
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 06 February 2024
Sec. Molecular Recognition
Volume 11 - 2024 | https://doi.org/10.3389/fmolb.2024.1268647
This article is part of the Research TopicIn Celebration of Women in Science: Molecular Recognition 2022View all 5 articles
 Claudia Michaelis1†
Claudia Michaelis1† Tamara M. I. Berger2†Kirill Kuhlmann2Rangina Ghulam1
Tamara M. I. Berger2†Kirill Kuhlmann2Rangina Ghulam1 Lukas Petrowitsch2Maria Besora Vecino2Bernd Gesslbauer3,4,5
Lukas Petrowitsch2Maria Besora Vecino2Bernd Gesslbauer3,4,5 Tea Pavkov-Keller2,4,5
Tea Pavkov-Keller2,4,5 Walter Keller2,4,5*
Walter Keller2,4,5* Elisabeth Grohmann1*
Elisabeth Grohmann1*Conjugation is a major mechanism that facilitates the exchange of antibiotic resistance genes among bacteria. The broad-host-range Inc18 plasmid pIP501 harbors 15 genes that encode for a type IV secretion system (T4SS). It is a membrane-spanning multiprotein complex formed between conjugating donor and recipient cells. The penultimate gene of the pIP501 operon encodes for the cytosolic monomeric protein TraN. This acts as a transcriptional regulator by binding upstream of the operon promotor, partially overlapping with the origin of transfer. Additionally, TraN regulates traN and traO expression by binding upstream of the PtraNO promoter. This study investigates the impact of nine TraN amino acids involved in binding to pIP501 DNA through site-directed mutagenesis by exchanging one to three residues by alanine. For three traN variants, complementation of the pIP501∆traN knockout resulted in an increase of the transfer rate by more than 1.5 orders of magnitude compared to complementation of the mutant with native traN. Microscale thermophoresis (MST) was used to assess the binding affinities of three TraN double-substituted variants and one triple-substituted variant to its cognate pIP501 double-stranded DNA. The MST data strongly correlated with the transfer rates obtained by biparental mating assays in Enterococcus faecalis. The TraN variants TraN_R23A-N24A-Q28A, TraN_H82A-R86A, and TraN_G100A-K101A not only exhibited significantly lower DNA binding affinities but also, upon complementation of the pIP501∆traN knockout, resulted in the highest pIP501 transfer rates. This confirms the important role of the TraN residues R23, N24, Q28, H82, R86, G100, and K101 in downregulating pIP501 transfer. Although TraN is not part of the mating pair formation complex, TraE, TraF, TraH, TraJ, TraK, and TraM were coeluted with TraN in a pull-down. Moreover, TraN homologs are present not only in Inc18 plasmids but also in RepA_N and Rep_3 family plasmids, which are frequently found in enterococci, streptococci, and staphylococci. This points to a widespread role of this repressor in conjugative plasmid transfer among Firmicutes.
The fundamental process of horizontal gene transfer (HGT) has greatly enhanced our understanding of microbial adaptation and the spread of antibiotic resistance genes (ARGs) as well as virulence factors among bacteria. The acquisition of new traits (drug resistance and metabolic and pathogenic capabilities) can confer selective advantages to bacteria and shape the evolution of microbial populations.
Conjugation is one of the major HGT mechanisms that enables the rapid transfer of ARGs from one bacterium to another. It is mediated by a membrane-embedded type IV secretion system (T4SS) that translocates single-stranded DNA from donor to recipient cells. Conjugation is a key mechanism in the development of multi-drug-resistant bacteria because it allows them to adapt to changing environments more efficiently (Li et al., 2019; Virolle et al., 2020; Costa et al., 2021). Conjugative T4SSs work in a contact-dependent manner: Gram-negative bacteria develop a pilus to initiate cell-to-cell contact between donor and recipient cells, while Gram-positive conjugation is thought to rely on adhesins on the donor cell surface. T4SSs have been studied in numerous Gram-negative bacterial species, but the understanding of Gram-positive T4SSs remains relatively limited (Grohmann et al., 2018; Costa et al., 2021). Mobile genetic elements (MGEs) such as conjugative plasmids and integrative and conjugative elements (ICEs) encode their own T4SS. One model plasmid used to study conjugative transfer in Gram-positive bacteria is the conjugative plasmid pIP501. This ∼30.6-kb plasmid belongs to incompatibility (Inc) group 18, whose representatives encode several resistance genes, such as linezolid or vancomycin resistance genes, or to the macrolide-lincosamide–streptogramine group of resistance genes (Kohler et al., 2018b; Cinthi et al., 2022; Tang et al., 2023). The plasmid can stably replicate and transfer into a variety of Gram-positive bacteria, including enterococci, streptococci, staphylococci, lactococci, Oenococcus oeni, pediococci, Listeria, Leuconostoc mesenteroides subsp. cremoris, lactobacilli, Bacillus subtilis, Streptomyces lividans, and Gram-negative Escherichia coli (Pérez-Díaz et al., 1982; Schaberg et al., 1982; Gonzalez and Kunka, 1983; Buu-Hoï et al., 1984; Pucci et al., 1988; Thompson and Collins, 1988; Langella and Chopin, 1989; Kurenbach et al., 2003; Zúñiga et al., 2003). Enterococci, particularly Enterococcus faecalis and E. faecium, are nosocomial pathogens of great clinical importance. They can cause severe healthcare-associated infections such as urinary tract infections, bloodstream infections, or endocarditis (Haliga et al., 2023; Mancuso et al., 2023). Owing to their high acquisition of ARGs and consequent resistance to a broad spectrum of antibiotics, these infections are challenging to treat (Khan et al., 2022; Murray et al., 2022).
The conjugative T4SS of the broad-host-range plasmid pIP501 is encoded on a ∼15-kb transfer (tra) operon and comprises 15 transfer genes (traA–traO). Transfer proteins are involved in DNA processing, relaxosome recruitment, mating pore formation (MPF), and the regulation of the expression of the tra operon. The last gene of the operon encodes for TraO, a putative surface adhesin, which enhances the cell-to-cell contact between donor and recipient cells prior to conjugative transfer of the plasmid. Transcriptional regulation is critical for bacteria to minimize the energy and metabolic costs associated with generating the multiprotein complex required for pIP501 transfer. Regulation of the transcription of conjugative transfer genes leads to tighter control of the conjugative process. The transcriptional regulation of Gram-positive T4SSs has been observed for many conjugative plasmids and other MGEs and has recently been reviewed (Kohler et al., 2019). The transcriptional regulation of transfer is primarily mediated by plasmid-encoded regulatory proteins that specifically bind to or in the vicinity of the promoter region, leading to repression of the transcription of the transfer genes. Quorum-sensing mechanisms for expression control of transfer genes have been identified, such as for the conjugative elements pLS20 or ICEBs1 from B. subtilis (Auchtung et al., 2005; Singh et al., 2013). In case of plasmid pCF10, the DNA binding activity of the repressor is further modulated by the ratio of two peptide sex pheromones, plasmid-encoded cCF10, and recipient pheromone iCF10 (Breuer et al., 2018).
The tra gene expression of pIP501 is regulated by the relaxase TraA, which binds to the main T4SS promoter, Ptra (Grohmann et al., 2016). The repressor TraN also acts as a transcriptional regulator by specifically binding to a preferred 34-bp sequence (original binding site [oBS]) located 69 bp upstream of the oriTpIP501; this serves as the origin of transfer where conjugative transfer is initiated. Furthermore, TraN binds to a second promoter, PtraNO, to regulate its own but also TraO expression (Kohler et al., 2018a).
The crystal structure of TraN has already been solved (Goessweiner-Mohr et al., 2012; Goessweiner-Mohr et al., 2014a). Co-crystallization of TraN was conducted to examine the interactions between TraN and its cognate pIP501 binding site (Kohler et al., 2018a). Both crystal structures show TraN in its monomeric form. Sequence-specific interactions between distinct amino acid side chains and bases of pIP501 DNA were identified as important recognition sites for the strong and sequence-specific binding of TraN. Analogous TraN-like functions have been discovered in other Inc18 and related conjugative plasmids (Kohler et al., 2018a). Furthermore, it was demonstrated that TraN exhibits structural similarities to transcriptional regulators of the MerR family as well as to excisionases from bacteriophages and the conjugative element Tn916 (Goessweiner-Mohr et al., 2014a).
This study aims to demonstrate the pivotal role of TraN key residues in sequence-specific DNA binding.
To determine the role of key amino acid residues of TraN in the regulation of pIP501 transfer, three single and four multiple alanine substitution variants were generated to complement the traN knockout in pIP501. The four TraN variants with the highest impact on pIP501 transfer were expressed in E. coli and purified. Microscale thermophoresis (MST) studies were performed to reveal the effect of TraN substitutions on DNA binding affinity. Furthermore, pull-down assays elucidated the potential T4SS interaction partners of TraN using immunoblotting and mass spectrometry (MS) to identify the coeluted Tra proteins.
A list of all strains and plasmids is provided in Supplementary Table S1. E. faecalis strains were routinely grown on BHI plates or in BHI medium with shaking at 37°C. E. coli was grown at 37°C in LB medium with shaking. Antibiotics were used at the concentrations given in Supplementary Table S1.
The pIP501∆traN in-frame knockout constructed by Kohler et al. (2018a) was complemented with the wild-type traN containing its native ribosomal binding site (RBS). TraN with its native RBS was thus amplified from pIP501 using the primers pEU327-BstYI_RBS traN fw and pEU327_SalI RBS traN rev. The PCR fragment was digested with SalI and BstYI and cloned into the shuttle vector pEU327. All primers used for cloning are listed in Supplementary Table S2.
Seven pEU327-RBS-traN variants were constructed for further complementation studies. They were designed using site-directed mutagenesis (New England Biolabs, Frankfurt am Main, Germany) according to the manufacturer’s instructions and transformed into competent E. coli DH5α cells. The sequence of the traN variants was verified by Sanger sequencing. For biparental mating assays, E. faecalis JH2-2 (pIP501∆traN) was transformed by electroporation with one of the seven pEU327-RBS-traN variants as described in Berger et al. (2022).
The biparental mating assays were performed as described previously (Berger et al., 2022). They were carried out in duplicates and repeated three times. pIP501 transfer rates are given as transconjugants per recipient cell. GraphPad Prism 8 statistical software was used for statistical analyses. Statistical significance was determined by using the unpaired two-tailed Mann–Whitney U test. Independent biological triplicates of knockout complementation by traN variants were compared with complementation by the native traN gene. p-values: p < 0.1 (*), p < 0.01 (**).
To add an N-terminal 7×His-tag in-frame to the traN sequence, the traN variants were subcloned into the 7×His-tag expression vector pQTEV. For improved readability, we refer to them without including the “His” prefix throughout the article. Thus, a Gibson Assembly Kit (New England Biolabs) was applied according to the manufacturer’s instructions. The primers for amplification of the pQTEV backbone (GA_pQTEV189-190 fw and GA_pQTEV189-190 rev) and traN (GA_pQTEV-traN fw and GA_pQTEV-traN rev) as well as those for sequencing are listed in Supplementary Table S2. The recombinant plasmids were transformed into E. coli DH5α (New England Biolabs). The sequence of the traN variants was confirmed by Sanger sequencing (Eurofins Genomics Germany). For expression of TraN and TraN variants (TraN_R23A-N24A-Q28A, TraN_G47A-G47A, TraN_H82A-R86A, and TraN_G100A-K101A), the recombinant plasmids were transformed into E. coli Lemo21 (DE3) cells (New England Biolabs).
To compare expression levels of the seven TraN variants with that of TraN, E. faecalis JH2-2 (pIP501∆traN) strains harboring pEU327-RBS-traN variants and pEU327-RBS-traN, respectively, were cultured in 4 mL BHI medium supplemented with respective antibiotics overnight under constant shaking at 37°C. E. faecalis JH2-2 (pEU327) and E. faecalis JH2-2 (pIP501∆traN) served as negative controls for verifying the antibody specificity. The overnight cultures were normalized to an optical density of 600 nm (OD600) of 2 in a volume of 1 mL and harvested by centrifugation at 8,000 ×g for 15 min at 4°C. The cell pellets were washed three times in 1 mL phosphate buffer (50 mM KH2PO4/K2HPO4, pH 7) and resuspended in 300 µL SDS-PAGE loading buffer as per standard protocol (Laemmli, 1970). They were lysed by sonication through two pulses of 8 s followed by boiling at 95°C for 10 min. Identical amounts of the protein lysates were loaded onto a 12% SDS polyacrylamide gel. Western blots were probed with polyclonal anti-TraN antibody (BioGenes GmbH, Berlin, Germany) at 1:10,000 dilution followed by secondary horseradish peroxidase conjugated IgG antibody (Promega GmbH, Germany) at a dilution of 1:10,000. To check whether the TraN mutations impaired the affinity of the TraN antibody, we performed a Western blot with the heterologously expressed purified variants TraN_R23A-N24A-Q28A, TraN_G47A-G48A, TraN_H82A-R86A, and TraN_G100A-K101A, and the heterologously expressed TraN. We loaded 0.75 µg of each protein onto a 12% SDS polyacrylamide gel followed by immunoblotting, as described above.
The same expression and purification protocol was applied to TraN and its variants. A multiple sequence alignment of all variants is shown in Supplementary Figure S1A.
We prepared 50 mL overnight cultures (ONC) supplemented with 0.1 mg/mL ampicillin either from a single colony of an agar plate or from a frozen cryostock. Three-liter LB broth supplemented with 0.1 mg/mL ampicillin was inoculated with the ONC to an initial OD600 of 0.05 and incubated under shaking at 37°C. Proteins were expressed after induction of the culture with 1 mM IPTG (final concentration) at an OD600 of 0.4–0.6. Incubation continued for 3 h at 37°C under shaking. The cells were harvested by centrifugation at 9,000 ×g (JLA 8.100, Beckman Coulter Inc., Brea, United States) and stored at −20°C.
The frozen cell pellets were resuspended in a triple amount of 50 mM HEPES pH 7.5, 100 mM (NH4)2SO4 supplemented with protease inhibitors (0.001 mg/mL pepstatin, 0.002 mg/mL antipain, and 0.02 mg/mL leupeptin final concentration; Merck KGaA, Darmstadt, Germany) in relation to the total net cell weight. The cell suspension was homogenized (T18 digital ULTRA-TURRAX®, IKA®-Werke GmbH & Co. KG, Staufen, Germany) and sonicated in a rosette cell on ice (T13 flat tip, 50% duty cycle, ∼60% amplitude, Sonopuls, UW/HD 2070, Bandelin electronic GmbH & Co. KG, Germany) twice for 10 min each. The cell debris were removed by centrifugation for 45 min at 4°C and 35,000 ×g.
The supernatants were filtered through 0.45 µm filters (Rotilabo® syringe filters, Carl-Roth GmbH + Co. KG, Karlsruhe, Germany) and loaded onto a HisTrap HP 5 mL column (Cytiva, Chalfont St. Giles, England) for affinity purification using the ÄKTA Pure system (Cytiva). The proteins were eluted with a 0.3 M imidazole step and a 0.3–1.5 M linear imidazole gradient. The quality of the samples was assessed by 12% SDS-PAGE, and the fractions containing most of the desired protein were pooled and loaded onto a 5-mL HeparinTrap column (Cytiva) to remove bound DNA. We applied 50 mM HEPES, 100 mM (NH4)2SO4 pH 7.5 as running buffer. The proteins were eluted with a NaCl gradient from 0 to 1.5 M. The eluting peak fractions containing most of the desired protein were pooled and concentrated using filter-insert spin tubes (Amicon® Ultra centrifugal filters, Ultracell® 3000 MWCO, Merck Millipore Ltd., Darmstadt, Germany).
Additional analytical gel filtration was performed with the ÄKTA avant system (Cytiva) to determine the purity and oligomerization state of TraN and its variants. For this, 500 μL protein samples were loaded onto a Sephadex 75 10/300 GL increase column (Cytiva) using a 2-mL loop and 50 mM HEPES, 100 mM (NH4)2SO4 pH 7.6 as running buffer. To check for DNA bound to the proteins, a chromatogram with two wavelengths was recorded at 280 nm for protein and 260 nm for DNA detection. Expression and purification were monitored by SDS-PAGE using 12% SDS polyacrylamide gels. To normalize the amount of loaded cell mass, we took samples equal to an OD600 of 2 in 1 mL and resuspended them in 20 µL SDS loading buffer.
Purified TraN and its variants were changed into a phosphate buffer (50 mM NaH2PO4 pH 7.0, 50 mM NaF) by filter-insert spin tubes (Amicon® Ultra centrifugal filters, Ultracell® 3000 MWCO, Merck Millipore Ltd.). Protein concentrations between 0.08 mg/mL and 0.13 mg/mL were used. CD measurements were performed on a J-1500 circular dichroism spectrophotometer (JASCO Corporation, Tokyo, Japan). Far-UV CD spectra were recorded at 20°C from 260 to 190 nm with a data pitch of 0.2 nm and a bandwidth of 2 nm applying a scan speed of 50 nm/min. Ten spectra were recorded and accumulated for each sample. The resulting spectra were baseline-corrected by subtracting the signal of the buffer. The CD signal was converted to mean residue ellipticity [θ]MRE, and the secondary structure was determined with DichroWeb using the CDSSTR algorithm (Compton and Johnson, 1986; Whitmore and Wallace, 2004; 2008; Miles et al., 2022) and SMP180t reference dataset (Abdul-Gader et al., 2011). A weighted-means smoothing algorithm over 25 datapoints was applied to all spectra.
Two pIP501-specific double-stranded DNA (dsDNA) oligonucleotides were created by annealing their complementary single strands ordered from Thermo Fisher Scientific. The melting temperature (Tm) of the oligonucleotides was calculated with SnapGene Viewer (SnapGene software, www.snapgene.com). The oligonucleotides corresponded to 1) a specific 34-mer (original TraN binding site (oBS), Tm = 56°C), and 2) a random 34-mer (oriTpIP501 region, to which no specific binding had been demonstrated (random DNA), Tm = 66°C) (Goessweiner-Mohr et al., 2014a).
The oligonucleotide sequences are given in Supplementary Table S2. The forward and reverse strands of the oligonucleotides were annealed before using them in MST. To this end, 30 µL of a 1 mM solution of each ss-oligonucleotide was combined. The mixture was kept at 95°C for 1 h in a heating block (Drybath Stdrd 1blck, Thermo Fisher Scientific) and then slowly cooled down in an enclosed Styrofoam box yielding 60 µL of 500 μM ds-oligonucleotide each.
Monolith NT.115 (NanoTemper Technologies GmbH, Munich, Germany) was used to measure the protein-DNA interactions between TraN and TraN variants with the oBS and the random DNA. The generation of the ds-oligonucleotides is described above in 2.8.
Purified TraN and its variants were changed into phosphate-buffered saline with tween (PBS-T) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.05% Tween 20 (v/v)) by filter-insert spin tubes (Amicon® Ultra Centrifugal Filters, Ultracell® 3000 MWCO, Merck Millipore Ltd.). Protein concentrations were determined using NanoDrop (ND1000, PEQLAB Biotechnologie GmbH, Erlangen, Germany). The proteins were diluted to 200 nM with PBS-T. As TraN and its variants featured an N-terminal 7×His-tag, a His-tag specific labeling method was used. The protein samples were mixed with RED-tris-NTA 2nd Generation dye (NanoTemper Technologies GmbH) in PBS-T at a molar ratio of 2:1 to avoid unbound dye and were incubated in the dark for 30 min at RT. The fluorescently labeled proteins were centrifuged at 15,000 × g for 10 min. The supernatants were used for the binding assays.
The ds-oligonucleotides (oBS and random DNA, chapter 2.8) were diluted to 25.6 µM with PBS-T. A two-fold serial dilution of the oligonucleotides was performed with PBS-T in 500 µL reaction tubes as per the manufacturer’s manual (NanoTemper Technologies GmbH) resulting in final DNA concentrations of 12.8 µM to 0.781 nM (Supplementary Table S3). To prepare 15 different dilutions with DNA:protein ratios of 256:1 to 0.0156:1, the fluorescently-labeled protein samples (100 nM) were added to each dilution, resulting in a final concentration of 50 nM protein per sample. As a negative control, PBS-T buffer and 25 nM RED-tris-NTA 2nd Generation dye was used. The samples were loaded into a capillary with a hydrophilic coating (NanoTemper Premium Capillaries, Cat# MO-K025, NanoTemper Technologies GmbH), placed into the capillary tray, and inserted into the Monolith NT.115 MST machine controlled by NTControl v2.1.31 software (NanoTemper Technologies GmbH). Capillary scans were performed at various LED power values before each measurement to ensure a sufficiently raw fluorescence signal (>200 counts). MST was performed at 60% LED and MST power with 5 s fluorescence detection before and after induction of local temperature gradient (Fluo before and Fluo after respectively) as well as with 30 s detection of the thermophoresis signal (MST On) and a 25 s delay between each measurement.
The data points were plotted using the MO. Affinity Analysis v.2.1.3 software (NanoTemper Technologies GmbH) and were fitted using the Kd model by setting the target concentration to 50 nM (final protein concentration). For calculation of the normalized fluorescence (Fnorm) value, the T-Jump + Thermophoresis preset with an automatically determined best signal-to-noise ratio was chosen. The curve fitting parameters including the Kd were estimated and automatically calculated by the software. The results are presented as plots of ΔFnorm against the ligand (dsDNA) concentration in nM.
The E. coli/P. megaterium (previously known as Bacillus megaterium) shuttle vectors pRBBm59-RBS-traN-Strep and pRBBm59-RBS-gfp-Strep were constructed using Gibson Assembly (New England Biolabs). pRBBm59-RBS-gfp-Strep not encoding any pIP501 tra gene was used as a negative control in the pull-down assays. The backbone of pRBBm59-RBS-Strep was amplified using pRBBm59-RBS-traB-Strep (Berger et al., 2022) as a template using the primers GA_pRBBm59-RBS-Strep fw and GA_pRBBm59-RBS-Strep rev. The traN gene was amplified from pIP501 with GA_pRBBm59-traN fw and GA_pRBBm59-traN rev, and gfp was amplified from pRBBm59 by using the primers pRBBm59-GFP fw and pRBBm59-GFP rev (Supplementary Table S2). We chose Priestia megaterium as the expression host because it is a Gram-positive expression system and thus is better suited for a Gram-positive T4SS. Expression trials using E. coli hosts showed very low Tra protein expression. This approach has already been used recently by Berger et al. (2022).
P. megaterium MS941 (pMBGm19-RBS-traB-traO) (Berger et al., 2022) was transformed with pRBBm59-RBS-traN-Strep and pRBBm59-RBS-gfp-Strep, respectively, according to the protocol in Biedendieck et al. (2011). Transformants were selected on LB agar containing 35 μg/mL chloramphenicol and 10 μg/mL tetracycline and were screened for the presence of the respective pRBBm59 plasmids by colony PCR.
Expression and purification of Strep-tagged TraN and its interaction partners from P. megaterium MS941 (pMGBm19-RBS-traB-traO, pRBBm59-RBS-traN-Strep) lysate were performed as per Berger et al. (2022) with some modifications.
The expression cultures (1.2 L in 5 L baffled flasks) were inoculated to an OD600 of 0.01 and incubated by shaking at 37°C to an OD600 of 0.3–0.4. The expression of TraB-TraO and Strep-tagged TraN was induced by the addition of 0.5% (w/v) xylose for pMGBm19 and 0.5% (w/v) sucrose for pRBBm59. After 5 hours, the cells were harvested by centrifugation at 9,000 × g at 4°C for 60 min. The cell pellet was resuspended in three times the cell wet weight of complex binding buffer (100 mM Tris/HCl, 300 mM NaCl, 1 mM EDTA, pH 8), supplemented with protease inhibitor (1 μg/mL pepstatin, 2 μg/mL antipain, 20 μg/mL leupeptin in DMSO). The cells were then lysed by sonication for 1 h on ice (50% duty cycle, 60% intensity; Sonopuls, UW/HD 2070; Bandelin electronic GmbH & Co. KG, Germany). After 60 min of centrifugation at 4°C and 38,000 × g, the supernatant was filtered through a MF-Millipore 0.45 µm MCE membrane (Merck, Darmstadt, Germany) to eliminate residual cell debris. The filtered lysate was loaded onto a 5-mL StrepTrap™ HP column pre-equilibrated with complex binding buffer using an ÄKTA pure 25-L chromatography system. The proteins were eluted using the complex binding buffer supplemented with 2.5 mM desthiobiotin. Peak fractions were loaded onto 12% SDS polyacrylamide gels and subjected to immunoblotting. Primary polyclonal anti-Tra antibodies were used against TraB, TraE, TraF, TraG, TraH, TraJ, TraK, TraM, and TraN (BioGenes GmbH) with a dilution of 1:10,000, and TraO with a dilution of 1:1,000, to probe for coeluted Tra proteins. We used as secondary antibody a horseradish peroxidase conjugated antibody against rabbit IgG (Promega GmbH), diluted to 1:10,000.
Bands containing the elution fractions of the pull-down were excised from a silver stained 12% SDS polyacrylamide gel and subjected to in-gel reduction, alkylation, and trypsinization as per Gesslbauer et al. (2018). Supplementary Table S6 shows the resulting peptides. Extracted peptides were purified and concentrated using C18 spin columns (Pierce™ C18 Spin Columns; Thermo Fisher Scientific) as per the manufacturer’s protocol.
The concentrated purified peptides were applied to liquid chromatography (LC) through an Ultimate 3,000 RSLCnano System (Thermo Fisher Scientific). Peptides were separated with a flow rate of 300 nL/min on a C18 Aurora UHPLC column (25 cm × 75 µm ID, 1.6 µm) with CaptiveSpray Insert (IonOpticks). The mobile phases were A) 0.1% (v/v) formic acid in water and B) 0.1% (v/v) formic acid in acetonitrile. The HPLC gradient for separation was 2% B for 6 min, 2%–25% B for 90 min, 25%–40% B for 10 min, 40%–80% B for 10 min, 80% B for 10 min, and 2% B for 12 min. The LC system was coupled online to a timsTOF Pro mass spectrometer (Bruker Corporation, Billerica, Massachusetts, United States) with a CaptiveSpray nano-electrospray ion source (Bruker); the mass spectrometer was operated in PASEF mode. TIMS, PASEF, and calibration settings were applied as described by Meier et al. (2018).
Mass spectrometry raw files were processed with MaxQuant version 2.0.1.0 (Cox and Mann, 2008). MS/MS spectra were searched against the UniProt P. megaterium (strain ATCC 12872/QMB1551) (B. megaterium) reference proteome databank (Proteome ID: UP000000935). The amino acid sequences of TraA, TraB, TraC, TraD, TraE, TraF, TraG, TraH, TraI, TraJ, TraK, TraL, TraM, TraN, and TraO were manually included in the P. megaterium FASTA file. Search parameters were set as follows: trypsin was set as enzyme; a maximum of two missed cleavages was allowed; the minimum peptide sequence length was seven amino acids; the maximum peptide mass was 4,600 Da. Carbamidomethylation of cysteine was set as a fixed modification; oxidation of methionine and acetylation of protein amino-termini were set as a variable modification. All other MaxQuant search parameters were equivalent to those described by Meier et al. (2018).
TraN homologs were identified with an iterative PSI-BLAST and tBLASTn search using the National Center of Biotechnology Information (NCBI) nucleotide database (https://www.ncbi.nlm.nih.gov/). The criteria selected were: 1) only putative functional proteins; 2) the presence of a putative oriT checked by oriTfinder server (https://bioinfo-mml.sjtu.edu.cn/oriTfinder/); 3) presence of a relaxase gene; 4) presence of an additional signature gene of conjugative plasmids; 5) only plasmid encoded TraN homologs; 6) exclusion of unnamed plasmid entries; 7) exclusion of artificial plasmids such as shuttle vectors. As all putative TraN homologs identified by an iterative PSI-BLAST using TraN as query were annotated as hypothetical proteins, we incorporated a tBLASTn search as an additional criterion for pre-selecting plasmid-encoded TraN homologs. The tBLASTn algorithm is used to compare protein query sequences against a nucleotide sequence database, allowing the detection of potential homologous DNA sequences that might not be annotated as protein.
The web server oriTfinder (Li et al., 2018) was used to rapidly detect putative oriTs and transfer-associated modules such as relaxase genes, type IV coupling protein (T4CP) genes, or T4SS gene clusters within the selected plasmid sequences. The oriT site is an essential module of self-transmissible elements. A multiple sequence alignment of selected TraN homologs was performed with T-Coffee (version 11.0) using the Expresso algorithm with default settings (Armougom et al., 2006; Di Tommaso et al., 2011). Additionally, all pIP501 transfer proteins (TraA to TraO) were selected as query sequences for tBLASTn searches to identify homologs in putative conjugative plasmids encoding traN homologous sequences.
PlasmidFinder 2.1 (Carattoli et al., 2014) was used to identify the incompatibility group of the plasmids selected according to the criteria above. FASTA-formatted plasmid DNA sequences from NCBI were uploaded to the website of the Center for Genomic Epidemiology (CGE; https://cge.food.dtu.dk/services/PlasmidFinder-2.0/) and analyzed using these settings: 75% minimum identity threshold; 60% minimum coverage threshold. The database for Gram-positive plasmid replicon sequences was selected. Replicon families/incompatibility groups of the plasmids were identified based on the detection of specific replication (rep) initiator genes. Plasmid sequences for which PlasmidFinder found no match were not further analyzed and were denoted “not characterized”.
The coLAB AlphaFold2 server (Mirdita et al., 2017; Mirdita et al., 2019; Mitchell et al., 2020; Jumper et al., 2021; Mirdita et al., 2022) was used for 3D structure prediction of TraN and its putative homologs. In addition, the structures of the TraN variants from pQTEV constructs (TraN, TraN_R23A-N24A-Q28A, TraN_G47A-G48A, TraN_H82A-R86A, and TraN_G100A-K101A) were predicted to check whether the amino acid substitutions had an influence on the protein fold. Further structural predictions were made for a set of conjugative transfer proteins from different Firmicutes conjugative plasmids (indicated in subscript) to identify shared secondary structure elements: TrsRpUC11B (GenBank accession number: WP_032398615.1), TrsRpUC08B (WP_032398615.1), TrsRpAF22 (WP_015062887.1), TrsRp001F (WP_095348652.1), Tra20pNP40 (WP_012477765.1), TraRpNP40 (WP_012477778.1), Orf5pMRC01 (WP_010890662.1), and TrsRpIBB477a (WP_010890662.1). To visualize and determine root mean square deviation (RMSD) values, we performed alignments of the predicted structures to the crystal structure of TraNpIP501 (PDB: 4P0Z, residues 1-117 are visible in the electron density) using PyMOL (PyMOL Molecular Graphics System, Version 2.6 Schrödinger, LLC). All alignments were performed using the “cealign” algorithm of PyMOL.
The transfer rate of the pIP501∆traN knockout has been shown to be two orders of magnitude higher than that of the original plasmid (Kohler et al., 2018a). However, this effect could not be restored to wild-type level by providing traN with the ribosomal binding site (RBS) of traJpIP501 in pEU327 in trans. In this study, we constructed the complementation plasmid pEU327-RBStraN-traN containing the original RBS of traN. Henceforth, pEU327-RBStraN-traN is referred to as pEU327-RBS-traN.
Providing pEU327-RBS-traN in trans to pIP501∆traN fully restored the wild-type pIP501 transfer rate in E. faecalis (Figures 1B,C). Thus, the polar effects of the traN knockout on the traO gene downstream in the tra operon can be excluded.
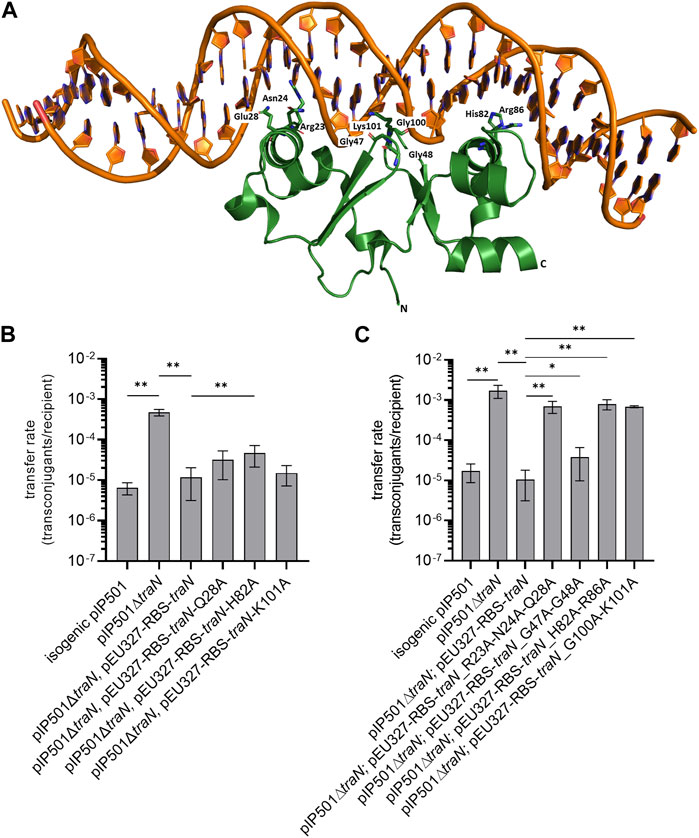
FIGURE 1. Biparental mating assays of pIP501, pIP501∆traN, and pIP501∆traN substituted with traN and traN carrying point mutations in the putative key residues listed below. E. faecalis JH2-2 (pIP501 isogenic wild type (obtained during generation of the traN knockout)), E. faecalis JH2-2 (pIP501∆traN) and E. faecalis JH2-2 (pIP501∆traN and pEU327-RBS-traN) were used as reference. (A) Crystal structure of TraN in complex (PDB: 6G1T) with original binding site (oBS). Mutated residues labeled and shown as sticks. (B) Complementation strains of traN knockout expressing single-substituted traN variants in trans (pEU327-RBS-traN_Q28A, pEU327-RBS-traN_H82A, pEU327-RBS-traN_K101A) and (C) complementation strains of traN knockout expressing multiple-substituted traN variants in trans (pEU327-RBS-traN_R23A-N24A-Q28A, pEU327-RBS-traN_G47A-G48A, pEU327-RBS-traN_H82A-R86A, and pEU327-RBS-traN_G100A-K101A) were applied as donors, E. faecalis OG1X as recipient. Transfer frequencies are presented as number of transconjugants per recipient cell. n = 3. Mean values are depicted with standard deviation. **p < 0.01, *p < 0.1 as determined by the Mann–Whitney U test.
To investigate the impact of single alanine substitutions in TraN on pIP501 transfer, putative key residues were chosen based on their ability to form direct protein-to-base interactions which are defined as class 1 interactions (Kohler et al., 2018a) and direct protein-to-DNA-backbone interactions, respectively. Residues forming only water-mediated interactions were excluded. TraN key amino acid residues were identified based on the TraN/pIP501 DNA complex crystal structure (Kohler et al., 2018a) (Figure 1A). TraN contains two-winged helix-turn-helix (wHTH) motifs. Motif 1 is comprised of the amino acids interacting with the major groove of the DNA—residues R23, N24, and Q28—within the N-terminal TraN domain. Motif 2 is comprised of the residues H82 and R86 of the C-terminal domain, respectively. In addition, interactions with the minor groove of the DNA are exerted by the two wings of the wHTH motifs—residues G47–G48 and G100–K101 (Kohler et al., 2018a). To observe the effect of single alanine substitutions in TraN on pIP501 transfer, three amino acids were selected, each belonging to one DNA interaction site (Q28, H82, K101). Here, we exchanged these putative key residues against alanine by site-directed mutagenesis to disrupt the direct base contacts to the original TraN binding site upstream of the Ptra promoter of the pIP501 tra operon (Figure 1A). In trans complementation of pIP501∆traN was assayed with these variants cloned in the expression plasmid pEU327. Complementation effects were tested in biparental mating assays with E. faecalis.
Transfer rates of E. faecalis JH2-2 (pIP501∆traN) complementation strains (pEU327-RBS-traN_Q28A, pEU327-RBS-traN_H82A and pEU327-RBS-traN_K101A) were compared with pIP501, the complemented knockout with traN in trans, and the pIP501∆traN knockout strain. The single TraN amino acid substitution (H82A) revealed a small but significant increase in transfer rate (4.66 × 10−5) compared to complementation with traN in trans (1.17 × 10−5). Q28 and K101 substitutions led to non-significant changes in transfer compared to complementation with traN (Figure 1B). All transfer rates (mean values and standard deviations) are given in Supplementary Table S4.
As single amino acid substitutions of putative TraN key residues had only a minor effect on pIP501 transfer, three double alanine substitutions (G47A-G48A, H82A-R86A, G100A-K101A) and one triple substitution (R23A-N24A-Q28A) were generated. These residues are located on the DNA-binding α-helix and the wing of the wHTH motif of both N- and C-terminal DNA binding motifs. Variants of pEU327-RBS-traN with multiple alanine substitutions (G47A-G48A, R23A-N24A-Q28A, H82A-H86A, G100A-K101A) were constructed. E. faecalis JH2-2 (pIP501∆traN) was complemented with those variants. They were applied to biparental mating assays as donors and E. faecalis OG1X as recipient. Three of four multiple substitutions (R23A-N24A-Q28A, H82A-R86A, and G100A-K101A) revealed a significantly enhanced transfer rate. In each case, the differences were larger than one order of magnitude. The transfer rate was similar to that of the ∆traN knockout strain with a transfer rate of 1.72 × 10−3 (Figure 1C, Supplementary Table S4). Complementation of pIP501∆traN with traN-G47A-G48A showed the lowest effect (3.79 × 10−5) in comparison to complementation with traN (1.06 × 10−5) and the pIP501 transfer rate (1.72 × 10−5). Mean values and standard deviation of all transfer rates are given in Supplementary Table S4.
To exclude the observed changes in transfer rate of pIP501∆traN complemented with traN variants cloned into pEU327 being due to lack of expression of the proteins, we verified their expression in E. faecalis JH2-2. Expression of TraN and seven TraN variants was investigated in E. faecalis JH2-2, harboring pIP501∆traN and pEU327 carrying traN or one of the traN variants.
TraN as well as all TraN variants were expressed in E. faecalis JH2-2 (Supplementary Figure S2). However, the level of expression appeared to differ. TraN_H82A-R86A and TraN_G100A-K101A particularly showed lower expression than TraN and the other variants (Supplementary Figure S2), so we verified whether the alanine substitutions changed the binding affinity of the anti-TraN antibody. The four multiple-substituted TraN variants and TraN (TraN, TraN_R23A-N24A-Q28A, TraN_G47A-G48A, TraN_H82A-R86, and TraN_G100A-K101A) were expressed in E. coli Lemo21 (DE3), purified as described in chapter 2.6 and applied to immunoblotting (Supplementary Figure S1C), resulting in signals with differing intensity for TraN and the variants.
Especially TraN_H82A-R86A, with the lowest signal intensity in the immunoblot checking the expression (Supplementary Figure S2), and also TraN_R23A-N24A-Q28A, and TraN_G100A-K101A yielded a weaker signal in the immunoblot than TraN. As we were using a polyclonal anti-TraN antibody, we assumed that it binds to different TraN epitopes. If one of these epitopes is changed due to alanine substitution(s), it may result in a weaker signal in the immunoblot. Thus, it can be argued that the lower signal intensity for these TraN variants in E. faecalis might be due to the mutation(s) of epitopes recognized by anti-TraN antibodies and not due to lower expression levels. In addition, we want to emphasize that, due to application of the overexpression plasmid pEU327, sufficient protein levels of the TraN variants TraN_H82A and TraN_H82A-R86 will be present, although expression levels of these two TraN variants seem to be lower—as evidenced in the immunoblot (Supplementary Figure S2).
TraN and the four variants (TraN_R23A-N24A-Q28A, TraN_G47A-G48A, TraN_H82A-R86A, TraN_G100A-K101A) were expressed in E. coli Lemo21 (DE3) and purified by HisTrap, followed by HeparinTrap affinity chromatography. To assess purity and oligomerization states, an analytical size-exclusion chromatography (SEC) was performed. All variants were well expressed and exhibited similar final yields of purified proteins. Corresponding SDS polyacrylamide gels of all purification steps are shown in Figure 2 and the chromatograms showing the elution profile of purified TraN and its variants in Supplementary Figure S1B. TraN and its variants showed a pronounced overexpression comparing the samples before induction with the samples at harvesting. The elution fractions of the analytical SEC runs showed that we retrieved TraN and all its variants in high purity. The 260/280 ratio indicates that TraN and its variants were all free from bound DNA, which is a crucial prerequisite for DNA binding studies (Supplementary Figure S1B).
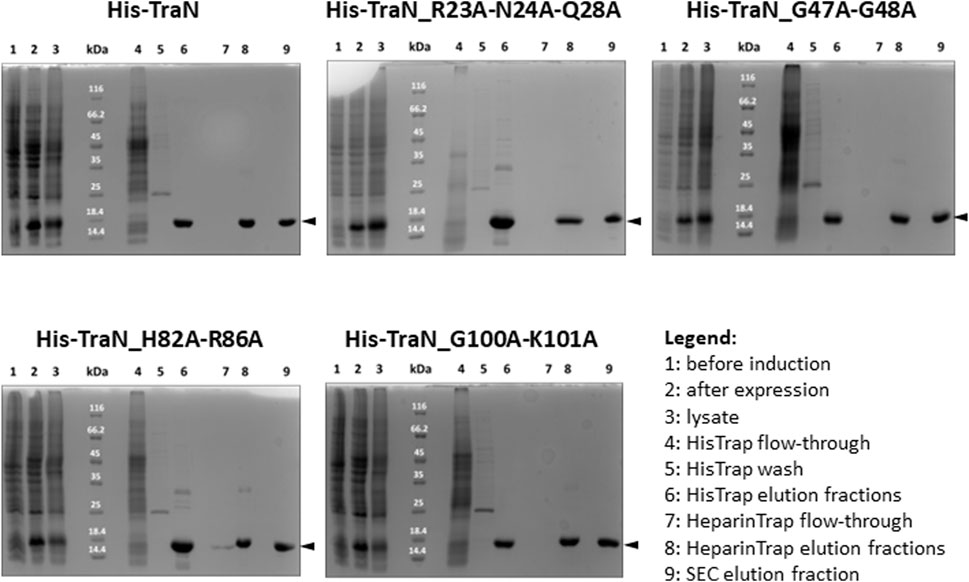
FIGURE 2. Coomassie-stained 12% SDS polyacrylamide gels of expression and purification of TraN and its variants. kDa: Pierce™ Unstained Protein molecular weight marker (Thermo Fisher). The expected molecular weight of all constructs ranges from 17.10 kDa (TraN_R23A-N24A-Q28A) to 17.32 kDa (TraN_G47A-G48A).
Circular dichroism (CD) measurements were performed to compare whether multiple alanine substitutions affect the fold and stability of TraN. All TraN variants showed almost identical CD spectra compared to TraN, displaying two minima at 207 nm and 216 nm. This suggests that there are no significant changes in secondary structure between the variants (Figure 3A).
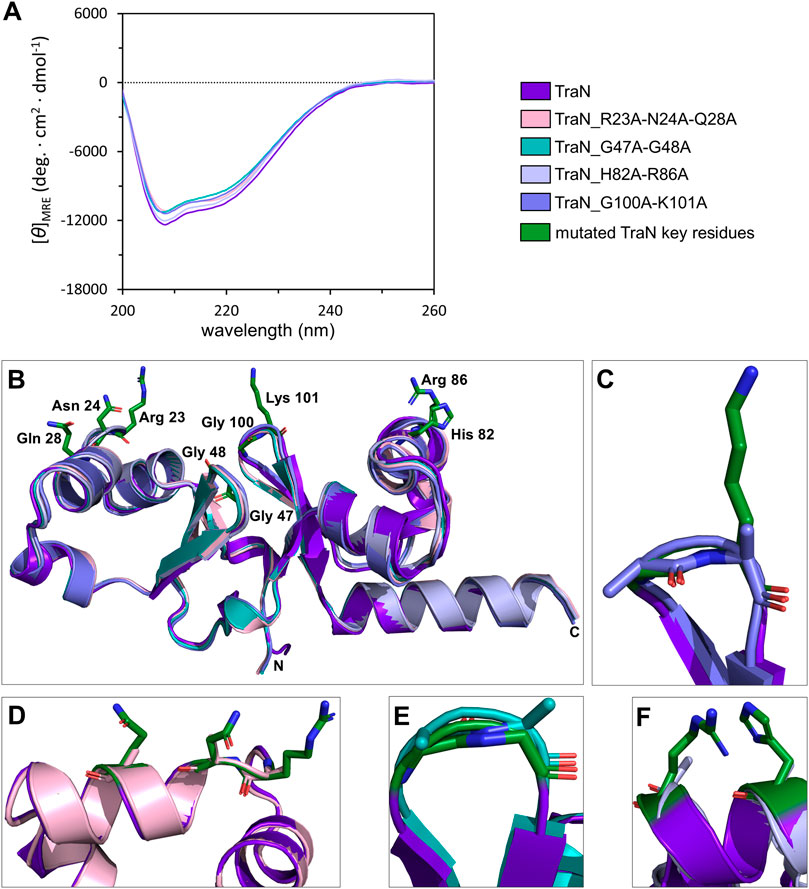
FIGURE 3. TraN fold seems unaffected in TraN variants, TraN_R23A-N24A-Q28A, TraN_G47A-G48A, TraN_H82A-R86A, and TraN_G100A-K101A. (A) Circular dichroism (CD) spectra of His-TraN and its variants. (B) Structural alignment of the crystal structure of TraN and the predicted structures of its variants showing that the substitutions do not have an impact on the protein fold. TraN is depicted in purple. The residues mutated in the variants are shown as green sticks. C- and N-termini are labeled. The color code for the TraN variants is shown in Supplementary Table S5. (C) Close-up of the wing motif of the C-terminal wHTH motif (TraN_G100A-K101A). The alanines likely protrude into the minor groove upon DNA binding. (D) Close-up of motif 1. The mutated residues of TraN_R23A-N24A-Q28A are shown as light pink sticks. (E) Close-up of the wing motif of the N-terminal wHTH motif (TraN_G47A-G48A). Introduced alanine residues likely protrude into the minor groove upon DNA binding. (F) Close-up of motif 2 (TraN_H82A-R86A).
We used the CoLab AlphaFold2 server for structure prediction of the TraN variants (TraN_Q28A, TraN_H82A, TraN_K101A, TraN_H82A-R86A, TraN_G47A-G48A, TraN_G100A-K101A, TraN_R23A-N24A-Q28A). The alignment of multiple-substituted TraN variants with the experimental TraN structure resulted in overall RMSD values between 0.588 Å (TraN_G47A-G48A) and 0.643 Å (TraN_H82A-R86A). The alignment of single-substituted variants to TraN gave even lower RMSD values of 0.519 Å to 0.540 Å (Supplementary Table S5). Alignments of the double- and triple-substituted variants are shown in Figure 3B. In addition, we provide close-up images of the regions showing the mutated residues (Figures 3C–F). The low RMSD values indicate that the mutations do not affect the protein fold in the predicted structures. In comparison, the alignment of the crystal structures of free TraN (PDB: 4P0Z) with TraN in complex with oBS (PDB: 6G1T) yielded a RMSD value of 0.837 Å.
To investigate the sequence-specific binding affinities of the TraN variants (TraN, TraN_R23A-N24A_Q28A, TraN_G47A-G48A, TraN_H82A-R86A, and TraN_G100A-K101A), we used MST with dsDNA substrates corresponding to the original TraN binding site (oBS) upstream of the pIP501 tra operon, and a random pIP501 sequence of the same size (34-mer). The oligonucleotides are identical to the 34-bp dsDNA (oBS) identified and used by Gössweiner-Mohr et al. (2014a) for isothermal titration calorimetry (ITC) measurements. Dose-response curves of all protein-DNA combinations are shown in Figure 4A. Additionally, the sites where mutations were introduced are shown on the TraN-DNA complex crystal structure (PDB: 6G1T) (Kohler et al., 2018a). Close-up images of the mutated sites are also depicted in Figure 3. The dissociation constants for the interaction of TraN and its variants with the ds-oligonucleotides are shown in Figure 4B. All fitted curves exhibit a sigmoidal shape from which the Kd was calculated. It was not possible for the interaction of the double-substituted proteins TraN_H82A-R86A and TraN_G100A-K101A with the random DNA to fit the data points by a sigmoidal curve, suggesting an extremely low binding affinity. In the presence of the random DNA, the Kd values for all other TraN variants were in the low micromolar range, suggesting a much lower affinity than the binding of the specific 34-mer (oBS).
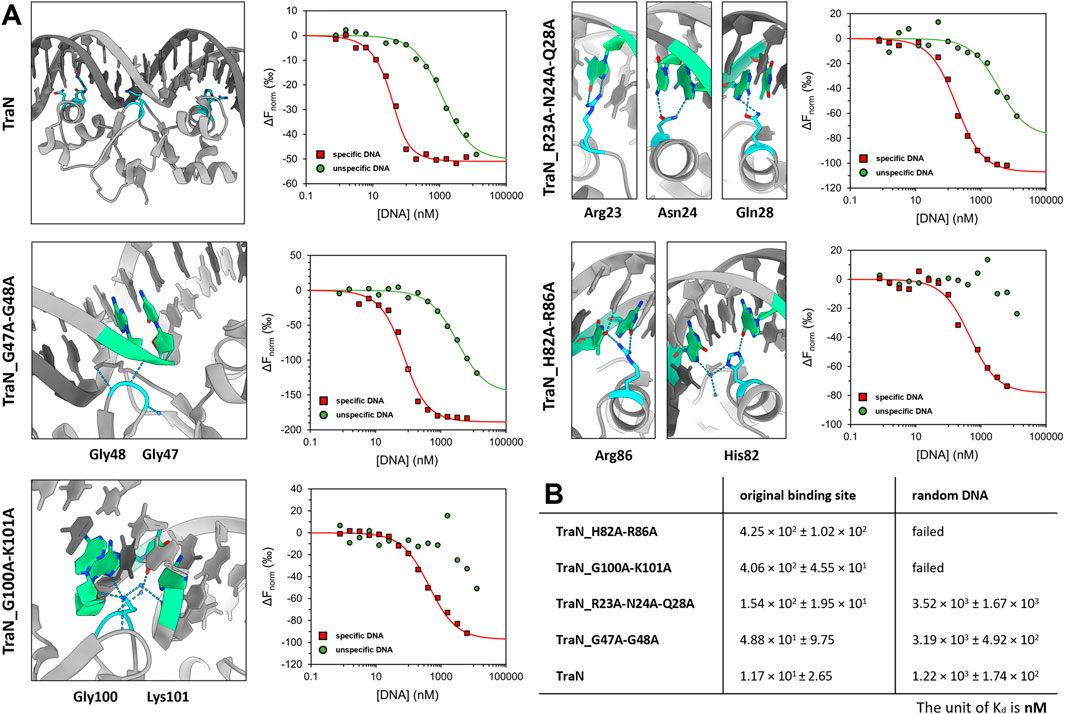
FIGURE 4. Graphical representation of TraN and its variants interacting with original binding site (oBS) and random ds-oligonucleotide (random DNA). (A) Cartoon representation of class (I) interactions between amino acid side chains and corresponding bases of oBS. The images were taken from the structure of TraN bound to oBS pIP501 DNA (PDB: 6G1T). Interacting residues are highlighted in cyan and respective bases of the oBS are highlighted in green. Water molecules are depicted in dark blue, hydrogen bonds in light blue. Normalized dose-response curves of TraN and its variants for interactions with oBS (red) and unspecific random DNA (green) acquired from MST binding assays. Wherever the fit was successful, the resulting curves exhibit a sigmoidal shape from which a dissociation constant (Kd) was calculated. (B) MST-derived dissociation constants (Kd) of TraN and its variants to oBS and random DNA.
TraN showed the strongest binding affinity to the oBS 34-mer sequence (1.17 × 101 ± 2.65 nM) followed by the double-substituted variant TraN_G47A-G48A (4.88 × 101 ± 9.75 nM). The triple-substituted variant TraN_R23A-N24A-Q28A showed an almost 13-fold increase in the Kd value (1.54 × 102 ± 1.95 × 101 nM) compared to TraN, suggesting a strong negative effect on DNA binding affinity. Both TraN_H82A-R86A (4.25 × 102 ± 1.02 × 102 nM) and TraN_G100A-K101A (4.06 × 102 ± 4.55 × 101 nM) variants showed approximately 36-fold higher Kd values yielding the lowest binding affinities to the oBS 34-mer of all variants compared to TraN.
The binding affinities determined through MST exhibit a strong correlation with the results obtained from the biparental mating assays. When the pIP501∆traN knockout strain was complemented with traN_G47A-G48A in trans (3.79 × 10−5 transconjugants/recipient), the impact on pIP501 transfer was minimal compared to complementation with traN-G100A-K101A (6.86 × 10−4) (Figure 1B). Consequently, the binding affinity of TraN_G47A-G48A was found to be higher than that of TraN_G100A-K101A, indicating its strongest capability in sequence-specific binding on pIP501 oBS amongst the tested variants.
Previously, yeast two-hybrid studies showed interactions of TraN with TraE, TraG, TraH, and TraL (Abajy et al., 2007). Therefore, we decided to perform a pull-down assay i) to confirm these interactions and ii) to elucidate further direct or indirect TraN interactions. Consequently, P. megaterium MS941 was co-transformed with pMGBm19-RBS-traB-traO and pRBBm59-RBS-traN-Strep. This resulted in co-expression of C-terminally Strep-tagged TraN (TraN-Strep) and pIP501 transfer proteins TraB to TraO in P. megaterium MS941.
Utilizing affinity chromatography, Strep-tagged TraN and the proteins interacting with it were purified from the clarified supernatant of P. megaterium MS941 (pMGBm19-RBS-traB-traO, pRBBm59-RBS-traN-Strep) lysate.
To detect coeluted Tra proteins, elution fractions from TraN pull-downs were subjected to immunoblotting with anti-Tra antibodies directed against TraB, TraE, TraF, TraG, TraH, TraJ, TraK, TraM, TraN, and TraO. The elution fraction was compared to the flow-through and lysate fraction. We could demonstrate the coelution of TraE, TraF, TraG, TraH, TraJ, TraK, TraM, and TraO, as evidenced by the presence of the respective bands (Figures 5A–J).
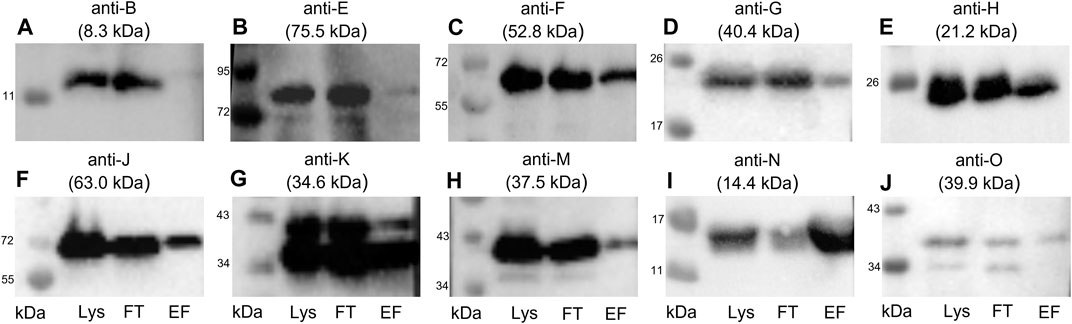
FIGURE 5. Western blots of the elution fractions of the pull-down with TraN-Strep. The following antibodies were used: (A) anti-TraB, (B) anti-TraE, (C) anti-TraF, (D) anti-TraG, (E) anti-TraH, (F) anti-TraJ, (G) anti-TraK, (H) anti-TraM, (I) anti-TraN, and (J) anti-TraO, targeting the respective Tra proteins. The MW of the respective protein is indicated above each blot. MW marker (Blue Prestained Protein Standard, New England Biolabs) was included to assess protein sizes (kDa). Three fractions of the TraN-based pull-down were analyzed: lysate (Lys) in lane 1, flow-through (FT) in lane 2, and elution fraction (EF) in lane 3. *14.4 kDa is the MW of native TraN. The fused Strep-tag adds 1 kDa; therefore, the protein appears slightly larger than in the immunoblots performed with native TraN.
TraB was not coeluted, suggesting no interaction with TraN. In contrast with the calculated molecular weight (MW) of 8.3 kDa, the expected MW on the SDS polyacrylamide gel and the corresponding immunoblot is around 12 kDa (Berger et al., 2022). The membrane incubated with anti-TraO antiserum showed two bands which most likely present two differently processed forms of the protein: one with its signal sequence and one without it. The cleavage results in a size difference of 2.5 kDa (Berger et al., 2022). The single band representing TraG had a MW of around 20 kDa instead of the calculated MW of 40.4 kDa. It is hypothesized that the T4SS protein complex includes only the CHAP domain of TraG, as the observed band corresponds to its calculated MW of 22 kDa (Arends et al., 2013; Berger et al., 2022). Likewise, TraF was detected at around 60 kDa, which was slightly above the calculated 52.8 kDa. TraE was coeluted in rather small amounts. The nitrocellulose membrane incubated with the anti-TraK antibody showed two bands (Figure 5G), indicating the presence of two TraK variants. This can be attributed to the existence of two different translational start codons for TraK (Goessweiner-Mohr et al., 2014b). The coelution of TraM might be because TraN and TraM are both DNA binding proteins. In contrast to its C-terminal domain, the N-terminal domain of TraM showed unspecific DNA binding (T. Berger and W. Keller, unpublished data).
To confirm that the coelution of proteins together with the Strep-tagged TraN is highly specific and not the result of unspecific binding, we conducted a negative control experiment. A GFP pull-down was performed followed by immunoblotting using anti-TraN or anti-TraF antibodies (e.g., for a Tra protein which was coeluted in the TraN pull-down). TraN and TraF could be detected only in the lysate and flow-through fraction but not in the elution fraction (Figure 6), suggesting that there is no unspecific binding of pIP501 proteins.
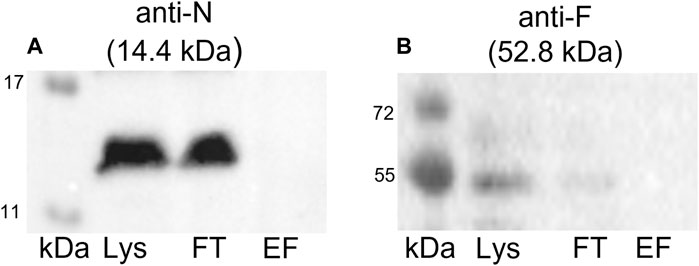
FIGURE 6. Western blots of the pull-down with GFP-Strep. The cell lysate (Lys) in lane 1, the flow-through (FT) in lane 2, and the elution fraction (EF) in lane 3 were analyzed. (A) Anti-TraN and (B) anti-TraF antibodies were used as primary antibodies. The used antibody and the MW of the respective Tra protein are given above each blot. MW marker (kDa) (Blue Prestained Protein Standard, New England Biolabs) was applied.
Next, we analyzed the elution fractions from TraN pull-downs with high-resolution mass spectrometry. An in-gel tryptic digest was performed from the elution fractions, and the resulting peptides were analyzed by nano-LC-MS/MS. For the pull-down elution fractions, the MS spectra yielded positive hits for all Tra proteins except for TraA (not included in the expression vector, negative control), TraC, TraG, TraL, and TraO. Looking at the LFQ intensity signals of the samples (blue labels in Supplementary Table S7), TraE, F, H, J, K, M, and N show strong signals. TraN occurs in a huge surplus over the other Tra proteins as it carries the Strep-tag and binds to the affinity column whether attached to other Tra proteins or as a monomeric protein. Notably, TraB and TraD are detected but not clearly quantifiable, indicating their low abundance in the pull-down fraction.
Homology searches with TraN were performed to find putative similar proteins in other bacterial species or even genera.
A previous search for TraN homologs was performed by Kohler et al. (2018a). Despite their common presence in mobile genetic elements from Firmicutes, TraN homologs have been rarely investigated. As many new plasmid sequences have been submitted to NCBI in the last 5 years, we conducted a new search for putative TraN homologs. Additionally, we not only used a larger dataset but also assigned the selected plasmid sequences to putative conjugative and non-conjugative plasmids. According to the criteria defined in chapter 2.12, 54 plasmid sequences and one transposon sequence were selected for further analysis. All protein sequences displaying over 87% identity on amino acid level to TraN originated from Firmicutes.
According to the tBLASTn alignments, 26 out of 54 homologs displayed 100% identity on protein level with TraN, and 39 out of 54 homologs exhibited 100% query coverage with protein identities between 88% and 99% (Supplementary Table S8).
An oriT was predicted for 44 out of the 54 plasmids (see Supplementary Table S8). For the remaining ten, a tBLASTn search with the sequence of the pIP501 relaxase TraA, the ATPase TraE, and the coupling protein TraJ also revealed no hits. A putative TraJpIP501 homolog was identified for the oriT-lacking plasmids pSM19035 (AY357120.1), pF104_1 (CP072892.1), pEGM181-1 (CP050484.1), and pGMrib (CP045673.1). Plasmid pEGM181-1 additionally encodes for a TraEpIP501 homolog but not for a TraApIP501 (Supplementary Table S9). Among the 54 plasmids with a predicted oriT, p2 (CP041740.1), plas1 (CP064340.1), and pVEF3 (AM931300.1) lack traA-/traE-/traJ-like sequences, with p2 being the only one encoding for a TraJpIP501 homolog (Supplementary Table S9).
Supplementary Table S8 provides additional information on the presence of a pIP501-like T4SS for the initially selected 54 plasmid sequences and one transposon sequence. The NCBI tBLASTn search employed the pIP501 transfer proteins TraA to TraO as queries. Of these MGEs, 65% encode a complete T4SS (traA—traO), underlining the conjugative potential of the TraN-encoding MGEs. Interestingly, four putative conjugative plasmids (p3-38, pEFS108_1, pL15-B, pW3) were identified as having a nearly complete T4SS cluster, lacking only a traO-like gene (Supplementary Table S8).
In total, at an amino acid level, 11 different TraN sequences were detected in 28 different plasmids.
Eleven plasmids, each representing a distinct TraNpIP501 homolog and containing a putative oriT, were selected from the initial set of 28 (Table 1). Each TraN homolog shares ≤99% protein identity with TraNpIP501. Table 1 displays homologs of three pIP501 signature proteins—TraApIP501, TraEpIP501, and TraJpIP501—in the 11 selected plasmid sequences, indicating the likely presence of a T4SS in these plasmids that encode traN homologous sequences. All plasmids encoding a TraNpIP501 homolog exhibited high-level protein identity (≥96%) to the pIP501 TraA, TraE, and TraJ T4SS proteins (Table 1). The 11 different TraN homologs were applied to a multiple sequence alignment using T-Coffee Expresso (Figure 7A).
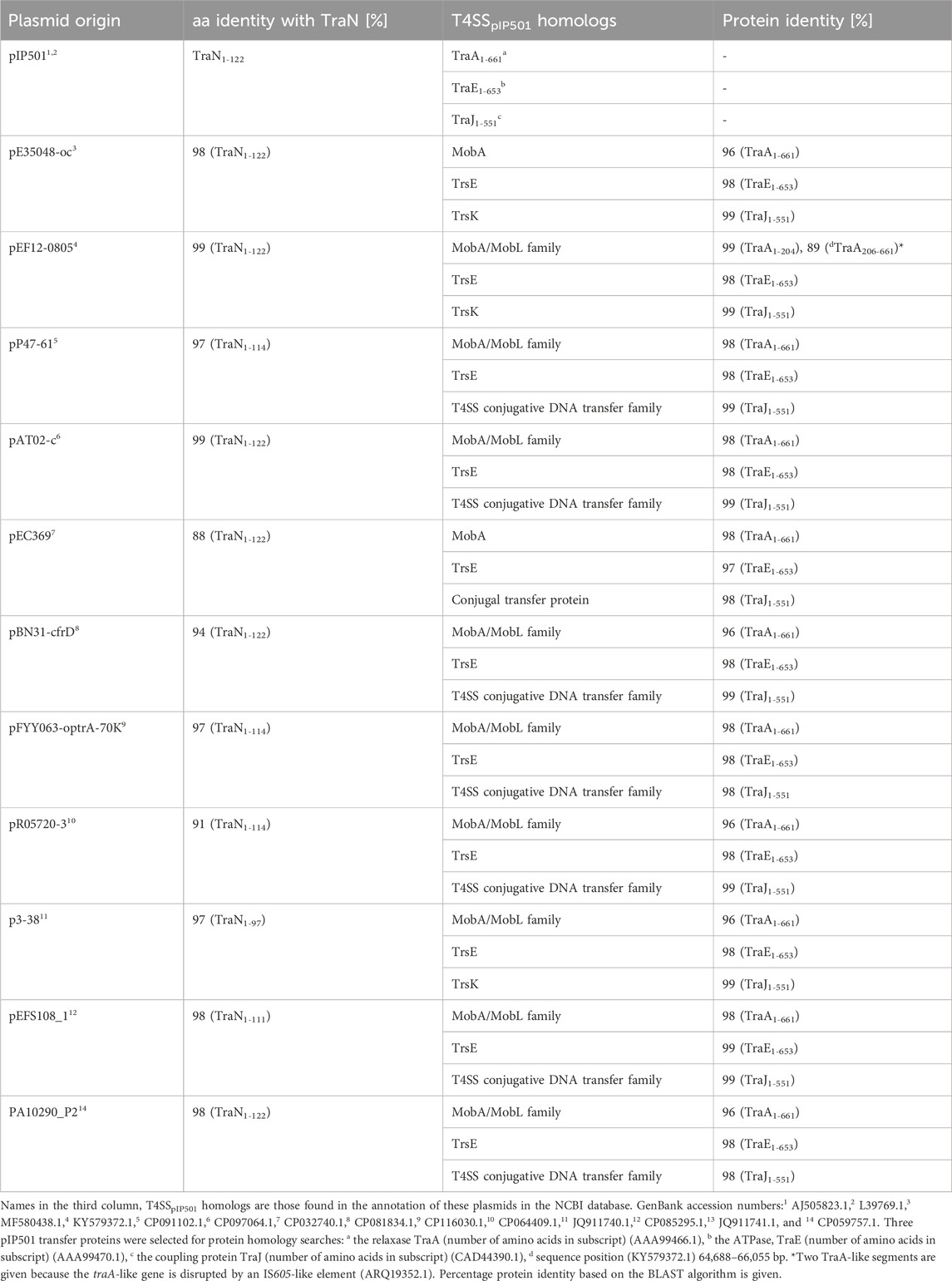
TABLE 1. TraApIP501, TraEpIP501, and TraJpIP501 homologs in putative conjugative plasmids encoding traN homologous sequences. All plasmids have a putative oriT predicted with oriTfinder.
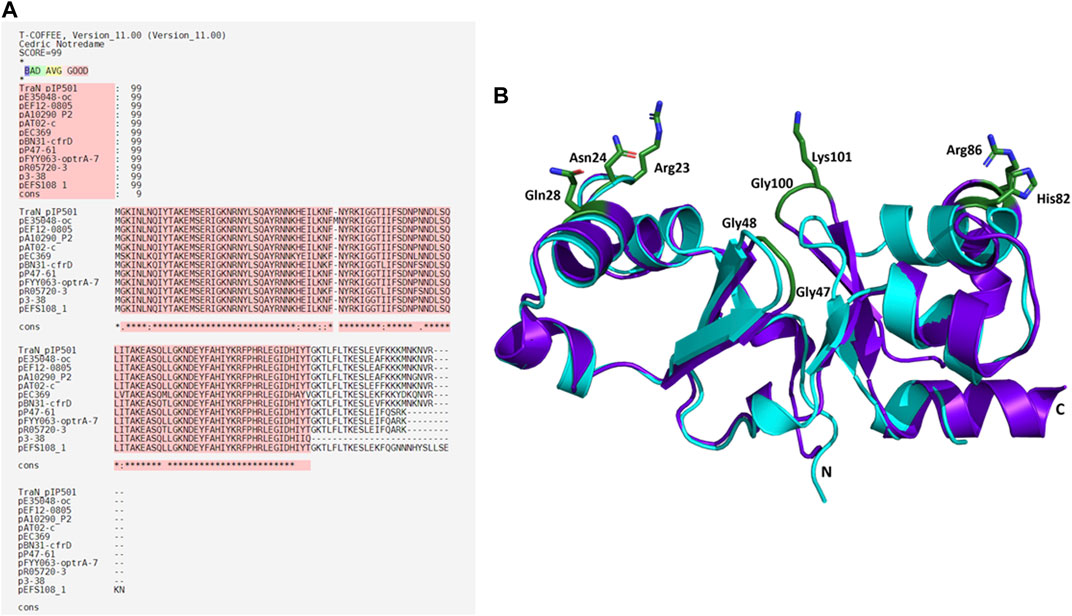
FIGURE 7. Alignments of TraNpIP501 and its homologs. (A) Multiple sequence alignment using Expresso algorithm implemented in T-Coffee. Homologs were identified by tBLASTn search. As these homologs are hypothetical, the names of the plasmids encoding the TraNpIP501 homolog are used instead of protein annotation. Asterisks (*) represent conserved amino acids in all aligned sequences, (:) indicates amino acids of high similarity, dots (.) indicate partial similarities. The top panel shows the evaluation scores from the T-Coffee Server (TCS) evaluation. The closer the score to 100, the better the alignment. The quality of the alignment is also represented in red. (B) Backbone alignment of TraNpIP501 (purple) with the homolog from p3-38 (cyan) using theoretical structures predicted with coLAB AlphaFold2 server. Residues (Arg23, Asn24, Gln28, Gly47, Gly48, His82, Arg86, Gly100, and Lys101) are shown as green sticks.
The PlasmidFinder tool was used for in silico prediction of plasmid replicon families of all 54 plasmids. The affiliations of the plasmids to a replicon family and their corresponding identity scores are listed in Supplementary Table S8. Of the 54 plasmids, 43 were assigned as Inc18 broad-host-range plasmids, three plasmids belonged to the RepA_N family, and one to the Rep_3 family. No replicon sequence matches were found for seven of the plasmids. TraN homologs were found within the T4SS region of putative conjugative plasmids belonging to Rep_3 and RepA_N incompatibility groups, including plasmids like pL9 (Rep_3) and pL15-B (RepA_N), with pL15-B lacking a traO-like gene. In essence, our findings reveal the presence of TraN across diverse plasmid replicon families, extending beyond Inc18. While traN, together with other putative T4SS genes, has been identified in many plasmids, conjugative transfer has been demonstrated experimentally for a only few, underlining the limited knowledge of Inc18 plasmids.
To the best of our knowledge, conjugative plasmid transfer has only been demonstrated for 11 of these plasmids: pRE25 (Schwarz et al., 2001), pAMβ1 (Clewell et al., 1974), pV386 (Cinthi et al., 2022), pPPM1000 (Davis et al., 2005), pWZ909 (Zhu et al., 2010), pW3, p3-38 (Liu et al., 2013), pBN31-cfrD (Zhu et al., 2022), pEC369 (Yin et al., 2018), pEFS108_1 (Kim et al., 2021), and pP47-61 (Li et al., 2022). Conjugation experiments using donor strains with pE34048-oc (Morroni et al., 2018), pEF12-0805 (Lazaris et al., 2017), and pStrcfr (Wang et al., 2013) failed. pSM19035 is annotated as non-conjugative (Behnke et al., 1981). Interestingly, an additional TraN homolog was found on the composite transposon Tn6349 located on the multiresistant Staphylococcus aureus strain AOUC-0915 (D’Andrea et al., 2019) encoding for erm(B), cfr, poxtA, and fexB resistance genes. However, the transposon was shown to be non-conjugative (D'Andrea et al., 2019).
Sequence alignments with T-Coffee Expresso were performed to determine the extent to which TraN homologous sequences deviate from TraNpIP501. As depicted in Table 1, five TraN homologs are truncated: that encoded on p3-38 (TraN1-97), the homolog from pEFS108_1 (TraN1-111), and those from pP47-61, pFYY063-optrA-70K and pR05720-3 (TraN1-114). The coLAB AlphaFold2 server was used to predict the structures of the 11 distinct TraNpIP501 homologs. The structures were aligned with the solved structure of TraN (PDB: 4P0Z). As the sequence identity is quite high, the overall structure of all homologs is likely very similar to TraNpIP501. The respective RMSD values are listed in Table 2. The most notable difference is observed in the homolog encoded on p3-38 (Figure 7B), where not all key residues are conserved. This discrepancy is attributed to its smaller size (99 aa instead of 122 aa), leading to the absence of Gly100 and Lys101. In all other homologs, the corresponding amino acids (Arg23, Asn24, Gln28, Gly47, Gly48, His82, Arg86, Gly100, and Lys101) were not altered, indicating that the mode of DNA recognition is conserved among all homologs compared.
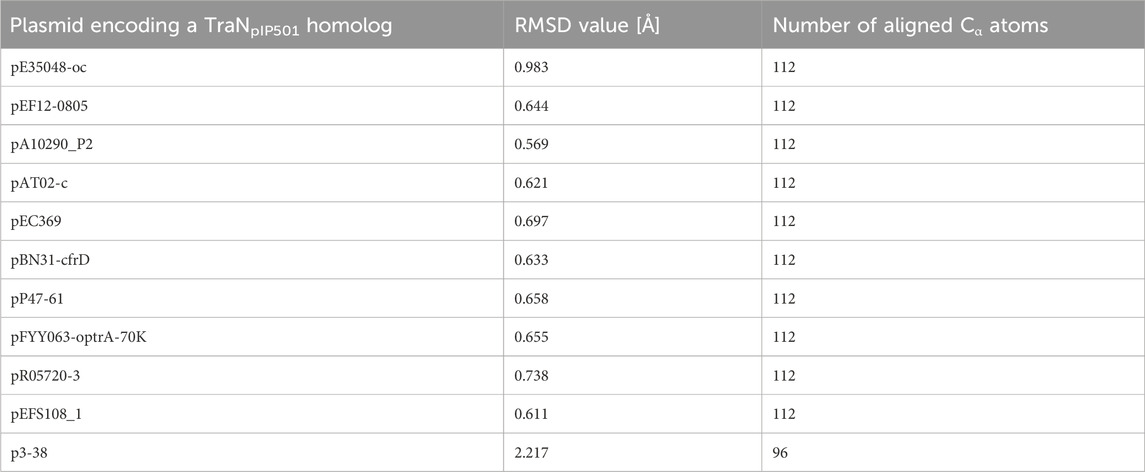
TABLE 2. TraN homologs aligned with the crystal structure of TraNpIP501 (PDB: 4P0Z) showing the root mean square deviation (RMSD) values for the backbone atoms. The number of the aligned amino acids is included.
The dissemination of ARGs or virulence factors through conjugative T4SSs across bacterial phylogenetic barriers is a global concern. The expression of T4SS proteins, the T4SS assembly, and DNA transfer require considerable energy, which emphasizes the need to regulate this energy-consuming process. The Inc18 group of plasmids, in this study represented by the prototype conjugative plasmid pIP501, contains multiple resistance plasmids with a broad host range among Gram-positive bacteria. Its repressor TraN has been proposed to be involved in regulation of the expression of the tra operon encoding the conjugative T4SS (Goessweiner-Mohr et al., 2014a; Kohler et al., 2018a).
Here, we validated the role of the putative TraN key residues R23, N24, Q28, G47, G48, H82, R86, G100, and K101 involved in specific TraN binding to its cognate pIP501 DNA. We performed in vivo biparental mating assays employing traN variants and in vitro MST measurements to decipher the sequence-specific DNA binding affinity of the TraN variants. Additionally, the study examined the putative functional role of TraN in the T4SS complex and its prevalence among plasmids from Gram-positive bacteria.
All nine residues were substituted by alanine to investigate the role of putative TraN key residues involved in binding to a specific DNA sequence (oBS) upstream of oriTpIP501 (Kohler et al., 2018a). For complementation studies, seven pEU327-RBS-traN variants with single (Q28A, H82A, and K101A) or multiple alanine substitutions (N23A-R24A-Q28A, G47A-G48A, H82A-R86A, and G100A-K101A) were generated. The pIP501∆traN complementation with TraN_R23A-N24A-Q28A, TraN_H82A-R86A, and TraN-G100A-K101A led to a significant increase of transfer by more than 1.8 log compared to the single amino acid substitutions, with the TraN_G100A-K101A variant exhibiting the highest transfer rate. The TraN double-substituted variant TraN_G47A-G48A only resulted in an increase of the transfer rate by less than one order of magnitude compared to original pIP501. The Gly47 and Gly48 only exhibit one direct protein-DNA interaction, which is potentially not affected by the Gly to Ala substitution. However, the alanine side chain might increase the sterical hindrance at the protein-DNA interface, leading to slightly weaker binding. We propose that the binding strength and specificity of TraN is mainly mediated by the residues forming direct base contacts in the major groove (Arg23, Asn24, Gln28, His82, and Arg86) or in the central minor groove (Lys101) of the dsDNA.
As the multiple-substituted TraN variants had the highest effect on pIP501 transfer frequency, MST was used to analyze their binding affinity to the specific TraN binding site (oBS) of the Ptra promoter. In general, the data from the in vitro DNA binding assays correlated well with the results from the mating assays, showing that TraN exhibited the lowest Kd value reflecting the strongest binding and thus proper regulation. The Kd of TraN bound to oBS DNA was similar to the Kd obtained with ITC measurements (Goessweiner-Mohr et al., 2014a). We found that residues H82, R86, G100, and K101 are most important for mediating high affinity binding of TraN to its oBS. The strong impact of the substitutions in the TraN_G100A-K101A variant on Kd is most likely due to the substitution of the Lys101 residue, which reaches deep into the minor groove of the dsDNA and forms a network of direct and indirect base contacts. The Gly100 residue enables the formation of a tight β-turn and is apparently necessary for the correct placement of Lys101 in the minor groove, as the effect of the double mutation is much more prominent than of the single mutation of TraN_K101A.
The His82 and Arg86 residues reach into the major groove of the oBS DNA and form an intricate network of direct and indirect base contacts. We observed similar DNA binding affinities for the TraN_H82A-R86A and the TraN_G100A-K101A variant. The key residues of the N-terminal recognition helix (motif 1), Arg23, Asn24, and Gln28 are positioned in the major groove of the second oBS half site and form an extended network of base interactions (Kohler et al., 2018a). However, the substitutions in the triple-substituted variant TraN_R23A-N24A-Q28A seem to affect the binding affinity less than the H82A-R86A double-substituted variant of the C-terminal recognition helix (motif 2). These differences in binding affinities between the N- and the C-terminal domains of TraN agree with previous observations that the C-terminal recognition helix (motif 2) contributes more to the thermal stability of the TraN-oBS complex than motif 1. In addition, calculations based on the complex structure yielded a higher free energy of assembly for the C-terminal domain than the N-terminal domain (Kohler et al., 2018a). The TraN_G47A-G48A variant showed only a small decrease in binding affinity compared to TraN. The crystal structure of TraN-oBS complex revealed a tight β-turn comprising residues G48 and G47, where the amide group of G48 forms a hydrogen bond to a phosphate group of the DNA backbone and the carboxyl group forms a hydrogen bond to Lys101, thus stabilizing the conformation of this minor groove binder. The substitutions of glycine by alanine do not compromise the hydrogen bonding pattern but may lead to a slight steric hindrance upon DNA binding, thus explaining the observed effects on binding affinity and transfer rates.
Studies on the expression levels of TraN variants compared to TraN showed that all variants are expressed to almost the same extent, except for TraN_H82A and TraN_H82A-R86A (Supplementary Figure S2), which also showed the lowest affinity to the anti-TraN antibody (Supplementary Figure S1C). Considering that pEU327 is an overexpression plasmid in contrast to pIP501, we are confident that the differences in transfer rate originate from the introduced mutation(s). This is supported by the results of the MST measurements, where the changes in binding affinity correlated well with the results of the mating assays. CD measurements showed similar spectra for all TraN variants and the native TraN, suggesting that the secondary structure content and the overall fold remain largely unchanged among all variants.
The pIP501-encoded T4SS consists of 15 Tra proteins, of which eight (TraB, TraC, TraF, TraH, TraI, TraK, TraL, and TraM) are likely involved in the buildup of the MPF complex, which serves as an envelope-spanning translocation channel. We investigated whether TraN can interact with other pIP501 T4SS components, as yeast two-hybrid studies had demonstrated interactions of TraN with several Tra proteins (Abajy et al., 2007). There, TraN has been reported to interact with itself, the ATPase TraE, the lytic transglycosylase TraG, and the membrane-associated VirB8-like TraH (Abajy et al., 2007). The pull-down assays in our study demonstrated coelution of TraE, TraF, TraG, TraH, TraJ, TraK, TraM, and TraO with TraN. Thus, they confirmed all these previously identified interactions. However, it should be mentioned that, for all tested proteins except TraN itself, the amount of protein in the elution fraction was significantly lower than in the flow-through fraction. We want to point out that not all Tra proteins pulled down necessarily interact directly with TraN, particularly those forming part of the MPF complex. Considering the exclusive presence of TraN in the cytosolic fraction (Goessweiner-Mohr et al., 2014a), we propose that TraN is not part of the MPF complex but rather forms direct or indirect interactions with components of the T4SS complex. This might clarify why certain putative MPF-forming Tra proteins, such as TraF, TraK, and TraM, were coeluted. In the case of TraM, coelution could occur since both proteins exhibit non-sequence-specific DNA binding properties, as recently demonstrated for the N-terminal domain of TraM (T. Berger and W. Keller, unpublished data). These results are corroborated by the high-resolution mass spectrometry analysis of the pull-down fractions, yielding significant signals for the TraN coelution of TraE, TraF, TraH, TraJ, TraK, and TraM.
In silico searches using tBLASTn identified plasmid-encoded TraN homologs exclusively in Firmicutes, with 81% of these plasmids containing an oriT. None of the putative TraN key amino acids which have been investigated in this study are affected in the TraN homologs, thus implying an important role for these residues.
The tBLASTn-based alignments using pIP501 T4SS proteins as query confirmed the presence of TraN homologs within putative T4SS gene clusters for over 65% of the selected 54 plasmids. Furthermore, plasmids were not exclusively found to belong to the Inc18 group but also to RepA_N and Rep_3 replicon families, underlining the widespread presence of pIP501-like T4SS. Most TraN homologs were found on multiple resistance Inc18 plasmids known to disseminate to a wide range of Gram-positive hosts (Kohler et al., 2018b; Nwaiwu, 2022; Zhu et al., 2022). Inc18 plasmids have been found in diverse Firmicutes isolated from food, healthcare facilities, livestock manure, or the environment (Biggel et al., 2021; Parra-Flores et al., 2021; Jozefíková et al., 2022; Tang et al., 2023). Three putative conjugative plasmids (plas1, pL15-b, and pW208) encoding a TraN homolog were affiliated to the RepA_N family. These multi-resistance plasmids are frequently found in staphylococci, enterococci, or streptococci (Weaver et al., 2009; Zhang et al., 2022). pL9 from the Rep_3 plasmid family also encodes a TraN homolog. These plasmids are frequently found in E. faecalis and E. faecium as well as in streptococci and staphylococci (Jensen et al., 2010; Mikalsen et al., 2015).
Structurally, the N-terminal domain of TraN shares similarities with the Xis excisionase from Tn916, and with a putative excisionase from Klebsiella pneumoniae (Goessweiner-Mohr et al., 2014a; Grohmann et al., 2016). Additionally, we identified a structurally similar putative DNA binding protein, BldC, from streptomycetes, which is related to MerR regulators (Savitzky and Golay, 1964; Schumacher et al., 2018) with an overall protein sequence identity of 18.3%. BldC (PDB: 6AMA) preferentially aligns with the C-terminal half of TraN (Supplementary Figure S3).
T4SS transcriptional regulators have been recently discovered in lactococcal plasmids which exhibit similarities to the MerR family of transcriptional regulators. Conjugative transfer of some of these plasmids—pNP40 (Harrington and Hill, 1991; O’Driscoll et al., 2006), pMRC01 (Coakley et al., 1997), pAF22 (Fallico et al., 2012), and pUC11B (Ortiz Charneco et al., 2023)—has been experimentally confirmed. Other lactococcal plasmids such as pUC08B, pIBB477a, and p001F have been recently discovered and categorized as pMRC01/pAF22-like conjugative plasmids (Ortiz Charneco et al., 2023). PlasmidFinder assigned pMRC01, pAF22, pUC08B, pUC11B, and pIBB477a to the Rep_3 replicon family and pNP40 to the RepA_N family. TraR and Tra20 from the conjugative Lactococcus lactis plasmid pNP40 were postulated as transcriptional repressors of the conjugative cluster (Ortiz Charneco et al., 2021). Both proteins exhibit low amino acid sequence identity to TraN (∼26% and ∼20%, respectively). However, structure prediction only detected a structural similarity with Tra20pNP40 (Figure 8A). TrsR was proposed as repressor of the tra operon of the conjugative pMRC01/pAF22-like pUC11B plasmid (Ortiz Charneco et al., 2023). A trsR- mutant led to a significant increase in pUC11B transfer in biparental mating assays. TrsRpUC11B only has low similarity with TraN on amino acid level (∼22%) but displays structural similarities to the C-terminal half of TraN (Figure 8B). In contrast to TraN, the predicted structures of these proteins show an additional elongated polypeptide stretch with low α-helix propensity (Figure 8). It is worth mentioning that the MPF channel protein TrsBpUC11B has been postulated as homolog of TraB (∼57% identity on amino acid level) (Berger et al., 2022). As TrsRpUC08B is 100% identical to TrsRpUC11B, only one protein is depicted here.
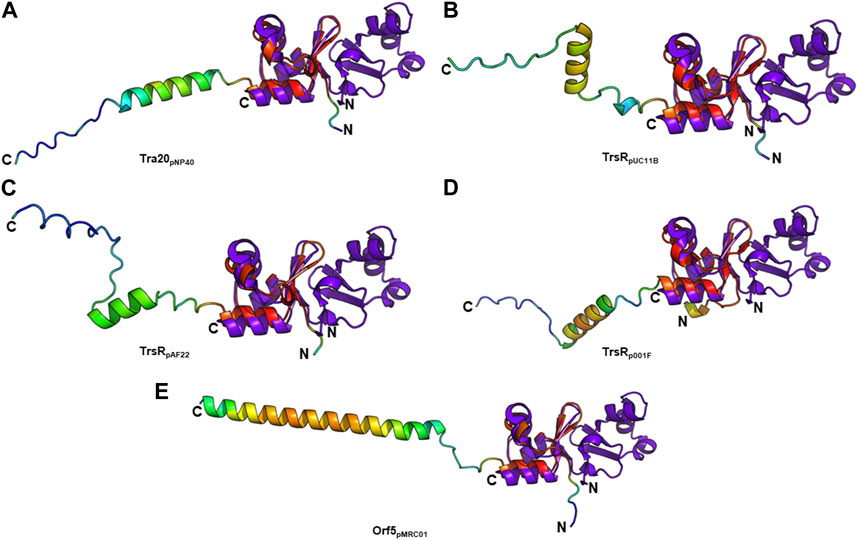
FIGURE 8. Structural alignment of TraNpIP501 (PDB: 4P0Z) with (A) Tra20pNP40, (B) TrsRpUC11B, (C) TrsRpAF22, (D) TrsRp001F, and (E) Orf5pMRC01. TrsRpUC08B is 100% identical to TrsRpUC11B, and TrsRpIBB477a to Orf5pMRC01. (A–E) Homologs are shown in rainbow, colored by pLDDT (per-residue local distance difference test) score (red = high pLDDT = high confidence; blue = low pLDDT = low confidence), TraNpIP501 in purple blue. All aligned proteins resemble the C-terminal half of TraNpIP501.
The TrsRpUC11B-like homologs of pAF22 (WP_015062887.1) and p001F (WP_095348652.1) from Lactococcus share ∼22% and ∼32% amino acid identity with TraNpIP501, respectively. As depicted in Figures 8C,D, both proteins also show high similarity to the C-terminal half of the TraN structure.
The tra region of pMRC01 is similar to that of the Inc18 plasmids pIP501 and pRE25, and the staphylococcal plasmids pSK41 and pG01 (Grohmann et al., 2003). A gene located downstream of the relaxase gene in the tra region of pMRC01 is also similar to members of the MerR family of transcriptional regulators (Ortiz Charneco et al., 2023). The respective Orf5 protein also structurally resembles the C-terminal TraNpIP501 half (Figure 8E). pIBB477a encodes a gene 100% identical to orf5pMRC01. The conjugative gene cluster of pIBB477a displays a notable level of sequence identity with pMRC01 (>85% amino acid identity) (Ortiz Charneco et al., 2023). All five predicted structures (Tra20pNP40, TrsRpUC11B, TrsRpAF22, TrsRp001F, and Orf5pMRC01) exhibit the same wHTH fold as the C-terminal domain of TraN consisting of three antiparallel β-strands as well as the helix-turn-helix motif with an additional C-terminal α-helix (Figure 8). Thus, structural features of the TraN fold are not only found in excisionases (Goessweiner-Mohr et al., 2014a) but also in plasmid-encoded proteins likely involved in the transcriptional regulation of conjugative transfer.
In conclusion, this study demonstrated the importance of distinct TraN amino acid residues responsible for effective sequence-specific binding of pIP501 DNA, thereby regulating tra operon expression and conjugative transfer. Based on the mating assays, residues Arg23, Asn24, Gln28, His82, Arg86, Gly100, and Lys101 contribute to the regulatory function of TraN, whereas Gly47 and Gly48 appear to play a minor role and likely contribute to binding strength rather than specificity.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
CM: conceptualization, investigation, methodology, writing–original draft, and writing–review & editing. TB: conceptualization, investigation, methodology, and writing–review and editing. KK: investigation and writing–review and editing. RG: investigation and writing–review and editing. LP: investigation and writing–review and editing. MV: investigation and writing–review and editing. BG: methodology, investigation, and writing–review and editing. TP-K: funding acquisition, methodology, supervision, and writing–review and editing. WK: conceptualization, funding acquisition, methodology, supervision, and writing–review and editing. EG: funding acquisition, methodology, supervision, conceptualization, writing–original draft, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Financial support of the Doctoral Academy of the University of Graz (Consortium BioMolStruct) and the Austrian Science Foundation FWF (DocFund DOC 130) is highly acknowledged. The Berliner Hochschule für Technik, Berlin, Germany, funded the doctoral thesis of CM. MS proteomics was supported by the Field of Excellence BioHealth—University of Graz. The authors acknowledge the financial support for open access publication by the University of Graz.
We thank Ulrich Stelzl and Sandra Fasching, Institute of Pharmaceutical Sciences, University of Graz, for providing access to the Monolith NT.115 machine for MST measurements.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were editorial board members of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2024.1268647/full#supplementary-material
Abajy, M. Y., Kopeć, J., Schiwon, K., Burzynski, M., Döring, M., Bohn, C., et al. (2007). A type IV-secretion-like system is required for conjugative DNA transport of broad-host-range plasmid pIP501 in gram-positive bacteria. J. Bacteriol. 189, 2487–2496. doi:10.1128/JB.01491-06
Abdul-Gader, A., Miles, A. J., and Wallace, B. A. (2011). A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy. Bioinformatics 27, 1630–1636. doi:10.1093/bioinformatics/btr234
Arends, K., Celik, E.-K., Probst, I., Goessweiner-Mohr, N., Fercher, C., Grumet, L., et al. (2013). TraG encoded by the pIP501 type IV secretion system is a two-domain peptidoglycan-degrading enzyme essential for conjugative transfer. J. Bacteriol. 195, 4436–4444. doi:10.1128/JB.02263-12
Armougom, F., Moretti, S., Poirot, O., Audic, S., Dumas, P., Schaeli, B., et al. (2006). Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 34, W604–W608. doi:10.1093/nar/gkl092
Auchtung, J. M., Lee, C. A., Monson, R. E., Lehman, A. P., and Grossman, A. D. (2005). Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 102, 12554–12559. doi:10.1073/pnas.0505835102
Behnke, D., Gilmore, M. S., and Ferretti, J. J. (1981). Plasmid pGB301, a new multiple resistance streptococcal cloning vehicle and its use in cloning of a gentamicin/kanamycin resistance determinant. Mol. Gen. Genet. 182, 414–421. doi:10.1007/BF00293929
Berger, T. M. I., Michaelis, C., Probst, I., Sagmeister, T., Petrowitsch, L., Puchner, S., et al. (2022). Small things matter: the 11.6-kDa TraB protein is crucial for antibiotic resistance transfer among enterococci. Front. Mol. Biosci. 9, 867136. doi:10.3389/fmolb.2022.867136
Biedendieck, R., Borgmeier, C., Bunk, B., Stammen, S., Scherling, C., Meinhardt, F., et al. (2011). Systems biology of recombinant protein production using Bacillus megaterium. Methods Enzymol. 500, 165–195. doi:10.1016/B978-0-12-385118-5.00010-4
Biggel, M., Nüesch-Inderbinen, M., Jans, C., Stevens, M. J. A., and Stephan, R. (2021). Genetic context of optrA and poxtA in florfenicol-resistant enterococci isolated from flowing surface water in Switzerland. Antimicrob. Agents Chemother. 65, e0108321. doi:10.1128/AAC.01083-21
Breuer, R. J., Hirt, H., and Dunny, G. M. (2018). Mechanistic features of the enterococcal pCF10 sex pheromone response and the biology of Enterococcus faecalis in its natural habitat. J. Bacteriol. 200, e00733-17. doi:10.1128/JB.00733-17
Buu-Hoï, A., Bieth, G., and Horaud, T. (1984). Broad host range of streptococcal macrolide resistance plasmids. Antimicrob. Agents Chemother. 25, 289–291. doi:10.1128/AAC.25.2.289
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi:10.1128/AAC.02412-14
Cinthi, M., Coccitto, S. N., Fioriti, S., Morroni, G., Simoni, S., Vignaroli, C., et al. (2022). Occurrence of a plasmid co-carrying cfr(D) and poxtA2 linezolid resistance genes in Enterococcus faecalis and Enterococcus casseliflavus from porcine manure, Italy. J. Antimicrob. Chemother. 77, 598–603. doi:10.1093/jac/dkab456
Clewell, D. B., Yagi, Y., Dunny, G. M., and Schultz, S. K. (1974). Characterization of three plasmid deoxyribonucleic acid molecules in a strain of Streptococcus faecalis: identification of a plasmid determining erythromycin resistance. J. Bacteriol. 117, 283–289. doi:10.1128/jb.117.1.283-289.1974
Coakley, M., Fitzgerald, G., and Ros, R. P. (1997). Application and evaluation of the phage resistance- and bacteriocin-encoding plasmid pMRC01 for the improvement of dairy starter cultures. Appl. Environ. Microbiol. 63 (4), 1434–1440. doi:10.1128/aem.63.4.1434-1440.1997
Compton, L. A., and Johnson, W. C. (1986). Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155, 155–167. doi:10.1016/0003-2697(86)90241-1
Costa, T. R. D., Harb, L., Khara, P., Zeng, L., Hu, B., and Christie, P. J. (2021). Type IV secretion systems: advances in structure, function, and activation. Mol. Microbiol. 115, 436–452. doi:10.1111/mmi.14670
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi:10.1038/nbt.1511
D'Andrea, M. M., Antonelli, A., Brenciani, A., Di Pilato, V., Morroni, G., Pollini, S., et al. (2019). Characterization of Tn6349, a novel mosaic transposon carrying poxtA, cfr and other resistance determinants, inserted in the chromosome of an ST5-MRSA-II strain of clinical origin. J. Antimicrob. Chemother. 74, 2870–2875. doi:10.1093/jac/dkz278
Davis, I. J., Roberts, A. P., Ready, D., Richards, H., Wilson, M., and Mullany, P. (2005). Linkage of a novel mercury resistance operon with streptomycin resistance on a conjugative plasmid in Enterococcus faecium. Plasmid 54, 26–38. doi:10.1016/j.plasmid.2004.10.004
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J.-M., et al. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17. doi:10.1093/nar/gkr245
Fallico, V., Ross, R. P., Fitzgerald, G. F., and McAuliffe, O. (2012). Novel conjugative plasmids from the natural isolate Lactococcus lactis subspecies cremoris DPC3758: a repository of genes for the potential improvement of dairy starters. J. Dairy. Sci. 95 (7), 3593–3593. doi:10.3168/jds.2011-5255
Gesslbauer, B., Kuerzl, D., Valpatic, N., and Bochkov, V. N. (2018). Unbiased identification of proteins covalently modified by complex mixtures of peroxidized lipids using a combination of electrophoretic mobility band shift with mass spectrometry. Antioxidants (Basel) 7, 116. doi:10.3390/antiox7090116
Goessweiner-Mohr, N., Eder, M., Hofer, G., Fercher, C., Arends, K., Birner-Gruenberger, R., et al. (2014a). Structure of the double-stranded DNA-binding type IV secretion protein TraN from Enterococcus. Acta Crystallogr. D. Biol. Crystallogr. 70, 2376–2389. doi:10.1107/S1399004714014187
Goessweiner-Mohr, N., Fercher, C., Abajy, M. Y., Grohmann, E., and Keller, W. (2012). Crystallization and first data collection of the putative transfer protein TraN from the Gram-positive conjugative plasmid pIP501. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 68, 1402–1405. doi:10.1107/S174430911204184X
Goessweiner-Mohr, N., Fercher, C., Arends, K., Birner-Gruenberger, R., Laverde-Gomez, D., Huebner, J., et al. (2014b). The type IV secretion protein TraK from the Enterococcus conjugative plasmid pIP501 exhibits a novel fold. Acta Crystallogr. D. Biol. Crystallogr. 70, 1124–1135. doi:10.1107/S1399004714001606
Gonzalez, C. F., and Kunka, B. S. (1983). Plasmid transfer in Pediococcus spp. intergeneric and intrageneric transfer of pIP501. Appl. Environ. Microbiol. 46, 81–89. doi:10.1128/aem.46.1.81-89.1983
Grohmann, E., Christie, P. J., Waksman, G., and Backert, S. (2018). Type IV secretion in gram-negative and gram-positive bacteria. Mol. Microbiol. 107, 455–471. doi:10.1111/mmi.13896
Grohmann, E., Goessweiner-Mohr, N., and Brantl, S. (2016). DNA-binding proteins regulating pIP501 transfer and replication. Front. Mol. Biosci. 3, 42. doi:10.3389/fmolb.2016.00042
Grohmann, E., Muth, G., and Espinosa, M. (2003). Conjugative plasmid transfer in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67, 277–301. doi:10.1128/MMBR.67.2.277-301.2003
Haliga, R. E., Sorodoc, V., Morarasu, B. C., Coman, A. E., Ceasovschih, A., Sirbu, O., et al. (2023). Native and prosthetic simultaneously double valve infective endocarditis with Enterococcus faecalis-Case-Based review. J. Pers. Med. 13, 300. doi:10.3390/jpm13020300
Harrington, A., and Hill, C. (1991). Construction of a Bacteriophage-Resistant Derivative of Lactococcus lactis subsp. lactis 425A by Using the Conjugal Plasmid pNP40. Appl. Environ. Microbiol. 57 (12), 3405–3409. doi:10.1128/aem.57.12.3405-3409.1991
Jensen, L. B., Garcia-Migura, L., Valenzuela, A. J. S., Løhr, M., Hasman, H., and Aarestrup, F. M. (2010). A classification system for plasmids from enterococci and other Gram-positive bacteria. J. Microbiol. Methods 80, 25–43. doi:10.1016/j.mimet.2009.10.012
Jozefíková, A., Valček, A., Šoltys, K., Nováková, E., and Bujdáková, H. (2022). Persistence and multi-ward dissemination of vancomycin-resistant Enterococcus faecium ST17 clone in hospital settings in Slovakia 2017-2020. Int. J. Antimicrob. Agents 59, 106561. doi:10.1016/j.ijantimicag.2022.106561
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Khan, A., Miller, W. R., Axell-House, D., Munita, J. M., and Arias, C. A. (2022). Antimicrobial susceptibility testing for enterococci. J. Clin. Microbiol. 60, e0084321. doi:10.1128/jcm.00843-21
Kim, E., Shin, S.-W., Kwak, H.-S., Cha, M.-H., Yang, S.-M., Gwak, Y.-S., et al. (2021). Prevalence and characteristics of phenicol-oxazolidinone resistance genes in Enterococcus faecalis and Enterococcus faecium isolated from food-producing animals and meat in korea. Int. J. Mol. Sci. 22, 11335. doi:10.3390/ijms222111335
Kohler, V., Goessweiner-Mohr, N., Aufschnaiter, A., Fercher, C., Probst, I., Pavkov-Keller, T., et al. (2018a). TraN: a novel repressor of an Enterococcus conjugative type IV secretion system. Nucleic Acids Res. 46, 9201–9219. doi:10.1093/nar/gky671
Kohler, V., Keller, W., and Grohmann, E. (2019). Regulation of gram-positive conjugation. Front. Microbiol. 10, 1134. doi:10.3389/fmicb.2019.01134
Kohler, V., Vaishampayan, A., and Grohmann, E. (2018b). Broad-host-range Inc18 plasmids: occurrence, spread and transfer mechanisms. Plasmid 99, 11–21. doi:10.1016/j.plasmid.2018.06.001
Kurenbach, B., Bohn, C., Prabhu, J., Abudukerim, M., Szewzyk, U., and Grohmann, E. (2003). Intergeneric transfer of the Enterococcus faecalis plasmid pIP501 to Escherichia coli and Streptomyces lividans and sequence analysis of its tra region. Plasmid 50, 86–93. doi:10.1016/s0147-619x(03)00044-1
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi:10.1038/227680a0
Langella, P., and Chopin, A. (1989). Conjugal transfer of plasmid pIP501 from Lactococcus lactis to Lactobacillus delbruckii subsp. bulgaricus and Lactobacillus helveticus. FEMS Microbiol. Lett. 60, 149–152. doi:10.1016/0378-1097(89)90498-9
Lazaris, A., Coleman, D. C., Kearns, A. M., Pichon, B., Kinnevey, P. M., Earls, M. R., et al. (2017). Novel multiresistance cfr plasmids in linezolid-resistant methicillin-resistant Staphylococcus epidermidis and vancomycin-resistant Enterococcus faecium (VRE) from a hospital outbreak: co-location of cfr and optrA in VRE. J. Antimicrob. Chemother. 72, 3252–3257. doi:10.1093/jac/dkx292
Li, P., Gao, M., Feng, C., Yan, T., Sheng, Z., Shi, W., et al. (2022). Molecular characterization of florfenicol and oxazolidinone resistance in Enterococcus isolates from animals in China. Front. Microbiol. 13, 811692. doi:10.3389/fmicb.2022.811692
Li, X., Xie, Y., Liu, M., Tai, C., Sun, J., Deng, Z., et al. (2018). oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 46, W229–W234. doi:10.1093/nar/gky352
Li, Y. G., Hu, B., and Christie, P. J. (2019). Biological and structural diversity of type IV secretion systems. Microbiol. Spectr. 7. doi:10.1128/microbiolspec.PSIB-0012-2018
Liu, Y., Wang, Y., Schwarz, S., Li, Y., Shen, Z., Zhang, Q., et al. (2013). Transferable multiresistance plasmids carrying cfr in Enterococcus spp. from swine and farm environment. Antimicrob. Agents Chemother. 57, 42–48. doi:10.1128/AAC.01605-12
Mancuso, G., Midiri, A., Gerace, E., Marra, M., Zummo, S., and Biondo, C. (2023). Urinary tract infections: the current scenario and future prospects. Pathogens 12, 623. doi:10.3390/pathogens12040623
Meier, F., Brunner, A.-D., Koch, S., Koch, H., Lubeck, M., Krause, M., et al. (2018). Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell Proteomics 17, 2534–2545. doi:10.1074/mcp.TIR118.000900
Mikalsen, T., Pedersen, T., Willems, R., Coque, T. M., Werner, G., Sadowy, E., et al. (2015). Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics 16, 282. doi:10.1186/s12864-015-1407-6
Miles, A. J., Ramalli, S. G., and Wallace, B. A. (2022). DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 31, 37–46. doi:10.1002/PRO.4153
Mirdita, M., von den Driesch, L., Galiez, C., Martin, M. J., Söding, J., and Steinegger, M. (2017). Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176. doi:10.1093/nar/gkw1081
Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., and Steinegger, M. (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. doi:10.1038/s41592-022-01488-1
Mirdita, M., Steinegger, M., and Söding, J. (2019). MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35, 2856–2858. doi:10.1093/bioinformatics/bty1057
Mitchell, A. L., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., et al. (2020). MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578. doi:10.1093/nar/gkz1035
Morroni, G., Brenciani, A., Antonelli, A., D'Andrea, M. M., Di Pilato, V., Fioriti, S., et al. (2018). Characterization of a multiresistance plasmid carrying the optrA and cfr resistance genes from an Enterococcus faecium clinical isolate. Front. Microbiol. 9, 2189. doi:10.3389/fmicb.2018.02189
Murray, C. J. L., Ikuta, K. S., Sharara, F., Swetschinski, L., Robles Aguilar, G., Gray, A., et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655. doi:10.1016/S0140-6736(21)02724-0
Nwaiwu, O. (2022). Comparative genome analysis of the first Listeria monocytogenes core genome multi-locus sequence types CT2050 AND CT2051 strains with their close relatives. AIMS Microbiol. 8, 61–72. doi:10.3934/microbiol.2022006
O’Driscoll, J., Glynn, F., Fitzgerald, G. F., and van Sinderen, D. (2006). Sequence analysis of the lactococcal plasmid pNP40: a mobile replicon for coping with environmental hazards. J. Bacteriol. 188 (18), 6629–6639. doi:10.1128/JB.00672-06
Ortiz Charneco, G., Kelleher, P., Buivydas, A., Dashko, S., de Waal, P. P., van Peij, N. N. M. E., et al. (2023). Delineation of a lactococcal conjugation system reveals a restriction-modification evasion system. Microb. Biotechnol. 16, 1250–1263. doi:10.1111/1751-7915.14221
Ortiz Charneco, G., Kelleher, P., Buivydas, A., Streekstra, H., Ver Themaat, E. L., de Waal, P. P., et al. (2021). Genetic dissection of a prevalent plasmid-encoded conjugation system in Lactococcus lactis. Front. Microbiol. 12, 680920. doi:10.3389/fmicb.2021.680920
Parra-Flores, J., Holý, O., Bustamante, F., Lepuschitz, S., Pietzka, A., Contreras-Fernández, A., et al. (2021). Virulence and antibiotic resistance genes in Listeria monocytogenes strains isolated from ready-to-eat foods in Chile. Front. Microbiol. 12, 796040. doi:10.3389/fmicb.2021.796040
Pérez-Díaz, J. C., Vicente, M. F., and Baquero, F. (1982). Plasmids in Listeria. Plasmid 8, 112–118. doi:10.1016/0147-619X(82)90049-X
Pucci, M. J., Monteschio, M. E., and Kemker, C. L. (1988). Intergeneric and intrageneric conjugal transfer of plasmid-encoded antibiotic resistance determinants in Leuconostoc spp. Appl. Environ. Microbiol. 54, 281–287. doi:10.1128/aem.54.2.281-287.1988
Savitzky, A., and Golay, M. J. E. (1964). Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 36, 1627–1639. doi:10.1021/ac60214a047
Schaberg, D. R., Clewell, D. B., and Glatzer, L. (1982). Conjugative transfer of R-plasmids from Streptococcus faecalis to Staphylococcus aureus. Antimicrob. Agents Chemother. 22, 204–207. doi:10.1128/AAC.22.2.204
Schumacher, M. A., den Hengst, C. D., Bush, M. J., Le, T. B. K., Tran, N. T., Chandra, G., et al. (2018). The MerR-like protein BldC binds DNA direct repeats as cooperative multimers to regulate Streptomyces development. Nat. Commun. 9, 1139. doi:10.1038/s41467-018-03576-3
Schwarz, F. V., Perreten, V., and Teuber, M. (2001). Sequence of the 50-kb conjugative multiresistance plasmid pRE25 from Enterococcus faecalis RE25. Plasmid 46, 170–187. doi:10.1006/plas.2001.1544
Singh, P. K., Ramachandran, G., Ramos-Ruiz, R., Peiró-Pastor, R., Abia, D., Wu, L. J., et al. (2013). Mobility of the native Bacillus subtilis conjugative plasmid pLS20 is regulated by intercellular signaling. PLoS Genet. 9, e1003892. doi:10.1371/journal.pgen.1003892
Tang, B., Zou, C., Schwarz, S., Xu, C., Hao, W., Yan, X.-M., et al. (2023). Linezolid-resistant Enterococcus faecalis of chicken origin harbored chromosome-borne optrA and plasmid-borne cfr, cfr(D), and poxtA2 genes. Microbiol. Spectr. 11, e0274122. doi:10.1128/spectrum.02741-22
Thompson, J. K., and Collins, M. A. (1988). Evidence for the conjugal transfer of the broad host range plasmid pIP501 into strains of Lactobacillus helveticus. J. Appl. Bacteriol. 65, 309–319. doi:10.1111/j.1365-2672.1988.tb01897.x
Virolle, C., Goldlust, K., Djermoun, S., Bigot, S., and Lesterlin, C. (2020). Plasmid transfer by conjugation in gram-negative bacteria: from the cellular to the community level. Genes (Basel) 11, 1239. doi:10.3390/genes11111239
Wang, Y., Li, D., Song, L., Liu, Y., He, T., Liu, H., et al. (2013). First report of the multiresistance gene cfr in Streptococcus suis. Antimicrob. Agents Chemother. 57, 4061–4063. doi:10.1128/AAC.00713-13
Weaver, K. E., Kwong, S. M., Firth, N., and Francia, M. V. (2009). The RepA_N replicons of Gram-positive bacteria: a family of broadly distributed but narrow host range plasmids. Plasmid 61, 94–109. doi:10.1016/j.plasmid.2008.11.004
Whitmore, L., and Wallace, B. A. (2004). DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673. doi:10.1093/nar/gkh371
Whitmore, L., and Wallace, B. A. (2008). Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400. doi:10.1002/bip.20853
Yin, M., Jiang, Y., Qian, C., Wu, F., Ying, Y., Wu, C., et al. (2018). Molecular characteristics and comparative genomics analysis of a clinical Enterococcus casseliflavus with a resistance plasmid. Infect. Drug Resist 11, 2159–2167. doi:10.2147/IDR.S180254
Zhang, E., Zong, S., Zhou, W., Zhou, J., Han, J., and Qu, D. (2022). Characterization and comparative genomics analysis of RepA_N multi-resistance plasmids carrying optrA from Enterococcus faecalis. Front. Microbiol. 13, 991352. doi:10.3389/fmicb.2022.991352
Zhu, W., Murray, P. R., Huskins, W. C., Jernigan, J. A., McDonald, L. C., Clark, N. C., et al. (2010). Dissemination of an Enterococcus Inc18-Like vanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54, 4314–4320. doi:10.1128/AAC.00185-10
Zhu, Y., Yang, W., Schwarz, S., Xu, Q., Yang, Q., Wang, L., et al. (2022). Characterization of the novel optrA-carrying pseudo-compound transposon Tn7363 and an Inc18 plasmid carrying cfr(D) in Vagococcus lutrae. J. Antimicrob. Chemother. 77, 921–925. doi:10.1093/jac/dkab478
Keywords: type IV secretion system, antibiotic resistance, conjugative transfer, pIP501, Enterococcus faecalis, transfer regulator, DNA binding proteins
Citation: Michaelis C, Berger TMI, Kuhlmann K, Ghulam R, Petrowitsch L, Besora Vecino M, Gesslbauer B, Pavkov-Keller T, Keller W and Grohmann E (2024) Effect of TraN key residues involved in DNA binding on pIP501 transfer rates in Enterococcus faecalis. Front. Mol. Biosci. 11:1268647. doi: 10.3389/fmolb.2024.1268647
Received: 28 July 2023; Accepted: 04 January 2024;
Published: 06 February 2024.
Edited by:
Sonsoles Martin-Santamaria, Spanish National Research Council (CSIC), SpainReviewed by:
Manuel Espinosa, Spanish National Research Council (CSIC), SpainCopyright © 2024 Michaelis, Berger, Kuhlmann, Ghulam, Petrowitsch, Besora Vecino, Gesslbauer, Pavkov-Keller, Keller and Grohmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Grohmann, ZWxpc2FiZXRoLmdyb2htYW5uQGJodC1iZXJsaW4uZGU=; Walter Keller, d2FsdGVyLmtlbGxlckB1bmktZ3Jhei5hdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.