 Alessandro T. Caputo1,2†
Alessandro T. Caputo1,2† Roberta Ibba1,3†
Roberta Ibba1,3† James D. Le Cornu1,4Benoit Darlot1Mario Hensen1Colette B. Lipp1Gabriele Marcianò5Snežana Vasiljević1Nicole Zitzmann1*
James D. Le Cornu1,4Benoit Darlot1Mario Hensen1Colette B. Lipp1Gabriele Marcianò5Snežana Vasiljević1Nicole Zitzmann1* Pietro Roversi6,7*
Pietro Roversi6,7*- 1Biochemistry Department, Oxford Glycobiology Institute, University of Oxford, Oxford, United Kingdom
- 2Commonwealth Scientific and Industrial Research Organisation, Clayton, VIC, Australia
- 3Department of Medicine, Surgery and Pharmacy, University of Sassari, Sassari, Italy
- 4Wellcome Trust Centre for Cell Biology, University of Edinburgh, Scotland, United Kingdom
- 5Biochemistry Department, University of Oxford, Oxford, United Kingdom
- 6IBBA-CNR Unit of Milano, Institute of Agricultural Biology and Biotechnology, Milano, Italy
- 7Department of Molecular and Cell Biology, Leicester Institute of Structural and Chemical Biology, University of Leicester, Leicester, United Kingdom
None of the current data processing pipelines for X-ray crystallography fragment-based lead discovery (FBLD) consults all the information available when deciding on the lattice and symmetry (i.e., the polymorph) of each soaked crystal. Often, X-ray crystallography FBLD pipelines either choose the polymorph based on cell volume and point-group symmetry of the X-ray diffraction data or leave polymorph attribution to manual intervention on the part of the user. Thus, when the FBLD crystals belong to more than one crystal polymorph, the discovery pipeline can be plagued by space group ambiguity, especially if the polymorphs at hand are variations of the same lattice and, therefore, difficult to tell apart from their morphology and/or their apparent crystal lattices and point groups. In the course of a fragment-based lead discovery effort aimed at finding ligands of the catalytic domain of UDP–glucose glycoprotein glucosyltransferase (UGGT), we encountered a mixture of trigonal crystals and pseudotrigonal triclinic crystals—with the two lattices closely related. In order to resolve that polymorphism ambiguity, we have written and described here a series of Unix shell scripts called CoALLA (crystal polymorph and ligand likelihood-based assignment). The CoALLA scripts are written in Unix shell and use autoPROC for data processing, CCP4-Dimple/REFMAC5 and BUSTER for refinement, and RHOFIT for ligand docking. The choice of the polymorph is effected by carrying out (in each of the known polymorphs) the tasks of diffraction data indexing, integration, scaling, and structural refinement. The most likely polymorph is then chosen as the one with the best structure refinement Rfree statistic. The CoALLA scripts further implement a likelihood-based ligand assignment strategy, starting with macromolecular refinement and automated water addition, followed by removal of the water molecules that appear to be fitting ligand density, and a final round of refinement after random perturbation of the refined macromolecular model, in order to obtain unbiased difference density maps for automated ligand placement. We illustrate the use of CoALLA to discriminate between H3 and P1 crystals used for an FBLD effort to find fragments binding to the catalytic domain of Chaetomium thermophilum UGGT.
1 Introduction
There is an urgent medical need to develop novel antiviral drugs, as exemplified by the recent Ebola, Zika, and SARS-CoV-2 outbreaks (Pardi and Weissman, 2020). In particular, an FDA-approved host-targeting broad-spectrum antiviral could revolutionize the treatment of existing and newly emerging viruses (Dwek et al., 2022). Recent works elucidating the structures of key endoplasmic reticulum (ER) enzymes assisting the folding of viral glycoproteins have opened new avenues for identifying novel antivirals (Caputo et al., 2016; Roversi et al., 2017; Warfield et al., 2020).
Host-targeting broad-spectrum antivirals are a possibility because many viruses hijack the same host enzymes during their life cycle (Oksenych and Kainov (2022). For example, the envelope glycoproteins of many viruses are exquisitely dependent on calnexin-mediated folding, a process enabled by the action of the ER endoplasmic reticulum enzyme UDP–Glc glycoprotein glucosyltransferase (UGGT) and the ER alpha glucosidases I and II (D’Alessio et al., 2010). ER alpha glucosidases I and II usher client proteins in and out of the calnexin cycle, which is part of the glycoprotein folding quality control in the ER, whilst UGGT plays a key role in retaining misfolded glycoproteins in the ER for a “second chance” at folding correctly (Hammond et al., 1994).
One of the more advanced strategies for host-targeting broad-spectrum antiviral drug development is focusing on iminosugars as active site inhibitors of the ER alpha glucosidases (Alonzi et al., 2017; Tyrrell et al., 2017). However, as carbohydrate mimics, iminosugar inhibitors of the ER alpha glucosidases have some undesired off-target effects, as they also inhibit certain other carbohydrate processing enzymes within the human host (Sayce et al., 2016). A new class of molecules inhibiting host glycoprotein folding enzymes that viruses depend upon would have great potential for antiviral therapy (Karade et al., 2021). To complement a programme of development of new allosteric ER alpha glucosidase inhibitors with fewer off-target effects, we endeavoured to investigate the potential of UGGT, the major calnexin cycle misfold sensor (Trombetta et al., 1989), as a novel antiviral target (Tax et al., 2019). Currently, no UGGT inhibitors are known other than the product UDP (Trombetta and Helenius, 1999) and the UDP–glucose analogue UDP-2-deoxy-2-fluoro-D-glucose (U2F), neither of which is selective for UGGT.
One effective strategy to broaden the knowledge of the chemical space of a given protein target is fragment-based lead discovery (FBLD), a sequence of experiments enabling the extraction of ligands of a chosen target macromolecule from a chemical library (Ciulli et al., 2006; Murray and Blundell, 2010; Chen et al., 2015; Müller et al., 2022). Whenever crystals of the target macromolecule reliably diffract to near-atomic resolution, single-crystal X-ray crystallography is one of the main techniques successfully used for FBLD (Ciulli et al., 2006; Murray and Blundell, 2010; Radoux et al., 2016; Müller et al., 2022).
We recently determined crystal structures of the ER glycoprotein folding quality control checkpoint enzyme, the UDP-Glc glycoprotein glucosyltransferase of Chaetomium thermophilum (CtUGGT) (Roversi et al., 2017). As well as a potential drug target against viruses (Dalziel et al., 2014), UGGT could be a target for compounds rescuing slightly misfolded and yet active glycoprotein mutants in certain instances of congenital rare disease (Amara et al., 1992) and against some cancers (Tax et al., 2019). We set out to find small-molecule ligands for this target via X-ray crystallography FBLD.
We could not crystallise the GT24 catalytic domain of human UGGT—so we used crystals of the GT24 catalytic domain of CtUGGT (hereinafter CtUGGTGT24) instead. The sequence of this fungal UGGT has about 70% similarity and 60% identity to the ones of the same domain of the two human UGGT isoforms (UGGT1 and UGGT2, see Figure 1) so that any ligands found with the crystals of CtUGGT would likely bind the human enzymes too—paving the way to a medicinal chemistry program towards modulators of human UGGT activity. In order to avoid fragments that would bind to the UDP–Glc pocket (and would then likely have some off-target affinity for a number of human glucosyltransferases using the same co-factor (Albesa-Jové and Guerin, 2016), the crystals used for the FBLD study were grown in the presence of Ca2+ and UDP–Glc (hereinafter CtUGGTGT24UDP−Glc).

FIGURE 1. Sequence alignment of GT24 domains of a few eukaryotic UGGTs. Ct: Chaetomium thermophilum; Hs: Homo sapiens; Mm: Mus musculus; At: Arabidopsis thaliana; Ce: Caenorhabditis elegans; Dm: Drosophila melanogaster; Sp: Schizosaccharomyces pombe; Ca: Candida albicans. The CtUGGT D1302 and D1304 residues coordinating the catalytic Ca2+ ion are completely conserved across these sequences. Red triangles mark the CtUGGT 1346WY1347 clamp. A blue square marks the position of CtUGGT Y1211 (coordinating the UDP–Glc uracyl ring). A green circle marks the position of CtUGGT D1435 (coordinating the Ca2+ ion).
The CtUGGTGT24UDP−Glc crystals turned out to belong to two different polymorphs. Crystal polymorphism refers to the growth of crystals of the same compound belonging to different crystal forms. Each of the crystal forms for the same compound is referred to as one of its crystal polymorphs. All polymorphs contain the same molecule, but each polymorph has its own distinct crystal lattice and/or space group symmetry and/or asymmetric unit contents (Buerger, 1936a; Buerger, 1936b). A number of different polymorphs can grow from a few related crystallisation conditions—or even from crystallisation solutions that are nominally the same but experience slight stochastic variations in variables such as temperature, rate of evaporation, and impurities (Jurnak, 1985; Carter et al., 1994; Zabara et al., 2011; Yekwa et al., 2017).
The occurrence of crystal polymorphism during FBLD efforts is not uncommon. For example, in a recent crystallographic screening project, 364 diffraction datasets were collected each from a crystal individually soaked with one compound from a library; of these, 16 crystals belonged to the orthorhombic P212121 space group instead of the common monoclinic P21 form; the two unit cells were also closely related (Schiebel et al., 2016). It is of note that systematic exploration of crystal polymorphism prior to FBLD can be of great advantage: the best diffracting polymorph can be selected, and various lattices expose different potential drug-binding sites (Vera et al., 2013). Unfortunately, implementation of polymorph assignment in automated FBLD data processing pipelines still leaves some to be desired.
For example, the XCE FBLD X-ray data processing pipeline used at Diamond Light Source (Krojer et al., 2017; Collins et al., 2018; Douangamath et al., 2021; Pearce et al., 2022) decides on the crystal polymorph using a comparison of point group symmetry and cell (ignoring the information encoded by the known polymorph atomic models), or it leaves polymorph attribution to manual intervention on the part of the user. The FBLD efforts at the BESSY and MAXIV synchrotrons use FragMAXapp for data processing (Lima et al., 2021); at EMBL Grenoble, the CRIMS suite is a large-scale, automated fragment screening pipeline enabling evaluation of libraries of over 1,000 fragments (Cornaciu et al., 2021); the IspyB system used at some synchrotrons allows for data integration and storage in alternative lattices in parallel, specifically to address the possibility of multiple polymorphs (Monaco et al., 2013); the FBLD efforts at the Swiss Light Source (SLS) rely on the FFCS processing pipeline (Kaminski et al., 2022). Regrettably, none of these FBLD data processing systems has a mechanism in place for automated polymorph assignment.
Overall, current implementations of the FBLD discovery process can be plagued by space group and cell ambiguity, especially if the polymorphs at hand are variations of the same lattice and, therefore, difficult to tell apart from their morphology and/or their apparent crystal lattices and point groups.
In order to expedite the analysis of the CtUGGTGT24UDP−Glc FBLD X-ray diffraction datasets, we have written and described here a series of Unix shell scripts called CoALLA (crystal polymorph and ligand likelihood-based Assignment). The CoALLA scripts are written in Unix shell and use autoPROC (Vonrhein et al., 2011) for data processing, CCP4-Dimple/REFMAC5 (Murshudov et al., 2011; Winn et al., 2011; Keegan et al., 2015) and BUSTER (Blanc et al., 2004; Bricogne et al., 2017) for refinement, and RHOFIT (Vonrhein et al., 2011) for ligand placement.
Unique to CoALLA is the implementation of the choice of the polymorph, which is effected by carrying out diffraction data indexing, integration, scaling, and structural refinement in each of the possible polymorphs. The most likely polymorph is then chosen based on the best structure refinement statistics.
2 CtUGGTGT24UDP−Glc crystal structure
The crystals of CtUGGTGT24UDP−Glc used for the FBLD effort belonged to the space group H3 (with cell edges a = b = 118.8 Å and c = 68.8 Å (cyan cell in Figures 2A,B, PDB ID 6FSN)) with one molecule per asymmetric unit. The crystals likely capture a conformation of UDP–Glc following initial binding to the protein: the co-factor’s ribose ring points towards the solvent (Figure 3A). The uracyl ring O4 atom accepts a hydrogen bond from the main chain NH of S1207, and its N3 atom donates one hydrogen bond to the main chain O of the same residue (top of Figure 3A). Half of the coordination sphere of the Ca2+ ion in the CtUGGTGT24 active site is occupied by the side chains from D1302 and D1304 belonging to the UGGT conserved 1302DAD1304 motif) and the side chain of the conserved D1435; the remaining three sites are occupied by an O atom from the β phosphate of UDP–Glc, the O2′ atom of the Glc ring, and a water molecule (Figure 3A).
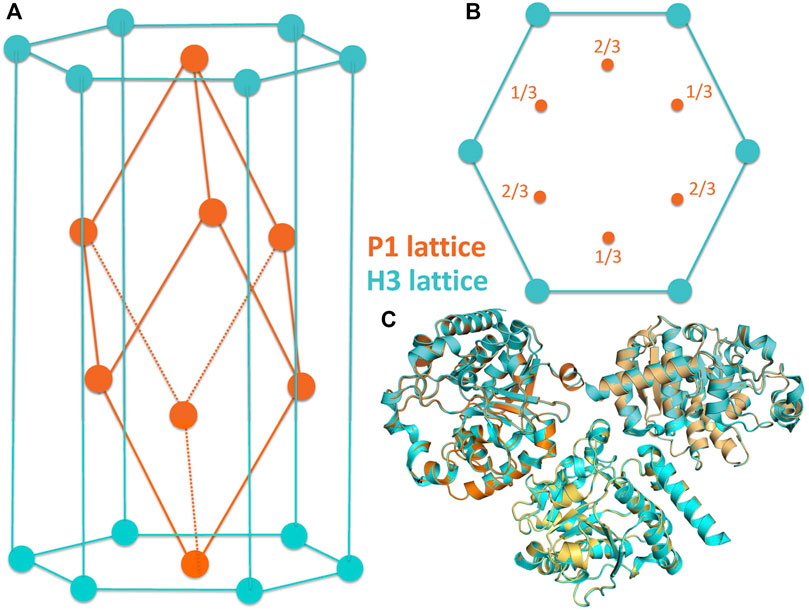
FIGURE 2. (A,B) Related crystal symmetries and lattices of the H3 (cyan) and P1 (orange) crystal forms of the CtUGGTGT24 crystals. (C)The molecule in the asymmetric unit of the H3 crystal is shown, together with two of its symmetry mates (cyan), in cartoon representation. This portion of the H3 lattice has been overlaid onto the asymmetric unit of the P1 crystals (three chains, painted orange, light orange, and yellow-orange; also in cartoon representation) by superposing the “A” of the H3 crystal to the “A” chain in the P1 crystal.
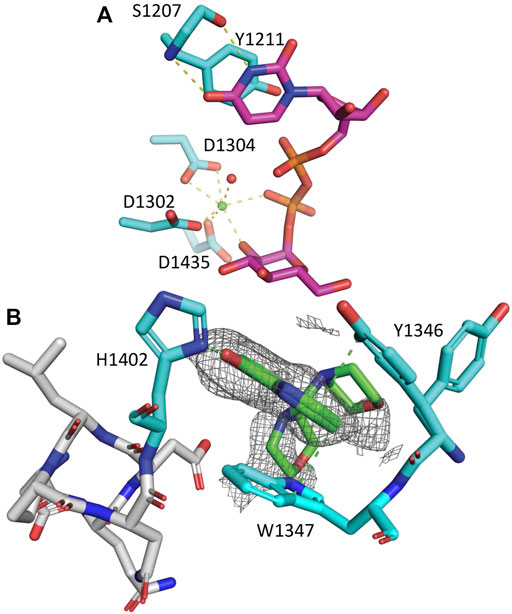
FIGURE 3. CtUGGTGT24UDP−Glc and CtUGGTGT245M−8OH−Q crystal structures. (A) CtUGGTGT24UDP−Glc (PDB ID 6FSN). Protein atoms in stick representation; C cyan (but UDP–Glc C magenta), O red, N blue, P orange. H-bonds and Ca-coordination bonds are in yellow dashed lines. At the top, the residues coordinating the uracyl ring: the side chain of CtUGGT Y1211 and the main chain of S1207. At the bottom, the Ca2+ ion is a green sphere, and its coordinating water molecules are red spheres. The side chains of residues D1302, D1304, and D1435 coordinate the Ca2+. Three more coordination sites are taken up by the β phosphate, the O2′ atom of its Glc ring, and a water molecule. The uracyl O4 atom accepts an H-bond from the S1207 main chain NH. (B) Zoom onto the CtUGGT 1346YW1347 clamp (C atoms in cyan) binding 5M-8OH-Q (C atoms in green). The 8OH-quinoline ring inserts and is sandwiched between the aromatic side chains of the conserved residues 1346YW1347. The two aromatic side chains stabilise the quinoline ring, forming an aromatic trimer; the 8-OH group of the quinoline also establishes an H-bond to the side chain of 1402H. Representative distances to interacting residues are in green dashed lines. Only two of the many morpholine ring placements are shown. The unbiased Fo–Fc map is represented as a grey mesh at a 2.0 σ contour level. PDB ID: 7ZXW.
2.1 Fragment-soaked CtUGGTGT24UDP−Glc crystal index in related P1 or H3 lattices
To discover CtUGGTGT24 ligands by FBLD, 768 CtUGGTGT24UDP−Glc crystals were soaked with as many compounds, and X-ray diffraction datasets were collected from each soaked crystal (see Table 1). During the initial analysis of these X-ray diffraction datasets, we discovered that, upon soaking, crystal symmetry was sometimes lowered to P1, with three molecules in the asymmetric unit of a pseudo-rhombohedral primitive cell (orange cell in Figures 2A,B, PDB ID 7ZLU) of dimensions a = 66.3 Å, b = 72.7 Å, c = 72.7 Å, α = 111.2°, β = 107.7°, and γ = 107.7°. This cell is a distortion of the R3 primitive rhombohedral cell, which for the CtUGGTGT24UDP−Glc H3 crystals used for the soaks, would have lattice parameters a = b = c = 72.32 Å and α = β = γ = 110.43°. The three molecules in the P1 crystal asymmetric unit are related by an NCS threefold axis equivalent to the H3 crystallographic threefold axis. The two packings are difficult to distinguish (see Figure 2C), and often a soaked CtUGGTGT24UDP−Glc X-ray diffraction dataset can index in both the H3 and P1 lattices, depending, for example, on the program used for indexing or the parameters of the automated indexing algorithm.

TABLE 1. Summary of the FBLD effort on CtUGGTGT24.
The CtUGGTGT24 FBLD datasets were initially processed through the XCE workflow tool (Krojer et al., 2017). Space group assignment in the XCE workflow relies on automated data processing taking place during automated data collection on the Diamond I04-1 beamline (Douangamath et al., 2021), but the decision about the possible space group and cell of a certain dataset is made without consulting available models. In essence, the initial XCE automated decision regarding the space group is based on X-ray diffraction scaling statistics and the cell and point group of the dataset. For example, XCE reads the PDB header of each declared polymorph and determines the point group and unit cell volume. Then, once processing each fragment-soaked dataset, if the MTZ point group is identical to the one of a reference polymorph and the unit cell volume is similar (within 12%, but this can be tuned), it assumes that they belong to the same crystal form. XCE also allows for the detection of unexpected crystal forms for a subset of the collected crystals: in the presence of different polymorphs, discrepancies between the reference files provided and some of the datasets arise—either in terms of the space groups and/or the unit cells’ volumes or because of high RRfree values after initial refinement. In the presence of crystal polymorphism, a certain degree of manual curation is, therefore, needed to run XCE successfully.
When faced with processing hundreds of datasets belonging to different and yet related crystal forms, we reasoned that from a Bayesian statistical standpoint (Bricogne, 1997), the best way of deciding on the correct crystal polymorph is the one that consults all the available data. In this conceptual framework, the likelihood of a certain dataset belonging to one of the known polymorphs can be best evaluated by refining, in turn, each known polymorphic structure against the dataset processed in that symmetry/cell and then choosing the one with the best refinement Rfree statistic.
3 Results on the CtUGGTGT24 FBLD effort
More than 1,200 CtUGGTGT24 crystals were grown, and 768 of them were soaked with as many compounds of the DSi-Poised Library (Diamond-SGC-iNext, ex DSPL, https://www.diamond.ac.uk/Instruments/Mx/Fragment-Screening/Fragment-Libraries/DSi-Poised-Library.html).
Each compound was at a concentration of 500 mM in deuterated DMSO; 40 nl of compound stock solution was dispensed by the Echo robot onto each 200-nl drop, making the final compound concentration 83 mM in 20% DMSO.
Automated data collection was carried out on 692 soaked crystals, but only 493 scaled datasets were obtained, spanning the resolution range of 1.72–11.4 Å. The majority of datasets diffracted to 3.5 Å resolution or better, see Figure 5A.
Only for 439 of these 493 datasets, a structure could be successfully refined in either H3 or P1 or both. Table 1 reports the overall counts, while Figures 5A,B report a few statistics for the 439 diffraction datasets that refined better either in H3 (390 datasets) or P1 (49 datasets).
On average, H3 crystals diffract better than P1 ones:
None of the 39 P1 crystals revealed any bound fragments. The P1 2.05 Å structure of a co-crystal of the CtUGGTGT24 domain with the UDP–Glc analogue (and UGGT inhibitor) UDP-2-deoxy-2-fluoro-D-glucose (U2F) was obtained after the FBLD effort and is available at the Protein Databank (https://www.rcsb.org) as PDB ID 7ZLU.
Three of the H3-soaked crystals turned out to contain density for a bound CtUGGTGT24 ligand.
3.1 Fragment x0441
A CtUGGTGT24UDP−Glc crystal soaked in (1-(1-ethyl-1H-pyrazol-5-yl)-N-methylmethanamine (SMILES string CCN1N=CC=C1CNC, “fragment x0441”) shows density for the ligand at a crystal contact, but the binding site is not particularly conserved: CtUGGT Y1350, H1402, Q1381, and M1403 (which in HsUGGT1 corresponds to H1406, N1458, Q1437, and M1459) (Figures 1, 4). The pose is rather ambiguous at this resolution (2.44 Å). This site, in the context of the full-length UGGT molecule, faces the UGGT central saddle and is proximal to the putative binding site of the first GlcNAc of the UGGT client’s glycan (close to CtUGGT H1402, which is HsUGGT1 N1508).
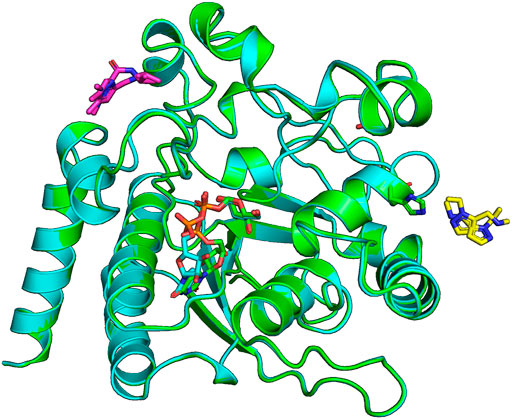
FIGURE 4. Ligands x0441 and x0763 bind to surface pockets of the CtUGGTGT24 domain. The crystal structures of CtUGGTGT24UDP−Glc soaked in compounds x0441 and x0763 are painted in green and cyan cartoon representation, respectively. The molecule of UDP–Glc in the catalytic pocket of each structure is represented in sticks (its C atoms also in green and cyan for the x0441 and x0763 soaked crystal structure, respectively; N atoms in blue, O atoms in red, and P atoms in orange). Two partially overlapping poses of compound x0673 are represented in sticks with magenta C atoms. Two partially overlapping poses of compound x0441 are represented in sticks with yellow C atoms, next to CtUGGTGT24 residue H1042, also represented in sticks.
3.2 Fragment x0763
The CtUGGTGT24UDP−Glc crystal soaked in (1,3-dimethyl-N-(propan-2-yl)-1H-pyrazole-5-carboxamide (SMILES string CC(C)NC(=O)C=1C=C(C)N(C)N1, “fragment x0763”) also showed residual electron density for the ligand (in two orientations), this time in a conserved pocket: CtUGGT T1442, M1441, R1333, R1452, and E1444 (which in HsUGGT1 corresponds to T1498, M1497, R1389, R1508, and E1500) (Figures 1, 4). The pocket is not far from the putative binding site for the C-branch of the client glycoprotein glycan (CtUGGT M1336, Y1339, and M1441 or HsUGGT1 M1392, Y1395, and M1507), but the interactions of the fragment with the GT domain are weak—perhaps because the binding site is at a crystal contact.
3.3 Fragment x0248: CtUGGTGT245M−8OH−Q crystal structure
The best hit of the FBLD effort was 5-[(morpholin-4-yl)methyl]quinolin-8-ol (SMILES string C1COCCN1CC2=C3C=CC=NC3=C(C=C2)O, “fragment x0248,” henceforth 5M-8OH-Q), a crystal soaked with which diffracted to 2.25 Å (PDB ID 7ZXW). Diffraction data from this crystal process in H3 with RmeasH3 = 0.323 and in P1 with RmeasP1 = 0.206. The CCP4-Dimple/REFMAC5 Rfrees are RfreeH3 = 0.227 and RfreeP1 = 0.2537.
The compound binds to a conserved patch on the surface of the CtUGGTGT24 domain, about 15 Å away from the UDP–Glc binding site (Figure 3B). The morpholine ring is partially disordered in the crystal, but one of its ring placements is 4.2 Å from the conserved 1396DQD1398 motif coordinating the Glc ring of UDP–Glc; the ligand also causes a displacement of the side chain of CtUGGTGT24 1346Y. Through this displacement, the 8OH-quinoline ring inserts and is sandwiched between the aromatic side chains of the conserved residues 1346YW1347—which we propose to call the “YW clamp.” The two aromatic side chains stabilise the quinoline ring, forming an aromatic trimer (Lanzarotti et al., 2020); the 8-OH group of the quinoline also establishes an H-bond to the side chain of 1402H (Figure 3B).
Tables 2, 3 report the crystallographic data and refinement statistics for the CtUGGTGT24UDP−Glc and CtUGGTGT245M−8OH−Q crystal structures.

TABLE 2. X-ray data collection parameters and data processing statistics for CtUGGTGT24 crystal structures.

TABLE 3. Refinement statistics for CtUGGTGT24 crystal structures. All structures contain a Ca+ ion coming from the protein solution.
4 Discussion
In the course of an X-ray crystallography FBLD effort aimed at the discovery of molecules binding the catalytic domain of Chaetomium thermophilum UGGT (CtUGGTGT24), we encountered crystal polymorphism: the majority of CtUGGTGT24 crystals belonged to space group H3, but some crystals lowered their symmetry to an equivalent P1 pseudo-rhombohedral lattice. The space group and lattice ambiguity was tricky to resolve by the automated FBLD data processing algorithm—which essentially did not commit to a specific polymorph and left the polymorph choice to the user. However, how is such a choice best implemented in an automated and reliable fashion?
First, if the coordinates of enough indexing spots are gathered, the reciprocal cell parameters can be rather precise. Errors in the reciprocal lattice choice can then be estimated by the differences between reciprocal cell parameters that would differ in the two reciprocal lattices being compared. For example, for the CtUGGTGT24 crystals described in this study, b* and c* would be identical in R3 but differ in P1.
In cases where alternative lattices are pseudo-equivalent and symmetry/cell changes are such that the volume of the asymmetric unit increases/decreases, a discriminating criterion between polymorphs can be based on indexing quality or average intensity of classes of reflections that are systematically extinct in one polymorph and allowed in another one. For example, if indexing and integration of a lattice-centred dataset are carried out in a primitive lattice, the data will appear pseudo-lattice-centred, with more reflections in the primitive than in the lattice-centred space group, and the question that will help discriminate the space groups is then how strong are the additional reflections only allowed in the primitive cell polymorph compared to those that are present in both polymorphs. The values of the fractional mean intensity of the additional reflections, perhaps as a function of resolution, would help discriminate between a truly centred lattice and a pseudo-centred one.
In the general case in which the polymorphs have equivalent lattices but different symmetries, or unrelated lattices altogether, a polymorph choice based on indexing quality (or average intensity of classes of reflections) may not be so straightforward.
A second class of statistics that may be conceivably used for polymorph discrimination are scaling statistics, but each commonly evaluated scaling statistic risks opening a different can of worms if chosen as the basis for polymorph discrimination. For example, Rmerge has been shown to privilege lower-symmetry space groups, and Rmeas was introduced to discriminate between space groups with different scaling multiplicities (Diederichs and Karplus, 1997; Karplus and Diederichs, 2015). Unfortunately, a robust estimation of Rmeas requires data multiplicity—and so does the estimation of CC1/2 (Karplus and Diederichs, 2015): both scaling statistics are strongly dependent on random (i.e., photon-counting) errors and may not discriminate very well between low-signal but correct symmetry and high-signal but wrong symmetry. Data multiplicity will be high enough for reliable polymorph choice (in the presence of polymorphs of low symmetry) only if more than half a reciprocal sphere of data is collected and/or a data collection strategy is followed—both of which are uncommon practices in most FBLD efforts. For example, in P1, two (or more) datasets of 180◦ each, with a large kappa offset, would be needed. Data of this kind may not be always available for low-symmetry space group FBLD datasets, thus ruling out the implementation of strategies based on statistics like Rmeas or CC1/2 or those where the error model comes into play, like average I/σ(I) and its sister statistic, asymptotic I/σ(I)asympt (ISa) (Diederichs, 2010). Supplementary Figure S1 illustrates scatter plots of outer shell I/σ(I), outer shell CC1/2, I/σ(I)asympt (ISa), and Rmeas separately for CtUGGTGT24 datasets that had lower Rfree in H3 or P1. None of these data processing-based statistics would lead to polymorph choice in overall agreement with the Rfree one.
We decided to test a polymorph choice strategy that would consult the known H3 and P1 atomic structures, and for each dataset use both of them individually in refinement against the diffraction data (processed in that symmetry), in order to resolve the H3 vs. P1 decision, which would then have the advantage of being based on all the prior information available (Jaynes, 1968).
Polymorph choice based on refinement statistics of course has its own risks/drawbacks. First, reliable refinements in all possible polymorphs depend on the availability of a structural model of good quality for each of them. Second, when the lattices of the polymorphs at hand are related, to properly compare Rfree values, the free set of reflections should be chosen consistently: for example, the free set of a lower-symmetry polymorph should be a symmetry expansion of the pseudo-equivalent higher-symmetry one. Finally, the standard error of an R value is (roughly) inversely related to the square root of the number of reflections. So how big does a difference of Rfree values have to be so that it is statistically significant? A statistical test would be needed to judge if the improvement on Rfree warrants the choice of the lower-symmetry space group over the higher-symmetry one.
More generally, we are aware that, strictly speaking, the use we make of Rfree is not what the statistic was invented for (Brunger, 1992), that is, discrimination between alternative models in view of one set of diffraction structure factor amplitudes. In the case of polymorph choice, the single set of data to account for are the diffraction images, while the alternative models to be tested against the diffraction data comprise lattice parameters (including the orientation matrix) and space group symmetry as well as the usual atomic model parameters.
The best approach to the problem would, therefore, require a single integrated piece of software that refines two classes of parameters against unprocessed X-ray diffraction images: the first class of parameters are the ones traditionally refined during X-ray data processing, and the second class are the parameters traditionally refined during macromolecular refinement. To be most useful for the purpose of polymorph choice, such a program would likely need to deal at least with some of the correlations between parameters belonging to either class (Roversi and Tronrud, 2021). Each refinement of the atomic structures of one of the possible polymorphs directly against the diffraction images would compute a Rfree, a free likelihood or another model comparison metric, (Babcock et al., 2018) and presumably enable the choice of the best polymorph as the one that uses only as few parameters as are needed to fit the signal but not the noise in the data, and no more.
Rather than a statistically solid (and likely time-consuming!) solution to the problem of polymorph choice in FBLD, we have aimed here at the implementation and testing of a simple protocol that would nevertheless choose polymorphs by consulting the information in the available atomic models. The scripts implementing the automated polymorph choice processed each of the 493 CtUGGTGT24 datasets both in H3 and in P1, enabling 439 structural refinements to take place in either or both space groups. For the 156 datasets that were refined in both H3 and P1, the polymorph choice was then based on the symmetry giving the lower Rfree. For each dataset, in each polymorph, our scripts inherit the free set of reflections from the reference dataset in that polymorph so that the choice of free sets for a pair of related-lattice polymorphs can be made consistently once and for all before running the refinements on which the polymorph choice is based. Unsurprisingly, analysis of the distributions of Rmeas and Rfree over these datasets reveals that in both polymorphs, the dispersion of the latter statistic is sharper than the one of the former (see Figure 5B), supporting the choice of the polymorph based on Rfree rather than Rmeas. The best hit was a CtUGGTGT24 2.2 Å H3 crystal soaked in 5M-8OH-Q. The molecule is bound to a conserved patch on the surface of the protein. The compound is now the starting point for a medicinal chemistry programme that will develop more potent and selective UGGT inhibitors.
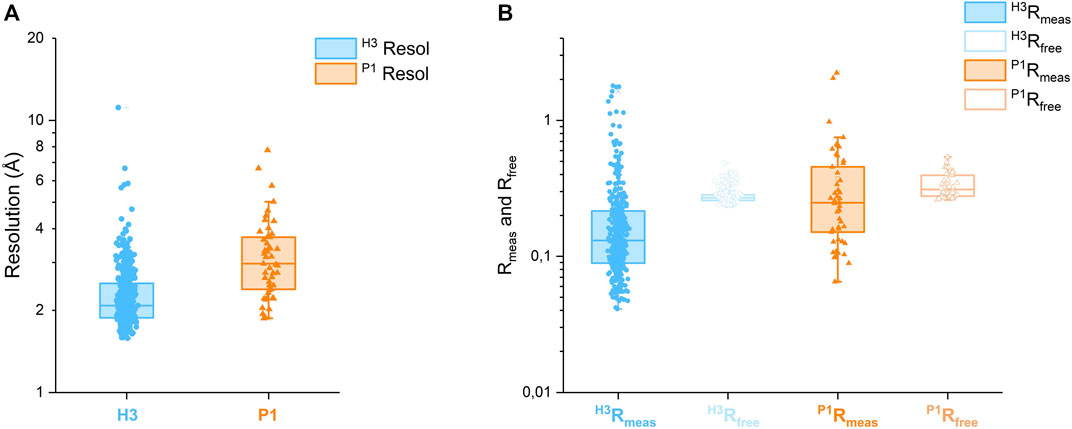
FIGURE 5. (A) Box plots of resolution for 390 CtUGGTGT24 FBLD datasets, which refined better in H3, and 49 datasets, which refined better in P1. (B) Box plots of Rmeas and Rfree for the same CtUGGTGT24 FBLD datasets. Both the resolution and R scales are logarithmic.
As to the generality/feasibility of our strategy, it is true that its computational requirements scale linearly with the number of possible polymorphs. For a given dataset and n possible polymorphs, all our approach requires is full data integration, scaling, and refinement in each polymorph. Synchrotron high-throughput automated data processing pipelines run such a series of steps many times per dataset already: for example, at some synchrotrons, the IspyB system (Monaco et al., 2013) enables data reduction and storage of each dataset at least three times: with xia2 and xia2. multiplex (Winter et al., 2013; Gildea et al., 2022), DIALS (Winter et al., 2022), and autoPROC (Vonrhein et al., 2011), run with a variety of defaults and making use of programs such as XDS (Kabsch, 2010) and/or toolkits such as CCTBX (Grosse-Kunstleve et al., 2002). Each of these data processing strands is then completed by a structural refinement. One of the reasons for this “redundancy” is indeed related to the possibility that different data processing algorithms may choose different polymorphs, thus alerting the user to the possibility of alternative symmetries/lattices. As we do not know of examples of fragment-based lead discovery efforts encountering more than n = 3 polymorphs, one possible solution to make a strategy equivalent to the one we suggest and yet retain generality and feasibility would be commitment to one of the currently implemented data processing pipelines and its repetition for each of n possible polymorphs. In most cases, if n is not large, such an approach would not add much extra time, and it would still systematically sample all possible polymorphs.
Whichever the best solution, implementation of automated polymorph assignment will be an important step towards the realisation of the full potential of crystal polymorphism in FBLD (Vera et al., 2013). It is our hope that with minimal tweaks, existing pipelines for FBLD data processing can be modified to implement ideas similar to the ones we have described here, when discriminating between polymorphs which are related and, therefore, difficult to distinguish on the basis of cell parameters, diffraction data apparent point group, and scaling statistics alone.
5 Materials and methods
5.1 CtUGGTGT24 cloning, protein expression, and purification
The C-terminally 6xHis-tagged CtUGGTGT24 construct corresponding to CtUGGT residues 1,187–1,473 was successfully amplified by PCR starting from the CtUGGT–pHLSec vector (Roversi et al., 2017) using primers CtUGGTGT24_Fwd: ggttgcgtagctgaaaccggtGAGGCAACCAAGTCCGTG and CtUGGTGT24_Rev: gatggtggtgcttggtaccTTCCCTCACTCTCCTCGC.
The amplified insert was identified by agarose gel electrophoresis (about 900 bp) and purified from the gel.
Following purification of the PCR products, the CtUGGTGT24 insert was assembled into the AgeI/KpnI linearised pHLSec vector (Aricescu et al., 2006) via ligation-independent cloning (aka Gibson assembly). After transformation and plating, E. coli colonies containing the desired construct were identified by colony PCR through identification by agarose gel electrophoresis of the correct size. CtUGGT–pHLsec DNA plasmid purification from the correctly identified colonies was carried out via DNA miniprep and the resulting plasmid DNA sent for sequencing for confirmation of the desired DNA construct.
The maxiprepped CtUGGT–pHLsec DNA plasmid was transfected into HEK293F cells following the protocol used for CtUGGT (Roversi et al., 2017). Purification was achieved by IMAC on an Åkta FPLC system, followed by gel filtration chromatography, after which the proteins were identified by SDS-PAGE. The final buffer was 20 mM HEPES pH 7.4, 50 mM NaCl, and 1 mM CaCl2.
5.2 CtUGGTGT24 co-crystallization with UDP-Glc
All crystals were grown at 18°C in sitting drops by the vapour diffusion method, set up with a mosquito liquid handling robot (TTP Labtech). Crystallisation drops had an initial volume of 200 nl. The volume ratio of protein to precipitant was either 1:1 or 2:1.
Crystals of CtUGGTGT24UDP−Glc grew in 1 week in a 1:1 mixture of CtUGGT at 6 mg/ml, 2 mM CaCl2, and 5 mM UDP–Glc and a number of Morpheus screen conditions (Gorrec, 2009, Gorrec, 2015).
The best crystals came from a crystal grown in Morpheus screen condition 2–9 containing 0.12 M ethylene glycol, 0.1 M buffer system 3 pH 8.5, and 30% v/v precipitant mix 1 (Gorrec, 2009; Gorrec, 2015).
5.3 CtUGGTGT24UDP−Glc crystal growth for FBLD
A 96-well deep well block was prepared with 500 μl in each well: 257.5 μl of Morpheus precipitant mix 1 (40% v/v PEG 500 MME; 20% w/v PEG 20000); 25.8 μl of 1 M Bicine (buffer system 3 acid component); 24.3 μl of 1 M Tris (buffer system 3 basic component); 192.4 μl of MilliQ Water. The Hydra robot at the Research Complex in Harwell was first used to transfer 25 μl of the crystallisation solution from the deep well block to each mother liquor well of six MRC 3-well crystallisation plates. Vapour diffusion experiments were set up at the Research Complex in Harwell in six MRC 3-well crystallisation plates—using a mosquito robot equipped with an anti-evaporation cover with 60% controlled humidity in order to avoid drying up of the crystallisation drops during deposition. A total of 6 × 96 × 3 = 1728 drops were set up. The CtUGGTGT24 protein at a concentration of 6.5 mg/ml, in the presence of 1 mM CaCl2 and 5 mM UDP–Glc), was mixed in protein: mother liquor proportions 1.35:1 (drops a and c) and 2:1 (drop d) in drops of total volume 200 nl, and the crystals were left to grow at 18°C. In less than a week, about two-thirds of the experiments yielded crystals.
5.4 CtUGGTGT24UDP−Glc crystal soaking for FBLD
Prior to the fragment-based lead discovery effort, 50 of the CtUGGTGT24UDP−Glc crystals were soaked in 0, 5, 10, 15, 20, and 30% DMSO and diffraction tested; no significant deterioration of the diffraction quality was observed. The space group prior to soaking is H3, one molecule per asymmetric unit, with cell edges a = b = 119 Å, and c = 69 Å.
All CtUGGTGT24UDP−Glc crystal drops were imaged, and the best crystals were marked for soaking with the Echo robot at the Xchem facility attached to beamline I04-1 at the Diamond Light Source (Collins et al., 2017; Douangamath et al., 2021). 768 compounds of the DSi-Poised Library (Diamond-SGC-iNext, ex DSPL https://www.diamond.ac.uk/Instruments/Mx/Fragment-Screening/Fragment-Libraries/DSi-Poised-Library.html) and compound stock solutions at a concentration of 500 mM in deuterated DMSO were each soaked into a crystal drop: 40 nl of the compound was dispensed by the Echo robot (Collins et al., 2017) onto each 200-nl drop, making the final compound concentration 83 mM in 20% DMSO. The soaked crystals were left incubating for a variable time between 2 and 4 h. 692 crystals were fished and cryocooled with the aid of the SGC Shifter Robot (Wright et al., 2021).
5.5 CtUGGTGT24UDP−Glc soaked crystal X-ray diffraction for FBLD
Automated data collection was carried out on 596 soaked crystals. Automated loop centring failed about 6% of the time, and about 50 crystals were re-measured with optical centring. The symmetry is sometimes lowered by soaking, the crystal can in this case index in space group P1 with three molecules in the asymmetric unit of a pseudo-rhombohedral cell of dimensions a = 66.3 Å b = 72.7 Å c = 72.7 Å, alpha = 111.2°, beta = 107.7°, and gamma = 107.7°. This cell is related to the one of the rhombohedral setting of the H3 crystals, a = b = c = 72.32 Å and α = β = γ = 110.43°.
5.6 CtUGGTGT24UDP−Glc X-ray data processing, model refinement, and ligand fitting for FBLD
Data processing, model refinement, and ligand fitting were carried out with the purpose-written shell script pipeline CoALLA. In order to decide on the correct symmetry, each dataset was indexed and integrated both in H3 and in P1 using the autoPROC suite of programmes; for each dataset, refinement of the protein model was carried out in both the H3 and P1 form in autoBUSTER, with the space group giving rise to the lower Rfree being chosen as the correct one for the calculation of the Fo–Fc map. The SMILES string for each compound was fed together with the refined model and phases in order to attempt docking the ligand using rhofit. The best hits were listed by ranking the rhofit score and/or CC and the hits inspected in Coot.
5.6.1 Data processing and polymorph assignment
In order to run CoALLA, all possible polymorphs must be known; moreover, two reference files must be available for each polymorph: one X-ray diffraction dataset with experimental structure factor amplitudes [in mtz format (Winn et al., 2011)] and its corresponding structure coordinates (in PDB format).
The CoALLA pipeline initially processes the diffraction data in each and every one of the polymorphs listed in input, using the data processing suite autoPROC Vonrhein et al. (2011).
The resolution of the data processed in each polymorph is initially chosen by autoPROC using the autoPROC command flag -M HighResCutOnCChalf, which by default sets the maximum resolution so that CC1/2 in the outer shell is no lower than 0.30.
At the indexing stage, in order to ensure consistent indexing across all FBLD datasets belonging to the same polymorph, the autoPROC command line flag -ref <reference dataset>—is used, enforcing the same indexing choice as the reference diffraction dataset for the polymorph being tested. If the reference cell dimensions differ significantly from any of the autoindexed ones, autoPROC may be unable to refine a reference-based indexing solution that fits the data: the reference dataset indexing choice is then enforced only after autoPROC indexing/integration by a run of CCP4-pointless (Evans, 2011) with the keyword TOLERANCE 101. At this stage, the reference dataset Rfree flags are also inherited for each polymorph (using CCP4-CAD) and are kept for all subsequent calculations for the dataset in that polymorph.
The PanDDA pipeline (Pearce et al., 2017a; Pearce et al., 2017b; Pearce et al., 2017c) in use at Diamond Light Source beamline I04-1 Douangamath et al. (2021)—where the data were collected—uses CCP4-Dimple/REFMAC5 (Murshudov et al., 2011; Winn et al., 2011; Keegan et al., 2015) as its refinement engine (see Figure 6). At the initial stage of deciding on the correct polymorph, it seemed natural to exploit the fast refinement capabilities of CCP4-Dimple. After data processing in all polymorphs, the hypothesis that the data belong to a certain polymorph is tested in CoALLA by running structural refinement in CCP4-Dimple (Winn et al., 2011; Keegan et al., 2015) against the data processed in each polymorph in turn.
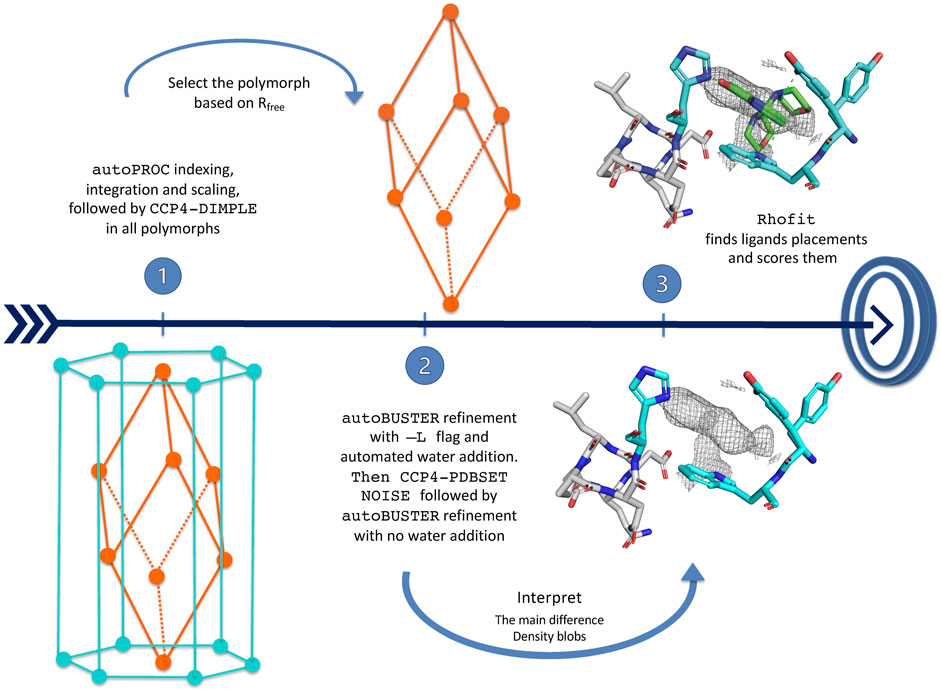
FIGURE 6. CoALLA flow diagram.
In each polymorph, the CoALLA scripts run two CCP4-Dimple refinements of the reference model against the dataset as processed in that polymorph: a rigid body refinement first, followed by a full atomic one. In order to be able to compare the Rfrees of models refined in different polymorphs, all CCP4-Dimple refinements are run in CoALLA up to the best common maximum resolution limit across all polymorphs, which the scripts obtain for each polymorph by harvesting the resolution limit from the relevant autoPROC output file (see Figure 6).
Once a CCP4-Dimple refinement of the structure has run for each polymorph, the polymorph with the best CCP4-Dimple refinement Rfree is chosen as the most likely one to continue the analysis. It is at this stage that CoALLA improves on automated polymorph choices based on data processing only: the available information about the structures of the polymorphs is added to the one present in the diffraction data to single out the most likely polymorph.
5.6.2 BUSTER structure refinements
The structures are then refined in BUSTER starting from the CCP4-Dimple output file, with automated NCS restraints and external restraints to the reference structure (Smart et al., 2012), command line keywords—target <ref.pdb>—autoncs).
The first BUSTER refinement is run with the -L command line flag: this turns on automated water updating. Waters initially also fill volumes where potential ligands are located, but the water atoms around residual difference density are then removed at the last BUSTER refinement cycle. This procedure enables water-building without deteriorating the quality of the difference density in putative ligand regions.
At this stage, CCP4-PDBSET is run on the BUSTER-refined model (with keyword NOISE) in order to introduce positional noise in the coordinates resulting from the refinement previously described—including any waters that survived the pruning at the end of the previous BUSTER refinement with automated water addition. The positional noise introduced by CCP4-PDBSETat this stage ought to wipe out any “memory” of the waters that may have been refined (and then were removed) in putative ligand pockets. A second BUSTER refinement (without water addition) is then run starting from the CCP4-PDBSET shaken model. The 2Fo–Fc and Fo–Fc density maps necessary for ligand identification and docking are computed at the end of this BUSTER refinement.
5.6.3 RHOFIT ligand placement
Automatic interpretation of crystal residual difference density fully automates the placement of ligands, providing an unbiased alternative to manual ligand placement, especially for data of low to medium resolution (Langer et al., 2008; Carolan and Lamzin, 2014; Echols et al., 2014; Wlodek et al., 2006). Algorithms can take into account protein–ligand interactions as well as fit to the map (Mooij et al., 2006).
The RHOFIT program (Smart et al., 2014) is used within the CoALLA pipeline to find the best placements in all the BUSTER refined maps.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: http://www.wwpdb.org/, 6fsn http://www.wwpdb.org/, 7zxw.
Author contributions
PR, ATC, and NZ conceived the study. PR, CBL, and SV cloned the CtUGGTGT24 construct. PR, JDLC, RI, and SV prepared the CtUGGTGT24 protein. PR, RI, and JDLC grew the CtUGGTGT24 crystals. PR, RI, ATC, BD, MH, and NZ carried out fragment-based lead discovery effort. ATC and PR wrote the CoALLA scripts. RI, ATC, BD, MH, GM, and PR analysed data. All authors contributed to the writing of the manuscript.
Funding
The work was funded by the Glycobiology Endowment and by two University of Oxford Confidence in Concept Schemes, grant references MRC–MC_PC_15029 and MRC–MC_PC_16056 (to NZ). PR was the recipient of an LISCB Wellcome Trust ISSF award, grant reference 204801/Z/16/Z and a Wellcome Trust Seed Award in Science, grant reference 214090/Z/18/Z. RI was the recipient of a Sardinian Regional Government PhD scholarship. AC was funded by a Wellcome Trust 4-Year Studentship reference 097300/Z/11/Z. NZ is a Fellow of Merton College, Oxford.
Acknowledgments
Tobias Krojer, Anthony Bradley, Patrick Collins, Alice Douangamath, Jose Brandao-Neto, Frank Von Delft, and all the staff of the I04-1 beamline at the Diamond Light Source helped with sample preparation and data collection. Conor Wild gave insightful comments on our choice of Rfree as a model comparison statistic. Constantina Foutinou helped with the preparation of the manuscript. Tobias Krojer, José A. Marquez, and Justyna A. Wojdyla kindly commented on the availability of automated polymorph choice algorithms in current implementations of FBLD data processing pipelines. Kay Diederichs kindly discussed the suitability of various statistics to compare scaling of a dataset in different symmetries. The authors thank the reviewers for helpful suggestions on the first submitted version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.960248/full#supplementary-material. The CoALLA shell script will be available from the corresponding authors upon request.
Footnotes
1Note that in Phenix-based FBLD pipelines, models are mapped to the same frame of reference as an isomorphous structure. The CoALLA algorithm choice of enforcing indexing to the polymorph’s reference dataset has the advantage that simple rigid-body refinement of the reference structure against the data processed in the chosen polymorph is enough to commit to consistent origin choices for all crystals belonging to a certain polymorph, without the need for any molecular replacement steps and/or origin reconciliation.
References
Albesa-Jové, D., and Guerin, M. E. (2016). The conformational plasticity of glycosyltransferases. Curr. Opin. Struct. Biol. 40, 23–32. Carbohydrate–protein interactions and glycosylation • Biophysical and molecular biological methods. doi:10.1016/j.sbi.2016.07.007
Alonzi, D. S., Scott, K. A., Dwek, R. A., and Zitzmann, N. (2017). Iminosugar antivirals: The therapeutic sweet spot. Biochem. Soc. Trans. 45, 571–582. doi:10.1042/BST20160182
Amara, J. F., Cheng, S. H., and Smith, A. E. (1992). Intracellular protein trafficking defects in human disease. Trends Cell. Biol. 2, 145–149. doi:10.1016/0962-8924(92)90101-r
Aricescu, A. R., Lu, W., and Jones, E. Y. (2006). A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D. Biol. Crystallogr. 62, 1243–1250. doi:10.1107/S0907444906029799
Babcock, N. S., Keedy, D. A., Fraser, J. S., and Sivak, D. A. (2018). Model selection for biological crystallography. bioRxiv. doi:10.1101/448795
Blanc, E., Roversi, P., Vonrhein, C., Flensburg, C., Lea, S. M., and Bricogne, G. (2004). Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D. Biol. Crystallogr. 60, 2210–2221. doi:10.1107/S0907444904016427
Bricogne, G., Blanc, E. M. B., Flensburg, C., Keller, P., Paciorek, W., et al. (2017). Transition metals in catalysis: The functional relationship. BUSTER 2.10.3.
Bricogne, G. (1997). “The bayesian statistical viewpoint on structure determination: Basic concepts and examples,” in Macromolecular crystallography. Editors C. W. Carter, and R. M. Sweet (San Diego, CA: Academic Press), 361–423. vol. 276 of Methods in Enzymology.
Brunger, A. T. (1992). Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. doi:10.1038/355472a0
Buerger, M. J. (1936a). The general rôle of composition in polymorphism. Proc. Natl. Acad. Sci. U. S. A. 22, 685–689. doi:10.1073/pnas.22.12.685
Buerger, M. J. (1936b). The kinetic basis of crystal polymorphism. Proc. Natl. Acad. Sci. U. S. A. 22, 682–685. doi:10.1073/pnas.22.12.682
Caputo, A. T., Alonzi, D. S., Marti, L., Reca, I.-B., Kiappes, J. L., Struwe, W. B., et al. (2016). Structures of mammalian ER α-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc. Natl. Acad. Sci. U. S. A. 113, E4630–E4638. doi:10.1073/pnas.1604463113
Carolan, C. G., and Lamzin, V. S. (2014). Automated identification of crystallographic ligands using sparse-density representations. Acta Crystallogr. D. Biol. Crystallogr. 70, 1844–1853. doi:10.1107/S1399004714008578
Carter, C. W., Doublié, S., and Coleman, D. E. (1994). Quantitative analysis of crystal growth. Tryptophanyl-tRNA synthetase crystal polymorphism and its relationship to catalysis. J. Mol. Biol. 238, 346–365. doi:10.1006/jmbi.1994.1297
Chen, X., Qin, S., Chen, S., Li, J., Li, L., Wang, Z., et al. (2015). A ligand-observed mass spectrometry approach integrated into the fragment based lead discovery pipeline. Sci. Rep. 5, 8361. doi:10.1038/srep08361
Ciulli, A., Williams, G., Smith, A. G., Blundell, T. L., and Abell, C. (2006). Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J. Med. Chem. 49, 4992–5000. doi:10.1021/jm060490r
Collins, P. M., Douangamath, A., Talon, R., Dias, A., Brandao-Neto, J., Krojer, T., et al. (2018). “Chapter eleven - achieving a good crystal system for crystallographic x-ray fragment screening,” in Modern approaches in drug discovery. Editor C. A. Lesburg (Academic Press), 251–264. vol. 610 of Methods in Enzymology. doi:10.1016/bs.mie.2018.09.027
Collins, P. M., Ng, J. T., Talon, R., Nekrosiute, K., Krojer, T., Douangamath, A., et al. (2017). Gentle, fast and effective crystal soaking by acoustic dispensing. Acta Crystallogr. D. Struct. Biol. 73, 246–255. doi:10.1107/S205979831700331X
Cornaciu, I., Bourgeas, R., Hoffmann, G., Dupeux, F., Humm, A.-S., Mariaule, V., et al. (2021). The automated crystallography pipelines at the EMBL HTX facility in Grenoble. J. Vis. Exp. (172), e62491. doi:10.3791/62491
D’Alessio, C., Caramelo, J. J., and Parodi, A. J. (2010). UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell. Dev. Biol. 21, 491–499. doi:10.1016/j.semcdb.2009.12.014
Dalziel, M., Crispin, M., Scanlan, C. N., Zitzmann, N., and Dwek, R. A. (2014). Emerging principles for the therapeutic exploitation of glycosylation. Science 343, 1235681. doi:10.1126/science.1235681
Diederichs, K., and Karplus, P. A. (1997). Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275. doi:10.1038/nsb0497-269
Diederichs, K. (2010). Quantifying instrument errors in macromolecular X-ray data sets. Acta Crystallogr. D. Biol. Crystallogr. 66, 733–740. doi:10.1107/S0907444910014836
Douangamath, A., Powell, A., Fearon, D., Collins, P. M., Talon, R., Krojer, T., et al. (2021). Achieving efficient fragment screening at XChem facility at Diamond light Source. J. Vis. Exp. (171), e62414 doi:10.3791/62414
Dwek, R. A., Bell, J. I., Feldmann, M., and Zitzmann, N. (2022). Host-targeting oral antiviral drugs to prevent pandemics. Lancet 399, 1381–1382. doi:10.1016/S0140-6736(22)00454-8
Echols, N., Moriarty, N. W., Klei, H. E., Afonine, P. V., Bunkóczi, G., Headd, J. J., et al. (2014). Automating crystallographic structure solution and refinement of protein–ligand complexes. Acta Crystallogr. D. Biol. Crystallogr. 70, 144–154. doi:10.1107/S139900471302748X
Evans, P. R. (2011). An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D. Biol. Crystallogr. 67, 282–292. doi:10.1107/S090744491003982X
Gildea, R. J., Beilsten-Edmands, J., Axford, D., Horrell, S., Aller, P., Sandy, J., et al. (2022). xia2.multiplex: a multi-crystal data-analysis pipeline. Acta Crystallogr. D. Struct. Biol. 78, 752–769. doi:10.1107/S2059798322004399
Gorrec, F. (2015). The MORPHEUS II protein crystallization screen. Acta Crystallogr. F. Struct. Biol. Commun. 71, 831–837. doi:10.1107/S2053230X1500967X
Gorrec, F. (2009). The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 42, 1035–1042. doi:10.1107/S0021889809042022
Grosse-Kunstleve, R. W., Sauter, N. K., Moriarty, N. W., and Adams, P. D. (2002). The computational crystallography toolbox: Crystallographic algorithms in a reusable software framework. J. Appl. Crystallogr. 35, 126–136. doi:10.1107/S0021889801017824
Hammond, C., Braakman, I., and Helenius, A. (1994). Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. U. S. A. 91, 913–917. doi:10.1073/pnas.91.3.913
Jaynes, E. T. (1968). Prior probabilities. IEEE Trans. Syst. Sci. Cyber. 4, 227–241. doi:10.1109/TSSC.1968.300117
Jurnak, F. (1985). Induction of elongation factor Tu-GDP crystal polymorphism by polyethylene glycol contaminants. J. Mol. Biol. 185, 215–217. doi:10.1016/0022-2836(85)90194-9
Kabsch, W. (2010). “XDS,” in Acta crystallographica. Section D, Biological crystallography, 66 (Chester, England: International Union of Crystallography), 125–132. doi:10.1107/S0907444909047337
Kaminski, J. W., Vera, L., Stegmann, D. P., Vering, J., Eris, D., Smith, K. M. L., et al. (2022). Fast fragment- and compound-screening pipeline at the Swiss Light Source. Acta Crystallogr. D. Struct. Biol. 78, 328–336. doi:10.1107/S2059798322000705
Karade, S. S., Hill, M. L., Kiappes, J. L., Manne, R., Aakula, B., Zitzmann, N., et al. (2021). N-substituted valiolamine derivatives as potent inhibitors of endoplasmic reticulum α-glucosidases I and II with antiviral activity. J. Med. Chem. 64, 18010–18024. doi:10.1021/acs.jmedchem.1c01377
Karplus, P. A., and Diederichs, K. (2015). Assessing and maximizing data quality in macromolecular crystallography. Curr. Opin. Struct. Biol. 34, 60–68. Carbohydrate-protein interactions • Biophysical and molecular biological methods. doi:10.1016/j.sbi.2015.07.003
Keegan, R., Wojdyr, M., Winter, G., and Ashton, A. (2015). Dimple: A difference map pipeline for the rapid screening of crystals on the beamline. Acta Crystallogr. A Found. Adv. 71, s18. doi:10.1107/S2053273315099702
Krojer, T., Talon, R., Pearce, N., Collins, P., Douangamath, A., Brandao-Neto, J., et al. (2017). The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallogr. D. Struct. Biol. 73, 267–278. doi:10.1107/S2059798316020234
Langer, G., Cohen, S. X., Lamzin, V. S., and Perrakis, A. (2008). Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179. doi:10.1038/nprot.2008.91
Lanzarotti, E., Defelipe, L. A., Marti, M. A., and Turjanski, A. G. (2020). Aromatic clusters in protein-protein and protein-drug complexes. J. Cheminform. 12, 30. doi:10.1186/s13321-020-00437-4
Lima, G. M. A., Jagudin, E., Talibov, V. O., Benz, L. S., Marullo, C., Barthel, T., et al. (2021). FragMAXapp: Crystallographic fragment-screening data-analysis and project-management system. Acta Crystallogr. D. Struct. Biol. 77, 799–808. doi:10.1107/S2059798321003818
Monaco, S., Gordon, E., Bowler, M. W., Delagenière, S., Guijarro, M., Spruce, D., et al. (2013). Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. J. Appl. Crystallogr. 46, 804–810. doi:10.1107/S0021889813006195
Mooij, W., Hartshorn, M., Tickle, I., Sharff, A., Verdonk, M., and Jhoti, H. (2006). Automated protein–ligand crystallography for structure-based drug design. ChemMedChem 1, 827–838. doi:10.1002/cmdc.200600074
Müller, J., Klein, R., Tarkhanova, O., Gryniukova, A., Borysko, P., Merkl, S., et al. (2022). Magnet for the needle in haystack: “crystal structure first” fragment hits unlock active chemical matter using targeted exploration of vast chemical spaces. J. Med. Chem. Epub ahead of print. doi:10.1021/acs.jmedchem.2c00813
Murray, C. W., and Blundell, T. L. (2010). Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 20, 497–507. doi:10.1016/j.sbi.2010.04.003
Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67, 355–367. doi:10.1107/S0907444911001314
Oksenych, V., and Kainov, D. E. (2022). Broad-spectrum antivirals and antiviral drug combinations. Viruses 14, 301. doi:10.3390/v14020301
Pardi, N., and Weissman, D. (2020). Development of vaccines and antivirals for combating viral pandemics. Nat. Biomed. Eng. 4, 1128–1133. doi:10.1038/s41551-020-00658-w
Pearce, N. M., Bradley, A. R., Krojer, T., Marsden, B. D., Deane, C. M., and von Delft, F. (2017a). Partial-occupancy binders identified by the Pan-Dataset Density Analysis method offer new chemical opportunities and reveal cryptic binding sites. Struct. Dyn. 4, 032104. doi:10.1063/1.4974176
Pearce, N. M., Krojer, T., Bradley, A. R., Collins, P., Nowak, R. P., Talon, R., et al. (2017b). A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 8, 15123. doi:10.1038/ncomms15123
Pearce, N. M., Krojer, T., and von Delft, F. (2017c). Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallogr. D. Struct. Biol. 73, 256–266. doi:10.1107/S2059798317003412
Pearce, N. M., Skyner, R., and Krojer, T. (2022). Experiences from developing software for large X-ray crystallography-driven protein-ligand studies. Front. Mol. Biosci. 9, 861491. doi:10.3389/fmolb.2022.861491
Radoux, C. J., Olsson, T. S. G., Pitt, W. R., Groom, C. R., and Blundell, T. L. (2016). Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325. doi:10.1021/acs.jmedchem.5b01980
Roversi, P., Marti, L., Caputo, A. T., Alonzi, D. S., Hill, J. C., Dent, K. C., et al. (2017). Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. U. S. A. 114, 8544–8549. doi:10.1073/pnas.1703682114
Roversi, P., and Tronrud, D. E. (2021). Ten things I `hate' about refinement. Acta Crystallogr. D. Struct. Biol. 77, 1497–1515. doi:10.1107/S2059798321011700
Sayce, A. C., Alonzi, D. S., Killingbeck, S. S., Tyrrell, B. E., Hill, M. L., Caputo, A. T., et al. (2016). Iminosugars inhibit dengue virus production via inhibition of ER alpha-glucosidases-not glycolipid processing enzymes. PLoS Negl. Trop. Dis. 10, e0004524. doi:10.1371/journal.pntd.0004524
Schiebel, J., Krimmer, S. G., Röwer, K., Knörlein, A., Wang, X., Park, A. Y., et al. (2016). High-throughput crystallography: Reliable and efficient identification of fragment hits. Structure 24, 1398–1409. doi:10.1016/j.str.2016.06.010
Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., et al. (2012). Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D. Biol. Crystallogr. 68, 368–380. doi:10.1107/S0907444911056058
Smart, O., Womack, T., Sharff, A., Flensburg, C., Keller, P., Paciorek, W., et al. (2014). RHOFIT, version 1.2.4. Cambridge, United Kingdom: Global Phasing Ltd.
Tax, G., Lia, A., Santino, A., and Roversi, P. (2019). Modulation of erqc and erad: A broad-spectrum spanner in the works of cancer cells? J. Oncol. 2019, 8384913. doi:10.1155/2019/8384913
Trombetta, E. S., and Helenius, A. (1999). Glycoprotein reglucosylation and nucleotide sugar utilization in the secretory pathway: Identification of a nucleoside diphosphatase in the endoplasmic reticulum. EMBO J. 18, 3282–3292. doi:10.1093/emboj/18.12.3282
Trombetta, S. E., Bosch, M., and Parodi, A. J. (1989). Glucosylation of glycoproteins by mammalian, plant, fungal, and trypanosomatid protozoa microsomal membranes. Biochemistry 28, 8108–8116. doi:10.1021/bi00446a022
Tyrrell, B. E., Sayce, A. C., Warfield, K. L., Miller, J. L., and Zitzmann, N. (2017). Iminosugars: Promising therapeutics for influenza infection. Crit. Rev. Microbiol. 43, 521–545. doi:10.1080/1040841X.2016.1242868
Vera, L., Antoni, C., Devel, L., Czarny, B., Cassar-Lajeunesse, E., Rossello, A., et al. (2013). Screening using polymorphs for the crystallization of protein–ligand complexes. Cryst. Growth & Des. 13, 1878–1888. doi:10.1021/cg301537n
Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., et al. (2011). Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. Biol. Crystallogr. 67, 293–302. doi:10.1107/S0907444911007773
Warfield, K. L., Alonzi, D. S., Hill, J. C., Caputo, A. T., Roversi, P., Kiappes, J. L., et al. (2020). Targeting endoplasmic reticulum α-glucosidase I with a single-dose iminosugar treatment protects against lethal influenza and dengue virus infections. J. Med. Chem. 63, 4205–4214. doi:10.1021/acs.jmedchem.0c00067
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242. doi:10.1107/S0907444910045749
Winter, G., Beilsten-Edmands, J., Devenish, N., Gerstel, M., Gildea, R. J., McDonagh, D., et al. (2022). DIALS as a toolkit. Protein Sci. 31, 232–250. doi:10.1002/pro.4224
Winter, G., Lobley, C. M. C., and Prince, S. M. (2013). Decision making in xia2. Acta Crystallogr. D. Biol. Crystallogr. 69, 1260–1273. doi:10.1107/S0907444913015308
Wlodek, S., Skillman, A. G., and Nicholls, A. (2006). Automated ligand placement and refinement with a combined force field and shape potential. Acta Crystallogr. D. Biol. Crystallogr. 62, 741–749. doi:10.1107/S0907444906016076
Wright, N. D., Collins, P., Koekemoer, L., Krojer, T., Talon, R., Nelson, E., et al. (2021). The low-cost Shifter microscope stage transforms the speed and robustness of protein crystal harvesting. Acta Crystallogr. D. Struct. Biol. 77, 62–74. doi:10.1107/S2059798320014114
Yekwa, E., Khourieh, J., Canard, B., Papageorgiou, N., and Ferron, F. (2017). Activity inhibition and crystal polymorphism induced by active-site metal swapping. Acta Crystallogr. D. Struct. Biol. 73, 641–649. doi:10.1107/S205979831700866X
Keywords: UGGT, crystal polymorphism, structure-based lead discovery, structure determination pipeline, [(morpholin-4yl)methyl]quinolin-8-ol
Citation: Caputo AT, Ibba R, Le Cornu JD, Darlot B, Hensen M, Lipp CB, Marcianò G, Vasiljević S, Zitzmann N and Roversi P (2022) Crystal polymorphism in fragment-based lead discovery of ligands of the catalytic domain of UGGT, the glycoprotein folding quality control checkpoint. Front. Mol. Biosci. 9:960248. doi: 10.3389/fmolb.2022.960248
Received: 02 June 2022; Accepted: 11 November 2022;
Published: 14 December 2022.
Edited by:
Anastassios C. Papageorgiou, University of Turku, FinlandReviewed by:
Manfred S. Weiss, Helmholtz Association of German Research Centers (HZ), GermanyMax Nanao, European Synchrotron Radiation Facility, France
Copyright © 2022 Caputo, Ibba, Le Cornu, Darlot, Hensen, Lipp, Marcianò, Vasiljević, Zitzmann and Roversi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Zitzmann, bmljb2xlLnppdHptYW5uQGJpb2NoLm94LmFjLnVr; Pietro Roversi, cGlldHJvLnJvdmVyc2lAaWJiYS5jbnIuaXQ=
†These authors have contributed equally to this work and share first authorship