 Hongyue Ma
Hongyue Ma Yan Dong
Yan Dong Yanhui Chu
Yanhui Chu Yanqin Guo
Yanqin Guo Luxin Li
Luxin Li- 1Department of Neurology, Hongqi Hospital of Mudanjiang Medical University, Mudanjiang, China
- 2College of Life Sciences, Mudanjiang Medical University, Mudanjiang, China
- 3Heilongjiang Key Laboratory of Tissue Damage and Repair, Mudanjiang Medical University, Mudanjiang, China
Alzheimer’s disease (AD) accounts for two-thirds of all dementia cases, affecting 50 million people worldwide. Only four of the more than 100 AD drugs developed thus far have successfully improved AD symptoms. Furthermore, these improvements are only temporary, as no treatment can stop or reverse AD progression. A growing number of recent studies have demonstrated that iron-dependent programmed cell death, known as ferroptosis, contributes to AD-mediated nerve cell death. The ferroptosis pathways within nerve cells include iron homeostasis regulation, cystine/glutamate (Glu) reverse transporter (system xc−), glutathione (GSH)/glutathione peroxidase 4 (GPX4), and lipid peroxidation. In the regulation pathway of AD iron homeostasis, abnormal iron uptake, excretion and storage in nerve cells lead to increased intracellular free iron and Fenton reactions. Furthermore, decreased Glu transporter expression leads to Glu accumulation outside nerve cells, resulting in the inhibition of the system xc− pathway. GSH depletion causes abnormalities in GPX4, leading to excessive accumulation of lipid peroxides. Alterations in these specific pathways and amino acid metabolism eventually lead to ferroptosis. This review explores the connection between AD and the ferroptosis signaling pathways and amino acid metabolism, potentially informing future AD diagnosis and treatment methodologies.
Introduction
Programmed cell death occurs by apoptosis, necroptosis, pyroptosis, ferroptosis, and cell death associated with autophagy and unprogrammed necrosis (Moujalled et al., 2021). In 2003, Dolma et al. (2003) revealed that the compound erastin could kill tumor cells via an RAS oncogene mutation, but cell death was not involved in changing the nucleus and caspase-3 activation. Yang et alexpanded on these results, discovering RAS-selective lethal compound 3, later shown to be the inducer of ferroptosis (Zhang et al., 2004; Yang and Stockwell, 2008). In 2012, Dixon et alwere the first to report ferroptosis as a type of cell death (Dixon et al., 2012). The occurrence of ferroptosis is related to the metabolism of iron, amino acids, GSH, reactive oxygen species (ROS), and lipid peroxides (LPOs). Fe3+ in the extracellular fluid is transported to cells via transferrin (Tf) and subsequently reduced to Fe2+. Excessive H2O2 reacts with the Fe2+ in cells to generate a large number of ROS through Fenton reactions, which promote the generation of intracellular LPOs and trigger ferroptosis. Electron microscope examination of cellular morphology during ferroptosis showed that the membrane ruptures, bubbles develop within mitochondria, which then atrophy, the mitochondrial cristae shrink or disappear, and the membrane density increases. Furthermore, although the nuclear shape appears normal, condensed chromatin is lacking (Xia et al., 2021; Ou et al., 2022). Biological activity is also altered. ROS and iron ions aggregate, the mitogen-activated protein kinase (MAPK) system is activated, cystine uptake is reduced, GSH is depleted, and system xc− is inhibited (Shin et al., 2018). A recent study found reduced iron accumulation, lipid peroxidation, and GSH and GPX4 in patients with neurodegenerative diseases. Magnetic resonance imaging showed that iron deposition is correlated with cognitive impairment, and this deposition is mainly observed in the hippocampus, cortex, and basal ganglia, where brain cells experience oxidative stress, lipid peroxidation, and increased cystine/Glu transporter expression, iron metabolism, and balance (Ghadery et al., 2015; Masaldan et al., 2019). Iron is deposited in the brain cells of AD patients, and excessive iron will exacerbate oxidative damage and cognitive deficits (Bao et al., 2021). Given this evidence an in-depth understanding of ferroptosis mechanisms involved in the occurrence and development of AD is needed to facilitate timely diagnosis and treatment before major brain damage occurs, improving the survival rate and quality of life of AD patients. The purpose of the present review is to summarize the mechanism of ferroptosis in nerve cells and further analyze the possible pathway of ferroptosis involved in AD, summarize the relationship between ferroptosis-related drugs and AD, and propose methods for future clinical practice.
Mechanisms of ferroptosis
Iron homeostasis and ferroptosis
The maintenance of iron homeostasis is essential for normal physiological function. There are two forms of iron in cells: Fe3+ and Fe2+. As a storage and transportation form of iron, Fe3+ is relatively stable. Fe2+ can transfer electrons, participate in various oxidation-reduction reactions and act as a reaction catalyst. An imbalance in iron homeostasis can result in lipid peroxidation and cellular oxidative stress, ultimately leading to ferroptosis. Therefore, the transfer in, transfer out, storage, and turnover of iron play important roles in the ferroptosis process.
Iron is transported by cells in two forms, Tf bound and non-Tf-bound iron. Tf is the primary protein responsible for iron transport. Under physiological conditions, Fe3+ is transferred into brain microvascular endothelial cells through endocytosis mediated by transferrin receptor 1 (TfR1) and Tf on the luminal side of the cells (Masaldan et al., 2019). In the acidic environment of the endosome, Fe3+ is reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate 3 (Derry et al., 2020; Li et al., 2020; Reichert et al., 2020; Jia et al., 2021). The divalent metal transporter 1 or zinc-iron regulatory protein family 8/14 can assist in moving the iron into the labile iron pool (LIP) (Derry et al., 2020; Li et al., 2020; Reichert et al., 2020; Jia et al., 2021; Qu et al., 2022). Under normal physiological conditions, oxidation–reduction activity of Fe2+ in the form of LIP is maintained at a low concentration (approximately 0.2–0.5 µM) to meet metabolic requirements (Petrat et al., 1999).
GSH has a high affinity with Fe2+ and the major component of LIP in the cytosol is presented as the GSH-Fe2+ conjugates (Lv and Shang, 2018). A decrease in intracellular GSH , increases the concentration of Fe2+ facilitating the Fenton reaction. The storage of labile iron in ferritin serves to circumvent its high reactivity, avoiding the generation of reactive species (Reichert et al., 2020). Ferritin is an intracellular complex of 24 subunits (composed of heavy and light ferritin chains) that stores up to 4,500 iron atoms in an inactive oxidized and reduced forms to protect cells and tissues from oxidative damage (De Domenico et al., 2009). The primary function of ferritin is to maintain the equilibrium between the reduced (Fe2+) and oxidized states (Fe3+) (Reichert et al., 2020). Ferritin can catalyze excessive intracellular Fe2+ into non-toxic Fe3+ in the presence of proteins related to iron metabolism, which are closely bound and stored in the ferritin complex to maintain iron homeostasis (Reichert et al., 2020). An iron metabolism imbalance—caused by abnormal ferritin—will induce ferroptosis (Mancias et al., 2015; Tang et al., 2018). Nuclear receptor coactivator 4 (NCOA4) is a selective carrier receptor that can perform selective autophagy of ferritin (ferritinophagy) when intracellular iron levels are low so that iron is released (Ling et al., 2017). Arginine on the surface of the heavy ferritin chain FTH1 binds to a C-terminal domain of NCOA4 when iron levels are low, thereby promoting the transfer of autophagosomes to lysosomes (Mancias et al., 2015; Tang et al., 2018; Cheng et al., 2021). Mancias et al. (2015). demonstrated that the amount of NCOA4 depends on whether it interacts with the HERC2 protein. NCOA4 on the autophagosome targets the HERC2 protein when intracellular iron levels are high and is degraded by the proteasome. NCOA4 degradation ultimately reduces the breakdown of ferritin. NCOA4-mediated ferritinophagy participates in some physiological processes associated with iron metabolism in the cell, including erythropoiesis. Recent evidence suggests that autophagy is a conserved catabolic cellular pathway. Moreover, the loss of HERC2 causes severe neurodevelopmental abnormalities, contributing to neurogenetic diseases (Morice-Picard et al., 2016; Tang et al., 2018). Therefore, it is thought that HERC2 deficiency would result in a malfunctioning response to elevated iron levels, resulting in ferritinophagy, free iron release, and neuronal cell damage.
Studies have shown that the intracellular LIP level is regulated by ferritin (Reichert et al., 2020), and in ferroptosis, ferritinophagy increases LIP (Dixon and Stockwell, 2014), which can activate the Fenton and Haber–Weiss reactions to generate ROS (Lane et al., 2018). Specifically, the ROS H2O2 oxidizes Fe2+ to Fe3+ via the Fenton reaction, forming the highly active hydroxyl radical (·OH), inducing oxidative stress and leading to ferroptosis, the accumulation of lipoid-OOH, and the oxidation of polyunsaturated fatty acids (PUFAs) (Kehrer, 2000; Qi et al., 2020).
To date, ferroportin 1 (FPN1, also known as solute carrier family 40 member 1) is the only nonheme cellular iron exporter identified in mammals. It transports iron from iron storage cells into the blood to optimize systemic iron homeostasis. In the central nervous system, FPN1 is distributed in most cell types, including neurons, astrocytes, oligodendrocytes, and brain microvascular endothelial cells (Bao et al., 2021).In neurons, amyloid precursor protein (APP) connects to FPN1 and stabilizes the expression level of FPN1. APP is also an iron oxidase that can oxidize Fe2+ to Fe3+ and transfer it out of cells (Duce et al., 2010). FPN1 is essential to embryonic development: mice with a global FPN1 deletion are embryonically lethal (Drakesmith et al., 2015).
Cystine/glutamate reverse transport system, GPX4 and ferroptosis
System xc− is a heterodimer composed of two solute carriers, solute carrier family 3A2 (SLC3A2) and solute carrier family 7A11 (SLC7A11). Through system xc−, Glu and cystine enter and leave the cell in equal amounts. Cystine, which is ingested, is then reduced to gamma-glutamylcysteine (γ-Glu-Cys) in the cell, which is involved in the synthesis of GSH. The excitatory amino acid Glu can induce the death of nerve cells, which is iron-dependent. It is speculated that Glu-induced death and ferroptosis may share the same signaling pathway. Studies have shown that system xc− is inhibited by ferroptosis (Sato et al., 2018; Li et al., 2020). Specifically, system xc− is inhibited by high extracellular Glu concentration, and decreasing intracellular GSH leads to peroxidase 4 (GPX4) inhibition and lipoxygenase (LOX) activation. Eventually, lipid peroxidation and cellular oxidative stress are generated, resulting in cell ferroptosis (Huang et al., 2020).
p53 plays a role in regulating ferroptosis via SLC7A11. The p53 tumor suppressor is “the guardian of the genome” that participates in the control of cell survival and division under various stresses. Beyond its effects on apoptosis, autophagy, and the cell cycle, p53 also regulates ferroptosis through a transcriptional or posttranslational mechanism. p53 can enhance ferroptosis by inhibiting the expression of SLC7A11 (Kang et al., 2019). A complete transactivation domain is necessary for p53 to regulate SLC7A11 and ferroptosis. Although mutant p53 inhibits the expression of SLC7A11, thereby promoting ferroptosis (Jiang et al., 2015a; Latunde-Dada, 2017). p53 mutants do not inhibit SLC7A11 (Jiang et al., 2015b; Liu and Gu, 2022). Recent reports indicate a more complex mechanism in which wild-type p53 may enhance survival advantage by promoting antioxidation in some cases, and p53-mediated activation of p21 (encoded by CDKN1a) inhibits phospholipid oxidation by protecting intracellular mercaptans (including GSH) (Tarangelo et al., 2018; Hu et al., 2020; Tang et al., 2021). In addition, p53 plays a crucial role in regulating dynamic ROS. Jiang et al. found that p53 regulates antioxidant response under short-term stress, assisting cell recovery. However, continued activation of p53 triggers a pro-oxidation reaction that induces cell death. An ROS increase in the late stage of p53 activation is partly due to the inhibition of SLC7A11 (Jiang et al., 2015b). Chu et al. showed that p53 could indirectly trigger arachidonate 12-lipoxygenase (ALOX12) function through transcriptional inhibition of SLC7A11, thus leading to ALOX12-dependent ferroptosis resulting from ROS stress (Chu et al., 2019).
Cells have several death escape mechanisms. In the ferroptotic process, one of the most important and most studied mechanisms involves the enzyme glutathione peroxidase 4 (GPX4) (Reichert et al., 2020). In mammals, the GPX family consists of eight members. GPX1–GPX4 are selenoproteins that contain selenocysteine in the catalytic center. GPX4 is the only enzyme known to reduce complex phospholipid hydroperoxides directly (Xia et al., 2021). Hydroperoxides can activate catalytic reactions in the presence of transition metals such as iron, which eventually leads to ferroptosis. Therefore, GPX4 is key to cell survival (Friedmann Angeli et al., 2019). GSH is part of an intracellular antioxidant system that plays an important role in free radical scavenging, anti-aging and antioxidation activities, and other major physiological functions. In ferroptosis, GPX4 uses GSH as a substrate to mediate the lipid-OOH conversion to lipid-OH, and the sulfhydryl group in GSH reduction is readily dehydrogenated to form oxidized glutathione disulfide (GSSG), which plays an antioxidant role. GSSG is reduced to GSH by GSH reductase in NADPH-participating reactions (Qiu et al., 2020; Lei et al., 2021). GSH is continuously produced by glutamate cysteine ligase (GCL) and glutathione synthetase (GSS), and GCL activity is the rate-limiting step in GSH synthesis (Thompson et al., 2009; Parpura et al., 2017; Stockwell et al., 2017; Abdalkader et al., 2018; Shi et al., 2021; Yan et al., 2021). When reduced GCL and GSS activity limits the synthesis of GSH, GPX4 is eventually inactivated, resulting in the accumulation of lipid peroxidation, further ROS production, and ultimately, ferroptosis.
Lipid metabolism and ferroptosis
Initiation of lipid peroxidation requires the removal of a bis-allylic hydrogen atom (located between two carbon–carbon double bonds) from polyunsaturated fatty acyl moieties in phospholipids (PUFA-PLs) incorporated into lipid bilayers. This process leads to the formation of a carbon-centered phospholipid radical (PL•) and subsequent reaction with molecular oxygen to yield a phospholipid peroxyl radical (PLOO•), which removes hydrogen from another PUFA, forming PLOOH. If not converted to the corresponding alcohol (PLOH) by GPX4, PLOOH and lipid free radicals—in particular, PLOO• and alkoxyl phospholipid radicals (PLO•)—will react with PUFA-PLs to propagate PLOOH production by removing more hydrogen atoms and reacting with molecular oxygen. This reaction eventually leads to the formation of lipid peroxide breakdown products. A consequence of this chain reaction is the eventual deterioration of membrane integrity and, ultimately, destabilization of organelles or cell membranes. Therefore, membranes with a high PUFA-PL content would be especially susceptible to peroxidation, as has been shown to occur in neurons (Jiang et al., 2021). Acyl-CoA synthetase long-chain family member 4 (ACSL4) determines a cell’s sensitivity to ferroptosis (Shin et al., 2018). Responsible for the esterification of PUFA into acyl-COA, ACSL4 promotes PUFA fatty acid activation. Activated the fatty acids under the action of Lysophosphatidylcholine Acyltransferase 3 (LPCAT3) transferred to inside and outside the cell membrane and esterification; the substrates can undergo peroxidation resulting in the formation of arachidonoyl (AA) and adrenoyl (AdA) acids. During this process, many LPOs and lipid ROS are formed, aggravating oxidative stress and contributing to ferroptosis (Friedmann Angeli et al., 2019; Reichert et al., 2020; Jia et al., 2021).
p53 also plays a role in lipid peroxidation. Cell membrane PUFAs can undergo peroxidation reactions via LOXs, iron-containing enzymes that also induce cell ferroptosis. p53 activates the LOX enzyme ALOX12 to induce ferroptosis in cells independent of GPX4 activity (Chu et al., 2019; Li and Li, 2020). Inhibition or knockdown of ALOX12 may be a new approach to interrupt ferroptosis (Ou et al., 2016). In contrast, loss of p53 prevents the accumulation of dipeptidyl-peptidase-4 (DPP4) in the nucleus, triggering membrane-associated DPP4-mediated lipid peroxidation. This peroxidation promotes the interaction between DPP4 and nicotinamide adenine dinucleotide phosphate oxidase 1 (NOX1), resulting in the formation of the NOX1–DPP4 complex, which mediates plasma membrane lipid peroxidation and ferroptosis (Xie et al., 2017; Li and Li, 2020). p53 inhibits ferroptosis by reducing the accumulation of toxic lipid ROS and inducing the expression of cyclin-dependent kinase inhibitor 1A (CDKN1A/p21) (Figure 1) (Li and Li, 2020; Song and Long, 2020).
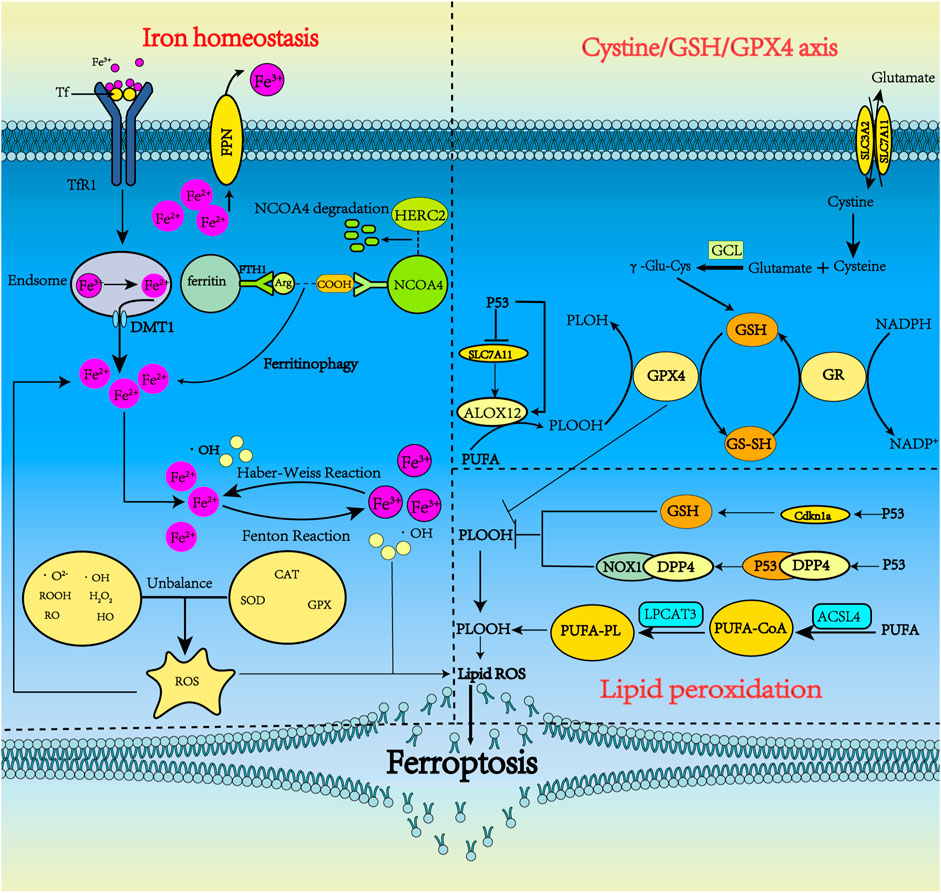
FIGURE 1. The metabolism pathways for ferroptosis. Ferroptosis can occur through three major pathways, iron homeostasis, the cystine/GSH/GPX4 axis, and lipid peroxidation. 1) Iron homeostasis. Tf carrying Fe3+ forms a complex with TfR1 and enters neurons via clathrin-mediated endocytosis. Fe3+ detaches from Tf and then is reduced by STEAP3. Fe2+ is pumped into the cytoplasm by DMT1 and is stored in ferritin in the form of Fe3+ when overloaded. Under some conditions, ferritin undergoes autophagy by binding with NCOA4, releasing iron, which subsequently leads to lethal iron levels and ferroptosis. NCOA4 can interact with HERC2, leading to NCOA4 degradation. Ferritinophagy increases LIP, which can activate the Fenton and Haber-Weiss reactions to generate ROS. 2) Cystine/GSH/GPX4 axis. System xc− includes two chains: a specific light chain, SLC7A11, and a heavy chain, SLC3A2. Through system xc−, Glu and cystine enter and leave the cell in equal amounts. Cystine, which is ingested, is then reduced to γ-Glu-Cys in the cell and becomes involved in the synthesis of GSH. GSH is continuously produced by GCL and GSS. In ferroptosis, GPX4 uses GSH as a substrate to mediate the lipid-OOH conversion to lipid-OH, and the sulfhydryl group in GSH reduction is readily dehydrogenated to form oxidized glutathione disulfide (GSSG), which plays an antioxidant role. P53 could indirectly trigger arachidonate 12-lipoxygenase (ALOX12) function through transcriptional inhibition of SLC7A11, thus leading to ALOX12-dependent ferroptosis resulting from ROS stress. 3) Lipid peroxidation. PUFA produces a large amount of lipid ROS through the continuous action of ACSl4 and LPCAT3. p53 can inhibit ferroptosis by inhibiting DPP4 activity or inducing CDKN1A expression.
Glutamate-storage, uptake and recycling
Glu, glutamine (Gln) and cysteine play vital roles in ferroptosis. The GSH molecule consists of glutamic acid, cysteine, and glycine (Gly), with cysteine being the limiting substrate in its formation. GSH plays an important role in cells and is the main low molecular weight antioxidant, regulating various important functions. Furthermore, Glu is not only one of the major excitatory amino acids in the brain, but also participates in the Glu–Gln cycle, which links glucose and amino acid metabolism to synaptic transmission, cellular homeostasis, and cellular energy metabolism (Figure 1). Therefore, Glu storage, synthesis, receptor signaling and transport, uptake and recycling are closely related to brain energy metabolism.
Glu transporters are mainly distributed in astrocyte synapses. The Glu bind to transporters which move it to the astrocyte cytoplasm. Glu transporters are co-transported into astrocytes by Na+ and Glu, and Na+ is transported to the extracellular space by Na+-K+-ATPase. Glu reacts with Gln synthetase (GS) to produce Gln; the ATP consumed in this process may be supplied by glycolysis, in which GS is only expressed in astrocytes (Parpura et al., 2017). Therefore, the Glu/Gln cycle plays a key role in maintaining Glu levels in the central nervous system (Andersen et al., 2021). The resulting Gln is released into the neuron, entering through the SLC1A5 receptor. The Gln absorbed by the neuron is converted into Glu under the action of glutaminase. GABA can also be produced by the action of Glu decarboxylase. In addition, Glu can be converted to α-ketoglutarate by Glu dehydrogenase or aminotransferase, then participate in the tricarboxylic acid (TCA) cycle, which provides citrate and oxaloacetate for lipid synthesis and converts Glu to aspartate (Haddad et al., 2021). Glu can be introduced into the TCA cycle when glucose supply is limited. p53 promotes Glu decomposition by up-regulating the expression of glutaminase 2 (Figure 2) (Liu and Gu, 2021).
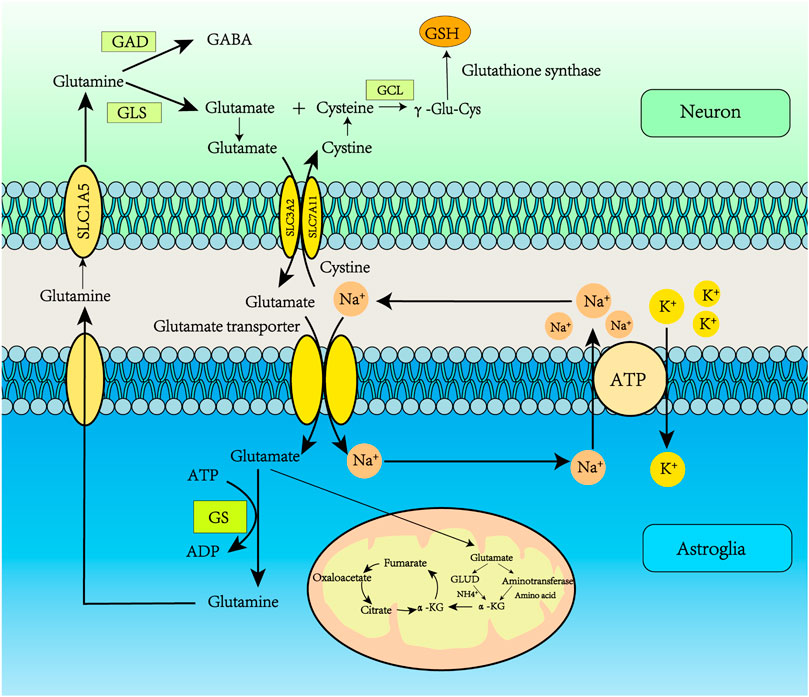
FIGURE 2. Glutamate recycling via the glutamate-glutamine cycle. Glu transporters are mainly distributed in astrocyte synapses. Glu binds to transporters which deliver it to the astrocyte cytoplasm. Glu transporters are co-transported into astrocytes by Na+ and Glu, and Na+ is transported to the extracellular space by Na+/K+-ATPase. Glu reacts with GS to produce Gln, and the ATP consumed in this process may be supplied by glycolysis. The resulting Gln is released into the neuron, and then Gln enters the neuron through the SLC1A5 receptor. The Gln absorbed by the neuron is converted into Glu under the action of glutaminase. GABA can also be produced by the action of Glu decarboxylase. In addition, Glu can be converted to α-ketoglutarate by Glu dehydrogenase or aminotransferase, participating in the tricarboxylic acid (TCA) cycle, which provides citrate and oxaloacetate for lipid synthesis and converts Glu to aspartate.
Glu receptors are roughly divided into ionic and metabolic types. Ionic Glu receptors include those of N-methyl-D-aspartate (NMDA), among the most important postsynaptic Glu receptors. Mediating the flow of Ca2+ (Liu et al., 2021), the NMDA receptor is a heterotetramer calcium channel, mainly composed of two NR1 subunits and two NR2 subunits (Furukawa et al., 2005). In the hippocampus, the NR2 subunit is mainly expressed as NR2A and NR2B. The synaptic NMDA receptors rich in NR2A subunits are primarily activated by the cAMP response element-binding protein, and brain-derived neurotrophic factor (BDNF) gene expression induces cell survival events (Leveille et al., 2008). BDNF is an important neurotrophic factor expressed in many brain regions such as the hypothalamus, cortex, brainstem and hippocampus. It plays a key role in the survival, differentiation, and growth of neuronal dendrites and axons, and the regulation of synaptic plasticity. Vesicular Glu transporters (VGLUT1) are located in the glutamatergic presynaptic vesicle plasma membrane, controlling Glu transport into the synaptic vesicle. The VGLUT1 quantity and the extracellular Glu concentration determine the speed and efficiency of transport (Chen et al., 2011).
The tripeptide GSH is the most abundant endogenous antioxidant in the body, removing free radicals and maintaining balance in the oxidative defense system. Several studies have shown that the first step in GSH synthesis involves the formation of γ-Glu-Cys in an ATP-dependent reaction catalyzed by GCL, which requires Mg2+ or Mn2+ as cofactors (Liu et al., 2021). γ-Glu-Cys can significantly increase GSH and the GSH/GSSG ratio (Liu et al., 2021). This step is rate-limiting because it depends on cysteine bioavailability and GCL activity. In the second step, γ-Glu-Cys and Gly form GSH via GSS activity (Stockwell et al., 2017). The cell’s ability to biosynthesize GSH is controlled by various factors, including intracellular substrate utilization (L-cysteine), GCL activity, rate-limiting enzymes in GSH synthesis, and GSH feedback inhibition of GCL (Haddad et al., 2021).
The most significant difference between ferroptosis and other types of programmed cell death is the change in mitochondrial morphology (Luo et al., 2021). Ferroptotic mitochondria are smaller with increased membrane density (Bao et al., 2021) and elevated cytoplasmic and lipid ROS radicals (Dixon et al., 2012; Johnson et al., 2021). ROS are partially reduced oxygen-containing molecules, including superoxide (O2•−), peroxides (H2O2 and ROOH), and free radicals (HO• and RO•) (Latunde-Dada, 2017; Chen et al., 2020; Foret et al., 2020). Superoxide is the most important free radical (Serviddio et al., 2015). Excessive ROS can be detoxified by antioxidants (enzymes and non-enzymes) and in reactions catalyzed by superoxide dismutase (Cu-SOD, Zn-SOD, and Mn-SOD), GPX and catalase. ROS are produced by glucose and glutamine (Gln) metabolism, which reduces GSH and GPX4 levels (Chen et al., 2020). An imbalance in ROS production and detoxification rates leads to oxidative stress, and the subsequent radicals generate damage DNA, proteins, and lipids (Chen et al., 2020). Under oxidative stress, high levels of superoxide can induce compounds including iron (4Fe-4S) clusters, heme, and ferritin to release Fe2+, which causes ferroptosis through Fenton and Haber–Weiss reactions. In ferroptosis, SLC7A11 and GSH depletion lead to iron-dependent ROS accumulation (Dixon and Stockwell, 2014).
Ferroptosis and alzheimer’s disease
Iron homeostasis and alzheimer’s disease
Regulation of iron homeostasis is important for maintaining normal brain function, and dysregulation of iron homeostasis in the brain can lead to oxidative stress and inflammatory responses, resulting in cell damage and ultimately neurodegenerative diseases. Typical neuropathological features of AD include the deposition of beta-amyloid (Aβ) into neuroinflammatory plaques, intracellular aggregates of Tau protein in neurofibrillary tangles, synaptic loss, neuroinflammation, and neuronal death. In addition to these typical pathologies, MRI data of AD patients have shown iron deposition in the hippocampus, cortex, and basal ganglia (Ghadery et al., 2015; Masaldan et al., 2019). Subsequent studies revealed increased iron, Tf, and ferritin in the brain (Ashraf et al., 2020; Bao et al., 2021). These findings suggest that neuronal cells in AD disease upregulate ferritin and downregulate FPN expression, increasing iron intake and reducing iron excretion. This process leads to increased free Fe2+ in cells and increased ferritin, manifested by iron deposition in the brain.
In AD, cytotoxicity induced by Aβ1-42 directly induces down-regulation of FPN in primary neurons and the hippocampus. Abnormal phosphorylation of the tau protein can lead to increased APP (Bao et al., 2021), a precursor of Aβ production and aggregation (Derry et al., 2020). APP is first broken down by either α-secretase or β-secretase and then by γ-secretase. In the physiological state, α-secretase is the first to cleave APP for the non-amyloidosis pathway. However, if APP is first cleaved by β-secretase, neurotoxic Aβ is produced (Tsatsanis et al., 2020). The protein furin plays a critical role in regulating the rate of proteolytic activation of α-secretase and β-secretase. Furin concentration is positively correlated with α-secretase activity but negatively correlated with β-secretase activity. Iron deposition results in reduced furin transcription and translation, thereby enhancing β-secretase activity by reducing furin protein expression. Enhanced β-secretase activity increases Aβ production through the amyloidosis pathway (Ward et al., 2014). The damaging cycle then continues with Aβ-induced downregulation of FPN and iron accumulation. Generally, the tau protein can mediate APP’s interaction with FPN on the cell surface to promote iron excretion. However, reduced tau protein is associated with AD, affecting FPN’s ability to excrete iron (Wang and Mandelkow, 2016; Ayton et al., 2020; Derry et al., 2020). Furthermore, increased FTH levels in AD are associated with lower FPN levels (Ashraf et al., 2020). Everett et al. found that amyloid plaques reduced Fe3+ to Fe2+ (Everett et al., 2014a), and Aβ could transform Fe3+ stored as ferrihydrite into redox-active biological substances containing Fe2+ (Everett et al., 2014b; Derry et al., 2020).
Due to increased iron intake and decreased iron excretion, increased intracellular liberation of Fe2+ activates the ferroptosis pathway. First, excessive Fe2+ enhances the Fenton reaction and produces oxhydryl radicals. Second, it promotes lipid peroxide production, which ultimately triggers ferroptosis.
GPX4 and alzheimer’s disease
GPX4, an antioxidant enzyme, is highly expressed in NCOA4 deficient mice (Bellelli et al., 2016). Yoo et alfound that GPX4 deletion in adult mice leads to mitochondrial damage, neurodegeneration in the hippocampus, and astrocyte proliferation (Yoo et al., 2012). Moreover, Hambright et al. (2017) found significant neurological deficits and cognitive impairment in GPX4-deficient mice. When Bao et al. (2021) injected Aβ into the brain of mice, they found elevated levels of iron and ferritin in the hippocampus and decreased levels of GPX4, suggesting that Aβ directly affects ferroptosis in neurons. Thus, GPX4 inhibition in ferroptosis offers protection against neurodegeneration (Cardoso et al., 2017). As a substrate of GPX4, GSH plays an antioxidant role. Studies have shown that γ-Glu-Cys can significantly increase GSH, increase the GSH/GSSG ratio, and decrease the generation of Aβ and oxidative stress (Liu et al., 2021). In AD patients, the GSH content is reduced (Ashraf et al., 2020), so effectively preventing GSH decrease is a promising new treatment strategy for AD occurrence and development.
Lipid metabolism and alzheimer’s disease
Recent studies have shown that lipid ROS in ferroptosis may cause AD. Although the highest PUFA content is found in adipose tissue, PUFA in brain tissue accounts for 30%–35% of the total fatty acid content, so the central nervous system is very vulnerable to lipid peroxidation (Peng et al., 2021). ACSL4 plays an important role in PUFA activation and determines ferroptosis sensitivity (Shin et al., 2018). Yan et al. (2022) analyzed the hippocampal transcriptome of the APP/PS1 mouse model and found elevated expression of ACSL4. Unfortunately, it is not known whether ACSL4 expression or activity is modified by AβO.
Dietary arachidonic acid (ARA) is the second most common type of PUFA in meninges phospholipids, where lipid peroxidation readily occurs, leading to lipid bilayer damage. Recent research shows that increased ARA intake induces cognitive alteration and increases the neurotoxicity of amyloid-β peptide (Aβ) (Thomas et al., 2017), which leads to AD. In addition to its involvement in synaptic plasticity and transmission, free ARA plays a crucial role in neuroinflammation through its conversion into various eicosanoids by cyclooxygenases, prostaglandin synthases, and lipoxygenases, the activities of which have been associated with neurodegenerative diseases (Czapski et al., 2016). As the brain’s consumption and metabolism of ARA are up-regulated in AD patients, suggesting that ARA is involved in the pathomechanism of this disease (Esposito et al., 2008), ARA consumption could constitute a risk factor for AD in humans and should be considered in future preventive strategies. ARA is specifically released from membrane phospholipids by cytosolic phospholipase A2 (cPLA2), which is translocated to the membranes in a cytosolic calciumdependent manner after its phosphorylation on Ser505 by MAPK. cPLA2 is activated by Aβoligomers. Its pharmacological inhibition or the suppression of its expression protects neuronal cells against the neurotoxicity of Aβ oligomers and preserves cognitive abilities (Czapski et al., 2016). The ARA released via cPLA2 induction can be metabolized by COX or LOXs (Chuang et al., 2015). Among LOX alterations, those of LOX12/15 can lead to oxidative stress, resulting in free radical-dependent DNA damage and poly (ADP-ribose) polymerase-1 overactivation, neuronal degeneration and death (Czapski et al., 2013). Therefore, treating AD with LOX has broad prospects.
Amino acid metabolism and alzheimer’s disease
Amino acids play an important role in the occurrence and development of AD. Glu is an important excitatory neurotransmitter in the body, helping to transmit information between nerve cells. During post-translational modification, many proteins undergo glycation reactions between their free reducing sugars and free amino groups. Some studies have shown increased levels of advanced glycation end-products (AGEs) in the brains of AD patients, suggesting that AGEs play an important role in activating microglia and Aβ deposition in AD (Byun et al., 2012). AGEs are irreversible adducts of the Maillard reaction that accumulate in the brain as we age. Glyoxal or methylglyoxal (MG) can contribute to AGE production (Currais and Maher, 2013). MG is primarily removed via the glyoxalase system, composed of Glo-1 and Glo-2. Glo-1 is the rate-limiting enzyme for the system and is dependent on GSH (Bijnen et al., 2018). Glo-1 activity also depends on the cellular redox state and the GSH/GSSG ratio (Haddad et al., 2021). AGE binding to albumin secreted by microglia results in toxicity and subsequent Aβ aggregation. Aβ can promote the release of Glu from vesicles into the synaptic cleft, leading to the activation of extrasynaptic N-methyl-D-aspartate receptors (NMDARs). Over-activation of NMDARs leads to calcium overload of postsynaptic neurons, inducing excitatory toxicity, neuronal apoptosis, and neurodegeneration. Kashani et al. (2008). found decreased Glu transporter expression in the cerebral cortex in patients with AD. VGLUT1is significantly decreased, possibly resulting in poor clearance of glutamic acid in the synaptic cleft, leading to excitatory toxicity. Furthermore, high extracellular Glu will inhibit system xc−, leading to ferroptosis.
The presence of amyloidoligomers (AβO) is closely correlated to the incidence of AD. Soluble AβO is currently considered the main source of brain neuron injury and central nervous system degeneration (Reiss et al., 2018). As an intermediate product of Aβ fibrosis, AβO is significantly more toxic than monomers and fibers. AβO can be classified as low molecular weight (<50 kDa) and high molecular weight (>50 kDa), and different sizes and morphologies of oligomers may produce different pathological effects. For example, in a mouse model, low molecular weight AβO (e.g., dimer and trimer) can significantly inhibit the long-term enhancement of hippocampal neurons and damage the spatial memory function. In contrast, high molecular weight soluble Aβ aggregates are more likely to induce microglial activation, resulting in neuroinflammatory responses (Figueiredo et al., 2013). AβO can interact with metabolic Glu receptor 5 to promote long-term depression and inhibit long-term potentiation in the hippocampus, leading to downstream responses and kinases activation. For example, p38-MAPK, the end of Jun N-terminal kinase (JNK), and cell-cycle dependent kinase affect the plasticity of gene transcription (Ittner et al., 2010). Furthermore, p38MAPK and JNK have been associated with AD-like lesions caused by diabetes mellitus (Kim and Song, 2020). Hyperphosphorylation of the tau protein can be mediated by activation of the p38MAPK/p53 signaling pathway (Sun et al., 2017). In addition, increased p53 expression inhibits the expression of the system xc− SLC7A11, resulting in reduced uptake of cystine, decreased GSH peroxidase activity, reduced cell antioxidant capacity, and increased sensitivity of cells to ferroptosis (Kang et al., 2019).
Neuropathological AD changes have been associated with impaired cerebral insulin signaling (Takeda et al., 2010; Martinez-Valbuena et al., 2019), and decreased insulin signaling in the brain can inhibit phosphatidylinositol 3-kinase/Akt and activate GSK-3β (Ma et al., 2015). Excessive iron in neurons can lead to tau hyperphosphorylation and NFT formation through the CDK5/P25 complex and GSK-3β kinase pathway (Yan and Zhang, 2019). GSK-3β phosphorylates Nrf2, leading to Nrf2 degradation (Chen et al., 2020; Qu et al., 2020). Kanninen et al. reviewed the neuroprotective role of Nrf2 in AD, with special emphasis on the role of GSK-3β in the Nrf2 pathway (Kanninen et al., 2011). Moreover, it was reported that GSK-3β inhibition in SAMP8 mice results in increased nuclear Nrf2 and total GST in the cortex (Farr et al., 2014; Qu et al., 2020). The complex roles played by ferroptosis in AD regulation are shown in Figure 3.
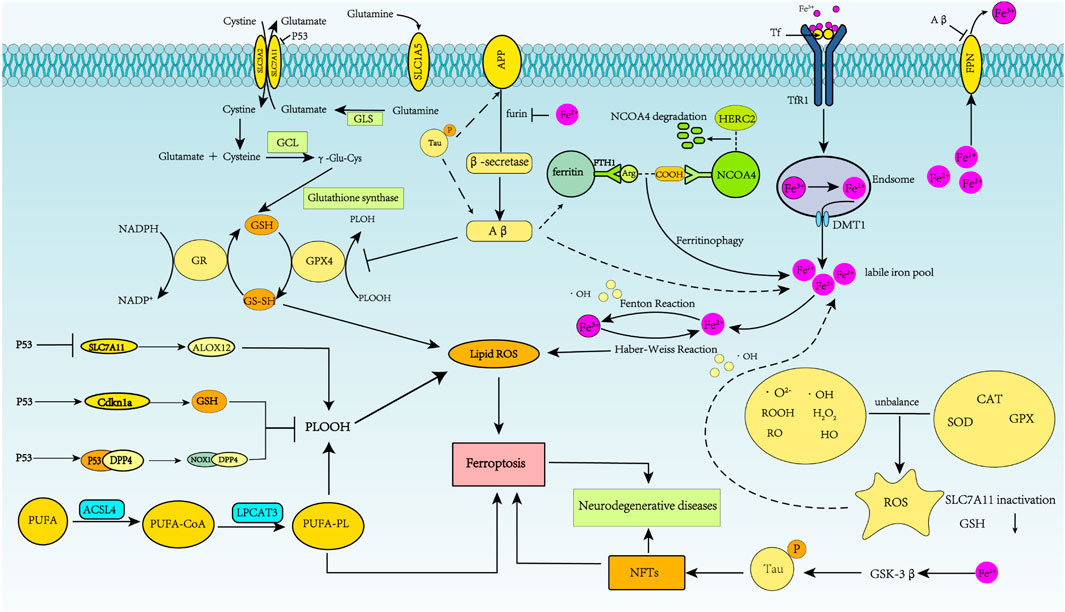
FIGURE 3. Schematic representation of ferroptosis regulation of Alzheimer’s disease. In AD, APP is first cleaved by β-secretase, and neurotoxic Aβ is produced. Abnormal phosphorylation of the tau protein can lead to increased APP and Aβ40 aggregation. Fe2+ enhances β-secretase activity by reducing furin protein expression, thereby increasing Aβ production through the amyloidosis pathway. Aβ1-42 directly induces down-regulation of FPN. Increased intracellular liberation of Fe2+ activates the ferroptosis pathway. Aβ can decrease levels of GPX4 and elevate levels of ferritin. Excessive iron in neurons can lead to tau hyperphosphorylation and NFT formation through the GSK-3β kinase pathway.
Ferroptosis inhibitors and clinical application
Ferroptosis inhibitors eliminate free radicals, inhibiting enzymes that produce lipids or LPOs and reducing free iron. Iron inhibitors are classified as aromatic amine antioxidants, α-tocopherol, nitroxides, natural polyphenol compounds, ACSL4 inhibitors, LOX inhibitors, or other types (Table 1). As patients with AD have iron deposits in their brain cells, and excess iron can exacerbate oxidative damage and cognitive deficits, ferroptosis inhibitors offer broad prospects for treating AD.
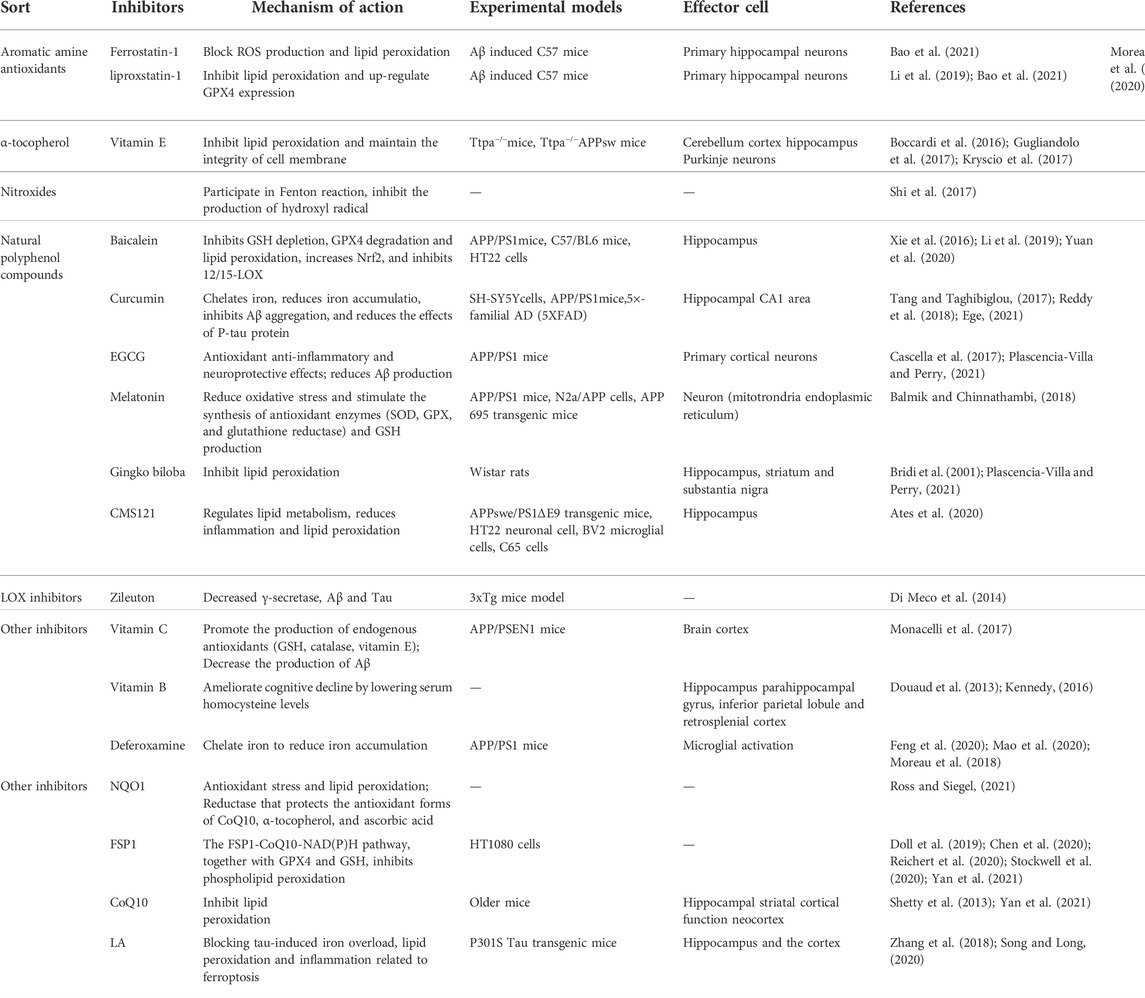
TABLE 1. Summary of the available ferroptosis Inhibitors in Alzheimer disease.
Aromatic amine antioxidants
Ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), aromatic amine antioxidants, are free radical scavengers that block ROS production and lipid peroxidation.
Fer-1 inhibits ferroptosis much more efficiently than phenolic antioxidants. The anti-ferroptotic activity of Fer-1 is due to the scavenging of initiating alkoxyl radicals produced, with other rearrangement products, by ferrous iron from lipid hydroperoxides. Fer-1 forms a complex with iron, confirmed in cells by calcein fluorescence which indicates decreased labile iron in the presence of Fer-1 (Miotto et al., 2020). In addition, Fer-1 significantly inhibits the production of cytoplasmic and lipid ROS and reverses Glu-induced suppression of GSH and Gpx in HT-22 cells, suggesting that Fer-1 protects HT-22 cells by blocking oxidative toxicity. Therefore, Nrf2 and Gpx4 up-regulation may be the basis of the cytoprotective mechanism of Fer-1 (Chu et al., 2020).
The aromatic amine Lip-1 is the foundation for the antioxidant activity of liproxstatin-1 analogs. It is an excellent radical-trapping antioxidant in phospholipid bilayers, using the bilayers of unilamellar liposomes originating from egg phosphatidylcholine. Lip-1 readily penetrates and remains within the lipid bilayer, permitting its active site to remain in close directional contact with the lipid peroxidation site and initiating the CH3OO• extraction of hydrogen atoms from aromatic amine sites (Sheng et al., 2017). Moreover, Lip-1 prevents BODIPY 581/591 C11 oxidation in Gpx4−/− cells but does not interfere with other classical types of cell death, such as TNFα-induced apoptosis and H2O2-induced necrosis (Friedmann Angeli et al., 2014).
Studies have shown that memory improves in Aβ-induced AD mice when Fer-1 and Lip-1 are administered, and Lip-1 has a more significant effect on memory (Bao et al., 2021). Therefore, aromatic amine antioxidants may offer significant AD treatment options.
α-Tocopherol
α-Tocopherol, the main type of Vitamin E in tissues, exerts its antioxidant capacity mainly by destroying the chain reaction of automatic oxidation (Zilka et al., 2017). α-Tocopherol transfer protein (TTP) is highly expressed in the brain and regulates the level and distribution of α-tocopherol. Vitamin E and TTP deficiency can lead to oxidative stress in the brain. It has been demonstrated that AD patients have low Vitamin E in the plasma, serum, and cerebrospinal fluid (Ashraf and So, 2020). Moreover, AD patients receiving vitamin E treatment experience slower declines in cognitive function and lower oxidative stress levels than patients receiving the placebo (Boccardi et al., 2016; Gugliandolo et al., 2017; Kryscio et al., 2017).
Nitroxides
Nitroxides can permeate the cell membranes and cross the blood–brain barrier (BBB). Fe2+ is the form of iron found in the LIP in vivo. The involvement of the iron (II)-citrate complex in Fenton-like reactions with H2O2 is considered an in vivo mechanism of the LIP that induces oxidative stress and many pathological conditions. The nitroxide Tempo combines with Fe2+-citrate to form a Tempo-Fe2+-citrate complex, which can effectively inhibit OH production. Nitroxides have significant therapeutic potential as antioxidants in oxidative stress-related diseases (Shi et al., 2017).
Natural polyphenol compounds
Baicalein is a natural polyphenol compound that inhibits LOXs by reducing oxidative stress and acts as an anti-inflammatory and neuroprotective agent. It inhibits GSH depletion, GPX4 degradation and lipid peroxidation, increases Nrf2, and inhibits 12/15-LOX (Xie et al., 2016; Li et al., 2019; Yuan et al., 2020). Activation of Nrf2 increases iron storage, reduces iron uptake by cells, and limits lipid ROS production (Hassannia et al., 2019; Xu et al., 2021). Baicalein-fed APP/PS1 mice show decreased BACE1 activity, decreased Aβ and p-tau levels, and superior behavioral test results. Another polyphenolic compound, curcumin chelates iron, reduces iron accumulation, scavenges ROS, increases the levels of SOD, Na+-K+-ATPase, catalase, GSH and mitochondrial complex enzyme (Ege, 2021), inhibits Aβ aggregation, and reduces the effects of P-tau protein. However, curcumin has poor water solubility and has demonstrated inadequate bioavailability in clinical trials. As a result, its application in the clinical treatment of AD is limited (Tang and Taghibiglou, 2017; Reddy et al., 2018; Ege, 2021).
Epigallocatechin gallate (EGCG) is another key polyphenol compound. Found in green tea, it has antioxidant, anti-inflammatory and neuroprotective effects (Plascencia-Villa and Perry, 2021). Treatment of AD mice with EGCG demonstrated that it exerted its protective effects by decreasing the expression of APP and Aβ in the hippocampus. Preclinical studies showed that EGCG has anti-inflammatory and neuroprotective effects against neuron injury and cerebral edema (Cascella et al., 2017).
Decreased melatonin, associated with decreased accumulation of polyphenols, is closely related to AD occurrence. As people age, the pineal gland calcifies, and melatonin secretion gradually decreases (Luo et al., 2020). AD patients experience decreased melatonin synthesis and secretion and abnormal secretion rhythms. Melatonin can reduce oxidative stress and stimulate the synthesis of antioxidant enzymes (SOD, GPX, and glutathione reductase) and GSH production (Balmik and Chinnathambi, 2018). Aβ plasma levels and deposition were found to be significantly reduced in APP/PS1 mice after 12 months of melatonin supplementation. However, clinical trials using melatonin (50–100 mg/day) for 10 days to 24 weeks showed that melatonin is safe but does not improve the cognitive ability of AD patients, only their sleep quality (Plascencia-Villa and Perry, 2021).
The ginkgo biloba tree (Gingko biloba) also produces polyphenols with antioxidant effects. After rats were injected with a standardized extract of ginkgo biloba, catalase and superoxide dismutase activities in the hippocampus, striatum, and substantia nigra were increased, lipid peroxidation decreased, and overall oxidative damage was reduced (Plascencia-Villa and Perry, 2021). Finally, the polyphenol derivative CMS121 acts as an antioxidant and inhibits fatty acid synthase to regulate lipid peroxidation levels. CMS121 was shown to improve memory, and cognitive function in APPswe/PS1 δ E9 double transgenic mice (Ates et al., 2020).
LOX inhibitors
5-Lipoxygenase (5LO) is widely expressed in central nervous system neurons, and its levels increase in an age-dependent manner in the hippocampus and cortex, two brain regions prone to neurodegenerative damage. Studies have shown that 5LO is up-regulated in AD. Zileuton, an anti-inflammatory compound, inhibits LOX5 and decreases γ-secretase, Aβ, and tau after three months of treatment in a 3xTg mouse AD model (Di Meco et al., 2014), demonstrating its broad prospects for clinical use.
Other inhibitors
APP/PS1 mice treated with the iron chelator deferoxamine demonstrate reduced Aβ and improved memory, but no significant improvement in cognition and memory is observed in AD patients (Feng et al., 2020). Furthermore, patients experience side effects such as loss of appetite and weight. NQO1, a reductase that can maintain antioxidant forms of CoQ10, α-tocopherol and ascorbic acid, plays an important role in maintaining antioxidant protection and inhibiting lipid peroxidation. It has long been associated with the early pathological changes of AD. However, NQO1 increases in the AD brain are limited to brain regions affected by AD pathology. Furthermore, NQO1 production is generally considered a protective response to oxidative stress, which has potential clinical significance in treating AD (Ross and Siegel, 2021).
The inhibition of ferroptosis by ferroptosis suppressor protein 1 (FSP1) is mediated by ubiquinone, also known as coenzyme Q10. U biquinone is converted on the cell membrane into its reduced prototype ubiquinol, which inhibits the peroxide reaction and prevents ferroptosis (Tang et al., 2021). FSP1 catalyzes the regeneration of CoQ10 through NAD(P)H, and the FSP1-CoQ10-NAD(P)H pathway, together with GPX4 and GSH, inhibits phospholipid peroxidation and ferroptosis, offering broad prospects for the treatment of degenerative diseases caused by ferroptosis (Doll et al., 2019; Chen et al., 2020; Mao et al., 2020; Reichert et al., 2020; Stockwell et al., 2020; Yan et al., 2021). GSH is a major antioxidant, combating oxidative stress. One study demonstrated that GSH levels in the hippocampus and cortex are significantly reduced in patients with mild cognitive impairment (MCI) and AD. Although GSH supplementation has been proposed as a therapeutic strategy for MCI and AD, it has not been evaluated in patients in clinical trials (Plascencia-Villa and Perry, 2021).
Vitamin C (ascorbic acid, AA) can increase GSH metabolism and improve cellular oxidative stress. Treatment with high concentrations of AA reduces amyloid plaque formation in the 5XFAD mouse model (Monacelli et al., 2017). Homocysteine is an important intermediate in methionine, folate, and onecarbon metabolism, and elevated homocysteine increases the risk of stroke, age-associated cognitive impairment, and AD. Randomized controlled trials and meta-analyses have indicated that homocysteine-lowering treatments may be recommended to prevent AD. Elevated homocysteine might promote post-stroke cognitive impairment (PSCI) through small vessel disease or AD pathology, which may explain our finding that homocysteine levels are associated with long-term incidence of PSCI (Li et al., 2021). Vitamin B can reduce cognitive decline by lowering serum homocysteine levels. Vitamins B1/B6/B9/B12 were found to improve brain metabolism, oxidative stress, inflammation, and cognition in patients with AD, and folic acid (1.25 mg/day, six months) reduces Aβ and inflammatory biomarkers (TNFα, IL6). Notably, high doses of Vitamin B (folic acid 0.8 mg, Vitamin B6 20 mg, Vitamin B12 0.5 mg) for 2 years was shown to slow the progression of brain atrophy significantly (Donnelly et al., 2008). Another antioxidant, α-lipoic acid, has been found to improve the cognitive function of AD patients by blocking tau-induced iron overload, lipid peroxidation and inflammation related to ferroptosis (Song and Long, 2020).
Targeted ferroptosis therapy for alzheimer’s disease
The endothelial cells of the BBB are essential in regulating brain iron uptake, and the Tf/TfR1 pathway is the major route for iron absorption in the brain (Yan and Zhang, 2019). However, the BBB presents challenges for the passage of some drug therapies into the brain. Therefore, developing effective nanomaterial carriers is crucial to improving drug delivery, release, and targeting efficiency. Nanomaterials that deliver drugs targeting ferroptosis have been extensively examined in recent years. Studies have shown that transferrin nanomaterials can penetrate the BBB and deliver drug molecules to the central nervous system (Luo et al., 2021; Zheng et al., 2021), thus providing new AD therapeutic options.
GSH levels decrease with age and possible development of AD. Few studies have focused on exploring the role of exosomes in the metastasis of GSH or its precursors to enhance and supplement intracellular GSH, especially in neuronal cells. More research is needed to understand the potential role of exosomes in oxidative stress and neuroprotection, including GSH transfer. GSH may also be used as a targeted ligand for translocation across the BBB via nanocarriers to treat various brain dysfunctions (Haddad et al., 2021). Moreover, therapy targeted to GSH degradation can effectively treat ferroptosis-mediated organ injury (Jiang et al., 2020).
Autophagosome accumulation is a significant feature in human AD patients and animal model neurons. Increased production and accumulation of Aβ in lysosomes has been observed in autophagy-deficient cells, suggesting that the turnover portion of Aβ is regulated by autophagy. As autophagy-related genes are highly expressed in early AD, enhanced autophagy may be a promising research area for achieving neuroprotection in AD patients (Moujalled et al., 2021).
Nrf2 target genes have been shown to be involved in GPX4 synthesis and function, intracellular iron homeostasis, and lipid peroxidation clearance. The Nrf2 protein regulates GSH and thioredoxin-based antioxidant systems (e.g.,TXN1, TXNRD1). Targeting the antioxidant transcription factor Nrf2 to inhibit ferroptosis is a promising new option for neurodegenerative control and significant in the study of human nervous system diseases and aging, especially neurodegenerative diseases such as Parkinson’s disease, AD, and Huntington’s disease (Song and Long, 2020).
The tau protein plays an important role in stabilizing microtubules. In AD pathology, if the protein is over-phosphorylated, it will separate from microtubules, leading to axonal microtubule disintegration (Wang and Mandelkow, 2016). Where microtubules are destroyed, and tau oligomers (tauO) are pathological, p53 cannot enter the nucleus. Over time, p53 outside the nucleus may become unstable and start to aggregate, and tauO near the nucleus interacts with p53 to form a p53 oligomer. Cell cycle arrest, DNA damage repair, apoptosis, and other crucial functions can be compromised when p53 is not allowed to enter the nucleus. Since the cell cannot be repaired and cell death cannot be controlled, conditions inside the cell will continue to deteriorate, promoting the accumulation of other disordered proteins. Targeting pathological tau proteins, especially tauO, may prevent p53 aggregation and destruction (Farmer et al., 2020). Since p53 controls many cellular functions, affecting this key transcription factor may lead to irreversible AD pathology. A deeper understanding of p53’s role in AD lesions is therefore warranted.
Conclusion
Compared with other tissues and organs of the human body, brain tissue is rich in PUFA and iron. It consumes substantial oxygen, so it is prone to lipid peroxidation, poor antioxidant capacity, and higher ferroptosis sensitivity. The occurrence of ferroptosis is closely related to the regulation of iron homeostasis, the ferroptosis signal pathway, and amino acid metabolism. AGEs are highly detectable in the blood and cerebrospinal fluid of patients with neurodegenerative diseases such as AD (Bär et al., 2003). As an important part of the ferroptosis antioxidant system, GSH helps eliminate AGEs, so increasing the level of GSH in the brain is a new strategy for treating AD. GCL is the rate-limiting enzyme of GSH synthesis, and when its activity decreases, GSH content can be reduced and GPX4 inactivated, leading to the accumulation of lipid peroxidation. This accumulation will further increase ROS and ultimately lead to ferroptosis. Therefore, the effective synthesis of GSH is a new direction for AD treatment.Mitochondria are the main source of ROS. When antioxidant factors are unbalanced within mitochondria, oxidative stress will lead to the release of Glu in neurons. High extracellular Glu will inhibit system xc−, resulting in ferroptosis. Moreover, Glu transporter expression in the cerebral cortex of AD patients and VGLUT1 significantly decreased, possibly resulting in poor clearance of glutamic acid in the synaptic cleft, leading to excitatory toxicity. Research on targeted therapy to reduce excitatory toxicity of Glu is promising for future AD treatment.
The mechanism of AD-mediated ferroptosis is gradually being clarified, and iron inhibitors have permitted some progress in AD treatment. This progress notwithstanding, the regulatory factors that regulate the ferroptosis signaling pathway vary and are dependent upon the AD stage. Therefore, the inducing factors and specific mechanisms of ferroptosis in AD remain to be elucidated and should be the focus of next studies. AD is a complex and multifactorial chronic disease. The clinical benefits of ferroptosis inhibitors in AD and their effects on other tissues are the focus of much current research. Whether a single drug or intervention targeting iron death can avoid, reduce or reverse AD requires substantial analysis. In-depth research on the different stages of AD involved in ferroptosis will help us develop a more comprehensive understanding of the AD onset and progression mechanisms and provide a more rigorous theoretical basis for prevention and treatment.
Author contributions
Conceptualization, LL and HM; original draft preparation, HM and YD; review and editing, LL and YG; supervision, YC.
Funding
This work was supported by Local Colleges and Universities Talent Development Funding from Heilongjiang Provincial Department of Finance (No. 2020GSP09), the Natural Science Foundation of Heilongjiang Province (No. H2017077), the Basic Scientific Research Project of University belongs to Heilongjiang (No. 2021-KYYWF-0519) and the Science and Technology Plan Project of Mudanjiang (No. HT2020JG070).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdalkader, M., Lampinen, R., Kanninen, K. M., Malm, T. M., and Liddell, J. R. (2018). Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front. Neurosci. 12, 466. doi:10.3389/fnins.2018.00466
Andersen, J. V., Markussen, K. H., Jakobsen, E., Schousboe, A., Waagepetersen, H. S., Rosenberg, P. A., et al. (2021). Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 196, 108719. doi:10.1016/j.neuropharm.2021.108719
Ashraf, A., Jeandriens, J., Parkes, H. G., and So, P. W. (2020). Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer's disease: Evidence of ferroptosis. Redox Biol. 32, 101494. doi:10.1016/j.redox.2020.101494
Ashraf, A., and So, P. W. (2020). Spotlight on ferroptosis: Iron-dependent cell death in Alzheimer's disease. Front. Aging Neurosci. 12, 196. doi:10.3389/fnagi.2020.00196
Ates, G., Goldberg, J., Currais, A., and Maher, P. (2020). CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer's disease. Redox Biol. 36, 101648. doi:10.1016/j.redox.2020.101648
Ayton, S., Wang, Y., Diouf, I., Schneider, J. A., Brockman, J., Morris, M. C., et al. (2020). Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 25 (11), 2932–2941. doi:10.1038/s41380-019-0375-7
Balmik, A. A., and Chinnathambi, S. (2018). Multi-faceted role of melatonin in neuroprotection and amelioration of tau aggregates in Alzheimer's disease. J. Alzheimers Dis. 62 (4), 1481–1493. doi:10.3233/JAD-170900
Bao, W. D., Pang, P., Zhou, X. T., Hu, F., Xiong, W., Chen, K., et al. (2021). Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell. Death Differ. 28 (5), 1548–1562. doi:10.1038/s41418-020-00685-9
Bär, K. J., Franke, S., Wenda, B., Muller, S., Kientsch-Engel, R., Stein, G., et al. (2003). Pentosidine and N(epsilon)-(carboxymethyl)-lysine in Alzheimer's disease and vascular dementia. Neurobiol. Aging 24 (2), 333–338. doi:10.1016/s0197-4580(02)00086-6
Bellelli, R., Federico, G., Matte', A., Colecchia, D., Iolascon, A., Chiariello, M., et al. (2016). NCOA4 deficiency impairs systemic iron homeostasis. Cell. Rep. 14 (3), 411–421. doi:10.1016/j.celrep.2015.12.065
Bijnen, M., Beelen, N., Wetzels, S., Gaar, J. v. d., Vroomen, M., Wijnands, E., et al. (2018). RAGE deficiency does not affect non-alcoholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr−/− mice. Sci. Rep. 8 (1), 15256. doi:10.1038/s41598-018-33661-y
Boccardi, V., Baroni, M., Mangialasche, F., and Mecocci, P. (2016). Vitamin E family: Role in the pathogenesis and treatment of Alzheimer's disease. Alzheimers Dement. 2 (3), 182–191. doi:10.1016/j.trci.2016.08.002
Bridi, R., Crossetti, F. P., Steffen, V. M., and Henriques, A. T. (2001). The antioxidant activity of standardized extract of Ginkgo biloba (EGb 761) in rats. Phytother. Res. 15 (5), 449–451. doi:10.1002/ptr.814
Byun, K., Bayarsaikhan, E., Kim, D., Kim, C. Y., Mook-Jung, I., Paek, S. H., et al. (2012). Induction of neuronal death by microglial AGE-albumin: Implications for Alzheimer's disease. PLoS One 7 (5), e37917. doi:10.1371/journal.pone.0037917
Cardoso, B. R., Hare, D. J., Bush, A. I., and Roberts, B. R. (2017). Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 22 (3), 328–335. doi:10.1038/mp.2016.196
Cascella, M., Bimonte, S., Muzio, M. R., Schiavone, V., and Cuomo, A. (2017). The efficacy of epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer's disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agent. Cancer 12, 36. doi:10.1186/s13027-017-0145-6
Chen, J., Wang, Y., Wu, J., Yang, J., Li, M., and Chen, Q. (2020). The potential value of targeting ferroptosis in early brain injury after acute CNS disease. Front. Mol. Neurosci. 13, 110. doi:10.3389/fnmol.2020.00110
Chen, K. H., Reese, E. A., Kim, H. W., Rapoport, S. I., and Rao, J. S. (2011). Disturbed neurotransmitter transporter expression in Alzheimer's disease brain. J. Alzheimers Dis. 26 (4), 755–766. doi:10.3233/JAD-2011-110002
Cheng, Y., Xie, Y., Chen, Y., and Liu, X. (2021). Epigenetic regulation and nonepigenetic mechanisms of ferroptosis drive emerging nanotherapeutics in tumor. Oxid. Med. Cell. Longev. 2021, 8854790. doi:10.1155/2021/8854790
Chu, B., Kon, N., Chen, D., Li, T., Liu, T., Jiang, L., et al. (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell. Biol. 21 (5), 579–591. doi:10.1038/s41556-019-0305-6
Chu, J., Liu, C. X., Song, R., and Li, Q. L. (2020). Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen. Res. 15 (3), 528–536. doi:10.4103/1673-5374.266060
Chuang, D. Y., Simonyi, A., Kotzbauer, P. T., Gu, Z., and Sun, G. Y. (2015). Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J. Neuroinflammation 12, 199. doi:10.1186/s12974-015-0419-0
Currais, A., and Maher, P. (2013). Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signal. 19 (8), 813–822. doi:10.1089/ars.2012.4996
Czapski, G. A., Adamczyk, A., Strosznajder, R. P., and Strosznajder, J. B. (2013). Expression and activity of PARP family members in the hippocampus during systemic inflammation: Their role in the regulation of prooxidative genes. Neurochem. Int. 62 (5), 664–673. doi:10.1016/j.neuint.2013.01.020
Czapski, G. A., Czubowicz, K., Strosznajder, J. B., and Strosznajder, R. P. (2016). The lipoxygenases: Their regulation and implication in Alzheimer's disease. Neurochem. Res. 41 (1-2), 243–257. doi:10.1007/s11064-015-1776-x
De Domenico, I., Ward, D. M., and Kaplan, J. (2009). Specific iron chelators determine the route of ferritin degradation. Blood 114 (20), 4546–4551. doi:10.1182/blood-2009-05-224188
Derry, P. J., Hegde, M. L., Jackson, G. R., Kayed, R., Tour, J. M., Tsai, A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer's disease from a ferroptosis perspective. Prog. Neurobiol. 184, 101716. doi:10.1016/j.pneurobio.2019.101716
Di Meco, A., Lauretti, E., Vagnozzi, A. N., and Pratico, D. (2014). Zileuton restores memory impairments and reverses amyloid and tau pathology in aged Alzheimer's disease mice. Neurobiol. Aging 35 (11), 2458–2464. doi:10.1016/j.neurobiolaging.2014.05.016
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10 (1), 9–17. doi:10.1038/nchembio.1416
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575 (7784), 693–698. doi:10.1038/s41586-019-1707-0
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 3 (3), 285–296. doi:10.1016/s1535-6108(03)00050-3
Donnelly, P. S., Caragounis, A., Du, T., Laughton, K. M., Volitakis, I., Cherny, R. A., et al. (2008). Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J. Biol. Chem. 283 (8), 4568–4577. doi:10.1074/jbc.M705957200
Douaud, G., Refsum, H., de Jager, C. A., Jacoby, R., Nichols, T. E., Smith, S. M., et al. (2013). Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. U. S. A. 110 (23), 9523–9528. doi:10.1073/pnas.1301816110
Drakesmith, H., Nemeth, E., and Ganz, T. (2015). Ironing out ferroportin. Cell. Metab. 22 (5), 777–787. doi:10.1016/j.cmet.2015.09.006
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 142 (6), 857–867. doi:10.1016/j.cell.2010.08.014
Ege, D. (2021). Action mechanisms of curcumin in Alzheimer's disease and its brain targeted delivery. Mater. (Basel) 14 (12), 3332. doi:10.3390/ma14123332
Esposito, G., Giovacchini, G., Liow, J. S., Bhattacharjee, A. K., Greenstein, D., Schapiro, M., et al. (2008). Imaging neuroinflammation in Alzheimer's disease with radiolabeled arachidonic acid and PET. J. Nucl. Med. 49 (9), 1414–1421. doi:10.2967/jnumed.107.049619
Everett, J., Cespedes, E., Shelford, L. R., Exley, C., Collingwood, J. F., Dobson, J., et al. (2014). Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer's disease peptide beta-amyloid. Inorg. Chem. 53 (6), 2803–2809. doi:10.1021/ic402406g
Everett, J., CespEdEs, E., Shelford, L. R., Exley, C., Collingwood, J. F., Dobson, J., et al. (2014). Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer's disease peptide beta-amyloid (1-42). J. R. Soc. Interface 11 (95), 20140165. doi:10.1098/rsif.2014.0165
Farmer, K. M., Ghag, G., Puangmalai, N., Montalbano, M., Bhatt, N., and Kayed, R. (2020). p53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer's disease. Acta Neuropathol. Commun. 8 (1), 132. doi:10.1186/s40478-020-01012-6
Farr, S. A., Ripley, J. L., Sultana, R., Zhang, Z., Niehoff, M. L., Platt, T. L., et al. (2014). Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic. Biol. Med. 67, 387–395. doi:10.1016/j.freeradbiomed.2013.11.014
Feng, H., Schorpp, K., Jin, J., Yozwiak, C. E., Hoffstrom, B. G., Decker, A. M., et al. (2020). Transferrin receptor is a specific ferroptosis marker. Cell. Rep. 30 (10), 3411–3423. doi:10.1016/j.celrep.2020.02.049
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33 (23), 9626–9634. doi:10.1523/JNEUROSCI.0482-13.2013
Foret, M. K., Lincoln, R., Do Carmo, S., Cuello, A. C., and Cosa, G. (2020). Connecting the "dots": From free radical lipid autoxidation to cell pathology and disease. Chem. Rev. 120 (23), 12757–12787. doi:10.1021/acs.chemrev.0c00761
Friedmann Angeli, J. P., Krysko, D. V., and Conrad, M. (2019). Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19 (7), 405–414. doi:10.1038/s41568-019-0149-1
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Biol. 16 (12), 1180–1191. doi:10.1038/ncb3064
Furukawa, H., Singh, S. K., Mancusso, R., and Gouaux, E. (2005). Subunit arrangement and function in NMDA receptors. Nature 438 (7065), 185–192. doi:10.1038/nature04089
Ghadery, C., Pirpamer, L., Hofer, E., Langkammer, C., Petrovic, K., Loitfelder, M., et al. (2015). R2* mapping for brain iron: Associations with cognition in normal aging. Neurobiol. Aging 36 (2), 925–932. doi:10.1016/j.neurobiolaging.2014.09.013
Gugliandolo, A., Bramanti, P., and Mazzon, E. (2017). Role of vitamin E in the treatment of Alzheimer's disease: Evidence from animal models. Int. J. Mol. Sci. 18 (12), E2504. doi:10.3390/ijms18122504
Haddad, M., Herve, V., Ben Khedher, M. R., Rabanel, J. M., and Ramassamy, C. (2021). Glutathione: An old and small molecule with great functions and new applications in the brain and in Alzheimer's disease. Antioxid. Redox Signal. 35 (4), 270–292. doi:10.1089/ars.2020.8129
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi:10.1016/j.redox.2017.01.021
Hassannia, B., Vandenabeele, P., and Vanden Berghe, T. (2019). Targeting ferroptosis to iron out cancer. Cancer Cell. 35 (6), 830–849. doi:10.1016/j.ccell.2019.04.002
Hu, Z., Mi, Y., Qian, H., Guo, N., Yan, A., Zhang, Y., et al. (2020). A potential mechanism of temozolomide resistance in glioma-ferroptosis. Front. Oncol. 10, 897. doi:10.3389/fonc.2020.00897
Huang, L., McClatchy, D. B., Maher, P., Liang, Z., Diedrich, J. K., Soriano-Castell, D., et al. (2020). Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell. Death Dis. 11 (10), 828. doi:10.1038/s41419-020-03020-9
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 142 (3), 387–397. doi:10.1016/j.cell.2010.06.036
Jia, M., Zhang, H., Qin, Q., Hou, Y., Zhang, X., Chen, D., et al. (2021). Ferroptosis as a new therapeutic opportunity for nonviral liver disease. Eur. J. Pharmacol. 908, 174319. doi:10.1016/j.ejphar.2021.174319
Jiang, L., Hickman, J. H., Wang, S. J., and Gu, W. (2015). Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell. Cycle 14 (18), 2881–2885. doi:10.1080/15384101.2015.1068479
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520 (7545), 57–62. doi:10.1038/nature14344
Jiang, M., Qiao, M., Zhao, C., Deng, J., Li, X., and Zhou, C. (2020). Targeting ferroptosis for cancer therapy: Exploring novel strategies from its mechanisms and role in cancers. Transl. Lung Cancer Res. 9 (4), 1569–1584. doi:10.21037/tlcr-20-341
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 22 (4), 266–282. doi:10.1038/s41580-020-00324-8
Johnson, J., Mercado-Ayon, E., Mercado-Ayon, Y., Dong, Y. N., Halawani, S., Ngaba, L., et al. (2021). Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 702, 108698. doi:10.1016/j.abb.2020.108698
Kang, R., Kroemer, G., and Tang, D. (2019). The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 133, 162–168. doi:10.1016/j.freeradbiomed.2018.05.074
Kanninen, K., White, A. R., Koistinaho, J., and Malm, T. (2011). Targeting glycogen synthase kinase-3β for therapeutic benefit against oxidative stress in Alzheimer's disease: Involvement of the nrf2-ARE pathway. Int. J. Alzheimers Dis. 2011, 985085. doi:10.4061/2011/985085
Kashani, A., Lepicard, E., Poirel, O., Videau, C., David, J. P., Fallet-Bianco, C., et al. (2008). Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 29 (11), 1619–1630. doi:10.1016/j.neurobiolaging.2007.04.010
Kehrer, J. P. J. T. (2000). The Haber–Weiss reaction and mechanisms of toxicity. Toxicology 149 (1), 43–50. doi:10.1016/s0300-483x(00)00231-6
Kennedy, D. O. (2016). B vitamins and the brain: Mechanisms, dose and efficacy--A review. Nutrients 8 (2), 68. doi:10.3390/nu8020068
Kim, O. Y., and Song, J. (2020). The importance of BDNF and RAGE in diabetes-induced dementia. Pharmacol. Res. 160, 105083. doi:10.1016/j.phrs.2020.105083
Kryscio, R. J., Abner, E. L., Caban-Holt, A., Lovell, M., Goodman, P., Darke, A. K., et al. (2017). Association of antioxidant supplement use and dementia in the prevention of Alzheimer's disease by vitamin E and selenium trial (PREADViSE). JAMA Neurol. 74 (5), 567–573. doi:10.1001/jamaneurol.2016.5778
Lane, D. J. R., Ayton, S., and Bush, A. I. (2018). Iron and Alzheimer's disease: An update on emerging mechanisms. J. Alzheimers Dis. 64 (1), S379–S395. doi:10.3233/JAD-179944
Latunde-Dada, G. O. (2017). Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta. Gen. Subj. 1861 (8), 1893–1900. doi:10.1016/j.bbagen.2017.05.019
Lei, P., Ayton, S., and Bush, A. I. (2021). The essential elements of Alzheimer's disease. J. Biol. Chem. 296, 100105. doi:10.1074/jbc.REV120.008207
Leveille, F., El GaamouchF., , Gouix, E., LecocqM., , Lobner, D., NicOle, O., et al. (2008). Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 22 (12), 4258–4271. doi:10.1096/fj.08-107268
Li, D., and Li, Y. (2020). The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 5 (1), 108. doi:10.1038/s41392-020-00216-5
Li, J., Cao, F., Yin, H. L., Huang, Z. J., Lin, Z. T., Mao, N., et al. (2020). Ferroptosis: Past, present and future. Cell. Death Dis. 11 (2), 88. doi:10.1038/s41419-020-2298-2
Li, Q., Li, Q. Q., Jia, J. N., Sun, Q. Y., Zhou, H. H., Jin, W. L., et al. (2019). Baicalein exerts neuroprotective effects in FeCl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 10, 638. doi:10.3389/fphar.2019.00638
Li, R., Weng, H., Pan, Y., Meng, X., Liao, X., Wang, M., et al. (2021). Relationship between homocysteine levels and post-stroke cognitive impairment in female and male population: From a prospective multicenter study. J. Transl. Int. Med. 9 (4), 264–272. doi:10.2478/jtim-2021-0035
Ling, Z., Kang, R., Zhu, S., Wang, X., Cao, L., Wang, H., et al. (2017). ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med. 9 (412), eaan5689. doi:10.1126/scitranslmed.aan5689
Liu, Y., Chen, Z., Li, B., Yao, H., Zarka, M., Welch, J., et al. (2021). Supplementation with gamma-glutamylcysteine (gamma-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 144, 104931. doi:10.1016/j.neuint.2020.104931
Liu, Y., and Gu, W. (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell. Death Differ. 29 (5), 895–910. doi:10.1038/s41418-022-00943-y
Liu, Y., and Gu, W. (2021). The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. doi:10.1016/j.semcancer.2021.03.010
Luo, F., Sandhu, A. F., Rungratanawanich, W., Williams, G. E., Akbar, M., Zhou, S., et al. (2020). Melatonin and autophagy in aging-related neurodegenerative diseases. Int. J. Mol. Sci. 21 (19), E7174. doi:10.3390/ijms21197174
Luo, L., Wang, H., Tian, W., Li, X., Zhu, Z., Huang, R., et al. (2021). Targeting ferroptosis-based cancer therapy using nanomaterials: Strategies and applications. Theranostics 11 (20), 9937–9952. doi:10.7150/thno.65480
Lv, H., and Shang, P. (2018). The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 10 (7), 899–916. doi:10.1039/c8mt00048d
Ma, D. L., Chen, F. Q., Xu, W. J., Yue, W. Z., Yuan, G., and Yang, Y. (2015). Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J. Neurochem. 135 (2), 301–308. doi:10.1111/jnc.13248
Mancias, J. D., Pontano Vaites, L., Nissim, S., Biancur, D. E., Kim, A. J., Wang, X., et al. (2015). Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4. doi:10.7554/eLife.10308
Mao, H., Zhao, Y., Li, H., and Lei, L. (2020). Ferroptosis as an emerging target in inflammatory diseases. Prog. Biophys. Mol. Biol. 155, 20–28. doi:10.1016/j.pbiomolbio.2020.04.001
Martinez-Valbuena, I., Valenti-Azcarate, R., Amat-Villegas, I., Riverol, M., Marcilla, I., de Andrea, C. E., et al. (2019). Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 86 (4), 539–551. doi:10.1002/ana.25570
Masaldan, S., Bush, A. I., Devos, D., Rolland, A. S., and Moreau, C. (2019). Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 133, 221–233. doi:10.1016/j.freeradbiomed.2018.09.033
Miotto, G., Rossetto, M., Di Paolo, M. L., Orian, L., Venerando, R., Roveri, A., et al. (2020). Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 28, 101328. doi:10.1016/j.redox.2019.101328
Monacelli, F., Acquarone, E., Giannotti, C., Borghi, R., and Nencioni, A. (2017). Vitamin C, aging and Alzheimer's disease. Nutrients 9 (7), E670. doi:10.3390/nu9070670
Moreau, C., Danel, V., Devedjian, J. C., Grolez, G., Timmerman, K., Laloux, C., et al. (2018). Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 29 (8), 742–748. doi:10.1089/ars.2017.7493
Morice-Picard, F., Benard, G., Rezvani, H. R., Lasseaux, E., Simon, D., Moutton, S., et al. (2016). Complete loss of function of the ubiquitin ligase HERC2 causes a severe neurodevelopmental phenotype. Eur. J. Hum. Genet. 25 (1), 52–58. doi:10.1038/ejhg.2016.139
Moujalled, D., Strasser, A., and Liddell, J. R. (2021). Molecular mechanisms of cell death in neurological diseases. Cell. Death Differ. 28 (7), 2029–2044. doi:10.1038/s41418-021-00814-y
Ou, M., Jiang, Y., Ji, Y., Zhou, Q., Du, Z., Zhu, H., et al. (2022). Role and mechanism of ferroptosis in neurological diseases. Mol. Metab. 61, 101502. doi:10.1016/j.molmet.2022.101502
Ou, Y., Wang, S. J., Li, D., Chu, B., and Gu, W. (2016). Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. U. S. A. 113 (44), E6806–E6812. doi:10.1073/pnas.1607152113
Parpura, V., Fisher, E. S., Lechleiter, J. D., Schousboe, A., Waagepetersen, H. S., Brunet, S., et al. (2017). Glutamate and ATP at the interface between signaling and metabolism in astroglia: Examples from pathology. Neurochem. Res. 42 (1), 19–34. doi:10.1007/s11064-016-1848-6
Peng, W., Zhu, Z., Yang, Y., Hou, J., Lu, J., Chen, C., et al. (2021). N2L, a novel lipoic acid-niacin dimer, attenuates ferroptosis and decreases lipid peroxidation in HT22 cells. Brain Res. Bull. 174, 250–259. doi:10.1016/j.brainresbull.2021.06.014
Petrat, F., Rauen, U., and Groot, H. D. J. H. (1999). Determ. chelatable iron pool Isol. rat hepatocytes by digital Fluoresc. Microsc. using Fluoresc. probe, phen green SK 29 (4).
Plascencia-Villa, G., and Perry, G. (2021). Preventive and therapeutic strategies in Alzheimer's disease: Focus on oxidative stress, redox metals, and ferroptosis. Antioxid. Redox Signal. 34 (8), 591–610. doi:10.1089/ars.2020.8134
Qi, X., Zhang, Y., Guo, H., Hai, Y., Luo, Y., and Yue, T. (2020). Mechanism and intervention measures of iron side effects on the intestine. Crit. Rev. Food Sci. Nutr. 60 (12), 2113–2125. doi:10.1080/10408398.2019.1630599
Qiu, Y., Cao, Y., Cao, W., Jia, Y., and Lu, N. (2020). The application of ferroptosis in diseases. Pharmacol. Res. 159, 104919. doi:10.1016/j.phrs.2020.104919
Qu, C., Peng, Y., and Liu, S. (2022). Ferroptosis biology and implication in cancers. Front. Mol. Biosci. 9, 892957. doi:10.3389/fmolb.2022.892957
Qu, Z., Sun, J., Zhang, W., Yu, J., and Zhuang, C. (2020). Transcription factor NRF2 as a promising therapeutic target for Alzheimer's disease. Free Radic. Biol. Med. 159, 87–102. doi:10.1016/j.freeradbiomed.2020.06.028
Reddy, P. H., Manczak, M., Yin, X., Grady, M. C., Mitchell, A., Tonk, S., et al. (2018). Protective effects of Indian spice curcumin against amyloid-beta in Alzheimer's disease. J. Alzheimers Dis. 61 (3), 843–866. doi:10.3233/JAD-170512
Reichert, C. O., de Freitas, F. A., Sampaio-Silva, J., Rokita-Rosa, L., Barros, P. d. L., Levy, D., et al. (2020). Ferroptosis mechanisms involved in neurodegenerative diseases. Int. J. Mol. Sci. 21 (22), E8765. doi:10.3390/ijms21228765
Reiss, A. B., Arain, H. A., Stecker, M. M., Siegart, N. M., and Kasselman, L. J. (2018). Amyloid toxicity in Alzheimer's disease. Rev. Neurosci. 29 (6), 613–627. doi:10.1515/revneuro-2017-0063
Ross, D., and Siegel, D. (2021). The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 41, 101950. doi:10.1016/j.redox.2021.101950
Sato, M., Kusumi, R., Hamashima, S., Kobayashi, S., Sasaki, S., Komiyama, Y., et al. (2018). The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci. Rep. 8 (1), 968. doi:10.1038/s41598-018-19213-4
Serviddio, G., Cassano, T., Pace, L., Bedse, G., Michele Lavecchia, A., De Marco, F., et al. (2015). Glutamate and mitochondria: Two prominent players in the oxidative stress-induced neurodegeneration. Curr Alzheimer Res. 13 (2), 185-97. doi:10.2174/1567205013666151218132725
Sheng, X., Shan, C., Liu, J., Yang, J., Sun, B., and Chen, D. (2017). Theoretical insights into the mechanism of ferroptosis suppression via inactivation of a lipid peroxide radical by liproxstatin-1. Phys. Chem. Chem. Phys. 19 (20), 13153–13159. doi:10.1039/c7cp00804j
Shetty, R. A., Forster, M. J., and Sumien, N. (2013). Coenzyme Q(10) supplementation reverses age-related impairments in spatial learning and lowers protein oxidation. Age (Dordr) 35 (5), 1821–1834. doi:10.1007/s11357-012-9484-9
Shi, F., Zhang, P., Mao, Y., Wang, C., Zheng, M., and Zhao, Z. (2017). The nitroxide Tempo inhibits hydroxyl radical production from the Fenton-like reaction of iron(II)-citrate with hydrogen peroxide. Biochem. Biophys. Res. Commun. 483 (1), 159–164. doi:10.1016/j.bbrc.2016.12.174
Shi, Z., Zhang, L., Zheng, J., Sun, H., and Shao, C. (2021). Ferroptosis: Biochemistry and biology in cancers. Front. Oncol. 11, 579286. doi:10.3389/fonc.2021.579286
Shin, D., Kim, E. H., Lee, J., and Roh, J. L. (2018). Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454–462. doi:10.1016/j.freeradbiomed.2018.10.426
Song, X., and Long, D. (2020). Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases. Front. Neurosci. 14, 267. doi:10.3389/fnins.2020.00267
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 171 (2), 273–285. doi:10.1016/j.cell.2017.09.021
Stockwell, B. R., Jiang, X., and Gu, W. (2020). Emerging mechanisms and disease relevance of ferroptosis. Trends Cell. Biol. 30 (6), 478–490. doi:10.1016/j.tcb.2020.02.009
Sun, Y., Xiao, Q., Luo, C., Zhao, Y., Pu, D., Zhao, K., et al. (2017). High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp. Cell. Res. 359 (2), 312–318. doi:10.1016/j.yexcr.2017.07.032
Takeda, S., Sato, N., Uchio-Yamada, K., Sawada, K., Kunieda, T., Takeuchi, D., et al. (2010). Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U. S. A. 107 (15), 7036–7041. doi:10.1073/pnas.1000645107
Tang, M., Chen, Z., Wu, D., and Chen, L. (2018). Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell. Physiol. 233 (12), 9179–9190. doi:10.1002/jcp.26954
Tang, M., and Taghibiglou, C. (2017). The mechanisms of action of curcumin in Alzheimer's disease. J. Alzheimers Dis. 58 (4), 1003–1016. doi:10.3233/JAD-170188
Tang, Z., Huang, Z., Huang, Y., Chen, Y., Huang, M., Liu, H., et al. (2021). Ferroptosis: The silver lining of cancer therapy. Front. Cell. Dev. Biol. 9, 765859. doi:10.3389/fcell.2021.765859
Tarangelo, A., Magtanong, L., Bieging-Rolett, K. T., Li, Y., Ye, J., Attardi, L. D., et al. (2018). p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell. Rep. 22 (3), 569–575. doi:10.1016/j.celrep.2017.12.077
Thomas, M. H., Paris, C., Magnien, M., Colin, J., Pelleieux, S., Coste, F., et al. (2017). Dietary arachidonic acid increases deleterious effects of amyloid-beta oligomers on learning abilities and expression of AMPA receptors: Putative role of the ACSL4-cPLA2 balance. Alzheimers Res. Ther. 9 (1), 69. doi:10.1186/s13195-017-0295-1
Thompson, J. A., White, C. C., Cox, D. P., Chan, J. Y., Kavanagh, T. J., Fausto, N., et al. (2009). Distinct Nrf1/2-independent mechanisms mediate as 3+-induced glutamate-cysteine ligase subunit gene expression in murine hepatocytes. Free Radic. Biol. Med. 46 (12), 1614–1625. doi:10.1016/j.freeradbiomed.2009.03.016
Tsatsanis, A., Wong, B. X., Gunn, A. P., Ayton, S., Bush, A. I., Devos, D., et al. (2020). Amyloidogenic processing of Alzheimer's disease beta-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 25 (9), 1958–1966. doi:10.1038/s41380-020-0762-0
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17 (1), 5–21. doi:10.1038/nrn.2015.1
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet. Neurol. 13 (10), 1045–1060. doi:10.1016/S1474-4422(14)70117-6
Xia, J., Si, H., Yao, W., Li, C., Yang, G., Tian, Y., et al. (2021). Research progress on the mechanism of ferroptosis and its clinical application. Exp. Cell. Res. 409 (2), 112932. doi:10.1016/j.yexcr.2021.112932
Xie, Y., Song, X., Sun, X., Huang, J., Zhong, M., Lotze, M. T., et al. (2016). Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 473 (4), 775–780. doi:10.1016/j.bbrc.2016.03.052
Xie, Y., Zhu, S., Song, X., Sun, X., Fan, Y., Liu, J., et al. (2017). The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell. Rep. 20 (7), 1692–1704. doi:10.1016/j.celrep.2017.07.055
Xu, G., Wang, H., Li, X., Huang, R., and Luo, L. (2021). Recent progress on targeting ferroptosis for cancer therapy. Biochem. Pharmacol. 190, 114584. doi:10.1016/j.bcp.2021.114584
Yan, H. F., Zou, T., Tuo, Q. Z., Xu, S., Li, H., Belaidi, A. A., et al. (2021). Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 6 (1), 49. doi:10.1038/s41392-020-00428-9
Yan, H., Yan, Y., Gao, Y., Zhang, N., Kumar, G., Fang, Q., et al. (2022). Transcriptome analysis of fasudil treatment in the APPswe/PSEN1dE9 transgenic (APP/PS1) mice model of Alzheimer's disease. Sci. Rep. 12 (1), 6625. doi:10.1038/s41598-022-10554-9
Yan, N., and Zhang, J. (2019). Iron metabolism, ferroptosis, and the links with Alzheimer's disease. Front. Neurosci. 13, 1443. doi:10.3389/fnins.2019.01443
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15 (3), 234–245. doi:10.1016/j.chembiol.2008.02.010
Yoo, S. E., Chen, L., Na, R., Liu, Y., Rios, C., Van Remmen, H., et al. (2012). Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52 (9), 1820–1827. doi:10.1016/j.freeradbiomed.2012.02.043
Yuan, Y., Men, W., Shan, X., Zhai, H., Qiao, X., Geng, L., et al. (2020). Baicalein exerts neuroprotective effect against ischaemic/reperfusion injury via alteration of NF-kB and LOX and AMPK/Nrf2 pathway. Inflammopharmacology 28 (5), 1327–1341. doi:10.1007/s10787-020-00714-6
Zhang, Y. H., Wang, D. W., Xu, S. F., Zhang, S., Fan, Y. G., Yang, Y. Y., et al. (2018). α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. doi:10.1016/j.redox.2017.11.001
Zhang, Z., Dmitrieva, N. I., Park, J. H., Levine, R. L., and Burg, M. B. (2004). High urea and NaCl carbonylate proteins in renal cells in culture and in vivo, and high urea causes 8-oxoguanine lesions in their DNA. Proc. Natl. Acad. Sci. U. S. A. 101 (25), 9491–9496. doi:10.1073/pnas.0402961101
Zheng, H., Jiang, J., Xu, S., Liu, W., Xie, Q., Cai, X., et al. (2021). Nanoparticle-induced ferroptosis: Detection methods, mechanisms and applications. Nanoscale 13 (4), 2266–2285. doi:10.1039/d0nr08478f
Keywords: Alzheimer’s disease, ferroptosis, oxidative stress, p53, lipid peroxidation
Citation: Ma H, Dong Y, Chu Y, Guo Y and Li L (2022) The mechanisms of ferroptosis and its role in alzheimer’s disease. Front. Mol. Biosci. 9:965064. doi: 10.3389/fmolb.2022.965064
Received: 09 June 2022; Accepted: 25 July 2022;
Published: 26 August 2022.
Edited by:
Guo Chen, China Pharmaceutical University, ChinaReviewed by:
Yi Wang, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, ChinaXin Wang, National Institutes of Health (NIH), United States
Copyright © 2022 Ma, Dong, Chu, Guo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqin Guo, eWFucWluZ3VvQHNpbmEuY29t; Luxin Li, bGlsdXhpbkBtZGptdS5lZHUuY24=