 Anupam Mondal
Anupam Mondal Arnab Bhattacherjee
Arnab Bhattacherjee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 05 December 2022
Sec. Biological Modeling and Simulation
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.953689
This article is part of the Research Topic Simulation of Protein Nucleic Acid Interaction View all 5 articles
DNA morphology is subjected to environmental conditions and is closely coupled with its function. For example, DNA experiences stretching forces during several biological processes, including transcription and genome transactions, that significantly alter its conformation from that of B-DNA. Indeed, a well-defined 1.5 times extended conformation of dsDNA, known as Σ-DNA, has been reported in DNA complexes with proteins such as Rad51 and RecA. A striking feature in Σ-DNA is that the nucleobases are partitioned into triplets of three locally stacked bases separated by an empty rise gap of
Protein–DNA interactions are central to several biological processes, such as DNA replication, transcription, repair, chromatin compaction, and gene regulation. Many of these processes can exert forces on DNA, such as the forces produced by DNA/RNA polymerases, as it moves over the template (Yin et al., 1995; Maier et al., 2000; Wuite et al., 2000), or the forces stemming from DNA itself for purposes including transport and tertiary structure manipulation (Nicklas, 1988). These pico-Newton (pN) scale forces exerted on DNA can affect the non-specific binding of proteins with the DNA. For instance, a range of forces applied to DNA has shown to greatly influence the binding of many DNA-binding proteins (Dahlke and Sing, 2018). Forces on DNA are also found to alter the dissociation rates of proteins from DNA (Xiao et al., 2010; Marko and Siggia, 1997; Marko, 2015) of ligands from receptors (Wu et al., 2007) and the DNA unwinding activity of hRPA protein (De Vlaminck et al., 2010). Furthermore, the mechanical properties of DNA under an applied force can potentially influence the DNA–protein interactions (Gore et al., 2006; Gemmen et al., 2006; Allemand et al., 2003). While these observations suggest that forces on DNA exist in a wide range of cellular processes, how it affects the protein–DNA recognition process remains completely unknown.
DNA can exist in various conformations under both in vivo and in vitro conditions, even in the absence of force (Alberts et al., 2008). The presence of external force alters the structure of DNA and exhibits multiple structural polymorphisms with a change in its helical parameters (Dans et al., 2012). Single-molecule pulling experiments (such as atomic force microscopy (AFM) (Clausen-Schaumann et al., 2000; Liu et al., 2010; Rief et al., 1999), optical/magnetic tweezers (Smith et al., 1996; Gosse and Croquette, 2002; Paik and Perkins, 2011), and molecular dynamics simulations (Lebrun and Lavery, 1996; Kosikov et al., 1999; Harris et al., 2005; Garai et al., 2017; Luan and Aksimentiev, 2008) revealed that when canonical B-form DNA is pulled mechanically by external stretching forces, the double-stranded DNA (dsDNA) is extended to almost 1.7 times beyond its original contour length. It has been observed that DNA undergoes an abrupt overstretching transition from B-DNA to an overstretched conformation when pulled with forces of 60–70 pN and eventually denatures at relatively higher forces (Smith et al., 1996; Bosaeus et al., 2012). The force-extension curve of dsDNA shows a characteristic plateau that indicates a highly cooperative transition from B-DNA to an overstretched ladder-like S-DNA conformation (Cluzel et al., 1996; Luan and Aksimentiev, 2008). The structure of S-DNA is usually a partly untwisted ladder in which the base-pairing is preserved, but the average rise per base-pair is increased as compared to unstretched B-DNA. The overstretched conformation of DNA may play an important role in the function of proteins that are able to make use of these structural properties of DNA. For example, the X-ray crystal structure of RecA protein bound to the genomic DNA reveals a superhelical topology in which the dsDNA is underwound and stretched to approximately 50% in length relative to canonical B-DNA (Chen et al., 2008). Such a nucleoprotein complex, thus, presents a situation where the conformational change in DNA can be coupled with an external force. A striking observation in it is that the extended DNA adopts a configuration where the nucleobases are partitioned into orderly stacked triplets of base-pairs separated by a large rise gap. This form of DNA structure is physically plausible when considered purely in static chemical terms (Bertucat et al., 1998) and is previously referred (Bosaeus et al., 2017; Taghavi et al., 2017) to as “Σ-DNA,” which may be considered a special case of overstretched S-form DNA. The degree of extension observed in Σ-form DNA has been identified even in the absence of recombinase proteins by using single-molecule force spectroscopy experiments (Bosaeus et al., 2012) and considered the most stable conformation without any cofactors. Even the presence of DNA-binding cofactors or intercalators has been observed to form a triplet kind of stable disproportionate DNA structure in solution through atomistic simulations of duplex DNA under an applied force (Taghavi et al., 2017). In a recent study, Bosaeus et al. (2017) analyzed the possible biological role of Σ-phase DNA and speculated that partitioning of bases into triplets separated by a stack-breaking gap may have a connection with the physical origin of the existence of three letters (codon/anti-codon) in the genetic code. Thus, one may ask whether the structural distortion in Σ-DNA is just a coincidence of protein binding or somehow the appearance of these structures is related to any biological function. Does this triplet structures have any connection in enhancing the recognition of base sequences by DNA-binding proteins? How do these structural changes in DNA under tension affect its interaction with the protein?
In order to understand these issues, we studied protein diffusion on force-induced stretched DNA using coarse-grained molecular dynamics simulations. Previously, many studies have probed the diffusion of a DNA-binding protein on unstretched DNA, in particular, the target search process on DNA by proteins. Several experimental (Blainey et al., 2009; Iwahara and Clore, 2006a, Iwahara and Clore, 2006b; Esadze and Iwahara, 2014; Elf et al., 2007; Clore, 2011), simulation (Marklund et al., 2013; Guardiani et al., 2014; Mondal and Bhattacherjee, 2015; Givaty and Levy, 2009; Bhattacherjee et al., 2016; Bhattacherjee and Levy, 2014a, Bhattacherjee and Levy, 2014b), and theoretical (Kolomeisky and Veksler, 2012; Mirny et al., 2009; von Hippel and Berg, 1989; Veksler and Kolomeisky, 2013; Mondal and Bhattacherjee, 2017) studies suggest that proteins associate their target sites orders of magnitude faster than a simple 3D diffusion-limited process. The mechanism behind this rapid translocation of proteins is due to the idea of facilitated diffusion, where the protein exhibits 1D diffusion along DNA in combination with 3D diffusion in the bulk. In 1D events, when the protein is associated non-specifically with DNA, it can move stochastically from one base to another along the contour of DNA (sliding). When detached from the DNA surface, protein can re-associate to a nearby site (hopping) after a short time period or can jump to a distant DNA segment (intersegmental transfer) (Mondal et al., 2022b). In our study, we pulled the DNA molecule by applying constant force to the 3′-3′ end of DNA and observed that DNA undergoes a conformational transition from B-DNA to an overstretched ladder-like S-DNA conformation via an intermediate Σ-DNA transition. Our analysis suggests that the resulting stretched DNA conformations feature non-uniform clusters of nucleotide bases such as doublets, triplets, and quadruplets separated by a large rise gap. Among these base-pair clusters, triplets are more stable than others, which protein favors to form during its 1D translocation along DNA, and the relative population of base-pair triplets governs the ruggedness of the protein–DNA binding energy landscape and thereby the ability of a protein to locate its target DNA sites. By analyzing different force regimes, we delineate the underlying translocation mechanism of a DNA-binding protein and underscore the significance of triplet formation in regulating the protein diffusion on stretched DNA.
In this work, we adopt coarse-grained models of protein and DNA to study protein diffusion along force-induced stretched DNA. The protein is represented by one bead per amino acid located at the respective Cα position (Bhattacherjee et al., 2016). The energetic of the protein is described by a structure-based Lennard–Jones potential that ensures the formation of native contacts found in the folded structure of the protein (Clementi et al., 2000b). Such a reduced model promises great advantages in studying the funnel-like energy landscape of protein folding (Clementi et al., 2000a), protein–protein interactions (Bhattacherjee and Wallin, 2012), and multiple basins free energy landscape for large-scale motion of proteins (Okazaki et al., 2006). For DNA, we use a 3SPN.2C coarse-grained model developed by de Pablo’s group in which each nucleotide is described by three beads and is positioned at the geometric centers of the phosphate, sugar, and base atoms (Freeman et al., 2014a). The 3SPN.2C model accurately estimates the correct structural properties of DNA such as helix width, base-pair rise, number of base-pair per turn, major and minor groove widths which are in good agreement with experimental results (Hinckley et al., 2013). The model successfully reproduces the mechanochemical properties of DNA such as sequence-dependent persistence length and flexibility of double-stranded DNA, prediction of melting temperature, and estimation of DNA hybridization rate constants for varying sequences under different ionic concentrations. Most importantly, the 3SPN.2C DNA model is developed by Freeman et al. (2014a) with the intention of studying interactions between protein and dsDNA only, and a special emphasis is placed on successfully capturing the effects of sequence-dependent DNA shape, local flexibilities, dsDNA persistence lengths, melting temperatures, and minor groove width profiles, which are extensively validated against experimental data. By incorporating these relevant DNA properties, the authors have recently identified the structural properties most critical to nucleosome formation (Freeman et al., 2014b) and found that histone– DNA binding affinity is encoded in its sequence-dependent shape, including subtle deviations from the ideal linear B-DNA geometry. Indeed, previous studies support that even nuances in the groove width and local variations in minor groove width play a significant role in its interaction with the proteins (Bhattacherjee and Levy, 2014a). A similar DNA model (3SPN.2C) has been extensively used previously by us (Mondal et al., 2022a, Mondal et al., 2022b) and other groups (Mondal et al., 2022a, Mondal et al., 2022b; Nagae et al., 2021; Brandani et al., 2021; Lin et al., 2021; Tan and Takada, 2020; Moller et al., 2019; Lequieu et al., 2017; Niina et al., 2017; Lequieu et al., 2016) to demonstrate the role of DNA shape dynamics and flexibility in regulating the interactions with proteins.
Interactions between protein and DNA molecules are treated at a non-specific level and are driven by electrostatic interactions and excluded volume effects. Excluded volume interactions are modeled by an r12 repulsive potential that penalizes the steric clashes during the non-specific encounter between them over the simulation. The electrostatic interactions between positively and negatively charged residues of protein and DNA are modeled by the Debye–Hückel potential. A negative charge of −0.6 is assigned to each phosphate atom to take the counterion condensation effect, and a net charge of +1 is assigned to lysine (Lys) and arginine (Arg) and − 1 for glutamate (Glu) and aspartate (Asp) residues. It is to note that the Debye–Hückel theory provides the advantage of introducing the effect of salt concentrations in the present model and thus enables us to investigate protein diffusion on double-stranded DNA in an ionic environment. Although the theory is limited to only dilute salt concentration, previously, the Debye–Hückel theory has been successfully applied to shed light on many crucial aspects of protein–DNA interactions (Mondal and Bhattacherjee, 2015; Bhattacherjee and Levy, 2014a, Bhattacherjee and Levy, 2014b; Dey and Bhattacherjee, 2018, Dey and Bhattacherjee, 2019a, Dey and Bhattacherjee, 2019b, Dey and Bhattacherjee, 2020; Mondal et al., 2022a, Mondal et al., 2022b).
The DNA molecule is subjected to constant stretching forces −F and +F applied to the 3′ termini. The stretching force is kept fixed during the simulation to investigate the large-scale motion of protein diffusion on a stretched DNA. The value of the constant stretching force varied in the range of 0–200 pN. A zero value of stretching force signifies that there is no external force acting on DNA. The DNA molecule is stretched along the Z-direction, and therefore, the energy function used for DNA stretching is given as follows:
To probe the dynamics of protein search on DNA, we choose Sap-1 protein from Protein Data Bank (PDB ID: 1BC8) as the searching protein. This DNA-binding protein is a 93-residue long globular protein consisting of 15 positively and six negatively charged amino acids and contains a winged-helix recognition region to scan DNA. The starting sequence-dependent structure of 100-bp B-DNA is generated from X3DNA (Zheng et al., 2009). The DNA sequence is given in Supplementary Figure S1 that contains 67% GC-content and 33% AT-content. The choice of this GC-rich sequence is inspired by the recent experimental studies (Bosaeus et al., 2012, Bosaeus et al., 2017). Both the molecules are placed at the center of a simulation box of dimension 350 Å × 350 Å × 800 Å. DNA is placed along the Z-axis, and the periodic boundary conditions are applied in all the directions. Such box dimensions are chosen in order to ensure that fully extended (
In order to understand the structure and dynamics of force-induced stretched DNA, we first probe the force–strain response of dsDNA. We apply constant stretching/pulling force on 3′ termini of the two strands of a randomly selected 100-bp GC-rich B-DNA molecule (see Figure 1A) containing 67% GC-content and 33% AT-content and perform coarse-grained molecular dynamics (MD) simulations. The constant stretching force is varied from 0 pN to 200 pN, and we present the force vs. strain curve in Figure 1B, where the strain is the ratio of the change in the end-to-end distance of the 3′-3′ ends of the two strands to its equilibrium contour length. We find that the stretching behavior of dsDNA in Figure 1B can be divided into three regimes. At forces F < 110 pN, DNA gets stretched by 10%–15% to its initial contour length. In this force regime, the slope of the curve has increased slightly, and DNA exists in its B-form conformation. However, there is a sharp increase in the slope from 110 pN up to about 150 pN where dsDNA starts to deform its helical structure (see Supplementary Video S1) and achieves 20%–60% extension compared to B-DNA and adopts a special kind of conformation referred to as Σ-DNA, which is stretched to about 50% in length compared to canonical B-DNA. Such a degree of extension is found in DNA complexed with recombinase proteins RecA and Rad51 (Chen et al., 2008; Xu et al., 2017). Indeed, a similar degree of extension is also observed in force-induced stretched GC-rich duplex DNA molecules (Bosaeus et al., 2012, Bosaeus et al., 2017). It is, however, known that the nature of the overstretching transition depends critically on the nature of the bp composition. For instance, in the experimental study of Bosaeus et al. (2012) and Bosaeus et al. (2017), the authors have chosen 60–64-bp short DNA molecules with base sequences containing 60% GC-content and 70% AT-content and observed that when the AT content is high (70%), the DNA molecule tends to overstretch via denaturation. Therefore, AT-rich sequences are not desirable for studying protein diffusion on stretched DNA. In contrast, GC-rich sequences (60% GC) are found to undergo a conformational transition from B-DNA to a distinct form of overstretched DNA that is characterized by a 51% extension and remains base-paired. Higher-level coarse-grained modeling studies (Manghi et al., 2012) tend to confirm this scenario. This observation suggests that the DNA sequence is an important parameter for controlling the transition from B-form to stretched and/or melted forms. In the present study, we consider a DNA sequence with the GC-content comparable to the one used in previous experimental studies to specifically investigate the protein diffusion on stretched DNA.
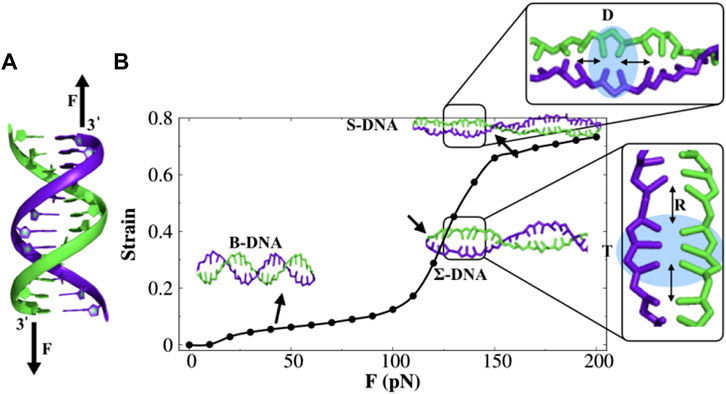
FIGURE 1. Double-stranded DNA (dsDNA) under constant stretching force. (A) Force is applied to the 3′ ends of both the strands, shown by black arrows. (B) Force–strain curve of dsDNA. With the increase in force, dsDNA undergoes a structural transition from B-DNA to S-DNA via an intermediate transition, referred to as Σ-DNA. The highlighted region in both the S-DNA and Σ-DNA conformations shows the appearance of stacked doublets (D) and triplets (T) of nucleobases separated by an empty gap of length R. The error bar for each symbol is defined as the standard error. The associated error bars are smaller than the point size.
Furthermore, increasing the force (above 150 pN) would lead to a plateau region, where the dsDNA extended 70%–80% from its initial contour length and emerged a ladder-like overstretched conformation, the so-called S-DNA. Previously, the existence of such an S-DNA structure has been found using experimental (Smith et al., 1996; Cluzel et al., 1996) and theoretical studies (Lai and Zhou, 2003). Some typical snapshots of different DNA structures at various stretching forces are provided in Supplementary Figure S2. We also monitored the changes in DNA length during the simulation and evaluated the kinetics of the structural transition between different forms of DNA at individual forces. The results are presented in Supplementary Figure S3. The result shows that the structural transition appears much faster with an increasing stretching force. For instance, the transition from B-DNA to Σ-DNA (50% extension) is ∼ 12.4 times faster at force F = 180 pN than that at force F = 140 pN, and the transition from Σ-DNA to S-DNA (70% extension) appears to be ∼ five times faster at F = 190 pN than that at F = 170 pN.
The overall behavior of the force–strain curve is consistent with the experimentally observed force–strain curve reported previously (Smith et al., 1996; Cluzel et al., 1996; Danilowicz et al., 2009). It is interesting to note that the bases in the overstretched conformation of DNA are organized in the form of doublets, triplets, quadruplets, and so on, followed by a rise gap of length R (see Figure 1B). Recently, the existence of a cluster of three bases (triplet) in Σ-DNA was reported, and the possible biological role of it was explained (Bosaeus et al., 2017). The formation of base triplets has been hypothesized to be correlated with the origin of a genetic code (Bosaeus et al., 2017). This raises a question of whether such triplets promote easier access to the DNA base-pairs by DNA-binding proteins (DBPs).
Before proceeding with investigating the same, we first characterize the structural details of the base-pair clusters under constant stretching force. In Figure 2A, we consider the two helical parameters, namely, the helical rise and DNA twist angle, and present their variation under constant stretching force. At forces F < 110 pN, DNA remains in its B-form structure, where the average rise (
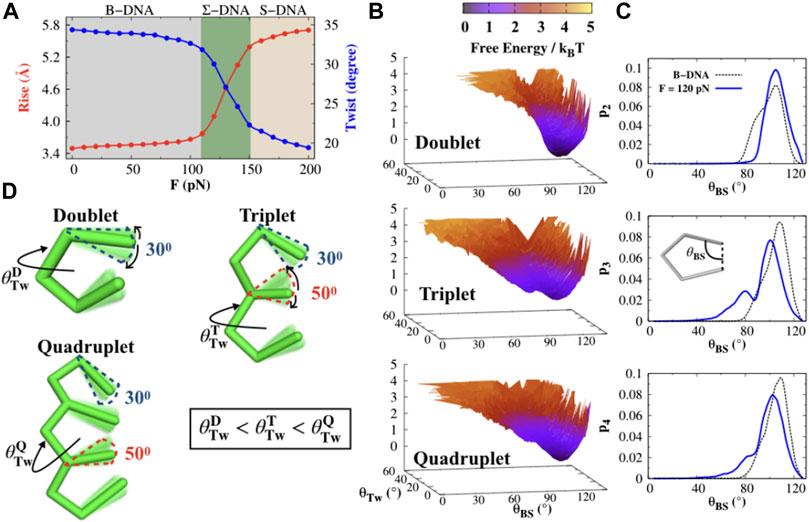
FIGURE 2. Structural characterization of the DNA bases. (A) DNA helical rise and twist angles are calculated as a function of stretching force F. The three shaded regions gray, green, and yellow correspond to three different DNA conformations, namely, B-, Σ-, and S-DNA that emerge due to stretching. The error bar for each symbol is defined as the standard error. The associated error bars are smaller than the point size. (B) Free energy profiles F(θTw, θBS) for the doublet, triplet, and quadruplet at force F = 120 pN, where θTw and θBS are the helical twist and base-stacking angles. (C) Probability histograms of the base-stacking angles (θBS) for the doublet (p2), triplet (p3), and quadruplet (p4) at a stretching force F = 120 pN (blue solid line) compared with B-DNA (dashed line). (D) Schematic representation of the structure of the doublet, triplet, and quadruplet. The twist angles for the doublet
Having seen the correct two-state transition, we observe that the main variation in the rise and twist angle arises when dsDNA adopts the Σ-form conformation. In this transition, the backbone of DNA gets extended by about 50% in length, which favors the formation of base-pair clusters such as doublets, triplets, and quadruplets. Due to the formation of such base-pair clusters, the consecutive groups of stacked bases are separated (by
We select Sap-1 protein as a representative DNA-binding protein since it has been widely studied both computationally (Bhattacherjee and Levy, 2014a; Mondal and Bhattacherjee, 2015; Dey and Bhattacherjee, 2018, 2019a, 2020) and experimentally (Mo et al., 1998; Guan et al., 2017) and investigate what effect does it bring on the formation of base-pair clusters while diffusing on stretched DNA. In Figure 3, we estimate the probability of forming doublets (p2), triplets (p3), and quadruplets (p4) in the presence and absence of Sap-1 protein as a function of constant stretching force F ranging from 0 to 200 pN. The same for higher-order base-pair clusters is shown in Supplementary Figure S6. In the presence of Sap-1 protein, the doublets (or triplets or quadruplets) are identified according to the following condition: first, we identify the closest DNA base-pair i from the recognition region of the Sap-1 protein when it diffuses one-dimensionally along the DNA surface and consider the segment (i − 6, i + 6). Within this segment, if the two (or three or four) consecutive nucleobases are separated by an empty gap of length 5 Å, then it will be considered forming a doublet (or triplet or quadruplet). The DNA segment (i − 6, i + 6) is particularly chosen, mainly because of the fact that a DNA segment spanning i − 6 to i + 6 around the protein-binding site covers a complete helical turn of 10.5 bp along DNA. Sap-1 protein binds specifically with a 9-bp short DNA stretch (almost a complete helical turn) through its recognition helix inside the DNA major groove. The choice, therefore, directly allows us to track the formation of base-pair clusters (from doublets to decuplets) along DNA when the protein scans the DNA bases through sliding/hopping modes, i.e., one-dimensionally. The same analysis performed on the protein-excluded region, i.e., the region other than the segment (i − 6, i + 6), is considered for analyzing the formation of different base-pair clusters in the absence of the protein. Following this procedure, we counted the number of doublets, triplets, and so on from the whole simulation trajectory, and the corresponding probability is calculated from the definition prescribed in the Supplementary Material. At forces F < 110 pN, protein favors the formation of doublets, triplets, and quadruplets compared to its unbound form (Figures 3A–C), and a reverse trend is observed for higher-order base-pair clusters (Supplementary Figure S6). When Σ-DNA and S-DNA emerge (F > 110 pN), we observe that only p2 and p3 increase with increasing F and then saturate (Figures 3A,B). Other probabilities (from p4 to p10) exhibit a decreasing trend with F (Figure 3C and Supplementary Figure S6), mainly because of the too-high backbone strain for higher-order base-pair clusters. Another important observation in this force regime is that the triplet formation propensity is facilitated by the presence of Sap-1 protein compared to its unbound form (Figure 3B). This feature is observed only for triplets (and not in doublets or quadruplets) and is the most important result of our study. Also, it is worthwhile to mention here that the doublet propensity at forces F > 110 pN is much higher than the triplet propensity; however, in comparison to its unbound form, the protein favors the base-pair triplets to form in this force regime while diffusing one-dimensionally along DNA. In Figure 3D, we have shown a plot that combines the probability of all different base-pair clusters (from p2 to p10) with respect to F. The color bar represents the differences in the probability in the presence and absence of the protein. Having seen the analysis clearly indicating that the formation of base triplets is favored by the diffusing protein relative to its unbound form under constant stretching force, we next move to explore the impact of such base clusters in regulating the accessibility of DNA bases to the searching protein.
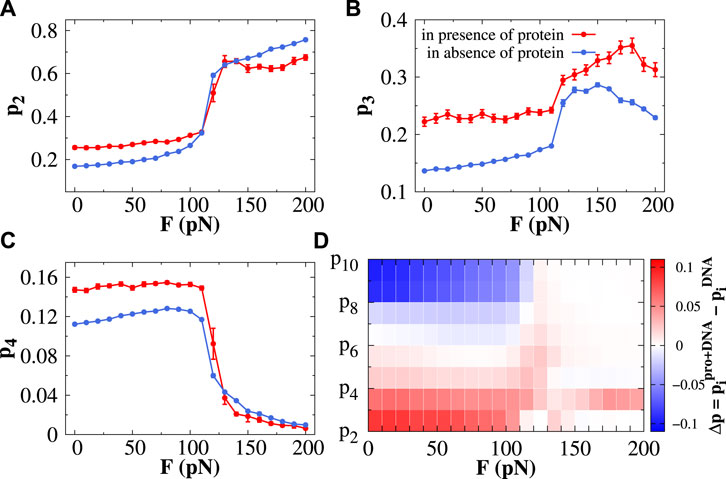
FIGURE 3. Effect of protein dynamics in the formation of base-pair clusters. The probability of forming the (A) doublet (p2), (B) triplet (p3), and (C) quadruplet (p4) is plotted as a function of stretching force in the presence (red line) and absence (blue line) of protein. The error bar for each symbol is defined as the standard error. (D) Presented contour plot combines the probability of forming all kinds of clusters starting from the doublet (p2) and triplet (p3) to decuplet (p10) with respect to stretching force, where the color bar represents the differences in the probabilities of forming different clusters in the presence and absence of protein.
To address the issue, we measure the heterogeneity of DNA base-pair clusters by estimating the Shannon entropy as a function of stretching force F from the associated probabilities of different clusters (see Figure 4A). The Shannon entropy (H) is defined as
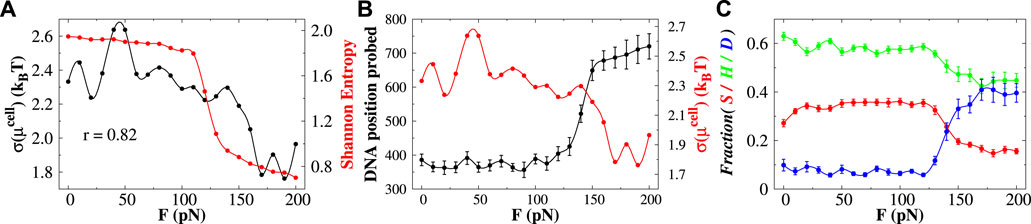
FIGURE 4. Effect of base-pair clusters on the diffusion of protein. (A) Correlations between the Shannon entropy H (denoted by the red line) and the roughness (standard deviation of μcell) of the chemical potential landscape (indicated by the black line) with the stretching force. The value of r indicates the correlation coefficient between them. (B) DNA position probed (black line) and the roughness of the chemical potential (red line) as a function of stretching force. (C) Effect of stretching force on the interplay between sliding (S), hopping (H), and 3D diffusion (D) by the searching protein. The error bar for each symbol is defined as the standard error. In (A) and the red curve in (B), the associated error bars are smaller than the point size.
Eq. 1 gives information about the distribution of the base-pair clusters—whether they occur with an equal probability (high entropy value) or whether some other clusters prevail (low entropy value). It, however, does not reflect the correlations between different base-pair clusters. At forces F < 110 pN, the Shannon entropy values are high and remain roughly constant, suggesting similar probability distribution of different base-pair clusters. At forces F > 110 pN (when both Σ-DNA and S-DNA arise), the H-value sharply decreases, indicating that the formation of some base-pair clusters prevails. Since the probability of doublets and triplets shows a higher probability (Figure 3) among which protein favors the formation of base triplets compared to its unbound form in this force regime, the information on the formation of base-pair triplets might have some connection with the diffusivity of the searching protein. To quantify the effect, we estimate the ruggedness of the potential energy landscape by measuring its roughness. We follow the method prescribed by Putzel et al. (2014) and consider the position of the center of mass of the recognition region of the Sap-1 protein as the order parameter. We partition the space into cubic cells of size 50 Å3 and measure the probability of the cell i to be occupied by the diffusing protein. As the searching protein moves between different cells, it will experience an effective potential equal to the excess contribution to the diffusing site’s chemical potential
where
Having seen the diffusion efficiency, we now move to understand the non-specific target search mechanism of the searching protein. By analyzing the structural details of each snapshot generated during the simulations, we estimate the propensities of different translocation modes such as the sliding, hopping, and 3D diffusion as a function of the applied force F, and the results are presented in Figure 4C. The detailed criteria of each of these search modes are described in the Supplementary Text. Our result suggests that at forces F < 110 pN, the propensity of each search mode remains constant. In this force regime, the Sap-1 protein mostly performs sliding and hopping dynamics on DNA and performs less 3D diffusion
In Figure 5A, we present the average number of sliding events
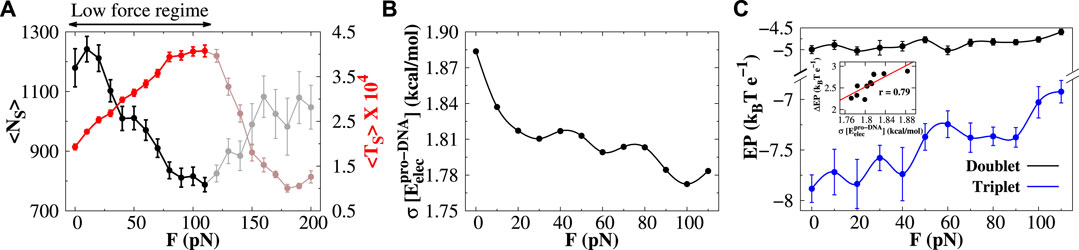
FIGURE 5. Protein diffusion under low stretching force. (A) Variations in the average number of sliding events (
In contrast to the low-force regime, the average number and duration of sliding events increase and decrease, respectively, in the high-force regime (highlighted portion in Figure 6A). Unlike the low-force regime, where the electrostatic potential energy surface for triplets regulates the sliding of protein, the mechanism for protein sliding in the high-force regime is not dependent on such potential. We find that the EP energy along the DNA contour for doublets and triplets is both invariant in the high-force regime (see Supplementary Figure S10). Rather we observe that with increasing force F, the protein–DNA electrostatic energy
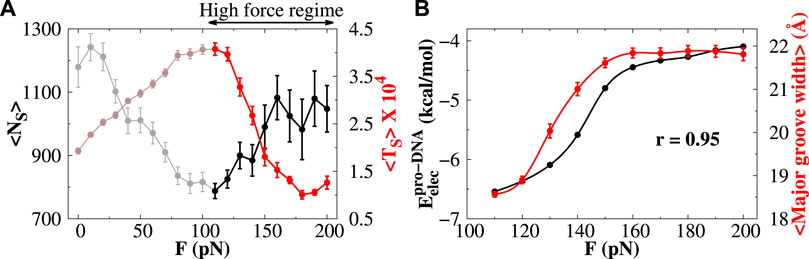
FIGURE 6. Protein diffusion under high stretching force. (A) Variations in the average number of sliding events (
While the recognition of DNA base sequences by proteins on unstretched DNA, in transcription and translation contexts, has been extensively studied in past decades, much remains to unveil about the role of extension of DNA in the nucleobase recognition by proteins. Several experimental and theoretical studies have shown that dsDNA exhibits a remarkable mechanical transition when it is stretched by an external force. When canonical B-form DNA is stretched mechanically at forces ranging 60–70 pN, its length gets elongated up to 70% to its initial contour length, and a structural transition from B-DNA into an overstretched S-DNA conformation was observed. During the transition, the existence of an overstretched Σ-DNA conformation is identified recently using a laser-tweezers force-spectroscopy experiment, where the degree of extension in Σ-form DNA is observed to be 50% extended compared to that in B-DNA. Such a Σ-DNA structure is also found in the presence of recombinase proteins. This Σ-form DNA may be considered a special case of stretched S-DNA, where the bases in Σ-DNA appear in a specific arrangement. Σ-DNA consists of stacked triplets of nucleobases, and these triplets are separated by a large rise gap. The appearance of these base triplets in stretched nucleic acids might have a biological role in enhancing the recognition of base sequences, as speculated and hypothesized recently (Bosaeus et al., 2017). To understand the connection between triplet formation and base recognition by proteins, we investigate the diffusion of a protein molecule on stretched DNA. We choose a DNA length of 100 bp, which is smaller than the DNA persistence length. For DNA longer than its persistence length, the flexibility of DNA and the partition of base-pairs are expected to be more prominent, although subjected to a detailed study. Here, we use the latest 3SPN.2C coarse-grained model of DNA to characterize the DNA dynamics, where the DNA is subjected to a constant stretching force applied to both of its 3′ ends. It is important to mention here that the 3SPN.2C DNA model has not been applied previously in the context of force-induced DNA stretching. Recently, the 3SPN.2C DNA model has been applied for estimating a small-magnitude force-dependent free energy profile of nucleosome unwrapping in which a constant force was applied to each end of DNA and nucleosomal DNA was pulled from both ends until the nucleosomal DNA unwraps from the histone surface (Lequieu et al., 2016). However, the applicability of the 3SPN.2C model is not restricted to the canonical B-DNA conformation. In a recent study, we used the same DNA model to investigate the diffusion of a protein molecule on supercoiled DNA structures with different degrees of supercoiling (Mondal et al., 2022b), suggesting the generality of the DNA model. Nevertheless, we validated the suitability of the DNA model for studying force-induced extension both qualitatively and quantitatively against the available experimental observations. The constant forces are varied between 0 and 200 pN. We find that when the dsDNA is stretched under constant force, the force–strain curve exhibits a structural transition from B-DNA to S-DNA conformation via an intermediate Σ-DNA state. The force–strain profile is reminiscent to the previously reported experimental observations. Our coarse-grained simulation results quantitatively describe the DNA helical parameters such as twist and rise in overstretched DNA conformations, which are consistent with the previous all-atom simulation results. Moreover, the DNA base-stacking angles in 50% stretched Σ-DNA conformation match quantitatively with the values obtained from the crystal structure of the RecA–dsDNA complex. Depending on the type of base-stacking and base-pairing interactions, different local conformations are observed which are in good agreement with the local conformations obtained from atomistic simulations. Furthermore, the resulting stretched DNA conformations show the clustering of stacked nucleobases such as doublets, triplets, and quadruplets, among which the triplet structures are found to be the most stable structure among doublets and quadruplets. This result is in qualitative agreement with the experimental measurements by Bosaeus et al. (2017). When the protein diffuses one-dimensionally along DNA, it favors the formation of base triplets in all force regimes compared to its unbound form. The relative population of base triplets reduces the ruggedness of the protein–DNA binding energy surface and results in the faster diffusion of proteins. Furthermore, we also underscore the mechanistic details of the diffusion dynamics of proteins in both low- and high-force regimes. In the low- and high-force regimes, we observed completely opposite behavior in the average number of sliding events and their time durations. In the low-force regime, the number of sliding events shows a decreasing pattern with force F. Analyzing the electrostatic potential in the triplet containing DNA structures, we find that the base-pair triplet reduces the ruggedness of the protein–DNA electrostatic potential surface on which the protein can smoothly perform sliding dynamics and spend higher time during sliding, which usually retards the target search efficiency. In contrast, the number of sliding events exhibits an increasing trend in the high-force regime, the rationale of which arises from the increased protein–DNA electrostatic energy due to the increased separation distance between phosphate groups, which helps the protein not to slide too long on DNA, and a right blend of the combination of 1D and 3D search modes accelerates the diffusing protein for the faster recognition of DNA bases on stretched DNA. Our study provides the plausible rationale behind the advantage of the formation of DNA base triplets that can be observed under in vitro conditions, the knowledge of which would be beneficial to understanding crucial DNA metabolic processes. Nevertheless, suitable experimental methods are warranted to validate our results and establish a more direct relationship between DNA base-pair partitions and flow of genetic information.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
AM and AB designed the research, performed the research, analyzed the data, and wrote the manuscript.
The research was supported by the Department of Science and Technology, India (DST SERB CRG/2019/001001 and MTR/2020/000664).
We gratefully acknowledge the financial support from DST India (DST SERB CRG/2019/001001 and MTR/2020/000664) and DBT BIC research grant. AM acknowledges the financial support from CSIR, India, in the form of a Senior Research Fellow (09/263(1119)/2017-EMR-I).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.953689/full#supplementary-material
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2008). Molecular biology of the cell. New York: Garland Science.
Allemand, J. F., Bensimon, D., and Croquette, V. (2003). Stretching DNA and RNA to probe their interactions with proteins. Curr. Opin. Struct. Biol. 13, 266–274. doi:10.1016/s0959-440x(03)00067-8
Bertucat, G., Lavery, R., and Prévost, C. (1998). A model for parallel triple helix formation by RecA: Single-single association with a homologous duplex via the minor groove. J. Biomol. Struct. Dyn. 16, 535–546. doi:10.1080/07391102.1998.10508268
Bhattacherjee, A., Krepel, D., and Levy, Y. (2016). Coarse-grained models for studying protein diffusion along DNA. WIREs. Comput. Mol. Sci. 6, 515–531. doi:10.1002/wcms.1262
Bhattacherjee, A., and Levy, Y. (2014a). Search by proteins for their DNA target site: 1. The effect of DNA conformation on protein sliding. Nucleic Acids Res. 42, 12404–12414. doi:10.1093/nar/gku932
Bhattacherjee, A., and Levy, Y. (2014b). Search by proteins for their DNA target site: 2. The effect of DNA conformation on the dynamics of multidomain proteins. Nucleic Acids Res. 42, 12415–12424. doi:10.1093/nar/gku933
Bhattacherjee, A., and Wallin, S. (2012). Coupled folding-binding in a hydrophobic/polar protein model: Impact of synergistic folding and disordered flanks. Biophys. J. 102, 569–578. doi:10.1016/j.bpj.2011.12.008
Blainey, P. C., Luo, G., Kou, S. C., Mangel, W. F., Verdine, G. L., Bagchi, B., et al. (2009). Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 16, 1224–1229. doi:10.1038/nsmb.1716
Bosaeus, N., El-Sagheer, A. H., Brown, T., Smith, S. B., Akerman, B., Bustamante, C., et al. (2012). Tension induces a base-paired overstretched DNA conformation. Proc. Natl. Acad. Sci. U. S. A. 109, 15179–15184. doi:10.1073/pnas.1213172109
Bosaeus, N., Reymer, A., Beke-Somfai, T., Brown, T., Takahashi, M., Wittung-Stafshede, P., et al. (2017). A stretched conformation of DNA with a biological role? Q. Rev. Biophys. 50, e11. doi:10.1017/S0033583517000099
Brandani, G. B., Tan, C., and Takada, S. (2021). The kinetic landscape of nucleosome assembly: A coarse-grained molecular dynamics study. PLoS Comput. Biol. 17, e1009253. doi:10.1371/journal.pcbi.1009253
Chen, Z., Yang, H., and Pavletich, N. P. (2008). Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453, 489–484. doi:10.1038/nature06971
Clausen-Schaumann, H., Rief, M., Tolksdorf, C., and Gaub, H. E. (2000). Mechanical stability of single DNA molecules. Biophys. J. 78, 1997–2007. doi:10.1016/S0006-3495(00)76747-6
Clementi, C., Jennings, P. A., and Onuchic, J. N. (2000a). How native-state topology affects the folding of dihydrofolate reductase and interleukin-1beta. Proc. Natl. Acad. Sci. U. S. A. 97, 5871–5876. doi:10.1073/pnas.100547897
Clementi, C., Nymeyer, H., and Onuchic, J. N. (2000b). Topological and energetic factors: What determines the structural details of the transition state ensemble and ”en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 298, 937–953. doi:10.1006/jmbi.2000.3693
Clore, G. M. (2011). Exploring translocation of proteins on DNA by NMR. J. Biomol. NMR 51, 209–219. doi:10.1007/s10858-011-9555-8
Cluzel, P., Lebrun, A., Heller, C., Lavery, R., Viovy, J. L., Chatenay, D., et al. (1996). Dna: An extensible molecule. Science 271, 792–794. doi:10.1126/science.271.5250.792
Dahlke, K., and Sing, C. E. (2018). Force-extension behavior of DNA in the presence of DNA-bending nucleoid associated proteins. J. Chem. Phys. 148, 084902. doi:10.1063/1.5016177
Danilowicz, C., Limouse, C., Hatch, K., Conover, A., Coljee, V. W., Kleckner, N., et al. (2009). The structure of DNA overstretched from the 5′5′ ends differs from the structure of DNA overstretched from the 3′3′ ends. Proc. Natl. Acad. Sci. U. S. A. 106, 13196–13201. doi:10.1073/pnas.0904729106
Dans, P. D., Pérez, A., Faustino, I., Lavery, R., and Orozco, M. (2012). Exploring polymorphisms in B-DNA helical conformations. Nucleic Acids Res. 40, 10668–10678. doi:10.1093/nar/gks884
De Vlaminck, I., Vidic, I., van Loenhout, M. T., Kanaar, R., Lebbink, J. H., and Dekker, C. (2010). Torsional regulation of hRPA-induced unwinding of double-stranded DNA. Nucleic Acids Res. 38, 4133–4142. doi:10.1093/nar/gkq067
Dey, P., and Bhattacherjee, A. (2019a). Disparity in anomalous diffusion of proteins searching for their target DNA sites in a crowded medium is controlled by the size, shape and mobility of macromolecular crowders. Soft Matter 15, 1960–1969. doi:10.1039/c8sm01933a
Dey, P., and Bhattacherjee, A. (2019b). Mechanism of facilitated diffusion of DNA repair proteins in crowded environment: Case study with human uracil DNA glycosylase. J. Phys. Chem. B 123, 10354–10364. doi:10.1021/acs.jpcb.9b07342
Dey, P., and Bhattacherjee, A. (2018). Role of macromolecular crowding on the intracellular diffusion of DNA binding proteins. Sci. Rep. 8, 844. doi:10.1038/s41598-017-18933-3
Dey, P., and Bhattacherjee, A. (2020). Structural basis of enhanced facilitated diffusion of DNA-binding protein in crowded cellular milieu. Biophys. J. 118, 505–517. doi:10.1016/j.bpj.2019.11.3388
Elf, J., Li, G. W., and Xie, X. S. (2007). Probing transcription factor dynamics at the single-molecule level in a living cell. Science 316, 1191–1194. doi:10.1126/science.1141967
Esadze, A., and Iwahara, J. (2014). Stopped-flow fluorescence kinetic study of protein sliding and intersegment transfer in the target DNA search process. J. Mol. Biol. 426, 230–244. doi:10.1016/j.jmb.2013.09.019
Freeman, G. S., Hinckley, D. M., Lequieu, J. P., Whitmer, J. K., and de Pablo, J. J. (2014a). Coarse-grained modeling of DNA curvature. J. Chem. Phys. 141, 165103. doi:10.1063/1.4897649
Freeman, G. S., Lequieu, J. P., Hinckley, D. M., Whitmer, J. K., and de Pablo, J. J. (2014b). DNA shape dominates sequence affinity in nucleosome formation. Phys. Rev. Lett. 113, 168101. doi:10.1103/PhysRevLett.113.168101
Garai, A., Mogurampelly, S., Bag, S., and Maiti, P. K. (2017). Overstretching of B-DNA with various pulling protocols: Appearance of structural polymorphism and S-DNA. J. Chem. Phys. 147, 225102. doi:10.1063/1.4991862
Gemmen, G. J., Millin, R., and Smith, D. E. (2006). Tension-dependent DNA cleavage by restriction endonucleases: Two-site enzymes are ”switched off” at low force. Proc. Natl. Acad. Sci. U. S. A. 103, 11555–11560. doi:10.1073/pnas.0604463103
Givaty, O., and Levy, Y. (2009). Protein sliding along DNA: Dynamics and structural characterization. J. Mol. Biol. 385, 1087–1097. doi:10.1016/j.jmb.2008.11.016
Gore, J., Bryant, Z., Stone, M. D., Nöllmann, M., Cozzarelli, N. R., and Bustamante, C. (2006). Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature 439, 100–104. doi:10.1038/nature04319
Gosse, C., and Croquette, V. (2002). Magnetic tweezers: Micromanipulation and force measurement at the molecular level. Biophys. J. 82, 3314–3329. doi:10.1016/S0006-3495(02)75672-5
Guan, L., He, P., Yang, F., Zhang, Y., Hu, Y., Ding, J., et al. (2017). Sap1 is a replication-initiation factor essential for the assembly of pre-replicative complex in the fission yeast Schizosaccharomyces pombe. J. Biol. Chem. 292, 6056–6075. doi:10.1074/jbc.M116.767806
Guardiani, C., Cencini, M., and Cecconi, F. (2014). Coarse-grained modeling of protein unspecifically bound to DNA. Phys. Biol. 11, 026003. doi:10.1088/1478-3975/11/2/026003
Harris, S. A., Sands, Z. A., and Laughton, C. A. (2005). Molecular dynamics simulations of duplex stretching reveal the importance of entropy in determining the biomechanical properties of DNA. Biophys. J. 88, 1684–1691. doi:10.1529/biophysj.104.046912
Hinckley, D. M., Freeman, G. S., Whitmer, J. K., and de Pablo, J. J. (2013). An experimentally-informed coarse-grained 3-site-per-nucleotide model of DNA: Structure, thermodynamics, and dynamics of hybridization. J. Chem. Phys. 139, 144903. doi:10.1063/1.4822042
Iwahara, J., and Clore, G. M. (2006a). Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature 440, 1227–1230. doi:10.1038/nature04673
Iwahara, J., and Clore, G. M. (2006b). Direct observation of enhanced translocation of a homeodomain between DNA cognate sites by NMR exchange spectroscopy. J. Am. Chem. Soc. 128, 404–405. doi:10.1021/ja056786o
Kolomeisky, A. B., and Veksler, A. (2012). How to accelerate protein search on DNA: Location and dissociation. J. Chem. Phys. 136, 125101. doi:10.1063/1.3697763
Kosikov, K. M., Gorin, A. A., Zhurkin, V. B., and Olson, W. K. (1999). DNA stretching and compression: Large-scale simulations of double helical structures. J. Mol. Biol. 289, 1301–1326. doi:10.1006/jmbi.1999.2798
Lai, P.-Y., and Zhou, Z. (2003). B- to s-form transition in double-stranded dna with basepair interactions. Phys. A Stat. Mech. its Appl. 321, 170–180. doi:10.1016/s0378-4371(02)01776-4
Lebrun, A., and Lavery, R. (1996). Modelling extreme stretching of DNA. Nucleic Acids Res. 24, 2260–2267. doi:10.1093/nar/24.12.2260
Lequieu, J., Córdoba, A., Schwartz, D. C., and de Pablo, J. J. (2016). Tension-dependent free energies of nucleosome unwrapping. ACS Cent. Sci. 2, 660–666. doi:10.1021/acscentsci.6b00201
Lequieu, J., Schwartz, D. C., and de Pablo, J. J. (2017). In silico evidence for sequence-dependent nucleosome sliding. Proc. Natl. Acad. Sci. U. S. A. 114, E9197–E9205. doi:10.1073/pnas.1705685114
Li, L., Li, C., Sarkar, S., Zhang, J., Witham, S., Zhang, Z., et al. (2012). DelPhi: A comprehensive suite for DelPhi software and associated resources. BMC Biophys. 5, 9. doi:10.1186/2046-1682-5-9
Lin, X., Leicher, R., Liu, S., and Zhang, B. (2021). Cooperative DNA looping by PRC2 complexes. Nucleic Acids Res. 49, 6238–6248. doi:10.1093/nar/gkab441
Liu, N., Bu, T., Song, Y., Zhang, W., Li, J., Zhang, W., et al. (2010). The nature of the force-induced conformation transition of dsDNA studied by using single molecule force spectroscopy. Langmuir 26, 9491–9496. doi:10.1021/la100037z
Luan, B., and Aksimentiev, A. (2008). Strain softening in stretched DNA. Phys. Rev. Lett. 101, 118101. doi:10.1103/PhysRevLett.101.118101
Maier, B., Bensimon, D., and Croquette, V. (2000). Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl. Acad. Sci. U. S. A. 97, 12002–12007. doi:10.1073/pnas.97.22.12002
Manghi, M., Destainville, N., and Palmeri, J. (2012). Mesoscopic models for DNA stretching under force: New results and comparison with experiments. Eur. Phys. J. E Soft Matter 35, 110. doi:10.1140/epje/i2012-12110-2
Marklund, E. G., Mahmutovic, A., Berg, O. G., Hammar, P., van der Spoel, D., Fange, D., et al. (2013). Transcription-factor binding and sliding on DNA studied using micro- and macroscopic models. Proc. Natl. Acad. Sci. U. S. A. 110, 19796–19801. doi:10.1073/pnas.1307905110
Marko, J. F. (2015). Biophysics of protein-DNA interactions and chromosome organization. Phys. A 418, 126–153. doi:10.1016/j.physa.2014.07.045
Marko, J. F., and Siggia, E. D. (1997). Driving proteins off DNA using applied tension. Biophys. J. 73, 2173–2178. doi:10.1016/S0006-3495(97)78248-1
Mirny, L., Slutsky, M., Wunderlich, Z., Tafvizi, A., Leith, J., and Kosmrlj, A. (2009). How a protein searches for its site on DNA: The mechanism of facilitated diffusion. J. Phys. A Math. Theor. 42, 434013. doi:10.1088/1751-8113/42/43/434013
Mo, Y., Vaessen, B., Johnston, K., and Marmorstein, R. (1998). Structures of SAP-1 bound to DNA targets from the E74 and c-fos promoters: Insights into DNA sequence discrimination by ets proteins. Mol. Cell. 2, 201–212. doi:10.1016/s1097-2765(00)80130-6
Moller, J., Lequieu, J., and de Pablo, J. J. (2019). The free energy landscape of internucleosome interactions and its relation to chromatin fiber structure. ACS Cent. Sci. 5, 341–348. doi:10.1021/acscentsci.8b00836
Mondal, A., and Bhattacherjee, A. (2015). Searching target sites on DNA by proteins: Role of DNA dynamics under confinement. Nucleic Acids Res. 43, 9176–9186. doi:10.1093/nar/gkv931
Mondal, A., and Bhattacherjee, A. (2017). Understanding the role of DNA topology in target search dynamics of proteins. J. Phys. Chem. B 121, 9372–9381. doi:10.1021/acs.jpcb.7b08199
Mondal, A., Mishra, S. K., and Bhattacherjee, A. (2022a). Nucleosome breathing facilitates cooperative binding of pluripotency factors Sox2 and Oct4 to DNA. Biophys. J. doi:10.1016/j.bpj.2022.10.039
Mondal, A., Sangeeta, , and Bhattacherjee, A. (2022b). Torsional behaviour of supercoiled DNA regulates recognition of architectural protein Fis on minicircle DNA. Nucleic Acids Res. 50, 6671–6686. doi:10.1093/nar/gkac522
Nagae, F., Brandani, G. B., Takada, S., and Terakawa, T. (2021). The lane-switch mechanism for nucleosome repositioning by DNA translocase. Nucleic Acids Res. 49, 9066–9076. doi:10.1093/nar/gkab664
Nicklas, R. B. (1988). The forces that move chromosomes in mitosis. Annu. Rev. Biophys. Biophys. Chem. 17, 431–449. doi:10.1146/annurev.bb.17.060188.002243
Niina, T., Brandani, G. B., Tan, C., and Takada, S. (2017). Sequence-dependent nucleosome sliding in rotation-coupled and uncoupled modes revealed by molecular simulations. PLoS Comput. Biol. 13, e1005880. doi:10.1371/journal.pcbi.1005880
Okazaki, K., Koga, N., Takada, S., Onuchic, J. N., and Wolynes, P. G. (2006). Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 103, 11844–11849. doi:10.1073/pnas.0604375103
Paik, D. H., and Perkins, T. T. (2011). Overstretching DNA at 65 pN does not require peeling from free ends or nicks. J. Am. Chem. Soc. 133, 3219–3221. doi:10.1021/ja108952v
Putzel, G. G., Tagliazucchi, M., and Szleifer, I. (2014). Nonmonotonic diffusion of particles among larger attractive crowding spheres. Phys. Rev. Lett. 113, 138302. doi:10.1103/PhysRevLett.113.138302
Rief, M., Clausen-Schaumann, H., and Gaub, H. E. (1999). Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 6, 346–349. doi:10.1038/7582
Smith, S. B., Cui, Y., and Bustamante, C. (1996). Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science 271, 795–799. doi:10.1126/science.271.5250.795
Taghavi, A., van der Schoot, P., and Berryman, J. T. (2017). DNA partitions into triplets under tension in the presence of organic cations, with sequence evolutionary age predicting the stability of the triplet phase - corrigendum. Q. Rev. Biophys. 50, e1. doi:10.1017/S0033583517000142
Tan, C., and Takada, S. (2020). Nucleosome allostery in pioneer transcription factor binding. Proc. Natl. Acad. Sci. U. S. A. 117, 20586–20596. doi:10.1073/pnas.2005500117
Veksler, A., and Kolomeisky, A. B. (2013). Speed-selectivity paradox in the protein search for targets on DNA: Is it real or not? J. Phys. Chem. B 117, 12695–12701. doi:10.1021/jp311466f
von Hippel, P. H., and Berg, O. G. (1989). Facilitated target location in biological systems. J. Biol. Chem. 264, 675–678. doi:10.1016/s0021-9258(19)84994-3
Wu, L., Xiao, B., Jia, X., Zhang, Y., Lü, S., Chen, J., et al. (2007). Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions. J. Biol. Chem. 282, 9846–9854. doi:10.1074/jbc.M609219200
Wuite, G. J., Smith, S. B., Young, M., Keller, D., and Bustamante, C. (2000). Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106. doi:10.1038/35003614
Xiao, B., Johnson, R. C., and Marko, J. F. (2010). Modulation of HU-DNA interactions by salt concentration and applied force. Nucleic Acids Res. 38, 6176–6185. doi:10.1093/nar/gkq435
Xu, J., Zhao, L., Xu, Y., Zhao, W., Sung, P., and Wang, H. W. (2017). Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat. Struct. Mol. Biol. 24, 40–46. doi:10.1038/nsmb.3336
Yin, H., Wang, M. D., Svoboda, K., Landick, R., Block, S. M., and Gelles, J. (1995). Transcription against an applied force. Science 270, 1653–1657. doi:10.1126/science.270.5242.1653
Keywords: DNA stretching, Σ-DNA, S-DNA, triplet formation, facilitated diffusion, protein−DNA interactions
Citation: Mondal A and Bhattacherjee A (2022) Understanding protein diffusion on force-induced stretched DNA conformation. Front. Mol. Biosci. 9:953689. doi: 10.3389/fmolb.2022.953689
Received: 26 May 2022; Accepted: 22 November 2022;
Published: 05 December 2022.
Edited by:
Qing-Chuan Zheng, Jilin University, ChinaReviewed by:
Hema Chandra Kotamarthi, Indian Institute of Technology Madras, IndiaCopyright © 2022 Mondal and Bhattacherjee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arnab Bhattacherjee, YXJuYWJAam51LmFjLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.