 Sneha Bheemireddy
Sneha Bheemireddy Sankaran Sandhya
Sankaran Sandhya Narayanaswamy Srinivasan
Narayanaswamy Srinivasan Ramanathan Sowdhamini
Ramanathan Sowdhamini- 1Molecular Biophysics Unit, Indian Institute of Science, Bangalore, India
- 2Department of Biotechnology, Faculty of Life and Allied Health Sciences, M.S. Ramaiah University of Applied Sciences, Bengaluru, India
- 3National Centre for Biological Sciences, TIFR, GKVK Campus, Bangalore, India
- 4Institute of Bioinformatics and Applied Biotechnology, Bangalore, India
RNA is the key player in many cellular processes such as signal transduction, replication, transport, cell division, transcription, and translation. These diverse functions are accomplished through interactions of RNA with proteins. However, protein–RNA interactions are still poorly derstood in contrast to protein–protein and protein–DNA interactions. This knowledge gap can be attributed to the limited availability of protein-RNA structures along with the experimental difficulties in studying these complexes. Recent progress in computational resources has expanded the number of tools available for studying protein-RNA interactions at various molecular levels. These include tools for predicting interacting residues from primary sequences, modelling of protein-RNA complexes, predicting hotspots in these complexes and insights into derstanding in the dynamics of their interactions. Each of these tools has its strengths and limitations, which makes it significant to select an optimal approach for the question of interest. Here we present a mini review of computational tools to study different aspects of protein-RNA interactions, with focus on overall application, development of the field and the future perspectives.
1 Introduction
The roles of RNA, as coding and non-coding RNA in transcription, translation, gene regulation, transport, catalysis, cell division and many other processes continues to expand with advancement in methods to study them (Crick, 1970; Pyle, 1993; Moore and Steitz, 2002; Sugiura et al., 2011; Barbieri and Kouzarides, 2020; Christopoulou and Granneman, 2022; Perez et al., 2022; Song et al., 2022). The growing repertoire of non-coding RNA types (ncRNA: ribosomal RNA, transfer RNA, miRNA, snRNA/small nuclear RNA, piRNA, catalytic RNA, small nucleolar RNA, etc.) shows their tremendous diversity in sequence, structure, subcellular localization, and function. Central to their role in fundamental biological processes are RNA-protein interactions (RPI) that primarily involve modular RNA-binding proteins (RBP), although exceptions exist (Castello et al., 2012). Typically, this recognition is effected through an RNA-binding domain (RBD) such as RNA recognition motif (RRM), hnRNP K homology (KH) domain, DEAD box helicase domain (DDX), pumilio homology domain (PUM-HD) etc. and may involve recognition of specific sequence motifs on RNA or such sequences in specific structural contexts (Jolma et al., 2020). Alternatively, this recognition is based on structures adopted by RNA such as in G-quadruplexes (Kharel et al., 2020). RBPs are also known to bind RNA through intrinsically disordered regions (IDRs), resulting in extended interaction interfaces and higher order assemblies (Calabretta and Richard, 2015). More recently, RNA interactome captures (Gerstberger et al., 2014; Hentze et al., 2018) have revealed that up to 10% of the entire proteome may be bound to RNA emphasizing their importance in function. Indeed, such studies are contributing to excellent resources for cataloguing the complete set of protein-RNA interactions (RPI) across various cell types and whole organisms (Baltz et al., 2012; Castello et al., 2012; Kwon et al., 2013; Mitchell et al., 2013; Beckmann et al., 2015; Matia-González et al., 2015; Liepelt et al., 2016; Sysoev et al., 2016; Wessels et al., 2016; Hentze et al., 2018; Trendel et al., 2019; Urdaneta et al., 2019). Perturbations of RNA-RBP interactions are known to result in cellular dysfunction and have been implicated in many diseases (Allerson et al., 1999; Batista and Chang, 2013). It is, therefore, important to characterize RNA-proteins interactions.
Several experimental approaches are available to study the physical association between individual proteins and RNA molecules and these have been described in excellent reviews (Darnell, 2010; Ramanathan et al., 2019; Licatalosi et al., 2020; Cozzolino et al., 2021; Gräwe et al., 2021). Broadly, these approaches are grouped into RNA-centric or protein-centric approaches. While the former attempts to study proteins that associate with an entire population of RNA or an RNA that is expressed in a specific cell type or tissue (Campbell and Wickens, 2015; Cook et al., 2015; Gerber, 2021), the latter aims to pull down all RNA that specifically interacts with a protein. These methods may involve antibody-based immunoprecipitation of RBP and interacting RNA or involve crosslinking between protein and RNA (Ramanathan et al., 2019). RNA associated with the protein is then isolated and further analysed by microarray, sequencing, hybridization or polymerase chain reaction-based methods. Here, not only can the actual RNA sequence be determined, but also its abundance in the immunoprecipitated sample can be estimated and this is useful to map the binding site of the RBP of interest to the RNA molecule (Gagliardi and Matarazzo, 2016). A definitive way to identify RNA-binding residues or study RNA-protein interfaces (i.e., amino acid residues that directly contact RNA) is to extract them from a high-resolution experimental structure of a protein–RNA complex. However, structures of more than 80% of the protein-RNA complexes are known (Bienert et al., 2017; Dimitrova-Paternoga et al., 2020) owing to challenges in biophysical techniques such as complicated measurement process, time, resolution limits, cost-intensive steps etc. (Ke and Doudna, 2004; Scott et al., 2008; Pai et al., 2017). Added challenges in the determination of structures of protein-RNA complexes include the recognition of RNA sequences of optimal size that will bind the protein specifically and stably and the inherent flexibility of such regions which can affect structural stability and structure determination efforts. The inherent diversity of RNA structures and proteins and the presence of intrinsically disordered regions presents an enormous challenge to the area as well (Dyson, 2012; Ottoz and Berchowitz, 2020; Vandelli et al., 2022). The cost and effort to experimentally capture and measure strengths of all biologically important RNA–protein complexes is challenging due to these ique complexities. Although many experimental methods are now available to characterize protein-RNA interactions, mechanistic details of these reactions are still known. However, the availability of protein–RNA complex data from diverse experiments serve as ideal candidates for computational data analysis that can develop knowledge-based trends and rules to characterize such interactions. Indeed, several reliable computational methods that have derived patterns from the analysis of large ensembles of data have gained traction in predicting protein–RNA interaction (RPI) sites. Such methods when complemented with experimental data have been useful to analyze large datasets, to generate various hypothesis on interactions that can be tested again through experiment (Jiang et al., 2020; Teimouri and Maali, 2020; Zhang et al., 2021a). Some of the promising outcomes include prediction of the potential RNA binding sites in SARS-CoV-2 nucleocapsid protein revealing potential drug targeting sites (Cubuk et al., 2021), or a more detailed derstanding of the catalytic core formation through studies of spliceosomes and the role of allostery in mRNA interaction with ribosomes (Bao et al., 2018; Bheemireddy et al., 2021).
In this mini review, we provide an overview of existing computational methods to accurately predict RPI and analyze such interfaces. Our review broadly categorises these tools into sequence-based predictions, that tap into features in protein or RNA sequence, and structure-based computational methods that derive from available crystal structures of protein-RNA complexes and enable prediction of protein-RNA interactions. We have broadly classified these approaches based on their application and relevance in specific analysis and briefly describe and highlight well cited methods in each sub-section. These subsections are grouped based on the nature of input data used for the various analyses and enable a new user to choose an appropriate tool, that is relevant to their specific application interests.
2 Computational methods to predict protein-RNA interactions
Over the past decade, computational methods to predict RPI have been developed using either protein/ RNA sequence features (such as residue identity or physicochemical properties) and structure-based methods that use structure-derived features (such as solvent-accessible surface area or secondary structure) to make predictions. Walia et al., have reviewed tools for sequence-based methods (Walia et al., 2017) while structure-based methods that use secondary and tertiary structure of RNA have been described elsewhere (Zhang et al., 2015). With the availability of diverse experimental data there is tremendous interest and increase in development of methods that can learn trends from the data and arrive at predictive models. Computational tools to predict RPI vary in complexity and are based on either a combination of a few features or involve the application of network-based approaches/ machine learning (ML)-based methods such as deep learning, that capture hierarchical representations of intrinsically hidden data features. Collectively, these independent approaches have significantly prompted the development of a variety of tools that (Figure 1) can be classified based on their applications and aspects of RPI that they predict. In the following sections, we have grouped these tools into broad functional categories based on the input data and present the major tools to computationally infer RPI (Supplementary Table S1).
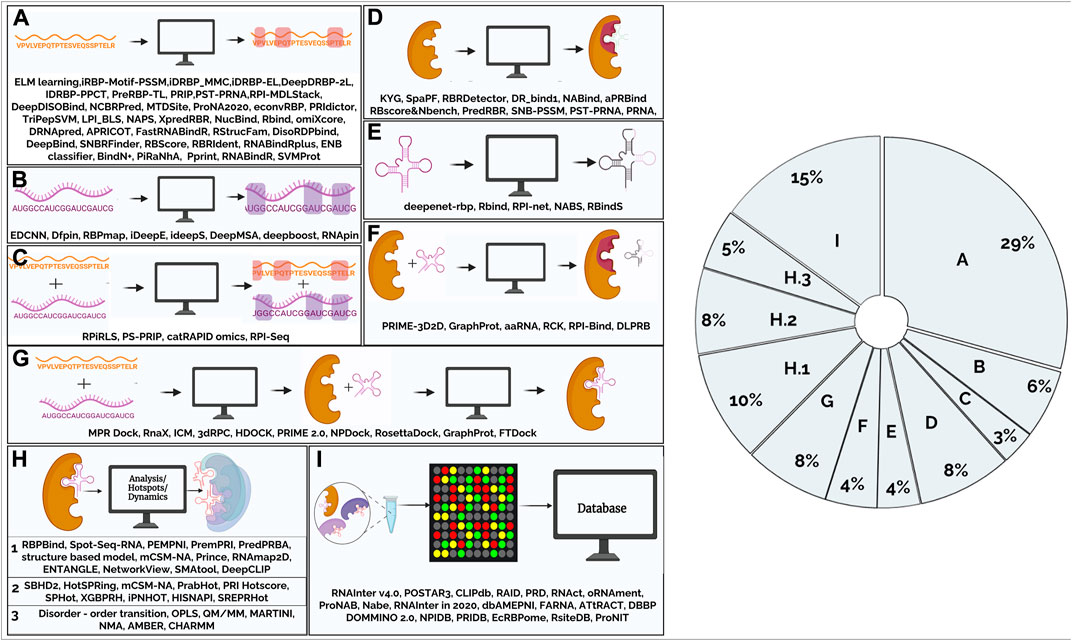
FIGURE 1. Figure summarizes the tools that are available to study protein-RNA interactions that have been classified based on their function. (A). Computational tools for predicting amino acid residues in protein-RNA interactions using protein sequence (2.1.1) (B). Computational tools for predicting nucleotides in protein-RNA interactions using RNA sequence (2.1.2) (C). Computational tools for predicting amino acid residues and nucleotides in protein-RNA interactions using sequence information. (2.1.3) (D). Computational tools for predicting amino acid residues in protein-RNA interactions using protein structural information (2.2.1) (E). Computational tools for predicting nucleotides in protein-RNA interactions using RNA structural information (2.2.2) (F). Computational tools for predicting amino acid residues and nucleotides in protein-RNA interactions using protein, RNA structural and sequence information (2.2.3) (G). Computational tools for modelling/docking of protein-RNA complexes (2.3) (H).1. Computational tools for analysis of protein-RNA complexes (2.4) (H).2 Computational tools for prediction of hotspots in protein-RNA complexes (2.5) (H).3. Computational tools to study dynamics using RNA-protein complex structure as input (2.6) (I). Databases (2.7). Pie-chart shows main areas in RNA-protein interaction methods. From the figure, it is evident that majority of the tools were developed for prediction of amino acid residues and very few tools for modelling protein-RNA interactions. Nearly, 30% of the tools are for predicting interactions in the protein-RNA complexes given a protein query sequence.
2.1 Methods that predict protein-RNA interactions using sequence information
2.1.1 Protein sequence as input
Available experimental datasets enable the mapping of sequence motifs to binding scores. This, in turn, results in large volumes of such labelled and training data from diverse experiments and acts as an appropriate input for deep learning methods such as Alipanahi et al., 2015, that can derive patterns and build predictive models based on the training dataset. The input data used for training are obtained from various high-throughput experiments such as protein-binding microarrays, RNAcompete assays, ChIP-seq and HT-SELEX. The method has been shown to efficiently predict binding specificities of in vivo data and to identify deleterious genomic variants. With over 2000 citations, this scalable and modular pattern discovery method is a popular application of ML-based learning methods with important applications in predicting protein-RNA interactions (Supplementary Table S1). PPrint (Kumar et al., 2008) is another notable application that uses support vector machines to predict such interactions. Prediction models in these studies were trained on RNA-binding protein chains that were extracted from available crystal structures of RNA-protein complexes. An added advantage is the inclusion of evolutionary information through position-specific scoring matrices (PSSMs) derived for the sequence homologues of the training dataset. Other platforms, such as RStrucfam (Ghosh et al., 2016) use Hidden Markov Models (HMM) of RNA binding protein families (derived from sequence and structure databases) to recognize such properties of a protein query starting from mere sequence information. Another popular method employs convolutional neural networks and an extreme learning machine (ELM) classifier, to extract features from RNA and protein interaction datasets that are obtained from structures of known complexes of proteins and ncRNA. Representations of such datasets through numerical matrices and application of a learning classifier have been shown to effectively predict RPI in model organisms (Wang et al., 2020).
2.1.2 RNA sequence as input
Just as the sequence features of proteins in RBP have contributed to the development of several methods to recognize such signatures in proteins, several methods are available to predict regions involved in such interactions in RNA as well. These motifs have been identified as targets of RBPs through experimental methods such as immunoprecipitation or cross-linking methods. Several tools have been developed to recognize RNA motifs/ sequences that bind proteins (Supplementary Table S1) by determining the short RNA sequence motifs that are known to occur at the interface of known protein-RNA complexes. Approaches such as RBPmap (Paz et al., 2014) allow the users to select motifs from a database of experimentally characterized motifs, that have been extracted from literature as a PSSM and use a Weighted-Rank (WR) approach to predict binding sites in query RNA sequences. A match score is computed for the motif per each position in the sequence in overlapping windows and its significance is evaluated taking into consideration the genome of interest. Properties of the motif environment, including the clustering propensity of binding sites and the overall tendency of such regions to be conserved provide additional advantages. Further, background model scores are employed to capture the significance of a match in the context that they occur in, in splice sites, 5′ and 3’ UTRs, non-coding RNAs and mid-intron/intergenic regions. RNApin (Panwar and Raghava, 2015) predicts such interaction sites using the trinucleotide composition profile of RNA and features extracted from RNA sequences using support vector machine (SVM). The method achieves accurate prediction and mapping of RBP binding sites on any input RNA sequence(s), provided by the users. While developed for organisms such as Drosophila, human and mouse, predictions can be made for other organisms as well.
The amount of experimentally verified RBP binding sites using CLIP-seq has exploded in recent times. Although variable between experiments, these data can serve as training set for machine learning models to predict missing RBP binding sites that may not be detected in some experiments. Methods such as Deepboost (Li et al., 2017) use a machine learning approach, called DeBooster, to accurately model the binding sequence preferences and identify the corresponding binding targets of RBPs from CLIP-seq data. ideepE (Pan and Shen, 2018) and ideepS (Pan et al., 2018) use a convolutional neural network-based approach that is trained on experimentally verified binding motifs from CLIP-seq data to identify RBP binding nucelotides. Here, known motifs were split into fixed length subsequences or padded into fixed length groups. These were employed to train convolutional neural networks (CNNs), to learn and extract high-level features that can identify sequence and structure binding motifs. Such methods were shown to achieve high accuracy to the order of 0.85.
Methods such as DFPin (Zhao et al., 2022) use the cascade structure of deep forest methods to extract key features based on RNA mono-nucleotide composition. Others such as EDCNN (Wang et al., 2022) address the issues of high-dimensionality, data sparsity and low model performance by combining evolutionary algorithms and different gradient descent models to optimize RNA-binding predictions. Integrated web servers such as RBPsuite (Pan et al., 2020) make use of deep learning, to predict nucleotides involved in protein-RNA interactions both in linear and circular RNA. Updates in the programs or training data are addressed at the server end. Such servers are not only useful to expand our knowledge of RBP-binding RNAs, but also, to investigate the effect of mutations on binding RBPs in RNA sequences, since they provide a binding score for a predicted interaction. Typically, these tools achieve an accuracy of 0.7–0.85.
2.1.3 Protein and RNA sequence as input
Apart from the above-mentioned tools, there are few tools which consider both amino-acid and RNA sequence information, to predict residues involved in RNA-protein interactions. catRAPIDomics (Armaos et al., 2021), which can be used for large scale data analysis, uses a HMM-based algorithm to combine secondary structure, hydrogen bonding and van der Waals contributions and predicts protein-RNA associations with great accuracy. RPISeq (Muppirala et al., 2011) is another method that employs machine learning classifiers for predicting such interactions using sequence information. Here, in addition to predicting if a protein and RNA interact, added functionalities of the web-based application include predictions of interactions between a protein sequence and up to 100 RNA user-provided sequences or an RNA sequence and up to 100 protein sequences provided by the user. Further, the server may also be probed to query RPIntDB (Muppirala et al., 2011), a database of known RNA-protein interactions, using a protein query sequence. Another method is PS-PRIP (Muppirala et al., 2016), a sequence motif-based method for “partner-specific” interfacial residue prediction. Here, short strings of amino acids or ribonucleotides that are composed of interacting residues are extracted as n-mer motifs from a dataset of 1,408 protein-RNA complex structures from the Protein Data Bank (PDB). These are stored in a look-up table that is scanned to identify such motifs in query protein or RNA sequences.
Many methods have been developed to predict residues involved in RNA interaction given an amino acid sequence (Figure 1, Supplementary Table S1). Of these, methods that focus on interface residue prediction use properties such as charge, amino acid composition, van der Waals volume, polarity, etc. These methods extract various features from amino acid residues and use them as the input to train the machine-learning models for classification. Several algorithms like SVM (Cai et al., 2003; Kumar et al., 2008; Murakami et al., 2010; Wang et al., 2010; Walia et al., 2014; Yang et al., 2015a; Bressin et al., 2019; Su et al., 2019; Qiu et al., 2020), neural networks (Alipanahi et al., 2015; Peng et al., 2017; Yan and Kurgan, 2017; Deng et al., 2018; Zhao and Du, 2020; Zhang et al., 2021b; Sun et al., 2021; Li and Liu, 2022; Zhang et al., 2022), naive bayes classifier (Sharan et al., 2017; Deng et al., 2021; Li et al., 2022) etc, as listed in Supplementary Table S1, have been successfully implemented. A common limitation faced by such ML-based methods is that the extracted features may be poorly representative of the physicochemical and environmental properties of amino acid residues, or their simplistic combination may introduce redundancy and affect overall prediction power of the approaches.
Structure-based methods to predict such residues are also popular and have benefitted from the availability of detailed structural information for more than 1,000 RNA-protein complexes in the PDB (Berman et al., 2000). In parallel, methods that address the partner prediction problem are also available (Muppirala et al., 2011). Such approaches derive from limited information on RNA-protein interaction partners in primary resources such as the PDB (Berman et al., 2000) and nucleic acid database/ NDB (Coimbatore Narayanan et al., 2014), and secondary resources such as PRIDB (Protein-RNA interface database) (Lewis et al., 2011) and BIPA (Biological Interaction database for Protein-nucleic Acid) (Lee and Blundell, 2009). They also use experimental data from in vivo or in vitro cross-linking studies that are focused on individual proteins or high-throughput RNA-binding microarray data, stored in repositories such as NPInter (noncoding RNAs and protein related biomacromolecules interaction database) (Wu et al., 2006; Teng et al., 2020), CLIPZ (database of post-transcriptional regulatory elements (RNA-binding proteins) built from cross-linking and immunoprecipitation data) (Khorshid et al., 2011) and RBPDB (database of RNA-binding protein specificities) (Cook et al., 2011). Other methods such as RBRIdent (identification of RNA-binding residues) (Xiong et al., 2015) use a genetic algorithm and integrate sequence and structure features by statistical analysis of interaction preferences between amino acid residues and their RNA partners from structure databases.
Tools like RBind (Binding sites on RNA) (Wang et al., 2018; Wang and Zhao, 2020), NAPS (network analysis of protein structures) (Chakrabarty et al., 2019) and PRIdictor (Protein-RNA Interaction predictor) (Tuvshinjargal et al., 2016) use a structural network approach for predicting such interactions. These methods tackle the challenge to predict not only RNA binding sites in proteins but also protein-binding sites in RNA. Several servers such as omiXcore (Armaos et al., 2017) use available CLIP data to predict amino acid residues. Here, a non-linear algorithm is trained on pooled RNA-protein interactions and accepts the proteins and large RNAs with a size between 500 and 20,000 as inputs. PRIP (protein-RNA interface predictor) uses a novel sequence semantics-based method to predict RPI (Li et al., 2022). Integrated classical machine and deep learning classifiers in methods such as RPI-MDLStack (RNA–protein interactions through deep learning with stacking strategy and LASSO) (Yu et al., 2022) have shown improved and robust performance in predicting such interactions. The accuracies of these programs range from 0.75 to 0.98.
2.2 Methods that predict protein-RNA interactions using structure information
Protein-RNA interactions can be predicted using structure-based information. These methods became possible as the number of protein-RNA complex structures deposited in the PDB increased in numbers. Here, either the availability of structures or structure predictions of protein, RNA or both as a complex have been employed to develop several approaches.
2.2.1 Protein structure as input
16% of the tools to predict RPI have been developed based on the structure of a protein (Figure 1). Initially, many combinations of either sequence-based or structure-based features were applied to obtain predictions of protein-RNA interaction residues. These included physicochemical features, side-chain environment, sequence conservation score, position-specific scoring matrices (PSSMs), relative accessible surface area (RASA), secondary structure (SS), interaction propensity and so on. PredRBR (Liu et al., 2017) is one of the noteworthy approaches that employs 189 features which are extracted from sequence, structural and energetic characteristics, as also two categories of Euclidian and Voronoi neighborhood features that are derived from protein-RNA complexes in the RBP170 dataset compiled by Lewis et al., 2010 (Lewis et al., 2011). It employs an mRMR-IFS (maximal relevance minimal redundancy) approach to select an optimal subset of 177 optimal features and a gradient tree boosting algorithm for regression and classification to derive a model, that is useful to predict such interaction sites in datasets that were not employed in the training of the algorithm. The use of such approaches was found to reduce the computational time and improve the performance overall. The results also highlight the benefits of basing RNA-binding residue prediction method on the Gradient tree boosting (GTB) algorithm and structural neighborhood characteristics (Euclidian and Voronoi).
Another popular method is PRNA (Liu et al., 2010) which employs a random forest method for predicting RNA binding sites in proteins, using features that have been extracted from representative protein-RNA complex structures derived from RsiteDB (Shulman-Peleg et al., 2009) (listed in Supplementary Table S1). The strength of the method is that these features are a combination of sequence and structure features. The authors have developed a method by considering the neighborhood of amino acids in the interaction sites since amino acids with different neighborhoods or in different local structures often exhibit preferences for their RNA partners. A combination of interaction propensity feature between the amino acid and its interacting nucleotides that considers neighborhood and individual residue properties defined by six descriptors including physicochemical characteristics, hydrophobic index, relative accessible surface area, secondary structure, sequence conservation score and side-chain environment was found to be a powerful combination and resulted in high accuracy in prediction of known and annotated protein-RNA interaction sites. Another method by Go and co-workers examines amino acid singlet and doublet residue propensity at known protein-RNA interfaces obtained from the (protein quaternary structure) PQS server (Kim et al., 2006) and PSSMs from homologous sequences to make predictions. Such approaches aim to capture not only the pairing preference of amino acid types through propensity calculations but also shed light on the co-operative contribution of various interactions that are known to lie at such interfaces (Kim et al., 2006). Trends such as the high preference for Arg, Lys or aromatic residues such as Tyr to occur at the interface and favoring interactions with RNA were gleaned through these studies. Prediction accuracy of these methods, while reasonably sensitive, would benefit extensively with the inclusion of more structures of such complexes as and when solved, since their analysis is primarily statistical in nature. Such methods can be applied to predict protein–RNA interface residues for query protein structures without biochemical or functional data. Other approaches include PST-PRNA (Li and Liu, 2022) that employs protein surface topography (PST), physicochemical characteristics, structural information, PSSM features and a deep residual network approach, SNB-PSSM (Liu et al., 2021a) that uses a spatial neighbor–based PSSM for extraction of evolutionary information and an SVM as a classifier, NABind (Sun et al., 2016) that includes novel features such as residue electrostatic surface potential and triplet interface propensity in a random forest algorithm. Other ML-based methods include RBscore and NBench (Miao and Westhof, 2016), RBRDetector (Yang et al., 2014), DR_bind1 (Chen et al., 2014), aPRBind (Liu et al., 2021b) and aaRNA (Li et al., 2014).
2.2.2 RNA structure as input
The shape and geometry of RNA can significantly influence RBP binding. Zhang and co-workers developed DeepNet-RBP (Zhang et al., 2015) which uses a deep learning framework to integrate RNA sequence, secondary and tertiary structural profiles, and constructs a ified representation to extract hidden structural features of RBP targets. The three main phases in their development include a data encoding phase of RNA sequences from CLIP-based experiments that were subjected to secondary structure prediction. Likewise, probable tertiary structural motifs were also derived for the sequences and encoded. In the training phase, a multimodal deep belief network (DBN) was employed to integrate the encoded sequence and structural profiles for available CLIP-seq datasets. The primary sequence and secondary structure automatically extract effective hidden structural features from the encoded raw sequence and structural profiles to predict RPI. Applications of this method were shown to effectively detect novel RBP binding sites on genomes and predict RBP binding sites in polypyrimidine tract-binding protein (PTB) and in internal ribosome entry site (IRES) segments and achieved an accuracy of 0.8–0.9. RBind and RBindS (Wang et al., 2018; Wang and Zhao, 2020) uses a network approach and RPI-net (Yan et al., 2021) which makes use of graph neural networks, are other powerful methods. NABS (Jiang et al., 2022) uses an integrative framework with both machine learning and template-based classifiers to predict binding nucleotides.
2.2.3 Prediction using both RNA and protein structures as input
GraphProt (Maticzka et al., 2014) is a flexible machine learning framework that is capable of deriving learning models of RBP binding preferences from high throughput experimental data such as CLIP-seq and RNAcompete (Ray et al., 2009). Here, RNAshapes (Steffen et al., 2006) tool is employed to predict the secondary structure of the bound RNA. This input is used to encode the bound RNA sequence and structure in a graph that preserves base-pairing information, which is fed into a support vector machine to classify RBP bound sites from bound sites using efficient graph kernels (Maticzka et al., 2014). When applied, both sequence and structure logos can be derived for an input query or to predict novel RBP sites. GraphProt can detect the binding sequence and structure preference of RBPs and further predict the RBP binding sites on any input RNAs. The machine leaning-based methods are trained in an RBP-specific manner since RBPs have different binding preferences. One of the main applications of this approach is the computational ability to detect targets of an RBP. Indeed, a user case scenario presented by the authors derives a model based on CLIP-seq data from kidney cells to identify potential targets in the entire transcriptome. Further, if affinity data from RNAcompete experiments are available, GraphProt can apply a regression approach that can distinguish target sites according to their binding strength as well. DLPRB (Ben-Bassat et al., 2018) uses both CNN and recurrent neural network (RNN) to predict protein-RNA interactions and could achieve area der curve (AUC) of 0.81. RCK (Orenstein et al., 2016) is an extension of RNAcontext by a k-mer sequence- and structure-based binding model (Luo et al., 2017). RPI-BIND (Luo et al., 2017) analyses solved RNA-protein structural complexes and employs protein local conformations (PLCs) and 12 classes of RNA local conformations (RLCs), to train a model and predict such interactions. PRIME-3D2D (Xie et al., 2020) is another tool that can predict protein-RNA complex structure and is amenable to genome-scale binding site prediction of proteins on RNA. It uses an alignment-based approach involving TM-align (Zhang and Skolnick, 2005) and LocARNA (multiple alignment of RNA) (Will et al., 2007), to model the protein-RNA complex from which interactions are inferred.
2.3 Tools for modelling or docking of Protein-RNA complexes
The availability of protein-RNA complex structure is important for an derstanding of the function of the complex at a molecular level. As of 2022, 4300 macromolecular complexes containing both protein and RNA (excluding RNA/DNA hybrids) were available in the Protein Data Bank (PDB) (Berman et al., 2000). However, due to the inherent limitations of experimental techniques, this is only a small fraction of all the structures deposited in PDB and all identified protein-RNA interactions. Computational modelling of protein-RNA complexes is one of the approaches by which complex structure can be predicted and employed further to study intermolecular interactions. Nithin and coworkers provide a detailed review on available bioinformatic tools for protein-RNA docking (Nithin et al., 2018). As in the modelling of protein-protein interactions, protein-RNA complexes also involve structural and physicochemical complementarity and have borrowed from existing protein-protein docking methods such as HDock, GRAMM, ZDock, RosettaDock etc. (Tovchigrechko and Vakser, 2006; Guilhot-Gaudeffroy et al., 2014; Pierce et al., 2014; Yan et al., 2017). Both template-based modelling and free docking are employed to predict binding modes of interactions in protein-RNA complexes. A recent study has highlighted some of the available tools for modelling such complexes (Madan et al., 2016). PRIME (Zheng et al., 2016) is a template-based comparative modelling program for protein-RNA interaction modelling in which TMalign (protein structural alignment) and RMalign (RNA structural alignment) are both employed for identifying a template for the protein and RNA sequence of interest. The output of the program is the modelled protein RNA-complex structure which is ranked based on a score like TMscore. A limitation faced by this approach is the inability to build a model in the absence of a reliable template. Further, the scoring function of the RNA alignment algorithm in PRIME is size-dependent, which limits its ability to detect good templates in some cases. The authors report that, like protein-protein complexes, the correlation of protein-RNA structural similarity with the binding mode is poorer on account of greater RNA flexibility. RStrucfam also offers three-dimensional models, once there is an association with a family of RBP (Ghosh et al., 2016).
An approach to model protein-RNA complexes in the absence of a structural template of the complex is to model the protein and RNA components individually and then perform docking and is referred to as free docking, as implemented in 3dRPC (Huang et al., 2018). Here, the docking method RPDOCK, a protein-RNA rigid docking protocol considers geometric and electrostatic factors such as atom packing, residue preferences, stacking interactions between bases and aromatic residues in generating the decoy poses that are evaluated using a specific scoring function. Despite these strengths, the inherent flexibility of RNA and the selection of the appropriate model from the large number of generated docking poses can present a computational challenge for the approach (Huang et al., 2013; Xie et al., 2020). One of the most cited methods for protein- RNA docking is HDOCK (Kim et al., 2014) which employs a hybrid docking algorithm of template-based modelling and free docking based on iterative knowledge-based scoring function. Here, both protein sequence and structure are accepted as inputs while for RNA only structure input is accepted due to inherent challenges in modelling RNA. If binding site information is available, this may be submitted prior to the analysis. A sequence similarity search is performed to find an ideal template from the PDB database using HHsearch (Fidler et al., 2016) for the protein sequence while FASTA is employed to find homologues for the RNA sequence. If the template identified is the same, the model for protein and RNA are derived using the template complex structure, else they are built individually based on different templates using Modeller (Šali and Blundell, 1993). A hierarchical Fast Fourier transform (FFT)-based docking program, HDOCKlite is then employed to sample binding orientations between the two models that are scored using a shape-based pairwise scoring function and over 100 binding models are generated for the user. The method may face limitations when the search algorithm is able to find a suitable homolog. Details on popular methods employed in docking are listed in Supplementary Table S1.
One of the challenges faced by rigid-docking methods involving RNA is the requirement for a suitable template. Several methods have become available to predict RNA 3D structure over the past few decades (Supplementary Table S1). Like proteins, 3D structure of RNA is also conserved and predictable. Therefore, alignment of RNA with previously determined RNA structures will help to predict their 3D structures and identify motifs that are significant for functions like ligand binding and active site. A popular method is EvoClustRNA (Magnus et al., 2019) which applies a multi-step modelling process and considers that RNA sequences from the same RNA family fold into similar and conserved structures. Such homologs are identified through searches in the Rfam database (Kalvari et al., 2021). Independent folding simulations are then performed, and the model selection is based on the most common structural arrangement of the common helical fragments. Current RNA folding algorithms can predict RNA structures of short to very long RNA sequences. Structure-based prediction algorithms (Das and Baker, 2007; Das et al., 2008; Parisien and Major, 2008; Das et al., 2010; Cao and Chen, 2011; Zhao et al., 2012; Magnus et al., 2019; Andrikos et al., 2022) achieve the highest accuracy with efficient alignments. A part of a well-aligned structure can be extracted and used as a fragment and many such fragments can be assembled on a template to form a 3D structure. Indeed, such methods aim to overcome limitations of template-based modelling which do not consider the flexibility of the RNA molecules, and the number of known RNA structures. Ab-initio tools (Das and Baker, 2007; Jossinet et al., 2010; Magnus et al., 2014; Xu et al., 2014; Mallet et al., 2022), on the other hand, face challenges such as the length of RNA, conformational sampling, evaluation of energies for the tertiary contacts, and knowledge-based energy functions. Coarse-grained approaches (Wang et al., 1999; Cao and Chen, 2005; Tan et al., 2006; Ding et al., 2008; Sharma et al., 2008; Jonikas et al., 2009; Pasquali and Derreumaux, 2010; Xia et al., 2010; Izzo et al., 2011; Xia et al., 2013; Kim et al., 2014; Shi et al., 2014; Oliver et al., 2022) model entire RNA structure using beads and these models are subjected to energy minimization, molecular dynamics and/or Monte Carlo simulations. These methods offer a way to derstand RNA folding and show improved efficiency in prediction of lengthy RNA molecules. Coarse-grained models, however, face limitations in entropy calculations and long-range tertiary interactions.
Future improvements in methods to model Protein-RNA complexes could focus on combinations of free docking and template-based algorithms because using either approach independently has a reduced accuracy of prediction.
2.4 Tools for studying and analysing protein-RNA interfaces
Evaluation of phenotypic impact of sequence variations is very important to derstand function. There are various tools for analysing the effect of mutations in protein-RNA complexes. ENTANGLE (Allers and Shamoo, 2001) is a structure-based analysis program, to analyze protein-RNA interactions that allows users to examine protein-RNA interactions in an available three-dimensional structure or model. Users can choose from a range of ways to study protein-RNA interactions and can examine the interactions through a graphics interface. In addition, the tool has been employed to build a protein-RNA interaction database that catalogs the various types of characteristic interactions of these complexes and considers electrostatic, hydrogen bonding and stacking interactions. Such studies and databases have provided useful insights to distinguish protein-RNA binding from other protein-DNA binding. mCSM-NA (Pires et al., 2017), DeepCLIP (Bjørnholt Grønning et al., 2020) and PremPRI (Zhang et al., 2020) are a few examples of tools which use machine learning to calculate binding affinity changes in a protein complex upon mutation. mCSM-NA employs graph-based signatures to represent protein-nucleic acid complexes and models the distance patterns in wild type proteins from the ProNIT database (Prabakaran et al., 2000). Residues in the vicinity of the mutated protein are labelled based on pharmacophore modelling which describes the geometry and physicochemical properties of the residue environment. A machine learning based model is then trained on the effects of such mutations to arrive at models that can predict changes in nucleic acid binding affinities. Input for the server is a protein-RNA complex structure and up to a maximum list of 20 single point mutations. The output includes predicted changes in binding affinity in Kcal/mol, the relative residue solvent accessibility of the mutated residue, predicted change in binding affinity and protein stability. Limited success of these techniques can be attributed to the intramolecular interactions and various conformations that are not considered in the physical models. Accurate prediction of RNA–protein binding affinities is very challenging, and a complete prediction framework for RNA–protein complexes is yet to be developed. Supplementary Table S1 lists and highlights features of various tools that are available to study interactions in these interfaces.
2.5 Tools for analysing hotspots in protein-RNA complexes
Conserved residues in binding sites that contribute to the strength of binding, and whose substitution to Alanine leads to an increase in the binding free energy (ΔG) of at least 2.0 kcal mol─1, are defined as hotspot residues (Clackson and Wells, 1995). Given the complex structure, many tools are available to predict hotspots in protein-RNA interfaces and predict the quantitative changes in free energy or a probability score for a hotspot residue. All these tools are built on thermodynamics data. PrabHot (Krüger et al., 2018), XGBPRH (Deng et al., 2019) and SREPRHot (Zhou et al., 2022) use ML algorithms and structural information to predict hotspot residues. Input for PrabHot, a webserver, is a protein-RNA complex structure. The method is based on 47 protein-RNA complexes from which various features such as network, exposure, sequence and structure determinants are extracted. An ensemble approach is employed to integrate SVM (Support Vector Machine), GTB (Gradient Tree Boosting) and ERT (Extremely Randomized Trees) based classification to predict the effect of mutations. Other methods include SPHot (Zhang et al., 2019) and iPNHOT (Zhu et al., 2020) that are based on SVM analyses of hotspot residues from protein sequences (Supplementary Table S1). SPHot uses only sequence information for the prediction. Although many tools are available, this area still has many limitations and challenges. These include lack of thermodynamic data, experimentally verified data sets and structural data. In future, we can expect that these tools can be improved to include the multiple conformational nature of protein-RNA complexes.
2.6 Tools for studying the structural dynamics of protein-RNA complexes
Functions of protein-RNA complexes are intimately linked to their dynamics. Long-timescale molecular dynamics (MD) simulations have been successfully used to characterize complex conformational transitions in proteins. In principle, MD simulation is a powerful tool for characterizing such conformational changes in RNA molecules, which depends on force-field parameters. AMBER force field (Tan et al., 2018) uses a combination of ab initio and empirical methods, modified electrostatic, van der Waals (vdW), and torsional parameters to accurately reproduce the energetics of nucleobase stacking, base pairing, and key torsional conformers. CHARMM modified its forcefield parameters to make it suitable for nucleic acids (Xu et al., 2016). OPLS-AA force field made changes in potential energy surfaces of the backbone α- and γ dihedral angles, for modelling of RNA (Robertson et al., 2019). Apart from this there are advanced quantum mechanics/molecular mechanics (QM/MM) computations which could be used to indirectly rationalize problems seen in MM-based MD simulations of protein–RNA complexes (Pokorná et al., 2018). Martini force field (Monticelli et al., 2008) helps in coarse-grained representation of protein-RNA complexes, and these models can be applied in MD simulations. Elastic network representation of protein-RNA (Pinamonti et al., 2015) complex can be employed in normal mode analysis which can be used to track flexibility and dynamics of a complex.
2.7 Databases for protein-RNA interactions
Various databases provide comprehensive repositories of RNA-protein interaction data that have been gathered from experiments, literature and other databases and computational predictions. PRD (Fujimori et al., 2012), NPInter (Wu et al., 2006; Teng et al., 2020), RNAct (Lang et al., 2019) and RAID (Zhang et al., 2014) help us integrate data from various platforms. CLIPdb (Yang et al., 2015b) and RPI- PRED RNA (Suresh et al., 2015) curate experimental information from literature and capture this in a table. PRIDB (Lewis et al., 2011) and RBPDB (Cook et al., 2011) characterize and provide data with the help of existing structural information. Protein family databases such as PFAM also contain RNA binding domain families. Nearly 3909 domain families are associated with the keyword RNA-binding. Such domain families that are represented as Hidden Markov models (HMM) are useful in sequence-based association of RNA binding function in a query protein sequence, using HMMPFAM (SR Eddy, 2004). There are also specialized databases, such as EcRBPOME (Ghosh et al., 2019) which offer putative RNA binding proteins in a genome-wide scale in around 600 E. coli strains. Among the above databases, NPInter, PRD, RNAct, CLIPdb and RBPDB provide and extract information from experimental techniques. In future, an exhaustive interaction database which could store detailed and multi-dimensional information about an interaction entry, such as a binding region/motif, structure detection method, interactions, etc. would add more value to such efforts. Furthermore, the ability to predict potential RPI is based on availability of such catalogued information, making such databases necessary and valuable.
3 Conclusion
This review provides a comprehensive survey of computational tools for the analysis of protein-RNA interactions. The availability of mathematical models and profiles, together with sensitive search algorithms, enable effective association of new genes into pre-existing families of RNA-binding proteins from mere sequence information. Repositories such as protein sequence families (PFAM) (Finn et al., 2016; Mistry et al., 2021) and in-built databases in servers such as RStrucFam (Ghosh et al., 2016) make focused searches easier. As seen in the pie-chart (Figure 1), majority of the tools that have been developed predict amino acid residues and very few tools are available for modelling protein-RNA interactions. Nearly 30% of the tools are for predicting interactions in the protein-RNA complexes given a protein query sequence. There is still room for better methods and approaches to evolve. In this section, we discuss some of the inherent challenges and outstanding issues.
RBPs are modular and may possess more than one RBD that are connected with flexible linkers or may occur as isoforms. Automatic tools are not available to provide accurate assignment of domain boundaries and nomenclature. Which of these domains bear sequence signatures of functionally important residues, how they co-operate in exhibiting function and their relative affinity for RNA still are open questions. Several proteins that contain RNA-binding domains also contain substantial stretches of disordered regions. Their structural and functional regulation are hard to capture and remain a treasure-house of knowns. Despite the choice of objective mathematical models, presence of spatial motifs of RNA-binding residues or RNA that acquire three-dimensional structure might escape detection. Some RBPs are known to bind both DNA and RNA and this might cause sufficient confusion in prediction since the results may be viewed as false positives.
The success of computational algorithms is usually measured using statistical parameters such as accuracy, sensitivity and specificity. The performance of algorithms to recognize RNA-binding proteins is challenged by the absence of a comprehensive and reliable gold-standard to look upon. It is now accepted that different databases of RNA-binding proteins that use different high-throughput experimental approaches and platforms do not agree very well with each other (Ghosh and Sowdhamini, 2016). It would be desirable to arrive at an ified experimental approach for the identification of RNA-binding proteins.
Finally, RNA-binding proteins play important roles in diverse biological roles such as developmental processes and ageing. They are also amenable to functional regulation such as phosphorylation switches and expression levels. In future, meta-analyses are required to assimilate such functional information.
Author contributions
SB, SS, NS and RS conceptualized and planned the review. SB did all preliminary review of literature and data presentation. SS, SB, and RS wrote and edited the paper. All authors approved the final version of the manuscript.
Funding
RS acknowledges infrastructural facilities of NCBS; the funding provided by JC Bose Fellowship (JBR/2021/000,006) from Science and Engineering Research Board, India, her Kiran Mazumdar Shaw Computational Biology Chair grant at Institute of Bioinformatics and Applied Biotechnology, Bangalore (IBAB/MSCB/182/2022) and Bioinformatics Centre Grant funded by Department of Biotechnology, India (BT/PR40187/BTIS/137/9/2021). This research is supported by Mathematical Biology program and FIST program sponsored by the Department of Science and Technology and by the Department of Biotechnology, Government of India in the form of IISc-DBT partnership programme. Support from the Bioinformatics and Computational biology Centre, DBT and support from UGC, India–Centre for Advanced Studies and Ministry of Human Resource Development, India is gratefully acknowledged. NS is a J. C. Bose National Fellow and SB is a CSIR senior research fellow.
Acknowledgments
SS acknowledges Dept of Biotechnology, MSRUAS for the seed grant and infrastructural support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.954926/full#supplementary-material
References
Alipanahi, B., Delong, A., Weirauch, M. T., and Frey, B. J. (2015). Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33 (8), 831–838. doi:10.1038/nbt.3300
Allers, J., and Shamoo, Y. (2001). Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 311 (1), 75–86. doi:10.1006/jmbi.2001.4857
Allerson, C. R., Cazzola, M., and Rouault, T. A. (1999). Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome. J. Biol. Chem. 274 (37), 26439–26447. doi:10.1074/jbc.274.37.26439
Andrikos, C., Makris, E., Kolaitis, A., Rassias, G., Pavlatos, C., and Tsanakas, P. (2022). Knotify: An efficient parallel platform for RNA pseudoknot prediction using syntactic pattern recognition. Methods Protoc. 5 (1). doi:10.3390/mps5010014
Armaos, A., Cirillo, D., and Gaetano Tartaglia, G. (2017). OmiXcore: A web server for prediction of protein interactions with large RNA. Bioinformatics 33 (19), 3104–3106. doi:10.1093/bioinformatics/btx361
Armaos, A., Colantoni, A., Proietti, G., Rupert, J., and Tartaglia, G. G. (2021). CatRAPID omics v2.0: Going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Res. 49 (1), W72–W79. doi:10.1093/nar/gkab393
Baltz, A. G., Munschauer, M., Schwanhäusser, B., Vasile, A., Murakawa, Y., Schueler, M., et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 46 (5), 674–690. doi:10.1016/j.molcel.2012.05.021
Bao, P., Boon, K. L., Will, C. L., Hartmuth, K., and Lührmann, R. (2018). Multiple RNA-RNA tertiary interactions are dispensable for formation of a functional U2/U6 RNA catalytic core in the spliceosome. Nucleic Acids Res. 46 (22), 12126–12138. doi:10.1093/nar/gky966
Barbieri, I., and Kouzarides, T. (2020). Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322. doi:10.1038/s41568-020-0253-2
Batista, P. J., and Chang, H. Y. (2013). Long noncoding RNAs: Cellular address codes in development and disease. Cell. 152, 1298–1307. doi:10.1016/j.cell.2013.02.012
Beckmann, B. M., Horos, R., Fischer, B., Castello, A., Eichelbaum, K., Alleaume, A. M., et al. (2015). The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 6, 10127. doi:10.1038/ncomms10127
Ben-Bassat, I., Chor, B., and Orenstein, Y. (2018). A deep neural network approach for learning intrinsic protein-RNA binding preferences. Bioinformatics 34, i638–i646. doi:10.1093/bioinformatics/bty600
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein Data Bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Bheemireddy, S., Sandhya, S., and Srinivasan, N. (2021). Comparative analysis of structural and dynamical features of ribosome upon association with mRNA reveals potential role of ribosomal proteins. Front. Mol. Biosci. 8, 757. doi:10.3389/fmolb.2021.654164
Bienert, S., Waterhouse, A., de Beer, T. A. P., Tauriello, G., Studer, G., Bordoli, L., et al. (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45 (D1), D313–D319. doi:10.1093/nar/gkw1132
Bjørnholt Grønning, A. G., Doktor, T. K., Larsen, S. J., Spangsberg Petersen, U. S., Holm, L. L., Bruun, G. H., et al. (2020). DeepCLIP: Predicting the effect of mutations on protein–RNA binding with deep learning. Nucleic Acids Res. 48 (13), 7099–7118.
Bressin, A., Schulte-Sasse, R., Figini, D., Urdaneta, E. C., Beckmann, B. M., and Marsico, A. (2019). TriPepSVM: De novo prediction of RNA-binding proteins based on short amino acid motifs. Nucleic Acids Res. 47 (9), 4406–4417. doi:10.1093/nar/gkz203
Cai, C. Z., Han, L. Y., Ji, Z. L., Chen, X., and Chen, Y. Z. (2003). SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 31 (13), 3692–3697. doi:10.1093/nar/gkg600
Calabretta, S., and Richard, S. (2015). Emerging roles of disordered sequences in RNA-binding proteins. Trends Biochem. Sci. 40, 662–672. doi:10.1016/j.tibs.2015.08.012
Campbell, Z. T., and Wickens, M. (2015). Probing RNA-protein networks: Biochemistry meets genomics. Trends Biochem. Sci. 40, 157–164. doi:10.1016/j.tibs.2015.01.003
Cao, S., and Chen, S. J. (2011). Physics-based de novo prediction of RNA 3D structures. J. Phys. Chem. B 115 (14), 4216–4226. doi:10.1021/jp112059y
Cao, S., and Chen, S. J. (2005). Predicting RNA folding thermodynamics with a reduced chain representation model. RNA 11 (12), 1884–1897. doi:10.1261/rna.2109105
Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B. M., Strein, C., et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 149 (6), 1393–1406. doi:10.1016/j.cell.2012.04.031
Chakrabarty, B., Naganathan, V., Garg, K., Agarwal, Y., and Parekh, N. (2019). NAPS update: Network analysis of molecular dynamics data and protein-nucleic acid complexes. Nucleic Acids Res. 47 (W1), W462–W470. doi:10.1093/nar/gkz399
Chen, Y. C., Sargsyan, K., Wright, J. D., Huang, Y. S., and Lim, C. (2014). Identifying RNA-binding residues based on evolutionary conserved structural and energetic features. Nucleic Acids Res. 42 (3), e15. doi:10.1093/nar/gkt1299
Christopoulou, N., and Granneman, S. (2022). The role of RNA-binding proteins in mediating adaptive responses in Gram-positive bacteria. FEBS J. 289, 1746–1764. doi:10.1111/febs.15810
Clackson, T., and Wells, J. A. (1995). A hot spot of binding energy in a hormone-receptor interface. Science 267 (5196), 383–386. doi:10.1126/science.7529940
Coimbatore Narayanan, B., Westbrook, J., Ghosh, S., Petrov, A. I., Sweeney, B., Zirbel, C. L., et al. (2014). The nucleic acid database: New features and capabilities. Nucleic Acids Res. 42 (D1), D114–D122. doi:10.1093/nar/gkt980
Cook, K. B., Hughes, T. R., and Morris, Q. D. (2015). High-throughput characterization of protein-RNA interactions. Briefings Funct. Genomics 14 (1), 74–89. doi:10.1093/bfgp/elu047
Cook, K. B., Kazan, H., Zuberi, K., Morris, Q., and Hughes, T. R. (2011). Rbpdb: A database of RNA-binding specificities. Nucleic Acids Res. 39 (Suppl. 1), D301–D308. doi:10.1093/nar/gkq1069
Cozzolino, F., Iacobucci, I., Monaco, V., and Monti, M. (2021). Protein-DNA/RNA interactions: An overview of investigation methods in the -omics era. J. Proteome Res. 20, 3018–3030. doi:10.1021/acs.jproteome.1c00074
Crick, F. (1970). Central dogma of molecular biology. Nature 227 (5258), 561–563. doi:10.1038/227561a0
Cubuk, J., Alston, J. J., Incicco, J. J., Singh, S., Stuchell-Brereton, M. D., Ward, M. D., et al. (2021). The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 12 (1), 1936. doi:10.1038/s41467-021-21953-3
Darnell, R. B. (2010). RNA regulation in neurologic disease and cancer. Cancer Res. Treat. 42 (3), 125. doi:10.4143/crt.2010.42.3.125
Das, R., and Baker, D. (2007). Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. U.S.A. 104 (37), 14664–14669. doi:10.1073/pnas.0703836104
Das, R., Karanicolas, J., and Baker, D. (2010). Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods 7 (4), 291–294. doi:10.1038/nmeth.1433
Das, R., Kudaravalli, M., Jonikas, M., Laederach, A., Fong, R., Schwans, J. P., et al. (2008). Structural inference of native and partially folded RNA by high-throughput contact mapping. Proc. Natl. Acad. Sci. U.S.A. 105 (11), 4144–4149. doi:10.1073/pnas.0709032105
Deng, L., Dong, Z., B, H. L., Bressin, A., Schulte-sasse, R., Figini, D., et al. (2021). FastRNABindR: Fast and accurate prediction of protein-RNA interface residues. Nucleic Acids Res. 34 (1), 1–14.
Deng, L., Dong, Z., and Liu, H. (2018). XPredRBR: Accurate and fast prediction of RNA-binding residues in proteins using eXtreme gradient boosting. Lect. Notes Comput. Sci. 10847 (61672541), 163–173. doi:10.1007/978-3-319-94968-0_14
Deng, L., Sui, Y., and Zhang, J. (2019). XGBPRH: Prediction of binding hot spots at Protein⁻RNA interfaces utilizing extreme gradient boosting. Genes. (Basel) 10 (3). doi:10.3390/genes10030242
Dimitrova-Paternoga, L., Jagtap, P. K. A., Chen, P. C., and Hennig, J. (2020). Integrative structural biology of protein-RNA complexes. Structure 28, 6–28. doi:10.1016/j.str.2019.11.017
Ding, F., Sharma, S., Chalasani, P., Demidov, V. V., Broude, N. E., and Dokholyan, N. V. (2008). Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 14 (6), 1164–1173. doi:10.1261/rna.894608
Dyson, H. J. (2012). Roles of intrinsic disorder in protein-nucleic acid interactions. Mol. Biosyst. 8, 97–104. doi:10.1039/c1mb05258f
Fidler, D. R., Murphy, S. E., Courtis, K., Antonoudiou, P., El-Tohamy, R., Ient, J., et al. (2016). Using HHsearch to tackle proteins of known function: A pilot study with PH domains. Traffic 17 (11), 1214–1226. doi:10.1111/tra.12432
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44 (D1), D279–D285. doi:10.1093/nar/gkv1344
Fujimori, S., Hino, K., Saito, A., Miyano, S., and Miyamoto-Sato, E. (2012). Prd: A protein-RNA interaction database. Bioinformation 8 (15), 729–730. doi:10.6026/97320630008729
Gagliardi, M., and Matarazzo, M. R. (2016). RIP: RNA immunoprecipitation. Methods Mol. Biol. 1480, 73–86. doi:10.1007/978-1-4939-6380-5_7
Gerber, A. P. (2021). Rna-centric approaches to profile the rna–protein interaction landscape on selected RNAs. Non-coding RNA 7, 1–15.
Gerstberger, S., Hafner, M., and Tuschl, T. (2014). A census of human RNA-binding proteins. Nat. Rev. Genet. 15 (12), 829–845. doi:10.1038/nrg3813
Ghosh, P., Joshi, A., Guita, N., Offmann, B., and Sowdhamini, R. (2019). EcRBPome: A comprehensive database of all known E. coli RNA-binding proteins. BMC Genomics 20 (1), 403. doi:10.1186/s12864-019-5755-5
Ghosh, P., Mathew, O. K., and Sowdhamini, R. (2016). RStrucFam: A web server to associate structure and cognate RNA for RNA-binding proteins from sequence information. BMC Bioinforma. 17 (1), 411. doi:10.1186/s12859-016-1289-x
Ghosh, P., and Sowdhamini, R. (2016). Genome-wide survey of putative RNA-binding proteins encoded in the human proteome. Mol. Biosyst. 12 (2), 532–540. doi:10.1039/c5mb00638d
Gräwe, C., Stelloo, S., van Hout, F. A. H., and Vermeulen, M. (2021). RNA-centric methods: Toward the interactome of specific RNA transcripts. Trends Biotechnol. 39 (9), 890–900. doi:10.1016/j.tibtech.2020.11.011
Guilhot-Gaudeffroy, A., Froidevaux, C., Azé, J., and Bernauer, J. (2014). Protein-RNA complexes and efficient automatic docking: Expanding rosettadock possibilities. PLoS One 9 (9), e108928. doi:10.1371/journal.pone.0108928
Hentze, M. W., Castello, A., Schwarzl, T., and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell. Biol. 19, 327–341. doi:10.1038/nrm.2017.130
Huang, Y., Li, H., and Xiao, Y. (2018). 3dRPC: A web server for 3D RNA-protein structure prediction. Bioinformatics 34 (7), 1238–1240. doi:10.1093/bioinformatics/btx742
Huang, Y., Liu, S., Guo, D., Li, L., and Xiao, Y. (2013). A novel protocol for three-dimensional structure prediction of RNA-protein complexes. Sci. Rep. 3, 1887. doi:10.1038/srep01887
Izzo, J. A., Kim, N., Elmetwaly, S., and Schlick, T. (2011). RAG: An update to the RNA-As-Graphs resource. BMC Bioinforma. 12, 219. doi:10.1186/1471-2105-12-219
Jiang, H., Bai, L., Ji, L., Bai, Z., Su, J., Qin, T., et al. (2020). Degradation of MicroRNA miR-466d-3p by Japanese encephalitis virus NS3 facilitates viral replication and interleukin-1β expression. J. Virol. 94 (15). doi:10.1128/JVI.00294-20
Jiang, Z., Xiao, S. R., and Liu, R. (2022). Dissecting and predicting different types of binding sites in nucleic acids based on structural information. Brief. Bioinform 23 (1), 1–19. doi:10.1093/bib/bbab411
Jolma, A., Zhang, J., Mondragón, E., Morgunova, E., Kivioja, T., Laverty, K. U., et al. (2020). Binding specificities of human RNA-binding proteins toward structured and linear RNA sequences. Genome Res. 30 (7), 962–973. doi:10.1101/gr.258848.119
Jonikas, M. A., Radmer, R. J., Laederach, A., Das, R., Pearlman, S., Herschlag, D., et al. (2009). Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 15 (2), 189–199. doi:10.1261/rna.1270809
Jossinet, F., Ludwig, T. E., and Westhof, E. (2010). Assemble: An interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 26 (16), 2057–2059. doi:10.1093/bioinformatics/btq321
Kalvari, I., Nawrocki, E. P., Ontiveros-Palacios, N., Argasinska, J., Lamkiewicz, K., Marz, M., et al. (2021). Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49 (D1), D192–D200. doi:10.1093/nar/gkaa1047
Ke, A., and Doudna, J. A. (2004). Crystallization of RNA and RNA?protein complexes. Methods 34 (3), 408–414. doi:10.1016/j.ymeth.2004.03.027
Kharel, P., Becker, G., Tsvetkov, V., and Ivanov, P. (2020). Properties and biological impact of RNA G-quadruplexes: From order to turmoil and back. Nucleic Acids Res. 48, 12534–12555. doi:10.1093/nar/gkaa1126
Khorshid, M., Rodak, C., and Zavolan, M. (2011). Clipz: A database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 39 (Suppl. 1), D245–D252. doi:10.1093/nar/gkq940
Kim, N., Laing, C., Elmetwaly, S., Jung, S., Curuksu, J., and Schlick, T. (2014). Graph-based sampling for approximating global helical topologies of RNA. Proc. Natl. Acad. Sci. U.S.A. 111 (11), 4079–4084. doi:10.1073/pnas.1318893111
Kim, O. T. P., Yura, K., and Go, N. (2006). Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 34 (22), 6450–6460. doi:10.1093/nar/gkl819
Krüger, D. M., Neubacher, S., and Grossmann, T. N. (2018). Protein–RNA interactions: Structural characteristics and hotspot amino acids. RNA 24 (11), 1457–1465.
Kumar, M., Gromiha, M. M., and Raghava, G. P. S. (2008). Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 71 (1), 189–194. doi:10.1002/prot.21677
Kwon, S. C., Yi, H., Eichelbaum, K., Föhr, S., Fischer, B., You, K. T., et al. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 20 (9), 1122–1130. doi:10.1038/nsmb.2638
Lang, B., Armaos, A., and Tartaglia, G. G. (2019). RNAct: Protein-RNA interaction predictions for model organisms with supporting experimental data. Nucleic Acids Res. 47 (D1), D601–D606. doi:10.1093/nar/gky967
Lee, S., and Blundell, T. L. (2009). Bipa: A database for protein-nucleic acid interaction in 3D structures. Bioinformatics 25 (12), 1559–1560. doi:10.1093/bioinformatics/btp243
Lewis, B. A., Walia, R. R., Terribilini, M., Ferguson, J., Zheng, C., Honavar, V., et al. (2011). Pridb: A protein-RNA interface database. Nucleic Acids Res. 39 (Suppl. 1), D277–D282. doi:10.1093/nar/gkq1108
Li, P., and Liu, Z. P. (2022). PST-PRNA: Prediction of RNA-binding sites using protein surface topography and deep learning. Bioinformatics 2022 (2), 1–7. doi:10.1093/bioinformatics/btac078
Li, S., Dong, F., Wu, Y., Zhang, S., Zhang, C., Liu, X., et al. (2017). A deep boosting based approach for capturing the sequence binding preferences of RNA-binding proteins from high-throughput CLIP-seq data. Nucleic Acids Res. 45 (14), E129. doi:10.1093/nar/gkx492
Li, S., Yamashita, K., Amada, K. M., and Standley, D. M. (2014). Quantifying sequence and structural features of protein-RNA interactions. Nucleic Acids Res. 42 (15), 10086–10098. doi:10.1093/nar/gku681
Li, Y., Lyu, J., Wu, Y., Liu, Y., and Huang, G. (2022). PRIP: A protein-RNA interface predictor based on semantics of sequences. Life 12 (2), 307. doi:10.3390/life12020307
Licatalosi, D. D., Ye, X., and Jankowsky, E. (2020). Approaches for measuring the dynamics of RNA–protein interactions. Wiley Interdiscip. Rev. RNA 11.
Liepelt, A., Naarmann-de Vries, I. S., Simons, N., Eichelbaum, K., Föhr, S., Archer, S. K., et al. (2016). Identification of RNA-binding proteins in macrophages by interactome capture. Mol. Cell. Proteomics 15 (8), 2699–2714. doi:10.1074/mcp.m115.056564
Liu, D., Tang, Y., Fan, C., Chen, Z., and Deng, L. (2017). “PredRBR: Accurate prediction of RNA-binding residues in proteins using gradient tree boosting,” in Proceedings - 2016 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2016 (Piscataway, NJ, USA: Institute of Electrical and Electronics Engineers Inc.), 47–52.
Liu, Y., Gong, W., Yang, Z., and Li, C. (2021). SNB-PSSM: A spatial neighbor-based PSSM used for protein–RNA binding site prediction. J. Mol. Recognit. 34 (6), 1–8. doi:10.1002/jmr.2887
Liu, Y., Gong, W., Zhao, Y., Deng, X., Zhang, S., and Li, C. (2021). APRBind: Protein-RNA interface prediction by combining sequence and I-TASSER model-based structural features learned with convolutional neural networks. Bioinformatics 37 (7), 937–942. doi:10.1093/bioinformatics/btaa747
Liu, Z. P., Wu, L. Y., Wang, Y., Zhang, X. S., and Chen, L. (2010). Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 26 (13), 1616–1622. doi:10.1093/bioinformatics/btq253
Luo, J., Liu, L., Venkateswaran, S., Song, Q., and Zhou, X. (2017). RPI-bind: A structure-based method for accurate identification of RNA-protein binding sites. Sci. Rep. 7 (1), 1–13. doi:10.1038/s41598-017-00795-4
Madan, B., Kasprzak, J. M., Tuszynska, I., Magnus, M., Szczepaniak, K., Dawson, W. K., et al. (2016). “Modeling of protein-RNA complex structures using computational docking methods,” in Methods in molecular biology (Totowa, NJ, USA: Humana Press), 353–372. doi:10.1007/978-1-4939-3569-7_21
Magnus, M., Kappel, K., Das, R., and Bujnicki, J. M. (2019). RNA 3D structure prediction guided by independent folding of homologous sequences. BMC Bioinforma. 20 (1), 512. doi:10.1186/s12859-019-3120-y
Magnus, M., Matelska, D., Łach, G., Chojnowski, G., Boniecki, M. J., Purta, E., et al. (2014). Computational modeling of RNA 3D structures, with the aid of experimental restraints. RNA Biol. 11. doi:10.4161/rna.28826
Mallet, V., Oliver, C., Broadbent, J., Hamilton, W. L., and Waldispühl, J. (2022). RNAglib: A python package for RNA 2.5 D graphs. Bioinformatics 38 (5), 1458–1459. doi:10.1093/bioinformatics/btab844
Matia-González, A. M., Laing, E. E., and Gerber, A. P. (2015). Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 22 (12), 1027–1033.
Maticzka, D., Lange, S. J., Costa, F., and Backofen, R. (2014). GraphProt: Modeling binding preferences of RNA-binding proteins. Genome Biol. 15 (1), R17. doi:10.1186/gb-2014-15-1-r17
Miao, Z., and Westhof, E. (2016). RBscore&NBench: A high-level web server for nucleic acid binding residues prediction with a large-scale benchmarking database. Nucleic Acids Res. 44 (W1), W562–W567. doi:10.1093/nar/gkw251
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–D419. doi:10.1093/nar/gkaa913
Mitchell, S. F., Jain, S., She, M., and Parker, R. (2013). Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 20 (1), 127–133. doi:10.1038/nsmb.2468
Monticelli, L., Kandasamy, S. K., Periole, X., Larson, R. G., Tieleman, D. P., and Marrink, S. J. (2008). The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 4 (5), 819–834. doi:10.1021/ct700324x
Moore, P. B., and Steitz, T. A. (2002). The involvement of RNA in ribosome function. Nature 418 (6894), 229–235. doi:10.1038/418229a
Muppirala, U., Lewis, B. A., Mann, C. M., and Dobbs, D. (2016). A motif-based method for predicting interfacial residues in both the RNA and protein components of protein-RNA complexes. Pac Symp. Biocomput 21, 445–455. doi:10.1142/9789814749411_0041
Muppirala, U. K., Honavar, V. G., and Dobbs, D. (2011). Predicting RNA-protein interactions using only sequence information. BMC Bioinforma. 12 (1), 489. doi:10.1186/1471-2105-12-489
Murakami, Y., Spriggs, R. v., Nakamura, H., and Jones, S. (2010). PiRaNhA: A server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res. 38 (Suppl. 2), W412–W416. doi:10.1093/nar/gkq474
Nithin, C., Ghosh, P., and Bujnicki, J. M. (2018). Bioinformatics tools and benchmarks for computational docking and 3D structure prediction of RNA-protein complexes. Genes. 9. doi:10.3390/genes9090432
Oliver, C., Mallet, V., Philippopoulos, P., Hamilton, W. L., and Waldispühl, J. (2022). Vernal: A tool for mining fuzzy network motifs in RNA. Bioinformatics 38 (4), 970–976. doi:10.1093/bioinformatics/btab768
Orenstein, Y., Wang, Y., and Berger, B. (2016). RCK: Accurate and efficient inference of sequence- and structure-based protein-RNA binding models from RNAcompete data. Bioinformatics 32 (12), i351–i359. doi:10.1093/bioinformatics/btw259
Ottoz, D. S. M., and Berchowitz, L. E. (2020). The role of disorder in RNA binding affinity and specificity. Open Biol. 10 (12), 200328. doi:10.1098/rsob.200328
Pai, A. A., Henriques, T., Mccue, K., Burkholder, A., Adelman, K., and Burge, C. B. (2017). The kinetics of pre-mRNA splicing in the Drosophila genome and the influence of gene architecture.
Pan, X., Fang, Y., Li, X., Yang, Y., and Shen, H. B. (2020). RBPsuite: RNA-protein binding sites prediction suite based on deep learning. BMC Genomics 21 (1), 884. doi:10.1186/s12864-020-07291-6
Pan, X., Rijnbeek, P., Yan, J., and Shen, H. B. (2018). Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics 19 (1), 511–11. doi:10.1186/s12864-018-4889-1
Pan, X., and Shen, H. B. (2018). Predicting RNA-protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 34 (20), 3427–3436. doi:10.1093/bioinformatics/bty364
Panwar, B., and Raghava, G. P. S. (2015). Identification of protein-interacting nucleotides in a RNA sequence using composition profile of tri-nucleotides. Genomics 105 (4), 197–203. doi:10.1016/j.ygeno.2015.01.005
Parisien, M., and Major, F. (2008). The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452 (7183), 51–55. doi:10.1038/nature06684
Pasquali, S., and Derreumaux, P. (2010). HiRE-RNA: A high resolution coarse-grained energy model for RNA. J. Phys. Chem. B 114 (37), 11957–11966. doi:10.1021/jp102497y
Paz, I., Kosti, I., Ares, M., Cline, M., and Mandel-Gutfreund, Y. (2014). RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 42 (W1), W361–W367. doi:10.1093/nar/gku406
Peng, Z., Wang, C., Uversky, V. N., and Kurgan, L. (2017). Prediction of disordered RNA, DNA, and protein binding regions using disoRDPbind. Methods Mol. Biol. 1484, 187–203. doi:10.1007/978-1-4939-6406-2_14
Perez, R. K., Gordon, M. G., Subramaniam, M., Kim, M. C., Hartoularos, G. C., Targ, S., et al. (2022). Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376 (6589), eabf1970. doi:10.1126/science.abf1970
Pierce, B. G., Wiehe, K., Hwang, H., Kim, B. H., Vreven, T., and Weng, Z. (2014). ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30 (12), 1771–1773. doi:10.1093/bioinformatics/btu097
Pinamonti, G., Bottaro, S., Micheletti, C., and Bussi, G. (2015). Elastic network models for RNA: A comparative assessment with molecular dynamics and shape experiments. Nucleic Acids Res. 43 (15), 7260–7269. doi:10.1093/nar/gkv708
Pires, D. E., Ascher, D. B., de Pesquisas RenéRen, C., Rachou, R., and Oswaldo Cruz, F. (2017). mCSM-NA: predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res. 45 (W1), W241–W246. doi:10.1093/nar/gkx236
Pokorná, P., Kruse, H., Krepl, M., and Šponer, J. (2018). QM/MM calculations on protein-RNA complexes: Understanding limitations of classical MD simulations and search for reliable cost-effective QM methods. J. Chem. Theory Comput. 14, 5419–5433. doi:10.1021/acs.jctc.8b00670
Prabakaran, P., An, J., Gromiha, M. M., Selvaraj, S., Uedaira, H., Kono, H., et al. (2000). ProNIT: Thermodynamic database for protein-nucleic acid interactions [internet]. Genome Inf. 11. Available at: http://www.rtc.riken.go.jp/jouhou/3dinsight/complexdb.html.
Pyle, A. M. (1993). Ribozymes: A distinct class of metalloenzymes. Science 261 (5122), 709–714. doi:10.1126/science.7688142
Qiu, J., Bernhofer, M., Heinzinger, M., Kemper, S., Norambuena, T., Melo, F., et al. (2020). ProNA2020 predicts protein-DNA, protein-RNA, and protein-protein binding proteins and residues from sequence. J. Mol. Biol. 432 (7), 2428–2443. doi:10.1016/j.jmb.2020.02.026
Ramanathan, M., Porter, D. F., and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat. Methods 16, 225–234. doi:10.1038/s41592-019-0330-1
Ray, D., Kazan, H., Chan, E. T., Castillo, L. P., Chaudhry, S., Talukder, S., et al. (2009). Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 27 (7), 667–670. doi:10.1038/nbt.1550
Robertson, M. J., Qian, Y., Robinson, M. C., Tirado-Rives, J., and Jorgensen, W. L. (2019). Development and testing of the OPLS-AA/M force field for RNA. J. Chem. Theory Comput. 15 (4), 2734–2742. doi:10.1021/acs.jctc.9b00054
Šali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815.
Scott, L. G., and Hennig, M. (2008). “RNA structure determination by NMR,” in Methods in molecular Biology™. Editor J. M. Keith (Totowa, NJ: Humana Press), Vol. 452, 29–61. doi:10.1007/978-1-60327-159-2_2
Sharan, M., Förstner, K. U., Eulalio, A., and Vogel, J. (2017). APRICOT: An integrated computational pipeline for the sequence-based identification and characterization of RNA-binding proteins. Nucleic Acids Res. 45 (11), e96. doi:10.1093/nar/gkx137
Sharma, S., Ding, F., and Dokholyan, N. V. (2008). IFoldRNA: Three-dimensional RNA structure prediction and folding. Bioinformatics 24 (17), 1951–1952. doi:10.1093/bioinformatics/btn328
Shi, Y. Z., Wang, F. H., Wu, Y. Y., and Tan, Z. J. (2014). A coarse-grained model with implicit salt for RNAs: Predicting 3D structure, stability and salt effect. J. Chem. Phys. 141 (10), 105102. doi:10.1063/1.4894752
Shulman-Peleg, A., Nussinov, R., and Wolfson, H. J. (2009). RsiteDB: A database of protein binding pockets that interact with RNA nucleotide bases. Nucleic Acids Res. 37 (Suppl. 1), D369–D373. doi:10.1093/nar/gkn759
Song, B., Shiromoto, Y., Minakuchi, M., and Nishikura, K. (2022). The role of RNA editing enzyme ADAR1 in human disease. Wiley Interdiscip. Rev. RNA 13, e1665. doi:10.1002/wrna.1665
Steffen, P., Voss, B., Rehmsmeier, M., Reeder, J., and Giegerich, R. (2006). RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 22 (4), 500–503. doi:10.1093/bioinformatics/btk010
Su, H., Liu, M., Sun, S., Peng, Z., and Yang, J. (2019). Improving the prediction of protein-nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics 35 (6), 930–936. doi:10.1093/bioinformatics/bty756
Sugiura, R., Satoh, R., Ishiwata, S., Umeda, N., and Kita, A. (2011). Role of RNA-binding proteins in MAPK signal transduction pathway. J. Signal Transduct. 2011, 1–8. doi:10.1155/2011/109746
Sun, M., Wang, X., Zou, C., He, Z., Liu, W., and Li, H. (2016). Accurate prediction of RNA-binding protein residues with two discriminative structural descriptors. BMC Bioinforma. 17 (1), 231. doi:10.1186/s12859-016-1110-x
Sun, Z., Zheng, S., Zhao, H., Niu, Z., Lu, Y., Pan, Y., et al. (2021). To improve the predictions of binding residues with DNA, RNA, carbohydrate, and peptide via multi-task deep neural networks. IEEE/ACM Trans. Comput. Biol. Bioinform 2021, 5963(c). doi:10.1109/tcbb.2021.3118916
Suresh, V., Liu, L., Adjeroh, D., and Zhou, X. (2015). RPI-pred: Predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 43 (3), 1370–1379. doi:10.1093/nar/gkv020
Sysoev, V. O., Fischer, B., Frese, C. K., Gupta, I., Krijgsveld, J., Hentze, M. W., et al. (2016). Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nat. Commun. 7, 12128. doi:10.1038/ncomms12128
Tan, D., Piana, S., Dirks, R. M., and Shaw, D. E. (2018). RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc. Natl. Acad. Sci. U. S. A. 115 (7), E1346–E1355. doi:10.1073/pnas.1713027115
Tan, R. K. Z., Petrov, A. S., and Harvey, S. C. (2006). YUP: A molecular simulation program for coarse-grained and multiscaled models. J. Chem. Theory Comput. 2 (3), 529–540. doi:10.1021/ct050323r
Teimouri, H., and Maali, A. (2020). Single-nucleotide polymorphisms in host pattern-recognition receptors show association with antiviral responses against SARS-CoV-2, in-silico trial. JoMMID 8 (2), 65–70. doi:10.29252/jommid.8.2.65
Teng, X., Chen, X., Xue, H., Tang, Y., Zhang, P., Kang, Q., et al. (2020). NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 48 (D1), D160–D165. doi:10.1093/nar/gkz969
Tovchigrechko, A., and Vakser, I. A. (2006). GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34 (WEB. SERV. ISS.), W310–W314. doi:10.1093/nar/gkl206
Trendel, J., Schwarzl, T., Horos, R., Prakash, A., Bateman, A., Hentze, M. W., et al. (2019). The human RNA-binding proteome and its dynamics during translational arrest. Cell. 176 (1–2), 391–e19. e19. doi:10.1016/j.cell.2018.11.004
Tuvshinjargal, N., Lee, W., Park, B., and Han, K. (2016). PRIdictor: Protein-RNA interaction predictor. BioSystems 139, 17–22. doi:10.1016/j.biosystems.2015.10.004
Urdaneta, E. C., Vieira-Vieira, C. H., Hick, T., Wessels, H. H., Figini, D., Moschall, R., et al. (2019). Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 10 (1), 990. doi:10.1038/s41467-019-08942-3
Vandelli, A., Cid Samper, F., Torrent Burgas, M., Sanchez de Groot, N., and Tartaglia, G. G. (2022). The interplay between disordered regions in RNAs and proteins modulates interactions within stress granules and processing bodies. J. Mol. Biol. 434 (1), 167159. doi:10.1016/j.jmb.2021.167159
Walia, R. R., el-Manzalawy, Y., Honavar, V. G., and Dobbs, D. (2017). “Sequence-based prediction of RNA-binding residues in proteins,” in Methods in molecular biology (Totowa, NJ, USA: Humana Press), 205–235. doi:10.1007/978-1-4939-6406-2_15
Walia, R. R., Xue, L. C., Wilkins, K., El-Manzalawy, Y., Dobbs, D., and Honavar, V. (2014). RNABindRPlus: A predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLoS One 9 (5), e97725. doi:10.1371/journal.pone.0097725
Wang, H., and Zhao, Y. (2020). RBinds: A user-friendly server for RNA binding site prediction. Comput. Struct. Biotechnol. J. 18, 3762–3765. doi:10.1016/j.csbj.2020.10.043
Wang, K., Jian, Y., Wang, H., Zeng, C., and Zhao, Y. (2018). RBind: Computational network method to predict RNA binding sites. Bioinformatics 34 (18), 3131–3136. doi:10.1093/bioinformatics/bty345
Wang, L., Huang, C., Yang, M. Q., and Yang, J. Y. (2010). BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol. 4 Suppl 1 (Suppl. 1), S3–S9. doi:10.1186/1752-0509-4-S1-S3
Wang, L., You, Z. H., Huang, D. S., and Zhou, F. (2020). Combining high speed ELM learning with a deep convolutional neural network feature encoding for predicting protein-RNA interactions. IEEE/ACM Trans. Comput. Biol. Bioinf. 17 (3), 972–980. doi:10.1109/tcbb.2018.2874267
Wang, R., Alexander, R. W., Vanloock, M., Vladimirov, S., Bukhtiyarov, Y., Harvey, S. C., et al. (1999). Three-dimensional placement of the conserved 530 loop of 16 S rRNA and of its neighboring components in the 30 S subunit [internet]. Available at: http://www.idealibrary.com.
Wang, Y., Yang, Y., Ma, Z., Wong, K. C., and Li, X. (2022). EDCNN: Identification of genome-wide RNA-binding proteins using evolutionary deep convolutional neural network. Bioinformatics 38 (3), 678–686. doi:10.1093/bioinformatics/btab739
Wessels, H. H., Imami, K., Baltz, A. G., Kolinski, M., Beldovskaya, A., Selbach, M., et al. (2016). The mRNA-bound proteome of the early fly embryo. Genome Res. 26 (7), 1000–1009. doi:10.1101/gr.200386.115
Will, S., Reiche, K., Hofacker, I. L., Stadler, P. F., and Backofen, R. (2007). Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 3 (4), e65–e91. doi:10.1371/journal.pcbi.0030065
Wu, T., Wang, J., Liu, C., Zhang, Y., Shi, B., Zhu, X., et al. (2006). NPInter: The noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res. 34 (1), D150–D152. doi:10.1093/nar/gkj025
Xia, Z., Bell, D. R., Shi, Y., and Ren, P. (2013). RNA 3D structure prediction by using a coarse-grained model and experimental data. J. Phys. Chem. B 117 (11), 3135–3144. doi:10.1021/jp400751w
Xia, Z., Gardner, D. P., Gutell, R. R., and Ren, P. (2010). Coarse-grained model for simulation of RNA three-dimensional structures. J. Phys. Chem. B 114 (42), 13497–13506. doi:10.1021/jp104926t
Xie, J., Zheng, J., Hong, X., Tong, X., and Liu, S. (2020). PRIME-3D2D is a 3D2D model to predict binding sites of protein-RNA interaction. Commun. Biol. 3 (1), 384. doi:10.1038/s42003-020-1114-y
Xiong, D., Zeng, J., and Gong, H. (2015). RBRIdent: An algorithm for improved identification of RNA-binding residues in proteins from primary sequences. Proteins 83 (6), 1068–1077. doi:10.1002/prot.24806
Xu, X., Zhao, P., and Chen, S. J. (2014). Vfold: A web server for RNA structure and folding thermodynamics prediction. PLoS One 9 (9), e107504. doi:10.1371/journal.pone.0107504
Xu, Y., Vanommeslaeghe, K., Aleksandrov, A., MacKerell, A. D., and Nilsson, L. (2016). AdditiveCHARMMforce field for naturally occurring modified ribonucleotides. J. Comput. Chem. 37 (10), 896–912. doi:10.1002/jcc.24307
Yan, J., and Kurgan, L. (2017). DRNApred, fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res. 45 (10), e84–e16. doi:10.1093/nar/gkx059
Yan, Y., Zhang, D., Zhou, P., Li, B., and Huang, S. Y. (2017). HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45 (W1), W365–W373. doi:10.1093/nar/gkx407
Yan, Z., Hamilton, W. L., and Blanchette, M. (2021). Graph neural representational learning of RNA secondary structures for predicting RNA-protein interactions. Bioinformatics 36 (1), I276–I284. doi:10.1093/bioinformatics/btaa456
Yang, X., Wang, J., Sun, J., and Liu, R. (2015). SNBRFinder: A sequence-based hybrid algorithm for enhanced prediction of nucleic acid-binding residues. PLoS One 10 (7), 1–23. doi:10.1371/journal.pone.0133260
Yang, X. X., Deng, Z. L., and Liu, R. (2014). RBRDetector: Improved prediction of binding residues on RNA-binding protein structures using complementary feature- and template-based strategies. Proteins 82 (10), 2455–2471. doi:10.1002/prot.24610
Yang, Y. C. T., Di, C., Hu, B., Zhou, M., Liu, Y., Song, N., et al. (2015). CLIPdb: A CLIP-seq database for protein-RNA interactions. BMC Genomics 16 (1), 51. doi:10.1186/s12864-015-1273-2
Yu, B., Wang, X., Zhang, Y., Gao, H., Wang, Y., Liu, Y., et al. (2022). RPI-MDLStack: Predicting RNA-protein interactions through deep learning with stacking strategy and LASSO. Appl. Soft Comput. 120, 108676. doi:10.1016/j.asoc.2022.108676
Zhang, F., Zhao, B., Shi, W., Li, M., and Kurgan, L. (2022). DeepDISOBind: Accurate prediction of RNA-, DNA- and protein-binding intrinsically disordered residues with deep multi-task learning. Brief. Bioinform 23 (1). doi:10.1093/bib/bbab521
Zhang, J., Chen, Q., and Liu, B. (2021). NCBRPred: Predicting nucleic acid binding residues in proteins based on multilabel learning. Brief. Bioinform 22 (5), 1–12. doi:10.1093/bib/bbaa397
Zhang, N., Lu, H., Chen, Y., Zhu, Z., Yang, Q., Wang, S., et al. (2020). PremPRI: Predicting the effects of Missense mutations on protein-RNA interactions. Int. J. Mol. Sci. 21 (15), 1–12. doi:10.3390/ijms21155560
Zhang, S., Zhao, L., and Xia, J. (2019). SPHot: Prediction of hot spots in protein-RNA complexes by protein sequence information and ensemble classifier. IEEE Access 7, 104941–104946. doi:10.1109/access.2019.2931552
Zhang, S., Zhou, J., Hu, H., Gong, H., Chen, L., Cheng, C., et al. (2015). A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 44 (4), e32. doi:10.1093/nar/gkv1025
Zhang, X., Wu, D., Chen, L., Li, X., Yang, J., Fan, D., et al. (2014). RAID: A comprehensive resource for human RNA-associated (RNA-RNA/RNA-protein) interaction. RNA 20 (7), 989–993. doi:10.1261/rna.044776.114
Zhang, Y., and Skolnick, J. (2005). TM-Align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33 (7), 2302–2309. doi:10.1093/nar/gki524
Zhang, Y., Zhu, Y., Xiao, M., Cheng, Y., He, D., Liu, J., et al. (2021). The long non-coding RNA TMPO-AS1 promotes bladder cancer growth and progression via OTUB1-induced E2F1 deubiquitination. Front. Oncol. 11, 519. doi:10.3389/fonc.2021.643163
Zhao, X., Zhang, Y., and Du, X. (2022). DFpin: Deep learning-based protein-binding site prediction with feature-based non-redundancy from RNA level. Comput. Biol. Med. 142 (1), 105216. doi:10.1016/j.compbiomed.2022.105216
Zhao, Y., and Du, X. (2020). econvRBP: Improved ensemble convolutional neural networks for RNA binding protein prediction directly from sequence. Methods 181-182, 15–23. doi:10.1016/j.ymeth.2019.09.008
Zhao, Y., Huang, Y., Gong, Z., Wang, Y., Man, J., and Xiao, Y. (2012). Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2, 734. doi:10.1038/srep00734
Zheng, J., Kundrotas, P. J., Vakser, I. A., and Liu, S. (2016). Template-based modeling of protein-RNA interactions. PLoS Comput. Biol. 12 (9), e1005120. doi:10.1371/journal.pcbi.1005120
Zhou, T., Rong, J., Liu, Y., Gong, W., and Li, C. (2022). An ensemble approach to predict binding hotspots in protein–RNA interactions based on SMOTE data balancing and Random Grouping feature selection strategies. Bioinformatics.
Zhu, X., Liu, L., He, J., Fang, T., Xiong, Y., and Mitchell, J. C. (2020). IPNHOT: A knowledge-based approach for identifying protein-nucleic acid interaction hot spots. BMC Bioinforma. 21 (1), 289. doi:10.1186/s12859-020-03636-w
Glossary
ncRNA non-coding RNA
miRNA microRNA
piRNA Piwi-interacting RNAl
ChIP-seq Chip sequencing
HT-SELEX High throughput systematic evolution of ligands by exponential enrichment
PPrint Prediction of Protein RNA-Interaction
CLIP cross-linking immunoprecipitation
RBPmap mapping binding sites of RNA-binding proteins
RNApin Protein Interacting Nucleotides (PINs) in RNA sequences
EDCNN Evolutionary Deep Convolutional Neural Network
catRAPIDomics computation of protein–RNA interaction propensities at the transcriptome-and RNA-binding proteome
RPISeq RNA-Protein Interaction Prediction
RPIntDB database of known RNA-protein interactions
PS-PRIP partner specific protein RNA interaction prediction
PredRBR predict RNA-binding residues
mRMR-IFS Minimum Redundancy Maximal Relevance (mRMR) method followed by incremental feature selection (IFS)
PRNA Promoter-associated RNAs
RsiteDB RNA binding sites database
PST-PRNA predicting RNA-binding sites (PRNA) based on protein surface topography (PST)
RBscore RNA-/DNA-binding residues in proteins and visualizes the prediction scores
NBench Name of a benchmark database
RBRDetector RNA-Binding Residue Detector
DR_bind1 a web server for predicting DNA-binding residues from the protein structure based on electrostatics, evolution and geometry
aPRBind ab initio algorithm ab initio Protein–RNA Binding site prediction
DeepNet-RBP An algorithm using Deep belief network (DBN) to predict RBP binding sites
aaRNA aaRNA is an RNA binding site predictor
RBind A computational network method to predict RNA binding sites
RBindS A user-friendly server for RNA binding site prediction
RPI-net RNA Protein Interaction prediction using Neural Networks
NABS An algorithm for Nucleic Acid Binding Site prediction
GraphProt a flexible machine-learning framework for learning models of RBP binding preferences
RNAcompete RNA compete is an in vitro protein-centric method used to analyze RNA-protein interactions
RNAshapes an integrated RNA analysis package based on abstract shapes
DLPRB A Deep Learning Approach for Predicting Protein-RNA Binding
RPI-BIND a structure-based method for accurate identification of RNA-protein binding sites
HDock Protein-protein and protein-DNA/RNA docking based on a hybrid algorithm of template-based modeling and ab initio free docking
HDOCKlite Another version of HDock
GRAMM The Global RAnge Molecular Matching
ZDock An initial-stage protein-docking algorithm
RosettaDock Docking algorithm developed as a part of Rosetta platform
PRIME Protein-RNA Interaction ModElling program
RStrucfam a web server to associate structure and cognate RNA for RNA-binding proteins from sequence information
3dRPC 3-D RNA_protein structure prediction
HHsearch profile‐profile tool that explicitly weights a proportion of an alignment to aligned (predicted) secondary structure. MSAs can be built by either PSI‐BLAST or HHblits
EvoClustRNA A clustering routine of evolutionary conserved regions (helical regions) for RNA fold prediction
Rfam an RNA family database
ENTANGLE a JAVA program that classifies and sorts potential protein-nucleic acid interactions
mCSM-NA mutation Cutoff Scanning Matrix applied to Nucleic Acids
DeepCLIP predicting the effect of mutations on protein–RNA binding with deep learning
PremPRI Predicting the Effects of Missense Mutations on Protein-RNA Interactions
ProNIT A database for protein nucleic acid interactions
PrabHot Prediction of Protein-RNA Binding Energy Hot Spots
XGBPRH an eXtreme Gradient Boosting (XGBoost) algorithm for Protein-RNA Hotspot prediction
SREPRHot a SMOTE and Random grouping strategies-based Ensemble learning model for Protein-RNA binding Hotspot prediction
SPHot Sequence-based Prediction of RNA-binding Hot Spots
iPNHOT identifying protein-nucleic acid interaction hot spots
AMBER Assisted Model Building with Energy Refinement
CHARMM Chemistry at Harvard Macromolecular Mechanics
PRD A protein-RNA interaction database
NPInter the noncoding RNAs and protein related biomacromolecules interaction database
RNAct protein–RNA interactome
RAID RNA-associated (RNA–RNA/RNA–protein) interaction Database
CLIPdb a CLIP-seq database for protein-RNA interactions
RPI PRED RNA—Another version of RNA-protein interaction prediction algorithm
PRIDB a protein—RNA interface database
RBPDB RNA-Binding Protein DataBase
Keywords: computational prediction, RNA-protein complex, RNA-protein interaction, RPI, interface prediction, machine learning, deep learning
Citation: Bheemireddy S, Sandhya S, Srinivasan N and Sowdhamini R (2022) Computational tools to study RNA-protein complexes. Front. Mol. Biosci. 9:954926. doi: 10.3389/fmolb.2022.954926
Received: 27 May 2022; Accepted: 20 September 2022;
Published: 07 October 2022.
Edited by:
Elena Evguenieva-Hackenberg, University of Giessen, GermanyReviewed by:
Fabrizio Ferrè, University of Bologna, ItalyCopyright © 2022 Bheemireddy, Sandhya, Srinivasan and Sowdhamini. This is an open-access article distributed der the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sankaran Sandhya, c2FuZGh5YS5idC5sc0Btc3J1YXMuYWMuaW4=; Ramanathan Sowdhamini, bWluaUBuY2JzLnJlcy5pbg==
†Deceased on 03/09/2021