
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 24 January 2023
Sec. Molecular Recognition
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1111598
This article is part of the Research TopicIn Celebration of Women in Science: Molecular Recognition 2022View all 5 articles
 Ángela Rodríguez1María Maneiro1
Ángela Rodríguez1María Maneiro1 Emilio Lence1José M. Otero1Mark J. van Raaij2Paul Thompson3Alastair R. Hawkins3
Emilio Lence1José M. Otero1Mark J. van Raaij2Paul Thompson3Alastair R. Hawkins3 Concepción González-Bello1*
Concepción González-Bello1*Irreversible inhibition of the enzyme type I dehydroquinase (DHQ1), a promising target for anti-virulence drug development, has been explored by enhancing the electrophilicity of specific positions of the ligand towards covalent lysine modification. For ligand design, we made use of the advantages offered by the intrinsic acid-base properties of the amino substituents introduced in the quinate scaffold, namely compounds 6–7 (R configuration at C3), to generate a potential leaving group, as well as the recognition pattern of the enzyme. The reactivity of the C2–C3 bond (Re face) in the scaffold was also explored using compound 8. The results of the present study show that replacement of the C3 hydroxy group of (–)-quinic acid by a hydroxyamino substituent (compound 6) provides a time-dependent irreversible inhibitor, while compound 7, in which the latter functionality was substituted by an amino group, and the introduction of an oxirane ring at C2–C3 bond, compound 8, do not allow covalent modification of the enzyme. These outcomes were supported by resolution of the crystal structures of DHQ1 from Staphylococcus aureus (Sa-DHQ1) and Salmonella typhi (St-DHQ1) chemically modified by 6 at a resolution of 1.65 and 1.90 Å, respectively, and of St-DHQ1 in the complex with 8 (1.55 Å). The combination of these structural studies with extensive molecular dynamics simulation studies allowed us to understand the molecular basis of the type of inhibition observed. This study is a good example of the importance of achieving the correct geometry between the reactive center of the ligand (electrophile) and the enzyme nucleophile (lysine residue) to allow selective covalent modification. The outcomes obtained with the hydroxyamino derivative 6 also open up new possibilities in the design of irreversible inhibitors based on the use of amino substituents.
The mechanism of action of most antibiotics in clinical use is based on inactivation of enzymes/proteins encoded by essential genes, thus preventing the synthesis and assembly of key components that are needed for bacterial growth (viability) (Cook and Wright 2022). The main metabolic pathways targeted by these drugs include cell-wall biosynthesis, DNA replication, RNA transcription, folate biosynthesis, and protein biosynthesis. Although this strategy is very effective and has given rise to a good arsenal of “life-saving” compounds, it causes substantial stress in the bacteria, which usually triggers the appearance of antibiotic-resistant strains. As such, to reduce the emergence and worldwide spread of multidrug-resistant bacteria, an innovative antibacterial approach that is increasingly being explored comprises inhibition of the bacterial capacity to produce the infection (pathogenesis) (Theuretzbacher and Piddock 2019; Hotinger et al., 2021; Krell and Matilla 2022). This “antibiotic-free” approach has several advantages over the traditional strategies dealing with bacterial viability, including: i) pathogenic bacteria are specifically targeted, with little or no impact on the normal human microbiota; ii) less selective pressure may be imposed to the development of bacterial resistance; and iii) natural clearing by the host immune system as a result of the in vivo scenario created, which resembles vaccination with a live attenuated strain (Dickey et al., 2017).
Several studies have identified the enzyme type I dehydroquinase (DHQ1, 3-dehydroquinate dehydratase, EC 4.2.1.10) as a promising target for anti-virulence drug development. The interesting features of this enzyme that have attracted our attention are: i) DHQ1 acts as a virulence factor in vivo as deletion of the aroD gene, which codes for this enzyme, has proven to afford satisfactory live oral vaccines (Tacket et al., 1992; Tacket et al., 1992; Kärnell et al., 1993; Dilts et al., 2000; Tacket and Levine 2007; Malcova et al., 2009; Revolledo and Ferreira 2012; Racz et al., 2013; Cunningham et al., 2020); ii) the aroD mutation is auxotrophic, affects bacterial cell wall integrity and blocks the bacterial ability to form biofilms (Cano et al., 2003; Nógrády et al., 2003; Sebkova et al., 2008); iii) DHQ1 is upregulated in small colony variants, a pathogenic form of bacteria that facilitates persistent and recurrent infections (Proctor et al., 2006; Zhang et al., 2017); iv) no mammalian homologues of the DHQ1 enzyme have been identified; and v) DHQ1 is present in several pathogenic bacteria, including Escherichia coli, Salmonella typhi and Staphylococcus aureus. DHQ1 catalyzes the reversible dehydratation of 3-dehydroquinic acid (1) to 3-dehydroshikimic acid (2), which is the third step in the shikimic acid pathway (Figure 1A) (González-Bello 2015). This conversion implies the overall syn elimination of water and removal of the pro-R hydrogen at C2 in 1. It follows a covalent catalysis mechanism with the formation of transient Schiff base species´ between the ketone moiety of the substrate and the ɛ-amino group of a catalytic lysine residue (K160/K170 in Sa-DHQ1 and St-DHQ1, respectively), which are finally hydrolyzed to regenerate the enzyme. An essential histidine residue (H133/H143 in Sa-DHQ1 and St-DHQ1, respectively), is also involved in the formation/hydrolysis of the Schiff base intermediates (Leech et al., 1995; Leech et al., 1998).
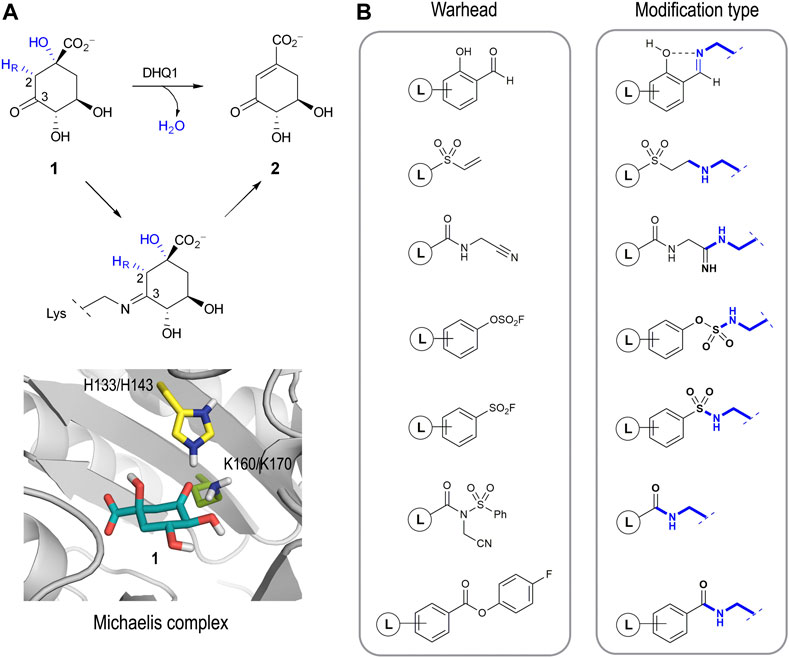
FIGURE 1. (A) Overall syn elimination of water from 3-dehydroquinic acid (1) catalyzed by DHQ1. The enzymatic process involves the formation of diverse Schiff base species via a catalytic lysine residue. Magnified view of the Michaelis complex obtained by MD simulation studies. Only the catalytic side-chain residues are shown. (B) Relevant reported electrophilic warheads for reversible/irreversible targeting of lysine residues and the covalent modification obtained.
The development of selective irreversible inhibitors is envisaged as an attractive approach for the inhibition of enzymes undergoing covalent catalysis as there is no dissociation equilibrium for competitive strategies. These types of inhibitors have additional benefits compared with non-covalent ligands, especially improved potency and prolonged drug effects (permanent inactivation of the target), and they are usually more robust against pharmacokinetic issues (Singh et al., 2011; Bauer 2015; Zhang et al., 2019). After years of overestimating their possible side effects, this type of inhibitor has become particularly important in drug-discovery programs, especially for non-chronic therapies such as those against infectious diseases and cancer (Sanderson 2013). The development of a selective irreversible inhibitor is not an easy task, because the introduction of overly reactive electrophiles in the scaffold will increase the chances of having undesired off-target effects. To overcome this issue, a booming strategy involves the use of barely reactive electrophilic groups that are only activated towards reaction upon binding to their target. In these cases, the correct arrangement of the ligand and the suitable geometry of the electrophilic group in the ligand relative to the enzyme/protein nucleophile are key for the success of the covalent modification. In recent years, the design of ligands capable of selectively modifying lysine residues is probably one of the most pursued and challenging goals (Hoyt et al., 2019). Although these residues are very abundant in proteins, in most cases they are not nucleophiles as the ɛ-amino group is protonated at physiological pH. As such, an ability to target protein pockets comprising residues that are able to generate transitory “reactive lysine residues” is crucial for this approach. Good examples of electrophilic warheads for reversible and irreversible covalent modification of lysine residues include salicylaldehyde-based probes (Yang et al., 2022), vinyl sulfones (Anscombe et al., 2015), nitriles (Smith et al., 2018), aryl fluorosulfates (Mortenson et al., 2018), aromatic sulfonyl fluorides (Grimster et al., 2013; Narayanan and Jones 2015; Zhao et al., 2017), N-acyl-N-alkyl sulfonamides (Tamura et al., 2018), and 4-fluorobenzoic esters (Dalton et al., 2018), amongst others (Figure 1B). In these examples, although the electrophilic character of the warhead is enhanced upon ligand binding, the reaction occurs at sp2/sp carbon atoms per se activated to a certain extent. An ability to get the reaction to occur at carbon atoms of the ligand with little electrophilic character (sp3), which are very common, appears a challenging task that has been little explored to date but which might expand our options for drug design.
Herein we study the possible activation of cyclic sp3-hybridized carbons for irreversible inhibition of the target. To this end, we make use of the intrinsic acid-base properties of amino substituents to generate a leaving group, as well as the recognition pattern of the targeted enzyme pocket. We reported recently that compounds 3 bearing a hydroxyaminomethyl group with an S configuration at C3 in the natural substrate (compound 1) are irreversible inhibitors of the enzyme DHQ1 (Figure 2) (Maneiro et al., 2019). A direct nucleophilic attack by the ɛ-amino group of the catalytic lysine at the exocyclic methylene group would take place with the formation of an amine linkage. We have also shown that the introduction of an aminomethyl and hydroxy substituent with an S configuration at C3 in 1 (compounds 4) also results in covalent modification of the catalytic lysine residue to afford a stable Schiff base as they are converted into reactive epoxides 5 (González-Bello et al., 2015; Lence et al., 2020). Herein we report the synthesis of compounds 6–7 in which enhancement of the electrophilicity of an endocyclic carbon atom in the ligand towards covalent modification is attempted by direct introduction of the amino functionality into the quinate scaffold (Figure 2). These compounds are substrate analogs in which the carbonyl group at C3 has been replaced by a hydroxyamino and an amino group with R configuration (compounds 6 and 7, respectively). The numbering of the natural substrate has been maintained for clarity. This stereochemistry was selected based on preliminary computational studies on the Michaelis complex of the DHQ1 enzymes from S. typhi (St-DHQ1) and S. aureus (Sa-DHQ1), which showed that the ketone group at C3 is activated by the catalytic histidine residue via its Re face towards nucleophilic attack by the catalytic lysine residue from the opposite site (Figure 1A). In addition, the reactivity of C3 in the quinate scaffold was also explored by introduction of an oxirane moiety with an S,S configuration at the C2-C3 bond (compound 8) to compare the geometry requirements of the C3 position towards nucleophilic attack. The crystal structures of Sa-DHQ1 and St-DHQ1 enzymes chemically modified by 6 at a resolution of 1.65 and 1.90 Å, respectively, and of St-DHQ1 in a complex with epoxide 8 (1.55 Å) are provided. These structural studies, in combination with various biochemical and molecular dynamics (MD) simulation studies, allowed us to understand the molecular basis of the type of inhibition observed.
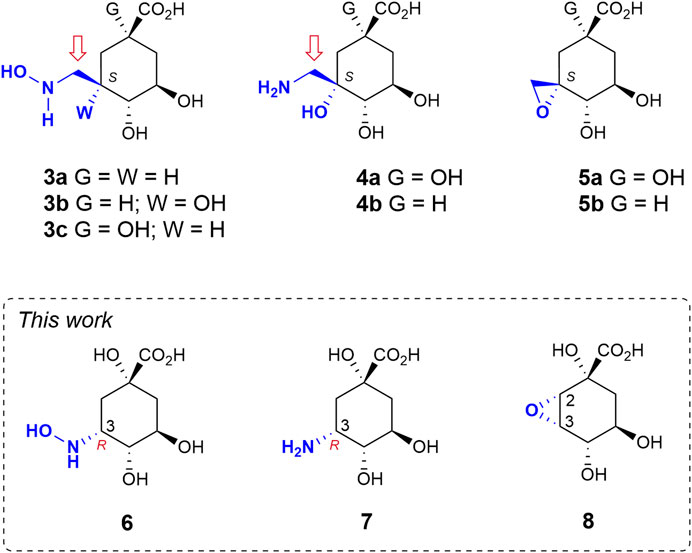
FIGURE 2. Relevant reported irreversible inhibitors and targeted compounds. The methylene group undergoing lysine modification is indicated with an arrow. The numbering corresponds to the natural substrate.
The strategy used to synthesise compounds 6–7 involved the initial preparation of ketone 10, which was synthesized in two steps from commercially available (–)-quinic acid (9) using previously reported protocols (Scheme 1) (Toscano et al., 2003). Treatment of ketone 10 with hydroxylamine gave oxime 11, in 89% yield, which was converted into compound 14 by regioselective reduction of the imino group with sodium cyanoborohydride (85% yield). The stereochemistry of the resulting new chiral center was determined by NOE experiments. Thus, irradiation of the H8ax signal (1.78 ppm) in compound 14 led to enhancement of the H6 and H7 signals by 6.2% and 4.2%, respectively. Finally, hydrolysis of the ester group with concomitant removal of the protecting groups by heating at 100°C with diluted HCl afforded the desired 3-hydroxyaminoquinic acid (6), as its hydrochloride salt, in 94% yield.
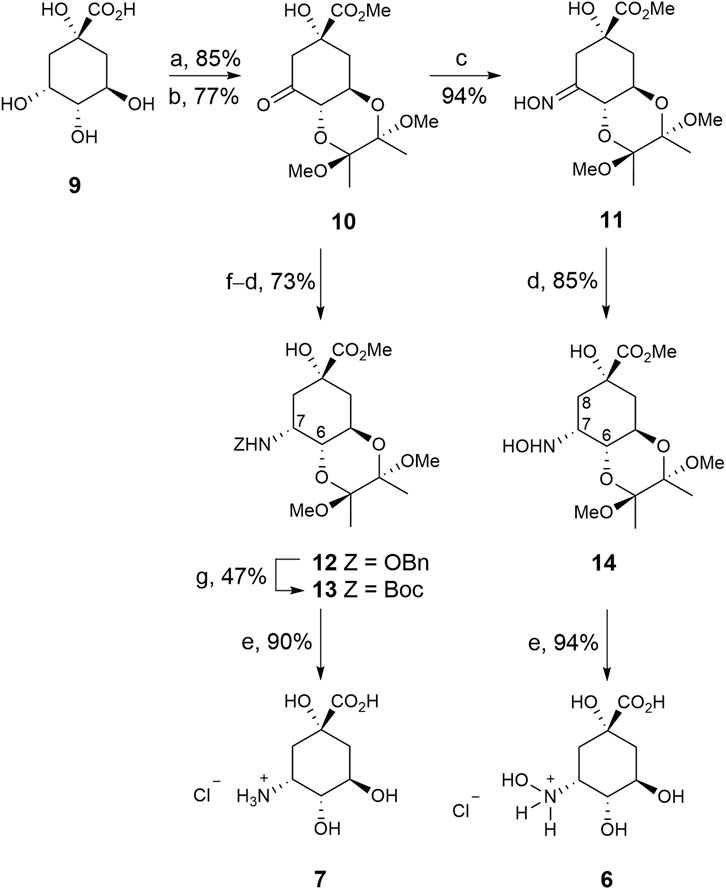
SCHEME 1. Synthesis of compounds 6–7. Reagents and conditions. (a) (MeCO)2, CH(OMe)3, MeOH, camphorsulfonic acid, 65°C. (b) PDC, MS 4 Å, CH2Cl2, RT. (c) NH2OH.HCl, NaOAc, MeCN, H2O, RT. (d) NaBH3CN, MeOH, HCl, RT. (e) HCl (0.3 M), 100°C. (f) NH2OBn.HCl, NaOAc, MeOH, MS 4 Å, RT. (g) 1. H2(g), Pd/C (10%), AcOH, MeOH, RT. 2. Boc2O, Et3N, DMF, RT.
3-Aminoquinic acid (7) was prepared by regioselective reductive amination of ketone 10 with benzylhydroxylamine to afford compound 12 in 73% yield. The benzyl group in 12 was removed by hydrogenolysis under acidic conditions and the resulting amine was converted into the Boc-amino protected derivative 13 by treatment with di-tert-butyl dicarbonate to facilitate its purification (47% yield). NOE experiments also confirmed the S configuration of the new chiral center as irradiation of the H7 signal (4.16 ppm) in 13 led to enhancement of the H6 and NH signals by 5.2% and 1.3%, respectively. Finally, compound 13 was transformed into the target compound 7, as its hydrochloride salt, in a similar manner to compound 6 from 14.
This compound was prepared from previously reported epoxide 18, which is synthesized in seven steps from (–)-quinic acid 9) (Scheme 2) (González et al., 2003). Briefly, quadruple protection of (–)-quinic acid (9) by reaction with benzaldehyde in acid medium, followed by treatment with N-bromosuccinimide in the presence of AIBN as radical initiator afforded bromobenzoate 15 (Bartlett et al., 1986). Treatment of 15 with tert-butyldimethylsilyl chloride and excess of DBU led to simultaneous formation of the silylether and the double bond. Treatment of benzoate 16 with potassium cyanide in methanol afforded hydroxycarbolactone 17 and the corresponding methyl ester, which was converted into 17 with sodium hydride. Epoxidation of allylic alcohol 17 by treatment with meta-chloroperbenzoic acid in the presence of sodium bicarbonate afforded the epoxy alcohol 18 as the sole diasteroisomer. Removal of the TBS-protecting group in 18 by treatment with aqueous TFA gave alcohol 19 in 68% yield. Finally, basic hydrolysis of lactone 19 and subsequent protonation with Amberlite IR-120 (H+) gave the desired oxirane derivative 8 in 97% yield.
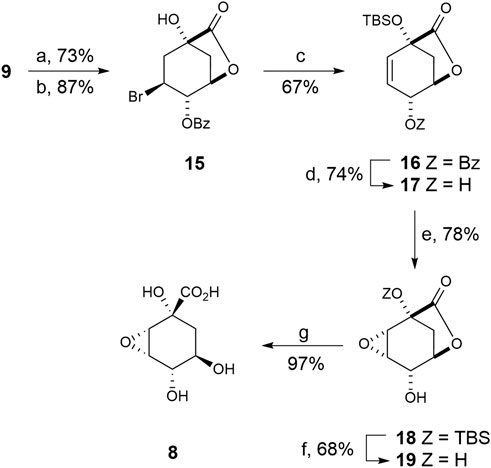
SCHEME 2. Synthesis of compound 8. Reagents and conditions. (a) PhCHO, DMF, PhMe, p-TsOH (cat), ∆. (b) NBS, AIBN (cat), PhH, ∆. (c) DBU, ClTBS, CH3CN, ∆. (d) 1. KCN, MeOH, RT. 2. NaH, THF, 0°C. (e) MCPBA, NaHCO3, CH2Cl2, ∆. (f) (20:1) TFA/H2O, RT. (g) 1. LiOH, RT, 2. Amberlite IR-120.
The inhibitory capacity of compounds 6–8 against DHQ1 enzymes from S. aureus and S. typhi was evaluated. For irreversible inhibition assays, the enzyme was incubated with the ligand and its remaining activity was progressively determined using aliquots from the incubation samples and the control. For reversible competitive inhibition assays, the activity of the enzyme in the presence of increasing amount of ligand was evaluated. The enzyme activity was determined by monitoring the increase in absorbance at 234 nm in the UV spectrum due to formation of the enone-carboxylate chromophore of the enzyme product, 3-dehydroshikimic acid 2). Compound 6 was found to be a time-dependent irreversible inhibitor of the two enzymes with KI values of 90 ± 8 and 130 ± 12 µM and kinact values of 8 ± 1 and 13 ± 1 ms−1 against Sa-DHQ1 and St-DHQ1, respectively (Supplementary Figure S1). In contrast, compounds 7 and 8 were found to be reversible competitive inhibitors of the two enzymes. For amino derivative 7, IC50 values of 482 ± 19 and 528 ± 33 µM against Sa-DHQ1 and St-DHQ1, respectively, were obtained. The inhibitory capacity of compound 8 was found to be poor, with IC50 values > 2 mM for both enzymes.
To obtain structural information on the binding mechanism of compounds 6–8, and further details of the covalent modification caused by 6, crystallization of DHQ1 from both S. aureus and S. typhi with the ligands reported herein was attempted. As a result, protein crystals with the appropriate diffracting properties complexed with compounds 6 and 8 were obtained. For epoxide 8, crystals were obtained by soaking using apo-St-DHQ1 crystals, while for hydroxylamine 6 they were achieved by co-crystallization. Crystals were mounted into cryoloops and were directly flash frozen by rapid immersion in liquid nitrogen. The crystallization conditions contained a sufficient amount of cryo-protectant (PEG) to directly freeze the protein crystals in liquid nitrogen without further manipulation. X-ray diffraction data were collected from crystals of Sa-DHQ1/6, St-DHQ1/6 and St-DHQ1/8 cryo-cooled in a stream of cold nitrogen gas (100 K) at ambient pressure using synchrotron radiation. The structures were determined by molecular replacement and refined. Full details are provided in the experimental section. A summary of the statistics following data reduction and processing is given in Table 1.
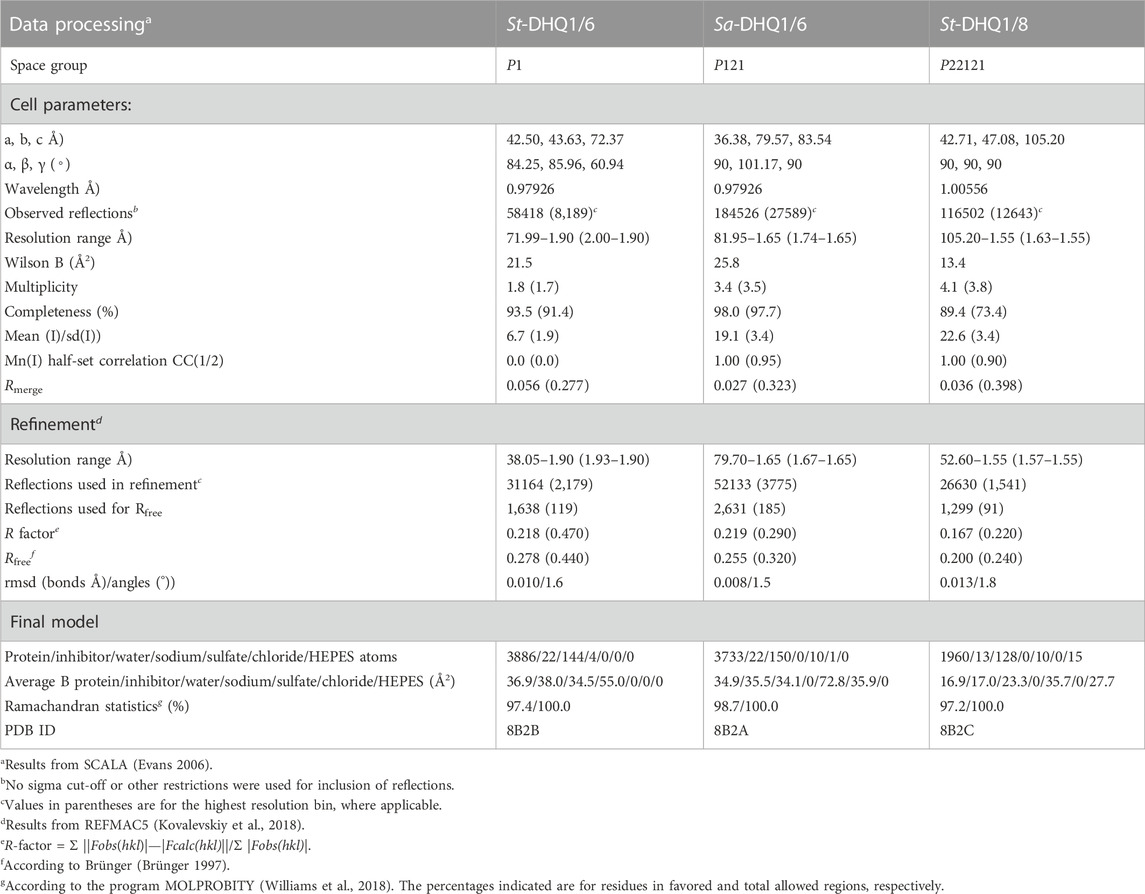
TABLE 1. Crystallographic data collection and refinement statistics for the St-DHQ1/6 and Sa-DHQ1/6enzyme adducts and St-DHQ1/8enzyme complexa.
The two adducts Sa-DHQ1/6 and St-DHQ1/6 crystallized as dimers with two molecules in the asymmetric unit (namely chains A and B) and were solved at a resolution of 1.65 and 1.90 Å, respectively. The two crystallographically independent copies superimpose well onto each other, with root-mean-square (rms) differences of 0.363 Å (Sa-DHQ1) and 0.117 Å (St-DHQ1) after superposition of Cα-atom pairs. Both chains are virtually identical.
Unbiased, calculated electron-density maps showed clear electron density for the ligand covalently bonded to the catalytic lysine residue (K160/K170 in Sa-DHQ1 and St-DHQ1, respectively) via an amine, which is visible in the two chains (Figure 3A). Remarkably, among all the lysine residues present in the two enzymes (13 for Sa-DHQ1 and 11 for St-DHQ1), only the catalytic lysine residue is modified by the ligand. Surprisingly, no electron density for the tertiary hydroxyl group in position C1 was identified in the two enzyme adducts (Figures 3B, C). Dehydration involving the C1–C2 bond took place to give a shikimate-like core, perhaps after covalent linkage at the C3 position in 6 via a similar mechanism to that observed for the natural substrate. Comparison of the structure of St-DHQ1 covalently modified by hydroxylamine 6 solved herein with the previously reported structure for the trapped enzyme-product Schiff base intermediate obtained by reduction with sodium borohydride (PDB ID 1QFE, 2.10 Å) (Gourley et al., 1999) showed that both structures are similar (rms = 0.3 between chains) (Supplementary Figure S2). Importantly, no cleavage of the amine bond promoted by the ligand was observed. Thus, compound 6 provides non-labile enzyme adducts by avoiding the intrinsic Schiff base hydrolysis performed by the enzyme.
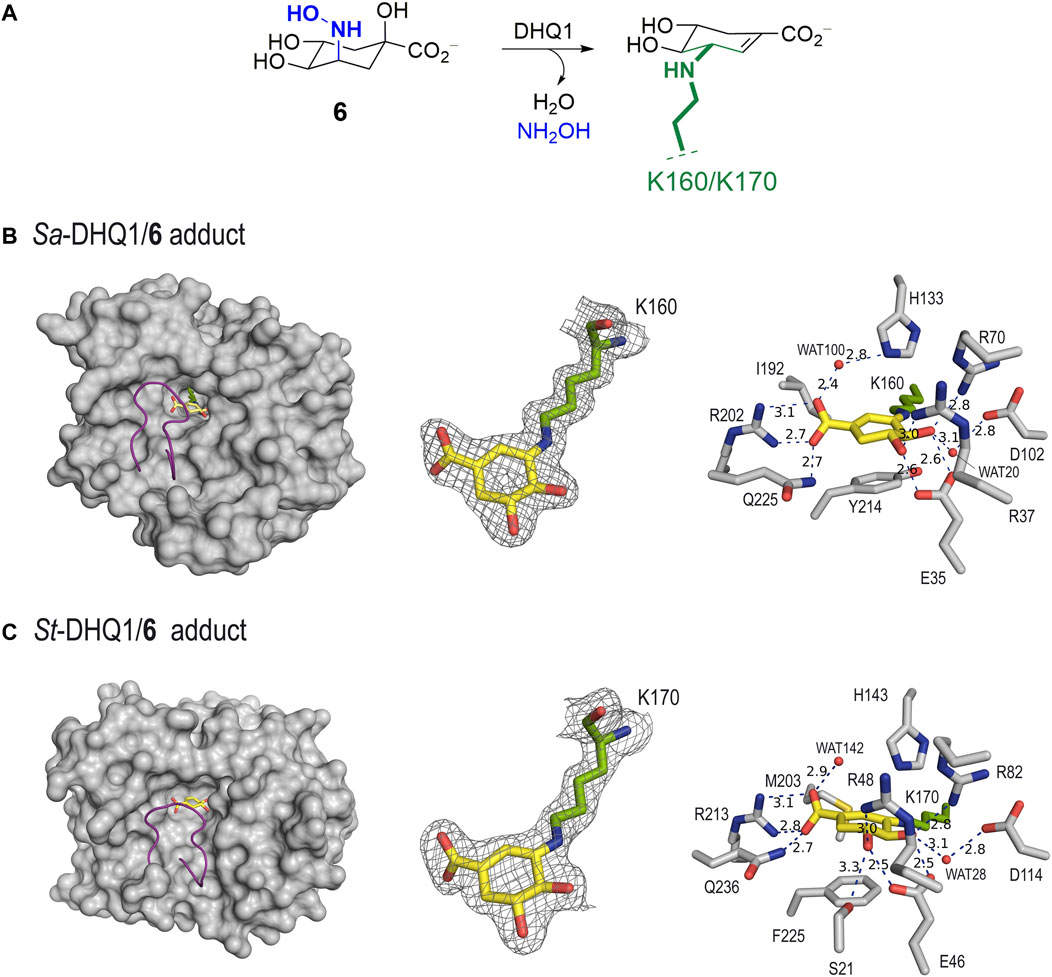
FIGURE 3. (A) Schematic representation of the chemical modification to the catalytic lysine residue of Sa-DHQ1 and St-DHQ1, as identified by X-ray crystallography. (B,C) Crystal structures of Sa-DHQ1 (B, PDB ID 8B2A, 1.65 Å, chain A) and St-DHQ1 (C, PDB ID 8B2B, 1.90 Å, chain A) covalently modified by hydroxylamine 6. Overall views of the enzyme adducts and interactions of the modified inhibitor (yellow) are shown. The substrate-covering loop (purple) is shown as a cartoon to visualize the active site. Hydrogen-bonding and electrostatic interactions between the ligands and both homologous enzymes are shown as dashed lines (blue). Relevant residues are shown and labeled. Unbiased electron density for inhibitor 6 and its covalent attachment to residues K160/K170 of Sa-DHQ1 and St-DHQ1, respectively. From the model obtained by molecular replacement, and before inclusion of the inhibitor molecule, refinement was performed to obtain unbiased density for the inhibitor molecule and other model changes. A maximum-likelihood weighted 2Fo − Fc map contoured at 1σ is shown up to 1.6 Å around the inhibitor molecule (yellow) and the catalytic lysine residue (green). The final model, including the inhibitor molecule, is superimposed onto the map. The distances are in angstroms.
In addition to the linkage to the catalytic lysine residue that blocks the enzyme active site, the enzyme-modified ligand is anchored to the enzyme via strong interactions with several conserved residues within the pocket. The latter comprises two main moieties in its chemical structure, which are similar for both structures (Figures 3B, C). Specifically: i) the carboxylate group in C1 establishes a salt-bridge with the guanidinium group of R202/R213, which has proven to be a key residue for recognition, along with a hydrogen bond with the amide side chain of Q225/Q236 (in Sa-DHQ1 and St-DHQ1, respectively), which is located on the substrate-covering loop that closes the active site for catalysis (Maneiro et al., 2014); and ii) the C4 and C5 hydroxyl groups establish a bidentate hydrogen bond with the carboxylate group of E35/E46, as well as hydrogen-bonding interactions with the guanidinium groups of R37/R48 and R70/R82 and with the carboxylate group of D102/D114 (in Sa-DHQ1 and St-DHQ1, respectively) via the structurally conserved water molecule, which is observed in all the reported crystal structures (such as PDB IDs 1SFJ (Nichols et al., 2004) and 1L9W (Lee et al., 2002)). Moreover, for both structures: i) a water molecule interacting with one of the oxygen atoms of the carboxylate group in the modified ligand via hydrogen bonding is observed [namely WAT100 (Sa-DHQ1) and WAT142 (St-DHQ1)]; and ii) the side chain of the catalytic histidine residue is located above the amino group of the modified lysine residue. For the St-DHQ1/6 enzyme adduct, a long hydrogen bond between the C5 hydroxyl group and the side chain of residue S21, which is located on the flexible substrate-covering loop, is also identified. As a result, this loop folds more above the modified ligand. Finally, the active site of both adducts is closed by the substrate-covering loop, thus protecting the catalytic pocket from the water environment.
The crystal structure of St-DHQ1 in complex with epoxide 8 crystallized with one molecule in the asymmetric unit and was solved at a resolution of 1.55 Å. The calculated maps revealed clear high electron density for the ligand molecule 8, which binds to the active site via a salt bridge between its C1 carboxylate group and the guanidinium group of the conserved R213, along with a network of attractive hydrogen-bonding interactions with several side-chain residues (Figure 4A). Thus, as for adduct St-DHQ1/6, the ligand establishes direct contacts with the residues S21, E46, R48, R82, R213, and Q236, as well as two water molecules (WAT36 and WAT71; Figure 4B).
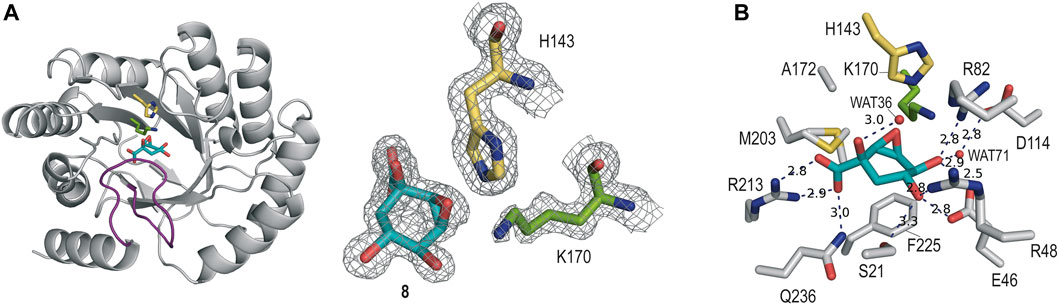
FIGURE 4. (A) Crystal structure of St-DHQ1 in complex with epoxide 8 (PDB ID 8B2C, 1.55 Å). Unbiased electron density for inhibitor 8 and the catalytic residues K170 and H143 of St-DHQ1. The substrate-covering loop is highlighted in purple. From the model obtained by molecular replacement and before inclusion of the inhibitor molecule, refinement was performed to obtain unbiased density for the inhibitor molecule and other model changes. A maximum-likelihood weighted 2Fo − Fc map contoured at 1σ is shown up to 1.6 Å around the inhibitor molecule (cyan) and residues K170 (green) and H143 (yellow). (B) Main contacts of epoxide 8 with St-DHQ1. Hydrogen-bonding and electrostatic interactions are shown as blue dashed lines. The distances are shown in angstrom. Only relevant residues are shown and labeled. Note how residue K170 shows a bent conformation.
The enzyme structure shows that the introduction of a three-membered ring at the C2 and C3 positions of the cyclohexane ring in the ligand causes a twist in the overall chair conformation and, as a result, the strength of the contacts of the C5 hydroxyl group, as well as the accessibility to the C3 carbon by the ɛ-amino group of K170, are altered. Thus, the hydrogen bond between the C5 hydroxyl group in 8 and the carboxylate group of E46 is 0.3 Å longer than in the St-DHQ1/6 adduct, while the interaction with residue R48 is shortened by 0.2 Å (Figures 4C vs 3C). More importantly, the CH group in the C3 position in 8 is twisted towards the bottom of the active site, thus hindering the reaction with the ɛ-amino group of K170, and the side chain of H143 is positioned close enough to the oxirane moiety in 8 (3.0 Å) for potential activation (Lewis acid). In addition, residue K170 shows a bent arrangement, and its ɛ-amino group is located pointing towards the carboxylate group of D114 (3.1 Å) and the structural water molecule (3.1 Å). The unsuitable orientation of the oxirane functional group within the cyclic compound might justify the lack of covalent modification observed experimentally.
The covalent modification of DHQ1 by compound 6 was also corroborated by mass spectrometry analysis of samples incubated with and without ligand. The MALDI spectra of St-DHQ1 enzyme incubated with 6 in ∼1:120 enzyme/ligand ratio for 24 h and at 25°C showed a peak for a covalently modified enzyme corresponding to an additional mass of 158 (theoretical value of the increased mass = 156; Supplementary Figure S3). No modifications were identified in similar studies carried out with compounds 7 and 8.
Of the two aminoquinic acid derivatives reported herein (compounds 6 and 7), only the former, which has a hydroxyamino group, causes covalent modification of the catalytic lysine residue. However, from a structural point of view, the two compounds have a nitrogen moiety with an R configuration in the C3 position, and the overall geometry for recognition appears to be similar. Intrigued by this finding, the molecular basis of these differences was explored by computational studies. To that end, models of the corresponding enzyme complexes (Michaelis) for subsequent covalent modification were generated, and the stability of the ligands, and interactions with the residues of the active site, were analyzed by performing MD simulation studies. The required DHQ1/6 and DHQ1/7 binary complexes were constructed using the enzyme geometries found in the structures reported herein, after manual replacement of the observed ligand by compounds 6 and 7. The resulting binary complexes were inmersed in a truncated octahedron box of water molecules and then subjected to 100 ns of dynamic simulation using the molecular mechanics force field AMBER ff14SB and GAFF (Wang et al., 2004).
Considering that the DHQ1 active site comprises various positively charged residues in the vicinity of the reactive pocket (Lys/His), specifically R70/R82 and R37/R48 (Sa-DHQ1/St-DHQ1, respectively), a key point in the recognition of compounds 6 and 7 appears to be the protonation state of its nitrogen substituent, as well as that for the catalytic histidine residue. For mechanistic considerations, the catalytic lysine residue was considered to be neutral. Thus, simulation studies with the two plausible protonation states (neutral and protonated) for the NH2 substituent in 7 and NHOH group in 6, along with the catalytic histidine residue [neutral (δ protonation) and dual (δ and ε protonation)], were carried out, and the stability of the ligand and its contacts within the pocket were analyzed during the whole simulation, as discussed below.
For compound 6, our simulation studies revealed that the ligand would be stable in the active site when either: 1) its NHOH substituent is in the neutral form and the catalytic histidine residue is protonated; or 2) the catalytic histidine residue is neutral and the ligand is protonated on its nitrogen atom, as no significant changes were observed during the whole simulation (Figures 5A and Supplementary Figure S4). Thus, analysis of the root-mean-square deviation (rmsd) plots for the protein backbone and the ligand calculated from the MD simulation of the complexes revealed low values (Supplementary Figure S5).
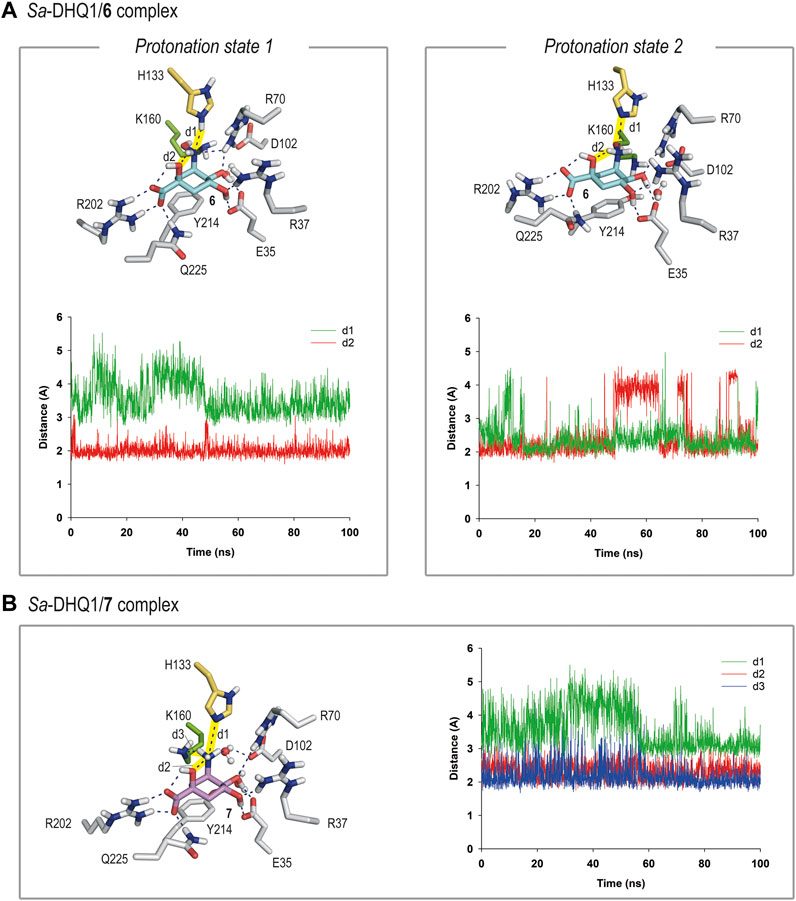
FIGURE 5. (A) Binding mode of compound 6 in the active site of Sa-DHQ1, as obtained by MD simulation studies, in which the two plausible protonation states of the NHOH moiety in 6 and residue H133 were considered. Snapshots taken after 80 (left) and 100 ns (right) are provided. Hydrogen-bonding and electrostatic interactions are shown as blue dashed lines. Only relevant residues are shown and labeled. Variation of the distances between H133 (HE2 or NE2 atoms) and the nitrogen atom in 6 (d1) and the NH group in 6 and its C1 hydroxyl group (oxygen atom, d2) in the Sa-DHQ1/6 binary complex during the whole simulation considering protonation states 1 and 2. The distances studied are highlighted with yellow shading in the protein figure. (B) Binding mode of compound 7 (ammonium salt) in the active site of Sa-DHQ1 obtained by MD simulation studies. Residue H133 was considered in its neutral form. Snapshot taken after 100 ns of simulation. Variation of the distances between the ammonium group in 7 (H or N atoms) and H133 (NE2 atom, d1), its tertiary hydroxyl group (oxygen atom, d2) and K170 (NZ atom, d3) in the Sa-DHQ1/7 binary complex during the whole simulation. The distances studied are highlighted with yellow shading in the protein figure. Note how the position of the catalytic lysine residue is frozen close to the C2 pocket by a strong hydrogen bond with the ammonium group in 7, with an average distance of 2.2 Å during the simulation.
In both cases, the arrangement of the NHOH substituent would be frozen by a defined network of hydrogen bonds. Thus, for option 1, the orientation of the NHOH group would be locked by three hydrogen bonds: one between the oxygen lone pair of the C1 hydroxyl group and the C3 aminic proton in the ligand, and two contacts involving the nitrogen lone pair in 6, one with the NH group ε) of the protonated histidine residue and other with one of the NH moieties of the guanidinium group of residue R70. These interactions were found to be very stable, as revealed by an analysis of the variation of the distances between the atoms involved during the simulation (Figure 5A). In addition, the lysine side-chain was allocated to the reactive pocket close to the C3 carbon atom in 6. As the estimated pKa values for hydroxylamine and a histidine residue are rather similar (about 6), it seems reasonable that both protonation states might exist (Boulet et al., 1999). On the other hand, the superposition of the reported crystal structures of the DHQ1/6 enzyme adducts with those obtained by MD simulations, in which the two plausible protonation states of the catalytic residues were considered, suggested that the modified lysine residue is likely to be protonated whereas the histidine residue is probably neutral (Supplementary Figure S6). Thus, other protonation scenarios would cause a significant motion in residues H133/H143 and D102/D114 (in Sa-DHQ1 and St-DHQ1, respectively) as well as the structural water molecule, all of which participate in stabilization of the adduct. On the other hand, the basicity of the amino substituent in 6 seems to be relevant for achieving lysine-covalent modification, since (–)-quinic acid (9), which possesses a hydroxyl group in the same position, does not form a covalent adduct with the enzyme, as revealed by the crystal structure of the Salmonella enterica DHQ1 enzyme in complex with 9 (PDB ID 4GUI, 1.78 Å) (Light et al., 2014).
In contrast, the results of the studies carried out with the DHQ1/7 binary complex revealed that only the ammonium salt of 7 would be stable in the active site, in agreement with the estimated pKa value for an amine (>9) (Figure 5B). More importantly, the ammonium group in 7 appears to freeze the position of the ε-amino group of K160 close to the C2 pocket by way of a strong hydrogen bond with the ammonium group in 7, with an average distance between the atoms involved of 2.2 Å during the whole simulation (Figure 5B). Similar outcomes were obtained with the S. aureus and S. typhi enzymes (Supplementary Figures S7, S8). As for compound 6, the ammonium substituent also establishes an internal hydrogen bond with the C1 hydroxyl group and an electrostatic interaction with H133. When the neutral form of the amino group was considered, large motion in the ligand, along with opening of the substrate-covering loop, was observed. To our knowledge, this is the first example of a secondary hydroxylamine derivative that cause the selective covalent modification of a sterically hindered lysine residue of an enzyme. Our outcomes support the potential of the hydroxyamino moiety as latent electrophile for lysine-targeted irreversible inhibition, when an exquisite anchoring of the ligand to the enzyme is achieved. The latter entails to study in-deep the enzyme recognition pattern, and its implementation into the scaffold to accomplish the required geometric perfection for reaction. More work must be done to improve inhibitor permeability into cells, for which aim an ester prodrug approch seems suitable as we demostrated before for inhibitors targeting the enzyme type II dehydroquinase (Tizón et al., 2011).
The results of the present study show that replacement of the C3 hydroxy group of (–)-quinic acid by a hydroxyamino substituent (compound 6) results in a time-dependent irreversible inhibitor, whereas compound 7, in which the latter functionality is replaced by an amino group, does not allow covalent modification of the enzyme. This finding was supported by resolution of the crystal structures for the DHQ1 from Staphylococcus aureus and Salmonella typhi, covalently modified by 6, at 1.65 and 1.90 Å, respectively, which revealed that the inhibitor selectively reacts with the catalytic lysine residue to form an amine. The modified ligand also undergoes dehydration at the tertiary hydroxyl group to afford a shikimate-like derivative. This structure is similar to that reported previously for the trapped enzyme-product Schiff-base intermediate obtained by reduction with sodium borohydride. Despite having a reactive electrophilic warhead, compound 8 proved to be a reversible competitive inhibitor of the enzyme and was very stable in the enzyme active site, as evidenced by resolution of the crystal structure of the Salmonella typhi DHQ1 enzyme in complex with 8 at 1.55 Å. The twisted arrangement of the cyclohexane ring in 8 towards residue F255 observed in the crystal structure, which is located on the bottom part of the active site, seems to hinder access of the essential lysine residue for nucleophilic attack.
The structures of the enzyme DHQ1 reported herein, in combination with MD simulation studies, provide valuable information relating to the binding modes of the targeted compounds, as well as insights into the molecular basis by which the quinate-based compounds explored are activated for lysine-covalent modification or not. Our simulation studies suggest that compound 7, the amino group of which would be protonated at the active site, could prevent covalent modification of the catalytic lysine residue by freezing the position of the enzyme nucleophile (ɛ-amino Lys group) on the same face as the leaving group (Re face). The catalytic histidine residue, which would be in its neutral form to avoid electrostatic repulsion with the ammonium group in 7, would also be anchored in the vicinity of the substituent by a strong hydrogen-bonding interaction. In contrast, the latter unreactive arrangement would be avoided, to some extent, by replacement of the amino group in 7 by the NHOH moiety, since in the latter case a neutral form of the leaving group in the ligand would be achieved upon binding, thus facilitating the required flexibility of the lysine residue for reaction. In addition, the neutral state of the NHOH substituent in 6 would allow the dual protonation state of the catalytic histidine residue, and the establishment of a strong hydrogen-bonding interaction between both moieties. Although additional computational studies are necessary to validate this hypothesis, the arrangement identified in the present study for the DHQ1/6 binary complex suggests a covalent modification mechanism consisting of nucleophilic attack of the lysine residue at C3 in 6 with release of NH2OH mediated by the essential histidine residue acting as a proton donor.
All starting materials and reagents were commercially available and were used without further purification. 1H (300 and 500 MHz) and 13C NMR spectra (75 and 125 MHz) were measured in deuterated solvents. J values are given in Hertz. NMR assignments were carried out by a combination of 1D, COSY, and DEPT-135 experiments. FTIR spectra were recorded in a PerkinElmer Two FTIR spectrometer with attenuated total reflection. Melting points were measured in a Büchi M-560 apparatus.
A suspension of ketone 10 (305 mg, 0.96 mmol) was prepared in two steps from commercially available (–)-quinic acid 9), as described previously (Toscano et al., 2003). Thus, a mixture of hydroxylamine hydrochloride (100 mg, 1.44 mmol) and sodium acetate trihydrate (196 mg, 1.44 mmol) in acetonitrile (8 mL) and water (3.2 mL) was stirred at room temperature for 24 h. The solvent was then removed under reduced pressure and the resulting residue was dissolved in dichloromethane and water. The organic layer was separated, dried (anh. Na2SO4), filtered and concentrated under reduced pressure. The resulting mixture was then purified by flash chromatography, eluting with diethyl ether, to yield compound 11 (286 mg, 89%) as a white foam.
A suspension of ketone 10 (500 mg, 1.57 mmol), O-benzylhydroxylamine hydrochloride (250.6 mg, 1.57 mmol), anhydrous sodium acetate (258 mg, 3.14 mmol) and 4 Å molecular sieves (500 mg) in dry methanol (8.0 mL) was stirred for 20 h at room temperature and under an inert atmosphere. The reaction mixture was filtered through a plug of Celite® and washed with methanol. The filtrate and washings were concentrated under reduced pressure. The resulting residue was dissolved in glacial acetic acid (3.9 mL), at room temperature and under an inert atmosphere, and then treated with sodium cyanoborohydride (197 mg, 3.14 mmol). After stirring for 1 h, the solvent was removed under reduced pressure and the resulting residue was dissolved in ethyl acetate and saturated NaHCO3. The aqueous layer was separated, dried (anh. Na2SO4), filtered and concentrated under reduced pressure. The reaction mixture was purified by flash chromatography, eluting with (50:50) diethyl ether/hexane, to yield compound 12 (489 mg, 73%) as a white foam.
A suspension of Pd/C (18 mg, 10%) in methanol (2.5 mL) and 5 drops of acetic acid was treated, under a hydrogen atmosphere and at room temperature, with a solution of the O-benzylhydroxylamine 12 (181 mg, 0.43 mmol) in methanol (2.5 mL) via canula. The resulting suspension was stirred at room temperature for 18 h. After removal of the hydrogen atmosphere, the suspension was filtered through a plug of Celite®. The filtrate and washings (methanol) were concentrated under reduced pressure. A solution of the resulting residue in dry DMF (4.5 mL) was treated successively with dry triethylamine (0.1 mL, 0.65 mmol) and di-tert-butyldicarbonate (113.5 mg, 0.52 mmol) and stirred for 3 h. The reaction mixture was diluted with ethyl acetate and water. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (×2). The combined organic extracts were dried (anh. Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with (80:20) diethyl ether/hexane, to yield compound 13 (85.5 mg, 47%) as a white foam.
NaBH3CN (27 mg, 0.42 mmol) and HCl (0.22 mL, 2.0 M) were added to a stirred solution of compound 11 (93 mg, 0.28 mmol) in methanol (0.45 mL) at room temperature and under an inert atmosphere. After stirring at room temperature for 3 h, powdered Na2CO3 (anh.) was added and the solvent was concentrated under reduced pressure. The reaction mixture was partitioned with ethyl acetate and water, the aqueous layer was separated, and the organic layer was washed with Na2CO3 (sat), dried (anh. Na2SO4), filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel, eluting with (90:10) diethyl ether/hexane, to afford compound 14 (80 mg, 85%) as a white foam.
A solution of compound 14 (71 mg, 0.21 mmol) in HCl (2.1 mL, 0.3 M) was heated at 100°C for 24 h. After cooling to room temperature, the solution was washed with ethyl acetate (×3). The aqueous layer was lyophilized to give compound 6 (41 mg, 94%) as a brown foam.
A solution of compound 13 (83.5 mg, 0.2 mmol) in an aqueous solution of HCl (2 mL, 0.3 M) was heated at 100 °C for 24 h. After cooling to room temperature, the solution was washed with ethyl acetate (×3). The aqueous layer was lyophilized to give compound 7 (45.5 mg, 90%) as a white foam.
A solution of silyl ether 18 (53 mg, 0.17 mmol) in (20:1) TFA/H2O (1.7 mL) was stirred at room temperature for 5 days. The solvents were then evaporated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel, eluting with diethyl ether, to give alcohol 19 (20 mg, 68%) as a colorless oil.
A solution of lactone 19 (30 mg, 0.17 mmol) in THF (1.7 mL) was treated with aqueous LiOH (0.86 mL, 0.5 M) and the resulting mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with Milli-Q water, and THF was evaporated under reduced pressure. The resulting aqueous solution was washed with diethyl ether (×3) and the aqueous extract was treated with Amberlite IR-120 (H+) until pH 6. The resin was filtered off and washed with milli-Q water. The filtrate and washings were lyophilized to give acid 8 (31 mg, 97%) as a white foam.
The St-DHQ1 and Sa-DHQ1 enzyme were purified as described previously (Moore et al., 1993; Nichols et al., 2004). Concentrated solutions of St-DHQ1 (0.85 mg mL−1, 30.74 µM) and Sa-DHQ1 (4 mg mL−1, 148.13 µM) were stored in potassium phosphate buffer (PPB) (50 mM) and DTT (1 mM) at pH 6.6 and −80°C. When required for assays, aliquots of the enzyme stocks were diluted in water and buffer and stored on ice. DHQ1 was assayed in the forward direction by monitoring the increase in absorbance at 234 nm in the UV spectrum due to the absorbance for the enone-carboxylate chromophore of 3-dehydroshikimic acid (ε/M−1 cm−1 12 000). Standard assay conditions were PPB (50 mM, pH 7.2) at 25°C. Each assay was initiated by addition of the substrate. Solutions of 3-dehydroquinic acid 1) were calibrated by equilibration with DHQ1 and measurement of the change in the UV absorbance at 234 nm due to formation of the enone-carboxylate chromophore of 3-dehydroshikimic acid. Under assay conditions, the kinetic parameters were: i) Sa-DHQ1: Km = 18 ± 3 μM; kcat = 255 ± 12 s−1; ii) St-DHQ1: Km = 24 ± 3 μM; kcat = 1.1 ± 0.1 s−1. The program GraFit 7.0.3. (Erithacus Software Ltd.). was used for data fitting.
Incubation of Sa-DHQ1 and St-DHQ1 (2.96 µM from a stock protein concentration of 0.85 mg mL−1 and 4 mg mL−1, respectively) with various aqueous solutions of compounds 6–8 (30–4,000 µM) was carried out in PPB (0.5 mL, 50 mM) at pH 7.2 at 25°C. The activity was determined progressively in duplicate over a 3 h period under the standard assay conditions (see above) using aliquots from the incubation samples and the control. Semilogarithmic plots of residual activity as a function of time with a range of inhibitor concentrations [I] were performed to obtain the first-order rate constant, kobs, calculated from the slope, and is related with kinact KI constants by Equation 1 implemented in GraFit (Plapp, 1982):
IC50 values were determined in duplicate by measuring the initial rates at fixed enzyme and substrate concentrations (14 µM) in the absence and in the presence of various compounds concentrations, which was adjusted to the Equation 2 implemented in GraFit:
were y is the observed effect at inhibitor concentration x, Emax corresponds to the maximum effect, IC50 is the concentration at which a 50% growth inhibition is obtained, and n is the slope of the curve.
St-DHQ1 (3.1 µM from a stock protein concentration of 0.85 mg mL−1) and Sa-DHQ1 (3.0 µM from a stock protein concentration of 4 mg mL−1) was incubated with various aqueous solutions of ligands 6–8 in PPB (0.5 mL, 50 mM) at pH 7.2, 25°C. The activity was determined progressively over a 24 h period under the standard assay conditions using aliquots from the incubation samples and the control. The activity was measured at a substrate concentration of 14 µM. The initial rate of each assay was measured. The samples were concentrated and washed (5 mM ammonium carbonate) for MALDI analysis by centrifugation at 4 °C using Amicon® centrifugal filters (Amicon Ultra-10). The samples were freeze-dried, diluted with 5 mM ammonium bicarbonate (5 µL) and analyzed by mass spectrometry using a MALDI TOF/TOF Mass Spectrometer (4,800 Analyzer, AbSciex). The ProteoMass™ MALDI calibration kit (Merck) was employed for calibration and sanipic acid as a matrix. Spectra were analyzed using the Data Explorer™ Software. Experiments were performed in triplicate.
St-DHQ1 (0.85 mg mL−1) and Sa-DHQ1 (4 mg mL−1) were concentrated to 11 and 20 mg mL−1, respectively, in 50 mM PPB pH 7.0, and 0.5 mM DDT. Ligands were dissolved at 250 mM in methanol and added to the protein solution at a ratio of 1:20 (v/v) to give a solution of approximately 10 equivalents of ligand per protein monomer. Crystals of up to: a) 0.2 mm × 0.2 mm of the St-DHQ1/6 (plate-shape prims); and b) 0.4 mm × 0.01 mm of the Sa-DHQ1/6 (needles) were obtained after 3 weeks of vapor diffusion in sitting drops comprised of 2.0 μL protein/ligand solution mixed with 2.0 μL reservoir solution against 0.15 mL reservoirs containing: i) for St-DHQ1: 12% PEG 2000 MME, 0.1 M MES pH 6.0; and ii) for Sa-DHQ1: 20% PEG 3350, 0.1 M Bis-Tris pH 5.5, 0.2 M Li2SO4.
Prism-shaped apo-St-DHQ1 crystals of up to 0.03 mm × 0.03 mm were obtained from solution of St-DHQ1 concentrated to 8 mg mL−1 in buffer A (10 mM Tris-HCl pH 7.4, 40 mM KCl) after 4 weeks of vapor diffusion in sitting drops comprised of 2.0 μL of protein solution mixed with 2.0 μL of reservoir solution and equilibrated against 0.15 mL reservoirs containing the crystallization mixture [26% PEG 2000 MME, 0.1 M Hepes pH 7.0]. St-DHQ1/8 complex crystals were obtained after soaking of apo-St-DHQ1 crystals in 10 mM solutions of epoxide 8 in the crystallization mixture for 48 h.
Crystals were mounted into cryoloops and directly flash-frozen by rapid immersion in liquid nitrogen. For the St-DHQ1/6 and Sa-DHQ1/6 enzyme adducts, X-ray diffraction data were collected on beamline BL13-Xaloc (Juanhuix et al., 2014) (Alba Synchrotron, Barcelona, Spain, detector Pilatus 6M-Dectris) from a crystal maintained at 100 K. For the St-DHQ1/8 enzyme complex, X-ray diffraction data were collected on beamline ID23–2 (ESRF; Grenoble, France) from a crystal maintained at 100 K. The diffraction data were processed, scaled, corrected for absorption effects and the crystal unit-cell parameters were calculated by global refinement with autoPROC (Vonrhein et al., 2011), which uses XDS (Kabsch 2010), AIMLESS (Evans 2006) and other programs from the CCP4 software suite (Winn et al., 2011).
The structures were solved by molecular replacement, using the program MOLREP (Vagin and Teplyakov 2010) with a search model generated from PDB entries 4CNN (St-DHQ1) and 6FSH (Sa-DHQ1), from which the ligand and solvent atoms were removed. The ligand structure and geometrical restraints, including the covalent link to the protein, were generated with the PRODRG2 (Schüttelkopf and van Aalten 2004) server and JLIGAND (Lebedev et al., 2012). The ligand was manually placed during model building, which was performed with COOT (Casañal et al., 2020). Reflections used to calculate Rfree (Brünger 1997) were selected randomly. Refinement of the models was performed with REFMAC5 (Kovalevskiy et al., 2018) and final structure validation was performed with MOLPROBITY (Williams et al., 2018) and the PDB validation server (Read et al., 2011). Data collection, refinement and model statistics are summarized in Table 1. Structure figures were prepared using PYMOL (DeLano 2002).
Minimization of the ligands, DHQ1/ligand adducts and binary complexes, and MD simulation of the resulting minimized adducts and complexes, were carried following our previously described protocol (Lence et al., 2020). The simulation time was 100 ns. The coordinates found in the crystal structures of Sa-DHQ1 and St-DHQ1 reported herein were employed. The stability and reliability of all the DHQ1/ligand adducts and complexes described herein were verified by analysing the root-mean-square deviation (rmsd) for the whole protein backbone (Cα, C, N and O atoms), the inhibitors and the modified ligands during the whole simulation by using the cpptraj module in AMBER 16.
Coordinates and structure factors are available from the Protein Data Bank with accession codes 8B2A, 8B2B and 8B2C.
AR and MM carried out the synthesis of the target compounds and the enzyme crystallization studies; EL performed the MD simulation studies; JO and MV solved the enzyme crystal structures; PT and AH produced and purified the DHQ1 enzymes; CG-B designed and coordinated the study and carried out the biochemical studies. The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.
Financial support from the Spanish State Agency of Research (PID2019-105512RB-I00/AEI/10.13039/501100011033, CG-B), the Xunta de Galicia (ED431C 2021/29 and Centro singular de investigación de Galicia accreditation 2019-2022 (ED431G 2019/03), CG-B), and the European Regional Development Fund (ERDF) is gratefully acknowledged.
We are grateful to the ALBA synchrotron (Barcelona, Spain) and ESRF (Grenoble, France) synchrotrons for the provision of beam time and to the Centro de Supercomputación de Galicia (CESGA) for use of the Finis Terrae computer. AR and MM acknowledge the Spanish Ministry of Science, Innovation and Universities and Xunta de Galicia for their fellowships (BES-2017-080946 and ED481D-2022-010, respectively).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1111598/full#supplementary-material
Anscombe, E., Meschini, E., Mora-Vidal, R., Martin, M. P., Staunton, D., Geitmann, M., et al. (2015). Identification and characterization of an irreversible inhibitor of CDK2. Chem. Biol. 22 (9), 1159–1164. doi:10.1016/j.chembiol.2015.07.018
Bartlett, P. A., Maitra, U., and Chouinard, P. M. (1986). Synthesis of "iso-EPSP" and evaluation of its interaction with chorismate synthase. J. Am. Chem. Soc. 108 (25), 8068–8071. doi:10.1021/ja00285a031
Bauer, R. A. (2015). Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 20 (9), 1061–1073. doi:10.1016/j.drudis.2015.05.005
Boulet, P., Gilardoni, F., Weber, J., Chermette, H., and Ellinger, Y. (1999). Theoretical study of interstellar hydroxylamine chemistry: Protonation and proton transfer mediated by H3+. Chem. Phys. 244 (2), 163–174. doi:10.1016/s0301-0104(99)00151-2
Brünger, A. T. (1997). Free R value: Cross-validation in crystallography. Methods Enzymol. 277, 366–396. doi:10.1016/s0076-6879(97)77021-6
Cano, D. A., Pucciarelli, M. G., Martínez-Moya, M., Casadesús, J., and García-del Portillo, F. (2003). Selection of small-colony variants of Salmonella enterica serovar typhimurium in nonphagocytic eucaryotic cells. Infect. Immun. 71 (7), 3690–3698. doi:10.1128/iai.71.7.3690-3698.2003
Casañal, A., Lohkamp, B., and Emsley, P. (2020). Current developments in coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29 (4), 1069–1078. doi:10.1002/pro.3791
Cook, M. A., and Wright, G. D. (2022). The past, present, and future of antibiotics. Sci. Transl. Med. 14 (657), eabo7793. doi:10.1126/scitranslmed.abo7793
Cunningham, A. L., Mann, B. J., Qin, A., Santiago, A. E., Grassel, C., Lipsky, M., et al. (2020). Characterization of Schu S4 aro mutants as live attenuated tularemia vaccine candidates. Virulence 11 (1), 283–294. doi:10.1080/21505594.2020.1746557
Dalton, S. E., Dittus, L., Thomas, D. A., Convery, M. A., Nunes, J., Bush, J. T., et al. (2018). Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition. J. Am. Chem. Soc. 140 (3), 932–939. doi:10.1021/jacs.7b08979
DeLano, W. L. (2002). The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific LLC. from http://www.pymol.org/.
Dickey, S. W., Cheung, G. Y. C., and Otto, M. (2017). Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 16 (7), 457–471. doi:10.1038/nrd.2017.23
Dilts, D. A., Riesenfeld-Orn, I., Fulginiti, J. P., Ekwall, E., Granert, C., Nonenmacher, J., et al. (2000). Phase I clinical trials of aroA aroD and aroA aroD htrA attenuated S. typhi vaccines; effect of formulation on safety and immunogenicity. Vaccine 18 (15), 1473–1484. doi:10.1016/s0264-410x(99)00424-7
Evans, P. (2006). Scaling and assessment of data quality. Acta Crystallogr. D. Biol. Crystallogr. 62, 72–82. doi:10.1107/S0907444905036693
González, C., Carballido, M., and Castedo, L. (2003). Synthesis of polyhydroxycyclohexanes and relatives from (-)-quinic acid. J. Org. Chem. 68 (6), 2248–2255. doi:10.1021/jo026692m
González-Bello, C. (2015). Specific chemical modification of bacterial type I dehydroquinase-opportunities for drug discovery. Future Med. Chem. 7 (17), 2371–2383. doi:10.4155/fmc.15.145
González-Bello, C., Tizón, L., Lence, E., Otero, J. M., van Raaij, M. J., Martinez-Guitian, M., et al. (2015). Chemical modification of a dehydratase enzyme involved in bacterial virulence by an ammonium derivative: Evidence of its active site covalent adduct. J. Am. Chem. Soc. 137 (29), 9333–9343. doi:10.1021/jacs.5b04080
Gourley, D. G., Shrive, A. K., Polikarpov, I., Krell, T., Coggins, J. R., Hawkins, A. R., et al. (1999). The two types of 3-dehydroquinase have distinct structures but catalyze the same overall reaction. Nat. Struct. Biol. 6 (6), 521–525. doi:10.1038/9287
Grimster, N. P., Connelly, S., Baranczak, A., Dong, J., Krasnova, L. B., Sharpless, K. B., et al. (2013). Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J. Am. Chem. Soc. 135 (15), 5656–5668. doi:10.1021/ja311729d
Hotinger, J. A., Morris, S. T., and May, A. E. (2021). The case against antibiotics and for anti-virulence therapeutics. Microorganisms 9 (10), 2049. doi:10.3390/microorganisms9102049
Hoyt, E. A., Cal, P. M. S. D., Oliveira, B. L., and Bernardes, G. J. L. (2019). Contemporary approaches to site-selective protein modification. Nat. Rev. Chem. 3 (3), 147–171. doi:10.1038/s41570-019-0079-1
Juanhuix, J., Gil-Ortiz, F., Cuní, G., Colldelram, C., Nicolás, J., Lidón, J., et al. (2014). Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 21, 679–689. doi:10.1107/S160057751400825X
Kabsch, W. (2010). Xds. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132. doi:10.1107/S0907444909047337
Kärnell, A., Cam, P. D., Verma, N., Lindberg, A. A., and Karnell, A. (1993). AroD deletion attenuates Shigella flexneri strain 2457T and makes it a safe and efficacious oral vaccine in monkeys. Vaccine 11 (8), 830–836. doi:10.1016/0264-410x(93)90358-5
Kovalevskiy, O., Nicholls, R. A., Long, F., Carlon, A., and Murshudov, G. N. (2018). Overview of refinement procedures within REFMAC5: Utilizing data from different sources. Acta Crystallogr. D. Struct. Biol. 74, 215–227. doi:10.1107/S2059798318000979
Krell, T., and Matilla, M. A. (2022). Antimicrobial resistance: Progress and challenges in antibiotic discovery and anti-infective therapy. Microb. Biotechnol. 15 (1), 70–78. doi:10.1111/1751-7915.13945
Lebedev, A. A., Young, P., Isupov, M. N., Moroz, O. V., Vagin, A. A., and Murshudov, G. N. (2012). JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D. Biol. Crystallogr. 68, 431–440. doi:10.1107/S090744491200251X
Lee, W. H., Perles, L. A., Nagem, R. A., Shrive, A. K., Hawkins, A., Sawyer, L., et al. (2002). Comparison of different crystal forms of 3-dehydroquinase from Salmonella typhi and its implication for the enzyme activity. Acta Crystallogr. D. Biol. Crystallogr. 58, 798–804. doi:10.1107/s0907444902003918
Leech, A. P., Boetzel, R., McDonald, C., Shrive, A. K., Moore, G. R., Coggins, J. R., et al. (1998). Re-evaluating the role of His-143 in the mechanism of type I dehydroquinase from Escherichia coli using two-dimensional 1H,13C NMR. J. Biol. Chem. 273 (16), 9602–9607. doi:10.1074/jbc.273.16.9602
Leech, A. P., James, R., Coggins, J. R., and Kleanthous, C. (1995). Mutagenesis of active site residues in type I dehydroquinase from Escherichia coli. Stalled catalysis in a histidine to alanine mutant. J. Biol. Chem. 270 (43), 25827–25836. doi:10.1074/jbc.270.43.25827
Lence, E., Maneiro, M., Sanz-Gaitero, M., van Raaij, M. J., Thompson, P., Hawkins, A. R., et al. (2020). Self-immolation of a bacterial dehydratase enzyme by its epoxide product. Chemistry 26 (36), 8035–8044. doi:10.1002/chem.202000759
Light, S. H., Antanasijevic, A., Krishna, S. N., Caffrey, M., Anderson, W. F., and Lavie, A. (2014). Crystal structures of type I dehydroquinate dehydratase in complex with quinate and shikimate suggest a novel mechanism of Schiff base formation. Biochemistry 53 (5), 872–880. doi:10.1021/bi4015506
Malcova, M., Karasova, D., and Rychlik, I. (2009). aroA and aroD mutations influence biofilm formation in Salmonella Enteritidis. FEMS Microbiol. Lett. 291 (1), 44–49. doi:10.1111/j.1574-6968.2008.01433.x
Maneiro, M., Lence, E., Sanz-Gaitero, M., Otero, J. M., Van Raaij, M. J., Thompson, P., et al. (2019). Hydroxylammonium derivatives for selective active-site lysine modification in the anti-virulence bacterial target DHQ1 enzyme. Org. Chem. Front. 6 (17), 3127–3135. doi:10.1039/c9qo00453j
Maneiro, M., Peón, A., Lence, E., Otero, J. M., Van Raaij, M. J., Thompson, P., et al. (2014). Insights into substrate binding and catalysis in bacterial type I dehydroquinase. Biochem. J. 462 (3), 415–424. doi:10.1042/BJ20140614
Moore, J. D., Hawkins, A. R., Charles, I. G., Deka, R., Coggins, J. R., Cooper, A., et al. (1993). Characterization of the type I dehydroquinase from Salmonella typhi. Biochem. J. 295, 277–285. doi:10.1042/bj2950277
Mortenson, D. E., Brighty, G. J., Plate, L., Bare, G., Chen, W., Li, S., et al. (2018). Inverse drug discovery" strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc. 140 (1), 200–210. doi:10.1021/jacs.7b08366
Narayanan, A., and Jones, L. H. (2015). Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 6 (5), 2650–2659. doi:10.1039/c5sc00408j
Nichols, C. E., Lockyer, M., Hawkins, A. R., and Stammers, D. K. (2004). Crystal structures of Staphylococcus aureus type I dehydroquinase from enzyme turnover experiments. Proteins 56 (3), 625–628. doi:10.1002/prot.20165
Nógrády, N., Imre, A., Rychlik, I., Barrow, P. A., and Nagy, B. (2003). Growth and colonization suppression of Salmonella enterica serovar Hadar in vitro and in vivo. FEMS Microbiol. Lett. 218 (1), 127–133. doi:10.1111/j.1574-6968.2003.tb11508.x
Plapp, B. V. (1982). Application of affinity labeling for studying structure and function of enzymes. Methods Enzymol. 87, 469–499. doi:10.1016/s0076-6879(82)87027-4
Proctor, R. A., von Eiff, C., Kahl, B. C., Becker, K., McNamara, P., Herrmann, M., et al. (2006). Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4 (4), 295–305. doi:10.1038/nrmicro1384
Racz, R., Chung, M., Xiang, Z., and He, Y. (2013). Systematic annotation and analysis of "virmugens"-virulence factors whose mutants can be used as live attenuated vaccines. Vaccine 31 (5), 797–805. doi:10.1016/j.vaccine.2012.11.066
Read, R. J., Adams, P. D., Arendall, W. B., Brunger, A. T., Emsley, P., Joosten, R. P., et al. (2011). A new generation of crystallographic validation tools for the protein data bank. Structure 19 (10), 1395–1412. doi:10.1016/j.str.2011.08.006
Revolledo, L., and Ferreira, A. J. P. (2012). Current perspectives in avian salmonellosis: Vaccines and immune mechanisms of protection. J. Appl. Poult. Res. 21 (2), 418–431. doi:10.3382/japr.2011-00409
Sanderson, K. (2013). Irreversible kinase inhibitors gain traction. Nat. Rev. Drug Discov. 12 (9), 649–651. doi:10.1038/nrd4103
Schüttelkopf, A. W., and van Aalten, D. M. (2004). Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D. Biol. Crystallogr. 60, 1355–1363. doi:10.1107/S0907444904011679
Sebkova, A., Karasova, D., Crhanova, M., Budinska, E., and Rychlik, I. (2008). Aro mutations in Salmonella enterica cause defects in cell wall and outer membrane integrity. J. Bacteriol. 190 (9), 3155–3160. doi:10.1128/JB.00053-08
Singh, J., Petter, R. C., Baillie, T. A., and Whitty, A. (2011). The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10 (4), 307–317. doi:10.1038/nrd3410
Smith, P. A., Koehler, M. F. T., Girgis, H. S., Yan, D., Chen, Y., Chen, Y., et al. (2018). Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 561 (7722), 189–194. doi:10.1038/s41586-018-0483-6
Tacket, C. O., Hone, D. M., Curtiss, R., Kelly, S. M., Losonsky, G., Guers, L., et al. (1992). Comparison of the safety and immunogenicity of delta aroC delta aroD and delta cya delta crp Salmonella typhi strains in adult volunteers. Infect. Immun. 60 (2), 536–541. doi:10.1128/IAI.60.2.536-541.1992
Tacket, C. O., Hone, D. M., Losonsky, G. A., Guers, L., Edelman, R., and Levine, M. M. (1992). Clinical acceptability and immunogenicity of CVD 908 Salmonella typhi vaccine strain. Vaccine 10 (7), 443–446. doi:10.1016/0264-410x(92)90392-w
Tacket, C. O., and Levine, M. M. (2007). CVD 908, CVD 908-htrA, and CVD 909 live oral typhoid vaccines: A logical progression. Clin. Infect. Dis. 45, S20–S23. doi:10.1086/518135
Tamura, T., Ueda, T., Goto, T., Tsukidate, T., Shapira, Y., Nishikawa, Y., et al. (2018). Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 9 (1), 1870. doi:10.1038/s41467-018-04343-0
Theuretzbacher, U., and Piddock, L. J. V. (2019). Non-traditional antibacterial therapeutic options and challenges. Cell Host Microbe 26 (1), 61–72. doi:10.1016/j.chom.2019.06.004
Tizón, L., Otero, J. M., Prazeres, V. F. V., Llamas-Saiz, A. L., Fox, G. C., van Raaij, M. J., et al. (2011). A prodrug approach for improving anti-tuberculosis activity of potent Mycobacterium tuberculosis type II dehydroquinase inhibitors. J. Med. Chem. 54 (17), 6063–6084. doi:10.1021/jm2006063
Toscano, M. D., Frederickson, M., Evans, D. P., Coggins, J. R., Abell, C., and González-Bello, C. (2003). Design, synthesis and evaluation of bifunctional inhibitors of type II dehydroquinase. Org. Biomol. Chem. 1 (12), 2075–2083. doi:10.1039/b301731a
Vagin, A., and Teplyakov, A. (2010). Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25. doi:10.1107/S0907444909042589
Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., et al. (2011). Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. Biol. Crystallogr. 67, 293–302. doi:10.1107/S0907444911007773
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25 (9), 1157–1174. doi:10.1002/jcc.20035
Williams, C. J., Headd, J. J., Moriarty, N. W., Prisant, M. G., Videau, L. L., Deis, L. N., et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27 (1), 293–315. doi:10.1002/pro.3330
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242. doi:10.1107/S0907444910045749
Yang, T., Cuesta, A., Wan, X., Craven, G. B., Hirakawa, B., Khamphavong, P., et al. (2022). Reversible lysine-targeted probes reveal residence time-based kinase selectivity. Nat. Chem. Biol. 18 (9), 934–941. doi:10.1038/s41589-022-01019-1
Zhang, P., Wright, J. A., Osman, A. A., and Nair, S. P. (2017). An aroD ochre mutation results in a Staphylococcus aureus small colony variant that can undergo phenotypic switching via two alternative mechanisms. Front. Microbiol. 8, 1001. doi:10.3389/fmicb.2017.01001
Zhang, T., Hatcher, J. M., Teng, M., Gray, N. S., and Kostic, M. (2019). Recent advances in selective and irreversible covalent ligand development and validation. Cell Chem. Biol. 26 (11), 1486–1500. doi:10.1016/j.chembiol.2019.09.012
Keywords: enzyme recognition, biomacromolecule simulations, irreversible inhibition, binding mode, organic synthesis, ligand activation, type I dehydroquinase, anti-virulence agents
Citation: Rodríguez Á, Maneiro M, Lence E, Otero JM, van Raaij MJ, Thompson P, Hawkins AR and González-Bello C (2023) Quinate-based ligands for irreversible inactivation of the bacterial virulence factor DHQ1 enzyme—A molecular insight†. Front. Mol. Biosci. 10:1111598. doi: 10.3389/fmolb.2023.1111598
Received: 29 November 2022; Accepted: 10 January 2023;
Published: 24 January 2023.
Edited by:
Mariapina D'Onofrio, University of Verona, ItalyReviewed by:
Madeline E. Kavanagh, The Scripps Research Institute, United StatesCopyright © 2023 Rodríguez, Maneiro, Lence, Otero, van Raaij, Thompson, Hawkins and González-Bello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Concepción González-Bello, Y29uY2VwY2lvbi5nb256YWxlei5iZWxsb0B1c2MuZXM=
†Dedicated to the memory of Prof. Gregorio Asensio
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.