 Congzhou M. Sha1,2†
Congzhou M. Sha1,2† Nikolay V. Dokholyan
Nikolay V. Dokholyan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 22 March 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.867241
This article is part of the Research Topic Machine Learning Assisted Biomolecular Discovery and Analysis for Next Generation Biosensing and Biomedicine View all 4 articles
Virtual screening is a cost- and time-effective alternative to traditional high-throughput screening in the drug discovery process. Both virtual screening approaches, structure-based molecular docking and ligand-based cheminformatics, suffer from computational cost, low accuracy, and/or reliance on prior knowledge of a ligand that binds to a given target. Here, we propose a neural network framework, NeuralDock, which accelerates the process of high-quality computational docking by a factor of 106, and does not require prior knowledge of a ligand that binds to a given target. By approximating both protein-small molecule conformational sampling and energy-based scoring, NeuralDock accurately predicts the binding energy, and affinity of a protein-small molecule pair, based on protein pocket 3D structure and small molecule topology. We use NeuralDock and 25 GPUs to dock 937 million molecules from the ZINC database against superoxide dismutase-1 in 21 h, which we validate with physical docking using MedusaDock. Due to its speed and accuracy, NeuralDock may be useful in brute-force virtual screening of massive chemical libraries and training of generative drug models.
Drug discovery as carried out by pharmaceutical companies requires an investment of years of research effort and billions of dollars (DiMasi et al., 2016). The preclinical pipeline for identifying a small molecule ligand for a protein target is: (1) biochemical screening of small molecules against a protein target or cellular assay, (2) medicinal chemistry optimization of candidate small molecules, and (3) validation of promising molecules in animals (Eder and Herrling, 2016). Step (1) is critical in identifying small molecules which bind tightly to the target (hits) and for their subsequent optimization in Step (2) (leads). Step (1) is expensive and time-consuming, taking several months to screen a small library of >105 compounds. Insufficient binding affinities of leads and hits from Steps (1) and (2) often lead to drug attrition in Step (3) and subsequent clinical trials, with attrition rates as high as 95% (Hutchinson and Kirk, 2011; Waring et al., 2015). Molecular dynamics and rational drug design can explore a larger part of the chemical space and potentially increase binding affinity of hits, but typical docking tools still only dock one compound every few minutes at moderate sampling accuracy [AutoDock Vina: 1.2 min (Trott and Olson, 2010), DOCK 6: 4.8 min (Allen et al., 2015), Glide 1.7 min (Schrödinger, 2020), MedusaDock: seconds to minutes (Fan et al., 2021)], while the chemical space of potential drugs may be as large as 1060 small molecules (Bohacek et al., 1996). Neural networks have shown significant promise in structural biology, accurately reproducing gold standard results in a fraction of the time (Jumper, 2020). Here, we accelerate the virtual docking process by 6 orders of magnitude, enabling docking of 109 compounds in a single day at low cost.
Modern methods of computational drug docking are implemented by tools such as MedusaDock (Ding et al., 2010; Wang and Dokholyan, 2019), AutoDock Vina (Trott and Olson, 2010; Forli et al., 2016; Goodsell et al., 2021), DOCK (Allen et al., 2015), and Glide (Friesner et al., 2004). These tools perform molecular docking using classical force fields to evaluate the binding energy or affinity of a small molecule to a protein pocket of interest. Here, we focus on MedusaDock because it performs fully flexible conformational sampling of both the protein and ligand, which mimics the induced fit model of protein-small molecule binding, whereas other tools generate ensembles of the protein which are then rigidly fixed and docked to the small molecule [AutoDock Vina (Evangelista et al., 2016), DOCK (Allen et al., 2015), Glide (Schrödinger, 2016)]. MedusaDock consists of two independent tasks: conformational sampling and scoring. Here, we show that both tasks can be well-approximated by a deep neural network at a fraction of the computational cost of traditional docking. Although we used MedusaDock to generate our data, the framework we have developed can be applied to the results of other docking tools.
The principal advantages of a neural network over traditional docking tools include differentiability (propagation of gradients in model training through automatic differentiation) and speed. Neural networks are also valuable for their composability, in which they can be used as subnetworks of larger neural networks while providing gradients for training (Rocktäschel and Riedel, 2017; Feng et al., 2020). Neural network inference is highly optimized on modern processors, and particularly on GPUs. One can achieve many orders of magnitude higher performance with neural network approximation than with traditional algorithms based on exact calculation (Basu et al., 2010). As molecular dynamics is already a significant approximation to a quantum mechanical and statistical reality, inaccuracies in neural network predictions may be acceptable for virtual docking purposes (He et al., 2014).
Deep neural networks for predicting binding affinities have been successful; however, there are drawbacks to specific approaches (Cang et al., 2018; Jiménez et al., 2018; Francoeur et al., 2020; Gentile et al., 2020). For example, Francoeur et al. (2020) proposed to approximate force fields using neural network-based scoring while still relying on extensive conformational sampling, hence retaining the major computational bottleneck of virtual docking. Gentile et al. (2020) used neural networks to aid in chemical screening, but the predictions were nonspecific: information about the protein pocket was not used in the screening (Gentile et al., 2020). KDEEP by Jiménez et al. (2018) uses computationally expensive convolutional architectures which limit inference speed, and was trained directly on protein-small molecule 3D crystal structures. Due to the inclusion of protein-small molecule crystal structures, KDEEP is biased and has limited generalizability to proteins not bound to small molecules. This issue of bias has been discussed in Francoeur et al. (2020) and arises from self-docking, in which the neural network is provided with a low conformational energy crystal structure as input, and therefore does not perform conformational sampling. Cang et al. (2018) use convolutional networks with manually constructed ligand features based on ligand topology but provide no forward validation with docking tools; instead, only binding affinity is predicted, increasing the risk of overfitting and self-docking bias.
In stark contrast to work such as in Francoeur et al. (2020), we do not explicitly train our neural network to predict the energy of a specific conformation. Rather, we train our network in a conformation-invariant manner by withholding conformational information in the inputs and by predicting population parameters like minimum energy and binding affinity. Unlike in work from Jiménez et al. (2018) and Cang et al. (2018), we limit bias from self-docking and overfitting. We train the neural network to directly predict the minimum binding energy evaluated by MedusaDock, based on a coarse 3D representation of the protein and a graph representation of the small molecule. The direct prediction of binding energy makes conformational sampling implicit in the neural network. The coarse protein representation and graph representation of the small molecule withholds the optimal orientation, alignment, and conformation of the small molecule in the protein pocket of interest from the neural network. Since we augment our training data with MedusaDock energies, we also reduce the likelihood of overfitting. With these design elements in our neural network NeuralDock, we achieve class-leading performance, as tested on the PDBbind 2013 core set. We perform a proof-of-concept docking for benchmarking and external validation purposes, using NeuralDock to dock nearly 109 molecules from the ZINC database against the enzyme superoxide dismutase-1 (SOD1), which is not present in the training, validation, or test sets. Finally, we validate the predicted energies using MedusaDock.
For NeuralDock inputs, we used crystal structures of 3875 known protein-ligand pairs from the PDBbind 2017 refined set (Wang et al., 2004), which were shuffled into a training set (N = 2,712, 70% of structures), and a validation set (N = 1,163, 30% of structures). One of these structures was not processable by MedusaDock 2.0, and 127 structures exceeded our computational resources. A further 496 ligands were not processable by rdkit 2020.09.327. This left a training set of N = 2,279 (84.0% of original) and a validation set of N = 972 (83.6% of original). The core set of PDBbind 2013 was used as the test set (N = 195); 154 of these were processable by rdkit. There is no overlap of proteins among the training, validation, and test sets.
We extracted the atoms in the protein which were within a cube of side length 20 angstroms, centered at the ligand. These protein pockets were encoded as 10 × 10 × 10, 2-angstrom resolution images with 8 channels corresponding to a one-hot encoding of (no atom, C, O, N, S, P, H, and other). If multiple atoms were contained in the same 2-angstrom cube, we took the maximum of each channel of the one-hot encodings. Using rdkit, we encoded the ligand as a length 36 atom type vectors with 7 channels (no atom, C, N, O, F, S, and other) and a 36 × 36 bond adjacency matrix with 5 channels (no bond, single, double, triple, aromatic/conjugated). For ligands with greater than 36 heavy atoms (hydrogens were excluded), we removed atoms with the least bond order until we were left with 36 total heavy atoms, thus attempting to preserve the important topologies present in the ligand. These input dimensions were chosen so that a comparable number of parameters in NeuralDock would be devoted to processing the protein and the ligand each (Figure 1A), as well as to prevent the ligand representation from becoming too sparse. We also chose a limit of 36 heavy atoms since molecules with greater than 36 heavy atoms are likely to exceed the 500 Dalton cutoff for Lipinski’s rule of five.
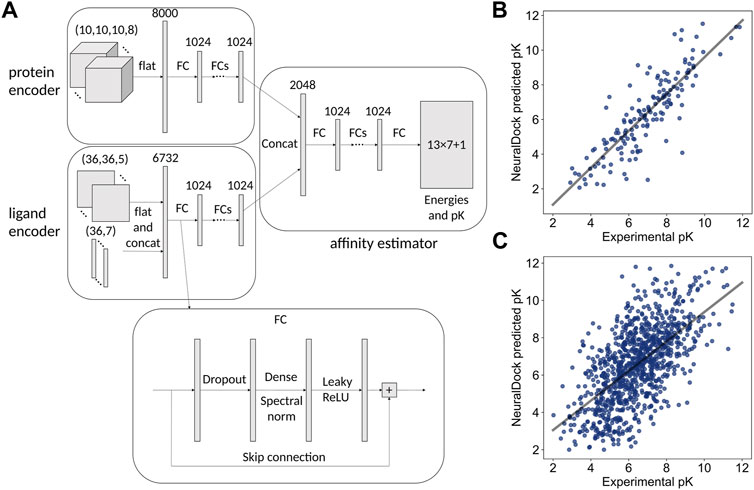
FIGURE 1. The neural network architecture on the left and performance comparison with MedusaDock on the right. (A) Inputs, hidden layers, and outputs are shown for the architecture. The protein pocket is flattened and fed into a subnetwork, and the ligand is processed similarly. The outputs of the two subnetworks are concatenated and fed into another subnetwork, which outputs 13 × 7 + 1 values representing the 7 summary statistics of the 13 energies output by MedusaDock, as well as the pK of the protein-ligand pair. The structure of each FC layer is shown at the bottom. (B) The 45 million parameter NeuralDock network achieves class-leading performance on the PDBbind 2013 core set (r = 0.85, p < 0.0001). (C) The 45 million parameter NeuralDock network achieves good agreement with experimentally determined pK on the validation set (r = 0.62, p < 0.0001).
For each protein-ligand pair, we ran MedusaDock for 24 h or 1000 iterations (whichever came first) on a single core of an Intel Xeon E5-2,680 v3 processor with 6 GB RAM. We collected summary statistics (mean, median, standard deviation, minimum, maximum, skew, and kurtosis) on the 13 interaction energies computed in MedusaDock’s force field, MedusaScore (Yin et al., 2008). Five of these energies were zero for most or all structures (see Supplementary Information). An interaction energy is defined as the total energy of the protein-ligand complex
In developing our models, we chose to focus on Ewithout VDWR, the interaction energy excluding repulsive van der Waals forces. Ewithout VDWR is the output of MedusaScore which is known to be the most highly correlated with experimental binding affinity (Yin et al., 2008).
The compounds (N = 997,402,117) were downloaded from the ZINC database (Irwin and Shoichet, 2005) (available in Tranches) and processed into the tensor input format described above. Of these compounds, 936,054,166 (94%) were processable by rdkit. We chose the 1UXM SOD1 PDB structure as a protein target (Berman et al., 2000). The interface between the A and B chains (Figure 2) was chosen as the binding pocket for 1UXM.
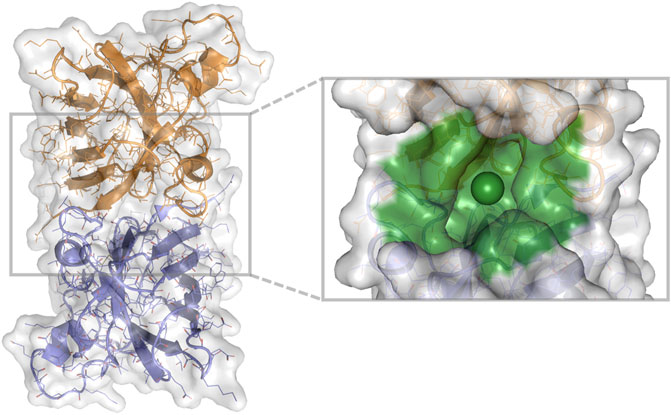
FIGURE 2. 1UXM A4V SOD1 dimer chains A (gold) and B (lilac), with the protein pocket of interest (green) in billion-molecule docking. A cartoon, stick, and water-accessible surface representation of 1UXM, an A4V mutant SOD1 dimer structure. The image was generated using PyMOL 2.4.0 (Schrödinger, 2021).
For NeuralDock, we chose a fully connected architecture, with spectral normalization (Miyato et al., 2018), dropout (Srivastava et al., 2014) with rate 0.2, LeakyReLU activation (Maas et al., 2013), and skip connections (He et al., 2016). These characteristics were chosen as standard methods of model regularization to prevent overfitting and vanishing or exploding gradients during training.
The NeuralDock architecture (Figure 1A) was implemented in TensorFlow 2.4.0 (Abadi et al., 2016) and Python 3.7. The inputs were the protein pocket and ligand topology, and the outputs were the 7 summary statistics of the 13 interaction energies computed by MedusaDock as well as the binding affinity pK = log10 K (dissociation/inhibitor constant KD/I with units of molar). We considered KD and KI to be the same for the training purposes. We chose 10 fully connected (FC) hidden layers for each of the three parts (protein encoder, ligand encoder, affinity predictor) of the network (Figure 1A), resulting in approximately 45 million trainable parameters. We varied the number of hidden layers as well as their widths during hyperparameter optimization (Table 1). The loss function was the L2 (squared difference) loss between the NeuralDock output energies and the MedusaDock output energies, as well as pKs. The Adam optimizer was used (Kingma and Ba, 2015), with a learning rate of 10−6. Training of each model took place on one NVIDIA Tesla T4 GPU, and the models were trained to convergence within a week. We trained a convolutional architecture, in which the FC blocks in the protein encoder (top left of Figure 1A) were replaced by spectrally normalized 3D inception modules (Szegedy et al., 2015) with a comparable number of trainable parameters (Table 1).

TABLE 1. Correlation coefficients of NeuralDock predicted minimum energy and MedusaDock output for a variety of architectures.
We used Lipinski’s rule of five (Lipinski et al., 2012) to evaluate small molecule lead quality and drug likeness, as well as the Quantitative Estimate of Drug likeness (QED) (Bickerton et al., 2012). Octanol-water partition coefficients (log P) were extracted from HTML files of the ZINC database, while all other quantities were computed using rdkit (RDKit, 2016).
Least squares regression was performed in Python 3.7 using SciPy 1.6.0 (Virtanen et al., 2020). Analysis of covariance (ANCOVA) was performed in Python 3.7 using Pingouin 0.3.12 (Vallat, 2018).
NeuralDock training on MedusaDock energies and experimental pKs converged, achieving agreement with experimental binding affinities for the test set, the PDBbind 2013 core set (Figure 1B), and the validation set taken from PDBbind 2017 (Figure 1C). As discussed by Francoeur et al. (2020), testing on the core set may offer a biased evaluation of binding affinity prediction performance. We provide the core set correlation for comparison with other methods which solely report that data (Table 2). Given our relatively small training set (2,331 structures) and the massive diversity of potential protein pockets and ligands, NeuralDock was able to learn the binding affinities accurately. We believe that the key to NeuralDock’s success is using high quality data produced by MedusaDock, which sampled thousands of conformations for each protein-ligand pair, while supplying coarse 3D protein information and only the small molecule’s topology. By using only the topology of the small molecule as input, we forced NeuralDock to approximate the effects of conformational sampling.

TABLE 2. Correlation coefficients for binding affinity prediction of a variety of neural networks.
To test the robustness, generalizability, and speed of NeuralDock, we performed virtual screening of a massive library of ligands (N = 936,054,166) against the pocket at the dimeric interface of SOD1 (Figure 2). We compared relationships among MedusaDock Ewithout VDWR, NeuralDock Ewithout VDWR, and experimental binding affinities (pK), and found that they agree (Figure 3). Ewithout VDWR is the component of MedusaScore which is most highly correlated with experimental pK (Yin et al., 2008). We demonstrate the correlation between NeuralDock and MedusaDock Ewithout VDWR, first on the validation set drawn from PDBbind 2017 refined set (r = 0.83, p < 0.0001), and then on 100 random small molecules from ZINC docked to 1UXM, an A4V mutant of the human superoxide dismutase-1 enzyme (r = 0.69, p < 0.0001) (Figure 3A). Therefore, NeuralDock was successful in learning to predict the minimum Ewithout VDWR in a single shot. Additionally, we performed 2-way ANCOVA, which measures the effects of a categorical variable. In this instance, it showed no statistically significant difference in the trends for the validation set and the external validation on 1UXM (F = 0.67, p = 0.41), which provides evidence that NeuralDock successfully generalizes from the training set to other protein-small molecule pairs, as well as achieving cross-docking success. Since the native structure 1UXM was not bound to any small molecule and MedusaDock performs fully flexible conformational sampling of both the protein and the small molecule, our results provide external validation of NeuralDock and demonstrate robustness under cross-docking, i.e., docking of small molecules to a protein as it appears in its native, unbound conformation.
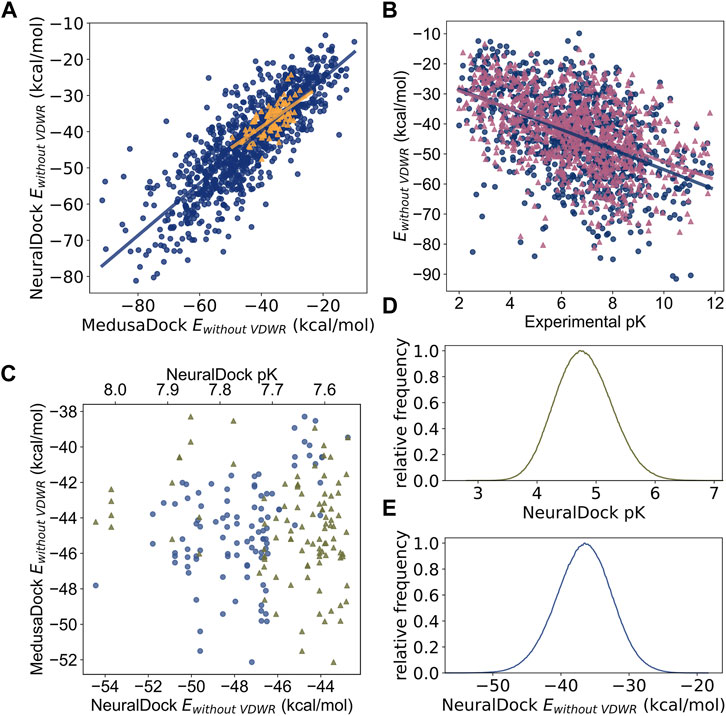
FIGURE 3. Comparisons among MedusaDock energies, NeuralDock predicted energies, and experimental binding affinity data. (A) The correlation between NeuralDock predicted Ewithout VDWR and MedusaDock Ewithout VDWR on the validation set (blue circles, r = 0.83, p < 0.0001) and 100 random small molecules docked to 1UXM (orange triangles, r = 0.69, p < 0.0001). NeuralDock performs well on the validation set in predicting MedusaDock energies, and the trend generalizes to 1UXM with no significant difference (2-way ANCOVA F = 0.67, p = 0.41). (B) The correlations of MedusaDock Ewithout VDWR (blue circles), NeuralDock predicted Ewithout VDWR (magenta triangles), and experimental binding affinity (pK) on the validation set (r = −0.48 for both data sets, p < 0.0001), with no significant difference (2-way ANCOVA F = 1.27, p = 0.26). (C) The 100 small molecules with maximum NeuralDock pK (green triangles), from docking of 936,054,166 small molecules from the ZINC library against 1UXM; the corresponding predicted Ewithout VDWR is plotted (lilac circles). The left is higher binding affinity (higher pK) and lower energy (lower Ewithout VDWR). (D) and (E) The relative frequency distributions (300 bins) of NeuralDock predicted pK (mean 4.07, std 0.47) and Ewithout VDWR (mean −36.6, std 4.1), respectively, on 8,099,176 (9% of total) randomly selected small molecules from the docking of 1UXM. The plots are centered at the means, the x-axis ranges are ± 5 standard deviations from the mean, and colors are repeated from (C). Note that both the Ewithout VDWR and pKs in (C) are drawn from the extreme tails of the distributions shown in (D).
We found agreement of MedusaDock Ewithout VDWR and NeuralDock Ewithout VDWR with experimental pK, with similar correlations for both tools (r = −0.48 for both, p < 0.0001), and no statistically significant difference in correlations with 2-way ANCOVA (F = 1.27, p = 0.26) (Figure 3B). The correlation of MedusaDock Ewithout VDWR with experimental pK (r = −0.48) is comparable to that of AutoDock Vina scoring (r = 0.41) as reported in Francoeur et al. (2020), with MedusaDock performing better, likely due to our extensive sampling and computational effort for each protein-ligand pair. NeuralDock predicts experimental pK better than MedusaDock on the validation set (Figures 1B, C, 3B), however, experimental validation of the pKs is needed to confirm that this result holds for SOD1 and other proteins. NeuralDock’s agreement with MedusaDock predictions demonstrates that it may be useful in replacing traditional docking tools such as MedusaDock and AutoDock Vina.
For the billion-molecule docking of 1UXM, we find that the 100 small molecules with the highest NeuralDock-predicted pKs also have low binding energies, with agreement of NeuralDock and MedusaDock Ewithout VDWR on those small molecules (Figure 3C). Furthermore, NeuralDock predicts that few other molecules have such high pK and low energy (Figure 3D). Combining these results with our validation set regression (Figure 1C), we predict that some of the 100 small molecules chosen have pK of approximately 8 (K ≈ 10 nanomolar). Ninety-five of the 100 compounds satisfy Lipinski’s rule of five, and all have reasonable QED scores (median 0.50, range 0.33–0.66). The drug likeness of these compounds can be explained as bias from the ZINC Tranches, in which only 14,744,513 (1.5% of the over 997 million) compounds have molecular weight exceeding 500 Daltons or log P exceeding 5.
Various architectures for NeuralDock yielded similar results (Table 1). We optimized the NeuralDock architecture based on Ewithout VDWR correlation, as Ewithout VDWR is most highly correlated with binding affinity (Yin et al., 2008). Optimizing the network based on Ewithout VDWR prevents overfitting the hyperparameters (i.e., number of layers and total number of trainable parameters), as MedusaDock Ewithout VDWR is an external source of training information. Including other MedusaDock energies in training also helped to prevent overfitting (Supplementary Table S2). Even a limited network such as the 4.9 million parameter model was able to generalize from the training set to the validation set (Table 1).
Benchmarking on a single Tesla T4 GPU was able to predict energies and pKs of 96,000 protein-small molecule pairs in 203.8 s, or 2 ms per protein-small molecule pair. Docking of the 937 million ZINC compounds with NeuralDock took 21 h on 25 GPUs, which matches our benchmarking of 2 ms per protein-small molecule pair per GPU. For 3875 structures (including those that exceeded our computational resources), it took 120 processors 4 weeks to perform extensive MedusaDock sampling (1000 iterations), or an average of 20 h per structure per processor. Note that the ability of MedusaDock to find low energy binding poses requires repeated conformational sampling, which can be controlled by running many iterations of MedusaDock, with each iteration taking seconds to minutes. Taking 10 h as a conservative minimum compute time for 1000 iterations of MedusaDock, NeuralDock performs 1.7 × 107 times faster. NeuralDock performs 105 times faster than DOCK 66, and 100 times faster than KDEEP (Jiménez et al., 2018). NeuralDock is also faster than the Def2018 General Ensemble by Francoeur et al. (2020), since we directly predict binding affinity in a single forward pass instead of requiring many conformational samples and forward passes through the network (Francoeur et al., 2020).
There are several potential improvements to NeuralDock. First, NeuralDock takes a coarse atomic image of the 20 Å protein pocket as input. Including inputs such as protein amino acid composition, secondary structure, and hydrophobicity may improve NeuralDock predictions. This potential change in inputs is supported by the fact that NeuralDock was able to learn local contributions to the energy well (i.e., EVWDA, Supplementary Tables S1, S2), while having difficulty with global contributions (e.g., Esolv, Supplementary Tables S1, S2). Second, NeuralDock is trained on a relatively small training set, and we did not use any small molecules which are off-target, i.e., do not bind to the given protein. However, our external validation with MedusaDock indicates that this potential source of bias was at least partially addressed by the variety of binding affinities present in our training set. This deficiency may be remedied by docking more targets with MedusaDock as a reference, such as the PDBbind general set, which incurs a significant computational cost in data preparation. Additionally, the PDBbind general set experimental pKs may be noisier than in the refined set. Third, the binding energies and pK are invariant under rotations of the protein pocket as well as under permutations in the atom order for the small molecule bond adjacency matrix and atom type vector. Newer neural network architectures which directly leverage these physical symmetries have been proposed (Finzi et al., 2020; Wu et al., 2021).
We posit that our small training set is not necessarily a major impediment to NeuralDock performance. The so-called double descent phenomenon occurs when the number of tunable parameters in a statistical model significantly exceeds the number of training samples (or degrees of freedom) (Belkin et al., 2019). The traditional tradeoff between bias and variance constitutes the first descent in test error, in which the optimal number of model parameters lies below the number necessary to precisely fit the data, a number known as the interpolation threshold. Contrary to conventional statistical wisdom, however, over-parametrizing may provide benefit in the model performance, causing a second descent in test error. In certain cases, additional training data may actually hurt model performance, whereas training past the point of 0 training set error (another instance of the interpolation threshold) provides a similar benefit to over-parametrization (Nakkiran et al., 2020). Although the double descent phenomenon has yet to be formally proven for deep neural networks, we observed the phenomenon in practice while training NeuralDock. With 46 million parameters and roughly 2000 data points for training, NeuralDock takes advantage of the second descent to achieve its high accuracy.
MolGAN is a generative adversarial approach to drug discovery, which does not take into account the protein pocket (Goodfellow et al., 2014; de Cao and Kipf, 2018). Since NeuralDock is end-to-end differentiable, one can augment MolGAN training with NeuralDock acting as a scoring function to create drugs targeting specific proteins. The addition of such guidance may improve the stability of MolGAN training. While automatically differentiable force fields are currently being developed for molecular dynamics (Schoenholz and Cubuk, 2019), the advantage of NeuralDock is it can immediately evaluate the quality of a generated small molecule, implicitly performing conformational sampling, whereas conformational sampling must still be done for a differentiable force field. NeuralDock does not predict the explicit minimum energy configuration for the protein-small molecule pair, however, once specific compounds are identified via NeuralDock, the candidates can be docked by MedusaDock or another docking software prior to experimental validation, as the computational cost of doing so is much less than the cost of traditional docking for the entire compound library.
NeuralDock is a robust neural network for predicting binding energies and affinities for protein-small molecule pairs. The key design elements that result in its class-leading performance include high quality scoring of thousands of protein-small molecule conformations, coarse 3D image representation of the protein pocket, and topological representation of the small molecule. Because NeuralDock is trained to output predicted energies and pK in a single shot, NeuralDock is faster than competing methods and can be used in billion-molecule virtual screening. NeuralDock has been validated using the fully flexible docking tool MedusaDock and is ready for experimental validation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
CMS implemented, trained, and validated the neural networks; performed the MedusaDock simulations; performed statistical and chemical calculations; created Figures 1, 3; and wrote the manuscript. JW wrote the protein pocket extraction software, aided with the MedusaDock simulations, and created Figure 2. All three authors contributed to discussions regarding architecture exploration and design, the cost function, and data analysis and interpretation.
We acknowledge support from the National Institutes for Health (R35 GM134864 and RF1 AG071675 to NVD), the National Science Foundation (2040667), and the Passan Foundation. The content in this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are thankful to the NSF Convergence Accelerator (award number 2040667) team for helpful discussions on machine learning.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.867241/full#supplementary-material
Abadi, M., Barham, P., Chen, J., Chen, Z., Davis, A., Dean, J., et al. (2016). “TensorFlow: A System for Large-Scale Machine Learning,” in Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation OSDI 2016. Berkeley: USENIX Association.
Allen, W. J., Balius, T. E., Mukherjee, S., Brozell, S. R., Moustakas, D. T., Lang, P. T., et al. (2015). DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 36, 1132–1156. doi:10.1002/jcc.23905
Basu, J. K., Bhattacharyya, D., and Kim, T. (2010). Use of Artificial Neural Network in Pattern Recognition. Int. J. Softw. Eng. Its Appl. 4.
Belkin, M., Hsu, D., Ma, S., and Mandal, S. (2019). Reconciling Modern Machine-Learning Practice and the Classical Bias-Variance Trade-Off. Proc. Natl. Acad. Sci. U. S. A. 116, 15849–15854. doi:10.1073/pnas.1903070116
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S., and Hopkins, A. L. (2012). Quantifying the Chemical beauty of Drugs. Nat. Chem. 4, 90–98. doi:10.1038/nchem.1243
Bohacek, R. S., McMartin, C., and Guida, W. C. (1996). The Art and Practice of Structure-Based Drug Design: A Molecular Modeling Perspective. Med. Res. Rev. 16, 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6
Cang, Z., Mu, L., and Wei, G. W. (2018). Representability of Algebraic Topology for Biomolecules in Machine Learning Based Scoring and Virtual Screening. Plos Comput. Biol. 14, e1005929. doi:10.1371/journal.pcbi.1005929
de Cao, N., and Kipf, T. (2018). MolGAN: An Implicit Generative Model for Small Molecular Graphs. Ithaca: arXiv.
DiMasi, J. A., Grabowski, H. G., and Hansen, R. W. (2016). Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J. Health Econ. 47, 20–33. doi:10.1016/j.jhealeco.2016.01.012
Ding, F., Yin, S., and Dokholyan, N. V. (2010). Rapid Flexible Docking Using a Stochastic Rotamer Library of Ligands. J. Chem. Inf. Model. 50, 1623–1632. doi:10.1021/ci100218t
Eder, J., and Herrling, P. L. (2016). Trends in Modern Drug Discovery. Handbook Exp. Pharmacol. 232, 3–22. doi:10.1007/164_2015_20
Evangelista, W., Weir, R. L., Ellingson, S. R., Harris, J. B., Kapoor, K., Smith, J. C., et al. (2016). Ensemble-based Docking: From Hit Discovery to Metabolism and Toxicity Predictions. Bioorg. Med. Chem. 24, 4928–4935. doi:10.1016/j.bmc.2016.07.064
Fan, M., Wang, J., Jiang, H., Feng, Y., Mahdavi, M., Madduri, K., et al. (2021). GPU-accelerated Flexible Molecular Docking. J. Phys. Chem. B 125, 1049–1060. doi:10.1021/acs.jpcb.0c09051
Feng, Y., Zheng, Z. o., Liu, Q., Greenspan, M., and Zhu, X. (2020). Exploring End-To-End Differentiable Natural Logic Modeling. Ithaca: arXiv. doi:10.18653/v1/2020.coling-main.101
Finzi, M., Stanton, S., Izmailov, P., and Wilson, A. G. (2020). Generalizing Convolutional Neural Networks for Equivariance to Lie Groups on Arbitrary Continuous Data. Ithaca: arXiv.
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., and Olson, A. J. (2016). Computational Protein-Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 11, 905–919. doi:10.1038/nprot.2016.051
Francoeur, P. G., Masuda, T., Sunseri, J., Jia, A., Iovanisci, R. B., Snyder, I., et al. (2020). Three-Dimensional Convolutional Neural Networks and a Cross-Docked Data Set for Structure-Based Drug Design. J. Chem. Inf. Model. 60, 4200–4215. doi:10.1021/acs.jcim.0c00411
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 47, 1739–1749. doi:10.1021/jm0306430
Gentile, F., Agrawal, V., Hsing, M., Ton, A. T., Ban, F., Norinder, U., et al. (2020). Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 6, 939–949. doi:10.1021/acscentsci.0c00229
Goodfellow, I. J., Pouget-Abadie, J., Mirza, M., Xu, B., Warde-Farley, D., Ozair, S., et al. (2014). “Generative Adversarial Nets,” in Advances in Neural Information Processing Systems. Montreal: NeurIPS, 3.
Goodsell, D. S., Sanner, M. F., Olson, A. J., and Forli, S. (2021). The AutoDock Suite at 30. Protein Sci. 30, 31–43. doi:10.1002/pro.3934
He, K., Zhang, X., Ren, S., and Sun, J. (2016). “Deep Residual Learning for Image Recognition,” in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition. Piscataway: IEEE. vols 2016-December. doi:10.1109/cvpr.2016.90
He, X., Zhu, T., Wang, X., Liu, J., and Zhang, J. Z. (2014). Fragment Quantum Mechanical Calculation of Proteins and its Applications. Acc. Chem. Res. 47, 2748–2757. doi:10.1021/ar500077t
Hutchinson, L., and Kirk, R. (2011). High Drug Attrition Rates-Wwhere Are We Going Wrong? Nat. Rev. Clin. Oncol. 8, 189–190. doi:10.1038/nrclinonc.2011.34
Irwin, J. J., and Shoichet, B. K. (2005). ZINC--a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 45, 177–182. doi:10.1021/ci049714+
Jiménez, J., Škalič, M., Martínez-Rosell, G., and De Fabritiis, G. (2018). KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 58, 287–296. doi:10.1021/acs.jcim.7b00650
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kingma, D. P., and Ba, J. (2015). “A Method for Stochastic Optimization,” in 3rd International Conference on Learning Representations (San Diego: ICLR 2015 - Conference Track Proceedings).
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2012). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 64. doi:10.1016/j.addr.2012.09.019
Maas, A. L., Hannun, A. Y., and Ng, A. Y. (2013). “Rectifier Nonlinearities Improve Neural Network Acoustic Models,” in ICML Workshop on Deep Learning for Audio, Speech and Language Processing. Atlanta: Proceedings of Machine Learning Research.
Miyato, T., Kataoka, T., Koyama, M., and Yoshida, Y. (2018). “Spectral Normalization for Generative Adversarial Networks,” in 6th International Conference on Learning Representations (Vancouver: ICLR 2018 - Conference Track Proceedings).
Nakkiran, P., Kaplun, G., Bansal, Y., Yang, T., Barak, B., Sutskever, I., et al. (2020). “Deep Double Descent: Where Bigger Models and More Data Hurt,” in 8th International Conference on Learning Representations. $\\{$ICLR$\\}$ 2020.
Rocktäschel, T., and Riedel, S. (2017). “End-to-end Differentiable Proving,” in Advances in Neural Information Processing Systems. Long Beach: NeurIPS. vols 2017-December.
Schoenholz, S. S., and Cubuk, E. D. (2019). End-to-end Differentiable, Hardware Accelerated, Molecular Dynamics in Pure Python. Ithaca: arXiv.
Schrödinger (2020). How Long Does it Take to Screen 10,000 Compounds with Glide? New York, NY: Schrödinger LLC. Available at: https://www.schrodinger.com/kb/1012.
Schrödinger (2016). What Is Ensemble Docking and How Can I Use it? . New York, NY: Schrödinger LLC. Available at: https://www.schrodinger.com/kb/28.
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I., and Salakhutdinov, R. (2014). Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 15, 1929–1958. Available at: https://jmlr.org/papers/v15/srivastava14a.html.
Szegedy, C., Liu, W., Jia, Y., Sermanet, P., Reed, S., Anguelov, D., et al. (2015). “Going Deeper with Convolutions,” in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, 7-12 June 2015 (IEEE), 1–19. doi:10.1109/cvpr.2015.7298594
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., et al. (2020). SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 17, 261–272. doi:10.1038/s41592-019-0686-2
Wang, J., and Dokholyan, N. V. (2019). MedusaDock 2.0: Efficient and Accurate Protein-Ligand Docking with Constraints. J. Chem. Inf. Model. 59, 2509–2515. doi:10.1021/acs.jcim.8b00905
Wang, R., Fang, X., Lu, Y., and Wang, S. (2004). The PDBbind Database: Collection of Binding Affinities for Protein-Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 47, 2977–2980. doi:10.1021/jm030580l
Waring, M. J., Arrowsmith, J., Leach, A. R., Leeson, P. D., Mandrell, S., Owen, R. M., et al. (2015). An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discov. 14, 475–486. doi:10.1038/nrd4609
Wu, Z., Pan, S., Chen, F., Long, G., Zhang, C., Yu, P. S, et al. (2021). A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Networks Learn. Syst. 32. doi:10.1109/tnnls.2020.2978386
Keywords: virtual docking, small molecule screening, drug screening, machine learning, binding affinity
Citation: Sha CM, Wang J and Dokholyan NV (2022) NeuralDock: Rapid and Conformation-Agnostic Docking of Small Molecules. Front. Mol. Biosci. 9:867241. doi: 10.3389/fmolb.2022.867241
Received: 31 January 2022; Accepted: 22 February 2022;
Published: 22 March 2022.
Edited by:
Feng Ding, Clemson University, United StatesReviewed by:
Binquan Luan, IBM Research, United StatesCopyright © 2022 Sha, Wang and Dokholyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolay V. Dokholyan, ZG9raEBwc3UuZWR1
†ORCID ID: Congzhou M. Sha, orcid.org/0000-0001-5301-9459; Jian Wang, orcid.org/0000-0001-7768-2802; Nikolay V. Dokholyan, orcid.org/0000-0002-8225-4025
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.