
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 09 February 2023
Sec. Biophysics
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1124563
This article is part of the Research TopicMolecular Level Atomistic and Structural Insights on Biological Macromolecules, Inhibition, and Dynamics StudiesView all 13 articles
 C. Kirubhanand1
C. Kirubhanand1 J. Merciline Leonora2S. Anitha3R. Sangeetha4
J. Merciline Leonora2S. Anitha3R. Sangeetha4 K. T. Nachammai5
K. T. Nachammai5 K. Langeswaran5*
K. Langeswaran5* S. Gowtham Kumar6*
S. Gowtham Kumar6*Introduction: Non-Small Cell Lung Cancer is the most prevalent type of cancer in lung cancer. Chemotherapy, radiation therapy, and other conventional cancer treatments have a low success rate. Thus, creating new medications is essential to halt the spread of lung cancer.
Methods: In this study bioactive nature of lochnericine against Non-Small Cell Lung Cancer (NSCLC) was analyzed using various computational approaches such as quantum chemical calculations, molecular docking, and molecular dynamic simulation. Furthermore, the MTT assay shows the anti-proliferation activity of lochnericine.
Results and Discussion: Using Frontier Molecular Orbital (FMO), the calculated band gap energy value associated with bioactive compounds and the molecule’s potential bioactivity is confirmed. The H38 hydrogen atom and O1 oxygen atom in the molecule are effectively electrophilic, and potential nucleophilic attack sites were confirmed through analysis of the Molecular electrostatic potential surface. Furthermore, the electrons within the molecule were delocalized, which confers bioactivity on the title molecule and was authorized through Mulliken atomic charge distribution analysis. A molecular docking study revealed that lochnericine inhibits non-small cell lung cancer-associated targeted protein. The lead molecule and targeted protein complex were stable during molecular dynamics simulation studies till the simulation period. Further, lochnericine demonstrated remarkable anti-proliferative and apoptotic features against A549 lung cancer cells. The current investigation powerfully suggests that lochnericine is a potential candidate for lung cancer.
Developed and developing nations consider cancer a significanthealth concern with increasing incidence and mortality rates. The tumor is caused by complex biological processes, namely uninhibited cell proliferation, cell death resistance, neo-angiogenesis, and metastasis (Hausman, 2019). Incursion and metastasis are the frequent causes of cancer-related mortality, resulting in additional invasion sites and severe organ damage. Cases of cancer are rapidly becoming a risk factor for various malignancies, most notably lung, liver, colorectal, and breast cancers. Because of the disease’s high notoriety, therapy has been a never-ending fight with few results (Mullard, 2020; Banaganapalli et al., 2022). One of the deadliest and most ubiquitous forms of cancer in people is lung cancer. The most prevalent type of cancer is NSCLC (non-small cell lung cancer), estimated as 85% among other cancers. Nearly 90% of non-small cell lung cancer occurrences are caused by smoking. Tumor-infiltrating lymphocytes (TILs), in general, and notably CD8+ TILs, are associated with a favourable prognosis in non-small cell lung cancer (NSCLC) (Gueguen et al., 2021). Traditional cancer treatments, such as surgery, radiation, and chemotherapy, have a low success rate. Thus, developing new drugs is crucial to stop lung cancer spread (Schabath and Cote, 2019).
In cancer treatment, natural products may be used in addition to traditional anticancer medications. Recently, researchers worldwide have focused on developing novel drugs with medicinal potential derived from natural sources such as plants. Plants and their phytoconstituents have been used for medical purposes since antiquity (Ekiert and Szopa, 2020). Natural-source chemicals attract the interest of scientists worldwide. Humans have extracted cancer chemotherapeutic chemicals from flora (Deng et al., 2020). Lochnericine is a significant monoterpene indole alkaloid (MIA), found in the roots of Madagascar periwinkle (Catharanthusroseus) and formed by the stereoselective C6, C7-epoxidation of tabersonine. The stereoselective C6 produces Lochnericine and C7-epoxidation of tabersonine and can be subsequently metabolized to produce more complex MIAs. Although the enzymes in charge of its downstream modifications have been identified, those in charge of lochnericine production have not.
Growth factors, namely hormones, mainly influence the migration of normal cells and the division of cells. Tyrosine kinase EGRF (Epidermal Growth Factor Receptor) influences cell proliferation, cell division, and tumour development. It is a representative transmembrane receptor which triggers signalling pathways via ligand-provoked dimerization (Kavitha et al., 2015; Zhou and Yao, 2016; Shaik et al., 2019). In human cancers, the most prevalently mutated oncogene is the Kirsten rat sarcoma (KRAS) gene involved in carcinogenesis, which accounts for more than twenty per cent of lung cancer, particularly NSCLC (Reck et al., 2021). There is a strong connotation between various solid tumours NTRK (Neurotrophic Tropomyosin Receptor Kinase) gene fusion, including NSCLC. In cancers covering NSCLC, ALK activation occurs through fusion gene formation, the preliminary actuating mutation in ALK. TRIM 1 is designated by cancer cells to encourage tumorigenic development and is upregulated in malignant cells. In tumour cells, it indorses elevatedproteasome activity (Wang et al., 2016). This study investigated a lochnericine molecule using DFT quantum chemical calculations, including molecular structure, vibrational (FMOs), and other vibrational analyses. The computational studieslikely docking analysis was also performedusing bioinformatics tools to check the molecule’s repressivebehaviour against lung cancer-associated targeted proteins. Computer-Aided Drug Design (CADD) has emerged as an effective method for determining potential lead compounds and improving the development of new treatments for various diseases. Molecular dynamics simulations determine the stability of protein-ligand complexes. Furthermore, in vitro cytotoxicity testing was carried out to confirm the anti-lung cancer activity.
The most stable molecular structure of lochnericine was optimized using the Gaussian 09 software, and the DFT/B3LYP method with LANL2DZ was used to optimize the structure. The Los Alamos National Laboratory 2 double zeta (LANL2DZ) for transition metals and all-electron basis sets for all other non-transition metal atoms are used more frequently in computations on systems including transition metals. (Frisch et al., 2009). The molecule’s vibrational wavenumbers were computed and assigned using the VEDA 4.0 software (Jamroz, 2004). The optimized molecular structure, vibrational wavenumbers, FMOs, MEP surface, and Mullikenatomic charge distributions of Lochnericine molecule were visualized using GaussView 05 software (Dennington et al., 2009). All the quantum chemical calculations were performed without regard for the potential energy surface to grasp the interception nature of the Lochnericine molecule.
In the present study, target receptor molecules, including EGFR [PDB ID: 2ITY], KRAS [PDB ID: 7LGI], Ntrk [PDB ID: 7VKO], and ALK [PDB ID: 2XP2], were obtained from the RCSB Protein Data Bank (Sussman et al., 1998). TRIM11 protein sequences were obtained from UniProt (UniProt ID: Q96F44) (UniProt, 2019). The 3D protein structure of the other protein, TRIM11, was modelled using the Swiss model (Schwede et al., 2003). Procheck in the SAVES server was used to verify the three-dimensional protein structure (Laskowski et al., 2016). All water molecules and irregular residues were purified from the primary protein structure. In the PyRx workspace folders, the protein receptor was saved as a PDBQT file after adding missing hydrogens and charges. Using the PubChem database, the lochnericine molecule was downloaded. The ligand was imported and prepared. These ligands were then converted to Auto Dock Ligand format (PDBQT).
Molecular docking was performed using Vina version 2.0 in PyRX (Dallakyan and Olson, 2015). In the PyRX interface, rigid docking was carried out for the targeted receptor molecules. The ligand and proteins were then subjected to docking to get the binding affinity with each other. Lamarckian genetic algorithm conformational search with the default parameters was utilized. Further, the grid on the targeted protein’s ligand-binding site was situated in the centre of the binding site. Protein-lead molecule interactions were analyzed using Discovery Studio Visualize software.
ADME prediction was performed in the SWISS ADME tool (Tripathi et al., 2019). This tool can forecast the pharmacokinetics and drug-like characteristics of active compounds. The numbers of rotatable bonds, hydrogen bond donors, acceptors, and molecular weight, were also predicted. The log p values represent the lipophilicity of the compounds. The logarithmic S value represents the solubility of water. Human gastrointestinal absorption (HIA) and blood-brain barrier (BBB) permeation were predicted pharmacokinetic properties in swiss adme.
In simulation studies using molecular dynamics (MD), the atomic and molecular motions within the protein structure were examined. Atoms and molecules can interact for 100 ns in the GROMACS (GROningenMAchine for Chemical Simulations) (Van Der Spoel et al., 2005). Macromolecular structure-to-function interactions may now be efficiently understood using an advanced technology known as molecular dynamics simulations (Awan et al., 2021; Bima et al., 2022). MD simulations were performed by using the GROMOS96 43a1 force field. Targeted receptor molecule and ligand complex topology was created by parametrizing the compound through the PRODRG server (Schüttelkopf and Van Aalten, 2004; Sangavi and Langeswaran, 2021). The simple point charge (SPC216) water model’s step for salvation is the cubic box centre to edges. To balance the MD simulations, counter ions (Na+ and Cl−) were also included in the simulation systems (Shaik et al., 2022). Adding counter-ions to the system to neutralize it. Two methods exist for adding ions: Solve the solution and then add ions to replace the solvent’s randomly distributed molecules. To solve a problem, distribute ions in accordance with the macromolecule’s electrostatic potential. The simulation box interacts with an endless number of its periodic images when periodic boundary conditions are utilized, and the Coulomb energy is calculated using grid-based techniques. As a result, if a simulation system is charged, the electrostatic energy will accumulate to infinity. We must neutralize the system by introducing counter-ions to address this problem and allow simulations to determine the electrostatic energy accurately. Using MD simulations at the atomistic level has the advantage of allowing for capturing and evaluating various motional contributions to the overall complex dynamics of the targeted receptor molecule. Potential steric obstacles between the atoms in the solvated system were removed using the energy minimization step and the 1,500 steepest descent followed by conjugate gradient techniques. The NVT and NPT ensemble’s two-step equilibration was carried out for 100 ps at constant volume, with gradual heating from 0 to 300 K, and at a pressure of 1 atm. The temperature is maintained with a Berendsen thermostat. The Parrinello-Rahman barostat is used to keep the system’s pressure constant. The simulation of each system took place for 100 ns? Xmgrace software was used to examine the flexibility and stability of the protein and ligand complex (Turner, 2005).
Sigma, St Louis, MO, United States, supplied the dimethyl sulfoxide (DMSO) and Lochnericine. All other chemicals and solvents were purchased from SISCO Research Laboratories (SRL). Lochnericine was synthesized in a serum-free RPMI medium after being dissolved in dimethyl sulfoxide (DMSO) at a final concentration of less than 0.1 percent (v/v). It did not affect cell viability) and filtered through a 0.045 mm syringe filter before being stored at 4°C.
The MTT assay was employed to assess the anti-proliferative efficacy of lochnericine against A549 lung cancer cells. Through mitochondrial enzymes, the translation of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to the formation of MTT is the critical mechanism involved in this cytotoxicity assay. NCCS Pune supplied the lung cancer cell line A549 for this investigation. In 24 well tissue culture plates, 2x 105 cells were cultured with 0.5 mL medium/well, 37°C temperature was maintained with CO2 5%, and relative humidity was about 95%. For 48 h, cells were treated with various concentrations of lochnericine. The cells were incubated at 37°C for 4 h after adding 200 µl of MTT solution (5 mg/mL), and cells were harvested. Through the gentle aspiration method, the medium and unread MTT were detached. At room temperature, the plates were shaken well for 5 min by adding 500 µl of DMSO. The crystallized dye was extracted by adding 1 mL of DMSO to the well. At 570 nm absorbance, the quantity of blue dye formed was estimated. The proliferation of the cells was examined by microscopic visualization at 48X magnification. The control and drug-treated cells were treated with 10% methanol for 5 min. Then cells were viewed under an inverted microscopeto confirm the anti-proliferative efficacy of the drug.
The mean and standard deviation data were displayed (SD). The noteworthyvariances were calculated using a one-way analysis of variance (ANOVA). A commercial software application (SPSS version 20) was employed for statistical analysis. SPSS offers data analysis for descriptive and bivariate statistics, numerical result forecasts, and predictions for classifying groups.
The Lochnericine molecule’s molecular structure was optimized using the DFT/B3LYP method with the cc-pVTZ basis set (Valarmathi et al., 2020a). Figure 1 depicts the optimized molecular structure of the lochnericine molecule, and its energy value was determined to be 1,111.43 a. u. Bond angle, bond length, and dihedral calculations collectively known as lochnericinestructural parameters were also displayed in Table 1. The C1 point group has been noticed in the molecular geometry of the lochnericine molecule. Lochnericine has been pointed to as a centrosymmetric structure due to its IR and Raman active vibrational modes. The optimized molecular structure of the Lochnericine molecule is located at a local minimum on the potential energy surface due to the absence of negative vibrational wavenumbers (Valarmathi et al., 2020b).
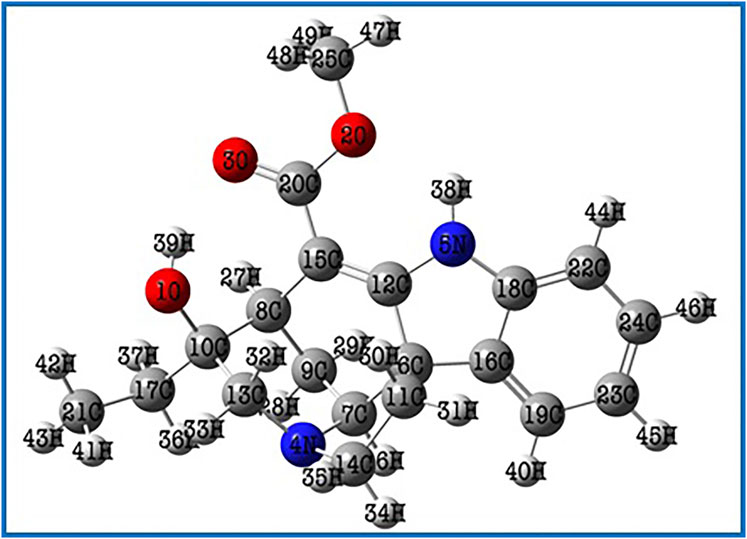
FIGURE 1. The optimized molecular structure of the Lochnericine molecule.

TABLE 1. The optimized structural parameters of Lochnericine molecule.
49 atoms and 141 normal vibration modes are present in the lochnericine, and all belong to the same symmetry species. The calculated values oflochnericinevibrational frequencies, IR intensity, and activity of Raman scattering have been represented in Yable 2. C1 point group symmetry exists in the Lochnericine molecule. Figure 2 depicts the theoretically simulated infrared and Raman spectra of the Lochnericine bioactive compound. The experimentally determined vibrational wavenumbers and theoretically calculated vibrational wavenumber values agreed well (Areans et al., 1988; Karabacak et al., 2008; Almansouret al., 2021). The molecule’s vibrational frequencies were calculated using the VEDA 4.0 programme, which many researchers have recognized as applicable (Zhou et al., 2021).
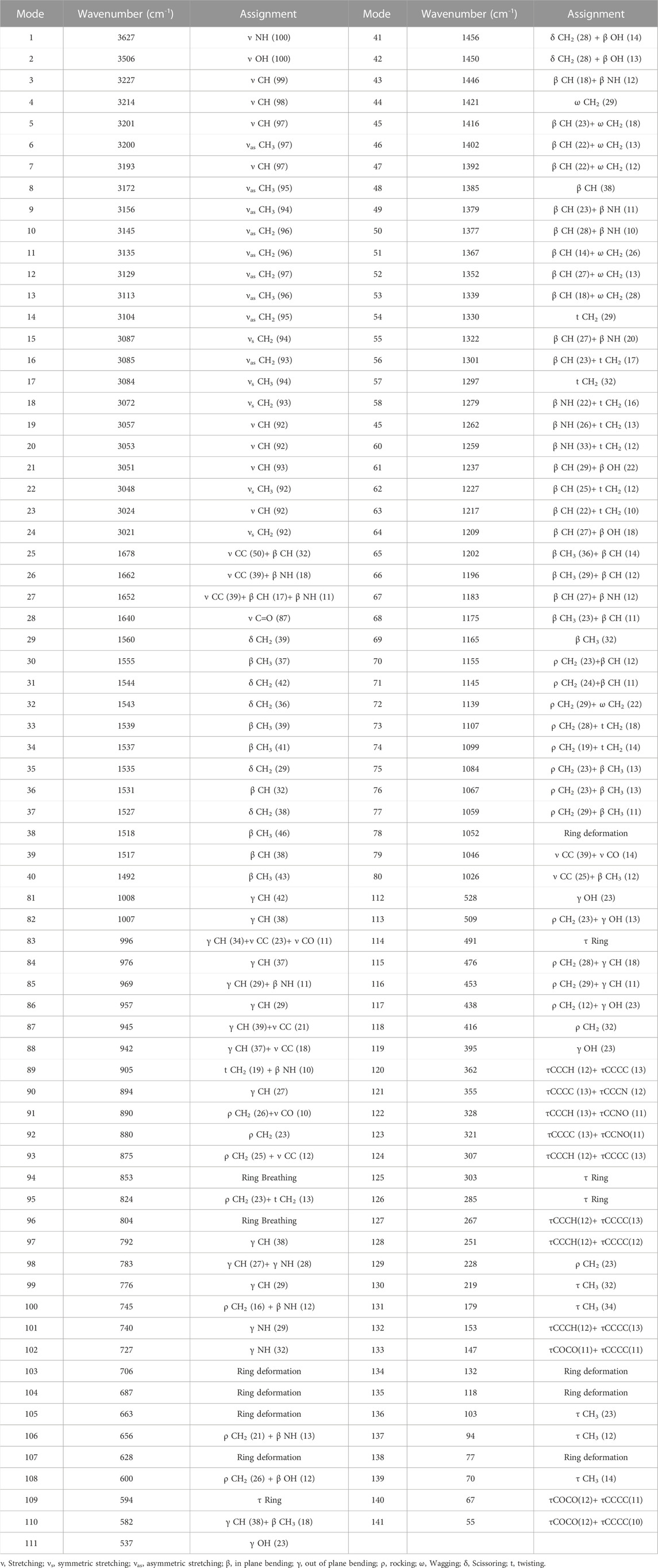
TABLE 2. Theoretical IR and Raman frequencies and their assignments for Lochnericine molecule.

FIGURE 2. Theoretically simulated infrared and Raman spectra of the Lochnericine molecule.
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are the two molecular orbitals that are used to analyse how a molecule interacts with other species (LUMO). The HOMO energy represents the capacity to provide electrons, whereas the LUMO energy represents the capacity to carry electrons (Koyambo-Konzapa et al., 2021). Electric and optical properties, UV-Vis spectra, and quantum chemistry all depend on FMOs. (Babu et al., 2016). The molecule’s kinetic stability and chemical reactivity—both crucial components in determining its electrical properties—are represented by the HOMO-LUMO gap. Small HOMO-LUMO gaps are linked to soft molecules that have strong chemical reactivity and limited kinetic stability. Figure 3 depicts the Lochnericine molecule’s FMOs. The FMOs’ related molecular features were calculated using Koopman’s theory (Mohamed Asath et al., 2017). Red and green are used to represent the positive and negative phases, respectively, in Figure 3. The calculated low energy gap value (4.18 eV), which also explains the intramolecular charge transfer interaction that affects the lochnericine molecule’s biological activity, supports the increased chemical reactivity of the compound (Saravanan and Blachandran, 2014). The electron affinity A) is the term used to describe the energy produced when an electron is introduced to an empty orbital, while the ionisation energy I) (5.49 eV) is used to describe the energy necessary to remove an electron from a full orbital (1.31 eV). The molecule is susceptible to electrophilic and nucleophilic reactions due to its anticipated high ionisation energy and low electron affinity. The chemical potentialμ = 3.40 eV, global softness, S = 2.09 eV, μ = 3.40 eV, global hardness, η = 0.47 eV, and global electrophilicity index, ψ = 12.29 eV, of the molecule were also computed. Based on the anticipated values of higher hardness and reduced softness, the molecule is probably stable. The molecule’s predicted chemical potential and electrophilicity index provide additional evidence that it has chemical stability comparable to that of compounds with potential bioactivity.
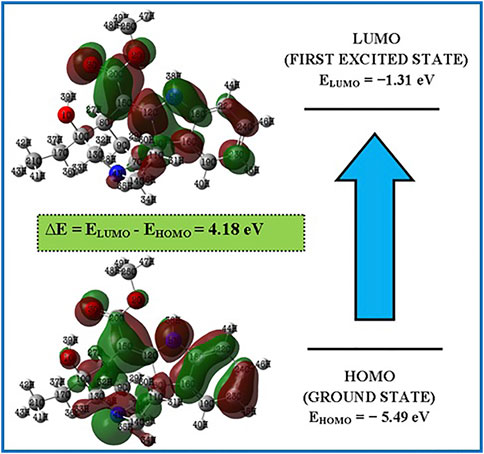
FIGURE 3. FMOs of the Lochnericine molecule.
Red, yellow, light blue, and blue on the MEP surface (Figure 4) represent, respectively, regions that are slightly electron-rich, slightly electron-rich, slightly electron-deficient, and slightly electron-deficient. It was revealed that the oxygen atoms lone pair electrons cause the regions around them to be electron-rich (red). Moreover, the oxygen atom O1 has a stronger electronegative potential, and all of the other oxygen atoms are classified as being in the slightly electron-rich zone. The slightly electron-deficient (light blue) portion of the molecule contained all of the hydrogen atoms. The molecule’s electron-deficient (blue) portion is identified as hydrogen atom H38. The hydrogen atom H38, which is shown as being electron-poor (blue), makes up the molecule. The neutral electrostatic potential envelopes were positioned around the molecule (green). Potential electrophilic and nucleophilic attack sites are the hydrogen atom H38 and the oxygen atom O1.
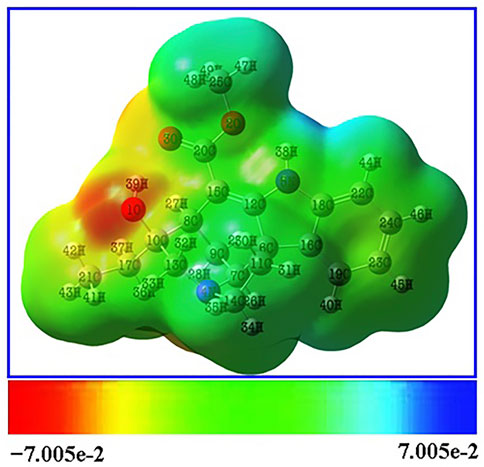
FIGURE 4. MEP surface of the Lochnericine molecule.
Mulliken’s atomic charge distribution influences the molecule’s dipole moment, electrostatic potential, electronegativity equalization, electronic structure, vibrational modes, and polarizability (Geetha et al., 2021). Because of its attachment to the two electronegative oxygen atoms, the carbon atom C20 has a higher positive charge value, as shown in Figure 5 representation of the predicted Mulliken atomic charge distribution values (O2 and O3). The highest electronegative charge values are found in the oxygen atom O1 and the nitrogen atom N5. The carbon atoms’ negative charge values significantly promote electron delocalization within the molecule.
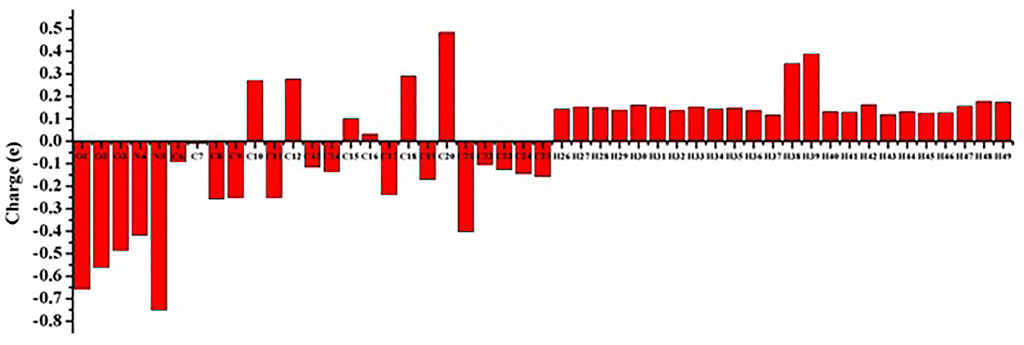
FIGURE 5. Mulliken atomic charge distribution of the Lochnericine molecule.
In drug designing, molecular docking is essential to determine the binding mode and efficacy of the active lead molecules. Molecular docking approaches were performed for all targeted receptor molecules. Receptor molecules docking score and interacting residues were tabulated in Table 3. EGFR receptor (PDB ID 2ITY) molecule interacts with the active lead molecules that lead to the formation of hydrogen bonds, Pi-Pi stacked, and Pi-Alkyl. Lead molecule interacts with glycine residue at the position of 724 and forms a conventional hydrogen bond. Leu 747 residue forms Pi-Alkyl interaction. Phenylalanine 723 amino acid forms the Pi-Pi Stacked. EGFR_ Lochnericine has good interaction, and thedocking score is −7.52 Kcal/Mol. 2 days and 3 days interactions between EGFR_Lochnericine were depicted in Figure 6A. A docking protocol was carried out between KRAS and Lochnericine, and the docking score between KRAS and lead molecules is −9.10 Kcal/Mol. Arginine amino acid at the position of 97 interacts with small molecules and forms the hydrogen bond. Another hydrogen bond is formed between Lysine 101 and Lochnericine. Pi-Alkyl formed by the Tyrosine 137 amino acid residues and 2 days and 3 days interactions between KRAS_Lochnericine were depicted in Figure 6B. Ntrk protein structure was downloaded from the RCSB PDB (PDB ID: 7VKO). The lead molecule with Glycine 667 amino acid residue forms the carbon-hydrogen bond. Further, Methionine 592 residue forms another carbon-hydrogen bond. Alkyl and Pi-Alkyl interactions were formed between lead molecule and amino acid residues (Valine 524, Tyrosine 591, Leucine 516, Alanine 542, Phenylalanine589 and Lysine 544). Ntrk_Lochnericine docking score is −8.24 Kcal/Mol, and the 2 days and 3 days interactions between Ntrk_Lochnericine are depicted in Figure 6C. ALK three-dimensional proteins with PDB ID:2XP2 were downloaded and prepared for docking. Alkyl and Pi-Alkyl interactions are formed by amino acid residue Leu 1,256, Ala 1,148, Val 1,130, Ile 1,171 and Lys 1,150, Glu 1,167, Asn 1,254 and Arg 1,253 amino acid residue interact with Lochnericine forms carbon-hydrogen bond. The docking score is −11.59 Kcal/Mol. 2 days and 3 days interactions between ALK_Lochnericine were depicted in Figure 6D. TRIM11 protein was modelled, and protein structure was assessed using the Saves server. TRIM11 forms Pi-Alkyl interaction with phenylalanine 407 amino acid residue. Tyrosine 343 forms a carbon-hydrogen bond, and the docking score between TRIM11_Lochnericine is −7.13 Kcal/Mol. 2 days and 3 days interactions between TRIM11_Lochnericine were depicted in Figure 6E. The docking score and interacting residues between the targeted receptor molecules and lead compound were tabulated in Table 4.
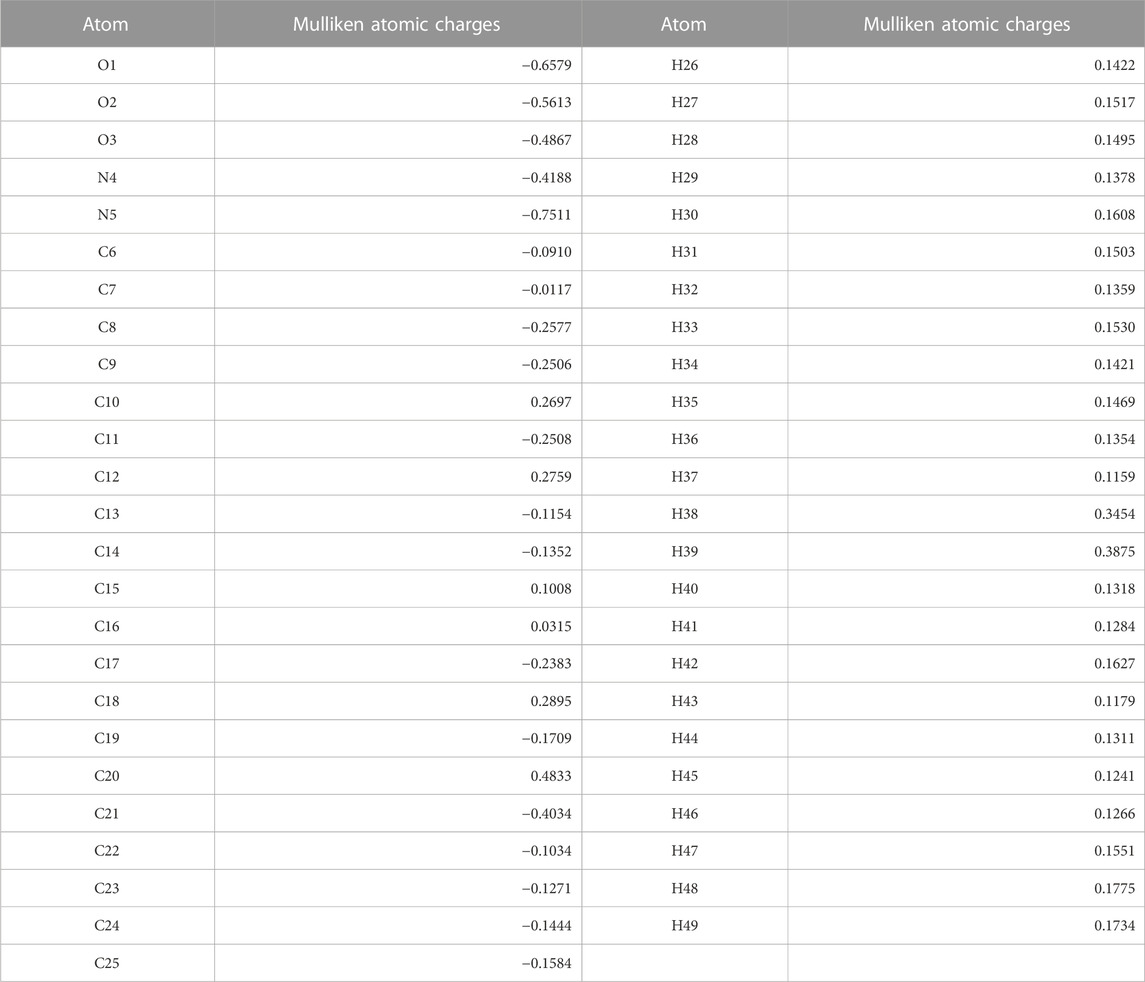
TABLE 3. Mulliken Atomic charges for optimized geometry.
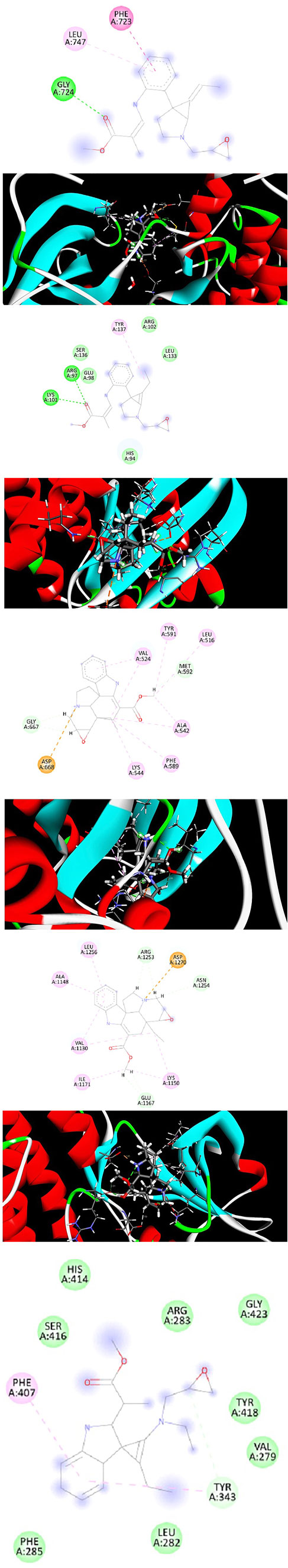
FIGURE 6. (A) 2d and 3d Interaction between EGFR_ Lochnericine. (B) 2d and 3d Interaction between KRAS_ Lochnericine. (C) 2d and 3d Interaction between Ntrk_ Lochnericine. (D) 2d and 3d Interaction between ALK_ Lochnericine. (E) 2d and 3d Interaction between TRIM11_ Lochnericine.
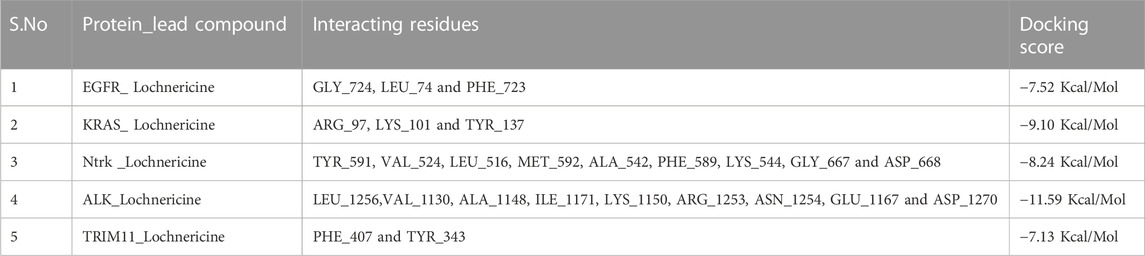
TABLE 4. Interacting residues between receptor molecules and lead compound.
In the drug-designing process, ADME prediction is essential for determining the lead molecule’s efficacy. These obey all the five rules of Lipinski. Several pharmacokinetic properties, pharmacokinetics, drug-likeness, and water solubility properties were tabulated in Table 5.Further, lead compound lipophilicity, size, insoluble, unsaturation, flexibility, and polar nature are shown in Figure 7.
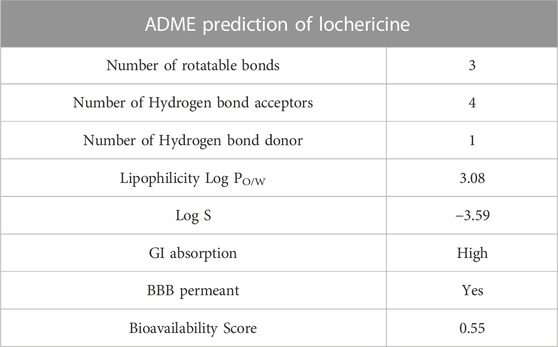
TABLE 5. ADME prediction of Lochnericine.
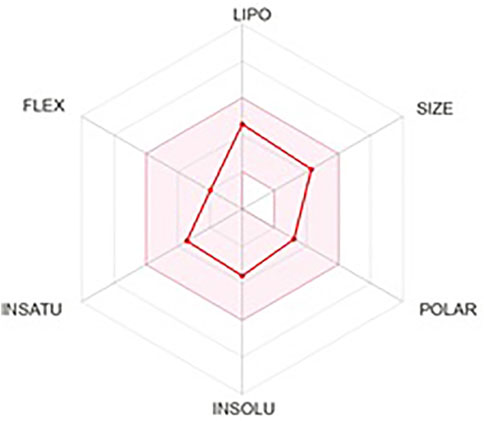
FIGURE 7. ADME prediction of Lochnericine.
Gromacs package’s molecular dynamics simulations were used to examine the stability of targeted protein-ligand complexes. Based on the docking score, interaction residues, and binding affinity between the targeted proteins and the Lochnericine. For 50 ns, molecular dynamics simulations were performed. Typically, MD simulation is used to predict the behaviour of macromolecules, and it relies on classical mechanics and Newton’s equation of motion to determine the speed and position of each atom in the system under consideration. As a result, MD performs a more concise structural assessment. The difference between the initial and final structural conformational positions was determined using the RMSD (Root Mean Square Deviation). The results of MDs of protein-ligand complexes are shown in Figure 8. The average range of RMSD deviation was observed between all the protein-ligand molecules. This indicates a good RMSD deviation. All protein receptor molecules indicate favourable RMSD deviation throughout the entire simulation period. RMSF (Root Mean Square Fluctuation) assesses and computes the average deviation of a targeted protein over time from an amino acid residue reference position. The average fluctuation rate between targeted protein-ligand complexes ranges between all the protein-ligand molecules. The fluctuation between the targeted receptor molecules and Lochnericineis depicted in Figure 8. The smallest fluctuation with good stability and the fluctuation rate that occurs in the loop or disallowed region have no implications on the complexes’ stability. Hydrogen bonds in protein-ligand complexes are calculated using cut-offs for the angle between the Hydrogen Donor-Acceptor and the distance between the Donor-Acceptor. Hydrogen bond interactions between targeted proteins and Lochnericine complexes are represented in Figure 8. EGFR (PDB ID-2ITY) with the lead compound represented in the black colour. 7LGI with the active compound represented in the green colour. The red colour indicates the 7VKO protein with lochnericine. TRIM11with lochnericine represent in blue colour. Throughout the simulation runs, the GROMACS command line gmx_rmsd was used to compute the RMSD values of each targeted protein-ligand complex’s stability profile. RMSD often interprets the deviation for a set of atoms (protein, ligand, or even ligand-protein complex) from the corresponding initial reference structure. Less RMSD values would therefore be associated with considerable stability due to changes in the examined molecule’s structure. Additionally, ligands show lower RMSD values corresponding to the ligand-protein complexes. A lower RMSD value denotes higher stability for the protein-ligand complexes. All targeted receptor molecules with lead compound lesser deviation within the range of 0.4 nm. All the potential receptors of Non-small cell lung cancer protein flexibility and target Residue Root-Mean-Square Fluctuation (RMSF) for each ligand-bound protein residue root-mean-square fluctuation (RMSF) profile were calculated in order to acquire more insights into the stability of the complex binding site. The GROMACS “gmx_rmsf” command line was used to calculate the specific backbone RMSF of each protein. This flexibility validation criterion shows the contribution of specific protein residues to the structural variations of the ligand/protein complex. The RMSF calculates the average deviation for each residue from its reference position inside the reduced initial structures over time. All the targeted receptor molecules show an acceptable fluctuation range except the disallowed region. The targeted protein-ligand complex fluctuates within the range of 0.50 nm.The lowest values for the deviation below 0.40 nm and the lesser deviation below 1 nm were found while considering the examined complexes. The above RMSD and RMSF analysis shows that the targeted protein-ligand complexes have good stability and flexibility. The stability of ligand-protein complexes and the corresponding conformational changes were thought to be best understood by examining the hydrogen bond network connections between the receptors and lochnericine. It was helpful to investigate the recognized ligand-protein hydrogen bond interactions and their relative frequency using the GROMACS command line gmxhbond, which analyses the hydrogen bonds (H-bonds) between all conceivable donors D and acceptors A. EGFR receptor molecule exhibit a maximum number of 4 hydrogen bonds between the targeted receptor molecule was observed. All other receptors possess the average number of 3 hydrogen bonds between the receptor and lochnericine. All the resultant complexes show better hydrogen bond interaction throughout the simulation period. Furthermore, lochnericine induced a stabilized favoured hydrogen bond association with the targeted receptor, which was sustained for significant MD simulation. This preferential hydrogen bond pair interaction with the targeted receptors introduces the potential activity of lochnericine to block the receptor molecules of non-small-cell lung cancer.
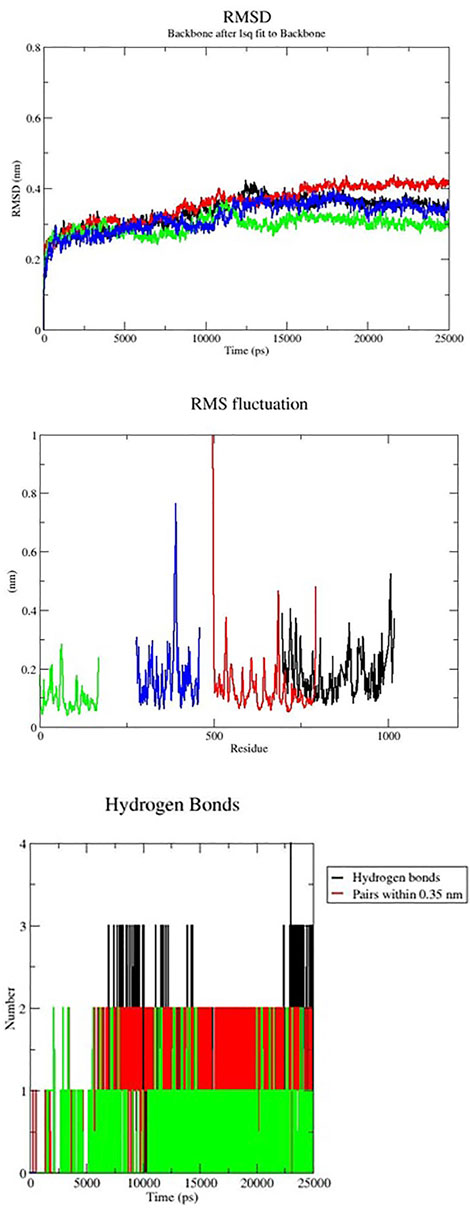
FIGURE 8. MD simulation of Nonsmall cell lung cancer targeted receptors with the lead compounds.
Several chemicals and natural compounds’ cytotoxic efficacy against malignant cells have been considered primary studies for the antitumor potential of the compounds. The conversion of MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) to MTT-formation using mitochondrial enzymes in the MTT assay is a notable assay for the valuation of the proliferation of cancer cells (Sangavi et al., 2022). Treatment Lochnericine for 24 and 48 h declinedcell proliferationin a dosage and time-dependent manner, as shown in Figure 9, 10. In the present study, Lochnericinecondensed cell feasibility in a concentration-dependent manner. Lochnericine inhibition of cell growth might be linked to the induction of cell death. Thus, Lochnericinesuppressive impact on A549 cells significantly supports Lochnericine’s anti-proliferation capability.
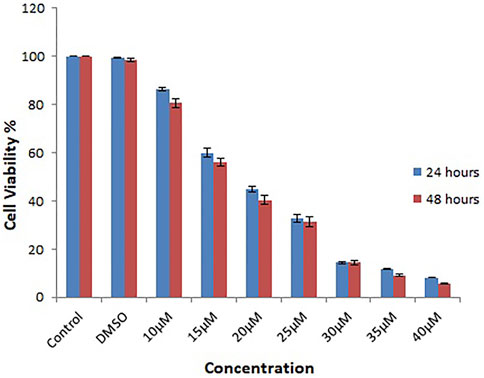
FIGURE 9. Depicts the Effect of Lochnericine on cell viability in the Human Lung Carcinoma cell line A549by MTT assay.
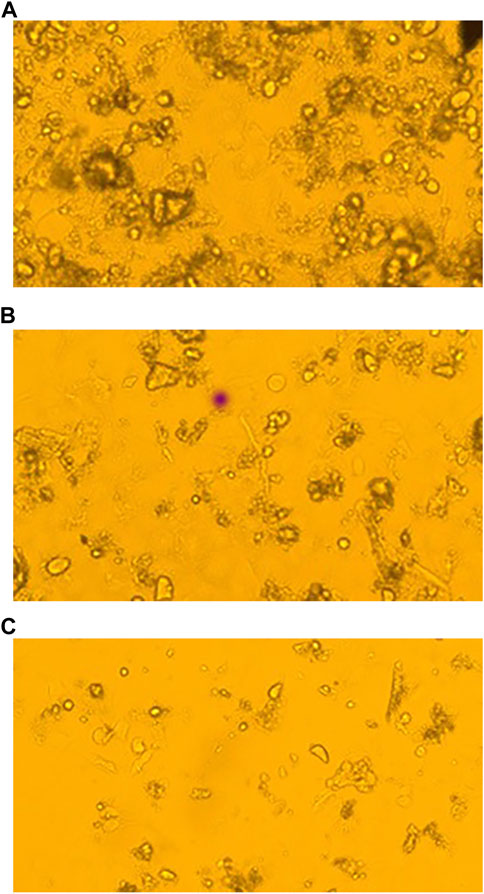
FIGURE 10. Fluorescence Microscopic images of Control and Lochnericine-treated A549 cells. (A) Control. (B) 25 µM Concentration of Lochnericine. (C) 30 µM Concentration of Lochnericine.
The optimization geometry for the lochnericine bioactive molecule was carried out using the DFT/B3LYP functional technique and the cc-PVTZ basis set. Simulated Lochnericine vibrational spectra, including infrared and Raman spectra, showed that the computed vibrational wavenumbers significantly correspond with those from earlier works of literature. The determined band gap energy value of 4.18 eV abutment lochnericine bioactivity. H38 hydrogen and O1 oxygen atom in the molecule are possible electrophilic and nucleophilic attack sites, according to a molecular electrostatic potential surface analysis. An examination of the atomic charge distribution of Mulliken verified the electron delocalization that led to the molecule’s bioactivity. non-small cell lung cancer potential targeted receptor molecules were selected and analyzed using a computational approach. During molecular docking studies, all the targeted receptor has a strong binding affinity for drugs containing lochnericine. Further targeted receptor and lochnericinedocked complexes showed good stability and lesser fluctuation during the simulation time. The protein-ligand complexes are more stable when their RMSD value is smaller. Every targeted receptor molecule has a lead compound deviation that seems to be smaller than 0.4 nm. Within a 0.50 nm range, the targeted protein-ligand complex fluctuates. While accounting for the studied complexes, the lowest values for the deviation below 0.40 nm and the lesser deviation below 1 nm were obtained. The targeted protein-ligand complexes exhibit good stability and flexibility, according to RMSD and RMSF analysis. Throughout the simulation, better hydrogen bond interaction can be seen in the resulting complexes with lochnericine.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
CK, SG, and KL designed the study, analyzed the data, and wrote the manuscript. JM, SA, and RS performed the experiments. KTN validated the data. All the authors have read and agreed to the published version of the manuscript.
Dr. KL thankfully acknowledges MHRD-RUSA 2.0 F.24/51/ 2014-U, Policy (TN Multi-Gen), Department of Education, Government of India for providing infrastructural facilities to carry out this research work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Almansour, A. I., Arumugam, N., Soliman, S. M., Krishnamoorthy, B. S., Halet, J. F., Vishnu Priya, R., et al. (2021). Stereoselective synthesis structure and DFT studies on fluoro- and nitro-substituted spirooxindole-pyrrolidine heterocyclic hybrids. J. Mol. Struct. 1237, 130396. doi:10.1016/j.molstruc.2021.130396
Areans, J. F., Lopez, I., Wooley, M. S., Otero, J. C., and Marcos, J. I. (1988). Vibrational spectrum and internal rotation in 2-methylpyrazine. J. Chem. Soc. Faraday Trans. Chem. Phys. 284, 53–65. doi:10.1039/f29888400053
Awan, Z. A., Bahattab, R., Kutbi, H. I., Noor, A. O. J., Al-Nasser, M. S., Shaik, N. A., et al. (2021). Structural and molecular interaction studies on familial hypercholesterolemia causative PCSK9 functional domain mutations reveals binding affinity alterations with LDLR. Int. J. Peptide Res. Ther. 27 (1), 719–733. doi:10.1007/s10989-020-10121-8
Babu, B., Chandrasekaran, J., Mohanbabu, B., Matsushita, Y., and Saravanakumar, M. (2016). Growth physicochemical and quantum chemical investigations on 2-amino 5-chloropyridinium 4-carboxybutanoate-an organic crystal for biological and optoelectronic device applications. RSC Adv. 6, 110884–110897. doi:10.1039/c6ra15791b
Banaganapalli, B., Mallah, B., Alghamdi, K. S., Albaqami, W. F., Alshaer, D. S., Alrayes, N., et al. (2022). Integrative weighted molecular network construction from transcriptomics and genome wide association data to identify shared genetic biomarkers for COPD and lung cancer. Plos one 17 (10), e0274629. doi:10.1371/journal.pone.0274629
Bima, A. I. H., Elsamanoudy, A. Z., Alghamdi, K. S., Shinawi, T., Mujalli, A., Kaipa, P. R., et al. (2022). Molecular profiling of melanocortin 4 receptor variants and agouti-related peptide interactions in morbid obese phenotype: A novel paradigm from molecular docking and dynamics simulations. Biologia 77 (5), 1481–1496. doi:10.1007/s11756-022-01037-3
Dallakyan, S., and Olson, A. J. (2015). Small-molecule library screening by docking with PyRx. Methods Mol. Biol. Clift. NJ) 1263, 243–250. doi:10.1007/978-1-4939-2269-7_19
Deng, L. J., Qi, M., Li, N., Lei, Y. H., Zhang, D. M., and Chen, J. X. (2020). Natural products and their derivatives: Promising modulators of tumor immunotherapy. JLeukoc Biol. 108 (2), 493–508. doi:10.1002/JLB.3MR0320-444R
Ekiert, H. M., and Szopa, A. (2020). Biological activities of natural products. Molecules 25 (23), 5769. doi:10.3390/molecules25235769
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09 revision C. 02. Wallingford CT: Gaussian Inc.
Geetha, R., Meera, M. R., Vijayakumar, C., Premkumar, R., and Milton Franklin Benial, A. (2021). Synthesis spectroscopic characterization molecular docking and in vitro cytotoxicity investigations on 8-amino-6-methoxy quinolinium picrate: A novel breast cancer drug. J. Biomol. Struct. Dyn. 2021, 1–14. doi:10.1080/07391102.2021.2024259
Gueguen, P., Metoikidou, C., Dupic, T., Lawand, M., Goudot, C., Baulande, S., et al. (2021). Contribution of resident and circulating precursors to tumor-infiltrating CD8+ T cell populations in lung cancer. Sci. Immunol. 6 (55), eabd5778. doi:10.1126/sciimmunol.abd5778
Hausman, D. M. (2019). What is cancer? Perspect. Biol. Med. 62 (4), 778–784. doi:10.1353/pbm.2019.0046
Jamroz, M. H. (2004). Vibrational energy distribution analysis VEDA 4.0 program. Warsaw: VEDA analysis.
Karabacak, M., Karagöz, D., and Kurt, M. (2008). Experimental (FT-IR and FT-Raman spectra) and theoretical (ab initio HF, DFT) study of 2-chloro-5-methylaniline. J. Mol. Struct. 892 (1-3), 25–31. doi:10.1016/j.molstruc.2008.04.054
Kavitha, M., Iravathy, G., Adi Maha, L. M., Ravi, V., Sridhar, K., Vijayanand, R. P., et al. (2015). Correlation between EGFR gene mutations and lung cancer: A hospital-based study. Asian Pac. J. Cancer Prev. 16 (16), 7071–7076. doi:10.7314/apjcp.2015.16.16.7071
Koyambo-Konzapa, S. J., MbesseKongbonga, G. Y., Premkumar, R., Duvalier RamlinaVamhindi, B. S., Nsangou, M., and Milton Franklin Benial, A. (2021). Spectroscopic quantum chemical molecular docking and molecular dynamics investigations of hydroxylic indole-3-pyruvic acid: A potent candidate for nonlinear optical applications and alzheimer's drug. J. Biomol. Struct. Dyn. 40, 10651–10664. doi:10.1080/07391102.2021.1947380
Laskowski, R. A., MacArthur, M. W., and Thornton, J. M. (2016). Procheck: Validation of protein-structure coordinates. Int. Tables Crystallogr. 21.4, 684–687. doi:10.1107/97809553602060000882
Mohamed Asath, R., Premkumar, R., Mathavan, T., and Milton Franklin Benial, A. (2017). Spectroscopic and molecular docking studies on NN-di-tert-butoxycarbonyl (Boc)-2-amino pyridine: A potential bioactive agent for lung cancer treatment. J. Mol. Struct. 1143, 415–423. doi:10.1016/j.molstruc.2017.04.117
Mullard, A. (2020). Addressing cancer's grand challenges. Nat. Rev. Drug Discov. 19 (12), 825–826. doi:10.1038/d41573-020-00202-0
Reck, M., Carbone, D. P., Garassino, M., and Barlesi, F. (2021). Targeting KRAS in non-small-cell lung cancer: Recent progress and new approaches. Ann. Oncol. 32 (9), 1101–1110. doi:10.1016/j.annonc.2021.06.001
Sangavi, P., and Langeswaran, K. (2021). Anti-tumorigenic efficacy of tangeretin in liver cancer-an in-silico approach. Curr. computer-aided drug Des. 17 (3), 337–343. doi:10.2174/1573409916666200219120254
Sangavi, P., Langeswaran, K., and Sangeetha, R. (2022). Identification and validation of hydroxychavicol from betel leaf as a promising breast cancer inhibitor: An in vitro and in silico analysis. Curr. Enzyme Inhib. 18 (2), 127–134. doi:10.2174/1573408018666220210141311
Saravanan, S., and Blachandran, V. (2014). Quantum mechanical study and spectroscopic (FT-IR FT-Raman UV–Visible) study potential energy surface scan Fukui function analysis and HOMO–LUMO analysis of 3-tert-butyl-4-methoxyphenol by DFT methods. Spectrochim. Acta 130, 604–620. doi:10.1016/j.saa.2014.04.058
Schabath, M. B., and Cote, M. L. (2019). Cancer progress and priorities: Lung cancer. Cancer Epidemiol. Biomarkers Prev. 28 (10), 1563–1579. doi:10.1158/1055-9965.EPI-19-0221
Schüttelkopf, A. W., and Van Aalten, D. M. (2004). Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60 (8), 1355–1363. doi:10.1107/S0907444904011679
Schwede, T., Kopp, J., Guex, N., and Peitsch, M. C. (2003). SWISS-MODEL: An automated protein homology-modeling server. Nucleic acids Res. 31 (13), 3381–3385. doi:10.1093/nar/gkg520
Shaik, N. A., Al-Kreathy, H. M., Ajabnoor, G. M., Verma, P. K., and Banaganapalli, B. (2019). Molecular designing, virtual screening and docking study of novel curcumin analogue as mutation (S769L and K846R) selective inhibitor for EGFR. Saudi J. Biol. Sci. 26 (3), 439–448. doi:10.1016/j.sjbs.2018.05.026
Shaik, N. A., Al-Saud, N. B. S., Aljuhani, T. A., Jamil, K., Alnuman, H., Aljeaid, D., et al. (2022). Structural characterization and conformational dynamics of alpha-1 antitrypsin pathogenic variants causing alpha-1-antitrypsin deficiency. Front. Mol. Biosci. 9, 1051511. doi:10.3389/fmolb.2022.1051511
Sussman, J. L., Lin, D., Jiang, J., Manning, N. O., Prilusky, J., Ritter, O., et al. (1998). Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D. Biol. Crystallogr. 54 (6-1), 1078–1084. doi:10.1107/s0907444998009378
Tripathi, P., Ghosh, S., and Talapatra, S. N. (2019). Bioavailability prediction of phytochemicals present in Calotropis procera (Aiton) R. Br. by using Swiss-ADME tool. World Sci. News 131, 147–163.
Turner, P. J. (2005). XMGRACE version 5.1. 19. Beaverton OR: Center for Coastal and Land-Margin Research Oregon Graduate Institute of Science and Technology.
UniProt (2019). UniProt: A worldwide hub of protein knowledge. Nucleic acids Res. 47 (1), D506–D515. doi:10.1093/nar/gky1049
Valarmathi, T., Premkumar, R., and Milton Franklin Benial, A. (2020b). Quantum chemical and molecular docking studies on 14-bis(methylamino)anthraquinone: A DFT approach. AIP Conf. Proc. 2270, 040001.
Valarmathi, T., Premkumar, R., and Milton Franklin Benial, A. (2020a). Spectroscopic and molecular docking studies on 1- hydroxyanthraquinone: A potent ovarian cancer drug. J. Mol. Struct. 1213, 128163. doi:10.1016/j.molstruc.2020.128163
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., and Berendsen, H. J. (2005). Gromacs: Fast flexible and free. J. Comput. Chem. 26 (16), 1701–1718. doi:10.1002/jcc.20291
Wang, X., Shi, W., Shi, H., Lu, S., Wang, K., Sun, C., et al. (2016). TRIM11 overexpression promotes proliferation, migration and invasion of lung cancer cells. J. Exp. Clin. Cancer Res. 35 (1), 100. doi:10.1186/s13046-016-0379-y
Zhou, C., and Yao, L. D. (2016). Strategies to improve outcomes of patients with EGRF-mutant non-small cell lung cancer: Review of the literature. JThorac Oncol. 11 (2), 174–186. doi:10.1016/j.jtho.2015.10.002
Keywords: lochnericine, quantum chemical calculations, molecular docking, molecular dynamics, anticancer study, non-small lung cancer
Citation: Kirubhanand C, Merciline Leonora J, Anitha S, Sangeetha R, Nachammai KT, Langeswaran K and Gowtham Kumar S (2023) Targeting potential receptor molecules in non-small cell lung cancer (NSCLC) using in silico approaches. Front. Mol. Biosci. 10:1124563. doi: 10.3389/fmolb.2023.1124563
Received: 15 December 2022; Accepted: 23 January 2023;
Published: 09 February 2023.
Edited by:
Chandrabose Selvaraj, Saveetha University, IndiaReviewed by:
Gunassekaran Gowri Rangaswamy, Kyungpook National University, Republic of KoreaCopyright © 2023 Kirubhanand, Merciline Leonora, Anitha, Sangeetha, Nachammai, Langeswaran and Gowtham Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Gowtham Kumar, Z293dGhhbV9waGRAeWFob28uY29t; K. Langeswaran, ZHIubGFuZ2Vzd2FyYW5AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.