 Jade S. Miller
Jade S. Miller Natalie E. Bennett1,3,4,5
Natalie E. Bennett1,3,4,5- 1Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Pharmacology Training Program, Department of Pharmacology, Vanderbilt University, Nashville, TN, United States
- 3Center for Bone Biology, Vanderbilt University Medical Center, Nashville, TN, United States
- 4Department of Veterans Affairs, Nashville VA Medical Center, Tennessee Valley Healthcare System, Nashville, TN, United States
- 5Program in Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN, United States
Due to the cellular plasticity that is inherent to cancer, the acquisition of resistance to therapy remains one of the biggest obstacles to patient care. In many patients, the surviving cancer cell subpopulation goes on to proliferate or metastasize, often as the result of dramatically altered cell signaling and transcriptional pathways. A notable example is the Hedgehog (Hh) signaling pathway, which is a driver of several cancer subtypes and aberrantly activated in a wide range of malignancies in response to therapy. This review will summarize the field’s current understanding of the many roles played by Hh signaling in drug resistance and will include topics such as non-canonical activation of Gli proteins, amplification of genes which promote tolerance to chemotherapy, the use of hedgehog-targeted drugs and tool compounds, and remaining gaps in our knowledge of the transcriptional mechanisms at play.
1 Introduction
The Hedgehog (Hh) signaling pathway was first described in 1980, and its roles in embryogenesis and cell polarity have been detailed in countless publications (Van Schmus et al., 1980). Targets of Hh signaling include several cyclins and regulators of mitosis including cyclin D1, cyclin D2, cyclin B, and p21 (Di Magliano and Hebrok, 2003). Several cancers including non-small cell lung cancers (NSCLC), gliomas, and breast carcinomas upregulate Hh signaling, thereby resisting cell cycle arrest (Fan and Khavari, 1999). Furthermore, mutations in the pathway are associated with a hereditary form of basal cell carcinoma (BCC) and overexpression of Shh is sufficient to cause spontaneous skin tumors (Oro et al., 1997; Yang et al., 2010).
Due to its significance in tumorigenesis, a handful of Hh pathway inhibitors have been FDA-approved for treatment of BCC, but both intrinsic and acquired resistance is prevalent due to receptor mutations (Atwood et al., 2015). Mechanisms of resistance to these targeted therapies will be discussed in further detail in Section 2 of this review.
Despite the advent of targeted therapies in cancer treatment, chemotherapies such as paclitaxel, cisplatin, and doxorubicin remain a standard of care for most cancer subtypes. These cytotoxic agents have revolutionized the field of oncology, yet an estimated 80%–90% of cancer-related mortalities are due to chemoresistance (Dan et al., 2020). This is due in par to the selective pressure that is inherent to chemotherapy, as a handful of surviving cells go on to proliferate, resulting in a malignancy that is newly resistant to treatment (Yu et al., 2017a). In this review, we will discuss the ways in which Hh signaling activates transcriptional programs which enable cancer cells to evade chemotherapy and targeted cancer therapies (summarized in Table 1).
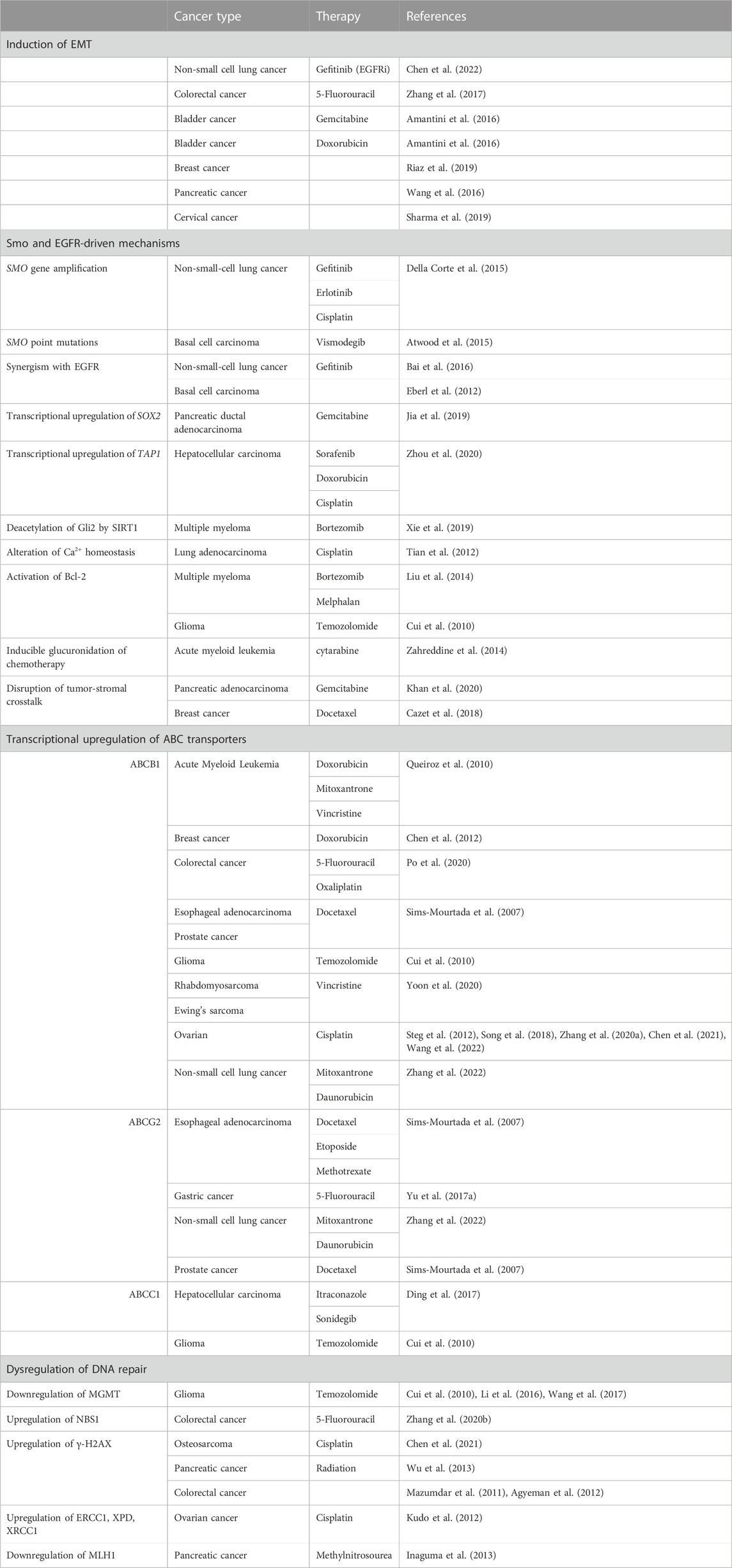
TABLE 1. Summary of known Hh pathway-mediated mechanisms of anticancer drug resistance.
1.1 The canonical hedgehog signaling pathway
There are three ligands present in vertebrates which may activate the Hh signaling pathway: Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh). In normal tissues, Dhh is primarily expressed in mammals during gonad development, Ihh is expressed in chondrocytes during endochondral ossification, and Shh is expressed in many developing tissues to regulate processes such as limb development, neural patterning, and proximal-distal patterning (Van Schmus et al., 1980; Vortkamp et al., 1996). Shh is also the ligand which is most often implicated in malignancies.
The Hh signaling pathway begins at the primary cilia (PC), a structure that protrudes from the cell and is formed by microtubules. The target for Hh ligands is Patched (encoded by the gene PTCH1), a transmembrane receptor which suppresses Smoothened (Smo) in the absence of ligand, preventing activation of downstream effectors of Hh signaling. Loss-of-function mutations in PTCH1 result in ligand-independent activation of SMO and constitutive Hh signaling, and these mutations are often a driver of malignancy. PTCH1 mutations are found in as many as 73% of basal cell carcinomas and are also associated with development of medulloblastoma and recurrence of breast cancer (Zurawel et al., 2000; Bonilla et al., 2016; Wang et al., 2019).
In the presence of ligand, the 7-transmembrane receptor SMO (discussed in section 2.1) is phosphorylated and translocates into the PC where it inhibits Suppressor of Fused (SUFU), releasing the Glioma-associated oncogene (Gli) proteins Gli1 and Gli2. The Gli proteins localize to the nucleus upon their activation, where they go on to bind to their transcriptional targets (Figure 1).
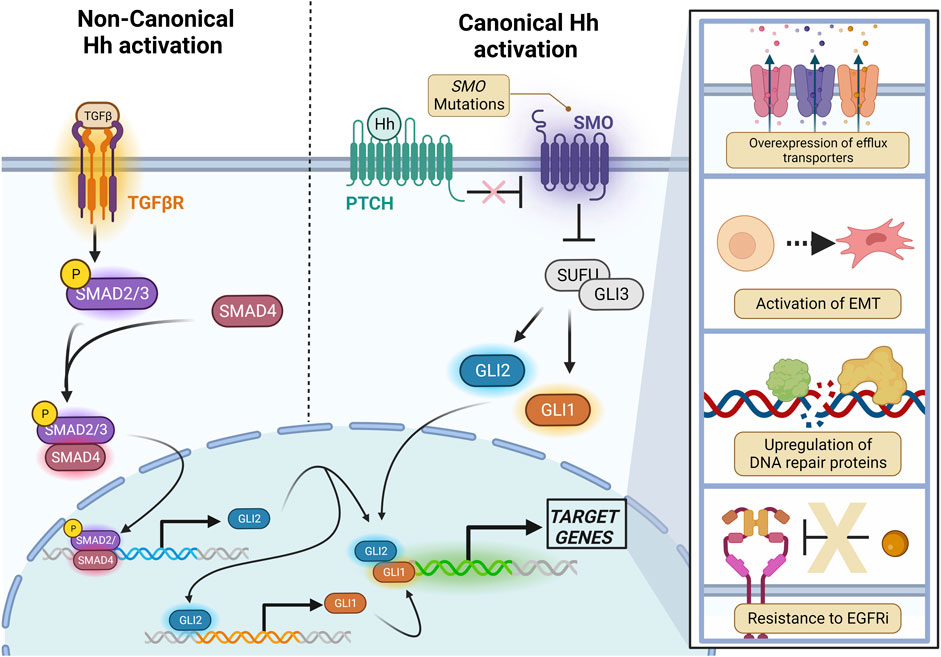
FIGURE 1. Canonical and Non-canonical Hh signaling converges on Gli proteins. Canonical (Smo-dependent) signaling is the result of a Hh ligand binding to Ptch, relieving its inhibition on Smo. Upon Smo activation, SUFU is inhibited, releasing the Gli1 and Gli2 transcription factors which then translocate into the nucleus to activate transcriptional targets. Gli2 may also be non-canonically activated by SMAD2/SMAD4 downstream of TGF-β. Created with BioRender.
Each of the Gli transcription factors (Gli1, Gli2, and Gli3) activate a diverse subset of genes, including several overlapping targets. Gli1 was the first discovered and is perhaps the most extensively characterized in the field of developmental biology, with high expression in mesenchymal stem cells (MSCs), neural stem cells (NSCs), and embryonic stem cells (ESCs) (Kinzler and Vogelstein, 1990). Gli1 and Gli2 share many transcriptional targets, and Gli2 can itself transcriptionally amplify Gli1 (Ali et al., 2019; Avery et al., 2021). Transcriptional targets of Gli2 also include NF-κB1, SMAD2, and Runx2 (Ali et al., 2019). In contrast to Gli1 and Gli2, Gli3 is active in the absence of Hh activation, and acts as a transcriptional repressor of Gli1 and Gli2 targets (Matissek and Elsawa, 2020).
The Gli transcription factors are also regulated through proteolytic processing. The GLI1 mRNA transcript may be alternatively spliced to generate an N-terminal deletion (GLI1ΔN) and a truncated variant (tGLI1), which are loss-of-function and gain-of-function mutations, respectively (Doheny et al., 2020). Unlike the other isoforms, tGli1 is expressed only in cancerous tissues and promotes angiogenesis in glioblastoma and breast cancer brain metastases (Doheny et al., 2020). Both Gli2 and Gli3 possess a processing determinant domain (PDD), and variations in this region contribute to differential proteolytic cleavage; Gli3 is proteolytically processed to a much greater extent than Gli2 in vivo (Pan and Wang, 2007). These cleaved forms of Gli2 and Gli3 act as transcriptional repressors of Gli1 and Gli2 targets and negatively regulate response to Hh signaling (Li et al., 2011; Matissek and Elsawa, 2020). Additionally, there are two other isoforms of Gli2 found in humans which may be differentially expressed in malignancy, but the functions of these variants are not well understood (Tojo et al., 2003).
There is some debate on whether drug resistance mechanisms are driven by Gli1 or Gli2. While the literature has indicated the importance of both transcription factors, it should be noted that Gli1 is a transcriptional target of Gli2, as overexpression of Gli2 results in upregulation of both Gli2 and Gli1, but the reverse is not true (Agyeman et al., 2012). Additionally, Gli2, but not Gli1, is required for aberrant activation of Hh signaling (Bai et al., 2002). Co-amplification of Gli1 and Gli2 occurs frequently in malignancy, but dependence on Gli1 or Gli2 varies between cancers, between subtypes, and between cell lines of the same subtype; Hh may even be activated heterogenously within the tumor microenvironment (Giammona et al., 2023). There are a multitude of publications indicating the importance of Hh signaling in the tumor immune microenvironment, from polarization of macrophages to T cell differentiation and immunosuppression, but the relationship between Hh signaling, cancer immunity, and resistance to therapy is still being established (Wang et al., 2023).
1.2 Non-canonical Hh signaling
Overexpression of Gli1 or Gli2 is observed in a wide array of chemoresistant malignancies, and this increase is often not accompanied by an increase in Shh ligands or Smo expression (Yu et al., 2017b). This is because Hh signaling may be activated independently of Smo as the result of several signaling cascades, most notably by Transforming growth factor β (TGF- β) (see below), or Epidermal growth factor (EGF) (discussed in Section 2.2) (Pietrobono et al., 2019).
1.2.1 Non-canonical Hh activation by TGF- β
Transforming Growth Factor-β (TGF-β) signals through the serine/threonine kinase receptors TGFbeta-RI and TGFbeta-RII, which both exist as dimers in their unbound form. Ligand binding to TGFbeta-RII induces its dimerization with TGFbeta-RI, forming a tetramer. The activated tetramer is autophosphorylated and recruits and phosphorylates receptor-regulated SMADs (r-SMADs) such as SMAD2 and SMAD3, which dissociate from the receptor complex and bind common mediator SMADs (co-SMADs) including SMAD4. The r-SMAD/co-SMAD complex translocates to the nucleus, inducing transcription of TGF-β-target genes, one of which is GLI2 (Dennler et al., 2007).
When discussing Hh-driven drug resistance, it is also important to acknowledge the role of non-canonical Hh signaling in response to anticancer therapies. TGF-β is often overexpressed in malignancy and its amplification has been linked to resistance to chemotherapy, immunotherapy, and targeted therapies (Zhang et al., 2021). Interestingly, large-scale transcriptomic analysis of 37 cancers has revealed a tighter correlation between TGFB and GLI2 gene expression than between SHH and GLI2 for prediction of an epithelial mesenchymal transition signature (de Reyniès et al., 2020). Additionally, activation of Gli2 and Gli2 is dependent on SMAD3 in several cell lines including keratinocytes and breast carcinomas, and activation of the Gli2 target gene PTHLH (encoding parathyroid hormone-related protein) occurs independently of canonical Hh signaling (Dennler et al., 2007; Johnson et al., 2011). In fact, TGF-β2 is itself a target of Gli2, indicating the presence of a positive feedback loop between the TGF-β and Hh signaling pathways (Javelaud et al., 2012; Xu et al., 2019). However, the molecular mechanisms and protein-protein interactions of TGF-β/Hh crosstalk are not fully understood.
1.3 Hedgehog-mediated activation of EMT
The process of epithelial to mesenchymal transition (EMT) occurs in healthy tissues during embryogenesis, allowing migration of mesenchymal cells through epithelial boundaries which would normally restrain them. This occurs through downregulation of adhesion proteins, separation of cell-cell junctions, cytoskeletal rearrangement, and secretion of matrix metalloproteinases (MMPs) which degrade the basement membrane, enabling movement through layers of tissue (Chen et al., 2011).
In the context of malignancy, cancer cells may undergo EMT in order to proliferate, invade, and metastasize. Cancers downregulate E-cadherin in favor of N-cadherin, which tethers with a lower affinity to the extracellular matrix (Their, 2002). This is accompanied by expression of transcription factors including Snail, Slug, ZEB1, ZEB2, and Twist as the result of enhanced growth factor signaling pathways including fibroblast growth factor (FGF) and TGF-β (Xu et al., 2009). Collectively, these transcription factors function to suppress apoptosis, prevent senescence, promote survival, and in some cases, confer resistance to a wide range of anticancer therapies.
One of the earliest observations of the correlation between EMT and chemoresistance was published in 1992 when Sommers et al. (1992) observed elevated expression of vimentin (a mesenchymal marker) in both Doxorubicin-resistant and Vinblastine-resistant breast cancer cell lines, correlating with enhanced invasive capacity. EMT may also drive resistance to targeted therapies such as EGFR inhibitors, as genetic silencing of TWIST1 sensitized EGFR-mutant lung cancer to osimertinib and erlotinib (Yochum et al., 2018).
While Hh signaling is certainly not the only mechanism in common between EMT and drug resistance, there are several studies in which Hh signaling has been implicated in cancer stemness or a mesenchymal phenotype. For example, Gli1 induction of Snail, nuclear localization of β-catenin and subsequent EMT has been documented in several cancers including ovarian, breast, and basal cell carcinoma (Riaz et al., 2019). Canonical Hh signaling also appears to drive a cancer stem-like phenotype in pancreatic cancer, as Smo knockdown resulted in reduction of EMT, self-renewal, and chemoresistance to gemcitabine (Wang et al., 2016). Non-canonical activation of Hh signaling is also a key component of EMT, as TGF-β signaling is frequently activated in the mesenchymal state (Xu et al., 2009). Interestingly, there appears to be a positive feedback loop between EMT and activation of Hh; loss of E-cadherin results in elevated expression of Shh, Ptch, Smo, Gli1, and Gli2. Conversely, pharmacological inhibition of Gli transcription results in reduction of sphere formation and invasive capacity (Sharma et al., 2019).
2 The role of hedgehog signaling in resistance to targeted therapy
Because Hh signaling activates such a broad range of transcriptional programs, it is a key mediator of resistance to targeted therapies. This section of the review will focus on the two drug classes whose resistance mechanisms are the most relevant to Hh signaling: Smo inhibitors and EGFR inhibitors.
2.1 Resistance to Smo inhibitors
There are currently three Smo inhibitors which have received FDA approval; Vismodegib (GDC-0449, Erivedge®) and sonidegib (ODOMZO®) were FDA-approved in 2012 and 2015, respectively, for treatment of patients with metastatic or locally advanced BCC. Additionally, Glasdegib (Daurismo®) is FDA-approved for the treatment of acute myeloid leukemia in combination with low-dose cytarabine (Nguyen and Cho, 2022). However, objective response rate to these therapies (47.6%, 38%, and 26.9%, respectively) remain hindered by both primary resistance (an absence of response to therapy) and secondary resistance (initial response followed by acquisition of resistance) (Cortes et al., 2019).
Most mutations in SMO fall into one of two categories. Mutations which occur in the ligand binding pocket disrupt chemical interactions with Smo inhibitors such as vismodegib. Alternatively, mutations may occur within the transmembrane helices of the receptor, resulting in constitutive activity and consequently, constitutive Gli activation. These variants often arise as the result of targeted therapy. Exome sequencing has revealed that as many as 50% of resistant basal cell carcinomas contain at least one mutation in SMO (Atwood et al., 2015). Mutations in other genes of the Hh pathway (including PTCH or SUFU), or amplification of Gli1 or Gli2 often occur independently of Smo, generating primary resistance to this drug class.
Fortunately, there may be a way to overcome these occurrences. There is overwhelming evidence (some of which is summarized in this review) that Gli inhibition may be an effective strategy for the evasion of Smo inhibitor-resistant cancers (Nguyen and Cho, 2022). Such Gli inhibitors do exist but are primarily in use as tool compounds with minimal translational or pre-clinical usage, highlighting a need for specific, potent inhibitors which function downstream of Smo.
2.2 Hedgehog activation in TKI resistance
Tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and cabozantinib are small-molecule compounds which specifically inhibit the activity of receptor tyrosine kinases such as the epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGFR) and platelet-derived growth factor receptor (PDGFR). However, prolonged exposure to these targeted therapies frequently results in resistance and relapse (Raez et al., 2022).
Surprisingly, one mechanism for resistance to TKIs may be amplification of Hh signaling. Cooperativity between downstream effectors of EGFR and Hh signaling was first observed in stem cells of the mammalian neocortex, and further evidence for EGFR-Hh synergism was presented in transcriptional profiling of immortalized keratinocytes, as EGF signaling modulated transcription of a subset of Gli target genes at the promoter level (Palma and Ruiz i Altaba, 2004; Kasper et al., 2023). This subset included pro-tumorigenic and metastatic markers such as JUN, FGF19, and CXCR4. Furthermore, an in vivo model of Gli2-driven basal cell carcinoma, EGFR deletion inhibited growth of Hh-driven lesions. This same study imitated these findings in a xenograft model of pancreatic cancer, which raises questions on the role of EGFR-Hh cooperativity in other malignancies with aberrantly activated Hh signaling (Eberl et al., 2012).
Beyond the synergism observed between EGFR and Hh downstream effectors, evidence also suggests that Hh signaling may itself promote EGFR activation through an MMP-mediated mechanism. Matrix metalloproteinases (MMPs) including MMP-2, MMP-7, and MMP-9 are known transcriptional targets of Hh signaling (Chen et al., 2013; Zhang et al., 2019). Upon their expression, MMPs are secreted into the microenvironment and proteolytically cleave targets in the extracellular matrix, promoting invasion and metastasis. These targets also include ligands such as heparin-binding EGF, or EGF precursors on the cell surface, resulting in ligand-dependent activation of EGFR (Reinchisi et al., 2013).
Currently, the EGFR inhibitor gefitinib is a first-line intervention for patients with advanced non-small cell lung cancer (NSCLC), but a prevalent problem is the occurrence of TKI resistance (Raez et al., 2022). There is considerable evidence for a Hh-driven mechanism of TKI resistance; high plasma levels of Shh in NSCLC patients with advanced disease correlates with poor objective response to several TKI therapies and a reduction in progression-free survival (210 vs. 342 days) (Takam Kamga et al., 2021). These outcomes have been explained by in vitro studies in which stimulation with exogenous Shh resulted in increased tolerance to Gefitinib in the NSCLC cell lines A549, PC9, and H1975. Additionally, gefitinib and the Smo antagonist SANT-1 synergistically inhibited proliferation and decreased expression of EMT markers such as E-Cad and Snail (Bai et al., 2016). Similarly, gefitinib and vismodegib (an FDA-approved Smo inhibitor) synergistically reduced NSLC tumors in mice in comparison to either drug as a monotherapy (Chen et al., 2022). Altogether, these studies make a promising argument for cooperativity between EGF and Hh and may inform the optimization of combined TKI and Smo inhibition in the clinic.
Interestingly, in their studies with gefitinib and SANT-1, Bai et al. (2016) observed increased expression of the efflux transporter ABCG2 as the result of exposure to Shh, and diminished ABCG2 after Hh inhibition in TKI-resistant cell lines. While transporter-mediated resistance to TKI is not extensively studied, gefitinib is a known inhibitor of ABCG2, and ABCG2 is a known transcriptional target of Hh signaling (Kitazaki et al., 2005). This implies that the role of Hh in gefitinib resistance is two-fold; first, downstream effectors of Hh signaling are upregulated to compensate for inhibition of EGFR, and second, Hh signaling is one potential mechanism for elevated drug efflux from the cell. The role of the Gli proteins in transcriptional regulation of drug efflux transporters such as ABCG2 will be discussed in further detail later in this review.
3 Gli-mediated transcription of ABC transporters
Another mechanism by which cancers acquire resistance is through elevated expression of transmembrane protein pumps known as drug efflux transporters, also known as drug efflux transporters, which results in multidrug resistance (MDR). The transporter family that is most often indicated in malignancy is the ATP-binding cassette (ABC) transporter family. In healthy tissues, ABC transporters serve a wide range of functions including ATP-mediated transport of peptides, xenobiotics, metabolites, and ions, and are expressed intracellularly or extracellularly depending on subfamily. Malignancies most often acquire MDR through transcriptionally upregulating expression of these transporters, preventing chemotherapy from reaching therapeutic concentrations intracellularly (Fletcher et al., 2010).
Drug transporter expression in resistant cell lines varies depending on the mechanism of transport for a given drug molecule. For example, paclitaxel is a known substrate of ABCB1 and ABCC3, as indicated by upregulation of these transporters in PTX-resistant cell lines (Němcová-Fürstová et al., 2016). Differential expression of ABC transporters also depends on subset of malignancy; for example, the HER2+ breast cancer cell line SK-BR-3 upregulates a subset of ABC transporters which is slightly different from MCF-7 (Luminal A) in response to PTX (Němcová-Fürstová et al., 2016).
3.1 ABCB1/MDR1/p-GP
The ABCB1 gene encodes for an efflux transporter protein known as p-Glycoprotein (also known as multidrug resistance gene 1, MDR1). ABCB1 is broadly expressed including in adrenal tissue, the gastrointestinal tract, and the liver, and is polyspecific for hundreds of hydrophobic endogenous and exogenous compounds, including a large proportion of chemotherapeutics (Uhlén et al., 2015). Notable substrates include doxorubicin and other anthracyclines, vinblastine, and taxol analogs such as paclitaxel and docetaxel (Robinson and Tiriveedhi, 2020). Therefore, ABCB1 is one of the most clinically significant efflux transporters which play a role in multidrug resistance.
The majority of research on the Gli-mediated transcription of ABCB1 has been done in models of ovarian cancer. Even between ovarian cancer cell lines, evidence exists for both Gli1 and Gli2-driven mechanisms, and it is unclear whether one or both are required for expression of ABCB1. In a 2014 study, a 34 base-pair segment of the ABCB1 promoter which contains a Gli-recognition site was cloned into a luciferase reporter vector and transfected into OVCAR3 and A2780 cell lines. Using electrophoretic mobility shift assays (EMSAs), the authors observed displacement of a DNA probe from the Gli-recognition site, which was reversed by the addition of an anti-Gli1 antibody, suggesting that Gli1 may directly bind to ABCB1, although this occurred to a greater extent with ABCG2 (see below) (Chen et al., 2014).
Some conflicting evidence was acquired in SKOV3 ovarian cancer cell lines, as transfection with Gli1 overexpressing plasmid did not result in an increase in ABCB1 mRNA (Zhang et al., 2020a; Zhang et al., 2020b). In addition to Gli1, a putative binding site for Gli2 was isolated in the promoter of ABCB1 by Wang et al. using point mutagenesis. This binding sequence is located at −875 to −867 of the ABCB1 promoter, and mutation resulted in significant reduction of luciferase reporter activity (Wang et al., 2022).
3.2 ABCG2/BCRP
ABCG2 (also known as breast cancer resistance protein, BCRP) is an efflux transporter which is most highly expressed in the GI tract, liver, mammary tissue, as well as in both male and female sex organs (Uhlén et al., 2015). Its expression in the liver canalicular membranes also contributes to its protective role against xenobiotics and toxic metabolites. Its substrates include several anticancer drugs including anthracyclines, camptothecin analogs, TKIs, and antimetabolites (Mo and Zhang, 2012).
Generation of 5FU-resistant gastric cancer cell lines resulted in a ∼3-fold increase in ABCG2 mRNA which was almost completely reversed by GLI2 shRNA knockdown (Yu et al., 2017b). Similarly, GLI1 knockdown resulted in significant downregulation of ABCG2 in OVCAR3 and A2780 ovarian cancer cell lines (Chen et al., 2014).
Recently, Hh-mediated ABCG2 expression has been linked to RUNX1, which is a downstream effector of TGF- β and bone morphogenetic protein (BMP) signaling. RUNX1 overexpression conferred resistance to 5-FU in three cell lines of colorectal cancer, which additionally correlated with expression of ABCG2 (Li et al., 2021). RUNX1 has been implicated in the resistance mechanisms of several cancer types including paclitaxel-resistant breast cancer and castration-resistant prostate cancer (Li et al., 2021; Fernández et al., 2023).
3.3 ABCC1/MRP1
ABCC1 (also known as Multidrug Resistance Protein 1, MRP1) is expressed nearly ubiquitously in epithelial and endothelial cells of the GI tract, gallbladder, kidney tubules, sex organs, and soft tissues (Uhlén et al., 2015). While there are a considerable number of studies indicating a correlation between Hh signaling and ABCB1 or ABCG2 expression, little work has been done on the relationship between Hh and ABCC1. A study using a subpopulation of stemlike Huh-7 hepatoma cell lines found elevation of Gli2 and ABCC1 in comparison to parental cell lines. Additionally, Gli2 expression correlated with mRNA levels of ABCC1 after pharmacological Smo inhibition (Sonidegib). Notably, Smo inhibition only partially reduced ABCC1 expression indicating the presence of non-canonical Gli activation (Ding et al., 2017). In this particular model of drug resistance, Gli2 did not correlate with ABCB1, ABCG2, or Gli1 expression even though this has been previously described in the literature.
3.4 Other ABC transporters
While ABCB1 and ABCG2 are the most studied ABC transporters in the context of chemoresistance, there are many other members of this superfamily which remain unexplored. A screen of protein expression revealed significant reductions in these two transporters after shGli2 knockdown, but significant changes were also observed in ABCA2, ABCB4, ABCC2, ABCC3, ABCC11, and ABCG1. And, somewhat paradoxically, a handful of ABC transporters were upregulated after Gli1 knockdown (Po et al., 2020). Of course, it would be a huge undertaking to study each of these proteins in the context of Hh signaling, but as the field progresses it should be noted that it will be highly unlikely that a unifying model of Gli-mediated efflux transporter expression emerges.
A major obstacle to overcome is the lack of ABC transporter-specific pharmacological inhibitors. In the 1990s and 2000s, clinical trials of inhibitors such as verapamil were terminated as the result of severe adverse events due to the presence of ABC transporters or structurally similar transmembrane pumps in processes such as calcium transport, action potentials, and clearance of metabolites (Stefan, 2019). Because the pumps themselves remain an elusive (if not impossible) drug target, an alternative approach may be the development of drugs which target the tissue or cancer-specific transcriptional mechanisms which result in their expression. Smo inhibitors such as sonidegib and vismodegib have yielded promising results as transcriptional inhibitors of ABCB1 and ABCG2, increasing uptake of substrates such as colchicine and daunorubicin (Zhang et al., 2009; Zhang et al., 2022). However, other potential drug targets in the Hh signaling pathway may reveal themselves as the transcriptional mechanisms of efflux transporter genes are resolved.
4 Gli-mediated upregulation of DNA repair proteins
Many chemotherapies incur DNA damage, which at therapeutic doses results in enough accumulation of damage to induce apoptosis. This may occur through formation of crosslinks between DNA strands (platinum agents), topoisomerase inhibition (doxorubicin and other anthracyclines), or formation of covalently bonded DNA adducts (temozolomide and other alkylating agents). Cancer cells often avoid the cytotoxic effects of these drugs through upregulation of efflux transporters (mentioned previously) or by overexpression of proteins involved in DNA damage repair so that DNA replication and mitosis may proceed (Meng et al., 2015).
There is growing evidence that non-canonical Hh signaling (and not Smo-dependent) is a key mechanism by which cancers activate the DNA damage response in order to evade chemotherapy. Upon treatment with cisplatin, ovarian cancer cell lines upregulated proteins associated with nucleotide excision repair, including ERCC1, XPD helicase, and XRCC1, a response which was blocked by shRNA knockdown of Gli1. Interestingly, treatment with cyclopamine had no such effect, suggesting that this particular DNA repair pathway activation occurs independently of canonical Hh signaling (Kudo et al., 2012).
Further evidence for the importance of non-canonical Gli activation was presented by Mazumdar et al. (2011) as the Gli antagonist GANT61, but not cyclopamine, induced growth arrest at G1-S phase and apoptosis. GANT61 treatment induced phosphorylation of H2AX (γH2AX), an early marker of the DNA damage response, as well as phosphorylation of ATM and its effector kinase Chk2, indicating the presence of double-stranded DNA breaks. Despite apparent activation of these proteins, COMET assay revealed significant DNA damage in HT29 cells treated with GANT61 in comparison to cyclopamine or DMSO control, indicating that either Gli1 or Gli2 partially inhibits the DNA damage response. Similar results were achieved by genetic overexpression of Gli3R, which has an inhibitory effect on Gli1 and Gli2 (Mazumdar et al., 2011). Furthermore, GANT61 treatment results in downregulation of genes involved in DNA replication such as thymidylate synthase, thymidine kinase, topoisomerase2, E2F, and DNA polymerase epsilon (Shi et al., 2010). GANT61 also displays synergistic lethality in ovarian cancer cells when combined with the Poly (ADP-ribose) polymerase (PARP) inhibitor Olaparib (Mani et al., 2021).
4.1 Regulation of MGMT
O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme which repairs methylated nucleotides and is often upregulated in response to alkylating agents. Hedgehog-mediated regulation of MGMT has been observed in gliomas which acquire resistance to temozolomide (TMZ), a first-line alkylating chemotherapeutic. In patient glioblastomas, MGMT expression is inversely correlated with response to TMZ therapy. Furthermore, Gli1 expression is negatively associated with prognosis and Gli1 nuclear localization correlates with MGMT expression (Wang et al., 2017).
Overexpression of Gli1 in glioblastoma cell lines results in increased tolerance to TMZ, whereas pharmacological inhibition of Hh signaling using GANT61 and cyclopamine resensitizes cells to chemotherapy, and treatment with cyclopamine reduced tumor burden in vivo (Li et al., 2016; Wang et al., 2017). In both cases, MGMT expression was reduced in a dose-dependent manner, suggesting that Hh signaling regulates MGMT expression in some way. However, Li et al. acknowledged in their research that GANT61 inhibits both Gli1 and Gli2. Bioinformatic analysis and chromatin immunoprecipitation of the MGMT promoter revealed a Gli1 recognition site, so while there is strong evidence for Gli1-driven expression of MGMT, whether this mechanism is Gli1 or Gli2-driven has not been fully explored.
5 Bone metastases: a potential case of Gli-mediated chemoresistance
The bone microenvironment is fertile soil for metastases; in the case of breast cancer, 60%–70% of patients have skeletal lesions at time of death (Coleman and Rubens, 1987). This is partially due to the presence of growth factors such as TGF-β and IGF, which are released from the bone matrix during bone turnover. This results in aberrant activation of Hh signaling in cancer cells: Gli2 is significantly overexpressed in bone metastases in comparison to healthy tissues and the primary site (Vanderburgh et al., 2019). A key transcriptional target of Gli2 is PTHLH, the gene which encodes Parathyroid Hormone-related Protein (PTHrP), an osteolytic factor which is secreted by cancer upon metastasis to the bone. Thus, Hh signaling is a significant driver of tumor-induced osteolytic bone destruction (Sterling et al., 2006).
There is a logical connection between metastasis and multidrug resistance; both metastatic and drug-resistant cancer are marked by a mesenchymal phenotype (see Section 1.3) and the ability to dramatically change their transcriptional programming. Clinical data has shown that when metastases acquire resistance to first-line chemotherapy, switching to an alternative chemotherapy does not improve overall survival (Smerage et al., 2014).
What if intrinsic resistance to chemotherapy is a byproduct of non-canonical Hh activation in these bone metastases? As described above, the transcriptional targets of Gli2 include several ABC transporters which are involved in chemoresistance (Po et al., 2020). Included in these transporters are ABCB1 and ABCC2, which were associated with increased risk of bone metastasis in a cohort of 73 patients (Sensorn et al., 2016). Thus, it may not be surprising that breast cancer cells grown on 3D bone-mimetic scaffolds display increased tolerance to chemotherapy (Kar et al., 2020).
While there is circumstantial evidence to support the idea of Gli-driven chemoresistance in bone metastases, to date no studies have explicitly detailed the molecular interactions which result in this phenotype. However, this is just one of many cases of aberrant Hh activation which merits further study.
6 Conclusion
There is a large body of in vitro and pre-clinical research on the function of Hh signaling in drug resistance, but due to the heterogeneity of cancer, there is most likely not a unifying model. Smo inhibitors have yielded promising results in clinical practice, but there are still several remaining obstacles to the therapeutic use of Hh inhibition.
A remaining problem with the use of Hh inhibitors to overcome resistance is that in their current form, they themselves are prone to resistance. The use of structural biology and genomics has illuminated the prevalence of polymorphisms in both SMO and PTCH1 which interfere with the use of Smo inhibitors in the clinic. Additionally, there is a growing body of evidence that Hh activation often occurs independently of its canonical receptors, which means that if Hh is to be targeted broadly in cancer therapy, there must be a focus on the Hh pathway downstream of Smo. Gli inhibitors such as GANT58 and GANT61 have the potential to sensitize a broad range of malignancies to several targeted and cytotoxic therapies, but there is considerable disagreement in the literature over their specificities for Gli1 or Gli2. In order to develop useful, target specific Gli inhibitors, the field must make a distinction between the transcriptional targets and roles of Gli1 and Gli2. Furthermore, these transcription factors are themselves regulated (or dysregulated) by a complex network of protein-protein interactions. Thus, the future of Hh-targeted therapies lies in the ability to identify and elucidate the molecular mechanisms which enable cancers to resist treatment.
Author contributions
JM: Conceptualization, Writing–original draft, Writing–review and editing. JR: Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Department of Veterans Affairs Merit Grant (1I01BX001957) and the National Institutes of Health Research Project Grant (1R01CA264508).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agyeman, A., Mazumdar, T., and Houghton, J. A. (2012). Regulation of DNA damage following termination of hedgehog (HH) survival signaling at the level of the GLI genes in human colon cancer. Oncotarget 3, 854–868. doi:10.18632/ONCOTARGET.586
Ali, S. A., Niu, B., Cheah, K. S. E., and Alman, B. (2019). Unique and overlapping GLI1 and GLI2 transcriptional targets in neoplastic chondrocytes. PLoS One 14, e0211333. doi:10.1371/journal.pone.0211333
Amantini, C., Morelli, M. B., Nabissi, M., Cardinali, C., Santoni, M., Gismondi, A., et al. (2016). Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 7, 50180–50194. doi:10.18632/ONCOTARGET.10326
Atwood, S. X., Sarin, K. Y., Whitson, R. J., Li, J. R., Kim, G., Rezaee, M., et al. (2015). Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27, 342–353. doi:10.1016/J.CCELL.2015.02.002
Avery, J. T., Zhang, R., and Boohaker, R. J. (2021). GLI1: a therapeutic target for cancer. Front. Oncol. 11, 673154. doi:10.3389/fonc.2021.673154
Bai, C., Auerbach, W., Lee, J., Stephen, D., and Joyner, A. (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761. doi:10.1242/dev.129.20.4753
Bai, X. Y., Zhang, X. C., Yang, S. Q., An, S. J., Chen, Z. H., Su, J., et al. (2016). Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One 11, e0149370. doi:10.1371/JOURNAL.PONE.0149370
Bonilla, X., Parmentier, L., King, B., Bezrukov, F., Kaya, G., Zoete, V., et al. (2016). Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 48 (4), 398–406. doi:10.1038/ng.3525
Cazet, A. S., Hui, M. N., Elsworth, B. L., Wu, S. Z., Roden, D., Chan, C. L., et al. (2018). Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 9, 2897. doi:10.1038/S41467-018-05220-6
Chen, D., Kang, X., Li, Z., Chen, L., Ma, Q., and Fan, P. (2021). Hedgehog/GLI1 signaling pathway regulates the resistance to cisplatin in human osteosarcoma. J. Cancer 12, 6676–6684. doi:10.7150/JCA.61591
Chen, H., Yang, D., Wang, Y., Tao, H., Luo, Y., Wu, A., et al. (2022). Activation of the Hedgehog pathway mediates resistance to epidermal growth factor receptor inhibitors in non-small cell lung cancer. J. Cancer 13, 987–997. doi:10.7150/JCA.63410
Chen, J. S., Huang, X. H., Wang, Q., Huang, J. Q., Zhang, L. J., Chen, X. L., et al. (2013). Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis 34, 10–19. doi:10.1093/CARCIN/BGS274
Chen, X., Lingala, S., Khoobyari, S., Nolta, J., Zern, M. A., and Wu, J. (2011). Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 55, 838–845. doi:10.1016/j.jhep.2010.12.043
Chen, Y., Bieber, M. M., and Teng, N. N. H. (2014). Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol. Carcinog. 53, 625–634. doi:10.1002/MC.22015
Chen, Y. J., Kuo, C. D., Chen, S. H., Chen, W. J., Huang, W. C., Chao, K. S. C., et al. (2012). Small-molecule synthetic compound norcantharidin reverses multi-drug resistance by regulating sonic hedgehog signaling in human breast cancer cells. PLoS One 7, e37006. doi:10.1371/JOURNAL.PONE.0037006
Coleman, R. E., and Rubens, R. D. (1987). The clinical course of bone metastases from breast cancer. Br. J. Cancer 55, 61–66. doi:10.1038/BJC.1987.13
Cortes, J. E., Heidel, F. H., Hellmann, A., Fiedler, W., Smith, B. D., Robak, T., et al. (2019). Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 33, 379–389. doi:10.1038/S41375-018-0312-9
Cui, D., Xu, Q., Wang, K., and Che, X. (2010). Gli1 is a potential target for alleviating multidrug resistance of gliomas. J. Neurol. Sci. 288, 156–166. doi:10.1016/j.jns.2009.09.006
Dan, V. M., Raveendran, R. S., and Baby, S. (2020). Resistance to intervention: paclitaxel in breast cancer. Mini-Reviews Med. Chem. 21, 1237–1268. doi:10.2174/1389557520999201214234421
de Reyniès, A., Javelaud, D., Elarouci, N., Marsaud, V., Gilbert, C., and Mauviel, A. (2020). Large-scale pan-cancer analysis reveals broad prognostic association between TGF-β ligands, not Hedgehog, and GLI1/2 expression in tumors. Sci. Rep. 10 (10), 14491–14498. doi:10.1038/s41598-020-71559-w
Della Corte, C. M., Bellevicine, C., Vicidomini, G., Vitagliano, D., Malapelle, U., Accardo, M., et al. (2015). SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 21, 4686–4697. doi:10.1158/1078-0432.CCR-14-3319
Dennler, S., André, J., Alexaki, I., Li, A., Magnaldo, T., ten Dijke, P., et al. (2007). Induction of sonic hedgehog mediators by transforming growth factor-beta: smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 67, 6981–6986. doi:10.1158/0008-5472.CAN-07-0491
Di Magliano, M. P., and Hebrok, M. (2003). Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 3 (3), 903–911. doi:10.1038/nrc1229
Ding, J., Zhou, X. T., Zou, H. Y., and Wu, J. (2017). Hedgehog signaling pathway affects the sensitivity of hepatoma cells to drug therapy through the ABCC1 transporter. Lab. Investig. 97, 819–832. doi:10.1038/labinvest.2017.34
Doheny, D., Manore, S. G., Wong, G. L., and Lo, H. W. (2020). Hedgehog signaling and truncated GLI1 in cancer. Cells 9, 2114. doi:10.3390/CELLS9092114
Eberl, M., Klingler, S., Mangelberger, D., Loipetzberger, A., Damhofer, H., Zoidl, K., et al. (2012). Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 4, 218–233. doi:10.1002/EMMM.201100201
Fan, H., and Khavari, P. A. (1999). Sonic hedgehog opposes epithelial cell cycle arrest. J. Cell Biol. 147, 71–76. doi:10.1083/JCB.147.1.71
Fernández, N. B., Sosa, S. M., Roberts, J. T., Recouvreux, M. S., Rocha-Viegas, L., Christenson, J. L., et al. (2023). RUNX1 is regulated by androgen receptor to promote cancer stem markers and chemotherapy resistance in triple negative breast cancer. Cells 12, 444. doi:10.3390/cells12030444
Fletcher, J. I., Haber, M., Henderson, M. J., and Norris, M. D. (2010). ABC transporters in cancer: more than just drug efflux pumps. Nat. Rev. Cancer 10 (10), 147–156. doi:10.1038/nrc2789
Giammona, A., Crivaro, E., and Stecca, B. (2023). Emerging roles of hedgehog signaling in cancer immunity. Int. J. Mol. Sci. 24, 1321. doi:10.3390/IJMS24021321
Inaguma, S., Riku, M., Hashimoto, M., Murakami, H., Saga, S., Ikeda, H., et al. (2013). GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through bhlhe41-mediated suppression of MLH1. Cancer Res. 73, 7313–7323. doi:10.1158/0008-5472.CAN-13-2008
Javelaud, D., Pierrat, M. J., and Mauviel, A. (2012). Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 586, 2016–2025. doi:10.1016/J.FEBSLET.2012.05.011
Jia, Y., Gu, D., Wan, J., Yu, B., Zhang, X., Chiorean, E. G., et al. (2019). The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene 38, 1764–1777. doi:10.1038/S41388-018-0553-0
Johnson, R. W., Nguyen, M. P., Padalecki, S. S., Grubbs, B. G., Merkel, A. R., Oyajobi, B. O., et al. (2011). TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 71, 822–831. doi:10.1158/0008-5472.CAN-10-2993
Kar, S., Katti, D. R., and Katti, K. S. (2020). Bone interface modulates drug resistance in breast cancer bone metastasis. Colloids Surf. B Biointerfaces 195, 111224. doi:10.1016/j.colsurfb.2020.111224
Kasper, M., Schnidar, H., Neill, G. W., Hanneder, M., Klingler, S., Blaas, L., et al. (2023). Selective modulation of hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell Biol. 26, 6283–6298. doi:10.1128/MCB.02317-05
Khan, M. A., Srivastava, S. K., Zubair, H., Patel, G. K., Arora, S., Moh’d, K., et al. (2020). Co-targeting of CXCR4 and hedgehog pathways disrupts tumor-stromal crosstalk and improves chemotherapeutic efficacy in pancreatic cancer. J. Biol. Chem. 295, 8413–8424. doi:10.1074/jbc.RA119.011748
Kinzler, K. W., and Vogelstein, B. (1990). The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 10, 634–642. doi:10.1128/mcb.10.2.634
Kitazaki, T., Oka, M., Nakamura, Y., Tsurutani, J., Doi, S., Yasunaga, M., et al. (2005). Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 49, 337–343. doi:10.1016/J.LUNGCAN.2005.03.035
Kudo, K., Gavin, E., Das, S., Amable, L., Shevde, L. A., and Reed, E. (2012). Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum–DNA adduct repair. Oncogene 31, 4718–4724. doi:10.1038/onc.2011.610
Li, J., Cai, J., Zhao, S., Yao, K., Sun, Y., Li, Y., et al. (2016). GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 35, 184–214. doi:10.1186/s13046-016-0463-3
Li, J., Wang, C., Pan, Y., Bai, Z., and Wang, B. (2011). Increased proteolytic processing of full-length Gli2 transcription factor reduces the Hedgehog pathway activity in vivo. Dev. Dyn. 240, 766–774. doi:10.1002/DVDY.22578
Li, Q., Lai, Q., He, C., Zhang, H., Pan, X., Li, H., et al. (2021). RUNX1 regulates the proliferation and chemoresistance of colorectal cancer through the Hedgehog signaling pathway. J. Cancer 12, 6363–6371. doi:10.7150/JCA.51338
Liu, Z., Xu, J., He, J., Zheng, Y., Li, H., Lu, Y., et al. (2014). A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 124, 2061–2071. doi:10.1182/BLOOD-2014-03-557298
Mani, C., Tripathi, K., Chaudhary, S., Somasagara, R. R., Rocconi, R. P., Crasto, C., et al. (2021). Hedgehog/GLI1 transcriptionally regulates FANCD2 in ovarian tumor cells: its inhibition induces HR-deficiency and synergistic lethality with PARP inhibition. Neoplasia 23, 1002–1015. doi:10.1016/J.NEO.2021.06.010
Matissek, S. J., and Elsawa, S. F. (2020). GLI3: a mediator of genetic diseases, development and cancer. Cell Commun. Signal. 18, 54–20. doi:10.1186/S12964-020-00540-X
Mazumdar, T., DeVecchio, J., Agyeman, A., Shi, T., and Houghton, J. A. (2011). Blocking hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 71, 5904–5914. doi:10.1158/0008-5472.CAN-10-4173
Meng, E., Hanna, A., Samant, R. S., and Shevde, L. A. (2015). The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers 7, 1333–1348. doi:10.3390/CANCERS7030839
Mo, W., and Zhang, J. T. (2012). Human ABCG2: structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 3, 1–27.
Němcová-Fürstová, V., Kopperová, D., Balušíková, K., Ehrlichová, M., Brynychová, V., Václavíková, R., et al. (2016). Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol. Appl. Pharmacol. 310, 215–228. doi:10.1016/J.TAAP.2016.09.020
Nguyen, N. M., and Cho, J. (2022). Hedgehog pathway inhibitors as targeted cancer therapy and strategies to overcome drug resistance. Int. J. Mol. Sci. 23, 1733. doi:10.3390/IJMS23031733
Oro, A. E., Higgins, K. M., Hu, Z., Bonifas, J. M., Epstein, E. H., and Scott, M. P. (1997). Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 276, 817–821. doi:10.1126/SCIENCE.276.5313.817
Palma, V., and Ruiz i Altaba, A. (2004). Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 131, 337–345. doi:10.1242/DEV.00930
Pan, Y., and Wang, B. (2007). A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 282, 10846–10852. doi:10.1074/JBC.M608599200
Pietrobono, S., Gagliardi, S., and Stecca, B. (2019). Non-canonical hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond smoothened. Front. Genet. 10, 556. doi:10.3389/fgene.2019.00556
Po, A., Citarella, A., Catanzaro, G., Besharat, Z. M., Trocchianesi, S., Gianno, F., et al. (2020). Hedgehog-GLI signalling promotes chemoresistance through the regulation of ABC transporters in colorectal cancer cells. Sci. Rep. 10, 1398–14014. doi:10.1038/s41598-020-70871-9
Queiroz, K. C. S., Ruela-De-Sousa, R. R., Fuhler, G. M., Aberson, H. L., Ferreira, C. V., Peppelenbosch, M. P., et al. (2010). Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 29, 6314–6322. doi:10.1038/onc.2010.375
Raez, L. E., Baca, Y., Nieva, J. J., Mamdani, H., Lopes, G., Borghaei, H., et al. (2022). Acquired EGFR-resistant mutations in non–small cell lung cancer (NSCLC). J. Clin. Oncol. 40, 9113. doi:10.1200/JCO.2022.40.16_SUPPL.9113
Reinchisi, G., Parada, M., Lois, P., Oyanadel, C., Shaughnessy, R., Gonzalez, A., et al. (2013). Sonic Hedgehog modulates EGFR dependent proliferation of neural stem cells during late mouse embryogenesis through EGFR transactivation. Front. Cell Neurosci. 7, 166. doi:10.3389/fncel.2013.00166
Riaz, S. K., Ke, Y., Wang, F., Kayani, M. A., and Malik, M. F. A. (2019). Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci. Rep. 9, 6620–6713. doi:10.1038/s41598-019-43093-x
Robinson, K., and Tiriveedhi, V. (2020). Perplexing role of P-glycoprotein in tumor microenvironment. Front. Oncol. 10, 265. doi:10.3389/fonc.2020.00265
Sensorn, I., Sukasem, C., Sirachainan, E., Chamnanphon, M., Pasomsub, E., Trachu, N., et al. (2016). ABCB1 and ABCC2 and the risk of distant metastasis in Thai breast cancer patients treated with tamoxifen. Onco Targets Ther. 9, 2121–2129. doi:10.2147/OTT.S100905
Sharma, A., De, R., Javed, S., Srinivasan, R., Pal, A., and Bhattacharyya, S. (2019). Sonic hedgehog pathway activation regulates cervical cancer stem cell characteristics during epithelial to mesenchymal transition. J. Cell Physiol. 234, 15726–15741. doi:10.1002/JCP.28231
Shi, T., Mazumdar, T., DeVecchio, J., Duan, Z. H., Agyeman, A., Aziz, M., et al. (2010). cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PLoS One 5, e13054. doi:10.1371/JOURNAL.PONE.0013054
Sims-Mourtada, J., Izzo, J. G., Ajani, J., and Chao, K. S. C. (2007). Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 26, 5674–5679. doi:10.1038/sj.onc.1210356
Smerage, J. B., Barlow, W. E., Hortobagyi, G. N., Winer, E. P., Leyland-Jones, B., Srkalovic, G., et al. (2014). Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. 32, 3483–3489. doi:10.1200/JCO.2014.56.2561
Sommers, C. L., Heckford, S. E., Skerker, J. M., Worland, P., Torri, J. A., Thompson, E. W., et al. (1992). Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 52, 5190–5197.
Song, X., Yan, L., Lu, C., Zhang, C., Zhu, F., Yang, J., et al. (2018). Activation of hedgehog signaling and its association with cisplatin resistance in ovarian epithelial tumors. Oncol. Lett. 15, 5569–5576. doi:10.3892/OL.2018.8008
Stefan, S. M. (2019). Multi-target ABC transporter modulators: what next and where to go? Future Med. Chem. 11, 2353–2358. doi:10.4155/FMC-2019-0185
Steg, A. D., Katre, A. A., Bevis, K. S., Ziebarth, A., Dobbin, Z. C., Shah, M. M., et al. (2012). Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 11, 1587–1597. doi:10.1158/1535-7163.MCT-11-1058
Sterling, J. A., Oyajobi, B. O., Grubbs, B., Padalecki, S. S., Munoz, S. A., Gupta, A., et al. (2006). The Hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 66, 7548–7553. doi:10.1158/0008-5472.CAN-06-0452
Takam Kamga, P., Swalduz, A., Costantini, A., Julié, C., Emile, J. F., Pérol, M., et al. (2021). High circulating sonic hedgehog protein is associated with poor outcome in EGFR-mutated advanced NSCLC treated with tyrosine kinase inhibitors. Front. Oncol. 11, 747692. doi:10.3389/fonc.2021.747692
Their, J. P. (2002). Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454. doi:10.1038/NRC822
Tian, F., Schroedl, K., Kiefl, R., Huber, R. M., and Bergner, A. (2012). The hedgehog pathway inhibitor GDC-0449 alters intracellular Ca2+ homeostasis and inhibits cell growth in cisplatin-resistant lung cancer cells. Anticancer Res. 32, 89–94.
Tojo, M., Kiyosawa, H., Iwatsuki, K., Nakamura, K., and Kaneko, F. (2003). Expression of the GLI2 oncogene and its isoforms in human basal cell carcinoma. Br. J. Dermatol 148, 892–897. doi:10.1046/J.1365-2133.2003.05284.X
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. doi:10.1126/science.1260419
Van Schmus, R. J., Wasson, J. T., Afiattalab, F., Wasson, J. T., Ribbe, R. H., Van Schmus, W. R., et al. (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801. doi:10.1038/287795a0
Vanderburgh, J. P., Kwakwa, K. A., Werfel, T. A., Merkel, A. R., Gupta, M. K., Johnson, R. W., et al. (2019). Systemic delivery of a Gli inhibitor via polymeric nanocarriers inhibits tumor-induced bone disease. J. Control. Release 311 (312), 257–272. doi:10.1016/j.jconrel.2019.08.038
Vortkamp, A., Lee, K., Lanske, B., Segre, G. V., Kronenberg, H. M., and Tabin, C. J. (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273, 613–622. doi:10.1126/SCIENCE.273.5275.613
Wang, C. Y., Chang, Y. C., Kuo, Y. L., Lee, K. T., Chen, P. S., Cheung, C. H. A., et al. (2019). Mutation of the PTCH1 gene predicts recurrence of breast cancer. Sci. Rep. 9, 16359. doi:10.1038/S41598-019-52617-4
Wang, F., Ma, L., Zhang, Z., Liu, X., Gao, H., Zhuang, Y., et al. (2016). Hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J. Cancer 7, 408–417. doi:10.7150/JCA.13305
Wang, J., Cui, B., Li, X., Zhao, X., Huang, T., and Ding, X. (2023). The emerging roles of Hedgehog signaling in tumor immune microenvironment. Front. Oncol. 13, 1171418. doi:10.3389/fonc.2023.1171418
Wang, K., Chen, D., Qian, Z., Cui, D., Gao, L., and Lou, M. (2017). Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 17, 117–212. doi:10.1186/s12935-017-0491-x
Wang, Q., Wei, X., Hu, L., Zhuang, L., Zhang, H., and Chen, Q. (2022). Hedgehog−Gli2 signaling promotes chemoresistance in ovarian cancer cells by regulating MDR1. Front. Oncol. 11, 794959. doi:10.3389/fonc.2021.794959
Wu, X. Y., Che, J., Sun, K. K., Shen, X. J., Yang, D., Zhong, N., et al. (2013). Cyclopamine increases the radiosensitivity of human pancreatic cancer cells by regulating the DNA repair signal pathway through an epidermal growth factor receptor-dependent pathway. Mol Med Rep 8, 979–983. doi:10.3892/mmr.2013.1605
Xie, Y., Liu, J., Jiang, H., Wang, J., Li, X., Wang, J., et al. (2019). Proteasome inhibitor induced SIRT1 deacetylates GLI2 to enhance hedgehog signaling activity and drug resistance in multiple myeloma. Oncogene 39, 922–934. doi:10.1038/s41388-019-1037-6
Xu, J., Lamouille, S., and Derynck, R. (2009). TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172. doi:10.1038/cr.2009.5
Xu, Y., Yu, P., Wang, S., Jiang, L., Chen, F., and Chen, W. (2019). Crosstalk between Hh and Wnt signaling promotes osteosarcoma progression. Int. J. Clin. Exp. Pathol. 12, 768–773.
Yang, L., Xie, G., Fan, Q., and Xie, J. (2010). Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 29, 469–481. doi:10.1038/onc.2009.392
Yochum, Z. A., Cades, J., Wang, H., Chatterjee, S., Simons, B. W., O’Brien, J. P., et al. (2018). Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 38, 656–670. doi:10.1038/s41388-018-0482-y
Yoon, J. W., Lamm, M., Chandler, C., Iannaccone, P., and Walterhouse, D. (2020). Up-regulation of GLI1 in vincristine-resistant rhabdomyosarcoma and Ewing sarcoma. BMC Cancer 20, 511–515. doi:10.1186/s12885-020-06985-0
Yu, B., Gu, D., Zhang, X., Li, J., Liu, B., Xie, J., et al. (2017a). GLI1-mediated regulation of side population is responsible for drug resistance in gastric cancer. Oncotarget 8, 27412–27427. doi:10.18632/ONCOTARGET.16174
Yu, B., Gu, D., Zhang, X., Liu, B., and Xie, J. (2017b). The role of GLI2-ABCG2 signaling axis for 5Fu resistance in gastric cancer. J. Genet. Genomics 44, 375–383. doi:10.1016/j.jgg.2017.04.008
Zahreddine, H. A., Culjkovic-Kraljacic, B., Assouline, S., Gendron, P., Romeo, A. A., Morris, S. J., et al. (2014). The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 511, 90–93. doi:10.1038/nature13283
Zhang, H., Hu, L., Cheng, M., Wang, Q., Hu, X., and Chen, Q. (2020a). The Hedgehog signaling pathway promotes chemotherapy resistance via multidrug resistance protein 1 in ovarian cancer. Oncol. Rep. 44, 2610–2620. doi:10.3892/OR.2020.7798
Zhang, H., Ma, J., Avery, J. T., Sambandam, V., Nguyen, T. H., Xu, B., et al. (2020b). GLI1 inhibitor SRI-38832 attenuates chemotherapeutic resistance by downregulating NBS1 transcription in BRAFV600E colorectal cancer. Front. Oncol. 10, 241. doi:10.3389/fonc.2020.00241
Zhang, H., Wang, Y., Chen, T., Zhang, Y., Xu, R., Wang, W., et al. (2019). Aberrant activation of hedgehog signalling promotes cell migration and invasion via matrix metalloproteinase-7 in ovarian cancer cells. J. Cancer 10, 990–1003. doi:10.7150/JCA.26478
Zhang, L., Song, R., Gu, D., Zhang, X., Yu, B., Liu, B., et al. (2017). The role of GLI1 for 5-Fu resistance in colorectal cancer. Cell Biosci. 7, 17–19. doi:10.1186/s13578-017-0145-7
Zhang, M., Zhang, Y. Y., Chen, Y., Wang, J., Wang, Q., and Lu, H. (2021). TGF-β signaling and resistance to cancer therapy. Front. Cell Dev. Biol. 9, 786728. doi:10.3389/fcell.2021.786728
Zhang, Y., Laterra, J., and Pomper, M. G. (2009). Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/pgp. Neoplasia 11, 96–101. doi:10.1593/NEO.81264
Zhang, Y., Vagiannis, D., Budagaga, Y., Sabet, Z., Hanke, I., Rozkoš, T., et al. (2022). Sonidegib potentiates the cancer cells’ sensitivity to cytostatic agents by functional inhibition of ABCB1 and ABCG2 in vitro and ex vivo. Biochem. Pharmacol. 199, 115009. doi:10.1016/J.BCP.2022.115009
Zhou, X. T., Ding, J., Li, H. Y., Zuo, J. L., Ge, S. Y., Jia, H. L., et al. (2020). Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J. Cell Mol. Med. 24, 4298–4311. doi:10.1111/JCMM.15090
Keywords: cancer, chemotherapy, resistance, hedgehog, Gli
Citation: Miller JS, Bennett NE and Rhoades JA (2023) Targeting hedgehog-driven mechanisms of drug-resistant cancers. Front. Mol. Biosci. 10:1286090. doi: 10.3389/fmolb.2023.1286090
Received: 30 August 2023; Accepted: 09 October 2023;
Published: 23 October 2023.
Edited by:
Bei Liu, The Ohio State University, United StatesReviewed by:
Kaushlendra Tripathi, University of Alabama at Birmingham, United StatesYong Jiang, East Tennessee State University, United States
Copyright © 2023 Miller, Bennett and Rhoades. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie A. Rhoades, anVsaWUucmhvYWRlc0B2dW1jLm9yZw==