 Ewud Agborbesong
Ewud Agborbesong Linda Xiaoyan Li
Linda Xiaoyan Li Lu Li
Lu Li Xiaogang Li
Xiaogang Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci., 29 June 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.922428
This article is part of the Research TopicMolecular Mechanisms Underlying Polycystic Kidney Disease: From the Smallest Bricks to the Big ScenarioView all 10 articles
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder, which is caused by mutations in the PKD1 and PKD2 genes, characterizing by progressive growth of multiple cysts in the kidneys, eventually leading to end-stage kidney disease (ESKD) and requiring renal replacement therapy. In addition, studies indicate that disease progression is as a result of a combination of factors. Understanding the molecular mechanisms, therefore, should facilitate the development of precise therapeutic strategies for ADPKD treatment. The roles of epigenetic modulation, interstitial inflammation, and regulated cell death have recently become the focuses in ADPKD. Different epigenetic regulators, and the presence of inflammatory markers detectable even before cyst growth, have been linked to cyst progression. Moreover, the infiltration of inflammatory cells, such as macrophages and T cells, have been associated with cyst growth and deteriorating renal function in humans and PKD animal models. There is evidence supporting a direct role of the PKD gene mutations to the regulation of epigenetic mechanisms and inflammatory response in ADPKD. In addition, the role of regulated cell death, including apoptosis, autophagy and ferroptosis, have been investigated in ADPKD. However, there is no consensus whether cell death promotes or delays cyst growth in ADPKD. It is therefore necessary to develop an interactive picture between PKD gene mutations, the epigenome, inflammation, and cell death to understand why inherited PKD gene mutations in patients may result in the dysregulation of these processes that increase the progression of renal cyst formation.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic disorder of the kidney, caused by mutations in the PKD1 and PKD2 genes that encode for transmembrane proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively (Grantham, 2008). The disease is characterized by the formation and enlargement of fluid filled cysts in the kidneys, and patients with ADPKD eventually develop renal insufficiency and end-stage kidney disease (ESKD), requiring dialysis or kidney transplants (Alam and Perrone, 2010; Grantham et al., 2011). Disease development is associated with hypertension, hematuria, and urinary tract infections (Bajwa et al., 2001; Ecder and Schrier, 2009; Torres and Harris, 2009). In addition, extra-renal complications include cyst formation in other epithelial organs, including the liver and pancreas (Harris et al., 1993). To date, there is only one Food and Drug Administration (FDA) approved treatment, Tolvaptan, for ADPKD (Torres et al., 2012). However, the long-term use of Tolvaptan causes side effects, such as thirst, polyuria, and liver injury (Torres et al., 2012). Therefore, there is an urgent need to develop more effective and safe treatments with the better understanding of molecular mechanisms of ADPKD.
Mutations in the PKD1 and PKD2 genes correlate with the development of ADPKD. Individuals with an inherited PKD gene mutation develop detectable renal cysts by 30 years of age (Nicolau et al., 1999). On average, PKD1 gene mutations lead to ESKD at ∼54 years compared to ∼74 years for PKD2 (Hateboer et al., 1999). A striking feature of ADPKD is the variability in the phenotype, with the disease severity, the on-set of ESKD, and the spectrum of extra-renal manifestations being highly variable between patients, and even within members of the same family (Milutinovic et al., 1992; Fick et al., 1993; Zerres et al., 1993). Owing to the research focus in our lab over the past 15 years, we will in this review, discuss three molecular mechanisms that may contribute to the disease variability and progression of ADPKD, including epigenetic mechanisms, inflammation, and cell death.
First, epigenetics is broadly defined as a genomic mechanism that reversibly influences gene expression without affecting the DNA sequence (Berger et al., 2009). Epigenetic regulation has been proposed as a potential mechanism to explain disease variability, including ADPKD (Villota-Salazar et al., 2016). We and others found an abnormal upregulation of epigenetic modifiers in kidneys from Pkd1 animal models and in ADPKD patients (Li, 2011; Bowden et al., 2021). Moreover, inhibition of specific epigenetic factors reduces cyst growth and improves kidney function in preclinical studies, enforcing the role of epigenetic mechanisms in ADPKD (Zhou et al., 2013; Zhou et al., 2014; Zhou et al., 2015; Li et al., 2017c).
Second, we found that the progression of PKD can be influenced by the presence of inflammatory factors such as tumor necrosis factor alpha (TNF-α) (Li et al., 2008) and macrophage migration inhibitory factor (MIF) (Chen et al., 2015) in the cyst fluid. Inhibiting or reducing inflammation by decreasing macrophages for example, has been demonstrated to reduce cyst burden and improve renal function, thereby displaying beneficial effects both on cyst burden and disease progression in preclinical PKD animal models (Swenson-Fields et al., 2013). The upregulation of genes associated with immune and inflammatory responses have also been identified by microarray analysis of ADPKD kidneys (Schieren et al., 2006; Song et al., 2009). Increased T cells (component of the adaptive immunity), specifically localized to cystic lesions, correlate with disease severity. In particular, the role of CD8+ T cells in inhibiting ADPKD disease progression has been demonstrated (Kleczko et al., 2018). These studies support the involvement of the inflammatory response, and to a broader scope, the innate and adaptive immune systems in the pathogenesis of ADPKD, and suggest that immunotherapy, such as the reactivation of T cells, might represent a novel therapeutic strategy. Third, we and others have reported that regulatory cell death, including apoptosis (Fan et al., 2013b), ferroptosis (Zhang et al., 2021c) and autophagy (Shillingford et al., 2006), plays a critical role in ADPKD animal models. However, there is a controversy as to whether regulated cell death promotes or delays cyst growth in ADPKD.
In this review, we discuss the roles and molecular mechanisms underlying epigenetics associated with DNA methylation and histone modifiers, inflammation, and programmed cell death in the regulation of disease progression in ADPKD. We debate on the short comings and controversies in the field and how these may impact the discovery of novel mechanisms and treatment options. In addition, we summarize the therapeutic implications and outcomes associated with the therapy of epigenetic, inflammation and cell death. Finally, we provide perspectives on how a better understanding of the diverse mechanisms involved in cyst growth may be applied for combined therapeutic strategies in ADPKD.
Epigenetic alterations which ultimately influence key signaling pathways, have recently been suggested to affect the pathogenesis of ADPKD (Li, 2015). Epigenetic mechanisms including, but not limited to DNA methylation and histone modification, act to regulate accessibility of the DNA by transcription factors to control gene expression (Stuppia et al., 2015). Epigenetic mechanisms play a role in cellular growth and differentiation during development, and as cells mature, these epigenetic modifications change to accommodate the role of the cell. These modifications, including any disease-causing epigenetic changes may be inherited (Robertson, 2005; Greer and Shi, 2012). In addition to regulating the chromatin state, histone modifiers are known to alter gene expression and protein function by post-translational modifications (Miller and Grant, 2013; Sadakierska-Chudy and Filip, 2015). Evidence for alterations in the epigenetic control of gene expression and protein function in ADPKD is accumulating (Li, 2011; Kerr et al., 2019; Li and Li, 2021) and emerging data regarding DNA methylation and histone/lysine modifiers in cystogenesis and ciliogenesis are discussed below.
DNA methylation is a stable and heritable epigenetic mark that involves the addition of a methyl group to cytosine residues on the genome by a group of enzymes, named DNA methyl transferases (DNMTs) (Bird, 2002). The DNMT family has four members, DNMT1, DNMT3A, DNMT3B, and DNMT3L. DNMT3A and DNMT3B are referred to as de novo methyl transferases, with DNMT3L acting as a stimulator of their catalytic activity (Jeltsch, 2006; Zhang and Xu, 2017). Together, they establish de novo methylation patterns which are maintained faithfully during cell replication by DNMT1, hence often referred to as maintenance methyl transferase (Schubeler, 2015). While this modification does not affect the DNA nucleotide sequence, it can modify the availability of the genome to the transcriptional machinery thereby affecting gene expression (Razin and Cedar, 1991). In general, methylation within the gene promoter is typically associated with gene repression and though not well understood, methylation within the gene body is typically associated with sustained or increased gene expression (Saxonov et al., 2006; Shen et al., 2007; Brenet et al., 2011). However, these could deviate from the norm as is the case of ADPKD, as discussed below. DNA methylation has been heavily implicated in human diseases. In cancers, for example, dysregulation of DNA methylation has been reported and inhibitors for DNA methyl transferases (DNMTs), have been developed and approved for the treatment of certain neoplasias, including chronic myelomonocytic leukemia (CMML) and myelodysplastic syndromes (MDS) (Jones et al., 2016). In ADPKD, cysts are believed to arise independently, however, the molecular alterations that underlie cyst formation are poorly understood. Recent studies have identified global methylation patterns of ADPKD patient kidneys and individual cysts, providing evidence of a role for DNA methylation in cystogenesis.
In a pioneering study, the global DNA methylation in ADPKD patient kidneys compared to non-ADPKD kidneys (Woo et al., 2014) was analyzed by methylated-CpG island recovery assay with parallel sequencing (MIRA-seq). This study found that 11,999 genomic fragments, out of the 15 million examined, were differentially hypermethylated, accounting for 91% of all methylation changes. However, only 1,228 genomic fragments were hypomethylated, accounting for 9% of all methylation changes. In addition, this study found that hypermethylation of the PKD1 gene body (exon 43) in ADPKD patient samples, negatively correlated with the PKD1 gene expression, suggesting that epigenetic silencing of the PKD1 gene is involved in kidney cyst development (Woo et al., 2014) (Table 1). As such, Woo et al. theorized that if hypermethylation of the ADPKD genome resulted in cyst growth, then inhibition of the DNMTs could be targeted for therapeutic purposes. In agreement with Woo et al., a second study, utilizing Reduced Representation Bisulfite Sequencing (RRBS) also reported that the PKD1 gene body was hypermethylated in ADPKD patient kidneys (Table 1). However, contrary to Woo et al., this study found that hypermethylation of the PKD1 gene body was associated with an increase in PKD1 gene expression rather than a decrease (Bowden et al., 2018). In addition, unlike Woo et al., this study showed a 2% difference in the methylation status of the genome, with ADPKD patient kidneys being hypomethylated (Bowden et al., 2018). Utilizing methylation-sensitive high-resolution melt (MS-HRM) analysis, a third study demonstrated that hypermethylation of the PKD1 promoter inversely correlated with gene expression in ADPKD patient blood (Hajirezaei et al., 2020) (Table 1). Fourth, analyzing global methylation patterns of individual cysts derived from the same ADPKD patient (Table 1) revealed that approximately 15% of analyzed fragments exhibited inter-cyst variation in DNA methylation pattern. While the CpG islands and gene body regions demonstrated elevated levels of methylation variation, the intergenic regions had comparatively stable methylation levels within cysts from the same ADPKD patient (Bowden et al., 2020).
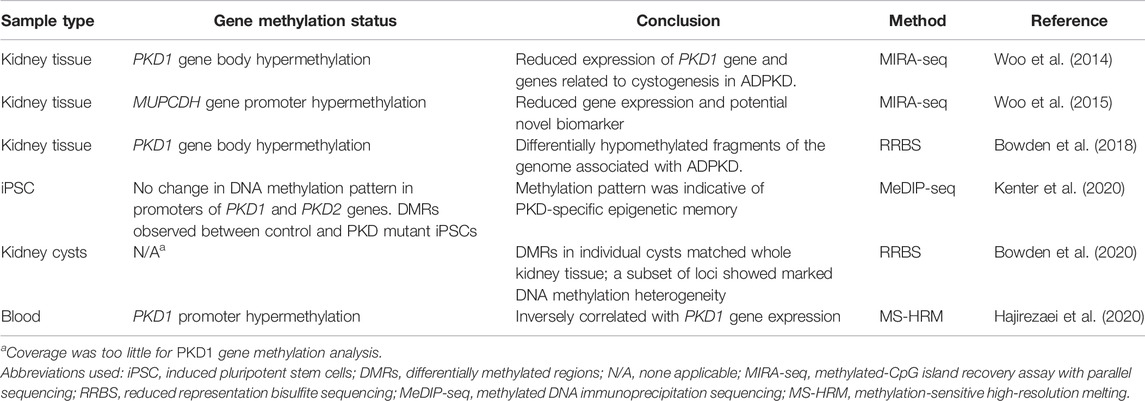
TABLE 1. Summary of DNA methylation studies in ADPKD.
The potential use of DNMT inhibitors (demethylating agents) for therapeutic purposes in cancers have been acknowledged and well documented. As such, the similarities, and associations between cancer cells and ADPKD suggest that DNMT inhibitors that slow the progress of tumors would have similar effects on cyst growth in ADPKD. At present, the demethylating agents used in clinics are cytotoxic, mutagenic and exhibit lack of specificity towards genes, limiting their clinical application. With the slow progression of ADPKD disease, the long-term use of such drugs may proof harmful. Furthermore, the effect of demethylating agents at pharmacological dosages may depend on the nature and/or extent of the epigenetic changes. ADPKD disease results from different mutations and presents with variable phenotypes, suggesting that the methylation status and subsequent molecular mechanisms may vary. Therefore, understanding epigenetics may help provide new mechanistic insights on cyst development and growth so that broad spectrum and tolerable epigenetic therapy may be developed for ADPKD disease. Advancements in genome-wide technologies have made it possible to analyze genomic methylation levels in ADPKD. Although these studies provide valuable information that point out changes in DNA methylation of the PKD1 gene, variations have been observed in the methylation status which might be caused by the differences in techniques used for analysis (Table 1). Therefore, use of Whole-Genome Bisulfite Sequencing (WGBS) may be more appropriate to provide a full, unbiased description of the extent of DNA methylation in ADPKD kidneys (Bowden et al., 2021). So far, majority of the methylation analysis studies conducted in ADPKD used kidney tissues from patients. It is important to mention that with the nature of the ADPKD disease (fluid-filled cystic kidneys), it is not practical to obtain kidney biopsies. This suggests that the data presented in the field, arises from kidney tissues obtained at ESKD. We speculate that from the initiation and on-set of cyst growth to ESKD, there may have been changes in the DNA methylation status that are not captured during these analyses. To overcome this problem, blood and/or urine samples from which genomic DNA may be obtained for analysis could be collected from ADPKD patients as the disease progresses. It is our belief that obtaining the DNA methylation status at multiple stages of the disease may provide a more comprehensive epigenetic landscape, which would lay out the foundation for future mechanistic insights and development of therapy in ADPKD. It is important to note that while blood and urine samples are readily accessible, the DNA methylation patterns identified in specific genes obtained from the blood and urine-derived genomic DNA, may not reflect the DNA methylation patterns in the genome of renal cystic epithelial cells and kidneys.
In the nucleus, DNA is organized and packaged around histone proteins, which control how accessible the DNA is to the transcription machinery (Cutter and Hayes, 2015). A range of post-translational modifications of these histone proteins (histone “tails”), play a vital role in gene expression. These post-translational modifications determine how tight or loose the histones are packaged, which in turn determines how freely DNA can be transcribed (Shen and Casaccia-Bonnefil, 2008). Several types of histone modifications are known including acetylation, and methylation, phosphorylation, ubiquitination, and sumoylation (Strahl and Allis, 2000; Lunyak and Rosenfeld, 2008; Tan et al., 2011; Graff and Tsai, 2013). Histone modifications at the N-terminal tails on amino acids such as lysine, arginine, serine, threonine, and tyrosine, are catalyzed by specific enzymes that act (Shen and Casaccia-Bonnefil, 2008). Acetylation of histones, catalyzed by histone acetyltransferases (HATs), results in active gene transcription, while deacetylation, catalyzed by histone deacetylases (HDACs) results in reduced levels of gene transcription (Vidali et al., 1988; Kuo and Allis, 1998; Turner, 2000). Histone methylation, regulated by histone methyl transferases (HMTs), results in either the activation or repression of gene transcription, depending on the targeted amino acid residue on the histone tail and/or the number of methyl groups added (mono-, di-, or tri-methylation) (Pedersen and Helin, 2010; Sawan and Herceg, 2010). Removal of methyl groups from the histone tails is catalyzed by histone demethylases (Pedersen and Helin, 2010). In ADPKD, there is accumulating evidence of the dysregulation of enzymes involved in histone acetylation/deacetylation and methylation/demethylation in cystic kidneys (Li, 2011; Li et al., 2017b; Bowden et al., 2021). To the best of our knowledge, there is little to no evidence regarding histone phosphorylation, ubiquitination, and sumoylation in ADPKD. Below, we summarize the role of histone modifying enzymes in ADPKD, with a focus on acetylation and methylation.
Growing evidence suggest that HDACs are important regulators of PKD genes and/or the signaling pathways that are involved in cystogenesis (Li, 2011). First, it has been proposed that polycystin signaling activates p53, which in turn, in cooperation with HDACs, controls PKD1 gene expression (Van Bodegom et al., 2006). This study found that the tumor suppressor protein/transcription factor, p53, was a negative regulator of PKD1, and that inhibition of HDAC activity rendered the PKD1 promoter overly sensitive. Second, HDAC5 was identified as a target of the PKD1-dependent fluid stress-sensing in renal epithelial cells (Xia et al., 2010). This study reported that polycystin-1 (PC1) facilitates calcium influx into the cell and subsequent phosphorylation of HDAC5 by protein kinase C. These studies suggested a role for HDACs in the regulation of cystogenesis in ADPKD. Subsequent studies further found that treatment with HDAC inhibitors decreased cyst growth in PKD mutants. In another study, the HDAC class I and II deacetylase inhibitor trichostatin A (TSA), and the Class I HDAC inhibitor, valproic acid, were found to effectively reduce cyst formation, body curvature and laterality in Pkd2 mutant zebra fish morphants (Cao et al., 2009). Valproic acid also reduced cyst growth in a Pkd1 mouse model. HDAC6 is upregulated in Pkd1 mutant mouse cells and was found to activate factors associated with cyst growth, such as EGFR (Liu et al., 2012). Inhibition of HDAC6 with tubacin attenuated cyst growth and improved kidney function through cAMP signaling by preventing Ca2+ efflux from the endoplasmic reticulum (Yanda et al., 2017). Additionally, the Class III HDAC SIRT1, was also upregulated in Pkd1 mutant mouse cells and kidneys to promote cyst growth and treatment with the SIRT1-specific inhibitor, EX-527, reduced cyst growth in Pkd1 mouse models (Zhou et al., 2013).
The primary cilium is a pivotal organelle for the pathogenesis of cystic kidney diseases, which presents on almost all eukaryotic cells (Eggenschwiler and Anderson, 2007). The cilium is a microtubule-based organelle that functions as a mechano- and chemo-sensor (Nauli et al., 2003). Signaling receptors expressed on the ciliary membrane mediate extracellular sensory signals, creating a response inside the cells (Eggenschwiler and Anderson, 2007; Gerdes et al., 2009; Hildebrandt et al., 2011). Polycystin-1 (PC1) and polycystin-2 (PC2) locate to the cilia and PC2 functions as a Ca2+ ion channel (Qian et al., 1997; Yoder et al., 2002; Wang et al., 2019). Furthermore, mutations in cilia-related genes or ablation of cilia result in cystic kidney diseases (Pazour and Rosenbaum, 2002; Yoder et al., 2002). In addition to their role in regulating cell proliferation associated signaling pathways in ADPKD, HDACs also regulate primary cilia structure. HDAC6 is reported to regulate primary cilia disassembly by deacetylating α-tubulin and subsequent studies demonstrated that inhibition of HDAC6 with tubacin, prevents primary cilia resorption (Pugacheva et al., 2007; Zilberman et al., 2009; Ran et al., 2015). The Class III HDAC SIRT2 was also found to regulate primary cilia disassembly and inhibition of SIRT2 by nicotinamide prevented this process (Zhou et al., 2014). Furthermore, inhibition of SIRT2 was found to reduce cyst growth in Pkd1 mouse kidneys (Zhou et al., 2014). Since HDAC6 and SIRT2 are increased in ADPKD, these studies suggest the involvement of HDAC6 and SIRT2 in the regulation of cystogenesis through cilia-dependent signaling in ADPKD. In sum, these studies suggest that HDACs contribute to ADPKD pathogenesis by regulating both PKD-mediated and cilia-dependent signaling pathways (Figure 1).
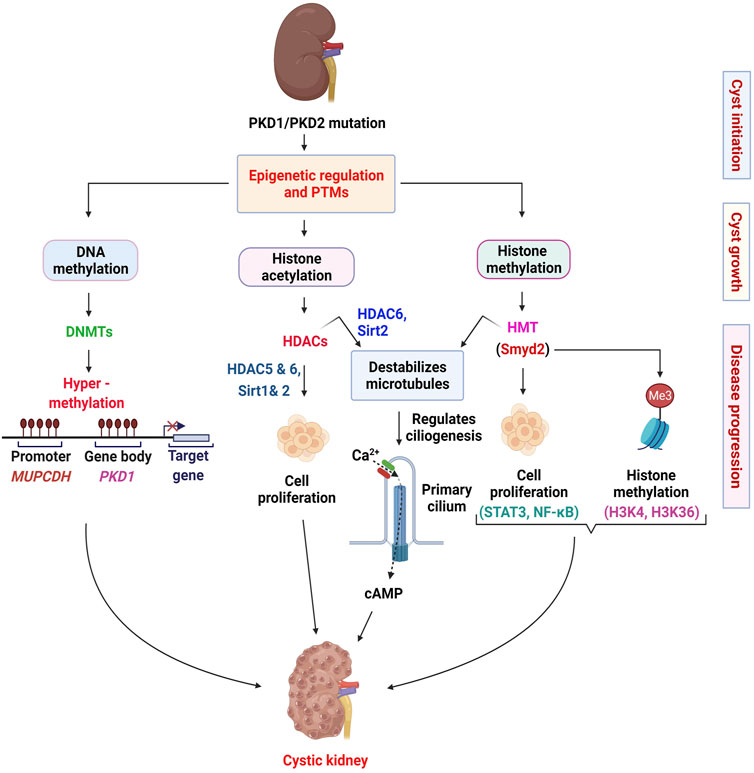
FIGURE 1. Epigenetic mechanisms implicated in the pathogenesis of ADPKD. In this scheme, we summarize the roles of DNA methyl transferases (DNMTs), histone deacetylases (HDACs), and histone methyl transferases (HMTs) in renal epithelial cells. We indicate the roles of DNMTs in regulating the transcription of PKD1 and MUPCDH genes. In general, we indicate the HDACs and the HMT involved in regulating cell proliferation associated pathways. We also depict the role of HDACs and HMTs in regulating ciliogenesis through deacetylation of α-tubulin (HDAC6 and SIRT2) and methylation of α-tubulin (Smyd2). The involvement of calcium signaling in these processes is possible but uncertain. The various stages of ADPKD (cyst initiation, cyst growth and disease progression) require different epigenetic controls and therefore may require different therapeutic approaches.
Histone methylation commonly occurs in specific arginine and lysine residues at the N- terminal tails of histones (Smith and Denu, 2009). Each arginine methylation can exist in either the mono-methylated, di-methylated symmetrical and asymmetrical methylated states (Byvoet et al., 1972; Bedford and Richard, 2005), whereas each lysine has three possible methylation states: mono-methylated, di-methylated, or tri-methylated states (Murray, 1964; Hempel et al., 1968; Nguyen et al., 2010). Differences in residue methylation and modification states correlate with either gene transcription activation or repression (Zhang and Reinberg, 2001; Li et al., 2007). Lysine methylation at H3 lysine 4 (H3K4) and H3K36 for example are associated with transcriptional activation (Ringrose and Paro, 2004). In contrast, methylation at H3K9 and H3K27 are associated with transcriptional repression (Heard et al., 2001; Schotta et al., 2002; Sims et al., 2003; Ringrose and Paro, 2004). To date, there is limited evidence to support a role for histone methylation in ADPKD pathogenesis. Recently, our group found that the histone/lysine methyl transferase SMYD2, one of the SET and MYND-containing lysine methyl transferases (SMYD), contributed to cyst growth in ADPKD (Li et al., 2017b). SMYD2 can methylate both H3K4 and H3K36 (Brown et al., 2006; Abu-Farha et al., 2008) and non-histone proteins, including p53/TP53 and RB1 (Huang et al., 2006; Cho et al., 2012).
First, we found that SMYD2 expression is increased in Pkd1 mutant mouse renal epithelial cells and kidneys as well as in ADPKD patient kidneys (Li et al., 2017b). Utilizing Pkd1 knockout mice and the SMYD2 inhibitor, AZ505, we showed that SMYD2 is a critical mediator of renal cyst growth in ADPKD. In addition, we found that SMYD2 promotes cyst growth in ADPKD via the methylation of H3K4 and H3K36. In particular, SMYD2 regulated cystic epithelial cell proliferation and survival through STAT3 and NF-κB. SMYD2-mediated methylation of STAT3 and NF-κB is important for the activation of these two pathways. We proposed that SMYD2, via two positive feedback loops: SMYD2/STAT3/SMYD2 and SMYD2/NF-κB/SMYD2, promotes cyst development in ADPKD (Figure 1). Second, we determined that SMYD2 is an α-tubulin methyl transferase that together with cyclin-dependent kinases 4 and 6 (CDK4/6), regulates ciliogenesis in renal epithelial cells (Li et al., 2020b). The cross-talk between CDK4/6 and SMYD2 is important for the regulation of ciliogenesis and targeting CDK4/6-SMYD2 signaling affects not only ciliogenesis but also cilia-dependent hedgehog signaling activation in Pkd1 mutant renal epithelial cells (Li et al., 2020b) (Figure 1). This was the first study to shed light on the contribution of an epigenetic regulator of histone methylation on cyst growth and cilia biogenesis, thereby linking the “tubulin code” (a concept that describes how post-translational modifications that mark subsets of microtubules in the cytoskeleton direct microtubule-based functions) (Park et al., 2016), and cilia-dependent signaling to histone methylation and cyst growth in ADPKD.
The study of epigenetics and associated post-translational modifications have increasingly become an area of interest in ADPKD. The culminative efforts from different research teams have led to the identification of an increasingly complex network of epigenetic mechanisms associated with cystogenesis (Figure 1). Thus far, studies in the field have focused on identifying dysregulated epigenetic modifiers and characterizing their roles and mechanisms in cystogenesis (Zhou et al., 2013; Zhou et al., 2014). Advances were made in the category of histone modifiers, with the identification of the first lysine methyl transferase, Smyd2 and how it is involved in the regulation of cystogenesis and ciliogenesis (Li et al., 2017b; Li et al., 2020b). These studies provided new molecular mechanisms of the disease and provided a novel target for therapeutic purposes. With the advancements made in technology, the field has witnessed an exponential burst in studies aimed at characterizing the epigenome of ADPKD kidneys (Woo et al., 2014; Woo et al., 2015; Bowden et al., 2018; Bowden et al., 2020). These studies have shed light on the role of DNA methylation in the pathogenesis of ADPKD. However, more studies are required to establish a consensus of DNA methylation markers and changes in ADPKD. In addition, how the PKD mutation affects epigenetic mechanisms remains unstudied. Also, the use of epigenetic patterns as markers for cell composition and origin of ADPKD cysts remains unclear. To address these questions, techniques such as whole-genome bisulfite sequencing (WGBS) and single-cell epigenomics sequencing could be applied. Though expensive, the use of WGBS would provide a complete and unbiased description of the extent of DNA methylation in ADPKD kidneys. Single-cell epigenomics sequencing on the other hand would be an effective way to identify the origin, composition, and differentially activated epigenetic mechanisms during the development and progression of ADPKD. Together, these techniques have the potential to identify specific molecular targets that would be more appropriate for ADPKD therapy.
The pharmacological control of epigenetic signatures has become a new frontier in different diseases, including cancer. However, the ubiquitous effects of epigenetic changes on pathways limit any potential clinical application in disease treatment. Hypo- and hyper-methylated states in DNA have been associated with ADPKD and thus they represent a potential therapeutic target. DNA methylation is catalyzed by the DNA methyl transferases (DNMTs), and this process potentially contributes to the suppression of gene transcription. This makes it challenging to design drugs whose mechanism of action relies on reactivation of abnormally silenced suppressor genes. There exist multiple classes of DNMT inhibitors (DNMTi) such as nucleoside analog inhibitors azacitidine, and decitabine, however, there is limited information on their efficacy in humans. Thus far, these inhibitors, in combination with chemotherapy have been employed for the treatment of cancers. However, with reports of their cytotoxicity, their use is short-term. With the slow progressive nature of ADPKD pathogenesis, long-term use of DNMTi may result in extensive cytotoxicity.
Changes in histone acetyl groups have also been recognized as epigenetic marks of ADPKD. For histone acetyltransferases (HATs) and histone deacetylases (HDACs), a correlation with cystic burden and severity has been demonstrated in ADPKD mouse models. The activity of HATs may be modulated by bromodomain and extra-terminal motif-containing proteins (BET). In this regard, Zhou et al. demonstrated that targeting the BET bromodomain (BRD) protein, Brd4 with its inhibitor JQ1 (a thieno-triazolo-1,4-diazapine) slows renal cyst growth in Pkd1 mutant mice (Table 2). With respect to potential modifiers of histone deacetylation, trichostatin A and valproic acid function as HDAC inhibitors (HDACi), and niacinamide acts as Sirtuin (SIRT) inhibitor. Even though HDACi are approved for the treatment of hematological malignancies, their beneficial application in ADPKD is limited to preclinical studies (Table 2). The Sirtuin inhibitor niacinamide on the other hand, is currently undergoing clinical trial for its potential use in the treatment of ADPKD (Table 2).
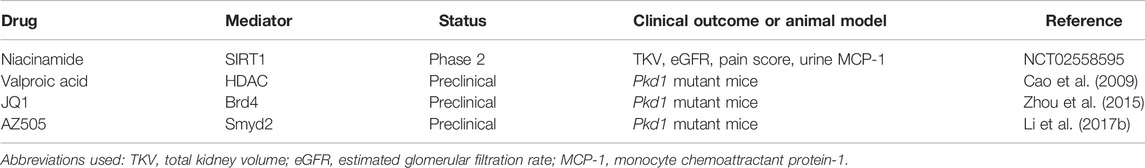
TABLE 2. Summary of clinical trials and preclinical studies targeting epigenetic factors in ADPKD.
Alterations in histone methylation patterns contribute to the epigenetic control of RNA transcription from DNA. Through the transfer of methyl group to lysine or arginine residues, histone methylation, like DNA methylation, is associated with transcriptional repression. However, exceptions exist depending on the methylated residues. Because of the ubiquitous function of the histone-lysine N-methyl transferase enzyme SMYD2, AZ505 may be a promising agent with histone methylation inhibitory properties. However, since SMYD2 plays diverse roles in different cells and organs by regulating distinct substrates, side effects may be unavoidable if SMYD2 inhibitors are used as therapeutic targets in ADPKD.
Despite compelling evidence, the role of epigenetic mechanisms in ADPKD remains unclear. In recent years, research from our lab and others have made substantial contributions towards understanding the mechanisms of epigenetic modifiers in ADPKD disease progression. DNA methyltransferase enzymes and histone modifiers are known to differentially affect the functioning of diverse pathways in cells and organs. One can speculate that oral or intravenous administration of drugs targeting these modifiers may have side effects. Therefore, the use of drug carriers with different affinities for target cells or organs such as kidneys in the case of ADPKD, for the delivery of inhibitors may be a direction of future research.
The roles of inflammatory response in the pathogenesis of ADPKD has become a central focus in the past decade (Li et al., 2008; Swenson-Fields et al., 2013; Chen et al., 2015; Sadasivam et al., 2019; Zimmerman et al., 2020). ADPKD patients are susceptible to exogenous pathogens due to multi-organ decline caused by loss of renal function, resulting in immune cells proliferation and cytokine secretion. As such, the renal inflammatory response in ADPKD patients has been recognized as a non-initial and secondary effect of cyst progression for a long time (Chen et al., 2015). However, because non-infectious inflammation is present in the early and progressive stages in most Pkd1 mutant mouse models and ADPKD patients, it is necessary to understand the roles and underlying mechanisms that drive inflammatory response in ADPKD, which may facilitate the development of novel therapeutic strategy for ADPKD treatment. In this section, we discuss how inflammatory response functions in the pathogenesis of ADPKD, and the therapeutic potential associated with these mechanisms.
The two immune systems, innate immune system, and adaptive immune system, which are unique in many aspects, are synergistically mobilized in response to endogenous or exogenous stimulus according to the patterns to recognize the pathogens and response timelines. Innate immune response, also termed non-specific and natural immune response, comes in as the first line of defense against invading pathogens or in response to altered endogenous molecules. This sometimes provides the initiating signal for adaptive immune response, composed of innate immune cells and innate immune associated molecules, including macrophages, dendritic cells, natural killer (NK) cells, natural killer T (NKT) cells, gamma-delta T cells (γδ T cells), eosinophil, neutrophil, mast cells and complement family proteins (Warrington et al., 2011; Netea et al., 2020). The adaptive immune response, also known as acquired or specific immune response, is composed of specialized, systemic cells and processes that eliminate pathogens or prevent their growth through antibodies and cytotoxic T cells. The adaptive immune system relies on the canonical T cells, and B cells by producing antibodies or effector T cells, upon stimulation with antigens presented by antigen-presenting cells (APC) and recognition of T cells receptor (TCR) or B cell receptor (BCR) (Lanzavecchia et al., 1999; Avalos and Ploegh, 2014; Gaudino and Kumar, 2019). NKT and γδ T cells also express TCRs and can also be categorized as adaptive immune cells. Based on their activation patterns, NKT and γδ T cells can also be characterized as innate immune cells. Due to the non-infectious environment during kidney development and the progression of polycystic kidney diseases, the categories of kidney resident immune cells are less complex. According to their origins, all the innate and adaptive immune cells are categorized into three main cell types—granulocytes, mononuclear phagocytes, and lymphocytes. Cells differentiated from granulocytes and mononuclear phagocytes are attributed to innate immune response, while lymphocytes, which include B cells and T cells engage in adaptive immune response. The granulocytes, including eosinophils, neutrophils, and mast cells, are responsible for allergic inflammation. As such, they are detected in peripheral blood and rarely detected in kidneys (Hume, 2008; Malech et al., 2014; Zimmerman et al., 2020). Thus, we will focus on the dominant immune cell types in the kidneys, including macrophages, NK cells, NKT cells, γδ T cells and canonical T cells, to understand their functional roles in the progression of ADPKD.
Kidney macrophages derive from circulating monocytes and resident macrophages. In general, the tissue-resident macrophage population is derived from the yolk sac and fetal liver during development but is complimented by circulating monocytes in response to stimulus (Epelman et al., 2014). Macrophages can be recruited by chemokines, such as monocyte chemoattractant protein-1 (MCP-1), to the damage sites or infectious tissues, switching from monocytes to macrophage. In different organs, macrophages may be attributed unique names, such as Kupffer cells in liver and alveolar macrophage or dust cell in lung (Laskin et al., 2001).
Accumulated evidence exists to support a role for interstitial macrophages in promoting cyst growth in human ADPKD and rodent cystic models (Karihaloo et al., 2011; Swenson-Fields et al., 2013). Treatment with liposomal clodronate to deplete phagocytic cells delayed cyst growth and improved renal function in Pkd1fl/fl:Pkhd1-Cre mice compared to vehicle treated animals (Karihaloo et al., 2011). Another study reported that M2-like macrophages are abundant in ADPKD patient and mouse kidneys, and depletion of these macrophages led to a milder cystic phenotype and an improved renal function in Pkd1 mutant mouse kidneys (Swenson-Fields et al., 2013). In general, classical M1-macrophage activation indicates oxidative and pro-apoptotic features, whereas the M2-macrophage activation has proliferative, remodeling and pro-fibrotic effects. This study emphasized a role of M2 macrophages in promoting the progression of ADPKD, highlighting the complexity and differences between M1 and M2 macrophages in this disease.
The renal interstitial macrophages are mainly recruited and derived from circulating monocytes to kidneys during the disease progression, supporting the hypothesis that chemokines and other factors that attract the infiltration of macrophages should also contribute to ADPKD progression. It has been reported that cultured Pkd1-deficient cells express elevated levels of macrophage chemoattractants, including Mcp1 and Cxcl16 (Figure 2), and both of these factors are able to stimulate macrophage migration, suggesting that they may contribute to the recruitment of macrophages to cystic kidneys (Karihaloo et al., 2011). To further understand the mechanisms involved in the recruitment of macrophage in PKD kidneys, we identified the role for the macrophage migration inhibitory factor (MIF) in the recruitment of macrophages to pericystic regions and MIF also regulated other signaling pathways to promote cyst growth in Pkd1 mutant mice (Chen et al., 2015).
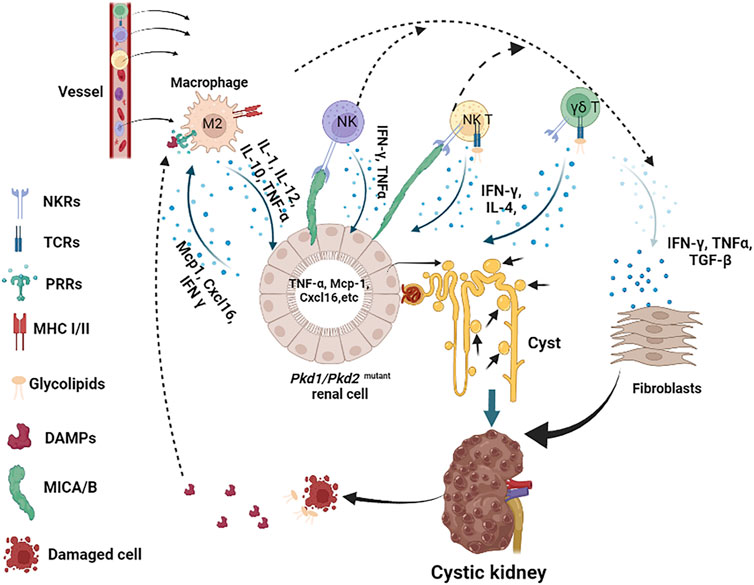
FIGURE 2. Innate immune response cells and molecules in the pathogenesis of ADPKD. In this scheme, we describe the major innate immune cells in the pathogenesis of ADPKD, including macrophages (referred to M2), NK, NKT and γδ T cells, and the major molecules that participate in this process, including DAMPs, glycolipids, and cytokines. We indicate the main receptors on immune cells, such as MHC I/II on macrophages, TCRs and NKRs on NK, NKT and γδ T cells. Pkd1 deficient renal cells release Mcp-1 and Cxcl16, which recruit macrophages to the kidney, other cytokines also attract NK, NKT and γδ T cells to infiltrate to kidney as well. Activated macrophages release TNF-α and other cytokines that stimulate renal cell proliferation and induce stress response, resulting in the accumulation of DAMPs in fluid or intestinal or induction of MICA/B on surface of renal cells. Upon recognition of DAMPs and glycolipids released by damaged or dying cells, or MICA/B presented on the surface of cystic cells, activated macrophages, NK, NKT and γδ T cells produce cytokines, such as TNF-α, IFN-ɣ, and TGF-β etc, to further promote cystic renal cell proliferation or renal fibrosis in ADPKD.
The activation of macrophages is a complicated process, dependent on synergistically coordinated signals from cytokines, ligands, and the corresponding receptors on macrophages (Mosser and Edwards, 2008). In the canonical pathway for macrophage activation and function in response to infection or injury, IFN-ɣ is the most potent macrophage-activating factor and is mainly triggered by viral or parasite infection and released by other immune cells or pathogen affected cells (Muller et al., 2018; Kang et al., 2019; Zhang et al., 2021). Macrophages can also be activated through pattern recognition receptors (PRRs) by an engagement with pathogen-associated molecular patterns (PAMPs) or the damage-associated molecular patterns (DAMPs). PAMPs are presented or released by pathogens, while DAMPs are derived from injured or dying cells (Amarante-Mendes et al., 2018; Roh and Sohn, 2018; Li and Wu, 2021). DAMPs are mainly related to altered self-molecules, including high-mobility group box 1 (HMGB1), S100 proteins, and heat shock proteins (HSPs), etc., reported to be abnormally expressed in diseases, but not or limited in normal situations (Roh and Sohn, 2018). Due to the non-infectious environment of the kidneys during early-stage progression of ADPKD, DAMPs may be the main stimulus for the activation of macrophages compared to PAMPs. The serum levels of HMGB1 are increased in ADPKD patients (Nakamura et al., 2011; Nakamura et al., 2012), while S100A8 and A9 were found to be remarkably increased in both ADPKD patients and mouse models (Lee et al., 2015). Many factors are known to induce the release of DAMPs, including cellular stressors, such as nitric oxide (NO), reactive oxygen species (ROS) and oxidized mitochondrial DNA. Under cellular stress, these factors are released into the cytosol and are responsible for the activation of macrophages (Malysheva et al., 2007; Muralidharan and Mandrekar, 2013; Minton, 2017).
Activated macrophages could either function against inflammatory (M1 macrophage) or further promote inflammatory response (M2 macrophage), respectively, to eliminate infected or injured cells, and assist in the activation of adaptive immune cells, or promote cell proliferation, cell remodeling and fibrosis through different cytokines (Mosser and Edwards, 2008; Arango Duque and Descoteaux, 2014; Krzyszczyk et al., 2018). Activated macrophages can produce cytokines, including IL-1, IL-12, IL-10, TNF-α, etc. (Figure 2). Among those cytokines, TNF-α, which is present in the cystic fluid of human ADPKD kidneys, can disrupt the localization of polycystin-2 to the plasma membrane and primary cilia through a TNF-α induced scaffold protein FIP2, to promote cyst formation in organ cultures and in Pkd2 mutant mice (Li et al., 2008). The cyst fluid TNF-α may be secreted by activated macrophages or PKD mutant cystic renal epithelial cells or both in ADPKD kidneys. These studies suggest that activated macrophage-mediated inflammation plays a role in promoting and/or inducing cystogenesis in the presence of cytokines during cyst expansion. In addition, DAMPs-PRRs activated macrophage may exert its effect through the formation of inflammasomes. Inflammasomes are a group of multimeric protein complexes that consist of a sensor molecule such as PRR, the adaptor protein ASC and caspase 1. Activated inflammasomes play a significant role by releasing IL-1β and IL-18, which are proteolytically activated by caspase 1 (Erlich et al., 2019; Zheng et al., 2020).
Natural killer (NK) cells, also called large granular lymphocytes (LGL), are the main innate immune cells that show strong cytolytic function against physiologically stressed cells (tumor and virus-infected cells) and represent 5%–20% of all circulating lymphocytes in humans (Perera Molligoda Arachchige, 2021). NKT and γδ T cells are very similar in many aspects, including: 1) NKT cells and γδ T cells both arise in the thymus, undergo T cell receptor (TCR) gene rearrangement and express CD3 molecule, either CD4 or CD8 molecules, or double negative of CD4/CD8, which is different from conventional T cells (Huang et al., 2014; Krijgsman et al., 2018), and 2) both NKT cells and γδ T cells acquire the expression of the natural killer receptor (NKR) NK1.1 during maturation, including inhibitory NKR (KIRs) and activating NKR (Sawa-Makarska et al.), which is the major difference between NKT cells, γδ T cells and conventional T cells (Krijgsman et al., 2018). Thus, NKT and γδ T cells are at the interface between the innate and adaptive immune system (Dranoff, 2004). The recognition patterns of NKT cells and γδ T cells are more like NK cells rather than conventional T cells, making them pass for innate immune cells instead of adaptive immune cells.
As innate immune cells, NK cells are major effectors of the innate immune system to kill target cells. The role of NK cells in renal fibrosis has been reported, where the accumulation of NK cells in the tubulointerstitial compartment of fibrotic kidneys was correlated with the severity of fibrosis (Law et al., 2017). There is no report regarding the roles of NK cells in the pathogenesis of ADPKD, however, the fact that renal fibrosis is one of the major features of ADPKD, suggests that NK cells may also contribute to renal fibrosis in ADPKD kidneys, and warrants investigation. NKT and γδ T contribute to the main portion of double negative T cell (CD4/CD8 negative, DN), reported to be increased in human ADPKD kidneys compared to controls (Sadasivam et al., 2019). The exact roles of NKT cells in ADPKD are not clear, however, it has been reported that kidney injury induces the activation of NKT cells, and causes hematuria and nephritic casts by damaging glomerular endothelial cells in a perforin-dependent manner through secretion of IFN-γ and other mechanisms and result in kidney dysfunction (Turner et al., 2018). This suggests a role for NKT cells during cyst expansion mediated by kidney injury. Taken together, NK cells, NKT cells and γδ T cells may play a vital role in the progression of ADPKD and need further investigation.
Next, we wanted to address the activation of NK cells, NKT cells and γδ T cells in ADPKD in the absence of pathogens. The fact that NKT cells and γδ T cells harbor two systems of receptors gives them the ability to activated via T cell-like mechanisms or NK cell-like mechanisms. TCRs on NKT and γδ T cells can recognize glycolipids in the context of CD1 family molecules (Liu and Huber, 2011; Pellicci et al., 2020), which is different from conventional T cells, mainly relying on MHC molecules to present peptides antigens. The representative molecule CD1d primarily expressed by antigen-presenting cells (APC), including macrophages, B cells and Dendritic cells, can present both exogenous and endogenous glycolipids in the context of CD1d to activate NKT cells and γδ T cells. Glycolipids include exogenous microbial- and non-microbial-derived glycolipids, and endogenous glycolipids. The latter is mainly released by apoptotic cells or damaged cells or expressed by malignant cells, but rarely detected on normal cells, including gangliosides and sulfatide, phospho-glycerolipids and sphingomyelin (Podbielska et al., 2011; Krijgsman et al., 2018). In ADPKD kidneys, metabolic glycerolipids derived from abnormally proliferative cells or released from DNA damage induced dead cells might be the main glycolipids antigens to stimulate the activation of NKT cells and γδ T cells. In addition to TCRs, NKT cells and γδ T cells also express NK cell receptors. As such, they are activated in a manner comparable to NK cells. Upon stimulation, the outcomes of NK cells, NKT cells and γδ T cells are dependent on the balance between inhibitory and activating signals obtained via the major inhibitory receptors, killer Ig-like receptors (KIRs), and killer cell activating receptors (Sawa-Makarska et al.), respectively (Long et al., 2013). KIRs, provide inhibitory signals upon binding with classical MHC molecules to maintain silence against normal cells (Kumar, 2018). KARs recognize a variety of MHC-like molecules, such as the canonical KAR, NKG2D, which recognizes MHC class I-like molecules A and B (MICA/B) and unique long-binding proteins, which are usually not expressed or lowly expressed in normal cells but robustly expressed on malignant or stress-induced cells, termed as “stress protein” (Long and Rajagopalan, 2002). Thus, these cells play a vital role to maintain homeostasis.
Although the expression of MIC-A/B or other ligands of NKT cells and γδ T cells receptors has not been reported on the surface of cystic cells in ADPKD, the fact that the MICA gene contains an NF-κB-binding site which is necessary and sufficient for transcriptional transactivation of MICA in response to TNFα in primary endothelial cells (ECs) (Lin et al., 2012), suggests a potential of MICA being expressed on cystic renal epithelial cells in ADPKD kidneys. In addition, it has been reported that the regulatory promoter module of MICA/B contains heat shock elements resembling those of HSP70 genes, suggesting that HSP70 and its family proteins also have the potential to stimulate the expression of MICA/B on stress-induced cells (Elsner et al., 2007; Schilling et al., 2015). As an important DAMPs, increased HSP70 also has the potential to induce the activation of NK cells, NKT and γδ T cells through the induction of MICA/B. Loss of self-MHC molecules or abnormal expression of MICA/B in cystic cells induced by HSPs and cyst fluid TNF-α, NK cells, NKT cells and γδ T cells might be activated and produce lots of cytokines. Similarly, upon activation via TCRs, NKT and γδ T cells could also rapidly expand and secrete a range of cytokines, mainly including IFN-ɣ and IL-4 (Coquet et al., 2008; Krijgsman et al., 2018), whereby IFN-ɣ could act as the most potent cytokine to stimulate the activation of macrophages. This would induce a feed-forward loop between these cells in ADPKD kidneys (Figure 2). Taken together, the engagement of receptors of innate immune cells and the potential ligands expressed on cystic cells or released by damaged cells may activate innate immune cells and contribute to the progression of ADPKD (Figure 2).
The adaptive immune response modulated by CD4 or CD8 T cells, and B cells, are responsible for cellular immunity and humoral immunity, respectively. The major difference between innate and adaptive immunity is the specificity of antigen recognition mediated by the TCRs or B cell receptor (BCR). The composition of conventional T cells (TCR αβ), referred to as T cells, are more complicated. T cells including Th1, Th2, CD4/CD25 regulator T cells, and Th17 cells, etc., all belong to CD4 T cells (Zhu et al., 2010). Activated CD8 T cells are mainly cytotoxic T cells, mediating a direct cell killing towards the target cells and releasing of cytokines, such as IFN-ɣ (Bhat et al., 2017; Nicolet et al., 2020). Activated CD4 T cells function significantly different from CD8 T cells, according to the specific phenotype, including the transcription activation or inhibition, and cytokines secretion (Luckheeram et al., 2012). Since the sterile-immune response is the main situation in ADPKD, there is no specific immunogen or pathogen to activate the adaptive immune cells, thus adaptive immune response is more likely a secondary response in ADPKD kidneys.
Reports indicate that both renal CD4+ and CD8+ T cell numbers are elevated, and correlate with disease severity in the Pkd1RC/RC mouse, but with selective activation of CD8+ T cells, as analyzed by flow cytometry analysis. In addition, immunodepletion of CD8+ T cells worsen ADPKD pathology in one to 3 months C57Bl/6 Pkd1RC/RC mice. Furthermore, the expression of T cell recruiting chemokines, CXCL9/CXCL10, which were secreted by cystic epithelial cells and renal interstitial cells, were significantly increased in kidneys of Pkd1RC/RC mice compared to those in kidneys from wildtype mice (Kleczko et al., 2018). These results suggested a protective role of CD8+ T cell in ADPKD and implied that the increase in T cells was as a result of extrarenal recruitment rather than the amplification of resident T cells. Besides CD8 T cells, there is no report defining the roles of CD4 T cells in ADPKD. A study found the CD4 T regulatory cells worsen chronic kidney disease (CKD) or end-stage kidney disease (ESKD) (Hartzell et al., 2020), suggested that CD4 T regulatory cells might also promote the progression of ADPKD.
Studies have reported increases in B cell numbers in ADPKD, however, little is known on the roles of B cells in disease progression. Furthermore, the mechanisms of activation of adaptive immune cells are still largely unknown. It has been proposed that DNA damage response mediated ubiquitous cell proliferation across the cystic kidneys, is the cause of the increases in T and B cells in ADPKD kidneys. Reports indicate that loss of polycystin-1 (PC1) impairs DNA damage response and induces cell proliferation of PC1 deficient cells in the kidneys (Zhang et al., 2021b). Because PC1 and PC2 are also expressed in lymphocytes, the intrinsic rate of DNA damage and the susceptibility to DNA damage agents are also increased in the peripheral blood lymphocytes from ADPKD patients (Aguiari et al., 2004; Li et al., 2013). Thus, PC1 deficiency may also induce cell proliferation in lymphocytes in an immune-recognition independent manner. In addition, activated innate immune cells could produce cytokines, such as IFN-ɣ, TNF-α TGF-β and IL-1β to regulate the amplification of adaptive cells and adaptive immune response, especially T cells. For example, by activating inflammasome, innate immune cells including macrophage, produce IL-1β, which is a pro-inflammatory cytokine that stimulates T cell activation, resulting in T cell differentiation under different conditions (Wan and Flavell, 2007; Croft, 2009; Van Den Eeckhout et al., 2020).
Immunotherapies to restore the dysregulated immune response in ADPKD by either the inactivation of the overactivated cell types or the activation of the protective cell types would benefit the patients. Several immunotherapeutic strategies have been evaluated in Pkd1 mutant mouse models (Table 3).
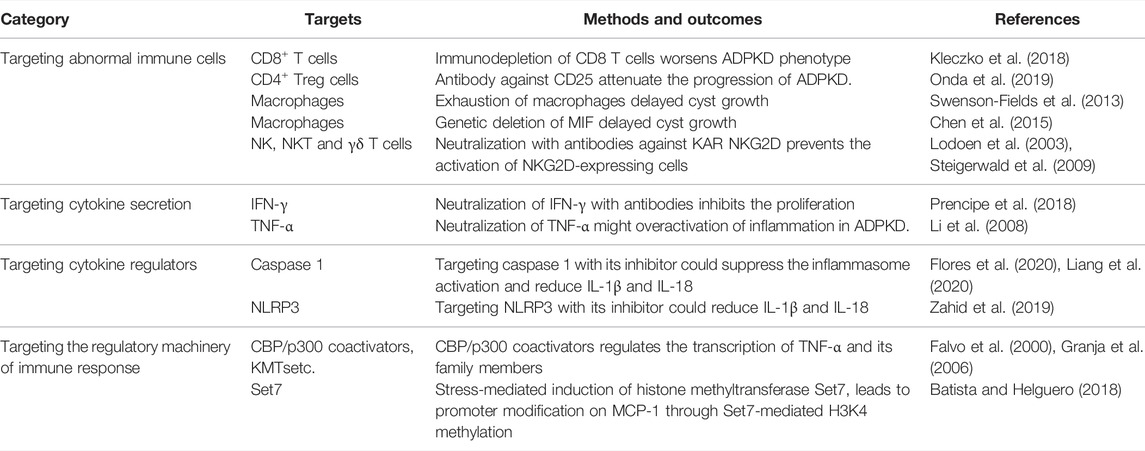
TABLE 3. Prospective immunotherapy strategies in the treatment of ADPKD.
(1) Targeting abnormal immune cells: First, it was reported that immunodepletion of CD8 T cells worsens ADPKD phenotype, suggesting that activation of CD8 T cells should attenuate the progression of ADPKD. Second, it has been found that CD4 Treg cells promote cyst growth, suggesting that specifically targeting CD4 Treg cells with antibody against CD25 rather than CD4 to delete Treg cells might be better than targeting other subsets of CD4 T cells, since CD25 was the first surface marker used to identify Tregs (Onda et al., 2019). Third, it has been confirmed that induced exhaustion of macrophages or genetic deletion of MIF delayed cyst growth and improved renal function (Swenson-Fields et al., 2013; Chen et al., 2015), supporting the hypothesis that targeting macrophages and factors associated with macrophage recruitment and function is a potential strategy for the treatment of ADPKD. With regards to other immune cell types such as NK, NKT and γδ T cells, the neutralization with antibodies against activating receptors may have beneficial effect. One possibility is to target KAR NKG2D with an antibody, which can prevent the activation of NKG2D-expressing cells (here referring to NK, NKT and γδ T cells) and cytokine secretion by inducing rapid internalization of antigen-antibody complex upon binding to NKG2D (Lodoen et al., 2003; Steigerwald et al., 2009).
(2) Targeting cytokine secretion: It has been reported that neutralization of IFN-γ with antibodies inhibits the proliferation and activation of immune cells in virus-infection model (Prencipe et al., 2018), and may be a strategy to be tested in ADPKD mouse models and patients. The neutralization of TNF-α might also provide another possibility to reduce overactivation of inflammation in ADPKD (Li et al., 2008).
(3) Targeting cytokine regulators: Caspase 1 is responsible for the release of both IL-1β and IL-18. Targeting caspase 1 with its inhibitor could suppress the inflammasome activation and downstream effects, which has already been tested in Alzheimer’s disease models and ischemia-associated blood-brain barrier dysfunction (Flores et al., 2020; Liang et al., 2020). In addition to the regulation of caspase 1, inhibitors targeting NLRP3, another component of inflammasome, have been evaluated in other disease associated cell models (Zahid et al., 2019).
(4) Targeting the regulatory machinery of immune response: As discussed above, epigenetic mechanisms contribute to ADPKD progression. Epigenetic regulation plays a role in renal inflammation in kidney diseases by regulating the expression of cytokines through chromatin modifications at the transcriptional levels. For example, the transcription of TNF-α and its family members is associated with several epigenetic regulators responsible for histone acetylation and methylation, including the CBP/p300 coactivators (Falvo et al., 2000; Granja et al., 2006). It has also been reported that stress-mediated induction of histone methyltransferase Set7, leads to promoter modification on MCP-1 through Set7-mediated H3K4 methylation (Batista and Helguero, 2018). These studies suggest that epigenetic regulation should also be involved in renal inflammation in PKD, providing an alternative strategy for the combination of immuno- and epigenetic therapy in ADPKD treatment.
In summary, both innate and adaptive immune responses participate in the pathogenesis of ADPKD through unique mechanisms. The roles of macrophages have been extensively investigated in PKD in the past decade. The roles of T cells in PKD have also been examined. So far there is not any clinical immunotherapeutic strategy for the treatment of ADPKD, all these immunotherapeutic strategies listed above are prospective immunotherapy according to the current studies in this field. (Table 3). Next, we may focus on the roles and mechanism of NK, NKT and γδ T cells in the regulation of PKD progression. In addition, the interaction of innate immune system and adaptive immune system should also be determined. Furthermore, the effect of immunotherapy should be assessed in pre-clinical and clinical trials. Overall, synergistical network of innate and adaptive immune cells, cytokines, immune related components, as well as the upstream regulatory factors and downstream effectors, make inflammation an important process in the pathogenesis of ADPKD. Epigenetic mechanisms contribute to the regulation of inflammatory response, and eventually regulate cell proliferation and cell death. In turn, the cytokines and other inflammatory factors regulate the expression and activity of these epigenetic factors, and regulate cell fates as well, such as proliferation and death. Thus, the integration of inflammation, epigenetic regulation and cell death should be investigated, in order to identify useful mechanisms to develop novel therapeutic strategies for the treatment of ADPKD in the future.
Cell death is an essential process to maintain homeostasis in the human body and is involved in diverse physiological processes including embryonic development and elimination of harmful or unnecessary cells (Norbury and Hickson, 2001; Fuchs and Steller, 2011; Tower, 2015). There are distinct types of regulated cell death, including apoptosis, autophagy, necrosis, and the most recently identified ferroptosis (Table 4). Thus far, three types of cell deaths, including apoptosis, autophagy and ferroptosis have been associated with the pathogenesis of ADPKD, and accumulated evidence suggests that targeting cell death pathways may be a potential therapeutic strategy for ADPKD treatment (Capuano et al., 2022).
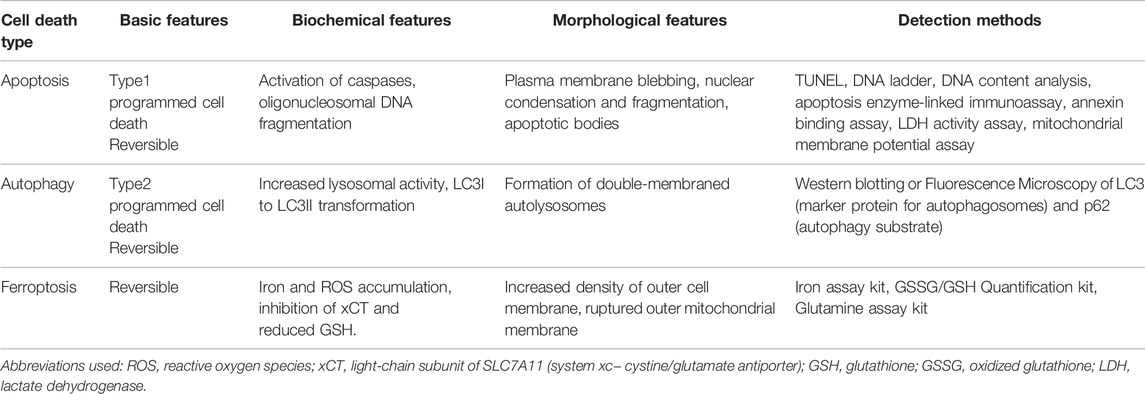
TABLE 4. Diverse types of cell death in ADPKD.
Apoptosis is the process of programmed cell death characterized by membrane shrinkage, chromatin condensation, nuclear fragmentation (pyknosis) and eventually the formation of apoptotic bodies (Elmore, 2007; Obeng, 2021). Apoptosis functions at all stages of human life, including embryonic development and aging to eliminate unwanted cells, and as a defense mechanism to remove injured cells that have been damaged beyond repair (Ke et al., 2018). There are two main pathways in apoptosis, the intrinsic and the extrinsic pathways (Figure 3), which are initiated and executed by two groups of caspases (cysteine-aspartic proteases), including the initiator caspases (caspases 2, 8, 9, 10) and the executioner caspases (caspases 3, 6, 7) (Zhou and Li, 2015). Caspases are a family of protease enzymes which can cleave their substrates at aspartic acid residues (Alnemri et al., 1996; Julien and Wells, 2017). The activation by initiator caspases can be induced by external death ligands or the release of cytochrome c from mitochondria, which initiate the apoptotic signals and directly cleave and activate executioner caspases for the execution of apoptotic program (Duclos et al., 2017).
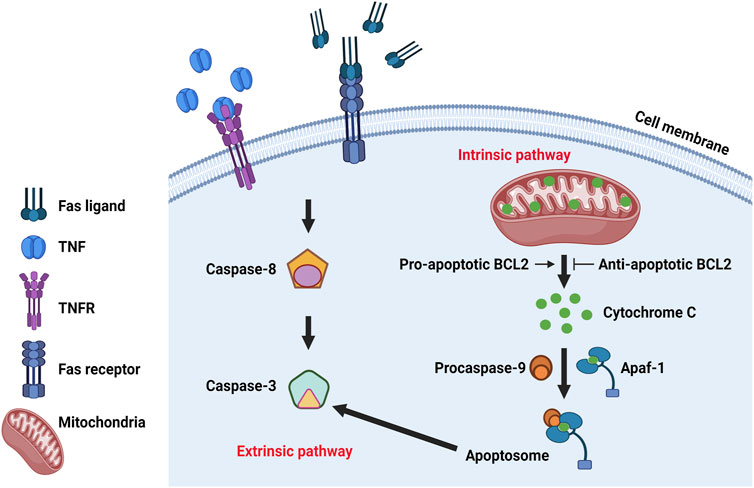
FIGURE 3. The molecular mechanism of apoptosis. Classic regulatory pathways of apoptosis include extrinsic pathway and intrinsic pathway. Intrinsic pathway is mitochondria dependent and regulated by BCL2 protein family which can influence the release of cytochrome C from the mitochondria. Cytochrome c can bind to the cytosolic protein Apaf-1 and promote the formation of an apoptosome to recruit and activate caspase-9, which in turn activates caspase-3 and leads to cell apoptosis. Extrinsic pathway is initiated by the transmission of death signals from the cell’s surface through the binding of ligands and the death receptors, resulting in the aggregation and recruitment of initiator caspases that subsequently activates executioner caspase 3, leading to apoptosis.
The intrinsic pathway is mitochondria-dependent, initiated when an injury occurs within the cell and mainly regulated by proteins of the BCL2 (B-cell lymphoma 2) family, which are evolutionarily conserved with shared Bcl-2 homology domains, including BH1, BH2, BH3, and BH4 (Boletta et al., 2000). BCL2 family proteins can be divided into three types: pro-apoptosis (Bcl-2, Bcl-xL, etc.), anti-apoptosis (BAX, BAK, etc.) and regulatory (BAD, BIK, BIM, etc.) members (Reed, 1998; Kale et al., 2018). Diverse types of BCL2 family contain different BH domains. Anti-apoptotic proteins usually contain BH1 and BH2 domains and pro-apoptotic proteins usually contain a BH3 domain which is essential for dimerization with other proteins of the Bcl-2 family and crucial for their killing activity. Some pro-apoptotic proteins also contain BH1 and BH2 domains (Bax and Bak). The BH3 domain may also be present in some anti-apoptotic proteins, such as Bcl-2 or Bcl-x(L) (Kale et al., 2018). The balance between pro- and anti-apoptotic Bcl-2 family members is essential to control the activity of caspases (Swanton et al., 1999; Marsden et al., 2002; Hatok and Racay, 2016; Roufayel, 2016). BCL2 family proteins are found in the mitochondrial membrane, where the pro-apoptotic proteins can promote the release of cytochrome c from mitochondria while anti-apoptotic proteins can inhibit its release (Adams and Cory, 1998; Korsmeyer, 1999; Morales-Cruz et al., 2014; Pena-Blanco and Garcia-Saez, 2018). Cytochrome c can bind to the cytosolic protein Apaf-1 and promote the formation of an apoptosome to recruit and activate caspase-9, which in turn can activate caspase-3 and leads to cell apoptosis (Bratton and Salvesen, 2010; Elena-Real et al., 2018).
The extrinsic pathway which begins outside the cell, is initiated by the transmission of death signals from the cell’s surface to intracellular signaling pathways through the binding of specific death receptors to their ligands. This results in the aggregation and recruitment of initiator caspases that subsequently activates executioner caspase 3, leading to apoptosis (Elmore, 2007; Green and Llambi, 2015). Several death ligands and their corresponding death receptors have been identified, including Fas ligand and Fas receptor (FasL/FasR), tumor necrosis factor (TNF) and its receptor 1 (TNF-α/TNFR1), Apo3L/DR3, Apo2L/DR4 and Apol2L/DR5 (Walczak, 2013). TNFRs are death receptors that belong to members of the TNF receptor superfamily with shared cysteine-rich extracellular domains and a cytoplasmic death domain (Micheau and Tschopp, 2003; Dostert et al., 2019). The classical extrinsic pathway is mediated by FasL/FasR and TNF-α/TNFR1 (Nagata, 1999; Rath and Aggarwal, 1999; Yanumula and Cusick, 2022). The binding of FasL with FasR results in the recruitment of the adaptor protein FADD (Fas-associated death domain) (Wajant, 2002; Caulfield and Lathem, 2014) which then associates with procaspase-8 to form the death-inducing signaling complex (DISC), leading to the activation of procaspase-8 (Kischkel et al., 1995; Wajant, 2002). Conversely, TNF binds to TNFR resulting in the transient recruitment of TRADD (TNF-related apoptosis inducing ligand), TNF receptor-associated factor 2 (TRAF2), TRAF5, cellular inhibitor of apoptosis 1 and 2 (cIAP1/2) and receptor interacting protein 1 (RIP1) to form pro-survival complex I (Micheau and Tschopp, 2003; Shi and Sun, 2018) which can activate nuclear factor κB (NF-κB) and JNK pathways to regulate the expression of pro-survival genes, including the cellular FLICE-like inhibitory protein (cFLIP) (Liu et al., 1996; Dhanasekaran and Reddy, 2008). With the deubiquitination of RIP1, TRADD and RIP1 become disassociated from complex I, then RIP1 associates with FADD and caspase-8 to form complex II, the so-called death complex to trigger cell death (Amin et al., 2018).
Autophagic cell death is another form of programmed cell death characterized by the large-scale accumulation of vacuolated-like structures called autophagosomes (Kroemer and Levine, 2008; Garg et al., 2015). Autophagy is an important physiological process that has been associated with ADPKD pathology and is regulated by known PKD associated signaling pathways. For example, the mammalian target of rapamycin (mTOR) inhibits autophagy, while the AMP-activated protein kinase (AMPK) is known to activate autophagy.
Autophagy process includes five steps: initiation, elongation, maturation, fusion, and degradation. The activation and phosphorylation of Unc-51 like autophagy activating kinase (ULK1) protein complex, inhibited by the mTOR pathway (including mTORC1 and mTORC2) and activated by the AMPK pathway, initiates autophagy. Under normal conditions, mTORC1 inhibits the activation of the ULK1 complex by phosphorylating Ser 757 of ULK1 and interrupting the interaction between ULK1 and AMPK. Upon stimulation by cellular or environmental stresses, inhibition of mTORC1 results in the dephosphorylation of ULK1. Meanwhile, AMPK directly activates ULK1 through phosphorylation of Ser 317 and Ser 777 in ULK1 (Kim et al., 2011). Phosphorylation of ULK1 initiates the process of autophagy as described above. Activated ULK1 then phosphorylates other components of the ULK1 complex (including FIP200, ATG13 and ATG101), and recruits the PI3KC3 complex (including BECN1, Vps15, Vps34, NRBF2, AMBRA1, Atg14) to coordinate the nucleation and biogenesis of autophagosome (Meijer and Codogno, 2006; Mizushima, 2010; Lin and Hurley, 2016). ATG9 is the only transmembrane protein in the ATG protein family that functions as a membrane carrier to deliver lipids to the forming autophagosome from several cellular membranes, including the plasma membrane, mitochondria, recycling endosomes and Golgi complex (Matoba et al., 2020; Sawa-Makarska et al., 2020). The process of expansion and maturation of the autophagosome membrane involves two ubiquitin-like conjugation systems: the conjugation of ATG12 to ATG5, and the conversion of LC3 I to LC3 II (Mizushima, 2020). The conjugation of Atg12 to Atg5 occurs at Lys130 through the activation of E1 enzyme Atg7 and the E2-like Atg10 (Mizushima et al., 1998; Otomo et al., 2013). The Atg12-Atg5 conjugate then forms a large protein complex with Atg16, acts as the E3 ligase for the conjugation of LC3 I to PE (phosphatidylethanolamine). Atg4 cleaves the C-terminal arginine of pro-LC3 to form LC3 I. The conversion of LC3 I to LC3 II is also a ubiquitin-like conjugation reaction. E1, E2, and E3-like enzymes are Atg7, Atg3, and Atg12-Atg5-Atg16 (Tanida et al., 2004). LC3 II is a characteristic marker of autophagic membranes and can recruit selective cargo to the autophagosome via its interaction with cargo receptors (Kabeya et al., 2000; Tanida et al., 2004). The maturation of the autophagosome leads to the autophagosome-lysosome fusion. The lysosome is a double-membrane cell organelle that contains digestive enzymes. The enzymes contained in the lysosome ensure the degradation of the cargo and cargo receptors which are recycled to be used again during cellular metabolism (Kriegenburg et al., 2018).
Ferroptosis is an iron dependent form of cell death which can be induced by small molecules such as erastin and Ras-selective lethal small molecule 3 (RSL3) (Dolma et al., 2003; Yang and Stockwell, 2008), and can be inhibited by specific inhibitors such as ferrostatin-1 (Fer-1), liproxstatin-1 and vitamin E. In addition, iron chelators and lipophilic antioxidants can prevent ferroptosis (Xie et al., 2016). The failure of glutathione-dependent antioxidant defenses initiates ferroptosis, leading to uncontrolled lipid peroxidation and eventually cell death (Li et al., 2020a; Zhang and Li, 2022). Ferroptosis is regulated by Glutathione/GPX4 (glutathione peroxidase 4) signaling pathways, iron metabolic signaling pathway and lipid metabolic signaling pathway as indicated in Figure 4 (Figure 4) (Li et al., 2020a). GPX4 is a selenoenzyme which can convert GSH (glutathione) into oxidized glutathione (GSSG) and reduces membrane cytotoxic lipid hydroperoxides to maintain cellular redox homeostasis and prevent the iron (Fe2+)-dependent formation of toxic lipid reactive oxygen species (ROS) (Seiler et al., 2008). Inhibition of GPX4 can lead to the accumulation of lipid peroxides and induction of ferroptosis (Yang and Stockwell, 2008; Yang et al., 2014).
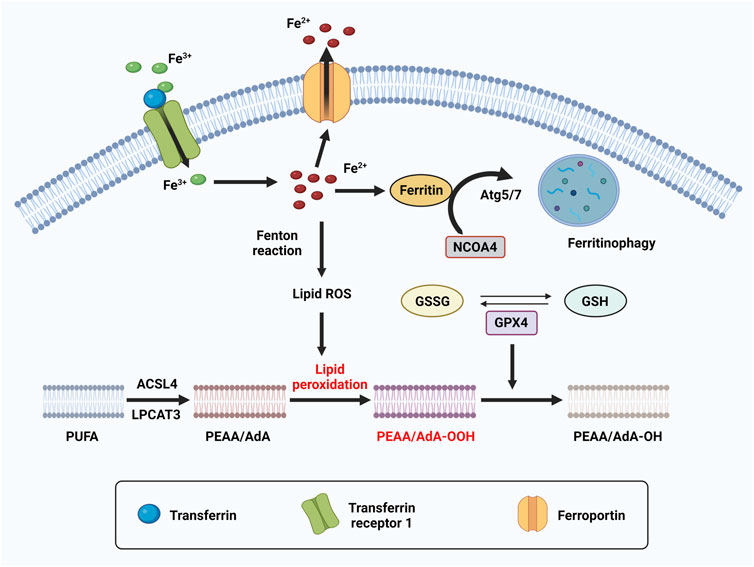
FIGURE 4. The molecular mechanism of ferroptosis. Ferroptosis is an iron dependent form of cell death. The metabolism of iron includes iron uptake (transferrin receptor), iron export (ferroportin), iron storing (ferritin), and ferritinophagy. Ferritinophagy is a selective autophagy of ferritin which mediated by Atg5/7 and NCOA4. Excess can directly induce ferroptosis through lipid peroxidation. GPX4, converts PEAA/AdA-OOH to PEAA/AdA-OH and inhibits ferroptosis. This reaction occurs through the use of glutathione (GSH) as a substrate.
Iron is one of the most abundant transition metals and is an essential element for many living organisms. Because iron-dependent oxidative damage is characteristic for ferroptosis, iron metabolism is controlled by ferroptosis in many aspects. The metabolism of iron includes iron uptake (transferrin receptor), iron export (ferroportin), iron storing (ferritin), and ferritinophagy, selective autophagy of ferritin mediated by lysosome and NCOA4 (nuclear receptor coactivator 4). Excess heme and non-heme iron can directly induce ferroptosis (Li Q. et al., 2017), and both heme and non-heme iron-containing enzymes, such as ALOXs, NOXs, and CYP can promote lipid peroxidation in ferroptosis. During ferroptosis, a phenomenon in which increased labile iron is released by the cell is referred to as ferritinophagy. NCOA4 is a selective cargo receptor which functions in ferritinophagy. The genetic inhibition of NCOA4 inhibited ferritin degradation and suppressed ferroptosis. In contrast, overexpression of NCOA4 increased ferritin degradation and promoted ferroptosis (Hou et al., 2016). Iron chelators are drugs that can remove extra iron from the body, blocking ferroptotic cell death both in vitro and in vivo. Ferritin is regulated by ATG5-ATG7 and NCOA4 pathways, as well as IREB2 (Iron Responsive Element Binding Protein 2). In addition, p62-Keap1-NRF2 and HSPB1 (Heat Shock Protein Family B Member 1) signaling pathways can also regulate iron metabolism.
Lipid peroxidation is a process of oxidative lipid degradation that eventually leads to ferroptosis. During this process, ROS such as oxygen free radicals attack lipids, especially polyunsaturated fatty acids (PUFAs). PUFAs are preferentially incorporated into phospholipids such as phosphatidylethanolamines (PEs) and transformed into PUFA-PEs by two enzymes, ACSL4 (Acyl-CoA synthetase long-chain family member 4) for synthesizing PEs and LPCAT3 (lysophosphatidylcholine acyltransferase 3) for lipid remodeling. Therefore, blocking the expression of ACSL4 and LPCAT3 results in the suppression of esterification of PEs reducing the accumulation of lipid peroxide substrates in cells, thus inhibiting ferroptosis (Kagan et al., 2017).
Aberrant apoptosis and apoptotic pathways were first detected in human ADPKD, the congenital mouse model of ARPKD (cpk), and the pcy mice (Woo, 1995). In the past decades, an increasing number of studies have demonstrated that apoptosis plays an essential role in the regulation of cystogenesis in ADPKD. However, whether apoptosis is increased or decreased in ADPKD kidneys, and whether induction of apoptotic cell death promotes, or delays cyst growth remains controversial.
First, studies found that apoptosis is elevated in rodent ADPKD models. In the Han:SPRD-Cy (Cy) rat model which closely resembles ADPKD, apoptosis is increased and correlates with the upregulation of caspase protein levels and activity and the downregulation of anti-apoptotic Bcl2 family proteins (Ecder et al., 2002; Tao et al., 2005b). In cpk mice, excessive apoptosis occurs in the interstitium while seldom evident in the cystic epithelium or noncystic tubules. The expression of various caspases, including bax and bcl-2, are also upregulated indicating that apoptotic cell death contributes to cyst formation (Ali et al., 2000). In Madin-Darby canine kidney (MDCK) cells, apoptosis was increased and overexpression of PC1 displayed resistance to thrombin/Gα12-stimulated apoptosis while PC1-silenced MDCK cells displayed enhanced thrombin-induced apoptosis (Boletta et al., 2000). In Pkd1 mutant mice, Pkd1 deletion in renal stromal cells by Foxd1-driven Cre showed epithelial changes and progressive cystogenesis accompanied by excessive apoptosis and proliferation (Nie and Arend, 2017). Pkd1 conditional knockout mice also showed increased expression levels of pro-apoptotic markers and downregulation of the anti-apoptotic marker (Rogers et al., 2016). In Pkd1-deleted MEFs, enhanced cytochrome c release and increased apoptosis were detected which caused increased sensitivity to ROS (Zhang et al., 2015). In Pkd2 mutant models, haploinsufficiency and loss of heterozygosity at the Pkd2 locus results in both increased proliferation and apoptosis (Belibi et al., 2004; Wilson and Goilav, 2007; Kim et al., 2009; Wegierski et al., 2009). In addition, Pkd2-overexpressing transgenic mice showed typical renal cyst growth with an increase in both proliferation and apoptosis (Park et al., 2009; Zhang et al., 2015). Besides, in Pkd1+/−pigs, both intrinsic and extrinsic apoptosis increased at ages of 1 month and 3 months. This provides more evidence for the correlation of apoptosis in ADPKD among different mammalian species (Wang et al., 2021).
Second, different studies have either reported no significant change or decreased apoptosis in rodent ADPKD models. In Pkd1flox/flox:Ksp-Cre mice, increased proliferation which correlated with increased cystic kidney, was observed, however, no significant changes in apoptosis were reported (Shibazaki et al., 2008). Meanwhile, apoptotic cells were rarely detected in kidneys of Pkd1 knockdown mice and in kidneys from Pkd1flox/flox:Ksp-Cre neonates. In proximal tubule derived cell lines lacking PC1 which spontaneously formed cysts, the overall number of apoptotic nuclei in kidney tissues was very low and did not differ significantly between cystic and normal kidneys (Wei et al., 2008). In the kidneys from Pkd1 conditional knockout and Pkd1 hypomorphic Pkd1nl/nl mice, apoptotic cells were rarely detected (Fan et al., 2013a). In Pkd1−/− E15.5 MEKs (mouse embryonic kidneys), apoptosis was rare and negligible as detected by TUNEL assay (Zhou et al., 2013). In Pkd1fl/fl:Smyd2+/+:Ksp-Cre neonates, apoptosis was rare in kidneys and knockout of Smyd2 induced cyst-lining epithelial cell death in kidneys from Pkd1fl/fl:Smyd2fl/fl:Ksp-Cre neonates (Li et al., 2017b). The controversial role for apoptosis in PKD is due to confounding factors including: 1) the use of variable animal models versus human PKD, 2) comparison at various stages of the disease, early versus late, 3) comparison between cyst lining epithelium versus normal appearing tubules. It is evident that both cell apoptosis and proliferation are dysregulated in ADPKD, and both may contribute to the general mechanism for cyst growth. Moreover, PKD kidneys are disproportionately enlarged. If apoptosis were therefore a predominant factor in the regulation of cyst growth, one would expect the kidneys to eventually involute. This therefore suggests that apoptosis is not the primary factor of cystogenesis (Zhou and Li, 2015).
Like the role of apoptosis in ADPKD, the role of autophagy in ADPKD is also controversial. The disruption of autophagy has been identified in diverse ADPKD animal models. However, both increased and decreased autophagy are reported. In cpk mice and Han:SPRD rats, autophagy is increased as autophagosomes were found by electron microscopy in the tubular cells lining the cysts, and enhanced autophagy components LC3-II and beclin-1 were also observed (Belibi et al., 2011). In PC1 deficient cells, basal autophagy was enhanced (Decuypere et al., 2021). However, in Pkd1 mutant mice, the expression level of autophagy genes was decreased although LC3-II protein level showed no change (Belibi et al., 2011; Chou et al., 2018). mTOR pathway is hyperactivated and suppressed autophagic flux is detected in the heart and kidneys of Pkd1 mutant mouse models (Atwood et al., 2020a; Atwood et al., 2020b). In the Pkd1 mutant zebrafish model, the mTOR pathway is abnormally activated and autophagy is inhibited (Zhu et al., 2017). In Pkd1flox/-:Ksp-Cre mice, autophagy-related protein ULK1 and the ratio of LC3 II/LC3 I decreased, indicating autophagy is inhibited (Liu et al., 2020). In Pkd1 transgenic mice, the expression levels of autophagy-related genes, including Atg5, Atg12, Ulk1, Beclin-1, and Sqstm1 (p62) were down-regulated. However, the p62 protein level in cystic lining cells was increased, indicating impaired degradation of the protein by the autophagy-lysosome pathway (Chou et al., 2018). In human ADPKD patient samples, autophagy did not change though LC3 was highly increased in cystic ADPKD patient kidneys. Meanwhile, the enhanced LC3 expression in PKD mouse models enlarged renal cysts and enhanced renal failure (Lee et al., 2020).
Dysregulation of the autophagy pathway has been associated with disease progression in ADPKD, however, the mechanisms involved in the PC1- and PC2-mediated regulation of autophagy remains unclear. Treatment with hyperosmolar concentrations of sorbitol or mannitol induces PC2 dependent autophagy and downregulation of PC2 prevents inhibition of hyperosmotic stress-induced mTOR pathway activation in HeLa and HCT116 cell lines (Pena-Oyarzun et al., 2017). PC2 can bind to BECN1, a component of PI3KC3 complex and is essential to initiate autophagy, through CC1 domain, which is in the carboxy-terminal tail of PC2. Note that the PC2-BECN1 complex is required for the induction of autophagy (Pena-Oyarzun et al., 2021). In Pkd2 knockout mice, the activation of autophagic flux was suppressed and overexpression of PC2 could increase autophagic flux (Criollo et al., 2018). While PC2 seems to be essential for stress-induced autophagy, PC1 also functions in the process of autophagy. Dysfunction of PC1 and/or PC2 in cell lines lead to reduced intracellular Ca2+ signaling, increased mTOR activity, increased cAMP levels and increased proliferation (Seeger-Nukpezah et al., 2015). PKD mutant cells can be more resistant against nutrient stress and delay cell death by maintaining autophagy modulated through PC1 in a PC2-dependent manner. In PC1 knockout mice, starvation-induced autophagic response is enhanced. Although knockdown of PC2 in PC1 knockout cells did not significantly alter autophagy level, PC2 knockout cells showed reduced protein level of PC1 and the PC1 band seemed to be at a lower molecular weight, suggesting that the stability of PC1 protein during starvation is dependent on PC2 expression (Decuypere et al., 2021).
The dysregulation of ferroptosis was detected in animal models of ADPKD and human ADPKD. The expression of antioxidant enzymes GPX and SOD and their activity were decreased in cpk mice and Han:SPRD rats (Maser et al., 2002) while lipid peroxidation is increased in human ADPKD patients (Schreiber et al., 2019), suggesting that ferroptosis plays an important role in ADPKD. Lipid peroxidation correlated with cyst growth and the activation of TMEM16A (anoctamin 1), a chloride bicarbonate transmembrane channel, inducing growth of renal cysts by lipid peroxidation while the direct inhibition of TMEM16A or inhibition of lipid peroxidation delayed cyst development in Pkd1 mutant mice (Schreiber et al., 2019). Treatment with two potent inhibitors of TMEM16A, niclosamide and benzbromarone, significantly reduced renal cyst size in Pkd1−/− mice compared to control mice (Cabrita et al., 2020). Recently, we reported the decreased expression of the negative regulator of ferroptosis GPX4, and the increased expression of iron importers TfR1 and DMT1 in Pkd1 mutant mouse models. This alteration in expression levels resulted in high iron levels, low GSH and GPX4 activity, increased lipid peroxidation, and a propensity for ferroptosis. 4HNE (4-hydroxynonnenal), a lipid peroxidation product, is also increased in Pkd1 null cells and is reportedly responsible for the promotion of proliferation of Pkd1 mutant cells through the activation of Akt, S6, Stat3, and Rb (Zhang and Li, 2022). These findings indicated that ferroptosis is highly associated with ADPKD progression.
In ADPKD, apoptosis may function as a double-edged sword. While apoptosis occurs in the normal-appearing, non-cystic tubules, loss of renal tissue may result in the functional progressive deterioration of kidney (Woo, 1995), increased apoptosis in ADPKD may inhibit hyperproliferation thus prevent the progression of PKD.
Targeting apoptosis to attenuate cyst growth is quite complex (Figure 5). First, the increase in apoptosis in ADPKD suggests that inhibition of apoptosis should delay cyst progression. Short-term treatment with the pan-caspase inhibitor IDN-8050 resulted in the inhibition of caspase-3 and caspase-7 activity in kidneys of Cy/Cy rats (Tao et al., 2005b) and decrease of apoptosis. This inhibited renal enlargement and cystogenesis and attenuated the loss of kidney function (Tao et al., 2005a). Deletion of caspase-3 resulted in the increased caspase-7 and decreased anti-apoptotic protein Bcl-2 levels, and prolonged the survival of cpk mice (Tao et al., 2008). In Han:SPRD rats, treatment with 2-hydroxyestradiol (2-OHE) reduced apoptosis and resulted in decreased cyst growth and preservation of kidney (Anderson et al., 2012). Furthermore, in two ARPKD mouse models (jck juvenile cystic kidney mice and cpk mice), treatment with cyclin-dependent kinase (CDK) inhibitor roscovitine resulted in long-lasting arrest of cystogenesis and decreased apoptosis. The decreased expression of pro-apoptotic protein Apaf1 and caspase-2, and increased expression of Bcl-2 and Bcl-xL were also detected. The may be caused by the inhibition of Cdk5, which is responsible for anti-apoptotic effects in neurodegenerative diseases, thereby resulting in effective reduction of apoptosis in cystic kidneys (Di Giovanni et al., 2005; Bukanov et al., 2006). The Glc-Cer synthase inhibitor, Genz-123346, also slowed cyst growth by decreasing the proliferation and apoptosis in jck mice (Natoli et al., 2010).
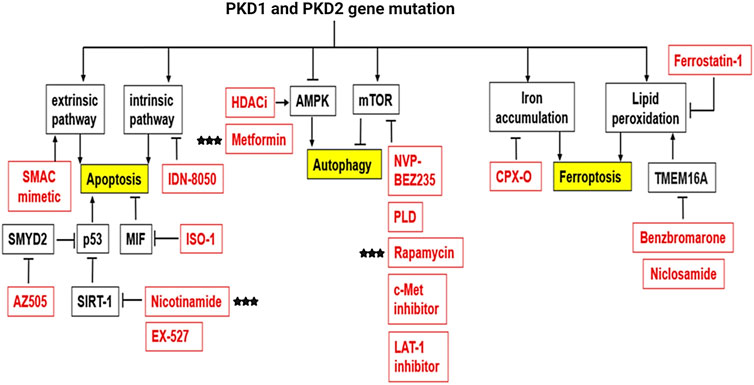
FIGURE 5. Cell death pathway and the drug targets in ADPKD. In ADPKD, mutation of PKD1/PKD2 causes dysregulation of cell death, including apoptosis, autophagy and ferroptosis. Apoptosis is regulated through extrinsic pathway and intrinsic pathway. Treatment with SMAC mimetic activates apoptosis through an extrinsic dependent pathway to delay cyst growth, while treatment with pan-caspases inhibitor IDN-8050 inhibits apoptosis through an intrinsic pathway which also reduces cyst size. Treatment with SIRT-1 inhibitors, including nicotinamide and EX-527, and Smyd2 inhibitor, AZ505, as well as MIF inhibitor, ISO-1, induced apoptosis in a p53 dependent manner and delay cyst growth. Autophagy is regulated through PKD associated AMPK and mTOR pathways. Treatment with inhibitors of mTOR pathway, including NVP-BEZ236, PLD and rapamycin as well as c-Met and LAT-1 inhibitors, delays cyst growth, and treatment with AMPK inducers, metformin and HDACi, also slows cyst growth. Ferroptosis is an iron dependent cell death characterized with the increase of lipid peroxidation. Treatment with CPX-O, a chelator of iron, inhibited cystogenesis. Treatment with ferroptosis inhibitor, Ferrostatin-1, delays cyst growth and cystic cell proliferation. TMEM16A can induce lipid peroxidation, and treatment with TMEM16A inhibitor, niclosamide and benzbromarone, significantly reduced renal cyst size in ADPKD animals (Three black stars indicate drugs in clinical trials).
Second, there is evidence to support that induction of apoptosis contributes to delayed cyst growth in ADPKD. As described above, TNF-α is an apoptosis ligand and accumulates at elevated levels in ADPKD cyst fluid (Li et al., 2008). While treatment with TNF-α alone did not induce apoptosis in either Pkd1 wild-type or null MEK cells even at high concentration (due to the activation of NF-κB which inhibited the activity of caspase 8) (Fan et al., 2013a), treatment with TNF-α combined with the SMAC (second mitochondria-derived activator of caspases) mimetic induced apoptosis in primary cultures of mural epithelial cells from human ADPKD cysts and Pkd1 null MEK cells, suggesting that SMAC-mimetic induces TNF-α–dependent cell death (Fan et al., 2013a). Importantly, treatment with SMAC-mimetic reduced cyst growth in Pkd1 mutant mice (Fan et al., 2013a). After this fundamental study, subsequent studies provided more evidence to support a beneficial role for the induction of cystic renal epithelial cell apoptosis. Treatment with SMAC-mimetic also showed cyst reduction in a drosophila model of ADPKD (Millet-Boureima et al., 2019). In Pkd1 mutant model, the deacetylase sirtuin 1 (SIRT1) is upregulated and treatment with pan-sirtuin inhibitor (nicotinamide) or a SIRT1-specific inhibitor (EX-527) delayed cyst growth through the deacetylation of p53, which released its inhibition on apoptosis in Pkd1 mutant mice (Zhou et al., 2013). A randomized double blinded small clinic trial with the enrollment of 36 patients was conducted to determine the effect of nicotinamide in ADPKD patients. Although nicotinamide is safe and well-tolerated in ADPKD patients, there was no beneficial effect in the inhibitor treated group compared to placebo treated group (El Ters et al., 2020). SMYD2, a lysine methyltransferase, also upregulated in Pkd1 mutant renal epithelial cells and kidneys, was shown to inhibit p53-dependent cystic renal epithelial cell apoptosis through the methylation of p53. Treatment with specific SMYD2 inhibitor AZ505 delayed cyst growth in Pkd1 mutant kidneys by increasing p53 mediated apoptosis (Li et al., 2017b). Macrophage migration inhibitory factor (MIF) reported to be upregulated in cyst-lining epithelial cells of Pkd1 mutant mouse kidneys, accumulates in cyst fluid of human ADPKD kidneys. Treatment with ISO-1, a MIF inhibitor, efficiently slowed cyst growth, accompanied with an increase in apoptosis (Chen et al., 2015). Genetic ablation of apoptosis regulatory genes also contributes to attenuation of cyst growth in ADPKD. Homozygous deletion of ILK (integrin-linked kinase) gene, whose product is a scaffold protein associated with multiple cellular functions including cell proliferation, resulted in increased caspase-3-mediated apoptosis and reduced cyst size of Pkd1fl/fl;Pkhd1-Cre mice (Raman et al., 2017). In cpk mice, haploinsufficiency of Pax2 attenuated progressing cyst growth via increased p53 mediated apoptosis. Pax2 is known to suppress p53 transcription by binding to cis-acting regulatory sequences (Stuart et al., 1995). Apoptosis in Pax2 heterozygous kidneys of cpk mice may be regulated in part by increased levels of p53 (Ostrom et al., 2000). MicroRNAs (miRNAs are short noncoding RNAs that acts as sequence-specific inhibitors of gene expression (O’Brien et al., 2018 #95). miR-21 is upregulated in renal cysts through cAMP signaling pathway and represses proapoptotic genes to inhibit cystic cell apoptosis and promotes PKD progression. Deletion of miR-21 showed attenuation of cyst growth and induction of apoptosis in kidneys from an orthologous model of ADPKD (Lakhia et al., 2016). These studies indicated a beneficial role for the induction of apoptosis in ADPKD treatment.
Direct targeting of dysfunctional autophagy in PKD animal models has been determined effective in slowing cyst growth. In Pkd1 mutant zebrafish, knocking down the core autophagy protein Atg5 caused inhibition of autophagy and promoted cystogenesis, while treatment with a specific inducer of Beclin-1 peptide activated autophagy and also slowed cyst growth (Zhu et al., 2017). However, oral supplement with trehalose, a natural autophagy enhancer, showed no effect on the progression of ADPKD in Pkd1 miRNA transgenic mice and could not restore impaired autophagy, suggesting that an oral supplement of trehalose may not affect the progression of ADPKD (Chou et al., 2018).
mTOR pathway plays a significant role in the regulation of autophagy and targeting mTOR pathway has been indicated effective for ADPKD (Zafar et al., 2010; Li et al., 2017a). Rapamycin has been used as an autophagy inducer that functions through inhibition of mTOR pathway. The effect of rapamycin in suppressing the aberrant epithelial proliferation of ADPKD kidney has been identified in rodent ADPKD animal models. Moreover, rapamycin was used in human studies, and while there was a reduction in TKV (total kidney volume) in rapamycin treated groups compared to placebo group, the decrease in TKV was not significant (Kim and Edelstein, 2012; Braun et al., 2014). Phospholipase D (PLD) is an enzyme of the phospholipase superfamily and its product phosphatidic acid (PA) regulate mTOR activity. The activity of PLD was elevated in PKD cells and targeting PLD with small molecule inhibitors could reduce cell proliferation and enhance the sensitivity of PKD cells to rapamycin which indicated that combining PLD inhibitors and rapamycin synergistically inhibited PKD cell proliferation (Liu et al., 2013). Two large randomized clinical trials with rapamycin in ADPKD were undertaken. However, the effect of rapamycin were unimpressive and the treatment presented an increased side-effect profile, which might be associated with the short-term administration of rapamycin and the lack of randomization (Kim and Edelstein, 2012). Other drugs targeting mTOR pathway such as NVP-BEZ235, a mTOR inhibitor, also inhibited proliferation and normalized kidney morphology in ADPKD (Liu et al., 2018). Defective ubiquitination of c-Met caused hyperactivation of mTOR in PKD and treatment with a c-Met inhibitor resulted in the inhibition of mTOR activity and blocked cystogenesis in Pkd1-null mice model of ADPKD (Qin et al., 2010). Branched-chain amino acids (BCAAs), including leucine, is an activator of mTOR pathway. Treatment with BCAA in Pkd1 conditional knockout mice accelerated disease progression and upregulation of mTOR pathway. L-type amino acid transporter 1 (LAT-1) is a transporter of neutral amino acids, including BCAAs. Treatment with LAT-1 inhibitor reduces mTOR signaling and attenuation of cyst growth indicated that LAT-1 inhibitor is a potential therapeutic agent for ADPKD (Yamamoto et al., 2017).
Targeting AMPK pathway is also involved in the treatment of ADPKD. Metformin is an AMPK inducer and treatment with metformin slowed cyst formation in both in vitro and ex vivo models of renal cystogenesis (Takiar et al., 2011; Chang et al., 2017). In a clinical trial, it was also determined that treatment with metformin improved disease progression. In this study, it was found that metformin effects on ADPKD progression are mediated through the activation of the AMPK pathway (Seliger et al., 2018). In addition, treatment with inhibitors of histone deacetylases (HDACi) delayed cyst growth in Pkd1−/− models, partially by activating AMPK pathway (Sun et al., 2019). Furthermore, inhibition of miR-25-3p enhanced autophagy by increasing ULK1 expression and the ratio of LC3 II/LC3 I in kidneys from Pkd1flox/−;Ksp-Cre mice (Liu et al., 2020). These studies further support that targeting specific autophagy regulators may attenuate cyst growth, as such, they provided novel targets for the treatment of ADPKD (Figure 5).
Lipid peroxidation correlated with cyst growth and the activation of TMEM16A (anoctamin 1), a chloride bicarbonate transmembrane channel, inducing growth of renal cysts by lipid peroxidation while the direct inhibition of TMEM16A or inhibition of lipid peroxidation delayed cyst development in Pkd1 mutant mice (Schreiber et al., 2019). Treatment with two potent inhibitors of TMEM16A, niclosamide and benzbromarone, significantly reduced renal cyst size in Pkd1−/− mice compared to control mice (Cabrita et al., 2020). Ferrostatin-1 is a synthetic antioxidant and also works as a lipid peroxidation inhibitor but much more efficiently. Treatment with Ferrostatin-1 inhibits ferroptosis and the proliferation of Pkd1 mutant renal epithelial cells and kidneys (Zhang et al., 2021c). Ciclopirox (CPX) or its olamine salt (CPX-O) inhibits the activity of iron-dependent enzymes through chelation of iron. Treatment with CPX-O in PKD mice inhibited cystogenesis as seen by the decrease of cyst index and cystic cell proliferation, and the improvement of renal function, suggesting that CPX-O may delay cyst growth through affecting ferroptosis process in Pkd1 mutant mice (Radadiya et al., 2021). In conclusion, targeting ferroptosis may be a new strategy for ADPKD treatment but still need clinical trials to identify (Figure 5).
In summary, the regulated cell death, including apoptosis, autophagy and ferroptosis, has been investigated in ADPKD, and there is evidence to suggest their involvement in cyst progression (Figure 5). In-spite of these studies, the role of regulated cell death in regulating cystogenesis in ADPKD remains unclear. Moreover, the controversial reports add to the complexity of programmed cell death in general, and how it associates to ADPKD. A major unresolved concern in the field is whether induction of regulated cell death promotes or delays cyst growth. Thus far, the answer to this question is based on two confounding factors including: 1) the type of cell death induced, and 2) the stage of the disease. While targeting regulated cell death appears to be an attractive approach for ADPKD therapy, we believe that further investigation is required to help identify more specific targets for the development of more precise therapy for ADPKD.
In the last decade, an understanding of common mechanisms involved in ADPKD disease, including epigenetic modifications, inflammation, and regulated cell death, have pointed to alternative approaches for potential therapeutic intervention, with promising results emerging from multiple pre-clinical studies. The elucidation of these different mechanisms has uncovered key concerns regarding disease progression. First, the disease model and experimental techniques used, and second, the stage of the disease during which samples are collected, determine the conclusions drawn and how the data obtained is extrapolated for therapeutic purposes. Third, there exits feedback mechanisms and cross-talks between dysregulated pathways in ADPKD which create a more complex network. For example, epigenetic modifications contribute to renal damage by regulating multiple cellular processes and signaling pathways including inflammation and cell death pathways, while inflammation and programmed cell death can contribute to the renal damage progression by inducing epigenetic modifications. Therefore, efforts towards creating a comprehensive picture of the epigenetic landscape and understanding how epigenetic mechanisms affect inflammation and regulated cell death pathways should lay out the foundation for future drug development against ADPKD.
The treatment of ADPKD is a long-term effort, owing to the progressive nature of the disease. Therefore, it is important and necessary to find more selective and less toxic drugs that will be effective for long-term treatment. As such, a better understanding of the mechanisms involved in ADPKD disease progression is essential. To achieve this goal, we should develop and apply the techniques of single cell epigenomics, including single-cell DNA methylome sequencing, single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq), single-cell ChIP-sequencing and single-cell Hi-C, in our future research to better understand how epigenetic mechanisms are involved in ADPKD progression. In addition, CRISPR–Cas9 screens may be applied to identify transcriptional or epigenetic factors that modulate the immune sensitivity of cystic cells or immune activity of immune cells. A better understanding of immunosuppression mechanisms in cystic kidneys should also facilitate the identification of novel immunotherapeutic targets and the development of an immunotherapeutic strategy for ADPKD treatment. Furthermore, to establish a connection of epigenetic mechanisms and renal inflammation with regulated cell death in ADPKD and to investigate the roles of other types of regulated cell death, such as pyroptosis (a highly inflammatory cell death usually caused by microbial infection), necroptosis (a regulated inflammatory mode of cell death that mimics features of both apoptotic and necrotic cell death) and oxeiptosis (a ROS-induced caspase-independent apoptosis-like cell death) in ADPKD should facilitate the identification of more specific targets for a more precise therapy for ADPKD. We believe that an effective and long-term treatment of ADPKD can be achieved by simultaneously or serially targeting different signaling pathways.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abu-Farha, M., Lambert, J.-P., Al-Madhoun, A. S., Elisma, F., Skerjanc, I. S., and Figeys, D. (2008). The Tale of Two Domains. Mol. Cell. Proteomics 7, 560–572. doi:10.1074/mcp.m700271-mcp200
Adams, J. M., and Cory, S. (1998). The Bcl-2 Protein Family: Arbiters of Cell Survival. Science 281, 1322–1326. doi:10.1126/science.281.5381.1322
Aguiari, G., Banzi, M., Gessi, S., Cai, Y., Zeggio, E., Manzati, E., et al. (2004). Deficiency of Polycystin‐2 Reduces Ca2+channel Activity and Cell Proliferation in ADPKD Lymphoblastoid Cells. FASEB J. 18, 884–886. doi:10.1096/fj.03-0687fje
Alam, A., and Perrone, R. D. (2010). Management of ESRD in Patients with Autosomal Dominant Polycystic Kidney Disease. Adv. Chronic Kidney Dis. 17, 164–172. doi:10.1053/j.ackd.2009.12.006
Ali, S. M., Wong, V. Y., Kikly, K., Fredrickson, T. A., Keller, P. M., Dewolf, W. E., et al. (2000). Apoptosis in Polycystic Kidney Disease: Involvement of Caspases. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 278, R763–R769. doi:10.1152/ajpregu.2000.278.3.r763
Alnemri, E. S., Livingston, D. J., Nicholson, D. W., Salvesen, G., Thornberry, N. A., Wong, W. W., et al. (1996). Human ICE/CED-3 Protease Nomenclature. Cell 87, 171. doi:10.1016/s0092-8674(00)81334-3
Amarante-Mendes, G. P., Adjemian, S., Branco, L. M., Zanetti, L. C., Weinlich, R., and Bortoluci, K. R. (2018). Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 9, 2379. doi:10.3389/fimmu.2018.02379
Amin, P., Florez, M., Najafov, A., Pan, H., Geng, J., Ofengeim, D., et al. (2018). Regulation of a Distinct Activated RIPK1 Intermediate Bridging Complex I and Complex II in TNFα-Mediated Apoptosis. Proc. Natl. Acad. Sci. U. S. A. 115, E5944–E5953. doi:10.1073/pnas.1806973115
Anderson, S., Oyama, T. T., Lindsley, J. N., Schutzer, W. E., Beard, D. R., Gattone, V. H., et al. (2012). 2-Hydroxyestradiol Slows Progression of Experimental Polycystic Kidney Disease. Am. J. Physiology-Renal Physiology 302, F636–F645. doi:10.1152/ajprenal.00265.2011
Arango Duque, G., and Descoteaux, A. (2014). Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 5, 491. doi:10.3389/fimmu.2014.00491
Atwood, D. J., Brown, C. N., Holditch, S. J., Pokhrel, D., Thorburn, A., Hopp, K., et al. (2020a). The Effect of Trehalose on Autophagy-Related Proteins and Cyst Growth in a Hypomorphic Pkd1 Mouse Model of Autosomal Dominant Polycystic Kidney Disease. Cell. Signal. 75, 109760. doi:10.1016/j.cellsig.2020.109760
Atwood, D. J., Pokhrel, D., Brown, C. N., Holditch, S. J., Bachu, D. M., Thorburn, A., et al. (2020b). Increased mTOR and Suppressed Autophagic Flux in the Heart of a Hypomorphic Pkd1 Mouse Model of Autosomal Dominant Polycystic Kidney Disease. Cell. Signal. 74, 109730. doi:10.1016/j.cellsig.2020.109730
Avalos, A. M., and Ploegh, H. L. (2014). Early BCR Events and Antigen Capture, Processing, and Loading on MHC Class II on B Cells. Front. Immunol. 5, 92. doi:10.3389/fimmu.2014.00092
Bajwa, Z. H., Gupta, S., Warfield, C. A., and Steinman, T. I. (2001). Pain Management in Polycystic Kidney Disease. Kidney Int. 60, 1631–1644. doi:10.1046/j.1523-1755.2001.00985.x
Batista, I. d. A. A., and Helguero, L. A. (2018). Biological Processes and Signal Transduction Pathways Regulated by the Protein Methyltransferase SETD7 and Their Significance in Cancer. Sig Transduct. Target Ther. 3, 19. doi:10.1038/s41392-018-0017-6
Bedford, M. T., and Richard, S. (2005). Arginine Methylation. Mol. Cell 18, 263–272. doi:10.1016/j.molcel.2005.04.003
Belibi, F. A., Reif, G., Wallace, D. P., Yamaguchi, T., Olsen, L., Li, H., et al. (2004). Cyclic AMP Promotes Growth and Secretion in Human Polycystic Kidney Epithelial Cells. Kidney Int. 66, 964–973. doi:10.1111/j.1523-1755.2004.00843.x
Belibi, F., Zafar, I., Ravichandran, K., Segvic, A. B., Jani, A., Ljubanovic, D. G., et al. (2011). Hypoxia-inducible Factor-1α (HIF-1α) and Autophagy in Polycystic Kidney Disease (PKD). Am. J. Physiology-Renal Physiology 300, F1235–F1243. doi:10.1152/ajprenal.00348.2010
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An Operational Definition of Epigenetics: Figure 1. Genes Dev. 23, 781–783. doi:10.1101/gad.1787609
Bhat, P., Leggatt, G., Waterhouse, N., and Frazer, I. H. (2017). Interferon-γ Derived from Cytotoxic Lymphocytes Directly Enhances Their Motility and Cytotoxicity. Cell Death Dis. 8, e2836. doi:10.1038/cddis.2017.67
Bird, A. (2002). DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 16, 6–21. doi:10.1101/gad.947102
Boletta, A., Qian, F., Onuchic, L. F., Bhunia, A. K., Phakdeekitcharoen, B., Hanaoka, K., et al. (2000). Polycystin-1, the Gene Product of PKD1 , Induces Resistance to Apoptosis and Spontaneous Tubulogenesis in MDCK Cells. Mol. Cell 6, 1267–1273. doi:10.1016/s1097-2765(00)00123-4
Bowden, S. A., Rodger, E. J., Chatterjee, A., Eccles, M. R., and Stayner, C. (2021). Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease. Int. J. Mol. Sci. 22, 13327. doi:10.3390/ijms222413327
Bowden, S. A., Stockwell, P. A., Rodger, E. J., Parry, M. F., Eccles, M. R., Stayner, C., et al. (2020). Extensive Inter-cyst DNA Methylation Variation in Autosomal Dominant Polycystic Kidney Disease Revealed by Genome Scale Sequencing. Front. Genet. 11, 348. doi:10.3389/fgene.2020.00348
Bowden, S. A., Rodger, E. J., Bates, M., Chatterjee, A., Eccles, M. R., and Stayner, C. (2018). Genome-Scale Single Nucleotide Resolution Analysis of DNA Methylation in Human Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 48, 415–424. doi:10.1159/000494739
Bratton, S. B., and Salvesen, G. S. (2010). Regulation of the Apaf-1-Caspase-9 Apoptosome. J. Cell Sci. 123, 3209–3214. doi:10.1242/jcs.073643
Braun, W. E., Schold, J. D., Stephany, B. R., Spirko, R. A., and Herts, B. R. (2014). Low-dose Rapamycin (Sirolimus) Effects in Autosomal Dominant Polycystic Kidney Disease: an Open-Label Randomized Controlled Pilot Study. Clin. J. Am. Soc. Nephrol. 9, 881–888. doi:10.2215/cjn.02650313
Brenet, F., Moh, M., Funk, P., Feierstein, E., Viale, A. J., Socci, N. D., et al. (2011). DNA Methylation of the First Exon Is Tightly Linked to Transcriptional Silencing. Plos One 6, e14524. doi:10.1371/journal.pone.0014524
Brown, M. A., Sims, R. J., Gottlieb, P. D., and Tucker, P. W. (2006). Identification and Characterization of Smyd2: a Split SET/MYND Domain-Containing Histone H3 Lysine 36-specific Methyltransferase that Interacts with the Sin3 Histone Deacetylase Complex. Mol. Cancer 5, 26. doi:10.1186/1476-4598-5-26
Bukanov, N. O., Smith, L. A., Klinger, K. W., Ledbetter, S. R., and Ibraghimov-Beskrovnaya, O. (2006). Long-lasting Arrest of Murine Polycystic Kidney Disease with CDK Inhibitor Roscovitine. Nature 444, 949–952. doi:10.1038/nature05348
Byvoet, P., Shepherd, G. R., Hardin, J. M., and Noland, B. J. (1972). The Distribution and Turnover of Labeled Methyl Groups in Histone Fractions of Cultured Mammalian Cells. Archives Biochem. Biophysics 148, 558–567. doi:10.1016/0003-9861(72)90174-9
Cabrita, I., Kraus, A., Scholz, J. K., Skoczynski, K., Schreiber, R., Kunzelmann, K., et al. (2020). Cyst Growth in ADPKD Is Prevented by Pharmacological and Genetic Inhibition of TMEM16A In Vivo. Nat. Commun. 11, 4320. doi:10.1038/s41467-020-18104-5
Cao, Y., Semanchik, N., Lee, S. H., Somlo, S., Barbano, P. E., Coifman, R., et al. (2009). Chemical Modifier Screen Identifies HDAC Inhibitors as Suppressors of PKD Models. Proc. Natl. Acad. Sci. U.S.A. 106, 21819–21824. doi:10.1073/pnas.0911987106
Capuano, I., Buonanno, P., Riccio, E., Amicone, M., and Pisani, A. (2022). Therapeutic Advances in ADPKD: the Future Awaits. J. Nephrol. 35, 397–415. doi:10.1007/s40620-021-01062-6
Caulfield, A. J., and Lathem, W. W. (2014). Disruption of Fas-Fas Ligand Signaling, Apoptosis, and Innate Immunity by Bacterial Pathogens. PLoS Pathog. 10, e1004252. doi:10.1371/journal.ppat.1004252
Chang, M.-Y., Ma, T.-L., Hung, C.-C., Tian, Y.-C., Chen, Y.-C., Yang, C.-W., et al. (2017). Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 7, 7161. doi:10.1038/s41598-017-07300-x
Chen, L., Zhou, X., Fan, L. X., Yao, Y., Swenson-Fields, K. I., Gadjeva, M., et al. (2015). Macrophage Migration Inhibitory Factor Promotes Cyst Growth in Polycystic Kidney Disease. J. Clin. Invest. 125, 2399–2412. doi:10.1172/jci80467
Cho, H.-S., Hayami, S., Toyokawa, G., Maejima, K., Yamane, Y., Suzuki, T., et al. (2012). RB1 Methylation by SMYD2 Enhances Cell Cycle Progression through an Increase of RB1 Phosphorylation. Neoplasia 14, 476. doi:10.1593/neo.12656
Chou, L. F., Cheng, Y. L., Hsieh, C. Y., Lin, C. Y., Yang, H. Y., Chen, Y. C., et al. (2018). Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease. Nutrients 11, 42. doi:10.3390/nu11010042
Coquet, J. M., Chakravarti, S., Kyparissoudis, K., Mcnab, F. W., Pitt, L. A., Mckenzie, B. S., et al. (2008). Diverse Cytokine Production by NKT Cell Subsets and Identification of an IL-17-producing CD4 − NK1.1 − NKT Cell Population. Proc. Natl. Acad. Sci. U.S.A. 105, 11287–11292. doi:10.1073/pnas.0801631105
Criollo, A., Altamirano, F., Pedrozo, Z., Schiattarella, G. G., Li, D. L., Rivera-Mejías, P., et al. (2018). Polycystin-2-dependent Control of Cardiomyocyte Autophagy. J. Mol. Cell. Cardiol. 118, 110–121. doi:10.1016/j.yjmcc.2018.03.002
Croft, M. (2009). The Role of TNF Superfamily Members in T-Cell Function and Diseases. Nat. Rev. Immunol. 9, 271–285. doi:10.1038/nri2526
Cutter, A. R., and Hayes, J. J. (2015). A Brief Review of Nucleosome Structure. Febs Lett. 589, 2914–2922. doi:10.1016/j.febslet.2015.05.016
Decuypere, J. P., Van Giel, D., Janssens, P., Dong, K., Somlo, S., Cai, Y., et al. (2021). Interdependent Regulation of Polycystin Expression Influences Starvation-Induced Autophagy and Cell Death. Int. J. Mol. Sci. 22, 13511. doi:10.3390/ijms222413511
Dhanasekaran, D. N., and Reddy, E. P. (2008). JNK Signaling in Apoptosis. Oncogene 27, 6245–6251. doi:10.1038/onc.2008.301
Di Giovanni, S., Movsesyan, V., Ahmed, F., Cernak, I., Schinelli, S., Stoica, B., et al. (2005). Cell Cycle Inhibition Provides Neuroprotection and Reduces Glial Proliferation and Scar Formation after Traumatic Brain Injury. Proc. Natl. Acad. Sci. U.S.A. 102, 8333–8338. doi:10.1073/pnas.0500989102
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell 3, 285–296. doi:10.1016/s1535-6108(03)00050-3
Dostert, C., Grusdat, M., Letellier, E., and Brenner, D. (2019). The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and beyond. Physiol. Rev. 99, 115–160. doi:10.1152/physrev.00045.2017
Dranoff, G. (2004). Cytokines in Cancer Pathogenesis and Cancer Therapy. Nat. Rev. Cancer 4, 11–22. doi:10.1038/nrc1252
Duclos, C. M., Champagne, A., Carrier, J. C., Saucier, C., Lavoie, C. L., and Denault, J.-B. (2017). Caspases Play in Traffic. Cell Death Dis. 8, e2636. doi:10.1038/cddis.2017.55
Ecder, T., Melnikov, V. Y., Stanley, M., Korular, D., Lucia, M. S., Schrier, R. W., et al. (2002). Caspases, Bcl-2 Proteins and Apoptosis in Autosomal-Dominant Polycystic Kidney Disease. Kidney Int. 61, 1220–1230. doi:10.1046/j.1523-1755.2002.00250.x
Ecder, T., and Schrier, R. W. (2009). Cardiovascular Abnormalities in Autosomal-Dominant Polycystic Kidney Disease. Nat. Rev. Nephrol. 5, 221–228. doi:10.1038/nrneph.2009.13
Eggenschwiler, J. T., and Anderson, K. V. (2007). Cilia and Developmental Signaling. Annu. Rev. Cell Dev. Biol. 23, 345–373. doi:10.1146/annurev.cellbio.23.090506.123249
El Ters, M., Zhou, X., Lepping, R. J., Lu, P., Karcher, R. T., Mahnken, J. D., et al. (2020). Biological Efficacy and Safety of Niacinamide in Patients with ADPKD. Kidney Int. Rep. 5, 1271–1279. doi:10.1016/j.ekir.2020.06.002
Elena-Real, C. A., Díaz-Quintana, A., González-Arzola, K., Velázquez-Campoy, A., Orzáez, M., López-Rivas, A., et al. (2018). Cytochrome C Speeds up Caspase Cascade Activation by Blocking 14-3-3ε-dependent Apaf-1 Inhibition. Cell Death Dis. 9, 365. doi:10.1038/s41419-018-0408-1
Elmore, S. (2007). Apoptosis: a Review of Programmed Cell Death. Toxicol. Pathol. 35, 495–516. doi:10.1080/01926230701320337
Elsner, L., Muppala, V., Gehrmann, M., Lozano, J., Malzahn, D., Bickeböller, H., et al. (2007). The Heat Shock Protein HSP70 Promotes Mouse NK Cell Activity against Tumors that Express Inducible NKG2D Ligands. J. Immunol. 179, 5523–5533. doi:10.4049/jimmunol.179.8.5523
Epelman, S., Lavine, K. J., and Randolph, G. J. (2014). Origin and Functions of Tissue Macrophages. Immunity 41, 21–35. doi:10.1016/j.immuni.2014.06.013
Erlich, Z., Shlomovitz, I., Edry-Botzer, L., Cohen, H., Frank, D., Wang, H., et al. (2019). Macrophages, rather Than DCs, Are Responsible for Inflammasome Activity in the GM-CSF BMDC Model. Nat. Immunol. 20, 397–406. doi:10.1038/s41590-019-0313-5
Falvo, J. V., Brinkman, B. M. N., Tsytsykova, A. V., Tsai, E. Y., Yao, T.-P., Kung, A. L., et al. (2000). A Stimulus-specific Role for CREB-Binding Protein (CBP) in T Cell Receptor-Activated Tumor Necrosis Factor α Gene Expression. Proc. Natl. Acad. Sci. U.S.A. 97, 3925–3929. doi:10.1073/pnas.97.8.3925
Fan, L. X., Zhou, X., Sweeney, W. E., Wallace, D. P., Avner, E. D., Grantham, J. J., et al. (2013a). Smac-mimetic-induced Epithelial Cell Death Reduces the Growth of Renal Cysts. J. Am. Soc. Nephrol. 24, 2010–2022. doi:10.1681/asn.2013020176
Fan, L. X., Zhou, X., Sweeney, W. E., Wallace, D. P., Avner, E. D., Grantham, J. J., et al. (2013b). Smac-Mimetic-Induced Epithelial Cell Death Reduces the Growth of Renal Cysts. J. Am. Soc. Nephrol. 24, 2010–2022. doi:10.1681/asn.2013020176
Fick, G. M., Johnson, A. M., Strain, J. D., Kimberling, W. J., Kumar, S., Manco-Johnson, M. L., et al. (1993). Characteristics of Very Early Onset Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 3, 1863–1870. doi:10.1681/asn.v3121863
Flores, J., Noël, A., Foveau, B., Beauchet, O., and Leblanc, A. C. (2020). Pre-symptomatic Caspase-1 Inhibitor Delays Cognitive Decline in a Mouse Model of Alzheimer Disease and Aging. Nat. Commun. 11, 4571. doi:10.1038/s41467-020-18405-9
Fuchs, Y., and Steller, H. (2011). Programmed Cell Death in Animal Development and Disease. Cell 147, 742–758. doi:10.1016/j.cell.2011.10.033
Garg, A. D., Galluzzi, L., Apetoh, L., Baert, T., Birge, R. B., Bravo-San Pedro, J. M., et al. (2015). Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 6, 588. doi:10.3389/fimmu.2015.00588
Gaudino, S. J., and Kumar, P. (2019). Cross-Talk between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 10, 360. doi:10.3389/fimmu.2019.00360
Gerdes, J. M., Davis, E. E., and Katsanis, N. (2009). The Vertebrate Primary Cilium in Development, Homeostasis, and Disease. Cell 137, 32–45. doi:10.1016/j.cell.2009.03.023
Gräff, J., and Tsai, L.-H. (2013). Histone Acetylation: Molecular Mnemonics on the Chromatin. Nat. Rev. Neurosci. 14, 97–111. doi:10.1038/nrn3427
Granja, A. G., Nogal, M. L., Hurtado, C., Del Aguila, C., Carrascosa, A. L., Salas, M. L., et al. (2006). The Viral Protein A238L Inhibits TNF-α Expression through a CBP/p300 Transcriptional Coactivators Pathway. J. Immunol. 176, 451–462. doi:10.4049/jimmunol.176.1.451
Grantham, J. J. (2008). Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 359, 1477–1485. doi:10.1056/nejmcp0804458
Grantham, J. J., Mulamalla, S., and Swenson-Fields, K. I. (2011). Why Kidneys Fail in Autosomal Dominant Polycystic Kidney Disease. Nat. Rev. Nephrol. 7, 556–566. doi:10.1038/nrneph.2011.109
Green, D. R., and Llambi, F. (2015). Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 7, a006080. doi:10.1101/cshperspect.a006080
Greer, E. L., and Shi, Y. (2012). Histone Methylation: a Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 13, 343–357. doi:10.1038/nrg3173
Hajirezaei, F., Ghaderian, S. M. H., Hasanzad, M., Nafar, M., Ghadiani, M. H., Biglari, S., et al. (2020). Methylation of the PKD1 Promoter Inversely Correlates with its Expression in Autosomal Dominant Polycystic Kidney Disease. rbmb.Net. 9, 193–198. doi:10.29252/rbmb.9.2.193
Harris, P. C., and Torres, V. E. (1993). “Polycystic Kidney Disease, Autosomal Dominant,” in GeneReviews(R). Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaaet al. (Seattle (WA).
Hartzell, S., Bin, S., Cantarelli, C., Haverly, M., Manrique, J., Angeletti, A., et al. (2020). Kidney Failure Associates with T Cell Exhaustion and Imbalanced Follicular Helper T Cells. Front. Immunol. 11, 583702. doi:10.3389/fimmu.2020.583702
Hateboer, N., v Dijk, M. A., Bogdanova, N., Coto, E., Saggar-Malik, A. K., Millan, J. L. S., et al. (1999). Comparison of Phenotypes of Polycystic Kidney Disease Types 1 and 2. Lancet 353, 103–107. doi:10.1016/s0140-6736(98)03495-3
Hatok, J., and Racay, P. (2016). Bcl-2 Family Proteins: Master Regulators of Cell Survival. Biomol. Concepts 7, 259–270. doi:10.1515/bmc-2016-0015
Heard, E., Rougeulle, C., Arnaud, D., Avner, P., Allis, C. D., and Spector, D. L. (2001). Methylation of Histone H3 at Lys-9 Is an Early Mark on the X Chromosome during X Inactivation. Cell 107, 727–738. doi:10.1016/s0092-8674(01)00598-0
Hempel, K., Lange, H. W., and Birkofer, L. (1968). E-N-Trimethyllysin, eine neue Aminosaure in Histonen. Naturwissenschaften 55, 37. doi:10.1007/bf00593411
Hildebrandt, F., Benzing, T., and Katsanis, N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543. doi:10.1056/nejmra1010172
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Huang, J., Perez-Burgos, L., Placek, B. J., Sengupta, R., Richter, M., Dorsey, J. A., et al. (2006). Repression of P53 Activity by Smyd2-Mediated Methylation. Nature 444, 629–632. doi:10.1038/nature05287
Huang, W., Huang, F., Kannan, A. K., Hu, J., and August, A. (2014). ITK Tunes IL-4-induced Development of Innate Memory CD8+ T Cells in a γδ T and Invariant NKT Cell-independent Manner. J. Leukoc. Biol. 96, 55–63. doi:10.1189/jlb.1ab0913-484rr
Hume, D. A. (2008). Differentiation and Heterogeneity in the Mononuclear Phagocyte System. Mucosal Immunol. 1, 432–441. doi:10.1038/mi.2008.36
Jeltsch, A. (2006). Molecular Enzymology of Mammalian DNA Methyltransferases. Curr. Top. Microbiol. Immunol. 301, 203–225. doi:10.1007/3-540-31390-7_7
Jones, P. A., Issa, J.-P. J., and Baylin, S. (2016). Targeting the Cancer Epigenome for Therapy. Nat. Rev. Genet. 17, 630–641. doi:10.1038/nrg.2016.93
Julien, O., and Wells, J. A. (2017). Caspases and Their Substrates. Cell Death Differ. 24, 1380–1389. doi:10.1038/cdd.2017.44
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 19, 5720–5728. doi:10.1093/emboj/19.21.5720
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P. F., Doll, S., Croix, C. S., et al. (2017). Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 13, 81–90. doi:10.1038/nchembio.2238
Kale, J., Osterlund, E. J., and Andrews, D. W. (2018). BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 25, 65–80. doi:10.1038/cdd.2017.186
Kang, K., Bachu, M., Park, S. H., Kang, K., Bae, S., Park-Min, K.-H., et al. (2019). IFN-γ Selectively Suppresses a Subset of TLR4-Activated Genes and Enhancers to Potentiate Macrophage Activation. Nat. Commun. 10, 3320. doi:10.1038/s41467-019-11147-3
Karihaloo, A., Koraishy, F., Huen, S. C., Lee, Y., Merrick, D., Caplan, M. J., et al. (2011). Macrophages Promote Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 22, 1809–1814. doi:10.1681/asn.2011010084
Ke, F. F. S., Vanyai, H. K., Cowan, A. D., Delbridge, A. R. D., Whitehead, L., Grabow, S., et al. (2018). Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell 173, 1217–1230. doi:10.1016/j.cell.2018.04.036
Kenter, A. T., Rentmeester, E., Riet, J., Boers, R., Boers, J., Ghazvini, M., et al. (2020). Cystic Renal-Epithelial Derived Induced Pluripotent Stem Cells from Polycystic Kidney Disease Patients. Stem Cells Transl. Med. 9, 478–490. doi:10.1002/sctm.18-0283
Kerr, K., Mcaneney, H., Flanagan, C., Maxwell, A. P., and Mcknight, A. J. (2019). Differential Methylation as a Diagnostic Biomarker of Rare Renal Diseases: a Systematic Review. BMC Nephrol. 20, 320. doi:10.1186/s12882-019-1517-5
Kim, H.-J., and Edelstein, C. L. (2012). Mammalian Target of Rapamycin Inhibition in Polycystic Kidney Disease: From Bench to Bedside. Kidney Res. Clin. Pract. 31, 132–138. doi:10.1016/j.krcp.2012.07.002
Kim, I., Ding, T., Fu, Y., Li, C., Cui, L., Li, A., et al. (2009). Conditional Mutation ofPkd2Causes Cystogenesis and Upregulates β-Catenin. J. Am. Soc. Nephrol. 20, 2556–2569. doi:10.1681/asn.2009030271
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi:10.1038/ncb2152
Kischkel, F. C., Hellbardt, S., Behrmann, I., Germer, M., Pawlita, M., Krammer, P. H., et al. (1995). Cytotoxicity-dependent APO-1 (Fas/CD95)-Associated Proteins Form a Death-Inducing Signaling Complex (DISC) with the Receptor. EMBO J. 14, 5579–5588. doi:10.1002/j.1460-2075.1995.tb00245.x
Kleczko, E. K., Marsh, K. H., Tyler, L. C., Furgeson, S. B., Bullock, B. L., Altmann, C. J., et al. (2018). CD8+ T Cells Modulate Autosomal Dominant Polycystic Kidney Disease Progression. Kidney Int. 94, 1127–1140. doi:10.1016/j.kint.2018.06.025
Korsmeyer, S. J. (1999). BCL-2 Gene Family and the Regulation of Programmed Cell Death. Cancer Res. 59, 1693s–1700s.
Kriegenburg, F., Ungermann, C., and Reggiori, F. (2018). Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 28, R512–R518. doi:10.1016/j.cub.2018.02.034
Krijgsman, D., Hokland, M., and Kuppen, P. J. K. (2018). The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol. 9, 367. doi:10.3389/fimmu.2018.00367
Kroemer, G., and Levine, B. (2008). Autophagic Cell Death: the Story of a Misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010. doi:10.1038/nrm2529
Krzyszczyk, P., Schloss, R., Palmer, A., and Berthiaume, F. (2018). The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 9, 419. doi:10.3389/fphys.2018.00419
Kumar, S. (2018). Natural Killer Cell Cytotoxicity and its Regulation by Inhibitory Receptors. Immunology 154, 383–393. doi:10.1111/imm.12921
Kuo, M.-H., and Allis, C. D. (1998). Roles of Histone Acetyltransferases and Deacetylases in Gene Regulation. Bioessays 20, 615–626. doi:10.1002/(sici)1521-1878(199808)20:8<615::aid-bies4>3.0.co;2-h
Lakhia, R., Hajarnis, S., Williams, D., Aboudehen, K., Yheskel, M., Xing, C., et al. (2016). MicroRNA-21 Aggravates Cyst Growth in a Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 27, 2319–2330. doi:10.1681/asn.2015060634
Lanzavecchia, A., Iezzi, G., and Viola, A. (1999). From TCR Engagement to T Cell Activation. Cell 96, 1–4. doi:10.1016/s0092-8674(00)80952-6
Laskin, D. L., Weinberger, B., and Laskin, J. D. (2001). Functional Heterogeneity in Liver and Lung Macrophages. J. Leukoc. Biol. 70, 163–170.
Law, B. M. P., Wilkinson, R., Wang, X., Kildey, K., Lindner, M., Rist, M. J., et al. (2017). Interferon-γ Production by Tubulointerstitial Human CD56bright Natural Killer Cells Contributes to Renal Fibrosis and Chronic Kidney Disease Progression. Kidney Int. 92, 79–88. doi:10.1016/j.kint.2017.02.006
Lee, E. J., Ko, J. Y., Oh, S., Jun, J., Mun, H., Lim, C. J., et al. (2020). Autophagy Induction Promotes Renal Cyst Growth in Polycystic Kidney Disease. EBioMedicine 60, 102986. doi:10.1016/j.ebiom.2020.102986
Lee, E. J., Park, E. Y., Mun, H., Chang, E., Ko, J. Y., Kim, D. Y., et al. (2015). Soluble Receptor for Advanced Glycation End Products Inhibits Disease Progression in Autosomal Dominant Polycystic Kidney Disease by Down-Regulating Cell Proliferation. FASEB J. 29, 3506–3514. doi:10.1096/fj.15-272302
Li, A., Fan, S., Xu, Y., Meng, J., Shen, X., Mao, J., et al. (2017a). Rapamycin Treatment Dose-Dependently Improves the Cystic Kidney in a New ADPKD Mouse Modelviathe mTORC1 and Cell-Cycle-Associated CDK1/cyclin axis. J. Cell. Mol. Med. 21, 1619–1635. doi:10.1111/jcmm.13091
Li, B., Carey, M., and Workman, J. L. (2007). The Role of Chromatin during Transcription. Cell 128, 707–719. doi:10.1016/j.cell.2007.01.015
Li, D., and Wu, M. (2021). Pattern Recognition Receptors in Health and Diseases. Sig Transduct. Target Ther. 6, 291. doi:10.1038/s41392-021-00687-0
Li, J., Cao, F., Yin, H.-l., Huang, Z.-j., Lin, Z.-t., Mao, N., et al. (2020a). Ferroptosis: Past, Present and Future. Cell Death Dis. 11, 88. doi:10.1038/s41419-020-2298-2
Li, L. X., Zhou, J. X., Wang, X., Zhang, H., Harris, P. C., Calvet, J. P., et al. (2020b). Cross-talk between CDK4/6 and SMYD2 Regulates Gene Transcription, Tubulin Methylation, and Ciliogenesis. Sci. Adv. 6, eabb3154. doi:10.1126/sciadv.abb3154
Li, L. X., Fan, L. X., Zhou, J. X., Grantham, J. J., Calvet, J. P., Sage, J., et al. (2017b). Lysine Methyltransferase SMYD2 Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. J. Clin. Invest 127, 2751–2764. doi:10.1172/jci90921
Li, L. X., Fan, L. X., Zhou, J. X., Grantham, J. J., Calvet, J. P., Sage, J., et al. (2017c). Lysine Methyltransferase SMYD2 Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. J. Clin. Investigation 127, 2751–2764. doi:10.1172/jci90921
Li, L. X., and Li, X. (2021). Epigenetically Mediated Ciliogenesis and Cell Cycle Regulation, and Their Translational Potential. Cells 10, 1662. doi:10.3390/cells10071662
Li, M., Qin, S., Wang, L., and Zhou, J. (2013). Genomic Instability in Patients with Autosomal-Dominant Polycystic Kidney Disease. J. Int. Med. Res. 41, 169–175. doi:10.1177/0300060513475956
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017d). Inhibition of Neuronal Ferroptosis Protects Hemorrhagic Brain. JCI Insight 2, e90777. doi:10.1172/jci.insight.90777
Li, X. (2011). Epigenetics and Autosomal Dominant Polycystic Kidney Disease. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1812, 1213–1218. doi:10.1016/j.bbadis.2010.10.008
Li, X. (2015). “Epigenetics in ADPKD: Understanding Mechanisms and Discovering Treatment,” in Polycystic Kidney Disease. Editor X. Li (Brisbane (AU). doi:10.15586/codon.pkd.2015.ch12
Li, X., Magenheimer, B. S., Xia, S., Johnson, T., Wallace, D. P., Calvet, J. P., et al. (2008). A Tumor Necrosis Factor-α-Mediated Pathway Promoting Autosomal Dominant Polycystic Kidney Disease. Nat. Med. 14, 863–868. doi:10.1038/nm1783
Liang, Y., Song, P., Chen, W., Xie, X., Luo, R., Su, J., et al. (2020). Inhibition of Caspase-1 Ameliorates Ischemia-Associated Blood-Brain Barrier Dysfunction and Integrity by Suppressing Pyroptosis Activation. Front. Cell Neurosci. 14, 540669. doi:10.3389/fncel.2020.540669
Lin, D., Lavender, H., Soilleux, E. J., and O'callaghan, C. A. (2012). NF-κB Regulates MICA Gene Transcription in Endothelial Cell through a Genetically Inhibitable Control Site. J. Biol. Chem. 287, 4299–4310. doi:10.1074/jbc.m111.282152
Lin, M. G., and Hurley, J. H. (2016). Structure and Function of the ULK1 Complex in Autophagy. Curr. Opin. Cell Biol. 39, 61–68. doi:10.1016/j.ceb.2016.02.010
Liu, G., Kang, X., Guo, P., Shang, Y., Du, R., Wang, X., et al. (2020). miR-25-3p Promotes Proliferation and Inhibits Autophagy of Renal Cells in Polycystic Kidney Mice by Regulating ATG14-Beclin 1. Ren. Fail. 42, 333–342. doi:10.1080/0886022x.2020.1745236
Liu, W., Fan, L. X., Zhou, X., Sweeney, W. E., Avner, E. D., and Li, X. (2012). HDAC6 Regulates Epidermal Growth Factor Receptor (EGFR) Endocytic Trafficking and Degradation in Renal Epithelial Cells. PLoS One 7, e49418. doi:10.1371/journal.pone.0049418
Liu, W., and Huber, S. A. (2011). Cross-talk between Cd1d-Restricted Nkt Cells and γδ Cells in T Regulatory Cell Response. Virol. J. 8, 32. doi:10.1186/1743-422x-8-32
Liu, Y., Pejchinovski, M., Wang, X., Fu, X., Castelletti, D., Watnick, T. J., et al. (2018). Dual mTOR/PI3K Inhibition Limits PI3K-dependent Pathways Activated upon mTOR Inhibition in Autosomal Dominant Polycystic Kidney Disease. Sci. Rep. 8, 5584. doi:10.1038/s41598-018-22938-x
Liu, Y., Käch, A., Ziegler, U., Ong, A. C. M., Wallace, D. P., Arcaro, A., et al. (2013). The Role of Phospholipase D in Modulating the MTOR Signaling Pathway in Polycystic Kidney Disease. PLoS One 8, e73173. doi:10.1371/journal.pone.0073173
Liu, Z.-g., Hsu, H., Goeddel, D. V., and Karin, M. (1996). Dissection of TNF Receptor 1 Effector Functions: JNK Activation Is Not Linked to Apoptosis while NF-Κb Activation Prevents Cell Death. Cell 87, 565–576. doi:10.1016/s0092-8674(00)81375-6
Lodoen, M., Ogasawara, K., Hamerman, J. A., Arase, H., Houchins, J. P., Mocarski, E. S., et al. (2003). NKG2D-mediated Natural Killer Cell Protection against Cytomegalovirus Is Impaired by Viral Gp40 Modulation of Retinoic Acid Early Inducible 1 Gene Molecules. J. Exp. Med. 197, 1245–1253. doi:10.1084/jem.20021973
Long, E. O., and Rajagopalan, S. (2002). Stress Signals Activate Natural Killer Cells. J. Exp. Med. 196, 1399–1402. doi:10.1084/jem.20021747
Long, E. O., Sik Kim, H., Liu, D., Peterson, M. E., and Rajagopalan, S. (2013). Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu. Rev. Immunol. 31, 227–258. doi:10.1146/annurev-immunol-020711-075005
Luckheeram, R. V., Zhou, R., Verma, A. D., and Xia, B. (2012). CD4⁺T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 925135. doi:10.1155/2012/925135
Lunyak, V. V., and Rosenfeld, M. G. (2008). Epigenetic Regulation of Stem Cell Fate. Hum. Mol. Genet. 17, R28–R36. doi:10.1093/hmg/ddn149
Malech, H. L., Deleo, F. R., and Quinn, M. T. (2014). The Role of Neutrophils in the Immune System: an Overview. Methods Mol. Biol. 1124, 3–10. doi:10.1007/978-1-62703-845-4_1
Malysheva, E. V., Kruglov, S. V., Nazarov, V. A., Manukhina, E. B., and Malyshev, I. Y. (2007). Intracellular and Extracellular NO Differentially Modulates the Stress Response and Apoptosis in Macrophages Exposed to the Influence of Biological or Physical Factors. Bull. Exp. Biol. Med. 143, 673–677. doi:10.1007/s10517-007-0211-y
Marsden, V. S., O'connor, L., O'reilly, L. A., Silke, J., Metcalf, D., Ekert, P. G., et al. (2002). Apoptosis Initiated by Bcl-2-Regulated Caspase Activation Independently of the Cytochrome c/Apaf-1/caspase-9 Apoptosome. Nature 419, 634–637. doi:10.1038/nature01101
Maser, R. L., Vassmer, D., Magenheimer, B. S., and Calvet, J. P. (2002). Oxidant Stress and Reduced Antioxidant Enzyme Protection in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 13, 991–999. doi:10.1681/asn.v134991
Matoba, K., Kotani, T., Tsutsumi, A., Tsuji, T., Mori, T., Noshiro, D., et al. (2020). Atg9 Is a Lipid Scramblase that Mediates Autophagosomal Membrane Expansion. Nat. Struct. Mol. Biol. 27, 1185–1193. doi:10.1038/s41594-020-00518-w
Meijer, A. J., and Codogno, P. (2006). Signalling and Autophagy Regulation in Health, Aging and Disease. Mol. Aspects Med. 27, 411–425. doi:10.1016/j.mam.2006.08.002
Micheau, O., and Tschopp, J. (2003). Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 114, 181–190. doi:10.1016/s0092-8674(03)00521-x
Miller, J. L., and Grant, P. A. (2013). The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell. Biochem. 61, 289–317. doi:10.1007/978-94-007-4525-4_13
Millet-Boureima, C., Chingle, R., Lubell, W. D., and Gamberi, C. (2019). Cyst Reduction in a Polycystic Kidney Disease Drosophila Model Using Smac Mimics. Biomedicines 7, 82. doi:10.3390/biomedicines7040082
Milutinovic, J., Rust, P. F., Fialkow, P. J., Agodoa, L. Y., Phillips, L. A., Rudd, T. G., et al. (1992). Intrafamilial Phenotypic Expression of Autosomal Dominant Polycystic Kidney Disease. Am. J. Kidney Dis. 19, 465–472. doi:10.1016/s0272-6386(12)80956-5
Minton, K. (2017). Stress-induced Macrophage Polarization. Nat. Rev. Immunol. 17, 277. doi:10.1038/nri.2017.41
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., et al. (1998). A Protein Conjugation System Essential for Autophagy. Nature 395, 395–398. doi:10.1038/26506
Mizushima, N. (2020). The ATG Conjugation Systems in Autophagy. Curr. Opin. Cell Biol. 63, 1–10. doi:10.1016/j.ceb.2019.12.001
Mizushima, N. (2010). The Role of the Atg1/ULK1 Complex in Autophagy Regulation. Curr. Opin. Cell Biol. 22, 132–139. doi:10.1016/j.ceb.2009.12.004
Morales-Cruz, M., Figueroa, C. M., González-Robles, T., Delgado, Y., Molina, A., Méndez, J., et al. (2014). Activation of Caspase-dependent Apoptosis by Intracellular Delivery of Cytochrome C-Based Nanoparticles. J. Nanobiotechnol 12, 33. doi:10.1186/s12951-014-0033-9
Mosser, D. M., and Edwards, J. P. (2008). Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 8, 958–969. doi:10.1038/nri2448
Müller, E., Speth, M., Christopoulos, P. F., Lunde, A., Avdagic, A., Øynebråten, I., et al. (2018). Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages when Combined with TLR Stimulation. Front. Immunol. 9, 2520. doi:10.3389/fimmu.2018.02520
Muralidharan, S., and Mandrekar, P. (2013). Cellular Stress Response and Innate Immune Signaling: Integrating Pathways in Host Defense and Inflammation. J. Leukoc. Biol. 94, 1167–1184. doi:10.1189/jlb.0313153
Murray, K. (1964). The Occurrence of Iε-N-Methyl Lysine in Histones. Biochemistry 3, 10–15. doi:10.1021/bi00889a003
Nagata, S. (1999). Fas Ligand-Induced Apoptosis. Annu. Rev. Genet. 33, 29–55. doi:10.1146/annurev.genet.33.1.29
Nakamura, T., Kawagoe, Y., Ueda, Y., Yamada, S., and Koide, H. (2011). Hemoperfusion Treatment in a Septic Shock Patient with Autosomal Dominant Polycystic Kidney Disease and Increased HMGB1 Protein Levels. Blood Purif. 32, 139–142. doi:10.1159/000325731
Nakamura, T., Sato, E., Fujiwara, N., Kawagoe, Y., Yamada, S., Ueda, Y., et al. (2012). Changes in Urinary Albumin Excretion, Inflammatory and Oxidative Stress Markers in ADPKD Patients with Hypertension. Am. J. Med. Sci. 343, 46–51. doi:10.1097/maj.0b013e31821f0552
Natoli, T. A., Smith, L. A., Rogers, K. A., Wang, B., Komarnitsky, S., Budman, Y., et al. (2010). Inhibition of Glucosylceramide Accumulation Results in Effective Blockade of Polycystic Kidney Disease in Mouse Models. Na.t Med. 16, 788–792.
Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., et al. (2003). Polycystins 1 and 2 Mediate Mechanosensation in the Primary Cilium of Kidney Cells. Nat. Genet. 33, 129–137. doi:10.1038/ng1076
Netea, M. G., Domínguez-Andrés, J., Barreiro, L. B., Chavakis, T., Divangahi, M., Fuchs, E., et al. (2020). Defining Trained Immunity and its Role in Health and Disease. Nat. Rev. Immunol. 20, 375–388. doi:10.1038/s41577-020-0285-6
Nguyen, D. P., Alai, M. M. G., Virdee, S., and Chin, J. W. (2010). Genetically Directing Ɛ-N, N-Dimethyl-L-Lysine in Recombinant Histones. Chem. Biol. 17, 1072–1076. doi:10.1016/j.chembiol.2010.07.013
Nicolau, C., Torra, R., Badenas, C., Vilana, R., Bianchi, L., Gilabert, R., et al. (1999). Autosomal Dominant Polycystic Kidney Disease Types 1 and 2: Assessment of US Sensitivity for Diagnosis. Radiology 213, 273–276. doi:10.1148/radiology.213.1.r99oc05273
Nicolet, B. P., Guislain, A., Van Alphen, F. P. J., Gomez-Eerland, R., Schumacher, T. N. M., Van Den Biggelaar, M., et al. (2020). CD29 Identifies IFN-γ-Producing Human CD8 + T Cells with an Increased Cytotoxic Potential. Proc. Natl. Acad. Sci. U.S.A. 117, 6686–6696. doi:10.1073/pnas.1913940117
Nie, X., and Arend, L. J. (2017). Deletion of Pkd1 in Renal Stromal Cells Causes Defects in the Renal Stromal Compartment and Progressive Cystogenesis in the Kidney. Lab. Invest 97, 1427–1438. doi:10.1038/labinvest.2017.97
Norbury, C. J., and Hickson, I. D. (2001). Cellular Responses to DNA Damage. Annu. Rev. Pharmacol. Toxicol. 41, 367–401. doi:10.1146/annurev.pharmtox.41.1.367
Obeng, E. (2021). Apoptosis (Programmed Cell Death) and its Signals - A Review. Braz. J. Biol. 81, 1133–1143. doi:10.1590/1519-6984.228437
Onda, M., Kobayashi, K., and Pastan, I. (2019). Depletion of Regulatory T Cells in Tumors with an Anti-CD25 Immunotoxin Induces CD8 T Cell-Mediated Systemic Antitumor Immunity. Proc. Natl. Acad. Sci. U.S.A. 116, 4575–4582. doi:10.1073/pnas.1820388116
O’brien, J., Hayder, H., Zayed, Y., and Peng, C. (2018). Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 9, 402.
Ostrom, L., Tang, M.-J., Gruss, P., and Dressler, G. R. (2000). Reduced Pax2 Gene Dosage Increases Apoptosis and Slows the Progression of Renal Cystic Disease. Dev. Biol. 219, 250–258. doi:10.1006/dbio.2000.9618
Otomo, C., Metlagel, Z., Takaesu, G., and Otomo, T. (2013). Structure of the Human ATG12∼ATG5 Conjugate Required for LC3 Lipidation in Autophagy. Nat. Struct. Mol. Biol. 20, 59–66. doi:10.1038/nsmb.2431
Park, E. Y., Sung, Y. H., Yang, M. H., Noh, J. Y., Park, S. Y., Lee, T. Y., et al. (2009). Cyst Formation in Kidney via B-Raf Signaling in the PKD2 Transgenic Mice. J. Biol. Chem. 284, 7214–7222. doi:10.1074/jbc.m805890200
Park, I. Y., Powell, R. T., Tripathi, D. N., Dere, R., Ho, T. H., Blasius, T. L., et al. (2016). Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell 166, 950–962. doi:10.1016/j.cell.2016.07.005
Pazour, G. J., and Rosenbaum, J. L. (2002). Intraflagellar Transport and Cilia-dependent Diseases. Trends Cell Biol. 12, 551–555. doi:10.1016/s0962-8924(02)02410-8
Pedersen, M. T., and Helin, K. (2010). Histone Demethylases in Development and Disease. Trends Cell Biol. 20, 662–671. doi:10.1016/j.tcb.2010.08.011
Pellicci, D. G., Koay, H.-F., and Berzins, S. P. (2020). Thymic Development of Unconventional T Cells: How NKT Cells, MAIT Cells and γδ T Cells Emerge. Nat. Rev. Immunol. 20, 756–770. doi:10.1038/s41577-020-0345-y
Peña-Blanco, A., and García-Sáez, A. J. (2018). Bax, Bak and beyond - Mitochondrial Performance in Apoptosis. FEBS J. 285, 416–431. doi:10.1111/febs.14186
Peña-Oyarzun, D., Rodriguez-Peña, M., Burgos-Bravo, F., Vergara, A., Kretschmar, C., Sotomayor-Flores, C., et al. (2021). PKD2/polycystin-2 Induces Autophagy by Forming a Complex with BECN1. Autophagy 17, 1714–1728. doi:10.1080/15548627.2020.1782035
Peña-Oyarzun, D., Troncoso, R., Kretschmar, C., Hernando, C., Budini, M., Morselli, E., et al. (2017). Hyperosmotic Stress Stimulates Autophagy via Polycystin-2. Oncotarget 8, 55984–55997. doi:10.18632/oncotarget.18995
Perera Molligoda Arachchige, A. S. (2021). Human NK Cells: From Development to Effector Functions. Innate Immun. 27, 212–229. doi:10.1177/17534259211001512
Podbielska, M., Levery, S. B., and Hogan, E. L. (2011). The Structural and Functional Role of Myelin Fast-Migrating Cerebrosides: Pathological Importance in Multiple Sclerosis. Clin. Lipidol. 6, 159–179. doi:10.2217/clp.11.8
Prencipe, G., Caiello, I., Pascarella, A., Grom, A. A., Bracaglia, C., Chatel, L., et al. (2018). Neutralization of IFN-γ Reverts Clinical and Laboratory Features in a Mouse Model of Macrophage Activation Syndrome. J. Allergy Clin. Immunol. 141, 1439–1449. doi:10.1016/j.jaci.2017.07.021
Pugacheva, E. N., Jablonski, S. A., Hartman, T. R., Henske, E. P., and Golemis, E. A. (2007). HEF1-Dependent Aurora a Activation Induces Disassembly of the Primary Cilium. Cell 129, 1351–1363. doi:10.1016/j.cell.2007.04.035
Qian, F., Germino, F. J., Cai, Y., Zhang, X., Somlo, S., and Germino, G. G. (1997). PKD1 Interacts with PKD2 through a Probable Coiled-Coil Domain. Nat. Genet. 16, 179–183. doi:10.1038/ng0697-179
Qin, S., Taglienti, M., Nauli, S. M., Contrino, L., Takakura, A., Zhou, J., et al. (2010). Failure to Ubiquitinate C-Met Leads to Hyperactivation of mTOR Signaling in a Mouse Model of Autosomal Dominant Polycystic Kidney Disease. J. Clin. Invest. 120, 3617–3628. doi:10.1172/jci41531
Radadiya, P. S., Thornton, M. M., Puri, R. V., Yerrathota, S., Dinh-Phan, J., Magenheimer, B., et al. (2021). Ciclopirox Olamine Induces Ferritinophagy and Reduces Cyst Burden in Polycystic Kidney Disease. JCI Insight 6, e141299. doi:10.1172/jci.insight.141299
Raman, A., Reif, G. A., Dai, Y., Khanna, A., Li, X., Astleford, L., et al. (2017). Integrin-Linked Kinase Signaling Promotes Cyst Growth and Fibrosis in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 28, 2708–2719. doi:10.1681/asn.2016111235
Ran, J., Yang, Y., Li, D., Liu, M., and Zhou, J. (2015). Deacetylation of α-tubulin and Cortactin Is Required for HDAC6 to Trigger Ciliary Disassembly. Sci. Rep. 5, 12917. doi:10.1038/srep12917
Rath, P. C., and Aggarwal, B. B. (1999). TNF-induced Signaling in Apoptosis. J. Clin. Immunol. 19, 350–364. doi:10.1023/a:1020546615229
Razin, A., and Cedar, H. (1991). DNA Methylation and Gene Expression. Microbiol. Rev. 55, 451–458. doi:10.1128/mr.55.3.451-458.1991
Ringrose, L., and Paro, R. (2004). Epigenetic Regulation of Cellular Memory by the Polycomb and Trithorax Group Proteins. Annu. Rev. Genet. 38, 413–443. doi:10.1146/annurev.genet.38.072902.091907
Robertson, K. D. (2005). DNA Methylation and Human Disease. Nat. Rev. Genet. 6, 597–610. doi:10.1038/nrg1655
Rogers, K. A., Moreno, S. E., Smith, L. A., Husson, H., Bukanov, N. O., Ledbetter, S. R., et al. (2016). Differences in the Timing and Magnitude of Pkd1 Gene Deletion Determine the Severity of Polycystic Kidney Disease in an Orthologous Mouse Model of ADPKD. Physiol. Rep. 4, e12846. doi:10.14814/phy2.12846
Roh, J. S., and Sohn, D. H. (2018). Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 18, e27. doi:10.4110/in.2018.18.e27
Roufayel, R. (2016). Regulation of Stressed-Induced Cell Death by the Bcl-2 Family of Apoptotic Proteins. Mol. Membr. Biol. 33, 89–99. doi:10.1080/09687688.2017.1400600
Sadakierska-Chudy, A., and Filip, M. (2015). A Comprehensive View of the Epigenetic Landscape. Part II: Histone Post-translational Modification, Nucleosome Level, and Chromatin Regulation by ncRNAs. Neurotox. Res. 27, 172–197. doi:10.1007/s12640-014-9508-6
Sadasivam, M., Noel, S., Lee, S. A., Gong, J., Allaf, M. E., Pierorazio, P., et al. (2019). Activation and Proliferation of PD-1+ Kidney Double-Negative T Cells Is Dependent on Nonclassical MHC Proteins and IL-2. J. Am. Soc. Nephrol. 30, 277–292. doi:10.1681/asn.2018080815
Sawa-Makarska, J., Baumann, V., Coudevylle, N., von Bülow, S., Nogellova, V., Abert, C., et al. (2020). Reconstitution of Autophagosome Nucleation Defines Atg9 Vesicles as Seeds for Membrane Formation. Science 369, 1206. doi:10.1126/science.aaz7714
Sawan, C., and Herceg, Z. (2010). Histone Modifications and Cancer. Adv. Genet. 70, 57–85. doi:10.1016/b978-0-12-380866-0.60003-4
Saxonov, S., Berg, P., and Brutlag, D. L. (2006). A Genome-wide Analysis of CpG Dinucleotides in the Human Genome Distinguishes Two Distinct Classes of Promoters. Proc. Natl. Acad. Sci. U.S.A. 103, 1412–1417. doi:10.1073/pnas.0510310103
Schieren, G., Rumberger, B., Klein, M., Kreutz, C., Wilpert, J., Geyer, M., et al. (2006). Gene Profiling of Polycystic Kidneys. Nephrol. Dial. Transplant. 21, 1816–1824. doi:10.1093/ndt/gfl071
Schilling, D., Tetzlaff, F., Konrad, S., Li, W., and Multhoff, G. (2015). A Hypoxia-Induced Decrease of Either MICA/B or Hsp70 on the Membrane of Tumor Cells Mediates Immune Escape from NK Cells. Cell Stress Chaperones 20, 139–147. doi:10.1007/s12192-014-0532-5
Schotta, G., Ebert, A., Krauss, V., Fischer, A., Hoffmann, J., Rea, S., et al. (2002). Central Role of Drosophila SU(VAR)3-9 in Histone H3-K9 Methylation and Heterochromatic Gene Silencing. Embo J. 21, 1121–1131. doi:10.1093/emboj/21.5.1121
Schreiber, R., Buchholz, B., Kraus, A., Schley, G., Scholz, J., Ousingsawat, J., et al. (2019). Lipid Peroxidation Drives Renal Cyst Growth In Vitro through Activation of TMEM16A. J. Am. Soc. Nephrol. 30, 228–242. doi:10.1681/asn.2018010039
Schübeler, D. (2015). Function and Information Content of DNA Methylation. Nature 517, 321–326. doi:10.1038/nature14192
Seeger-Nukpezah, T., Geynisman, D. M., Nikonova, A. S., Benzing, T., and Golemis, E. A. (2015). The Hallmarks of Cancer: Relevance to the Pathogenesis of Polycystic Kidney Disease. Nat. Rev. Nephrol. 11, 515–534. doi:10.1038/nrneph.2015.46
Seiler, A., Schneider, M., Förster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 8, 237–248. doi:10.1016/j.cmet.2008.07.005
Seliger, S. L., Abebe, K. Z., Hallows, K. R., Miskulin, D. C., Perrone, R. D., Watnick, T., et al. (2018). A Randomized Clinical Trial of Metformin to Treat Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 47, 352–360. doi:10.1159/000488807
Shen, L., Kondo, Y., Guo, Y., Zhang, J., Zhang, L., Ahmed, S., et al. (2007). Genome-wide Profiling of DNA Methylation Reveals a Class of Normally Methylated CpG Island Promoters. PLoS Genet. 3, 2023–2036. doi:10.1371/journal.pgen.0030181
Shen, S., and Casaccia-Bonnefil, P. (2008). Post-translational Modifications of Nucleosomal Histones in Oligodendrocyte Lineage Cells in Development and Disease. J. Mol. Neurosci. 35, 13–22. doi:10.1007/s12031-007-9014-x
Shi, J.-H., and Sun, S.-C. (2018). Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 9, 1849. doi:10.3389/fimmu.2018.01849
Shibazaki, S., Yu, Z., Nishio, S., Tian, X., Thomson, R. B., Mitobe, M., et al. (2008). Cyst Formation and Activation of the Extracellular Regulated Kinase Pathway after Kidney Specific Inactivation of Pkd1. Hum. Mol. Genet. 17, 1505–1516. doi:10.1093/hmg/ddn039
Shillingford, J. M., Murcia, N. S., Larson, C. H., Low, S. H., Hedgepeth, R., Brown, N., et al. (2006). The mTOR Pathway Is Regulated by Polycystin-1, and its Inhibition Reverses Renal Cystogenesis in Polycystic Kidney Disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5466–5471. doi:10.1073/pnas.0509694103
Sims, R. J., Nishioka, K., and Reinberg, D. (2003). Histone Lysine Methylation: a Signature for Chromatin Function. Trends Genet. 19, 629–639. doi:10.1016/j.tig.2003.09.007
Smith, B. C., and Denu, J. M. (2009). Chemical Mechanisms of Histone Lysine and Arginine Modifications. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1789, 45–57. doi:10.1016/j.bbagrm.2008.06.005
Song, X., Di Giovanni, V., He, N., Wang, K., Ingram, A., Rosenblum, N. D., et al. (2009). Systems Biology of Autosomal Dominant Polycystic Kidney Disease (ADPKD): Computational Identification of Gene Expression Pathways and Integrated Regulatory Networks. Hum. Mol. Genet. 18, 2328–2343. doi:10.1093/hmg/ddp165
Steigerwald, J., Raum, T., Pflanz, S., Cierpka, R., Mangold, S., Rau, D., et al. (2009). Human IgG1 Antibodies Antagonizing Activating Receptor NKG2D on Natural Killer Cells. MAbs 1, 115–127. doi:10.4161/mabs.1.2.7630
Strahl, B. D., and Allis, C. D. (2000). The Language of Covalent Histone Modifications. Nature 403, 41–45. doi:10.1038/47412
Stuart, E. T., Haffner, R., Oren, M., and Gruss, P. (1995). Loss of P53 Function through PAX-Mediated Transcriptional Repression. EMBO J. 14, 5638–5645. doi:10.1002/j.1460-2075.1995.tb00251.x
Stuppia, L., Franzago, M., Ballerini, P., Gatta, V., and Antonucci, I. (2015). Epigenetics and Male Reproduction: the Consequences of Paternal Lifestyle on Fertility, Embryo Development, and Children Lifetime Health. Clin. Epigenetics 7, 120. doi:10.1186/s13148-015-0155-4
Sun, L., Hu, C., and Zhang, X. (2019). Histone Deacetylase Inhibitors Reduce Cysts by Activating Autophagy in Polycystic Kidney Disease. Kidney Dis. 5, 163–172. doi:10.1159/000499368
Swanton, E., Savory, P., Cosulich, S., Clarke, P., and Woodman, P. (1999). Bcl-2 Regulates a Caspase-3/caspase-2 Apoptotic Cascade in Cytosolic Extracts. Oncogene 18, 1781–1787. doi:10.1038/sj.onc.1202490
Swenson-Fields, K. I., Vivian, C. J., Salah, S. M., Peda, J. D., Davis, B. M., Van Rooijen, N., et al. (2013). Macrophages Promote Polycystic Kidney Disease Progression. Kidney Int. 83, 855–864. doi:10.1038/ki.2012.446
Takiar, V., Nishio, S., Seo-Mayer, P., King, J. D., Li, H., Zhang, L., et al. (2011). Activating AMP-Activated Protein Kinase (AMPK) Slows Renal Cystogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 2462–2467. doi:10.1073/pnas.1011498108
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 146, 1016–1028. doi:10.1016/j.cell.2011.08.008
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518. doi:10.1016/j.biocel.2004.05.009
Tao, Y., Kim, J., Faubel, S., Wu, J. C., Falk, S. A., Schrier, R. W., et al. (2005a). Caspase Inhibition Reduces Tubular Apoptosis and Proliferation and Slows Disease Progression in Polycystic Kidney Disease. Proc. Natl. Acad. Sci. U.S.A. 102, 6954–6959. doi:10.1073/pnas.0408518102
Tao, Y., Kim, J., Stanley, M., He, Z., Faubel, S., Schrier, R. W., et al. (2005b). Pathways of Caspase-Mediated Apoptosis in Autosomal-Dominant Polycystic Kidney Disease (ADPKD). Kidney Int. 67, 909–919. doi:10.1111/j.1523-1755.2005.00155.x
Tao, Y., Zafar, I., Kim, J., Schrier, R. W., and Edelstein, C. L. (2008). Caspase-3 Gene Deletion Prolongs Survival in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 19, 749–755. doi:10.1681/asn.2006121378
Torres, V. E., Chapman, A. B., Devuyst, O., Gansevoort, R. T., Grantham, J. J., Higashihara, E., et al. (2012). Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 367, 2407–2418. doi:10.1056/nejmoa1205511
Torres, V. E., and Harris, P. C. (2009). Autosomal Dominant Polycystic Kidney Disease: the Last 3 Years. Kidney Int. 76, 149–168. doi:10.1038/ki.2009.128
Tower, J. (2015). Programmed Cell Death in Aging. Ageing Res. Rev. 23, 90–100. doi:10.1016/j.arr.2015.04.002
Turner, B. M. (2000). Histone Acetylation and an Epigenetic Code. Bioessays 22, 836–845. doi:10.1002/1521-1878(200009)22:9<836::aid-bies9>3.0.co;2-x
Turner, J.-E., Becker, M., Mittrücker, H.-W., and Panzer, U. (2018). Tissue-Resident Lymphocytes in the Kidney. J. Am. Soc. Nephrol. 29, 389–399. doi:10.1681/asn.2017060599
Van Bodegom, D., Saifudeen, Z., Dipp, S., Puri, S., Magenheimer, B. S., Calvet, J. P., et al. (2006). The Polycystic Kidney Disease-1 Gene Is a Target for P53-Mediated Transcriptional Repression. J. Biol. Chem. 281, 31234–31244. doi:10.1016/s0021-9258(19)84036-x
Van Den Eeckhout, B., Tavernier, J., and Gerlo, S. (2020). Interleukin-1 as Innate Mediator of T Cell Immunity. Front. Immunol. 11, 621931. doi:10.3389/fimmu.2020.621931
Vidali, G., Ferrari, N., and Pfeffer, U. (1988). Histone Acetylation: a Step in Gene Activation. Adv. Exp. Med. Biol. 231, 583–596. doi:10.1007/978-1-4684-9042-8_49
Villota-Salazar, N. A., Mendoza-Mendoza, A., and González-Prieto, J. M. (2016). Epigenetics: from the Past to the Present. Front. Life Sci. 9, 347–370. doi:10.1080/21553769.2016.1249033
Wajant, H. (2002). The Fas Signaling Pathway: More Than a Paradigm. Science 296, 1635–1636. doi:10.1126/science.1071553
Walczak, H. (2013). Death Receptor-Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 5, a008698. doi:10.1101/cshperspect.a008698
Wan, Y. Y., and Flavell, R. A. (2007). Regulatory T Cells, Transforming Growth Factor- , and Immune Suppression. Proc. Am. Thorac. Soc. 4, 271–276. doi:10.1513/pats.200701-020aw
Wang, R., Li, W., Zhang, S., Song, Y., Dai, H., Tan, T., et al. (2021). The Effects of Intrinsic Apoptosis on Cystogenesis in PKD1-Deficient ADPKD Pig Model. Gene 798, 145792. doi:10.1016/j.gene.2021.145792
Wang, Z., Ng, C., Liu, X., Wang, Y., Li, B., Kashyap, P., et al. (2019). The Ion Channel Function of Polycystin-1 in the Polycystin-1/polycystin-2 Complex. EMBO Rep. 20, e48336. doi:10.15252/embr.201948336
Warrington, R., Watson, W., Kim, H. L., and Antonetti, F. R. (2011). An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 7 (Suppl. 1), S1. doi:10.1186/1710-1492-7-S1-S1
Wegierski, T., Steffl, D., Kopp, C., Tauber, R., Buchholz, B., Nitschke, R., et al. (2009). TRPP2 Channels Regulate Apoptosis through the Ca2+ Concentration in the Endoplasmic Reticulum. Embo J. 28, 490–499. doi:10.1038/emboj.2008.307
Wei, F., Karihaloo, A., Yu, Z., Marlier, A., Seth, P., Shibazaki, S., et al. (2008). Neutrophil Gelatinase-Associated Lipocalin Suppresses Cyst Growth by Pkd1 Null Cells In Vitro and In Vivo. Kidney Int. 74, 1310–1318. doi:10.1038/ki.2008.395
Wilson, P. D., and Goilav, B. (2007). Cystic Disease of the Kidney. Annu. Rev. Pathol. Mech. Dis. 2, 341–368. doi:10.1146/annurev.pathol.2.010506.091850
Woo, D. (1995). Apoptosis and Loss of Renal Tissue in Polycystic Kidney Diseases. N. Engl. J. Med. 333, 18–25. doi:10.1056/nejm199507063330104
Woo, Y. M., Shin, Y., Hwang, J. A., Hwang, Y. H., Lee, S., Park, E. Y., et al. (2015). Epigenetic Silencing of the MUPCDH Gene as a Possible Prognostic Biomarker for Cyst Growth in ADPKD. Sci. Rep. 5, 15238. doi:10.1038/srep15238
Woo, Y. M., Bae, J.-B., Oh, Y.-H., Lee, Y.-G., Lee, M. J., Park, E. Y., et al. (2014). Genome-wide Methylation Profiling of ADPKD Identified Epigenetically Regulated Genes Associated with Renal Cyst Development. Hum. Genet. 133, 281–297. doi:10.1007/s00439-013-1378-0
Xia, S., Li, X., Johnson, T., Seidel, C., Wallace, D. P., and Li, R. (2010). Polycystin-dependent Fluid Flow Sensing Targets Histone Deacetylase 5 to Prevent the Development of Renal Cysts. Development 137, 1075–1084. doi:10.1242/dev.049437
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: Process and Function. Cell Death Differ. 23, 369–379. doi:10.1038/cdd.2015.158
Yamamoto, J., Nishio, S., Hattanda, F., Nakazawa, D., Kimura, T., Sata, M., et al. (2017). Branched-chain Amino Acids Enhance Cyst Development in Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 92, 377–387. doi:10.1016/j.kint.2017.01.021
Yanda, M. K., Liu, Q., Cebotaru, V., Guggino, W. B., and Cebotaru, L. (2017). Histone Deacetylase 6 Inhibition Reduces Cysts by Decreasing cAMP and Ca2+ in Knock-Out Mouse Models of Polycystic Kidney Disease. J. Biol. Chem. 292, 17897–17908. doi:10.1074/jbc.m117.803775
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331. doi:10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic Lethal Screening Identifies Compounds Activating Iron-dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 15, 234–245. doi:10.1016/j.chembiol.2008.02.010
Yanumula, A., and Cusick, J. K. (2022). “Biochemistry, Extrinsic Pathway of Apoptosis,” in StatPearls (Treasure Island (FL).
Yoder, B. K., Hou, X., and Guay-Woodford, L. M. (2002). The Polycystic Kidney Disease Proteins, Polycystin-1, Polycystin-2, Polaris, and Cystin, Are Co-localized in Renal Cilia. J. Am. Soc. Nephrol. 13, 2508–2516. doi:10.1097/01.asn.0000029587.47950.25
Zafar, I., Ravichandran, K., Belibi, F. A., Doctor, R. B., and Edelstein, C. L. (2010). Sirolimus Attenuates Disease Progression in an Orthologous Mouse Model of Human Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 78, 754–761. doi:10.1038/ki.2010.250
Zahid, A., Li, B., Kombe, A. J. K., Jin, T., and Tao, J. (2019). Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 10, 2538. doi:10.3389/fimmu.2019.02538
Zerres, K., Rudnik-Schoneborn, S., and Deget, F. (1993). Childhood Onset Autosomal Dominant Polycystic Kidney Disease in Sibs: Clinical Picture and Recurrence Risk. German Working Group on Paediatric Nephrology (Arbeitsgemeinschaft Fur Padiatrische Nephrologie. J. Med. Genet. 30, 583–588. doi:10.1136/jmg.30.7.583
Zhang, F., Mears, J. R., Mears, J. R., Shakib, L., Beynor, J. I., Shanaj, S., et al. (2021a). IFN-γ and TNF-α Drive a CXCL10+ CCL2+ Macrophage Phenotype Expanded in Severe COVID-19 Lungs and Inflammatory Diseases with Tissue Inflammation. Genome Med. 13, 64. doi:10.1186/s13073-021-00881-3
Zhang, J. Q. J., Saravanabavan, S., and Rangan, G. K. (2021b). Effect of Reducing Ataxia-Telangiectasia Mutated (ATM) in Experimental Autosomal Dominant Polycystic Kidney Disease. Cells 10, 532. doi:10.3390/cells10030532
Zhang, T., Sell, P., Braun, U., and Leitges, M. (2015). PKD1 Protein Is Involved in Reactive Oxygen Species-Mediated Mitochondrial Depolarization in Cooperation with Protein Kinase Cδ (PKCδ). J. Biol. Chem. 290, 10472–10485. doi:10.1074/jbc.m114.619148
Zhang, W., and Xu, J. (2017). DNA Methyltransferases and Their Roles in Tumorigenesis. Biomark. Res. 5, 1. doi:10.1186/s40364-017-0081-z
Zhang, X., Li, L. X., Ding, H., Torres, V. E., Yu, C., and Li, X. (2021c). Ferroptosis Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease Mouse Models. J. Am. Soc. Nephrol. 32, 2759–2776. doi:10.1681/asn.2021040460
Zhang, X., and Li, X. (2022). Abnormal Iron and Lipid Metabolism Mediated Ferroptosis in Kidney Diseases and Its Therapeutic Potential. Metabolites 12. doi:10.3390/metabo12010058
Zhang, Y., and Reinberg, D. (2001). Transcription Regulation by Histone Methylation: Interplay between Different Covalent Modifications of the Core Histone Tails. Genes Dev. 15, 2343–2360. doi:10.1101/gad.927301
Zheng, D., Liwinski, T., and Elinav, E. (2020). Inflammasome Activation and Regulation: toward a Better Understanding of Complex Mechanisms. Cell Discov. 6, 36. doi:10.1038/s41421-020-0167-x
Zhou, J. X., and Li, X. (2015). “Apoptosis in Polycystic Kidney Disease: From Pathogenesis to Treatment,” in Polycystic Kidney Disease. Editor X. Li (Brisbane (AU). doi:10.15586/codon.pkd.2015.ch9
Zhou, X., Fan, L. X., Li, K., Ramchandran, R., Calvet, J. P., and Li, X. (2014). SIRT2 Regulates Ciliogenesis and Contributes to Abnormal Centrosome Amplification Caused by Loss of Polycystin-1. Hum. Mol. Genet. 23, 1644–1655. doi:10.1093/hmg/ddt556
Zhou, X., Fan, L. X., Peters, D. J. M., Trudel, M., Bradner, J. E., and Li, X. (2015). Therapeutic Targeting of BET Bromodomain Protein, Brd4, Delays Cyst Growth in ADPKD. Hum. Mol. Genet. 24, 3982–3993. doi:10.1093/hmg/ddv136
Zhou, X., Fan, L. X., Sweeney, W. E., Denu, J. M., Avner, E. D., and Li, X. (2013). Sirtuin 1 Inhibition Delays Cyst Formation in Autosomal-Dominant Polycystic Kidney Disease. J. Clin. Invest. 123, 3084–3098. doi:10.1172/jci64401
Zhu, J., Yamane, H., and Paul, W. E. (2010). Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 28, 445–489. doi:10.1146/annurev-immunol-030409-101212
Zhu, P., Sieben, C. J., Xu, X., Harris, P. C., and Lin, X. (2017). Autophagy Activators Suppress Cystogenesis in an Autosomal Dominant Polycystic Kidney Disease Model. Hum. Mol. Genet. 26, 158–172. doi:10.1093/hmg/ddw376
Zilberman, Y., Ballestrem, C., Carramusa, L., Mazitschek, R., Khochbin, S., and Bershadsky, A. (2009). Regulation of Microtubule Dynamics by Inhibition of the Tubulin Deacetylase HDAC6. J. Cell Sci. 122, 3531–3541. doi:10.1242/jcs.046813
Keywords: ADPKD, epigenetics, post-translational modifications, inflammation, cell death
Citation: Agborbesong E, Li LX, Li L and Li X (2022) Molecular Mechanisms of Epigenetic Regulation, Inflammation, and Cell Death in ADPKD. Front. Mol. Biosci. 9:922428. doi: 10.3389/fmolb.2022.922428
Received: 18 April 2022; Accepted: 07 June 2022;
Published: 29 June 2022.
Edited by:
Annarita Di Mise, University of Bari Aldo Moro, ItalyReviewed by:
Cherie Stayner, University of Otago, New ZealandCopyright © 2022 Agborbesong, Li, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaogang Li, bGkueGlhb2dhbmdAbWF5by5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.