
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 30 January 2023
Sec. Cellular Biochemistry
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1129435
This article is part of the Research Topic Transient Receptor Potential Channels (TRP): Signal Transduction View all 5 articles
 Michelle N. Sullivan1†
Michelle N. Sullivan1† Pratish Thakore2†
Pratish Thakore2† Vivek Krishnan2
Vivek Krishnan2 Sushma Alphonsa2Wencheng Li3
Sushma Alphonsa2Wencheng Li3 Yumei Feng Earley2,4
Yumei Feng Earley2,4 Scott Earley2*
Scott Earley2*Introduction: Hypoxia-induced dilation of cerebral arteries orchestrated by Ca2+-permeable transient receptor potential ankyrin 1 (TRPA1) cation channels on endothelial cells is neuroprotective during ischemic stroke, but it is unknown if the channel has a similar impact during hemorrhagic stroke. TRPA1 channels are endogenously activated by lipid peroxide metabolites generated by reactive oxygen species (ROS). Uncontrolled hypertension, a primary risk factor for the development of hemorrhagic stroke, is associated with increased ROS production and oxidative stress. Therefore, we hypothesized that TRPA1 channel activity is increased during hemorrhagic stroke.
Methods: Severe, chronic hypertension was induced in control (Trpa1fl/fl) and endothelial cell-specific TRPA1 knockout (Trpa1-ecKO) mice using a combination of chronic angiotensin II administration, a high-salt diet, and the addition of a nitric oxide synthase inhibitor to drinking water. Blood pressure was measured in awake, freely-moving mice using surgically placed radiotelemetry transmitters. TRPA1-dependent cerebral artery dilation was evaluated with pressure myography, and expression of TRPA1 and NADPH oxidase (NOX) isoforms in arteries from both groups was determined using PCR and Western blotting techniques. In addition, ROS generation capacity was evaluated using a lucigenin assay. Histology was performed to examine intracerebral hemorrhage lesion size and location.
Results: All animals became hypertensive, and a majority developed intracerebral hemorrhages or died of unknown causes. Baseline blood pressure and responses to the hypertensive stimulus did not differ between groups. Expression of TRPA1 in cerebral arteries from control mice was not altered after 28 days of treatment, but expression of three NOX isoforms and the capacity for ROS generation was increased in hypertensive animals. NOX-dependent activation of TRPA1 channels dilated cerebral arteries from hypertensive animals to a greater extent compared with controls. The number of intracerebral hemorrhage lesions in hypertensive animals did not differ between control and Trpa1-ecKO animals but were significantly smaller in Trpa1-ecKO mice. Morbidity and mortality did not differ between groups.
Discussion: We conclude that endothelial cell TRPA1 channel activity increases cerebral blood flow during hypertension resulting in increased extravasation of blood during intracerebral hemorrhage events; however, this effect does not impact overall survival. Our data suggest that blocking TRPA1 channels may not be helpful for treating hypertension-associated hemorrhagic stroke in a clinical setting.
Stroke is the second-leading cause of mortality worldwide and the fifth-leading cause of death in the United States (Donkor, 2018; Tsao et al., 2022). Strokes are classified into two categories: ischemic and hemorrhagic. Ischemic stroke is more common and caused by regional interruptions of the blood supply to the brain, a sudden loss of function, and neuronal death. Hemorrhagic stroke results from the rupture of cerebral blood vessels and bleeding into the brain tissue (Bamford et al., 1991). Although hemorrhagic strokes are less common, rates of morbidity and long-term disability following an event are much higher compared with ischemic strokes (Virani et al., 2021). We previously demonstrated that selective knockout of transient receptor potential ankyrin 1 (TRPA1) cation channels from vascular endothelial cells exacerbated neuronal damage following ischemic stroke, and treatment with a TRPA1 agonist improved outcomes (Pires and Earley, 2018). Thus, we concluded that endothelial cell TRPA1 activity is neuroprotective following ischemic stroke. The effects of endothelial cell TRPA1 channels on the incidence or severity of hemorrhagic stroke events have not been previously reported. Here, we investigated how outcomes associated with hypertension-induced hemorrhagic stroke are impacted by cerebral vascular TRPA1 channel activity.
Uncontrolled systemic hypertension is a primary risk factor for hemorrhagic stroke (Hägg-Holmberg et al., 2019). Increased arterial intraluminal pressure enhances smooth muscle cell contractility (Harder et al., 1985; Pratt et al., 2002; Amberg et al., 2003; Bannister et al., 2012) elevating cerebral vascular resistance and augmenting stress on the vascular wall (Hayashi and Naiki, 2009). Greater cerebral perfusion pressure and tangential stress also damages the endothelium over time, weakening the vessel wall to the point of rupture (Izzard et al., 2005; De Ciuceis et al., 2007). Cerebral autoregulation, the process by which cerebral blood flow is maintained during changes in intraluminal pressure, is often impaired during hypertension which contributes to the development of stroke (Strandgaard et al., 1973; Lammie, 2002; Faraco and Iadecola, 2013). Loss of cerebral autoregulation increases perfusion pressure in the capillaries and damages the blood-brain barrier, leading to cerebral edema, inflammation, and neuronal degeneration (Lammie, 2002; Faraco and Iadecola, 2013; Shekhar et al., 2017). Hypertension also enhances the expression of reactive oxygen species (ROS)-generating NADPH oxidase (NOX) enzymes and ROS production in the vasculature (Rajagopalan et al., 1996; Zalba et al., 2001; Landmesser et al., 2003; Adler and Huang, 2004; Kazama et al., 2004; Paravicini et al., 2004; Matsuno et al., 2005). Although ROS are critical for a variety of physiological responses (Knock et al., 2009; Xiao et al., 2009), excessive ROS accumulation, also called oxidative stress, causes intramural damage and subsequent recruitment of inflammatory cells, increased lipid peroxidation, activation of matrix metalloproteases, and deposition of extracellular matrix. However, the molecular pathogenesis of ROS production and accumulation during hypertension-associated hemorrhagic stroke is unclear.
TRPA1 channels are present in the endothelium of cerebral arteries but aren’t found in endothelial cells from the mesenteric, coronary, renal, and dermal vascular beds (Sullivan et al., 2015). Endothelial cell TRPA1 channels dilate cerebral arteries and orchestrate neurovascular coupling in response to electrophilic compounds and ROS metabolites (Earley et al., 2009; Qian et al., 2013; Sullivan et al., 2015; Pires and Earley, 2018; Thakore et al., 2021). We have previously shown that ROS generated by the NOX2 enzyme indirectly activate TRPA1 channels in the cerebral artery endothelium by a pathway that requires the peroxidation of membrane phospholipids (Sullivan et al., 2015). TRPA1 channels activated in this manner produce subcellular Ca2+ signals that stimulate intermediate conductance Ca2+-activated K+ (IK) channels to evoke vasodilation (Sullivan et al., 2015). As NOX expression and ROS generation are enhanced in the vascular wall during hypertension (Rajagopalan et al., 1996; Zalba et al., 2001; Landmesser et al., 2003; Adler and Huang, 2004; Kazama et al., 2004; Paravicini et al., 2004; Matsuno et al., 2005), endothelial cell TRPA1 channel activity may also be elevated.
Here, we tested the hypothesis that increased ROS generation associated with severe hypertension increases endothelial cell TRPA1 activity to dilate cerebral arteries. We also investigated the role of TRPA1 during hypertension-associated hemorrhagic stroke. We induced hemorrhagic stroke by severely increasing blood pressure through combined angiotensin II (Ang II) administration, a high-salt (HS) diet ad libitum, and drinking water supplemented with the nitric oxide synthase (NOS) inhibitor Nitro-L-arginine methyl ester hydrochloride (L-NAME) in control (Trpa1fl/fl) and endothelial cell-specific TRPA1 knockout (Trpa1-ecKO). All animals developed hypertension, and most that completed the protocol had intracerebral hemorrhages. Cerebral artery Nox1, Nox2, and Nox4 expression and ROS generation capacity were enhanced during hypertension. TRPA1-dependent dilation of cerebral arteries in response to NOX activation was also elevated in hypertensive mice. Histological analysis revealed similar numbers of intracerebral hemorrhages in the brains of control and Trpa1-ecKO mice, but the lesions were significantly smaller in Trpa1-ecKO mice. However, overall morbidity and mortality didn’t differ between groups. Together, these findings suggest that enhanced ROS production during hypertension increases cerebral artery endothelial cell TRPA1 activity leading to vasodilation and expansion of hemorrhagic lesions.
Blood pressure (BP) and heart rate were measured in conscious, freely moving Trpa1-ecKO and control Trpa1fl/fl mice through surgical implantation of radiotelemetry transmitters as previously described (Feng et al., 2010; Li et al., 2014b; Thakore et al., 2020; Krishnan et al., 2022); mice were allowed to recover for 2 weeks. Animals were then chronically infused with Ang II (1.2 μg/kg/min) using osmotic minipumps, surgically inserted into the subcutaneous space, and fed a HS (8% NaCl) chow diet (Figure 1A). After 2 weeks of chronic Ang II/HS treatment, the NOS inhibitor L-NAME was introduced into the drinking water (120 mg/kg/day) to increase systemic BP further. Treatment was maintained for 28 days after minipump implantation, and BP, heart rate (HR), and locomotor activity were monitored daily (Figure 1A). Baseline systolic, diastolic, and mean arterial pressures, HR, and locomotor activity didn’t differ between Trpa1fl/fl and Trpa1-ecKO mice. Ang II/HS treatment increased systolic, diastolic, and mean arterial pressure in control and Trpa1-ecKO mice, and BP was further elevated after adding L-NAME to the drinking water (Figures 1B–D). These responses didn’t differ between Trpa1fl/fl and Trpa1-ecKO mice. The treatment didn’t alter HR or locomotor activity for either group (Figures 1E, F). Increased cardiac weight, suggestive of cardiac hypertrophy, was observed in Trpa1fl/fl and Trpa1-ecKO mice treated with Ang II/HS/L-NAME compared to their untreated counterparts (Figure 1G). However no differences between Trpa1fl/fl and Trpa1-ecKO mice were observed, suggesting that endothelial cell TRPA1 expression isn’t directly involved in cardiac hypertrophy associated with hypertension. These data indicate that Ang II/HS/L-NAME treatment induced severe hypertension and that genetic deletion of endothelial cell TRPA1 channels didn’t alter basal hemodynamics nor increases in BP in response to the hypertensive stimuli.
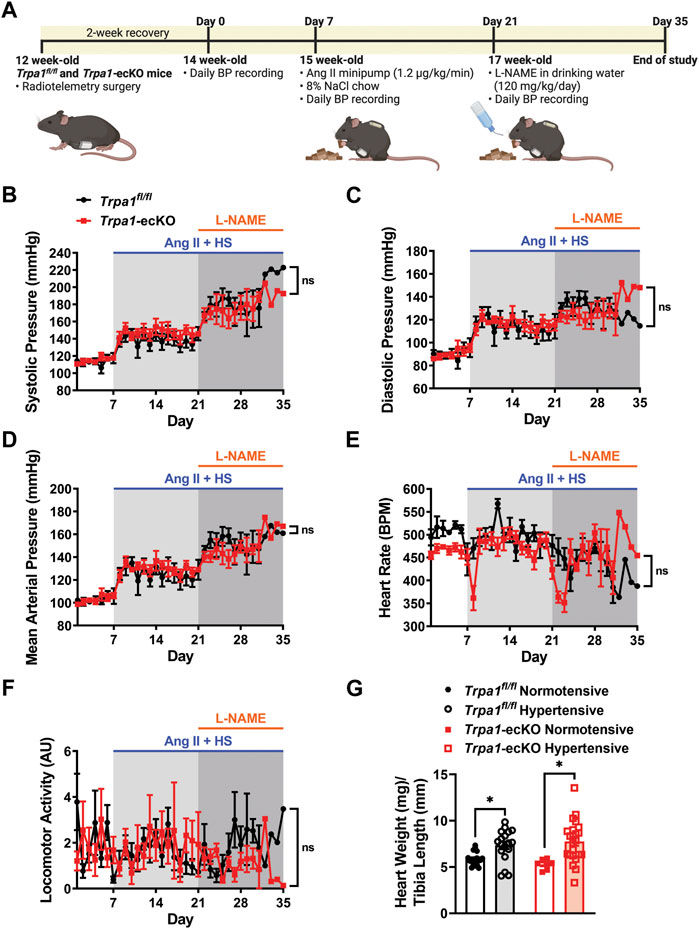
FIGURE 1. Endothelial cell Trpa1 knockout does not affect resting BP or responses to hypertensive stimuli. (A) Schematic representation of the hypertension protocol. 12-week-old Trpa1fl/fl (control) and Trpa1-ecKO mice were surgically implanted with radiotelemetry probes to measure blood pressure (BP). Following a 2-week recovery, baseline BP was recorded for 7 days. These mice were then implanted with subcutaneous Ang II minipumps (1.2 μg/kg/min) and fed an 8% high-salt (HS) diet. After 2 weeks, the drinking water was supplemented with the NOS inhibitor L-NAME and treatment was continued for another 2 weeks. (B–F) Systolic BP (B), diastolic BP (C), mean arterial pressure (D), heart rate (E), and locomotor activity (F) were recorded from control and Trpa1-ecKO mice before and after Ang II/HS/L-NAME treatment (n = 8 for control, n = 6 for Trpa1-ecKO, two-way ANOVA, ns = not significant). (G) Heart weight normalized to tibia length was assessed from normotensive and hypertensive control and Trpa1-ecKO mice (n = 18 for normotensive control, n = 18 for hypertensive control, n = 7 for normotensive Trpa1-ecKO, n = 23 for hypertensive Trpa1-ecKO; two-way ANOVA, *p < 0.05).
After the end of the treatment (28 days after minipump placement), a cohort of Trpa1fl/fl mice was euthanized, and cerebral arteries were harvested. Trpa1 mRNA transcript (Figure 2A) and TRPA1 protein levels (Figure 2B) didn’t differ between normotensive and hypertensive animals. However, we found that mRNA levels of the ROS-producing enzymes Nox1, Nox2, and Nox4 were enhanced in cerebral arteries from hypertensive mice (Figure 2C). Using a chemiluminescent detection method, we also found that the capacity for superoxide anion (O2−) production was elevated in cerebral arteries from hypertensive mice compared with normotensive animals (Figure 2D). We previously reported that administration of the NOX substrate NADPH dilated cerebral arteries by activating TRPA1 channels through lipid peroxidation (Sullivan et al., 2015). Here, we found that NADPH dilated cerebral arteries from hypertensive animals to a greater extent compared with normotensive mice (Figures 2E, F). The selective TRPA1 blocker HC-030031 attenuated NADPH-induced dilation (Figures 2E, F). These data suggest that the capacity for NOX-induced activation of TRPA1 channels and subsequent dilation of cerebral arteries are enhanced during hypertension.
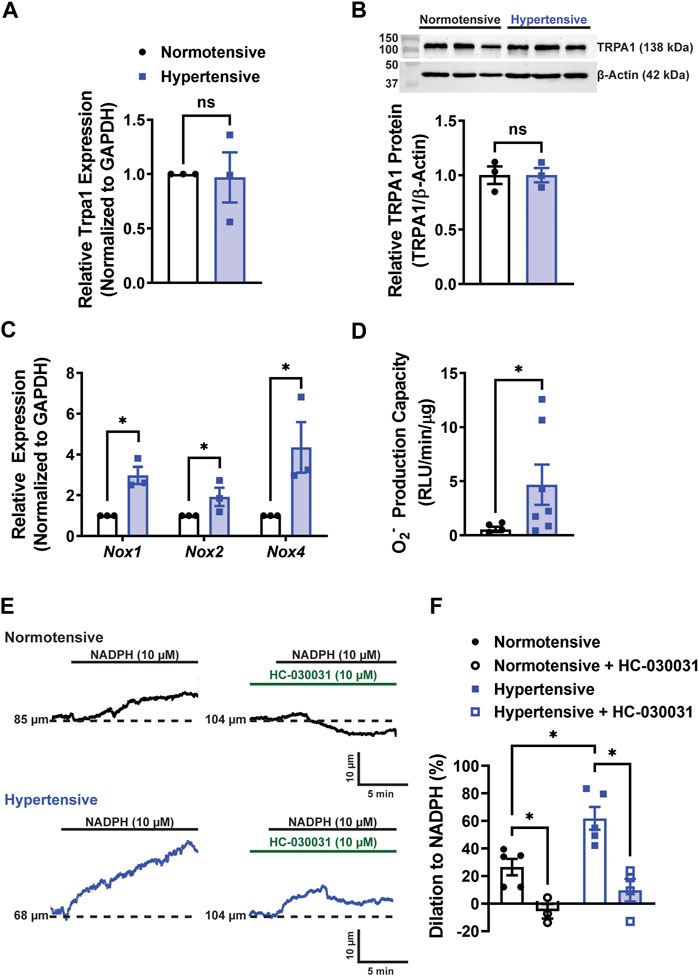
FIGURE 2. ROS production and TRPA1-dependent cerebral artery dilation are enhanced during hypertension. (A) Summary data showing relative Trpa1 mRNA expression levels in cerebral arteries from normotensive and hypertensive Trpa1fl/fll mice (n = 3 per group, unpaired t-test, ns = not significant). (B) Representative Western blots and summary data showing relative TRPA1 protein expression levels in cerebral arteries of normotensive and hypertensive Trpa1fl/fll mice (n = 3 per group, unpaired t-test, ns = not significant). (C) Summary data showing the relative mRNA expression levels of Nox1, Nox2, and Nox4 in cerebral arteries of normotensive and hypertensive Trpa1fl/fll mice (n = 3 per group, unpaired t-test, *p < 0.05). (D) Summary data showing the O2− production as measured using a lucigenin assay in cerebral arteries of normotensive and hypertensive Trpa1fl/fll mice (n = 5 for normotensive, n = 7 for hypertensive, Mann-Whitney test, *p < 0.05). (E) Representative traces of changes in lumen diameter of isolated cerebral arteries from normotensive and hypertensive Trpa1fl/fll mice in response to NADPH (10 µM) before and after TRPA1 inhibition with HC-030031 (10 µM). (F) Summary data showing the vasodilation in response to NADPH in isolated cerebral arteries from normotensive and hypertensive Trpa1fl/fll mice, with and without the TRPA1 inhibitor HC-030031 (n = 5 for normotensive, n = 3 for normotensive + HC-030031, n = 5 for hypertensive, n = 3 for hypertensive + HC-030031, two-way ANOVA, *p < 0.05).
Treatment with Ang II/HS/L-NAME was maintained for 28 days (after minipump implantation) or until mice exhibited signs of morbidity (hunched posture, squinted eyes, ruffled haircoat, dehydration, etc.), including clinical signs suggestive of stroke (circling behavior, head tilt, mobility impairment, muscular fasciculations, other motor dysfunction) (Iida et al., 2005) (Supplementary Videos S1, S2). Approximately 20% of mice from both groups completed the protocol without apparent morbidity (Figures 3A, B). Of the mice that didnt finish the study, about 40% were found deceased in the cage (mortality of unknown cause), 26%–31% displayed neurological dysfunction suggestive of stroke, and between 8% and 15% suffered from generalized, severe subcutaneous edema (Supplementary Figure S1) (Figure 3B). No differences in morbidities were observed between Trpa1fl/fl and Trpa1-ecKO animals.
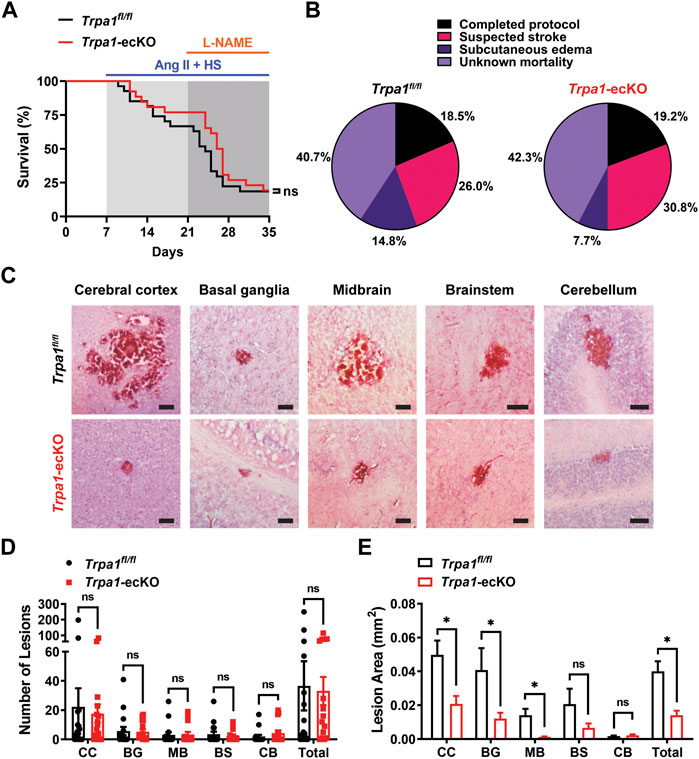
FIGURE 3. Endothelial cell TRPA1 knockout reduces the size of intracerebral hemorrhages during severe hypertension but doesn’t improve other outcomes. (A, B) Kaplan-Meier plot (A) and pie charts indicating the distribution of outcomes (B) of Trpa1fl/fl and Trpa1-ecKO mice treated with Ang II/HS/L-NAME (n = 27 for control and n = 26 for Trpa1-ecKO, Log-rank (Mantel-Cox) test, ns = not significant). (C) Representative images of H and E stained brain sections from Trpa1fl/fll and Trpa1-ecKO mice treated with Ang II/HS/L-NAME. Lesions were found in the cerebral cortex (CC), basal ganglia (BG), midbrain (MB), brain stem (BS), and cerebellum (CB) in brains of mice from both groups. Scale bar = 100 µm. (D) Summary data showing the number of lesions per brain from Trpa1fl/fll and Trpa1-ecKO mice treated with Ang II/HS/L-NAME (n = 16 for control, n = 15 for Trpa1-ecKO, unpaired t-test, ns = not significant). (E) Summary data showing the lesion area in brains from Trpa1fl/fll and Trpa1-ecKO mice treated with Ang II/HS/L-NAME (CC: n = 357 for Trpa1fl/fll and n = 265 for Trpa1-ecKO; BG: n = 90 for Trpa1fl/fll and n = 79 for Trpa1-ecKO; MB: n = 50 for Trpa1fl/fll and n = 51 for Trpa1-ecKO; BS: n = 57 for Trpa1fl/fll and n = 36 for Trpa1-ecKO; CB: n = 31 for Trpa1fl/fll and n = 65 for Trpa1-ecKO; total: n = 585 for control and n = 496 for Trpa1-ecKO, unpaired t-test, *p < 0.05, ns = not significant).
The volume of intracerebral hemorrhage lesions is commonly used as a prognostic indicator in stroke patients and is obtained using advanced diagnostic imaging methods, such as MRI (Broderick et al., 1993; Linfante et al., 1999; Kidwell et al., 2004). Due to the technical difficulties associated with high-resolution imaging of mouse brains in vivo and the acute onset of morbidity and mortality, lesion size was measured using a histological approach. Surviving mice and those removed from the protocol because of suspected stroke or subcutaneous edema were perfusion-fixed for histologic analysis. Their brains were removed, sectioned, and stained with hematoxylin and eosin (H and E) to identify sites of intracerebral hemorrhage (Figure 3C). Lesions were detected in 12 of 16 Trpa1fl/fl mice and 13 of 15 Trpa1-ecKO animals (no significant difference between Trpa1fl/fl and Trpa1-ecKO mice). Lesions were predominantly found in the cerebral cortex but were also detected in the basal ganglia, midbrain, brain stem, and cerebellum in brains of mice from both groups (Figures 3C and D). The number of lesions per brain did not differ between groups (Figure 3D), but the mean area of individual lesions was significantly smaller in the brains of Trpa1-ecKO mice compared to controls (Figure 3E). Lesions in the cerebral cortex, basal ganglia, and midbrain were significantly smaller in the brains of Trpa1-ecKO mice compared to control Trpa1fl/fl mice. These data suggest that endothelial cell TRPA1 knockout reduces the size of intracerebral hemorrhages in hypertensive mice, but this effect doesn’t improve survival.
The current study investigated the involvement of endothelial cell TRPA1 channels in the pathogenesis of hemorrhagic stroke associated with severe hypertension. Mice were subjected to a protocol that significantly increased systemic BP to levels that caused intracerebral hemorrhage in a majority of the animals studied. Induction of severe hypertension increased the capacity for ROS generation and TRPA1-dependent dilation of cerebral arteries by increasing the expression of NOX enzymes. Intracerebral hemorrhages were smaller in Trpa1-ecKO mice compared to controls, suggesting that TRPA1 channel activity-associated vasodilation and subsequent increase in cerebral blood flow may lead to hematoma expansion during intracerebral hemorrhage events (Figure 4). However, we didn’t observe any differences in survival or other outcomes between control and Trpa1-ecKO mice, suggesting that interventions targeting TRPA1 channels in the clinical setting may have minimal impact on hemorrhagic stroke patients.
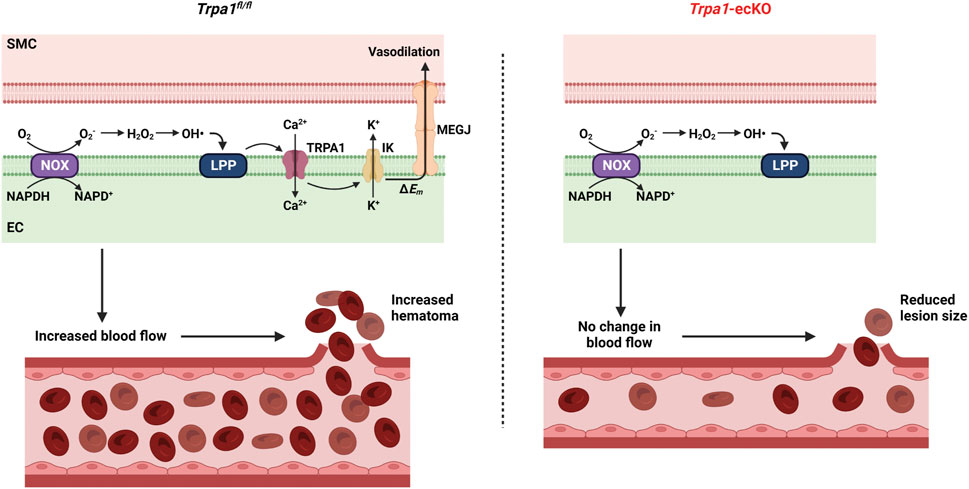
FIGURE 4. Hypothetical model of TRPA1-induced hematoma expansion. Severe hypertension increases NOX-dependent superoxide (O2−) generation in endothelial cells. O2− is dismutased to hydrogen peroxide (H2O2), which undergoes the Fenton reaction to produce hydroxyl radicals (OH•). OH• oxidizes membrane lipids to generate lipid peroxidation products (LPP) which activate TRPA1 channels. Ca2+ influx through TRPA1 channels stimulates K+ efflux through intermediate conductance Ca2+-activated K+ (IK) channels leading to plasma membrane hyperpolarization (ΔEm). Hyperpolarization spreads to smooth muscle cells through myoendothelial gap junctions (MEGJ), resulting in vasodilation and increased blood flow, leading to a larger hematoma when vascular wall integrity is compromised. Trpa1-ecKO don’t experience ROS-induced vasodilation, resulting in smaller lesions.
Spontaneous hemorrhagic stroke is challenging to study in mice. The previously described models require daily, invasive interventions or are unwieldy to use with transgenic mice. For example, Wakisaka et al. (2010b) chronically infused mice with Ang II and supplied L-NAME in the drinking water, followed by twice daily subcutaneous injections of Ang II or norepinephrine to transiently increase blood pressure. This method increased systemic BP, but only the mice chronically infused and acutely injected with Ang II consistently developed stroke. Approximately 73% of mice injected with Ang II developed signs of stroke, compared to ∼40% of mice injected with norepinephrine (Wakisaka et al., 2010b). A study from Iida et al. (2005) described a genetic model of spontaneous hemorrhagic stroke using double transgenic mice overexpressing human renin and angiotensinogen (R+/A+) treated with an HS diet and L-NAME in the drinking water. R+/A+ mice are innately hypertensive but don’t spontaneously develop stroke. Upon treatment with HS and L-NAME, BP increases substantially, which correlates with the development of intracerebral hemorrhages in 100% of the mice. However, combining a double transgenic mouse line like R+/A+ mice with other transgenic lines (e.g., Trpa1-ecKO) requires extensive breeding to obtain animals with the four required transgenes. The Ang II/HS/L-NAME protocol consistently elevated BP in all mice can be readily applied to transgenic animals and doesn’t require daily procedures (e.g., injections). Approximately 81% of mice that completed this protocol developed hemorrhagic lesions, similar to the model reported by Wakisaka et al. However, about 40% of the animals undergoing this hypertensive treatment regimen died of unknown causes, somewhat limiting utility. The precise causes of death for these animals were unclear. It is possible that the animals may have had a fatal stroke, myocardial infarction, or aneurysm due to the severity of the blood pressure increase. However, these animals were found after rigor mortis had set in, making it difficult to perform the necessary investigations to identify the precise cause.
Hemorrhagic strokes cause higher rates of mortality and long-term disability in survivors compared with ischemic strokes (Virani et al., 2021). Brain damage secondary to intracerebral hemorrhage is multifactorial, including increased intracranial pressure due to hematoma formation, neuroinflammation, excitotoxicity, and subsequent edema and neuronal cell death (Xi et al., 2001). ROS produced by the breakdown of extravasated blood and the associated inflammatory response drive secondary brain damage after intracerebral hemorrhage. (Duan et al., 2016; Qu et al., 2016). In addition, expression of NOX1, NOX2, and NOX4 significantly increases in brain tissue and the cerebral vasculature after stroke, and these enzymes are a substantial source of pathological ROS following intracerebral hemorrhage (Wakisaka et al., 2008; Wakisaka et al., 2010a; Zhang et al., 2016). Genetic deletion or pharmacological inhibition of NOX2 and NOX4 reduces brain tissue damage and improves neurological outcomes following hemorrhagic stroke (Shin et al., 2002; Tang et al., 2005). Excessive ROS peroxidizes plasma membrane lipids and increases intracellular Ca2+ levels (Ermak and Davies, 2002; Van et al., 2016). Ca2+ overload triggers several downstream pathways that damage cells and organelles, ultimately leading to cell death (Eigel et al., 2004; Gu et al., 2011; Li et al., 2014a; Duan et al., 2016). However, the precise mechanisms downstream of NOX-generated ROS that drive Ca2+ influx in the context of hemorrhagic stroke are unknown. We have previously shown that ROS metabolites endogenously activate TRPA1 in cerebral artery endothelial cells to induce vasodilation (Sullivan et al., 2015; Pires and Earley, 2018). This mechanism was neuroprotective following ischemic stroke by redirecting blood to hypoxic areas of the brain, ensuring an adequate supply of oxygen and nutrients, and reducing overall infarct size (Pires and Earley, 2018). Interestingly, the current study shows that not only does this protection not extend to cases of hemorrhagic stroke, but instead, TRPA1 channel activity appears to exacerbate the size of the hemorrhagic lesions. We propose ROS-activated endothelial cell TRPA1 channels enhance cerebral blood flow, resulting in more profound blood extravasation during intracerebral hemorrhage events. Removal of this process by knocking out TRPA1 channels resulted in smaller hemorrhagic lesions within the parenchyma. A prior study reported that hematoma expansion is a significant factor for the acute progression of neuronal damage associated with intracerebral hemorrhages (Chen et al., 2014). Interestingly, although the hemorrhagic lesion area was significantly reduced in animals devoid of endothelial TRPA1 channels, we didn’t observe any differences in morbidity or mortality between control Trpa1fl/fl and Trpa1-ecKO mice. Thus, our data suggest that blocking TRPA1 channels may not be helpful for treating hypertension-associated hemorrhagic stroke.
All animal procedures used in the study complied with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of the University of Nevada, Reno School of medicine, and Colorado State University. Endothelial cell-specific deletion of TRPA1 was achieved by initially crossing mice homozygous for loxP sequences flanking S5/S6 transmembrane domains of TRPA1 (floxed TRPA1; Strain number: 008,654; The Jackson Laboratory, Bar Harbor, Maine) with heterozygous TekCre mice which express cre recombinase (cre) driven by the promoter for the endothelial cell-specific receptor tyrosine kinase TEK (Strain number: 008,863; The Jackson Laboratory, Bar Harbor, Maine) to produce intermediate heterozygote mice which were used to generate Trpa1-ecKO mice, as previously described (Sullivan et al., 2015; Thakore et al., 2021). Mice homozygous for floxed TRPA1 and positive for cre were considered Trpa1-ecKO, and littermates homozygous for floxed TRPA1 but negative for cre were used as controls (Trpa1fl/fl). Mice were kept on a 12-h light/dark cycle and were fed standard chow and regular water ad libitum unless otherwise specified. Our prior study demonstrated that TRPA1 expression is undetectable by Western blotting in cerebral arteries from Trpa1-ecKO mice (Sullivan et al., 2015).
At 12 weeks of age, mice underwent telemetry probe implantation as previously described (Feng et al., 2010; Li et al., 2014b; Thakore et al., 2020; Krishnan et al., 2022). Briefly, anesthesia was induced by 4%–5% isoflurane carried in 100% O2 (flow rate 1 L/min), after which anesthesia was maintained by adjusting isoflurane to 1.5%–2%; pre-operative analgesia was provided by a single subcutaneous injection of 0.05 mg/kg buprenorphine (Zoopharm, Windsor, CO). The neck was shaved and then sterilized with iodine. Under aseptic conditions, an incision (∼1 cm) was made to separate the oblique and tracheal muscles and expose the left common carotid artery. The catheter of a radio telemetry transmitter (PA-C10; Data Science International, Harvard Bioscience, Inc., Minneapolis, MN) was surgically implanted in the left common carotid artery and secured using non-absorbable silk suture threads. The body of the transmitter was placed in a subcutaneous pocket caudal to the right forelimb. After a 14-day recovery period, baseline BP, heart rate, and locomotor activity were recorded in conscious mice for 7 days. Data were collected daily every 10 s for 2 h (from 2 p.m. to 4 p.m.) for the duration of the study protocol and analyzed.
At 15 weeks of age, mice were surgically implanted with an osmotic minipump filled with Ang II (1.2 μg/kg/min; Sigma-Aldrich, St. Louis, MO) and fed 8% NaCl (HS; Envigo, Indianapolis, IN) chow ad libitum. After 2 weeks of Ang II and HS treatment, L-NAME (120 mg/kg/day; Sigma-Aldrich) was added to the drinking water. Treatment was continued for two additional weeks to induce severe hypertension and hemorrhagic strokes. Mice were monitored at least once daily. At the end of the treatment protocol, or sooner if mice showed behavioral signs of a stroke or other morbidity, mice were euthanized and perfusion-fixed in ice-cold 4% paraformaldehyde (PFA) solution for brain histology. Non-fixed brains were also collected from a cohort of Trpa1fl/fl mice for ex vivo analysis.
Cerebral arteries from three animals were isolated and pooled together from control normotensive and hypertensive animals. RNA was extracted and purified using a RNeasy mini kit (Qiagen, Hilden, Germany). A reverse transcriptase reaction was performed using 200 ng RNA per group with a QuantiTect RT kit (Qiagen) to generate complementary DNA (cDNA) samples. Quantitative PCR was performed using a QuantiTect SYBR Green kit (Qiagen). Primers for mouse Gapdh (QT01658692, Qiagen), Trpa1 (QT01595937, Qiagen), Nox1 (QT00140091, Qiagen), Nox2 (QT00139797, Qiagen), and Nox4 (QT00126042, Qiagen) were used to determine respective mRNA levels. This process was repeated twice to generate results from three independent biological samples. The relative expression levels were calculated using the Pfaffl method (Pfaffl, 2001) as previously described (Crnich et al., 2010).
Cerebral arteries were isolated from control normotensive and hypertensive animals and snap-frozen in liquid nitrogen. RIPA lysis buffer (50 µl) (Thermo Fisher Scientific, Waltham, MA) containing a protease inhibitor cocktail (Thermo Fisher Scientific) was added to each artery sample and homogenized by sonication (20 x 1-s pulses) and mechanical disruption by a Fisher Scientific Tissuemiser (10 s) on ice. Samples were centrifuged at 13,000 rpm for 10 min, and the supernatant was transferred to a new tube. Protein concentration for each sample was determined using a BCA Protein assay (Thermo Fisher Scientific). Protein samples (10 ng) were added to SDS sample buffer and heated at 70°C for 10 min. Immediately after denaturation, proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk and 1% BSA in PBS containing 0.1% Tween and 0.02% sodium azide (PBS-TA) for 30 min at room temperature on a rocker and then exposed to a rabbit anti-TRPA1 antibody (1:500, ACC-037, Alomone Labs, Jerusalem, Israel) in 5% milk, 1% BSA (PBS-TA) overnight at room temperature on a rocker. The same blot was probed independently with a rabbit β-actin antibody (1:1,000, ab8227, Abcam, Cambridge, United Kingdom). The membranes were then washed with PBS-T for 3 × 5 min and exposed to a goat anti-rabbit secondary antibody (1:10,000, Invitrogen) in 5% milk, 1% BSA (PBS-T) for 2 h at room temperature on a rocker, followed by 5-min washes with PBS-T, incubated in Supersignal ECL substrate (Thermo Fisher Scientific) for 1–3 min, and imaged. Protein bands were quantified using ImageJ software (version 1.52n, National Institutes of Health, Bethesda, MD).
Cerebral arteries were isolated from control normotensive and hypertensive animals. RIPA lysis buffer (50 µL) containing a protease inhibitor cocktail was added to each artery sample and homogenized by sonication (20 × 1-s pulses) and mechanical disruption by a Fisher Scientific Tissuemiser (10 s) on ice. Samples were centrifuged at 13,000 rpm for 10 min, and the supernatant was transferred to a new tube. Protein concentration for each sample was determined using a BCA Protein assay (Thermo Fisher Scientific). The isolated protein (∼50–60 µl) was diluted to 400 µL with HEPES-buffered saline, and lucigenin reagent (5 μM; Cayman Chemicals, Ann Arbor, MI) was added. At this concentration lucigenin doesn’t produce excess superoxide and provides an accurate assessment of the rate of superoxide production (Skatchkov et al., 1999; Munzel et al., 2002; Guzik and Channon, 2005). 200 µL was added to each of two wells in a 96-well plate. Apocynin (100 µM) was added to one of the wells, and the other was treated with vehicle (DMSO). Basal luminescence was measured in relative light units (RLU) every minute for 10 min using a BioTek Synergy H1 microplate reader (Biotek, Winooski, VT). NADPH (200 μM, Sigma-Aldrich) was then dispensed into each well, and luminescence was measured every minute for 50 min. O2− production was calculated as the change in RLU/minute per µg protein. This ex vivo assay measures the capacity for O2− production, but not O2− level in cerebral arteries in vivo.
Isolated vessel experiments were performed as previously described (Sullivan et al., 2015; Wenceslau et al., 2021). Briefly, mouse middle cerebral arteries were isolated and placed in ice-cold MOPS-buffered saline. Arterial segments were then cannulated between two glass cannulas in a chamber (Living Systems Instrumentation, St. Albans City, VT) and secured with single-filament silk. Arteries were superfused with a physiological saline solution (PSS; 119 mM NaCl, 4.7 mM KCl, 21 mM NaHCO3, 1.17 mM MgSO4, 1.8 mM CaCl2, 1.18 mM KH2PO4, 5 mM glucose, 0.03 mM EDTA) warmed to 37°C and bubbled with 21% O2, 6% CO2, and balanced N2. Vessels were pressurized to 60 mmHg and gently stretched to simulate physiological conditions. Intraluminal pressure was lowered 20 mmHg and arteries were equilibrated for 15 min. Viability was assessed by superfusing a high [K+] PSS (60 mM KCl, 63.7 mM NaCl) to induce vasoconstriction. Follow a 15 min wash, intraluminal pressure was increased to 60 mmHg, and arteries were allowed to develop spontaneous myogenic tone. The % dilation was calculated as the % change in myogenic tone before and after the addition of NADPH.
Brains were removed from perfusion-fixed mice and stored overnight in 4% PFA. Brains were then serially dehydrated in 7.5%, 15%, and 30% sucrose (Sigma-Aldrich) solutions at 4°C and frozen in OCT sectioning medium at −80°C. The tissue was sectioned with a cryostat (Leica Biosystems, Wetzlar, Germany), generating 35 µm thick sections collected every 500 µm. Sections were mounted on glass slides (Superfrost Plus, VWR International, Radnor, PA) and refrigerated overnight. These sections were then stained with hematoxylin and eosin using the H&E staining kit (Abcam, Cambridge, United Kingdom), per the manufacturer’s instructions. Stained sections were sealed using the Cytoseal XYL mounting medium (Thermo Fisher Scientific) and allowed to dry for a minimum of 24 h. Sections were imaged using a bright field microscope (BZ-X710, Keyence Corporation, Osaka, Japan) at ×4 magnification.
Intracerebral hemorrhage lesions were identified as an abnormal appearance of blood within the brain tissue (appears red by eosin staining). Serial coronal sections of brains were viewed by light microscopy, and the number of hemorrhagic lesions within each of the following brain regions was recorded: cerebral cortex, basal ganglia, midbrain, brainstem, or cerebellum. ImageJ image analysis software was used to measure each lesion’s maximum area (mm2).
All summary data are presented as means ± SEM. Statistical analyses and graphical presentations were performed using GraphPad Prism software (version 9.5.0, GraphPad Software, San Diego, CA). The value of n refers to the number of mice per group used for radiotelemetry experiments, survival assays, lesion number quantification, qRT-PCR and Western blots. For lesion area quantification, n refers to the number of lesions from each mouse, and for isolated vessel experiments, n is the number of arteries per group. Statistical analyses were performed using Students paired or unpaired two-tailed t-test with or without Welch’s correction as appropriate, repeated measures or non-repeated measures two-way analysis of variance (ANOVA) with a Šidák correction for multiple comparisons, or Log-rank (Mantel-Cox) test. A value of p < 0.05 was considered as statistically significant.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committees of the University of Nevada, Reno School of medicine, and Colorado State University.
SE initiated and supervised the project. SE, MS, and YF designed the experiments. MS, WL, and YF performed radiotelemetry experiments. MS and PT performed minipump implantation surgeries. MS, PT, VK, and SA monitored mice after surgery. MS performed qRT-PCR, western blot, and arterial myography experiments. MS and VK performed histology experiments. MS, PT, VK, SA, YF, and SE analyzed the data. MS, PT, VK, and SE wrote the manuscript, prepared the figures, and revised the manuscript.
This study was supported by grants from the National Institutes of Health (NHLBI R35155008 and NIGMS P20GM130459 to SE, R01HL122770 to YF, NINDS RF1NS110044, and R33NS115132 to SE, and AHA 15PRE22670024 to MS). The Transgenic Genotyping and Phenotyping Core and the High Spatial and Temporal Resolution Imaging Core at the COBRE Center for Molecular and Cellular Signaling in the Cardiovascular System, University of Nevada, Reno, are maintained by grants from NIH/NIGMS (P20GM130459 Sub#5451 and P20GM130459 Sub#5452).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1129435/full#supplementary-material
Adler, S., and Huang, H. (2004). Oxidant stress in kidneys of spontaneously hypertensive rats involves both oxidase overexpression and loss of extracellular superoxide dismutase. Am. J. Physiol. Ren. Physiol. 287, F907–F913. doi:10.1152/ajprenal.00060.2004
Amberg, G. C., Bonev, A. D., Rossow, C. F., Nelson, M. T., and Santana, L. F. (2003). Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J. Clin. Invest. 112, 717–724. doi:10.1172/JCI18684
Bamford, J., Sandercock, P., Dennis, M., Burn, J., and Warlow, C. (1991). Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 337, 1521–1526. doi:10.1016/0140-6736(91)93206-o
Bannister, J. P., Bulley, S., Narayanan, D., Thomas-Gatewood, C., Luzny, P., Pachuau, J., et al. (2012). Transcriptional upregulation of α2δ-1 elevates arterial smooth muscle cell voltage-dependent Ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension 60, 1006–1015. doi:10.1161/HYPERTENSIONAHA.112.199661
Broderick, J. P., Brott, T. G., Duldner, J. E., Tomsick, T., and Huster, G. (1993). Volume of intracerebral hemorrhage. A powerful and easy-to-use predictor of 30-day mortality. Stroke 24, 987–993. doi:10.1161/01.str.24.7.987
Chen, S., Zeng, L., and Hu, Z. (2014). Progressing haemorrhagic stroke: Categories, causes, mechanisms and managements. J. Neurol. 261, 2061–2078. doi:10.1007/s00415-014-7291-1
Crnich, R., Amberg, G. C., Leo, M. D., Gonzales, A. L., Tamkun, M. M., Jaggar, J. H., et al. (2010). Vasoconstriction resulting from dynamic membrane trafficking of TRPM4 in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 299, C682–C694. doi:10.1152/ajpcell.00101.2010
De Ciuceis, C., Porteri, E., Rizzoni, D., Rizzardi, N., Paiardi, S., Boari, G. E., et al. (2007). Structural alterations of subcutaneous small-resistance arteries may predict major cardiovascular events in patients with hypertension. Am. J. Hypertens. 20, 846–852. doi:10.1016/j.amjhyper.2007.03.016
Donkor, E. S. (2018). Stroke in the 21(st) century: A snapshot of the burden, epidemiology, and quality of life. Stroke Res. Treat. 2018, 3238165. doi:10.1155/2018/3238165
Duan, X., Wen, Z., Shen, H., Shen, M., and Chen, G. (2016). Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell Longev. 2016, 1203285. doi:10.1155/2016/1203285
Earley, S., Gonzales, A. L., and Crnich, R. (2009). Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 104, 987–994. doi:10.1161/CIRCRESAHA.108.189530
Eigel, B. N., Gursahani, H., and Hadley, R. W. (2004). ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated Guinea pig ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 286, H955–H963. doi:10.1152/ajpheart.00721.2003
Ermak, G., and Davies, K. J. (2002). Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 38, 713–721. doi:10.1016/s0161-5890(01)00108-0
Faraco, G., and Iadecola, C. (2013). Hypertension: A harbinger of stroke and dementia. Hypertension 62, 810–817. doi:10.1161/HYPERTENSIONAHA.113.01063
Feng, Y., Xia, H., Cai, Y., Halabi, C. M., Becker, L. K., Santos, R. A., et al. (2010). Brain-selective overexpression of human Angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ. Res. 106, 373–382. doi:10.1161/CIRCRESAHA.109.208645
Gu, Y., Dee, C. M., and Shen, J. (2011). Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. Sch. Ed. 3, 1216–1231. doi:10.2741/222
Guzik, T. J., and Channon, K. M. (2005). Measurement of vascular reactive oxygen species production by chemiluminescence. Methods Mol. Med. 108, 73–89. doi:10.1385/1-59259-850-1:073
Hägg-Holmberg, S., Dahlström, E. H., Forsblom, C. M., Harjutsalo, V., Liebkind, R., Putaala, J., et al. (2019). The role of blood pressure in risk of ischemic and hemorrhagic stroke in type 1 diabetes. Cardiovasc Diabetol. 18, 88. doi:10.1186/s12933-019-0891-4
Harder, D. R., Smeda, J., and Lombard, J. (1985). Enhanced myogenic depolarization in hypertensive cerebral arterial muscle. Circ. Res. 57, 319–322. doi:10.1161/01.res.57.2.319
Hayashi, K., and Naiki, T. (2009). Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J. Mech. Behav. Biomed. Mater 2, 3–19. doi:10.1016/j.jmbbm.2008.05.002
Iida, S., Baumbach, G. L., Lavoie, J. L., Faraci, F. M., Sigmund, C. D., and Heistad, D. D. (2005). Spontaneous stroke in a genetic model of hypertension in mice. Stroke 36, 1253–1258. doi:10.1161/01.str.0000167694.58419.a2
Izzard, A. S., Rizzoni, D., Agabiti-Rosei, E., and Heagerty, A. M. (2005). Small artery structure and hypertension: Adaptive changes and target organ damage. J. Hypertens. 23, 247–250. doi:10.1097/00004872-200502000-00002
Kazama, K., Anrather, J., Zhou, P., Girouard, H., Frys, K., Milner, T. A., et al. (2004). Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ. Res. 95, 1019–1026. doi:10.1161/01.RES.0000148637.85595.c5
Kidwell, C. S., Chalela, J. A., Saver, J. L., Starkman, S., Hill, M. D., Demchuk, A. M., et al. (2004). Comparison of MRI and CT for detection of acute intracerebral hemorrhage. JAMA 292, 1823–1830. doi:10.1001/jama.292.15.1823
Knock, G. A., Snetkov, V. A., Shaifta, Y., Connolly, M., Drndarski, S., Noah, A., et al. (2009). Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic. Biol. Med. 46, 633–642. doi:10.1016/j.freeradbiomed.2008.11.015
Krishnan, V., Ali, S., Gonzales, A. L., Thakore, P., Griffin, C. S., Yamasaki, E., et al. (2022). STIM1-dependent peripheral coupling governs the contractility of vascular smooth muscle cells. Elife 11, e70278. doi:10.7554/eLife.70278
Lammie, G. A. (2002). Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 12, 358–370. doi:10.1111/j.1750-3639.2002.tb00450.x
Landmesser, U., Dikalov, S., Price, S. R., Mccann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi:10.1172/JCI14172
Li, Q., Pogwizd, S. M., Prabhu, S. D., and Zhou, L. (2014a). Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in Guinea pig cardiomyocytes. PLoS One 9, e93928. doi:10.1371/journal.pone.0093928
Li, W., Peng, H., Mehaffey, E. P., Kimball, C. D., Grobe, J. L., Van Gool, J. M., et al. (2014b). Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension. Hypertension 63, 316–323. doi:10.1161/HYPERTENSIONAHA.113.02041
Linfante, I., Llinas, R. H., Caplan, L. R., and Warach, S. (1999). MRI features of intracerebral hemorrhage within 2 hours from symptom onset. Stroke 30, 2263–2267. doi:10.1161/01.str.30.11.2263
Matsuno, K., Yamada, H., Iwata, K., Jin, D., Katsuyama, M., Matsuki, M., et al. (2005). Nox1 is involved in angiotensin II-mediated hypertension: A study in nox1-deficient mice. Circulation 112, 2677–2685. doi:10.1161/CIRCULATIONAHA.105.573709
Munzel, T., Afanas'Ev, I. B., Kleschyov, A. L., and Harrison, D. G. (2002). Detection of superoxide in vascular tissue. Arterioscler. Thromb. Vasc. Biol. 22, 1761–1768. doi:10.1161/01.atv.0000034022.11764.ec
Paravicini, T. M., Chrissobolis, S., Drummond, G. R., and Sobey, C. G. (2004). Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke 35, 584–589. doi:10.1161/01.STR.0000112974.37028.58
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. doi:10.1093/nar/29.9.e45
Pires, P. W., and Earley, S. (2018). Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 7, e35316. doi:10.7554/eLife.35316
Pratt, P. F., Bonnet, S., Ludwig, L. M., Bonnet, P., and Rusch, N. J. (2002). Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 40, 214–219. doi:10.1161/01.hyp.0000025877.23309.36
Qian, X., Francis, M., Solodushko, V., Earley, S., and Taylor, M. S. (2013). Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 20, 138–148. doi:10.1111/micc.12004
Qu, J., Chen, W., Hu, R., and Feng, H. (2016). The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxid. Med. Cell Longev. 2016, 2592935. doi:10.1155/2016/2592935
Rajagopalan, S., Kurz, S., Munzel, T., Tarpey, M., Freeman, B. A., Griendling, K. K., et al. (1996). Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 97, 1916–1923. doi:10.1172/JCI118623
Shekhar, S., Wang, S., Mims, P. N., Gonzalez-Fernandez, E., Zhang, C., He, X., et al. (2017). Impaired cerebral autoregulation-A common neurovascular pathway in diabetes may play a critical role in diabetes-related alzheimer's disease. Curr. Res. Diabetes Obes. J. 2, 555587. doi:10.19080/crdoj.2017.2.555587
Shin, H. K., Lee, J. H., Kim, K. Y., Kim, C. D., Lee, W. S., Rhim, B. Y., et al. (2002). Impairment of autoregulatory vasodilation by NAD(P)H oxidase-dependent superoxide generation during acute stage of subarachnoid hemorrhage in rat pial artery. J. Cereb. Blood Flow. Metab. 22, 869–877. doi:10.1097/00004647-200207000-00012
Skatchkov, M. P., Sperling, D., Hink, U., Mulsch, A., Harrison, D. G., Sindermann, I., et al. (1999). Validation of lucigenin as a chemiluminescent probe to monitor vascular superoxide as well as basal vascular nitric oxide production. Biochem. Biophys. Res. Commun. 254, 319–324. doi:10.1006/bbrc.1998.9942
Strandgaard, S., Olesen, J., Skinhoj, E., and Lassen, N. A. (1973). Autoregulation of brain circulation in severe arterial hypertension. Br. Med. J. 1, 507–510. doi:10.1136/bmj.1.5852.507
Sullivan, M. N., Gonzales, A. L., Pires, P. W., Bruhl, A., Leo, M. D., Li, W., et al. (2015). Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal 8, ra2. doi:10.1126/scisignal.2005659
Tang, J., Liu, J., Zhou, C., Ostanin, D., Grisham, M. B., Neil Granger, D., et al. (2005). Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J. Neurochem. 94, 1342–1350. doi:10.1111/j.1471-4159.2005.03292.x
Thakore, P., Alvarado, M. G., Ali, S., Mughal, A., Pires, P. W., Yamasaki, E., et al. (2021). Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 10, e63040. doi:10.7554/eLife.63040
Thakore, P., Pritchard, H. A. T., Griffin, C. S., Yamasaki, E., Drumm, B. T., Lane, C., et al. (2020). TRPML1 channels initiate Ca(2+) sparks in vascular smooth muscle cells. Sci. Signal 13, eaba1015. doi:10.1126/scisignal.aba1015
Tsao, C. W., Aday, A. W., Almarzooq, Z. I., Alonso, A., Beaton, A. Z., Bittencourt, M. S., et al. (2022). Heart disease and stroke statistics-2022 update: A report from the American heart association. Circulation 145, e153–e639. doi:10.1161/CIR.0000000000001052
Van, D. P. J., Neyts, E. C., Verlackt, C. C. W., and Bogaerts, A. (2016). Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 7, 489–498. doi:10.1039/c5sc02311d
Virani, S. S., Alonso, A., Aparicio, H. J., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., et al. (2021). Heart disease and stroke statistics-2021 update: A report from the American heart association. Circulation 143, e254–e743. doi:10.1161/CIR.0000000000000950
Wakisaka, Y., Chu, Y., Miller, J. D., Rosenberg, G. A., and Heistad, D. D. (2010a). Critical role for copper/zinc-superoxide dismutase in preventing spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. Stroke 41, 790–797. doi:10.1161/STROKEAHA.109.569616
Wakisaka, Y., Chu, Y., Miller, J. D., Rosenberg, G. A., and Heistad, D. D. (2010b). Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J. Cereb. Blood Flow. Metab. 30, 56–69. doi:10.1038/jcbfm.2009.183
Wakisaka, Y., Miller, J. D., Chu, Y., Baumbach, G. L., Wilson, S., Faraci, F. M., et al. (2008). Oxidative stress through activation of NAD(P)H oxidase in hypertensive mice with spontaneous intracranial hemorrhage. J. Cereb. Blood Flow. Metab. 28, 1175–1185. doi:10.1038/jcbfm.2008.7
Wenceslau, C. F., Mccarthy, C. G., Earley, S., England, S. K., Filosa, J. A., Goulopoulou, S., et al. (2021). Guidelines for the measurement of vascular function and structure in isolated arteries and veins. Am. J. Physiol. Heart Circ. Physiol. 321, H77–h111. doi:10.1152/ajpheart.01021.2020
XI, G., Hua, Y., Bhasin, R. R., Ennis, S. R., Keep, R. F., and Hoff, J. T. (2001). Mechanisms of edema formation after intracerebral hemorrhage: Effects of extravasated red blood cells on blood flow and blood-brain barrier integrity. Stroke 32, 2932–2938. doi:10.1161/hs1201.099820
Xiao, Q., Luo, Z., Pepe, A. E., Margariti, A., Zeng, L., and Xu, Q. (2009). Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am. J. Physiol. Cell Physiol. 296, C711–C723. doi:10.1152/ajpcell.00442.2008
Zalba, G., San Jose, G., Moreno, M. U., Fortuno, M. A., Fortuno, A., Beaumont, F. J., et al. (2001). Oxidative stress in arterial hypertension: Role of NAD(P)H oxidase. Hypertension 38, 1395–1399. doi:10.1161/hy1201.099611
Keywords: endothelium, TRPA1, stroke, hypertension, hemorrhage
Citation: Sullivan MN, Thakore P, Krishnan V, Alphonsa S, Li W, Feng Earley Y and Earley S (2023) Endothelial cell TRPA1 activity exacerbates cerebral hemorrhage during severe hypertension. Front. Mol. Biosci. 10:1129435. doi: 10.3389/fmolb.2023.1129435
Received: 21 December 2022; Accepted: 17 January 2023;
Published: 30 January 2023.
Edited by:
Mark Stephen Taylor, University of South Alabama, United StatesReviewed by:
Alexander A. Mongin, Albany Medical College, United StatesCopyright © 2023 Sullivan, Thakore, Krishnan, Alphonsa, Li, Feng Earley and Earley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott Earley, c2VhcmxleUBtZWQudW5yLmVkdQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.