 Christopher J. Requejo Cier
Christopher J. Requejo Cier Nicolas Valentini
Nicolas Valentini Caroline Lamarche
Caroline Lamarche- 1Department of Microbiology, Infectiology and Immunology, Hôpital Maisonneuve-Rosemont Research Institute, Université de Montréal, Montreal, QC, Canada
- 2Department of Medicine, Hôpital Maisonneuve-Rosemont Research Institute, Université de Montréal, Montreal, QC, Canada
Regulatory T cells (Tregs) adoptive immunotherapy is emerging as a viable treatment option for both autoimmune and alloimmune diseases. However, numerous challenges remain, including limitations related to cell number, availability of target-specific cells, stability, purity, homing ability, and safety concerns. To address these challenges, cell engineering strategies have emerged as promising solutions. Indeed, it has become feasible to increase Treg numbers or enhance their stability through Foxp3 overexpression, post-translational modifications, or demethylation of the Treg-specific demethylated region (TSDR). Specificity can be engineered by the addition of chimeric antigen receptors (CARs), with new techniques designed to fine-tune specificity (tandem chimeric antigen receptors, universal chimeric antigen receptors, synNotch chimeric antigen receptors). The introduction of B-cell targeting antibody receptor (BAR) Tregs has paved the way for effective regulation of B cells and plasma cells. In addition, other constructs have emerged to enhance Tregs activation and function, such as optimized chimeric antigen receptors constructs and the use of armour proteins. Chimeric antigen receptor expression can also be better regulated to limit tonic signaling. Furthermore, various opportunities exist for enhancing the homing capabilities of CAR-Tregs to improve therapy outcomes. Many of these genetic modifications have already been explored for conventional CAR-T therapy but need to be further considered for CAR-Tregs therapies. This review highlights innovative CAR-engineering strategies that have the potential to precisely and efficiently manage immune responses in autoimmune diseases and improve transplant outcomes. As these strategies are further explored and optimized, CAR-Treg therapies may emerge as powerful tools for immune intervention.
Introduction
Autoimmune and alloimmune diseases are major public health challenges. Autoimmune diseases affect millions of individuals worldwide and can lead to severe disability and even death (Jacobson et al., 1997). More than 80 different types of autoimmune diseases have been identified, ranging from lupus and rheumatoid arthritis to type 1 diabetes and multiple sclerosis (Jacobson et al., 1997; Maahs et al., 2010; Wahren-Herlenius and Dörner, 2013). Alloimmune reactions lead to solid organ rejection after a transplantation and graft-versus-host disease (GVHD) after a hematopoietic stem cell transplantation (HCT). Indeed, despite modern immunosuppressive therapy, approximately 8% of kidney transplant recipients will develop acute rejection within 1 year (Lentine et al., 2023), and between 9% and 50% of patients will develop acute GVHD after HCT (Filipovich et al., 2005; Jagasia et al., 2012; Lee et al., 2013).
Immunosuppressive drugs are currently the standard of care for managing these diseases (Sparks, 2019). While these medications can be effective, they also carry significant side effects, including an increased risk of infection, cardiovascular disease, and cancer (Marcén, 2009; Downey, 2016; Salzer et al., 2016). Given these limitations, innovative therapeutic approaches are needed to treat autoimmune diseases and alloimmune responses.
Immunotherapy, an approach that harnesses the ability of the immune system to combat disease, holds great promise for the treatment of autoimmune and alloimmune diseases. Within the complex network of immune cells, regulatory T cells (Tregs) have emerged as key players in the maintenance of immune tolerance (Karim et al., 2005; Waldmann et al., 2006; Sakaguchi et al., 2008; Kassan et al., 2011; Hou et al., 2019). Tregs are a specialized subset of CD4+ T lymphocytes characterized by high expression of IL-2Rα (CD25) and the transcription factor forkhead box p3 (Foxp3) (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003), as well as low expression of IL-7Rα (CD127) (Liu et al., 2006). Tregs use a variety of mechanisms to effectively suppress immune responses, including direct cell-to-cell contact and indirect pathways involving the secretion of interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) secretion (reviewed in (Valentini et al., 2023)).
Preclinical models and early-phase clinical trials using adoptive Treg immunotherapy to promote tolerance have yielded promising results, underscoring the safety and potential effectiveness of this approach (Brunstein et al., 2011; Sarkar et al., 2014; Bluestone et al., 2015; Leclerc and Lamarche, 2021). However, challenges remain in the development and implementation of this therapeutic approach. These challenges arise as early as the isolation and selection of Tregs, further exacerbating their limited availability. Additionally, they affect the potential impact on the specificity, survival, and activation of the acquired Tregs. The use of chimeric antigen receptors (CARs) to redirect the specificity of Tregs has emerged as a promising strategy to improve the efficacy of Treg-based cell therapy (Fransson et al., 2012; Boardman et al., 2017; Noyan et al., 2017; Sicard et al., 2020). CARs are synthetic receptors that redirect cellular specificity independently of major histocompatibility complexes (MHC). CAR-Tregs showed significant potential in preclinical studies in transplantation, autoimmune diseases, and other diseases (Table 1). Further advancements in CAR constructs for Tregs and gene editing technologies hold substantial promise for the development of effective cell therapies. Nevertheless, even though this approach presents a potential solution to the aforementioned challenges, standardization, activation, and the accurate induction of a regulatory phenotype continue to pose hurdles for research teams.
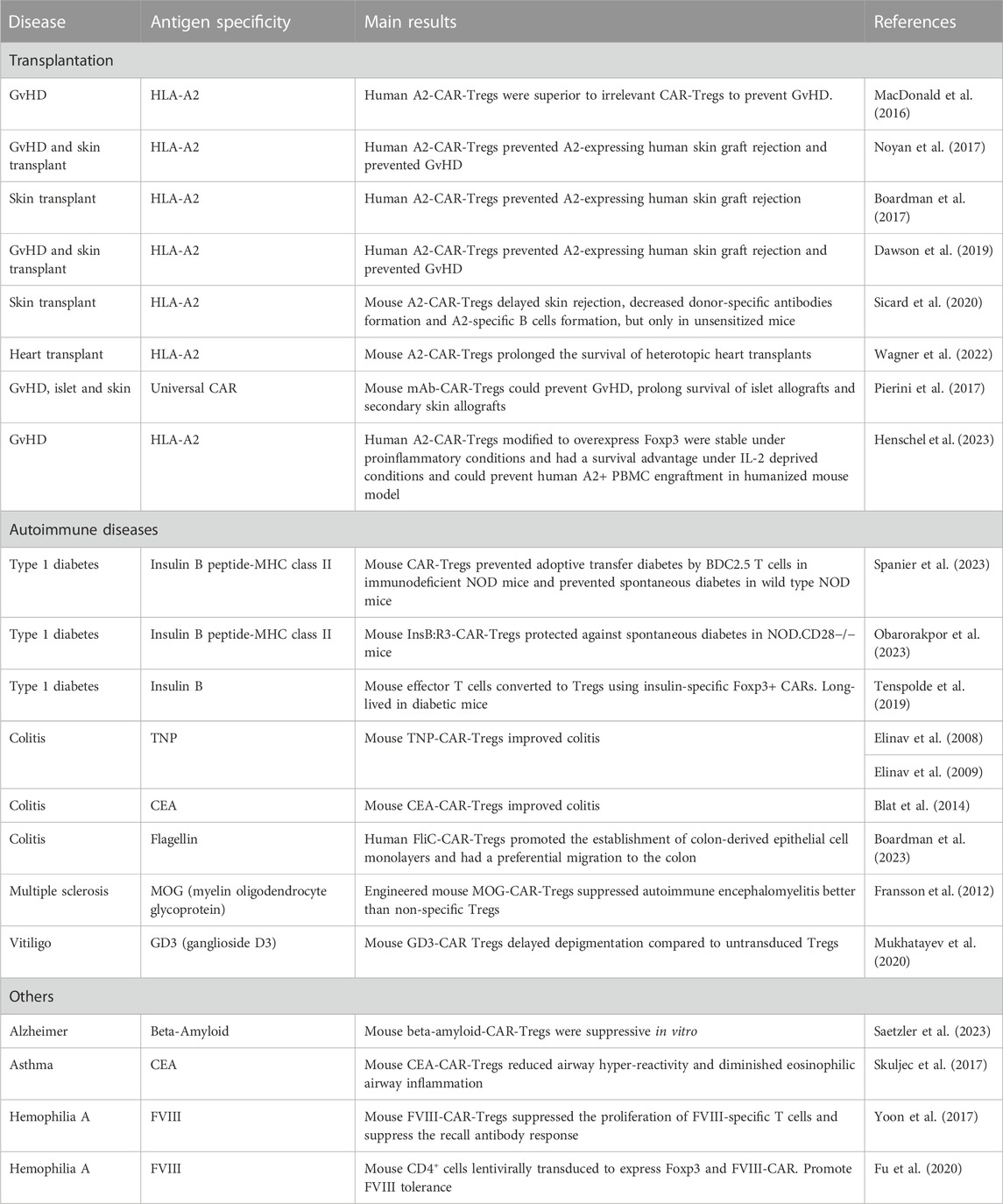
TABLE 1. Pre-clinical studies using CAR Tregs.
In contrast to effector CAR T cells, CAR constructs designed for Tregs (CAR Tregs) represent a distinct branch within the field of CAR T cell therapies. While effector CAR T cells are primarily engineered to target and eliminate specific disease-related cells, such as cancer cells, by inducing cytotoxicity and releasing pro-inflammatory cytokines (reviewed in (Sterner and Sterner, 2021)), CAR Tregs adopt an immunosuppressive approach by regulating immune responses. Rather than enhancing immune responses, as effector CAR T cells do, CAR Tregs focus on modulating and suppressing immune activity to maintain equilibrium and prevent pathological immune responses. A comprehensive understanding of these distinctions is paramount for tailoring CAR T cell therapies to specific clinical applications and optimizing their outcomes. This review will first address the challenges with the starting material (i.e., selection, purity, quantity), and the later sections will address the challenges associated with CAR constructs.
Treg selection
Tregs are not a homogeneous group of cells; instead, they encompass diverse subpopulations with varying immunosuppressive capabilities, phenotypes, and stability (Figure 1). When using antigen-specific Tregs for cell therapy, ensuring purity and stability is of paramount importance. Indeed, the simultaneous proliferation of antigen-specific conventional T cells alongside Tregs in adoptive immunotherapy could be detrimental, and the maximum ratio of conventional T cells that can be inhibited or contained by Tregs is unknown. One method to enhance cell purity is to select naïve Tregs based on CD45RA expression. This was shown more than 15 years ago by Hoffmann et al. (Hoffmann et al., 2006). More recently, this finding has been validated in CAR Tregs (Imura et al., 2020). When compared to CD45RO+ Tregs, only CD45RA+ CD19 CAR Tregs have demonstrated the ability to maintain substantial levels of Foxp3, Helios, and Ctla-4 expression (Imura et al., 2020). It is worth noting that following activation, conventional T cells can upregulate CD25 expression but will express the CD45RO isoform rather than the CD45RA.
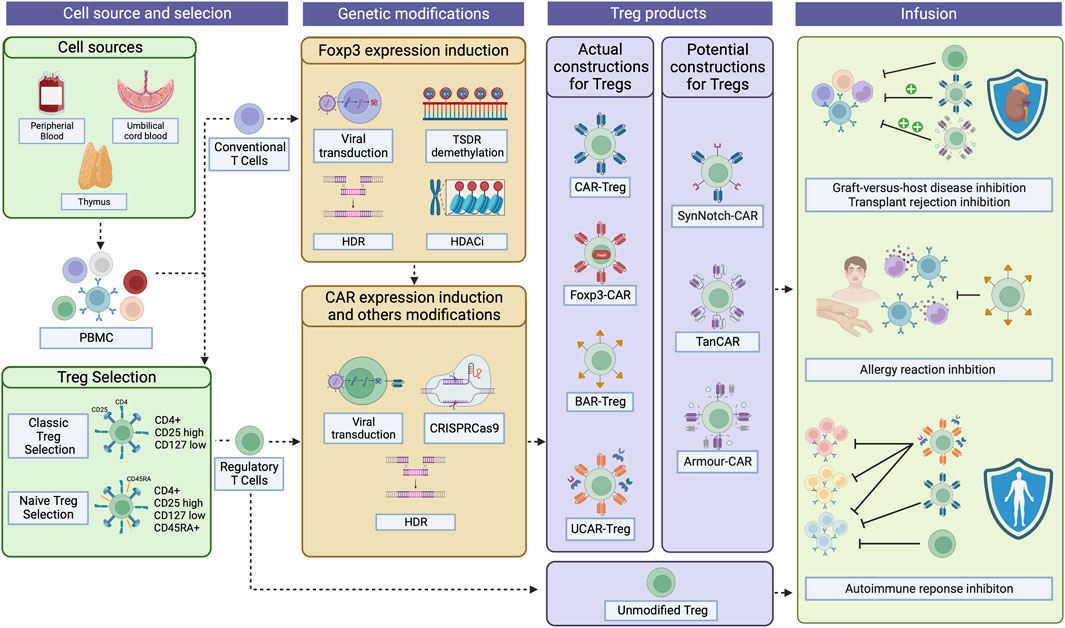
FIGURE 1. CAR-Treg development workflow. For CAR-Treg development, peripheral blood mononuclear cells (PBMC)s can be retrieved from various sources, including the peripheral blood, umbilical cord blood, or even the thymus. Tregs are then isolated through magnetic enrichment or sorted as CD4+CD25hiCD127low cells. For naïve Tregs, CD45RAhi cells are then selected. Given that obtaining Tregs fromPBMCs can yield limited quantities due to their low proportion, conventional CD4+ T cells overexpressing Foxp3 emerge as a possible substrate for CAR-Treg production. Foxp3 can be expressed in those cells after viral transduction, TSDR demethylation, HDR, or the use of HDAC inhibitors. Then, CAR constructs can be integrated into the cells by viral transduction, HDR, or CRISPR-Cas9 technology, among others. To date, four types of CAR constructs have been tested in Tregs: classical CAR, Foxp3-CAR, BAR, and UCAR. Other constructs have, until now, been exclusively tested in effector T cells but holds potential as CAR-Treg therapies: SynNotch-CAR, TanCAR, and Armour-CAR. Then, the desired CAR-Tregs can be infused into patients to treat and/or prevent graft-versus-host disease, transplant rejection, allergies, and autoimmune diseases. CAR, Chimeric Antigen Receptor; TSDR, Treg-specific demethylated region; HDR, Homology-Directed Repair; HDAC, Histone deacetylases; BAR, B cell-targeting Ab receptors; UCAR, Universal CAR; SynNotch, Synthetic Notch CAR; TanCAR, Tandem CAR. Created with BioRender.com.
Exploring new sources of Treg
Tregs, which comprise a mere 5% of CD4+ T cells, represent a scarcity that poses a challenge for adoptive immunotherapy. The magnitude of this challenge is amplified in autologous treatments, where certain autoimmune and alloimmune disorders are associated with diminished Treg populations (Demirkiran et al., 2006). Moreover, the administration of immunosuppressive drugs can affect both the quantity and quality of Tregs (Demirkiran et al., 2008). For instance, in the ARTEMIS trial, Tang et al. highlight the intricacies involved in Treg manufacturing in liver transplant recipients. In the ARTEMIS trial is a phase I/II trial of autologous donor alloantigen-reactive Tregs in liver transplant recipients, 2–7 years after transplantation, 4/10 were excluded from receiving Treg infusions due to the inability to meet the minimum threshold of 100 × 106 cells. The decrease in donor-specific Treg expansion in liver transplant recipients can be attributed to a lower absolute peripheral blood Treg count compared to participants in other Treg adoptive immunotherapy trials, along with a decrease in the number of donor-reactive Tregs. Furthermore, their analysis suggested a progressive process of Treg activation, exhaustion, senescence, or deletion after liver transplantation (Tang et al., 2022). In an effort to overcome this hurdle, numerous research teams have directed their efforts toward generating novel sources of Tregs, including induced Tregs generation, in vivo Treg expansion, or via the genetic reprogramming of conventional CD4+ T cells (Figure 1). These innovative approaches aim to augment the pool of available Tregs for therapeutic applications, thereby opening new pathways in the field of adoptive immunotherapy.
Induced Treg generation
The first approach involves the induction of Tregs in vitro. Indeed, under certain circumstances and factors such as TGF-β and IL-2, naïve CD4+ T cells can express Foxp3 and develop a regulatory phenotype (Zheng et al., 2002; Chen et al., 2003). This approach was first tested in HCT recipients in a phase I safety study (MacMillan et al., 2021). Human CD4+CD25− conventional T cells were cultured with IL-2, rapamycin and TGF- β with anti-CD3, and artificial antigen-presenting cells to generate induced Tregs, which were then infused into HCT from HLA-identical sibling donors. However, a major concern with induced Tregs is the potential for instability, particularly under inflammatory conditions (Koenecke et al., 2009; Beres et al., 2011). Although patients from this first study did not show an increase in GVHD after infusion, alternative approaches are being developed.
In vivo Treg expansion
Tregs do not produce IL-2 themselves, but express the high affinity IL-2 receptor CD25. Therefore, the administration of low doses of IL-2 can selectively expand Tregs without conventional T cell activation and expansion (reviewed in (Ye et al., 2018)). However, IL-2 can also activate conventional T cells and NK cells. Consequently, numerous strategies have been developed to selectively increase Tregs and prevent adverse effects, such as Il-2 muteins with greater Treg specificity (Khoryati et al., 2020), anti-IL-2 monoclonal antibodies, or IL-2/anti-IL-2 complexes to modify binding to the IL-2 receptor (Boyman et al., 2006; Polhill et al., 2012; Trotta et al., 2018). Other strategies aim to combine IL-2 with cytokine pathway blockade, such as an anti-IL-6 receptor (Ye et al., 2018), or to modify IL-2 to prolong its half-life, allowing for the use of an ultra-low doses (Bell et al., 2015).
Treg generation through Foxp3 overexpression
The first approach to generate Tregs from conventional cells is to overexpress Foxp3 (Figure 1). Indeed, Foxp3 is a master transcription factor for Treg development and function. Sakaguchi et al. pioneered this approach, inducing Foxp3 expression in CD25−, CD45RO− CD4+ lymphocytes by retroviral transduction. The transduced cells exhibited a regulatory phenotype similar to CD25+ CD4+ Tregs, demonstrated in vitro suppressive capabilities, decreased proliferation, cytokine production reduction, and heightened expression of Treg-associated molecules, such as CD25 and CTLA4 (Yagi et al., 2004). While this initial study offers valuable and promising insights into the involvement of Foxp3 in Treg development, it solely showed that the transduced cells displayed a Treg-like phenotype. However, they also exhibited unconventional Treg features, including reduced production of IL-10 (Yagi et al., 2004). Subsequently, Allan et al. developed a third-generation lentivirus-based strategy to ectopically express high levels of Foxp3 under the control of the human elongation factor 1α promoter, which does not fluctuate with T cell activation status (Allan et al., 2008). This method facilitated the development of suppressor cells from both naïve and memory CD4+ T cells. Nevertheless, despite their demonstrated in vitro suppressive capacity, the expected significant production of IL-10 and TGF-β was not observed, suggesting that Foxp3 alone might not be sufficient to induce Treg production (Allan et al., 2008).
Bacchetta et al. also tested a third-generation lentivirus-mediated FOXP3 gene transfer in conventional CD4+ T cells, confirming its in vitro suppressive capacity. Notably, they were the first to test the suppressive ability in vivo, demonstrating its effectiveness in a xenogeneic graft-versus-host disease model (xGvHD) (Passerini et al., 2013). However, they did not evaluate IL-10 and TGF-β production in their cells, a notable omission given previous observations. While IL-10 and TGF-β production may not be essential for mediating in vitro suppressive function (Allan et al., 2008), their role in the xGvHD model remains unexplored. Finally, Honaker et al. used gene editing based on homology-directed repair (HDR) to induce Foxp3 expression (Honaker et al., 2020). The targeted insertion of a robust enhancer-promoter proximal to the first coding exon bypassed epigenetic silencing and ensured stable and strong expression of endogenous Foxp3. The HDR-edited T cells expressed canonical Treg markers and exhibited suppressive capabilities both in vitro and in vivo (Honaker et al., 2020). However, these cells exhibited lower suppressive potential than normal Tregs and displayed transcriptomic differences from thymic Tregs, such as IKZF2 (Honaker et al., 2020). These observations collectively suggest that Foxp3 expression alone is insufficient to fully recapitulate the Treg function. Given that the transcription factor Helios is implicated in Treg differentiation, stability and suppression, researchers explored the possibility of co-expressing Foxp3 and Helios in conventional cells. However, this co-expression led to significantly reduced cell proliferation and survival, limiting its clinical application (Seng et al., 2020).
Foxp3 overexpression in conventional CD4+ T cells has also been combined with CAR expression (Fransson et al., 2012; Martin et al., 2021). This combination enabled the expression of a Treg phenotype, resulting in a reduced IL-2 and TNF-ɑ secretion and increased in IL-10 secretion. Additionally, these cells were shown to be effective in an xGvHD model (Martin et al., 2021) and an autoimmune encephalomyelitis mouse model (Fransson et al., 2012). While this approach is promising, further validation is required before clinical application can be considered.
Strategies aiming at increasing Foxp3 expression may not only serve to convert conventional T cells into Tregs, but also to increase Treg stability. Indeed, Henschel et al. developed a specialized CAR vector incorporating Foxp3 into CAR-Tregs (Henschel et al., 2023). They observed significant improvements in both the safety and efficacy of the resulting CAR-Treg product. Notably, in a challenging microenvironment characterized by pro-inflammatory conditions and limited IL-2 availability, Foxp3-CAR-Tregs exhibited sustained Foxp3 expression, surpassing the expression levels observed in control CAR-Tregs. Furthermore, the introduction of additional exogenous Foxp3 expression did not induce any discernible phenotypic alterations or functional impairments, such as cellular exhaustion, loss of Treg characteristics, or aberrant cytokine secretion (Henschel et al., 2023). Overall, these advancements offer potential pathways for the development of more accessible and safe immunotherapies targeting limited Treg numbers. However, further optimization is needed to generate Tregs from conventional CD4+ T cells.
Treg generation through post-translational modifications
Another method to induce Tregs from conventional CD4+ T cells involves the use of epigenetic regulators (Figure 1). Histone/protein deacetylase (HDAC)s can regulate chromatin remodeling, gene expression, and transcription factor expression by removing acetyl groups from histones. This deacetylation process by HDACs typically results in chromatin condensation, restricting access by the transcription machinery and consequently repressing gene expression. Additionally, HDACs function as post-transcriptional modifiers that regulate protein acetylation (reviewed in (Kim and Bae, 2011)). Indeed, HDAC inhibition therapy can induce Foxp3 expression. This therapy increases histone acetylation, which leads to the relaxation of the chromatin structure surrounding the FOXP3 gene. This chromatin decondensation facilitates easier access for transcription factors to interact with the FOXP3 gene, thereby enhancing its transcription and subsequent protein synthesis. Consequently, cells become more proficient at suppressing immune responses and promoting transplant tolerance (Tao et al., 2007; Lucas et al., 2009). There are 18 known mammalian HDACs divided into four classes sometimes with subclasses (I, IIa, IIb, III, IV) and the use of pan-HDAC inhibitors may have various side effects. Therefore, not all HDAC inhibitors promote Treg functions (Wang et al., 2009). Pharmacological targeting and deletion experiments have shown a positive impact on Tregs for HDAC2, HDAC7, HDAC9, HDAC6, HDAC10, HDAC11 and Sirtuin-1. HDAC6, in particular, stands out as the most promising candidate due to the availability of a specific inhibitor and the fact that HDAC6 knockout mice exhibit normal health and behaviour. Hence, the HDAC6 isoform is the primary target for enhancing the immunosuppressive activity of Tregs (de Zoeten et al., 2011; Kalin et al., 2012; Kurohara et al., 2021; Lee et al., 2022). However, HDAC10 could also be of interest, as Wayne Hancock’s team demonstrated an increased suppressive ability of HDAC10−/− mouse Tregs, both in vitro and in vivo (Dahiya et al., 2020). There is a lot of potential for HDAC inhibitors to induce Tregs in vivo (reviewed in (Wang et al., 2018)). However, their use in adoptive immunotherapies and Treg engineering has not yet been explored.
Treg generation through Treg-specific demethylation region (TSDR) demethylation
Three conserved non-coding DNA sequence (CNS) elements located at the FOXP3 locus have significant implications for the fate of Tregs. Specifically, CNS1 serves as a TGF-β-sensitive enhancer element crucial for induced Treg generation, CNS2 plays a critical role in stabilizing Foxp3 expression within dividing Treg cells, and CSN3 can modulate Treg frequency in the thymus and periphery but becomes dispensable once Foxp3 is expressed (Zheng et al., 2010). Notably, CSN2, also known as Treg-specific demethylation region (TSDR), exhibits demethylation in stable Treg populations (Feng et al., 2014), in contrast to T cells with transient Foxp3 expression (Floess et al., 2007; Polansky et al., 2008). Recent scientific advances have enabled the selective de-methylation of the TSDR within the endogenous chromatin environment. This epigenetic editing resulted in an increased Foxp3 expression in Jurkat cells (Wilk et al., 2022) and human T cells (Kressler et al., 2020). However, this increase was not associated with phenotypic Treg induction (Kressler et al., 2020). Selective demethylation of the TSDR represents a significant scientific breakthrough. However, it requires further investigation and may need to be combined with other strategies to achieve the desired outcomes.
Enhancing tregs specificity
Currently, three clinical trials are registered using monospecific CAR-Tregs in humans (Table 1). However, enhancing the specificity of CAR-Tregs, meaning the number of antigens they can recognize, holds great potential to significantly improve their therapeutic efficacy. The use of monospecific CAR-T effector cells for the treatment of diseases such as cancer has been limited by antigen escape, a phenomenon where the target antigen’s expression is lost, rendering the CAR-T effector cells unable to perform their intended function. In response, several research teams have explored solutions and developed alternative CAR designs (Figure 2), that have shown success in enhancing the effector response (Hegde et al., 2013; Kloss et al., 2013; Roybal et al., 2016; Ruella et al., 2016). Currently, there is no evidence that antigen escape is an concern with CAR-Treg immunotherapies. However, some of the following approaches may prove beneficial.
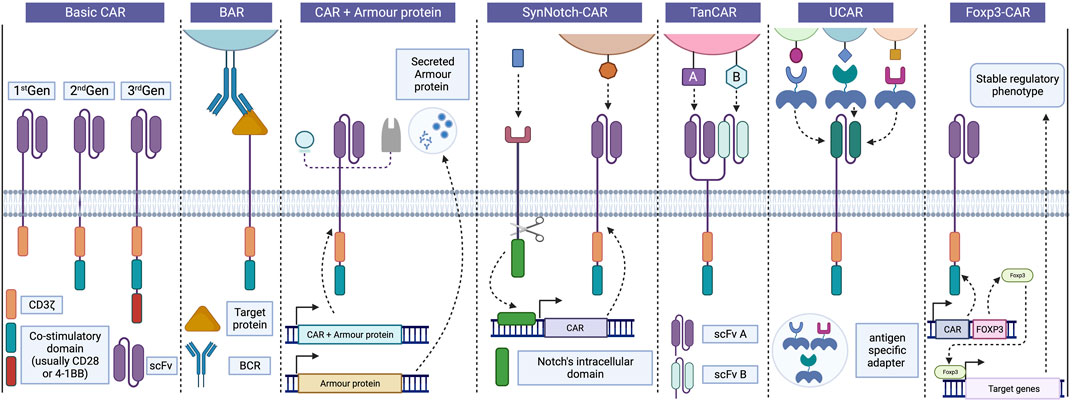
FIGURE 2. Progress in CAR design and strategies. Basic CARs are divided usually into three generations based on the number of co-stimulatory domains associated with CD3z, CD28 and 4-1BB being the most common. In Tregs, BAR receptors replace the scFv portion for a target protein recognized by the BCR, enabling interaction between Tregs and B cells and inhibiting antibody production. Incorporating “armour proteins” into CAR-T cells improve their function and provides precise control over their activity, enhancing the safety and efficacy of CAR-T therapies in various diseases. These strategies can also be applied to CAR-Tregs to enhance their suppressive capacity and survival. SynNotch CAR strategy employs a positive feedback receptor to regulate CAR-T cell activity, releasing a transcription factor that induces the expression of a CAR targeting a second antigen following recognition of the initial antigen. Bispecific TanCAR cells can recognize two different antigens using distinct scFv portions, but only one of them is required for cell activation, thereby avoiding antigen escape. UCARs enable the binding to target cells through independent and antigen-specific switch modules, allowing for adaptability to different tissue antigens by exchanging adapters. Finally, Foxp3-CAR allows for an increase and stable expression of Foxp3 in Tregs, leading to significant improvements in the safety and efficacy of CAR-Treg products. BAR, B cell-targeting Ab receptors; BCR, B cell receptor; CAR, Chimeric antigen receptor; Foxp3, Forkhead box 3; scFv, Single-chain variable fragment; SynNotch CAR, Synthetic Notch CAR; TanCAR, Tandem CAR; Treg, Regulatory T cells; UCAR, Universal CAR. Created with BioRender.com.
Tandem CAR (TanCAR)
One strategy to expand the diversity of recognized antigens involves the use of bispecific TanCARs, capable of targeting two different antigens (Figure 2). Grada et al. developed this novel receptor and demonstrated that TanCARs retain the cytolytic capacity of T cells, even when one of the target molecules is lost. This approach also results in a more controlled response against tumors by recognizing both targets in an animal disease model (Grada et al., 2013). Other studies have adopted a similar strategy, yielding comparable results (Jensen and Riddell, 2014; Zah et al., 2016; Jia et al., 2019). However, this approach has not yet been applied to Tregs.
Universal CAR (UCAR)
The application of CAR-Tregs and CAR-T cells is limited by wide range of target antigens dependent on the patient’s condition and by the currently limited catalog of available CAR receptors. To address this challenge, universal CAR platforms (UCAR) were developed (Figure 2). In summary, T cells are engineered with a signaling module that binds to a specific epitope on a switching molecule. The switching module includes an antigen-binding domain and a switching epitope that is recognized by the signaling module (Figure 2). Crosslinking with target cells is thus mediated by these independent antigen-specific switching modules that contain the peptide epitope recognized by the UCAR. These adapters can be easily exchanged to target different tissue antigens, Many UCARs have been engineered and tested in conventional T cells and are reviewed in (Liu et al., 2019). To the best of our knowledge, two groups have tested them in Tregs. Meyer et al. tested a CAR that binds antibodies conjugated in the Fc region with FITC. They showed that these CAR-Tregs could prolong mice pancreatic islet allografts and promote tolerance to secondary skin grafts (Batista et al., 2017). Bachmann et al. tested a UCAR that recognizes a sequence of 10 amino acids (5B9 tag) of the nuclear protein LA/SS-B (Koristka et al., 2014; Koristka et al., 2018; Kegler et al., 2019), and the transduction procedure itself did not affect the phenotype and activity of the Tregs (Koristka et al., 2014; Koristka et al., 2018). The advantages of UCARs are their high versatility and easy reprogrammability. They could also be safer, as they do not have an effect without the infusion of the switching module. However, as they rely on the infusion of exogenous sequences or epitopes, they require more regulatory examination for safety and efficacy.
B cell-targeting Ab receptors (BAR)
The BAR strategy involves replacing the scFv of the CAR with the antigen itself to regulate B-cells and plasma cells that are antigen-specific (Figure 2). It was initially used by Ellebrecht et al., in cytotoxic T cells, to destroy autoantigen-specific hybridoma in a pemphigus vulgaris model (Ellebrecht et al., 2016). This concept was further tested using immunodominant FVIII domains (A2 and C2) to destroy anti-FVIII hybridomas, which are problematic in hemophilia patients (Baaten et al., 2018). The FVIII A2 and C2 domains of BAR-CD8 T cells were assessed in vitro (human) and in vivo (mouse). The same team then tested their FVIII BAR in Tregs and demonstrated their ability to suppress the memory antibody response in spleen cultures from FVIII-immunized mice in vitro and completely prevented the development of anti-FVIII antibodies following FVIII immunization (Zhang et al., 2018). The use of BAR-Tregs also demonstrated promising potential in providing clinical protection against severe allergic reactions. In their study, Abdeladhim et al. developed a BAR using ovalbumin (OVA) as the targeted protein (Abdeladhim et al., 2019) and observed a reduction in hypothermia (an indicator of anaphylaxis) following OVA exposure. However, in the in vivo setting, no discernible reduction in the circulating levels of OVA-specific IgE was detected (Abdeladhim et al., 2019). Nonetheless, these results have opened the door to the use of BAR-T cells and BAR-Tregs in autoimmune diseases and transplantation.
SynNotch CAR
To address the issue of hyperactivity associated with CAR-T cells and the subsequent toxicity concerns, researchers have developed SynNotch receptors incorporating a two-step positive feedback circuit (Figure 2). This circuit comprises a priming signaling receptor devoid of activating-signaling capacity. Upon antigen recognition, these receptors trigger a trans-membrane cleavage, akin to a notch receptor, releasing a transcription factor. This transcription factor then initiates the expression of a CAR targeting another antigen (Figure 2). Both antigens must be present to activate the cell. This technology has been extensively tested in various cancer models using conventional T cells (reviewed in (Stock et al., 2023)) and it holds potential to enhance the effectiveness and safety of CAR-T cell therapies. Thus far it has not been explored in Tregs, but it could prove valuable for targeting combinations of antigens when one or both are widely distributed throughout the body.
Enhancing activation and function
Optimization of the CAR construct
Typically, CARs consist of four domains: an extracellular antigen-binding domain responsible for recognizing the target antigen, a spacer or hinge region that provides the necessary receptor length, a transmembrane domain anchoring the receptor to the plasma membrane, and an intracellular signaling domain that transmits both the first and second co-stimulatory signals (Bridgeman et al., 2010) (Figure 2). Interestingly, different intracellular co-stimulatory domains in second-generation CAR-T cells exert distinct effects on cell differentiation, metabolism, and function. For Tregs, the CD28 co-stimulation domain appears to be the most effective in maintaining Treg function. Initially, Maus et al. compared the CD28 co-stimulation domain to 4-1BB, and only CD28 CAR-Tregs demonstrated efficacy in vivo (Boroughs et al., 2019). Subsequently, Dawson et al. designed a total of 10 anti-HLA-A2 CAR receptors differing in their co-receptor signaling domains. In an xGvHD model, CARs encoding TNFR family co-receptors (OX40, GITR, 4-1BB, and TNFR2) were ineffective in promoting CAR-Tregs function, while CD28-CAR-Tregs exhibited highest effectiveness. PD-1-CAR Tregs displayed ineffective activation, and TNFR2 CARs resulted in a marked increase in TSDR methylation (Dawson et al., 2020). These results were further corroborated by Zuber et al., confirming the negative impact of 4-1BB signaling in Tregs (Lamarthée et al., 2021). These findings offer valuable insights for the future design and optimization of CAR-Treg therapies.
Armour proteins
The genetic engineering of CAR-T cells has paved the way for a wide range of innovative approaches aimed at enhancing their efficacy and anti-tumoral function. One particularly intriguing strategy involves the incorporation of “armour” proteins through modifications in CAR genes. These proteins, whether secreted or expressed on the cell surface, possess the potential to augment T cell activity and positively influence the tumor microenvironment (Figure 2). By finely tuning each component of CAR genetic constructs, precise control can be exerted over the functionality, capacity, and safety of the resulting CAR-T cells.
An example of such advancement can be found in Cherkassky et al.‘s study, which developed anti-mesothelin CAR-T cells transduced with a truncated dominant-negative PD-1 receptor. This receptor, devoid of intracellular signaling domains, binds to PD-L1 but fails to transmit inhibitory signals. Consequently, it enables the CAR-T cells to resist PD-L1-induced exhaustion, thereby prolonging survival in xenograft models of pleural mesotheliomas (Cherkassky et al., 2016). Another notable study by Yeku et al. focused on the design of CAR-T cells capable of constitutive secretion of IL-12. In a model of ovarian peritoneal carcinomatosis, these CAR-T cells exhibited enhanced expansion and greater anti-tumoral efficacy (Yeku et al., 2017).
In the field of adoptive immunotherapy, the use of armour proteins holds great potential for enhancing the suppressive and survival capabilities of CAR-Tregs. Employing secreted armour proteins such as IL-2 could potentially improve proliferation and survival, while TGF-β or IL-10 (Mohseni et al., 2021) could foster a more immunosuppressive environment. However, it should be noted that IL-10 overexpression alone may not suffice to maintain tolerance if cells lose other immunosuppressive abilities (Rana et al., 2021). The co-expression of membrane molecules like Pd-1 and Ctla-4 with the CAR could enhance contact-mediated inhibitory capacities. Furthermore, the expression of transgenic receptors can also be combined with small-molecule therapeutics to promote efficacy and/or survival. For example, the incorporation of an orthogonal IL-2 receptor, selectively responsive to orthogonal IL-2 injection yet retaining retains native IL-2 signalling through STAT5, can facilitate the selective expansion of the transduced Tregs (Sasaki et al., 2014; Hirai et al., 2021). Given these significant advancements, CAR-Tregs designed with armour proteins emerge as a promising immunotherapy modality applicable to various disorders.
Fine-tuning CAR expression
In most studies, CARs have been introduced into Tregs through lentiviral or retroviral transduction. This method, however, leads to continuous expression of the endogenous TCR and poses the risk of random integration of the CAR construct into the genome, resulting in varying levels of CAR expression, transcriptional silencing, or unintended disruption of crucial genes. Strong expression can also lead to clustering of the CARs, leading to ligand-independent tonic signaling, and consequently, to CAR-T cell (Long et al., 2015) and Treg exhaustion (Lamarche et al., 2023).
To circumvent these consequences, Eyquem et al. devised a precise integration system employing CRISPR-Cas9 to introduce an anti-CD19 CAR at the constant locus of the T cell receptor alpha (TRAC) in effector T cells. Their strategy involved designing a gRNA targeting the 5′end of the first exon of TRAC and employing an adeno-associated virus (AAV)-based repair vector encoding a self-cleaving P2A peptide followed by the CAR-CD19 cDNA. The resulting CAR-T cells not only demonstrated uniform CAR-CD19 expression, but also exhibited a reduction in tonic signaling, exhaustion and an increase in antitumour efficacy (Eyquem et al., 2017). TRAC-CAR-T cells also surpassed conventionally edited CAR-T cells in an acute lymphoblastic leukemia murine model. Additionally, the insertion at the TRAC locus prevented tonic signaling and facilitated effective CAR internalization and subsequent re-expression following single or repeated antigen exposure, effectively delaying effector T cell differentiation and exhaustion (Eyquem et al., 2017).
Building upon this approach, Muller et al. generated human anti-HLA-A2 CAR-Tregs into the TRAC locus after TCR expression elimination. They employed CRISPR-Cas9 to eliminate TCR expression and achieved the introduction of the anti-HLA-A2 CAR construct through direct integration into the TRAC locus using homology-directed repair (HDR). The resulting cells demonstrated uniform CAR expression, retained their phenotype (Foxp3+, Helios+), and exhibited A2-specific suppressive functionality in vitro and in vivo. These cells demonstrated a delay in GvHD development, exclusively in the presence of HLA-A2, whether expressed by simultaneously injected peripheral blood mononuclear cells or the recipient mice themselves. These outcomes effectively illustrated that these genetically engineered CAR-Tregs display antigen-dependent in vivo suppression, independent of TCR expression (Muller et al., 2021).
Another approach is to create an alternative TCR fusion construct (TRuC), which fuses the scFv to the N terminus of the TCRε subunit (Baeuerle et al., 2019; Ding et al., 2023; Lesch et al., 2023). This approach can limit the CAR density on the cell surface and better replicates physiological TCR signaling. It has been applied to Tregs and has demonstrated great immunosuppressive potential (Rana et al., 2021).
Increasing Treg homing
Shaping Tregs for better homing abilities to specific inflammation sites has been a topic of interest since the early 2000s. Studies investigating homing patterns of Tregs using UV irradiation showed migration to lymph nodes upon injection (Wilk et al., 2022). The induction of conventional T cells could then be reduced as T-cell priming occurs within the lymph nodes. To efficiently suppress inflammation in the periphery, local injection of Tregs could circumvent this limitation for specific external area-targeted immunotherapy (Schwarz et al., 2004). The alternative is to use integrin or chemokine expression to facilitate specific homing (reviewed in (Lamarche and Levings, 2018)). Their potential in homing-target specific-CARs have yet to be fully established, but preclinical studies on CD4 T cells have lead the way for some of our current models (Table 2).
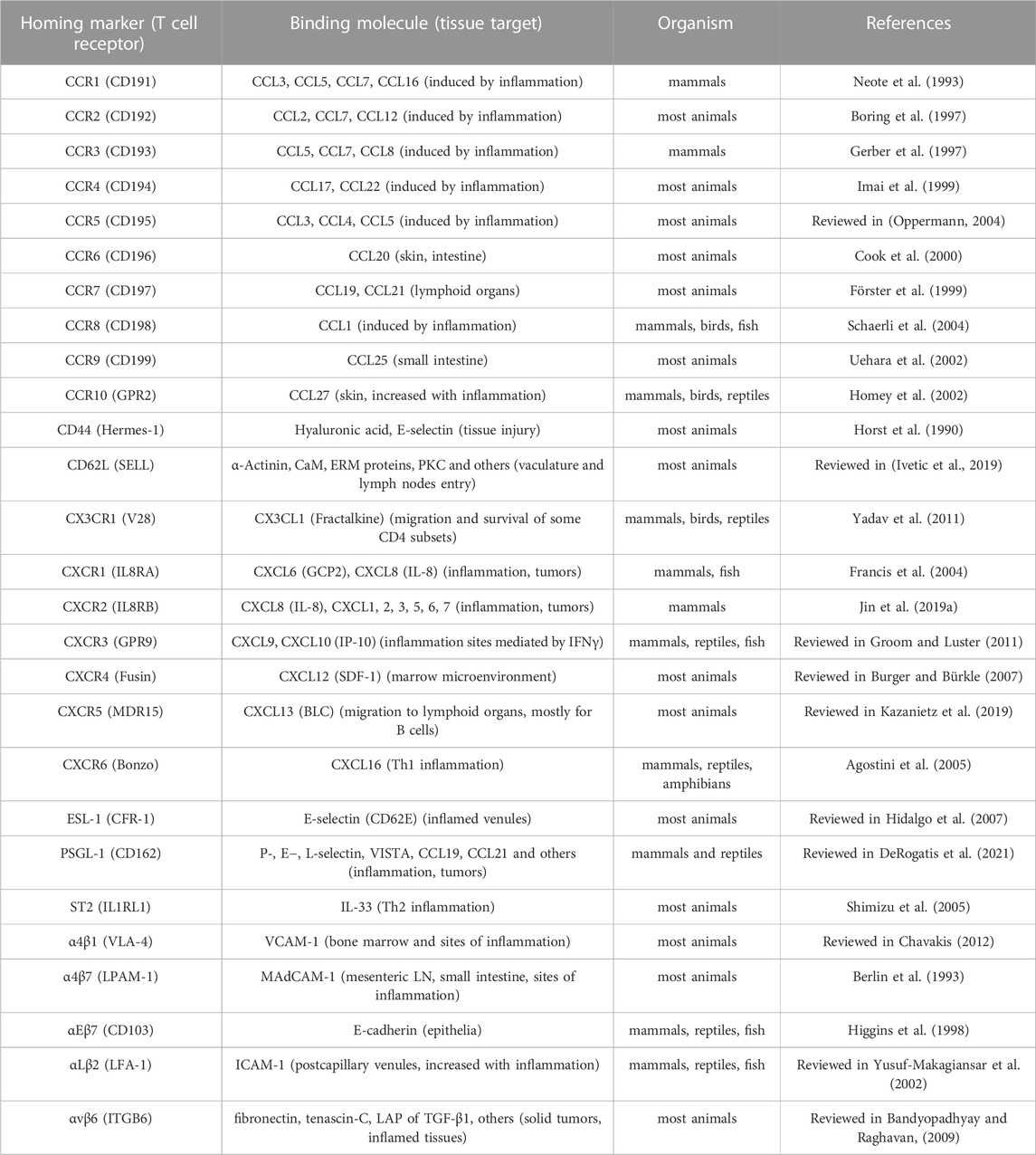
TABLE 2. Homing markers on CD4 T cells for increased target-specificity.
It should also be considered that antigen-specific Tregs inherently exhibit more efficient homing abilities than expanded Tregs generated from polyclonal pools (Lam et al., 2017). The homing abilities of CAR-Tregs surpass those of TCR-engineered Tregs because they do not rely on MHC-presentation and are less dependent on IL-2 (as reviewed in (Campbell, 2015)). Adding supplemental factors to these engineering and expansion protocols for CAR-Tregs, such as RA, transduced KLF2, or other Th-mediated response elements, may lead to a newer generation of Tregs that are even more tailored and efficient in their immunosuppressive capabilities and specifically suited for diverse contexts.
Studies in conventional CAR-T cell therapy have demonstrated that chemokine-specific CARs can more efficiently inhibit the growth of solid and immunosuppressive cancers. Human solid tumors xenograft models have been evaluated for chemokines such as CXCL1-2. The intra-tumoral presence of CAR-T cells increased when modified for CXCR1-2 surface expression (Jin et al., 2019b). Transduction of CXCR2 in CAR-T cells targeting αvβ6, an integrin strongly expressed in pancreatic cancer, controlled tumor growth in a xenograft mouse model (Whilding et al., 2019). Known mechanisms of Treg homing in breast cancer have also been studied such as the CCL1-CCR8 axis. To improve trafficking and resistance to immunosuppression, CCR8-modified CAR-T cells were transduced for a TGFβ-insensitive receptor (DNRII) and showed consistent tumor regression compared to CCR8-only expressing CAR-T cells (Cadilha et al., 2021).
Such CAR modifications in Tregs could lead to inflammation-specific immunosuppression. Expression of α4β7-integrin, CCR9 and CCR6 in Foxp3+ Tregs enhances recruitment to sites of intestinal inflammation (Elias et al., 2008; Menning et al., 2010). CXCR3 in Tregs, such as found in Tbet+Foxp3+ T helper (Th)1-Tregs, improves migration towards CXCL9, 10 and 11 (Hoeppli et al., 2019). The transduction of these chemokine receptors in CAR-Tregs requires further study to evaluate their efficiency. For a more Th2-like mediated response in humoral dysregulations, IL-33 receptor (ST2) and CCR4, which have been shown to be involved in tissue-resident Treg maintenance and better graft retention, could also be considered for chemokine-modified CAR-Tregs (Vasanthakumar et al., 2015). The expression of CCR2, CCR5 and CCR7 expression on CARs Tregs may reduce graft rejection due to better migration to lymph nodes (Zhang et al., 2009). The stability of these newly engineered CAR-Tregs under in vitro and in vivo inflammatory expansion conditions requires further investigation. However, it is worth noting that natural Tregs have demonstrated the ability to maintain their suppressive phenotype (Dijke et al., 2016).
Safety
CAR-Tregs are likely to be safer than conventional CAR-T therapies because they should not cause problems such as tumor lysis syndrome, hematotoxicity and cytokine release syndrome. Theoretically, there is a risk of off-target effects resulting from their immunosuppressive effects, such as an increased risk of infection or cancer. However, all phase I/II clinical studies using Treg adoptive immunotherapy have demonstrated the safety of this therapy, with no evidence of increased risk of infection or cancer (Santegoets et al., 2015; Abhishek et al., 2023). Another potential concern would be the contamination of Treg products with conventional T cells, which could exacerbate autoimmune or alloimmune diseases. In the event of an infection or cancer, elimination of Tregs with immunosuppressive drugs could worsen the condition. Alternatively, a reversible or permanent “OFF” switch could be used to eliminate CAR-Tregs without disturbing the immune system. As the first clinical trials of CAR-Treg therapy in humans are currently underway, their safety has yet to be demonstrated (Table 3).
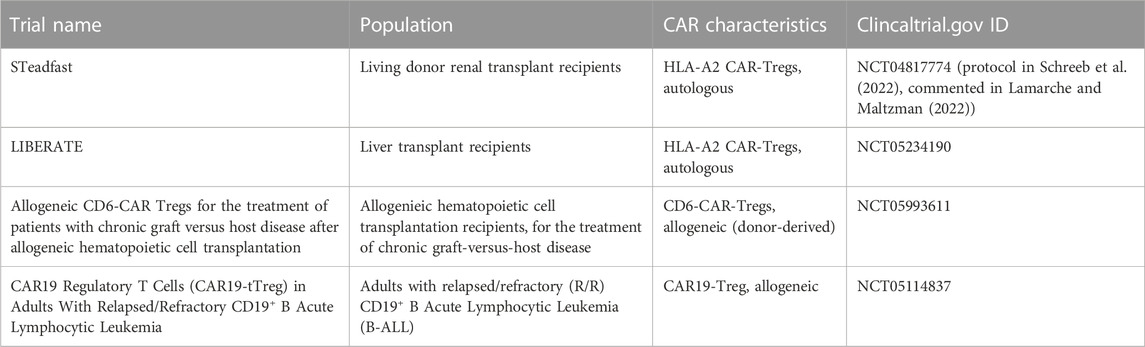
TABLE 3. Registered clinical trials using CAR-Treg adoptive immunotherapy.
One approach that has been developed for conventional CAR-T cell therapy involves the use of kinase inhibitors, such as dasatinib, to induce a functional “OFF” state (reviewed in (Stock et al., 2023)). However, this approach has not been studied in Tregs, and the differential impact on Tregs compared to conventional T cells has not been assessed. This approach could also be detrimental if it leads to immune hyporesponsiveness due to increased off-target effects arose.
Another strategy involves the use of enzyme inhibitors, which can reversibly lead to CAR degradation. This requires the addition of a bromodomain or protease domain on the CAR, which can be modulated using a proteolysis-targeting chimera compound or a protease inhibitor (reviewed in (Stock et al., 2023)). None of these strategies have been studied in CAR-Tregs thus far.
Permanent elimination of CAR-T cells can be achieved by integrating suicide genes into these cells. For example, metabolic suicide gene systems can be employed with a non-toxic prodrug therapy administered systematically. Genetically-modified cells possess the gene necessary to convert the prodrug into a toxic compound (reviewed in (Stock et al., 2023)). Alternatively, a chemical inducer of dimerization could also be administered to induce dimerization and activation of components of the apoptotic pathway (reviewed in (Stock et al., 2023)). Cells can also be engineered to co-express a target marker alongside the CAR. These options include CD52, CD20, the truncated epidermal growth factor receptor (EGFRt), or an EGFR-FOLR1 fusion receptor. However, the impact of administering anti-CD52, anti-CD20 or anti-EGFR antibodies to already immunocompromised patients must be carefully considered. In the case of an infection or cancer, these drugs might even have detrimental effects.
Conclusion
The development of advanced generations of CARs, such as TanCARs, UCARs, SynNotch CARs, or BARs, holds great promise for CAR-Treg therapy by overcoming challenges related to immunogenicity and production limitations. Furthermore, the incorporation of armour proteins and safety mechanisms like suicide genes, as well as enhancements of homing abilities through chemokine receptors, have the potential to revolutionize the targeting precision and stability of CAR-Tregs. The use of these innovative constructs brings us closer to achieving precise, safe, and efficient treatments for autoimmune and alloimmune diseases. However, further progress requires a rigorous evaluation of these novel CAR-Tregs in preclinical models prior to their introduction into clinical trials.
Author contributions
CL: Supervision, Writing–review and editing. CR: Conceptualization, Writing–original draft. NV: Writing–original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and of this article. The authors’ own work in this area was supported by the Cole Foundation and the KRESCENT program of the Kidney Foundation of Canada. CL is supported by a junior 1 Salary Award from the Fonds de Recherche en Santé du Québec—Santé (FRQS).
Conflict of interest
CL is an inventor on pending patents related to HLA-A2-specific CARs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdeladhim, M., Zhang, A. H., Kropp, L. E., Lindrose, A. R., Venkatesha, S. H., Mitre, E., et al. (2019). Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin. Immunol. 207, 49–54. doi:10.1016/j.clim.2019.07.009
Abhishek, K., Nidhi, M., Chandran, S., Shevkoplyas, S. S., and Mohan, C. (2023). Manufacturing regulatory T cells for adoptive cell therapy in immune diseases: A critical appraisal. Clin. Immunol. 251, 109328. doi:10.1016/j.clim.2023.109328
Agostini, C., Cabrelle, A., Calabrese, F., Bortoli, M., Scquizzato, E., Carraro, S., et al. (2005). Role for CXCR6 and its ligand CXCL16 in the pathogenesis of T-cell alveolitis in sarcoidosis. Am. J. Respir. Crit. Care Med. 172 (10), 1290–1298. doi:10.1164/rccm.200501-142OC
Allan, S. E., Alstad, A. N., Merindol, N., Crellin, N. K., Amendola, M., Bacchetta, R., et al. (2008). Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 16 (1), 194–202. doi:10.1038/sj.mt.6300341
Baaten, C., Swieringa, F., Misztal, T., Mastenbroek, T. G., Feijge, M. A. H., Bock, P. E., et al. (2018). Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects. Blood Adv. 2 (18), 2320–2331. doi:10.1182/bloodadvances.2017011544
Baeuerle, P. A., Ding, J., Patel, E., Thorausch, N., Horton, H., Gierut, J., et al. (2019). Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 10 (1), 2087. doi:10.1038/s41467-019-10097-0
Bandyopadhyay, A., and Raghavan, S. (2009). Defining the role of integrin alphavbeta6 in cancer. Curr. Drug Targets 10 (7), 645–652. doi:10.2174/138945009788680374
Batista, E. P. A., Ngowo, H. S., Opiyo, M., Shubis, G. K., Meza, F. C., Okumu, F. O., et al. (2017). Semi-field assessment of the BG-Malaria trap for monitoring the African malaria vector, Anopheles arabiensis. PLoS One 12 (10), e0186696. doi:10.1371/journal.pone.0186696
Bell, C. J., Sun, Y., Nowak, U. M., Clark, J., Howlett, S., Pekalski, M. L., et al. (2015). Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J. Autoimmun. 56, 66–80. doi:10.1016/j.jaut.2014.10.002
Beres, A., Komorowski, R., Mihara, M., and Drobyski, W. R. (2011). Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin. Cancer Res. 17 (12), 3969–3983. doi:10.1158/1078-0432.Ccr-10-3347
Berlin, C., Berg, E. L., Briskin, M. J., Andrew, D. P., Kilshaw, P. J., Holzmann, B., et al. (1993). Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74 (1), 185–195. doi:10.1016/0092-8674(93)90305-A
Blat, D., Zigmond, E., Alteber, Z., Waks, T., and Eshhar, Z. (2014). Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 22 (5), 1018–1028. doi:10.1038/mt.2014.41
Bluestone, J. A., Buckner, J. H., Fitch, M., Gitelman, S. E., Gupta, S., Hellerstein, M. K., et al. (2015). Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 7 (315), 315ra189. doi:10.1126/scitranslmed.aad4134
Boardman, D. A., Philippeos, C., Fruhwirth, G. O., Ibrahim, M. A., Hannen, R. F., Cooper, D., et al. (2017). Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am. J. Transpl. 17 (4), 931–943. doi:10.1111/ajt.14185
Boardman, D. A., Wong, M. Q., Rees, W. D., Wu, D., Himmel, M. E., Orban, P. C., et al. (2023). Flagellin-specific human CAR Tregs for immune regulation in IBD. J. Autoimmun. 134, 102961. doi:10.1016/j.jaut.2022.102961
Boring, L., Gosling, J., Chensue, S. W., Kunkel, S. L., Farese, R. V., Broxmeyer, H. E., et al. (1997). Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Investigation 100 (10), 2552–2561. doi:10.1172/JCI119798
Boroughs, A. C., Larson, R. C., Choi, B. D., Bouffard, A. A., Riley, L. S., Schiferle, E., et al. (2019). Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight 5 (8), e126194. doi:10.1172/jci.insight.126194
Boyman, O., Kovar, M., Rubinstein, M. P., Surh, C. D., and Sprent, J. (2006). Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311 (5769), 1924–1927. doi:10.1126/science.1122927
Bridgeman, J. S., Hawkins, R. E., Hombach, A. A., Abken, H., and Gilham, D. E. (2010). Building better chimeric antigen receptors for adoptive T cell therapy. Curr. Gene Ther. 10 (2), 77–90. doi:10.2174/156652310791111001
Brunstein, C. G., Miller, J. S., Cao, Q., McKenna, D. H., Hippen, K. L., Curtsinger, J., et al. (2011). Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117 (3), 1061–1070. doi:10.1182/blood-2010-07-293795
Burger, J. A., and Bürkle, A. (2007). The CXCR4 chemokine receptor in acute and chronic leukaemia: A marrow homing receptor and potential therapeutic target. Br. J. Haematol. 137 (4), 288–296. doi:10.1111/j.1365-2141.2007.06590.x
Cadilha, B. L., Benmebarek, M. R., Dorman, K., Oner, A., Lorenzini, T., Obeck, H., et al. (2021). Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 7 (24), eabi5781. doi:10.1126/sciadv.abi5781
Campbell, D. J. (2015). Control of regulatory T cell migration, function, and homeostasis. J. Immunol. 195 (6), 2507–2513. doi:10.4049/jimmunol.1500801
Chavakis, T. (2012). Leucocyte recruitment in inflammation and novel endogenous negative regulators thereof. Eur. J. Clin. Investigation 42 (6), 686–691. doi:10.1111/j.1365-2362.2012.02677.x
Chen, W., Jin, W., Hardegen, N., Lei, K. J., Li, L., Marinos, N., et al. (2003). Conversion of peripheral CD4+CD25-naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198 (12), 1875–1886. doi:10.1084/jem.20030152
Cherkassky, L., Morello, A., Villena-Vargas, J., Feng, Y., Dimitrov, D. S., Jones, D. R., et al. (2016). Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 126 (8), 3130–3144. doi:10.1172/jci83092
Cook, D. N., Prosser, D. M., Forster, R., Zhang, J., Kuklin, N. A., Abbondanzo, S. J., et al. (2000). CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity 12 (5), 495–503. doi:10.1016/S1074-7613(00)80201-0
Dahiya, S., Beier, U. H., Wang, L., Han, R., Jiao, J., Akimova, T., et al. (2020). HDAC10 deletion promotes Foxp3(+) T-regulatory cell function. Sci. Rep. 10 (1), 424. doi:10.1038/s41598-019-57294-x
Dawson, N. A. J., Rosado-Sánchez, I., Novakovsky, G. E., Fung, V. C. W., Huang, Q., McIver, E., et al. (2020). Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci. Transl. Med. 12 (557), eaaz3866. doi:10.1126/scitranslmed.aaz3866
Dawson, N. A., Lamarche, C., Hoeppli, R. E., Bergqvist, P., Fung, V. C., McIver, E., et al. (2019). Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight 4 (6), e123672. doi:10.1172/jci.insight.123672
de Zoeten, E. F., Wang, L., Butler, K., Beier, U. H., Akimova, T., Sai, H., et al. (2011). Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell Biol. 31 (10), 2066–2078. doi:10.1128/mcb.05155-11
Demirkiran, A., Hendrikx, T. K., Baan, C. C., and van der Laan, L. J. (2008). Impact of immunosuppressive drugs on CD4+CD25+FOXP3+ regulatory T cells: does in vitro evidence translate to the clinical setting? Transplantation 85 (6), 783–789. doi:10.1097/TP.0b013e318166910b
Demirkiran, A., Kok, A., Kwekkeboom, J., Kusters, J. G., Metselaar, H. J., Tilanus, H. W., et al. (2006). Low circulating regulatory T-cell levels after acute rejection in liver transplantation. Liver Transpl. 12 (2), 277–284. doi:10.1002/lt.20612
DeRogatis, J. M., Viramontes, K. M., Neubert, E. N., and Tinoco, R. (2021). PSGL-1 immune checkpoint inhibition for CD4+ T cell cancer immunotherapy. Front. Immunol. 12, 636238. doi:10.3389/fimmu.2021.636238
Dijke, I. E., Hoeppli, R. E., Ellis, T., Pearcey, J., Huang, Q., McMurchy, A. N., et al. (2016). Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am. J. Transpl. 16 (1), 58–71. doi:10.1111/ajt.13456
Ding, J., Guyette, S., Schrand, B., Geirut, J., Horton, H., Guo, G., et al. (2023). Mesothelin-targeting T cells bearing a novel T cell receptor fusion construct (TRuC) exhibit potent antitumor efficacy against solid tumors. Oncoimmunology 12 (1), 2182058. doi:10.1080/2162402x.2023.2182058
Downey, C. (2016). Serious infection during etanercept, infliximab and adalimumab therapy for rheumatoid arthritis: A literature review. Int. J. Rheum. Dis. 19 (6), 536–550. doi:10.1111/1756-185x.12659
Elias, K. M., Laurence, A., Davidson, T. S., Stephens, G., Kanno, Y., Shevach, E. M., et al. (2008). Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111 (3), 1013–1020. doi:10.1182/blood-2007-06-096438
Elinav, E., Adam, N., Waks, T., and Eshhar, Z. (2009). Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 136 (5), 1721–1731. doi:10.1053/j.gastro.2009.01.049
Elinav, E., Waks, T., and Eshhar, Z. (2008). Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology 134 (7), 2014–2024. doi:10.1053/j.gastro.2008.02.060
Ellebrecht, C. T., Bhoj, V. G., Nace, A., Choi, E. J., Mao, X., Cho, M. J., et al. (2016). Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353 (6295), 179–184. doi:10.1126/science.aaf6756
Eyquem, J., Mansilla-Soto, J., Giavridis, T., van der Stegen, S. J., Hamieh, M., Cunanan, K. M., et al. (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543 (7643), 113–117. doi:10.1038/nature21405
Feng, Y., Arvey, A., Chinen, T., van der Veeken, J., Gasteiger, G., and Rudensky, A. Y. (2014). Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 158 (4), 749–763. doi:10.1016/j.cell.2014.07.031
Filipovich, A. H., Weisdorf, D., Pavletic, S., Socie, G., Wingard, J. R., Lee, S. J., et al. (2005). National Institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol. Blood Marrow Transpl. 11 (12), 945–956. doi:10.1016/j.bbmt.2005.09.004
Floess, S., Freyer, J., Siewert, C., Baron, U., Olek, S., Polansky, J., et al. (2007). Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5 (2), e38. doi:10.1371/journal.pbio.0050038
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4 (4), 330–336. doi:10.1038/ni904
Förster, R., Schubel, A., Breitfeld, D., Kremmer, E., Renner-Müller, I., Wolf, E., et al. (1999). CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99 (1), 23–33. doi:10.1016/S0092-8674(00)80059-8
Francis, J. N., Jacobson, M. R., Lloyd, C. M., Sabroe, I., Durham, S. R., and Till, S. J. (2004). CXCR1+CD4+ T cells in human allergic disease. J. Immunol. 172 (1), 268–273. doi:10.4049/jimmunol.172.1.268
Fransson, M., Piras, E., Burman, J., Nilsson, B., Essand, M., Lu, B., et al. (2012). CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 9, 112. doi:10.1186/1742-2094-9-112
Fu, R. Y., Chen, A. C., Lyle, M. J., Chen, C. Y., Liu, C. L., and Miao, C. H. (2020). CD4(+) T cells engineered with FVIII-CAR and murine Foxp3 suppress anti-factor VIII immune responses in hemophilia a mice. Cell Immunol. 358, 104216. doi:10.1016/j.cellimm.2020.104216
Gerber, B. O., Zanni, M. P., Uguccioni, M., Loetscher, M., Mackay, C. R., Pichler, W. J., et al. (1997). Functional expression of the eotaxin receptor CCR3 in T lymphocytes co-localizing with eosinophils. Curr. Biol. 7 (11), 836–843. doi:10.1016/S0960-9822(06)00371-X
Grada, Z., Hegde, M., Byrd, T., Shaffer, D. R., Ghazi, A., Brawley, V. S., et al. (2013). TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2 (7), e105. doi:10.1038/mtna.2013.32
Groom, J. R., and Luster, A. D. (2011). CXCR3 in T cell function. Exp. Cell Res. 317 (5), 620–631. doi:10.1016/j.yexcr.2010.12.017
Homey, B., Alenius, H., Müller, A., Soto, H., Bowman, E. P., Yuan, W., et al. (2002). CCL27–CCR10 interactions regulate T cell–mediated skin inflammation. Nat. Med. 8 (2), 157–165. doi:10.1038/nm0202-157
Hegde, M., Corder, A., Chow, K. K., Mukherjee, M., Ashoori, A., Kew, Y., et al. (2013). Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 21 (11), 2087–2101. doi:10.1038/mt.2013.185
Henschel, P., Landwehr-Kenzel, S., Engels, N., Schienke, A., Kremer, J., Riet, T., et al. (2023). Supraphysiological FOXP3 expression in human CAR-Tregs results in improved stability, efficacy, and safety of CAR-Treg products for clinical application. J. Autoimmun. 138, 103057. doi:10.1016/j.jaut.2023.103057
Hidalgo, A., Peired, A. J., Wild, M. K., Vestweber, D., and Frenette, P. S. (2007). Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 26 (4), 477–489. doi:10.1016/j.immuni.2007.03.011
Higgins, J. M. G., Mandlebrot, D. A., Shaw, S. K., Russell, G. J., Murphy, E. A., Chen, Y.-T., et al. (1998). Direct and regulated interaction of integrin alphaEbeta7 with E-cadherin. J. Cell Biol. 140 (1), 197–210. doi:10.1083/jcb.140.1.197
Hirai, T., Ramos, T. L., Lin, P. Y., Simonetta, F., Su, L. L., Picton, L. K., et al. (2021). Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J. Clin. Invest. 131 (8), e139991. doi:10.1172/jci139991
Hoeppli, R. E., MacDonald, K. N., Leclair, P., Fung, V. C. W., Mojibian, M., Gillies, J., et al. (2019). Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am. J. Transpl. 19 (1), 62–76. doi:10.1111/ajt.14936
Hoffmann, P., Eder, R., Boeld, T. J., Doser, K., Piseshka, B., Andreesen, R., et al. (2006). Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood 108 (13), 4260–4267. doi:10.1182/blood-2006-06-027409
Honaker, Y., Hubbard, N., Xiang, Y., Fisher, L., Hagin, D., Sommer, K., et al. (2020). Gene editing to induce FOXP3 expression in human CD4(+) T cells leads to a stable regulatory phenotype and function. Sci. Transl. Med. 12 (546), eaay6422. doi:10.1126/scitranslmed.aay6422
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299 (5609), 1057–1061. doi:10.1126/science.1079490
Horst, E., Meijer, C. J., Radaskiewicz, T., van Dongen, J. J., Pieters, R., Figdor, C. G., et al. (1990). Expression of a human homing receptor (CD44) in lymphoid malignancies and related stages of lymphoid development. Leukemia 4 (5), 383–389.
Hou, T. Z., Qureshi, O. S., and Sansom, D. M. (2019). Measuring CTLA-4-dependent suppressive function in regulatory T cells. Methods Mol. Biol. 1899, 87–101. doi:10.1007/978-1-4939-8938-6_7
Imai, T., Nagira, M., Takagi, S., Kakizaki, M., Nishimura, M., Wang, J., et al. (1999). Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 11 (1), 81–88. doi:10.1093/intimm/11.1.81
Imura, Y., Ando, M., Kondo, T., Ito, M., and Yoshimura, A. (2020). CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight 5 (14), e136185. doi:10.1172/jci.insight.136185
Ivetic, A., Hoskins Green, H. L., and Hart, S. J. (2019). L-Selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 10, 1068. doi:10.3389/fimmu.2019.01068
Jacobson, D. L., Gange, S. J., Rose, N. R., and Graham, N. M. (1997). Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 84 (3), 223–243. doi:10.1006/clin.1997.4412
Jagasia, M., Arora, M., Flowers, M. E., Chao, N. J., McCarthy, P. L., Cutler, C. S., et al. (2012). Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood 119 (1), 296–307. doi:10.1182/blood-2011-06-364265
Jensen, M. C., and Riddell, S. R. (2014). Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol. Rev. 257 (1), 127–144. doi:10.1111/imr.12139
Jia, H., Wang, Z., Wang, Y., Liu, Y., Dai, H., Tong, C., et al. (2019). Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 12 (1), 57. doi:10.1186/s13045-019-0741-6
Jin, L., Tao, H., Karachi, A., Long, Y., Hou, A. Y., Na, M., et al. (2019a). CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 10 (1), 4016. doi:10.1038/s41467-019-11869-4
Jin, L., Tao, H., Karachi, A., Long, Y., Hou, A. Y., Na, M., et al. (2019b). CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 10 (1), 4016. doi:10.1038/s41467-019-11869-4
Kalin, J. H., Butler, K. V., Akimova, T., Hancock, W. W., and Kozikowski, A. P. (2012). Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J. Med. Chem. 55 (2), 639–651. doi:10.1021/jm200773h
Karim, M., Feng, G., Wood, K. J., and Bushell, A. R. (2005). CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood 105 (12), 4871–4877. doi:10.1182/blood-2004-10-3888
Kassan, M., Galan, M., Partyka, M., Trebak, M., and Matrougui, K. (2011). Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 31 (11), 2534–2542. doi:10.1161/atvbaha.111.233262
Kazanietz, M. G., Durando, M., and Cooke, M. (2019). CXCL13 and its receptor CXCR5 in cancer: inflammation, immune response, and beyond. Front. Endocrinol. 10, 471. doi:10.3389/fendo.2019.00471
Kegler, A., Koristka, S., Bergmann, R., Berndt, N., Arndt, C., Feldmann, A., et al. (2019). T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology 8 (9), e1621676. doi:10.1080/2162402x.2019.1621676
Khattri, R., Cox, T., Yasayko, S. A., and Ramsdell, F. (2003). An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4 (4), 337–342. doi:10.1038/ni909
Khoryati, L., Pham, M. N., Sherve, M., Kumari, S., Cook, K., Pearson, J., et al. (2020). An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 5 (50), eaba5264. doi:10.1126/sciimmunol.aba5264
Kim, H. J., and Bae, S. C. (2011). Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 3 (2), 166–179.
Kloss, C. C., Condomines, M., Cartellieri, M., Bachmann, M., and Sadelain, M. (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 31 (1), 71–75. doi:10.1038/nbt.2459
Koenecke, C., Czeloth, N., Bubke, A., Schmitz, S., Kissenpfennig, A., Malissen, B., et al. (2009). Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur. J. Immunol. 39 (11), 3091–3096. doi:10.1002/eji.200939432
Koristka, S., Cartellieri, M., Feldmann, A., Arndt, C., Loff, S., Michalk, I., et al. (2014). Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood 124 (21), 3494. doi:10.1182/blood.V124.21.3494.3494
Koristka, S., Kegler, A., Bergmann, R., Arndt, C., Feldmann, A., Albert, S., et al. (2018). Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J. Autoimmun. 90, 116–131. doi:10.1016/j.jaut.2018.02.006
Kressler, C., Gasparoni, G., Nordström, K., Hamo, D., Salhab, A., Dimitropoulos, C., et al. (2020). Targeted de-methylation of the FOXP3-TSDR is sufficient to induce physiological FOXP3 expression but not a functional Treg phenotype. Front. Immunol. 11, 609891. doi:10.3389/fimmu.2020.609891
Kurohara, T., Tanaka, K., Takahashi, D., Ueda, S., Yamashita, Y., Takada, Y., et al. (2021). Identification of novel histone deacetylase 6-selective inhibitors bearing 3,3,3-trifluorolactic amide (TFLAM) motif as a zinc binding group. Chembiochem 22 (22), 3158–3163. doi:10.1002/cbic.202100255
Lam, A. J., Hoeppli, R. E., and Levings, M. K. (2017). Harnessing advances in T regulatory cell biology for cellular therapy in transplantation. Transplantation 101 (10), 2277–2287. doi:10.1097/tp.0000000000001757
Lamarche, C., and Levings, M. K. (2018). Guiding regulatory T cells to the allograft. Curr. Opin. Organ Transpl. 23 (1), 106–113. doi:10.1097/mot.0000000000000483
Lamarche, C., and Maltzman, J. S. (2022). Chimeric antigen receptor regulatory T cell in transplantation: the future of cell therapy?. Kidney Int. Rep. 7 (6), 1149–1152. doi:10.1016/j.ekir.2022.04.003
Lamarche, C., Ward-Hartstonge, K., Mi, T., Lin, D. T. S., Huang, Q., Brown, A., et al. (2023). Tonic-signaling chimeric antigen receptors drive human regulatory T cell exhaustion. Proc. Natl. Acad. Sci. U. S. A. 120 (14), e2219086120. doi:10.1073/pnas.2219086120
Lamarthée, B., Marchal, A., Charbonnier, S., Blein, T., Leon, J., Martin, E., et al. (2021). Transient mTOR inhibition rescues 4-1BB CAR-Tregs from tonic signal-induced dysfunction. Nat. Commun. 12 (1), 6446. doi:10.1038/s41467-021-26844-1
Leclerc, S., and Lamarche, C. (2021). Cellular therapies in kidney transplantation. Curr. Opin. Nephrol. Hypertens. 30 (6), 584–592. doi:10.1097/mnh.0000000000000737
Lee, J. H., Kim, H. S., Jang, S. W., and Lee, G. R. (2022). Histone deacetylase 6 plays an important role in TGF-β-induced murine Treg cell differentiation by regulating cell proliferation. Sci. Rep. 12 (1), 22550. doi:10.1038/s41598-022-27230-7
Lee, S. E., Cho, B. S., Kim, J. H., Yoon, J. H., Shin, S. H., Yahng, S. A., et al. (2013). Risk and prognostic factors for acute GVHD based on NIH consensus criteria. Bone Marrow Transpl. 48 (4), 587–592. doi:10.1038/bmt.2012.187
Lentine, K. L., Smith, J. M., Miller, J. M., Bradbrook, K., Larkin, L., Weiss, S., et al. (2023). OPTN/SRTR 2021 annual data report: kidney. Am. J. Transpl. 23 (2), S21–s120. doi:10.1016/j.ajt.2023.02.004
Lesch, S., Nottebrock, A., Rataj, F., Heise, C., Endres, S., and Kobold, S. (2023). PD-1-CD28 fusion protein strengthens mesothelin-specific TRuC T cells in preclinical solid tumor models. Cell Oncol. (Dordr) 46 (1), 227–235. doi:10.1007/s13402-022-00747-9
Liu, D., Zhao, J., and Song, Y. (2019). Engineering switchable and programmable universal CARs for CAR T therapy. J. Hematol. Oncol. 12 (1), 69. doi:10.1186/s13045-019-0763-0
Liu, W., Putnam, A. L., Xu-Yu, Z., Szot, G. L., Lee, M. R., Zhu, S., et al. (2006). CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 203 (7), 1701–1711. doi:10.1084/jem.20060772
Long, A. H., Haso, W. M., Shern, J. F., Wanhainen, K. M., Murgai, M., Ingaramo, M., et al. (2015). 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 21 (6), 581–590. doi:10.1038/nm.3838
Lucas, J. L., Mirshahpanah, P., Haas-Stapleton, E., Asadullah, K., Zollner, T. M., and Numerof, R. P. (2009). Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol. 257 (1-2), 97–104. doi:10.1016/j.cellimm.2009.03.004
Maahs, D. M., West, N. A., Lawrence, J. M., and Mayer-Davis, E. J. (2010). Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. North Am. 39 (3), 481–497. doi:10.1016/j.ecl.2010.05.011
MacDonald, K. G., Hoeppli, R. E., Huang, Q., Gillies, J., Luciani, D. S., Orban, P. C., et al. (2016). Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 126 (4), 1413–1424. doi:10.1172/jci82771
MacMillan, M. L., Hippen, K. L., McKenna, D. H., Kadidlo, D., Sumstad, D., DeFor, T. E., et al. (2021). First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv. 5 (5), 1425–1436. doi:10.1182/bloodadvances.2020003219
Marcén, R. (2009). Immunosuppressive drugs in kidney transplantation: impact on patient survival, and incidence of cardiovascular disease, malignancy and infection. Drugs 69 (16), 2227–2243. doi:10.2165/11319260-000000000-00000
Martin, A., Daris, M., Johnston, J. A., and Cui, J. (2021). HLA-A*02:01-directed chimeric antigen receptor/forkhead box P3-engineered CD4+ T cells adopt a regulatory phenotype and suppress established graft-versus-host disease. Cytotherapy 23 (2), 131–136. doi:10.1016/j.jcyt.2020.10.002
Menning, A., Loddenkemper, C., Westendorf, A. M., Szilagyi, B., Buer, J., Siewert, C., et al. (2010). Retinoic acid-induced gut tropism improves the protective capacity of Treg in acute but not in chronic gut inflammation. Eur. J. Immunol. 40 (9), 2539–2548. doi:10.1002/eji.200939938
Mohseni, Y. R., Saleem, A., Tung, S. L., Dudreuilh, C., Lang, C., Peng, Q., et al. (2021). Chimeric antigen receptor-modified human regulatory T cells that constitutively express IL-10 maintain their phenotype and are potently suppressive. Eur. J. Immunol. 51 (10), 2522–2530. doi:10.1002/eji.202048934
Mukhatayev, Z., Dellacecca, E. R., Cosgrove, C., Shivde, R., Jaishankar, D., Pontarolo-Maag, K., et al. (2020). Antigen specificity enhances disease control by Tregs in vitiligo. Front. Immunol. 11, 581433. doi:10.3389/fimmu.2020.581433
Muller, Y. D., Ferreira, L. M. R., Ronin, E., Ho, P., Nguyen, V., Faleo, G., et al. (2021). Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front. Immunol. 12, 686439. doi:10.3389/fimmu.2021.686439
Neote, K., DiGregorio, D., Mak, J. Y., Horuk, R., and Schall, T. J. (1993). Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 72 (3), 415–425. doi:10.1016/0092-8674(93)90118-a
Noyan, F., Zimmermann, K., Hardtke-Wolenski, M., Knoefel, A., Schulde, E., Geffers, R., et al. (2017). Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am. J. Transpl. 17 (4), 917–930. doi:10.1111/ajt.14175
Obarorakpor, N., Patel, D., Boyarov, R., Amarsaikhan, N., Cepeda, J. R., Eastes, D., et al. (2023). Regulatory T cells targeting a pathogenic MHC class II: insulin peptide epitope postpone spontaneous autoimmune diabetes. Front. Immunol. 14, 1207108. doi:10.3389/fimmu.2023.1207108
Oppermann, M. (2004). Chemokine receptor CCR5: insights into structure, function, and regulation. Cell. Signal. 16 (11), 1201–1210. doi:10.1016/j.cellsig.2004.04.007
Passerini, L., Rossi Mel, E., Sartirana, C., Fousteri, G., Bondanza, A., Naldini, L., et al. (2013). CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci. Transl. Med. 5 (215), 215ra174. doi:10.1126/scitranslmed.3007320
Pierini, A., Iliopoulou, B. P., Peiris, H., Pérez-Cruz, M., Baker, J., Hsu, K., et al. (2017). T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight 2 (20), e92865. doi:10.1172/jci.insight.92865
Polansky, J. K., Kretschmer, K., Freyer, J., Floess, S., Garbe, A., Baron, U., et al. (2008). DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 38 (6), 1654–1663. doi:10.1002/eji.200838105
Polhill, T., Zhang, G. Y., Hu, M., Sawyer, A., Zhou, J. J., Saito, M., et al. (2012). IL-2/IL-2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J. Am. Soc. Nephrol. 23 (8), 1303–1308. doi:10.1681/asn.2011111130
Rana, J., Perry, D. J., Kumar, S. R. P., Muñoz-Melero, M., Saboungi, R., Brusko, T. M., et al. (2021). CAR- and TRuC-redirected regulatory T cells differ in capacity to control adaptive immunity to FVIII. Mol. Ther. 29 (9), 2660–2676. doi:10.1016/j.ymthe.2021.04.034
Roybal, K. T., Rupp, L. J., Morsut, L., Walker, W. J., McNally, K. A., Park, J. S., et al. (2016). Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164 (4), 770–779. doi:10.1016/j.cell.2016.01.011
Ruella, M., Barrett, D. M., Kenderian, S. S., Shestova, O., Hofmann, T. J., Perazzelli, J., et al. (2016). Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 126 (10), 3814–3826. doi:10.1172/jci87366
Saetzler, V., Riet, T., Schienke, A., Henschel, P., Freitag, K., Haake, A., et al. (2023). Development of beta-amyloid-specific CAR-tregs for the treatment of alzheimer's disease. Cells 12 (16), 2115. doi:10.3390/cells12162115
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133 (5), 775–787. doi:10.1016/j.cell.2008.05.009
Salzer, J., Svenningsson, R., Alping, P., Novakova, L., Björck, A., Fink, K., et al. (2016). Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology 87 (20), 2074–2081. doi:10.1212/wnl.0000000000003331
Santegoets, S. J., Dijkgraaf, E. M., Battaglia, A., Beckhove, P., Britten, C. M., Gallimore, A., et al. (2015). Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol. Immunother. 64 (10), 1271–1286. doi:10.1007/s00262-015-1729-x
Sarkar, D., Biswas, M., Liao, G., Seay, H. R., Perrin, G. Q., Markusic, D. M., et al. (2014). Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol. Ther. Methods Clin. Dev. 1, 14030-. doi:10.1038/mtm.2014.30
Sasaki, Y., Horiuchi, H., Kawashima, H., Mukai, T., and Yamamoto, Y. (2014). NADH oxidase of Streptococcus thermophilus 1131 is required for the effective yogurt fermentation with lactobacillus delbrueckii subsp. bulgaricus 2038. Biosci. Microbiota Food Health 33 (1), 31–40. doi:10.12938/bmfh.33.31
Schaerli, P., Ebert, L., Willimann, K., Blaser, A., Roos, R. S., Loetscher, P., et al. (2004). A skin-selective homing mechanism for human immune surveillance T cells. J. Exp. Med. 199 (9), 1265–1275. doi:10.1084/jem.20032177
Schreeb, K., Culme-Seymour, E., Ridha, E., Dumont, C., Atkinson, G., Hsu, B., et al. (2022). Study design: human leukocyte antigen class I molecule A(*)02-Chimeric antigen receptor regulatory T cells in renal transplantation. Kidney Int. Rep. 7 (6), 1258–1267. doi:10.1016/j.ekir.2022.03.030
Schwarz, A., Maeda, A., Wild, M. K., Kernebeck, K., Gross, N., Aragane, Y., et al. (2004). Ultraviolet radiation-induced regulatory T cells not only inhibit the induction but can suppress the effector phase of contact hypersensitivity. J. Immunol. 172 (2), 1036–1043. doi:10.4049/jimmunol.172.2.1036
Seng, A., Krausz, K. L., Pei, D., Koestler, D. C., Fischer, R. T., Yankee, T. M., et al. (2020). Coexpression of FOXP3 and a Helios isoform enhances the effectiveness of human engineered regulatory T cells. Blood Adv. 4 (7), 1325–1339. doi:10.1182/bloodadvances.2019000965
Shimizu, M., Matsuda, A., Yanagisawa, K., Hirota, T., Akahoshi, M., Inomata, N., et al. (2005). Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum. Mol. Genet. 14 (19), 2919–2927. doi:10.1093/hmg/ddi323
Sicard, A., Lamarche, C., Speck, M., Wong, M., Rosado-Sánchez, I., Blois, M., et al. (2020). Donor-specific chimeric antigen receptor Tregs limit rejection in naive but not sensitized allograft recipients. Am. J. Transpl. 20 (6), 1562–1573. doi:10.1111/ajt.15787
Skuljec, J., Chmielewski, M., Happle, C., Habener, A., Busse, M., Abken, H., et al. (2017). Chimeric antigen receptor-redirected regulatory T cells suppress experimental allergic airway inflammation, a model of asthma. Front. Immunol. 8, 1125. doi:10.3389/fimmu.2017.01125
Spanier, J. A., Fung, V., Wardell, C. M., Alkhatib, M. H., Chen, Y., Swanson, L. A., et al. (2023). Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J. Clin. Invest. 133, e168601. doi:10.1172/jci168601
Sparks, J. A. (2019). Rheumatoid arthritis. Ann. Intern Med. 170 (1), Itc1–itc16. doi:10.7326/aitc201901010
Sterner, R. C., and Sterner, R. M. (2021). CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11 (4), 69. doi:10.1038/s41408-021-00459-7
Stock, S., Klüver, A. K., Fertig, L., Menkhoff, V. D., Subklewe, M., Endres, S., et al. (2023). Mechanisms and strategies for safe chimeric antigen receptor T-cell activity control. Int. J. Cancer 153, 1706–1725. doi:10.1002/ijc.34635
Tang, Q., Leung, J., Peng, Y., Sanchez-Fueyo, A., Lozano, J. J., Lam, A., et al. (2022). Selective decrease of donor-reactive T(regs) after liver transplantation limits T(reg) therapy for promoting allograft tolerance in humans. Sci. Transl. Med. 14 (669), eabo2628. doi:10.1126/scitranslmed.abo2628
Tao, R., de Zoeten, E. F., Ozkaynak, E., Chen, C., Wang, L., Porrett, P. M., et al. (2007). Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13 (11), 1299–1307. doi:10.1038/nm1652
Tenspolde, M., Zimmermann, K., Weber, L. C., Hapke, M., Lieber, M., Dywicki, J., et al. (2019). Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 103, 102289. doi:10.1016/j.jaut.2019.05.017
Trotta, E., Bessette, P. H., Silveria, S. L., Ely, L. K., Jude, K. M., Le, D. T., et al. (2018). A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat. Med. 24 (7), 1005–1014. doi:10.1038/s41591-018-0070-2
Uehara, S., Grinberg, A., Farber, J. M., and Love, P. E. (2002). A role for CCR9 in T lymphocyte development and migration. J. Immunol. 168 (6), 2811–2819. doi:10.4049/jimmunol.168.6.2811
Valentini, N., Requejo Cier, C. J., and Lamarche, C. (2023). Regulatory T-cell dysfunction and its implication for cell therapy. Clin. Exp. Immunol. 213 (1), 40–49. doi:10.1093/cei/uxad051
Vasanthakumar, A., Moro, K., Xin, A., Liao, Y., Gloury, R., Kawamoto, S., et al. (2015). The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16 (3), 276–285. doi:10.1038/ni.3085
Wagner, J. C., Ronin, E., Ho, P., Peng, Y., and Tang, Q. (2022). Anti-HLA-A2-CAR Tregs prolong vascularized mouse heterotopic heart allograft survival. Am. J. Transpl. 22 (9), 2237–2245. doi:10.1111/ajt.17063
Wahren-Herlenius, M., and Dörner, T. (2013). Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 382 (9894), 819–831. doi:10.1016/s0140-6736(13)60954-x
Waldmann, H., Adams, E., Fairchild, P., and Cobbold, S. (2006). Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol. Rev. 212, 301–313. doi:10.1111/j.0105-2896.2006.00406.x
Wang, L., Beier, U. H., Akimova, T., Dahiya, S., Han, R., Samanta, A., et al. (2018). Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am. J. Transpl. 18 (7), 1596–1603. doi:10.1111/ajt.14749
Wang, L., de Zoeten, E. F., Greene, M. I., and Hancock, W. W. (2009). Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat. Rev. Drug Discov. 8 (12), 969–981. doi:10.1038/nrd3031
Whilding, L. M., Halim, L., Draper, B., Parente-Pereira, A. C., Zabinski, T., Davies, D. M., et al. (2019). CAR T-cells targeting the integrin αvβ6 and Co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel) 11 (5), 674. doi:10.3390/cancers11050674
Wilk, C., Effenberg, L., Abberger, H., Steenpass, L., Hansen, W., Zeschnigk, M., et al. (2022). CRISPR/Cas9-mediated demethylation of FOXP3-TSDR toward Treg-characteristic programming of Jurkat T cells. Cell Immunol. 371, 104471. doi:10.1016/j.cellimm.2021.104471
Yadav, A. K., Lal, A., and Jha, V. (2011). Association of circulating fractalkine (CX3CL1) and CX3CR1+CD4+ T cells with common carotid artery intima-media thickness in patients with chronic kidney disease. J. Atheroscler. Thrombosis 18 (11), 958–965. doi:10.5551/jat.8722
Yagi, H., Nomura, T., Nakamura, K., Yamazaki, S., Kitawaki, T., Hori, S., et al. (2004). Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int. Immunol. 16 (11), 1643–1656. doi:10.1093/intimm/dxh165
Ye, C., Brand, D., and Zheng, S. G. (2018). Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct. Target Ther. 3, 2. doi:10.1038/s41392-017-0002-5
Yeku, O. O., Purdon, T. J., Koneru, M., Spriggs, D., and Brentjens, R. J. (2017). Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 7 (1), 10541. doi:10.1038/s41598-017-10940-8
Yoon, J., Schmidt, A., Zhang, A. H., Königs, C., Kim, Y. C., and Scott, D. W. (2017). FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood 129 (2), 238–245. doi:10.1182/blood-2016-07-727834
Yusuf-Makagiansar, H., Anderson, M. E., Yakovleva, T. V., Murray, J. S., and Siahaan, T. J. (2002). Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med. Res. Rev. 22 (2), 146–167. doi:10.1002/med.10001
Zah, E., Lin, M. Y., Silva-Benedict, A., Jensen, M. C., and Chen, Y. Y. (2016). T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 4 (6), 498–508. doi:10.1158/2326-6066.Cir-15-0231
Zhang, A. H., Yoon, J., Kim, Y. C., and Scott, D. W. (2018). Targeting antigen-specific B cells using antigen-expressing transduced regulatory T cells. J. Immunol. 201 (5), 1434–1441. doi:10.4049/jimmunol.1701800
Zhang, N., Schröppel, B., Lal, G., Jakubzick, C., Mao, X., Chen, D., et al. (2009). Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 30 (3), 458–469. doi:10.1016/j.immuni.2008.12.022
Zheng, S. G., Gray, J. D., Ohtsuka, K., Yamagiwa, S., and Horwitz, D. A. (2002). Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25-precursors. J. Immunol. 169 (8), 4183–4189. doi:10.4049/jimmunol.169.8.4183
Keywords: regulatory T cells, tolerance, chimeric antigen receptor (CAR), immunotherapy, genetic engineering, cell therapy
Citation: Requejo Cier CJ, Valentini N and Lamarche C (2023) Unlocking the potential of Tregs: innovations in CAR technology. Front. Mol. Biosci. 10:1267762. doi: 10.3389/fmolb.2023.1267762
Received: 27 July 2023; Accepted: 20 September 2023;
Published: 12 October 2023.
Edited by:
Alfonso Rodríguez Gil, IBIS (Universidad de Sevilla, CSIC, HUVR, Junta de Andalucía), SpainReviewed by:
Takashi MaruYama, National Institutes of Health (NIH), United StatesJennifer Whangbo, Harvard Medical School, United States
Copyright © 2023 Requejo Cier, Valentini and Lamarche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Lamarche, Q2Fyb2xpbmUubGFtYXJjaGUuMUB1bW9udHJlYWwuY2E=