 Masaki Saito1†
Masaki Saito1† Wataru Otsu
Wataru Otsu Yuhei Nishimura
Yuhei Nishimura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci., 14 September 2023
Sec. Lipids, Membranes and Membranous Organelles
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1232188
This article is part of the Research TopicPrimary Cilia as Therapeutic TargetsView all 8 articles
The primary cilium is a single immotile microtubule-based organelle that protrudes into the extracellular space. Malformations and dysfunctions of the cilia have been associated with various forms of syndromic and non-syndromic diseases, termed ciliopathies. The primary cilium is therefore gaining attention due to its potential as a therapeutic target. In this review, we examine ciliary receptors, ciliogenesis, and ciliary trafficking as possible therapeutic targets. We first discuss the mechanisms of selective distribution, signal transduction, and physiological roles of ciliary receptors. Next, pathways that regulate ciliogenesis, specifically the Aurora A kinase, mammalian target of rapamycin, and ubiquitin-proteasome pathways are examined as therapeutic targets to regulate ciliogenesis. Then, in the photoreceptors, the mechanism of ciliary trafficking which takes place at the transition zone involving the ciliary membrane proteins is reviewed. Finally, some of the current therapeutic advancements highlighting the role of large animal models of photoreceptor ciliopathy are discussed.
The primary cilium is a single immotile microtubule-based organelle protruding into the extracellular space. The ciliary membrane abundantly contains specific membrane receptors and ion channels in its limited surface region, which enables cells to sense the extracellular circumstances. Ciliopathies are a group of inherited diseases involving dysfunction of the cilia arising from variants in genes encoding proteins that localize to the cilia or centrosomes. As most cells possess primary cilia, ciliopathies may exhibit pleiotropic effects resulting in a wide range of anomalies in multiple organ systems. Some of the common features include retinitis pigmentosa, renal cystic disease, polydactyly, situs inversus, mental retardation, hypoplasia of corpus callosum, dandy-walker malformation, posterior encephalocele, and hepatic disease, among others (Baker and Beales, 2009). Nearly 200 ciliopathy genes have been documented [reviewed in Wheway et al. (2019)], some of which encoding proteins that are part of complexes involved in cilia formation, function, and trafficking. As such, phenotypic overlaps may be observed between variants in genes encoding components of protein complexes associated in disease such as Bardet-Biedl syndrome, Meckel syndrome, and nephronophthisis.
The primary cilium is therefore gaining attention due to its potential as a drug discovery target. In this review, we provide an introduction to the cilia, the molecules involved in their functions, and an outlook of the current findings about the primary cilium. Specifically, we will examine ciliary receptors, ciliogenesis, and ciliary trafficking as possible therapeutic targets for ciliopathies. First, the mechanisms of selective distribution, signal transduction, and physiological roles of membrane receptors in primary cilia is reviewed to examine the ciliary receptors as therapeutic targets. The next target is ciliogenesis which is a fundamental mechanism that regulates the structural formation of primary cilia, while its dysregulation is associated with various forms of ciliopathy and cancer. We specifically review the Aurora A kinase, mammalian target of rapamycin, and ubiquitin-proteasome pathways as therapeutic targets to regulate ciliogenesis. Ciliary trafficking is critical in cilia maintenance and involves the ciliary membrane proteins for the transport and turnover of critical proteins at the transition zone. Specifically, we examine the trafficking pathway of photoreceptors to review the machinery and molecules involved in intracellular trafficking as possible therapeutic targets. Finally, we review some of the current therapeutic advancements highlighting the role of large animal models of photoreceptor ciliopathy in developing AAV gene therapy for future translational application in patients.
The surface area of the primary cilia is only 1/1,000 of the entire cell surface. The cilia function as sensory organelles because they harbor selective G protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), and ion channels on their surfaces. This selective distribution is enabled by a diffusion barrier composed of the transition fibers and Y-links in the transition zone (Reiter et al., 2012; Takao and Verhey, 2016). By sensing extracellular cues, including autacoids, hormones, lights, lipids, mechanical stress, and neurotransmitters, cilia regulate cell fate determination and homeostasis. Dysregulation of ciliary signaling leads to hereditary dysfunction and malformations of various organs, known as ciliopathies, with manifestations of brain malformations, mental disability, skeletal malformations, polycystic kidney disease, and retinal degeneration (Reiter and Leroux, 2017). In this section, by introducing the mechanisms of selective distribution, signal transduction, and physiological roles of membrane receptors in primary cilia, we discuss the prospects of ciliary receptors as therapeutic targets for ciliopathies.
Limited types of RTKs and GPCRs have been identified in primary cilia (Table 1). For example, insulin-like growth factor-1 receptor (IGF-1R), platelet-derived growth factor receptor α (PDGFRα), and transforming growth factor-β receptor (TGF-βR) are ciliary RTKs (Schneider et al., 2005; Clement et al., 2013; Yeh et al., 2013). Dopamine receptors (D1R, D2R, D5R), free-fatty acid receptor 4 (FFAR4), melanin-concentrating hormone receptor 1 (MCHR1), neuropeptide Y receptors 2 and 5 (NPY2, NPY5), rhodopsin, serotonin receptor 6 (5-HT6), smoothened (SMO), and somatostatin receptor 3 (SSTR3), in addition to olfactory GPCRs, are ciliary GPCRs (Schou et al., 2015; Wachten and Mick, 2021). In general, GPCRs transduce signals of the Gαq/11, Gαs, Gαi, and Gα12/13 signaling pathways. Among these, Gαq/11 activates phospholipase C, which further induces the inositol 1,4,5-trisphosphate/Ca2+ and diacylglycerol/protein kinase C pathways. Gαs activates adenylyl cyclases (ACs), which produce cyclic AMP (cAMP) and promote the activation of the protein kinase A/Epac pathway. In contrast, Gαi inhibits ACs and suppresses cAMP production. It has been considered that ciliary GPCRs activate Gα protein(s) consistent with the receptors in the extracilia. Intriguingly, recent studies have revealed the possibility that certain GPCRs can mediate cilia-specific signaling pathways that are distinct from extracilia pathways.
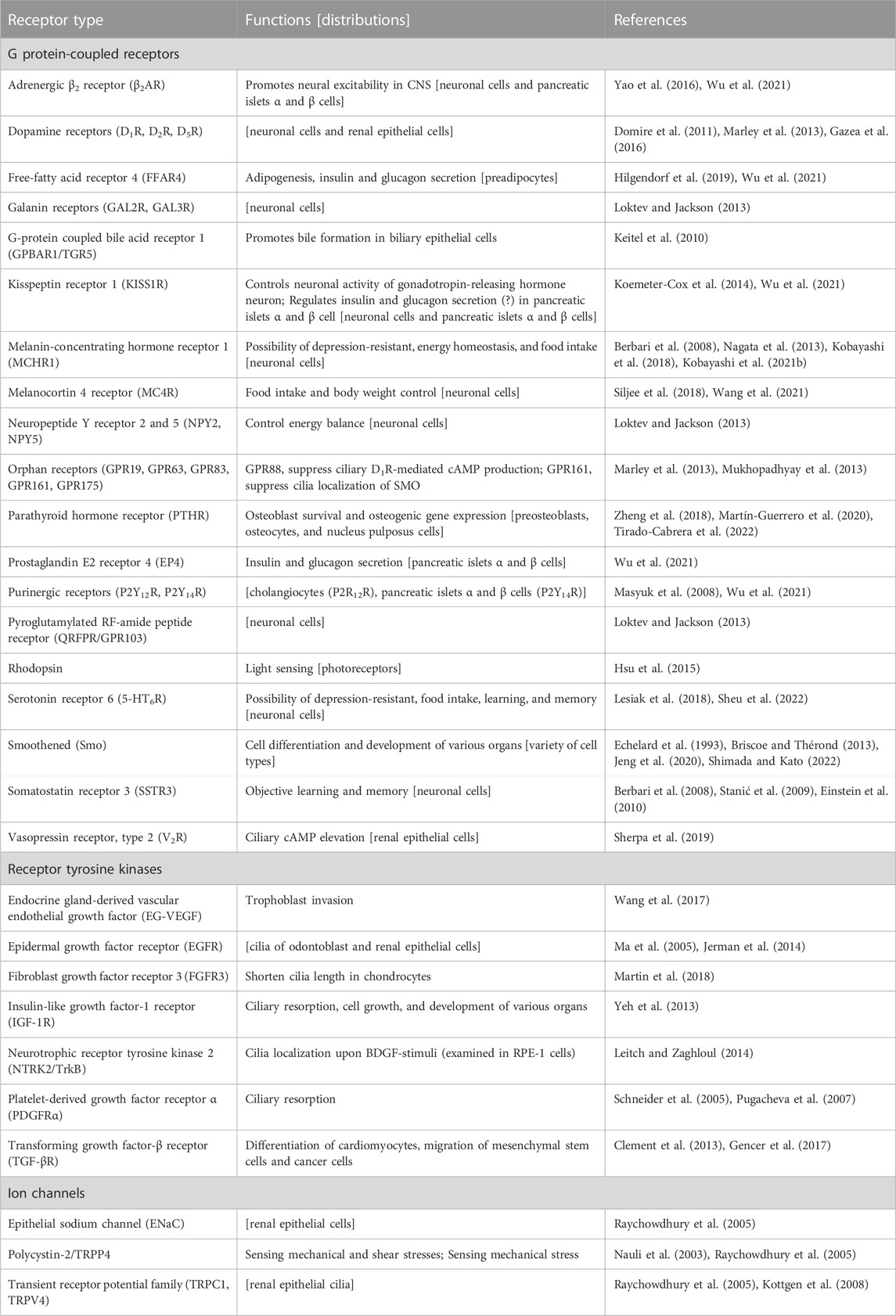
TABLE 1. List of ciliary membrane receptors.
Ciliary GPCRs are trafficked from the cytosol to primary cilia with the aid of intraflagellar transport complex A (IFT-A) and tubby-like protein 3 (TULP3) (Mukhopadhyay et al., 2010). Knockdown of TULP3 decreases ciliary localization of various rhodopsin family (class A) GPCRs (Badgandi et al., 2017). Ciliary localization of GPCRs is also achieved by the presence of ciliary targeting sequences (CTS) in the intracellular third loop and the carboxy-terminus of GPCRs. The Ax[S/A]xQ sequence in the third intracellular loop of 5-HT6R, MCHR1, and SSTR3 is essential for the ciliary localization of the receptors (Berbari et al., 2008; Nagata et al., 2013). This consensus sequence is also present in other ciliary GPCRs, including D1R, rhodopsin, opsin, and olfactory receptors 52N1, 52N4, and 6V1 (Berbari et al., 2008; Domire et al., 2011). Intriguingly, TGF-βR type 1, an RTK, harbors the A31TALQ35 sequence (Gencer et al., 2017). However, because the sequence is located in the extracellular region, the mechanism of ciliary targeting remains unclear. An [R/K][I/L]W sequence is located in the third loop of NPY2R and GPR83, whereas the [I/V]KARK sequence is in the intracellular third loop of GPR161 (Loktev and Jackson, 2013; Mukhopadhyay et al., 2013). The VxPx sequence of rhodopsin and the LPG motif of 5-HT6R have been identified at the carboxy terminus (Mazelova et al., 2009; Barbeito and Garcia-Gonzalo, 2021). Mechanistically, TULP3 interacts with the Ax[S/A]xQ and [I/V]KARK sequences, together with the KTRKIKP motif of fibrocystin (Badgandi et al., 2017). 5-HT6R interacts with RABL2 via the CTS for ciliary localization (Dateyama et al., 2019; Barbeito and Garcia-Gonzalo, 2021). The VxPx sequence comprises a protein complex of small G proteins, Arf4 and Rab11, a Rab11 effector FIP3, and an Arf GTPase-activating protein, ASAP1, which regulates the trafficking of rhodopsin from the trans-Golgi membrane to cilia (Mazelova et al., 2009).
The ciliary GPCRs exit from the cilia upon exposure to their agonists, as demonstrated by D1R, GPR161, NPY2R, SMO, and SSTR3 (Domire et al., 2011; Nager et al., 2017; Shinde et al., 2020). The BBSome, a stable protein complex composed of nine Bardet-Biedl syndrome (BBS) proteins, BBS1, BBS2, BBS3/ARL6, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18/BBIP10, is an adaptor between the IFT-B complexes and cargo molecules (Nachury et al., 2007; Liew et al., 2014). Agonist-induced K63-linked ubiquitination (UbK63) and retrograde transport by IFT-B and BBSome contribute to the exit of ciliary GPCRs (Nozaki et al., 2018; Shinde et al., 2020; Zhou et al., 2022). Interestingly, vesicle release from the ciliary tip (exocytosis) is reported as another mechanism of BBSome-dependent exit of ciliary GPCRs (Nager et al., 2017).
The mechanisms underlying the ciliary localization of GPCRs and RTKs have remained largely unresolved. Uncovering these mechanisms would be helpful in understanding the pathogenic mechanisms of ciliopathies.
The Hedgehog signal is a typical ciliary signal and its functions and regulatory mechanisms have been extensively studied. Hedgehog participates in embryonic organogenesis and tissue homeostasis by controlling the differentiation and proliferation of tissue stem/progenitor cells; it controls the development of various organs at the appropriate time and location, including the brain, heart, lung, esophagus, stomach, breast, liver, gallbladder, pancreas, intestines, and blood vessels (Echelard et al., 1993; Briscoe and Thérond, 2013; Jeng et al., 2020; Shimada and Kato, 2022). Three hedgehog proteins in mammals, Sonic, Indian, and Desert hedgehog, target different cell types via their common receptor, SMO (Ingham and McMahon, 2001). In the absence of Hedgehog, a cilia-localizing 12 transmembrane receptor patched-1 (PTCH1) suppresses the ciliary localization of SMO; hence, SMO is distributed in the extraciliary region (Han et al., 2009; Wong et al., 2009). In this case, the full-length GLI is phosphorylated by protein kinase A (PKA) and subsequently ubiquitinated and proteolyzed to form a GLI repressor (GLI-R) (Pan et al., 2006). The cilia-localizing Gαs-coupled receptor, GPR161, represses basal SMO activity through Gαs/AC/PKA-dependent GLI-R formation (Mukhopadhyay et al., 2013). In contrast, when Hedgehog binds to PTCH1, it is internalized from the ciliary membrane and degraded in lysosomes, and thus, SMO translocates into the cilium (Rohatgi et al., 2007; Yue et al., 2014). Ciliary SMO activates GLI by dephosphorylating the PKA phosphorylation site via an unidentified phosphatase(s) and phosphorylating other sites through unc-51 like kinase 3 (ULK3) and serine/threonine protein kinase 36 (STK36) (Niewiadomski et al., 2014; Han et al., 2019). Finally, active GLI binds to the consensus sequence GACCACCCA and induces the expression of specific genes that regulate the cell fate of target cells (Hallikas et al., 2006). Defect in ciliogenesis by depleting IFT proteins or membrane-associated cytoskeletal protein 4.1G abolishes cilia-mediated hedgehog signaling, highlighting the physiological importance of hedgehog signaling in vivo and in vitro (Gazea et al., 2016; Yuan et al., 2016; Eguether et al., 2018; Saito et al., 2022).
Interestingly, extraciliary SMO transduces the Gαi signal. In cilia-deficient preosteoblasts, SMO is widespread in the extraciliary region and inhibits osteoblast differentiation by activating the Gαi/RhoA pathway [34]. The ability of SMO to activate Gαi signaling has also been reported in mammary epithelial cells, where SMO promotes cell proliferation via pertussis toxin-sensitive Gαi2 signaling (Villanueva et al., 2015). This evidence demonstrates the presence of dual functions of Hedgehog through ciliary and extraciliary SMO, although insufficient evidence has been obtained regarding whether extraciliary SMO activates Gαi signaling in ciliated cells. Similar to SMO, parathyroid hormone receptor (PTHR), a Gαs- and Gαq-coupled GPCR, translocates into primary cilia upon PTH-related protein treatment and shear stress stimuli; ciliary PTHR activates canonical GLI-dependent cell survival and osteogenic gene expression in osteoblastic and osteoclastic cells (Zheng et al., 2018; Martín-Guerrero et al., 2020; Tirado-Cabrera et al., 2022). Based on these studies, it is presumed that primary cilia have a specialized condition(s) of coupling between GPCRs and their effector proteins, which is distinct from that of the extraciliary region.
RTKs and GPCRs coordinate to control brain development and function. Neural progenitor cells (radial glial cells) of the cortex display primary cilia on the ventricular surface during the G0/G1 phases of the cell cycle at the embryonic stage. Growth factor stimuli, such as IGF-1, primes ciliary resorption and re-entry of cells into the G2/M phases, which leads to the proliferation and differentiation of cells, initiating corticogenesis (Li et al., 2011; Yeh et al., 2013) Mechanistically, Aurora A kinase (AURKA) has been identified as one of the major molecules; a growth factor stimulus elicits HEF1-dependent activation of AURKA, which in turn promotes histone deacetylase 6 (HDAC6) activation and ciliary axonemal destabilization (Pugacheva et al., 2007). Calmodulin, NIMA-related protein kinase 2 (NEK2), and polo-like kinase 1 (PLK1) regulate the AURKA pathway (Lee et al., 2012; Plotnikova et al., 2012; Spalluto et al., 2012). AURKA also assembles actin in cilia and causes decapitation of the ciliary tip membrane to execute ciliary resorption (Phua et al., 2017). In contrast, Tctex-1 is a light chain component of the dynein complex. It plays a dynein-independent role when phosphorylated at Thr94 (phospho-(T94)Tctex-1) and is released from the dynein complex. Activation of ciliary IGF-1R accumulates phospho-(T94)Tctex-1 at the ciliary transition zone, and subsequently, phospho-(T94)Tctex-1 primes Cdc42- and Arp2/3-dependent branched actin organization and clathrin-dependent endocytosis at the ciliary pocket membrane (Li et al., 2011; Yeh et al., 2013; Saito et al., 2017). Currently, there are no reports connecting AURKA- and Tctex-1-mediated mechanisms. In addition to IGF-1R, PDGFRα, a cilia-localizing RTK, also promotes ciliary resorption, indicating a possible contribution of this receptor to brain development (Schneider et al., 2005).
SSTR3, a Gαi-coupled receptor, is highly expressed in the CA1 and CA3 regions of the hippocampus and the granular layer of the dentate gyrus in postnatal stages (Stanić et al., 2009). It is considered that CA1 cilia-localizing SSTR3 regulates object learning and memory by activating neuronal cilia-specific AC type 3 (AC3) (Einstein et al., 2010; Wang et al., 2011). MCHR1, a Gαi-coupled receptor that is extensively expressed in the brain, controls food intake and energy homeostasis (Shimada et al., 1998; Chen et al., 2002; Marsh et al., 2002). It has been reported that ciliary MCHR1 in the hippocampal CA1 region regulates these functions, as well as depression-resistant behavior (Kobayashi et al., 2018). Shortening of hippocampal cilia through the MCHR1/Gαi/Akt pathway is considered to present a possible mechanism of feeding and mood (Kobayashi et al., 2021a). In contrast, 5-HT6R in cilia is considered to contribute to depression and eating defects, and learning and memory (Fisas et al., 2006; Svenningsson et al., 2007; King et al., 2008; Sheu et al., 2022). Physiological roles are executed by modulation of the epigenetic state in neuronal cells, stabilization of the neuronal ciliary structure, changes in neuronal dendritic morphology, and physical contact of the 5-HT6R-positive cilia with synapses of neuronal axons (the axo-ciliary synapse) (Lesiak et al., 2018; Sheu et al., 2022). Overexpression of 5-HT6R can elongate primary cilia, hense careful consideration is required when exogenously tagged 5-HT6R is used as a ciliary marker (Lesiak et al., 2018). Moreover, neuropeptide Y receptor 2 (NPY2R) controls the energy balance by inhibiting the ciliary AC through activation of its coupling Gi/o protein in hypothalamic neuronal cilia (Loktev and Jackson, 2013). The adrenergic β2 receptor (β2AR) increases the ciliary cAMP level via Gs protein and enhances neuronal excitability in the central nervous system (CNS) (Yao et al., 2016). These functional studies of neuronal ciliary GPCRs suggest that the GPCRs transduce ciliary signaling through trimeric G protein(s). However, it remains unclear how do the neural ciliary GPCRs transduce ciliary signaling differently from extraciliary GPCR signaling. Functions and/or distributions of other neural ciliary GPCRs are listed in Table 1. Further studies are required to understand the physiological roles and signal transduction of neuronal ciliary GPCRs.
Among the four free fatty acid receptors (FFARs), FFAR1/GPR40, FFAR2/GPR43, FFAR3/GPR41, and FFAR4/GPR120, FFAR4/GPR120 are distributed in primary cilia in preadipocytes (Hilgendorf et al., 2019). FFAR4/GPR120 is recognized as a Gαq-coupled GPCR, and it is activated by various saturated fatty acids, unsaturated fatty acids, omega-3 fatty acids, and omega-6 fatty acids (Hirasawa et al., 2005; Wu et al., 2013; Kimura et al., 2020). Omega-3 fatty acid docosahexaenoic acid (DHA) activates ciliary FFAR4/GPR120, increases ciliary cAMP production, and promotes the differentiation of preadipocytes to mature adipocytes (adipogenesis), highlighting the role of this receptor in white adipose tissue generation. FFAR4/GPR120 and EP4 increase insulin and glucagon secretion by augmenting ciliary cAMP levels. A possibility that FFAR4/GPR120 transduces Gαs signaling is supported by a recent study which is performed in HEK293 cells (Mao et al., 2023). These studies collectively suggest a possibility that FFAR4/GPR120 couples with Gαs in cilia. β2AR, purinergic receptor P2Y14R, and KISS1R are also expressed in pancreatic islets α and β cells (Wu et al., 2021). The functions and/or distributions of these receptors are listed in Table 1.
Polycystin-2 (PC2) is a six-transmembrane protein that can permeate Ca2+ into the cilium by sensing flow and shear stress. PC2, a nonselective Ca2+ channel, is also known as TRPP1 and a member of the transient receptor potential (TRP) subfamily. Mutation in the PKD2 gene, which encodes PC2, is causative of 15% of the autosomal dominant polycystic kidney disease (ADPKD) (Heyer et al., 2016). PC2 functions by forming a heterodimer with polycystin-1 (PC1), an eleven-transmembrane protein, through their coiled-coil domains. In renal epithelial cells isolated from PC1-knockout mice, or wild type renal epithelial cells treated with anti-PC1 or -PC2 neutralizing antibodies, PC2 failed to induce ciliary Ca2+-influx, showing the importance of heterodimer formation in PC2 function (Nauli et al., 2003). PC2 also forms a heterodimer with TRPV4 and is distributed on the primary cilia of canine kidney epithelial cell line (MDCK; Madin-Darby canine kidney) (Kottgen et al., 2008). Knockdown of TRPV4 failed to induce the mechanical stimulation-dependent elevation of intracellular Ca2+. In contrast, knockdown of PC2 abolished normal cyst formation in the zebrafish pronephros, but knockdown of TRPV4 did not disturb the cyst formation (Kottgen et al., 2008). Further study is required to fully elucidate the function of ciliary TRPV4 in kidney cells.
While Hedgehog controls the proliferation of normal tissue stem/progenitor cells, aberrant activation of Hedgehog signaling promotes stem cell maintenance, self-renewal, and regeneration of cancer stem cells and drives basal cell carcinoma, bladder cancer, breast cancer, chondrosarcoma, gastric cancer medulloblastoma, pancreatic cancer, and rhabdomyosarcoma (Han et al., 2009; Wong et al., 2009; Jeng et al., 2020; Yang et al., 2020). Vismodegib, sonidegib, and glasdegib are small-molecule SMO antagonists that have been approved by the US Food and Drug Administration (FDA). Vismodegib and sonidegib are used for basal cell carcinoma (Gould et al., 2014; Burness, 2015) and glasdegib is used for acute myeloid leukemia (Norsworthy et al., 2019). In addition, no approved drug targeting ciliary GPCRs has been identified to date. However, it is plausible that some of the drugs act on ciliary GPCRs because agonists/antagonists of certain GPCRs, which are also distributed in primary cilia, have been developed (e.g., β2AR agonists and antagonists, PTHR agonist teriparatide, EP4 antagonist grapiprant). Interestingly, tolvaptan, a selective vasopressin V2 receptor (V2R) antagonist, is an FDA-approved ADPKD drug (Raina et al., 2022). It slows the decline in estimated glomerular filtration rate of ADPKD (Torres, 2019). V2R is distributed in the primary cilia of two renal epithelial cells from the proximal tubule (LLC-KP1 cells) and inner medullary collecting duct (IMCD cells) (Sherpa et al., 2019). Although the ciliary V2R can increase the cAMP levels, its physiological roles are unresolved and remain to be investigated in future studies. Numerous antibody drugs recognize RTKs. Among these, teprotumumab, an anti-IGF-1R monoclonal antibody, has been approved for the treatment of active thyroid-eye disease (Douglas et al., 2020), and olaratumab, an anti-PDGFRα monoclonal antibody, has been used for the treatment of advanced soft tissue sarcoma (Shirley, 2017).
It is anticipated that diagnostic, preventive, or therapeutic agents against ciliary receptors for ciliopathies will be developed in the near future. For this, it is important that we understand the physiological roles and regulatory mechanisms of ciliary receptors in more detail.
The structure of primary cilia changes dynamically to regulate the proliferation and differentiation of cells spatiotemporally (Nishimura et al., 2019; Kasahara and Inagaki, 2021). Ciliogenesis is a fundamental mechanism to regulate the structure of primary cilia (Patel and Tsiokas, 2021). Ciliogenesis involves several steps including the transportation of small cytoplasmic vesicles from the Golgi apparatus to the mother centriole which is converted the basal body (Lee et al., 2018; Wu et al., 2018), anchoring the basal body to the plasma membrane via the distal appendage (Pitaval et al., 2017), removal of coiled-coil protein 110 (CP110) from the basal body to initiate axoneme elongation (Spektor et al., 2007; Schmidt et al., 2009; Goetz et al., 2012; Xu et al., 2016), the fusion of the ciliary vesicle with the plasma membrane and transportation of tubulin with various modification such as acetylation and glutamylation to increase axoneme length (Ishikawa and Marshall, 2017; Wloga et al., 2017). Dysregulation of ciliogenesis is associated with various diseases, including cancer and ciliopathy (Shiromizu et al., 2020; Zhao et al., 2023). Modulation of ciliogenesis in non-tumor cells can also be used to regulate the differentiation in response to pathological stimulation, becoming less prone to diseases caused by the stimulation (Nishimura et al., 2021a; Yamakawa et al., 2021). Therefore, targeting of ciliogenesis can be therapeutic to these diseases (Nishimura et al., 2021b; Duong Phu et al., 2021). In this section, we focus on Aurora A kinase (AURKA), mammalian target of rapamycin (mTOR), and ubiquitin-proteasome (UPS) pathways as therapeutic targets to regulate ciliogenesis (Table 2).
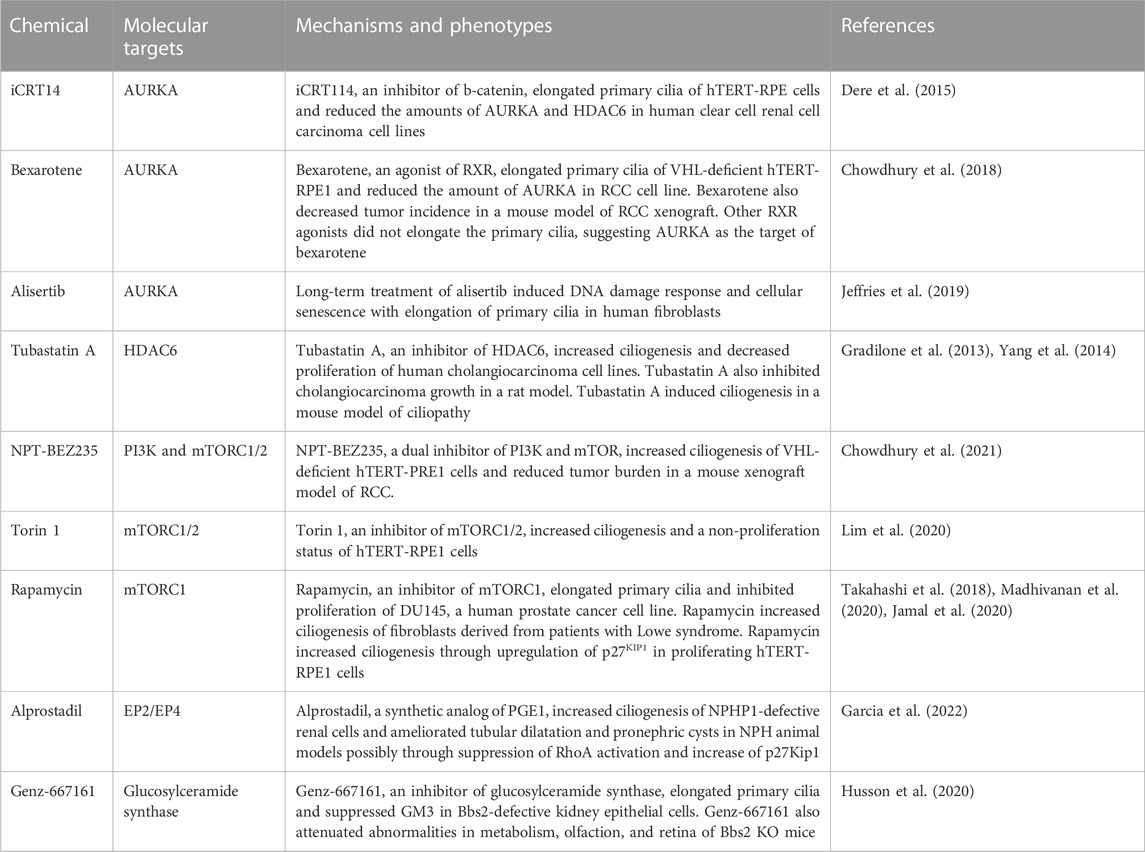
TABLE 2. Pharmacological agents to stimulate ciliogenesis.
AURKA is a key player in the inhibition of ciliogenesis and is located at the basal body. It is activated by various proteins including neural precursor cells expressed, developmentally downregulated protein 9 (NEDD9) (Pugacheva et al., 2007), centrosomal protein 55 (CEP55) (Zhang et al., 2021), and trichoplein (TCHP) (Inoko et al., 2012). Activated AURKA inhibits ciliogenesis through the phosphorylation of substrates such as histone deacetylase 6 (HDAC6) (Pugacheva et al., 2007; Kim et al., 2014; Sánchez de Diego et al., 2014). Phosphorylated HDAC6 deacetylates α-tubulin and reduces the stability of axoneme microtubules (Pugacheva et al., 2007; Sánchez de Diego et al., 2014). Activation of AURKA and suppression of ciliogenesis are observed in various cancers, including epithelial ovarian cancer (Egeberg et al., 2012), prostate cancer (Qie et al., 2020), pancreatic ductal adenocarcinoma (Li et al., 2003; Kobayashi et al., 2017), and glioblastoma (Duncan et al., 2010; Álvarez-Satta and Matheu, 2018). Inhibition of HDAC6 restores ciliogenesis and suppresses the proliferation of cancer cells, including glioblastoma (Urdiciain et al., 2019), cholangiocarcinoma (Gradilone et al., 2013), and chondrosarcoma (Xiang et al., 2017). These findings suggest that inhibition of AURKA signaling may suppress the proliferation of these tumor cells through stimulation of ciliogenesis (Nishimura et al., 2019; Peixoto et al., 2020).
Inhibition of AURKA signaling can be done in different ways, including inhibition of the kinase activity, decreasing the expression, and targeting proteins that can activate or be activated by AURKA (Bertolin and Tramier, 2020; Nishimura et al., 2021b). Alisertib, also known as MLN8237, is an ATP-competitive inhibitor of AURKA (Sells et al., 2015). iCRT14, a βcatenin-responsive transcription inhibitor, and bexarotene, a retinoid X receptor agonist, can decrease the expression of AURKA (Dere et al., 2015; Chowdhury et al., 2018). These chemicals induce ciliogenesis and may be used as novel therapies for diseases associated with the loss of primary cilia. However, ciliogenesis induced by the inhibition of AURKA may cause premature senescence by preventing the formation of the mitotic spindle in non-tumor cells (Jeffries et al., 2019). Targeting proteins that can bind and activate AURKA in selective cells may be desirable to maximize therapeutic efficacy and minimize side effects. TCHP may be a candidate for this approach because the knockout mice are viable and show resistance to high-fat diet-induced obesity and increased regeneration following skeletal muscle injuries (Yamakawa et al., 2021; Yamakawa et al., 2022). Chemicals can be developed that inhibit the interaction between AURKA and the binding partner or degrade ciliary proteins by proteolysis-targeting chimeras (Janeček et al., 2016; Bagka et al., 2022). Tubastatin-A, an inhibitor of HDAC6, can also be used to treat diseases associated with ciliary defects by inducing ciliogenesis (Gradilone et al., 2013; Yang et al., 2014).
mTOR signaling is also a major player in ciliogenesis. Activation of class I phosphatidylinositol-3 kinase (PI3K) phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) at the plasma membrane to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3) (Sugiyama et al., 2019; Margaria et al., 2020). PIP3 binds to the Pleckstrin-homology (PH) domain of the serine/threonine kinase AKT, which recruits AKT to the plasma membrane to be phosphorylated on Thr308 and Ser473 by phosphatidylinositol-dependent protein 1 (PDK1) and mTOR complex 2 (mTORC2), respectively (Manning and Toker, 2017). The phosphorylated AKT phosphorylates various substrates, including tuberous sclerosis complex (TSC) 1/2 and glycogen synthase kinase 3β (GSK3β) (Margaria et al., 2020). The phosphorylated GSK3β suppresses ciliogenesis and the dysregulation of this pathway contributes to various ciliopathy phenotypes (Thoma et al., 2007; Beurel et al., 2015; Conduit and Vanhaesebroeck, 2020). mTOR complex 1 (mTORC1) is also involved in ciliogenesis (Lai and Jiang, 2020). Overexpression of Rheb, an activator of mTORC1, suppressed ciliogenesis induced by glucose deprivation, whereas inhibition of mTORC1 by rapamycin increased ciliogenesis through upregulation of p27KIP1, a cyclin-dependent kinase inhibitor (Takahashi et al., 2018). These studies suggest that inhibition of mTOR signaling may also be therapeutic by stimulating ciliogenesis.
NPT-BEZ235, a dual inhibitor of PI3K and mTOR, increased ciliogenesis of von Hippel Lindau (VHL)-deficient human telomerase reverse transcriptase (hTERT)-immortalized retinal pigment epithelial (hTERT-RPE1) cells (Chowdhury et al., 2021). NPT-BEZ235 also reduced tumor burden in a mouse xenograft model of VHL-null renal cell carcinoma (RCC) (Chowdhury et al., 2021). Torin 1, an inhibitor of mTORC1/2, increased ciliogenesis and a non-proliferation status of hTERT-RPE1 cells (Lim et al., 2020). Rapamycin, an inhibitor of mTORC1, increased the ciliogenesis of fibroblasts derived from patients with Lowe syndrome, a ciliopathy caused by mutation of OCRL1 (Madhivanan et al., 2020). Rapamycin also elongated primary cilia and inhibited proliferation of DU145, a human prostate cancer cell line (Jamal et al., 2020). The combination of rapamycin and rosuvastatin alleviated the abnormal phenotypes of the patient-derived fibroblasts (Madhivanan et al., 2020). Rapamycin increased ciliogenesis through the upregulation of p27KIP1, a cyclin-dependent kinase inhibitor, in proliferating hTERT-RPE1 cells (Takahashi et al., 2018). p27KIP1 stimulates ciliogenesis through the regulation of the docking of preciliary vesicles to the distal appendage of the basal body (Yukimoto et al., 2020). p27KIP1 is also involved in the ciliogenesis stimulated by alprostadil, a synthetic analog of prostaglandin E1 (Garcia et al., 2022). mTORC2 increases the synthesis of glycosylceramide (Guri et al., 2017). Genz-667161, an inhibitor of glycosylceramide synthase, alleviated multi-organ pathology in Bardet-Biedl syndrome mice through increasing ciliogenesis (Husson et al., 2020).
The ubiquitin-proteasome pathway also regulates ciliogenesis through the control of the proteolysis of ciliary proteins (Hossain and Tsang, 2019; Shiromizu et al., 2020; Habeck and Schweiggert, 2022). Protein ubiquitination occurs in three steps, including binding ubiquitin to ubiquitin-activating enzymes (E1), transfer of the ubiquitin to ubiquitin-conjugating enzymes (E2), and ligation of the ubiquitin to lysine residues on the target protein. The selectivity of target protein ubiquitination is conferred by the combination of E2 and E3 enzymes. Deubiquitinases (DUB) remove the ubiquitin moieties from ubiquitinated proteins (Leznicki and Kulathu, 2017). For example, TCHP is ubiquitinated and deubiquitinated by CRL3-KCTD17 and ubiquitin-specific peptidase 8 (USP8), respectively (Kasahara et al., 2014; Kasahara et al., 2018). Knockdown of KCTD17 shortened primary cilia of hTERT-RPE1 cells through inhibition of proteolysis of TCHP, whereas knockdown of USP8 increased the ciliogenesis through stimulation of proteolysis of TCHP (Kasahara et al., 2014; Kasahara et al., 2018). IQ motif containing B1 (IQCB1), also known as NPHP5, is ubiquitinated by membrane-associated ring-CH-type finger 7 (MARCHF7) and tripartite motif containing 32 (TRIM32) and deubiquitinated by ubiquitin-specific peptidase 9X (USP9X) (Das et al., 2017a). CP110 is ubiquitinated and deubiquitinated by SCF-CyclinF and ubiquitin-specific peptidase 33 (USP33), respectively (D'Angiolella et al., 2010; Li et al., 2013). These findings suggest that targeting E3 ubiquitin ligases and DUBs that regulate ciliary protein may be used to treat cilia-related diseases (Shiromizu et al., 2020).
Scientific advances in the mechanisms for cell type-specific ciliogenesis increase the possibility to develop therapeutic drugs for diseases caused by the impairment of ciliogenesis.
The cilium contains a distinct composition of lipids, transmembrane, and membrane-associated proteins which are regulated by the transition zone at the base of the cilium. Since the primary cilia do not have a translational system, ciliary membrane proteins are supplied from the cytoplasm. Mutations in approximately 200 genes have been identified to be linked to ciliopathies (Reiter and Leroux, 2017), and a variety of ciliopathies are caused by the mistargeting of ciliary membrane proteins. Thus, the transport and turnover of receptors located on the cilia have been investigated as clinical interests. While the mechanisms of ciliary transport have been reviewed extensively by others (Malicki and Avidor-Reiss, 2014; Mukhopadhyay et al., 2017; Blacque et al., 2018; Witzgall, 2018; Long and Huang, 2019; Sánchez-Bellver et al., 2021), herein we describe the recent findings from the ciliary transport studies and provide insights into the therapeutic approaches.
The secretory pathway via vesicles that carry ciliary membrane proteins has been well-studied in rhodopsin transport (Wang and Deretic, 2014). Rhodopsin is a GPCR responsible for visual signals in photoreceptors. The photoreceptor is an excellent model for studying ciliary trafficking as the rods have exaggerated cilia, termed the outer segment (OS). OS-specific proteins such as the rhodopsin reaches the destined localization through the connecting cilium, which is equivalent to the transition zone of the primary cilium. The C-terminus of rhodopsin retains a CTS motif sequence (VxPx) that is sufficient for its targeting to OS. The general idea is that membrane-integrated proteins are newly synthesized at the endoplasmic reticulum (ER) and transported through the Golgi apparatus and the trans-Golgi network (TGN). The trafficking of newly synthesized rhodopsin from the TGN to the cilium is facilitated by nucleating the formation of a series of protein complexes called rhodopsin transport carriers (RTCs) including Arf4. Arf4-GTP binds to the VxPx motif of rhodopsin (Deretic et al., 2005) and coordinates the transport of rhodopsin in transport vesicles together with ankyrin repeat and PH domain 1 (ASAP1), Rab11-GTP, and Rab11 family interacting protein3 (FIP3) (Wang et al., 2012; Wang and Deretic, 2014). After the release of Arf4 from the complex, the C-terminal cytoplasmic tail of rhodopsin associates with the dynein light chain Tctex-1, which allows RTCs to traffic from the Golgi apparatus to the base of the cilium (Tai et al., 1999). These post-Golgi rhodopsins transit through Rab11-positive recycling endosomes, and preferentially enter the OS in the dark (Hsu et al., 2015). The C-terminal OS targeting signal of rhodopsin partially overlaps with the binding site for visual arrestin, thus the interaction between photoexcited rhodopsin and visual arrestin could contribute to retaining OS entry under light stimulation. Further investigation is required to establish this proposed model on the molecular mechanism underlying the rhodopsin OS entry regulated by light.
On the other hand, some other integral membrane proteins utilize an unconventional intracellular trafficking pathway that bypasses the Golgi apparatus on their way to the primary cilium (Witzgall, 2018). One example is peripherin-2/rds (PRPH2), a photoreceptor-specific tetraspanin protein concentrated in the OS that is essential for its development and structure (Travis et al., 1989). N-glycans attached with PRPH2 isolated from rod OS is still sensitive to endoglycosidase H (Travis et al., 1991; Tian et al., 2014). Pharmacological inhibition of the transport via the Golgi apparatus had no effect on the distribution of PRPH2 at the primary cilium (Tian et al., 2014). The C-terminus of PRPH2 contains the lysine residues for ubiquitination and binding to Hrs, a component of the endosomal sorting complex required for transport (ESCRT) complex which functions to be targeted to the late endosomal, and then to cilia (Otsu et al., 2019). Of note, the murine rod OS exhibits a complementary periodic pattern composed of PRPH2-rich discs and rhodopsin-rich discs (Hsu et al., 2015), suggesting that these proteins could be transported to the OS independently. Another example is polycystin-2 (PKD2), whose mutations are linked to autosomal dominant polycystic kidney diseases characterized by the lifelong formation of fluid-filled cysts originating from parts of the nephron and collecting ducts, leading to renal failure. PKD2 mutations account for 15% of the autosomal dominant polycystic kidney diseases whereas 85% of the disease is caused by PKD1 mutations (Mochizuki et al., 1996). The structure of PKD2 is similar to that of the TRP (transient receptor potential) channel family and is distributed to the ER and primary cilia. Interestingly, polycystin-2 bypasses the TGN on its way to the cilium while retaining the sensitivity to endoglycosidase H (Cai et al., 1999; Hoffmeister et al., 2011). In addition, the endosomal network regulates the intracellular transport of polycystin-2 via the interaction of a retromer component with the N-terminus of polycystin-2 (Feng et al., 2017), suggesting that the endocytic pathway has some roles in this unconventional transport to the primary cilia. These unconventional pathways can be a therapeutic target to modulate the ciliary expression of specific cargos.
As described in the previous section, the CTS has been found in ciliary proteins including rhodopsin (Deretic et al., 1998), polycystin-2 (Geng et al., 2006), the cyclic nucleotide-gated channel CNGB1 (Jenkins et al., 2006) as well as soluble protein ADP-ribosylation factor-like 13B (ARL13B) (Nozaki et al., 2017) and inositol polyphosphate-5-phosphatase (INPP5E) (Humbert et al., 2012). The interaction of CTS with binding partners has been investigated and attracted attention as a potential therapeutic target. Rab GTPase and Rab-like membrane trafficking proteins play a crucial role in endomembrane organization and have been linked to cilia-related processes (Blacque et al., 2018). Ciliary membrane trafficking of RTCs is mediated by the Arf4-Rab8-Rab11 cascade (Mazelova et al., 2009; Wang et al., 2012). Recently, Mahajan et al. (2023) reported that ARL13B, an ARF/Arl-family GTPase, has the ciliary targeting sequence at the C-terminal stretch of 17 amino acids containing the RVEP motif, which binds to Rab8-GDP and TNPO1 simultaneously. It has been reported that Rab29 interacts with Rab8, Rab11 and IFT20, and is required for TCR recycling in Jurkat T cells (Onnis et al., 2015). Rab23 is involved in the lateral transport of the D1 dopaminergic receptor from the plasma membrane into the ciliary membrane together with IFT-B and KIF17 (Leaf and Von Zastrow, 2015). The mutations in Rab28 have been implicated in the degenerative eye disease known as autosomal recessive cone-rod dystrophy characterized by an early onset progressive photoreceptor loss (Roosing et al., 2013). Rab28 is a conserved cilium-associated component that has been linked to ciliary transport machinery and is associated with the periciliary membrane, behaving with the IFT (Jensen et al., 2016). Rab28 is also proposed to serve cell non-autonomous functions as a regulator of releasing ciliary ectosomes carrying glial cell morphogenic factors in nematodes (Wang et al., 2014; Jensen et al., 2016; Blacque et al., 2018). Turn et al. (2022) identified new roles of ARF GAPs, ELMOD1, and ELMOD3, in protein trafficking from the Golgi to cilia.
IFT complexes are important for the anterograde and retrograde movement of proteins in primary cilia. IFT-A is critical for retrograde transport driven by a dynein-2 motor, while IFT-B complexes are required for anterograde transport driven by a kinesin-2 motor. IFT interacts with DGKδ (Ding et al., 2017). In Bardet-Biedl syndrome, it has been shown that BBSome plays a crucial role in ciliary trafficking (Jin et al., 2010; Wiens et al., 2010). It mediates the ciliary entry of SSTR3, while it is required forthe signal-dependent exit of the dopamine receptor (Drd1), SSTR3, Gpr161, and PTCH1 from cilia. TULP3, a ciliary protein (Mukhopadhyay et al., 2010), is essential for the transport of cargos including polycystin-2 (Badgandi et al., 2017). Chemicals with the activities which modify these types of machinery can be therapeutic targets.
The lipid composition of primary cilia is substantially different from the other part of the cell body. The lipid compositions in the ciliary membrane were regulated by the recruitment of ciliary phosphoinositide phosphatase, INPP5E. INPP5E maintains the phosphoinositide PI(4,5)P2 at a lower level at the ciliary membrane in mammalian cells. INPP5E localizes to the photoreceptor IS and CC, but not to the OS, and retina-specific KO for INPP5E exhibits a rapid rod-cone generation resembling Leber congenital amaurosis (LCA) (Sharif et al., 2021). The interaction between INPP5E and ARL13B is essential for their ciliary membrane retention but is dispensable for its entry into cilia (Qiu et al., 2021). Moreover, ARL13B is required for retinogenesis and the morphogenesis of photoreceptor OS discs (Dilan et al., 2019). Recently, Palicharla et al. (2023) revealed that the tubby domain of TULP3 directly interacts with the amphipathic helix structure of ARL13B, which mediates the trafficking of ARL13B itself as well as lipidated cargos. On the other hand, this important role of ARL13B in the delivery of ciliary cargos can be uncoupled from either the ciliary localization of ARL13B and its regulation of Shh signal transduction (Gigante et al., 2020), suggesting that ARL13B may be involved in the ciliary transport outside of the cilium. Ciliary membrane-associated proteins contain the covalent attachment of a lipid-like farnesyl or a geranylgeranyl group, in which post-translational modifications occur at the ER surface. The transport of lipidated cargo proteins to the cilium is assisted by lipid-binding proteins. These proteins function as trafficking chaperones in which a hydrophobic pocket binding to the lipid fraction enables the lipidated protein to be delivered into the cilium. Two proteins, PDE6D, and UNC119B, have been identified. Once the lipidated proteins undergo post-translational modification at the ER, they form a soluble complex with the trafficking chaperones and transport toward the cilium. ARL3-GTP which is activated by ARL13B is recruited to the complex, and the lipidated cargo is released and associated with the ciliary membrane (Fansa et al., 2016; Hanke-Gogokhia et al., 2016). These interactions can be a therapeutic target to facilitate or block the ciliary transport of specific cargos.
Lateral diffusion is one of the ways for ciliary targeting after the transport vesicle with ciliary membrane protein fused with the somatic plasma membrane and moves up to the primary cilia in case of smoothened (Milenkovic et al., 2009). Early transmission electron microscopy (TEM) studies of vertebrate photoreceptors have revealed a specialized structure between the inner segment and ciliary OS named “connecting cilium” by Eduardo De Robertis (1956) (de Robertis, 1956). The connections between microtubule doublets and the ciliary membrane exhibit Y-shaped structures (Tokuyasu and Yamada, 1959; Gilula and Satir, 1972). The mouse model clarified that retinitis pigmentosa GTPase regulator (RPGR) is localized to the photoreceptor transition zone, and mandatory for the distribution of rhodopsin in the OS. The diffusion barrier is the membrane portion of the ciliary transition zone, which plays a crucial role in maintaining the specific compositions of proteins and lipids in the ciliary membrane from the plasma membrane (Hu and Nelson, 2011). The final step of lateral transition remains to be elucidated. Septin 2 (SEPT2), a member of the septin family of guanosine triphosphatases, is localized at the base of the ciliary membrane and plays a crucial role in retaining receptors in the ciliary membrane (Hu et al., 2010). Chemicals for the cytoskeleton would remove these barriers and can improve ciliary transport.
In recent years, gene mutations involved in ciliopathies have been found, and many ciliary motif sequences and associated molecular machinery have been identified. Even though recent findings provide valuable insight into the molecular and cell biological mechanism underlying the trafficking of ciliary membrane proteins, the mechanisms are still not fully understood. Since the event of trafficking occurs intracellularly, only small molecules that penetrate the cell membrane are potentially available as therapeutic candidates. Moreover, in case of loss of function gene mutation, modifying the transport itself does not guarantee the functionality of the mutants. Gene therapy, especially the replacement of a faulty gene seems to be a promising approach to the treatment of ciliopathies. We will discuss the strategies of gene therapies for ciliopathies and their recent progress in the next section.
Animal models, both naturally occurring and transgenic, have informed on the molecular basis, pathogenesis, and hence into potential therapeutic targets of a multitude of hereditary diseases. Mammalian models, in particular larger models such as canine, porcine, and primates that better mimic human organs and conditions, have been instrumental in bringing new therapies to patients. Inherited retinal diseases (IRDs) which typically manifest as progressive blinding conditions are prime examples of successful translational development in recent years. While IRDs are broadly recognized to affect vision through the dysfunction of certain cell types, primarily the photoreceptors and the retinal pigment epithelia, certain subcategories of IRDs have been recognized to be ciliopathies. Herein we will review the preclinical and clinical developments of photoreceptor ciliopathy IRDs.
The landscape of ciliopathy IRD genes has been reviewed previously (Estrada-Cuzcano et al., 2012; Chen et al., 2021), and our knowledge regarding the pool of known genes and their genotype-phenotype association continues to expand. To date, 281 genes have been associated with IRDs (RetNet, accessed 8/18/23, https://web.sph.uth.edu/RetNet/) of which a considerable fraction is understood to be photoreceptor ciliopathy genes. Clinically, they manifest as broad and often overlapping category of non-syndromic IRDs encompassing retinitis pigmentosa (RP), cone-rod dystrophy (CRD), cone degeneration (CD), and LCA as well as syndromic IRDs such as Bardet-Biedl syndrome (BBS). In most non-syndromic IRDs, primary deficiency associated with the cilia leading to photoreceptor degeneration and hence vision loss in the patient was not suggested until the gene was identified and the localization or function of its protein product was suggested in photoreceptor cilia. While the IRD phenotype in syndromic ciliopathy is explained by the expression in multiple cell types of the disease-associated gene product, the non-syndromic ciliopathy IRDs suggest a cell-type specific role of the disease-associated gene product or that of its modifier, if present.
While simple organisms such as C. elegans have contributed to the understanding of the biology and pathophysiology of cilia, therapeutic advancement which targets ciliopathy has been further accelerated through the development of mammalian models. Generation and/or characterization of murine models of IRDs most of which are transgenic have allowed perhaps the most comprehensive understanding of genetic association with IRD phenotypes. Meanwhile, murine models of IRDs often disadvantaged themselves by poorly recapitulating the human phenotype. There was also variable therapeutic relevance due to anatomical differences such as the globe size and the distribution of photoreceptor cell types across the retina. Phenotypic consistency of the murine models is an advantage allowing for the reliable evaluation of therapeutic outcomes. Still, there exceptions of some forms of IRD in which phenotypic variability has been documented to be affected by the genomic background of the murine strain carrying the primary causal genetic variant (Mollema et al., 2011).
Large animal models of IRD, the most notable of which may be naturally occurring canine models which have contributed to the understanding of the molecular basis (Miyadera et al., 2012), pathogenesis, and therapeutic development. To date, at least 53 different forms of canine IRDs have been identified at the molecular level, of which 18 are found to be ciliopathy IRDs (Table 3).
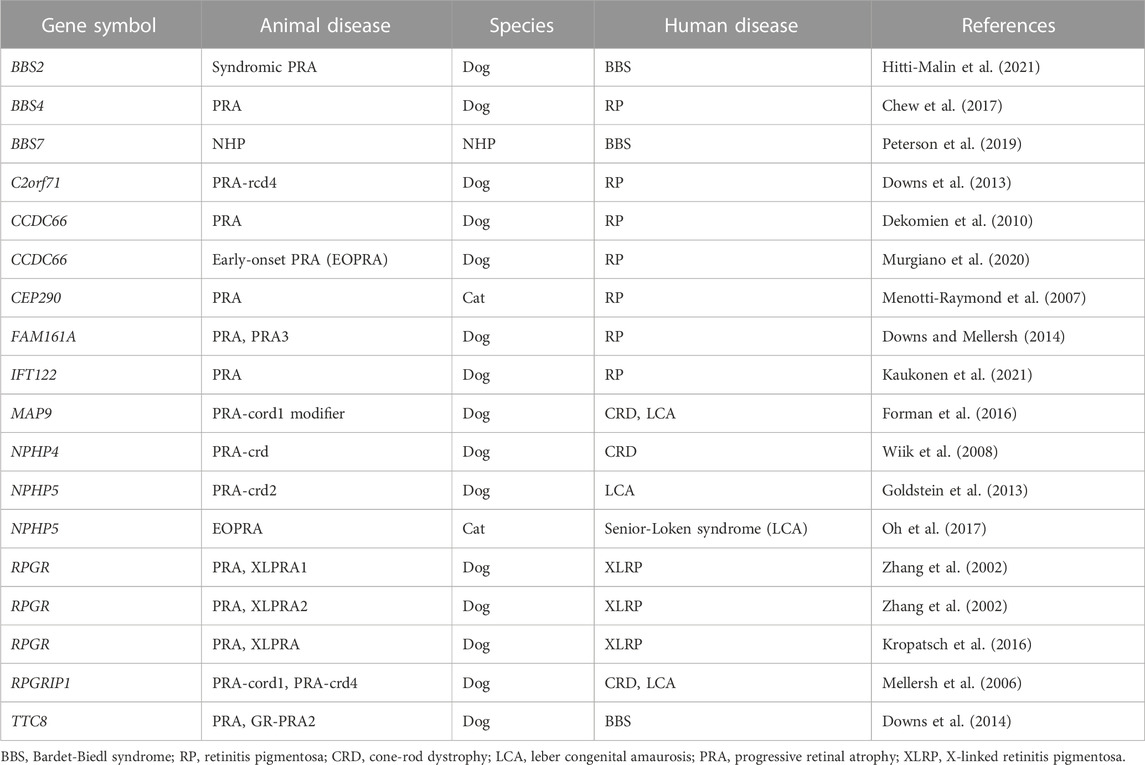
TABLE 3. Ciliopathy IRDs in large animals and their associated genes/human diseases.
Therapeutically, canine models have paved the way for new AAV-based gene therapies to be translated into clinical applications of ciliopathy IRDs. Beltran et al. (2012) evaluated gene augmentation therapy in canine models of X-linked RP (XLRP) caused by variants in RPGR which encodes a photoreceptor ciliary protein (Beltran et al., 2012). Subretinal injection of AAV-RPGR in the canine models resulted in structural and functional photoreceptor preservation. AAV-RPGR therapy in XLRP patients is now in phase 3 clinical trials (NCT04671433, NCT04794101). Lhériteau et al. (2014) evaluated AAV gene therapy in the RPGRIP1-deficient dog model of CRD/LCA. Subretinal injection of AAV-RPGRIP1 improved photoreceptor survival and functional rescue. AAV-RPGRIP1 therapies have since been optimized further now with a late-stage preclinical product that utilizes an ancestral AAV serotype with broad tissue tropism (Wassmer et al., 2017). More recently, Aguirre et al. showed that AAV-NPHP5 therapy in NPHP5 mutant dogs stably restores photoreceptor structure, function, and vision (Aguirre et al., 2021). The safety and efficacy of gene therapy in these large animal ciliopathy models of IRDs provide a path for translation to human treatment.
AAV-based gene delivery approach has been studied extensively in the emerging molecular therapies against photoreceptor ciliopathies. Clinical trials are ongoing, including those aimed at augmenting the genes such as RPGR, RPGRIP1, NPHP4, or NPHP5. Notably, these examples are monogenic IRDs where a single gene defect is causal hence its augmentation was expected to be curative. Increasingly, IRDs arising from defects in more than one gene are being recognized and molecularly characterized among patients as well as in animal models (Appelbaum et al., 2020). In a canine model of RPGRIP1-CRD, multiple loci, two of which correspond to genes encoding ciliary proteins RPGRIP1 and MAP9, have been associated with disease expression (Das et al., 2017b), potentially obscuring the optimal therapeutic target. Detailed phenotypic evaluation of mutant canines variably affected by the loci confirmed the primary disease loci as RPGRIP1 (Ripolles-Garcia et al., 2023) indicating that it would be the solely sufficient therapeutic target, rather than necessitating multiple genes to be augmented. The complexes formed by ciliary proteins at each functional site of the ciliary structure make the corresponding disease phenotypes susceptible to modifications by changes in their interacting proteins. Phenotypic and future preclinical studies in animal models provide insights into identifying and refining the optimal therapeutic target molecules.
The significance of primary cilia in normal physiology and disease pathology is increasingly being recognized as our understanding of the unique characteristics of primary cilia continues to expand. Of particular interest is the localization and physiologic role of ciliary receptors that are critical in primary cilia function as sensory organelles. In addition to the US FDA-approved SMO antagonists, the development of drugs that target other receptors is desired as diagnostic, preventive, and therapeutic agents in the future. Ongoing clarification of the molecular mechanistic basis of ciliary function including ciliogenesis, ciliary trafficking, and ciliary maintenance is leading to possible targets and windows for therapeutic intervention to be developed. For example, functional characterization of PJA2, a ubiquitin ligase, revealed that ubiquitylation of BBS1 by PJA2 regulates ciliary trafficking of GPR161 in hTERT-RPE1 cells (Chiuso et al., 2023). Cilia-specific ubiquitinome analysis identified proteins that could regulate ESCRT-dependent clathrin-mediated endocytosis and caveolin 1-mediated cilia formation in murine inner medullary collecting duct 3 (IMCD3) cells and hTERT-RPE1 cells, respectively (Aslanyan et al., 2023). In silico approach using the International Mouse Phenotype Consortium data and STRING, a database of known and predicted protein-protein interactions, have successfully found novel ciliopathy genes (Higgins et al., 2022). Chemicals to regulate these ciliary genes could be generated using technologies that lead to targeted protein degradation, such as proteolysis-targeting chimeras and small-molecule hydrophobic tagging (Bhole et al., 2023; Xie et al., 2023). In vitro phenotypic screening can also be used for repositioning clinical drugs to treat ciliopathies (Benmerah et al., 2023). For example, eupatilin, a drug used to treat gastritis and peptic ulcers, was identified as a positive hit that could induce ciliogenesis in a drug screening using CEP290-null cells (Kim et al., 2018). Recent studies have also revealed that primary cilia are involved in a wide variety of cellular functions such as the regulation of spliceosome and the facilitation of signaling (Cerulo et al., 2023; Macarelli et al., 2023), suggesting that chemicals targeting primary cilia may be applicable to treat an array of diseases than previously thought. To date, multiple examples of ciliopathy IRD gene therapy in large animal models of retinal degeneration have shown that ciliopathy phenotypes may be reversed. At present, the therapeutic strategy for the retinal ciliiopathies remain largely aimed at rescuing or restoring the function of the target cell as a whole, rather than focusing on a particular mechanistic process or subceullar structure associated specifically with the cilia (Figure 1). Owing to the considerable advances in the field of viral gene delivery and model systems, development of gene therapies that augment molecules critical in the function and structure of photoreceptor cilia such as CEP290 and RPGR have exploded in recent years (Table 4). Further advances in both in vitro and in vivo studies are expected to bring targeted therapy to patients suffering from various forms of ciliopathies.
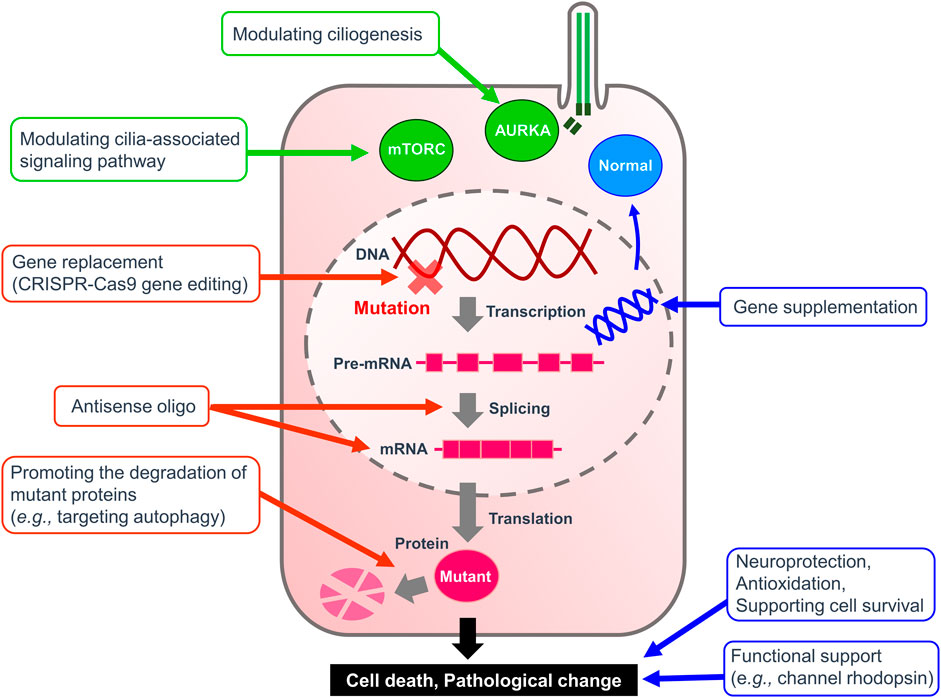
FIGURE 1. Therapeutic strategy for ciliopathies. A diagram illustrating the therapeutic targets to alleviate ciliopathies. Green arrows highlight targeting ciliogenesis and cilia-related pathways summarized in Table 2. Red arrows indicate therapies to remedy the causal mutation itself or its products. Blue arrows show the treatments to replace the malfunctioning system or to assist the survival of affected cells.
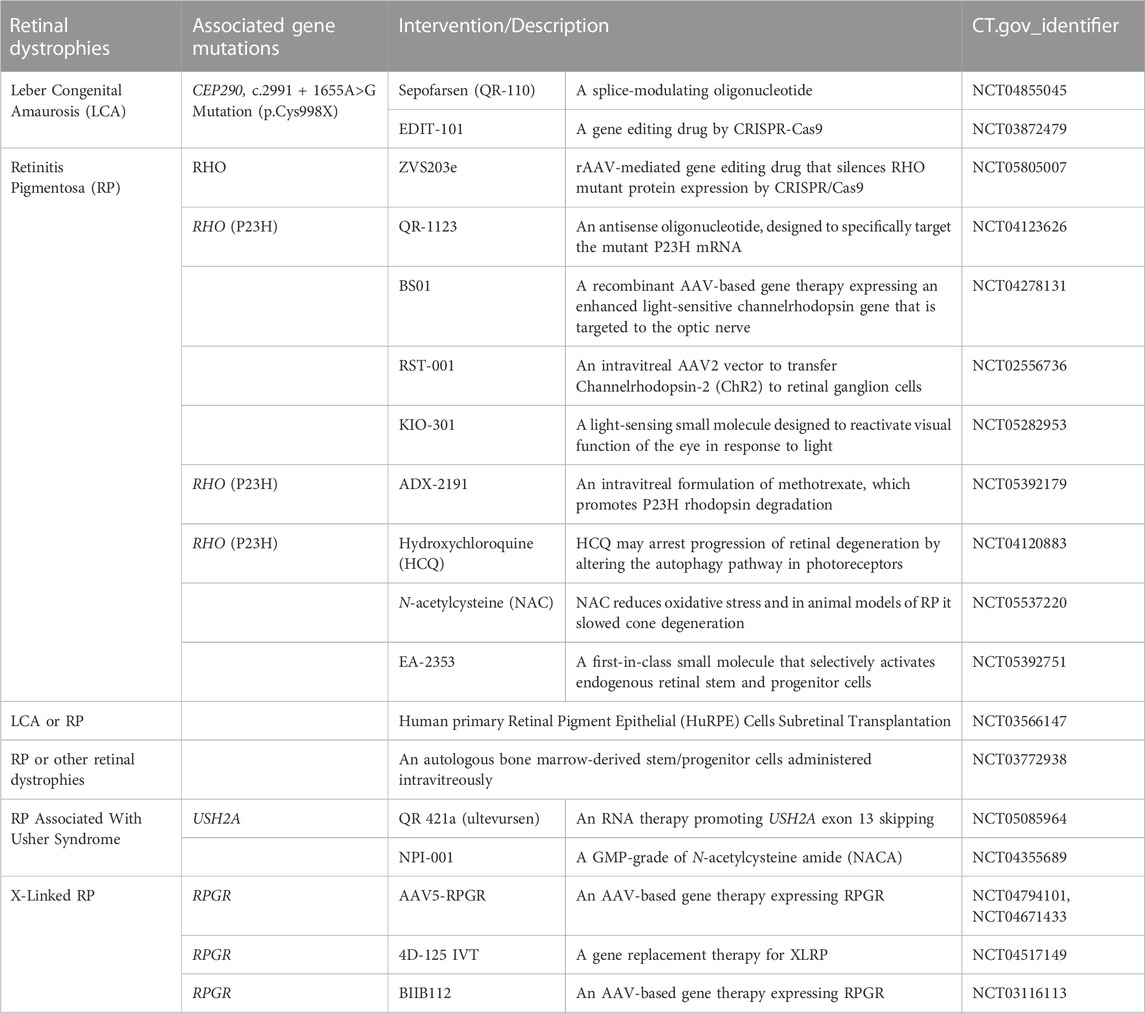
TABLE 4. Clinical trials in ciliopathies featuring retinal dystrophies.
MS, WO, KM, and YN wrote the draft. All authors contributed to the article and approved the submitted version.
This work was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for Promotion of Science (Nos 21K06059 to MS and 23K06355 to YN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aguirre, G. D., Cideciyan, A. V., Dufour, V. L., Ripolles-García, A., Sudharsan, R., Swider, M., et al. (2021). Gene therapy reforms photoreceptor structure and restores vision in NPHP5-associated Leber congenital amaurosis. Mol. Ther. 29 (12), 3528. doi:10.1016/j.ymthe.2021.10.021
Álvarez-Satta, M., and Matheu, A. (2018). Primary cilium and glioblastoma. Ther. Adv. Med. Oncol. 10, 1758835918801169. doi:10.1177/1758835918801169
Appelbaum, T., Murgiano, L., Becker, D., Santana, E., and Aguirre, G. D. (2020). Candidate genetic modifiers for RPGR retinal degeneration. Invest. Ophthalmol. Vis. Sci. 61 (14), 20. doi:10.1167/iovs.61.14.20
Aslanyan, M. G., Doornbos, C., Diwan, G. D., Anvarian, Z., Beyer, T., Junger, K., et al. (2023). A targeted multi-proteomics approach generates a blueprint of the ciliary ubiquitinome. Front. Cell Dev. Biol. 11, 1113656. doi:10.3389/fcell.2023.1113656
Badgandi, H. B., Hwang, S.-H., Shimada, I. S., Loriot, E., and Mukhopadhyay, S. (2017). Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell Biol. 216 (3), 743–760. doi:10.1083/jcb.201607095
Bagka, M., Choi, H., Heritier, M., Scapozza, L., Wu, Y., and Hoogendoorn, S. (2022). Targeted protein degradation reveals BET bromodomains as the cellular target of Hedgehog Pathway Inhibitor-1. bioRxiv. doi:10.1101/2022.08.16.504103
Baker, K., and Beales, P. L. (2009). Making sense of cilia in disease: the human ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 151C (4), 281–295. doi:10.1002/ajmg.c.30231
Barbeito, P., and Garcia-Gonzalo, F. R. (2021). HTR6 and SSTR3 targeting to primary cilia. Biochem. Soc. Trans. 49 (1), 79–91. doi:10.1042/BST20191005
Beltran, W. A., Cideciyan, A. V., Lewin, A. S., Iwabe, S., Khanna, H., Sumaroka, A., et al. (2012). Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 109 (6), 2132–2137. doi:10.1073/pnas.1118847109
Benmerah, A., Briseno-Roa, L., Annereau, J. P., and Saunier, S. (2023). Repurposing small molecules for nephronophthisis and related renal ciliopathies. Kidney Int. 104 (2), 245–253. doi:10.1016/j.kint.2023.04.027
Berbari, N. F., Johnson, A. D., Lewis, J. S., Askwith, C. C., and Mykytyn, K. (2008). Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol. Biol. Cell 19 (4), 1540–1547. doi:10.1091/mbc.e07-09-0942
Bertolin, G., and Tramier, M. (2020). Insights into the non-mitotic functions of aurora kinase A: more than just cell division. Cell. Mol. Life Sci. 77 (6), 1031–1047. doi:10.1007/s00018-019-03310-2
Beurel, E., Grieco, S. F., and Jope, R. S. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. doi:10.1016/j.pharmthera.2014.11.016
Bhole, R. P., Kute, P. R., Chikhale, R. V., Bonde, C. G., Pant, A., and Gurav, S. S. (2023). Unlocking the potential of PROTACs: A comprehensive review of protein degradation strategies in disease therapy. Bioorg Chem. 139, 106720. doi:10.1016/j.bioorg.2023.106720
Blacque, O. E., Scheidel, N., and Kuhns, S. (2018). Rab GTPases in cilium formation and function. Small GTPases 9 (1-2), 76–94. doi:10.1080/21541248.2017.1353847
Briscoe, J., and Thérond, P. P. (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14 (7), 416–429. doi:10.1038/nrm3598
Burness, C. B. (2015). Sonidegib: sonidegib: first global ApprovalGlobal approval. Drugs 75 (13), 1559–1566. doi:10.1007/s40265-015-0458-y
Cai, Y., Maeda, Y., Cedzich, A., Torres, V. E., Wu, G., Hayashi, T., et al. (1999). Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 274 (40), 28557–28565. doi:10.1074/jbc.274.40.28557
Cerulo, L., Pezzella, N., Caruso, F. P., Parente, P., Remo, A., Giordano, G., et al. (2023). Single-cell proteo-genomic reveals a comprehensive map of centrosome-associated spliceosome components. iScience 26 (5), 106602. doi:10.1016/j.isci.2023.106602
Chen, H. Y., Kelley, R. A., Li, T., and Swaroop, A. (2021). Primary cilia biogenesis and associated retinal ciliopathies. Semin. Cell Dev. Biol. 110, 70–88. doi:10.1016/j.semcdb.2020.07.013
Chen, Y., Hu, C., Hsu, C.-K., Zhang, Q., Bi, C., Asnicar, M., et al. (2002). Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology 143 (7), 2469–2477. doi:10.1210/endo.143.7.8903
Chew, T., Haase, B., Bathgate, R., Willet, C. E., Kaukonen, M. K., Mascord, L. J., et al. (2017). A coding variant in the gene bardet-biedl syndrome 4 (BBS4) is associated with a novel form of canine progressive retinal atrophy. G3 (Bethesda) 7 (7), 2327–2335. doi:10.1534/g3.117.043109
Chiuso, F., Delle Donne, R., Giamundo, G., Rinaldi, L., Borzacchiello, D., Moraca, F., et al. (2023). Ubiquitylation of BBSome is required for ciliary assembly and signaling. EMBO Rep. 24 (4), e55571. doi:10.15252/embr.202255571
Chowdhury, P., Perera, D., Powell, R. T., Talley, T., Tripathi, D. N., Park, Y. S., et al. (2021). Therapeutically actionable signaling node to rescue AURKA driven loss of primary cilia in VHL-deficient cells. Sci. Rep. 11 (1), 10461. doi:10.1038/s41598-021-89933-7
Chowdhury, P., Powell, R. T., Stephan, C., Uray, I. P., Talley, T., Karki, M., et al. (2018). Bexarotene - a novel modulator of AURKA and the primary cilium in VHL-deficient cells. J. Cell Sci. 131 (24), jcs219923. doi:10.1242/jcs.219923
Clement, C. A., Ajbro, K. D., Koefoed, K., Vestergaard, M. L., Veland, I. R., Henriques de Jesus, M. P. R., et al. (2013). TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 3 (6), 1806–1814. doi:10.1016/j.celrep.2013.05.020
Conduit, S. E., and Vanhaesebroeck, B. (2020). Phosphoinositide lipids in primary cilia biology. Biochem. J. 477 (18), 3541–3565. doi:10.1042/BCJ20200277
D'Angiolella, V., Donato, V., Vijayakumar, S., Saraf, A., Florens, L., Washburn, M. P., et al. (2010). SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466 (7302), 138–142. doi:10.1038/nature09140
Das, A., Qian, J., and Tsang, W. Y. (2017a). USP9X counteracts differential ubiquitination of NPHP5 by MARCH7 and BBS11 to regulate ciliogenesis. PLoS Genet. 13 (5), e1006791. doi:10.1371/journal.pgen.1006791
Das, R. G., Marinho, F. P., Iwabe, S., Santana, E., McDaid, K. S., Aguirre, G. D., et al. (2017b). Variabilities in retinal function and structure in a canine model of cone-rod dystrophy associated with RPGRIP1 support multigenic etiology. Sci. Rep. 7 (1), 12823. doi:10.1038/s41598-017-13112-w
Dateyama, I., Sugihara, Y., Chiba, S., Ota, R., Nakagawa, R., Kobayashi, T., et al. (2019). RABL2 positively controls localization of GPCRs in mammalian primary cilia. J. Cell Sci. 132 (2), jcs224428. doi:10.1242/jcs.224428
de Robertis, E. (1956). Electron microscope observations on the submicroscopic organization of the retinal rods. J. Biophys. Biochem. Cytol. 2 (3), 319–330. doi:10.1083/jcb.2.3.319
Dekomien, G., Vollrath, C., Petrasch-Parwez, E., Boeve, M. H., Akkad, D. A., Gerding, W. M., et al. (2010). Progressive retinal atrophy in schapendoes dogs: mutation of the newly identified CCDC66 gene. Neurogenetics 11 (2), 163–174. doi:10.1007/s10048-009-0223-z
Dere, R., Perkins, A. L., Bawa-Khalfe, T., Jonasch, D., and Walker, C. L. (2015). β-catenin links von Hippel-Lindau to aurora kinase A and loss of primary cilia in renal cell carcinoma. J. Am. Soc. Nephrol. 26 (3), 553–564. doi:10.1681/ASN.2013090984
Deretic, D., Schmerl, S., Hargrave, P. A., Arendt, A., and McDowell, J. H. (1998). Regulation of sorting and post-Golgi trafficking of rhodopsin by its C-terminal sequence QVS(A)PA. Proc. Natl. Acad. Sci. U. S. A. 95 (18), 10620–10625. doi:10.1073/pnas.95.18.10620
Deretic, D., Williams, A. H., Ransom, N., Morel, V., Hargrave, P. A., and Arendt, A. (2005). Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc. Natl. Acad. Sci. U. S. A. 102 (9), 3301–3306. doi:10.1073/pnas.0500095102
Dilan, T. L., Moye, A. R., Salido, E. M., Saravanan, T., Kolandaivelu, S., Goldberg, A. F. X., et al. (2019). ARL13B, a joubert syndrome-associated protein, is critical for retinogenesis and elaboration of mouse photoreceptor outer segments. J. Neurosci. 39 (8), 1347–1364. doi:10.1523/JNEUROSCI.1761-18.2018
Ding, J., Shao, L., Yao, Y., Tong, X., Liu, H., Yue, S., et al. (2017). DGKδ triggers endoplasmic reticulum release of IFT88-containing vesicles destined for the assembly of primary cilia. Sci. Rep. 7 (1), 5296. doi:10.1038/s41598-017-05680-8
Domire, J. S., Green, J. A., Lee, K. G., Johnson, A. D., Askwith, C. C., and Mykytyn, K. (2011). Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell. Mol. Life Sci. 68 (17), 2951–2960. doi:10.1007/s00018-010-0603-4
Douglas, R. S., Kahaly, G. J., Patel, A., Sile, S., Thompson, E. H. Z., Perdok, R., et al. (2020). Teprotumumab for the treatment of active thyroid eye disease. N. Engl. J. Med. 382 (4), 341–352. doi:10.1056/NEJMoa1910434
Downs, L. M., Bell, J. S., Freeman, J., Hartley, C., Hayward, L. J., and Mellersh, C. S. (2013). Late-onset progressive retinal atrophy in the Gordon and Irish Setter breeds is associated with a frameshift mutation in C2orf71. Anim. Genet. 44 (2), 169–177. doi:10.1111/j.1365-2052.2012.02379.x
Downs, L. M., and Mellersh, C. S. (2014). An Intronic SINE insertion in FAM161A that causes exon-skipping is associated with progressive retinal atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS One 9 (4), e93990. doi:10.1371/journal.pone.0093990
Downs, L. M., Wallin-Hakansson, B., Bergstrom, T., and Mellersh, C. S. (2014). A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever. Canine Genet. Epidemiol. 1, 4. doi:10.1186/2052-6687-1-4
Duncan, C. G., Killela, P. J., Payne, C. A., Lampson, B., Chen, W. C., Liu, J., et al. (2010). Integrated genomic analyses identify ERRFI1 and TACC3 as glioblastoma-targeted genes. Oncotarget 1 (4), 265–277. doi:10.18632/oncotarget.137
Duong Phu, M., Bross, S., Burkhalter, M. D., and Philipp, M. (2021). Limitations and opportunities in the pharmacotherapy of ciliopathies. Pharmacol. Ther. 225, 107841. doi:10.1016/j.pharmthera.2021.107841
Echelard, Y., Epstein, D. J., St-Jacques, B., Shen, L., Mohler, J., McMahon, J. A., et al. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75 (7), 1417–1430. doi:10.1016/0092-8674(93)90627-3
Egeberg, D. L., Lethan, M., Manguso, R., Schneider, L., Awan, A., Jørgensen, T. S., et al. (2012). Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia 1 (1), 15. doi:10.1186/2046-2530-1-15
Eguether, T., Cordelieres, F. P., and Pazour, G. J. (2018). Intraflagellar transport is deeply integrated in hedgehog signaling. Mol. Biol. Cell 29 (10), 1178–1189. doi:10.1091/mbc.E17-10-0600
Einstein, E. B., Patterson, C. A., Hon, B. J., Regan, K. A., Reddi, J., Melnikoff, D. E., et al. (2010). Somatostatin signaling in neuronal cilia is criticalfor object recognition memory. J. Neurosci. 30 (12), 4306–4314. doi:10.1523/JNEUROSCI.5295-09.2010
Estrada-Cuzcano, A., Roepman, R., Cremers, F. P. M., den Hollander, A. I., and Mans, D. A. (2012). Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet. 21 (R1), R111–R124. doi:10.1093/hmg/dds298
Fansa, E. K., Kösling, S. K., Zent, E., Wittinghofer, A., and Ismail, S. (2016). PDE6δ-mediated sorting of INPP5E into the cilium is determined by cargo-carrier affinity. Nat. Commun. 7, 11366. doi:10.1038/ncomms11366
Feng, S., Streets, A. J., Nesin, V., Tran, U., Nie, H., Onopiuk, M., et al. (2017). The sorting nexin 3 retromer pathway regulates the cell surface localization and activity of a wnt-activated polycystin channel complex. J. Am. Soc. Nephrol. 28 (10), 2973–2984. doi:10.1681/ASN.2016121349
Fisas, A., Codony, X., Romero, G., Dordal, A., Giraldo, J., Mercé, R., et al. (2006). Chronic 5-HT6 receptor modulation by E-6837 induces hypophagia and sustained weight loss in diet-induced obese rats. Br. J. Pharmacol. 148 (7), 973–983. doi:10.1038/sj.bjp.0706807
Forman, O. P., Hitti, R. J., Boursnell, M., Miyadera, K., Sargan, D., and Mellersh, C. (2016). Canine genome assembly correction facilitates identification of a MAP9 deletion as a potential age of onset modifier for RPGRIP1-associated canine retinal degeneration. Mamm. Genome 27 (5-6), 237–245. doi:10.1007/s00335-016-9627-x
Garcia, H., Serafin, A. S., Silbermann, F., Porée, E., Viau, A., Mahaut, C., et al. (2022). Agonists of prostaglandin E2 receptors as potential first in class treatment for nephronophthisis and related ciliopathies. Proc. Natl. Acad. Sci. U. S. A. 119 (18), e2115960119. doi:10.1073/pnas.2115960119
Gazea, M., Tasouri, E., Tolve, M., Bosch, V., Kabanova, A., Gojak, C., et al. (2016). Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 409 (1), 55–71. doi:10.1016/j.ydbio.2015.10.033
Gencer, S., Oleinik, N., Kim, J., Panneer Selvam, S., De Palma, R., Dany, M., et al. (2017). TGF-β receptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci. Signal. 10 (502), eaam7464. doi:10.1126/scisignal.aam7464
Geng, L., Okuhara, D., Yu, Z., Tian, X., Cai, Y., Shibazaki, S., et al. (2006). Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J. Cell Sci. 119 (7), 1383–1395. doi:10.1242/jcs.02818
Gigante, E. D., Taylor, M. R., Ivanova, A. A., Kahn, R. A., and Caspary, T. (2020). ARL13B regulates Sonic hedgehog signaling from outside primary cilia. Elife 9, e50434. doi:10.7554/eLife.50434
Gilula, N. B., and Satir, P. (1972). The ciliary necklace. A ciliary membrane specialization. J. Cell Biol. 53 (2), 494–509. doi:10.1083/jcb.53.2.494
Goetz, S. C., Liem, K. F., and Anderson, K. V. (2012). The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 151 (4), 847–858. doi:10.1016/j.cell.2012.10.010
Goldstein, O., Mezey, J. G., Schweitzer, P. A., Boyko, A. R., Gao, C., Bustamante, C. D., et al. (2013). IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds. Invest. Ophthalmol. Vis. Sci. 54 (10), 7005–7019. doi:10.1167/iovs.13-12915
Gould, S. E., Low, J. A., Marsters, J. C., Robarge, K., Rubin, L. L., de Sauvage, F. J., et al. (2014). Discovery and preclinical development of vismodegib. Expert Opin. Drug Discov. 9 (8), 969–984. doi:10.1517/17460441.2014.920816
Gradilone, S. A., Radtke, B. N., Bogert, P. S., Huang, B. Q., Gajdos, G. B., and LaRusso, N. F. (2013). HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 73 (7), 2259–2270. doi:10.1158/0008-5472.CAN-12-2938
Guri, Y., Colombi, M., Dazert, E., Hindupur, S. K., Roszik, J., Moes, S., et al. (2017). mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32 (6), 807–823. doi:10.1016/j.ccell.2017.11.011
Habeck, G., and Schweiggert, J. (2022). Proteolytic control in ciliogenesis: temporal restriction or early initiation? Bioessays 44 (9), e2200087. doi:10.1002/bies.202200087
Hallikas, O., Palin, K., Sinjushina, N., Rautiainen, R., Partanen, J., Ukkonen, E., et al. (2006). Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124 (1), 47–59. doi:10.1016/j.cell.2005.10.042
Han, Y.-G., Kim, H. J., Dlugosz, A. A., Ellison, D. W., Gilbertson, R. J., and Alvarez-Buylla, A. (2009). Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 15 (9), 1062–1065. doi:10.1038/nm.2020
Han, Y., Wang, B., Cho, Y. S., Zhu, J., Wu, J., Chen, Y., et al. (2019). Phosphorylation of ci/gli by fused family kinases promotes hedgehog signaling. Dev. Cell 50 (5), 610–626. doi:10.1016/j.devcel.2019.06.008
Hanke-Gogokhia, C., Wu, Z., Gerstner, C. D., Frederick, J. M., Zhang, H., and Baehr, W. (2016). Arf-like protein 3 (ARL3) regulates protein trafficking and ciliogenesis in mouse photoreceptors. J. Biol. Chem. 291 (13), 7142–7155. doi:10.1074/jbc.M115.710954
Heyer, C. M., Sundsbak, J. L., Abebe, K. Z., Chapman, A. B., Torres, V. E., Grantham, J. J., et al. (2016). Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27 (9), 2872–2884. doi:10.1681/ASN.2015050583
Higgins, K., Moore, B. A., Berberovic, Z., Adissu, H. A., Eskandarian, M., Flenniken, A. M., et al. (2022). Analysis of genome-wide knockout mouse database identifies candidate ciliopathy genes. Sci. Rep. 12 (1), 20791. doi:10.1038/s41598-022-19710-7
Hilgendorf, K. I., Johnson, C. T., Mezger, A., Rice, S. L., Norris, A. M., Demeter, J., et al. (2019). Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell 179 (6), 1289–1305. doi:10.1016/j.cell.2019.11.005
Hirasawa, A., Tsumaya, K., Awaji, T., Katsuma, S., Adachi, T., Yamada, M., et al. (2005). Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11 (1), 90–94. doi:10.1038/nm1168
Hitti-Malin, R. J., Burmeister, L. M., Lingaas, F., Kaukonen, M., Pettinen, I., Lohi, H., et al. (2021). A missense variant in the bardet-biedl syndrome 2 gene (BBS2) leads to a novel syndromic retinal degeneration in the shetland sheepdog. Genes (Basel) 12 (11), 1771. doi:10.3390/genes12111771
Hoffmeister, H., Babinger, K., Gürster, S., Cedzich, A., Meese, C., Schadendorf, K., et al. (2011). Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 192 (4), 631–645. doi:10.1083/jcb.201007050
Hossain, D., and Tsang, W. Y. (2019). The role of ubiquitination in the regulation of primary cilia assembly and disassembly. Semin. Cell Dev. Biol. 93, 145–152. doi:10.1016/j.semcdb.2018.09.005
Hsu, Y.-C., Chuang, J.-Z., and Sung, C.-H. (2015). Light regulates the ciliary protein transport and outer segment disc renewal of mammalian photoreceptors. Dev. Cell 32 (6), 731–742. doi:10.1016/j.devcel.2015.01.027
Hu, Q., Milenkovic, L., Jin, H., Scott, M. P., Nachury, M. V., Spiliotis, E. T., et al. (2010). A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329 (5990), 436–439. doi:10.1126/science.1191054
Hu, Q., and Nelson, W. J. (2011). Ciliary diffusion barrier: the gatekeeper for the primary cilium compartment. Cytoskeleton 68 (6), 313–324. doi:10.1002/cm.20514
Humbert, M. C., Weihbrecht, K., Searby, C. C., Li, Y., Pope, R. M., Sheffield, V. C., et al. (2012). ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. U. S. A. 109 (48), 19691–19696. doi:10.1073/pnas.1210916109
Husson, H., Bukanov, N. O., Moreno, S., Smith, M. M., Richards, B., Zhu, C., et al. (2020). Correction of cilia structure and function alleviates multi-organ pathology in Bardet-Biedl syndrome mice. Hum. Mol. Genet. 29 (15), 2508–2522. doi:10.1093/hmg/ddaa138
Ingham, P. W., and McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15 (23), 3059–3087. doi:10.1101/gad.938601
Inoko, A., Matsuyama, M., Goto, H., Ohmuro-Matsuyama, Y., Hayashi, Y., Enomoto, M., et al. (2012). Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J. Cell Biol. 197 (3), 391–405. doi:10.1083/jcb.201106101
Ishikawa, H., and Marshall, W. F. (2017). Intraflagellar transport and ciliary dynamics. Cold Spring Harb. Perspect. Biol. 9 (3), a021998. doi:10.1101/cshperspect.a021998
Jamal, M. H., Nunes, A. C. F., Vaziri, N. D., Ramchandran, R., Bacallao, R. L., Nauli, A. M., et al. (2020). Rapamycin treatment correlates changes in primary cilia expression with cell cycle regulation in epithelial cells. Biochem. Pharmacol. 178, 114056. doi:10.1016/j.bcp.2020.114056
Janeček, M., Rossmann, M., Sharma, P., Emery, A., Huggins, D. J., Stockwell, S. R., et al. (2016). Allosteric modulation of AURKA kinase activity by a small-molecule inhibitor of its protein-protein interaction with TPX2. Sci. Rep. 6, 28528. doi:10.1038/srep28528
Jeffries, E. P., Di Filippo, M., and Galbiati, F. (2019). Failure to reabsorb the primary cilium induces cellular senescence. FASEB J. 33 (4), 4866–4882. doi:10.1096/fj.201801382R
Jeng, K.-S., Chang, C.-F., and Lin, S.-S. (2020). Sonic hedgehog signaling in organogenesis, tumors, and tumor microenvironments. Int. J. Mol. Sci. 21 (3), 758. doi:10.3390/ijms21030758
Jenkins, P. M., Hurd, T. W., Zhang, L., McEwen, D. P., Brown, R. L., Margolis, B., et al. (2006). Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Curr. Biol. 16 (12), 1211–1216. doi:10.1016/j.cub.2006.04.034
Jensen, V. L., Carter, S., Sanders, A. A. W. M., Li, C., Kennedy, J., Timbers, T. A., et al. (2016). Whole-organism developmental expression profiling identifies RAB-28 as a novel ciliary GTPase associated with the BBSome and intraflagellar transport. PLoS Genet. 12 (12), e1006469. doi:10.1371/journal.pgen.1006469
Jerman, S., Ward, H. H., Lee, R., Lopes, C. A., Fry, A. M., MacDougall, M., et al. (2014). OFD1 and flotillins are integral components of a ciliary signaling protein complex organized by polycystins in renal epithelia and odontoblasts. PLoS One 9 (9), e106330. doi:10.1371/journal.pone.0106330
Jin, H., White, S. R., Shida, T., Schulz, S., Aguiar, M., Gygi, S. P., et al. (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141 (7), 1208–1219. doi:10.1016/j.cell.2010.05.015
Kasahara, K., Aoki, H., Kiyono, T., Wang, S., Kagiwada, H., Yuge, M., et al. (2018). EGF receptor kinase suppresses ciliogenesis through activation of USP8 deubiquitinase. Nat. Commun. 9 (1), 758. doi:10.1038/s41467-018-03117-y
Kasahara, K., and Inagaki, M. (2021). Primary ciliary signaling: links with the cell cycle. Trends Cell Biol. 31 (12), 954–964. doi:10.1016/j.tcb.2021.07.009
Kasahara, K., Kawakami, Y., Kiyono, T., Yonemura, S., Kawamura, Y., Era, S., et al. (2014). Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun. 5, 5081. doi:10.1038/ncomms6081
Kaukonen, M., Pettinen, I. T., Wickstrom, K., Arumilli, M., Donner, J., Juhola, I. J., et al. (2021). A missense variant in IFT122 associated with a canine model of retinitis pigmentosa. Hum. Genet. 140 (11), 1569–1579. doi:10.1007/s00439-021-02266-3
Keitel, V., Ullmer, C., and Haussinger, D. (2010). The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol. Chem. 391 (7), 785–789. doi:10.1515/BC.2010.077
Kim, M., Kim, M., Lee, M.-S., Kim, C.-H., and Lim, D.-S. (2014). The MST1/2-SAV1 complex of the Hippo pathway promotes ciliogenesis. Nat. Commun. 5, 5370. doi:10.1038/ncomms6370
Kim, Y. J., Kim, S., Jung, Y., Jung, E., Kwon, H. J., and Kim, J. (2018). Eupatilin rescues ciliary transition zone defects to ameliorate ciliopathy-related phenotypes. J. Clin. Invest. 128 (8), 3642–3648. doi:10.1172/JCI99232
Kimura, I., Ichimura, A., Ohue-Kitano, R., and Igarashi, M. (2020). Free fatty acid receptors in health and disease. Physiol. Rev. 100 (1), 171–210. doi:10.1152/physrev.00041.2018
King, N., Westbrook, M. J., Young, S. L., Kuo, A., Abedin, M., Chapman, J., et al. (2008). The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451 (7180), 783–788. doi:10.1038/nature06617
Kobayashi, T., Nakazono, K., Tokuda, M., Mashima, Y., Dynlacht, B. D., and Itoh, H. (2017). HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma. EMBO Rep. 18 (2), 334–343. doi:10.15252/embr.201541922
Kobayashi, Y., Okada, T., Miki, D., Sekino, Y., Koganezawa, N., Shirao, T., et al. (2021a). Properties of primary cilia in melanin-concentrating hormone receptor 1-bearing hippocampal neurons in vivo and in vitro. Neurochem. Int. 142, 104902. doi:10.1016/j.neuint.2020.104902
Kobayashi, Y., Takemoto, R., Yamato, S., Okada, T., Iijima, M., Uematsu, Y., et al. (2018). Depression-resistant phenotype in mice overexpressing regulator of G protein signaling 8 (RGS8). Neuroscience 383, 160–169. doi:10.1016/j.neuroscience.2018.05.005
Kobayashi, Y., Tomoshige, S., Imakado, K., Sekino, Y., Koganezawa, N., Shirao, T., et al. (2021b). Ciliary GPCR-based transcriptome as a key regulator of cilia length control. FASEB Bioadv 3 (9), 744–767. doi:10.1096/fba.2021-00029
Koemeter-Cox, A. I., Sherwood, T. W., Green, J. A., Steiner, R. A., Berbari, N. F., Yoder, B. K., et al. (2014). Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc. Natl. Acad. Sci. U. S. A. 111 (28), 10335–10340. doi:10.1073/pnas.1403286111
Kottgen, M., Buchholz, B., Garcia-Gonzalez, M. A., Kotsis, F., Fu, X., Doerken, M., et al. (2008). TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 182 (3), 437–447. doi:10.1083/jcb.200805124
Kropatsch, R., Akkad, D. A., Frank, M., Rosenhagen, C., Altmuller, J., Nurnberg, P., et al. (2016). A large deletion in RPGR causes XLPRA in Weimaraner dogs. Canine Genet. Epidemiol. 3, 7. doi:10.1186/s40575-016-0037-x
Lai, Y., and Jiang, Y. (2020). Reciprocal regulation between primary cilia and mTORC1. Genes 11 (6), 711. doi:10.3390/genes11060711
Leaf, A., and Von Zastrow, M. (2015). Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4, e06996. doi:10.7554/eLife.06996
Lee, K. H., Johmura, Y., Yu, L.-R., Park, J.-E., Gao, Y., Bang, J. K., et al. (2012). Identification of a novel Wnt5a-CK1ɛ-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J. 31 (14), 3104–3117. doi:10.1038/emboj.2012.144
Lee, S.-H., Joo, K., Jung, E. J., Hong, H., Seo, J., and Kim, J. (2018). Export of membrane proteins from the Golgi complex to the primary cilium requires the kinesin motor, KIFC1. FASEB J. 32 (2), 957–968. doi:10.1096/fj.201700563R
Leitch, C. C., and Zaghloul, N. A. (2014). BBS4 is necessary for ciliary localization of TrkB receptor and activation by BDNF. PLoS One 9 (5), e98687. doi:10.1371/journal.pone.0098687
Lesiak, A. J., Brodsky, M., Cohenca, N., Croicu, A. G., and Neumaier, J. F. (2018). Restoration of physiological expression of 5-HT6 receptor into the primary cilia of null mutant neurons lengthens both primary cilia and dendrites. Mol. Pharmacol. 94 (1), 731–742. doi:10.1124/mol.117.111583
Leznicki, P., and Kulathu, Y. (2017). Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 130 (12), 1997–2006. doi:10.1242/jcs.201855
Lhériteau, E., Petit, L., Weber, M., Le Meur, G., Deschamps, J.-Y., Libeau, L., et al. (2014). Successful gene therapy in the RPGRIP1-deficient dog: A large model of cone–rod dystrophy. Mol. Ther. 22 (2), 265–277. doi:10.1038/mt.2013.232
Li, A., Saito, M., Chuang, J.-Z., Tseng, Y.-Y., Dedesma, C., Tomizawa, K., et al. (2011). Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat. Cell Biol. 13 (4), 402–411. doi:10.1038/ncb2218
Li, D., Zhu, J., Firozi, P. F., Abbruzzese, J. L., Evans, D. B., Cleary, K., et al. (2003). Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin. Cancer Res. 9 (3), 991–997.
Li, J., D'Angiolella, V., Seeley, E. S., Kim, S., Kobayashi, T., Fu, W., et al. (2013). USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495 (7440), 255–259. doi:10.1038/nature11941
Liew, G. M., Ye, F., Nager, A. R., Murphy, J. P., Lee, J. S., Aguiar, M., et al. (2014). The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 31 (3), 265–278. doi:10.1016/j.devcel.2014.09.004
Lim, J., Son, J., Ryu, J., and Kim, J.-E. (2020). SIRT2 affects primary cilia formation by regulating mTOR signaling in retinal pigmented epithelial cells. Int. J. Mol. Sci. 21 (6), 2240. doi:10.3390/ijms21062240
Loktev, A. V., and Jackson, P. K. (2013). Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep. 5 (5), 1316–1329. doi:10.1016/j.celrep.2013.11.011
Long, H., and Huang, K. (2019). Transport of ciliary membrane proteins. Front. Cell Dev. Biol. 7, 381. doi:10.3389/fcell.2019.00381
Ma, R., Li, W. P., Rundle, D., Kong, J., Akbarali, H. I., and Tsiokas, L. (2005). PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol. Cell Biol. 25 (18), 8285–8298. doi:10.1128/MCB.25.18.8285-8298.2005
Macarelli, V., Leventea, E., and Merkle, F. T. (2023). Regulation of the length of neuronal primary cilia and its potential effects on signalling. Trends Cell Biol. doi:10.1016/j.tcb.2023.05.005
Madhivanan, K., Ramadesikan, S., Hsieh, W.-C., Aguilar, M. C., Hanna, C. B., Bacallao, R. L., et al. (2020). Lowe syndrome patient cells display mTOR- and RhoGTPase-dependent phenotypes alleviated by rapamycin and statins. Hum. Mol. Genet. 29 (10), 1700–1715. doi:10.1093/hmg/ddaa086
Mahajan, D., Madugula, V., and Lu, L. (2023). Rab8 and TNPO1 are ciliary transport adaptors for GTPase Arl13b by interacting with its RVEP motif-containing ciliary targeting sequence. J. Biol. Chem. 299, 104604. doi:10.1016/j.jbc.2023.104604
Malicki, J., and Avidor-Reiss, T. (2014). From the cytoplasm into the cilium: bon voyage. Organogenesis 10 (1), 138–157. doi:10.4161/org.29055
Manning, B. D., and Toker, A. (2017). AKT/PKB signaling: aKT/PKB signaling: navigating the networkthe network. Cell 169 (3), 381–405. doi:10.1016/j.cell.2017.04.001
Mao, C., Xiao, P., Tao, X. N., Qin, J., He, Q. T., Zhang, C., et al. (2023). Unsaturated bond recognition leads to biased signal in a fatty acid receptor. Science 380 (6640), eadd6220. doi:10.1126/science.add6220
Margaria, J. P., Campa, C. C., De Santis, M. C., Hirsch, E., and Franco, I. (2020). The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell. Signal. 66, 109468. doi:10.1016/j.cellsig.2019.109468
Marley, A., Choy, R. W., and von Zastrow, M. (2013). GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross-talk. PLoS One 8 (8), e70857. doi:10.1371/journal.pone.0070857
Marsh, D. J., Weingarth, D. T., Novi, D. E., Chen, H. Y., Trumbauer, M. E., Chen, A. S., et al. (2002). Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc. Natl. Acad. Sci. U. S. A. 99 (5), 3240–3245. doi:10.1073/pnas.052706899
Martin, L., Kaci, N., Estibals, V., Goudin, N., Garfa-Traore, M., Benoist-Lasselin, C., et al. (2018). Constitutively-active FGFR3 disrupts primary cilium length and IFT20 trafficking in various chondrocyte models of achondroplasia. Hum. Mol. Genet. 27 (1), 1–13. doi:10.1093/hmg/ddx374
Martín-Guerrero, E., Tirado-Cabrera, I., Buendía, I., Alonso, V., Gortázar, A. R., and Ardura, J. A. (2020). Primary cilia mediate parathyroid hormone receptor type 1 osteogenic actions in osteocytes and osteoblasts via Gli activation. J. Cell. Physiol. 235 (10), 7356–7369. doi:10.1002/jcp.29636
Masyuk, A. I., Gradilone, S. A., Banales, J. M., Huang, B. Q., Masyuk, T. V., Lee, S. O., et al. (2008). Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 295 (4), G725–G734. doi:10.1152/ajpgi.90265.2008
Mazelova, J., Astuto-Gribble, L., Inoue, H., Tam, B. M., Schonteich, E., Prekeris, R., et al. (2009). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 28 (3), 183–192. doi:10.1038/emboj.2008.267
Mellersh, C. S., Boursnell, M. E., Pettitt, L., Ryder, E. J., Holmes, N. G., Grafham, D., et al. (2006). Canine RPGRIP1 mutation establishes cone-rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics 88 (3), 293–301. doi:10.1016/j.ygeno.2006.05.004
Menotti-Raymond, M., David, V. A., Schaffer, A. A., Stephens, R., Wells, D., Kumar-Singh, R., et al. (2007). Mutation in CEP290 discovered for cat model of human retinal degeneration. J. Hered. 98 (3), 211–220. doi:10.1093/jhered/esm019
Milenkovic, L., Scott, M. P., and Rohatgi, R. (2009). Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 187 (3), 365–374. doi:10.1083/jcb.200907126
Miyadera, K., Acland, G. M., and Aguirre, G. D. (2012). Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies. Mamm. Genome 23 (1-2), 40–61. doi:10.1007/s00335-011-9361-3
Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S. L., Veldhuisen, B., Saris, J. J., et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272 (5266), 1339–1342. doi:10.1126/science.272.5266.1339
Mollema, N. J., Yuan, Y., Jelcick, A. S., Sachs, A. J., von Alpen, D., Schorderet, D., et al. (2011). Nuclear receptor Rev-erb alpha (Nr1d1) functions in concert with Nr2e3 to regulate transcriptional networks in the retina. PLoS One 6 (3), e17494. doi:10.1371/journal.pone.0017494
Mukhopadhyay, S., Badgandi, H. B., Hwang, S.-H., Somatilaka, B., Shimada, I. S., and Pal, K. (2017). Trafficking to the primary cilium membrane. Mol. Biol. Cell 28 (2), 233–239. doi:10.1091/mbc.E16-07-0505
Mukhopadhyay, S., Wen, X., Chih, B., Nelson, C. D., Lane, W. S., Scales, S. J., et al. (2010). TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24 (19), 2180–2193. doi:10.1101/gad.1966210
Mukhopadhyay, S., Wen, X., Ratti, N., Loktev, A., Rangell, L., Scales, S. J., et al. (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152 (1-2), 210–223. doi:10.1016/j.cell.2012.12.026
Murgiano, L., Becker, D., Spector, C., Carlin, K., Santana, E., Niggel, J. K., et al. (2020). CCDC66 frameshift variant associated with a new form of early-onset progressive retinal atrophy in Portuguese Water Dogs. Sci. Rep. 10 (1), 21162. doi:10.1038/s41598-020-77980-5
Nachury, M. V., Loktev, A. V., Zhang, Q., Westlake, C. J., Peranen, J., Merdes, A., et al. (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129 (6), 1201–1213. doi:10.1016/j.cell.2007.03.053
Nagata, A., Hamamoto, A., Horikawa, M., Yoshimura, K., Takeda, S., and Saito, Y. (2013). Characterization of ciliary targeting sequence of rat melanin-concentrating hormone receptor 1. Gen. Comp. Endocrinol. 188, 159–165. doi:10.1016/j.ygcen.2013.02.020
Nager, A. R., Goldstein, J. S., Herranz-Perez, V., Portran, D., Ye, F., Garcia-Verdugo, J. M., et al. (2017). An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell 168 (1-2), 252–263. doi:10.1016/j.cell.2016.11.036
Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33 (2), 129–137. doi:10.1038/ng1076
Niewiadomski, P., Kong, J. H., Ahrends, R., Ma, Y., Humke, E. W., Khan, S., et al. (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 6 (1), 168–181. doi:10.1016/j.celrep.2013.12.003
Nishimura, Y., Kasahara, K., Shiromizu, T., Watanabe, M., and Inagaki, M. (2019). Primary cilia as signaling hubs in health and disease. Adv. Sci. 6 (1), 1801138. doi:10.1002/advs.201801138
Nishimura, Y., Yamakawa, D., Shiromizu, T., and Inagaki, M. (2021b). Aurora A and AKT kinase signaling associated with primary cilia. Cells 10 (12), 3602. doi:10.3390/cells10123602
Nishimura, Y., Yamakawa, D., Uchida, K., Shiromizu, T., Watanabe, M., and Inagaki, M. (2021a). Primary cilia and lipid raft dynamics. Open Biol. 11 (8), 210130. doi:10.1098/rsob.210130
Norsworthy, K. J., Luo, L., Hsu, V., Gudi, R., Dorff, S. E., Przepiorka, D., et al. (2019). FDA approval summary: ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutationfor relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 25 (11), 3205–3209. doi:10.1158/1078-0432.CCR-18-3749
Nozaki, S., Katoh, Y., Kobayashi, T., and Nakayama, K. (2018). BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS One 13 (3), e0195005. doi:10.1371/journal.pone.0195005
Nozaki, S., Katoh, Y., Terada, M., Michisaka, S., Funabashi, T., Takahashi, S., et al. (2017). Regulation of ciliary retrograde protein trafficking by the Joubert syndrome proteins ARL13B and INPP5E. J. Cell Sci. 130 (3), 563–576. doi:10.1242/jcs.197004
Oh, A., Pearce, J. W., Gandolfi, B., Creighton, E. K., Suedmeyer, W. K., Selig, M., et al. (2017). Early-onset progressive retinal atrophy associated with an IQCB1 variant in african black-footed cats (Felis nigripes). Sci. Rep. 7, 43918. doi:10.1038/srep43918
Onnis, A., Finetti, F., Patrussi, L., Gottardo, M., Cassioli, C., Spanò, S., et al. (2015). The small GTPase Rab29 is a common regulator of immune synapse assembly and ciliogenesis. Cell Death Differ. 22 (10), 1687–1699. doi:10.1038/cdd.2015.17
Otsu, W., Hsu, Y.-C., Chuang, J.-Z., and Sung, C.-H. (2019). The late endosomal pathway regulates the ciliary targeting of tetraspanin protein peripherin 2. J. Neurosci. 39 (18), 3376–3393. doi:10.1523/JNEUROSCI.2811-18.2019
Palicharla, V. R., Hwang, S.-H., Somatilaka, B. N., Legué, E., Shimada, I. S., Familiari, N. E., et al. (2023). Interactions between TULP3 tubby domain and ARL13B amphipathic helix promote lipidated protein transport to cilia. Mol. Biol. Cell 34 (3), ar18. doi:10.1091/mbc.E22-10-0473
Pan, Y., Bai, C. B., Joyner, A. L., and Wang, B. (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26 (9), 3365–3377. doi:10.1128/MCB.26.9.3365-3377.2006
Patel, M. M., and Tsiokas, L. (2021). Insights into the regulation of ciliary disassembly. Cells 10 (11), 2977. doi:10.3390/cells10112977
Peixoto, E., Richard, S., Pant, K., Biswas, A., and Gradilone, S. A. (2020). The primary cilium: the primary cilium: its role as a tumor suppressor organellerole as a tumor suppressor organelle. Biochem. Pharmacol. 175, 113906. doi:10.1016/j.bcp.2020.113906
Peterson, S. M., McGill, T. J., Puthussery, T., Stoddard, J., Renner, L., Lewis, A. D., et al. (2019). Bardet-biedl syndrome in rhesus macaques: A nonhuman primate model of retinitis pigmentosa. Exp. Eye Res. 189, 107825. doi:10.1016/j.exer.2019.107825
Phua, S. C., Chiba, S., Suzuki, M., Su, E., Roberson, E. C., Pusapati, G. V., et al. (2017). Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 168 (1-2), 264–279. doi:10.1016/j.cell.2016.12.032
Pitaval, A., Senger, F., Letort, G., Gidrol, X., Guyon, L., Sillibourne, J., et al. (2017). Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J. Cell Biol. 216 (11), 3713–3728. doi:10.1083/jcb.201610039
Plotnikova, O. V., Nikonova, A. S., Loskutov, Y. V., Kozyulina, P. Y., Pugacheva, E. N., and Golemis, E. A. (2012). Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol. Biol. Cell 23 (14), 2658–2670. doi:10.1091/mbc.E11-12-1056
Pugacheva, E. N., Jablonski, S. A., Hartman, T. R., Henske, E. P., and Golemis, E. A. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129 (7), 1351–1363. doi:10.1016/j.cell.2007.04.035
Qie, Y., Wang, L., Du, E., Chen, S., Lu, C., Ding, N., et al. (2020). TACC3 promotes prostate cancer cell proliferation and restrains primary cilium formation. Exp. Cell Res. 390 (2), 111952. doi:10.1016/j.yexcr.2020.111952
Qiu, H., Fujisawa, S., Nozaki, S., Katoh, Y., and Nakayama, K. (2021). Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol. Open 10 (1), bio057653. doi:10.1242/bio.057653
Raina, R., Houry, A., Rath, P., Mangat, G., Pandher, D., Islam, M., et al. (2022). Clinical utility and tolerability of tolvaptan in the treatment of autosomal dominant polycystic kidney disease (ADPKD). Drug Healthc. Patient Saf. 14, 147–159. doi:10.2147/DHPS.S338050
Raychowdhury, M. K., McLaughlin, M., Ramos, A. J., Montalbetti, N., Bouley, R., Ausiello, D. A., et al. (2005). Characterization of single channel currents from primary cilia of renal epithelial cells. J. Biol. Chem. 280 (41), 34718–34722. doi:10.1074/jbc.M507793200
Reiter, J. F., Blacque, O. E., and Leroux, M. R. (2012). The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 13 (7), 608–618. doi:10.1038/embor.2012.73
Reiter, J. F., and Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18 (9), 533–547. doi:10.1038/nrm.2017.60
Ripolles-Garcia, A., Murgiano, L., Ziolkowska, N., Marinho, F. P., Roszak, K., Iffrig, S., et al. (2023). Natural disease history of a canine model of oligogenic RPGRIP1-cone-rod dystrophy establishes variable effects of previously and newly mapped modifier loci. Hum. Mol. Genet. 32, 2139–2151. doi:10.1093/hmg/ddad046
Rohatgi, R., Milenkovic, L., and Scott, M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317 (5836), 372–376. doi:10.1126/science.1139740
Roosing, S., Rohrschneider, K., Beryozkin, A., Weisschuh, N., Kohl, S., Wissinger, B., et al. (2013). The farnesylated small GTPase RAB28 is mutated in autosomal recessive cone-rod dystrophy. Invest. Ophthalmol. Vis. Sci. 54 (15), 1228.
Saito, M., Hirano, M., Izumi, T., Mori, Y., Ito, K., Saitoh, Y., et al. (2022). Cytoskeletal protein 4.1G is essential for the primary ciliogenesis and osteoblast differentiation in bone formation. Int. J. Mol. Sci. 23 (4), 2094. doi:10.3390/ijms23042094
Saito, M., Otsu, W., Hsu, K.-S., Chuang, J.-Z., Yanagisawa, T., Shieh, V., et al. (2017). Tctex-1 controls ciliary resorption by regulating branched actin polymerization and endocytosis. EMBO Rep. 18 (8), 1460–1472. doi:10.15252/embr.201744204
Sánchez de Diego, A., Alonso Guerrero, A., Martínez-A, C., and van Wely, K. H. M. (2014). Dido3-dependent HDAC6 targeting controls cilium size. Nat. Commun. 5, 3500. doi:10.1038/ncomms4500
Sánchez-Bellver, L., Toulis, V., and Marfany, G. (2021). On the wrong track: alterations of ciliary transport in inherited retinal dystrophiesof ciliary transport in inherited retinal dystrophies. Front. Cell Dev. Biol. 9, 623734. doi:10.3389/fcell.2021.623734
Schmidt, T. I., Kleylein-Sohn, J., Westendorf, J., Le Clech, M., Lavoie, S. B., Stierhof, Y.-D., et al. (2009). Control of centriole length by CPAP and CP110. Curr. Biol. 19 (12), 1005–1011. doi:10.1016/j.cub.2009.05.016
Schneider, L., Clement, C. A., Teilmann, S. C., Pazour, G. J., Hoffmann, E. K., Satir, P., et al. (2005). PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 15 (20), 1861–1866. doi:10.1016/j.cub.2005.09.012
Schou, K. B., Pedersen, L. B., and Christensen, S. T. (2015). Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 16 (9), 1099–1113. doi:10.15252/embr.201540530
Sells, T. B., Chau, R., Ecsedy, J. A., Gershman, R. E., Hoar, K., Huck, J., et al. (2015). MLN8054 and alisertib (MLN8237): MLN8054 and alisertib (MLN8237): discovery of selective oral aurora A inhibitorsof selective oral aurora A inhibitors. ACS Med. Chem. Lett. 6 (6), 630–634. doi:10.1021/ml500409n
Sharif, A. S., Gerstner, C. D., Cady, M. A., Arshavsky, V. Y., Mitchell, C., Ying, G., et al. (2021). Deletion of the phosphatase INPP5E in the murine retina impairs photoreceptor axoneme formation and prevents disc morphogenesis. J. Biol. Chem. 296, 100529. doi:10.1016/j.jbc.2021.100529
Sherpa, R. T., Mohieldin, A. M., Pala, R., Wachten, D., Ostrom, R. S., and Nauli, S. M. (2019). Sensory primary cilium is a responsive cAMP microdomain in renal epithelia. Sci. Rep. 9 (1), 6523. doi:10.1038/s41598-019-43002-2
Sheu, S.-H., Upadhyayula, S., Dupuy, V., Pang, S., Deng, F., Wan, J., et al. (2022). A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 185 (18), 3390–3407.e18. doi:10.1016/j.cell.2022.07.026
Shimada, I. S., and Kato, Y. (2022). Ciliary signaling in stem cells in health and disease: ciliary signaling in stem cells in health and disease: hedgehog pathway and beyondpathway and beyond. Semin. Cell Dev. Biol. 129, 115–125. doi:10.1016/j.semcdb.2022.04.011
Shimada, M., Tritos, N. A., Lowell, B. B., Flier, J. S., and Maratos-Flier, E. (1998). Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396 (6712), 670–674. doi:10.1038/25341
Shinde, S. R., Nager, A. R., and Nachury, M. V. (2020). Ubiquitin chains earmark GPCRs for BBSome-mediated removal from cilia. J. Cell Biol. 219 (12), e202003020. doi:10.1083/jcb.202003020
Shirley, M. (2017). Olaratumab: first global ApprovalGlobal approval. Drugs 77 (1), 107–112. doi:10.1007/s40265-016-0680-2
Shiromizu, T., Yuge, M., Kasahara, K., Yamakawa, D., Matsui, T., Bessho, Y., et al. (2020). Targeting E3 ubiquitin ligases and deubiquitinases in ciliopathy and cancer. Int. J. Mol. Sci. 21 (17), 5962. doi:10.3390/ijms21175962
Siljee, J. E., Wang, Y., Bernard, A. A., Ersoy, B. A., Zhang, S., Marley, A., et al. (2018). Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet. 50 (2), 180–185. doi:10.1038/s41588-017-0020-9
Spalluto, C., Wilson, D. I., and Hearn, T. (2012). Nek2 localises to the distal portion of the mother centriole/basal body and is required for timely cilium disassembly at the G2/M transition. Eur. J. Cell Biol. 91 (9), 675–686. doi:10.1016/j.ejcb.2012.03.009
Spektor, A., Tsang, W. Y., Khoo, D., and Dynlacht, B. D. (2007). Cep97 and CP110 suppress a cilia assembly program. Cell 130 (4), 678–690. doi:10.1016/j.cell.2007.06.027
Stanić, D., Malmgren, H., He, H., Scott, L., Aperia, A., and Hökfelt, T. (2009). Developmental changes in frequency of the ciliary somatostatin receptor 3 protein. Brain Res. 1249, 101–112. doi:10.1016/j.brainres.2008.10.024
Sugiyama, M. G., Fairn, G. D., and Antonescu, C. N. (2019). Akt-ing up just about everywhere: compartment-specific akt activation and function in receptor tyrosine kinase signaling-specific akt activation and function in receptor tyrosine kinase signaling. Front. Cell Dev. Biol. 7, 70. doi:10.3389/fcell.2019.00070
Svenningsson, P., Tzavara, E. T., Qi, H., Carruthers, R., Witkin, J. M., Nomikos, G. G., et al. (2007). Biochemical and behavioral evidence for antidepressant-like effects of 5-HT6 receptor stimulation. J. Neurosci. 27 (15), 4201–4209. doi:10.1523/JNEUROSCI.3110-06.2007
Tai, A. W., Chuang, J. Z., Bode, C., Wolfrum, U., and Sung, C. H. (1999). Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell 97 (7), 877–887. doi:10.1016/s0092-8674(00)80800-4
Takahashi, K., Nagai, T., Chiba, S., Nakayama, K., and Mizuno, K. (2018). Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell Sci. 131 (1), jcs208769. doi:10.1242/jcs.208769
Takao, D., and Verhey, K. J. (2016). Gated entry into the ciliary compartment. Cell. Mol. Life Sci. 73 (1), 119–127. doi:10.1007/s00018-015-2058-0
Thoma, C. R., Frew, I. J., Hoerner, C. R., Montani, M., Moch, H., and Krek, W. (2007). pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 9 (5), 588–595. doi:10.1038/ncb1579
Tian, G., Ropelewski, P., Nemet, I., Lee, R., Lodowski, K. H., and Imanishi, Y. (2014). An unconventional secretory pathway mediates the cilia targeting of peripherin/rds. J. Neurosci. 34 (3), 992–1006. doi:10.1523/JNEUROSCI.3437-13.2014
Tirado-Cabrera, I., Martin-Guerrero, E., Heredero-Jimenez, S., Ardura, J. A., and Gortázar, A. R. (2022). PTH1R translocation to primary cilia in mechanically-stimulated ostecytes prevents osteoclast formation via regulation of CXCL5 and IL-6 secretion. J. Cell. Physiol. 237 (10), 3927–3943. doi:10.1002/jcp.30849
Tokuyasu, K., and Yamada, E. (1959). The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J. Biophys. Biochem. Cytol. 6 (2), 225–230. doi:10.1083/jcb.6.2.225
Torres, V. E. (2019). Pro: tolvaptan delays the progression of autosomal dominant polycystic kidney diseasedelays the progression of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transpl. 34 (1), 30–34. doi:10.1093/ndt/gfy297
Travis, G. H., Brennan, M. B., Danielson, P. E., Kozak, C. A., and Sutcliffe, J. G. (1989). Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature 338 (6210), 70–73. doi:10.1038/338070a0
Travis, G. H., Sutcliffe, J. G., and Bok, D. (1991). The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron 6 (1), 61–70. doi:10.1016/0896-6273(91)90122-g
Turn, R. E., Hu, Y., Dewees, S. I., Devi, N., East, M. P., Hardin, K. R., et al. (2022). The ARF GAPs ELMOD1 and ELMOD3 act at the Golgi and cilia to regulate ciliogenesis and ciliary protein traffic. Mol. Biol. Cell 33 (7), cor1. doi:10.1091/mbc.E21-09-0443_corr
Urdiciain, A., Erausquin, E., Meléndez, B., Rey, J. A., Idoate, M. A., and Castresana, J. S. (2019). Tubastatin A, an inhibitor of HDAC6, enhances temozolomide-induced apoptosis and reverses the malignant phenotype of glioblastoma cells. Int. J. Oncol. 54 (5), 1797–1808. doi:10.3892/ijo.2019.4739
Villanueva, H., Visbal, A. P., Obeid, N. F., Ta, A. Q., Faruki, A. A., Wu, M.-F., et al. (2015). An essential role for Gα(i2) in Smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci. Signal. 8 (394), ra92. doi:10.1126/scisignal.aaa7355
Wachten, D., and Mick, D. U. (2021). Signal transduction in primary cilia - analyzing and manipulating GPCR and second messenger signaling. Pharmacol. Ther. 224, 107836. doi:10.1016/j.pharmthera.2021.107836
Wang, C. Y., Tsai, H. L., Syu, J. S., Chen, T. Y., and Su, M. T. (2017). Primary cilium-regulated EG-VEGF signaling facilitates trophoblast invasion. J. Cell Physiol. 232 (6), 1467–1477. doi:10.1002/jcp.25649
Wang, J., and Deretic, D. (2014). Molecular complexes that direct rhodopsin transport to primary cilia. Prog. Retin. Eye Res. 38, 1–19. doi:10.1016/j.preteyeres.2013.08.004
Wang, J., Morita, Y., Mazelova, J., and Deretic, D. (2012). The Arf GAP ASAP1 provides a platform to regulate Arf4-and Rab11-Rab8-mediated ciliary receptor targeting. EMBO J. 31 (20), 4057–4071. doi:10.1038/emboj.2012.253
Wang, J., Silva, M., Haas, L. A., Morsci, N. S., Nguyen, K. C. Q., Hall, D. H., et al. (2014). C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol. 24 (5), 519–525. doi:10.1016/j.cub.2014.01.002
Wang, Y., Bernard, A., Comblain, F., Yue, X., Paillart, C., Zhang, S., et al. (2021). Melanocortin 4 receptor signals at the neuronal primary cilium to control food intake and body weight. J. Clin. Invest. 131 (9), e142064. doi:10.1172/JCI142064
Wang, Z., Phan, T., and Storm, D. R. (2011). The type 3 adenylyl cyclase is required for novel object learning and extinction of contextual memory: role of cAMP signaling in primary cilia. J. Neurosci. 31 (15), 5557–5561. doi:10.1523/JNEUROSCI.6561-10.2011
Wassmer, S., Pawlyk, B., Blake, J., Sandberg, M. A., Pierce, E. A., and Vandenberghe, L. H. (2017). Evaluation of in silico reconstructed ancestral adeno-associated virus for gene augmentation therapy in a mouse model of LCA. Invest. Ophthalmol. Vis. Sci. 58 (8), 4086.
Wheway, G., Mitchison, H. M., and Genomics England Research, C. (2019). Opportunities and challenges for molecular understanding of ciliopathies-the 100,000 genomes project. Front. Genet. 10, 127. doi:10.3389/fgene.2019.00127
Wiens, C. J., Tong, Y., Esmail, M. A., Oh, E., Gerdes, J. M., Wang, J., et al. (2010). Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J. Biol. Chem. 285 (21), 16218–16230. doi:10.1074/jbc.M109.070953
Wiik, A. C., Wade, C., Biagi, T., Ropstad, E. O., Bjerkas, E., Lindblad-Toh, K., et al. (2008). A deletion in nephronophthisis 4 (NPHP4) is associated with recessive cone-rod dystrophy in standard wire-haired dachshund. Genome Res. 18 (9), 1415–1421. doi:10.1101/gr.074302.107
Witzgall, R. (2018). Golgi bypass of ciliary proteins. Semin. Cell Dev. Biol. 83, 51–58. doi:10.1016/j.semcdb.2018.03.010
Wloga, D., Joachimiak, E., Louka, P., and Gaertig, J. (2017). Posttranslational modifications of tubulin and cilia. Cold Spring Harb. Perspect. Biol. 9 (6), a028159. doi:10.1101/cshperspect.a028159
Wong, S. Y., Seol, A. D., So, P.-L., Ermilov, A. N., Bichakjian, C. K., Epstein, E. H., et al. (2009). Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 15 (9), 1055–1061. doi:10.1038/nm.2011
Wu, C.-T., Chen, H.-Y., and Tang, T. K. (2018). Myosin-Va is required for preciliary vesicle transportation to the mother centriole during ciliogenesis. Nat. Cell Biol. 20 (2), 175–185. doi:10.1038/s41556-017-0018-7
Wu, C. T., Hilgendorf, K. I., Bevacqua, R. J., Hang, Y., Demeter, J., Kim, S. K., et al. (2021). Discovery of ciliary G protein-coupled receptors regulating pancreatic islet insulin and glucagon secretion. Genes Dev. 35 (17-18), 1243–1255. doi:10.1101/gad.348261.121
Wu, Q., Wang, H., Zhao, X., Shi, Y., Jin, M., Wan, B., et al. (2013). Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene 32 (49), 5541–5550. doi:10.1038/onc.2013.264
Xiang, W., Guo, F., Cheng, W., Zhang, J., Huang, J., Wang, R., et al. (2017). HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia. Oncol. Rep. 38 (1), 229–236. doi:10.3892/or.2017.5694
Xie, S., Zhu, J., Li, J., Zhan, F., Yao, H., Xu, J., et al. (2023). Small-molecule hydrophobic tagging: A promising strategy of druglike technology for targeted protein degradation. J. Med. Chem. 66, 10917–10933. doi:10.1021/acs.jmedchem.3c00736
Xu, Q., Zhang, Y., Wei, Q., Huang, Y., Hu, J., and Ling, K. (2016). Phosphatidylinositol phosphate kinase PIPKIγ and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat. Commun. 7, 10777. doi:10.1038/ncomms10777
Yamakawa, D., Katoh, D., Kasahara, K., Shiromizu, T., Matsuyama, M., Matsuda, C., et al. (2021). Primary cilia-dependent lipid raft/caveolin dynamics regulate adipogenesis. Cell Rep. 34 (10), 108817. doi:10.1016/j.celrep.2021.108817
Yamakawa, D., Tsuboi, J., Kasahara, K., Matsuda, C., Nishimura, Y., Kodama, T., et al. (2022). Cilia-mediated insulin/akt and ST2/JNK signaling pathways regulate the recovery of muscle injury. Adv. Sci. 10 (1), e2202632. doi:10.1002/advs.202202632
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020). Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 5 (1), 8. doi:10.1038/s41392-020-0110-5
Yang, Y., Ran, J., Liu, M., Li, D., Li, Y., Shi, X., et al. (2014). CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 24 (11), 1342–1353. doi:10.1038/cr.2014.136
Yao, G., Luo, C., Harvey, M., Wu, M., Schreiber, T. H., Du, Y., et al. (2016). Disruption of polycystin-L causes hippocampal and thalamocortical hyperexcitability. Hum. Mol. Genet. 25 (3), 448–458. doi:10.1093/hmg/ddv484
Yeh, C., Li, A., Chuang, J.-Z., Saito, M., Cáceres, A., and Sung, C.-H. (2013). IGF-1 activates a cilium-localized noncanonical Gβγ signaling pathway that regulates cell-cycle progression. Dev. Cell 26 (4), 358–368. doi:10.1016/j.devcel.2013.07.014
Yuan, X., Cao, J., He, X., Serra, R., Qu, J., Cao, X., et al. (2016). Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat. Commun. 7, 11024. doi:10.1038/ncomms11024
Yue, S., Tang, L.-Y., Tang, Y., Tang, Y., Shen, Q.-H., Ding, J., et al. (2014). Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. Elife 3, e02555. doi:10.7554/eLife.02555
Yukimoto, H., Miyamoto, T., Kiyono, T., Wang, S., Matsuura, S., Mizoguchi, A., et al. (2020). A novel CDK-independent function of p27Kip1 in preciliary vesicle trafficking during ciliogenesis. Biochem. Biophys. Res. Commun. 527 (3), 716–722. doi:10.1016/j.bbrc.2020.05.048
Zhang, Q., Acland, G. M., Wu, W. X., Johnson, J. L., Pearce-Kelling, S., Tulloch, B., et al. (2002). Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet. 11 (9), 993–1003. doi:10.1093/hmg/11.9.993
Zhang, Y.-C., Bai, Y.-F., Yuan, J.-F., Shen, X.-L., Xu, Y.-L., Jian, X.-X., et al. (2021). CEP55 promotes cilia disassembly through stabilizing Aurora A kinase. J. Cell Biol. 220 (2), e202003149. doi:10.1083/jcb.202003149
Zhao, H., Khan, Z., and Westlake, C. J. (2023). Ciliogenesis membrane dynamics and organization. Semin. Cell Dev. Biol. 133, 20–31. doi:10.1016/j.semcdb.2022.03.021
Zheng, L., Cao, Y., Ni, S., Qi, H., Ling, Z., Xu, X., et al. (2018). Ciliary parathyroid hormone signaling activates transforming growth factor-β to maintain intervertebral disc homeostasis during aging. Bone Res. 6, 21. doi:10.1038/s41413-018-0022-y
Zhou, Z., Katoh, Y., and Nakayama, K. (2022). CEP19-RABL2-IFT-B axis controls BBSome-mediated ciliary GPCR export. Mol. Biol. Cell 33 (13), ar126. doi:10.1091/mbc.E22-05-0161

Keywords: ciliary receptors, ciliogenesis, ciliary transport, ciliopathy, inherited retinal dystrophies, photoreceptor
Citation: Saito M, Otsu W, Miyadera K and Nishimura Y (2023) Recent advances in the understanding of cilia mechanisms and their applications as therapeutic targets. Front. Mol. Biosci. 10:1232188. doi: 10.3389/fmolb.2023.1232188
Received: 31 May 2023; Accepted: 24 August 2023;
Published: 14 September 2023.
Edited by:
Anna Sansone, Consiglio Nazionale delle Ricerche (Bologna), ItalyReviewed by:
Rossella Venditti, Telethon Institute of Genetics and Medicine (TIGEM), ItalyCopyright © 2023 Saito, Otsu, Miyadera and Nishimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiko Miyadera, a21peWFAdXBlbm4uZWR1; Yuhei Nishimura, eXVoZWlAbWVkLm1pZS11LmFjLmpw
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.