 Debora Gentile
Debora Gentile Marianna Esposito
Marianna Esposito Paolo Grumati
Paolo Grumati
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 03 August 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.930223
This article is part of the Research Topic Regulatory Networks in Cancer Cell Metabolism View all 4 articles
Autophagy is an evolutionary conserved catabolic pathway that uses a unique double-membrane vesicle, called autophagosome, to sequester cytosolic components, deliver them to lysosomes and recycle amino-acids. Essentially, autophagy acts as a cellular cleaning system that maintains metabolic balance under basal conditions and helps to ensure nutrient viability under stress conditions. It is also an important quality control mechanism that removes misfolded or aggregated proteins and mediates the turnover of damaged and obsolete organelles. In this regard, the idea that autophagy is a non-selective bulk process is outdated. It is now widely accepted that forms of selective autophagy are responsible for metabolic rewiring in response to cellular demand. Given its importance, autophagy plays an essential role during tumorigenesis as it sustains malignant cellular growth by acting as a coping-mechanisms for intracellular and environmental stress that occurs during malignant transformation. Cancer development is accompanied by the formation of a peculiar tumor microenvironment that is mainly characterized by hypoxia (oxygen < 2%) and low nutrient availability. Such conditions challenge cancer cells that must adapt their metabolism to survive. Here we review the regulation of autophagy and selective autophagy by hypoxia and the crosstalk with other stress response mechanisms, such as UPR. Finally, we discuss the emerging role of ER-phagy in sustaining cellular remodeling and quality control during stress conditions that drive tumorigenesis.
The term autophagy, derived from the Greek “self-eating,” describes a catabolic process through which cytoplasmic cargos are delivered to lysosomes for degradation and recycling (He and Klionsky, 2009). Autophagy, together with the ubiquitin-proteasome system (UPS), are the two major protein degradative pathways in the cells and are essential for maintaining cellular proteostasis. However, while the UPS is mainly devoted to the rapid turnover of short-lived proteins, autophagy degrades long-lived proteins and organelles (Cohen-Kaplan et al., 2016). Under basal conditions, autophagy contributes to the metabolic balance of cells thus ensuring cell viability. Following lysosomal degradation, the breakdown products are released into the cytosol and recycled to generate energy (Kaur and Debnath, 2015). However, under stressful conditions such as nutrient starvation, hypoxia, genotoxic stress, and pathogen infection, autophagy is dramatically induced to preserve cellular homeostasis and to ensure proper quality control by removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria and ER and eliminating intracellular pathogens (Tolkovsky, 2009; Galluzzi et al., 2017; Grumati et al., 2018; Reggio et al., 2020). Yet, insufficient, or excessive autophagy may lead to cell death. Thus, defective autophagy has been implicated in the pathogenesis of various diseases like cancer and neurodegenerative disorders (Mizushima et al., 2008). In cancer biology, autophagy is considered a double-edged sword since it can work both as a tumor suppressor and a tumor-promoting mechanism depending on the stage of cancer progression (Corcelle et al., 2009). It acts as a tumor-suppressive mechanism during cancer initiation and malignant transformation by removing damaged organelles, thereby limiting cell proliferation and genomic instability. On the other hand, autophagy is required for tumor progression and maintenance once malignant transformation is established, as cancer cells use autophagy for continuous supply of energy and nutrients (Corcelle et al., 2009).
It is now becoming clear that cancer cells are essentially characterized by a unique metabolic environment in which many stress-response pathways are activated and finely coordinated to promote tumor growth. The main driving force behind this metabolic rewiring is the establishment of a unique milieu within the tumor, known as tumor microenvironment (TME) (Yang, 2017). TME negatively affects normal cell physiology and facilitates cancerous transformation. Indeed, hyperproliferation of malignant cells causes loss of normal tissue architecture, accompanied by an abnormal vascularization, resulting in a dysfunctional distribution of nutrients, growth factors and oxygen within the tumor (Hanahan and Weinberg, 2011). Low oxygen availability, commonly referred to as hypoxia, is a hallmark of TME and forces cancer cells to adapt to a condition of constant stress. To cope with such stress, cancer cells activate a series of hypoxia-associated pathways comprised of HIFs, mTOR, and the unfolded protein response (UPR) which ultimately converge on autophagy (Wouters and Koritzinsky, 2008).
Different types of autophagy have been described in mammalian cells: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy (CMA) (Parzych and Klionsky, 2014). The three types of autophagy differ in the specificity of the substrate, the delivery mechanism of the cargo, their regulation, and the conditions in which they are activated.
The CMA is a selective form of autophagy in which cytosolic proteins, equipped with a lysosomal targeting motif (KFER), are selectively recognized by the chaperone protein Hsp70, and delivered to the surface of the lysosome. At the lysosome the chaperone-protein complex interacts with the lysosomal associated membrane protein 2 (LAMP-2), promoting its translocation into lysosomal lumen for degradation (Cuervo and Wong, 2014).
In contrast to CMA, both micro- and macro-autophagy involve dynamic membrane rearrangement. During micro-autophagy, cytoplasmic components translocate into the lysosome through a direct invagination, protrusion, or septation of the lysosomal membrane (Li et al., 2012).
Macro-autophagy is the most well-characterized form of autophagy and, is generally referred to as autophagy. The main feature that characterizes and differentiates macro-autophagy - simply autophagy hereafter - from CMA and micro-autophagy is the delivery of cytosolic cargo proteins to lysosomes via a unique double-membrane vesicle structure called autophagosome (Feng et al., 2014).
The formation of autophagosomes is a dynamic process that consists of several steps such as initiation, elongation, closure, and fusion with lysosomes. Each of these steps is finely regulated by a hierarchical interplay of autophagy-related proteins (ATGs) and signaling pathways that are differently activated upon a plethora of stimuli (Yang and Klionsky, 2010).
The initial step of autophagy is the surrounding and sequestering of cytoplasmic cargo and organelles within an isolation membrane called phagophore. This steps typically occurs in a specific structure, closely associated with the ER, called omegasome (Axe et al., 2008). In particular, upon autophagy induction, the Unc-51-like kinase 1/2 (ULK1/2), consisting of the serine/threonine kinase ULK1/2, ATG13, FIP200 (focal adhesion kinase family interacting protein of 200 kDa), and ATG101, translocates to the omegasome and regulates the recruitment of a second complex, the PI3KC3 (class III phosphatidylinositol 3-kinase) complex I, consisting of the VPS34, VPS15, Beclin-1and several binding proteins such as AMBRA1 (Di Bartolomeo et al., 2010; Nazio et al., 2013). The PI3KC3 complex I produces phosphatidylinositol 3-phosphate (PI3P) that acts as a platform for autophagosome biogenesis (Axe et al., 2008).
The main players in the next step are the two ubiquitin-like proteins, ATG12 and ATG8/LC3, that cooperate with their conjugation system to sustain the elongation of the phagophore membrane. ATG12 is conjugated to ATG5 to form the ATG12-ATG5 complex in a reaction mediated by ATG7 and ATG10 (E1 and E2-like enzymes, respectively). Then, the ATG12–ATG5 non-covalently interacts with ATG16L, which oligomerizes to form a large multimeric complex called the ATG16L complex. At this point, ATG5–ATG12–ATG16L complex promotes ATG3-mediated conjugation of activated LC3/GABARAP family members to the carboxyl glycine of phosphatidylethanolamine (PE) that is integrated into the growing phagophore (Kabeya et al., 2000; Nath et al., 2014). LC3 and its family members then mediate membrane tethering and hemi fusion, a function which is crucial for the expansion and fusion of autophagosomes with lysosomes (Nakatogawa et al., 2007). Elongation of phagophore requires membrane input from other organelles and ATG9 is the best candidate for this role. ATG9 is the only transmembrane ATG protein, identified thus far, that assists the growth of autophagic membranes by a poorly defined mechanism that may involve the trafficking of ATG9-containing vesicles between the trans-Golgi network, plasma membrane, recycling endosomes (REs), and autophagic membranes (Feng and Klionsky, 2017). Once the expanding ends of the phagophore fuse, the autophagosome is formed. At this point, newly formed autophagosomes undergo a maturation process. They fuse with early and late endosomes to form intermediate structures, called amphisomes, which, ultimately, fuse with lysosomes (He and Klionsky, 2009).
Autophagy has long been considered a non-selective bulk degradation process in which cytoplasmic cargo is randomly encapsulated in nascent autophagosomes. However, many forms of selective autophagy, which lead to the degradation of specific organelles, proteins and pathogens, have been described (Tolkovsky, 2009; Galluzzi et al., 2017; Grumati et al., 2018; Reggio et al., 2020).
Selective forms of autophagy share the same structural and molecular features as bulk autophagy, but are mediated by specific receptors (Stolz et al., 2014). Selective autophagy receptors, display exclusive binding regions which allow them to act as a bridge between their cargo and the autophagic machinery through the interaction with LC3/GABARAP family members (Johansen and Lamark, 2011; Johansen and Lamark, 2020; Gatica et al., 2018). These domains, known as AIM, LIR, or GIM (i.e., Atg8-interacting motif, LC3-interacting region, and GABARAP-interaction motif) are usually characterized by sequence resembling [W/F/Y] XX [L/V/I] where X can be any amino acid (Birgisdottir et al., 2013; Johansen and Lamark, 2020). Historically, the first autophagic receptor with a LIR consensus sequence to be identified was p62/SQSTM1. In addition to the LC3/GABARAP family, p62 can also bind ubiquitin (Ub) via a C-terminal UBA domain, thus allowing the degradation of ubiquitinated cargo by selective autophagy (Kirkin et al., 2009).
Given its importance, autophagy machinery is regulated by a plethora of signaling pathways. The principal player is the mammalian Target of Rapamycin (mTOR) (the mammalian ortholog of yeast TOR), which is a signaling control hub downstream of nutrients, growth receptors, hypoxia and ATP levels (Wullschleger et al., 2006). mTOR is a Serine/Threonine kinase that belongs to the phosphatidylinositol kinase-related kinase (PIKK) family (Abraham, 2004) and was first described as the physiological target of the immunosuppressant drug rapamycin (Sabers et al., 1995). In mammals, mTOR is sensitive to amino acids and glucose levels which, under normal conditions, keep mTOR active. Under conditions of nutrient depletion mTOR activity is inhibited, thus leading to the induction of autophagy (Jung et al., 2010). The mTOR pathway is composed of two functional complexes, that are differentially involved in autophagy modulation: the rapamycin-sensitive mTOR complex 1 (mTORC1), and the mTOR complex 2 (mTORC2) that is unaffected by rapamycin (Laplante and Sabatini, 2009). Under optimal growth conditions, mTORC1 directly inhibits autophagy through hyperphosphorylation of ATG13 and phosphorylation of ULK1 (Ser638 and Ser758). These modifications prevent the interaction between ATG13 and ULK1 and the formation of the initiation complex ULK1-ATG13-FIP200-ATG101 (Hosokawa et al., 2009). Nutrient deprivation, or rapamycin treatment, inactivates mTORC1 leading to the dephosphorylation of ATG13 and the formation of the initiation complex which translocates to the autophagy initiation sites and regulates the recruitment of the others ATG proteins (Jung et al., 2009). Inhibition of mTORC1 is associated with reduced phosphorylation of the ribosomal protein S6 kinase (also known as p70S6K) and the translation initiation factor 4E binding proteins-1 (4E-BP1), intended to slow down protein synthesis and meet the changing nutrient needs of the cells (Ma and Blenis, 2009).
As part of the energy-sensing cascade, mTORC1 senses changes in the extracellular energy state via AMP-dependent protein kinase (AMPK). Once activated, AMPK stimulates catabolic processes, ATP generating pathways and inhibits anabolic processes such as the synthesis of lipids, carbohydrates, and proteins to assure cell survival (Villanueva-Paz et al., 2016). In accordance with its physiological role, AMPK is linked to the regulation of autophagy. In particular, AMPK plays a dual role in activating autophagy by 1) inactivating mTORC1 and 2) directly activating ULK1, both via phosphorylation (Kim et al., 2011). Since mTORC1 is a key regulator of autophagy, one of the mechanisms of AMPK-dependent induction of autophagy is through mTORC1 inhibition. AMPK inhibits mTORC1 1) directly, by phosphorylating RAPTOR (Gwinn et al., 2008) and 2) indirectly, by phosphorylating Tuberous Sclerosis Complex 2 (TSC2) (Inoki et al., 2003). Moreover, AMPK can also activate autophagy via ULK1 phosphorylation. Under glucose starvation, AMPK directly phosphorylates of ULK1 at Ser317 and Ser777. Under nutrient rich conditions, on the other hand, active mTORC1 prevents ULK1 activation through the phosphorylation of Ser757, which disrupts the interaction between AMPK and ULK1 (Kim et al., 2011). Besides its role as a nutrient sensor, mTOR integrates numerous upstream stimuli including growth-factor signals (insulin or IGF-1), Toll-like receptor (TLR) ligands and cytokines, all of which converge on the tuberous sclerosis complex 1 and 2 (TSC1 and TSC2), the principle upstream inhibitor of mTOR. TSC2 forms a functional complex with TSC1 to inhibit the two key regulators of mTOR mediated translation: S6K and 4EBP1 (Huang and Manning, 2008).
As a central pathway involved in cellular metabolism, autophagy has been largely implicated in cancer biology. However, its role in cancer is complex since it can act as a tumor suppressor as well as a tumor-promoting mechanism (Corcelle et al., 2009; Kimmelman, 2011). Nowadays, is widely accepted that the role of autophagy in cancer is dynamic. Indeed, during the early stages of malignant transformation autophagy works as a tumor-suppressive mechanism limiting cellular proliferation by removing toxic proteins and organelles. Thus, preventing the accumulation of chronic cellular damage that could favor malignant transformation. In contrast, once malignant transformation has occurred, autophagy sustains cancer cell growth by supplying building blocks for macromolecule biosynthesis (Kimmelman, 2011).
Direct evidence for the role autophagy plays in tumor suppression come from mouse genetic studies employing Atg7, Atg5, and Beclin-1 knockout animals. The absence of these genes impairs autophagy flux and promotes tumor initiation (Qu et al., 2003; Takamura et al., 2011). For instance, the Beclin-1 locus is deleted in up to 75% of ovarian cancers and in up to 50%–70% of breast cancers (Liang et al., 1999). However, an important observation is that all of these tumors remain benign, indicating that even though autophagy depletion contributes to tumor initiation, functional autophagy machinery is required for tumor progression to a malignant stage (Takamura et al., 2011).
Additionally, the accumulation of p62, as a result of autophagy inhibition, is important in the promotion of tumorigenesis through a variety of mechanisms, including deregulation of NF-κB signaling, accumulation of ROS and increased DNA damage (Mathew et al., 2009). In particular, the accumulation of p62, in autophagy-deficient cells, inhibits the degradation of p53 and β-Catenin by sequestering ubiquitinated proteins and preventing their proteasomal degradation (Korolchuk et al., 2009).
On the other hand, other evidence suggest that autophagy plays a protective role in cancer cells and promotes tumor growth in advanced cancers (Luo et al., 2016; Liu et al., 2018). Indeed, rapidly developing cancer cells require cellular building blocks to support energy production. In accordance with this, a high basal level of autophagy was found in Ras mutated cancer cells (Su, 2018; Zhu et al., 2019). Moreover, even in the presence of copious nutrients, human cell lines with mutations in H-Ras or K-Ras, have elevated basal levels of autophagy (Guo et al., 2011).
Regions with very low oxygen levels are found in many solid tumors and likely occur because of inadequate vascular formation due to the uncontrolled proliferation of malignant cells. Those regions where oxygen concentration is <2%, are named the hypoxic zone and represent one of the hallmark of cancer (Hanahan and Weinberg, 2011).
The main transcription factor that regulates hypoxic response is HIF-1. HIF-1 is a heterodimeric complex consisting of the hypoxia-induced subunit HIF-1α and the constitutively expressed subunit HIF-1β that transcriptionally regulates the expression of several genes (Semenza, 2010). Because HIF-1β is constitutively expressed, HIF-1α, which contains an oxygen-dependent degradation domain (ODDD) and is tightly regulated by oxygen, is considered the major regulatory subunit of the HIF-1 complex (Semenza, 2007). Under normoxia, HIF-1α protein is rapidly degraded, resulting in minimal transcriptional activity of the HIF-1 complex. When cells are subjected to hypoxic conditions, HIF-1α is stabilized and translocates from the cytosol to the nucleus, where it interacts with HIF-1β to promote transcriptional activity (Semenza, 2007, 2010) (Figure 1). Autophagy is one of the pathways regulated by HIF-1. In particular, HIF-1 is involved in the regulation of key genes involved in the initiation and progression of autophagosome formation including BNIP3, BNIP3L/NIX, ATG7, ATG5, and ATG9A (Zhang and Ney, 2009; Gui et al., 2016; Abdul Rahim et al., 2017).
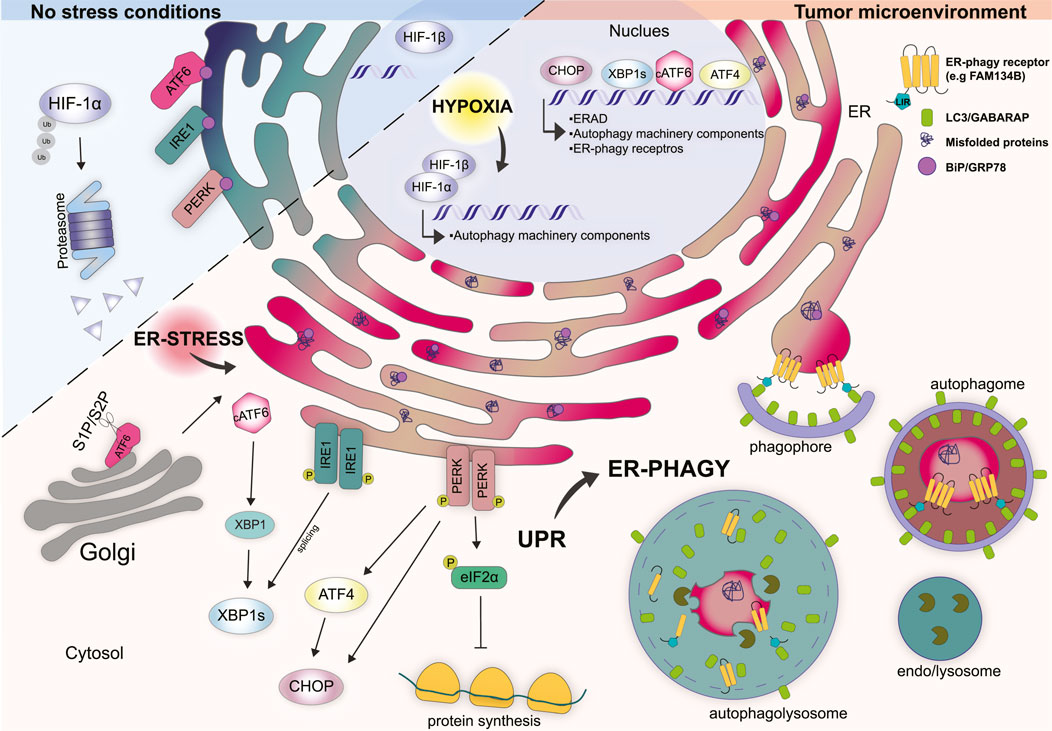
FIGURE 1. Cellular response mechanisms in non-stress conditions vs. tumor microenvironment. Under physiological conditions, HIF-1α is localized in the cytosol and rapidly degraded by the proteasome. The UPR stressors ATF6, IRE1, and PERK are kept inactive by the binding to the ER chaperone BiP/GRP78. Tumor microenvironment is characterized by hypoxia which induces the translocation of HIF-1α into the nucleus where it forms a stable complex with HIF-1β. HIF-1α/β complex regulates the transcription of several genes involved in the function and regulation of autophagy machinery. Hypoxia impairs the folding capacity of the ER and induces a rapid accumulation of misfolded proteins thus determining ER stress. Under ER stress conditions BiP/GRP78 bind misfolded proteins and dissociate from the UPR sensors. ATF6 translocates to the Golgi where is cleaved by S1P and S2P proteases that generate cytosolic transcription factor cATF6. cATF6 interact con XBP1 and regulates the transcription of genes involved in protein folding, trafficking and ERAD. Dimerization and phosphorylation of IRE1 activate the splicing of XBP1. PERK activation induces the phosphorylation of eIF2α preventing mRNA translation. Moreover, PERK induces the transcription factors ATF4 and CHOP. In the nucleus CHOP, ATF4, and XBP1s coordinate the transcription of several genes involved in the UPR, ERAD, and autophagy-related genes including some ER-phagy receptor genes. Together hypoxia, ER stress and UPR trigger ER-phagy to restore ER homeostasis.
Bcl2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L), also known as NIX (BNIP3L/NIX) are two pro-apoptotic proteins that localize to the outer mitochondria membrane (OMM) and share several common features (Ney, 2015). Their expression is transcriptionally regulated by HIF-1α during hypoxia and both play an important role in the modulation of hypoxia induced-autophagy (Zhang and Ney, 2009). In particular, BNIP3/BNIP3L influence autophagy by modulating the Bcl2-Beclin-1 and BclXL-Beclin-1 complexes (Bellot et al., 2009). Under non-stressful conditions, both Bcl2 and BclXL are bound to Beclin-1 thus inhibiting phagophore formation. The upregulation of BNIP3/BNIP3L upon hypoxia induces the release of Beclin-1 from Bcl2 and BclXL which bind with higher affinity to BNIP3 and BNIP3L via their BH3 domain (Bellot et al., 2009). The free form of Beclin-1 is then able to interact with the VPS34 complex and induce autophagy (Bellot et al., 2009). Beyond their contribution to modulating general autophagy, BNIP3 and BNIP3L can also act as mitophagy receptors. Indeed, they harbor a LIR motif that is exposed to the cytoplasm, through which they interact with LC3/GABARAP and mediate clearance of mitochondria via selective autophagy (Novak et al., 2010; Hanna et al., 2012). The precise mechanism of BNIP3/BNIP3L induced-mitophagy is not completely clear, but it seems that several phosphorylation events are crucial for the function of these proteins as mitophagy receptors. In particular, the phosphorylation of BNIP3L at Ser81 is necessary for mitophagy induction under ischemic conditions (Yuan et al., 2017). Moreover, BNIP3 phosphorylation at Ser17 and Ser24 strongly increases its interaction with LC3, thus promoting mitophagy (Yuan et al., 2017). However, the precise mechanism by which these two receptors are modulated in response to hypoxia remains to be elucidated and warrants further investigation.
In addition to BNIP3/BNIP3L, FUNDC1 is another integral OMM protein containing a LIR domain that is implicated in hypoxia-induced mitophagy (Liu et al., 2012). Indeed, FUNDC1 mediated-mitophagy is appears to be specifically triggered under hypoxic conditions, since FUNDC1 knockdown in HeLa cells does not affect starvation-induced mitophagy, and only plays a moderate role in FCCP-induced mitophagy (Liu et al., 2012). From a mechanistic point of view, during normoxia FUNDC1 is phosphorylated by SRC kinase at Tyr18, and by casein kinase 2 (CK2) at Ser13, on its LIR domain. These phosphorylation events decrease FUNDC1’s binding affinity for LC3, thus preventing cargo recognition within the autophagosome (Liu et al., 2012; Kuang et al., 2016). On the contrary, upon hypoxia, both SRC and CK2 are inactivated and FUNDC1 phosphorylation is significantly reduced, thus allowing it to strongly interact with LC3B-II, resulting in the removal of damaged mitochondria by mitophagy. In particular, CK2 is phosphorylated under normoxia but dephosphorylated in response to hypoxia by the mitochondrial phosphatase PGAM5 (Chen et al., 2017). PGAM5 is normally inhibited by the interaction with BclXL which blocks its phosphatase activity. When cells are under hypoxic conditions, the degradation of BclXL induces PGAM5 activation which, in turn, dephosphorylates FUNDC1 at Ser13 (Wu et al., 2014). The inactivation of SRC under hypoxic conditions is mediated by a single phosphorylation event at Tyr416 (Ozkirimli and Post, 2006). As a consequence, phosphorylation at this site blocks FUNDC1 phosphorylation at Tyr18 (Ozkirimli and Post, 2006). This two-part system allows the fine-tuning of mitophagy during hypoxia.
The ER is the largest organelle in the cell consisting of a large membrane network that originates from the nuclear envelope and spreads throughout the cytosol. Structurally, it can be divided into sheets and tubules. The sheets are covered by ribosomes and form the rough ER, which is the primary site of protein synthesis and folding. The tubules form smooth ER whose function is to synthesize lipids and steroids and buffer cytosolic calcium (Schwarz and Blower, 2016). Moreover, the ER has the important task of interacting with other organelles such as the Golgi apparatus, mitochondria, lysosomes, endosomes, and even the plasma membrane, at highly specialized membrane contact sites (Phillips and Voeltz, 2016). These contacts regulate organellar functions such as Ca2+ homeostasis, lipid composition, fission, trafficking, and participation in signal transduction events (Phillips and Voeltz, 2016).
Several cellular stressors, such as Ca2+ levels, redox imbalance or altered protein glycosylation, can disrupt ER homeostasis and lead to a condition known as ER stress. Low oxygen availability and glucose shortage, which characterize the tumor microenvironment, are well-characterized ER stress stimuli. Under such conditions, the ability of the ER to form disulfide bonds is impaired, with the consequent accumulation of unfolded/misfolded proteins in the ER lumen (Chipurupalli et al., 2019). To overcome this stressful condition, cells have developed an integrated system of stress-responsive pathways composed of the UPR, ERAD, and autophagy, to restore homeostasis and normal ER function.
The unfolded protein response (UPR) is triggered by the accumulation of unfolded/misfolded proteins with the aim to re-establish ER homeostasis (Corazzari et al., 2017). UPR makes use of three ER-transmembrane proteins that act as stressor sensors: the inositol-requiring enzyme 1α (IRE 1α), the pancreatic endoplasmic reticulum kinase (PERK), and the activating transcription factor 6 (ATF6) (Gardner et al., 2013) (Figure 1). Mechanistically, during non-stress conditions the activation of these sensors is inhibited by the binding with the chaperon protein BiP - binding immunoglobulin protein - (also known as GRP78 - 78 kDa glucose-regulated protein), which is the most represented ER chaperone (Bertolotti et al., 2000). Conversely, upon ER stress BiP dissociates from the ER stress sensor, due to its capacity to bind misfolded proteins with higher affinity, thus leading to the activation of the above-mentioned sensors and their downstream signaling (Figure 1) (Bertolotti et al., 2000).
PERK is a transmembrane type I protein characterize by a cytosolic serine/threonine kinase domain which is enriched at the mitochondria-associated ER membrane sites (MAMs) (Verfaillie et al., 2012). Upon ER stress, BiP dissociates from PERK thus inducing its activation through homodimerization and autophosphorylation (Bertolotti et al., 2000). The best-characterized substrate of activated PERK is the eukaryotic translation initiation factor 2α (eIF2α) (Harding et al., 1999). PERK-dependent phosphorylation of eIF2α, at Ser 51, attenuates protein synthesis by global inhibition of 5′ cap-dependent translation while selectively increasing the cap-independent translation of many mRNAs, such as ATF4 - activating transcription factor 4 (Harding et al., 2000; Scheuner et al., 2001). ATF4, in turn, induces the transcription factor C/EBP (CHOP) and several other genes involved in the regulation of autophagy, amino acid metabolism, antioxidant response, and even cell death (Figure 1) (Lu et al., 2004; Vattem and Wek, 2004; Liu et al., 2015).
IRE1α is the most evolutionarily conserved arm of the UPR and consists of a type I transmembrane protein equipped with a cytosolic Serine/Threonine kinase domain (Cox et al., 1993). Upon activation, which occurs because of oligomerization and autophosphorylation, IRE1α goes through a conformational change that activates its RNAase activity. Activation of IRE1α promotes the unusual splicing of XBP1 - X-box-binding protein - mRNA, thus generating a modified mRNA isoform, indicated as XBP1s (where “s” means spliced). The spliced mRNA encodes for a functional XBP1s protein that acts as a transcription factor and regulates the expression of genes involved in protein folding, trafficking as well as components of the ER-associated protein degradation program (ERAD) (Figure 1) (Yoshida et al., 2001).
ATF6 is a type II transmembrane protein that is characterized by a cAMP-responsive element-binding protein and an ATF basic leucine zipper domain. During ER stress ATF6 translocates to the Golgi apparatus where it is cleaved by S1P and S2P proteases (known as MBTPS1 and MBTPS2) generating a cytosolic transcription factor (Haze et al., 1999). Cleaved ATF6α forms a heterodimer with XPB1 and regulates the transcription of unspliced XBP1 and other genes involved in protein folding and degradation in the ER such as chaperones, foldases and ERAD components (Lee et al., 2002; Lee et al., 2003). The overall outcome, from the activation of these three branches of the UPR, is to slow down protein synthesis, enhance the ER’s post-translation modification capacity and increase the degradation of unfolded/misfolded proteins by the ERAD system and autophagy (Figure 1).
Considering its vital role in ER stress mitigation, it is not surprising that UPR activation is a hallmark of several human cancers. Indeed, the UPR enables cancer cell survival and enhances their ability to adapt to adverse environmental conditions (Romero-Ramirez et al., 2004; Fels and Koumenis, 2006). In support of this notion, accumulating evidence demonstrate that all the molecular actors of the UPR are involved in tumor growth in one way or another. For example, IRE1 signaling seems to be crucial for hepatocellular carcinoma (HCC) initiation, whereas PERK activation is essential in the later stage of tumor progression (Vandewynckel et al., 2015). Moreover, both BiP and XBP1 are implicated in sustaining tumor cell survival and in mediating the tumor cells’ response to glucose deprivation (Spiotto et al., 2010). Furthermore, all three UPR signaling pathways have been shown to be activate during prostate cancer progression (Liu et al., 2017).
Nowadays, it is widely accepted that UPR does not act alone to counteract ER stress but is rather closely associated with autophagy. Indeed, all three branches of the UPR positively modulate autophagy induction upon ER stress (Verfaillie et al., 2010).
IRE1 actively modulates ER stress induced autophagy. In particular, active IRE1 mediates mitogen-activated protein kinase 8 (MAPK8) activation which, in turn, induces autophagy (Ogata et al., 2006). MAPK8 is considered a major stress-regulated protein kinase that controls stress-induced autophagy and apoptosis (Xia et al., 1995; Ogata et al., 2006). Mechanistically, MAPK8 interacts with c-Jun N-terminal kinase (JNK) that phosphorylates the two autophagy inhibitory proteins, BcL-2 and BcL-XL, which dissociate from the key autophagy inducer Beclin-1, resulting in autophagy induction (Wei et al., 2008; Corazzari et al., 2015). In addition, the IRE1-XBP1s axis has also been involved in the induction of autophagy (Margariti et al., 2013). In particular, XBP1s directly binds the Beclin-1 promoter in the nucleus, thus enhancing autophagy induction via the transcriptional upregulation of Beclin-1 (Suzuki et al., 2009; Margariti et al., 2013).
In addition, the ectopic expression of aggregated polyglutamine proteins (poliQ) triggers autophagy in a PERK-dependent manner, further supporting the link between UPR and autophagy. In particular, genetic substitution of Ser51 with Ala on eIF2α, prevented polyQ protein clearance by autophagy, suggesting a prominent role for the PERK-eIF2α axis in the activation of autophagy in response to the accumulation of unfolded/aggregate proteins (Kouroku et al., 2007). PERK activation also results in the downstream expression of both ATF4 and CHOP. ATF4 induces autophagy by increasing the expression of ATG12 (Kouroku et al., 2007) and others important autophagy genes like MAP1LC3, BECN1, ATG3, ATG12, and ATG16L1 (B’chir et al., 2013), while CHOP positively regulates the expression of Tribbles Homolog 3 (TRB3) resulting in the downstream inhibition of the mTOR complex through the inhibition of AKT activity, thus further stimulating autophagy (Salazar et al., 2009).
It has been suggested that the activation of the ATF6 branch of UPR, also triggers autophagy. Indeed, upon ER stress cATF6 induces the expression of Death-Associated Protein Kinase 1 (DAPK1) (Kalvakolanu and Gade, 2012; Gade et al., 2014), which phosphorylates Beclin-1 and promotes its dissociation from its negative regulator Bcl2, thus promoting autophagosome formation. Moreover, cATF6 also indirectly regulates autophagy via CHOP and XBP1 (Zalckvar et al., 2009).
ER stress is often accompanied by the release of calcium into the cytosol. Acting as a second messenger, calcium activates several Ca2+-dependent signaling pathways, including autophagy. In particular, the release of Ca2+ from ER induced by thapsigargin, ionomicyn, and vitamin D analogues, activates the Ca2+/calmodulin-dependent kinase β, which in turn activates AMPK and inhibits mTOR, thereby inducing autophagy (Høyer-Hansen et al., 2007).
Moreover, it has been shown that the activation of protein kinase C theta (PRKCQ/PKCθ) in response to ER stress can modulate autophagy in a Ca2+-dependent manner (Sakaki and Kaufman, 2008). Indeed, Ca2+ is an essential component of PRKCQ activation under ER stress conditions, which is supported by the observation that cells treated with the intracellular Ca2+ chelator BAPTA-AM deactivate PRKCQ with a consequent blockage of autophagy flux (Sakaki et al., 2008). Altogether, these reports support a crucial function of cytosolic Ca2+ mobilization, and modulation of Ca2+-related pathways upon ER stress in autophagy induction.
In this scenario, emerging data indicate an important role for mitochondria associated ER-membranes (MAMs). MAMs are flexible ER membrane-derived structures in which ER and mitochondria are physically and functionally connected (Csordás et al., 2006). MAMs serve as structural microdomains that allow the transfer of Ca2+ between the ER and mitochondria to regulate mitochondria bioenergetics and likely contribute to enhancing mitochondrial ATP production to satisfy increased energy demands resulting from stress. Consistently, alterations in the interaction between ER and mitochondria leads to the disruption of Ca2+ transfer between the two organelles, and to ER stress (Barazzuol et al., 2021). Moreover, MAM-mediated mitochondrial Ca2+ overload leads to mitochondrial depolarization and eventually triggers cell death (Walter and Hajnóczky, 2005).
The cross talk between ER and mitochondria is modulated in different ways and by numerous players. Among them, Mitofusin 2 (MFN2), a dynamin-related GTPase localized to the surface of both ER and mitochondria. The homologous interaction between ER-associated MFN2 and mitochondrial MFN2 facilitates interorganelle tethering (de Brito and Scorrano, 2008). Ablation of MFN2 results in an increased distance between ER and mitochondria and a disturbed Ca2+ transfer between the two organelles (de Brito and Scorrano, 2008). MFN2 is an important regulator of UPR and mitochondrial functions. Indeed, MFN2 interacts with PERK, at MAMs, to suppress its activation under normal conditions. Loss of function of MFN2 causes an increase in the production of reactive oxygen species (ROS), mitochondrial calcium overload, and impaired mitochondrial morphology through the sustained activation of PERK. Consistently, PERK inhibition rescues mitochondrial integrity in MFN2 mutant cells, thus identifying the MFN2-PERK axis as an important regulator of mitochondrial processes (Muñoz et al., 2013). Accordingly, PERK is also crucial for regulating MAMs integrity (Verfaillie et al., 2012). These data point out the important role of UPR and MAMs in regulating ER and mitochondrial dynamics and maintaining cellular homeostasis.
A growing body of evidence suggests that, in cancer cells, chronic activation of the ER stress response influences malignant progression by altering the function of the immune cells that coexist in the tumor microenvironment. The immune response is a key element implicated in cancer development. During early stages of tumorigenesis, the immune system is able to recognize and suppress tumor cells that express neoantigens in major histocompatibility complex class I (MHC-I) molecules, through natural killer (NK) cells and cytotoxic lymphocytes (CTL) (Dunn et al., 2002). Early evidence suggests that the induction of ER stress and activation of the UPR may inhibit the surface expression of MCH-I molecules via XBP1 and ATF6 overexpression (de Almeida et al., 2007). Moreover, increased ER stress triggered by glucose withdrawal in EL4 lymphoma cells, caused eIF2α-mediated inhibition of protein synthesis with a consequent impairment of MHC-I peptide presentation (Granados et al., 2009). Disruption of MHC-I antigen presentation has also been linked to the XBP1 branch of the UPR. In epithelial cells treated with the proteasome inhibitor ALLN and tunicamycin, to induce ER stress, XBP1-dependent induction of miR-346 negatively regulated the expression of the ER transporter involved in antigen processing 1 (TAP1) that is implicated in proper ER peptide influx and MCH-I antigen presentation (Bartoszewski et al., 2011). However, whether chronic induction of UPR in cancer cells facilitates immune evasion toward the above-mentioned mechanism needs to be further examined. Of note, some evidence demonstrate that autophagy may also play a role in the immune evasion of cancer cells. Autophagy promotes the degradation of MHC-I molecules with a consequent reduction in their surface expression in pancreatic ductal adenocarcinoma (Yamamoto et al., 2020). In particular, MHC-I molecules are targeted for selective autophagic degradation mediated by NBR1 and pharmacological and genetic inhibition of autophagy increases the surface expression of those molecules, thus restoring the susceptibility of pancreatic cells to elimination by CTL cells (Yamamoto et al., 2020).
ER stress can influence the immune response within the TME through the so-called transmissible ER stress - TERS. Soluble factors released from prostate, lung or melanoma cancer cells undergoing ER stress, can induce similar ER stress and UPR activation in bone marrow-derived myeloid cells, that is accompanied by the induction of pro-tumorigenic and immunosuppressive functions (Mahadevan and Zanetti, 2011). Additional studies further confirmed that TERS facilitates the communication between ER stressed cancers cells and other cancer cells or tumor-infiltrating immune cells such as tumor-associated macrophages (TAMs), (Rodvold et al., 2017) and that this phenomena reduced antigen processing and presentation and diminished T cell proliferation (Mahadevan et al., 2012).
Lastly, ER stress has been involved in the modulation of the inflammatory response within the TME. Indeed, all three UPR-branches induce the translocation of NF-κB to the nucleus with consequent inflammatory gene transcription, albeit via different mechanisms. PERK leads to NF-κB activation via a peculiar mechanism in which translational inhibition reduces the IκB/NF-κB ratio, therefore allowing the nuclear translocation of NF-κB and the transcription of inflammatory genes (Deng et al., 2004). Upon activation, IRE1α forms a complex with TNF-αR–associated factor 2 (TRAF2) at its cytosolic domain, that directly mediates IκB phosphorylation via IκB kinase (IKK), thus leading to NF-κB activation (Hu et al., 2006). Moreover, ATF6 was shown to participate in NF-κB activation in an AKT-dependent manner (Yamazaki et al., 2009).
These data support the idea that the ER stress response is a key regulator of cellular inflammation. However, a mechanistic link between ER stress induced autophagy, immunomodulation, and chronic inflammation within the tumor microenvironment remains to be better characterized.
The term ER-phagy refers to the selective degradation of a discrete portion of ER within an autophagolysosome (Grumati et al., 2018). Given its complexity, the ER is a highly dynamic organelle that undergoes continuous remodeling to accommodate the changing demands in cellular protein homeostasis. The selective autophagic degradation of the ER, known as ER-phagy, substantially contributes to the ER remodeling process (Wilkinson, 2019; Gubas and Dikic, 2022).
ER degradation can occur either via micro- or macro-ER-phagy. Micro-ER-phagy occurs through direct invagination of ER portions into the endo-lysosomes, whereas macro-ER-phagy relies on a subset of specific cargo receptors and depends on the core molecular autophagy machinery (Reggiori and Molinari, 2022). Mechanistically, these receptors promote ER fragmentation and engage luminal, membrane-bound, and cytosolic factors that drive lysosomal clearance of select ER domains along with their content (Grumati et al., 2018).
To date, eight ER-resident proteins have been characterized as ER-phagy receptors in mammals: the family with sequence similarity 134 members (FAM134A, B, C) (Khaminets et al., 2015; Reggio et al., 2021), Reticulon 3 (RTN3) (Grumati et al., 2017), Translocation protein SEC62 (SEC62) (Fumagalli et al., 2016), Cell-Cycle Progression Gene 1 (CCPG1) (Smith et al., 2018), Atlastin 3 (ATL3) (Chen et al., 2019), and Testis-Expressed protein 264 (TEX264) (An et al., 2019; Chino et al., 2019). As selective autophagy receptors, they harbor a functional LIR domain, that allows their binding with the LC3/GABARAP, and are degraded inside lysosomes along with the ER.
FAM134B, also known as RETREG1 or JK1, was the first ER-phagy receptor to be identified (Khaminets et al., 2015). Structurally, it is an intra-membrane protein that localizes to ER sheets and has a LIR domain at the C-terminal end (Figure 2) (Khaminets et al., 2015). Cells depleted of FAM134B show a substantial increase in ER volume and its deletion in vivo causes severe sensory neuropathy (Kurth et al., 2009; Khaminets et al., 2015). Conversely, overexpression of FAM134B causes an increased ER fragmentation and re-shuffling of ER membranes into autophagosomes (Khaminets et al., 2015). This indicates the fundamental role of FAM134B in maintaining ER shape and homeostasis. On the contrary, FAM134A and FAM134C do not have a similar role under basal conditions but are mainly activated upon stress (Reggio et al., 2021).
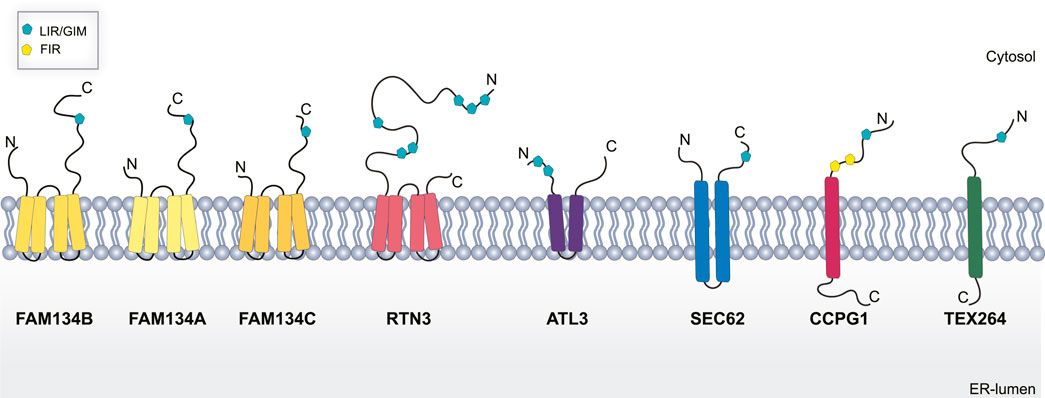
FIGURE 2. Schematic representation of ER-phagy receptors. Abbreviations: LIR/GIR, LC3/GABARAP interacting region; FIR, FIP200 interacting region; C, C terminal domain; N, N-terminal domain.
FAM134B was first described as an oncogene in esophageal squamous cell carcinoma (ESCC) (Tang et al., 2001). However, it is now widely accepted that it has a dual role in cancer, acting as a tumor-promoter in ESCC and in hepatocellular carcinoma (HCC), or a tumor suppressor in colorectal cancer (CRC) and breast cancer (BC) (Tang et al., 2007; Dai et al., 2017; Islam et al., 2017; Zhang et al., 2019; Table 1). The precise mechanisms behind these dual roles are far from being understood, even though several lines of evidence indicate that it may regulate cancer cell death and apoptosis through its role as an ER-phagy receptor (Mo et al., 2020).
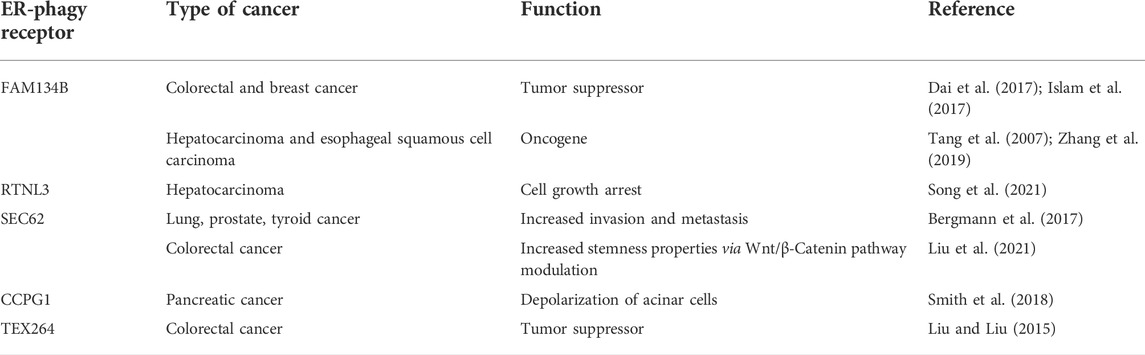
TABLE 1. ER-phagy receptors in different cancer subset.
RTN3L is devoted to the selective degradation of ER tubules (Grumati et al., 2017). It is part of the RTNs family which includes RTN1, RTN2, RTN3, and RTN4. Each of them contains a highly homologous RHD domain but they significantly differ in their N-terminal domain (Grumati et al., 2017), have several isoforms with different lengths (Yang and Strittmatter, 2007). The only member of this family characterized as an ER-phagy receptor is the long isoform of RTN3 (RTN3L). It harbors six functional LIR domains in the N terminal portion of the protein (Figure 2), and forms dimers to fragment ER tubules into discrete portions to be delivered lysosomes (Grumati et al., 2017). RTN3 has also been implicated in cancer. It acts as a tumor suppressor in hepatocellular carcinoma (HCC) inducing cell growth arrest, both in vitro and in vivo, by facilitating p53 phosphorylation, at Ser392, via Chk2 (Song et al., 2021; Table 1). However, the precise role of RTN3 in cancer needs to be further elucidated.
ATL3 is another ER-phagy receptor belonging to the Atlastin protein family, which includes ATL1, and ATL2 (Rismanchi et al., 2008). These are dynamin-like proteins that favor ER membrane fusion through a conformational change driven by GTP hydrolysis (Bian et al., 2011). ALT3 has two GIM motifs (Figure 2) that specifically interact with GABARAP and promote selective degradation of ER during starvation (Chen et al., 2019). Interestingly, mutations in ALT3 are associated with hereditary sensory and autonomic neuropathy type I (HSAN I). Mutations at Y192C and P338R affect ATL3 binding avidity to GABARAP, therefore, hampering its function as an ER-phagy receptor (Chen et al., 2019).
SEC62 together with SEC61 and SEC63 form the multiprotein translocon complex which imports newly nascent polypeptides from ribosomes into the ER (Linxweiler et al., 2017); however, SEC62 is the only one among them to be linked to ER-phagy. As an ER-phagy receptor, it is equipped with a LIR domain at the C-terminal (Figure 2). SEC62 mediated ER-phagy is activated in eukaryotic cells recovering from an ER stress inducing condition (Fumagalli et al., 2016). Indeed, mammalian cells tend to expand their ER surface in response to stress. Therefore, SEC62-mediate ER-phagy aims to reshape the ER to a pre-stress state, thus it is also called recov-ER-phagy (Fumagalli et al., 2016).
According to these features, in non-small cell lung, thyroid, and prostate cancer, upregulation of SEC62 confers tolerance to ER stress and contributes to invasion and metastasis (Bergmann et al., 2017). Moreover, in colorectal cancer, increased expression of SEC62 enhances stemness properties by modulating the Wnt/β-Catenin pathway, and this is associated with poor prognosis in patients (Liu et al., 2021; Table 1).
CCPG1 is a transmembrane ER-phagy receptor that contains a LIR domain and a FIP200 interacting region (FIR) (Figure 2) indicating a potential role of this receptor in the initiation step of autophagosome formation (Smith et al., 2018). Of note, CCPG1 is upregulated in response to UPR stress induced by DTT, tunicamycin and thapsigargin. Moreover, in these conditions, CCPG1-mediated ER-phagy depends on the binding of both ATG8 and FIP200 (Smith et al., 2018). Consistent with its role in proteostasis, CCPG1 knockout mice display depolarization of acinar cells of the exocrine pancreas that was accompanied by the accumulation of ER-produced secretory proteins (Smith et al., 2018; Table 1).
Finally, TEX264 is a single-pass transmembrane protein (Figure 2) individuated as an ER-phagy receptor in two independent proteomics-based studies (An et al., 2019; Chino et al., 2019). TEX264 localizes, in punctate structures, at the three-way junctions of the ER. Moreover, through a proximity labeling approach, other proteins involved in autophagosome formation, such as VPS34 complex proteins and WIPI2, have been detected in proximity with TEX264. This finding led to the hypothesis that TEX264 acts as a zipper-like helper allowing the ER membrane to enter into close contact with the phagophore membrane through a trans interaction (An et al., 2019). TEX264 is a marker protein for colorectal cancer, but its precise contribution in tumor development need to be further elucidated (Liu and Liu, 2015; Table 1).
Autophagy induction triggered by ER stress, has a dual role in re-establishing general cellular homeostasis and to restoring internal ER-homeostasis by reestablishing its physiological size. To do so, the UPR induces the selective degradation of ER membranes and luminal proteins via ER-phagy (Molinari, 2021).
The first evidence that supported the idea of UPR-induced ER remodeling, comes from study from Bernales and colleagues. They treated wild-type cells with dithiothreitol to induce UPR, and then carried out an ultrastructural analysis of the ER. They found that folding stress significantly increased the volume of ER in yeast and that ER expansion was followed by the appearance of large double membrane vesicles filled with stacked membrane cisternae in many cells. They examined flash-frozen/freeze-substituted sections stained with osmium and found the delimiting outer membranes were densely decorated with ribosomes, suggesting that those were ER-derived membranes. They therefore referred to these vesicles as ER-containing autophagosomes, or ERAs. To further confirm this observation, they performed immunogold labeling of an ER marker protein, and observed selective labeling of clearly identifiable ER structures, as well as selective labeling of ERAs. Therefore both the sequestered and the sequestering membranes come from the ER, indicating that the formation of ERAs involves an engulfment of the ER by itself, and this process is termed “ER-phagy” (Bernales et al., 2006).
ER turnover via autophagy can be elicited by several stimuli including nutrient deprivation and chemicals agents (e.g., thapsigargin and dithiothreitol -DTT-) that are also able to induce general autophagy, ER stress and UPR. In some cases, these pleiotropic signals may increase the level of ER-phagy receptors. FAM134B was shown to be transcriptionally regulated by TFEB/TFE3 transcription factors, in MEFs and HeLa cells upon prolonged starvation (Cinque et al., 2020). Activation of the TFEB/TFE3 axis also increases FAM134B expression in chondrocytes exposed to fibroblast growth factor 18 (FGF18) (Cinque et al., 2020). In glioblastoma cells, FAM134B and TEX264 transcription is highly induced upon treatment with loperamide, a compound that induces ER stress resulting in autophagy-induced cell death (Liao et al., 2019; Koenig et al., 2020; Zielke et al., 2021). SEC62 expression and the following SEC62-mediated ER-phagy is enhanced by globular adiponectin treatment of cardiomyocytes subjected to chronic intermittent hypoxia, which protects them from ER stress-induced cell death (Zhang et al., 2020). CCPG1 expression is increased in cells treated with DTT, tunicamycin, or thapsigargin (Smith et al., 2018). In particular, evidence suggests that transcriptional regulation of this ER-phagy receptor depends on PERK (Adamson et al., 2016). These data support the idea that ER-phagy acts as a stress response mechanism that works in concert with the UPR to resolve the stressful condition. Consistently, MCF-7 cells (breast cancer) survive hypoxia induced ER stress by promoting FAM134B mediated ER-phagy (Chipurupalli et al., 2022).
As part of the recovery process, ER-phagy ensures proper ER fitness by eliminating excess ER membranes, created in response to stress, and may contribute to the elimination of protein aggregates. FAM134B binds misfolded procollagen with the aid of the ER chaperone Calnexin (CANX), which acts as a co-receptor. In particular, CANX recognizes and binds misfolded procollagen in the ER lumen and interacts with FAM134B which drives the autophagic degradation of both procollagen and CANX (Forrester et al., 2019). However, it is not clear whether ER-phagy contributes clearing out of UPR substrates when the UPR is defective.
Other observations further support the crosstalk between the UPR and ER-phagy. In particular: 1) chemical inducers of the UPR also trigger ER-phagy (Bernales et al., 2006; Ogata et al., 2006; Moretti et al., 2017; Liu et al., 2019), 2) ER-phagy protects from cell death upon the induction of the UPR (Ogata et al., 2006; Zhao et al., 2020), 3) ER-phagy receptors such as FAM134B, TEX264 (Zielke et al., 2021), and CCPG1 are induced upon the activation of the UPR and promote ER turnover (Smith et al., 2018) and 4) ER-phagy receptors, such as SEC62, are activated during recovery from ER stress and play a crucial role in resuming the physiological size and function of the ER (Fumagalli et al., 2016).
Ultimately, it can be affirmed that, by working in a coordinated manner, UPR and ER-phagy ensure ER plasticity, which is essential for a proper response to the metabolic changes of the cell. However, the mechanistic details of this relationship have yet to be fully understood (Figure 1).
In response to the stressful conditions within the tumor microenvironment, cancer cells must adapt their metabolism by activating a series of metabolic pathways that ensure their survival. Hypoxia is a stressful condition that drives such metabolic changes. It triggers autophagy and UPR which, in turn, sustain the induction of autophagy, creating a feedback loop of signaling pathways that are differentially modulated. Under stress conditions, both bulk and selective autophagy are actively regulated implicating ER-phagy as an important piece of this complicated puzzle. Although a direct link between ER-phagy and cancer progression is still missing, several lines of evidence support this hypothesis. Indeed, operating in concert with the UPR, and being itself induced upon hypoxia, ER-phagy may contribute to cancer cell homeostasis by maintaining ER fitness. In this scenario, hypoxia acts as an ER stress inducer while UPR and ER-phagy are prompted to resolve the stress and promote cell survival. In a tumorigenic environment this is favoring cancer cell adaptation and proliferation. However, the ER-phagy field is still in its infancy and much more needs to be understood. Although the number of ER-phagy receptors identified and characterized in recent years has exponentially increased, very little is known regarding the regulatory mechanisms that lead to their activation. Understanding these pathways will help uncover the role of ER-phagy in cancer progression, paving the way to the discovery of new druggable targets.
DG, ME, and PG wrote and edited the manuscript.
The authors acknowledge AIRC (MFAG 24856) and Telethon Foundation (TMPGCBX16TT). We acknowledge Anna Vainshtein (Craft Science Inc.) for critical reading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdul Rahim, S. A., Dirkse, A., Oudin, A., Schuster, A., Bohler, J., Barthelemy, V., et al. (2017). Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 117, 813–825. doi:10.1038/bjc.2017.263
Abraham, R. T. (2004). PI 3-kinase related kinases: “big” players in stress-induced signaling pathways. DNA Repair 3, 883–887. doi:10.1016/j.dnarep.2004.04.002
Adamson, B., Norman, T. M., Jost, M., Cho, M. Y., Nuñez, J. K., Chen, Y., et al. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882. doi:10.1016/j.cell.2016.11.048
An, H., Ordureau, A., Paulo, J. A., Shoemaker, C. J., Denic, V., Harper, J. W., et al. (2019). TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol. Cell 74, 891–908. doi:10.1016/j.molcel.2019.03.034
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi:10.1083/jcb.200803137
Barazzuol, L., Giamogante, F., and Calì, T. (2021). Mitochondria associated membranes (MAMs): Architecture and physiopathological role. Cell Calcium 94, 102343. doi:10.1016/j.ceca.2020.102343
Bartoszewski, R., Brewer, J. W., Rab, A., Crossman, D. K., Bartoszewska, S., Kapoor, N., et al. (2011). The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 286, 41862–41870. doi:10.1074/jbc.M111.304956
B’chir, W., Maurin, A.-C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. doi:10.1093/nar/gkt563
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouysségur, J., et al. (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581. doi:10.1128/MCB.00166-09
Bergmann, T. J., Fumagalli, F., Loi, M., and Molinari, M. (2017). Role of SEC62 in ER maintenance: A link with ER stress tolerance in SEC62-overexpressing tumors? Mol. Cell. Oncol. 4, e1264351. doi:10.1080/23723556.2016.1264351
Bernales, S., McDonald, K. L., and Walter, P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4, e423. doi:10.1371/journal.pbio.0040423
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi:10.1038/35014014
Bian, X., Klemm, R. W., Liu, T. Y., Zhang, M., Sun, S., Sui, X., et al. (2011). Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. U. S. A. 108, 3976–3981. doi:10.1073/pnas.1101643108
Birgisdottir, Å. B., Lamark, T., and Johansen, T. (2013). The LIR motif - crucial for selective autophagy. J. Cell Sci. 126, 3237–3247. doi:10.1242/jcs.126128
Chen, Q., Xiao, Y., Chai, P., Zheng, P., Teng, J., Chen, J., et al. (2019). ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol. 29, 846–855. doi:10.1016/j.cub.2019.01.041
Chen, Z., Siraj, S., Liu, L., and Chen, Q. (2017). MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 13, 1244–1245. doi:10.1080/15548627.2017.1310789
Chino, H., Hatta, T., Natsume, T., and Mizushima, N. (2019). Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 74, 909–921. doi:10.1016/j.molcel.2019.03.033
Chipurupalli, S., Kannan, E., Tergaonkar, V., D’Andrea, R., and Robinson, N. (2019). Hypoxia induced ER stress response as an adaptive mechanism in cancer. Int. J. Mol. Sci. 20, 749. doi:10.3390/ijms20030749
Chipurupalli, S., Ganesan, R., Martini, G., Mele, L., Reggio, A., Esposito, M., et al. (2022). Cancer cells adapt FAM134B/BiP mediated ER-phagy to survive hypoxic stress. Cell Death Dis. 13, 357. doi:10.1038/s41419-022-04813-w
Cinque, L., De Leonibus, C., Iavazzo, M., Krahmer, N., Intartaglia, D., Salierno, F. G., et al. (2020). MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B. EMBO J. 39, e105696. doi:10.15252/embj.2020105696
Cohen-Kaplan, V., Livneh, I., Avni, N., Cohen-Rosenzweig, C., and Ciechanover, A. (2016). The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell Biol. 79, 403–418. doi:10.1016/j.biocel.2016.07.019
Corazzari, M., Gagliardi, M., Fimia, G. M., and Piacentini, M. (2017). Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 7, 78. doi:10.3389/fonc.2017.00078
Corazzari, M., Rapino, F., Ciccosanti, F., Giglio, P., Antonioli, M., Conti, B., et al. (2015). Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 22, 946–958. doi:10.1038/cdd.2014.183
Corcelle, E. A., Puustinen, P., and Jäättelä, M. (2009). Apoptosis and autophagy: Targeting autophagy signalling in cancer cells -’trick or treats. FEBS J. 276, 6084–6096. doi:10.1111/j.1742-4658.2009.07332.x
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. doi:10.1016/0092-8674(93)90648-a
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi:10.1083/jcb.200604016
Cuervo, A. M., and Wong, E. (2014). Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 24, 92–104. doi:10.1038/cr.2013.153
Dai, X., Hua, T., and Hong, T. (2017). Integrated diagnostic network construction reveals a 4-gene panel and 5 cancer hallmarks driving breast cancer heterogeneity. Sci. Rep. 7, 6827. doi:10.1038/s41598-017-07189-6
de Almeida, S. F., Fleming, J. V., Azevedo, J. E., Carmo-Fonseca, M., and de Sousa, M. (2007). Stimulation of an unfolded protein response impairs MHC class I expression. J. Immunol. 178, 3612–3619. doi:10.4049/jimmunol.178.6.3612
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi:10.1038/nature07534
Deng, J., Lu, P. D., Zhang, Y., Scheuner, D., Kaufman, R. J., Sonenberg, N., et al. (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24, 10161–10168. doi:10.1128/MCB.24.23.10161-10168.2004
Di Bartolomeo, S., Corazzari, M., Nazio, F., Oliverio, S., Lisi, G., Antonioli, M., et al. (2010). The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 191, 155–168. doi:10.1083/jcb.201002100
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998. doi:10.1038/ni1102-991
Fels, D. R., and Koumenis, C. (2006). The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol. Ther. 5, 723–728. doi:10.4161/cbt.5.7.2967
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi:10.1038/cr.2013.168
Feng, Y., and Klionsky, D. J. (2017). Autophagic membrane delivery through ATG9. Cell Res. 27, 161–162. doi:10.1038/cr.2017.4
Forrester, A., De Leonibus, C., Grumati, P., Fasana, E., Piemontese, M., Staiano, L., et al. (2019). A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 38, e99847. doi:10.15252/embj.201899847
Fumagalli, F., Noack, J., Bergmann, T. J., Cebollero, E., Pisoni, G. B., Fasana, E., et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18, 1173–1184. doi:10.1038/ncb3423
Gade, P., Manjegowda, S. B., Nallar, S. C., Maachani, U. B., Cross, A. S., Kalvakolanu, D. V., et al. (2014). Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 34, 4033–4048. doi:10.1128/MCB.00397-14
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo‐San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi:10.15252/embj.201796697
Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169. doi:10.1101/cshperspect.a013169
Gatica, D., Lahiri, V., and Klionsky, D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242. doi:10.1038/s41556-018-0037-z
Granados, D. P., Tanguay, P.-L., Hardy, M.-P., Caron, E., de Verteuil, D., Meloche, S., et al. (2009). ER stress affects processing of MHC class I-associated peptides. BMC Immunol. 10, 10. doi:10.1186/1471-2172-10-10
Grumati, P., Dikic, I., and Stolz, A. (2018). ER-phagy at a glance. J. Cell Sci. 131, jcs217364. doi:10.1242/jcs.217364
Grumati, P., Morozzi, G., Hölper, S., Mari, M., Harwardt, M.-L. I., Yan, R., et al. (2017). Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6, e25555. doi:10.7554/eLife.25555
Gubas, A., and Dikic, I. (2022). ER remodeling via ER-phagy. Mol. Cell 82, 1492–1500. doi:10.1016/j.molcel.2022.02.018
Gui, L., Liu, B., and Lv, G. (2016). Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp. Ther. Med. 11, 2233–2239. doi:10.3892/etm.2016.3190
Guo, J. Y., Chen, H.-Y., Mathew, R., Fan, J., Strohecker, A. M., Karsli-Uzunbas, G., et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. doi:10.1101/gad.2016311
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi:10.1016/j.molcel.2008.03.003
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., Gustafsson, Å. B., et al. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104. doi:10.1074/jbc.M111.322933
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi:10.1016/s1097-2765(00)00108-8
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi:10.1038/16729
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi:10.1091/mbc.10.11.3787
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi:10.1146/annurev-genet-102808-114910
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1–atg13–fip200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi:10.1091/mbc.e08-12-1248
Høyer-Hansen, M., Bastholm, L., Szyniarowski, P., Campanella, M., Szabadkai, G., Farkas, T., et al. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205. doi:10.1016/j.molcel.2006.12.009
Hu, P., Han, Z., Couvillon, A. D., Kaufman, R. J., and Exton, J. H. (2006). Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26, 3071–3084. doi:10.1128/MCB.26.8.3071-3084.2006
Huang, J., and Manning, B. D. (2008). The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 412, 179–190. doi:10.1042/BJ20080281
Inoki, K., Zhu, T., and Guan, K.-L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi:10.1016/s0092-8674(03)00929-2
Islam, F., Gopalan, V., Wahab, R., Smith, R. A., Qiao, B., Lam, A. K.-Y., et al. (2017). Stage dependent expression and tumor suppressive function of FAM134B (JK1) in colon cancer. Mol. Carcinog. 56, 238–249. doi:10.1002/mc.22488
Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. doi:10.4161/auto.7.3.14487
Johansen, T., and Lamark, T. (2020). Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 432, 80–103. doi:10.1016/j.jmb.2019.07.016
Jung, C. H., Jun, C. B., Ro, S.-H., Kim, Y.-M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. doi:10.1091/mbc.e08-12-1249
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M., and Kim, D.-H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi:10.1016/j.febslet.2010.01.017
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi:10.1093/emboj/19.21.5720
Kalvakolanu, D. V., and Gade, P. (2012). IFNG and autophagy: A critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 8, 1673–1674. doi:10.4161/auto.21403
Kaur, J., and Debnath, J. (2015). Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472. doi:10.1038/nrm4024
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. doi:10.1038/nature14498
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi:10.1038/ncb2152
Kimmelman, A. C. (2011). The dynamic nature of autophagy in cancer. Genes Dev. 25, 1999–2010. doi:10.1101/gad.17558811
Kirkin, V., McEwan, D. G., Novak, I., and Dikic, I. (2009). A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269. doi:10.1016/j.molcel.2009.04.026
Koenig, U., Robenek, H., Barresi, C., Brandstetter, M., Resch, G. P., Gröger, M., et al. (2020). Cell death induced autophagy contributes to terminal differentiation of skin and skin appendages. Autophagy 16, 932–945. doi:10.1080/15548627.2019.1646552
Korolchuk, V. I., Mansilla, A., Menzies, F. M., and Rubinsztein, D. C. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527. doi:10.1016/j.molcel.2009.01.021
Kouroku, Y., Fujita, E., Tanida, I., Ueno, T., Isoai, A., Kumagai, H., et al. (2007). ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239. doi:10.1038/sj.cdd.4401984
Kuang, Y., Ma, K., Zhou, C., Ding, P., Zhu, Y., Chen, Q., et al. (2016). Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12, 2363–2373. doi:10.1080/15548627.2016.1238552
Kurth, I., Pamminger, T., Hennings, J. C., Soehendra, D., Huebner, A. K., Rotthier, A., et al. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 41, 1179–1181. doi:10.1038/ng.464
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594. doi:10.1242/jcs.051011
Lee, A.-H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi:10.1128/mcb.23.21.7448-7459.2003
Lee, K., Tirasophon, W., Shen, X., Michalak, M., Prywes, R., Okada, T., et al. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466. doi:10.1101/gad.964702
Li, W.-W., Li, J., and Bao, J.-K. (2012). Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 69, 1125–1136. doi:10.1007/s00018-011-0865-5
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676. doi:10.1038/45257
Liao, Y., Duan, B., Zhang, Y., Zhang, X., and Xia, B. (201920009–20023). Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. J. Biol. Chem. 294, 20009–20023. doi:10.1074/jbc.RA119.008709
Linxweiler, M., Schick, B., and Zimmermann, R. (2017). Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2, 17002. doi:10.1038/sigtrans.2017.2
Liu, D., Wu, H., Wang, C., Li, Y., Tian, H., Siraj, S., et al. (2019). STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749. doi:10.1038/s41418-018-0251-z
Liu, J., Xiao, M., Li, J., Wang, D., He, Y., He, J., et al. (2017). Activation of UPR signaling pathway is associated with the malignant progression and poor prognosis in prostate cancer. Prostate 77, 274–281. doi:10.1002/pros.23264
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi:10.1038/ncb2422
Liu, M., Jiang, L., Fu, X., Wang, W., Ma, J., Tian, T., et al. (2018). Cytoplasmic liver kinase B1 promotes the growth of human lung adenocarcinoma by enhancing autophagy. Cancer Sci. 109, 3055–3067. doi:10.1111/cas.13746
Liu, X.-X., and Liu, F.-J. (2015). Novel bioinformatic identification of differentially expressed tissue-specific and cancer-related proteins from the Human Protein Atlas for biomarker discovery. Genet. Mol. Res. 14, 4557–4565. doi:10.4238/2015.May.4.14
Liu, X., Su, K., Sun, X., Jiang, Y., Wang, L., Hu, C., et al. (2021). Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 40, 132. doi:10.1186/s13046-021-01934-6
Liu, Z., Lv, Y., Zhao, N., Guan, G., and Wang, J. (2015). Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 6, e1822. doi:10.1038/cddis.2015.183
Lu, P. D., Harding, H. P., and Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33. doi:10.1083/jcb.200408003
Luo, T., Fu, J., Xu, A., Su, B., Ren, Y., Li, N., et al. (2016). PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 12, 1355–1371. doi:10.1080/15548627.2015.1034405
Mahadevan, N. R., Anufreichik, V., Rodvold, J. J., Chiu, K. T., Sepulveda, H., Zanetti, M., et al. (2012). Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8+ T cell priming. PLoS One 7, e51845. doi:10.1371/journal.pone.0051845
Ma, X. M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318. doi:10.1038/nrm2672
Mahadevan, N. R., and Zanetti, M. (2011). Tumor stress inside out: Cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment. J. Immunol. 187, 4403–4409. doi:10.4049/jimmunol.1101531
Margariti, A., Li, H., Chen, T., Martin, D., Vizcay-Barrena, G., Alam, S., et al. (2013). XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 288, 859–872. doi:10.1074/jbc.M112.412783
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H.-Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075. doi:10.1016/j.cell.2009.03.048
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi:10.1038/nature06639
Mo, J., Chen, J., and Zhang, B. (2020). Critical roles of FAM134B in ER-phagy and diseases. Cell Death Dis. 11, 983. doi:10.1038/s41419-020-03195-1
Molinari, M. (2021). ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev. Cell 56, 949–966. doi:10.1016/j.devcel.2021.03.005
Moretti, J., Roy, S., Bozec, D., Martinez, J., Chapman, J. R., Ueberheide, B., et al. (2017). STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell 171, 809–823. doi:10.1016/j.cell.2017.09.034
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Mart\’\inez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi:10.1038/emboj.2013.168
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178. doi:10.1016/j.cell.2007.05.021
Nath, S., Dancourt, J., Shteyn, V., Puente, G., Fong, W. M., Nag, S., et al. (2014). Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat. Cell Biol. 16, 415–424. doi:10.1038/ncb2940
Nazio, F., Strappazzon, F., Antonioli, M., Bielli, P., Cianfanelli, V., Bordi, M., et al. (2013). mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416. doi:10.1038/ncb2708
Ney, P. A. (2015). Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 1853, 2775–2783. doi:10.1016/j.bbamcr.2015.02.022
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi:10.1038/embor.2009.256
Ogata, M., Hino, S.-I., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231. doi:10.1128/MCB.01453-06
Ozkirimli, E., and Post, C. B. (2006). Src kinase activation: A switched electrostatic network. Protein Sci. 15, 1051–1062. doi:10.1110/ps.051999206
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473. doi:10.1089/ars.2013.5371
Phillips, M. J., and Voeltz, G. K. (2016). Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82. doi:10.1038/nrm.2015.8
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, 1809–1820. doi:10.1172/JCI20039
Reggio, A., Buonomo, V., Berkane, R., Bhaskara, R. M., Tellechea, M., Peluso, I., et al. (2021). Role of FAM134 paralogues in endoplasmic reticulum remodeling, ER-phagy, and Collagen quality control. EMBO Rep. 22, e52289. doi:10.15252/embr.202052289
Reggio, A., Buonomo, V., and Grumati, P. (2020). Eating the unknown: Xenophagy and ER-phagy are cytoprotective defenses against pathogens. Exp. Cell Res. 396, 112276. doi:10.1016/j.yexcr.2020.112276
Reggiori, F., and Molinari, M. (2022). ER-Phagy: Mechanisms, regulation and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiol. Rev.
Rismanchi, N., Soderblom, C., Stadler, J., Zhu, P.-P., and Blackstone, C. (2008). Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 17, 1591–1604. doi:10.1093/hmg/ddn046
Rodvold, J. J., Chiu, K. T., Hiramatsu, N., Nussbacher, J. K., Galimberti, V., Mahadevan, N. R., et al. (2017). Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci. Signal. 10, eaah7177. doi:10.1126/scisignal.aah7177
Romero-Ramirez, L., Cao, H., Nelson, D., Hammond, E., Lee, A.-H., Yoshida, H., et al. (2004). XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 64, 5943–5947. doi:10.1158/0008-5472.CAN-04-1606
Sabers, C. J., Martin, M. M., Brunn, G. J., Williams, J. M., Dumont, F. J., Wiederrecht, G., et al. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270, 815–822. doi:10.1074/jbc.270.2.815
Sakaki, K., and Kaufman, R. J. (2008). Regulation of ER stress-induced macroautophagy by protein kinase C. Autophagy 4, 841–843. doi:10.4161/auto.6607
Sakaki, K., Wu, J., and Kaufman, R. J. (2008). Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J. Biol. Chem. 283, 15370–15380. doi:10.1074/jbc.M710209200
Salazar, M., Carracedo, A., Salanueva, I. J., Hernández-Tiedra, S., Lorente, M., Egia, A., et al. (2009). Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 119, 1359–1372. doi:10.1172/jci37948
Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., et al. (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176. doi:10.1016/s1097-2765(01)00265-9
Schwarz, D. S., and Blower, M. D. (2016). The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 73, 79–94. doi:10.1007/s00018-015-2052-6
Semenza, G. L. (2010). HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56. doi:10.1016/j.gde.2009.10.009
Semenza, G. L. (20072007). Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE, cm8. doi:10.1126/stke.4072007cm8
Smith, M. D., Harley, M. E., Kemp, A. J., Wills, J., Lee, M., Arends, M., et al. (2018). CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217–232. doi:10.1016/j.devcel.2017.11.024
Song, S., Shi, Y., Wu, W., Wu, H., Chang, L., Peng, P., et al. (2021). Reticulon 3-mediated Chk2/p53 activation suppresses hepatocellular carcinogenesis and is blocked by Hepatitis B virus. Gut 70, 2159–2171. doi:10.1136/gutjnl-2020-321386
Spiotto, M. T., Banh, A., Papandreou, I., Cao, H., Galvez, M. G., Gurtner, G. C., et al. (2010). Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 70, 78–88. doi:10.1158/0008-5472.CAN-09-2747
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi:10.1038/ncb2979
Su, C.-C. (2018). Tanshinone IIA can inhibit MiaPaCa-2 human pancreatic cancer cells by dual blockade of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways. Oncol. Rep. 40, 3102–3111. doi:10.3892/or.2018.6670
Suzuki, H., Kanekura, K., Levine, T. P., Kohno, K., Olkkonen, V. M., Aiso, S., et al. (2009). ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 108, 973–985. doi:10.1111/j.1471-4159.2008.05857.x
Takamura, A., Komatsu, M., Hara, T., Sakamoto, A., Kishi, C., Waguri, S., et al. (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800. doi:10.1101/gad.2016211
Tang, J. C., Lam, K. Y., Law, S., Wong, J., and Srivastava, G. (2001). Detection of genetic alterations in esophageal squamous cell carcinomas and adjacent normal epithelia by comparative DNA fingerprinting using inter-simple sequence repeat PCR. Clin. Cancer Res. 7, 1539–1545.
Tang, W. K., Chui, C. H., Fatima, S., Kok, S. H. L., Pak, K. C., Ou, T. M., et al. (2007). Oncogenic properties of a novel gene JK-1 located in chromosome 5p and its overexpression in human esophageal squamous cell carcinoma. Int. J. Mol. Med. 19, 915–923.
Tolkovsky, A. M. (2009). Mitophagy. Biochim. Biophys. Acta - Mol. Cell Res. 1793, 1508–1515. doi:10.1016/j.bbamcr.2009.03.002
Vandewynckel, Y.-P., Laukens, D., Bogaerts, E., Paridaens, A., den Bussche, A., Verhelst, X., et al. (2015). Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 9, 93–104. doi:10.1007/s12072-014-9582-0
Vattem, K. M., and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 101, 11269–11274. doi:10.1073/pnas.0400541101
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J.-P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi:10.1038/cdd.2012.74
Verfaillie, T., Salazar, M., Velasco, G., and Agostinis, P. (2010). Linking ER stress to autophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 930509. doi:10.1155/2010/930509
Villanueva-Paz, M., Cotán, D., Garrido-Maraver, J., Oropesa-Ávila, M., de la Mata, M., Delgado-Pavón, A., et al. (2016). AMPK regulation of cell growth, apoptosis, autophagy, and bioenergetics. Exp. Suppl. 107, 45–71. doi:10.1007/978-3-319-43589-3_3
Walter, L., and Hajnóczky, G. (2005). Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J. Bioenerg. Biomembr. 37, 191–206. doi:10.1007/s10863-005-6600-x
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. doi:10.1016/j.molcel.2008.06.001
Wilkinson, S. (2019). ER-Phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 286, 2645–2663. doi:10.1111/febs.14932
Wouters, B. G., and Koritzinsky, M. (2008). Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 8, 851–864. doi:10.1038/nrc2501
Wu, H., Xue, D., Chen, G., Han, Z., Huang, L., Zhu, C., et al. (2014). The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10, 1712–1725. doi:10.4161/auto.29568
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi:10.1016/j.cell.2006.01.016
Xia, Z., Dickens, M., Raingeaud, J., Davis, R. J., and Greenberg, M. E. (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Sci. (80-. ) 270, 1326–1331. doi:10.1126/science.270.5240.1326
Yamamoto, K., Venida, A., Yano, J., Biancur, D. E., Kakiuchi, M., Gupta, S., et al. (2020). Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105. doi:10.1038/s41586-020-2229-5
Yamazaki, H., Hiramatsu, N., Hayakawa, K., Tagawa, Y., Okamura, M., Ogata, R., et al. (2009). Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183, 1480–1487. doi:10.4049/jimmunol.0900017
Yang, L. V. (2017). Tumor microenvironment and metabolism. Int. J. Mol. Sci. 18, E2729. doi:10.3390/ijms18122729
Yang, Y. S., and Strittmatter, S. M. (2007). The reticulons: A family of proteins with diverse functions. Genome Biol. 8, 234. doi:10.1186/gb-2007-8-12-234
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi:10.1016/j.ceb.2009.11.014
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi:10.1016/s0092-8674(01)00611-0
Yuan, Y., Zheng, Y., Zhang, X., Chen, Y., Wu, X., Wu, J., et al. (2017). BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 13, 1754–1766. doi:10.1080/15548627.2017.1357792
Zalckvar, E., Berissi, H., Mizrachy, L., Idelchuk, Y., Koren, I., Eisenstein, M., et al. (2009). DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 10, 285–292. doi:10.1038/embor.2008.246
Zhang, J., and Ney, P. A. (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 16, 939–946. doi:10.1038/cdd.2009.16
Zhang, X., Ding, X., Marshall, R. S., Paez-Valencia, J., Lacey, P., Vierstra, R. D., et al. (2020). Reticulon proteins modulate autophagy of the endoplasmic reticulum in maize endosperm. Elife 9, e51918. doi:10.7554/eLife.51918
Zhang, Z.-Q., Chen, J., Huang, W.-Q., Ning, D., Liu, Q.-M., Wang, C., et al. (2019). FAM134B induces tumorigenesis and epithelial-to-mesenchymal transition via Akt signaling in hepatocellular carcinoma. Mol. Oncol. 13, 792–810. doi:10.1002/1878-0261.12429
Zhao, D., Zou, C.-X., Liu, X.-M., Jiang, Z.-D., Yu, Z.-Q., Suo, F., et al. (2020). A UPR-induced soluble ER-phagy receptor acts with VAPs to confer ER stress resistance. Mol. Cell 79, 963–977. e3. doi:10.1016/j.molcel.2020.07.019
Zhu, D., Zhou, J., Zhao, J., Jiang, G., Zhang, X., Zhang, Y., et al. (2019). ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. J. Cell. Physiol. 234, 8899–8907. doi:10.1002/jcp.27551
Keywords: cancer, autophagy, ER-phagy, hypoxia, ER stress, UPR, endoplasmic reticulum
Citation: Gentile D, Esposito M and Grumati P (2022) Metabolic adaption of cancer cells toward autophagy: Is there a role for ER-phagy?. Front. Mol. Biosci. 9:930223. doi: 10.3389/fmolb.2022.930223
Received: 27 April 2022; Accepted: 04 July 2022;
Published: 03 August 2022.
Edited by:
Emiliano Maiani, Saint Camillus International University of Health and Medical Sciences, ItalyReviewed by:
Giacomo Milletti, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2022 Gentile, Esposito and Grumati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Grumati, cC5ncnVtYXRpQHRpZ2VtLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.