 Charles B. Trelford
Charles B. Trelford Lina Dagnino
Lina Dagnino Gianni M. Di Guglielmo
Gianni M. Di Guglielmo- 1Department of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 2Department of Oncology, Children’s Health Research Institute and Lawson Health Research Institute, London, ON, Canada
Transforming growth factor-β (TGFβ) is a ubiquitous cytokine essential for embryonic development and postnatal tissue homeostasis. TGFβ signalling regulates several biological processes including cell growth, proliferation, apoptosis, immune function, and tissue repair following injury. Aberrant TGFβ signalling has been implicated in tumour progression and metastasis. Tumour cells, in conjunction with their microenvironment, may augment tumourigenesis using TGFβ to induce epithelial-mesenchymal transition, angiogenesis, lymphangiogenesis, immune suppression, and autophagy. Therapies that target TGFβ synthesis, TGFβ-TGFβ receptor complexes or TGFβ receptor kinase activity have proven successful in tissue culture and in animal models, yet, due to limited understanding of TGFβ biology, the outcomes of clinical trials are poor. Here, we review TGFβ signalling pathways, the biology of TGFβ during tumourigenesis, and how protein quality control pathways contribute to the tumour-promoting outcomes of TGFβ signalling.
Introduction
Transforming growth factor-β (TGFβ), a central modulator of development, growth, proliferation, immune function, apoptosis, and homeostasis, plays key roles in cellular communication (Hajek et al., 2012). TGFβ is secreted as a latent cytokine that is sequestered by extracellular matrix (ECM) proteins (Isogai et al., 2003). Following enzymatic or allosteric-mediated release and subsequent activation of TGFβ, TGFβ ligands bind to ubiquitously expressed cell surface receptors (Horiguchi et al., 2012). Autocrine or paracrine TGFβ signalling modulates cell function by regulating transcription, translation, and post-translational modifications of several proteins (Massagué, 2012). Alterations in TGFβ signalling pathways have been implicated in numerous pathologies, including congenital diseases, fibrotic disorders, immune dysfunction, and tumourigenesis (Massagué, 2008; Neuzillet et al., 2015). The regulation of TGFβ signalling in cancer is complex, as it generally plays a tumour suppressive role in normal tissues and early tumour development (Principe et al., 2014). In contrast, mutations or abnormalities in the tumour suppressive arms of TGFβ signalling are common in advanced cancers (Harradine and Akhurst, 2006). In tumour cells, this cytokine drives tumourigenesis by inducing epithelial-mesenchymal transition (EMT), metastasis, angiogenesis, autophagy, and immune supression (Bierie and Moses, 2006). In this review, we will discuss TGFβ signalling pathways and how TGFβ may progress tumourigenesis.
Transforming growth factor-β pathways
The TGFβ superfamily consists of 33-members of secreted cytokines that are ubiquitously expressed in vertebrates and invertebrates. This superfamily includes TGFβ proteins, bone morphogenetic proteins (BMPs), activins, inhibins, nodal, lefty1, lefty2, anti-muellerian hormone (AMH), growth differentiation factors (GDFs), myostatin, and glial cell-derived neurotrophic factor (GDNF) (Lichtman et al., 2016). On the basis of their biological functions and mature protein structure, these members can be subclassified into four subfamilies (David and Massagué, 2018). In humans, the TGFβ subfamily consists of TGFβ1, TGFβ2, TGFβ3, the activin/inhibin/nodal subfamily consists of activinA, activinB, nodal, lefty1, lefty2, inhibinα, inhibinβ, the BMP/GDF subfamily consists of nine BMPs, and nine GDFs, and the fourth subfamily that has no defined relationship includes AMH, BMP15, GDF9, GDF15, and GDNF (Mueller and Nickel, 2012).
As homodimers or heterodimers, TGFβ superfamily members signal through heteromeric TGFβ receptor complexes. Seven different type I receptors, five type II receptors, and betaglycan and endoglin type III receptors have been described in vertebrates and invertebrates (Weiss and Attisano, 2013). Receptor activation leads to signalling cascades modulated by several classes of Sma-mothers against decapentaplegic (Smad) proteins, such as receptor regulated Smads (R-Smads), common Smads (co-Smads), and inhibitory Smads (I-Smads) (Massagué, 2012) as well as non-Smad signalling proteins (Mu et al., 2012). Although an extensive number of TGFβ superfamily members activate specific subsets of receptors and signalling molecules, this review will focus on the TGFβ subfamily.
Synthesis and post-translational modifications of TGFβ
In most metazoans, three genes encoding TGFβ isoforms have been described, and in humans the TGFB1, TGFB2, and TGFB3 genes are located on chromosomes 19, 1, and 14, respectively (Nishimura et al., 1993; Cruts et al., 1995; Green et al., 2001). Although TGFB1, TGFB2, and TGFB3 genes are highly conserved across species, there are some exceptions. For instance, TGFB4 has been identified in avian species; however, genetic mapping of chicken TGFB4 suggested that it is orthologous to human TGFB1 (Halper et al., 2004). Moreover, some South African frogs (Xenopus laevis) express a tgfb5 gene (Kondaiah et al., 1990). Translation of the TGFB1, TGFB2, and TGFB3 mRNA generates precursor polypeptides termed pre-pro-TGFβ, which are composed, respectively of 390, 412, and 412 amino acid residues (Khalil, 1999). The pre-pro-TGFβ species are composed of a signal peptide, a large amino-terminal latency-associated peptide (LAP), which ensures proper folding and transportation through the Golgi complex, and the residues of the mature ligand (Principe et al., 2014). Following signal peptide removal, disulfide isomerase catalyzes the formation of three disulfide bonds between two pre-pro-TGFβ monomers, linking cysteine residues at two positions in the LAP and one position in what will become the mature ligand. This modification gives rise to pro-TGFβ (Gentry et al., 1988). Within the Golgi complex membrane, furin and other convertases cleave LAP to generate small latent TGFβ complexes. Non-covalent bonds tether LAP to TGFβ, rendering the latter inactive (Poniatowski et al., 2015). Small latent TGFβ complexes, composed of a mature 25 kDa TGFβ dimer and two LAP moieties, are subsequently packaged into secretory vesicles in the Golgi complex (Dubois et al., 1995). Once secreted from the cell, the small latent TGFβ complexes are retained in the extracellular matrix (ECM), bound to latent TGFβ binding proteins (LTBPs) to form large latent TGFβ complexes (Massagué, 2012; Principe et al., 2014). TGFβ dimers can subsequently be released from the large latent TGFβ complexes through various enzymatic reactions or allosteric mechanisms (Figure 1) (Wipff et al., 2007; Tatti et al., 2008).
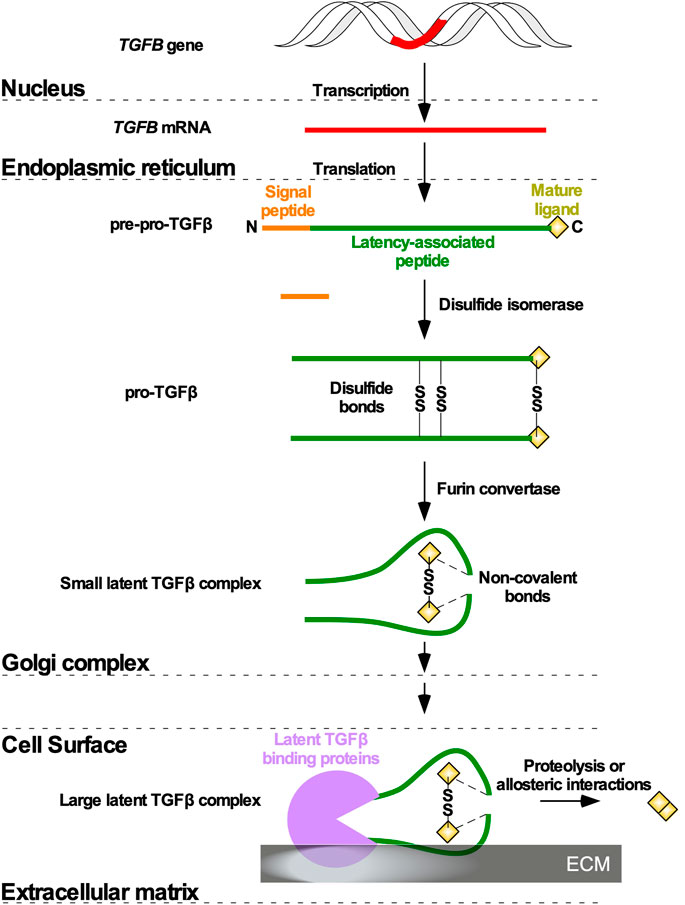
FIGURE 1. TGFβ ligand maturation. Following Transforming Growth Factor-β (TGFB) gene (red) transcription and TGFB mRNA translation in the nucleus and endoplasmic reticulum, respectively, TGFβ is synthesized as a precursor pro-TGFβ (pre-pro-TGFβ). Pre-pro-TGFβ contains an amino (N)-terminal signal peptide latency-associated peptide, and mature ligand. The N terminal signal peptide ensures transportation to the Golgi complex. In the Golgi complex, the signal peptide is cleaved, and disulfide isomerases catalyze disulfide bonds (SS) between two pre-pro-TGFβ monomers to generate pro-TGFβ. Furin convertases modify the latency-associated peptides, which non-covalently associate with mature ligands to generate a small latent TGFβ complex. The small latent TGFβ complex is secreted from the cell and attaches to latent TGFβ binding proteins in the extracellular matrix to form a large latent TGFβ complex. Mature ligands are released from the large latent TGFβ complexes via allosteric interactions or proteolysis mediated by enzymes.
The enzymatic activation of TGFβ through proteolysis requires matrix metalloproteinases (MMPs), plasmin, and other proteases (Kobayashi et al., 2014; Korol et al., 2014). MMP2 and MMP9 are Ca2+-dependent Zn+2-containing endopeptidases that target the LAP-binding domains of LTBPs, releasing TGFβ from the large latent TGFβ complexes. Plasmin generated at the cell surface, following plasminogen cleavage by urokinase plasminogen, also contributes to TGFβ release from LAPs (Yee et al., 1993; Yu and Stamenkovic, 2000). Alternatively, allosteric activation of TGFβ is dependent on several LAP-binding cell surface proteins, such as thrombospondin-1, mannose 6-phosphate receptors, and integrins, which induce conformational rearrangements of LAP (Dennis and Rifkin, 1991; Schultz-Cherry and Murphy-Ullrich, 1993; Sarrazy et al., 2014; Takasaka et al., 2018). Modifications of LAP are also induced by reactive oxygen species (Pociask et al., 2004) as well as acidic (pH < 2) or basic (pH > 12) environments (Lyons et al., 1988). Since these diverse LAP conformers no longer favour binding to TGFβ, the latter is released from the large latent TGFβ complexes.
Smad-dependent TGFβ signalling
After TGFβ ligands are released from large latent TGFβ complexes, they bind to cognate cell surface receptors. The Type I and II TGFβ receptors (TGFβRI and TGFβRII) exhibit serine-threonine kinase activity, and initiate signalling cascades upon ligand stimulation (Wrana et al., 1994). Type III TGFβ receptors (TGFβRIIIs) do not exhibit catalytic activity, but may facilitate the interaction between TGFβ ligands and TGFβRII (López-Casillas et al., 1994; Mclean and Di Guglielmo, 2010). TGFβ signalling is initiated when TGFβ binds to TGFβRII, triggering the association and phosphorylation of the glycine/serine domain of TGFβRI (Massagué, 2012). TGFβRI in turn phosphorylates downstream intracellular signalling molecules to induce canonical Smad-dependent and non-canonical Smad-independent TGFβ signalling, respectively (Massagué et al., 2000; Gunaratne and DiGuglielmo, 2013; McLean et al., 2013; Gunaratne et al., 2014).
All three classes of Smad proteins, R-Smads (Smad2/3), co-Smad (Smad4), and I-Smads (Smad6/7), temporally regulate TGFβ signalling (Massagué et al., 2005). Signal initiation begins when TGFβRI phosphorylates Smad2 or Smad3 on the carboxyl (C) terminus serine-serine-x-serine (SSXS) motif. Phosphorylated Smad2/3 is then released from the Smad anchor for receptor activation (SARA) protein into the cytoplasm (Tsukazaki et al., 1998; Qin et al., 2002), where it can form hetero-dimeric or hetero-trimeric complexes with Smad4 (Massague and Wotton, 2000; David and Massagué, 2018). These complexes subsequently translocate into the nucleus, where they regulate gene expression directly, by activating transcription, or indirectly by modulating the activity of other transcription factors (Finnson et al., 2013). Smad targeted genes include I-Smads (Chen et al., 1999), cyclin-dependent kinase 4 (CDK4) (Ewen et al., 1995), and EMT-transcription factors, including Snail Family Transcriptional Repressor one and 2 (SNAIL and SLUG), Zinc Finger E-box Binding Homeobox 1 and 2 (ZEB1 and ZEB2), Twist-related Protein 1 (TWIST1), Forkhead box C2 (FOXC2), Forkhead box A1 (FOXA1), Forkhead box A2 (FOXA2), Paired-related Homeobox 1 (PRX1), and High Mobility Group AT-hook 2 (HMGA2; Figure 2A) (Hajek et al., 2012; Katsuno et al., 2013). Through negative feedback mechanisms, Smad6 and Smad7 terminate TGFβ pathway activation (Figure 2A). I-Smads block R-Smad access to TGFβRI or recruit phosphatases (Iyengar, 2017; Kim and Baek, 2018), leading to dephosphorylation of active receptors (Shi et al., 2004). I-Smads also form complexes with E3 ubiquitin ligases, such as Smad ubiquitination regulatory factor 1 or 2 (Smurf1 or Smurf2), resulting in the degradation of TGFβ receptors (Kim and Baek, 2018; Miller et al., 2018).
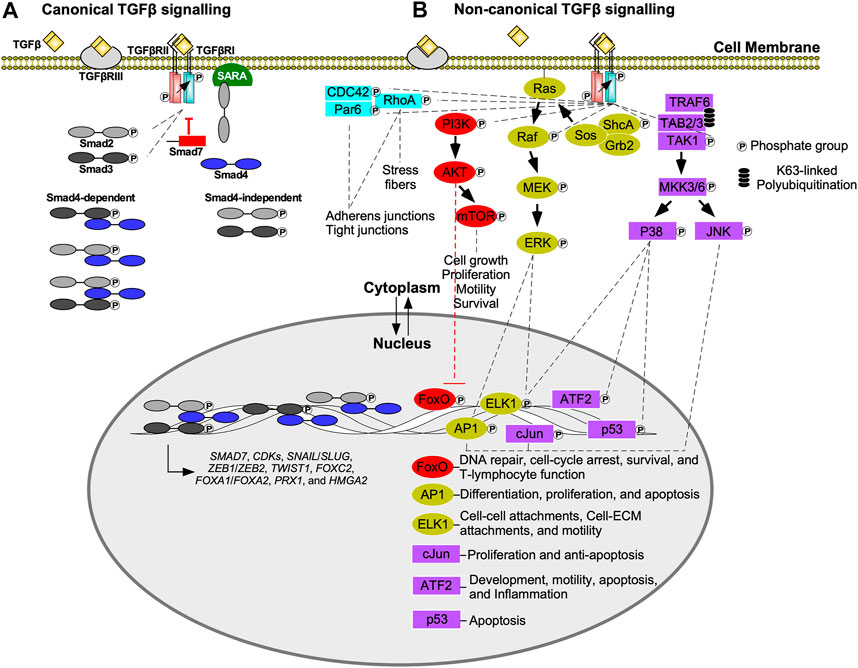
FIGURE 2. Canonical (Smad-dependent) and non-canonical (Smad-independent) TGFβ signalling. (A) Transforming growth factor-β (TGFβ) receptor type III (TGFβRIII) presents TGFβ to the type II receptor (TGFβRII). The TGFβ-TGFβRII complex phosphorylates TGFβ receptor type I (TGFβRI), which in turn phosphorylates R-Smads, Smad2 or Smad3. Phosphorylated Smad2/3 are released from the Smad anchor for receptor activation (SARA) protein, and translocate into the nucleus or form heterodimers/heterotrimers with Smad4 prior to nuclear translocation. Once in the nucleus, Smads function as transcription factors or interact with other transcription factors to regulate gene expression. Examples of genes regulated by Smads include, cyclin-dependent kinases (CDKs), Snail Family Transcriptional Repressor 1 and 2 (SNAIL/SLUG), Zinc Finger E-box Binding Homeobox one and 2 (ZEB1/ZEB2), Twist-related Protein 1 (TWIST1), Forkhead box C2 (FOXC2), Forkhead box A1 (FOXA1), Forkhead box A2 (FOXA2), Paired-related Homeobox 1 (PRX1), High Mobility Group AT-hook 2 (HMGA2), and SMAD7—which in turn dampens TGFβ signal transduction. (B) In non-canonical transforming growth factor-β (TGFβ) signalling, TGFβ receptor type I (TGFβRI) phosphorylates numerous downstream signalling molecules including TGFβ-activated kinase 1 (TAK1), src homology domain containing protein A (ShcA), and phosphoinositide 3-kinase (PI3K). Although partitioning defective six homolog (Par6) binds to TGFβRI, it is phosphorylated by TGFβRII. TGFβRI kinase activity is also important for Ras homolog family member A (RhoA) and cell division control protein 42 (CDC42) activation. The Par6/CDC42/RhoA pathway regulates adherens junctions, tight junctions, and stress fiber formation. PI3K phosphorylates protein kinase B (AKT), which inhibits Forkhead box O (FoxO) transcription factors that regulate genes responsible for DNA repair, cell cycle arrest, survival, and T-lymphocyte function. AKT also regulates cell growth, proliferation, motility, and survival by activating mechanistic target of rapamycin (mTOR). After ShcA is phosphorylated, it forms a complex with growth factor receptor bound 2 (Grb2) and sons of sevenless (Sos) to phosphorylate membrane bound Ras. This initiates a signalling cascade involving mitogen-activated protein kinase kinase (Raf), mitogen-activated protein kinase (MEK), and extracellular signal-regulated kinase 1 (ERK1). ERK1 upregulates activator protein 1 (AP1) and E-twenty-six Like-1 Protein (ELK1) transcription factors. AP1 upregulates genes that regulate differentiation, proliferation, and apoptosis, whereas ELK1 upregulates genes involved with cell-cell attachments, cell-extracellular matrix (ECM) attachments, and motility. TGFβRI phosphorylation promotes lysine (K)63-linked polyubiquitination of tumour necrosis factor receptor-associated factor 6 (TRAF6). TRAF6 forms a complex with TAK1 binding protein two and 3 (TAB2 and TAB3) to recruit TAK1. TGFβRI phosphorylates TAK1, which initiates signalling cascades that phosphorylate mitogen-activated protein kinase 3/6 (MKK3/6). MKK3/6 phosphorylates c-Jun amino-terminal kinase (JNK) and p38 MAPK. JNK regulates c-Jun and AP1 transcription factors, whereas p38 MAPK regulates activating transcription factor 2 (ATF2), p53, and ELK1 transcription factors. cJun upregulates genes involved with proliferation and survival, whereas ATF2 upregulates genes that modulate development, motility, apoptosis, and inflammation.
Structure of Smad proteins
Smad structure accounts for differences in Smad function. Structurally, Smad proteins have a Mad Homology 1 (MH1) domain, separated by a flexible linker region from a MH2 domain (Shi et al., 1998; Macias et al., 2015). MH1 domains contain a nuclear localization signal and β-hairpin loop that mediates interactions with glycine cysteine-rich Smad-binding elements on DNA (Jonk et al., 1998; Shi et al., 1998), whereas MH2 domains interact with TGFβ receptors and mediate binding to other Smad proteins, transcription factors, and co-activators or co-repressors of transcription (Wu et al., 2001). Among the three regions, the greatest variability is observed within the linker region. The linker region of R-Smads contain phosphorylation sites for multiple kinases, such as CDKs and mitogen-activated protein kinases (MAPKs) (Massagué et al., 2005). Furthermore, within the linker region, both R-Smads and I-Smads, but not Smad4, have a proline-proline-x-tyrosine (PPXY) motif to bind to E3 ubiquitin ligases (Qin et al., 1999; Macias et al., 2015). Although MH1 and MH2 domains are highly conserved, there are some notable differences. I-Smads are missing the MH1 domain, therefore, cannot bind to DNA (Miyazawa and Miyazono, 2017). The MH2 domains of R-Smads have a β1-strand, L3 loop, and α-helix five structure that together mediates binding to TGFβRI or SARA (Shi et al., 1998; Wu et al., 2001; Macias et al., 2015). Although the structure of Smad2 and Smad3 are similar, there are notable differences. For instance, Smad2 has two inserts in its MH1 domain (Shi et al., 1998). One of these inserts, known as the E3 insert, was once believed to disrupt the β-hairpin loop, preventing Smad2 from binding DNA (Dennler et al., 1998; Dennler et al., 1999). Further analysis indicated that different conformations of the E3 insert regulate MH1 domain structure, which explains why in some instances Smad2 has been shown to bind to DNA (Aragón et al., 2019).
Although Smad4 is essential to many TGFβ-dependent changes in gene expression, Smad4 is not essential for R-Smad nuclear translocation nor is it necessary for some TGFβ-dependent transcriptional functions (Ten Dijke and Hill, 2004). Smad4 also performs TGFβ-independent functions that include silencing the expression of TGFβ target genes in T-lymphocytes (T-cells) (Igalouzene et al., 2022), upregulating genes that promote natural killer (NK) cell maturation (Wang et al., 2018), and tumour suppression by mediating Aurora A kinase degradation (Jia et al., 2014). Although the roles of Smad4 remain incompletely understood, Smad4 is the only Smad with a nuclear export signal and a Smad activation domain (SAD) within its linker region. The SAD region is recognized by the chromatin modifiers p300 and CREB-binding protein co-activators (Pouponnot et al., 1998). Although Smad4 SAD deletion cells are still able to bind p300 and CREB co-activators, these Smad4-p300 and Smad4-CREB complexes are unable to activate transcription (De Caestecker et al., 2000). In this manner, Smad4 contributes to the regulation of gene expression through p300 and CREB-binding protein co-activator complexes.
Smad-independent TGFβ signalling
Smad-independent TGFβ signalling occurs through various pathways (Figure 2B) (Zhang, 2009). One involves the MAPK cascade via tumour necrosis factor receptor-associated factor 6 (TRAF6). Upon stimulation by TGFβ, TGFβRI associates with TRAF6, leading to lysine (K)63 polyubiquitination of this protein. K63-linked polyubiquitination provides a scaffold that subsequently recruits TGFβ-activated kinase 1 (TAK1), as well as TAK1-binding proteins. After TAK1-dependent phosphorylation, MAPK kinase 3/6 phosphorylates c-Jun amino-terminal kinase (JNK) and p38 MAPK. JNK and p38 MAPK translocate into the nucleus, where they phosphorylate several targets, including p53, activator protein 1 (AP1), E-twenty-six like-1 protein (ELK1), activating transcription factor 2 (ATF2), and cJun (Yamashita et al., 2008). These transcription factors regulate the expression of genes involved in apoptosis, inflammation, motility, development, cell-cell attachments, cell-ECM attachments, and proliferation (De Borst et al., 2006).
The protein kinase B (AKT) pathway is activated by TGFβRI phosphorylation of phosphoinositide 3-kinase (PI3K), which in turn activates AKT (Suwanabol et al., 2012). Downstream targets of AKT include mechanistic target of rapamycin (mTOR), a regulator of cell growth, proliferation, motility, survival, autophagy, transcription, and protein synthesis (Zhang et al., 2013). Additionally, AKT inhibits Forkhead box O (FoxO) transcription factors, which are important regulators of CDKs, survival, DNA repair, and T-cell activity (Zhang et al., 2005; Zhang et al., 2011).
Smad-independent TGFβ signalling also leads to modulation of small GTPase activity (Edlund et al., 2002). Specifically, TGFβRII can phosphorylate partitioning defective six homolog (Par6) (Ozdamar et al., 2005), whereas Ras homolog family member A (RhoA), and cell division control protein 42 (CDC42) activation relies on TGFβRI activity (Fleming et al., 2009; Kim et al., 2016). These proteins modulate cell-cell and cell-ECM attachments by regulating the function, stability, and organization of proteins essential to adherens and tight junctions. RhoA also promotes cell migration by inducing stress fiber formation (Warner et al., 2010; Nunes de Almeida et al., 2019). Stress fibers are contractile actomyosin bundles found in non-muscle cells composed of filamentous actin, α-actinin, and non-muscle myosin II filaments that may aid in cell movement (Hakkinen et al., 2011; Lehtimäki et al., 2021).
Tyrosine residues on the src homology domain containing protein A (ShcA) was also reported to be phosphorylated by TGFβRI (Lee et al., 2007). ShcA forms a complex containing growth factor receptor bound 2 (Grb2) and sons of sevenless (Sos) to activate Ras. The latter initiates downstream MAPK cascades that ultimately phosphorylates extracellular signal-regulated kinase (ERK) (Derynck and Zhang, 2003). ERK phosphorylates transcription factors, such as AP1 and ELK1, that regulate the expression of genes essential for cell-cell attachments, cell-ECM attachments, motility, differentiation, proliferation, and apoptosis (Zhang, 2009; Mu et al., 2012).
TGFβ receptor endocytosis regulates signalling strength and duration
Endocytosis of TGFβRI, TGFβRII, and TGFβ-TGFβRII complexes are mediated via clathrin- or caveolae-dependent mechanisms (Figure 3) (Le Roy and Wrana, 2005). Clathrin-dependent endocytosis allows TGFβ signalling to continue following receptor internalization and is associated with signal amplification (Yakymovych et al., 2018). Clathrin-coated pits sequester TGFβ receptors via the clathrin coat adaptor complex 2 (AP2) (Yao et al., 2002). AP2 is a hetero-tetramer that binds to clathrin and consists of four adaptins (β2, µ2, α, and σ2) (Kovtun et al., 2020). Unlike many receptors within the plasma membrane that bind to µ2-adaptin, TGFβ receptors directly bind to β2-adaptin (Yao et al., 2002). Next, several proteins facilitate budding and fission of clathrin-coated pits that are internalized as clathrin-coated vesicles. Clathrin-coated vesicles subsequently shed AP2 and fuse with the early endosome membrane compartment in a Rab5-dependent manner (Semerdjieva et al., 2008). Early endosome membrane compartments are enriched in phosphatidylinositol 3-phosphate (PI3P), which serve as recruitment sites for FYVE domain-containing proteins, such as early endosome antigen 1 (EEA1), endofin, and SARA (Lee et al., 2005). By associating with SARA on early-endosomal membranes, the R-Smads, Smad2/3, are poised to interact with TGFβ receptors (Itoh et al., 2002). Since regions involved in clathrin-dependent internalization are enriched in SARA, these routes of subcellular trafficking promote TGFβRI-dependent R-Smad phosphorylation (Macias et al., 2015). SARA also amplifies TGFβ signalling because SARA overexpression leads to endosomal swelling, which delays receptor recycling/degradation (Hu et al., 2002). In support of this, when the localization of SARA and EEA1-positive early endosomes was disrupted, there was a decrease in both TGFβ-induced Smad2 phosphorylation and Smad2 nuclear translocation (Tsukazaki et al., 1998; Hayes et al., 2002). Finally, endofin facilitates TGFβ signalling because it binds to TGFβRI and Smad4, which brings Smad4 in close proximity to phosphorylated R-Smads. Indeed, endofin knockdown reduced transcriptional responses to TGFβ and impaired TGFβ-dependent apoptosis (Chen et al., 2007). Therefore, clathrin-dependent trafficking of TGFβ receptors enables R-Smad phosphorylation in the early endosome and prolongs the duration in which ligands, receptors, and downstream signalling molecules are in close proximity. The early endosome is primarily responsible for sorting endocytosed TGFβ receptors, which may either recycle back to the plasma membrane in Rab11-positive vesicles (Yin et al., 2013) or be degraded in Rab7-positive late endosomes and lysosomes (Feng et al., 1995) (Figure 3A).
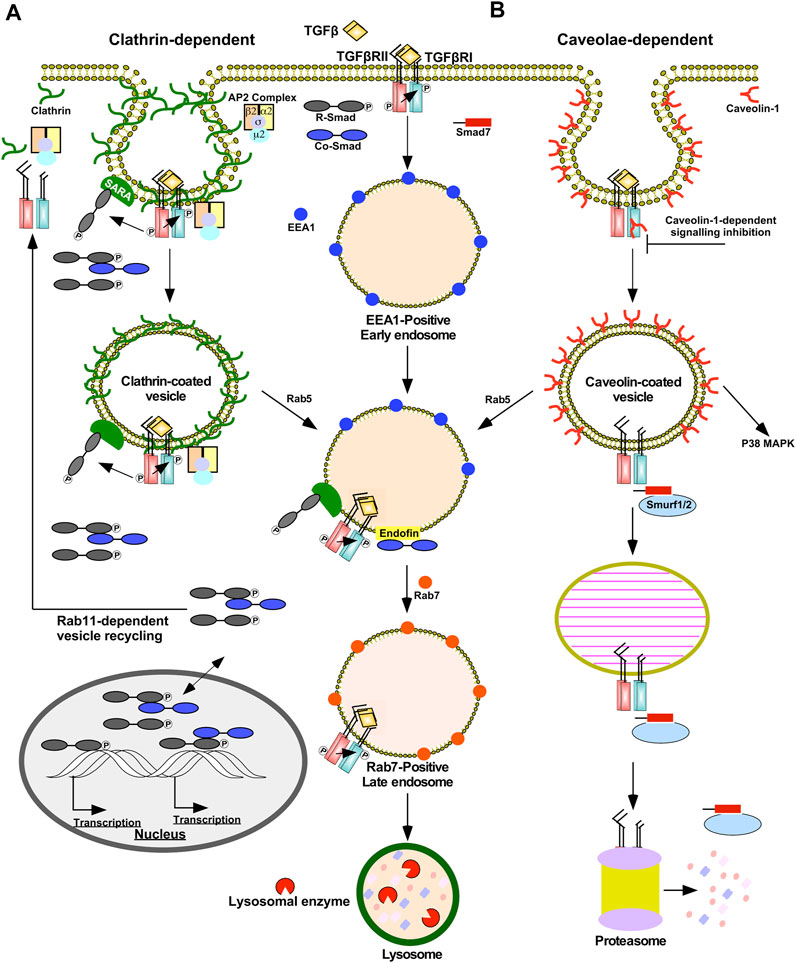
FIGURE 3. Clathrin- and caveolae-dependent endocytosis regulates the duration and strength of TGFβ signalling. (A) Clathrin-dependent receptor trafficking is mediated by triskelion shaped clathrin proteins (green). Clathrin tethers transforming growth factor-β (TGFβ) receptors to clathrin-coated pits via the β2 adaptin of the clathrin coat adaptor complex 2 (AP2). Clathrin-coated pits pinch off the plasma membrane to form clathrin-coated vesicles that fuse with early endosome membrane compartments by a Rab5-dependent process. In the presence of TGFβ, TGFβ receptors within clathrin-coated vesicles are active and phosphorylate downstream signalling molecules, such as Smads. Clathrin-coated pits and vesicles are enriched in Smad anchor for receptor activation (SARA) proteins that bind to R-Smads, which augments TGFβ signalling. Early endosomes bind to FYVE domain-containing proteins, such as endofin and SARA. Endofin enhances TGFβ signalling in early endosomes by tethering Smad4 to early endosomes. Clathrin-dependent receptor trafficking promotes R-Smad phosphorylation, which subsequently enters the nucleus with and without Smad4 to regulate transcription. The fates of the TGFβ receptors subjected to clathrin-dependent receptor trafficking involve recycling back to the plasma membrane in Rab11-positive vesicles or lysosomal degradation. Lysosomal degradation occurs after early endosomes mature into Rab7-positive late endosomes, which eventually fuse with lysosomes. (B) Caveolae-dependent receptor trafficking is facilitated by caveolin-1 proteins (red). Caveolae-coated vesicles are associated with dampening TGFβ signalling; however, non-canonical p38 MAPK signalling requires caveolae-coated vesicles. Caveolin-1 may bind to TGFβ receptor type I (TGFβRI) directly and attenuate its kinase activity. Caveolae-coated vesicles are enriched with Smad7-Smurf2 complexes that target TGFβ receptors to proteasome-dependent degradation. Prior to degradation, caveolae-coated vesicles may fuse with early endosomes in a Rab5-dependent manner or mature into caveolin-1-positive endosomes known as caveosomes.
Caveolae are plasma membrane invaginations enriched with caveolin-1 that are localized in membrane rafts, plasma membrane subdomains rich in cholesterol and glycosphingolipids (Golub et al., 2004). Caveolin-positive vesicles may mature into or fuse with pre-existing caveosomes or early endosomes in a Rab5-independent or -dependent manner, respectively (Pelkmans et al., 2004). Caveolin-dependent endocytosis is associated with dampening and disrupting TGFβ signalling. Unlike clathrin-coated vesicles, SARA localizes away from membrane rafts and Smad7-Smurf2 complexes are commonly associated with caveolin-positive vesicles. Due to the association with Smad7-Smurf2, TGFβRII/TGFβRI complexes within caveolin-positive vesicles are targeted for proteasomal degradation (Guglielmo et al., 2003; Le Roy and Wrana, 2005). Caveolin-1 also has been shown to directly bind to TGFβRI following stimulation, which suppresses Smad2 phosphorylation possibly by antagonizing TGFβRI kinase activity (Razani et al., 2001) (Figure 3B). Caveolin-1 also disrupts TGFβ signalling through association with CD109, a TGFβ co-receptor. In the presence of ligands, CD109 promotes the localization of TGFβ receptors in caveolae and increases receptor degradation (Bizet et al., 2011). Indeed, after the TGFβRII/TGFβRI complexes are endocytosed in caveolin-positive vesicles, TGFβ signalling is inhibited (Guglielmo et al., 2003). However, the activation of some non-Smad signalling pathways, such as p38 MAPK, rely on the localization of TGFβ receptors in caveolae (Zuo and Chen, 2009).
In summary, the route of TGFβ receptor subcellular trafficking regulates signalling duration, strength, and receptor fate (McLean and Di Guglielmo, 2014). Although some TGFβ signalling occurs in the absence of receptor internalization, clathrin- or caveolae-dependent endocytosis can enhance or dampen TGFβ signal transduction pathways (Yakymovych et al., 2018).
The role of the ubiquitin-proteasome pathway in TGFβ signalling
The ubiquitin-proteasome pathway (UPP) also regulates the strength and duration of TGFβ signalling (Wang, 2003). The polyubiquitination of TGFβ receptors, R-Smads, and downstream effectors is dependent on E1 (activating), E2 (conjugating), and E3 (ubiquitin ligase) enzymes (Kim and Baek, 2018). E1 enzymes hydrolyze ATP to activate the C terminus of ubiquitin. Activated ubiquitin is then transferred to an E2 enzyme. E3 enzymes subsequently bind to E2-ubiquitin conjugates and transfers ubiquitin to K residues on TGFβ receptors, R-Smads or downstream effectors (Komander, 2009). K48-linked polyubiquitin chains target TGFβ receptors, R-Smads, and downstream effectors to 26S proteasomes, which are multi subunit proteases (Finley et al., 2016). Deubiquitinating enzymes decrease proteasome-dependent degradation by removing ubiquitin (Kim and Baek, 2018) (Figure 4). Although ubiquitination is important for proteasome-dependent degradation, it is also necessary to facilitate signalling (Adhikari et al., 2007). For instance, K63-linked polyubiquitination functions as a scaffold to recruit and activate protein kinase complexes (Yamashita et al., 2008). As previously discussed, ubiquitin ligases catalyze K63-linked polyubiquitin chains on TRAF6 to recruit TAK1 to facilitate Smad-independent TGFβ signalling (Landström, 2010).
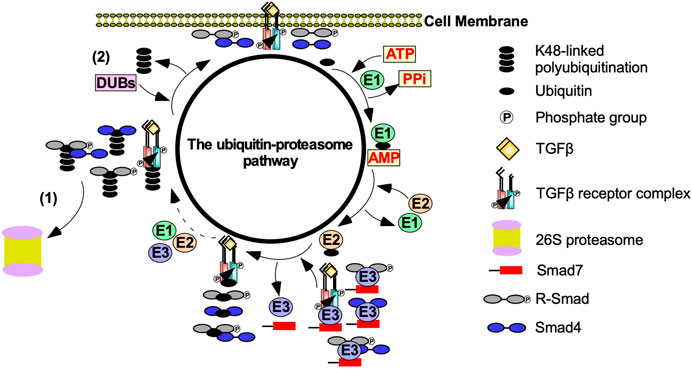
FIGURE 4. The effect of the ubiquitin-proteasome pathway on TGFβ signalling. Transforming growth factor-β (TGFβ) signalling is tightly regulated by the ubiquitin-proteasome pathway. After TGFβ binds to the TGFβ-receptors, the ubiquitin-proteasome pathway is activated to prevent uncontrolled TGFβ signalling. E1 activating enzymes hydrolases ATP to bind to ubiquitin. Ubiquitin is then transferred to an E2 conjugating enzyme. Smad7 binds to E3 ubiquitin ligases, which conjugates ubiquitin to TGFβ receptors, receptor Smads (R-Smads), Smad4, and R-Smad-Smad4 complexes. This process is repeated until TGFβ receptors, R-Smads, Smad4 or R-Smad-Smad4 complexes are polyubiquitinated. Polyubiquitinated components of the TGFβ pathway are then subject to (1) proteasome-dependent degradation or (2) the removal of the ubiquitin-linked chains mediated by deubiquitinating enzymes (DUBs).
Given that TGFβ signalling regulates a diverse set of cellular processes, modulating TGFβ signalling through a balance of ubiquitin ligase and deubiquitinating enzyme activity is important (Ten Dijke and Hill, 2004). By degrading TGFβ receptors, R-Smads, and downstream effectors, E3 ubiquitin ligases, protects cells from aberrant TGFβ signalling (Gao et al., 2009). However, there are numerous examples where ubiquitin ligases prolong TGFβ signalling. For instance, Smad2-Smurf2 complexes lead to the destruction of Ski-related protein N (SnoN) and Ski, which are protooncogenes that impede TGFβ signalling (Sun et al., 1999; Bonni et al., 2001). Arkadia, an E3 ubiquitin ligase, amplifies TGFβ signalling by ubiquitinating I-Smads (Koinuma et al., 2003). Paradoxically, if deubiquitinating enzymes remove K48-linked polyubiquitin chains on SnoN, Ski or Smad7, TGFβ signalling is dampened (Zhao et al., 2011). Therefore, ubiquitin ligases and deubiquitinating enzymes may both antagonize or promote TGFβ signalling depending on the function of the ubiquitinated protein.
Mutations in genes involved in TGFβ signalling
Alterations in the TGFβ signalling pathway due to genetic mutations are the underlying cause of various hereditary congenital malformations, as well as diseases that arise later in life (Wang et al., 2012; Saito et al., 2018). Germline mutations impair embryonic development, whereas increased susceptibility to develop cancer is associated with somatic mutations (Harradine and Akhurst, 2006). The clinical consequences of mutations in the TGFβ signalling pathway are complex, because the tumour microenvironment and TGFβ signalling vary among patients and among different tissues within the same individual (Massagué, 2008).
Germline mutations in the TGFβ signalling pathway
Genetically engineered mouse models with targeted inactivation of various TGFβ ligands have been generated to investigate the importance of TGFβ on development and viability (Glick, 2012). Tgfb1−/− mice can either succumb during mid-gestation as a result of vascular and hematopoiesis defects, or a few weeks after as a consequence of systemic inflammation (Shull et al., 1992; Kulkarni et al., 1993; Dickson et al., 1995). Death occurs shortly before, during or within minutes of birth in Tgfb2−/− mice, due to impaired cardiovascular function. These animals exhibit cardiac, craniofacial, limb, eye, inner ear, and urogenital defects (Sanford et al., 1997; Dünker and Krieglstein, 2002). Tgfb3−/− mice exhibit cleft palates that interfere with feeding, eventually resulting in death (Dünker and Krieglstein, 2002; Aluwihare et al., 2009). The majority of Smad-null mice die in utero, indicating that Smad proteins are required for proper embryonic development as previously reviewed (Datto and Wang, 2000). Specifically, Smad2−/− and Smad4−/− mice die early in embryogenesis, due to defects in the organization of the primitive germ layers and extensive mesodermal defects (Nomura and Li, 1998; Chu et al., 2004). Smad3−/− mice are viable, but exhibit impaired local inflammatory responses and accelerated wound healing (Ashcroft et al., 1999; Ling and Robinson, 2002).
In patients, familial juvenile polyposis, which increases the risk of gastrointestinal cancer, is correlated with SMAD4 mutants that produce truncated proteins with a loss or partial loss of function (Howe et al., 1998; Johansson et al., 2015). Although juvenile polyposis patients have been screened for SMAD2 and SMAD3 mutations, only SMAD4 germline mutants are identified as an underlying cause of juvenile polyposis (Bevan et al., 1999). However, screening colorectal adenoma patients revealed that mutations to the SMAD4 loci are rare (Lipton et al., 2003). SMAD4 mutations in patients with juvenile polyposis syndrome may also develop hereditary hemorrhagic telangiectasia, which results in abnormal vascular structures (Heald et al., 2015).
Somatic mutations in the TGFβ signalling pathway
Frameshift and missense mutations in TGFBRI are common in several tumour types (Moore-Smith and Pasche, 2011). For example, the TGFBRI*6A mutation in exon one is a loss of three Alanine residues in a 9-Alanine repeat region that increases cancer susceptibility associated with impaired anti-proliferative TGFβ signalling (Liao et al., 2010). Inactivating mutations in TGFBR2 are frequently present in tumours that exhibit microsatellite instability (Vincent et al., 1996), such as those found in subsets of colon carcinomas, which express truncated mutant forms of TGFβR2 (Ogino et al., 2007). SMAD4 is the most common Smad family gene mutated in malignant tumours (Sarshekeh et al., 2017). Inactivating SMAD4 mutations have been found in approximately 50% of pancreatic adenocarcinomas (Howe et al., 1998), 20% of colorectal carcinomas (Chu et al., 2004), and 5% of head and neck squamous cell carcinomas (Lin et al., 2019a). Smad4 mutations also correlate with tumour formation (Lin et al., 2019b) and may predict poor prognosis and aggressive tumour phenotypes (Fang et al., 2021). For instance, mice with conditional targeted inactivation of Smad4 in the oral epithelium developed spontaneous squamous cell carcinomas (Bornstein et al., 2009). Although somatic mutations of the TGFβ pathway may promote tumour formation, similar mutations in cancerous cells that rely on TGFβ can decrease tumour growth (Pino et al., 2010). Since somatic mutations of the TGFβ pathway may promote or block tumourigenesis depending on the stage of the disease, this is important to bear in mind when assessing the benefits and risks of using TGFβ signalling inhibitors in cancer treatment (Khoshakhlagh et al., 2019).
TGFβ signalling in tumourigenesis
Cells escape the tumour suppressing arms of TGFβ signalling through mutations that impede specific TGFβ pathways or abnormalities in processes that dampen TGFβ signalling (David and Massagué, 2018). Functional inactivation of the tumour suppressing arms of TGFβ signalling can contribute to carcinogenesis through various mechanisms (Massagué, 2008; David and Massagué, 2018). Major mechanisms that contribute to the pro-tumourigenic effects of TGFβ include inhibition of immune function, activation of angiogenesis/lymphangiogenesis, and the initiation of EMT (Ferrari et al., 2009; Flavell et al., 2010; Batlle and Massagué, 2019).
Inhibition of anti-cancer immune responses
As prolonged activation of the immune system can induce inflammation and tissue damage, the immune system is modulated through inhibitory mechanisms (Sitkovsky and Ohta, 2005). Cells in the tumour and its microenvironment benefit from these immunological safeguards by producing excessive amounts of immunosuppressive cytokines, such as TGFβ (Flavell et al., 2010; Batlle and Massagué, 2019). TGFβ inhibits many components of both the innate and adaptive immune systems, which creates an environment favourable for tumour growth (Moo-Young et al., 2009).
Tumour cells are targeted for destruction by cells of the innate immune system, which include monocytes, macrophages, dendritic cells, neutrophils, basophils, eosinophils, and NK cells (Gajewski et al., 2013). Through phagocytosis, macrophages, neutrophils, and dendritic cells engulf tumour cell debris and tumour cells missing essential cell surface proteins or expressing danger signals (Chan and Housseau, 2008; Sarode and Sarode, 2014; Zhou et al., 2021). Macrophages, neutrophils, and dendritic cells also attach antigens to their major histocompatibility complexes (MHCs) to activate T- and B- lymphocytes (T- and B-cells) of the adaptive immune system (Figure 5) (Gajewski et al., 2013). The effects of TGFβ on dendritic cells include interference with antigen presenting activity, immobilization, and upregulation of TGFβ production, creating a positive feedback loop to maintain a decrease in immune responses against the tumour (Esebanmen and Langridge, 2017). Furthermore, by interfering with dendritic cell antigen presenting activity, TGFβ blocks naive T-cell and B-cell differentiation into anti-tumour phenotypes (Liu et al., 2018). TGFβ within the tumour microenvironment may manipulate macrophages and neutrophils to differentiate into phenotypes that contributes to tumour growth rather than destroy tumour cells. These macrophages and neutrophils are typically referred to as tumour-associated macrophages (TAMs) and tumour-associated neutrophils (TANs), respectively (Fridlender et al., 2009; Danhier et al., 2017). TGFβ-recruited TAMs can phagocytose antigen-containing particles prior to their recognition by dendritic cells. Therefore, TAMs suppress the antigen presenting abilities of dendritic cells, hindering activation of the adaptive immune system (Liu et al., 2018; Batlle and Massagué, 2019). TGFβ recruited TANs have decreased cytotoxicity and secrete extensive quantities of MMPs to free TGFβ from large latent TGFβ complexes, which increases the concentration of active TGFβ ligands in the tumour microenvironment, contributing to a positive feedback loop (Figure 6) (Germann et al., 2020).
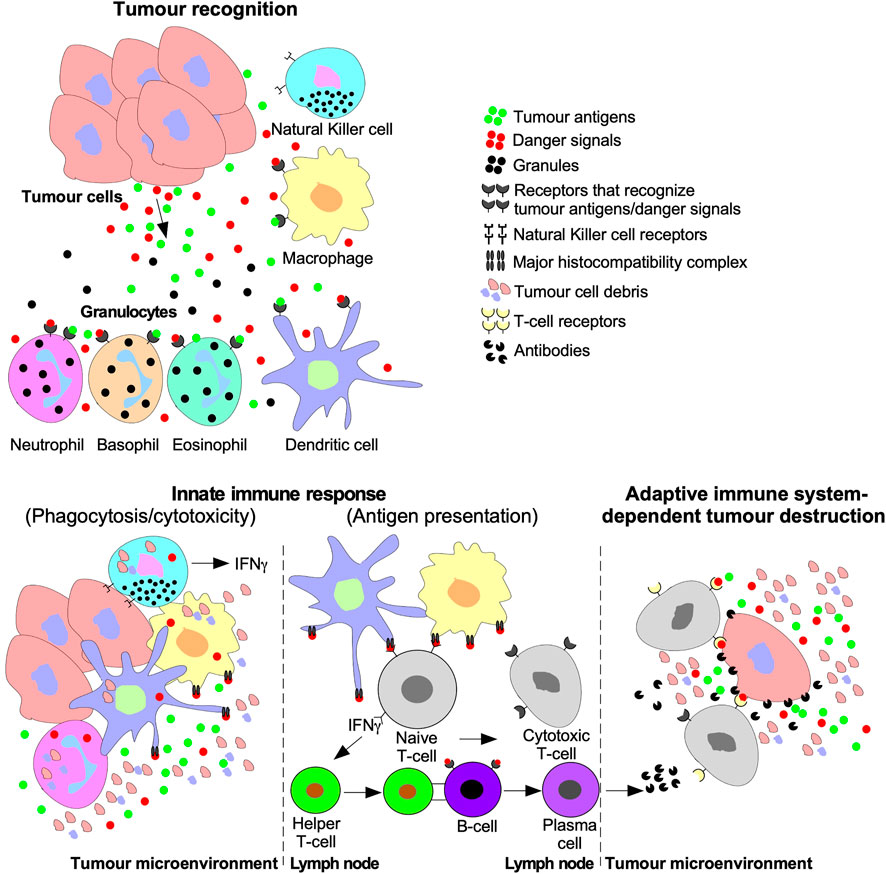
FIGURE 5. Tumour recognition and destruction mediated by the innate and adaptive immune systems. Tumour cells release antigens and danger signals that serve as a chemotactic gradient to recruit cells of the innate immune system ((Natural Killer (NK) cells, macrophages, dendritic cells, and granulocytes (neutrophils, basophils, and eosinophils)). Cells of the innate immune system may destroy tumours using cytolytic/phagocytic functions or activate the adaptive immune system. The adaptive immune system is activated by humoral signals, such as interferon-γ (IFNγ), which is released by NK cells, dendritic cells, and macrophages. Furthermore, antigen presenting macrophages and dendritic cells deliver tumour antigens using the major histocompatibility complex to Naive T-lymphocytes (T-cells) or B-lymphocytes (B-cells). Naive T-cells are stimulated to differentiate into Cytotoxic T-cells and Helper T-cells. B-cell differentiation into cytotoxic antibody-producing plasma cells is triggered by B-cell receptors binding to Helper T-cells or tumour antigens. The adaptive immune system facilitates tumour destruction via Cytotoxic T-cells releasing enzymes into tumour cells or antibodies produced by plasma cells.
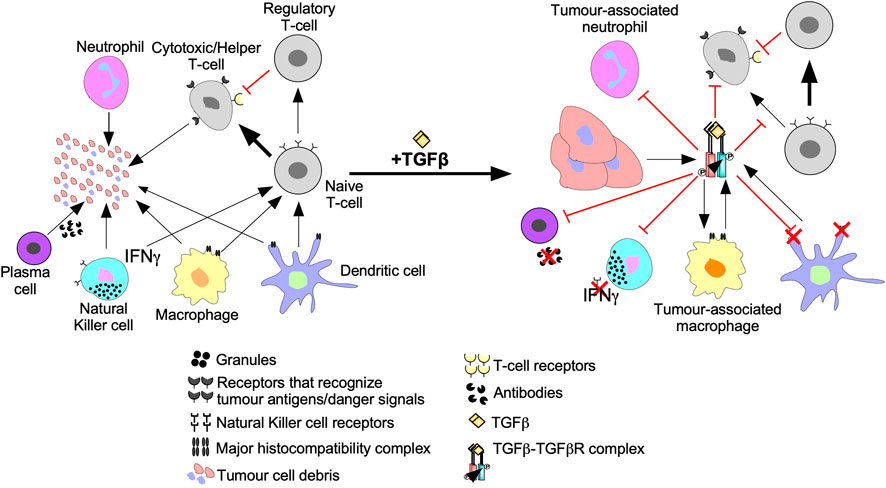
FIGURE 6. The inhibition of anti-cancer immune responses by TGFβ. In the absence of immunosuppressive cytokines, cells of the innate and adaptive immune system destroy tumour cells as described in Figure 7. However, the addition of transforming growth factor-β (TGFβ) suppresses tumour recognition and cytotoxic functions of the innate and adaptive immune systems. For instance, TGFβ suppresses antigen presenting function of macrophages, neutrophils, and dendritic cells by downregulation the major histocompatibility complex. TGFβ decreases Natural Killer cell receptors, which spares tumour cells from Natural Killer cell-mediated destruction. TGFβ dampens immune cell recruitment by disrupting interferon-γ (IFNγ) production in Natural Killer cells, dendritic cells, and macrophages. TGFβ induces macrophages and neutrophils to differentiate into tumour-associated macrophages and tumour-associated neutrophils, respectively, which augment tumourigenesis. TGFβ disrupts B-cell differentiation into plasma cells and attenuates antibody production. TGFβ also alters Naive T-cell differentiation to favour tumour promoting Regulatory T-cells instead of Cytotoxic T-cells or Helper T-cells that mediate tumour cell destruction. Regulatory T-cells promote tumourigenesis by suppressing Cytotoxic T-cell function.
NK cells are specialized leukocytes that do not rely on MHCs or humoral signals to recognize tumour cells (Abel et al., 2018). Instead, NK cells recognize tumour cells using cell surface receptors. Upon binding to tumour cells, NK cells release interferon-γ (IFNγ) into the tumour microenvironment and cytolytic antibodies directly into the tumour cell (Castro et al., 2018). Thus, NK cells eliminate tumour cells by triggering an antibody-dependent cell-mediated cytotoxic response and activate other leukocytes using IFNγ (Figure 5) (Abel et al., 2018). TGFβ blocks NK cell-mediated adaptive immune system activation by downregulating the transcription factor T-bet, leading to reduced IFNγ expression (Hayashi et al., 2003; Mohammadzadeh et al., 2014). The TGFβ-dependent loss of IFNγ decreases the activity of leukocytes, downregulates antigen presenting MHCs in antigen presenting leukocytes, and impedes chemotaxis (Castro et al., 2018). TGFβ also downregulates NK receptors responsible for recognizing and destroying tumour cells (Figure 6) (Castriconi et al., 2003).
Like the innate immune system, the adaptive immune system facilitates tumour cell death using humoral immunity and cell-mediated immunity. Cell-mediated immunity and humoral immunity is facilitated by T-cells. Following antigen presentation, naive T-cells differentiate into effector T-cells, such as cytotoxic T-cells and helper T-cells (Fazilleau et al., 2009; Farhood et al., 2019). Cytotoxic T-cells specifically eliminate cells expressing the antigen presented whereas helper T-cells release humoral signals to activate other leukocytes (Figure 5) (Belardelli and Ferrantini, 2002; Fazilleau et al., 2009). In tumour microenvironments with elevated TGFβ levels, decreased numbers and limited anti-tumour cytolytic activity of cytotoxic T-cells have been observed, through mechanisms that include induction of T-cell apoptosis (Thomas and Massagué, 2005; Flavell et al., 2010; Liu et al., 2018). TGFβ also disrupts T-cell anti-tumourigenic activity by upregulating genes that promote naive T-cell differentiation into less cytotoxic phenotypes, such as Tregs (Figure 6) (Zhang et al., 2018). Plasma cells are adaptive immune system cells that mediate humoral immunity. Upon antigen presentation, B-cells differentiate into plasma cells that produce antibodies to eliminate tumour cells (Figure 5) (Kurosaki et al., 2015). TGFβ attenuates the anti-tumourigenic capacity of B-cells by interfering with their differentiation into plasma cells, antibody production, and proliferation (Figure 6) (Schwartz et al., 2016).
Activation of angiogenesis and lymphangiogenesis
Angiogenesis promotes tumour growth and invasion because as tumours grow, blood carrying oxygen and nutrients is blocked from reaching interior tumour cells (Nishida et al., 2006). To bypass this, tumour microenvironments are enriched with cytokines, such as TGFβ, that alter cellular processes within endothelial cells and mural cells to generate new vessels (Figure 7A) (Ferrari et al., 2009). The effects of TGFβ on angiogenesis, endothelial cells, and on mural cells are complex. Although in normal vessels TGFβ supports vascular development by recruiting mural cells toward endothelial cells (Walshe et al., 2009), TGFβ in tumour vasculature induces the differentiation of endothelial cells into mural cells (Hirschi et al., 2003). Then, mural cells secrete angiogenic factors and form defective interactions with endothelial cells resulting in disorganized vasculature (Sun et al., 2021). In endothelial cells, binding of TGFβ to TGFBRII leads to the activation of two distinct type I receptors: endothelial cell-specific activin receptor-like kinase 1, which signals through Smad1/5/8, as well as the ubiquitous TGFβRI, which signals through Smad2/3 (COLLETTA et al., 1988; Goumans et al., 2002; Mallet et al., 2006; Ito et al., 2009). Smad1/5/8 signalling induces endothelial cell proliferation and migration (Ray et al., 2010), whereas Smad2/3 signalling induces endothelial cell differentiation into mesenchymal-like mural cells (Hirschi et al., 2003; Jiang et al., 2018). TGFβ can promote angiogenesis through TGFβRI, but inhibits growth factor-induced endothelial sprouting/branching through mechanisms that involve cross-talk with Notch-activated pathways (Mallet et al., 2006; Aspalter et al., 2015). In mural cells and endothelial cells, TGFβ also induces Smad-dependent expression of vascular endothelial growth factor (VEGF), thrombospondin-4 (TSP-4), MMPs, microRNA-29a, and other genes that stimulate endothelial cell proliferation and migration (Massagué, 2008; Ferrari et al., 2009).
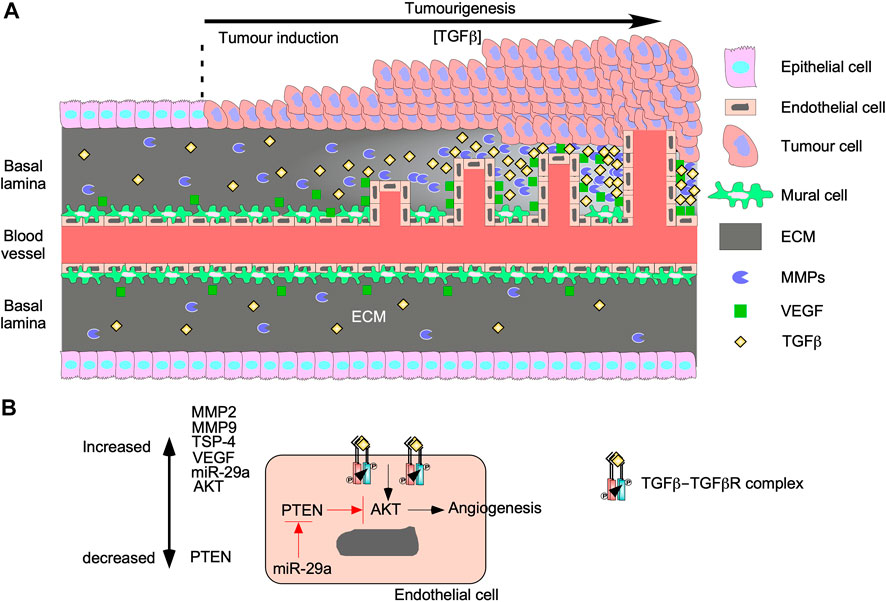
FIGURE 7. TGFβ augments tumourigenesis by inducing angiogenesis. (A) As tumours grow, the concentration of transforming growth factor-β (TGFβ) in the tumour microenvironment increases. TGFβ upregulates genes involved with proliferation and migration in mural cells, which results in endothelial cell migration and leaky vessels. TGFβ binds to TGFβ receptors on endothelial cells to upregulate vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs). Both proteins are secreted into the basal lamina and increase proportionally to TGFβ. VEGF binds to endothelial cells and stimulates proliferation and migration. MMPs breakdown proteins in the basal lamina to remodel the extracellular matrix (ECM) to carve out space for vessel formation. The new vessels grow and become more organized as time passes. (B) TGFβ binds to TGFβ receptors on endothelial cells and upregulate MMP2, MMP9, thrombospondin-4 (TSP-4), microRNA-29s (miR-29a), VEGF, and protein kinase B (AKT). miR-29a blocks the translation of phosphatase and tensin homolog (PTEN), which is a known AKT inhibitor. Since the AKT pathway has been linked to angiogenesis, TGFβ signalling may induce angiogenesis through the AKT pathway.
VEGF enhances endothelial cell migration, proliferation, and resistance to apoptosis (Ferrari et al., 2009; Suzuki et al., 2012) by activating two tyrosine kinase VEGF receptors (VEGFR1 and VEGFR2). VEGFR1 activation is involved with migration whereas VEGFR2 activation regulates proliferation and survival (Wang et al., 2017). Interestingly, TGFβ activates apoptosis, which suggests that VEGF and TGFβ have opposing roles on endothelial cell survival. However, many studies suggest that pro-apoptotic TGFβ signalling is necessary for angiogenesis because it ensures less branching and increases vasculature organization (Haque and Morris, 2017). Furthermore, TGFβ upregulates ECM remodelling proteins in endothelial cells, such as TSP-4 and MMPs (Tirino et al., 2013; Muppala et al., 2017). By a Smad3-dependent mechanism, TGFβ activates post-translation processes that increase TSP-4 protein levels (Muppala et al., 2017). The importance of TSP-4 on endothelial cell proliferation and migration during angiogenesis was verified when TGFβ-induced angiogenesis was attenuated in Tsp-4−/− mouse models (Muppala et al., 2017). Additionally, TGFβ upregulates the expression of MMP2 and MMP9 in endothelial cells and cells of the tumour microenvironment, thus facilitating ECM remodelling and releasing ECM-sequestered cytokines (Yu and Stamenkovic, 2000). Therefore, MMPs play a role in TGFβ-mediated angiogenesis by releasing latent TGFβ from LAP and LTBP (Tatti et al., 2008) as well as generating the space required for endothelial cell migration, proliferation, and microvessel formation (Park et al., 2018). Finally, microRNA-29a silences phosphatase and tensin homolog (PTEN) RNA expression (Wang et al., 2013), leading to increased AKT pathway activity and activation of TGFβ-induced angiogenesis (Chen et al., 2020). Since blocking PTEN activity increases the activity of the AKT pathway (Chen et al., 2020), the Smad-independent PI3K/AKT TGFβ signalling pathway may play a major role in TGFβ-induced angiogenesis (Figure 7B).
Tumour cells primarily metastasize through the lymphatic system due to the thinner walls and increased permeability of lymphatic vessels, relative to blood vasculature (Chaffer et al., 2016). Furthermore, cancer cells may drain directly into the lymphatic system if they break free from tumours (Karlsson et al., 2017). Two mechanisms for TGFβ contribution to metastasis through the lymphatic system have been proposed. Due to the greater representation of leukocytes in the lymphatic system, lymph node metastasis requires immune suppression (Liu and Cao, 2016). Therefore, the inhibitory effects of TGFβ on leukocytes present in the lymphatic system may promote tumour cell survival and increases dissemination (Liu and Cao, 2016). Additionally, Smad-dependent and -independent TGFβ signalling induces lymphangiogenesis, formation of new lymphatic vessels from pre-existing lymphatic vessels (García-Caballero et al., 2017), by upregulating VEGF-C, which in turn promotes growth, proliferation, migration, and survival of endothelial cells bordering lymphatic vessels (Pak et al., 2019). Cells of the tumour microenvironment that respond to TGFβ, such as TAMs, may also mediate lymphangiogenesis via a VEGF receptor 3-dependent process (Alishekevitz et al., 2016).
Epithelial-mesenchymal transition (EMT)
Epithelial-mesenchymal transition (EMT), a biological process whereby cells of epithelial origin acquire characteristics of mesenchymal cells, is essential for embryogenesis and wound healing (Tan et al., 2015; Chaffer et al., 2016). EMT is involved in the ability of carcinoma cells to acquire motile and invasive phenotypes, thus contributing to tumour progression and metastasis (Craene and Berx, 2013). During EMT, there is a loss of epithelial properties, such as apical/basolateral polarity, cytoskeleton polarization, cell-cell adhesions (adherens junctions, tight junctions, and gap junctions), and attachment to the basal lamina. Subsequently, the cells acquire spindle-shaped morphology, transient focal point cell-cell attachments, lamellipodia/filopodia formation, front-back polarity, stress fibers, and increased motility (Figure 8) (Chaffer et al., 2016; Karlsson et al., 2017).
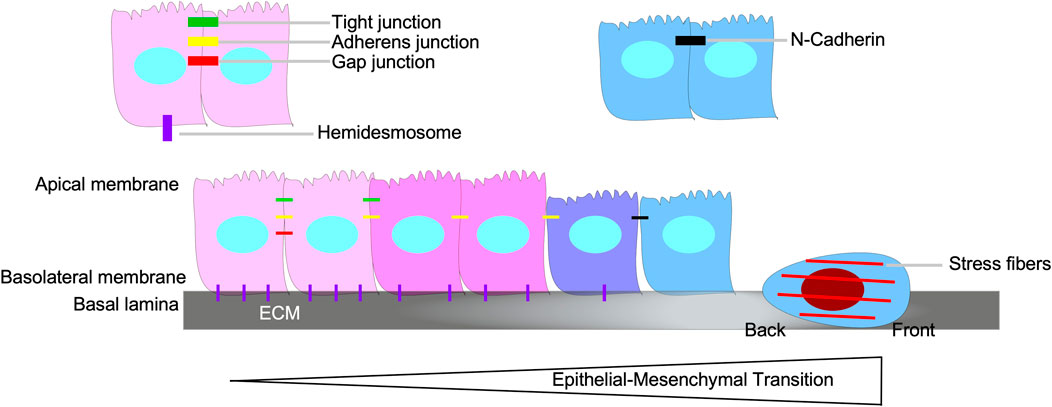
FIGURE 8. Epithelial-mesenchymal transition. Epithelial-mesenchymal transition (EMT) is the biological process of an epithelial cell loses its epithelial properties, such as apical/basolateral polarity, tight junctions, gap junctions, adherens junctions, and hemidesmosomes, and develop mesenchymal properties, which includes the capacity to breakdown the basal lamina, assert back/front polarity, spindle-shaped morphology, induce stress fiber formation, and N-Cadherin-dependent cell-cell attachments.
The profound phenotypical and morphological characteristics observed during EMT are amplified by signals that tumour cells receive from the tumour microenvironment, such as TGFβ (Kawata et al., 2012). TGFβ contributes to the initiation of the EMT program, via transcription-dependent and -independent mechanisms (Gunaratne and DiGuglielmo, 2013; Tirino et al., 2013; Ganesan et al., 2016; Tripathi et al., 2019). TGFβ upregulates various EMT-transcription factors (SNAIL, SLUG, TWIST, ZEB1, ZEB2, FOXC2, FOXA1, FOXA2, PRX1, and HMGA2), which decrease the expression of epithelial genes, whilst increasing that of mesenchymal genes (Figure 9) (Barrallo-Gimeno and Nieto, 2005; Kokudo et al., 2008; Kume, 2008; Miyazono, 2009; Xu et al., 2009; Mikheeva et al., 2010; Lee and Yutzey, 2011; Wu et al., 2011; Kaufhold and Bonavida, 2014; Ganesan et al., 2016; Niu et al., 2016; Katsura et al., 2017; Vu and Datta, 2017; Maturi et al., 2018; Atala, 2019; Stemmler et al., 2019). For example, SNAIL, SLUG, and ZEB1 downregulate the expression of E-Cadherin, a protein required for strong adherens junctions observed in epithelial cells, whereas TWIST upregulates the expression of N-Cadherin, a mesenchymal protein that forms weak transient cell-cell interactions (Barrallo-Gimeno and Nieto, 2005; Mikheeva et al., 2010; Dhasarathy et al., 2011; Lee and Yutzey, 2011; Kaufhold and Bonavida, 2014; Maturi et al., 2018). An in-depth analysis of genes targeted by EMT-transcription factors that mediate the transition of epithelial to mesenchymal phenotypes are outlined in previous reviews (Wrana, 2013; Batlle and Massagué, 2019).
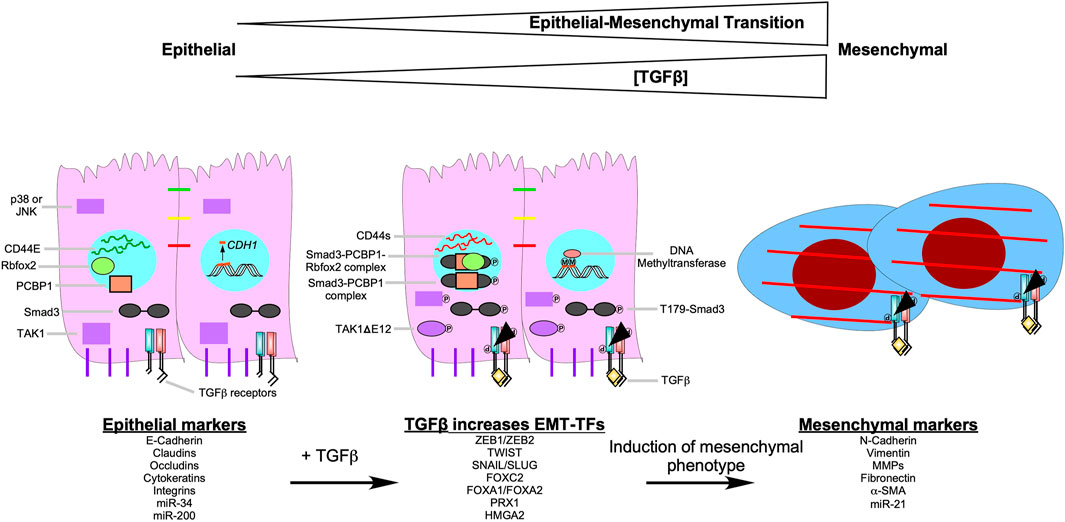
FIGURE 9. TGFβ signalling pathways that induce epithelial-mesenchymal transition. As the concentration of transforming growth factor-β (TGFβ) increases, the epithelial-mesenchymal transition (EMT) program becomes more pronounced. After TGFβ binds to the TGFβ receptors, it upregulates EMT-transcription factors (EMT-TFs), such as Snail Family Transcriptional Repressor one and 2 (SNAIL/SLUG), Zinc Finger E-box Binding Homeobox one and 2 (ZEB1/ZEB2), Twist-related Protein 1 (TWIST1), Forkhead box C2 (FOXC2), Forkhead box A1 (FOXA1), Forkhead box A2 (FOXA2), Paired-related Homeobox 1 (PRX1), and High Mobility Group AT-hook 2 (HMGA2). EMT-TFs downregulate epithelial markers ((E-Cadherin, claudins, occludins, cytokeratins, integrins, microRNA (miR)-34, and miR-200)) and upregulate mesenchymal markers ((N-Cadherin, vimentin, matrix metalloproteinases (MMPs), fibronectin, α-smooth muscle actin (α-SMA), and miR-21)). TGFβ induces EMT by increasing DNA methyltransferase activity. In the presence of TGFβ, DNA methyltransferase methylates (M) the promoters of epithelial genes, such as Cadherin 1(CDH1). Also, when TGFβ receptor type I phosphorylates Smad3 at threonine 179 (T179-Smad3), it may associate with the RNA-binding protein poly (RC) binding protein 1 (PCBP1). Smad3-PCBP1 complexes alter CD44 splicing from CD44E, which is found in epithelial cells, to CD44s. CD44s splice variants modulate cell-cell adhesion to promote EMT. The Smad3-PCBP1 complex associated with Rbfox2 that mediates alternative splicing of TGFβ-activated kinase 1 (TAK1) to favour TAK1ΔGlu 12 (TAK1ΔE12) variants. TAK1ΔE12 is constitutively active, which leads to the constitutive phosphorylation of p38 MAPK (p38) and cJun N-terminal Kinase (JNK). P38 and JNK upregulate genes that promote EMT.
TGFβ can promote EMT through non-canonical, Smad3-dependent regulation of RNA splicing. Phosphorylation of Smad3 on Thr179, subsequent to TGFβ receptor stimulation, impairs binding to Smad4 and to DNA (Gao et al., 2009; Inui et al., 2011; Tang et al., 2011), but induces Smad3 association with the RNA-binding protein poly (RC) binding protein 1 (PCBP1) in the nucleus (Tripathi and Zhang, 2017). The Smad3-PCBP1 species catalyzes alternative splicing of myriad transcripts involved in EMT, including RNAs encoding the CD44 glycoprotein, which modulates cell-cell adhesion (Ponta et al., 2003). Multiple CD44 splice variants exist. CD44E is preferentially expressed in normal epithelial cells, whereas the mesenchymal isoform CD44s is ubiquitous. In epithelial carcinoma cells, Smad3-PCBP1 complexes induce a splicing switch from CD44E to CD44s, resulting in activation of EMT and invasion (Thomas and Massagué, 2005). Similarly, complex formation between Smad3, PCBP1, and the RNA-binding protein Rbfox2 mediates expression of the alternative TAK1 splice variant TAK1ΔGlu 12 (TAK1ΔE12) (Braeutigam et al., 2014). TAK1ΔE12 is constitutively active, which means downstream signalling kinases, such as p38 MAPK and JNK, are constitutively phosphorylated (Yamashita et al., 2008; Tripathi et al., 2019). Transcription factors regulated by p38 MAPK and JNK are involved with upregulating genes that promote proliferation and EMT (Figure 9) (Zhao et al., 2017).
Finally, TGFβ can also promote EMT by upregulating DNA methyltransferases, which hypermethylate promoters of various genes involved in the regulation of the cell cycle, apoptosis, cell-cell attachments, ECM production, and cell movement (Lu et al., 2017). For example, in ovarian carcinoma cells, reduced transcription of CDH1, which encodes E-Cadherin, is associated with hypermethylation in the presence of TGFβ (Figure 9) (Cardenas et al., 2014).
Similar to EMT, endothelial-mesenchymal transition (EndMT) occurs when endothelial cells lose tight junctions and downregulate various endothelial cell markers, such as VE-Cadherin, to acquire mesenchymal properties, including expression of α-smooth muscle actin and N-Cadherin (Hong et al., 2018). EndMT is important during cardiac development and wound healing, and is believed to be an important contributor to certain pathologies (Lin et al., 2012). EndMT has been described in cardiovascular pathologies, such as atherosclerosis, cardiac fibrosis, and pulmonary hypertension (Jimenez and Piera-Velazquez, 2016). Recently, evidence has emerged that some cancer-associated fibroblasts (CAFs) have an endothelial origin (Zeisberg et al., 2007). These CAFs express α-smooth muscle actin and type I collagen, which are markers associated with excessive scarring and ECM remodelling (Yeon et al., 2018). A pathway linking TGFβ to EndMT involves TGFβ-mediated upregulation of SNAIL, which in turn induces downregulation of VE-Cadherin (Platel et al., 2019). Additionally, when TGFβ-dependent ERK phosphorylation was blocked, TGFβ-dependent EndMT was attenuated (Wylie-Sears et al., 2014).
There are several factors involved with TGFβ-dependent EMT/EndMT regulation. First, the chromatin structure and epigenetics of a cell dictate if SNAIL and other transcription factors can access genes subject to their regulation (Millanes-Romero et al., 2013; Kaufhold and Bonavida, 2014). Second, miRNAs block the expression of EMT/EndMT-transcription factors. For instance, microRNA-34 and microRNA-200 prevent the translation of SNAIL and ZEB1, respectively (Chaffer et al., 2016; Imani et al., 2017; Title et al., 2018). Finally, each cell type has different intracellular signalling configurations. Therefore, the rate in which different cell types conduct Smad-dependent or -independent signalling is not the same (Wu et al., 2016). In conclusion, cells that upregulate microRNAs that block EMT/EndMT-transcription factor translation, contain DNA methylation in the promoters of genes regulated by EMT/EndMT-transcription factors, and favour tumour suppressive TGFβ pathways are less likely to undergo TGFβ-dependent EMT/EndMT.
The relationship between autophagy and the tumour promoting properties of TGFβ
Immunosuppression, increased angiogenesis, and EMT are the most widely studied mechanisms whereby TGFβ promotes tumourigenesis. However, the pro-tumourigenic activity of TGFβ likely includes additional biological processes, such as autophagy (Suzuki et al., 2010). Autophagy, Greek for self-devouring, is a catabolic process where cells degrade and recycle their own macromolecules and organelles primarily via lysosomes (Kaur and Debnath, 2015). Autophagy is essential for recycling the building blocks of lipids, carbohydrates, and proteins as well as eliminating invading pathogens, protein aggregates, and damaged organelles (Bernard and Klionsky, 2013). Although autophagy is primarily facilitated by lysosomes, which are acidic organelles that contain luminal degradative hydrolases, other acidic vesicles, such as late endosomes, contribute to autophagic degradation (Lawrence and Zoncu, 2019).
The idea that TGFβ-dependent tumourigenesis may rely on autophagy is supported by the extensive roles that autophagy plays in tumour development, maintenance, and metastasis (Mathew et al., 2007). Similar to TGFβ, the tumour regulatory consequences of autophagy are context dependent, as autophagy can result in either tumour suppression or promotion, depending on the stage of tumour development (Kiyono et al., 2009; Glick et al., 2010). In non-cancerous tissues, autophagy functions as a homeostatic safeguard by removing protein aggregates, damaged organelles, and other metabolic stressors, all of which protects against neoplastic transformation (Mathew et al., 2009; Klionsky et al., 2016). However, autophagy participates in the survival of established tumour cells under conditions of hypoxia, oxidative damage, metabolic stress, and starvation. Furthermore, cancer cells with elevated rates of autophagy tend to grow more rapidly and are prone to metastasize (Kiyono et al., 2009; Rebecca and Amaravadi, 2016; Alizadeh et al., 2018). Autophagy has been linked to EMT, MMP secretion, angiogenesis, evasion of immune surveillance, promigratory cytokine secretion, anoikis resistance, and stemness in tumour cells (Mowers et al., 2017). Autophagy has also been implicated in resistance to chemotherapeutic agents that target rapidly dividing cells, because it promotes tumour cell dormancy (Table 1) (O’Donovan et al., 2011). Accordingly, silencing of autophagic proteins can increase the efficacy of chemotherapeutic agents (Zhang et al., 2015). Autophagy can also improve survival of circulating tumour cells and establishment of the pre-metastatic niche (Mowers et al., 2017), as well as increase tumour cell survival after metastasis (Pavlides et al., 2012; Rebecca and Amaravadi, 2016). Overall, autophagy plays important roles in the regulation of EMT, immune surveillance, and angiogenesis (Suzuki et al., 2010; Tuloup-Minguez et al., 2013; Alizadeh et al., 2018; Wu et al., 2018; Losier et al., 2019).
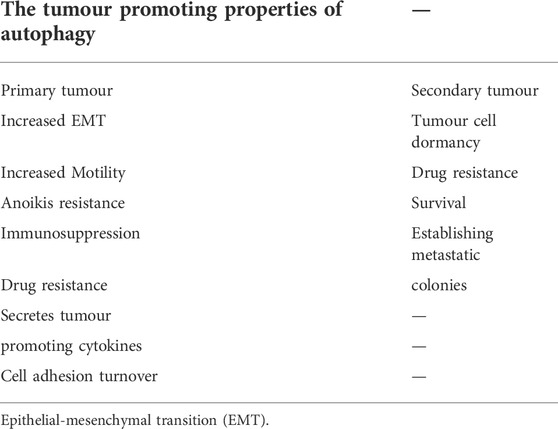
TABLE 1. The tumour promoting properties of autophagy.
Mechanism of TGFβ-induced autophagy
Both Smad-dependent and -independent TGFβ signalling can contribute to increases in the rate of autophagy (i.e. autophagic flux). Smad-dependent signalling activates transcription of genes essential to autophagy, such as autophagy-related gene (ATG)5, ATG7, BECLIN1, and DAPK1 (Figure 10A) (Suzuki et al., 2010; Ma et al., 2017). TGFβ can also increase steady-state levels of beclin1, autophagy-related protein (Atg)7, Atg5, uncoordinated 51-like autophagy activating kinase 1 (ULK1), and microtubule-associated protein light chain 3-II (LC3-II) (Xu et al., 2012; Trelford and Guglielmo, 2020). Non-canonical TAK1-mediated TGFβ signalling has also been implicated in regulation of autophagy. Specifically, TGFβ induces phosphorylation and activation of 5’ adenosine monophosphate-activated protein kinase (AMPK) by TAK1 (Herrero-Martín et al., 2009), thereby increasing autophagy as AMPK activates ULK1 and suppresses mTOR (Mcalpine et al., 2013). mTOR antagonizes autophagy through the addition of an inhibitory phosphate to ULK1, which prevents the formation of the autophagy initiating ULK1 complex (Makhov et al., 2014). TAK1 and JNK signalling have also been linked to increased steady-state levels of LC3 and beclin1. LC3 and beclin1 steady-state levels are correlated to the number of autophagosomes, double membrane vesicles that sequester cellular cargo prior to fusing with lysosomes, and increased lysosomal degradation (Figure 10B) (Shin et al., 2013). In support of this, TGFβ increases autophagosomes production, LC3 co-localization with autophagosomes or lysosomes, and autophagosome-lysosome fusion in a variety of cell types (Figure 10C&D) (Alizadeh et al., 2018; Trelford and Guglielmo, 2020).
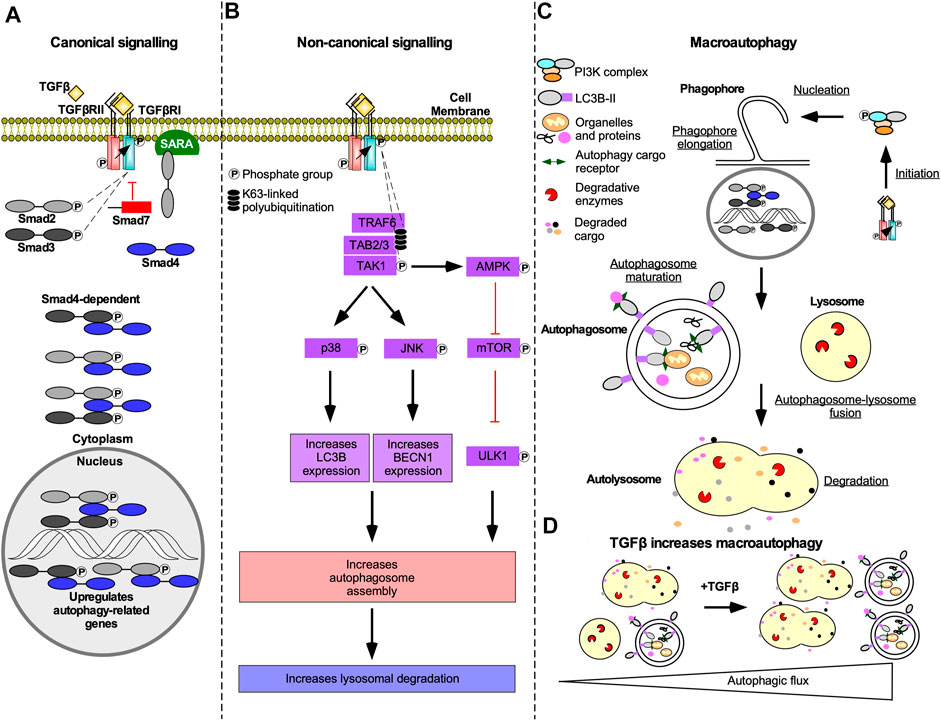
FIGURE 10. The mechanism of TGFβ-dependent autophagy. (A) In Smad-dependent transforming growth factor-β (TGFβ) signalling described in Figure 2, phosphorylated receptor Smads (R-Smads) enter the nucleus with Smad4 and upregulate genes essential to autophagy. Although R-Smad transcription factors may function independently of Smad4, Smad4 knockdown blocked TGFβ-dependent autophagy. (B) In Smad-independent TGFβ signalling described in Figure 4, polyubiquitination of tumour necrosis factor receptor-associated factor 6 (TRAF6) recruits TGFβ-activated kinase 1 (TAK1) binding proteins two and 3 (TAB2/3), which leads to TAK1 phosphorylation. Phosphorylated TAK1 activates p38 mitogen-activated protein kinase (p38) and c-Jun amino-terminal kinase (JNK) that phosphorylate several transcription factors that upregulate microtubule-associated protein light chain 3B (LC3B) and beclin1 (BECN1) expression, respectively. TAK1 also phosphorylates 5’ adenosine monophosphate-activated protein kinase (AMPK), which is an inhibitor of an autophagy suppressor called mechanistic target of rapamycin (mTOR). mTOR suppresses autophagy by adding an inhibitory phosphate to uncoordinated-51-like autophagy activating protein kinase 1 (ULK1). LC3B, BECN1, and ULK1 promote autophagosome assembly, which may increase lysosomal-dependent degradation. (C) Both Smad-dependent and -independent TGFβ signalling induces macroautophagy. Macroautophagy is initiated when complexes containing ULK1 phosphorylate beclin1. Beclin1 is then primed to form protein complexes that are recruited to the rough endoplasmic reticulum membrane to nucleate phagophores. As the phagophore membranes are elongated with lipids and LC3B, cargo proteins, and organelles, such as mitochondria, are sequestered within autophagosomes. Once phagophore assembly is complete, it forms a mature double membrane vesicle called an autophagosomes. Autophagosomes fuse with lysosomes to generate autolysosomes. The autophagosomes and cargo are degraded by lysosomal proteases. (D) Schematic illustrating that in the absence of TGFβ there are few autophagosomes and autolysosomes. In the presence of TGFβ, the number of autophagosomes and autolysosomes is increased.
In non-small cell lung cancer cells transfected with a pMRX-IP-green fluorescent protein (GFP)-LC3-red fluorescent protein (RFP)-LC3ΔGly construct, TGFβ decreased the GFP/RFP ratio, which verified that TGFβ upregulated autophagic flux (Trelford and Guglielmo, 2020). However, the TGFβ-dependent increase in autophagic flux was attenuated by Smad4 knockdown or TAK1/TRAF6/p38 MAPK pathway disruption (Trelford and Di Guglielmo, 2021). In the same cell line system, TGFβ increased the proportion of phosphorylated ULK1 mediated by AMPK and further investigation showed that ULK1 inhibition blocked TGFβ-dependent autophagy (Trelford and Di Guglielmo, 2021; Trelford and Guglielmo, 2021). In summary, Smad-dependent and -independent TGFβ signalling activate autophagy in a ULK1-dependent manner (Trelford and Di Guglielmo, 2021).
The activation of autophagy through TGFβ augments tumourigenesis
Autophagy and TGFβ signalling are reciprocally regulated. In fact, autophagy inhibition blocks Smad-dependent TGFβ signalling by impairing TGFβ receptor endocytosis (Trelford and Di Guglielmo, 2022). Also, siRNA targeting of ATGs disrupt TGFβ-induced apoptosis and cell cycle arrest (Irimie et al., 2015). TGFβ-induced autophagy has been implicated in EMT, angiogenesis, and immune suppression (Figure 11A). For instance, TGFβ signalling pathways that activate autophagy regulate pro-tumourigenic TGFβ outcomes. Indeed, disrupting Smad4 and TAK1/TRAF6/p38 MAPK signalling pathways blocked TGFβ-dependent E-Cadherin to N-Cadherin shift and stress fiber formation (Trelford and Di Guglielmo, 2022). Attenuation of TGFβ-induced migration has also been reported following inhibition of autophagy (Alizadeh et al., 2018). In pancreatic ductal adenocarcinoma cells, autophagy is required for TGFβ-induced migration, proliferation, and invasion (He et al., 2019; Li et al., 2021). TGFβ-induced autophagy also decreases the expression of proinflammatory cytokines in macrophages (Pokharel et al., 2016). Furthermore, genomic analysis of colon cancer revealed that autophagy upregulates immune checkpoint molecules that dampen the immune response, whereas EMT, TGFβ, and angiogenic pathways were enhanced (Zhu et al., 2020). In vivo xenograft models of breast cancer demonstrate that TGFβ-induced autophagy protected fibroblasts from cell death-mediated by nutrient starvation and increased CAF phenotypes (Liu et al., 2016). Although the research of the effect of TGFβ-induced autophagy in tumourigenesis is scarce, data shows that as TGFβ signalling and autophagy are upregulated, angiogenesis and EMT increase whereas the immune response is dampened (Figure 11B) (Bustos et al., 2020).
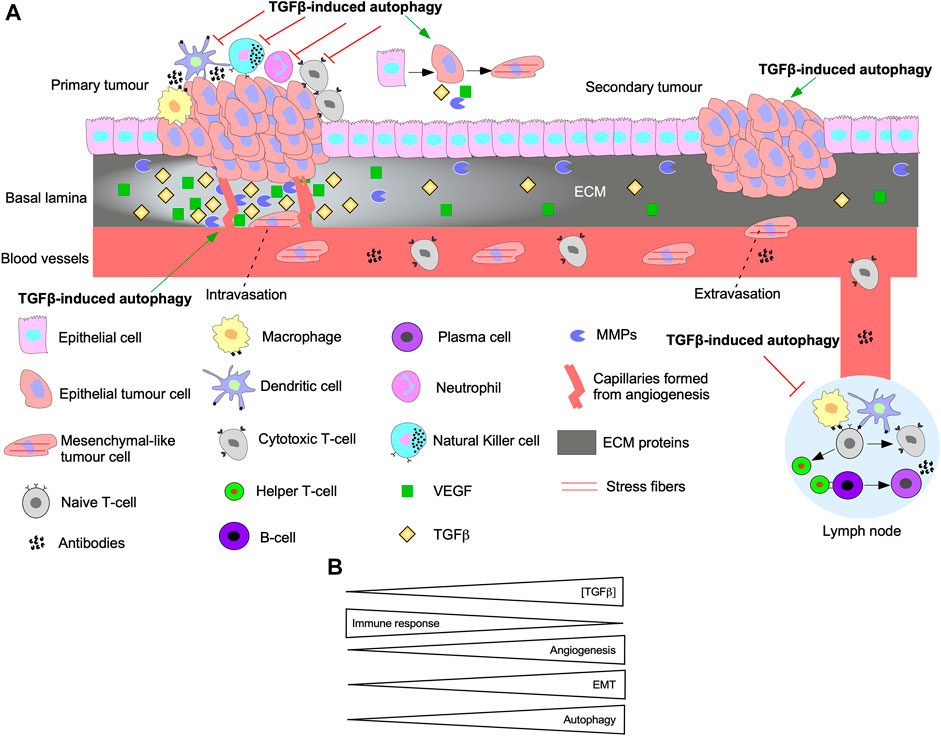
FIGURE 11. The interplay between autophagy and TGFβ signalling in tumourigenesis. (A) A schematic summarizing the effect of TGFβ-induced autophagy on EMT, immune surveillance, angiogenesis, and metastasis. Epithelial cells acquire mutations to the TGFβ pathway until they become cancerous and proliferate rapidly to form the primary tumour. TGFβ-induced autophagy protects tumour cells from the innate immune system (macrophages, dendritic cells, neutrophils, Natural Killer cells) and cells of the adaptive immune system (Naive T-cell, Cytotoxic T-cells, Helper T-cells, B-cells, and plasma cells). Furthermore, TGFβ and autophagy can prevent activation of immune cells that reside in lymph nodes. TGFβ-induced autophagy promotes the release of vascular endothelial growth factor (VEGF) that stimulate angiogenesis. Over time, cells acquire a mesenchymal-like phenotype and release matrix metalloproteinases (MMPs) to breakdown the basal lamina and intravasate into the bloodstream. TGFβ-induced autophagy promotes intravasation because it protects cells that detach from the basal lamina against anoikis-dependent cell death. The mesenchymal-like tumour cells extravasate from the blood vessel at a distant site from the primary tumour. Autophagy is critical for promoting phenotypes to help tumour cells adapt to new environments and establish secondary tumour sites. (B) As the concentration of transforming growth factor-β (TGFβ) increases, the immune response is inhibited, whereas angiogenesis, epithelial-mesenchymal transition (EMT), and autophagy are activated.
Autophagy cargo receptors bridge autophagy and TGFβ signalling
Although there are several catabolic processes that regulate protein quality control in mammalian cells, the UPP and autophagy/lysosome pathway are the two central processes (Wojcik, 2013). Due to difference in substrate selectivity, preparation for degradation, and degradative organelles, the UPP and autophagy do not necessarily compete with one another. Instead, their relationship may be described as compensatory. For instance, when autophagy or the UPP are disrupted, the other major route of protein degradation increases protein turnover to compensate for the disruption (Wojcik, 2013). One explanation is that both lysosome and proteosome-dependent degradation rely on ubiquitination to identify proteins destined for degradation (Lecker et al., 2006; Pankiv et al., 2007; Kirkin et al., 2009). Also, both autophagy and the UPP depend on cargo adaptor proteins such as protein 62/sequestosome 1 (p62/SQSTM1) to deliver substrate proteins (Cohen-Kaplan et al., 2016). Currently, the mechanism of how p62/SQSTM1 decides which pathway receives the ubiquitinated protein remains unknown. Thus far, what has been shown is that p62/SQSTM1 is an autophagy cargo receptor protein that functions in autophagic degradation, regulates EMT, binds to ubiquitin, and is important for TGFβ signalling (Puissant et al., 2012a; Moscat and Diaz-Meco, 2012; Bitto et al., 2014).
P62/SQSTM1 is composed of several domains including a phox bem1 (PB1) domain, ZZ-type zinc finger (ZZ) domain, TRAF binding (TB) domain, LC3-interacting region (LIR), and ubiquitin-associated (UBA) domain. The UBA domain allows p62/SQSTM1 to functions as a ubiquitin receptor protein that targets ubiquitinated proteins to proteasomes (Puissant et al., 2012b; Cohen-Kaplan et al., 2016). In addition to regulating autophagy and the proteasome, p62/SQSTM1 can sequester several downstream TGFβ signalling molecules, including p38 MAPK, TRAF6, and aPKC using the ZZ, TB, and PB1 domains, respectively. These proteins have been implicated in modulating autophagy induction and TGFβ receptor trafficking (Sanz et al., 1999). Furthermore, using the PB1 domain, p62/SQSTM1 self-oligomerizes to sequester intracellular cargo during cell stress or disruption to protein turnover pathways (Lippai and Low, 2014). Also, between the ZZ and TB domains, there is a region of p62/SQSTM1 that interacts with Raptor, a component of mechanistic target of rapamycin complex 1, which is an additional link between p62/SQSTM1 and autophagy (Figure 12).
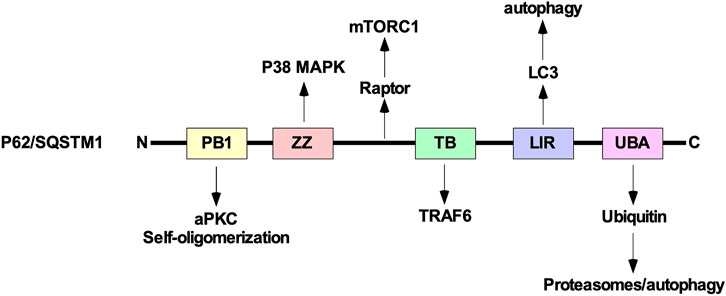
FIGURE 12. The structure of p62/SQSTM1. From the amino (N)-terminal to carboxyl (C)-terminal, p62/SQSTM1 is comprised of the phox bem1 (PB1), ZZ-type zinc finger (ZZ), tumour necrosis factor receptor-associated factor (TRAF) binding (TB), microtubule-associated protein light chain 3 (LC3)-interacting region (LIR), and ubiquitin-associated (UBA) domains. The PB1 domain allows protein 62/sequestosome 1 (p62/SQSTM1) to interact with atypical protein kinase C (aPKC) and self-oligomerize. The ZZ and TB domain have been shown to interact with downstream transforming growth factor-β (TGFβ) signalling molecules, such as p38 mitogen-activated protein kinase (MAPK) and TRAF6, respectively. Between the ZZ and TB domains, p62/SQSTM1 associates with Raptor, which is a component of mechanistic target of rapamycin complex 1 (mTORC1). The LIR binds to LC3 and is necessary to facilitate selective autophagy. The UBA domain recognizes ubiquitin prior to delivering ubiquitin-conjugated proteins to proteasomes or lysosomes.
An image based genome wide small interfering RNA screen in mammalian cells identified Smurf1 as a mediator of selective autophagy (Orvedahl et al., 2011). Since we know that Smurf1 also mediates the UPP, this suggests that TGFβ-specific signalling modulators also have the potential to regulate protein degradation pathways. Therefore, there is evidence of crosstalk between TGFβ signal transduction pathways, autophagy, and the UPP. Given that autophagy, proteasomes, and p62/SQSTM1 regulate TGFβ-dependent EMT (Bertrand et al., 2015; Moon et al., 2017; Alizadeh et al., 2018) and are altered by TGFβ treatment (Bonni et al., 2001; Liang et al., 2020), proteins such as p62/SQSTM1 may be important to understanding the crosstalk between protein degradation pathways and TGFβ signalling. Although the role of p62/SQSTM1 in tumourigenesis is context dependent, it may be an important pharmacological target for regulating TGFβ signalling transduction in cancer (Yuan et al., 2013).
Targeting TGFβ signalling in cancer therapy
Due to the abnormal TGFβ signalling in tumour cells and elevated TGFβ ligand concentrations in tumour microenvironments, modern adjuvant therapies aim to antagonize TGFβ signalling (Yingling et al., 2004). Although TGFβ antagonists are ineffective at treating tumourigenesis as monotherapies, antagonizing TGFβ as part of combination therapies is promising (Teixeira et al., 2020). Current strategies employed to mitigate pro-tumourigenic TGFβ signalling have been extensively reviewed elsewhere (Sheen et al., 2013; Kim et al., 2021). As such, this review will summarize therapeutic strategies undergoing clinical investigations.
Modern adjuvant therapies antagonize pro-tumourigenic TGFβ signalling by targeting TGFβ ligand production, TGFβ-TGFβ receptor interactions, and TGFβ receptor kinase activity (Kim et al., 2021). Antisense oligodeoxynucleotides, such as Trabedersen (AP12009), AP11014, and AP15012 attenuate the mRNA expression of TGFβ2, TGFβ1, and TGFβ1, respectively. Although AP11014 and AP15012 are in pre-clinical development (Sheen et al., 2013), Trabedersen has proven to be safe and effective and is undergoing phase III clinical trials (Bogdahn et al., 2011). TGFβ-TGFβ receptor interactions are pharmacologically blocked using ligand traps or neutralizing antibodies against TGFβ ligands or TGFβ receptors. AVID200, a TGFβ trap comprised of TGFβRII ectodomains fused to human fragment crystallizable domains, has demonstrated high affinity for TGFβ1 and TGFβ3 in clinical trials (Yap et al., 2020). Furthermore, the success of pre-clinical studies of soluble TGFβRII and betaglycan receptors verify that ligand trapping is an effective approach at antagonizing TGFβ signalling in vivo (Bandyopadhyay et al., 2002). As for neutralizing antibodies, Fresolimumab, a pan TGFβ human monoclonal antibody, is in clinical trials for malignant melanoma (Morris et al., 2014). TGFβRI kinase inhibitors, such as Vactosertib and Galunisertib, are safe and effective antagonists of TGFβ signalling and clinical trials assessing their potential in combination therapies are in progress (Figure 13) (Herbertz et al., 2015; Song et al., 2019).
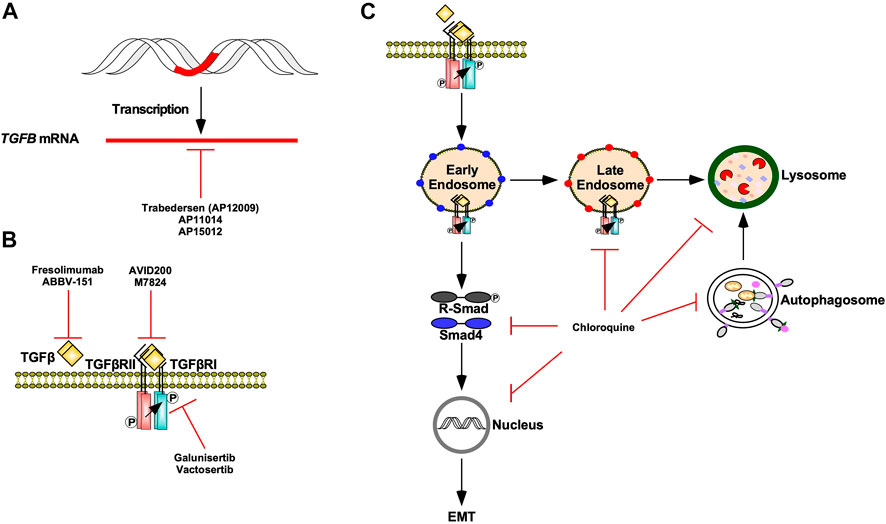
FIGURE 13. TGFβ signalling targeted therapies. (A) Trabedersen (AP12009), AP11014, and AP15012 are antisense oligodeoxynucleotides that decrease TGFB expression via mRNA targeting. (B) Fresolimumab and ABBV-151 are monoclonal antibodies against TGFβ ligands that block TGFβ from binding to TGFβ receptor type II (TGFβRII). AVID200 and M7824 are ligand traps that compete with TGFβRII for TGFβ ligands. Galunisertib and Vactosertib are TGFβ receptor type I (TGFβRI) kinase antagonists. (C) Chloroquine is an autophagy inhibitor that blocks autophagosomes and endosomes from fusing with lysosomes as well as lysosomal-dependent degradation. Chloroquine impedes TGFβ receptor internalization and trafficking through early endosome, late endosome, and lysosome membrane compartments. Chloroquine also decreases receptor regulated Smad (R-Smad) phosphorylation, R-Smad nuclear translocation, and TGFβ-dependent epithelial-mesenchymal transition (EMT).
Given that TGFβ protects tumour cells from the immune system and cancer cells stimulate immune checkpoint inhibitory receptors, anti-tumourigenic immunotherapies are being developed to stimulate immune-mediated destruction of tumour cells (Bai et al., 2019). As such, numerous clinical trials are assessing the efficacy of combining immune checkpoint inhibitors alongside TGFβ signalling antagonists (Maruyama et al., 2022). For instance, ABBV-151 and Budigalimab (formerly known as ABBV-181), anti-TGFβ1 and anti-programmed cell death receptor one antibodies, respectively, have begun phase I clinical trials for advanced solid tumours (Powderly et al., 2020). Likewise, the safety and efficacy of Vactosertib or Galunisertib in conjunction with Durvalumab, a monoclonal programed cell death ligand 1 (PD-L1) antibody, are under investigation in lung, pancreatic, colorectal, and gastric cancer clinical trials (Bai et al., 2019). Finally, M7824, a bifunctional fusion protein containing an extracellular TGFβRII domain and antibody against PD-L1, localizes to tumour microenvironments, sequesters TGFβ ligands, and stimulates T-cell immune activity (Figure 13) (Knudson et al., 2018; Paz-Ares et al., 2018; Lind et al., 2020).
Although the dual blockage of immune checkpoint inhibitors and TGFβ signalling is promising, several obstacles with respect to antagonizing TGFβ signalling in tumourigenesis remain. For instance, targeting TGFβ signalling has been successful in vitro and in pre-clinical studies; however, these outcomes fail to translate in clinical trials (Teixeira et al., 2020). Limited understanding of the interplay between the numerous proteins involved in TGFβ synthesis, activation, signalling, and signalling crosstalk are among the shortcoming of utilizing modern TGFβ inhibitors in adjuvant combination therapies (Kim et al., 2021). Indeed, the combination of the ubiquitous expression of TGFβ ligands, lack of dosing regimens, and its dual role in tumourigenesis pose a challenge to utilizing TGFβ antagonists in cancer therapy (Sheen et al., 2013).
To date, few autophagy inhibitors have been approved for clinical trials for anticancer therapy. Among those approved, diprotic weak bases, such as chloroquine and hydroxychloroquine, and the proton pump inhibitor, pantoprazole, antagonize autophagy by limiting endosomal and/or lysosomal acidification, which blunts lysosomal fusion and lysosomal hydrolase activity (Beil et al., 1992; Halcrow et al., 2021). However, anti-tumourigenic properties of chloroquine, hydroxychloroquine, and pantoprazole rely on both autophagy inhibition and decreasing glycolysis, lactate production, and cytosolic pH (Halcrow et al., 2021). Despite there being no clinical trials investigating autophagy inhibitors in combination with TGFβ signalling antagonist, in vitro studies suggest that chloroquine can disrupt TGFβ signalling (Wu et al., 2018). In Mv1Lu cells, chloroquine antagonized TGFβRII internalization and decreased co-localization with EEA1, Rab7, and LAMP1-positive membrane compartments. Furthermore, R-Smad phosphorylation, R-Smad nuclear translocation, and mesenchymal phenotypes in NSCLC cells treated with TGFβ1 were suppressed by chloroquine (Figure 13) (Trelford and Di Guglielmo, 2022). As such, autophagy inhibitors may be applicable in targeting tumourigenesis driven by aberrant TGFβ signalling without the need to utilize a direct inhibitor of the TGFβ pathway.
Concluding remarks
This review highlights TGFβ signalling pathways that contribute to homeostasis and tumour biology. TGFβ enhances tumourigenesis by promoting proliferation, immune suppression, angiogenesis, lymphangiogenesis, EMT, EndMT, and autophagy. Components of the TGFβ pathway pharmaceutically targeted in clinical trials are limited to TGFβ synthesis, TGFβ-TGFβ receptor interactions, and TGFβRI kinase activity. Although some combination therapies may improve patient prognosis, the efficacy of TGFβ signalling antagonists are underwhelming. Based on the existing literature, there is an abundance of studies exploring TGFβ-dependent EMT, angiogenesis, and immune suppression. Even though there is still much to be learned about these processes and how they interact with each other to promote tumourigenesis, studies exploring the impact that TGFβ has on other tumour promoting biological processes are scarce. Indeed, further work is needed to explore the relationship between TGFβ and autophagy as well as other processes involved with protein quality control, which may yield new therapeutic approaches in targeting TGFβ-dependent tumourigenesis.
Author contributions
CT composed the figures and developed the first manuscript draft. GDG edited the figures, revised the manuscript, and prepared the final manuscript draft for submission. LD assisted with the manuscript draft and helped prepare the final version of the manuscript for submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abel, A. M., Yang, C., Thakar, M. S., and Malarkannan, S. (2018). Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 9, 1869–1923. doi:10.3389/fimmu.2018.01869
Adhikari, A., Xu, M., and Chen, Z. J. (2007). Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26, 3214–3226. doi:10.1038/sj.onc.1210413
Alishekevitz, D., Gingis-Velitski, S., Kaidar-Person, O., Gutter-Kapon, L., Scherer, S. D., Raviv, Z., et al. (2016). Macrophage-induced lymphangiogenesis and metastasis following paclitaxel chemotherapy is regulated by VEGFR3. Cell. Rep. 17, 1344–1356. doi:10.1016/j.celrep.2016.09.083
Alizadeh, J., Glogowska, A., Thliveris, J., Kalantari, F., Shojaei, S., Hombach-Klonisch, S., et al. (2018). Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta. Mol. Cell. Res. 1865, 749–768. doi:10.1016/j.bbamcr.2018.02.007
Aluwihare, P., Mu, Z., Zhao, Z., Yu, D., Weinreb, P. H., Horan, G. S., et al. (2009). Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell. Sci. 122, 227–232. doi:10.1242/jcs.035246
Aragón, E., Wang, Q., Zou, Y., Morgani, S. M., Ruiz, L., Kaczmarska, Z., et al. (2019). Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes. Dev. 33, 1506–1524. doi:10.1101/gad.330837.119
Ashcroft, G. S., Yang, X., Glick, A. B., WeinsteinM., , Letterio, J. L., Mizel, D. E., et al. (1999). Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell. Biol. 1, 260–266. doi:10.1038/12971
Aspalter, I. M., Gordon, E., Dubrac, A., Ragab, A., Narloch, J., Vizan, P., et al. (2015). Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat. Commun. 6, 7264. doi:10.1038/ncomms8264
Atala, A. (2019). Re: Targeting FOXA1-mediated repression of TGF-β signaling suppresses castration-resistant prostate cancer progression. J. Urology 202, 36. doi:10.1097/01.ju.0000557730.48240.ec
Bai, X., Yi, M., Jiao, Y., Chu, Q., and Wu, K. (2019). Blocking TGF-β signaling to enhance the efficacy of immune checkpoint inhibitor. Onco. Targets. Ther. 12, 9527–9538. doi:10.2147/OTT.S224013
Bandyopadhyay, A., Lopez-Casillas, F., Malik, S. N., Montiel, J. L., Mendoza, V., Yang, J., et al. (2002). Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. 62, 4690–4695.
Barrallo-Gimeno, A., and Nieto, M. A. (2005). The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 132, 3151–3161. doi:10.1242/dev.01907
Batlle, E., and Massagué, J. (2019). Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940. doi:10.1016/j.immuni.2019.03.024
Beil, W., Staar, U., and Sewing, K. F. (1992). Pantoprazole: A novel H+/K+-ATPase inhibitor with an improved pH stability. Eur. J. Pharmacol. 218, 265–271. doi:10.1016/0014-2999(92)90178-7
Belardelli, F., and Ferrantini, M. (2002). Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 23, 201–208. doi:10.1016/s1471-4906(02)02195-6
Bernard, A., and Klionsky, D. J. (2013). Autophagosome formation : Tracing the source. Dev. Cell. 25, 116–117. doi:10.1016/j.devcel.2013.04.004
Bertrand, M., Petit, V., Jain, A., Amsellem, R., Johansen, T., Larue, L., et al. (2015). SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell. Cycle 14, 364–374. doi:10.4161/15384101.2014.987619
Bevan, S., Woodford-Richens, K., Rozen, P., Eng, C., Young, J., DunlopM., , et al. (1999). Screening SMAD1, SMAD2, SMAD3, and SMAD5 for germline mutations in juvenile polyposis syndrome. Gut 45, 406–408. doi:10.1136/gut.45.3.406
Bierie, B., and Moses, H. L. (2006). Tumour microenvironment: TGFbeta: The molecular jekyll and hyde of cancer. Nat. Rev. Cancer 6, 506–520. doi:10.1038/nrc1926
Bitto, A., Lerner, C. A., Nacarelli, T., Crowe, E., Torres, C., and Sell, C. (2014). p62/SQSTM1 at the interface of aging, autophagy, and disease. Age 36, 9626–1137. doi:10.1007/s11357-014-9626-3
Bizet, A. A., Liu, K., Tran-Khanh, N., Saksena, A., Vorstenbosch, J., Finnson, K. W., et al. (2011). The TGF-β co-receptor, CD109, promotes internalization and degradation of TGF-β receptors. Biochim. Biophys. Acta 1813, 742–753. doi:10.1016/j.bbamcr.2011.01.028
Bogdahn, U., Hau, P., Stockhammer, G., Venkataramana, N. K., Mahapatra, A. K., Suri, A., et al. (2011). Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro. Oncol. 13, 132–142. doi:10.1093/neuonc/noq142
Bonni, S., Wang, H. R., Causing, C. G., Kavsak, P., Stroschein, S. L., Luo, K., et al. (2001). TGF-β induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell. Biol. 3, 587–595. doi:10.1038/35078562
Bornstein, S., White, R., Malkoski, S., Oka, M., Han, G., Cleaver, T., et al. (2009). Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Invest. 119, 3408–3419. doi:10.1172/JCI38854
Braeutigam, C., Rago, L., Rolke, A., WaLdmeier, L., Christofori, G., and Winter, J. (2014). The RNA-binding protein Rbfox2: An essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 33, 1082–1092. doi:10.1038/onc.2013.50
Bustos, S. O., Antunes, F., Rangel, M. C., and Chammas, R. (2020). Emerging autophagy functions shape the tumor microenvironment and play a role in cancer progression - implications for cancer therapy. Front. Oncol. 10, 1–17. doi:10.3389/fonc.2020.606436
Cardenas, H., Vieth, E., Lee, J., Segar, M., Liu, Y., Nephew, K. P., et al. (2014). TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 9, 1461–1472. doi:10.4161/15592294.2014.971608
Castriconi, R., Cantoni, C., Della Chiesa, M., Vitale, M., Marcenaro, E., Conte, R., et al. (2003). Transforming growth factor β1 inhibits expression of NKP30 and NKG2d receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 100, 4120–4125. doi:10.1073/pnas.0730640100
Castro, F., Cardoso, A. P., Gonçalves, R. M., Serre, K., and Oliveira, M. J. (2018). Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847–919. doi:10.3389/fimmu.2018.00847
Chaffer, C. L., San Juan, B. P., Lim, E., and Weinberg, R. A. (2016). EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 35, 645–654. doi:10.1007/s10555-016-9648-7
Chan, C. W., and Housseau, F. (2008). The ‘kiss of death’ by dendritic cells to cancer cells. Cell. Death Differ. 15, 58–69. doi:10.1038/sj.cdd.4402235
Chen, S. J., Yuan, W., Mori, Y., Levenson, A., TrojanowskaM., , and Varga, J. (1999). Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: Involvement of smad 3. J. Invest. Dermatol. 112, 49–57. doi:10.1046/j.1523-1747.1999.00477.x
Chen, X., Jiang, C., Sun, R., Yang, D., and Liu, Q. (2020). Circular noncoding RNA NR3C1 acts as a miR-382-5p sponge to protect RPE functions via regulating PTEN/AKT/mTOR signaling pathway. Mol. Ther. 28, 929–945. doi:10.1016/j.ymthe.2020.01.010
Chen, Y. G., Wang, Z., Ma, J., Zhang, L., and Lu, Z. (2007). Endofin, a FYVE domain protein, interacts with Smad4 and facilitates transforming growth factor-beta signaling. J. Biol. Chem. 282, 9688–9695. doi:10.1074/jbc.M611704200
Chu, G. C., Dunn, N. R., Anderson, D. C., Oxburgh, L., and Robertson, E. J. (2004). Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development 131, 3501–3512. doi:10.1242/dev.01248
Cohen-Kaplan, V., Ciechanover, A., and Livneh, I. (2016). P62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 7, 83833–83834. doi:10.18632/oncotarget.13805
Colletta, G., Cirafici, A. M., Imbriaco, M., and Vecchio, G. (1988). Inhibitory action of transforming growth factor beta on thyroid cells. Ann. N. Y. Acad. Sci. 551, 372–373. doi:10.1111/j.1749-6632.1988.tb22367.x
Craene, B. D., and Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 13, 97–110. doi:10.1038/nrc3447
Cruts, M., BackHovens, H., Wang, S. Y., Van Gassen, G., Theuns, J., De Jonghe, C. D., et al. (1995). Molecular genetic analysis of familial early-onset alzheimer’s disease linked to chromosome 14q24.3. Hum. Mol. Genet. 4, 2363–2371. doi:10.1093/hmg/4.12.2363
Danhier, P., Banski, P., Payen, V. L., Grasso, D., Ippolito, L., Sonveaux, P., et al. (2017). Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta. Bioenerg. 1858, 556–572. doi:10.1016/j.bbabio.2017.02.001
Datto, M., and Wang, X. F. (2000). The Smads: Transcriptional regulation and mouse models. Cytokine Growth Factor Rev. 11, 37–48. doi:10.1016/s1359-6101(99)00027-1
David, C. J., and Massagué, J. (2018). Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 19, 419–435. doi:10.1038/s41580-018-0007-0
De Borst, M. H., Wassef, L., Kelly, D. J., Van Goor, H., and Navis, G. (2006). Mitogen activated protein kinase signaling in the kidney: Target for intervention? Signal Transduct. 6, 32–53. doi:10.1002/sita.200500063
De Caestecker, M. P., YahaTa, T., Wang, D., Parks, W. T., Huang, S., Hill, C. S., et al. (2000). The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J. Biol. Chem. 275, 2115–2122. doi:10.1074/jbc.275.3.2115
Dennis, P. A., and Rifkin, D. B. (1991). Cellular activation of latent transforming growth factor β requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc. Natl. Acad. Sci. U. S. A. 88, 580–584. doi:10.1073/pnas.88.2.580
Dennler, S., Huet, S., and Gauthier, J. M. (1999). A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 18, 1643–1648. doi:10.1038/sj.onc.1202729
Dennler, S., Itoh, S., Vivien, D., ten Dijke, P., Huet, S., and Gauthier, J. M. (1998). Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100. doi:10.1093/emboj/17.11.3091
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi:10.1038/nature02006
Dhasarathy, A., Phadke, D., Mav, D., Shah, R. R., and Wade, P. A. (2011). The transcription factors snail and slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One 6, e26514. doi:10.1371/journal.pone.0026514
Dickson, M. C., Martin, J. S., Cousins, F. M., Kulkarni, A. B., KarlSSon, S., and Akhurst, R. J. (1995). Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development 121, 1845–1854. doi:10.1242/dev.121.6.1845
Dubois, C., Maprise, M.-H., Blanchette, F., Gentry, L., and Leduc, R. (1995). Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem. 270, 10618–10624. doi:10.1074/jbc.270.18.10618
Dünker, N., and Krieglstein, K. (2002). Tgfbeta2 -/- Tgfbeta3 -/- double knockout mice display severe midline fusion defects and early embryonic lethality. Anat. Embryol. 206, 73–83. doi:10.1007/s00429-002-0273-6
Edlund, S., Landström, M., Heldin, C. H., and Aspenström, P. (2002). Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell. 13, 902–914. doi:10.1091/mbc.01-08-0398
Esebanmen, G. E., and Langridge, W. H. R. (2017). The role of TGF-beta signaling in dendritic cell tolerance. Immunol. Res. 65, 987–994. doi:10.1007/s12026-017-8944-9
Ewen, M. E., Oliver, C. J., Sluss, H. K., Miller, S. J., and Peeper, D. S. (1995). p53-Dependent repression of CDK4 translation in TGF-β-induced G1 cell- cycle arrest. Genes. Dev. 9, 204–217. doi:10.1101/gad.9.2.204
Fang, T., Liang, T., Wang, Y., Wu, H., Liu, S., Xie, L., et al. (2021). Prognostic role and clinicopathological features of SMAD4 gene mutation in colorectal cancer: A systematic review and meta-analysis. BMC Gastroenterol. 21, 297–312. doi:10.1186/s12876-021-01864-9
Farhood, B., Najafi, M., and Mortezaee, K. (2019). CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 234, 8509–8521. doi:10.1002/jcp.27782
Fazilleau, N., McHeyzer-Williams, L. J., Rosen, H., and McHeyzer-Williams, M. G. (2009). The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 10, 375–384. doi:10.1038/ni.1704
Feng, Y., Press, B., and Wandinger-Ness, A. (1995). Rab 7: An important regulator of late endocytic membrane traffic. J. Cell. Biol. 131, 1435–1452. doi:10.1083/jcb.131.6.1435
Ferrari, G., Cook, B. D., Terushkin, V., Pintucci, G., and Mignatti, P. (2009). Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 219, 449–458. doi:10.1002/jcp.21706
Finley, D., Chen, X., and Walters, K. J. (2016). Gates, channels, and switches: Elements of the proteasome machine. Trends biochem. Sci. 41, 77–93. doi:10.1016/j.tibs.2015.10.009
Finnson, K. W., McLean, S., Di Guglielmo, G. M., and Philip, A. (2013). Dynamics of transforming growth factor beta signaling in wound healing and scarring. Adv. Wound Care 2, 195–214. doi:10.1089/wound.2013.0429
Flavell, Richard, Sanjabi, Shomyseh, Stephen Wrzesinski, P. L.-L., and Licona-Limon, P. (2010). The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 10, 554–567. doi:10.1038/nri2808
Fleming, Y. M., Ferguson, G. J., Spender, L. C., Larsson, J., KarlSSon, S., Ozanne, B. W., et al. (2009). TGF-β-mediated activation of RhoA signalling is required for efficient V12HaRas and V600EBRAF transformation. Oncogene 28, 983–993. doi:10.1038/onc.2008.449
Fridlender, Z. G., Sun, J., Kim, S., Kapoor, V., Cheng, G., Ling, L., et al. (2009). Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell. 16, 183–194. doi:10.1016/j.ccr.2009.06.017
Gajewski, T. F., Schreiber, H., and Fu, Y. X. (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022. doi:10.1038/ni.2703
Ganesan, R., Mallets, E., and Gomez-Cambronero, J. (2016). The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 10, 663–676. doi:10.1016/j.molonc.2015.12.006
Gao, S., Alarcon, C., Sapkota, G., Rahman, S., Chen, P. Y., Goerner, N., et al. (2009). Ubiquitin ligase Nedd4L targets activated smad2/3 to limit TGF-β signaling. Mol. Cell. 36, 457–468. doi:10.1016/j.molcel.2009.09.043
García-Caballero, M., Paupert, J., Blacher, S., Van de Velde, M., Quesada, A. R., Medina, M. A., et al. (2017). Targeting VEGFR-3/-2 signaling pathways with AD0157: A potential strategy against tumor-associated lymphangiogenesis and lymphatic metastases. J. Hematol. Oncol. 10, 122. doi:10.1186/s13045-017-0484-1
Gentry, L. E., Lioubin, M. N., Purchio, A. F., and Marquardt, H. (1988). Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol. Cell. Biol. 8, 4162–4168. doi:10.1128/mcb.8.10.4162
Germann, M., Zangger, N., Sauvain, M. O., Sempoux, C., Bowler, A. D., Wirapati, P., et al. (2020). Neutrophils suppress tumor‐infiltrating T cells in colon cancer via matrix metalloproteinase‐mediated activation of TGF β. EMBO Mol. Med. 12, 106811–e10716. doi:10.15252/emmm.201910681
Glick, A. B. (2012). The role of TGFβSignaling in squamous cell cancer: Lessons from mouse models. J. Skin. Cancer 2012, 1–12. doi:10.1155/2012/249063
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy : Cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi:10.1002/path.2697
Golub, T., Wacha, S., and Caroni, P. (2004). Spatial and temporal control of signaling through lipid rafts. Curr. Opin. Neurobiol. 14, 542–550. doi:10.1016/j.conb.2004.08.003
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Rosendahl, A., Sideras, P., and ten Dijke, P. (2002). Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 21, 1743–1753. doi:10.1093/emboj/21.7.1743
Green, A. J., Barcellos, L. F., Rimmler, J. B., Garcia, M. E., Caillier, S., Lincoln, R. R., et al. (2001). Sequence variation in the transforming growth factor-beta1 (TGFB1) gene and multiple sclerosis susceptibility. J. Neuroimmunol. 116, 116–124. doi:10.1016/s0165-5728(01)00283-1
Guglielmo, G. M. Di, Roy, C. Le, Goodfellow, A. F., and Wrana, J. L. (2003). Distinct endocytic pathways regulate TGF- β receptor signalling and turnover. Nat. Cell. Biol. 5, 410–421. doi:10.1038/ncb975
Gunaratne, A., Chan, E., El-chabib, T., Carter, D., and Di Guglielmo, G. M. (2014). aPKC alters TGFβ response in NSCLC cells via both Smad-dependent and Smad- independent pathways. J. Cell. Sci. 128, 487–498. doi:10.1242/jcs.155440
Gunaratne, A., and DiGuglielmo, G. M. (2013). Par6 is phosphorylated by aPKC to facilitate EMT. Cell. adh. Migr. 7, 357–361. doi:10.4161/cam.25651
Hajek, R., Sevcikova, S., Kubiczkova, L., and Sedlarikova, L. (2012). TGF-Beta: An excellent servant but a bad master. J. Transl. Med. 10.
Hakkinen, K. M., Harunaga, J. S., Doyle, A. D., and Yamada, K. M. (2011). Direct comparisons of the morphology, migration, cell adhesions, and actin cytoskeleton of fibroblasts in four different three-dimensional extracellular matrices. Tissue Eng. Part A 17, 713–724. doi:10.1089/ten.TEA.2010.0273
Halcrow, P. W., Geiger, J. D., and Chen, X. (2021). Overcoming chemoresistance: Altering pH of cellular compartments by chloroquine and hydroxychloroquine. Front. Cell. Dev. Biol. 9, 1–14. doi:10.3389/fcell.2021.627639
Halper, J., Burt, D. W., and Romanov, M. N. (2004). On reassessment of the chicken TGFB4 gene as TGFB1. Growth factors. 22, 121–122. doi:10.1080/08977190410001712878
Haque, S., and Morris, J. C. (2017). Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother. 13, 1741–1750. doi:10.1080/21645515.2017.1327107
Harradine, K. A., and Akhurst, R. J. (2006). Mutations of TGFbeta signaling molecules in human disease. Ann. Med. 38, 403–414. doi:10.1080/07853890600919911
Hayashi, H., Inoue, Y., Tsutsui, H., Okamura, H., Nakanishi, K., and Onozaki, K. (2003). TGFbeta down-regulates IFN-gamma production in IL-18 treated NK cell line LNK5E6. Biochem. Biophys. Res. Commun. 300, 980–985. doi:10.1016/s0006-291x(02)02939-x
Hayes, S., Chawla, A., and Corvera, S. (2002). TGFβ receptor internalization into EEA1-enriched early endosomes: Role in signaling to Smad2. J. Cell. Biol. 158, 1239–1249. doi:10.1083/jcb.200204088
He, R., Wang, M., Zhao, C., Shen, M., Yu, Y., He, L., et al. (2019). TFEB-driven autophagy potentiates TGF-β induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 38, 340–414. doi:10.1186/s13046-019-1343-4
Heald, B., Rigelsky, C., Moran, R., LaGuardia, L., O'Malley, M., Burke, C. A., et al. (2015). Prevalence of thoracic aortopathy in patients with juvenile Polyposis Syndrome-Hereditary Hemorrhagic Telangiectasia due to SMAD4. Am. J. Med. Genet. A 167, 1758–1762. doi:10.1002/ajmg.a.37093
Herbertz, S., Sawyer, J. S., Stauber, A. J., Gueorguieva, I., Driscoll, K. E., Estrem, S. T., et al. (2015). Clinical development of galunisertib (Ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. devel. Ther. 9, 4479–4499. doi:10.2147/DDDT.S86621
Herrero-Martín, G., Hoyer-Hansen, M., Garcia-Garcia, C., Fumarola, C., Farkas, T., Lopez-Rivas, A., et al. (2009). TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 28, 677–685. doi:10.1038/emboj.2009.8
Hirschi, K. K., Burt, J. M., Hirschi, K. D., and Dai, C. (2003). Gap junction communication mediates transforming growth factor-β activation and endothelial-induced mural cell differentiation. Circ. Res. 93, 429–437. doi:10.1161/01.RES.0000091259.84556.D5
Hong, L., Du, X., Li, W., Mao, Y., Sun, L., and Li, X. (2018). EndMT: A promising and controversial field. Eur. J. Cell. Biol. 97, 493–500. doi:10.1016/j.ejcb.2018.07.005
Horiguchi, M., Ota, M., and Rifkin, D. B. (2012). Matrix control of transforming growth factor-β function. J. Biochem. 152, 321–329. doi:10.1093/jb/mvs089
Howe, J. R., Roth, S., Ringold, J. C., Summers, R. W., Jarvinen, H. J., Sistonen, P., et al. (1998). Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Sci. (80) 280, 1086–1088. doi:10.1126/science.280.5366.1086
Hu, Y., Chuang, J. Z., Xu, K., McGraw, T. G., and Sung, C. H. S. A. R. A. (2002). SARA, a FYVE domain protein, affects Rab5-mediated endocytosis. J. Cell. Sci. 115, 4755–4763. doi:10.1242/jcs.00177
Igalouzene, R., Hernandez-Vargas, H., Benech, N., Guyennon, A., Bauche, D., Barrachina, C., et al. (2022). SMAD4 TGF-β – independent function preconditions naive CD8 + T cells to prevent severe chronic intestinal inflammation. J. Clin. Invest. 132. doi:10.1172/jci151020
Imani, S., Wei, C., Cheng, J., Khan, M. A., Fu, S., Yang, L., et al. (2017). MicroRNA-34a targets epithelial to mesenchymal transitioninducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget 8, 21362–21379. doi:10.18632/oncotarget.15214
Inui, M., Manfrin, A., Mamidi, A., Martello, G., Morsut, L., Soligo, S., et al. (2011). USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell. Biol. 13, 1368–1375. doi:10.1038/ncb2346
Irimie, A. I., Braicu, C., Zanoaga, O., Pileczki, V., Soritau, O., Berindan-Neagoe, I., et al. (2015). Inhibition of tumor necrosis factor alpha using RNA interference in oral squamous cell carcinoma. J. B.U.ON. 20, 1107–1114.
Isogai, Z., Ono, R. N., Ushiro, S., Keene, D. R., Chen, Y., Mazzieri, R., et al. (2003). Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278, 2750–2757. doi:10.1074/jbc.M209256200
Ito, C., Akimoto, T., Ioka, T., Kobayashi, T., and Kusano, E. (2009). TGF-β inhibits vascular sprouting through TGF-β type I receptor in the mouse embryonic aorta. Tohoku J. Exp. Med. 218, 63–71. doi:10.1620/tjem.218.63
Itoh, F., Divecha, N., Brocks, L., Oomen, L., Janssen, H., Calafat, J., et al. (2002). The FYVE domain in Smad anchor for receptor activation (SARA) is sufficient for localization of SARA in early endosomes and regulates TGF-beta/Smad signalling. Genes. cells. 7, 321–331. doi:10.1046/j.1365-2443.2002.00519.x
Iyengar, P. V. (2017). Regulation of ubiquitin enzymes in the TGF-β pathway. Int. J. Mol. Sci. 18, 877. doi:10.3390/ijms18040877
Jia, L., Lee, H. S., Wu, C. F., Kundu, J., Park, S. G., Kim, R. N., et al. (2014). SMAD4 suppresses AURKA-induced metastatic phenotypes via degradation of AURKA in a TGFβ-independent manner. Mol. Cancer Res. 12, 1779–1795. doi:10.1158/1541-7786.MCR-14-0191
Jiang, Y., Zhou, X., Hu, R., and Dai, A. (2018). TGF-β1-induced SMAD2/3/4 activation promotes RELM-β transcription to modulate the endothelium-mesenchymal transition in human endothelial cells. Int. J. Biochem. Cell. Biol. 105, 52–60. doi:10.1016/j.biocel.2018.08.005
Jimenez, S. A., and Piera-Velazquez, S. (2016). Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 51, 26–36. doi:10.1016/j.matbio.2016.01.012
Johansson, J., Sahin, C., Pestoff, R., Ignatova, S., Forsberg, P., Edsjo, A., et al. (2015). A novel SMAD4 mutation causing severe juvenile polyposis syndrome with protein losing enteropathy, immunodeficiency, and hereditary haemorrhagic telangiectasia. Case Rep. Gastrointest. Med. 2015, 140616–140625. doi:10.1155/2015/140616
Jonk, L. J. C., Itoh, S., Heldin, C. H., Ten Dijke, P., and Kruijer, W. (1998). Identification and functional characterization of a smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 273, 21145–21152. doi:10.1074/jbc.273.33.21145
Karlsson, M. C., Gonzalez, S. F., Welin, J., and Fuxe, J. (2017). Epithelial-mesenchymal transition in cancer metastasis through the lymphatic system. Mol. Oncol. 11, 781–791. doi:10.1002/1878-0261.12092
Katsuno, Y., Lamouille, S., and Derynck, R. (2013). TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 25, 76–84. doi:10.1097/CCO.0b013e32835b6371
Katsura, A., Tamura, Y., Hokari, S., Harada, M., Morikawa, M., Sakurai, T., et al. (2017). ZEB1-regulated inflammatory phenotype in breast cancer cells. Mol. Oncol. 11, 1241–1262. doi:10.1002/1878-0261.12098
Kaufhold, S., and Bonavida, B. (2014). Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 33, 62. doi:10.1186/s13046-014-0062-0
Kaur, J., and Debnath, J. (2015). Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell. Biol. 16, 461–472. doi:10.1038/nrm4024
Kawata, M., Koinuma, D., Ogami, T., Umezawa, K., Iwata, C., Watabe, T., et al. (2012). TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. 151, 205–216. doi:10.1093/jb/mvr136
Khalil, N. (1999). TGF-Beta: From latent to active. Microbes Infect. 1, 1255–1263. doi:10.1016/s1286-4579(99)00259-2
Khoshakhlagh, M., Soleimani, A., Binabaj, M. M., Avan, A., Ferns, G. A., Khazaei, M., et al. (2019). Therapeutic potential of pharmacological TGF-β signaling pathway inhibitors in the pathogenesis of breast cancer. Biochem. Pharmacol. 164, 17–22. doi:10.1016/j.bcp.2019.03.031
Kim, B. G., Malek, E., Choi, S. H., Ignatz-Hoover, J. J., and Driscoll, J. J. (2021). Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 14, 55. doi:10.1186/s13045-021-01053-x
Kim, S. Y., and Baek, K. H. (2018). TGF-β signaling pathway mediated by deubiquitinating enzymes. Cell. Mol. Life Sci. 76, 653–665. doi:10.1007/s00018-018-2949-y
Kim, W., Kim, E., Lee, S., Kim, D., Chun, J., Park, K. H., et al. (2016). TFAP2C-mediated upregulation of TGFBR1 promotes lung tumorigenesis and epithelial-mesenchymal transition. Exp. Mol. Med. 48, e273. doi:10.1038/emm.2016.125
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., et al. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 33, 505–516. doi:10.1016/j.molcel.2009.01.020
Kiyono, K., Suzuki, H. I., Matsuyama, H., Morishita, Y., Komuro, A., Kano, M. R., et al. (2009). Autophagy is activated by TGF- β and potentiates TGF- β – mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 5, 8844–8852. doi:10.1158/0008-5472.can-08-4401
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi:10.1080/15548627.2015.1100356
Knudson, K. M., Hicks, K. C., Luo, X., Chen, J. Q., Schlom, J., and Gameiro, S. R. (2018). M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 7, e1426519. doi:10.1080/2162402X.2018.1426519
Kobayashi, T., Kim, H., Liu, X., Sugiura, H., Kohyama, T., Fang, Q., et al. (2014). Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, 1006–1015. doi:10.1152/ajplung.00015.2014
Koinuma, D., Shinozaki, M., Komuro, A., Goto, K., Saitoh, M., Hanyu, A., et al. (2003). Arkadia amplifies TGF-β superfamily signalling through degradation of Smad7. EMBO J. 22, 6458–6470. doi:10.1093/emboj/cdg632
Kokudo, T., Suzuki, Y., Yoshimatsu, Y., Yamazaki, T., Watabe, T., and Miyazono, K. (2008). Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell. Sci. 121, 3317–3324. doi:10.1242/jcs.028282
Komander, D. (2009). The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953. doi:10.1042/BST0370937
Kondaiah, P., Sands, M. J., Smith, J. M., Fields, A., Roberts, A. B., Sporn, M. B., et al. (1990). Identification of a novel transforming growth factor-beta (TGF-beta 5) mRNA in Xenopus laevis. J. Biol. Chem. 265, 1089–1093. doi:10.1016/s0021-9258(19)40162-2
Korol, A., Pino, G., Dwivedi, D., Robertson, J. V., Deschamps, P. A., and West-Mays, J. A. (2014). Matrix metalloproteinase-9-null mice are resistant to TGF-β-induced anterior subcapsular cataract formation. Am. J. Pathol. 184, 2001–2012. doi:10.1016/j.ajpath.2014.03.013
Kovtun, O., Kane Dickson, V., Kelly, B. T., Owen, D. J., and Briggs, J. A. G. (2020). Architecture of the AP2: Clathrin coat on the membranes of clathrin-coated vesicles. Sci. Adv. 6, eaba8381. doi:10.1126/sciadv.aba8381
Kulkarni, A. B., Huh, C. G., Becker, D., Geiser, A., LyghtM., , Flanders, K. C., et al. (1993). Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U. S. A. 90, 770–774. doi:10.1073/pnas.90.2.770
Kume, T. (2008). Foxc2 transcription factor: A newly described regulator of angiogenesis. Trends cardiovasc. Med. 18, 224–228. doi:10.1016/j.tcm.2008.11.003
Kurosaki, T., Kometani, K., and Ise, W. (2015). Memory B cells. Nat. Rev. Immunol. 15, 149–159. doi:10.1038/nri3802
Landström, M. (2010). The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell. Biol. 42, 585–589. doi:10.1016/j.biocel.2009.12.023
Lawrence, R. E., and Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell. Biol. 21, 133–142. doi:10.1038/s41556-018-0244-7
Le Roy, C., and Wrana, J. L. (2005). Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell. Biol. 6, 112–126. doi:10.1038/nrm1571
Lecker, S. H., Goldberg, A. L., and Mitch, W. E. (2006). Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819. doi:10.1681/ASN.2006010083
Lee, M. K., Pardoux, C., Hall, M. C., Lee, P. S., Warburton, D., Qing, J., et al. (2007). TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967. doi:10.1038/sj.emboj.7601818
Lee, M. P., and Yutzey, K. E. (2011). Twist1 directly regulates genes that promote cell proliferation and migration in developing heart valves. PLoS One 6, e29758. doi:10.1371/journal.pone.0029758
Lee, S. A., Eyeson, R., Cheever, M. L., Geng, J., Verkhusha, V. V., Burd, C., et al. (2005). Targeting of the FYVE domain to endosomal membranes is regulated by a histidine switch. Proc. Natl. Acad. Sci. U. S. A. 102, 13052–13057. doi:10.1073/pnas.0503900102
Lehtimäki, J. I., Rajakylä, E. K., Tojkander, S., and Lappalainen, P. (2021). Generation of stress fibers through myosin-driven reorganization of the actin cortex. Elife 10, e60710. doi:10.7554/eLife.60710
Li, J., Chen, X., Kang, R., Zeh, H., Klionsky, D. J., and Tang, D. (2021). Regulation and function of autophagy in pancreatic cancer. Autophagy 17, 3275–3296. doi:10.1080/15548627.2020.1847462
Liang, C., Xu, J., Meng, Q., Zhang, B., Liu, J., Hua, J., et al. (2020). TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy 16, 486–500. doi:10.1080/15548627.2019.1628540
Liao, R. Y., Mao, C., Qiu, L. X., Ding, H., Chen, Q., and Pan, H. F. (2010). TGFBR1*6A/9A polymorphism and cancer risk: A meta-analysis of 13, 662 cases and 14, 147 controls. Mol. Biol. Rep. 37, 3227–3232. doi:10.1007/s11033-009-9906-7
Lichtman, M. K., Otero-Vinas, M., and Falanga, V. (2016). Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 24, 215–222. doi:10.1111/wrr.12398
Lin, F., Wang, N., and Zhang, T. C. (2012). The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 64, 717–723. doi:10.1002/iub.1059
Lin, L. H., Chang, K. W., Cheng, H. W., and Liu, C. J. (2019). SMAD4 somatic mutations in head and neck carcinoma are associated with tumor progression. Front. Oncol. 9, 1379. doi:10.3389/fonc.2019.01379
Lin, L. H., Chang, K. W., Cheng, H. W., and Liu, C. J. (2019). SMAD4 somatic mutations in head and neck carcinoma are associated with tumor progression. Front. Oncol. 9, 1379–1412. doi:10.3389/fonc.2019.01379
Lind, H., Gameiro, S. R., Jochems, C., Donahue, R. N., Strauss, J., Gulley, J. L., et al. (2020). Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 8, 0004333–e510. doi:10.1136/jitc-2019-000433
Ling, E. M., and Robinson, D. S. (2002). Transforming growth factor-beta1: Its anti-inflammatory and pro-fibrotic effects. Clin. Exp. Allergy 32, 175–178. doi:10.1046/j.1365-2222.2002.01287.x
Lippai, M., and Low, P. (2014). The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 832704. doi:10.1155/2014/832704
Lipton, L., Sieber, O. M., Thomas, H. J. W., Hodgson, S. V., Tomlinson, I. P. M., and Woodford-Richens, K. (2003). Germline mutations in the TGF-beta and Wnt signalling pathways are a rare cause of the ‘multiple’ adenoma phenotype. J. Med. Genet. 40, 355–e44. doi:10.1136/jmg.40.4.e35
Liu, F. L., Mo, E. P., Yang, L., Du, J., Wang, H. S., Zhang, H., et al. (2016). Autophagy is involved in TGF-β1-induced protective mechanisms and formation of cancer-associated fibroblasts phenotype in tumor microenvironment. Oncotarget 7, 4122–4141. doi:10.18632/oncotarget.6702
Liu, M., Li, S., and Li, M. O. (2018). TGF-β control of adaptive immune tolerance: A break from treg cells. BioEssays 40, e1800063–e1800066. doi:10.1002/bies.201800063
Liu, Y., and Cao, X. (2016). Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 94, 509–522. doi:10.1007/s00109-015-1376-x
López-Casillas, F., Payne, H. M., Andres, J. L., and Massagué, J. (1994). Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: Mapping of ligand binding and GAG attachment sites. J. Cell. Biol. 124, 557–568. doi:10.1083/jcb.124.4.557
Losier, T. T., Akuma, M., McKee-Muir, O. C., LeBlond, N. D., Suk, Y., Alsaadi, R. M., et al. (2019). AMPK promotes xenophagy through priming of autophagic kinases upon detection of bacterial outer membrane vesicles. Cell. Rep. 26, 2150–2165.e5. doi:10.1016/j.celrep.2019.01.062
Lu, Y., Wang, L., Li, H., Li, Y., Ruan, Y., Lin, D., et al. (2017). SMAD2 inactivation inhibits CLDN6 methylation to suppress migration and invasion of breast cancer cells. Int. J. Mol. Sci. 18, E1863. doi:10.3390/ijms18091863
Lyons, R. M., Keski-Oja, J., and Moses, H. L. (1988). Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J. Cell. Biol. 106, 1659–1665. doi:10.1083/jcb.106.5.1659
Ma, C. L., Qiao, S., Li, Y. C., Wang, X. F., Sun, R. J., Zhang, X., et al. (2017). TGF-β1 promotes human hepatic carcinoma HepG2 cells invasion by upregulating autophagy. Eur. Rev. Med. Pharmacol. Sci. 21, 2604–2610.
Macias, M. J., Martin-Malpartida, P., and Massagué, J. (2015). Structural determinants of Smad function in TGF-β signaling. Trends biochem. Sci. 40, 296–308. doi:10.1016/j.tibs.2015.03.012
Makhov, P., Golovine, K., TepEr, E., Kutikov, A., MehRazin, R., CorcorAn, A., et al. (2014). Piperlongumine promotes autophagy via inhibition of Akt/mTOR signalling and mediates cancer cell death. Br. J. Cancer 110, 899–907. doi:10.1038/bjc.2013.810
Mallet, C., Vittet, D., Feige, J.-J., and Bailly, S. (2006). TGFbeta1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: Respective contribution of ALK1 and ALK5. Stem Cells 24, 2420–2427. doi:10.1634/stemcells.2005-0494
Maruyama, T., Chen, W., and Shibata, H. (2022). TGF-Β and cancer immunotherapy. Biol. Pharm. Bull. 45, 155–161. doi:10.1248/bpb.b21-00966
Massagué, J., Blain, S. W., and Lo, R. S. (2000). TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 103, 295–309. doi:10.1016/s0092-8674(00)00121-5
Massagué, J., Seoane, J., and Wotton, D. (2005). Smad transcription factors. Genes. Dev. 19, 2783–2810. doi:10.1101/gad.1350705
Massagué, J. (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell. Biol. 13, 616–630. doi:10.1038/nrm3434
Massague, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754. doi:10.1093/emboj/19.8.1745
Mathew, R., Karantza-wadsworth, V., and White, E. (2007). Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. doi:10.1038/nrc2254
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell. 137, 1062–1075. doi:10.1016/j.cell.2009.03.048
Maturi, V., Enroth, S., Heldin, C. H., and Moustakas, A. (2018). Genome–wide binding of transcription factor ZEB1 in triple-negative breast cancer cells. J. Cell. Physiol. 233, 7113–7127. doi:10.1002/jcp.26634
Mcalpine, F., Williamson, L. E., Tooze, S. A., and Chan, E. Y. W. (2013). Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 9, 361–373. doi:10.4161/auto.23066
McLean, S., Bhattacharya, M., and Di Guglielmo, G. M. (2013). βarrestin2 interacts with TβRII to regulate Smad-dependent and Smad-independent signal transduction. Cell. Signal. 25, 319–331. doi:10.1016/j.cellsig.2012.10.001
McLean, S., and Di Guglielmo, G. M. (2014). TGFβ in endosomal signaling. Methods Enzymol. 535, 39–54. doi:10.1016/B978-0-12-397925-4.00003-1
Mclean, S., and Di Guglielmo, G. M. (2010). TGFβ (transforming growth factor β) receptor type III directs clathrin-mediated endocytosis of TGFβ receptor types I and II. Biochem. J. 429, 137–145. doi:10.1042/BJ20091598
Mikheeva, S. A., Mikheev, A. M., Petit, A., Beyer, R., Oxford, R. G., Khorasani, L., et al. (2010). TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 9, 194. doi:10.1186/1476-4598-9-194
Millanes-Romero, A., Herranz, N., Perrera, V., Iturbide, A., Loubat-Casanovas, J., Gil, J., et al. (2013). Regulation of heterochromatin transcription by snail1/LOXL2 during epithelial-to-mesenchymal transition. Mol. Cell. 52, 746–757. doi:10.1016/j.molcel.2013.10.015
Miller, D. S. J., Bloxham, R. D., Jiang, M., Gori, I., Saunders, R. E., Das, D., et al. (2018). The dynamics of TGF-β signaling are dictated by receptor trafficking via the ESCRT machinery. Cell. Rep. 25, 1841–1855. doi:10.1016/j.celrep.2018.10.056
Miyazawa, K., and Miyazono, K. (2017). Regulation of TGF-β family signaling by inhibitory smads. Cold Spring Harb. Perspect. Biol. 9, 0220955–a22125. doi:10.1101/cshperspect.a022095
Miyazono, K. (2009). Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 314–323. doi:10.2183/pjab.85.314
Mohammadzadeh, A., Pourfathollah, A. A., Shahrokhi, S., Hashemi, S. M., Moradi, S. L. A., and Soleimani, M. (2014). Immunomodulatory effects of adipose-derived mesenchymal stem cells on the gene expression of major transcription factors of T cell subsets. Int. Immunopharmacol. 20, 316–321. doi:10.1016/j.intimp.2014.03.003
Moo-Young, T. A., Larson, J. W., Belt, B. A., Tan, M. C., Hawkins, W. G., Eberlein, T. J., et al. (2009). Tumor derived TGF-Beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J. Immunother. 32, 12–21. doi:10.1097/CJI.0b013e318189f13c
Moon, K., Lee, H. G., Baek, W. K., Lee, Y., Kim, K. S., Jun, J. H., et al. (2017). Bortezomib inhibits proliferation, migration, and TGF-β1-induced epithelial-mesenchymal transition of RPE cells. Mol. Vis. 23, 1029–1038.
Moore-Smith, L., and Pasche, B. (2011). TGFBR1 signaling and breast cancer. J. Mammary Gland. Biol. Neoplasia 16, 89–95. doi:10.1007/s10911-011-9216-2
Morris, J. C., Tan, A. R., Olencki, T. E., Shapiro, G. I., Dezube, B. J., Reiss, M., et al. (2014). Phase I study of GC1008 (Fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One 9, e90353. doi:10.1371/journal.pone.0090353
Moscat, J., and Diaz-Meco, M. T. (2012). P62: A versatile multitasker takes on cancer. Trends biochem. Sci. 37, 230–236. doi:10.1016/j.tibs.2012.02.008
Mowers, E. E., Sharifi, M. N., and Macleod, K. F. (2017). Autophagy in cancer metastasis. Oncogene 36, 1619–1630. doi:10.1038/onc.2016.333
Mu, Y., Gudey, S. K., and Landström, M. (2012). Non-Smad signaling pathways. Cell. Tissue Res. 347, 11–20. doi:10.1007/s00441-011-1201-y
Mueller, T. D., and Nickel, J. (2012). Promiscuity and specificity in BMP receptor activation. FEBS Lett. 586, 1846–1859. doi:10.1016/j.febslet.2012.02.043
Muppala, S., Xiao, R., Krukovets, I., Verbovetsky, D., YendamuRi, R., HabibN., , et al. (2017). Thrombospondin-4 mediates TGF-β-induced angiogenesis. Oncogene 36, 5189–5198. doi:10.1038/onc.2017.140
Neuzillet, C., Tijeras-Raballand, A., Cohen, R., Cros, J., Faivre, S., Raymond, E., et al. (2015). Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 147, 22–31. doi:10.1016/j.pharmthera.2014.11.001
Nishida, N., Yano, H., Nishida, T., Kamura, T., and Kojiro, M. (2006). Angiogenesis in cancer. Vasc. Health Risk Manag. 2, 213–219. doi:10.2147/vhrm.2006.2.3.213
Nishimura, D. , Purchio, A. , and Murray, C. (1993). Linkage localization of TGFB2 and the human homeobox gene hlx1 to chromosome 1q. Genomics 15, 357–364. doi:10.1006/geno.1993.1068
Niu, W., Zhang, M., Chen, H., Wang, C., Shi, N., Jing, X., et al. (2016). Peroxiredoxin 1 promotes invasion and migration by regulating epithelial-to-mesenchymal transition during oral carcinogenesis. Oncotarget 7, 47042–47051. doi:10.18632/oncotarget.9705
Nomura, M., and Li, E. (1998). Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393, 786–790. doi:10.1038/31693
Nunes de Almeida, F., Walther, R. F., Pressé, M. T., Vlassaks, E., and Pichaud, F. (2019). Cdc42 defines apical identity and regulates epithelial morphogenesis by promoting apical recruitment of Par6-aPKC and Crumbs. Development 146. doi:10.1242/dev.175497
O’Donovan, T. R., O’Sullivan, G. C., and McKenna, S. L. (2011). Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 7, 509–524. doi:10.4161/auto.7.6.15066
Ogino, S., Kawasaki, T., Ogawa, A., Kirkner, G. J., Loda, M., and Fuchs, C. S. (2007). TGFBR2 mutation is correlated with CpG island methylator phenotype in microsatellite instability-high colorectal cancer. Hum. Pathol. 38, 614–620. doi:10.1016/j.humpath.2006.10.005
Orvedahl, A., Sumpter, R., Xiao, G., Ng, A., Zou, Z., Tang, Y., et al. (2011). Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. doi:10.1038/nature10546
Ozdamar, B., Bose, R., Barrios-Rodiles, M., Wang, H. R., Zhang, Y., and Wrana, J. L. (2005). Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Sci. (80) 307, 1603–1609. doi:10.1126/science.1105718
Pak, K. H., Park, K. C., and Cheong, J. H. (2019). VEGF-C induced by TGF- β1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 19, 799. doi:10.1186/s12885-019-5972-y
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi:10.1074/jbc.M702824200
Park, J. Y., Park, D. H., Jeon, Y., Kim, Y. J., Lee, J., Shin, M. S., et al. (2018). Eupatilin inhibits angiogenesis-mediated human hepatocellular metastasis by reducing MMP-2 and VEGF signaling. Bioorg. Med. Chem. Lett. 28, 3150–3154. doi:10.1016/j.bmcl.2018.08.034
Pavlides, S., Vera, I., Gandara, R., Sneddon, S., Pestell, R. G., Mercier, I., et al. (2012). Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 16, 1264–1284. doi:10.1089/ars.2011.4243
Paz-Ares, L., Kim, T., Vicente, D., Felip, E., Lee, D., Lee, K., et al. (2018). Updated results of M7824 (MSB0011359C): A bifunctional fusion protein targeting TGF-β and PD-L1, in second-line (2L) NSCLC. Ann. Oncol. 29, viii529. doi:10.1093/annonc/mdy292.085
Pelkmans, L., Bürli, T., Zerial, M., and Helenius, A. (2004). Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 118, 767–780. doi:10.1016/j.cell.2004.09.003
Pino, M. S., Kikuchi, H., Zeng, M., Herraiz, M., Sperduti, I., Berger, D., et al. (2010). Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterology 138, 1406–1417. doi:10.1053/j.gastro.2009.12.010
Platel, V., Faure, S., Corre, I., and Clere, N. (2019). Endothelial-to-mesenchymal transition (EndoMT): Roles in tumorigenesis, metastatic extravasation and therapy resistance. J. Oncol. 2019, 8361945. doi:10.1155/2019/8361945
Pociask, D. A., Sime, P. J., and Brody, A. R. (2004). Asbestos-derived reactive oxygen species activate TGF-beta1. Lab. Invest. 84, 1013–1023. doi:10.1038/labinvest.3700109
Pokharel, S. M., Shil, N. K., and Bose, S. (2016). Autophagy, TGF-β, and SMAD-2/3 signaling regulates interferon-β response in respiratory syncytial virus infected macrophages. Front. Cell. Infect. Microbiol. 6, 174–179. doi:10.3389/fcimb.2016.00174
Poniatowski, L. A., Wojdasiewicz, P., Gasik, R., and Szukiewicz, D. (2015). Transforming growth factor beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 137823. doi:10.1155/2015/137823
Ponta, H., Sherman, L., and Herrlich, P. A. (2003). CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell. Biol. 4, 33–45. doi:10.1038/nrm1004
Pouponnot, C., Jayaraman, L., and Massagué, J. (1998). Physical and functional interaction of SMADS and p300/CBP. J. Biol. Chem. 273, 22865–22868. doi:10.1074/jbc.273.36.22865
Powderly, J., Shimizu, T., Lorusso, P., Razak, A., Miller, K., Balar, A., et al. (2020). 596TiP Phase I first-in-human study of ABBV-151 as monotherapy or in combination with budigalimab in patients with locally advanced or metastatic solid tumours. Ann. Oncol. 31, S499. doi:10.1016/j.annonc.2020.08.710
Principe, D. R., Doll, J. A., Bauer, J., Jung, B., Munshi, H. G., Bartholin, L., et al. (2014). Tgf-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 106, djt369–16. doi:10.1093/jnci/djt369
Puissant, A., Fenouille, N., and Auberger, P. (2012). When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2, 397–413.
Puissant, A., Fenouille, N., and Auberger, P. (2012). When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2, 397–413.
Qin, B., Lam, S. S. W., and Lin, K. (1999). Crystal structure of a transcriptionally active smad4 fragment. Structure 7, 1493–1503. doi:10.1016/s0969-2126(00)88340-9
Qin, B. Y., Lam, S. S., Correia, J. J., and Lin, K. (2002). Smad3 allostery links TGF-β receptor kinase activation to transcriptional control. Genes. Dev. 16, 1950–1963. doi:10.1101/gad.1002002
Ray, B. N., Lee, N. Y., How, T., and Blobe, G. C. (2010). ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 31, 435–441. doi:10.1093/carcin/bgp327
Razani, B., Zhang, X. L., BitzerM., , von Gersdorff, G., Bottinger, E. P., and Lisanti, M. P. (2001). Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J. Biol. Chem. 276, 6727–6738. doi:10.1074/jbc.M008340200
Rebecca, V. W., and Amaravadi, R. K. (2016). Emerging strategies to effectively target autophagy in cancer. Oncogene 35, 1–11. doi:10.1038/onc.2015.99
Saito, A., Horie, M., and Nagase, T. (2018). TGF-β signaling in lung health and disease. Int. J. Mol. Sci. 19, 2460–2518. doi:10.3390/ijms19082460
Sanford, L. P., Ormsby, I., Gittenberger-de Groot, A. C., Sariola, H., Friedman, R., Boivin, G. P., et al. (1997). TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659–2670. doi:10.1242/dev.124.13.2659
Sanz, L., Sanchez, P., and Moscat, J. (1999). The interaction of p62 with RIP links the atypical PKCs to NF- κ B activation. EMBO 18, 3044–3053. doi:10.1093/emboj/18.11.3044
Sarode, S. C., and Sarode, G. S. (2014). Neutrophil-tumor cell cannibalism in oral squamous cell carcinoma. J. Oral Pathol. Med. 43, 454–458. doi:10.1111/jop.12157
Sarrazy, V., Koehler, A., Chow, M. L., Zimina, E., Li, C. X., Kato, H., et al. (2014). Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 102, 407–417. doi:10.1093/cvr/cvu053
Sarshekeh, A. M., Advani, S., Overman, M. J., Manyam, G., Kee, B. K., Fogelman, D. R., et al. (2017). Association of SMAD4 mutation with patient demographics, tumor characteristics, and clinical outcomes in colorectal cancer. PLoS One 12, 01733455–e173414. doi:10.1371/journal.pone.0173345
Schultz-Cherry, S., and Murphy-Ullrich, J. E. (1993). Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J. Cell. Biol. 122, 923–932. doi:10.1083/jcb.122.4.923
Schwartz, M., Zhang, Y., and Rosenblatt, J. D. (2016). B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 4, 40. doi:10.1186/s40425-016-0145-x
Semerdjieva, S., Shortt, B., Maxwell, E., Singh, S., Fonarev, P., Hansen, J., et al. (2008). Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J. Cell. Biol. 183, 499–511. doi:10.1083/jcb.200806016
Sheen, Y. Y., Kim, M. J., Park, S. A., Park, S. Y., and Nam, J. S. (2013). Targeting the transforming growth factor-β signaling in cancer therapy. Biomol. Ther. Seoul. 21, 323–331. doi:10.4062/biomolther.2013.072
Shi, W., Sun, C., He, B., Xiong, W., Shi, X., Yao, D., et al. (2004). GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J. Cell. Biol. 164, 291–300. doi:10.1083/jcb.200307151
Shi, Y., Wang, Y. F., Jayaraman, L., Yang, H., Massague, J., and Pavletich, N. P. (1998). Crystal structure of a Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-β signaling. Cell. 94, 585–594. doi:10.1016/s0092-8674(00)81600-1
Shin, J. H., Min, S. H., Kim, S. J., Kim, Y. I., Park, J., Lee, H. K., et al. (2013). TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1. Sci. Rep. 3, 1561–1611. doi:10.1038/srep01561
Shull, M. M., Ormsby, I., Kier, A. B., PawlowSki, S., Diebold, R. J., YinM., , et al. (1992). Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359, 693–699. doi:10.1038/359693a0
Sitkovsky, M. V., and Ohta, A. (2005). The ‘danger’ sensors that STOP the immune response: The A2 adenosine receptors? Trends Immunol. 26, 299–304. doi:10.1016/j.it.2005.04.004
Song, K. M., Chung, D. Y., Choi, M. J., Ghatak, K., Minh, N. N., Limanjaya, A., et al. (2019). Vactosertib, a novel, orally bioavailable activin receptor-like kinase 5 inhibitor, promotes regression of fibrotic plaques in a rat model of Peyronie’s disease. World J. Mens. Health 37, 552. doi:10.5534/wjmh.190071
Stemmler, M. P., Eccles, R. L., Brabletz, S., and Brabletz, T. (2019). Non-redundant functions of EMT transcription factors. Nat. Cell. Biol. 21, 102–112. doi:10.1038/s41556-018-0196-y
Sun, R., Kong, X., Qiu, X., Huang, C., and Wong, P. P. (2021). The emerging roles of pericytes in modulating tumor microenvironment. Front. Cell. Dev. Biol. 9, 676342–676410. doi:10.3389/fcell.2021.676342
Sun, Y., Liu, X., Ng-Eaton, E., Lodish, H. F., and Weinberg, R. A. (1999). SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc. Natl. Acad. Sci. U. S. A. 96, 12442–12447. doi:10.1073/pnas.96.22.12442
Suwanabol, P. A., Seedial, S. M., Zhang, F., Shi, X., Si, Y., Liu, B., et al. (2012). TGF-β and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 302, 2211–2219. doi:10.1152/ajpheart.00966.2011
Suzuki, H. I., Kiyono, K., and Miyazono, K. (2010). Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy 6, 645–647. doi:10.4161/auto.6.5.12046
Suzuki, Y., Ito, Y., Mizuno, M., Kinashi, H., Sawai, A., Noda, Y., et al. (2012). Transforming growth factor-Β induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int. 81, 865–879. doi:10.1038/ki.2011.464
Takasaka, N., Seed, R. I., Cormier, A., Bondesson, A. J., Lou, J., Elattma, A., et al. (2018). Integrin αvβ8-expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI insight 3, 1–17. doi:10.1172/jci.insight.122591
Tan, E. J., Olsson, A. K., and Moustakas, A. (2015). Reprogramming during epithelial to mesenchymal transition under the control of TGFβ. Cell. adh. Migr. 9, 233–246. doi:10.4161/19336918.2014.983794
Tang, L. Y., Yamashita, M., Coussens, N. P., Tang, Y., Wang, X., Li, C., et al. (2011). Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 30, 4777–4789. doi:10.1038/emboj.2011.393
Tatti, O., Vehviläinen, P., Lehti, K., and Keski-Oja, J. (2008). MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell. Res. 314, 2501–2514. doi:10.1016/j.yexcr.2008.05.018
Teixeira, A. F., ten Dijke, P., and Zhu, H. J. (2020). On-target anti-TGF-β therapies are not succeeding in clinical cancer treatments: What are remaining challenges? Front. Cell. Dev. Biol. 8, 605. doi:10.3389/fcell.2020.00605
Ten Dijke, P., and Hill, C. S. (2004). New insights into TGF-β-Smad signalling. Trends biochem. Sci. 29, 265–273. doi:10.1016/j.tibs.2004.03.008
Thomas, D. A., and Massagué, J. (2005). TGF- β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 8, 369–380. doi:10.1016/j.ccr.2005.10.012
Tirino, V., CameRlingo, R., Bifulco, K., Irollo, E., Montella, R., PainoF., , et al. (2013). TGF-β1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell. Death Dis. 4, e620. doi:10.1038/cddis.2013.144
Title, A. C., Hong, S. J., Pires, N. D., Hasenohrl, L., Godbersen, S., Stokar-Regenscheit, N., et al. (2018). Genetic dissection of the miR-200–Zeb1 axis reveals its importance in tumor differentiation and invasion. Nat. Commun. 9, 4671. doi:10.1038/s41467-018-07130-z
Trelford, C. B., and Di Guglielmo, G. M. (2022). Autophagy regulates transforming growth factor β signaling and receptor trafficking. Biochim. Biophys. acta. Mol. Cell. Res. 1869, 119284. doi:10.1016/j.bbamcr.2022.119284
Trelford, C. B., and Di Guglielmo, G. M. (2021). Canonical and non-canonical TGFβ signalling activate autophagy in a ULK1- dependent manner. Front. Cell. Dev. Biol. 9, 712124. doi:10.3389/fcell.2021.712124
Trelford, C. B., and Guglielmo, G. M. Di. (2020). Assessing methods to quantitatively validate TGFβ-dependent autophagy. Biol. Open 9. doi:10.1242/bio.055103
Trelford, C. B., and Guglielmo, G. M. Di. (2021). Molecular mechanisms of mammalian autophagy. Biochem. J. 478, 3395–3421. doi:10.1042/bcj20210314
Tripathi, V., Shin, J. H., Stuelten, C. H., and Zhang, Y. E. (2019). TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 38, 3185–3200. doi:10.1038/s41388-018-0655-8
Tripathi, V., and Zhang, Y. E. (2017). Redirecting RNA splicing by SMAD3 turns TGF-β into a tumor promoter. Mol. Cell. Oncol. 4, 12656999–e1265703. doi:10.1080/23723556.2016.1265699
Tsukazaki, T., Chiang, T. A., Davison, A. F., Attisano, L., and Wrana, J. L. S. A. R. A. (1998). SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 95, 779–791. doi:10.1016/s0092-8674(00)81701-8
Tuloup-Minguez, V., Hamai, A., Greffard, A., Nicolas, V., Codogno, P., and Botti, J. (2013). Autophagy modulates cell migration and β1 integrin membrane recycling. Cell. Cycle 12, 3317–3328. doi:10.4161/cc.26298
Vincent, F., Hagiwara, K., Ke, Y., Stoner, G. D., Demetrick, D. J., and Bennett, W. P. (1996). Mutation analysis of the transforming growth factor β type II receptor in sporadic human cancers of the pancreas, liver, and breast. Biochem. Biophys. Res. Commun. 223, 561–564. doi:10.1006/bbrc.1996.0934
Vu, T., and Datta, P. K. (2017). Regulation of EMT in colorectal cancer: A culprit in metastasis. Cancers (Basel) 9, 1711–E222. doi:10.3390/cancers9120171
Walshe, T. E., Saint-Geniez, M., Maharaj, A. S. R., Sekiyama, E., Maldonado, A. E., and D'Amore, P. A. (2009). TGF-β is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS One 4, 51499–e5216. doi:10.1371/journal.pone.0005149
Wang, J., Wang, Y., Wang, Y., Ma, Y., Lan, Y., and Yang, X. (2013). Transforming growth factor β-regulated microrna-29a promotes angiogenesis through targeting the phosphatase and tensin homolog in endothelium. J. Biol. Chem. 288, 10418–10426. doi:10.1074/jbc.M112.444463
Wang, T. (2003). The 26S proteasome system in the signaling pathways of TGF-beta superfamily. Front. Biosci. 8, 1057–1127. doi:10.2741/1057
Wang, X., Freire Valls, A., Schermann, G., Shen, Y., Moya, I. M., Castro, L., et al. (2017). YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev. Cell. 42, 462–478. doi:10.1016/j.devcel.2017.08.002
Wang, Y., Chu, J., Yi, P., Dong, W., Saultz, J., Wang, Y., et al. (2018). SMAD4 promotes TGF-β-independent NK cell homeostasis and maturation and antitumor immunity. J. Clin. Invest. 128, 5123–5136. doi:10.1172/JCI121227
Wang, Y., Qiang, Q., Wei, X., Wang, F., Jiang, J., and Guo, Q. (2012). Nan Association between TGFBR1 polymorphisms and cancer risk: A meta-analysis of 35 case-control studies. PLoS One 7, e42899. doi:10.1371/journal.pone.0042899
Warner, S. J., Yashiro, H., and Longmore, G. D. (2010). The cdc42/par6/aPKC polarity complex regulates apoptosis-induced compensatory proliferation in epithelia. Curr. Biol. 20, 677–686. doi:10.1016/j.cub.2010.03.025
Weiss, A., and Attisano, L. (2013). The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2, 47–63. doi:10.1002/wdev.86
Wipff, P. J., Rifkin, D. B., Meister, J. J., and Hinz, B. (2007). Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell. Biol. 179, 1311–1323. doi:10.1083/jcb.200704042
Wojcik, S. (2013). Crosstalk between autophagy and proteasome protein degradation systems: Possible implications for cancer therapy. Folia histochem. Cytobiol. 51, 249–264. doi:10.5603/FHC.2013.0036
Wrana, J. L., Attisano, L., Wieser, R., Ventura, F., and Massague, J. (1994). Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. doi:10.1038/370341a0
Wrana, J. L. (2013). Signaling by the TGF superfamily. Cold Spring Harb. Perspect. Biol. 5, a011197. doi:10.1101/cshperspect.a011197
Wu, J., Chen, X., Liu, X., Huang, S., He, C., Chen, B., et al. (2018). Autophagy regulates TGF-β2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Mol. Med. Rep. 17, 3607–3614. doi:10.3892/mmr.2017.8360
Wu, J., Liu, Z., Shao, C., Gong, Y., Hernando, E., Lee, P., et al. (2011). HMGA2 overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT genes. Cancer Res. 71, 349–359. doi:10.1158/0008-5472.CAN-10-2550
Wu, J. W., HuM., , Chai, J., Seoane, J., HuseM., , Li, C., et al. (2001). Crystal structure of a phosphorylated Smad2: Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-β signaling. Mol. Cell. 8, 1277–1289. doi:10.1016/s1097-2765(01)00421-x
Wu, M., Chen, G., and Li, Y. P. (2016). TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009. doi:10.1038/boneres.2016.9
Wylie-Sears, J., Levine, R. A., and Bischoff, J. (2014). Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-β-induced phosphorylation of ERK. Biochem. Biophys. Res. Commun. 446, 870–875. doi:10.1016/j.bbrc.2014.03.014
Xu, J., Lamouille, S., and Derynck, R. (2009). TGF-Β-induced epithelial to mesenchymal transition. Cell. Res. 19, 156–172. doi:10.1038/cr.2009.5
Xu, Y., Yang, S., Huang, J., Ruan, S., Zheng, Z., and Lin, J. (2012). TGF-β1 induces autophagy and promotes apoptosis in renal tubular epithelial cells. Int. J. Mol. Med. 29, 781–790. doi:10.3892/ijmm.2012.911
Yakymovych, I., Yakymovych, M., and Heldin, C. H. (2018). Intracellular trafficking of transforming growth factor β receptors. Acta Biochim. Biophys. Sin. 50, 3–11. doi:10.1093/abbs/gmx119
Yamashita, M., Fatyol, K., Jin, C., Wang, X., Liu, Z., and Zhang, Y. E. (2008). TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell. 31, 918–924. doi:10.1016/j.molcel.2008.09.002
Yao, D., Ehrlich, M., Henis, Y. I., and Leof, E. B. (2002). Transforming growth factor-beta receptors interact with AP2 by direct binding to beta2 subunit. Mol. Biol. Cell. 13, 4001–4012. doi:10.1091/mbc.02-07-0104
Yap, T. A., O'Carrigan, B., Penney, M. S., Lim, J. S., Brown, J. S., de Miguel Luken, M. J., et al. (2020). AVID200, First-in-class TGF-beta1 and beta3 selective inhibitor: Results of a phase I monotherapy dose escalation study in solid tumours and evidence of target engagement in patients. J. Clin. Oncol. 38, 3195–3204. doi:10.1200/JCO.19.02404
Yee, J. A., Yan, L., Dominguez, J. C., Allan, E. H., and Martin, T. J. (1993). Plasminogen‐dependent activation of latent transforming growth factor beta (TGFβ) by growing cultures of osteoblast‐like cells. J. Cell. Physiol. 157, 528–534. doi:10.1002/jcp.1041570312
Yeon, J. H., Jeong, H. E., Seo, H., Cho, S., Kim, K., Na, D., et al. (2018). Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 76, 146–153. doi:10.1016/j.actbio.2018.07.001
Yin, X., Murphy, S. J., Wilkes, M. C., Ji, Y., and Leof, E. B. (2013). Retromer maintains basolateral distribution of the type II TGF-β receptor via the recycling endosome. Mol. Biol. Cell. 24, 2285–2298. doi:10.1091/mbc.E13-02-0093
Yingling, J. M., Blanchard, K. L., and Sawyer, J. S. (2004). Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 3, 1011–1022. doi:10.1038/nrd1580
Yu, Q., and Stamenkovic, I. (2000). Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes. Dev. 14, 163–176. doi:10.1101/gad.14.2.163
Yuan, Z.-Y., Li, M., Xi, S. Y., Fu, J., and He, J. (2013). Accumulation of p62 is associated with poor prognosis in patients with triple-negative breast cancer. Onco. Targets. Ther. 6, 883–888. doi:10.2147/OTT.S46222
Zeisberg, E. M., Potenta, S., Xie, L., Zeisberg, M., and Kalluri, R. (2007). Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128. doi:10.1158/0008-5472.CAN-07-3127
Zhang, H., Caudle, Y., Wheeler, C., Zhou, Y., Stuart, C., Yao, B., et al. (2018). TGF-β1/Smad2/3/Foxp3 signaling is required for chronic stress-induced immune suppression. J. Neuroimmunol. 314, 30–41. doi:10.1016/j.jneuroim.2017.11.005
Zhang, H. S., Cao, E. H., and Qin, J. F. (2005). Homocysteine induces cell cycle G1 arrest in endothelial cells through the PI3K/Akt/FOXO signaling pathway. Pharmacology 74, 57–64. doi:10.1159/000083684
Zhang, L., Zhou, F., and ten Dijke, P. (2013). Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends biochem. Sci. 38, 612–620. doi:10.1016/j.tibs.2013.10.001
Zhang, W., Li, Q., Song, C., and Lao, L. (2015). Knockdown of autophagy-related protein 6, Beclin-1, decreases cell growth, invasion, and metastasis and has a positive effect on chemotherapy-induced cytotoxicity in osteosarcoma cells. Tumour Biol. 36, 2531–2539. doi:10.1007/s13277-014-2868-y
Zhang, X., Tang, N., Hadden, T. J., and Rishi, A. K. (2011). Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1813, 1978–1986. doi:10.1016/j.bbamcr.2011.03.010
Zhang, Y. E. (2009). Non-Smad pathways in TGF-β signaling. Cell. Res. 19, 128–139. doi:10.1038/cr.2008.328
Zhao, M., Howard, E. W., Parris, A. B., Guo, Z., Zhao, Q., and Yang, X. (2017). Alcohol promotes migration and invasion of triple-negative breast cancer cells through activation of p38 MAPK and JNK. Mol. Carcinog. 56, 849–862. doi:10.1002/mc.22538
Zhao, Y., Thornton, A. M., Kinney, M. C., Ma, C. A., Spinner, J. J., Fuss, I. J., et al. (2011). The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor β (TGF-β) signaling and the development of regulatory T cells. J. Biol. Chem. 286, 40520–40530. doi:10.1074/jbc.M111.292961
Zhou, X., Liu, X., and Huang, L. (2021). Macrophage-mediated tumor cell phagocytosis: Opportunity for nanomedicine intervention. Adv. Funct. Mat. 31, 2006220. doi:10.1002/adfm.202006220
Zhu, S., Wu, Q., Zhang, B., Wei, H., Li, B., Shi, W., et al. (2020). Autophagy-related gene expression classification defines three molecular subtypes with distinct clinical and microenvironment cell infiltration characteristics in colon cancer. Int. Immunopharmacol. 87, 106757. doi:10.1016/j.intimp.2020.106757
Keywords: transforming growth factor-b (TGFb), Smad, receptor trafficking, epithelial-mesenchymal transition (EMT), autophagy, protein 62/sequestosome 1 (p62/SQSTM1)
Citation: Trelford CB, Dagnino L and Di Guglielmo GM (2022) Transforming growth factor-β in tumour development. Front. Mol. Biosci. 9:991612. doi: 10.3389/fmolb.2022.991612
Received: 11 July 2022; Accepted: 15 September 2022;
Published: 04 October 2022.
Edited by:
Birija Sankar Patro, Bhabha Atomic Research Centre (BARC), IndiaReviewed by:
Bing Shen, Shanghai General Hospital, ChinaSujit Kumar Bhutia, National Institute of Technology Rourkela, India
Copyright © 2022 Trelford, Dagnino and Di Guglielmo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianni M. Di Guglielmo, Sm9obi5kaWd1Z2xpZWxtb0BzY2h1bGljaC51d28uY2E=