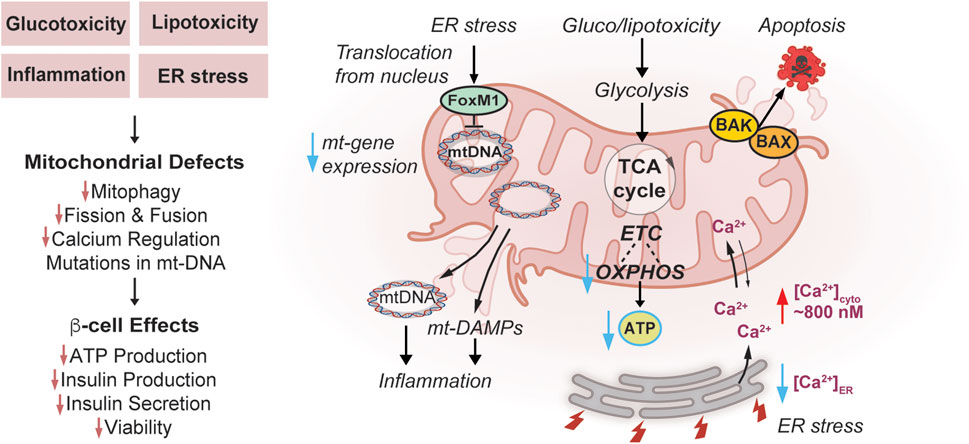
FIGURE 2. Effects of Mitochondrial Dysfunction on β-cells in Diabetes. Diabetogenic stress, such as glucotoxicity, lipotoxicity, inflammation, and endoplasmic reticulum (ER) stress impair mitochondrial turnover/mitophagy, dynamics, and calcium regulation. In addition, ER stress induced by diabetogenic stress causes FOXM1 to translocate from the nucleus to mitochondria, bind mtDNA and impair replication potentially contributing to mtDNA mutations. Inflammation can be amplified through release of mt-DNA and mt-DAMPs. Diabetogenic stresses enhance mitochondrial apoptosis pathways through Bcl-2 Associated X-protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) activation. Gluco-/lipotoxic stress impairs mitochondrial ETC and OXPHOS, and lowers coupling to ATP synthesis. These stress pathways culminate in a loss of glucose-responsiveness including reduced ATP synthesis, insulin production and secretion, and diminished viability of β-cells.
3.1 Mitochondrial metabolism and bioenergetics in gluco- and lipotoxicity
Dysregulated mitochondrial bioenergetics, metabolism, and OXPHOS are frequently observed in models of T2D. A common approach is to use glucotoxic and lipotoxic culture conditions to model the chronically elevated glucose and free fatty acid (FFA) levels in T2D. This metabolic stress leads to defective insulin secretion, apoptosis, and dedifferentiation of β-cells owing to molecular causes involving a combination of mitochondrial dysfunction, autophagy impairment, inflammation, ER, and oxidative stress (Lytrivi et al., 2020). Non-diabetic human donor islets cultured in chronic hyperglycemic conditions elevated mitochondrial metabolism and ATP levels even when switched back to low glucose (Chareyron et al., 2020). This overactivity at low glucose resulted in blunted OXPHOS/ATP responses to high glucose (Figure 2), and accumulation of both PPP intermediates and glycerol-3-phosphate (Chareyron et al., 2020). Another study on human islets in culture similarly found that hyperglycemia/glucotoxicity (3 days; 25 mM glucose) initially increased the expression of mitochondrial carriers, ETC components, and mitochondrial import machinery (Jimenez-Sanchez et al., 2020). Direct effects of inhibiting mitochondrial pyruvate metabolism using 10 µM UK5099 rescued the metabolic defects and glucose induced Ca2+ influx, supporting a theory that enhanced early mitochondrial metabolism contributes to islet dysfunction in hyperglycemia (Chareyron et al., 2020). Unfortunately, whether these effects were MPC-dependent, and if this dose also restored insulin secretion was not evaluated, this is a relevant question because at higher doses (100 µM) it decreases insulin secretion (Patterson et al., 2014). Interestingly, lipotoxic culture conditions (3 days; 0.4 mM oleate/palmitate) have opposing effects and downregulate mitochondrial ETC gene expression (Jimenez-Sanchez et al., 2020). Lipotoxicity modeled through chronic exposure to 0.5 mM palmitate increases superoxide/mtROS production, and decreases enzymes related to carbon metabolism such as citrate synthase (Maris et al., 2013). Single-cell transcriptomic studies on rat β-cells cultured in lipotoxic conditions revealed potential upregulation in glycolysis (Aldoa, Gapdh, Pgk1, and Eno1), OXPHOS (ATP5b, Cox4i2, Cox5b, Cycs, Ndufs3, and Ndufa5), and antioxidant (Sod2 or Prdx1) pathways (Vivoli et al., 2023).
Additional gluco/lipotoxic mechanisms of β-cell death include lipid peroxidation contributing to increased iron uptake and ferroptosis (Hansen et al., 2018). Islets from patients with T2D have increased lipid peroxidation adducts (MacDonald et al., 2013) and these lipid peroxides impair mitochondrial function by damaging mtDNA, RNA, and ETC machinery (Lang et al., 2011; Ademowo et al., 2017). Pancreatic β-cells are particularly low in superoxide dismutase and catalase activity, resulting in their increased sensitivity to ROS, and rely heavily on GSH, NADPH, and thioredoxin to reduce ROS (Shalev, 2014; Miki et al., 2018; Stancill et al., 2019). Supplementation with antioxidants or TCA cycle intermediates prevented β-cell death and restored insulin secretion impaired with lipotoxic stress in INS1 cells (Boucher et al., 2004; Lee J. H. et al., 2014). However, in prolonged gluco/lipotoxic exposure (10 weeks) an elevated antioxidant response is suggested to contribute to loss of GSIS (Fu et al., 2017). These studies emphasize the importance of ROS and mitochondrial metabolism in the protective and damaging adaptive aspects of gluco/lipotoxicity-induced β-cell death.
Apoptotic cell death pathways have also been implicated, in part in gluco/lipotoxicity in β-cells. In terms of pro-apoptotic proteins both Bcl-2 Associated X-protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) are expressed in β-cells and have overlapping capacities to activate cell death (White et al., 2020) (Figure 2). BAX and BAK can also amplify the unfolded protein response (UPR) to further exacerbate the effects of gluco/lipotoxicity. The pro-survival proteins BCL2 and BCL-xL are also both expressed in β-cells and when depleted, glucose-stimulated ROS increases via UCP-2-independent proton leak and enhances permeability transition pore (White et al., 2020).
The non-obese βV59M mouse, carrying an inducible Kir6.2-V59M activating mutation in the KATP channel specifically in pancreatic β-cells, also effectively models chronic hyperglycemia/hypoinsulinemia and is a tool for studying the effects of T2D-related stress. Within the islets of these mice, genes related to glycolysis, polyol, and PPP are increased, whereas inner and outer mitochondrial membrane, mitochondrial ribosomal, and ETC proteins were diminished (Haythorne et al., 2019). Metabolic studies revealed that ATP and OXPHOS were decreased alongside upregulation of PPP and accumulation of glycogen, all of which correlated with hyperglycemia in INS1 cells (Haythorne et al., 2019). While gluco/lipotoxic culture conditions clearly impair mitochondrial metabolism, function, and dynamics, the precise molecular mechanisms and causal role of mitochondrial dysfunction in diabetes in this model remains uncertain.
It is important to note that in vitro gluco/lipotoxicity studies have significant limitations. They are intended to model the chronic and progressive hyperglycemia and hyperlipidemia in T2D but are conducted in much shorter acute settings (years versus days). Additional limitations arise from concentrations used for glucose and FFAs. Higher concentrations of glucose (8–33 mM) may hinder β-oxidation and metabolism of FFAs. The concentrations of glucose used in cell culture (5–33 mM glucose) often surpass postprandial levels experienced in vivo in mice (8–15 mM), non-diabetic individuals (6–10 mM), and T2D humans (>10 mM glucose) (Bruce et al., 2021). FFAs are observed to range from 100 µM to >1 mM in human plasma (Huber and Kleinfeld, 2017). Ideally, in vitro lipotoxicity models would reflect FFA concentrations experienced by islets in vivo. However, measuring these concentrations is challenging due to confounding factors including release of FFA from cells, lipoprotein activity, and local delivery of FFA to islets (Lytrivi et al., 2020). Additional variability arises from technical aspects of FFAs treatment in cell culture, which include variations in the purity of mixtures and methods complexing to bovine serum albumin at differing ratios with changing affinities based on the length of carbon chains and saturation of FFAs (Lytrivi et al., 2020). In the absence of high glucose FFAs do not impair insulin production or β-cell survival (Jacqueminet et al., 2000; El-Assaad et al., 2003). In INS1 cells, lipotoxicity does not exert an effect on its own but requires high glucose (11 and 25 mM) to increase triglyceride accumulation, proton leak, and MMP (Oberhauser et al., 2020). Oleate (C18:1) has the strongest effects compared to other FFAs (palmitate; C16:0, linoleate; C18:2, and linolenate; C18:3), and all FFAs except palmitate reduce insulin secretion regardless of glucose concentration (Oberhauser et al., 2020). In this study, oleate was protective against chronic high glucose, and only high glucose increased cleaved caspase 3 (Oberhauser et al., 2020). Interestingly, β-oxidation did not increase with overloading of FFAs at any glucose concentration, suggesting that glucose oxidation is favored and presides over lipid metabolism in INS1 cells (Oberhauser et al., 2020). Human islets may recover from palmitate (2 days; 0.5 mM) or high glucose (2 days; 22.2 mM) treatment alone after a washout period returning to 5.5 mM glucose, but combined glucolipotoxicity has irreversible detrimental effects (Marselli et al., 2020). In the absence of human or clinical data, the importance of lipotoxicity, independent of glucose levels, has been challenged in the field (Weir, 2020). While FFAs have acute effects on insulin secretion, there is overall less evidence that alone they impair β-cell survival in acute in vitro culture settings.
3.2 Dysregulation of mitochondrial calcium in diabetes
As noted in Section 2.3 mitochondrial Ca2+ is necessary for glucose-induced insulin release. MCU depletion in mouse islets exacerbates the effects of glucolipotoxicity, an acute model of metabolic stress related to T2D, by severely diminishing mitochondrial Ca2+ and cytosolic ATP/ADP (Tarasov et al., 2012). MCU also helps regulate cytosolic Ca2+ levels and may draw Ca2+ into the mitochondria to alleviate Ca2+ overload in the cytosol which can result from lipotoxicity in T2D (Ly et al., 2020). The effects of MCU on cytosolic Ca2+ are likely generalized as exemplified in studies on cardiomyocytes in which MCU overexpression drastically depletes cytosolic Ca2+ (Drago et al., 2012). In MIN6 cells, exposure to lipotoxic stress increases expression of MCU at ambient glucose concentrations and silencing of MCU alleviates lipotoxic-induced superoxide/mtROS formation and yet accelerates cytosolic Ca2+ overload (Ly et al., 2020). These studies linking mitochondrial Ca2+ to insulin secretion indicate that the MCU complex is essential for metabolism-insulin secretion coupling, and it may protect β-cells against gluco/lipotoxic stress and improve their capacity to dissipate cytosolic Ca2+ overload.
3.3 Mitochondria and diabetogenic ER stress
Mitochondrial dysfunction in diabetes is associated with ER stress (Halban et al., 2014; Hasnain et al., 2016; Zhang et al., 2020). ER stress contributes to excessive and damaging accumulation of Ca2+ in the mitochondrial matrix leading to inner membrane permeabilization and apoptosis (Deniaud et al., 2008). Gluco/lipotoxicity associated with diabetes also induces ER stress and excess Ca2+ release into the cytosol, further impairing mitochondria and insulin secretion. Interestingly, MCU is upregulated by lipotoxic stress, which may promote detoxification of cytosolic Ca2+ overload whereas knockdown of MCU exacerbates Ca2+ overload, autophagy inhibition, and cytotoxicity in the MIN6 β-cell line (Ly et al., 2020). It was proposed that ER stress-mediated overload of cytosolic Ca2+ may impair autophagy and therefore mitophagy and is another route by which ER stress could impair mitochondrial function (Ly et al., 2020). ER Ca2+ levels are also regulated by the sarco/endoplasmic reticulum calcium ATPase (SERCA), which pumps Ca2+ into the ER and is dependent on ATP for activity. In healthy β-cells, ER Ca2+ can change from 200 to 500 µM in low (5 mM) to high (20 mM) glucose, respectively, and this increase and range of concentrations are required for proper protein folding in the ER (Tengholm et al., 1999; Xu et al., 2015; Idevall-Hagren and Tengholm, 2020). Pharmacologic ER stress increases cytosolic Ca2+ to ∼800 nM, at the expense of ER Ca2+ and can inactivate SERCA (Zhang et al., 2020) (Figure 2). Pharmacologic activation of SERCA with CDN1163 increased ER and mitochondrial Ca2+ content, MMP, OXPHOS, and ATP synthesis, effects that were recapitulated with overexpression of SERCA2a and b (Nguyen et al., 2023). Augmented SERCA activation protected MIN6 cells from lipotoxic-induced cytosolic and mitochondrial ROS (Nguyen et al., 2023). Furthermore, enhanced organellar Ca2+ levels are associated with increased peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) and TFAM expression, suggesting an improved potential for mitochondrial biogenesis (Nguyen et al., 2023). Nature provides additional evidence of the impact of ER-Ca2+ on β-cell dysfunction in diabetes since mutations in the gene wolframin causes diabetes mellitus and non-autoimmune insulin/β-cell-deficiency in Wolfram syndrome Type 1 (Morikawa and Urano, 2022). Interestingly, these mutations also associate with secondary mitochondrial dysfunction although their distinct contribution to diabetes development is uncertain in this context of protein misfolding (La Morgia et al., 2020).
ER stress can impact the cells in non-Ca2+-dependent manners as well, including the UPR. β-cells are uniquely reliant on and sensitive to ER stress (Sharma et al., 2021) because they are the only cells that produce and process high levels of insulin prohormone (Sun et al., 2015; Yong et al., 2021). Although highly associated with both T1 and T2D, it remains uncertain whether proinsulin processing defects induce or result from disease (Sharma et al., 2021). The importance of insulin processing to β-cell function is evident from the numerous mutations in the human insulin gene leading to hormone misfolding, that cause mutant INS-gene-induced diabetes of youth (MIDY) (Haataja et al., 2021). Importantly, serine/threonine-protein kinase/endoribonuclease IRE1 (IRE1), a regulator of UPR, is required for enhancing insulin biosynthesis in the ER in response to transient glucose, however, in chronic glucotoxic conditions it becomes hyperactive and enhances ER stress (Lipson et al., 2006). Blocking the UPR by overexpressing a novel regulator of IRE1, EDEM1, improves glycemia and insulin secretion in rats treated with streptozotocin as a model of β-cell loss (Flintoaca Alexandru et al., 2023). The transcription factor FoxM1 is activated by ER stress in β-cells, whereby it translocates from the nucleus to the mitochondria, directly binds mtDNA, and impairs mitochondrial gene transcription, respiration, and ATP synthesis (Kobiita et al., 2023) (Figure 2). FoxM1 high-expressing β-cells are protected from ER and glucotoxic stress by an overall reduction in protein synthesis and mitochondrial activity and are associated with increased cell cycle progression and cell survival (Kobiita et al., 2023). Taken together, these studies reveal that ER stress can impair mitochondrial function and contribute to diabetes pathogenesis.
3.4 Defective mitophagy in diabetes
Diabetes, particularly T2D, can be referred to as a disease of aging, in that cumulative dysfunction over time can impair β-cells, culminating in an increased prevalence of 1 in 3 people over 65 years old developing T2D (Kalyani et al., 2017). In addition, certain aspects of aging may enhance the risk of developing T2D in elderly populations, such as sarcopenia (Mesinovic et al., 2019; Chen et al., 2023), which may be dependent on declining mitochondrial function with age. Mitophagy has the clearest connection to diseases of aging since its impacts are less pronounced acutely, and rather develop over time as damaged mitochondria accumulate. In human T2D islets, GSIS and ATP are diminished and associated with loss of mitochondrial hyperpolarization, swollen cristae, and increased mitochondrial density volume (Anello et al., 2005), and these functional and structural defects imply reduced mitophagy. In response to HFD, enlarged islets have elevated mitophagy due to hypoxia and the hypoxia-inducible factor 1-ɑ (HIF-1ɑ)/BCL2 interacting protein 3 (BNIP3) axis (Aoyagi et al., 2023). Despite the increased mitophagy, damaged mitochondria still accumulated more in enlarged islets, likely due to increased ROS. Another regulator of mitophagy via Parkin is the mitochondrial Rho protein 1 (MIRO1), since its depletion in β-cells leads to mitochondrial dysfunction, reduced insulin release, and reduced mitophagy (Chen et al., 2017). Expression of MIRO1 was reduced in islets from T2D patients and db/db diabetic mice, supporting a possible role of mitophagy deficiency in the pathogenicity of T2D. Transcription factor EB (TFEB) may regulate mitophagy via its established role in autophagy gene expression and lysosome formation (Oh et al., 2023). In β-cells treated with rotenone or oligomycin, TFEB translocates to mitochondria and correlates with lysosomal Ca2+ release and mitophagy (Park et al., 2022). TFEBKO mice were sensitized to HFD-induced glucose intolerance and impaired insulin secretion, and had reduced mitophagy, cytochrome c release, and oxygen consumption (Park et al., 2022). The role of mitochondrial transcription factor B2 (TFB2M), another regulator of mitochondrial gene expression, was observed in mouse TFB2M-KO β-cells which had impaired mitophagy via defects in autophagosome-lysosome fusion, and this was associated with mitochondrial dysfunction, and deficient insulin release (Nicholas et al., 2017).
The T1D susceptibility gene, C-type lectin domain containing 16a (Clec16a) plays a prominent role in mitophagy in β-cells through the PINK1 target Nrdp1 E3 ubiquitin ligase (Sidarala et al., 2020). Clec16a KO islets have defective mitochondria with impaired oxygen consumption and ATP levels and reduced insulin secretion (Soleimanpour et al., 2014). Pro-inflammatory cytokines induce mitophagy as a protective response, and mitophagy-deficient β-cells accumulated dysfunctional mitochondria and had increased cell death, whereas overexpression of Clec16 rescued viability and mitophagy in human β-cells (Sidarala et al., 2020). Furthermore, β-cell-specific Clec16a-KO mice developed more severe diabetes after multiple low-dose streptozotocin treatments, a model of β-cell loss in T1D (Sidarala et al., 2020).
Another potential player is the protein Nor1, a member of the Nr4a subfamily of nuclear receptors, whose expression is increased in T2D islets and correlated with increased β-cell apoptosis (Close et al., 2020). Upon pro-inflammatory cytokine treatment, Nor1 translocates to mitochondria and is associated with decreased GSIS, glucose oxidation, and ATP synthesis, possibly due to increased mitochondrial fragmentation and mitophagy (Close et al., 2020). Mitophagy driven through these pathways may be independent of Parkin since ParkinKO β-cells display no phenotype (Corsa et al., 2019), however, Parkin may yet be relevant under diabetogenic stress conditions inducing β-cell loss (Hoshino et al., 2014).
4 The interplay of inflammation and mitochondria in diabetes
Autoimmunity in T1D involves a complex interplay of innate and adaptive immune cell effectors contributing to β-cell inflammation at different stages of disease. Cytotoxic T-cells have the capacity to directly eliminate β-cells through mechanisms involving perforin and granzyme B, engaging mitochondrial cell death pathways (Thomas et al., 2010; Chen et al., 2018). MtROS in both β-cells and immune effector cells can also play a pivotal role in inflammation and β-cell death in T1D (Chen et al., 2018). For instance, autoreactive T-cell antigen-specific activation is dependent on mtROS which subsequently hyperpolarizes MMP and can impair mitochondrial function (Chen et al., 2018). Scavenging excess mtROS in diabetogenic T-cells reduces their shift to aerobic glycolysis and impairs their ability to induce diabetes following adoptive transfer (Previte et al., 2017). Recently, tryptophan metabolism was found to protect against in vitro T-cell mediated β-cell killing through PD-L1 agonism (Rachdi et al., 2023). This protection may involve mitochondrial metabolism, as exogenous tryptophan may enhance NAD and mitochondrial function via the kynurenine pathway (Castro-Portuguez and Sutphin, 2020). In addition, the mitochondrial gene NADH dehydrogenase 2 (mt-ND2), a T1D-associated SNP, demonstrates a resistant allele that prevents diabetes in NOD mice, a model of T1D, following adoptive transfer of diabetogenic T-cells, and reduces T-cell mediated killing of a β-cell line in vitro (Chen et al., 2011). Macrophages also contribute to β-cell inflammation in T1D, potentially by presenting β-cell antigens, releasing mtROS, and amplifying proinflammatory cytokines signals leading to β-cell destruction (Padgett et al., 2013; Burg and Tse, 2018; Chen et al., 2018; Zirpel and Roep, 2021).
In T2D, autoimmunity-like inflammation termed ‘sterile inflammation’ has recently emerged as an etiology. This refers to inflammation occurring in the absence of overt pathogen or infection. Mitochondrial dysfunction, mtDNA mutations, and ROS have been implicated in the process of ‘inflammaging’ a state of chronic inflammation associated with aging and involved in T2D development (Zuo et al., 2019; Dabravolski et al., 2021). Another term used to describe chronic inflammation in T2D is ‘metaflammation’, a state of inflammation stimulated by an excess of nutrients and aging (Prattichizzo et al., 2018). Factors and cytokines derived from adipose in obesity, such as TNF and IL-6, may amplify β-cell sterile inflammation and NFkB-signaling contributing to T2D development (Biondi et al., 2022). On the other hand, cellular senescence, and senescence-associated secretory phenotype (SASP) cytokines (IL-1ɑ/β, IL-6, and TNF) are chronically elevated in diabetes patients, suggesting that senescence also influences inflammation associated with T2D and excess nutrients (Prattichizzo et al., 2016). Islet-resident macrophages are the primary immune cell effector implicated in T2D-related β-cell inflammation (Ying et al., 2019). We anticipate that macrophages may respond to metabolic signals originating from β-cells, including mitochondrially-derived metabolites and factors. Efferocytosis of dying β-cells in mouse models of β-cell loss stimulates macrophages to secrete insulin growth factor 1 (IGF-1) potentially enhancing β-cell renewal and playing a role in β-cell mass expansion in prediabetes and early phases of T2D (Nackiewicz et al., 2020). Islet-resident macrophages increase mitochondrial OXPHOS gene expression following β-cell loss in mice (Nackiewicz et al., 2020).
Mitochondria can directly stimulate inflammation through the release of damage-associated molecular patterns (mtDAMPs) (Wang et al., 2023). Cytosolic free mtDNA can act as a potential DAMP (Figure 2), as it is particularly sensitive to damage by ROS because it lacks histones for mtDNA protection and has no apparent mtDNA repair response (Yakes and Van Houten, 1997). Increasing studies have documented that release of mtDNA activates the cyclic GMP-AMP synthase - stimulator of interferon genes (cGAS-STING) pathway triggering adipose inflammation in obesity and T2D (Bai et al., 2017; Bai and Liu, 2019; Bai et al., 2020). In mice fed HFD, mt-DNA release induced β-cell senescence and was dependent on cGAS-STING activation (Yakes and Van Houten, 1997). However, STING may play additional roles in β-cell GSIS, as STING knockout mice, although protected from insulin resistance exhibited diminished insulin responses (Qiao et al., 2022). Mitochondrially-encoded peptides are another means by which mitochondria may influence inflammation. The mitochondrial peptide MOTS-c (mitochondrial open reading frame of the 12S rRNA type-c) is suggested to have anti-inflammatory properties, protecting NOD mice against T-cell-mediated β-cell destruction through a mechanism involving T cell receptor (TCR) and mammalian target of rapamycin (mTOR) signaling (Kong et al., 2021). Circulating levels of MOTS-c may also be reduced in humans with T2D in correlation with poor glycemic control (Du et al., 2018; Kong et al., 2023).
Mitochondria can also play a role in molecular signaling of inflammation-induced β-cell death although a precise singular molecular mechanism remains elusive. Pro-inflammatory cytokines are frequently used to model β-cell death due to inflammation in T1 and T2D. Blocking apoptosome formation with knockdown of apoptotic protease activating factor 1 (APAF-1) does not protect INS1 cells from cytokine-mediated cell death (Collier et al., 2011). However, depletion of mt-DNA and thus mitochondria, from a human β-cell line completely abolishes cytokine-induced cell death (Lightfoot et al., 2011). In another human β-cell line, cytokines were similarly found to induce caspase-mediated apoptosis (Frorup et al., 2023). Inflammation-induced β-cell death may involve species-specific mechanisms. Currently, the precise mechanism is not fully defined, and the in vivo relevance is undefined (Usmani-Brown et al., 2019). Pro-inflammatory cytokines stimulate inflammatory signaling via mitochondria, impair OXPHOS, induce mitophagy and increase mtROS (Lambelet et al., 2018; Sidarala et al., 2020). Human islets treated with pro-inflammatory cytokines (IL1β, TNF, IFNγ) have altered mitochondrial metabolite pathways (reduced pyruvate carboxylation and arginine; increased citrulline and kynurenine), diminished mitochondrial protein turnover, and increased mitochondrial oxidation of glutathione (Garcia-Contreras et al., 2017; Fu et al., 2020; Fu et al., 2021). In INS1 cells, proinflammatory cytokines restrict GSIS by impairing pyruvate oxidation, but this occurs independently of coupling to ATP synthesis (Barlow et al., 2018). Collectively, the evidence described in this section indicates that mitochondria influence intrinsic β-cell demise and immune effector activity thereby influencing β-cell inflammation in both T1 and T2D. Our understanding is still preliminary and more detailed molecular analyses of the role of mitochondria in diabetic inflammation are needed in future studies.
5 Other pathways influencing mitochondria in β-cells
Few mitochondrial proteins, other than metabolic enzymes/respiratory complex components, have been tested in loss of function studies in pancreatic β-cells. Table 1 summarizes the mitochondrial and non-mitochondrial genes/proteins described in this section and their roles in β-cells. Deficiency in the expression of the mitochondrial inner membrane protein, MPV17, has been linked to ROS accumulation, mtDNA mutations, and cell death in different cell types. In contrast, β-cell-specific MPV17 knockout mice are protected from apoptosis induced by diabetogenic paradigms (STZ, INS2Akita mutation) associated with T1D, thus it may have a proapoptotic role in β-cells (Tang et al., 2023). Several mitoribosomal proteins (MRPs) are downregulated in T2D islets, and depletion of MRPL59 in haploinsufficient mice led to insulin secretion defects and exacerbated hyperglycemia in response to HFD (Hong et al., 2022). The transcription factor B1 mitochondrial (TFB1M) is linked to the development of T2D by reducing insulin secretion and promoting hyperglycemia (Koeck et al., 2011). Knockdown of a nuclear TFB1M homolog, dimethyladenosine transferase 1 homolog (DIMT1), a ribosomal RNA methyltransferase, reduces levels of OXPHOS, ATP synthase, ETC genes, and also disrupts mitochondrial function, MMP hyperpolarization, and insulin secretion in response to glucose (Verma et al., 2022).
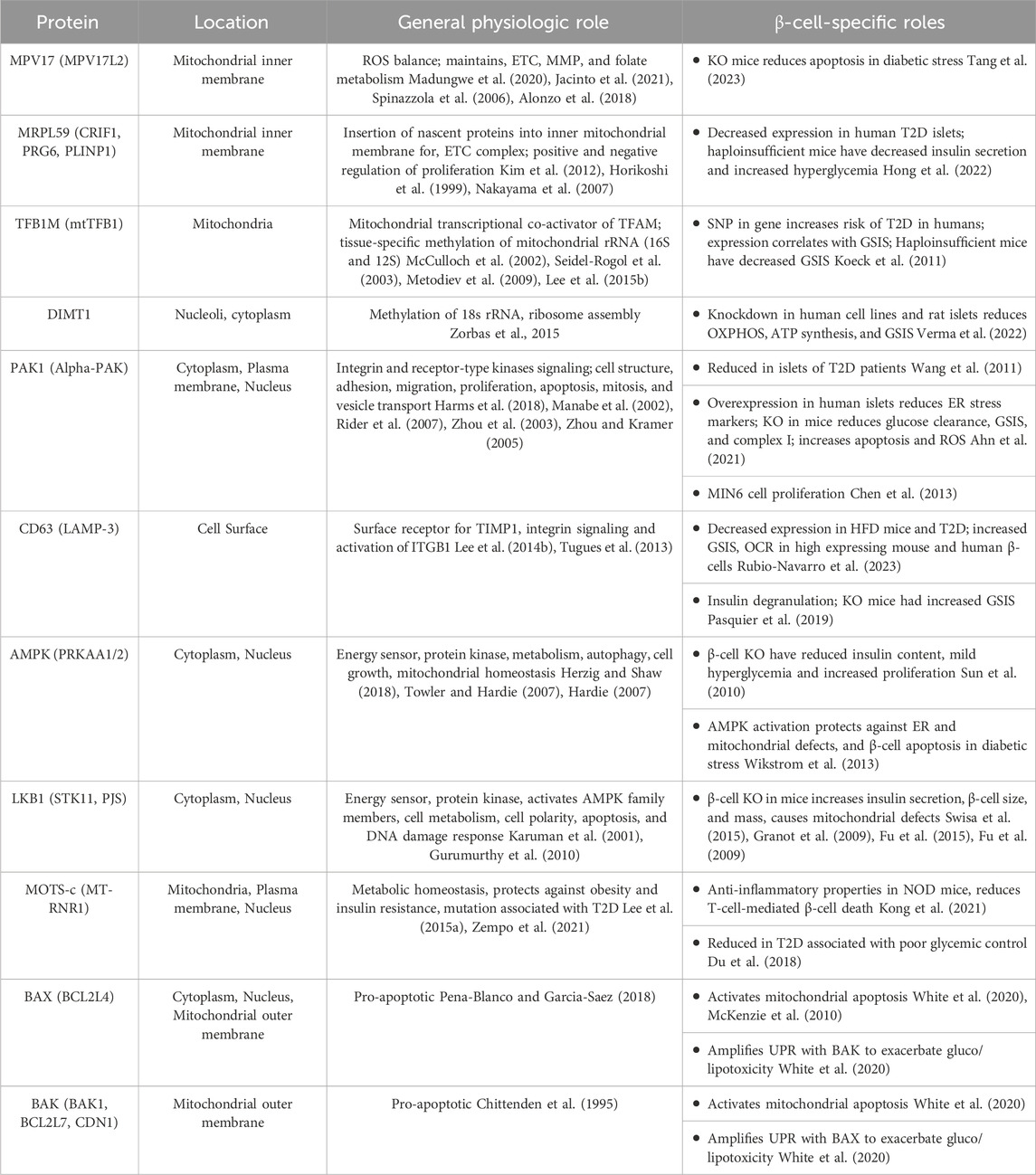
TABLE 1. Other mitochondrial and non-mitochondrial proteins impacting β-cell mitochondrial function. Protein name and localization verified on Uniprot, general physiologic roles and β-cell-specific roles are described.
The serine/threonine p21-activated kinase 1, PAK1, is required for β-cell replication mass and is reduced in T2D islets (Wang et al., 2011; Chen et al., 2013). β-cell-specific depletion of PAK1 impaired glucose tolerance and elevated β-cell apoptosis in mice. PAK1KO β-cells have fewer mitochondria, reduced GSIS, reduced mitochondrial ETC (complex I), and elevated ROS, whereas overexpression of PAK1 in human T2D islets reduces markers of ER stress (Ahn et al., 2021).
Single-cell transcriptomics studies on islets revealed a β-cell subset expressing high levels of CD63 and mitochondrial genes (Rubio-Navarro et al., 2023). This population was diminished in islets from HFD mice, db/db mice, and T2D human islets (Rubio-Navarro et al., 2023). Pseudo-islets generated from FACS-purified CD63 high cells had increased GSIS, enhanced mitochondrial activity, and restored glycemia when transplanted in NOD SCID diabetic mice compared to CD63 low-expressing pseudo-islets (Rubio-Navarro et al., 2023). On the other hand, lysosomal CD63 may contribute to insulin degranulation in ob/ob diabetic mice (Pasquier et al., 2019). In INS1 cells, in vitro stress-induced degranulation required CD63, and islets from CD63 knockout mice had reduced degranulation and increased GSIS (Pasquier et al., 2019). It would be helpful to know in future studies if CD63 localization determines differential effects on β-cell health, it is possible that CD63 plays a role outside of insulin granules and/or lysosomes producing beneficial effects.
Adenosine monophosphate–activated protein kinase (AMPK) is a master regulator of cellular energy sensing and as such plays homeostatic roles in regulating mitochondrial structure and function (Hardie et al., 2016; Herzig and Shaw, 2018). In β-cells, AMPK attenuates insulin release by regulating β-cell mass and insulin content via its ability to inhibit mTOR and yet has limited effects on acute insulin secretion during high glucose when AMPK is inactivated (Fu et al., 2013; Rourke et al., 2018). Double knockout of AMPK alpha 1 and 2 catalytic subunits revealed that AMPK depletion leads to mild hyperglycemia and insulin deficiency, increased replication but reduced β-cell size (Sun et al., 2010). Loss of energy sensing LKB1, an upstream AMPK kinase, increases insulin secretion, β-cell size and mass, and yet also leads to profound mitochondrial defects (Fu et al., 2009; Granot et al., 2009; Fu et al., 2015; Swisa et al., 2015). The defects in mitochondrial metabolism and respiration are dependent on a block in pyruvate metabolism, as respiration was restored with pyruvate supplementation (Fu et al., 2015). It is unlikely that the mitochondrial effects of LKB1 are due to AMPK given the lack of overlapping phenotypes between their respective knockout models. Interestingly, AMPK activation can prevent defects in ER and mitochondrial morphology and β-cell apoptosis during lipotoxic stress in a manner dependent on DRP1 (Wikstrom et al., 2013). Postnatal developmental studies found that β-cell maturation during weaning relies on switching from mTOR to AMPK signaling that promotes a shift to oxidative metabolism and enhanced mitochondrial biogenesis as β-cells mature (Jaafar et al., 2019). In db/db T2D-like diabetic mouse islets, the mTOR to AMPK switch is reversed and associated with a loss of oxidative metabolism and de-differentiation of β-cells (Jaafar et al., 2019). In addition, chronic hyperglycemia enhances mTOR and inhibits AMPK which is associated with decreased PDH activity and may underlie reduced oxidative metabolism during T2D-related diabetogenic metabolic stress (Haythorne et al., 2022). The role of AMPK, LKB1, and mTOR in regulating β-cell mitochondria is still under development with unanswered questions on the contribution of potential direct mitochondrial targets or cellular energy sensing resulting in these effects.
6 Monogenic forms of mitochondrial diabetes
Additional evidence of the impact of mitochondria on human β-cell function is evident from mutations or depletions in mtDNA that cause mitochondrial diabetes. This form of diabetes can also result from mutations of genes encoding mitochondrial proteins in the nuclear DNA. It is estimated that over 80% of pathogenic mutations in mtDNA occur in the MT-TL1 gene, wherein the m.3243A>G mutation is associated with maternally inherited diabetes and deafness (MIDD) and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) (Ryytty and Hamalainen, 2023). Patients with MIDD have a higher risk of developing diabetes, and the m.3243A>G mutation is a biomarker that follows a maternal inheritance pattern (Zhang et al., 2015). Another pathogenic mutation can occur in MRPs, specifically, patients with mutations in MRPL9, MRPL27, and MRPL45 have an increased risk of developing diabetes (Sylvester et al., 2004; Hong et al., 2022). In general, in these diseases, symptoms manifest as mutations in mtDNA accumulate in tissues with a high reliance on oxidative metabolism, such as brain/central nervous system, muscle, pancreas and kidneys. Diabetes is the most common endocrinopathy, however clinical symptoms vary widely, likely due to heteroplasmic mutations with the presence of wild-type and mutated mtDNA (Russell et al., 2020). The heterogeneity makes it difficult to isolate precise disease mechanisms, and even the same mutations have high clinical heterogeneity. Disease onset is thought to result when a critical threshold of mtDNA mutation is reached on a per-cell basis (Russell et al., 2020). Experimental therapeutic strategies are exploring the reversal of mitochondrial defects (increasing OXPHOS, mitophagy, mitochondrial biogenesis, and antioxidants) rather than treating the clinical symptoms. Mitochondrial diabetes development is primarily due to β-cell deficiencies and unlike T2D, it less often involves insulin resistance and is frequently treated with insulin supplementation (Karaa and Goldstein, 2015).
7 Mitochondria in stem cell-derived β-cells/islets
The low availability of human islets is a major roadblock for islet cell transplantation therapy, which is the only cellular strategy to replenish insulin supply in T1D patients. The development of stem cell (SC) -derived pancreatic islets and β-cells provides a promising solution for therapeutic challenges (Pagliuca et al., 2014; Rezania et al., 2014). Additional benefits of this system include the generation of a universal donor, the potential for genetically modifying these cells to evade attack by the immune system, improved functional capacity, and a safety switch that can prevent aberrant replication or outcomes. SC-islets generated in vitro remain as immature β-cells and only fully mature in vivo after transplantation (Pagliuca et al., 2014; Rezania et al., 2014). SC-islet maturity is evaluated in vitro by assessing GSIS, which is significantly reduced and lacking in biphasic pattern compared to primary islets. Studies on SC-islet responses to glucose revealed that SC-islets have a significantly lower capacity for OXPHOS in response to glucose, likely due to a block in glycolysis that reduces pyruvate synthesis and impairs mitochondrial metabolism and supplementation with mitochondrial substrates pyruvate and succinate improves in vitro GSIS (Davis et al., 2020). Another study found metabolic deficits were notable in glucose-derived TCA intermediates and glutathione in SC-islets compared to primary islets (Balboa et al., 2022). Cell clustering in vitro seems to improve oxygen consumption, mitochondrial biogenesis, and insulin secretion, implying enhanced SC-derived β-cell maturation (Nair et al., 2019). Whether enhancing mitochondrial metabolism and OXPHOS can actively improve β-cell differentiation in SC-derived islets or if reduced mitochondrial function is simply a consequence of immature β-cells is an interesting open question.
8 The ugly: are mitochondria a cause or effect of diabetes?
It is exceptionally challenging to separate mitochondrial defects that are consequential versus causative in diabetes. Numerous observations of mitochondrial defects co-segregate with pancreatic β-cell dysfunction in diabetes, but few have tested whether improving the mitochondrial defects alone can ameliorate or reverse diabetes. We emphasize that the following aspects of mitochondria are supported by strong evidence in playing a direct, causal role in β-cell health and dysfunction in diabetes as opposed to merely being a consequence of it: Ca2+ uptake (Alam et al., 2012; Tarasov et al., 2012; Vishnu et al., 2021), bioenergetics (Zhang et al., 2011; Inoue et al., 2022; Lang et al., 2023), metabolism (Patterson et al., 2014; Vigueira et al., 2014; Fu et al., 2020; Lewandowski et al., 2020; Fu et al., 2021), mitophagy (Soleimanpour et al., 2014; Chen et al., 2017; Sidarala et al., 2020), and fission/fusion (Twig et al., 2008; Molina et al., 2009; Reinhardt et al., 2016; Hennings et al., 2018; Georgiadou et al., 2022; Sidarala et al., 2022) and mitoribosomes/translation (Hong et al., 2022; Verma et al., 2022). In gluco/lipotoxic stress, β-cell death is preventable with antioxidants and cellular damage is dependent on ROS, which is mainly generated in mitochondria, thus mitochondria can be attributed a causal role in this in vitro model of metabolic stress.
Cause versus effect is difficult to assess in mitochondrial bioenergetics and metabolism due to the numerous intermediate steps and bidirectional pathways. One of the key biochemistry tools that is lacking in the β-cell field is the ability to test predictions on purified mitochondria. The challenges include low starting material when isolating β-cells out of whole islets, high potential for insulin granules to contaminate mitochondrial preps, and of contamination from other islet cell types. In addition, an integrated understanding of the role of mitochondria in GSIS would benefit from more electrophysiologists working together with mitochondrial metabolism and bioenergetics experts (Nicholls, 2016). There is more evidence for the role of mitochondria in T2D, however, in the context of T1D, the heavy importance of β-cell loss implicates mitochondrial cell death pathways, for more on these aspects we refer the reader to other reviews (Johnson and Luciani, 2010; Vig et al., 2021). It is our opinion that the evidence generally supports that mitochondrial cell death contributes to mechanisms of β-cell death in diabetes.
9 Conclusion and additional considerations
In conclusion, the intricate interplay between mitochondria and diabetes, particularly in the context of pancreatic β-cells, underscores the pivotal role these organelles play in maintaining cellular homeostasis and function. Mitochondrial dysfunction is synonymous with β-cell dysfunction in diabetes, it impairs insulin secretion and sensitizes β-cells to an environment high in metabolic/nutrient stress and cell death. Understanding the molecular mechanisms governing mitochondrial function in pancreatic β-cells is essential for developing targeted therapeutic interventions aimed at preserving or restoring their vital role in glucose homeostasis. One promising candidate is Metformin, which improved mitochondrial function/fitness in peripheral blood mononuclear cells and reduced hyperglycemia in naïve T2D patients treated for 3 months (Bhansali et al., 2020). Observations in this study suggested that Metformin increased mitophagy and reduced mtROS and MMP (Bhansali et al., 2020).
Improvements in technology and the development of new tools to study mitochondria should improve our understanding of their causal roles in β-cell health and demise. An example is advances in Cryo-EM-Tomography allow us to visualize the entire β-cell mitochondrial network and it would be interesting to see how/if it changes in diabetes and in response to diabetogenic stress (hyperglycemia, inflammation, ER, oxidative). It would also be interesting to test how the mitochondrial network structurally impacts insulin granule arrangement, as well as lipid droplet dynamics, a feature more prominent in human β-cells. Mitochondrial heterogeneity is another important consideration relevant to β-cell health, metabolism, function, and survival, as we did not describe this area, we refer the reader to a recent review (Ngo et al., 2021). Much less is known about the role of mitochondria in other islet cell types, however, these are likely to be important as other islet cells are responsive to glucose, have alternate hormonal action to β-cells, and can impact β-cells activity through a paracrine- and glucose-dependent manner (Capozzi et al., 2019). Another area of interest is the study of inter-organ crosstalk (Evans and Wei, 2022; Langlois et al., 2022), for example, understanding how the gut microbiome impacts β-cell function, survival and inflammation and the role of mitochondria and related-secreted factors/exosomes is an understudied area (Fernandez-Millan and Guillen, 2022; Wei et al., 2023). Overall, our current understanding supports that mitochondrial bioenergetics, metabolism, structure, and biomass are determinants of β-cell health and their dysfunction likely plays a causal role in diabetes development. Future studies using new technologies to study mitochondria in primary β-cells will greatly enhance our knowledge of the importance of these dynamic organelles in β-cell health and diabetes.
Author contributions
AR: Writing–original draft, Writing–review and editing. BW: Writing–original draft, Writing–review and editing. AF: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the Juvenile Diabetes Research Foundation (3-COE-2023-1308-A-N).
Acknowledgments
Several icons and graphics in our figures were sourced from Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abulizi, A., Cardone, R. L., Stark, R., Lewandowski, S. L., Zhao, X., Hillion, J., et al. (2020). Multi-Tissue acceleration of the mitochondrial phosphoenolpyruvate cycle improves whole-body metabolic health. Cell Metab. 32, 751–766. doi:10.1016/j.cmet.2020.10.006
Ademowo, O. S., Dias, H. K. I., Burton, D. G. A., and Griffiths, H. R. (2017). Lipid (per) oxidation in mitochondria: an emerging target in the ageing process? Biogerontology 18, 859–879. doi:10.1007/s10522-017-9710-z
Affourtit, C., and Brand, M. D. (2006). Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem. J. 393, 151–159. doi:10.1042/BJ20051280
Ahn, M., Oh, E., Mccown, E. M., Wang, X., Veluthakal, R., and Thurmond, D. C. (2021). A requirement for PAK1 to support mitochondrial function and maintain cellular redox balance via electron transport chain proteins to prevent β-cell apoptosis. Metabolism 115, 154431. doi:10.1016/j.metabol.2020.154431
Alam, M. R., Groschner, L. N., Parichatikanond, W., Kuo, L., Bondarenko, A. I., Rost, R., et al. (2012). Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J. Biol. Chem. 287, 34445–34454. doi:10.1074/jbc.M112.392084
Alonzo, J. R., Venkataraman, C., Field, M. S., and Stover, P. J. (2018). The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J. Biol. Chem. 293, 20285–20294. doi:10.1074/jbc.RA118.004788
Anello, M., Lupi, R., Spampinato, D., Piro, S., Masini, M., Boggi, U., et al. (2005). Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48, 282–289. doi:10.1007/s00125-004-1627-9
Aoyagi, K., Yamashita, S. I., Akimoto, Y., Nishiwaki, C., Nakamichi, Y., Udagawa, H., et al. (2023). A new beta cell-specific mitophagy reporter mouse shows that metabolic stress leads to accumulation of dysfunctional mitochondria despite increased mitophagy. Diabetologia 66, 147–162. doi:10.1007/s00125-022-05800-8
Ashcroft, F. M., and Rorsman, P. (2013). K(ATP) channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol. 9, 660–669. doi:10.1038/nrendo.2013.166
Attane, C., Peyot, M. L., Lussier, R., Poursharifi, P., Zhao, S., Zhang, D., et al. (2016). A beta cell ATGL-lipolysis/adipose tissue axis controls energy homeostasis and body weight via insulin secretion in mice. Diabetologia 59, 2654–2663. doi:10.1007/s00125-016-4105-2
Bai, J., Cervantes, C., He, S., He, J., Plasko, G. R., Wen, J., et al. (2020). Mitochondrial stress-activated cGAS-STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun. Biol. 3, 257. doi:10.1038/s42003-020-0986-1
Bai, J., Cervantes, C., Liu, J., He, S., Zhou, H., Zhang, B., et al. (2017). DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. U. S. A. 114, 12196–12201. doi:10.1073/pnas.1708744114
Bai, J., and Liu, F. (2019). The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes 68, 1099–1108. doi:10.2337/dbi18-0052
Balboa, D., Barsby, T., Lithovius, V., Saarimaki-Vire, J., Omar-Hmeadi, M., Dyachok, O., et al. (2022). Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 40, 1042–1055. doi:10.1038/s41587-022-01219-z
Barlow, J., Solomon, T. P. J., and Affourtit, C. (2018). Pro-inflammatory cytokines attenuate glucose-stimulated insulin secretion from INS-1E insulinoma cells by restricting mitochondrial pyruvate oxidation capacity - novel mechanistic insight from real-time analysis of oxidative phosphorylation. PLoS One 13, e0199505. doi:10.1371/journal.pone.0199505
Berger, C., and Zdzieblo, D. (2020). Glucose transporters in pancreatic islets. Pflugers Arch. 472, 1249–1272. doi:10.1007/s00424-020-02383-4
Bhansali, S., Bhansali, A., Dutta, P., Walia, R., and Dhawan, V. (2020). Metformin upregulates mitophagy in patients with T2DM: a randomized placebo-controlled study. J. Cell Mol. Med. 24, 2832–2846. doi:10.1111/jcmm.14834
Biondi, G., Marrano, N., Borrelli, A., Rella, M., Palma, G., Calderoni, I., et al. (2022). Adipose tissue secretion pattern influences β-cell wellness in the transition from obesity to type 2 diabetes. Int. J. Mol. Sci. 23, 5522. doi:10.3390/ijms23105522
Blagov, A. V., Summerhill, V. I., Sukhorukov, V. N., Popov, M. A., Grechko, A. V., and Orekhov, A. N. (2023). Type 1 diabetes mellitus: inflammation, mitophagy, and mitochondrial function. Mitochondrion 72, 11–21. doi:10.1016/j.mito.2023.07.002
Boucher, A., Lu, D., Burgess, S. C., Telemaque-Potts, S., Jensen, M. V., Mulder, H., et al. (2004). Biochemical mechanism of lipid-induced impairment of glucose-stimulated insulin secretion and reversal with a malate analogue. J. Biol. Chem. 279, 27263–27271. doi:10.1074/jbc.M401167200
Bruce, C. R., Hamley, S., Ang, T., Howlett, K. F., Shaw, C. S., and Kowalski, G. M. (2021). Translating glucose tolerance data from mice to humans: insights from stable isotope labelled glucose tolerance tests. Mol. Metab. 53, 101281. doi:10.1016/j.molmet.2021.101281
Burg, A. R., and Tse, H. M. (2018). Redox-sensitive innate immune pathways during macrophage activation in type 1 diabetes. Antioxid. Redox Signal 29, 1373–1398. doi:10.1089/ars.2017.7243
Campbell, J. E., and Newgard, C. B. (2021). Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 22, 142–158. doi:10.1038/s41580-020-00317-7
Capozzi, M. E., Svendsen, B., Encisco, S. E., Lewandowski, S. L., Martin, M. D., Lin, H., et al. (2019). β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4, e126742. doi:10.1172/jci.insight.126742
Castro-Portuguez, R., and Sutphin, G. L. (2020). Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 132, 110841. doi:10.1016/j.exger.2020.110841
Chareyron, I., Christen, S., Moco, S., Valsesia, A., Lassueur, S., Dayon, L., et al. (2020). Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 63, 2628–2640. doi:10.1007/s00125-020-05275-5
Chen, H., Huang, X., Dong, M., Wen, S., Zhou, L., and Yuan, X. (2023). The association between sarcopenia and diabetes: from pathophysiology mechanism to therapeutic strategy. Diabetes Metab. Syndr. Obes. 16, 1541–1554. doi:10.2147/DMSO.S410834
Chen, J., Gusdon, A. M., Piganelli, J., Leiter, E. H., and Mathews, C. E. (2011). mt-Nd2(a) Modifies resistance against autoimmune type 1 diabetes in NOD mice at the level of the pancreatic β-cell. Diabetes 60, 355–359. doi:10.2337/db10-1241
Chen, J., Stimpson, S. E., Fernandez-Bueno, G. A., and Mathews, C. E. (2018). Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid. Redox Signal 29, 1361–1372. doi:10.1089/ars.2017.7346
Chen, L., Liu, C., Gao, J., Xie, Z., Chan, L. W. C., Keating, D. J., et al. (2017). Inhibition of Miro1 disturbs mitophagy and pancreatic β-cell function interfering insulin release via IRS-Akt-Foxo1 in diabetes. Oncotarget 8, 90693–90705. doi:10.18632/oncotarget.20963
Chen, Y. C., Fueger, P. T., and Wang, Z. (2013). Depletion of PAK1 enhances ubiquitin-mediated survivin degradation in pancreatic β-cells. Islets 5, 22–28. doi:10.4161/isl.24029
Chittenden, T., Flemington, C., Houghton, A. B., Ebb, R. G., Gallo, G. J., Elangovan, B., et al. (1995). A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 14, 5589–5596. doi:10.1002/j.1460-2075.1995.tb00246.x
Close, A. F., Dadheech, N., Lemieux, H., Wang, Q., and Buteau, J. (2020). Disruption of beta-cell mitochondrial networks by the orphan nuclear receptor nor1/nr4a3. Cells 9 (1), 168. doi:10.3390/cells9010168
Collier, J. J., Burke, S. J., Eisenhauer, M. E., Lu, D., Sapp, R. C., Frydman, C. J., et al. (2011). Pancreatic β-cell death in response to pro-inflammatory cytokines is distinct from genuine apoptosis. PLoS One 6, e22485. doi:10.1371/journal.pone.0022485
Corsa, C. A. S., Pearson, G. L., Renberg, A., Askar, M. M., Vozheiko, T., Macdougald, O. A., et al. (2019). The E3 ubiquitin ligase parkin is dispensable for metabolic homeostasis in murine pancreatic β cells and adipocytes. J. Biol. Chem. 294, 7296–7307. doi:10.1074/jbc.RA118.006763
Dabravolski, S. A., Orekhova, V. A., Baig, M. S., Bezsonov, E. E., Starodubova, A. V., Popkova, T. V., et al. (2021). The role of mitochondrial mutations and chronic inflammation in diabetes. Int. J. Mol. Sci. 22, 6733. doi:10.3390/ijms22136733
Davis, J. C., Alves, T. C., Helman, A., Chen, J. C., Kenty, J. H., Cardone, R. L., et al. (2020). Glucose response by stem cell-derived β cells In Vitro is inhibited by a bottleneck in glycolysis. Cell Rep. 31, 107623. doi:10.1016/j.celrep.2020.107623
Deas, E., Piipari, K., Machhada, A., Li, A., Gutierrez-Del-Arroyo, A., Withers, D. J., et al. (2014). PINK1 deficiency in β-cells increases basal insulin secretion and improves glucose tolerance in mice. Open Biol. 4, 140051. doi:10.1098/rsob.140051
De Marchi, U., Thevenet, J., Hermant, A., Dioum, E., and Wiederkehr, A. (2014). Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J. Biol. Chem. 289, 9182–9194. doi:10.1074/jbc.M113.513184
Deniaud, A., Sharaf El Dein, O., Maillier, E., Poncet, D., Kroemer, G., Lemaire, C., et al. (2008). Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299. doi:10.1038/sj.onc.1210638
De Vos, A., Heimberg, H., Quartier, E., Huypens, P., Bouwens, L., Pipeleers, D., et al. (1995). Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 96, 2489–2495. doi:10.1172/JCI118308
Drago, I., De Stefani, D., Rizzuto, R., and Pozzan, T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 109, 12986–12991. doi:10.1073/pnas.1210718109
Du, C., Zhang, C., Wu, W., Liang, Y., Wang, A., Wu, S., et al. (2018). Circulating MOTS-c levels are decreased in obese male children and adolescents and associated with insulin resistance. Pediatr. Diabetes 19, 1058–1064. doi:10.1111/pedi.12685
El-Assaad, W., Buteau, J., Peyot, M. L., Nolan, C., Roduit, R., Hardy, S., et al. (2003). Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144, 4154–4163. doi:10.1210/en.2003-0410
Eto, K., Tsubamoto, Y., Terauchi, Y., Sugiyama, T., Kishimoto, T., Takahashi, N., et al. (1999). Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283, 981–985. doi:10.1126/science.283.5404.981
Evans, R. M., and Wei, Z. (2022). Interorgan crosstalk in pancreatic islet function and pathology. FEBS Lett. 596, 607–619. doi:10.1002/1873-3468.14282
Ferdaoussi, M., Dai, X., Jensen, M. V., Wang, R., Peterson, B. S., Huang, C., et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest. 125, 3847–3860. doi:10.1172/JCI82498
Fernandez-Millan, E., and Guillen, C. (2022). Multi-organ crosstalk with endocrine pancreas: a focus on how gut microbiota shapes pancreatic beta-cells. Biomolecules 12, 104. doi:10.3390/biom12010104
Flintoaca Alexandru, P. R., Chiritoiu, G. N., Lixandru, D., Zurac, S., Ionescu-Targoviste, C., and Petrescu, S. M. (2023). EDEM1 regulates the insulin mRNA level by inhibiting the endoplasmic reticulum stress-induced IRE1/JNK/c-Jun pathway. iScience 26, 107956. doi:10.1016/j.isci.2023.107956
Foster, H. R., Ho, T., Potapenko, E., Sdao, S. M., Huang, S. M., Lewandowski, S. L., et al. (2022). β-cell deletion of the PKm1 and PKm2 isoforms of pyruvate kinase in mice reveals their essential role as nutrient sensors for the KATP channel. Elife 11, e79422. doi:10.7554/eLife.79422
Frorup, C., Gerwig, R., Svane, C. A. S., Mendes Lopes De Melo, J., Henriksen, K., Floyel, T., et al. (2023). Characterization of the functional and transcriptomic effects of pro-inflammatory cytokines on human EndoC-βH5 beta cells. Front. Endocrinol. (Lausanne) 14, 1128523. doi:10.3389/fendo.2023.1128523
Fu, A., Alvarez-Perez, J. C., Avizonis, D., Kin, T., Ficarro, S. B., Choi, D. W., et al. (2020). Glucose-dependent partitioning of arginine to the urea cycle protects β-cells from inflammation. Nat. Metab. 2, 432–446. doi:10.1038/s42255-020-0199-4
Fu, A., Eberhard, C. E., and Screaton, R. A. (2013). Role of AMPK in pancreatic beta cell function. Mol. Cell Endocrinol. 366, 127–134. doi:10.1016/j.mce.2012.06.020
Fu, A., Ng, A. C., Depatie, C., Wijesekara, N., He, Y., Wang, G. S., et al. (2009). Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab. 10, 285–295. doi:10.1016/j.cmet.2009.08.008
Fu, A., Robitaille, K., Faubert, B., Reeks, C., Dai, X. Q., Hardy, A. B., et al. (2015). LKB1 couples glucose metabolism to insulin secretion in mice. Diabetologia 58, 1513–1522. doi:10.1007/s00125-015-3579-7
Fu, A., Van Rooyen, L., Evans, L., Armstrong, N., Avizonis, D., Kin, T., et al. (2021). Glucose metabolism and pyruvate carboxylase enhance glutathione synthesis and restrict oxidative stress in pancreatic islets. Cell Rep. 37, 110037. doi:10.1016/j.celrep.2021.110037
Fu, J., Cui, Q., Yang, B., Hou, Y., Wang, H., Xu, Y., et al. (2017). The impairment of glucose-stimulated insulin secretion in pancreatic β-cells caused by prolonged glucotoxicity and lipotoxicity is associated with elevated adaptive antioxidant response. Food Chem. Toxicol. 100, 161–167. doi:10.1016/j.fct.2016.12.016
Garcia-Contreras, M., Tamayo-Garcia, A., Pappan, K. L., Michelotti, G. A., Stabler, C. L., Ricordi, C., et al. (2017). Metabolomics study of the effects of inflammation, hypoxia, and high glucose on isolated human pancreatic islets. J. Proteome Res. 16, 2294–2306. doi:10.1021/acs.jproteome.7b00160
Georgiadou, E., Haythorne, E., Dickerson, M. T., Lopez-Noriega, L., Pullen, T. J., Da Silva Xavier, G., et al. (2020). The pore-forming subunit MCU of the mitochondrial Ca(2+) uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 63, 1368–1381. doi:10.1007/s00125-020-05148-x
Georgiadou, E., Muralidharan, C., Martinez, M., Chabosseau, P., Akalestou, E., Tomas, A., et al. (2022). Mitofusins Mfn1 and Mfn2 are required to preserve glucose- but not incretin-stimulated β-cell connectivity and insulin secretion. Diabetes 71, 1472–1489. doi:10.2337/db21-0800
Gerencser, A. A. (2018). Metabolic activation-driven mitochondrial hyperpolarization predicts insulin secretion in human pancreatic beta-cells. Biochim. Biophys. Acta Bioenerg. 1859, 817–828. doi:10.1016/j.bbabio.2018.06.006
Gerencser, A. A., Mookerjee, S. A., Jastroch, M., and Brand, M. D. (2016). Measurement of the absolute magnitude and time courses of mitochondrial membrane potential in primary and clonal pancreatic beta-cells. PLoS One 11, e0159199. doi:10.1371/journal.pone.0159199
Gheni, G., Ogura, M., Iwasaki, M., Yokoi, N., Minami, K., Nakayama, Y., et al. (2014). Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 9, 661–673. doi:10.1016/j.celrep.2014.09.030
Graciano, M. F., Valle, M. M., Kowluru, A., Curi, R., and Carpinelli, A. R. (2011). Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets 3, 213–223. doi:10.4161/isl.3.5.15935
Granot, Z., Swisa, A., Magenheim, J., Stolovich-Rain, M., Fujimoto, W., Manduchi, E., et al. (2009). LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metab. 10, 296–308. doi:10.1016/j.cmet.2009.08.010
Gurumurthy, S., Xie, S. Z., Alagesan, B., Kim, J., Yusuf, R. Z., Saez, B., et al. (2010). The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 468, 659–663. doi:10.1038/nature09572
Haataja, L., Arunagiri, A., Hassan, A., Regan, K., Tsai, B., Dhayalan, B., et al. (2021). Distinct states of proinsulin misfolding in MIDY. Cell Mol. Life Sci. 78, 6017–6031. doi:10.1007/s00018-021-03871-1
Habarou, F., Brassier, A., Rio, M., Chretien, D., Monnot, S., Barbier, V., et al. (2015). Pyruvate carboxylase deficiency: an underestimated cause of lactic acidosis. Mol. Genet. Metab. Rep. 2, 25–31. doi:10.1016/j.ymgmr.2014.11.001
Halban, P. A., Polonsky, K. S., Bowden, D. W., Hawkins, M. A., Ling, C., Mather, K. J., et al. (2014). β-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37, 1751–1758. doi:10.2337/dc14-0396
Hansen, J. B., Dos Santos, L. R. B., Liu, Y., Prentice, K. J., Teudt, F., Tonnesen, M., et al. (2018). Glucolipotoxic conditions induce β-cell iron import, cytosolic ROS formation and apoptosis. J. Mol. Endocrinol. 61, 69–77. doi:10.1530/JME-17-0262
Hardie, D. G. (2007). AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785. doi:10.1038/nrm2249
Hardie, D. G., Schaffer, B. E., and Brunet, A. (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201. doi:10.1016/j.tcb.2015.10.013
Harms, F. L., Kloth, K., Bley, A., Denecke, J., Santer, R., Lessel, D., et al. (2018). Activating mutations in PAK1, encoding p21-activated kinase 1, cause a neurodevelopmental disorder. Am. J. Hum. Genet. 103, 579–591. doi:10.1016/j.ajhg.2018.09.005
Hasnain, S. Z., Prins, J. B., and Mcguckin, M. A. (2016). Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 56, R33–R54. doi:10.1530/JME-15-0232
Haythorne, E., Lloyd, M., Walsby-Tickle, J., Tarasov, A. I., Sandbrink, J., Portillo, I., et al. (2022). Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat. Commun. 13, 6754. doi:10.1038/s41467-022-34095-x
Haythorne, E., Rohm, M., Van De Bunt, M., Brereton, M. F., Tarasov, A. I., Blacker, T. S., et al. (2019). Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 10, 2474. doi:10.1038/s41467-019-10189-x
Hennings, T. G., Chopra, D. G., Deleon, E. R., Vandeusen, H. R., Sesaki, H., Merrins, M. J., et al. (2018). In vivo deletion of β-cell Drp1 impairs insulin secretion without affecting islet oxygen consumption. Endocrinology 159, 3245–3256. doi:10.1210/en.2018-00445
Henquin, J. C., Dufrane, D., Kerr-Conte, J., and Nenquin, M. (2015). Dynamics of glucose-induced insulin secretion in normal human islets. Am. J. Physiol. Endocrinol. Metab. 309, E640–E650. doi:10.1152/ajpendo.00251.2015
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi:10.1038/nrm.2017.95
Hong, H. J., Joung, K. H., Kim, Y. K., Choi, M. J., Kang, S. G., Kim, J. T., et al. (2022). Mitoribosome insufficiency in β cells is associated with type 2 diabetes-like islet failure. Exp. Mol. Med. 54, 932–945. doi:10.1038/s12276-022-00797-x
Horikoshi, N., Cong, J., Kley, N., and Shenk, T. (1999). Isolation of differentially expressed cDNAs from p53-dependent apoptotic cells: activation of the human homologue of the Drosophila peroxidasin gene. Biochem. Biophys. Res. Commun. 261, 864–869. doi:10.1006/bbrc.1999.1123
Hoshino, A., Ariyoshi, M., Okawa, Y., Kaimoto, S., Uchihashi, M., Fukai, K., et al. (2014). Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. U. S. A. 111, 3116–3121. doi:10.1073/pnas.1318951111
Huber, A. H., and Kleinfeld, A. M. (2017). Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 58, 578–585. doi:10.1194/jlr.M074260
Idevall-Hagren, O., and Tengholm, A. (2020). Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 103, 20–30. doi:10.1016/j.semcdb.2020.01.008
Inoue, R., Tsuno, T., Togashi, Y., Okuyama, T., Sato, A., Nishiyama, K., et al. (2022). Uncoupling protein 2 and aldolase B impact insulin release by modulating mitochondrial function and Ca(2+) release from the ER. iScience 25, 104603. doi:10.1016/j.isci.2022.104603
Jaafar, R., Tran, S., Shah, A. N., Sun, G., Valdearcos, M., Marchetti, P., et al. (2019). mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J. Clin. Invest. 129, 4124–4137. doi:10.1172/JCI127021
Jacinto, S., Guerreiro, P., De Oliveira, R. M., Cunha-Oliveira, T., Santos, M. J., Grazina, M., et al. (2021). MPV17 mutations are associated with a quiescent energetic metabolic profile. Front. Cell Neurosci. 15, 641264. doi:10.3389/fncel.2021.641264
Jacqueminet, S., Briaud, I., Rouault, C., Reach, G., and Poitout, V. (2000). Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism 49, 532–536. doi:10.1016/s0026-0495(00)80021-9
Jesinkey, S. R., Madiraju, A. K., Alves, T. C., Yarborough, O. H., Cardone, R. L., Zhao, X., et al. (2019). Mitochondrial GTP links nutrient sensing to β cell health, mitochondrial morphology, and insulin secretion independent of OxPhos. Cell Rep. 28, 759–772 e10. doi:10.1016/j.celrep.2019.06.058
Jeyarajan, S., Zhang, I. X., Arvan, P., Lentz, S. I., and Satin, L. S. (2023). Simultaneous measurement of changes in mitochondrial and endoplasmic reticulum free calcium in pancreatic beta cells. Biosens. (Basel) 13, 382. doi:10.3390/bios13030382
Jimenez-Sanchez, C., Brun, T., and Maechler, P. (2020). Mitochondrial carriers regulating insulin secretion profiled in human islets upon metabolic stress. Biomolecules 10, 1543. doi:10.3390/biom10111543
Jitrapakdee, S., Wutthisathapornchai, A., Wallace, J. C., and Macdonald, M. J. (2010). Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia 53, 1019–1032. doi:10.1007/s00125-010-1685-0
Johnson, J. D., and Luciani, D. S. (2010). Mechanisms of pancreatic beta-cell apoptosis in diabetes and its therapies. Adv. Exp. Med. Biol. 654, 447–462. doi:10.1007/978-90-481-3271-3_19
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. U. S. A. 96, 13807–13812. doi:10.1073/pnas.96.24.13807
Kabra, U. D., Moruzzi, N., Berggren, P. O., and Jastroch, M. (2022). Drp1 overexpression decreases insulin content in pancreatic MIN6 cells. Int. J. Mol. Sci. 23, 12338. doi:10.3390/ijms232012338
Kalyanaraman, B. (2020). “Pitfalls of reactive oxygen species (ROS) measurements by fluorescent probes and mitochondrial superoxide determination using MitoSOX,” in Measuring oxidants and oxidative stress in biological systems. Editors L. J. Berliner,, and N. L. Parinandi (Cham (CH): Springer Nature).
Kalyani, R. R., Golden, S. H., and Cefalu, W. T. (2017). Diabetes and aging: unique considerations and goals of care. Diabetes Care 40, 440–443. doi:10.2337/dci17-0005
Karaa, A., and Goldstein, A. (2015). The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr. Diabetes 16, 1–9. doi:10.1111/pedi.12223
Karuman, P., Gozani, O., Odze, R. D., Zhou, X. C., Zhu, H., Shaw, R., et al. (2001). The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol. Cell 7, 1307–1319. doi:10.1016/s1097-2765(01)00258-1
Kaufman, B. A., Li, C., and Soleimanpour, S. A. (2015). Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol. Asp. Med. 42, 91–104. doi:10.1016/j.mam.2015.01.004
Khan, A., Ling, Z. C., and Landau, B. R. (1996). Quantifying the carboxylation of pyruvate in pancreatic islets. J. Biol. Chem. 271, 2539–2542. doi:10.1074/jbc.271.5.2539
Kim, S. J., Kwon, M. C., Ryu, M. J., Chung, H. K., Tadi, S., Kim, Y. K., et al. (2012). CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 16, 274–283. doi:10.1016/j.cmet.2012.06.012
Klec, C., Madreiter-Sokolowski, C. T., Stryeck, S., Sachdev, V., Duta-Mare, M., Gottschalk, B., et al. (2019). Glycogen synthase kinase 3 beta controls presenilin-1-mediated endoplasmic reticulum Ca²⁺ leak directed to mitochondria in pancreatic islets and β-cells. Cell Physiol. Biochem. 52, 57–75. doi:10.33594/000000005
Kobiita, A., Silva, P. N., Schmid, M. W., and Stoffel, M. (2023). FoxM1 coordinates cell division, protein synthesis, and mitochondrial activity in a subset of β cells during acute metabolic stress. Cell Rep. 42, 112986. doi:10.1016/j.celrep.2023.112986
Koeck, T., Olsson, A. H., Nitert, M. D., Sharoyko, V. V., Ladenvall, C., Kotova, O., et al. (2011). A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab. 13, 80–91. doi:10.1016/j.cmet.2010.12.007
Kong, B. S., Lee, C., and Cho, Y. M. (2023). Mitochondrial-encoded peptide MOTS-c, diabetes, and aging-related diseases. Diabetes Metab. J. 47, 315–324. doi:10.4093/dmj.2022.0333
Kong, B. S., Min, S. H., Lee, C., and Cho, Y. M. (2021). Mitochondrial-encoded MOTS-c prevents pancreatic islet destruction in autoimmune diabetes. Cell Rep. 36, 109724. doi:10.1016/j.celrep.2021.109724
Lagundzin, D., Hu, W. F., Law, H. C. H., Krieger, K. L., Qiao, F., Clement, E. J., et al. (2019). Delineating the role of FANCA in glucose-stimulated insulin secretion in β cells through its protein interactome. PLoS One 14, e0220568. doi:10.1371/journal.pone.0220568
Lambelet, M., Terra, L. F., Fukaya, M., Meyerovich, K., Labriola, L., Cardozo, A. K., et al. (2018). Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic β-cell apoptosis. Cell Death Dis. 9, 96. doi:10.1038/s41419-017-0121-5
La Morgia, C., Maresca, A., Amore, G., Gramegna, L. L., Carbonelli, M., Scimonelli, E., et al. (2020). Calcium mishandling in absence of primary mitochondrial dysfunction drives cellular pathology in Wolfram Syndrome. Sci. Rep. 10, 4785. doi:10.1038/s41598-020-61735-3
Lang, A. L., Nissanka, N., Louzada, R. A., Tamayo, A., Pereira, E., Moraes, C. T., et al. (2023). A defect in mitochondrial complex III but not in complexes I or IV causes early β-cell dysfunction and hyperglycemia in mice. Diabetes 72, 1262–1276. doi:10.2337/db22-0728
Lang, F., Ullrich, S., and Gulbins, E. (2011). Ceramide formation as a target in beta-cell survival and function. Expert Opin. Ther. Targets 15, 1061–1071. doi:10.1517/14728222.2011.588209
Langlois, A., Dumond, A., Vion, J., Pinget, M., and Bouzakri, K. (2022). Crosstalk communications between islets cells and insulin target tissue: the hidden face of iceberg. Front. Endocrinol. (Lausanne) 13, 836344. doi:10.3389/fendo.2022.836344
Lee, C., Zeng, J., Drew, B. G., Sallam, T., Martin-Montalvo, A., Wan, J., et al. (2015a). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. doi:10.1016/j.cmet.2015.02.009
Lee, J. H., Jung, I. R., Choi, S. E., Lee, S. M., Lee, S. J., Han, S. J., et al. (2014a). Toxicity generated through inhibition of pyruvate carboxylase and carnitine palmitoyl transferase-1 is similar to high glucose/palmitate-induced glucolipotoxicity in INS-1 beta cells. Mol. Cell Endocrinol. 383, 48–59. doi:10.1016/j.mce.2013.12.002
Lee, S., Rose, S., Metodiev, M. D., Becker, L., Vernaleken, A., Klopstock, T., et al. (2015b). Overexpression of the mitochondrial methyltransferase TFB1M in the mouse does not impact mitoribosomal methylation status or hearing. Hum. Mol. Genet. 24, 7286–7294. doi:10.1093/hmg/ddv427
Lee, S. Y., Kim, J. M., Cho, S. Y., Kim, H. S., Shin, H. S., Jeon, J. Y., et al. (2014b). TIMP-1 modulates chemotaxis of human neural stem cells through CD63 and integrin signalling. Biochem. J. 459, 565–576. doi:10.1042/BJ20131119
Lee, Y. H., Kim, J., Park, K., and Lee, M. S. (2019). β-cell autophagy: mechanism and role in β-cell dysfunction. Mol. Metab. 27S, S92–S103. doi:10.1016/j.molmet.2019.06.014
Leloup, C., Tourrel-Cuzin, C., Magnan, C., Karaca, M., Castel, J., Carneiro, L., et al. (2009). Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 58, 673–681. doi:10.2337/db07-1056
Levi-D'ancona, E., Sidarala, V., and Soleimanpour, S. A. (2023). Complementary approaches to interrogate mitophagy flux in pancreatic beta-cells. J. Vis. Exp. doi:10.3791/65789
Lewandowski, S. L., Cardone, R. L., Foster, H. R., Ho, T., Potapenko, E., Poudel, C., et al. (2020). Pyruvate kinase controls signal strength in the insulin secretory pathway. Cell Metab. 32, 736–750. doi:10.1016/j.cmet.2020.10.007
Lightfoot, Y. L., Chen, J., and Mathews, C. E. (2011). Role of the mitochondria in immune-mediated apoptotic death of the human pancreatic β cell line βLox5. PLoS One 6, e20617. doi:10.1371/journal.pone.0020617
Lipson, K. L., Fonseca, S. G., Ishigaki, S., Nguyen, L. X., Foss, E., Bortell, R., et al. (2006). Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 4, 245–254. doi:10.1016/j.cmet.2006.07.007
Ly, L. D., Ly, D. D., Nguyen, N. T., Kim, J. H., Yoo, H., Chung, J., et al. (2020). Mitochondrial Ca(2+) uptake relieves palmitate-induced cytosolic Ca(2+) overload in MIN6 cells. Mol. Cells 43, 66–75. doi:10.14348/molcells.2019.0223
Lytrivi, M., Castell, A. L., Poitout, V., and Cnop, M. (2020). Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 432, 1514–1534. doi:10.1016/j.jmb.2019.09.016
Macdonald, M. J. (1993). Estimates of glycolysis, pyruvate (de)carboxylation, pentose phosphate pathway, and methyl succinate metabolism in incapacitated pancreatic islets. Arch. Biochem. Biophys. 305, 205–214. doi:10.1006/abbi.1993.1413
Macdonald, M. J., Langberg, E. C., Tibell, A., Sabat, G., Kendrick, M. A., Szweda, L. I., et al. (2013). Identification of ATP synthase as a lipid peroxide protein adduct in pancreatic islets from humans with and without type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 98, E727–E731. doi:10.1210/jc.2012-4203
Madungwe, N. B., Feng, Y., Imam Aliagan, A., Tombo, N., Kaya, F., and Bopassa, J. C. (2020). Inner mitochondrial membrane protein MPV17 mutant mice display increased myocardial injury after ischemia/reperfusion. Am. J. Transl. Res. 12, 3412–3428. doi:10.1161/res.121.suppl_1.143
Maechler, P., Carobbio, S., and Rubi, B. (2006). In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int. J. Biochem. Cell Biol. 38, 696–709. doi:10.1016/j.biocel.2005.12.006
Maechler, P., and Wollheim, C. B. (2000). Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. J. Physiol. 529 (1), 49–56. doi:10.1111/j.1469-7793.2000.00049.x
Mailloux, R. J., Fu, A., Robson-Doucette, C., Allister, E. M., Wheeler, M. B., Screaton, R., et al. (2012). Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J. Biol. Chem. 287, 39673–39685. doi:10.1074/jbc.M112.393538
Manabe, R., Kovalenko, M., Webb, D. J., and Horwitz, A. R. (2002). GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 115, 1497–1510. doi:10.1242/jcs.115.7.1497
Maris, M., Robert, S., Waelkens, E., Derua, R., Hernangomez, M. H., D'hertog, W., et al. (2013). Role of the saturated nonesterified fatty acid palmitate in beta cell dysfunction. J. Proteome Res. 12, 347–362. doi:10.1021/pr300596g
Marselli, L., Piron, A., Suleiman, M., Colli, M. L., Yi, X., Khamis, A., et al. (2020). Persistent or transient human β cell dysfunction induced by metabolic stress: specific signatures and shared gene expression with type 2 diabetes. Cell Rep. 33, 108466. doi:10.1016/j.celrep.2020.108466
Martinez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. doi:10.1038/s41467-019-13668-3
Matschinsky, F. M., and Wilson, D. F. (2019). The central role of glucokinase in glucose homeostasis: a perspective 50 Years after demonstrating the presence of the enzyme in islets of langerhans. Front. Physiol. 10, 148. doi:10.3389/fphys.2019.00148
Mcculloch, L. J., Van De Bunt, M., Braun, M., Frayn, K. N., Clark, A., and Gloyn, A. L. (2011). GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 104, 648–653. doi:10.1016/j.ymgme.2011.08.026
Mcculloch, V., Seidel-Rogol, B. L., and Shadel, G. S. (2002). A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol. Cell Biol. 22, 1116–1125. doi:10.1128/mcb.22.4.1116-1125.2002
Mckenzie, M. D., Jamieson, E., Jansen, E. S., Scott, C. L., Huang, D. C., Bouillet, P., et al. (2010). Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes. 59, 644–52.
Merrins, M. J., Corkey, B. E., Kibbey, R. G., and Prentki, M. (2022). Metabolic cycles and signals for insulin secretion. Cell Metab. 34, 947–968. doi:10.1016/j.cmet.2022.06.003
Mesinovic, J., Zengin, A., De Courten, B., Ebeling, P. R., and Scott, D. (2019). Sarcopenia and type 2 diabetes mellitus: a bidirectional relationship. Diabetes Metab. Syndr. Obes. 12, 1057–1072. doi:10.2147/DMSO.S186600
Metodiev, M. D., Lesko, N., Park, C. B., Camara, Y., Shi, Y., Wibom, R., et al. (2009). Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 9, 386–397. doi:10.1016/j.cmet.2009.03.001
Miki, A., Ricordi, C., Sakuma, Y., Yamamoto, T., Misawa, R., Mita, A., et al. (2018). Divergent antioxidant capacity of human islet cell subsets: a potential cause of beta-cell vulnerability in diabetes and islet transplantation. PLoS One 13, e0196570. doi:10.1371/journal.pone.0196570
Molina, A. J., Wikstrom, J. D., Stiles, L., Las, G., Mohamed, H., Elorza, A., et al. (2009). Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58, 2303–2315. doi:10.2337/db07-1781
Morikawa, S., and Urano, F. (2022). The role of ER stress in diabetes: exploring pathological mechanisms using Wolfram syndrome. Int. J. Mol. Sci. 24, 230. doi:10.3390/ijms24010230
Mukai, E., Fujimoto, S., and Inagaki, N. (2022). Role of reactive oxygen species in glucose metabolism disorder in diabetic pancreatic β-cells. Biomolecules 12, 1228. doi:10.3390/biom12091228
Munhoz, A. C., Riva, P., Simoes, D., Curi, R., and Carpinelli, A. R. (2016). Control of insulin secretion by production of reactive oxygen species: study performed in pancreatic islets from fed and 48-hour fasted wistar rats. PLoS One 11, e0158166. doi:10.1371/journal.pone.0158166
Nackiewicz, D., Dan, M., Speck, M., Chow, S. Z., Chen, Y. C., Pospisilik, J. A., et al. (2020). Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience 23, 100775. doi:10.1016/j.isci.2019.100775
Nair, G. G., Liu, J. S., Russ, H. A., Tran, S., Saxton, M. S., Chen, R., et al. (2019). Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 21, 263–274. doi:10.1038/s41556-018-0271-4
Nakayama, K., Nakayama, N., Wang, T. L., and Shih Ie, M. (2007). NAC-1 controls cell growth and survival by repressing transcription of Gadd45GIP1, a candidate tumor suppressor. Cancer Res. 67, 8058–8064. doi:10.1158/0008-5472.CAN-07-1357
Newsholme, P., Keane, K. N., Carlessi, R., and Cruzat, V. (2019). Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 317, C420–C433. doi:10.1152/ajpcell.00141.2019
Ngo, J., Osto, C., Villalobos, F., and Shirihai, O. S. (2021). Mitochondrial heterogeneity in metabolic diseases. Biol. (Basel) 10, 927. doi:10.3390/biology10090927
Nguyen, H. T., Noriega Polo, C., Wiederkehr, A., Wollheim, C. B., and Park, K. S. (2023). CDN1163, an activator of sarco/endoplasmic reticulum Ca2+ ATPase, up-regulates mitochondrial functions and protects against lipotoxicity in pancreatic β-cells. Br. J. Pharmacol. 180, 2762–2776. doi:10.1111/bph.16160
Nicholas, L. M., Valtat, B., Medina, A., Andersson, L., Abels, M., Mollet, I. G., et al. (2017). Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol. Metab. 6, 651–663. doi:10.1016/j.molmet.2017.05.005
Nicholls, D. G. (2016). The pancreatic β-cell: a bioenergetic perspective. Physiol. Rev. 96, 1385–1447. doi:10.1152/physrev.00009.2016
Nita, , Hershfinkel, M., Fishman, D., Ozeri, E., Rutter, G. A., Sensi, S. L., et al. (2012). The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS One 7, e46649. doi:10.1371/journal.pone.0046649
Noda, M., Yamashita, S., Takahashi, N., Eto, K., Shen, L. M., Izumi, K., et al. (2002). Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J. Biol. Chem. 277, 41817–41826. doi:10.1074/jbc.M207690200
Oberhauser, L., Granziera, S., Colom, A., Goujon, A., Lavallard, V., Matile, S., et al. (2020). Palmitate and oleate modify membrane fluidity and kinase activities of INS-1E β-cells alongside altered metabolism-secretion coupling. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118619. doi:10.1016/j.bbamcr.2019.118619
Oh, S. J., Park, K., Sonn, S. K., Oh, G. T., and Lee, M. S. (2023). Pancreatic β-cell mitophagy as an adaptive response to metabolic stress and the underlying mechanism that involves lysosomal Ca2+ release. Exp. Mol. Med. 55, 1922–1932. doi:10.1038/s12276-023-01055-4
Padgett, L. E., Broniowska, K. A., Hansen, P. A., Corbett, J. A., and Tse, H. M. (2013). The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N. Y. Acad. Sci. 1281, 16–35. doi:10.1111/j.1749-6632.2012.06826.x
Pagliuca, F. W., Millman, J. R., Gurtler, M., Segel, M., Van Dervort, A., Ryu, J. H., et al. (2014). Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439. doi:10.1016/j.cell.2014.09.040
Park, K., Lim, H., Kim, J., Hwang, Y., Lee, Y. S., Bae, S. H., et al. (2022). Lysosomal Ca2+-mediated TFEB activation modulates mitophagy and functional adaptation of pancreatic β-cells to metabolic stress. Nat. Commun. 13, 1300. doi:10.1038/s41467-022-28874-9
Pasquier, A., Vivot, K., Erbs, E., Spiegelhalter, C., Zhang, Z., Aubert, V., et al. (2019). Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat. Commun. 10, 3312. doi:10.1038/s41467-019-11170-4
Patterson, J. N., Cousteils, K., Lou, J. W., Manning Fox, J. E., Macdonald, P. E., and Joseph, J. W. (2014). Mitochondrial metabolism of pyruvate is essential for regulating glucose-stimulated insulin secretion. J. Biol. Chem. 289, 13335–13346. doi:10.1074/jbc.M113.521666
Pena-Blanco, A., and Garcia-Saez, A. J. (2018). Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J. 285, 416–431. doi:10.1111/febs.14186
Pi, J., Bai, Y., Zhang, Q., Wong, V., Floering, L. M., Daniel, K., et al. (2007). Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56, 1783–1791. doi:10.2337/db06-1601
Plecita-Hlavata, L., Engstova, H., Holendova, B., Tauber, J., Spacek, T., Petraskova, L., et al. (2020). Mitochondrial superoxide production decreases on glucose-stimulated insulin secretion in pancreatic β cells due to decreasing mitochondrial matrix NADH/NAD+ ratio. Antioxid. Redox Signal 33, 789–815. doi:10.1089/ars.2019.7800
Prattichizzo, F., De Nigris, V., La Sala, L., Procopio, A. D., Olivieri, F., and Ceriello, A. (2016). "Inflammaging" as a druggable target: a senescence-associated secretory phenotype-centered view of type 2 diabetes. Oxid. Med. Cell Longev. 2016, 1810327. doi:10.1155/2016/1810327
Prattichizzo, F., De Nigris, V., Spiga, R., Mancuso, E., La Sala, L., Antonicelli, R., et al. (2018). Inflammageing and metaflammation: the yin and yang of type 2 diabetes. Ageing Res. Rev. 41, 1–17. doi:10.1016/j.arr.2017.10.003
Prentki, M., Corkey, B. E., and Madiraju, S. R. M. (2020). Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 63, 10–20. doi:10.1007/s00125-019-04976-w
Prentki, M., Matschinsky, F. M., and Madiraju, S. R. (2013). Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185. doi:10.1016/j.cmet.2013.05.018
Previte, D. M., O'connor, E. C., Novak, E. A., Martins, C. P., Mollen, K. P., and Piganelli, J. D. (2017). Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS One 12, e0175549. doi:10.1371/journal.pone.0175549
Pullen, T. J., Khan, A. M., Barton, G., Butcher, S. A., Sun, G., and Rutter, G. A. (2010). Identification of genes selectively disallowed in the pancreatic islet. Islets 2, 89–95. doi:10.4161/isl.2.2.11025
Qiao, J., Zhang, Z., Ji, S., Liu, T., Zhang, X., Huang, Y., et al. (2022). A distinct role of STING in regulating glucose homeostasis through insulin sensitivity and insulin secretion. Proc. Natl. Acad. Sci. U. S. A. 119, e2101848119. doi:10.1073/pnas.2101848119
Rachdi, L., Zhou, Z., Berthault, C., Lourenco, C., Fouque, A., Domet, T., et al. (2023). Tryptophan metabolism promotes immune evasion in human pancreatic β cells. EBioMedicine 95, 104740. doi:10.1016/j.ebiom.2023.104740
Rebelato, E., Abdulkader, F., Curi, R., and Carpinelli, A. R. (2011). Control of the intracellular redox state by glucose participates in the insulin secretion mechanism. PLoS One 6, e24507. doi:10.1371/journal.pone.0024507
Reinhardt, F., Schultz, J., Waterstradt, R., and Baltrusch, S. (2016). Drp1 guarding of the mitochondrial network is important for glucose-stimulated insulin secretion in pancreatic beta cells. Biochem. Biophys. Res. Commun. 474, 646–651. doi:10.1016/j.bbrc.2016.04.142
Rezania, A., Bruin, J. E., Arora, P., Rubin, A., Batushansky, I., Asadi, A., et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133. doi:10.1038/nbt.3033
Rider, L., Shatrova, A., Feener, E. P., Webb, L., and Diakonova, M. (2007). JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J. Biol. Chem. 282, 30985–30996. doi:10.1074/jbc.M701794200
Robson-Doucette, C. A., Sultan, S., Allister, E. M., Wikstrom, J. D., Koshkin, V., Bhattacharjee, A., et al. (2011). Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 60, 2710–2719. doi:10.2337/db11-0132
Rorsman, P., and Braun, M. (2013). Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 75, 155–179. doi:10.1146/annurev-physiol-030212-183754
Rourke, J. L., Hu, Q., and Screaton, R. A. (2018). AMPK and friends: central regulators of β cell biology. Trends Endocrinol. Metab. 29, 111–122. doi:10.1016/j.tem.2017.11.007
Rubio-Navarro, A., Gomez-Banoy, N., Stoll, L., Dundar, F., Mawla, A. M., Ma, L., et al. (2023). A beta cell subset with enhanced insulin secretion and glucose metabolism is reduced in type 2 diabetes. Nat. Cell Biol. 25, 565–578. doi:10.1038/s41556-023-01103-1
Russell, O. M., Gorman, G. S., Lightowlers, R. N., and Turnbull, D. M. (2020). Mitochondrial diseases: hope for the future. Cell 181, 168–188. doi:10.1016/j.cell.2020.02.051
Rutter, G. A., Sidarala, V., Kaufman, B. A., and Soleimanpour, S. A. (2023). Mitochondrial metabolism and dynamics in pancreatic beta cell glucose sensing. Biochem. J. 480, 773–789. doi:10.1042/BCJ20230167
Ryytty, S., and Hamalainen, R. H. (2023). The mitochondrial m.3243A>G mutation on the dish, lessons from in vitro models. Int. J. Mol. Sci. 24, 13478. doi:10.3390/ijms241713478
Sakai, K., Matsumoto, K., Nishikawa, T., Suefuji, M., Nakamaru, K., Hirashima, Y., et al. (2003). Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem. Biophys. Res. Commun. 300, 216–222. doi:10.1016/s0006-291x(02)02832-2
Schuit, F., De Vos, A., Farfari, S., Moens, K., Pipeleers, D., Brun, T., et al. (1997). Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J. Biol. Chem. 272, 18572–18579. doi:10.1074/jbc.272.30.18572
Seidel-Rogol, B. L., Mcculloch, V., and Shadel, G. S. (2003). Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat. Genet. 33, 23–24. doi:10.1038/ng1064
Sekine, N., Cirulli, V., Regazzi, R., Brown, L. J., Gine, E., Tamarit-Rodriguez, J., et al. (1994). Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 269, 4895–4902. doi:10.1016/s0021-9258(17)37629-9
Shalev, A. (2014). Minireview: thioredoxin-interacting protein: regulation and function in the pancreatic β-cell. Mol. Endocrinol. 28, 1211–1220. doi:10.1210/me.2014-1095
Sharma, R. B., Landa-Galvan, H. V., and Alonso, L. C. (2021). Living dangerously: protective and harmful ER stress responses in pancreatic β-cells. Diabetes 70, 2431–2443. doi:10.2337/dbi20-0033
Sidarala, V., Pearson, G. L., Parekh, V. S., Thompson, B., Christen, L., Gingerich, M. A., et al. (2020). Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 5, e141138. doi:10.1172/jci.insight.141138
Sidarala, V., Zhu, J., Levi-D'ancona, E., Pearson, G. L., Reck, E. C., Walker, E. M., et al. (2022). Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat. Commun. 13, 2340. doi:10.1038/s41467-022-29945-7
Soleimanpour, S. A., Gupta, A., Bakay, M., Ferrari, A. M., Groff, D. N., Fadista, J., et al. (2014). The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 157, 1577–1590. doi:10.1016/j.cell.2014.05.016
Spinazzola, A., Viscomi, C., Fernandez-Vizarra, E., Carrara, F., D'adamo, P., Calvo, S., et al. (2006). MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 38, 570–575. doi:10.1038/ng1765
Stancill, J. S., Broniowska, K. A., Oleson, B. J., Naatz, A., and Corbett, J. A. (2019). Pancreatic β-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 294, 4843–4853. doi:10.1074/jbc.RA118.006219
Stark, R., Pasquel, F., Turcu, A., Pongratz, R. L., Roden, M., Cline, G. W., et al. (2009). Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem. 284, 26578–26590. doi:10.1074/jbc.M109.011775
Stewart, J. B. (2021). Current progress with mammalian models of mitochondrial DNA disease. J. Inherit. Metab. Dis. 44, 325–342. doi:10.1002/jimd.12324
Sugden, M. C., and Holness, M. J. (2011). The pyruvate carboxylase-pyruvate dehydrogenase axis in islet pyruvate metabolism: going round in circles? Islets 3, 302–319. doi:10.4161/isl.3.6.17806
Sun, G., Tarasov, A. I., Mcginty, J., Mcdonald, A., Da Silva Xavier, G., Gorman, T., et al. (2010). Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia 53, 924–936. doi:10.1007/s00125-010-1692-1
Sun, J., Cui, J., He, Q., Chen, Z., Arvan, P., and Liu, M. (2015). Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 42, 105–118. doi:10.1016/j.mam.2015.01.001
Swisa, A., Granot, Z., Tamarina, N., Sayers, S., Bardeesy, N., Philipson, L., et al. (2015). Loss of liver kinase B1 (LKB1) in beta cells enhances glucose-stimulated insulin secretion despite profound mitochondrial defects. J. Biol. Chem. 290, 20934–20946. doi:10.1074/jbc.M115.639237
Sylvester, J. E., Fischel-Ghodsian, N., Mougey, E. B., and O'brien, T. W. (2004). Mitochondrial ribosomal proteins: candidate genes for mitochondrial disease. Genet. Med. 6, 73–80. doi:10.1097/01.gim.0000117333.21213.17
Taddeo, E. P., Alsabeeh, N., Baghdasarian, S., Wikstrom, J. D., Ritou, E., Sereda, S., et al. (2020). Mitochondrial proton leak regulated by cyclophilin D elevates insulin secretion in islets at nonstimulatory glucose levels. Diabetes 69, 131–145. doi:10.2337/db19-0379
Taddeo, E. P., Stiles, L., Sereda, S., Ritou, E., Wolf, D. M., Abdullah, M., et al. (2018). Individual islet respirometry reveals functional diversity within the islet population of mice and human donors. Mol. Metab. 16, 150–159. doi:10.1016/j.molmet.2018.07.003
Tang, Q., Shi, W., Liu, M., Tang, L., Ren, W., and Shi, S. (2023). Mitochondrial protein MPV17 promotes β-cell apoptosis in diabetogenesis. Clin. Sci. (Lond) 137, 1195–1208. doi:10.1042/CS20230164
Tarasov, A. I., Semplici, F., Li, D., Rizzuto, R., Ravier, M. A., Gilon, P., et al. (2013). Frequency-dependent mitochondrial Ca(2+) accumulation regulates ATP synthesis in pancreatic β cells. Pflugers Arch. 465, 543–554. doi:10.1007/s00424-012-1177-9
Tarasov, A. I., Semplici, F., Ravier, M. A., Bellomo, E. A., Pullen, T. J., Gilon, P., et al. (2012). The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One 7, e39722. doi:10.1371/journal.pone.0039722
Tengholm, A., Hellman, B., and Gylfe, E. (1999). Glucose regulation of free Ca(2+) in the endoplasmic reticulum of mouse pancreatic beta cells. J. Biol. Chem. 274, 36883–36890. doi:10.1074/jbc.274.52.36883
Thomas, H. E., Trapani, J. A., and Kay, T. W. (2010). The role of perforin and granzymes in diabetes. Cell Death Differ. 17, 577–585. doi:10.1038/cdd.2009.165
Towler, M. C., and Hardie, D. G. (2007). AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 100, 328–341. doi:10.1161/01.RES.0000256090.42690.05
Tugues, S., Honjo, S., Konig, C., Padhan, N., Kroon, J., Gualandi, L., et al. (2013). Tetraspanin CD63 promotes vascular endothelial growth factor receptor 2-β1 integrin complex formation, thereby regulating activation and downstream signaling in endothelial cells in vitro and in vivo. J. Biol. Chem. 288, 19060–19071. doi:10.1074/jbc.M113.468199
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi:10.1038/sj.emboj.7601963
Usmani-Brown, S., Perdigoto, A. L., Lavoie, N., Clark, P., Korah, M., Rui, J., et al. (2019). β cell responses to inflammation. Mol. Metab. 27S, S104–S113. doi:10.1016/j.molmet.2019.06.013
Vais, H., Payne, R., Paudel, U., Li, C., and Foskett, J. K. (2020). Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca(2+) uptake. Proc. Natl. Acad. Sci. U. S. A. 117, 21731–21739. doi:10.1073/pnas.2005976117
Verma, G., Bowen, A., Gheibi, S., Hamilton, A., Muthukumar, S., Cataldo, L. R., et al. (2022). Ribosomal biogenesis regulator DIMT1 controls β-cell protein synthesis, mitochondrial function, and insulin secretion. J. Biol. Chem. 298, 101692. doi:10.1016/j.jbc.2022.101692
Vetterli, L., Carobbio, S., Pournourmohammadi, S., Martin-Del-Rio, R., Skytt, D. M., Waagepetersen, H. S., et al. (2012). Delineation of glutamate pathways and secretory responses in pancreatic islets with β-cell-specific abrogation of the glutamate dehydrogenase. Mol. Biol. Cell 23, 3851–3862. doi:10.1091/mbc.E11-08-0676
Vig, S., Lambooij, J. M., Zaldumbide, A., and Guigas, B. (2021). Endoplasmic reticulum-mitochondria crosstalk and beta-cell destruction in type 1 diabetes. Front. Immunol. 12, 669492. doi:10.3389/fimmu.2021.669492
Vigueira, P. A., Mccommis, K. S., Schweitzer, G. G., Remedi, M. S., Chambers, K. T., Fu, X., et al. (2014). Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 7, 2042–2053. doi:10.1016/j.celrep.2014.05.017
Vishnu, N., Hamilton, A., Bagge, A., Wernersson, A., Cowan, E., Barnard, H., et al. (2021). Mitochondrial clearance of calcium facilitated by MICU2 controls insulin secretion. Mol. Metab. 51, 101239. doi:10.1016/j.molmet.2021.101239
Vivoli, A., Ghislain, J., Filali-Mouhim, A., Angeles, Z. E., Castell, A. L., Sladek, R., et al. (2023). Single-cell RNA sequencing reveals a role for reactive oxygen species and peroxiredoxins in fatty acid-induced rat β-cell proliferation. Diabetes 72, 45–58. doi:10.2337/db22-0121
Waldeck-Weiermair, M., Alam, M. R., Khan, M. J., Deak, A. T., Vishnu, N., Karsten, F., et al. (2012). Spatiotemporal correlations between cytosolic and mitochondrial Ca(2+) signals using a novel red-shifted mitochondrial targeted cameleon. PLoS One 7, e45917. doi:10.1371/journal.pone.0045917
Wang, Y., Wang, J., Tao, S. Y., Liang, Z., Xie, R., Liu, N. N., et al. (2023). Mitochondrial damage-associated molecular patterns: a new insight into metabolic inflammation in type 2 diabetes mellitus. Diabetes Metab. Res. Rev., e3733. doi:10.1002/dmrr.3733
Wang, Z., Oh, E., Clapp, D. W., Chernoff, J., and Thurmond, D. C. (2011). Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 286, 41359–41367. doi:10.1074/jbc.M111.291500
Wei, J., Wang, Z., Han, T., Chen, J., Ou, Y., Wei, L., et al. (2023). Extracellular vesicle-mediated intercellular and interorgan crosstalk of pancreatic islet in health and diabetes. Front. Endocrinol. (Lausanne) 14, 1170237. doi:10.3389/fendo.2023.1170237
Weir, G. C. (2020). Glucolipotoxicity, β-cells, and diabetes: the emperor has No clothes. Diabetes 69, 273–278. doi:10.2337/db19-0138
White, S. A., Zhang, L. S., Pasula, D. J., Yang, Y. H. C., and Luciani, D. S. (2020). Bax and Bak jointly control survival and dampen the early unfolded protein response in pancreatic β-cells under glucolipotoxic stress. Sci. Rep. 10, 10986. doi:10.1038/s41598-020-67755-3
Wiederkehr, A., Szanda, G., Akhmedov, D., Mataki, C., Heizmann, C. W., Schoonjans, K., et al. (2011). Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 13, 601–611. doi:10.1016/j.cmet.2011.03.015
Wikstrom, J. D., Israeli, T., Bachar-Wikstrom, E., Swisa, A., Ariav, Y., Waiss, M., et al. (2013). AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol. Endocrinol. 27, 1706–1723. doi:10.1210/me.2013-1109
Wikstrom, J. D., Sereda, S. B., Stiles, L., Elorza, A., Allister, E. M., Neilson, A., et al. (2012). A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoS One 7, e33023. doi:10.1371/journal.pone.0033023
Xu, H., Martinoia, E., and Szabo, I. (2015). Organellar channels and transporters. Cell Calcium 58, 1–10. doi:10.1016/j.ceca.2015.02.006
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 94, 514–519. doi:10.1073/pnas.94.2.514
Ying, W., Lee, Y. S., Dong, Y., Seidman, J. S., Yang, M., Isaac, R., et al. (2019). Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 29, 457–474 e5. doi:10.1016/j.cmet.2018.12.003
Yong, J., Johnson, J. D., Arvan, P., Han, J., and Kaufman, R. J. (2021). Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat. Rev. Endocrinol. 17, 455–467. doi:10.1038/s41574-021-00510-4
Zempo, H., Kim, S. J., Fuku, N., Nishida, Y., Higaki, Y., Wan, J., et al. (2021). A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c. Aging (Albany NY) 13, 1692–1717. doi:10.18632/aging.202529
Zhang, C. Y., Baffy, G., Perret, P., Krauss, S., Peroni, O., Grujic, D., et al. (2001). Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745–755. doi:10.1016/s0092-8674(01)00378-6
Zhang, G. F., Jensen, M. V., Gray, S. M., El, K., Wang, Y., Lu, D., et al. (2021). Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metab. 33, 804–817 e5. doi:10.1016/j.cmet.2020.11.020
Zhang, I. X., Raghavan, M., and Satin, L. S. (2020). The endoplasmic reticulum and calcium homeostasis in pancreatic beta cells. Endocrinology 161, bqz028. doi:10.1210/endocr/bqz028
Zhang, J., Guo, J., Fang, W., Jun, Q., and Shi, K. (2015). Clinical features of MELAS and its relation with A3243G gene point mutation. Int. J. Clin. Exp. Pathol. 8, 13411–13415.
Zhang, M., Goforth, P., Bertram, R., Sherman, A., and Satin, L. (2003). The Ca2+ dynamics of isolated mouse beta-cells and islets: implications for mathematical models. Biophys. J. 84, 2852–2870. doi:10.1016/S0006-3495(03)70014-9
Zhang, Z., Wakabayashi, N., Wakabayashi, J., Tamura, Y., Song, W. J., Sereda, S., et al. (2011). The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 22, 2235–2245. doi:10.1091/mbc.E10-12-0933
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44, 3–15. doi:10.3892/ijmm.2019.4188
Zhao, S., Mugabo, Y., Iglesias, J., Xie, L., Delghingaro-Augusto, V., Lussier, R., et al. (2014). α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 19, 993–1007. doi:10.1016/j.cmet.2014.04.003
Zhou, G. L., Zhuo, Y., King, C. C., Fryer, B. H., Bokoch, G. M., and Field, J. (2003). Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol. Cell Biol. 23, 8058–8069. doi:10.1128/mcb.23.22.8058-8069.2003
Zhou, H., and Kramer, R. H. (2005). Integrin engagement differentially modulates epithelial cell motility by RhoA/ROCK and PAK1. J. Biol. Chem. 280, 10624–10635. doi:10.1074/jbc.M411900200
Zirpel, H., and Roep, B. O. (2021). Islet-resident dendritic cells and macrophages in type 1 diabetes: in search of bigfoot's print. Front. Endocrinol. (Lausanne) 12, 666795. doi:10.3389/fendo.2021.666795
Zorbas, C., Nicolas, E., Wacheul, L., Huvelle, E., Heurgue-Hamard, V., Lafontaine, D. L., et al. (2015). The human 18S rRNA base methyltransferases DIMT1L and WBSCR22-TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol Biol Cell, 26, 2080–95.
Zuo, L., Prather, E. R., Stetskiv, M., Garrison, D. E., Meade, J. R., Peace, T. I., et al. (2019). Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. Int. J. Mol. Sci. 20, 4472. doi:10.3390/ijms20184472
Glossary

Keywords: Type 1 diabetes, Type 2 Diabetes, mitochondria, pancreatic beta cells, bioenergetics, metabolism, calcium, insulin secretion
Citation: Rivera Nieves AM, Wauford BM and Fu A (2024) Mitochondrial bioenergetics, metabolism, and beyond in pancreatic β-cells and diabetes. Front. Mol. Biosci. 11:1354199. doi: 10.3389/fmolb.2024.1354199
Received: 12 December 2023; Accepted: 17 January 2024;
Published: 09 February 2024.
Edited by:
Hans-Georg Sprenger, Dana–Farber Cancer Institute, United StatesReviewed by:
Manuel Gutierrez-Aguilar, National Autonomous University of Mexico, MexicoJonathan Barlow, University of Birmingham, United Kingdom
Copyright © 2024 Rivera Nieves, Wauford and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Accalia Fu, YWNjYWxpYS5mdTFAdW1hc3NtZWQuZWR1
†These authors have contributed equally to this work