
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Mol. Biosci., 16 May 2022
Sec. Structural Biology
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.890862
This article is part of the Research TopicStructure-Function Metrology of ProteinsView all 13 articles
 Tristan O. C. Kwan1
Tristan O. C. Kwan1 Stefan A. Kolek2
Stefan A. Kolek2 Amy E. Danson1
Amy E. Danson1 Rosana I. Reis1
Rosana I. Reis1 Ines S. Camacho1
Ines S. Camacho1 Patrick D. Shaw Stewart2*
Patrick D. Shaw Stewart2* Isabel Moraes1*
Isabel Moraes1*Structure-function relationships of biological macromolecules, in particular proteins, provide crucial insights for fundamental biochemistry, medical research and early drug discovery. However, production of recombinant proteins, either for structure determination, functional studies, or to be used as biopharmaceutical products, is often hampered by their instability and propensity to aggregate in solution in vitro. Protein samples of poor quality are often associated with reduced reproducibility as well as high research and production expenses. Several biophysical methods are available for measuring protein aggregation and stability. Yet, discovering and developing means to improve protein behaviour and structure-function integrity remains a demanding task. Here, we discuss workflows that are made possible by adapting established biophysical methods to high-throughput screening approaches. Rapid identification and optimisation of conditions that promote protein stability and reduce aggregation will support researchers and industry to maximise sample quality, stability and reproducibility, thereby reducing research and development time and costs.
Human health is continuously challenged not only by external toxic substances but also by microorganisms such as pathogenic bacteria and viruses. Thus, understanding biology at a cellular and molecular level is paramount to human existence. Proteins are sophisticated molecular machines essential in all cellular processes. However, because of their complex intrinsic three-dimensional (3D) molecular network of hydrogen bonds, Van der Waals forces, hydrophobic interactions and disulfide links, proteins have a natural tendency to denature (unfold) or aggregate (formation of non-physiological dimers and higher-order aggregates). The pathways and mechanisms by which proteins unfold and aggregate are not yet fully understood. However, external factors such as temperature, pH, ionic strength, different buffer systems, mechanical stress or even the presence of “foreign bodies” like dust, glass particles and microscopic oil droplets can lead to structural and behavioural changes of the protein in solution.
Currently, there are many critical reasons why scientists need to monitor aggregation and stability of proteins in solution. Firstly, pure and stable protein material is always required for structural studies for any of the three most popular methods—X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy (cryo-EM) (Chari et al., 2015; Kozak et al., 2016; Kotov et al., 2019; Kwan et al., 2019). Protein purification strategies and protocols are particularly dependent on the target protein that frequently misbehaves when isolated in solution. Therefore, the ability to quickly search chemical spaces that increase protein stability with minimal amounts of sample is critical to such biophysical applications. Over the last few decades, however, developments in gene editing tools, molecular biology, instrumentation, and light sources, along with many other multidisciplinary approaches, have enabled a new age in protein structure determination (Adli, 2018). Consequently, structural biologists have pursued more challenging targets such as integral membrane proteins and protein-protein complexes. Moreover, the structural biology community is also now strongly focused on time-resolved approaches to study protein dynamics and conformational changes over time (Kwan et al., 2020; Bonvin, 2021; Cohen, 2021). Secondly, critical to biopharmaceutical industry and biomedicine, protein unfolding and aggregation also poses many challenges in the development of biologics (therapeutic agents manufactured in living systems such as a bacteria, plant or animal cells) because of its impact on the final product quality in terms of safety, clinical efficacy, and immunogenicity (Ratanji et al., 2014; Roberts, 2014; Krause and Sahin, 2019). Presently, the most popular biologics in the market are monoclonal antibodies, cytokines, enzymes, and peptide hormones. Although these may offer many clinical advantages when compared with small molecule drugs, they are far more complex to develop and produce at a larger scale (Plant et al., 2014; Puetz and Wurm, 2019; Jovčevska and Muyldermans, 2020). By their intrinsic nature, protein-based drugs are extremely sensitive to physical and chemical environmental factors such as temperature, shear-forces, light, pH, glycosylation, and enzymatic action Rajan et al., 2021. Hence, probing protein aggregation during the development and manufacturing of protein-based drugs is essential to achieve a final product that is not only stable for long-term storage but also harbors fewer impurities that stimulate an anti-drug immune response (Ratanji et al., 2014; Svilenov et al., 2018; Ribeiro et al., 2019). Finally, production of high-quality protein is also important in many other industrial applications such as agriculture, food and beverage processing, production of cosmetics and biofuels, and in biotechnology applications such as biosensors and optogenetics (Zhang and Cohen, 2017).
Nevertheless, inherent to all these approaches is the protein sample quality in terms of purity and stability. Currently, a variety of biophysical techniques are available to study protein behaviour in solution. These range from simple approaches to complex methods that require both technical expertise and costly instruments. For example, Makowski et al. (2022) reviewed the use of a set of key bionanotechnologies for preparing functional and stable membrane proteins in diverse types of lipoparticles in combination with several biophysical assays, including affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS). In a seminal study by Jain et al. (2017), 137 isotypematched IgG1 antibodies were evaluated with a dozen biophysical property assays to define the boundaries of drug-like behaviour for future studies. Screening multiple macromolecular constructs is distinct from the exploration of chemical space with a single target, and will not be considered further here.
In this perspective, we have summarised simple and readily available approaches protein aggregation and stability within a high-throughput (HTP) setting. These biophysical techniques are also capable of screening and probing the addition of specific ligands, additives or excipients to the protein of interest that can reduce its propensity to unfold or aggregate, as well as its susceptibility to proteolysis (Senisterra and Finerty, 2009).
Automation and miniaturisation combined with multi-well microplates have enabled the development of HTP approaches where many samples (up to thousands) are tested in parallel under given conditions. Introduced by pharmaceutical companies in the early 1990s for the massive screening of potential “drug-like” compounds, HTP approaches are extensively used by scientists today in academic laboratories, research institutes and industry in a variety of applications (Macarron et al., 2011; Kwan et al., 2019; Kiriiri, et al., 2020).
Recently, researchers have started to use HTP approaches to explore chemical space by testing protein samples against premixed sets of buffers, excipients, additives, salts, osmolytes, and co-factors (often referred as stability screens). Suitable stability screens are commercially available in 96- and 384-well format, or they can be assembled in-house. To maximise the throughput towards evaluation of protein behaviour in the presence of such stability screens or ligands, HTP approaches were also applied to simple but robust biophysical methods such as dynamic light scattering (DLS), differential scanning fluorimetry (DSF) and circular dichroism (CD). Each of these biophysical methods provides different types of information and, whether used as a standalone or integrated, they have proven to be noteworthy screening tools when probing protein aggregation or to identify conditions that promote protein integrity and stability in solution (Figure 1).
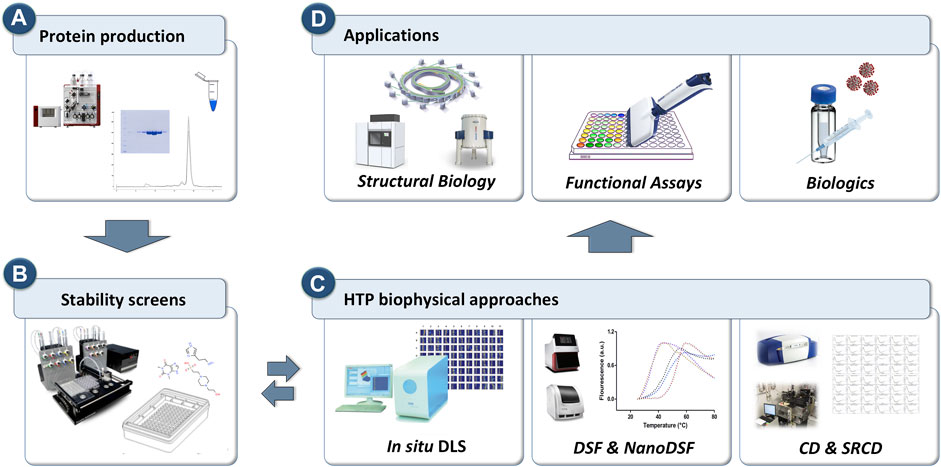
FIGURE 1. Schematic representation of the workflow for the HTP biophysical approaches to probe protein behavior for downstream and upstream applications. (A) Proteins are purified using standard purification strategies. (B) HTP screens are prepared to explore chemical space. (C) in situ Dynamic Light Scattering (DLS), differential scanning fluorimetry (DSF) and circular dichroism (CD) that can operate in HTP mode are used as primary screening tools to probe protein stability. (D) Three key applications where protein monodispersity and stability is crucial.
DLS is a common technique employed to assess the behaviour of proteins in solution. This simple yet powerful and non-invasive biophysical measurement correlates the random motion of particles in a fluid with their sizes. During a DLS experiment, particles in solution are illuminated by a visible and monochromatic laser light while a detector records the light scattered by the particles. The intensity of light fluctuates at a frequency that is determined by the particles’ size. Because proteins and other macromolecules in solution are not always spherical in shape and are always solvated, the size calculated by a DLS instrument is referred to as the “hydrodynamic radius”. The physical principles of light scattering and DLS have been extensively described in the literature (International Organization for Standardization, 1996; Lorber et al., 2012; Stetefeld et al., 2016; Kwan et al., 2019). For DLS experiments, the data are usually presented in terms of sample polydispersity. If the sample remains monodisperse and shows a peak at the expected size, the protein can be assumed to be stable in the chemical and physical conditions (temperature) used (Shiba et al., 2010). With the development of HTP approaches to DLS, the behaviour of proteins can now be determined in the presence of ligands, substrates, additives, and buffers in parallel at microlitre volumes in plate formats (Meyer et al., 2015; Kwan et al., 2019; Dauer et al., 2021) Moreover, the DLS measurement can be run over several time points, allowing degradation or changes in aggregation to be monitored over time. For example, we found that a bacterial ABC transporter, which is a membrane protein, tended to aggregate after purification. We used a 96-component screen to monitor aggregation over 9 h. The screen contained a set of conditions that has previously increased solubility and stability of proteins. We were able to identify 12 reagents that reduced aggregation of the ABC transporter (Supplementary Figure S1). Note that DLS provides extra information about the degree of aggregation, including the size of aggregates and the number of peaks, whereas other methods such as DSF give only a single numerical value corresponding to the “melting point” of the protein. HTP DLS provides a practical and simple probe for stability that can be implemented easily in the protein production workflow and downstream applications in structural biology such as crystallography and/or cryoEM. Finally, for the production of biologics, HTP DLS measurements can also be used to determine sample quality and be implemented as a quality control measurement between production batches (Hulse et al., 2013; Yu et al., 2013; Bhirde et al., 2018; Dauer et al., 2021).
DSF, also commonly referred to as thermal-shift assay or ThermoFluor assay, is an inexpensive and highly sensitive biophysical technique that can be used to determine the thermal stability of a target protein in the presence or absence of ligands, substrates, and buffers. The DSF principle measures the protein thermal denaturation over time (typically about 1°C min−1) and is based on the relationship between protein stability and its Gibbs free energy of unfolding (ΔGu). When the amount of unfolded protein equals the amount of folded protein, the value of ΔGu becomes zero and the system has reached what is known as the protein “melting point” temperature (Tm). Proteins with higher stability have larger ΔGu values and therefore a higher Tm. The DSF experimental setup is very simple and typically does not require expensive equipment. The protein sample, in the presence of a highly sensitive fluorescent probe, also known as a “reporter” or extrinsic dye, is gradually heated over time (in order to trigger denaturation) from its stable baseline temperature. This eventually results in exposure of the hydrophobic regions that are usually buried within the protein’s folded structure. As more of the protein’s hydrophobic regions become exposed, an increasing number of dye molecules bind to the hydrophobic sites, resulting in an increase of the fluorescence signal. Once the protein reaches its maximum unfolded state, it starts to aggregate and precipitate, and consequently, the dye molecules dissociate and the fluorescence signal drops (Bai et al., 2019). In the majority of the instruments, the DSF output is displayed as a series of sigmoidal curves, also known as “melt curves”, where the point of inflection gives the Tm value (Supplementary Figure S2A). Although there are many fluorescent dyes available on the market, the most frequently used for soluble proteins is SYPRO Orange, which is inexpensive, highly sensitive and has low interference with small molecules. Its excitation/emission wavelength (480 nm/569 nm) is usually compatible with most filters available in fluorescent plate readers and RT-PCR machines. However, because SYPRO Orange also binds to the hydrophobic regions of lipids and detergent micelles, it is unsuitable for membrane protein studies. Instead, a reactive thiol-specific fluorochrome, N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl] maleimide (CPM), was introduced in 2008 (Alexadrov et al., 2008), which specifically reacts with the cysteine side chains that are usually buried in the interior of membrane proteins. Therefore, as the target membrane protein unfolds, the cysteines become exposed and the CPM fluorescence signal increases. Supplementary Figure S2B shows DSF results obtained for the membrane protein LacY with CPM dye in combination with five detergents that were identified from a detergent screen. Note, however, that the use of the CPM dye is associated with two limitations. One is its excitation/emission wavelength (387 nm/463 nm) that requires specific filters that usually are only available by request. The other limitation is that the protein of study must have cysteine residues in the transmembrane domains. More recently, a label-free DSF method known as nanoDSF was developed (Alexander et al., 2014; Gao et al., 2020; Real-Hohn et al., 2020). In this approach the protein’s intrinsic fluorescence, based on signals mainly from the native tryptophan residues, is measured at both 330 and 350 nm in low-volume capillaries, which produces more robust data in comparison to detection at a single wavelength. NanoDSF is especially useful for studying membrane proteins (Kwan et al., 2019; Gao et al., 2020) and viral particles (Real-Hohn, et al., 2020).
Either DSF or nanoDSF approaches are also used to optimise protein buffers for long-term storage, and to determine optimal conditions for protein refolding (Biter et al., 2016; Wang et al., 2017) and protein stability in solution, greatly aiding structural studies such as crystallisation and cryo-EM (Martinez et al., 2021). Finally, the DSF approach is also extensively used in early drug discovery platforms. Many proteins can be stabilised by the binding of low molecular weight ligands, usually resulting in an increase of the Tm and therefore, DSF is regarded as a simple and inexpensive, and popular method to assess protein-ligand interactions (Huynh and Partch, 2015).
The multi-well design of thermal cycling machines and fluorescent plate readers, including instruments that make use of small volume glass capillaries, allow DSF to be used as a HTP tool requiring relatively low (microliter range) sample volumes. A downside of DSF, however, is that larger macromolecular complexes may not unfold cooperatively (thus having multiple transition states), making stabilising conditions difficult to identify (Stark and Chari, 2016). This is particularly problematic if cryo-EM is the intended downstream process, as typically this is the method of choice for the structural elucidation of large protein complexes.
CD is a biophysical technique that measures the differential absorption between left and right circularly polarised light by a chiral molecule. Polypeptide backbone information can be obtained from measurements taken in the far-UV region of 180–250 nm, due to the amide electronic transitions, and can provide estimates on the protein secondary structure content such as alpha-helices and beta-sheets (Kwan et al., 2019; Miles et al., 2021). The near-UV region of 260–300 nm is used to characterise the aromatic side chain transitions of tyrosine, tryptophan, and phenylalanine, which are often situated at binding sites, and thus measurements in this region provide valuable information regarding ligand interactions (Kwan et al., 2019).
The application of CD can be significantly broadened by the elevated photon flux of a synchrotron, which improves the signal-to-noise levels considerably, giving rise to a technique known as synchrotron radiation CD (SRCD). SRCD is capable of measuring at lower wavelengths, greatly improving the analysis of beta-sheet content and fold motifs, as well as at lower sample volumes and concentrations (Wallace et al., 2011; Hussain and Siligardi, 2016), which is a significant advantage when studying membrane proteins, for which high concentrations and volumes are difficult and costly to produce. Additionally, higher ionic solutions can be measured (up to 500 mM NaCl) due to the higher flux, which extends the buffer screening capability of conventional CD (Hussain and Siligardi, 2016). For example, beamline B23 at Diamond Light Source operates in a HTP manner (also referred to as HT-CD), with 96 or 384-well suprasil quartz plates available for data collection, in which protein samples of just 15 ul at 0.5 mg/ml can be measured allowing users to screen a wide range of buffers and/or ligands without the laborious task of changing and washing each cuvette (Hussain et al., 2016). Supplementary Figure S3 shows results obtained with HT-CD for two membrane proteins in combination with 12 detergents. CD data analysis is also swift due to the introduction of software that enables users to remotely process results quickly and easily (Hussain et al., 2015). Recent advances have led to the development of time-resolved SRCD (TR-SRCD), which is capable of subpicosecond temporal resolutions, providing insight into biological mechanisms at a molecular level (Auvray et al., 2019).
Purified proteins are critical tools in a variety of research and industrial applications. However, these vital molecular machines are inherently unstable and prone to aggregation both in vivo and in vitro. Protein aggregates vary from small dimers to large assemblies that can be specifically formed during protein production and the storage process, including prolonged storage. Unfortunately, protein aggregates are usually irreversible and exceptionally stable. Therefore, effective strategies to quickly probe and quantify protein aggregation including protein stability in vitro are urgently needed. However, one of the major challenges is the absence of a single analytic approach that is able to cover the whole spectrum of protein behaviour in vitro, and therefore several different biophysical approaches are usually required.
Over the years, with the development of HTP technology, many traditional biophysical techniques such as DLS, DSF, and CD have evolved from standalone approaches, where samples used to be measured one at a time, to robust and sensitive methods able to analyse dozens of samples in parallel using minimal amounts of material and in standard workflows that can be applied to many targets. Each of these HTP biophysical methods has its own strengths and weaknesses (see Table 1), thus their applicability will depend on the information required.
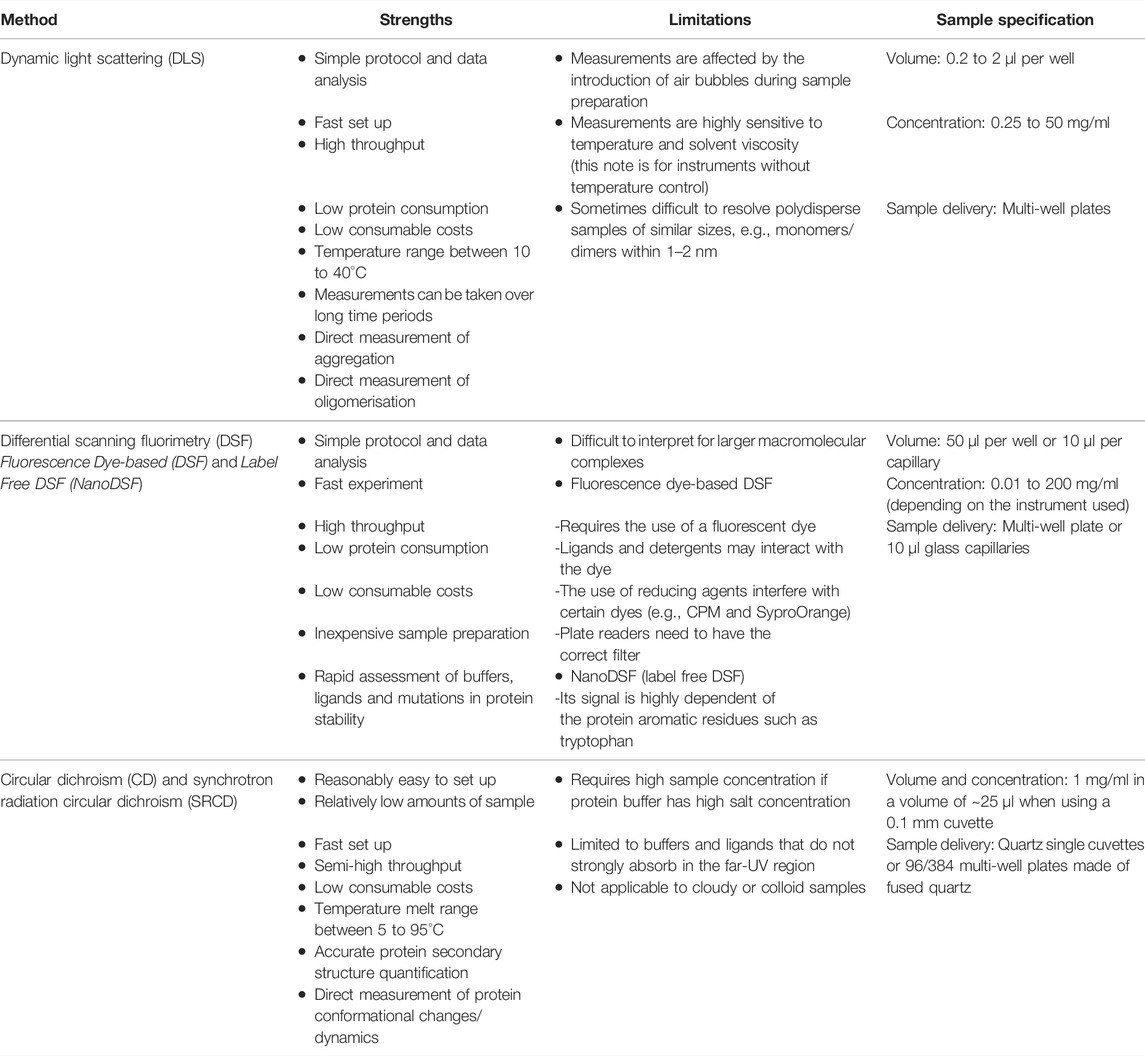
TABLE 1. Summary of the strengths and limitations of the HTP biophysical methods presented in this perspective.
One of the major advances in drug discovery and development has been its synergy with a wide variety of such biophysical methods (Raynal et al., 2014; Renaud et al., 2016; Holdgate et al., 2019; Kiriiri, et al., 2020). Today, modern medicine relies heavily on the use of conventional drugs (synthetic small chemical substances) and biologics (e.g., proteins, antibodies and small peptides) as therapeutic agents. While genomic and proteomic approaches have uncovered an increasing number of potential druggable proteins, compelling Pharma businesses to adopt target-based drug discovery strategies, many challenges still remain. These include poor understanding of the mode of action of the target protein and its interaction with the therapeutic agent, as well as difficulties associated with the manufacturing of high-quality protein-based biologics in terms of purity and stability (Davis 2020; Emmerich et al., 2021). In this perspective, we sought to direct the reader’s attention to the application of conventional and simple biophysical methods within automated HTP settings that are able not only to assess protein stability and integrity in solution but also to probe protein behaviour in the presence of multi-parametric screens of buffers, excipients, additives, salts, osmolytes, and co-factors. Moreover, these HTP biophysical approaches can make significant contributions to downstream applications such as X-ray crystallography, NMR and cryo-EM, as well as to the upstream applications in drug discovery, development and formulation.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
TK, SK, AD, IC, PS, and IM conceptualised, drafted and finalized the manuscript. TK, SK, AD, and RR executed the experiments. All authors approved the submitted version.
SK, PS were employed by the Douglas Instruments Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge funding from the United Kingdom’s Department of Business, Energy and Industrial Strategy (BEIS). We are grateful for support from the Membrane Protein Laboratory under Wellcome Trust grant number 20289/Z16/Z. We thank the award of experimental time (SM123352) on the B23 Beamline at Diamond Light Source (United Kingdom) and we acknowledge the support of Dr. Giuliano Siligardi and Dr. Rohanah Hussain.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.890862/full#supplementary-material
Adli, M. (2018). The CRISPR Tool Kit for Genome Editing and beyond. Nat. Commun. 9, 1–13. doi:10.1038/s41467-018-04252-2
Alexander, C. G., Wanner, R., Johnson, C. M., Breitsprecher, D., Winter, G., Duhr, S., et al. (2014). Novel Microscale Approaches for Easy, Rapid Determination of Protein Stability in Academic and Commercial Settings. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1844, 2241–2250. doi:10.1016/j.bbapap.2014.09.016
Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A., and Stevens, R. C. (2008). Microscale Fluorescent Thermal Stability Assay for Membrane Proteins. Structure 16, 351–359. doi:10.1016/j.str.2008.02.004
Auvray, F., Dennetiere, D., Giuliani, A., Jamme, F., Wien, F., Nay, B., et al. (2019). Time Resolved Transient Circular Dichroism Spectroscopy Using Synchrotron Natural Polarization. Struct. Dyn. 6, 054307. doi:10.1063/1.5120346
Bai, N., Roder, H., Dickson, A., and Karanicolas, J. (2019). Isothermal Analysis of ThermoFluor Data Can Readily Provide Quantitative Binding Affinities. Sci. Rep. 9, 1–15. doi:10.1038/s41598-018-37072-x
Bhirde, A. A., Chiang, M.-J., Venna, R., Beaucage, S., and Brorson, K. (2018). High-Throughput In-Use and Stress Size Stability Screening of Protein Therapeutics Using Algorithm-Driven Dynamic Light Scattering. J. Pharm. Sci. 107, 2055–2062. doi:10.1016/j.xphs.2018.04.017
Biter, A. B., de la Peña, A. H., Thapar, R., Lin, J. Z., and Phillips, K. J. (2016). DSF Guided Refolding as a Novel Method of Protein Production. Sci. Rep. 6, 1–9. doi:10.1038/srep18906
Bonvin, A. M. J. J. (2021). 50 Years of PDB: a Catalyst in Structural Biology. Nat. Methods 18, 448–449. doi:10.1038/s41592-021-01138-y
Chari, A., Haselbach, D., Kirves, J.-M., Ohmer, J., Paknia, E., Fischer, N., et al. (2015). ProteoPlex: Stability Optimization of Macromolecular Complexes by Sparse-Matrix Screening of Chemical Space. Nat. Methods 12, 859–865. doi:10.1038/nmeth.3493
Cohen, A. E. (2021). A New Era of Synchrotron-Enabled Macromolecular Crystallography. Nat. Methods 18, 433–434. doi:10.1038/s41592-021-01146-y
Dauer, K., Pfeiffer-Marek, S., Kamm, W., and Wagner, K. G. (2021). Microwell Plate-Based Dynamic Light Scattering as a High-Throughput Characterization Tool in Biopharmaceutical Development. Pharmaceutics 13, 172. doi:10.3390/PHARMACEUTICS13020172
Davis, R. L. (2020). Mechanism of Action and Target Identification: a Matter of Timing in Drug Discovery. iScience 23, 101487. doi:10.1016/j.isci.2020.101487
Emmerich, C. H., Gamboa, L. M., Hofmann, M. C. J., Bonin-Andresen, M., Arbach, O., Schendel, P., et al. (2021). Improving Target Assessment in Biomedical Research: the GOT-IT Recommendations. Nat. Rev. Drug Discov. 20, 64–81. doi:10.1038/s41573-020-0087-3
Gao, K., Oerlemans, R., and Groves, M. R. (2020). Theory and Applications of Differential Scanning Fluorimetry in Early-Stage Drug Discovery. Biophys. Rev. 12, 85–104. doi:10.1007/s12551-020-00619-2
Holdgate, G., Embrey, K., Milbradt, A., and Davies, G. (2019). Biophysical Methods in Early Drug Discovery. ADMET DMPK 7, 222–241. doi:10.5599/admet.733
Hulse, W. L., Gray, J., and Forbes, R. T. (2013). Evaluating the Inter and Intra Batch Variability of Protein Aggregation Behaviour Using Taylor Dispersion Analysis and Dynamic Light Scattering. Int. J. Pharm. 453, 351–357. doi:10.1016/J.IJPHARM.2013.05.062
Hussain, R., Benning, K., Javorfi, T., Longo, E., Rudd, T. R., Pulford, B., et al. (2015). CDApps: Integrated Software for Experimental Planning and Data Processing at Beamline B23, Diamond Light Source. J. Synchrotron Radiat. 22, 465–468. doi:10.1107/S1600577514028161
Hussain, R., Jávorfi, T., Rudd, T. R., and Siligardi, G. (2016). High-Throughput SRCD Using Multi-Well Plates and its Applications. Sci. Rep. 6, 1–6. doi:10.1038/srep38028
Hussain, R., and Siligardi, G. (2016). Characterisation of Conformational and Ligand Binding Properties of Membrane Proteins Using Synchrotron Radiation Circular Dichroism (SRCD). Adv. Exp. Med. Biol. 922, 43–59. doi:10.1007/978-3-319-35072-1_4
Huynh, K., and Partch, C. L. (2015). Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. Curr. Protoc. Protein Sci. 79, 28–29. doi:10.1002/0471140864.ps2809s79
International Organization for Standardization, (1996). 13321 Particle Size Analysis - Photon Correlation Spectroscopy. Available at: https://www.iso.org/obp/ui/#iso:std:iso:13321 (Accessed November 9, 2021).
Jain, T., Sun, T., Durand, S., Hall, A., Houston, N. R., Nett, J. H., et al. (2017). Biophysical Properties of the Clinical-Stage Antibody Landscape. Proc. Natl. Acad. Sci. U.S.A. 114, 944–949. doi:10.1073/pnas.1616408114
Jovčevska, I., and Muyldermans, S. (2020). The Therapeutic Potential of Nanobodies. BioDrugs 34, 11–26. doi:10.1007/s40259-019-00392-z
Kiriiri, G. K., Njogu, P. M., and Mwangi, A. N. (2020). Exploring Different Approaches to Improve the Success of Drug Discovery and Development Projects: a Review. Futur J. Pharm. Sci. 6, 1–12. doi:10.1186/s43094-020-00047-9
Kotov, V., Bartels, K., Veith, K., Josts, I., Subhramanyam, U. K. T., Günther, C., et al. (2019). High-throughput Stability Screening for Detergent-Solubilized Membrane Proteins. Sci. Rep. 9, 1–19. doi:10.1038/s41598-019-46686-8
Kozak, S., Lercher, L., Karanth, M. N., Meijers, R., Carlomagno, T., and Boivin, S. (2016). Optimization of Protein Samples for NMR Using Thermal Shift Assays. J. Biomol. NMR 64, 281–289. doi:10.1007/s10858-016-0027-z
Krause, M. E., and Sahin, E. (2019). Chemical and Physical Instabilities in Manufacturing and Storage of Therapeutic Proteins. Curr. Opin. Biotechnol. 60, 159–167. doi:10.1016/j.copbio.2019.01.014
Kwan, T. O. C., Axford, D., and Moraes, I. (2020). Membrane Protein Crystallography in the Era of Modern Structural Biology. Biochem. Soc. Trans. 48, 2505–2524. doi:10.1042/BST20200066
Kwan, T. O. C., Reis, R., Siligardi, G., Hussain, R., Cheruvara, H., and Moraes, I. (2019). Selection of Biophysical Methods for Characterisation of Membrane Proteins. Ijms 20, 2605. doi:10.3390/ijms20102605
Lorber, B., Fischer, F., Bailly, M., Roy, H., and Kern, D. (2012). Protein Analysis by Dynamic Light Scattering: Methods and Techniques for Students. Biochem. Mol. Biol. Educ. 40, 372–382. doi:10.1002/bmb.20644
Macarron, R., Banks, M. N., Bojanic, D., Burns, D. J., Cirovic, D. A., Garyantes, T., et al. (2011). Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov. 10, 188–195. doi:10.1038/nrd3368
Makowski, E. K., Schardt, J. S., and Tessier, P. M. (2022). Improving Antibody Drug Development Using Bionanotechnology. Curr. Opin. Biotechnol. 74, 137–145. doi:10.1016/j.copbio.2021.10.027
Martinez, S. E., Singh, A., De Wijngaert, B., Sultana, S., Dharia, C., Vanbuel, H., et al. (2021). Assembly and Cryo-EM Structure Determination of Yeast Mitochondrial RNA Polymerase Initiation Complex Intermediates. Star. Protoc. 2, 100431. doi:10.1016/j.xpro.2021.100431
Meyer, A., Dierks, K., Hussein, R., Brillet, K., Brognaro, H., and Betzel, C. (2015). Systematic Analysis of Protein-Detergent Complexes Applying Dynamic Light Scattering to Optimize Solutions for Crystallization Trials. Acta Cryst. Sect. F. 71, 75–81. doi:10.1107/S2053230X14027149
Miles, A. J., Janes, R. W., and Wallace, B. A. (2021). Tools and Methods for Circular Dichroism Spectroscopy of Proteins: A Tutorial Review. Chem. Soc. Rev. 50, 8400–8413. doi:10.1039/D0CS00558D
Plant, A. L., Locascio, L. E., May, W. E., and Gallagher, P. D. (2014). Improved Reproducibility by Assuring Confidence in Measurements in Biomedical Research. Nat. Methods 11, 895–898. doi:10.1038/nmeth.3076
Puetz, J., and Wurm, F. M. (2019). Recombinant Proteins for Industrial versus Pharmaceutical Purposes: a Review of Process and Pricing. Processes 7, 476. doi:10.3390/pr7080476
Rajan, R., Ahmed, S., Sharma, N., Kumar, N., Debas, A., and Matsumura, K. (2021). Review of the Current State of Protein Aggregation Inhibition from a Materials Chemistry Perspective: Special Focus on Polymeric Materials. Mat. Adv. 2, 1139–1176. doi:10.1039/d0ma00760a
Ratanji, K. D., Derrick, J. P., Dearman, R. J., and Kimber, I. (2014). Immunogenicity of Therapeutic Proteins: Influence of Aggregation. J. Immunotoxicol. 11, 99–109. doi:10.3109/1547691X.2013.821564
Raynal, B., Lenormand, P., Baron, B., Hoos, S., and England, P. (2014). Quality Assessment and Optimization of Purified Protein Samples: Why and How? Microb. Cell Fact. 13, 1–10. doi:10.1186/s12934-014-0180-6
Real-Hohn, A., Groznica, M., Löffler, N., Blaas, D., and Kowalski, H. (2020). NanoDSF: In Vitro Label-free Method to Monitor Picornavirus Uncoating and Test Compounds Affecting Particle Stability. Front. Microbiol. 11, 1442. doi:10.3389/fmicb.2020.01442
Renaud, J.-P., Chung, C.-W., Danielson, U. H., Egner, U., Hennig, M., Hubbard, R. E., et al. (2016). Biophysics in Drug Discovery: Impact, Challenges and Opportunities. Nat. Rev. Drug Discov. 15, 679–698. doi:10.1038/nrd.2016.123
Ribeiro, R., Abreu, T. R., Silva, A. C., Gonçalves, J., and Moreira, J. N. (2019). Insights on the Formulation of Recombinant Proteins. Curr. Pharm. Biotechnol. 171, 23–54. doi:10.1007/10_2019_119
Roberts, C. J. (2014). Protein Aggregation and its Impact on Product Quality. Curr. Opin. Biotechnol. 30, 211–217. doi:10.1016/j.copbio.2014.08.001
Senisterra, G. A., and Finerty, P. J. (2009). High Throughput Methods of Assessing Protein Stability and Aggregation. Mol. Biosyst. 5, 217–223. doi:10.1039/B814377C
Shiba, K., Niidome, T., Katoh, E., Xiang, H., Han, L., Mori, T., et al. (2010). Polydispersity as a Parameter for Indicating the Thermal Stability of Proteins by Dynamic Light Scattering. Anal. Sci. 26, 659–663. doi:10.2116/ANALSCI.26.659
Stark, H., and Chari, A. (2016). Sample Preparation of Biological Macromolecular Assemblies for the Determination of High-Resolution Structures by Cryo-Electron Microscopy. Microsc. (Tokyo) 65, 23–34. doi:10.1093/jmicro/dfv367
Stetefeld, J., McKenna, S. A., and Patel, T. R. (2016). Dynamic Light Scattering: a Practical Guide and Applications in Biomedical Sciences. Biophys. Rev. 8, 409–427. doi:10.1007/S12551-016-0218-6
Svilenov, H., Markoja, U., and Winter, G. (2018). Isothermal Chemical Denaturation as a Complementary Tool to Overcome Limitations of Thermal Differential Scanning Fluorimetry in Predicting Physical Stability of Protein Formulations. Eur. J. Pharm. Biopharm. 125, 106–113. doi:10.1016/j.ejpb.2018.01.004
Wallace, B. A., Gekko, K., Vrønning Hoffmann, S., Lin, Y.-H., Sutherland, J. C., Tao, Y., et al. (2011). Synchrotron Radiation Circular Dichroism (SRCD) Spectroscopy: An Emerging Method in Structural Biology for Examining Protein Conformations and Protein Interactions. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 649:177–178. doi:10.1016/j.nima.2010.10.135
Wang, Y., van Oosterwijk, N., Ali, A. M., Adawy, A., Anindya, A. L., Dömling, A. S. S., et al. (2017). A Systematic Protein Refolding Screen Method Using the DGR Approach Reveals that Time and Secondary TSA Are Essential Variables. Sci. Rep. 7, 9355. doi:10.1038/s41598-017-09687-z
Yu, Z., Reid, J. C., and Yang, Y.-P. (2013). Utilizing Dynamic Light Scattering as a Process Analytical Technology for Protein Folding and Aggregation Monitoring in Vaccine Manufacturing. J. Pharm. Sci. 102, 4284–4290. doi:10.1002/jps.23746
Keywords: protein aggregation, DLS, DSF, CD, crystallography, cryo-EM, drug discovery, high-throughput
Citation: Kwan TOC, Kolek SA, Danson AE, Reis RI, Camacho IS, Shaw Stewart PD and Moraes I (2022) Measuring Protein Aggregation and Stability Using High-Throughput Biophysical Approaches. Front. Mol. Biosci. 9:890862. doi: 10.3389/fmolb.2022.890862
Received: 06 March 2022; Accepted: 25 April 2022;
Published: 16 May 2022.
Edited by:
Qian Han, Hainan University, ChinaReviewed by:
Jim Warwicker, The University of Manchester, United KingdomCopyright © 2022 Kwan, Kolek, Danson, Reis, Camacho, Shaw Stewart and Moraes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick D. Shaw Stewart, cGF0cmlja0Bkb3VnbGFzLmNvLnVr; Isabel Moraes, aXNhYmVsLm1vcmFlc0BucGwuY28udWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.