 Selina M. GarciaTracy R. Yellowhair
Selina M. GarciaTracy R. Yellowhair Neil D. DetweilerRosstin AhmadianLindsay M. Herbert
Neil D. DetweilerRosstin AhmadianLindsay M. Herbert Laura V. Gonzalez Bosc
Laura V. Gonzalez Bosc Thomas C. Resta
Thomas C. Resta Nikki L. Jernigan*
Nikki L. Jernigan*- Department of Cell Biology and Physiology, University of New Mexico School of Medicine, Albuquerque, NM, United States
Acid-sensing ion channel 1a (ASIC1a) is a voltage-independent, non-selective cation channel that conducts both Na+ and Ca2+. Activation of ASIC1a elicits plasma membrane depolarization and stimulates intracellular Ca2+-dependent signaling pathways in multiple cell types, including vascular smooth muscle (SM) and endothelial cells (ECs). Previous studies have shown that increases in pulmonary vascular resistance accompanying chronic hypoxia (CH)-induced pulmonary hypertension requires ASIC1a to elicit enhanced pulmonary vasoconstriction and vascular remodeling. Both SM and EC dysfunction drive these processes; however, the involvement of ASIC1a within these different cell types is unknown. Using the Cre-LoxP system to generate cell-type-specific Asic1a knockout mice, we tested the hypothesis that SM-Asic1a contributes to CH-induced pulmonary hypertension and vascular remodeling, whereas EC-Asic1a opposes the development of CH-induced pulmonary hypertension. The severity of pulmonary hypertension was not altered in mice with specific deletion of EC-Asic1a (TekCre-Asic1afl/fl). However, similar to global Asic1a knockout (Asic1a−/-) mice, mice with specific deletion of SM-Asic1a (MHCCreER-Asic1afl/fl) were protected from the development of CH-induced pulmonary hypertension and right heart hypertrophy. Furthermore, pulmonary hypertension was reversed when deletion of SM-Asic1a was initiated in conditional MHCCreER-Asic1afl/fl mice with established pulmonary hypertension. CH-induced vascular remodeling was also significantly attenuated in pulmonary arteries from MHCCreER-Asic1afl/fl mice. These findings were additionally supported by decreased CH-induced proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) from Asic1a−/- mice. Together these data demonstrate that SM-, but not EC-Asic1a contributes to CH-induced pulmonary hypertension and vascular remodeling. Furthermore, these studies provide evidence for the therapeutic potential of ASIC1a inhibition to reverse pulmonary hypertension.
1 Introduction
Under normal physiological conditions, the pulmonary circulation is maintained in a low-pressure, low-resistance state, with little or no resting vascular tone. During pathological conditions, a sustained increase in pulmonary vascular resistance leads to the development of pulmonary hypertension. Pulmonary hypertension is a progressive and often fatal pulmonary vascular disease defined by a mean pulmonary arterial pressure >20 mmHg (Simonneau et al., 2019). Over time, the elevated vascular resistance and pulmonary arterial pressure increase right ventricular afterload. When the adaptive mechanisms of right ventricular dilation and hypertrophy can no longer compensate for the high vascular resistance in the lung, right heart failure occurs and is associated with a poor prognosis.
Although pulmonary hypertension stems from different underlying causes, the increase in pulmonary vascular resistance in all forms of pulmonary hypertension can be attributed to a combination of sustained pulmonary vasoconstriction and vascular remodeling. Enhanced vasoconstriction is linked to pulmonary arterial endothelial cell (PAEC) dysfunction and hyperreactivity of pulmonary arterial smooth muscle cells (PASMCs) (Budhiraja et al., 2004; Lin et al., 2004; Nagaoka et al., 2004; Jernigan et al., 2008; Broughton et al., 2010; Weise-Cross et al., 2018). The impact of remodeling on pulmonary vascular resistance is primarily due to thickening of the intimal and/or medial layer of small muscular arteries and distal neomuscularization, depicted by the appearance of cells expressing smooth muscle (SM)-specific markers in normally non-muscular precapillary, intra-acinar vessels. This complex pathogenesis is thought to be initiated by endothelial cell (EC) injury and apoptosis followed by the emergence of excessive proliferation and migration of apoptosis-resistant PAECs and PASMCs, and cellular trans-differentiation in the form of EC-mesenchymal transition and SM phenotypic transformations (Voelkel & Tuder, 2000; Shimoda & Laurie, 2013; Gao et al., 2016). Metabolic derangements that promote aerobic glycolysis and inhibition of mitochondrial oxidative respiration have been shown to drive the extensive right ventricular and vascular remodeling in both animal models and patients with pulmonary hypertension (McMurtry et al., 2004; Bonnet et al., 2006; Xu et al., 2007; Sutendra et al., 2010; Fessel et al., 2012; Dromparis et al., 2013; Pak et al., 2013). The shift in cellular metabolism to lactic acid fermentation leads to pathological increases in extracellular acidity. Several ion channels are either directly gated or their activity modulated by alterations in intracellular and extracellular pH including acid-sensing ion channels (ASIC), transient receptor potential vanilloid receptor 1 (TRPV1), the transient receptor potential ankyrin repeat receptor 1 (TRPA1), some two-pore domain (K2P) channels, inwardly rectifying K+ channels (Kir), and voltage-gated Na+, Ca2+, and K+ channels (Harguindey et al., 2017).
Acid-sensing ion channels (ASICs) constitute a subfamily of the amiloride-sensitive, degenerin/epithelial Na+ channel (Deg/ENaC) superfamily that form H+-gated, voltage-insensitive cation channels. Similar to ENaCs, ASICs are highly selective for Na+ over other ions; except ASIC1a which additionally conducts Ca2+ (Waldmann et al., 1997; Xiong et al., 2004; Yermolaieva et al., 2004). The influx of Na+ and Ca2+ contributes to membrane depolarization, activation of Ca2+–calmodulin-dependent mechanisms, and other second-messenger pathways signifying the diverse roles played by ASIC1a in intracellular signaling and excitability under both normal and pathological conditions. ASICs have been primarily studied in neurons due to their ubiquitous expression throughout the central and peripheral nervous systems. Consequently, it is less well recognized that ASICs are expressed in a variety of other cell types including oligodendrocytes, mesenchymal, epithelial, endothelial, muscle, adipose/endocrine, and immune cells where they have been implicated in a range of pathologies (Foster et al., 2021; Karlsson et al., 2021). Although the expression of ASIC1 has been reported in vascular SM and ECs (Grifoni et al., 2008; Jernigan et al., 2009; Chung et al., 2010; Akanji et al., 2019; Garcia et al., 2020; Redd et al., 2021), less is known about the functional role of ASIC1 to regulate vascular homeostasis in disease states.
Previous studies from our laboratory have identified a novel role for ASIC1a in the development of chronic hypoxia (CH)-induced pulmonary hypertension (Nitta et al., 2014). ASIC1 is expressed in both PASMCs and PAECs; however, the contribution of ASIC1a to the pathological mechanisms leading to pulmonary artery remodeling and the development of pulmonary hypertension in these two vascular cell types is unclear. While prior studies indicate PASMC ASIC1a mediates pulmonary vasoconstriction (Jernigan et al., 2012), the functional role of ASIC1 in PAECs is unknown. Based on studies showing EC ASIC1 in mesenteric arteries contributes to endothelial-dependent vasodilation (Garcia et al., 2018), we speculate PAEC ASIC1a may be protective against the development of pulmonary hypertension. To test the hypotheses that SM-Asic1a contributes to CH-induced pulmonary hypertension and vascular remodeling and EC-Asic1a opposes the development of CH-induced pulmonary hypertension we will use the Cre-loxP system to generate mice with selective EC-Asic1a deletion (TekCre-Asic1afl/fl) or inducible SM-Asic1a deletion (MHCCreER-Asic1afl/fl).
Materials and methods
Ethical approval
All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Protocol #19-200899-HSC) and abide by the National Institutes of Health guidelines for animal use. All animals were anesthetized with an overdose of pentobarbital sodium (200 mg/kg, i.p.) and immediately euthanized by exsanguination after the loss of consciousness.
Animals
Studies were completed in adult male wildtype (Asic1a+/+) or various transgenic mice (12–16 weeks old) as shown in Table 1. To selectively delete Asic1a in ECs or SM, Asic1afl/fl mice were crossed with TekCre or MHCCreER transgenic mice, respectively. Homozygote and/or heterozygote mice were bred and Cre transgene expression and deletion of the Asic1a gene were confirmed by PCR and agarose gel electrophoresis (Table 1). Animals were housed one to five per cage in a specific pathogen-free (SPF) animal care facility and maintained on a 12:12-h light-dark cycle. Standard chow (Teklad soy protein-free diet #2920, Envigo) and water were provided ad libitum. Animals were randomly allocated to experimental groups and when possible, genotype and treatment assignments were blinded to the investigators. Male mice were studied exclusively since the expression of iCreERT2 under the control of the SM promoter is inserted on the Y chromosome. Furthermore, our previous data showed no significant interaction between sex and the development of hypoxic pulmonary hypertension (Nitta et al., 2014). For induction of Cre activity, MHCCreER-Asic1afl/fl mice were injected with 75 mg/kg tamoxifen (TAM; Sigma-Aldrich, CAS #10540-29–1) in corn oil, once daily for five consecutive days. Following 14 days, Cre recombinase was assessed from tail DNA using the 5′ LoxP forward and 3′ LoxP reverse primer pair (Table 1). Tamoxifen was administered to MHCCreER-Asic1afl/fl mice to induce SM-Asic1a knockout either 1) 2 weeks before CH as a preventative (pTAM) protocol or 2) following 3 weeks CH as a therapeutic (tTAM) protocol to assess reversal of established pulmonary hypertension. We have previously shown that mice develop pulmonary hypertension following 3-weeks CH exposure (Sheak et al., 2020). Asic1a disruption was assessed from total RNA (1 µg) in the brain (positive control) and isolated pulmonary arteries by RT-PCR. Total RNA was extracted using TRIzol and reversed transcribed to cDNA (Transcription First-Strand cDNA Synthesis kit, Roche, 04379012001). Amplification of Asic1a was achieved by PCR (iCycler, Bio-Rad) using REDExtract-N_Amp PCR Ready Mix (Sigma-Aldrich, XNAT) and Asic1 primers: forward: 5′ CACATGCCAGGGGATGCCCC 3′ and reverse: 5′ AGCCGGTGCTTAATGACCTC 3’ (410 bp). The PCR product was separated using gel electrophoresis on a 3% agarose gel and stained with ethidium bromide for visualization under UV light.
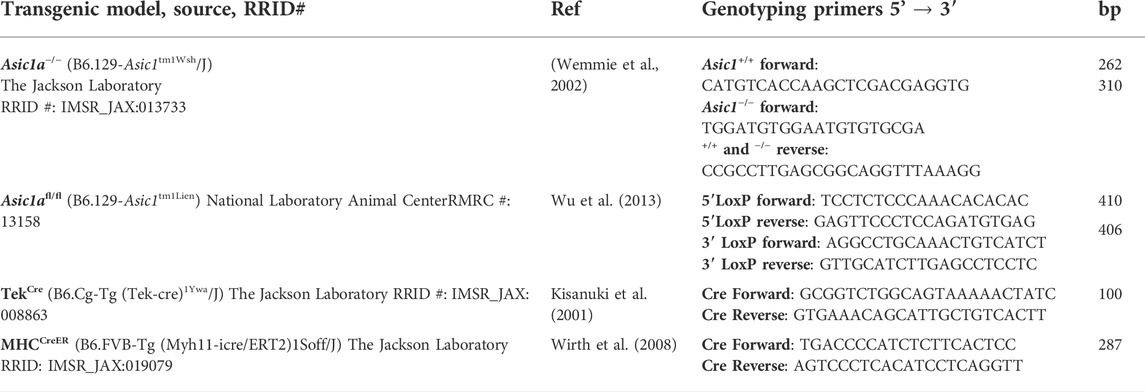
TABLE 1. Transgenic mouse models, source, reference, primers, and expected base pairs to identify each genotype.
Assessment of Systemic Mean arterial Blood Pressure
Blood pressure and heart rate were recorded in mice using radiotelemetry devices (PA-C10 implant; Data Systems International). Telemetry transmitters were surgically implanted under sterile conditions with inhaled isoflurane anesthesia (2% isoflurane and 98% O2 gas mixture). The analgesic Buprenex (buprenorphine; 0.1 mg/kg, IM) was administered before the start of surgery to provide effective recovery and preemptive pain management. Using sterile techniques, a midline incision was made to expose the carotid artery by blunt dissection. A small incision was made in the carotid artery between two silk sutures and the end of the catheter of a PA-C10 small implantable telemetry probe was inserted and advanced toward the heart. The tip was tied in place and the body of the telemeter was secured subcutaneously in the mid-flank region of the carotid artery. The wound was closed with sterile suture and the mouse was allowed to recover 5 days before blood pressure measurements. Blood pressure was recorded for 72 h (every 15 min for 10s-intervals) and data were presented as 24 h averages
Exposure to CH
CH is a common complication of chronic lung diseases and a key stimulus in the development of pulmonary hypertension. Animals designated for exposure to CH were housed in a clear-plexiglass hypobaric chamber (∼0.5 m3) with barometric pressure maintained at ∼380 mmHg for 6 weeks. The hypobaric chamber was partially evacuated with a vacuum pump allowing for continuous airflow of 30 L/min through the chamber. The chamber was opened 2 times a week to change bedding and provide fresh water and food. Age-matched control animals were housed at ambient barometric pressure (∼630 mmHg in Albuquerque, NM). We have previously demonstrated that this mouse model mimics many of the cardiopulmonary changes observed in human pulmonary hypertension including increased right ventricular systolic pressure, right ventricular hypertrophy, enhanced vasoconstriction, and arterial remodeling (Nitta et al., 2014; Detweiler et al., 2019; Sheak et al., 2020).
Immunofluorescence from paraffin-embedded lung tissue
Mice were anesthetized with pentobarbital sodium (200 mg/kg i. p.). After a median sternotomy, heparin (100 U/20 g body wt) was injected directly into the RV, and the pulmonary artery was cannulated with a 22-gauge feeding needle. The preparation was immediately perfused with 0.1 M PBS containing 10–4 M papaverine to maximally dilate the vasculature and flush the circulation of blood. The lungs were then perfused with 25 ml fixative (0.1 M PBS containing 4% sucrose, 4% paraformaldehyde, and 10–4 M papaverine) at a pressure of 50 cm H2O above the hilum, and the trachea inflated to a pressure of 25 cm H2O creating a transmural distending pressure of 25 cm H2O during fixation to ensure vessels were fully dilated. The trachea was ligated with 4–0 silk, and the lungs were immersed in fixative overnight, dehydrated, and then mounted in paraffin.
Sections were cut (5 μm thick) and mounted onto Superfrost Plus slides (Fisher Scientific). Antibody-antigen binding was enhanced by heat-mediated antigen retrieval using either Tris-EDTA Buffer (10 mM Tris, 1 mM EDTA, 0.05% Tween-20, pH 9) for 15 min at 100°C (for Ki-67) or Citric-Acid-Sodium Citrate Buffer (pH 6, 0.05% Tween-20) for 25 min at 100°C (for remodeling and ASIC1 expression). Sections were incubated with primary (24 h at 4°C) and secondary antibodies (24 h at 4°C) as indicated in Table 2. We have previously determined the specificity of goat anti-ASIC1 using wild-type and knockout mice (Nitta et al., 2014). Sections were mounted with FluoroGel (Electron Microscopy Sciences), and cross-section images of pulmonary arterioles (<100 µm) were acquired sequentially by confocal microscopy (TCS SP5, Leica) using Argon (488 nm/∼20 mW, HeNe (543 nm/∼1 mW), and HeNe (633 nm/∼10 mW) class IIIb lasers and a ×63 objective.
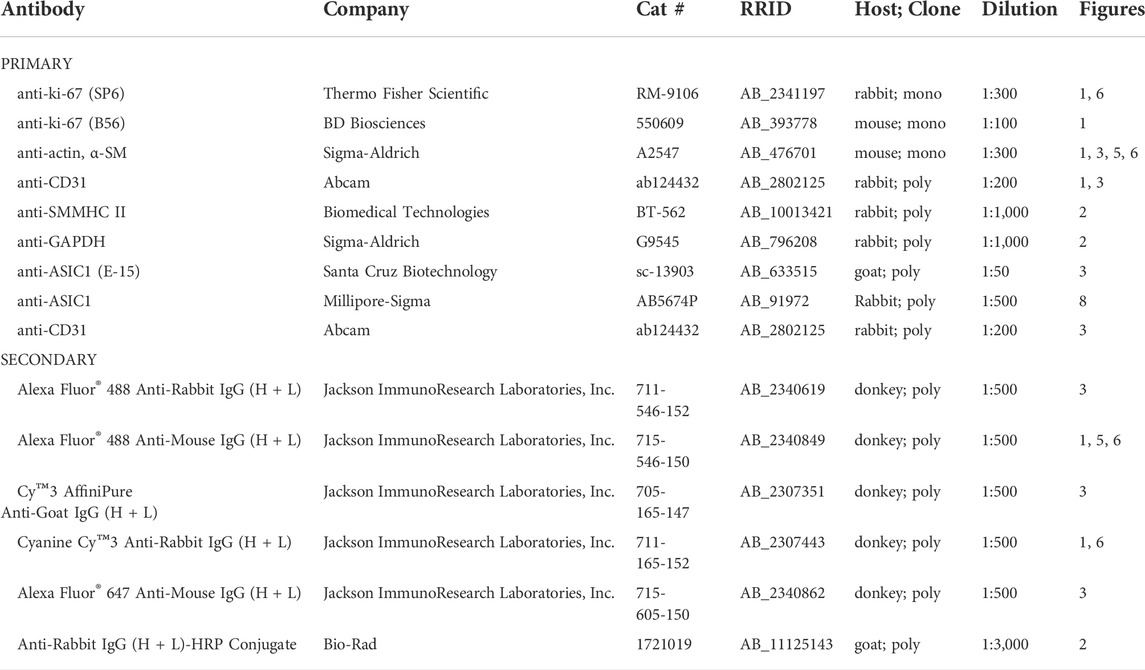
TABLE 2. List of primary and secondary antibodies used for immunofluorescence and western blot analysis.
Assessment of cellular proliferation using Ki-67
Lung sections were incubated with anti-Ki-67 (Table 2) and the percent Ki-67 positive SM and ECs were calculated from ∼15-20 vessels per animal (5 animals/group) using ImageJ software (National Institutes of Health). Vessels were identified by morphology and SMA or CD31 immunofluorescence and nuclei were stained with TO-PRO™-3 iodide (1:1,000; Invitrogen, T3605) for 15 min at room temperature before mounting the sections.
Assessment of cell-specific ASIC1 deletion
Lung sections were incubated with antibodies against ASIC1, SMA, and CD31 (Table 2). Images were taken of five pulmonary arteries per group. Using ImageJ software (NIH), a mask was made of either SMA or CD31 immunofluorescence and the mean intensity of ASIC1 was determined in each mask.
Assessment of arterial remodeling using α-SM actin immunofluorescence
Images were thresholded using ImageJ software. Regions of interest (ROIs) were drawn around each fully muscularized artery. The percent thresholded area to total ROI area was calculated for each artery and multiplied by 100 to get the percent muscularization. Arterial diameter was calculated based on the circumference of the ROI and analysis was conducted by arterial diameter: <25 μm, 25–50 μm, or 50–100 μm. Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence.
Western blot analysis
SM myosin heavy chain (MHC) and GAPDH protein expression were determined by western blot analysis. The whole lung was homogenized in Tris-HCl homogenization buffer (containing 225 mM sucrose, 2 mM Tris-HCL, 2 mM EDTA, 12 µM leupeptin, 1 µM pepstatin A, and 0.3 µM aprotinin) with a glass homogenizer and centrifuged at 10,000 g for 10 min at 4°C to remove insoluble debris. Sample protein concentrations were determined by the Qubit Protein Assay (Life Technologies). Samples were boiled for 5 min in sample buffer and 20 µg of protein was separated by SDS-PAGE (7.5% Tris/glycine) and transferred to a polyvinylidene difluoride membrane. The blot was blocked at room temperature for 1 h with 5% nonfat dry milk then incubated overnight at 4°C in primary antibodies followed by 1 h at room temperature in secondary antibodies (Table 2). Proteins were then detected by autoradiography film (GeneMate) following chemiluminescence labeling (ECL; Pierce, 32209). Quantification of protein expression was done using ImageJ software and MHC expression was normalized to GAPDH. GAPDH was probed subsequently to MHC.
Assessment of pulmonary hypertension
Following 6 weeks CH, mice were anesthetized (2% isoflurane and 98% O2 gas mixture) and right ventricular systolic pressure (RVSP) and heart rate were measured via transdiaphragmatic direct cardiac puncture as previously described (Nitta et al., 2014). An upper transverse laparotomy was performed to expose the diaphragm. A 25-gauge needle, connected to a pressure transducer (model APT300, Harvard Apparatus) through a saline-filled catheter, was inserted into the RV via a closed-chest transdiaphragmatic approach, and the output amplified using a TAM-A bridge amplifier (Hugo Saks Electronik; Harvard Apparatus) and recorded using Powerlab data acquisition and LabChart software (ADInstruments). The derivative of max RV pressure over time (dP/dtmax) provides an index of RV contractility. The pressure-time index is the area under the systolic pressure curve and is indicative of RV workload and oxygen consumption. Right ventricular hypertrophy in response to CH was assessed by measuring the mass ratio of the right ventricle to left ventricle plus septum (Fulton’s index).
Proliferation/migration in PASMCs
Generation of mouse PASMCs (mPASMC)
Animals were anesthetized with pentobarbital sodium (200 mg/kg body weight, IP), and the heart and lungs were removed by midline thoracotomy. Intrapulmonary arteries (∼second–fifth order) were dissected from surrounding lung parenchyma and enzymatically digested by incubating in reduced-Ca2+ Hank’s Balanced Salt Solution (HBSS) containing papain (9.5 U/ml), type-I collagenase (2 mg/ml), dithiothreitol (1 mg/ml), and BSA (2 mg/ml) at 37°C for 20 min. PASMCs were dispersed by gentle trituration with a fire-polished pipette in Ca2+-free HBSS. Freshly dispersed PASMCs were plated on gelatin-coated dishes and cultured in SM Cell Medium (Cell Biologics) containing 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2-95% air at 37°C. Before experiments, PASMCs were cultured for at least 48 h in a serum-free SMC Medium containing insulin, EGF, hydrocortisone, l-glutamine, and 1% penicillin/streptomycin (M2268SF Cell Biologics). Cellular purity was >95%, as assessed by morphological appearance under phase-contrast microscopy and immunofluorescence staining for SM 22α as previously described (Detweiler et al., 2019).
mPASMC Proliferation
To determine the involvement of ASIC1a in proliferation, mPASMCs from Asic1a+/+ and Asic1a−/− mice were incubated with bromodeoxyuridine (BrdU; 10 μM) for 24, 48, and 72 h hypoxia (2% O2, 5% CO2) using a hypoxic incubator subchamber (Biospherix C-Chamber). mPASMCs were fixed and labeled with a conjugated Anti-BrdU FITC antibody (BD Biosciences) to measure BrdU incorporation in newly synthesized DNA of 20,000 events per sample by flow cytometric analysis (LSR-Fortessa flow cytometer with FACSDiva, version 3.0 software; BD Biosciences). PDGF-BB (20 ng/ml, Millipore) was added for 72 h to generate a positive staining control.
mPASMC migration
mPASMC migration was assessed using a modified Boyden chamber (Costar Transwell inserts 6.5 mm diameter, 8.0 µm pore size). mPASMCs were counted using a standard grid assay and plated on the insert at 1 × 105 cells/well in basal media (plus 1% FBS). Basal media was also added to the lower well of the Boyden chamber and the cells were incubated 24 h in normoxia (95% air, 5% CO2) or hypoxia (2% O2, 5% CO2) to stimulate migration. After 24 h, mPASMCs were fixed with 2% paraformaldehyde for 15 min and then stained with Coomassie Blue for 5 min. Cells were washed several times to remove excess Coomassie and images were taken with a ×20 objective on an Eclipse E400 microscope with a DS-Fi1 camera and analyzed using NIS-Elements F 3.0 software (Nikon). Five random brightfield images were taken per well for the total number of cells before the un-migrated PASMCs from the top of the filters were removed with a cotton swab and an additional five images were taken to obtain the number of migrated cells. The Coomassie-stained mPASMCs were used to determine the area of migrated to total mPASMCs, which was multiplied by 100 to get the percent migrated mPASMC (ImageJ).
Human PASMCs (hPASMC; Cascade Biologics, #C-009-5C) were grown on poly-l-lysine-coated plates in Media 231 (Invitrogen, #M231500) with SM growth supplement (Invitrogen, #S00725) in a humidified atmosphere of 5% CO2-95% air at 37°C. hPASMC were used at passage three to five and were treated with vehicle (H2O), amiloride (30 μM, Enzo Life Sciences), or psalmotoxin 1 (PcTX1; 20 nM, Phoenix Pharmaceuticals) for the duration of the following experiments.
ASIC1 expression in hPASMC
Total and cell surface expression of ASIC1 was determined by western blot analysis (see above). hPASMC were grown until ∼90% confluent in 75-cm2 flasks and exposed to 12 h normoxia (95% air, 5% CO2) or hypoxia (2% O2, 5% CO2). To determine plasma membrane localization of ASIC1, we used a cell surface protein isolation kit (Pierce, Thermo Fisher Scientific) as previously described (Herbert et al., 2016, 2018). hPASMCs were incubated with Sulfo-NHS-SS-Biotin (Pierce) for 30 min at 4°C. The reaction was quenched and hPASMCs were harvested and lysed with 10 mM TrisHCl homogenization buffer and spun at 10,000 g for 2 min. The clarified supernatant was added to NeutrAvidin Agarose resin columns for 1 h a room temperature. The flow-through was collected as the cytosolic protein fraction, and surface protein was collected by elution with 5× sample buffer. We previously demonstrated the specificity of cell surface assay to fractionate cell surface vs intracellular proteins (Herbert et al., 2016). Surface protein (25 μl) or cytosolic protein lysates (20 μg) were separated by SDS-PAGE (7.5% Tris/glycine) and transferred to PVDF membranes. ASIC1 was detected in cell surface and cytosolic fractions by exposure of the blot to chemiluminescence-sensitive film (GeneMate). Quantification of ASIC1 bands was accomplished by densitometric analysis of scanned images (ImageJ) and expressed as the ratio of plasma membrane to cytosolic densitometric units.
hPASMC migration
An in vitro scratch assay was performed on confluent monolayers of hPASMCs. Monolayers were manually scraped with a 100 µL pipette tip and then gently washed twice with PBS to remove non-adherent cells. Images of the wounded area were captured immediately after the scratch (time zero) and following a 12 h exposure to normoxia (95% air, 5% CO2) or hypoxia (2% O2, 5% CO2). Images were taken with a ×20 objective on an Eclipse E400 microscope with a DS-Fi1 camera and analyzed using NIS-Elements F 3.0 software (Nikon). A grid attached to the bottom of the cell culture plate was used as a reference point to capture images of the same location at each time interval. The wounded area was determined using ImageJ (National Institutes of Health). Healing was quantified as % Reinvasion = (AreaI–AreaT)/AreaI × 100%, where: AreaI = Initial area, and AreaT = Area at time (T) 12 h after injury.
hPASMC proliferation
hPASMCs were trypsinized and the cell suspension was mixed with equal parts Trypan blue solution to a final concentration of 0.4% to assess cell viability. A homogenous mixture was loaded into a disposable chamber side and the cell number was determined using the Countess automated cell counter (Invitrogen).
Statistics
All data are expressed as means ± standard error. Percentage data were converted to normal distributions by arcsine transforms before parametric analysis. Normal distribution was tested using the Shapiro-Wilks Normality Test (p > 0.05). Values of n and statistical tests are specified in the figure legends and were made using Prism 9 (GraphPad Software). A probability of ≤ 0.05 with a power level of 0.80 was accepted as statistically significant for all comparisons.
Results
CH-induced vascular cell proliferation and phenotypic switch is ASIC1a dependent
Ki-67 is a nuclear protein that is expressed during cellular proliferation. To examine in vivo proliferation of vascular cells over the development of CH-induced pulmonary hypertension we determined the percent Ki-67 positive PAECs and PASMCs cells following a 0-, 3-, 7-, and 28-day exposure to CH in Asic1a+/+ mice (Figure 1A). The number of proliferating PAECs and PASMCs was highest following 3 days CH (Figure 1B). The percent of proliferating PAECs and PASMCs was still elevated by 7 days, but PASMC proliferation decreased by half. The percent proliferating PAECs and PASMCs at 28 days was not significantly different compared to baseline (Figure 1B), as the majority of proliferating cells at 28 days were extravascular cells. Based on these data, we then examined PAEC and PASMC proliferation in Asic1a−/- mice following 3 days CH. The percent of proliferating PAECs was significantly reduced in Asic1a−/- mice but was elevated compared to controls (Figure 1C). Moreover, there was no effect of CH to induce proliferation of PASMCs in Asic1a−/- mice (Figure 1D).
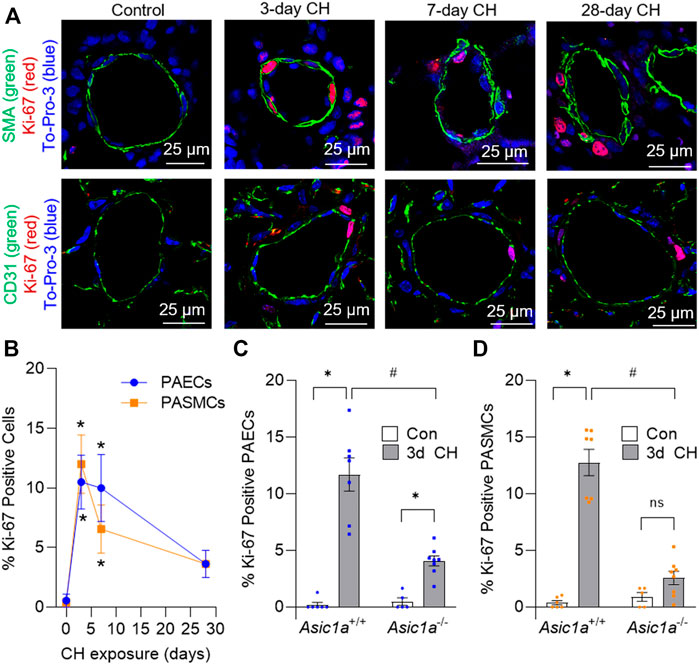
FIGURE 1. CH-induced proliferation of vascular cells is ASIC1a dependent. (A) Representative immunofluorescence images showing Ki-67 (red), α-SMA (green, top row), CD31 (green, bottom row), and To-Pro-3 (blue) in small pulmonary arteries (<100 µm) from Asic1a+/+ mice and (B) summary data showing the percent Ki-67 positive pulmonary arterial endothelial cells (PAECs, blue circles) and pulmonary arterial smooth muscle cells (PASMCs, orange squares) under control conditions or following exposure to CH (3-, 7-, or 28-days). n = 3 animals per group (∼20 vessels were averaged for each animal); analyzed as one-way ANOVA for each cell type and individual groups compared with Šídák’s multiple comparisons tests. Summary data showing percent Ki-67 positive (C) PAECs and (D) PASMCs in small pulmonary arteries (<100 µm) from Asic1a+/+ and Asic1a−/- mice under control conditions or following 3-days CH exposure. n = five to eight animals (∼15 vessels were averaged for each animal); analyzed by two-way ANOVA. Significant interactions between the individual groups (p < 0.0001 for both PAECs and PASMCs) were compared with Šídák’s multiple comparisons tests; *p < 0.05 vs. control; #p < 0.05 vs. corresponding 3-days CH; ns = not significant.
We next determined if the CH-induced increase in PASMC proliferation is associated with a loss in contractile phenotype by analyzing the protein expression of SM myosin heavy chain (MHC) in lung tissue at 0- (Con), 1-, 2-, 3-, 5-, and 7-days CH (Figure 2A). Consistent with the greatest percent of Ki-67 positive PASMCs following 3 days CH (Figure 1B), MHC was significantly decreased after 3-days CH in Asic1a+/+ animals (Figures 2B,C). After 7 days of CH, MHC was increased compared to control levels (Figure 2B). CH did not decrease MHC levels in Asic1a−/- mice (Figure 2C). Hypoxia did not change expression levels of GAPDH (p = 0.2082). Together, these data suggest ASIC1a contributes to PAEC and PASMC proliferation and PASMC phenotypic change that is seen in pulmonary arteries in response to CH exposure.
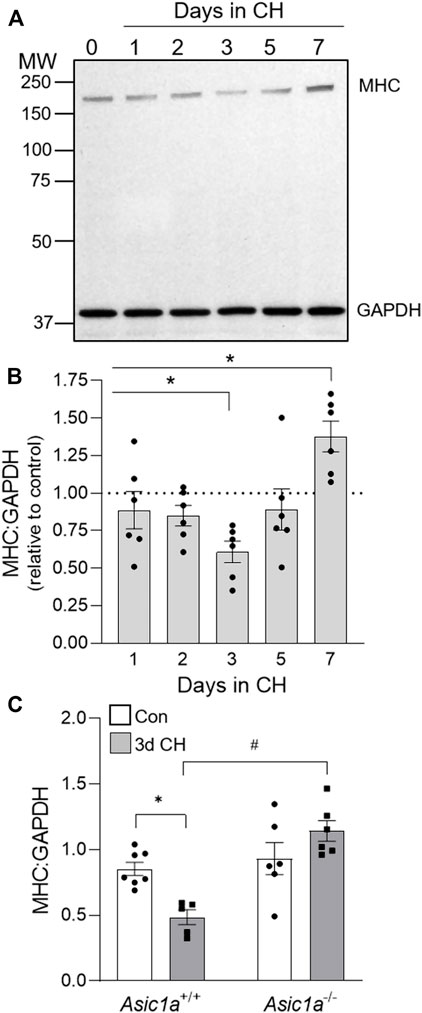
FIGURE 2. CH-induced loss of contractile protein, MHC, is ASIC1a dependent. (A) Representative western blot and (B) the effect of CH exposure (1–7 days) on MHC to GAPDH protein expression in whole lung homogenates in Asic1a+/+ animals. The scanned image of the film was converted to greyscale and adjusted for brightness/contrast. n = 6/group; analyzed by one-way ANOVA and individual groups compared with Šídák’s multiple comparisons tests. (C) Summary data for MHC to GAPDH in whole lungs from Asic1a+/+ and Asic1a−/- mice under control conditions or following 3-days CH exposure. n = 5-7 animals/group; analyzed by two-way ANOVA. Significant interactions between the individual groups (p = 0.0022) were compared with the Šídák’s multiple comparisons tests; *p < 0.05 vs. control; #p < 0.05 vs. Asic1a+/+ mice.
SM-specific knockout of Asic1a protects against the development of and reverses hypoxic pulmonary hypertension
To determine the specific role of ASIC1a in PAEC and PASMC remodeling in pulmonary hypertension, we generated mice with either EC (TekCre-Asic1afl/fl) or conditional SM (MHCCreER-Asic1afl/fl) specific deletion of Asic1a. As demonstrated previously in Asic1a+/+ and Asic1a−/- mice (Nitta et al., 2014), ASIC1 was detected as punctate fluorescence within the PASMCs and PAECs from Asic1afl/fl and MHCCreER-Asic1afl/fl (without TAM-induced Cre recombinase) mice (Figures 3A,B). Line profile through the vessel wall shows that TekCre-Asic1afl/fl mice lack expression of ASIC1 in ECs but retain PASMC expression, whereas MHCCreER-Asic1afl/fl (TAM) (with TAM-induced Cre recombinase) lack expression of ASIC1 in PASMCs but retain PAECs expression (Figures 3A–D). Figure 3E shows TAM-induced Cre recombination between loxP sites and loss of the intervening genomic sequence (exons 2-3) in tail DNA before and after TAM in the same animal. ASIC1 is highly expressed in the central nervous system and Figure 3F shows that TAM-induced Cre recombinase did not significantly alter Asic1a mRNA levels in brain tissue, but there was no detectable expression in isolated pulmonary arteries (PA).
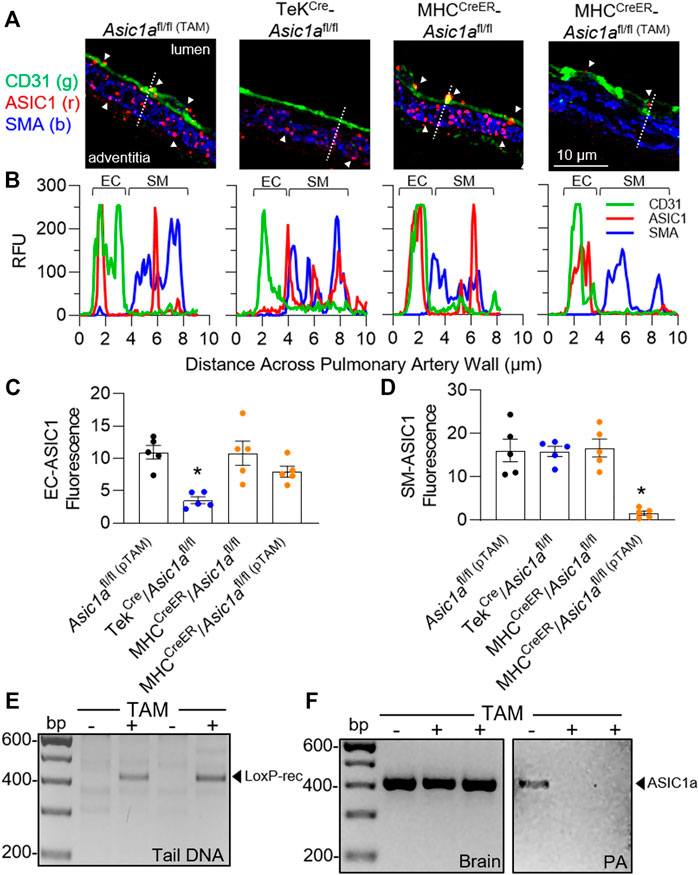
FIGURE 3. Characterization of transgenic mice. (A) Representative immunofluorescence for ASIC1 (red), CD31 (green), and SMA (blue) in pulmonary arteries from lung sections of Asic1afl/fl (TAM), TekCre-Asic1afl/fl, and MHCCreER-Asic1afl/fl mice treated with vehicle or tamoxifen (TAM). White arrowheads show punctate immunofluorescence of ASIC1 in EC or SM. The dotted line in each image indicates the line profile shown in (B) of relative fluorescence units across the artery wall from lumen to adventitia. Summary analysis of ASIC1 expression in either (C) EC or (D) SM of pulmonary arteries from lung sections. n = 5 animals per group; analyzed by one-way ANOVA and individual groups compared with Šídák’s multiple comparisons tests. (E) PCR of tail DNA showing Cre-mediated LoxP recombination and excision of targeted Asic1a in the same MHCCreER-Asic1afl/fl mouse before (-) and after (+) tamoxifen (TAM) treatment. (F) Asic1a mRNA expression in brain tissue and intrapulmonary arteries (PA) in MHCCreER-Asic1afl/fl mice with vehicle (-) or TAM (+). Scanned images of the blots were inverted and adjusted for brightness/contrast.
To determine the role of PAEC and PASMC ASIC1a in CH-induced pulmonary hypertension we developed three different CH treatment paradigms represented in Figures 4A, 1) vehicle: Asic1a+/+, Asic1a−/-, TekCre-Asic1afl/fl, and MHCCreER-Asic1afl/fl mice were treated with vehicle (corn oil) and 2 weeks later exposed to control or CH for 6 weeks; 2) preventative (pTAM): Asic1afl/fl (pTAM) and MHCCreER-Asic1afl/fl (pTAM) mice were treated with TAM (5 days) and 2 weeks later exposed to control or CH for 6 weeks; 3) therapeutic (tTAM): MHCCreER-Asic1afl/fl (tTAM) mice were exposed to control or CH for 3 weeks to establish pulmonary hypertension. After 3 weeks CH, mice were treated with TAM (5 days with con/CH exposure) and then continued in con/CH for an additional 2 weeks. Table 3 demonstrates that selective deletion of SM- or EC-Asic1a did not significantly alter mean arterial blood pressure or heart rate in conscious mice and is similar to what we have previously recorded in wildtype mice (Detweiler et al., 2019).
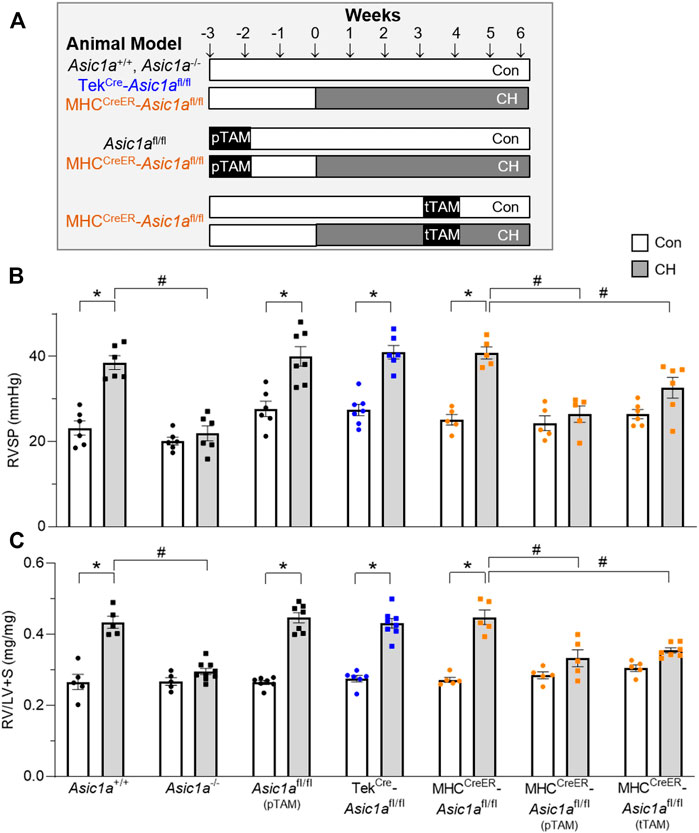
FIGURE 4. SM-specific knockout of Asic1a protects against development of pulmonary hypertension and reverses established hypoxic pulmonary hypertension. (A) Experimental design showing treatments of no TAM, preventative (pTAM, administered before CH), and therapeutic (tTAM, administered following established pulmonary hypertension). (B) Right ventricular systolic pressure (RVSP, mmHg) and (C) Fulton’s Index (RV/LV + S) in Asic1a+/+, Asic1a−/-, Asic1afl/fl (pTAM), TekCre-Asic1afl/fl, MHCCreER-Asic1afl/fl, MHCCreER-Asic1afl/fl (pTAM), or MHCCreER-Asic1afl/fl (tTAM) mice under control conditions (white bars, circles) or following 6 weeks CH (grey bars, squares). SM-Asic1a knockout was induced by treatment with tamoxifen as a preventive (pTAM; before exposure to CH) or therapeutic approach (tTAM; following establishment of CH-induced pulmonary hypertension). n = 5-8/group; analyzed by two-way ANOVA. Significant interactions between the individual groups (p < 0.0001 for both RVSP and RV/LV + S) were compared with Šídák’s multiple comparisons tests; *p < 0.05 vs. control; #p < 0.05 vs. respective genetic control.

TABLE 3. Mean arterial blood pressure (MABP) and heart rate in genetically-modified mice treated with or without tamoxifen (pTAM).
Similar to our previous reports, exposure to CH significantly increased right ventricular systolic pressure (RVSP; Figure 4B) and right heart hypertrophy (Figure 4C) in Asic1a+/+, but not Asic1a−/- mice (Nitta et al., 2014). CH led to a similar increase in RVSP and RV hypertrophy in Asic1afl/fl (pTAM), TekCre-Asic1afl/fl, and MHCCreER-Asic1afl/fl mice (Figure 4). Along with serving as a control for TekCre-Asic1afl/fl and MHCCreER-Asic1afl/fl mice, the Asic1afl/fl (pTAM) mice also provide evidence that TAM does not affect the development of pulmonary hypertension. Table 4 shows that CH does not affect body mass or heart rate in any of the transgenetic animals. However, the increase in RVSP and RV hypertrophy in Asic1afl/fl (pTAM), TekCre-Asic1afl/fl, and MHCCreER-Asic1afl/fl mice is associated with greater RV contractility and workload as indicated by increased dP/dtmax and pressure time index, respectively. These data suggest EC-specific deletion of Asic1 does not contribute to increased RVSP and RV hypertrophy following CH. In contrast, SM-specific deletion of Asic1a in MHCCreER-Asic1afl/fl (pTAM) mice by TAM-induced Cre recombinase before CH prevented any CH-induced increases in RVSP, RV hypertrophy, dP/dtmax, and pressure time index (Figure 4 and Table 3). Additionally, treating MHCCreER-Asic1afl/fl (tTAM) with TAM at week three of the 6-week CH exposure reversed increases in RVSP, RV hypertrophy, dP/dtmax, and pressure-time index to similar levels as control mice (Figure 4 and Table 3). These data demonstrate that deletion of SM-, but not EC-Asic1a both prevents and reverses CH-induced pulmonary hypertension, and RV hypertrophy and dysfunction.
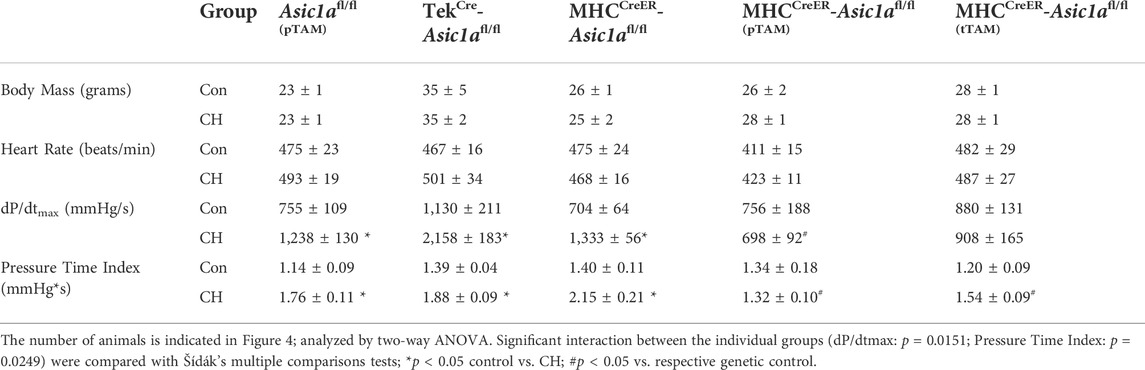
TABLE 4. Body mass, heart rate, cardiac contractility, and pressure-time index in anesthetized control and CH genetically-modified mice treated with or without tamoxifen (TAM).
SM-specific knockout of Asic1a attenuates vascular remodeling
Vascular remodeling was assessed using immunofluorescence of SM α-actin (SMA) in vessels ranging from <25 μm, 25–50 μm, and 50–100 μm, as shown in Figure 5A. CH caused a significant increase in % muscularization in each artery size from TekCre-Asic1afl/fl mice. Some vessels (∼5–10%) from TekCre-Asic1afl/fl mice exposed to CH displayed hypercellular lesions projecting outward from the medial and adventitial layers into the adjacent lung parenchyma (Figure 5A). Although this adventitial remodeling was not analyzed as part of the medial thickness, the cells within these areas were observed to express SMA at a lower fluorescence intensity compared to the medial layer, likely representing (myo)fibroblasts. This outward remodeling was not present in wildtype or other transgenic mouse models (Nitta et al., 2014; Detweiler et al., 2019; Sheak et al., 2020), suggesting specific EC deletion of Asic1a may facilitate vascular remodeling. CH increased % muscularization in MHCCreER-Asic1afl/fl mice (no TAM-induced Cre recombinase) that was attenuated by both pTAM and tTAM treatments (Figure 5B). SM-specific deletion of Asic1a had a greater effect to reduce (neo)muscularization in arteries <25 µm as there was not a significant difference compared to control arteries. Furthermore, deletion of SM-Asic1a therapeutically was more effective in reducing arterial muscularization than preventative SM-Asic1a deletion. In 50–100 µm arteries from MHCCreER-Asic1afl/fl (tTAM) mice, muscularization was not significantly different between control and CH (Figure 5B).
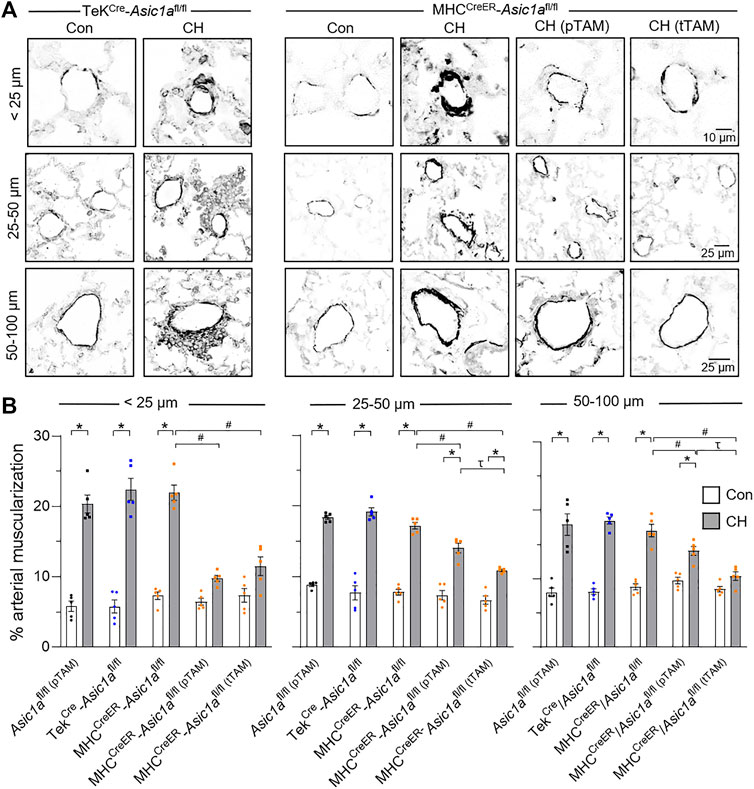
FIGURE 5. SM ASIC1a contributes to vascular remodeling following CH. (A) Representative SMA immunofluorescence (black) images of small pulmonary arteries in lung sections from TekCre-Asic1afl/fl, MHCCreER-Asic1afl/fl, MHCCreER-Asic1afl/fl (pTAM), or MHCCreER-Asic1afl/fl (tTAM) mice under control conditions (white bars) or following 6 weeks CH (filled bars). Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence. (B) Percent muscularization calculated as percent thresholded SMA area divided by total arterial area based on arterial diameter: <25 μm (n = ∼30 vessels from four animals/group), 25–50 μm (n = ∼100 vessels from four animals/group), or 50–100 μm (n = ∼50 vessels from four animals/group); analyzed by two-way ANOVA. Significant interactions between the individual groups (p < 0.0001 for all vessel diameter ranges) compared with Šídák’s multiple comparisons tests. *p < 0.05 vs. control; #p < 0.05 vs. (-) TAM; and τ p < 0.05 pTAM vs. tTAM.
PAEC and PASMC proliferation in MHCCreER-Asic1afl/fl mice was additionally evaluated using immunofluorescence to identify Ki-67 positive cells (Figure 6). Deletion of SM-Asic1a did not significantly affect the % of positive Ki-67 nuclei in PAECs, but significantly reduced the percent of proliferating PASMCs (Figure 6B). Together these data show that SM-Asic1a contributes to CH-induced vascular remodeling by contributing to both muscularization and PASMC proliferation.
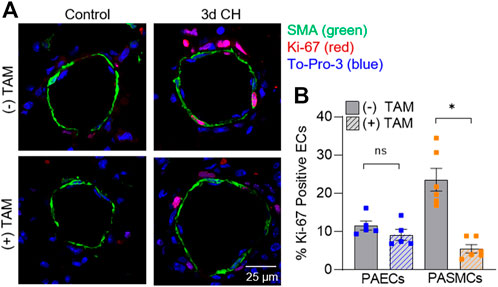
FIGURE 6. SM ASIC1a contributes to CH-induced PASMC proliferation. (A) Representative immunofluorescence images showing Ki-67 (red), α-SMA (green), and Sytox (blue) in small pulmonary arteries (<100 µm) from control and 3 days CH MHCCreER-Asic1afl/fl mice following treatment with vehicle or tamoxifen (TAM). (B) Summary data for percent Ki-67 positive cells in endothelial cells (EC, blue) and smooth muscle cells (SMC, orange). Summary data for control mice are not shown since there were no detectable Ki-67 positive cells. n = 75 vessels from five animals (15 vessels each) per group; analyzed by unpaired t-test; #p < 0.05 vs. (-) TAM 3-days CH; ns = not significant.
ASIC1 contributes to PASMC migration and proliferation
PASMCs were exposed to in vitro hypoxia, followed by assessing migration and proliferation via transwell assays and flow cytometry for BrdU-positive cells, respectively. Hypoxia significantly increased the percent of migrating mPASMCs from Asic1a+/+, but not Asic1a−/- mice (Figures 7A,B). Proliferation, as assessed by BrdU incorporation, was significantly less in mPASMCs from Asic1a−/- mice under normoxia and following exposure to 24 and 48 h hypoxia (Figure 7C).
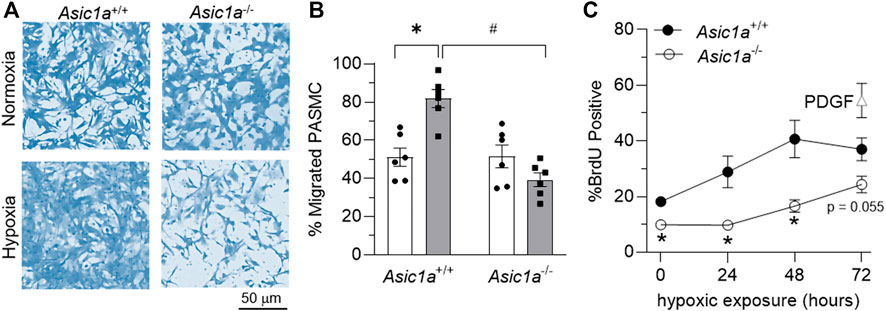
FIGURE 7. ASIC1a contributes to mPASMC migration and proliferation. (A) Representative brightfield images of coomassie-stained mPASMC and (B) summary data for % of migrated mPASMC from Asic1a+/+ and Asic1a−/- mice exposed to normoxia (5% CO2, 95% air; orange circles) or hypoxia (5% CO2, 2% O2; blue squares) for 24 h n = 6 animals/group; *p < 0.05 vs. hypoxia and #p < 0.05 Asic1a+/+; analyzed by two-way ANOVA. Significant interactions between the individual groups (p = 0.0003) compared using Šídák’s multiple comparisons tests. (C) Flow cytometry analysis of percent of mPASMC from Asic1a+/+ and Asic1a−/- mice with BrdU incorporation under normoxia (0 h; 5% CO2, 95% air) or hypoxia (5% CO2, 2% O2) for 24, 48, or 72 h. Normoxic mPASMC from Asic1a+/+ were incubated with PDGF-BB (20 ng/ml) for 72 h as a positive control. n = five to eight animals/group, analyzed by unpaired t-tests at each time point, *p < 0.05.
To determine if these findings translate to human PASMCs (hPASMCs) we assessed the effects of hypoxia on ASIC1 expression, migration, and proliferation in hPASMCs in the absence or presence of ASIC1 inhibition. Although 12 h hypoxia did not alter the total expression of ASIC1 (Figures 8A,B), it significantly increased the plasma membrane (cell surface) compared to cytosolic expression (Figure 8C). This is consistent with previous studies in 4 weeks CH-exposed rats where total expression of ASIC1 is unchanged but hypoxia causes a subcellular translocation of ASIC1 to the plasma membrane (Herbert et al., 2016, 2018). The percent reinvasion of hPASMCs was greater following 12 h hypoxia compared to normoxia and this was prevented by pre-treatment with amiloride or PcTX1 (Figures 8D,E). Hypoxia-induced proliferation was additionally blocked by amiloride and PcTX1 in hPASMCs (Figure 8F). Taken together, these data suggest ASIC1 involvement in hypoxia-mediated hPASMC proliferation and migration.
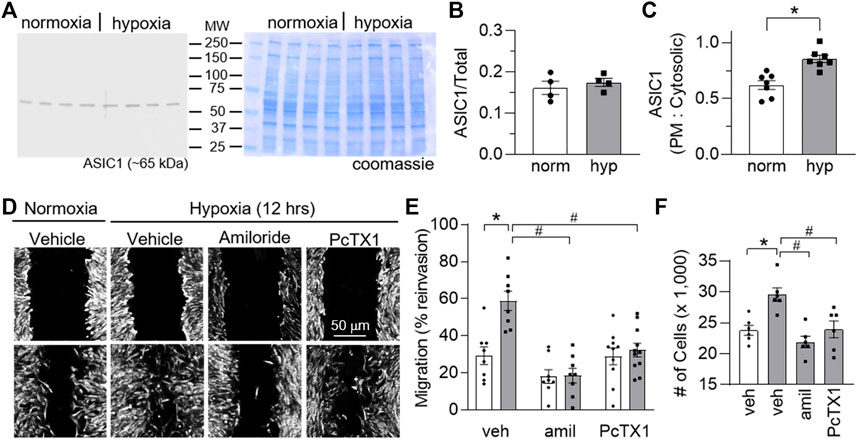
FIGURE 8. ASIC1 contributes to human PASMC migration and proliferation. (A) Representative Western blots of ASIC1 (predicted ∼65 kDa) and corresponding coomassie-stained blot and (B) summary data showing ASIC1 expression (normalized to entire lane on coomassie-stained blot) in human PASMC (hPASMC) exposed to normoxia (norm, 5% CO2, 95% air; white bars/circles) or 12 h hypoxia (hyp, 5% CO2, 2% O2; grey bars/squares). Scanned images of the film were converted to greyscale and adjusted for brightness/contrast. (C) Summary data showing Western blot analysis for biotinylated (plasma membrane) and cytosolic ASIC1 protein expression in hPASMC exposed to normoxia or hypoxia. (D) Brightfield images of hPASMC immediately following the scratch (baseline time 0; top row of images) or 12 h post-exposure (bottom row of images) to normoxia (5% CO2, 95% air; orange circles) or hypoxia (5% CO2, 2% O2; blue squares) and (E) summary data showing percent reinvasion of hPASMC into the wounded area in the presence of vehicle, amiloride or PcTX1. n = 8-11/group; analyzed by two-way ANOVA. Significant interactions between the individual groups (p = 0.0032) were compared using Šídák’s multiple comparisons tests. (F) increase in the number of cells after 24 h of normoxia or hypoxia in the presence of vehicle, amiloride, or PcTX1. hPASMC were initially plated at a density of 15,000 cells/well. n = 6/group; analyzed by one-way ANOVA and individual groups compared using Šídák’s multiple comparisons tests. *p < 0.05 vs. normoxic group; #p < 0.05 vs. corresponding vehicle group.
Discussion
Our laboratory has previously demonstrated that ASIC1a contributes to the development of CH-induced pulmonary hypertension by contributing to enhanced agonist-induced vasoconstriction and vascular remodeling (Nitta et al., 2014). In the pulmonary circulation, ASIC1 is expressed in both PASMCs and PAECs and the goal of the current study was to determine if there is a differential contribution of EC and SM ASIC1 to the development of pulmonary hypertension. We found that specific deletion of EC-Asic1a did not affect the development of pulmonary hypertension; whereas deletion of SM-Asic1a prevented CH-induced pulmonary hypertension and reduced medial vascular remodeling. This reduction in remodeling was associated with decreased proliferation and migration of PASMCs from Asic1a−/- mice. We further demonstrate that deletion of SM-Asic1a in mice with established pulmonary hypertension effectively reversed increases in RVSP, RV hypertrophy, and vascular remodeling, signifying ASIC1 as a potential therapeutic target for pulmonary hypertension.
In the pulmonary circulation, ASIC1 is activated in response to various vasoactive factors (endothelin-1, UTP) and alveolar hypoxia resulting in PASMC Ca2+ influx and pulmonary arterial constriction (Jernigan et al., 2012; Nitta et al., 2014). Inhibition of ASIC1 or Asic1a gene deletion abolishes the enhanced agonist-induced vasoconstriction following CH. The activation of ASIC1a in PASMCs following stimulation of G-protein coupled receptors appears to be independent of pH changes. Although we do not know the exact mechanism leading to non-proton activation of ASIC1, our previous work demonstrates ASIC1a is activated secondary to store-depletion of the sarcoplasmic reticulum, a mechanism referred to as store-operated Ca2+ entry (Jernigan et al., 2009, 2012). We have also demonstrated that ASIC1 contributes to acute hypoxic pulmonary vasoconstriction (Nitta et al., 2014) and the persistent PASMC membrane depolarization following CH exposure (Jernigan et al., 2021). Despite the requirement for ASIC1 in the development of pulmonary hypertension, this response is not dependent on an increase in total ASIC1 protein expression. Rather hypoxia causes subcellular relocalization of ASIC1 to the plasma membrane (Nitta et al., 2014; Herbert et al., 2018), a response that occurs in hPASMC as early as 12 h of hypoxic exposure (Figure 8). Furthermore, we have recently demonstrated that primary-cultures of PASMC from pulmonary hypertensive animals show a shift in cellular metabolism that promotes glycolysis and lactic acid fermentation leading to extracellular acidification (Tuineau et al., 2022). Further research is necessary to determine the importance of this pH shift to activate ASIC1 in pulmonary hypertension.
Although Asic1a−/− mice are also protected from CH-induced vascular remodeling and right ventricular hypertrophy (Nitta et al., 2014), it is unclear if ASIC1a is directly involved in the remodeling process or whether ASIC1a indirectly promotes remodeling by increasing vasoconstriction and pulmonary vascular resistance. The current findings that ASIC1a contributes to hypoxia-induced PASMC proliferation and migration support a direct contribution of ASIC1a to vascular remodeling. These data corroborate several studies showing that ASIC1, expressed in a variety of cancers, plays a role in regulating multiple malignant processes including proliferation, migration, epithelial-mesenchymal transition, and cell cycle progression (Kapoor et al., 2009; Rooj et al., 2012; Jin et al., 2015; Wu et al., 2017; Zhu et al., 2017; Chen et al., 2018; Ding et al., 2021; Zhu et al., 2021). Conversely, prevention of right ventricular hypertrophy following deletion of SM-Asic1a suggests cardiomyocyte remodeling in this mouse model of hypoxic pulmonary hypertension largely occurs due to the role of ASIC1a to increase pulmonary vascular resistance.
Early studies proposed that the initial increase in pulmonary vascular resistance in response to hypoxic exposure is largely due to hypoxic pulmonary vasoconstriction; whereas the structural changes in the pulmonary vascular bed following sustained exposure to hypoxia are the major determinant of elevated vascular resistance with disease progression (Sime et al., 1971; Lockhart et al., 1976; Fried et al., 1983). Interestingly, however, studies show no active PASMC proliferation in end-stage lung tissue from idiopathic and hereditary pulmonary arterial hypertensive patients (Majka et al., 2008) suggesting active proliferation occurs early in the disease process as we observed in mice. Although the degree of CH-induced pulmonary hypertension, right ventricular hypertrophy, and pulmonary vascular remodeling (mainly medial muscularization) in mice is modest compared to some other species, the same cellular processes seem to be involved and genetically modified mice allow us to investigate the function of specific proteins in pulmonary hypertension. Furthermore, our data is consistent with other studies, showing actively proliferating PASMCs and PAECs within the first 3–5 days of hypoxic exposure that subsides by 4 weeks CH (Meyrick & Reid, 1979; Quinlan et al., 2000; Paddenberg et al., 2007; Nozik-Grayck et al., 2008; Bierer et al., 2011). This increase in proliferating vascular cells at 3-days CH corresponds with a significant decrease in lung SM MHC expression suggesting PASMC phenotypic switching–the transition from the quiescent contractile to the proliferative synthetic phenotype (Owens, 2007). Previous research in our laboratory demonstrates that the Ca2+/calcineurin-dependent transcription factor known as nuclear factor of activated T cells isoform-3 (NFATc3) is required for CH-induced pulmonary arterial remodeling. This process involves an initial proliferation of PASMC (dedifferentiation) followed by differentiation (upregulation of differentiation marker soluble guanylyl cyclase α1) and hypertrophy of PASMC (upregulation of SMA) (de Frutos et al., 2007, 2009; Bierer et al., 2011). Importantly, we showed that ASIC1-dependent Ca2+ influx stimulates NFATc3 activation following 5-days CH, providing an essential link between activation of ASIC1a and transcriptional regulation of PASMC phenotypic transformation (Gonzalez Bosc et al., 2016). Whether ASIC1a regulates other transcription factors essential to pulmonary vascular remodeling, like FOXM1 (Dai J et al., 2018; Dai Z et al., 2018), requires further investigation.
PASMCs play a central role in vascular remodeling due to the remarkable ability to dynamically modulate their phenotype to ensure contractile and synthetic functions (Wang et al., 2015; Roostalu et al., 2018). TAM-induced deletion of SM-Asic1a normalized RVSP in CH-exposed MHCCreER-Asic1afl/fl mice to near control levels in both preventative and therapeutic protocols. Although knockdown of SM-Asic1a effectively eliminated remodeling in intra-acinar vessels (<25 µm), there was still a considerable degree of muscularization in small arteries (25–100 µm). This could signify a different contribution of ASIC1a to hypertrophy, hyperplasia (proliferation), and migration and how these remodeling processes differ in pre-capillary (intra-acinar) versus small arteries. Hypertrophy plays a large role in overall medial thickening and we have previously shown that PASMCs from Asic1a−/- mice do not exhibit CH-induced hypertrophy (Jernigan et al., 2021). Using [3H]-thymidine uptake as a marker of proliferation, previous studies in hypoxic-exposed rats have demonstrated that SM cell proliferation doubles in large pulmonary arteries while intra-acinar arteries do not show evidence of [3H]-thymidine uptake (Meyrick & Reid, 1979). Rather, the remodeling of the intra-acinar arteries involves the appearance of cells expressing SM-specific markers in normally non-muscular vessels. This is thought to be mediated mainly by distal migration of nonproliferative SM cells and differentiation of existing precursor SM cells and/or pericytes (Meyrick & Reid, 1979). ASIC1a may contribute to migration more than proliferation, as suggested by the in vitro assay in which BrdU incorporation at 72 h hypoxia was not statistically different between PASMCs from Asic1a+/+ and Asic1a−/- mice (p = 0.055; Figure 7).
Mice develop moderate PH and right ventricular hypertrophy when exposed to CH and this is associated with modest pulmonary vascular remodeling (mainly medial muscularization). As such, it is worth noting the limitations of assessing the role of PASMC ASIC1a in CH-induced remodeling in MHCCreER-Asic1afl/fl mice. First, other SM precursors, pericytes, and/or (myo)fibroblasts contribute to vascular remodeling following CH. These “dedifferentiated” SM-like cells lack expression of MHC, which drives Asic1a deletion in MHCCreER-Asic1afl/fl mice. This may explain why remodeling is more effectively inhibited in global Asic1a−/- (Nitta et al., 2014) compared to MHCCreER-Asic1afl/fl mice. Indeed, evaluation of SM cell profile using lineage tracing shows very few mature medial MYH11+ SM cells within the cell population of atherosclerotic lesions (Chappell et al., 2016; Jacobsen et al., 2017; Misra et al., 2018). Second, deletion of SM-Asic1a therapeutically (tTAM) was more effective in reducing arterial muscularization than preventative SM-Asic1a deletion (pTAM). Although MHC expression is decreased following 3 days of CH when cells are proliferating, MHC and SMA (de Frutos et al., 2007) expression are upregulated at 7 days of CH exposure. This suggests the newly proliferated cells are transitioning to contractile SM leading to more PASMC in the medial layer and a greater number of PASMC to target with MHC-driven gene recombinase. Importantly, our data demonstrate that deletion of SM-Asic1a in established pulmonary hypertension not only halts remodeling but leads to reversal of the remodeling process. Further studies are necessary to determine the mechanism of this reversal.
Interactions between PASMCs and PAECs are essential for the maintenance of PASMC phenotype, and PAEC dysfunction in pulmonary hypertension leads to proliferation and migration of resident vascular cells and induces a PASMC phenotypic switch (Humbert et al., 2008). Although the functional role of ASIC1a in PAECs is unknown; mesenteric endothelial cell ASIC1a contributes to endothelial-dependent vasodilation through activation of intermediate- and small-conductance Ca2+-activated K+ channels (Garcia et al., 2018). Based on these studies, we anticipated selective loss of EC-Asic1a would lead to endothelial dysfunction and exacerbate pulmonary hypertension. On the contrary, TekCre-Asic1afl/fl mice develop pulmonary hypertension comparable to Asic1a+/+ (or Asic1afl/fl) mice. Despite a similar RVSP, however, we observed that remodeled pulmonary arteries from TekCre-Asic1afl/fl mice had a more advanced outward remodeling than typically observed in mice. Similar prominent outward remodeling has been noted in rats exposed to SU5416/CH (Jernigan et al., 2017), cows with Brisket’s disease [hypoxic pulmonary hypertension in cattle residing at high altitudes] (Davie et al., 2006; Newman et al., 2011), and patients with pulmonary arterial hypertension (Stacher et al., 2012). As with human lung samples, we were unable to quantitate the outward adventitial remodeling due to methodological limitations. First, the pronounced outward remodeling was only observed in ∼5–10% of arteries analyzed. Although these cells express SMA, it is sparse and lower intensity than PASMCs in the medial layer, and migration into the parenchyma makes the precise boundaries difficult to demark. Furthermore, this adventitial remodeling that occurred in a small proportion of arteries is likely not sufficient to raise RVSP since the increase in vessel wall thickness predominantly occurred in an outward direction without encroachment on the lumen. Therefore, although the overall effect on RVSP and RV hypertrophy was minimal with the loss of EC-Asic1a, we currently cannot discount a possible role of EC ASIC1a to mitigate pulmonary vascular medial remodeling.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Protocol #19-200899-HSC).
Author contributions
All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. SG, TY, LH, ND, RA, and NJ contributed to the acquisition, analysis, interpretation, and drafting/revising of the work. SG, LB, TR, and NJ contributed to the concept, design, interpretation, and critically revising of the work for important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL-111084 (to N.L. Jernigan) and American Heart Association 18TPA34110281 (to N.L. Jernigan). Trainee support of this work was supported by NIH grants T32 HL-007736 (to T.C. Resta), K12-GM-088021 (to A. Wandinger-Ness), F31 HL-145836 (to S.M. Garcia), and R25-GM-060201 (to M. Werner-Washburne).
Acknowledgments
The authors would like to thank Tamara Howard, MS (University of New Mexico Health Sciences Center, Albuquerque, NM) for assistance with the preparation of lung sections for immunofluorescence.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akanji, O., Weinzierl, N., Schubert, R., and Schilling, L. (2019). Acid sensing ion channels in rat cerebral arteries: Probing the expression pattern and vasomotor activity. Life Sci. 227, 193–200. doi:10.1016/j.lfs.2019.04.054
Bierer, R., Nitta, C. H., Friedman, J., Codianni, S., de Frutos, S., Dominguez-Bautista, J. A., et al. (2011). NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 301 (6), L872–L880. doi:10.1152/ajplung.00405.2010
Bonnet, S., Michelakis, E. D., Porter, C. J., Andrade-Navarro, M. A., Thébaud, B., Bonnet, S., et al. (2006). An abnormal mitochondrial-hypoxia inducible factor-1alpha-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 113 (22), 2630–2641. doi:10.1161/circulationaha.105.609008
Broughton, B. R., Jernigan, N. L., Norton, C. E., Walker, B. R., and Resta, T. C. (2010). Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am. J. Physiol. Lung Cell. Mol. Physiol. 298 (2), L232–L242. doi:10.1152/ajplung.00276.2009
Budhiraja, R., Tuder, R. M., and Hassoun, P. M. (2004). Endothelial dysfunction in pulmonary hypertension. Circulation 109 (2), 159–165. doi:10.1161/01.CIR.0000102381.57477.50
Chappell, J., Harman, J. L., Narasimhan, V. M., Yu, H., Foote, K., Simons, B. D., et al. (2016). Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 119 (12), 1313–1323. doi:10.1161/CIRCRESAHA.116.309799
Chen, X., Sun, X., Wang, Z., Zhou, X., Xu, L., Li, F., et al. (2018). Involvement of acid-sensing ion channel 1a in gastric carcinoma cell migration and invasion. Acta Biochim. Biophys. Sin. 50 (5), 440–446. doi:10.1093/abbs/gmy026
Chung, W.-S., Farley, J. M., Swenson, A., Barnard, J. M., Hamilton, G., Chiposi, R., et al. (2010). Extracellular acidosis activates ASIC-like channels in freshly isolated cerebral artery smooth muscle cells. Am. J. Physiol. Cell. Physiol. 298 (5), C1198–C1208. doi:10.1152/ajpcell.00511.2009
Dai, J, J., Zhou, Q., Tang, H., Chen, T., Li, J., Raychaudhuri, P., et al. (2018). Smooth muscle cell-specific FoxM1 controls hypoxia-induced pulmonary hypertension. Cell. Signal. 51, 119–129. doi:10.1016/j.cellsig.2018.08.003
Dai, Z, Z., Zhu, M. M., Peng, Y., Jin, H., Machireddy, N., Qian, Z., et al. (2018). Endothelial and smooth muscle cell interaction via FoxM1 signaling mediates vascular remodeling and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 198 (6), 788–802. doi:10.1164/rccm.201709-1835OC
Davie, N. J., Gerasimovskaya, E. V., Hofmeister, S. E., Richman, A. P., Jones, P. L., Reeves, J. T., et al. (2006). Pulmonary artery adventitial fibroblasts cooperate with vasa vasorum endothelial cells to regulate vasa vasorum neovascularization: A process mediated by hypoxia and endothelin-1. Am. J. Pathol. 168 (6), 1793–1807. doi:10.2353/ajpath.2006.050754
de Frutos, S., Nitta, C. H., Caldwell, E., Friedman, J., and González Bosc, L. V. (2009). Regulation of soluble guanylyl cyclase-alpha1 expression in chronic hypoxia-induced pulmonary hypertension: Role of NFATc3 and HuR. Am. J. Physiol. Lung Cell. Mol. Physiol. 297 (3), L475–L486. doi:10.1152/ajplung.00060.2009
de Frutos, S., Spangler, R., Alò, D., and Bosc, L. V. G. (2007). NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha-actin up-regulation. J. Biol. Chem. 282 (20), 15081–15089. doi:10.1074/jbc.M702679200
Detweiler, N. D., Herbert, L. M., Garcia, S. M., Yan, S., Vigil, K. G., Sheak, J. R., et al. (2019). Loss of acid-sensing ion channel 2 enhances pulmonary vascular resistance and hypoxic pulmonary hypertension. J. Appl. Physiol. 127 (2), 393–407. doi:10.1152/japplphysiol.00894.2018
Ding, J., Zhang, R., Li, H., Ji, Q., Cheng, X., Thorne, R. F., et al. (2021). ASIC1 and ASIC3 mediate cellular senescence of human nucleus pulposus mesenchymal stem cells during intervertebral disc degeneration. Aging 13 (7), 10703–10723. doi:10.18632/aging.202850
Dromparis, P., Paulin, R., Sutendra, G., Qi, A. C., Bonnet, S., and Michelakis, E. D. (2013). Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ. Res. 113 (2), 126–136. doi:10.1161/circresaha.112.300699
Fessel, J. P., Hamid, R., Wittmann, B. M., Robinson, L. J., Blackwell, T., Tada, Y., et al. (2012). Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ. 2 (2), 201–213. PMC. doi:10.4103/2045-8932.97606
Foster, V. S., Rash, L. D., King, G. F., and Rank, M. M. (2021). Acid-sensing ion channels: Expression and function in resident and infiltrating immune cells in the central nervous system. Front. Cell. Neurosci. 15, 738043. doi:10.3389/fncel.2021.738043
Fried, R., Meyrick, B., Rabinovitch, M., and Reid, L. (1983). Polycythemia and the acute hypoxic response in awake rats following chronic hypoxia. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 55 (4), 1167–1172. doi:10.1152/jappl.1983.55.4.1167
Gao, Y., Chen, T., and Raj, J. U. (2016). Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 54, 451–460. doi:10.1165/rcmb.2015-0323TR
Garcia, S. M., Herbert, L. M., Walker, B. R., Resta, T. C., and Jernigan, N. L. (2020). Coupling of store-operated calcium entry to vasoconstriction is acid-sensing ion channel 1a dependent in pulmonary but not mesenteric arteries. PloS One 15 (7), e0236288. doi:10.1371/journal.pone.0236288
Garcia, S., Naik, J. S., Resta, T. C., and Jernigan, N. L. (2018). Acid sensing ion channel 1 contributes to endothelium‐derived hyperpolarizing factor induced vasodilation in small mesenteric arteries. FASEB J. 32 (S1). doi:10.1096/fasebj.2018.32.1_supplement.902.9
Gonzalez Bosc, L. V., Plomaritas, D. R., Herbert, L. M., Giermakowska, W., Browning, C., and Jernigan, N. L. (2016). ASIC1-mediated calcium entry stimulates NFATc3 nuclear translocation via PICK1 coupling in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 311 (1), L48–L58. doi:10.1152/ajplung.00040.2016
Grifoni, S. C., Jernigan, N. L., Hamilton, G., and Drummond, H. A. (2008). ASIC proteins regulate smooth muscle cell migration. Microvasc. Res. 75 (2), 202–210. doi:10.1016/j.mvr.2007.08.003
Harguindey, S., Stanciu, D., Devesa, J., Alfarouk, K., Cardone, R. A., Polo Orozco, J. D., et al. (2017). Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 43, 157–179. doi:10.1016/j.semcancer.2017.02.003
Herbert, L. M., Nitta, C. H., Yellowhair, T. R., Browning, C., Gonzalez Bosc, L. V., Resta, T. C., et al. (2016). PICK1/calcineurin suppress ASIC1-mediated Ca2+ entry in rat pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell. Physiol. 310 (5), C390–C400. doi:10.1152/ajpcell.00091.2015
Herbert, L. M., Resta, T. C., and Jernigan, N. L. (2018). RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am. J. Physiol. Cell. Physiol. 314 (2), C166-C176–C176. doi:10.1152/ajpcell.00159.2017
Humbert, M., Montani, D., Perros, F., Dorfmüller, P., Adnot, S., and Eddahibi, S. (2008). Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vasc. Pharmacol. 49 (4–6), 113–118. doi:10.1016/j.vph.2008.06.003
Jacobsen, K., Lund, M. B., Shim, J., Gunnersen, S., Füchtbauer, E.-M., Kjolby, M., et al. (2017). Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2 (19), 95890. doi:10.1172/jci.insight.95890
Jernigan, N. L., Herbert, L. M., Walker, B. R., and Resta, T. C. (2012). Chronic hypoxia upregulates pulmonary arterial ASIC1: A novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am. J. Physiol. Cell. Physiol. 302 (6), C931–C940. doi:10.1152/ajpcell.00332.2011
Jernigan, N. L., Naik, J. S., and Resta, T. C. (2021). Acid-sensing ion channel 1 contributes to pulmonary arterial smooth muscle cell depolarization following hypoxic pulmonary hypertension. J. Physiol. 599 (21), 4749–4762. doi:10.1113/JP282231
Jernigan, N. L., Naik, J. S., Weise-Cross, L., Detweiler, N. D., Herbert, L. M., Yellowhair, T. R., et al. (2017). Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLOS ONE 12 (6), e0180455. doi:10.1371/journal.pone.0180455
Jernigan, N. L., Paffett, M. L., Walker, B. R., and Resta, T. C. (2009). ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 297 (2), L271–L285. doi:10.1152/ajplung.00020.2009
Jernigan, N. L., Walker, B. R., and Resta, T. C. (2008). Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 295 (3), L515–L529. doi:10.1152/ajplung.00355.2007
Jin, C., Ye, Q.-H., Yuan, F.-L., Gu, Y.-L., Li, J.-P., Shi, Y.-H., et al. (2015). Involvement of acid-sensing ion channel 1α in hepatic carcinoma cell migration and invasion. Tumour Biol. 1, 4309–4317. doi:10.1007/s13277-015-3070-6
Kapoor, N., Bartoszewski, R., Qadri, Y. J., Bebok, Z., Bubien, J. K., Fuller, C. M., et al. (2009). Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 284 (36), 24526–24541. doi:10.1074/jbc.M109.037390
Karlsson, M., Zhang, C., Méar, L., Zhong, W., Digre, A., Katona, B., et al. (2021). A single-cell type transcriptomics map of human tissues. Sci. Adv. 7 (31), eabh2169. doi:10.1126/sciadv.abh2169
Kisanuki, Y. Y., Hammer, R. E., Miyazaki, J., Williams, S. C., Richardson, J. A., and Yanagisawa, M. (2001). Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230 (2), 230–242. doi:10.1006/dbio.2000.0106
Lin, M.-J., Leung, G. P. H., Zhang, W.-M., Yang, X.-R., Yip, K.-P., Tse, C.-M., et al. (2004). Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ. Res. 95 (5), 496–505. doi:10.1161/01.RES.0000138952.16382.ad
Lockhart, A., Zelter, M., Mensch-Dechene, J., Antezana, G., Paz-Zamora, M., Vargas, E., et al. (1976). Pressure-flow-volume relationships in pulmonary circulation of normal highlanders. J. Appl. Physiol. 41 (4), 449–456. doi:10.1152/jappl.1976.41.4.449
Majka, S. M., Skokan, M., Wheeler, L., Harral, J., Gladson, S., Burnham, E., et al. (2008). Evidence for cell fusion is absent in vascular lesions associated with pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 295 (6), L1028–L1039. doi:10.1152/ajplung.90449.2008
McMurtry, M. S., Bonnet, S., Wu, X., Dyck, J. R. B., Haromy, A., Hashimoto, K., et al. (2004). Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 95 (8), 830–840. doi:10.1161/01.RES.0000145360.16770.9f
Meyrick, B., and Reid, L. (1979). Hypoxia and incorporation of 3H-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am. J. Pathol. 96 (1), 51.
Misra, A., Feng, Z., Chandran, R. R., Kabir, I., Rotllan, N., Aryal, B., et al. (2018). Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat. Commun. 9 (1), 2073. doi:10.1038/s41467-018-04447-7
Nagaoka, T., Morio, Y., Casanova, N., Bauer, N., Gebb, S., McMurtry, I., et al. (2004). Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 287 (4), L665–L672. doi:10.1152/ajplung.00050.2003
Newman, J. H., Holt, T. N., Hedges, L. K., Womack, B., Memon, S. S., Willers, E. D., et al. (2011). High-altitude pulmonary hypertension in cattle (brisket disease): Candidate genes and gene expression profiling of peripheral blood mononuclear cells. Pulm. Circ. 1 (4), 462–469. doi:10.4103/2045-8932.93545
Nitta, C. H., Osmond, D. A., Herbert, L. M., Beasley, B. F., Resta, T. C., Walker, B. R., et al. (2014). Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 306 (1), H41–H52. doi:10.1152/ajpheart.00269.2013
Nozik-Grayck, E., Suliman, H. B., Majka, S., Albietz, J., Van Rheen, Z., Roush, K., et al. (2008). Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 295 (3), L422–L430. doi:10.1152/ajplung.90293.2008
Ortega-Ramírez, A., Vega, R., and Soto, E. (2017). Acid-sensing ion channels as potential therapeutic targets in neurodegeneration and neuroinflammation. Mediators of Inflammation, e3728096. doi:10.1155/2017/3728096
Owens, G. K. (2007). Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found. Symp. 283, 174–191. doi:10.1002/9780470319413.ch14
Paddenberg, R., Stieger, P., von Lilien, A.-L., Faulhammer, P., Goldenberg, A., Tillmanns, H. H., et al. (2007). Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir. Res. 8, 15. doi:10.1186/1465-9921-8-15
Pak, O., Sommer, N., Hoeres, T., Bakr, A., Waisbrod, S., Sydykov, A., et al. (2013). Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell. Mol. Biol. 49 (3), 358–367. doi:10.1165/rcmb.2012-0361OC
Quinlan, T. R., Li, D., Laubach, V. E., Shesely, E. G., Zhou, N., and Johns, R. A. (2000). ENOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 279 (4), L641–L650. doi:10.1152/ajplung.2000.279.4.L641
Redd, M. A., Scheuer, S. E., Saez, N. J., Yoshikawa, Y., Chiu, H. S., Gao, L., et al. (2021). Therapeutic inhibition of acid-sensing ion channel 1a recovers heart function after ischemia-reperfusion injury. Circulation 144 (12), 947–960. doi:10.1161/CIRCULATIONAHA.121.054360
Rooj, A. K., McNicholas, C. M., Bartoszewski, R., Bebok, Z., Benos, D. J., and Fuller, C. M. (2012). Glioma-specific cation conductance regulates migration and cell cycle progression. J. Biol. Chem. 287 (6), 4053–4065. doi:10.1074/jbc.M111.311688
Roostalu, U., Aldeiri, B., Albertini, A., Humphreys, N., Simonsen-Jackson, M., Wong, J. K. F., et al. (2018). Distinct cellular mechanisms underlie smooth muscle turnover in vascular development and repair. Circ. Res. 122 (2), 267–281. doi:10.1161/CIRCRESAHA.117.312111
Sheak, J. R., Jones, D. T., Lantz, B. J., Maston, L. D., Vigil, D., Resta, T. C., et al. (2020). NFATc3 regulation of collagen V expression contributes to cellular immunity to collagen type V and hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 319 (6), L968–L980. doi:10.1152/ajplung.00184.2020
Shimoda, L. A., and Laurie, S. S. (2013). Vascular remodeling in pulmonary hypertension. J. Mol. Med. 91 (3), 297–309. PubMed. doi:10.1007/s00109-013-0998-0
Sime, F., Pealoza, D., and Ruiz, L. (1971). Bradycardia, increased cardiac output, and reversal of pulmonary hypertension in altitude natives living at sea level. Br. Heart J. 33 (5), 647–657. doi:10.1136/hrt.33.5.647
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53 (1), 1801913. doi:10.1183/13993003.01913-2018
Stacher, E., Graham, B. B., Hunt, J. M., Gandjeva, A., Groshong, S. D., McLaughlin, V. V., et al. (2012). Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186 (3), 261–272. doi:10.1164/rccm.201201-0164OC
Sutendra, G., Bonnet, S., Rochefort, G., Haromy, A., Folmes, K. D., Lopaschuk, G. D., et al. (2010). Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2 (44), 44ra58. doi:10.1126/scitranslmed.3001327
Tuineau, M. N., Herbert, L. M., Resta, T. C., and Jernigan, N. L. (2022). Pulmonary arterial smooth muscle cell monocarboxylate transporter 1/4 contributes to extracellular acidosis following chronic hypoxia-induced pulmonary hypertension. FASEB J. 36 (S1). doi:10.1096/fasebj.2022.36.S1.R4010
Voelkel, N. F., and Tuder, R. M. (2000). Hypoxia-induced pulmonary vascular remodeling: A model for what human disease? J. Clin. Investig. 106 (6), 733–738. PubMed. doi:10.1172/JCI11144
Waldmann, R., Champigny, G., Bassilana, F., Heurteaux, C., and Lazdunski, M. (1997). A proton-gated cation channel involved in acid-sensing. Nature 386 (6621), 173–177. doi:10.1038/386173a0
Wang, G., Jacquet, L., Karamariti, E., and Xu, Q. (2015). Origin and differentiation of vascular smooth muscle cells. J. Physiol. 593 (14), 3013–3030. doi:10.1113/JP270033
Weise-Cross, L., Sands, M. A., Sheak, J. R., Broughton, B. R. S., Snow, J. B., Gonzalez Bosc, L. V., et al. (2018). Actin polymerization contributes to enhanced pulmonary vasoconstrictor reactivity after chronic hypoxia. Am. J. Physiol. Heart Circ. Physiol. 314 (5), H1011-H1021–H1021. doi:10.1152/ajpheart.00664.2017
Wemmie, J. A., Taugher, R. J., and Kreple, C. J. (2013). Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 14 (7), 461–471. doi:10.1038/nrn3529
Wemmie, J., Chen, J., Askwith, C., Hruska-Hageman, A., Price, M., Nolan, B., et al. (2002). The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477. doi:10.1016/s0896-6273(02)00661-x
Wirth, A., Benyó, Z., Lukasova, M., Leutgeb, B., Wettschureck, N., Gorbey, S., et al. (2008). G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14 (1), 64–68. doi:10.1038/nm1666
Wu, P.-Y., Huang, Y.-Y., Chen, C.-C., Hsu, T.-T., Lin, Y.-C., Weng, J.-Y., et al. (2013). Acid-sensing ion channel-1a is not required for normal hippocampal LTP and spatial memory. J. Neurosci. 33 (5), 1828–1832. doi:10.1523/jneurosci.4132-12.2013
Wu, Y., Gao, B., Xiong, Q.-J., Wang, Y.-C., Huang, D.-K., and Wu, W.-N. (2017). Acid-sensing ion channels contribute to the effect of extracellular acidosis on proliferation and migration of A549 cells. Tumour Biol. 39 (6), 1010428317705750. doi:10.1177/1010428317705750
Xiong, Z.-G., Zhu, X.-M., Chu, X.-P., Minami, M., Hey, J., Wei, W.-L., et al. (2004). Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 118 (6), 687–698. doi:10.1016/j.cell.2004.08.026
Xiong, Z. G., Chu, X. P., and Simon, R. P. (2007). Acid sensing ion channels—novel therapeutic targets for ischemic brain injury. Front. Biosci. 12, 1376–1386. doi:10.2741/2154
Xu, W., Koeck, T., Lara, A. R., Neumann, D., DiFilippo, F. P., Koo, M., et al. (2007). Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 104 (4), 1342–1347. PMC. doi:10.1073/pnas.0605080104
Yermolaieva, O., Leonard, A. S., Schnizler, M. K., Abboud, F. M., and Welsh, M. J. (2004). Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U. S. A. 101 (17), 6752–6757. doi:10.1073/pnas.0308636100
Zhu, L., Yin, J., Zheng, F., Ji, L., Yu, Y., and Liu, H. (2021). ASIC1 inhibition impairs the proliferation and migration of pancreatic stellate cells induced by pancreatic cancer cells. Neoplasma 68 (1), 174–179. doi:10.4149/neo_2020_200803N811
Keywords: vascular remodeling, endothelium, proliferation, migration, right heart hypertrophy
Citation: Garcia SM, Yellowhair TR, Detweiler ND, Ahmadian R, Herbert LM, Gonzalez Bosc LV, Resta TC and Jernigan NL (2022) Smooth muscle Acid-sensing ion channel 1a as a therapeutic target to reverse hypoxic pulmonary hypertension. Front. Mol. Biosci. 9:989809. doi: 10.3389/fmolb.2022.989809
Received: 08 July 2022; Accepted: 15 September 2022;
Published: 05 October 2022.
Edited by:
Elena Petroff, Montclair State University, United StatesReviewed by:
Leah Reznikov, University of Florida, United StatesZhiyu Dai, University of Arizona, United States
Heather Drummond, University of Mississippi Medical Center School of Dentistry, United States
Copyright © 2022 Garcia, Yellowhair, Detweiler, Ahmadian, Herbert, Gonzalez Bosc, Resta and Jernigan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikki L. Jernigan, bmplcm5pZ2FuQHNhbHVkLnVubS5lZHU=