
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 15 June 2022
Sec. Structural Biology
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.865743
This article is part of the Research Topic Insights in Structural Biology: 2021 View all 10 articles
 Sofia S. Mariasina1,2
Sofia S. Mariasina1,2 Chi-Fon Chang3
Chi-Fon Chang3 Tsimafei L. Navalayeu4Anastasia A. Chugunova4
Tsimafei L. Navalayeu4Anastasia A. Chugunova4 Sergey V. Efimov5
Sergey V. Efimov5 Viktor G. Zgoda6
Viktor G. Zgoda6 Vasily A. Ivlev7
Vasily A. Ivlev7 Olga A. Dontsova4,8
Olga A. Dontsova4,8 Petr V. Sergiev2,4,8
Petr V. Sergiev2,4,8 Vladimir I. Polshakov1*
Vladimir I. Polshakov1*Williams-Beuren syndrome (WBS) is a genetic disorder associated with the hemizygous deletion of several genes in chromosome 7, encoding 26 proteins. Malfunction of these proteins induce multisystemic failure in an organism. While biological functions of most proteins are more or less established, the one of methyltransferase WBSCR27 remains elusive. To find the substrate of methylation catalyzed by WBSCR27 we constructed mouse cell lines with a Wbscr27 gene knockout and studied the obtained cells using several molecular biology and mass spectrometry techniques. We attempted to pinpoint the methylation target among the RNAs and proteins, but in all cases neither a direct substrate has been identified nor the protein partners have been detected. To reveal the nature of the putative methylation substrate we determined the solution structure and studied the conformational dynamic properties of WBSCR27 in apo state and in complex with S-adenosyl-L-homocysteine (SAH). The protein core was found to form a canonical Rossman fold common for Class I methyltransferases. N-terminus of the protein and the β6–β7 loop were disordered in apo-form, but binding of SAH induced the transition of these fragments to a well-formed substrate binding site. Analyzing the structure of this binding site allows us to suggest potential substrates of WBSCR27 methylation to be probed in further research.
Williams-Beuren syndrome (WBS) is a complex developmental disorder, induced by haploinsufficiency of 24–26 genes in the chromosome region 7q11 (Schubert, 2009). Multisystem disorders associated with this disease include aortic stenosis, hypercalcemia, impaired glucose metabolism, thyroid dysfunction, growth retardation, characteristic facial appearance, mental deficiency, and “friendly” personality which is usually considered as hyper-friendliness (Jones et al., 2000; Pober, 2010; Masserini et al., 2013).
For some phenotypic features, the impact of a specific gene deletion is already well-established. For instance, the ELN gene, being a part of WBS deletion, encodes the protein elastin, a component of vascular walls, and its insufficiency leads to aortic stenosis (Ewart et al., 1993). Deletion of gene LIMK1, encoding LIM-kinase 1, brings about impaired visuospatial constructive cognition (Frangiskakis et al., 1996). There is also solid evidence on the insufficiency of BAZ1B contributing to hypercalcemia through interaction with the vitamin D receptor (Kitagawa et al., 2003). However, the impact of other genes lost in case of WBS manifestation remains unclear.
Uncovering the physiological consequences of gene loss on the behavior characteristic phenotype of WBS patients is among the most complicated directions in WBS studies. There is no direct evidence, but the decreased expression level of some gene products from the WBS chromosome region is likely to be involved. Interestingly, domestic dogs exhibit some of the behavioral traits typical of humans with WBS (vonHoldt et al., 2017): as compared to their ancestor, the gray wolf, domestic dogs have heightened propensity to initiate social contacts showing “hyper-sociability.” Comparing the dog genome to the Yellowstone gray wolf one revealed mobile element insertions affecting transcriptional regulation in the genes responsible for WBS (vonHoldt et al., 2018). Transcriptome sequencing confirmed that the expression levels of six genes placed in WBS chromosome region (WBSCR17, LIMK1, GTF2I, WBSCR27, BAZ1B, and BCL7B) differ between these animals accounting for different behavior patterns.
One of the plausible candidate proteins associated with the behavioral aspects of WBS is WBSCR27. Human and chimpanzee genome sequences were compared and nine human-specific frameshift mutations were identified (Hahn and Lee, 2005). One of these mutations is placed within the WBSCR27 gene coding sequence: there is an 11 bp insertion in human WBSCR27. The insertion occurred specifically in the human lineage and probably could somehow affect the functioning of the protein; and thereby directly or indirectly alter human social behavior in comparison with chimpanzees. However, there is no direct experimental proof of this hypothesis yet.
There is a dearth of information about the biological function of WBSCR27. To outline some functional role of this protein only differential gene expression was measured. The expression level of WBSCR27 was reported to change in response to different external conditions. The overexpression of WBSCR27 was found in three tumor types: esophageal carcinoma, stomach adenocarcinoma, and kidney renal papillary cell carcinoma (Campeanu et al., 2021) by bioinformatic analysis of data available in TCGA (The Cancer Genome Atlas). WBSCR27 is overexpressed also in colon cancer and can be used as a prognostic marker of this desease (Wang et al., 2022).
Salvianolic acid B treatment was recently studied as a potential therapeutic approach for obesity (An et al., 2019). To examine the differential gene expression in mouse white adipose tissue caused by treating with Salvianolic acid B RNA-Seq was performed demonstrating that 234 lncRNAs, 19 circRNAs, and 132 mRNAs were differentially expressed. Among the mRNAs, the upregulated expression of WBSCR27 was the highest, with a fold change of 2.053. These results were confirmed by the qPCR. The other upregulated protein-coding genes were involved in the insulin resistance pathway, while the downregulated genes mainly participated in the IL-17 signaling pathway.
Nevertheless, these data do not shed any light on the possible role of WBSCR27 in WBS, as well as on its biological function in general. Metzger and colleagues (Metzger et al., 2019) previously identified C21orf127 MTase (later renamed to KMT9) to be responsible for histone lysine methylation. Surprisingly, this protein turned out to be a seven-β-stranded methyltransferase (MTase), while all histone lysine methylating proteins known before belonged to SET-domain family (Husmann and Gozani, 2019). To find out other histone lysine MTases within the seven-β-stranded MTase family, cluster analysis on multiple amino acid sequence alignments of putative seven-β-stranded MTase domains was performed. WBSCR27 was found among the seven closest homologs of C21orf127 and was tested for histone methylation in the in vitro assay, but did not show any methylation activity.
In our previous work (Mariasina et al., 2020) we demonstrated that WBSCR27 effectively interacts with the cofactor S-(5′-adenosyl)-L-methionine (SAM) and has a canonical Rossman fold, typical of Class I MTases. This information supports the bioinformatic assignment of WBSCR27 to MTases; however, the substrate of methylation catalyzed by this enzyme is still unknown. Here we determined the solution structure of WBSCR27 in apo-state and in complex with the cofactor. This work may shed light on the possible biological role of this protein making the complete mapping of gene deletions in WBS and physiological consequences one step closer.
The uniformly 15N and 15N/13C enriched protein was expressed in E. coli cells grown on 15N or 15N/13C M9 minimal media using the glucose-13C (2 g/L) and/or ammonium sulphate-15N (1 g/L) as a source of stable isotopes. The protein selectively 13C-labelled in the methyl groups of Thr and Met residues was expressed in 15N M9 media containing 100% D2O and ISOGRO®-D supplemented by Met-ε-13СH3 and Thr-γ-13СH3 as well as fully deuterated 2-ketobutyrate and Gly-d2 to prevent cross-labelling (Kerfah et al., 2015).
Protein samples were purified as described in Mariasina et al. (2018). Refolding was an important step in the purification procedure of the apo-form of the protein. In this case after affinity chromatography on the Ni-NTA column and subsequent His-tag cleavage, the protein samples were denatured in 6 M urea, washed from endogenous SAH by dialysis, and refolded back to the native form (Mariasina et al., 2018). Samples of the WBSCR27-SAH complex with identical content of the 13C and 15N isotopes in both protein and ligand were purified without refolding. In all other cases samples for NMR structural studies were prepared by adding the corresponding ligand to the apo-form of WBSCR27 followed by the dialysis against the buffer containing 50 mM NaCl, 50 mM sodium phosphate (pH 7.0), 10 mM DTT and 0.02% NaN3.
[Methyl 13C]-labelled SAM was synthesized from SAH (Sigma) and 13CH3I (Cambridge Isotope Laboratories) according to the described method (Huber et al., 2016). 15 mg of SAH were dissolved in 500 µl of deuterated formic acid. 300 µl of 13CH3I were added to the resulting solution. The mixture was vortexed for 2 h and then stirred at room temperature in the dark. The completeness of the reaction was monitored by 1H NMR. After 5 days, 1 ml of water was added and the unreacted 13C methyl iodide was extracted with 2 ml of diethyl ether twice. The pH of the aqueous phase was adjusted to 7.15, after which the sample was applied to an ion exchange chromatographic column (732-0003 BioRad cartridge) preliminarily equilibrated with 0.01 M sodium phosphate buffer solution (pH 7.15). The column was washed with 55 ml of the same buffer solution (at first, the uncharged unreacted SAH and subproduct MTA were washed off, then the positively charged SAM was washed off). Next, the column was washed first with 20 ml of 0.1 M acetic acid, then with 20 ml of 4 M acetic acid. The main part of the product came off the column in the interval between 13 and 26 ml.
The SAM-containing fractions were evaporated in SpeedVac and dried in a freeze-dryer. A mixture of (S,S)- and (R,S)-SAM diastereomers was obtained, which is in agreement with the earlier described results (Bennett et al., 2017). The purity of the obtained product was controlled by NMR spectroscopy, the concentration was determined by UV spectrophotometry (ε260 = 16,000 M−1 cm−1). The overall conversion rate can be estimated at 50% of the initial SAH. The yield of pure substance after purification was 10%.
13C, 15N uniformly labelled SAH was obtained from E. coli cell line overexpressing the WBSCR27 protein and grown on 13C,15N M9 medium. The method was based on the propensity of WBSCR27 to be co-purified with the cofactor and isolated in the form of a complex with SAH. The details of this protocol will be published elsewhere. The quality of the obtained 13C,15N-SAH was confirmed by 1D and 2D NMR spectra (Supplementary Figure S1). The sample contained DTT as an impurity, which does not interfere with the following procedure and was not removed from the product. The concentration of the obtained SAH was measured using UV absorbance at 260 nm (ε260 = 16,000 M−1 cm−1). In total, we obtained 3.2 µmol of 13C,15N-SAH from 2 L of 13C,15N M9 medium.
Mouse embryonic fibroblast cells NIH3T3 were cultured in DMEM/F12 medium (Gibco), supplemented with 10% FBS (Gibco), 1% Penicillin/Streptomycin (Gibco), and 1% Glutamax (Gibco) at 37°С, 5% CO2 and used for all genetic manipulations.
The cell lines created for this study are schematically shown in Supplementary Figure S2. The cell lines A and B were prepared for studying intercellular localization of WBSCR27. The cell line A ectopically expressed fusion of WBSCR27 with far-red fluorescent protein mKate2 on N-terminal (Shemiakina et al., 2012). Similarly, the cell line B ectopically expressed hemagglutinin epitope YPYDVPDYA known as an HA tag (Field et al., 1988). The cell line B together with an endogenous C-terminal WBSCR27-HA fusion (cell line C) were used in co-immunoprecipitation experiments aimed at finding the possible macromolecular partners of WBSCR27. The knockout line (D) containing point mutations in the 2nd exon was used to study the phenotypic consequences of WBSCR27 depletion. To confirm WBSCR27 depletion on the protein level an HA-tag was added to the C-terminus of WBSCR27 in the knockout line (E). Two cell lines F and G were created from the knockout line for proximity labelling in the BioID experiment (Roux et al., 2018). These cell lines ectopically expressed prokaryotic biotin ligase BirA mutant (R118G) from E.coli (designated as BirA*), fused to HA and WBSCR27 (HA-BirA*-WBSCR27) or only to HA as the control (HA-BirA*). The details of cloning and constructing these cell lines are provided in Supplementary Data.
The intercellular localization of WBSCR27 was verified using cells with ectopic expression of HA or mKate2 (Shemiakina et al., 2012) fusions with WBSCR27. mKate2-WBSCR27 and HA-WBSCR27 expressing cells were seeded on coverslips and kept in the incubator overnight. The next day, the cells were washed 3 times for 5 min with PBST (PBS + 0.1% Triton X-100) and fixed with freshly-prepared 4% paraformaldehyde (in PBS) for 10 min at room temperature. The coverslips were rinsed with PBST (3 times for 5 min), followed by permeabilization with 1% Triton X-100 (in PBS) for 15 min at room temperature and subsequent PBST wash (3 times for 5 min).
The permeabilized coverslips with mKate2-WBSCR27 cells were incubated with 100 mM DAPI in PBS for 5 min at room temperature, followed by washing with PBST (2 times for 7 min). The coverslips were mounted with Mowiol (Sigma) and dried overnight.
The permeabilized coverslips with HA-WBSCR27 cells were blocked with 3% BSA (in PBST) for 1 h at room temperature, followed by incubation with primary anti-HA antibodies (Sigma, 3F10) and then with secondary goat anti-rat Alexa555 conjugated antibodies (Thermo Fisher Scientific) overnight at 4°С in PBST. After washing with PBST (3 times for 5 min), the coverslips were subjected to DAPI staining and mounting as described for mKate2-WBSCR27. Imaging was done with the Nikon Ti-E fluorescence microscope.
Cells were cultured in five 15 cm plates to 95% confluency in a DMEM-F12 medium supplemented with 10% FBS, Glutamax, penicillin and streptomycin, and doxycycline hyclate (Sigma) at a concentration of 1 μg/ml at 37°C, 5% CO2. The cells were harvested by trypsin, washed twice with 5 ml of PBS and kept frozen at −80°C prior to the immunoprecipitation (IP) experiment.
The frozen cells were resuspended in a lysis buffer [100 mM Hepes-KOH pH 7.5, 150 mM NaCl, 0.05% Triton X-100, 1 mM DTT and complete protease inhibitor cocktail (Roche)]. After centrifugation at 13,000 g for 30 min, the supernatant was transferred to a new tube. For IP, 100 µl of anti HA-beads (Sigma Aldrich) were added to the lysate obtained from 1 g of wet cell mass and incubated overnight at 4°С. After five washes with the lysis buffer, proteins were eluted with a PAGE loading buffer for 5 min at 95°С. The protein eluates were analyzed by PAGE followed by silver staining and Western-blotting. Several bands present exclusively in the samples corresponding to the HA-tagged WBSCR27 were analyzed using MALDI according to the standard protocol (Chugunova et al., 2019).
For cross-linking experiments, the cells were resuspended in 1% formaldehyde (in PBS) and mixed for 7 min at room temperature. The cells were pelleted (500 g, 3 min) and washed twice with 1.25 M glycine (in PBS) to quench the remaining formaldehyde. Next, the cells were lysed as described in the paragraph above. The control sample was subjected to the same protocol, but omitting the formaldehyde addition step.
The BioID experiment was performed in accordance with the published protocol (Roux et al., 2018). Three cell lines based on NIH3T3 ΔWBSCR27 were used in the experiment: expressing HA-BirA*-WBSCR27, HA-BirA*, and parental ΔWBSCR27.
Ten 15 cm dishes for each cell line were seeded. Biotin labelling was performed when the cells reached approximately 80% confluency, the medium was changed to a fresh complete medium containing 50 μM biotin and incubated for 16–18 h. In the next stage the medium was completely removed by aspiration, the cells were rinsed twice with 5 ml/dish of PBS, and treated by 600 µl of lysis buffer/dish. The cells were harvested by gentle scraping. After centrifugation we obtained 1–2 g of the cells. The affinity purification of biotinylated proteins was performed using Dynabeads M-280 Streptavidin (Thermo Fisher Scientific).
The eluates from streptavidin beads were treated by trypsin and subsequently analyzed by shotgun proteomics (a technique for identifying proteins in complex mixtures such as cell lysates using a combination of high performance liquid chromatography and tandem mass spectrometry). Mass spectroscopy analysis was performed in triplicates with a Q Exactive HF-X mass spectrometer (Q Exactive HF-X Hybrid Quadrupole-Orbitrap™ Mass spectrometer, Thermo Fisher Scientific, Rockwell, IL, United States). The experimental details were published earlier (Laptev et al., 2020). The obtained raw data were processed using SearchGui (Barsnes and Vaudel, 2018) and PeptideShaker (Vaudel et al., 2015) programs with built-in search engines X! Tandem, MS Amanda, OMSSA, and Comet. Protein sequences of the complete mouse proteome provided by Uniprot (August 2019) were used for protein identification. N-terminal acetylation as well as the oxidation of methionine residues were set as variable modifications for the peptide search. Up to two missed cleavages were allowed for trypsin digestion. The false discovery rates for peptide and protein identifications were set to 1%.
The experiment was performed as described earlier (Lesnyak et al., 2006). Total RNA was purified from NIH3T3 cell lines (WT and ΔWBSCR27) using Trizol reagent (ThermoFisher). The reverse transcription was performed with Maxima Reverse Transcriptase (ThermoFisher) using 32P-labelled oligonucleotide complementary to the 28 S rRNA fragment 4,537–4,551. The products of the reverse transcription were separated by electrophoresis in the 10% (w/v) denaturing polyacrylamide gel and visualized by phosphorimagery.
NMR samples at a concentration of 0.2–0.6 mM for 13C and/or 15N-labelled WBSCR27 and its complex with SAH were prepared in 95% H2O/5% D2O, 50 mM NaCl, 50 mM sodium phosphate buffer (pH 7.0), 10 mM DTT, and 0.02% NaN3. NMR spectra were recorded at 308 K on Bruker AVANCE 600, 700, 800, and 850 MHz spectrometers equipped with a triple resonance (1H, 13C, 15N) room temperature probe (600 MHz), Prodigy probe (700 MHz), and CryoProbe (800 and 850 MHz), or a quadruple resonance (1H, 13C, 15N, 31P) CryoProbe (700 MHz). 1D NMR spectra were processed and analyzed using Mnova software (Mestrelab Research, Spain). 2D and 3D spectra were processed by NMRPipe (Delaglio et al., 1995) and analyzed using NMRFAM-Sparky (Lee et al., 2015).
Earlier we reported the backbone and side chain signal assignments for the complex SAH-WBSCR27 (BMRB-27417, Mariasina et al., 2018) and for the apo form of the protein (BMRB-27578, Mariasina et al., 2020) and deposited these data in BioMagResBank (https://bmrb.io). This information was used to determine NMR restraints. Backbone φ and ψ dihedral angle restraints were determined from the chemical shift values of the backbone atoms 13Cα, 13Cβ, 13CO, 1Hα, 1HN, and 15N using TALOS+ software (Shen et al., 2009). Two independent sets of residual dipolar coupling (RDC) constants were measured in the nematic phase of a colloidal suspension of filamentous Pf1 phages (Hansen et al., 1998) and in a dilute liquid crystalline medium, consisting of DMPC/DHPC bicelles (Ottiger and Bax, 1999). The RDC values were calculated as a difference of the 15N-1H splitting values measured in the IPAP-HSQC spectrum (Ottiger et al., 1998) acquired in anisotropic and isotropic conditions. Hydrogen bond restraints were assigned to the amide groups having slow H/D exchange rates and located near carbonyl groups, as identified in the initial set of structures. NOE distance restraints were determined from the 1H,13C HSQC-NOESY and 1H,15N HSQC-NOESY spectra measured with a 100 ms mixing time. The initial set of NOE restraints (mainly intra-residue and sequential correlations) was selected manually. The rest of the cross-peaks were assigned using the automatic iterative procedure of spectra assignment/structure calculation implemented in ARIA 2.3 software (Bardiaux et al., 2012). The assignments were further manually verified by multiple steps of structure refinement using the simulated annealing protocol of the CNS 1.21 software package (Brunger et al., 1998). Database values of conformational torsion angle pseudopotentials (Kuszewski et al., 1997) were used during the final cycles of the structure refinement to improve the quality of protein backbone conformation. The structure quality and restraint violations have been analyzed using the CNS tools, Procheck-NMR (Laskowski et al., 1996) and an in-house written NMRest program (Ivanova et al., 2007). The final families of 20 NMR structures of the SAH-WBSCR27 complex and apo-WBSCR27 were selected from 200 calculated conformers in accordance with the lowest-energy criterion and the absence of the residues in the disallowed regions of the Ramachandran map. The restraints used in structure calculations and statistics for the obtained NMR structures are presented in Supplementary Tables S1, S2. Additional details of structure calculations are provided in Supplementary Material. Structure visualization and analysis were carried out using PyMOL (Schrodinger LLC) and Discovery Studio Visualizer (Dassault Systemes Biovia Corp.).
R1 and R2 relaxation rates and 1H-15N heteronuclear NOEs for 15N uniformly labelled WBSCR27-SAH complex and the apo-form of WBSCR27 were measured at 308 K on a Bruker AVANCE III HD 700 MHz spectrometer. The measured experimental values were analyzed with a model-free formalism using the program RelaxFit written in-house (Polshakov et al., 1999). All the details of the NMR relaxation data collection and analysis are provided in Supplementary Material.
Amide H/D exchange rates in both 15N labelled apo-WBSCR27 and complex WBSCR27-SAH were measured using heteronuclear 15N–1H NMR spectroscopy (at 35°C and pH 7.0) on a Bruker AVANCE Neo 700 MHz spectrometer. The details of the H/D exchange rate measurements and the calculation of the protection factors of the amide HN atoms are given in the Supplementary Data.
NMR experiments were carried out to test the binding of amino acids, nucleosides, and short DNA fragments to WBSCR27. The WBSCR27-SAM complex was prepared using 15N-WBSCR27 (0.3 mM) and SAM (1.2 mM) in 320 µl of 90% H2O/10% D2O.
To test possible binding of amino acids to WBSCR27, a mixture of seven amino acids (Thr, Ser, Arg, Tyr, Cys, Glu, and Lys) in equimolar ratio was prepared. This mixture was added to WBSCR27-SAM samples to obtain molar ratios WBSCR27:SAM:mixture of 1:4:5 and 1:4:10. To test possible interactions of WBSCR27 with the fragments of nucleic acids, we mixed each of nucleosides (guanosine, uridine, cytidine, thymidine) with WBSCR27-SAM separately to obtain molar ratios WBSCR27:SAM:nucleoside = 1:4:10. We additionally prepared a mixture of desoxyoligonucleotide AAACCTCGCATTACGAACGGCTCC with the WBSCR27-SAM sample with a ratio of WBSCR27:SAM:DNA = 1:4:1. The purpose of testing an interaction of this arbitrary oligonucleotide with WBSCR27 was to check the possibility of oligonucleotide chain binding by protein. For each sample the 15N,1H HSQC spectrum was measured at 308 K and 600 MHz.
NMR spectroscopy was used to determine the binding ability of SAM epimers towards WBSCR27. The [methyl 13C]-SAM obtained via a chemical synthesis from SAH in a concentration of 0.1 mM was used for this purpose. Since the synthetic product is an equimolar mixture of (S,S)- and (R,S)-SAM diastereomers, the concentration of individual components was 0.05 mM. 13C,1H HSQC spectra were recorded at 308 K and 600 MHz 1H frequency for four samples: 1) a free SAM [a mixture of 0.05 mM (S,S)-SAM and 0.05 mM (R,S)-SAM], 2) a free apo-WBSCR27 (protein concentration 0.05 mM), 3) a mixture of 0.05 mM (S,S)-SAM, 0.05 mM (R,S)-SAM, and 0.05 mM WBSCR27 (1:1:1 ratio), and 4) a mixture of 0.05 mM (S,S)-SAM, 0.05 mM (R,S)-SAM, and 0.1 mM WBSCR27 (1:1:2 ratio).
To determine the intracellular localization of WBSCR27 we inserted HA-WBSCR27 and mKate2-WBSCR27 fusion protein genes under a doxycycline inducible promoter into the NIH3T3 cell line via Sleeping Beauty transposase (Mátés et al., 2009). Visualizing both fusion proteins by anti-HA epitope immunocytochemical staining of fixed and permeabilized cells for HA-WBSCR27, and fluorescent microscopy of cells expressing mKate2-WBSCR27 allowed us to reveal both the cytoplasmic and nucleus distribution of WBSCR27 (Figure 1).
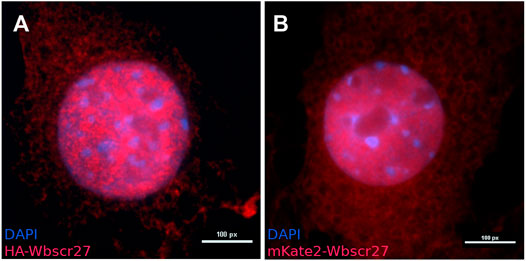
FIGURE 1. The intracellular localization of WBSCR27 in NIH3T3 cells. (A) HA-WBSCR27; (B) mKate2-WBSCR27. The signal is present in both the cytoplasm and nucleus. The nucleus was visualized with DAPI.
To outline the putative partners WBSCR27 protein interacts with, we applied a NIH3T3 cell line with ectopic expression of HA-WBSCR27. After inducing the fusion gene expression by doxycycline, HA-WBSCR27 was immunoprecipitated by an immobilized anti-HA antibody (Supplementary Figure S3A). While a protein of 27 kDa mass identified as HA-WBSCR27 by immunoblotting (Supplementary Figure S3B) was clearly present in the immunoprecipitate, no bands of its putative protein partners were observed. To exclude the possibility that the lack of identifiable partner proteins results from HA-WBSCR27 overexpression (which should lead to a significant decrease in the portion of the complex of WBSCR27 with its potential partner against the background of free protein), or that an N-terminally located HA tag prevents interaction with partner proteins, we used CRISPR/Cas9 directed cleavage and subsequent homologous recombination to create an NIH3T3 cell line with the natural Wbscr27 gene C-terminally appended with an HA coding part. Immunoprecipitation of WBSCR27-HA from the extracts of the latter cell line (Supplementary Figure S3C) did not lead to the identification of potential WBSCR27 partner proteins.
The homolog of WBSCR27, rRNA MTase WBSCR22, and a number of other MTases form a complex with TRMT112. To specifically address the possibility that TRMT112 might coprecipitate with WBSCR27, we analyzed the eluate after HA-WBSCR27 immunoprecipitation by TRMT112 specific antibodies (data not shown) and found no evidence favoring the interaction between WBSCR27 and TRMT112.
The WBSCR27 MTase might establish only transient contacts with its possible substrates, being disengaged in the process of immunopurification. To this end, we immunopurified ectopically expressed HA-WBSCR27 following the formaldehyde treatment of the cells. Application of the formaldehyde cross-linking to isolate WBSCR27 partner proteins also did not help to identify the interacting proteins due to the absence or negligible yield of covalently crosslinked products (Supplementary Figure S4).
An alternative approach to address the short-lived protein-protein interactions is BioID, a proximity-dependent protein biotinylation in vivo by mutant promiscuously active biotin ligase BirA* followed by biotin-affinity capture (Roux et al., 2018). To apply BioID to search for WBSCR27 protein partners we created a cell line expressing HA-BirA*-WBSCR27 fusion and a control cell line expressing HA-BirA*. After labelling was induced by supplementing the cell cultures with biotin, the biotinylated proteins were purified from the cell extracts via streptavidine affinity capture and analyzed by shotgun proteomics. While both HA-BirA*-WBSCR27 and HA-BirA* fusion proteins were successfully expressed (Supplementary Figure S5A) and biotinylated endogenous proteins (Supplementary Figure S5B), panoramic proteome analysis of the biotinylated proteins (Supplementary Table S3) did not identify proteins modified specifically by the WBSCR27 protein fusion.
To identify potential RNA partners of WBSCR27 protein we applied PAR-CLIP protocol as described by Gopanenko and co-authors (Gopanenko et al., 2017). The cells of the NIH3T3 line expressing HA-WBSCR27 protein grown in the presence of 4-thiouridine were subsequently subjected to mild UV irradiation to induce RNA-protein cross-linking. After RNA fragmentation and HA-WBSCR27 immunopurification, cross-linked RNA was labelled with γ-[32P]ATP and analyzed by gel electrophoresis and autoradiography (Supplementary Figure S6). Despite our efforts, the PAR-CLIP method did not allow identifying an RNA partner/substrate of the WBSCR27 enzyme.
To search for potential WBSCR27 substrates we used CRISPR/Cas9 guided WBSCR27 gene inactivation. Biallelic mutations disrupting the WBSCR27 reading frame were introduced to the second exon of the gene. Due to the absence of sufficient specificity of anti-WBSCR27 antibodies (data not shown), we verified the lack of WBSCR27 protein in the knockout cell line by biallelic extension of the Wbscr27 reading frame with the HA coding region. Disrupting the reading frame of the Wbscr27-HA gene resulted in the disappearance of the band stained by anti-HA antibodies (Supplementary Figure S7).
For most methylated rRNA nucleotides, the enzymes responsible for their modification are known (Sergiev et al., 2018). The only methylated nucleotide of mammalian ribosomal RNA for which the enzyme responsible for the modification has not yet been identified is the m3U4530 of the 28S rRNA (human rRNA numbering). This nucleotide is located in the peptidyl transferase center of the large ribosomal subunit (Sergiev et al., 2018). To validate whether this modification is due to the enzymatic activity of WBSCR27, we carried out a reverse transcription experiment similar to that used to identify a bacterial MTase modifying G2445 of the 23 S rRNA (Lesnyak et al., 2006). The method is based on m3U inducing reverse transcription arrest. The experiment showed that the reverse transcription arrest is observed in rRNA from both the WT and WBSCR27 knockout cells (Supplementary Figure S8). These data clearly indicate that WBSCR27 is not responsible for modifying U4530 and, therefore, rRNA cannot be a methylation substrate for this enzyme.
The fragment-based lead discovery approach was used to probe the interactions of the possible WBSCR27 substrate fragments with protein. NMR techniques are usually able to detect highly specific interactions of small fragments of a larger ligand with a protein, even in the case of weak binding (Polshakov et al., 2019). We investigated the interactions of small compounds mimicking the fragments of macromolecules, namely: amino acids (Thr, Ser, Arg, Tyr, Cys, Glu, and Lys), nucleosides (guanosine, uridine, cytidine, timidine), and desoxyoligonucleotides with 15N-labelled WBSCR27-SAM complex using the methods of heteronuclear NMR spectroscopy. In none of the studied fragments was specific binding observed, leading to a change of the 1H and/or 15N chemical shifts of the amide groups of certain WBSCR27 amino acids upon adding ligands to the protein.
The sulfonium atom of SAM represents a chiral center, and both (R) and (S)-epimers are stable (Figure 2A). In living cells, the natural SAM (S,S-epimer) is biosynthesized from L-methionine and ATP by methionine adenosyltransferase (Zhang and Zheng, 2016). Chemically synthesized SAM contains both stereoisomers in equal amounts. We demonstrated by 2D NMR using synthesized [methyl 13C]-labelled racemic (S,S/R,S)-SAM that WBSCR27 preferably binds the (S,S) stereoisomer. With an excess of WBSCR27, only the (S,S)-SAM isomer is bound, while the (R,S)-epimer remains in the free form (Figure 2B, left panel). With an increase in WBSCR27 content, the signals of the bound form (R,S)-SAM appear, but their intensity is significantly lower than that of the signals of the (S,S)-SAM-WBSCR27 complex (Figure 2B, right panel). This indicates a significantly lower affinity of the (R,S)-epimer compared with the (S,S)-epimer.
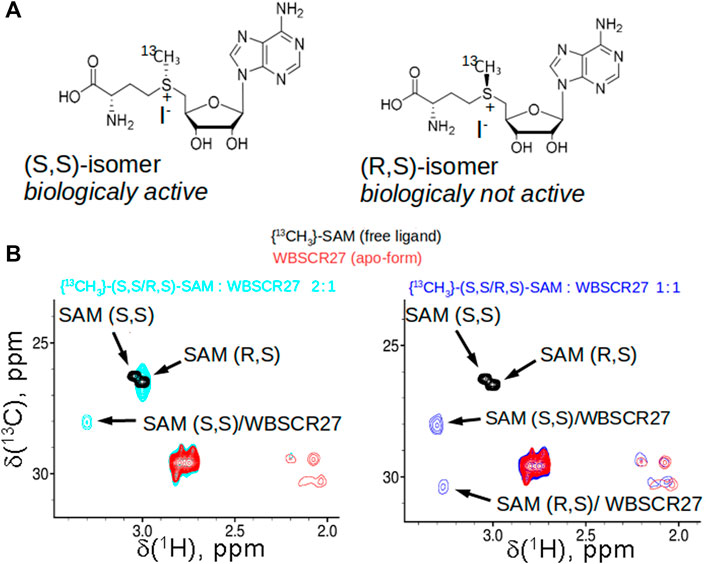
FIGURE 2. SAM stereoisomers and their binding to WBSCR27. (A) Two stable epimers of SAM: biologically active (S,S)-SAM, synthesized enzymatically from ATP and methionine in cells, and (R,S)-SAM, which is obtained as a by-product synthetically. (B) Overlay of the fragments of 13C,1H HSQC spectra of racemic 13CH3-labelled (R,S/S,S)-SAM (black), apo-WBSCR27 (red) and the mixture of SAM and protein in the ratio 2:1 (cyan, shown in the left panel) or 0.9:1 (blue, shown in the right panel).
Earlier, we found that both SAM and SAH strongly bind to WBSCR27 (Mariasina et al., 2020). Relatively small changes in the chemical shifts of the signals of the residues in the binding site of these two ligands indicate that the structure of WBSCR27-SAM and WBSCR27-SAH complexes is similar (Mariasina et al., 2020). At the same time, bound SAM rapidly (several hours) decomposes to SAH; as a result, only the WBSCR27-SAH complex remains sufficiently stable to measure a series of heteronuclear NMR spectra. To determine the structures of the complex WBSCR27-SAH and the protein in apo-form traditional heteronuclear NMR and restrained molecular dynamics techniques were used. To assign the signals of bound SAH and protein-ligand NOEs, the NMR spectra for the complexes of 15N-WBSCR27 with 13C-labelled and unlabelled SAH were measured and compared. Several protein-ligand NOEs were also identified in NOESY spectra of the complex of 13C,15N-WBSCR27 with unlabelled SAH. In total, 21 protein-ligand NOEs were used in structure calculation (Supplementary Table S1).
The solution structure of the WBSCR27 apo-form (Figure 3A) shows that the first 51 amino acid residues of the protein as well as the loop between residues 204 and 228 (loop67 in Figure 3C) are unstructured. SAH binding to the protein puts in order the N-terminal protein fragment by forming three well-structured α-helices (α1–α3) and a short helical part in the first nine residues (Figure 3B). The structure of WBSCR27 has a canonical Rossman fold, typical of most of the Class I MTases (Figure 3C). The protein core consists of seven β-strands (β1–β7) surrounded by five α-helices (α4–α8, Figure 3C). SAH binds to the residues on the tips of three β-strands (β1–β3) and strengthens these interactions by several hydrophobic and electrostatic contacts with the amino acid residues in the helices α1, α2, and α3 (Figure 4). The purine fragment of SAH binds predominantly to the amino acid residues of the protein β-core, while the methionine fragment interacts exclusively with the residues of helices α1–α3. The position of SAH methionine fragment is determined less precisely. The pairwise RMSD of the coordinates of the heavy atoms of the methionine fragment of SAH in the final family of structures is 2.3 ± 0.5 Å. For the adenosine fragment of SAH this value is 2.0 ± 0.6 Å. This may be due to higher mobility of the first three α-helices relative to the protein core.
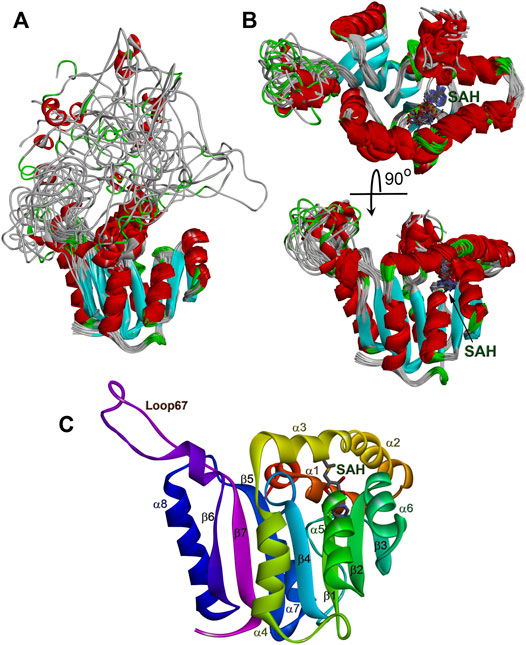
FIGURE 3. Structure of WBSCR27 in apo-form (A) and in complex with SAH (B,C). Families of 20 NMR conformers (A,B) and rainbow colored structure of WBSCR27-SAH complex with labelled elements of the protein structure are shown.
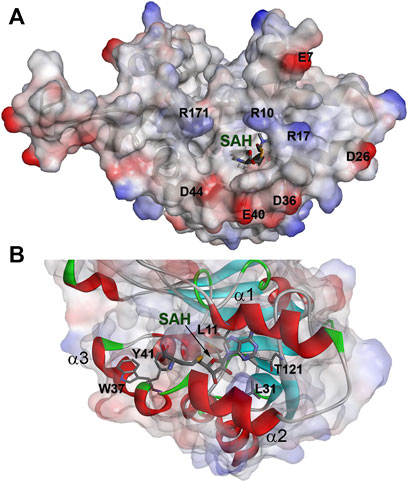
FIGURE 4. The binding site of SAH in complex with WBSCR27. (A) Electrostatic surface potential of the protein. (B) Amino acids of WBSCR27 participating in the interaction with SAH.
The protein backbone dynamics of WBSCR27 in both the apo-form and the complex with SAH was investigated by analyzing 15N relaxation experiments and hydrogen-to-deuterium (H/D) exchange rates of the amide protons. The values of the overall rotational correlation time τc, calculated from the 15N T1 and T2 data measured at 308 K (Kay et al., 1989), are 12.5 ± 0.2 and 10.4 ± 0.3 ns for the apo-form and the WBSCR27-SAH complex, respectively. The differences in the τc values for the apo-form and the complex appear to reflect the distinctions in the shape of the protein molecule. The protein globule is apparently more compact in the case of the WBSCR27-SAH complex, while the long unstructured N-terminal tail in the apo-form slows down protein tumbling. The experimentally measured 15N relaxation parameters were interpreted using the model-free formalism (Lipari and Szabo, 1982) with extension to include chemical exchange contributions Rex to the transverse relaxation rates (Clore et al., 1990) (Supplementary Material for details). Figure 5 (for the apo-form of WBSCR27) and Figure 6 (for WBSCR27-SAH) show the measured 15N relaxation parameters R1, R2, and NOE with the calculated order parameters S2 and Rex values, plotted against the corresponding residue numbers. The mobility of the WBSCR27 backbone in the apo-form is significantly higher than that in the complex with SAH, which agrees well with the observed results of the structural studies.
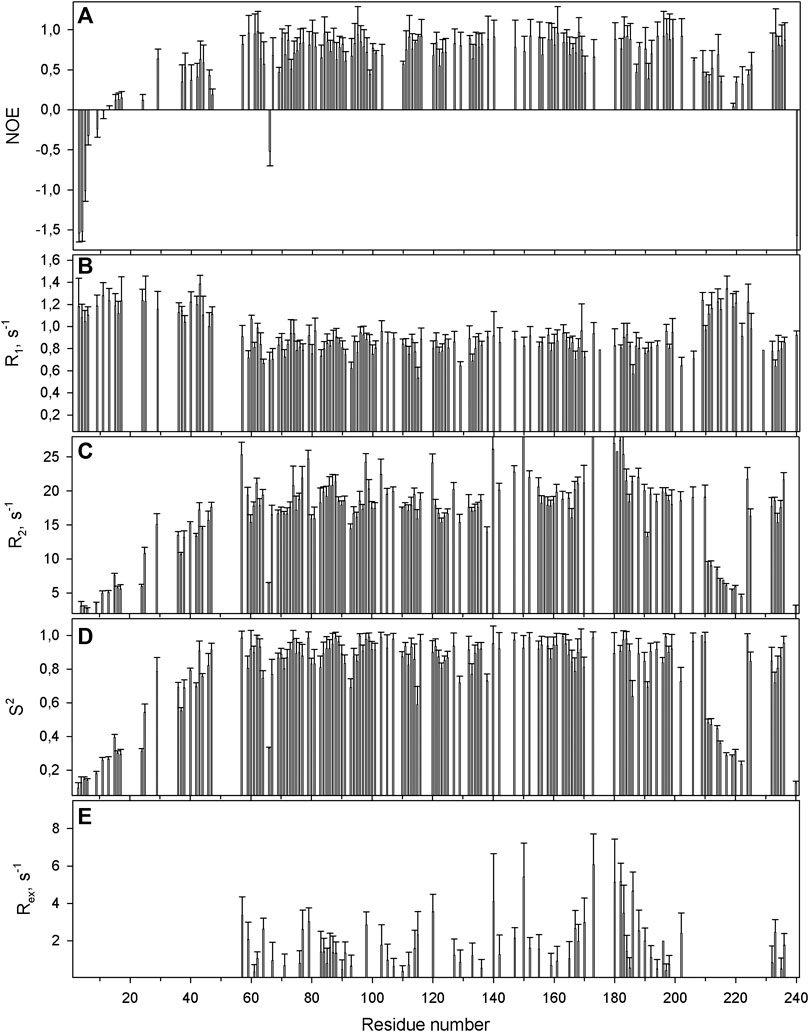
FIGURE 5. The relaxation parameters of the amide 15N nuclei of each residue of the apo-form of WBSCR27, measured at 16.3 T (700 MHz proton resonance frequency) and 308 K. (A) The heteronuclear 15N,1H-steady-state NOE values. (B) The longitudinal relaxation rate R1 (s−1). (C) The transverse relaxation rate R2 (s−1). (D) The order parameter S2 determined by model-free analysis. (E) Chemical exchange Rex contributions to the transverse relaxation rates (s−1).
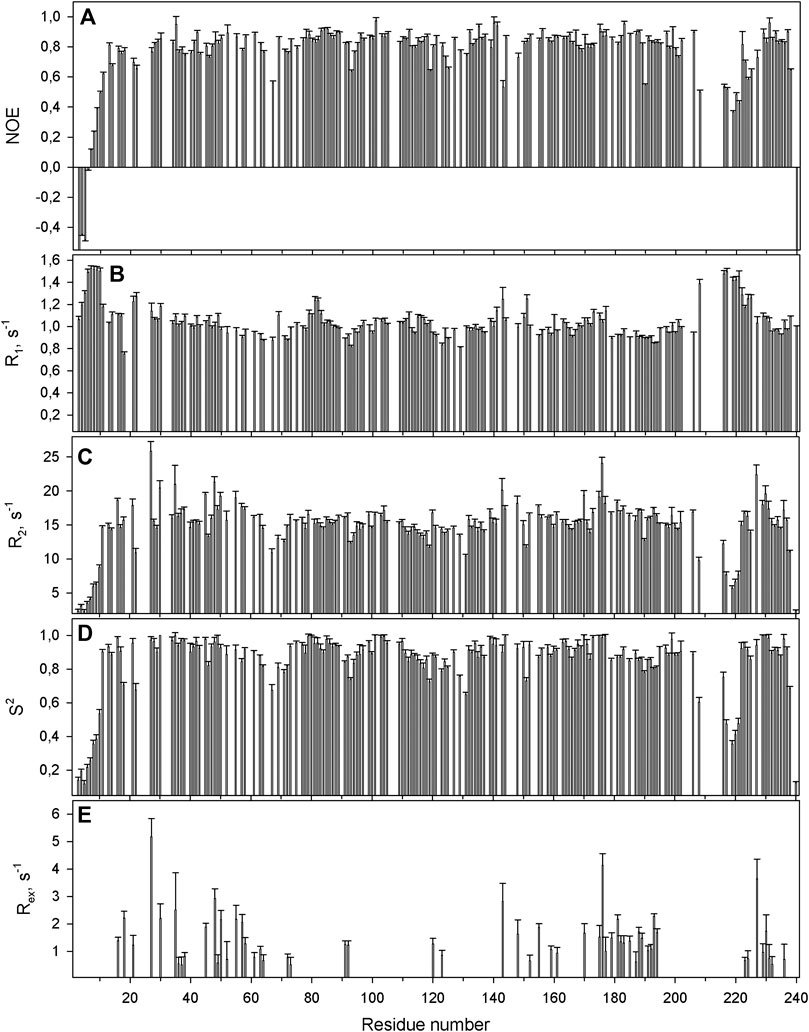
FIGURE 6. The relaxation parameters of the amide 15N nuclei of each residue of the SAH-WBSCR27 complex, measured at 16.3 T (700 MHz proton resonance frequency) and 308 K. (A) The heteronuclear 15N,1H-steady-state NOE values. (B) The longitudinal relaxation rate R1 (s−1). (C) The transverse relaxation rate R2 (s−1). (D) The order parameter S2 determined by model-free analysis. (E) Chemical exchange Rex contributions to the transverse relaxation rates (s−1).
Figure 7 shows the distribution of the protection factors PF for the amino acid residues of the apo-form of WBSCR27 and the WBSCR27-SAH complex, as analyzed from the measured proton-to-deuterium exchange rates. Binding of SAH leads to a significant increase in the PF of most amino acid residues of WBSCR27, which reflects the slowing down of high-amplitude protein backbone motions upon the ligand binding and the strengthening of the hydrogen bond network within the protein molecule.
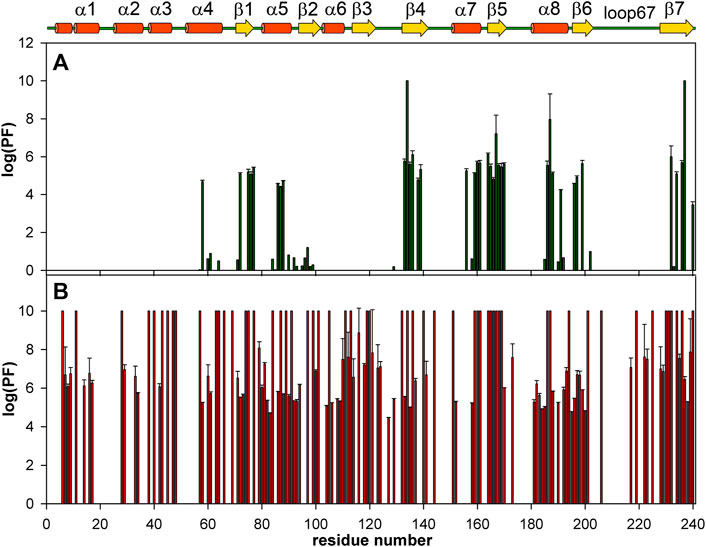
FIGURE 7. Bar charts showing the amide HN protection factors of the apo-WBSCR27 (A) and complex WBSCR27-SAH (B). The elements of protein secondary structure identified for WBSCR27-SAH solution structure are shown on the top.
Dissecting the WBSCR27 structure and delineating its functions may pave the way to understanding the molecular mechanisms underlying the clinical manifestations of WBS. In its turn, this may contribute to developing clinical interventions aiming to compensate for the symptoms of this genetic disease.
Comparing structures of the WBSCR27 apo-form and its complex with SAH indicates the formation of three additional α-helices at the N-terminal tail of the protein upon the cofactor binding (Figure 3). In the apo-form the first 50 residues forming these helices turn out to be disordered. These structural observations are clearly confirmed by the results of the protein backbone dynamics studies (Figures 5–8). For many residues of the apo-WBSCR27 the order parameters of amide NH bonds, determined via the 15N relaxation measurements, are much lower than the corresponding values for the WBSCR27-SAH complex (Figures 5, 6). These results indicate a high amplitude backbone motion of the apo-form of the protein in a time scale from ps to ns. There are also significant differences in the rates of backbone motions occurring in the ms time scale and characterized by the conformational exchange. The protein fragments which contain residues participating in conformational exchange are colored in orange in Figure 8.
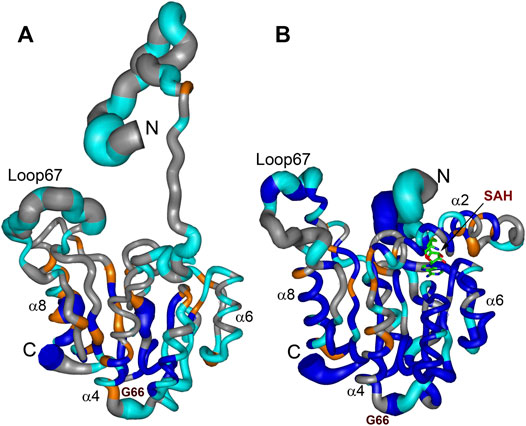
FIGURE 8. Summary of dynamic and conformational behaviour of the WBSCR27 in apo-form (A) and in complex with SAH (B). Representative NMR models of apo-WBSCR27 and complex SAH-WBSCR27 are shown according to protein mobility in a broad time scale. The thickness of the chain is proportional to the value (1 − S2) representing the extent of the local amplitude of protein backbone motion in ps-ns time scale. Fragments of the protein backbone containing amino acids undergoing conformational exchange in ms time scale (with values Rex exceeding 2 s−1) are colored orange. Protein backbone fragments with residues having high values of protection factors determined from the analysis of hydrogen-to-deuterium HN exchange rates are colored blue. Residues for which 15N relaxation data could not be obtained (proline residues and those with overlapped HN signals) are shown in gray. Representative secondary structure elements and SAH molecule are labelled.
The amplitude of the fast protein backbone motions, as well as the conformational transitions occurring in the ms timescale, are much greater for the case of the apo-form than for the WBSCR27-SAH complex. This difference in protein dynamics is observed not only for the first 50 amino acid residues unstructured in the apo-form, but also for the well-structured protein core. For example, the fragments of β4, β5, and α8 are highly mobile in the apo-form. Loop 67 remains highly mobile, both in the apo-form and in the complex. This loop is likely to be involved in recognizing the substrate molecule, and following its binding with helices α1–α3, loop 67 fixes this interaction, after which its mobility should disappear. Notably, the mobility of the G66 residue is high in both forms: the apo-form and the WBSCR27-SAH complex, the magnitude of the 15N-1H NOE of the amide group of this residue is negative. However, the mobility of this residue located at the apex of the loop between α1 and β1 is unlikely to play any functional role.
The differences in the amplitude of the slow protein backbone motions, occurring on a time scale from minutes to hours and determined from analyzing the H/D exchange, are even more obvious when we compare the apo-form and the protein-ligand complex. Relatively high values of amide NH protection factors for the apo-form of WBSCR27 are observed only for the very central part of the protein core (Figures 7, 8). Interestingly, all the amide groups of the residues from outer β-strand β3 and helices α4 and α6 are unprotected in the apo-form of the protein indicating a high mobility of these elements of the secondary structure. After SAH binding, almost the whole protein molecule, except for loop regions, turns out to be well protected from exchanging amide protons with water. This follows from large values of the protection factors of the corresponding NH groups (Figure 8).
Helices α1–α3 in the WBSCR27-SAH complex surround the SAH molecule and partially form its binding site. These three helices also form a binding site for the potential substrate of the methylation reaction, catalyzed by WBSCR27. The adenosine fragment of SAH binds to the tips of the three strands β1, β2, and β3, while the methionine chain is positioned between the helices α2 and α3. The backbone carbonyl groups and amide hydrogens of the residues 100, 101, and 121 form several hydrogen bonds with the adenosine fragment. The side chains of the residues L11, L31, A77, and T121 form hydrophobic interactions with the adenine moiety of the SAH. In the region of the methionine fragment, there are also two aromatic residues, Y41 and W37. If the role of Y41 is most likely related to interacting with the carboxyl or amino group of the SAH methionine fragment, then W37 may participate in the interaction with a potential substrate fragment. It may, for instance, hold the aromatic base of RNA or DNA by stacking interaction in the position favorable for methylating this nucleotide or the neighboring one. This possibility is evidenced by the outward orientation of the side chain of W37 from the protein core, and its proximity to the sulfur atom of the SAH methionine residue.
The structure of WBSCR27 represents a classical Rossmann fold (Figure 3C), typical of all Class I SAM-dependent MTases. More than 120 various members of this enzyme family were classified (Martin and McMillan, 2002). They have different methylation substrates and very little sequence identity, but a highly conserved structural fold (Figure 9) and a β-sheet core, formed by seven β-strands (Figure 9). There are some variations in the number, length and orientations of α-helices surrounding this β-sheet, but they are still rather conservative in their structure. The greatest differences, as expected, are observed in the structure of the substrate-binding regions (colored purple in Figure 9), although the substrate molecule can also interact with the residues in the core region of the protein. The substrate-binding domain should ensure the selectivity of the substrate molecule binding and its correct positioning relative to the methyl group of the co-factor. However, the variability in the structure of the substrate-binding domain is great even for one type of substrate. Figures 9B,D,E show the structures of RNA MTases with significantly different substrate-binding fragments. Notably, there are two or three α-helices in the proximity to the SAM binding site in RNA MTases. A similar topology is observed in the case of DNA MTases (Figure 9C). The topology of the substrate-binding domain for small molecule methylation MTase (the structure of glycine MTase is shown in Figure 9F as an example, Luka et al., 2007) is markedly different. Based on these structural considerations, a nucleic acid would be the most likely substrate for WBSCR27, but other options cannot be ruled out.
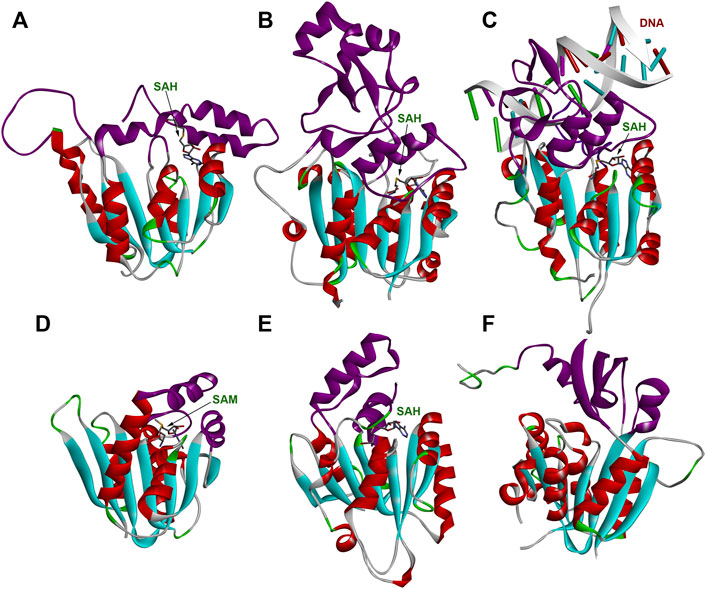
FIGURE 9. Structures of several MTases. (A) WBSCR27 in complex with SAH (PDB id 7QCB); (B) S. pombe tRNAAsp MTase DNMT2 in complex with SAH (PDB id 6FDF); (C) human DNA MTase DNMT3A in complex with SAH and DNA fragment (PDB id 6BRR); (D) S. cerevisiae rRNA MTase Bud23 (PDB id 4QTU); (E) human tRNAHis MTase BCDIN3D in complex with SAH (PDB id 6L8U); (F) human glycine MTase (PDB id 2AZT). Rossmann-fold of the enzymes’ core is colored in cyan (β-strands) and red (α-helices). Substrate-binding domains are colored purple.
One of the closest sequence homologs of WBSCR27 is a human protein WBSCR22 and its yeast ortholog Bud23 (Mariasina et al., 2018). WBSCR22 is a 18S rRNA MTase involved in pre-rRNA processing and ribosome 40 S subunit biogenesis (Haag et al., 2015). WBSCR22 has an interaction partner—the protein TRMT112 which is vital for the functional activity of this MTase in mammalian cells (Õunap et al., 2015). The known 3D structure of the complex of Bud23 with TRMT112 (Létoquart et al., 2014) allows comparing the TRMT112-binding interface on the surface of Bud23 with the similar area of the molecular surface of WBSCR27 (Supplementary Figure S9). The patterns of electrostatic potentials on the surface of Bud23 and WBSCR27 are quite different, and it is unlikely that TRMT112 can be the functional partner of WBSCR27. However, we tested this hypothesis using co-immunoprecipitation and antibody staining for TRMT112. The experimental data obtained confirm the conclusion that these proteins do not interact with each other.
The protein folding topology and three-dimensional structure of WBSCR27 are similar to those of RNA MTase GidB (Romanowski et al., 2002). Notably, for GidB, as well as for WBSCR27, the enzyme function and methylation substrate were initially unknown. At the same time, establishing the three-dimensional structure of this MTase accelerated identifying the methylation substrate. This enzyme (alias RsmG) was recently shown to be responsible for N7 methylation in G527 of 16 S bacterial rRNA (Abedeera et al., 2020).
While our work has not yielded WBSCR27 substrates and partners, the negative result of this kind is also informative potentially narrowing down the range of possibilities for future research. First and foremost, WBSCR27 seems not to form any stable interaction with a substrate, unlike MTases functioning as the molecular switches in ribosome assembly, i.e., bacterial KsgA (Connolly et al., 2008) or mammalian METTL15 (Laptev et al., 2020). A number of MTases responsible for modifying the translation apparatus components and other substrates form a stable complex with the TRMT112 protein (Zorbas et al., 2015; Metzger et al., 2019; van Tran et al., 2019; Yang et al., 2021), while mRNA specific MTase METTL3 forms a stable complex with METTL14 (Liu et al., 2014; Wang et al., 2014) and WTAP (Ping et al., 2014). The results of this study disfavor the scenario that WBSCR27 forms a stable functional complex with other proteins. WBSCR27 is likely to be a standalone MTase only transiently interacting with its substrate. Moreover, WBSCR27 is unlikely to catalyze protein methylation, otherwise there must be a target protein that would be found in one of the experiments described above.
Our results do not exclude that WBSCR27 possibly participates in the methylation of a small molecule, whose chemical properties do not allow it to be identified in cell lysates by NMR methods. An overarching theory unpacking WBSCR27 functions is still ahead, and we hope that determining the three-dimensional structure of this enzyme in the apo-form and in the form of a complex with SAH will help achieve this goal.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
SM, OD, PS, and VP designed the study. SM, C-FC, AC, TN, VZ, SE, and VI performed experiments and acquired data. SM and VP carried out structure calculations. SM, PS, and VP analyzed and interpreted data and drafted the manuscript. All authors contributed to the article and approved the submitted version.
The Russian Foundation for Basic Research (grant 20-04-00318) supported the NMR and structural studies. Gene editing experiments were supported by the Russian Science Foundation (grant 21-64-00006). Search for potential substrates was supported by the Russian Foundation for Basic Research (grant 20-04-00736). This work was done also with support of the Interdisciplinary Scientific and Educational School of Moscow University «Molecular Technologies of the Living Systems and Synthetic Biology». The 800 and 850 MHz NMR experiments were carried out using the equipment of the High-Field Nuclear Magnetic Resonance Center (HFNMRC) supported by Academia Sinica (AS-CFII-108-112), Taipei, Taiwan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to Moscow State University (Russia) for the opportunity to use the NMR facilities and the supercomputer SKIF Lomonosov. The authors are grateful to Advanced Isotope Labelling for NMR School in Grenoble (France, 2019) for the opportunity to prepare specifically labelled WBSCR27 samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.865743/full#supplementary-material
Abedeera, S. M., Hawkins, C. M., and Abeysirigunawardena, S. C. (2020). RsmG Forms Stable Complexes with Premature Small Subunit rRNA during Bacterial Ribosome Biogenesis. RSC Adv. 10 (38), 22361–22369. doi:10.1039/d0ra02732d
An, T., Zhang, J., Lv, B., Liu, Y., Huang, J., Lian, J., et al. (2019). Salvianolic Acid B Plays an Anti-obesity Role in High Fat Diet-Induced Obese Mice by Regulating the Expression of mRNA, circRNA, and lncRNA. PeerJ. 7, e6506. doi:10.7717/peerj.6506
Bardiaux, B., Malliavin, T., and Nilges, M. (2012). “ARIA for Solution and Solid-State NMR,” in Protein NMR Techniques. Editors A. Shekhtman, and D.S. Burz (Totowa, NJ: Humana Press), 453–483. doi:10.1007/978-1-61779-480-3_23
Barsnes, H., and Vaudel, M. (2018). SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and De Novo Engines. J. Proteome Res. 17 (7), 2552–2555. doi:10.1021/acs.jproteome.8b00175
Bennett, M. R., Shepherd, S. A., Cronin, V. A., and Micklefield, J. (2017). Recent Advances in Methyltransferase Biocatalysis. Curr. Opin. Chem. Biol. 37, 97–106. doi:10.1016/j.cbpa.2017.01.020
Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., et al. (1998). Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Cryst. D. 54 (5), 905–921. doi:10.1107/S0907444998003254
Campeanu, I. J., Jiang, Y., Liu, L., Pilecki, M., Najor, A., Cobani, E., et al. (2021). Multi-omics Integration of Methyltransferase-like Protein Family Reveals Clinical Outcomes and Functional Signatures in Human Cancer. Sci. Rep. 11 (1), 14784. doi:10.1038/s41598-021-94019-5
Chugunova, A., Loseva, E., Mazin, P., Mitina, A., Navalayeu, T., Bilan, D., et al. (2019). LINC00116 Codes for a Mitochondrial Peptide Linking Respiration and Lipid Metabolism. Proc. Natl. Acad. Sci. U.S.A. 116 (11), 4940–4945. doi:10.1073/pnas.1809105116
Clore, G. M., Driscoll, P. C., Wingfield, P. T., and Gronenborn, A. M. (1990). Analysis of the Backbone Dynamics of interleukin-1.Beta. Using Two-Dimensional Inverse Detected Heteronuclear Nitrogen-15-Proton NMR Spectroscopy. Biochemistry 29 (32), 7387–7401. doi:10.1021/bi00484a006
Connolly, K., Rife, J. P., and Culver, G. (2008). Mechanistic Insight into the Ribosome Biogenesis Functions of the Ancient Protein KsgA. Mol. Microbiol. 70 (5), 1062–1075. doi:10.1111/j.1365-2958.2008.06485.x
Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 6 (3), 277–293. doi:10.1007/BF00197809
Ewart, A. K., Morris, C. A., Atkinson, D., Jin, W., Sternes, K., Spallone, P., et al. (1993). Hemizygosity at the Elastin Locus in a Developmental Disorder, Williams Syndrome. Nat. Genet. 5, 11–16. doi:10.1038/ng0993-11
Field, J., Nikawa, J., Broek, D., MacDonald, B., Rodgers, L., Wilson, I. A., et al. (1988). Purification of a RAS-Responsive Adenylyl Cyclase Complex from Saccharomyces cerevisiae by Use of an Epitope Addition Method. Mol. Cell Biol. 8 (5), 2159–2165. doi:10.1128/mcb.8.5.2159-2165.1988
Frangiskakis, J. M., Ewart, A. K., Morris, C. A., Mervis, C. B., Bertrand, J., Robinson, B. F., et al. (1996). LIM-kinase1 Hemizygosity Implicated in Impaired Visuospatial Constructive Cognition. Cell 86 (1), 59–69. doi:10.1016/S0092-8674(00)80077-X
Gopanenko, A. V., Malygin, A. A., Tupikin, A. E., Laktionov, P. P., Kabilov, M. R., and Karpova, G. G. (2017). Human Ribosomal Protein eS1 Is Engaged in Cellular Events Related to Processing and Functioning of U11 snRNA. Nucleic Acids Res. 45 (15), 9121–9137. doi:10.1093/nar/gkx559
Haag, S., Kretschmer, J., and Bohnsack, M. T. (2015). WBSCR22/Merm1 Is Required for Late Nuclear Pre-ribosomal RNA Processing and Mediates N7-Methylation of G1639 in Human 18S rRNA. RNA 21 (2), 180–187. doi:10.1261/rna.047910.114
Hahn, Y., and Lee, B. (2005). Identification of Nine Human-specific Frameshift Mutations by Comparative Analysis of the Human and the Chimpanzee Genome Sequences. Bioinformatics 21 (Suppl. l_1), i186–i194. doi:10.1093/bioinformatics/bti1000
Hansen, M. R., Mueller, L., and Pardi, A. (1998). Tunable Alignment of Macromolecules by Filamentous Phage Yields Dipolar Coupling Interactions. Nat. Struct. Mol. Biol. 5 (12), 1065–1074. doi:10.1038/4176
Huber, T. D., Johnson, B. R., Zhang, J., and Thorson, J. S. (2016). AdoMet Analog Synthesis and Utilization: Current State of the Art. Curr. Opin. Biotechnol. 42, 189–197. doi:10.1016/j.copbio.2016.07.005
Husmann, D., and Gozani, O. (2019). Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 26 (10), 880–889. doi:10.1038/s41594-019-0298-7
Ivanova, E. V., Kolosov, P. M., Birdsall, B., Kelly, G., Pastore, A., Kisselev, L. L., et al. (2007). Eukaryotic Class 1 Translation Termination Factor eRF1 − the NMR Structure and Dynamics of the Middle Domain Involved in Triggering Ribosome-dependent Peptidyl-tRNA Hydrolysis. FEBS J. 274 (16), 4223–4237. doi:10.1111/j.1742-4658.2007.05949.x
Jones, W., Bellugi, U., Lai, Z., Chiles, M., Reilly, J., Lincoln, A., et al. (2000). II. Hypersociability in Williams Syndrome. J. Cogn. Neurosci. 12 (Suppl. 1), 30–46. doi:10.1162/089892900561968
Kay, L. E., Torchia, D. A., and Bax, A. (1989). Backbone Dynamics of Proteins as Studied by 15N Inverse Detected Heteronuclear NMR Spectroscopy: Application to Staphylococcal Nuclease. Biochemistry 28 (23), 8972–8979. doi:10.1021/bi00449a003
Kerfah, R., Plevin, M. J., Sounier, R., Gans, P., and Boisbouvier, J. (2015). Methyl-specific Isotopic Labeling: a Molecular Tool Box for Solution NMR Studies of Large Proteins. Curr. Opin. Struct. Biol. 32, 113–122. doi:10.1016/j.sbi.2015.03.009
Kitagawa, H., Fujiki, R., Yoshimura, K., Mezaki, Y., Uematsu, Y., Matsui, D., et al. (2003). RETRACTED: The Chromatin-Remodeling Complex WINAC Targets a Nuclear Receptor to Promoters and Is Impaired in Williams Syndrome. Cell 113 (7), 905–917. doi:10.1016/S0092-8674(03)00436-7
Kuszewski, J., Gronenborn, A. M., and Clore, G. M. (1997). Improvements and Extensions in the Conformational Database Potential for the Refinement of NMR and X-Ray Structures of Proteins and Nucleic Acids. J. Magnetic Reson. 125 (1), 171–177. doi:10.1006/jmre.1997.1116
Laptev, I., Shvetsova, E., Levitskii, S., Serebryakova, M., Rubtsova, M., Zgoda, V., et al. (2020). METTL15 Interacts with the Assembly Intermediate of Murine Mitochondrial Small Ribosomal Subunit to Form m4C840 12S rRNA Residue. Nucl. Acids Res. 48 (14), 8022–8034. doi:10.1093/nar/gkaa522
Laskowski, R., Rullmann, J. A., MacArthur, M., Kaptein, R., and Thornton, J. (1996). AQUA and PROCHECK-NMR: Programs for Checking the Quality of Protein Structures Solved by NMR. J. Biomol. NMR 8 (4), 477–486. doi:10.1007/BF00228148
Lee, W., Tonelli, M., and Markley, J. L. (2015). NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 31 (8), 1325–1327. doi:10.1093/bioinformatics/btu830
Lesnyak, D. V., Sergiev, P. V., Bogdanov, A. A., and Dontsova, O. A. (2006). Identification of Escherichia coli m2G Methyltransferases: I. The ycbY Gene Encodes a Methyltransferase Specific for G2445 of the 23 S rRNA. J. Mol. Biol. 364 (1), 20–25. doi:10.1016/j.jmb.2006.09.009
Létoquart, J., Huvelle, E., Wacheul, L., Bourgeois, G., Zorbas, C., Graille, M., et al. (2014). Structural and Functional Studies of Bud23-Trm112 Reveal 18S rRNA N 7 -G1575 Methylation Occurs on Late 40S Precursor Ribosomes. Proc. Natl. Acad. Sci. U.S.A. 111 (51), E5518–E5526. doi:10.1073/pnas.1413089111
Lipari, G., and Szabo, A. (1982). Model-Free Approach to the Interpretation of Nuclear Magnetic-Resonance Relaxation in Macromolecules .1. Theory and Range of Validity. J. Am. Chem. Soc. 104 (17), 4546–4559. doi:10.1021/ja00381a009
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 Complex Mediates Mammalian Nuclear RNA N6-Adenosine Methylation. Nat. Chem. Biol. 10 (2), 93–95. doi:10.1038/nchembio.1432
Luka, Z., Pakhomova, S., Luka, Y., Newcomer, M. E., and Wagner, C. (2007). Destabilization of Human glycine N-Methyltransferase by H176N Mutation. Protein Sci. 16 (9), 1957–1964. doi:10.1110/ps.072921507
Mariasina, S. S., Chang, C. F., Petrova, O. A., Efimov, S. V., Klochkov, V. V., Kechko, O. I., et al. (2020). Williams–Beuren Syndrome-Related Methyltransferase WBSCR27: Cofactor Binding and Cleavage. Febs J. 287 (24), 5375–5393. doi:10.1111/febs.15320
Mariasina, S. S., Petrova, O. A., Osterman, I. A., Sergeeva, O. V., Efimov, S. V., Klochkov, V. V., et al. (2018). NMR Assignments of the WBSCR27 Protein Related to Williams-Beuren Syndrome. Biomol. NMR Assign. 12 (2), 303–308. doi:10.1007/s12104-018-9827-2
Martin, J., and McMillan, F. M. (2002). SAM (Dependent) I AM: the S-adenosylmethionine-dependent Methyltransferase Fold. Curr. Opin. Struct. Biol. 12 (6), 783–793. doi:10.1016/S0959-440X(02)00391-3
Masserini, B., Bedeschi, M. F., Bianchi, V., Scuvera, G., Beck-Peccoz, P., Lalatta, F., et al. (2013). Prevalence of Diabetes and Pre-diabetes in a Cohort of Italian Young Adults with Williams Syndrome. Am. J. Med. Genet. 161 (4), 817–821. doi:10.1002/ajmg.a.35655
Mátés, L., Chuah, M. K. L., Belay, E., Jerchow, B., Manoj, N., Acosta-Sanchez, A., et al. (2009). Molecular Evolution of a Novel Hyperactive Sleeping Beauty Transposase Enables Robust Stable Gene Transfer in Vertebrates. Nat. Genet. 41 (6), 753–761. doi:10.1038/ng.343
Metzger, E., Wang, S., Urban, S., Willmann, D., Schmidt, A., Offermann, A., et al. (2019). KMT9 Monomethylates Histone H4 Lysine 12 and Controls Proliferation of Prostate Cancer Cells. Nat. Struct. Mol. Biol. 26 (5), 361–371. doi:10.1038/s41594-019-0219-9
Ottiger, M., and Bax, A. (1999). Bicelle-based Liquid Crystals for NMR-Measurement of Dipolar Couplings at Acidic and Basic pH Values. J. Biomol. NMR 13 (2), 187–191. doi:10.1023/a:1008395916985
Ottiger, M., Delaglio, F., and Bax, A. (1998). Measurement ofJand Dipolar Couplings from Simplified Two-Dimensional NMR Spectra. J. Magnetic Reson. 131 (2), 373–378. doi:10.1006/jmre.1998.1361
Õunap, K., Leetsi, L., Matsoo, M., and Kurg, R. (2015). The Stability of Ribosome Biogenesis Factor WBSCR22 Is Regulated by Interaction with TRMT112 via Ubiquitin-Proteasome Pathway. PLOS One 10 (7), e0133841. doi:10.1371/journal.pone.0133841
Ping, X.-L., Sun, B.-F., Wang, L., Xiao, W., Yang, X., Wang, W.-J., et al. (2014). Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res. 24 (2), 177–189. doi:10.1038/cr.2014.3
Pober, B. R. (2010). Williams-beuren Syndrome. N. Engl. J. Med. 362 (3), 239–252. doi:10.1056/NEJMra0903074
Polshakov, V. I., Batuev, E. A., and Mantsyzov, A. B. (2019). NMR Screening and Studies of Target - Ligand Interactions. Russ. Chem. Rev. 88 (1), 59–98. doi:10.1070/rcr4836
Polshakov, V. I., Birdsall, B., Frenkiel, T. A., Gargaro, A. R., and Feeney, J. (1999). Structure and Dynamics in Solution of the Complex of Lactobacillus Casei Dihydrofolate Reductase with the New Lipophilic Antifolate Drug Trimetrexate. Protein Sci. 8 (3), 467–481. doi:10.1110/ps.8.3.467
Romanowski, M. J., Bonanno, J. B., and Burley, S. K. (2002). Crystal Structure of the Escherichia coli Glucose-Inhibited Division Protein B (GidB) Reveals a Methyltransferase Fold. Proteins 47 (4), 563–567. doi:10.1002/prot.10121
Schubert, C. (2009). The Genomic Basis of the Williams–Beuren Syndrome. Cell. Mol. Life Sci. 66 (7), 1178–1197. doi:10.1007/s00018-008-8401-y
Roux, K. J., Kim, D. I., Burke, B., and May, D. G. (2018). BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 91 (1), 19.23.1–19.23.15. doi:10.1002/cpps.51
Sergiev, P. V., Aleksashin, N. A., Chugunova, A. A., Polikanov, Y. S., and Dontsova, O. A. (2018). Structural and Evolutionary Insights into Ribosomal RNA Methylation. Nat. Chem. Biol. 14, 226–235. doi:10.1038/nchembio.2569
Shemiakina, I. I., Ermakova, G. V., Cranfill, P. J., Baird, M. A., Evans, R. A., Souslova, E. A., et al. (2012). A Monomeric Red Fluorescent Protein with Low Cytotoxicity. Nat. Commun. 3 (1), 1204. doi:10.1038/ncomms2208
Shen, Y., Delaglio, F., Cornilescu, G., and Bax, A. (2009). TALOS Plus : a Hybrid Method for Predicting Protein Backbone Torsion Angles from NMR Chemical Shifts. J. Biomol. NMR 44 (4), 213–223. doi:10.1007/s10858-009-9333-z
van Tran, N., Ernst, F. G. M., Hawley, B. R., Zorbas, C., Ulryck, N., Hackert, P., et al. (2019). The Human 18S rRNA m6A Methyltransferase METTL5 Is Stabilized by TRMT112. Nucl. Acids Res. 47 (15), 7719–7733. doi:10.1093/nar/gkz619
Vaudel, M., Burkhart, J. M., Zahedi, R. P., Oveland, E., Berven, F. S., Sickmann, A., et al. (2015). PeptideShaker Enables Reanalysis of MS-derived Proteomics Data Sets. Nat. Biotechnol. 33 (1), 22–24. doi:10.1038/nbt.3109
vonHoldt, B. M., Ji, S. S., Aardema, M. L., Stahler, D. R., Udell, M. A. R., and Sinsheimer, J. S. (2018). Activity of Genes with Functions in Human Williams-Beuren Syndrome Is Impacted by Mobile Element Insertions in the Gray Wolf Genome. Genome Biol. Evol. 10 (6), 1546–1553. doi:10.1093/gbe/evy112
vonHoldt, B. M., Shuldiner, E., Koch, I. J., Kartzinel, R. Y., Hogan, A., Brubaker, L., et al. (2017). Structural Variants in Genes Associated with Human Williams-Beuren Syndrome Underlie Stereotypical Hypersociability in Domestic Dogs. Sci. Adv. 3 (7), e1700398. doi:10.1126/sciadv.1700398
Wang, K., Zhang, J., Deng, M., Ju, Y., and Ouyang, M. (2022). METTL27 is a Prognostic Biomarker of Colon Cancer and Associated With Immune Invasion. J. South. Med. Univ. 42 (4), 486–497. doi:10.12122/j.issn.1673-4254.2022.04.04
Wang, Y., Li, Y., Toth, J. I., Petroski, M. D., Zhang, Z., and Zhao, J. C. (2014). N6-methyladenosine Modification Destabilizes Developmental Regulators in Embryonic Stem Cells. Nat. Cell Biol. 16 (2), 191–198. doi:10.1038/ncb2902
Yang, W.-Q., Xiong, Q.-P., Ge, J.-Y., Li, H., Zhu, W.-Y., Nie, Y., et al. (2021). THUMPD3-TRMT112 Is a m2G Methyltransferase Working on a Broad Range of tRNA Substrates. Nucl. Acids Res. 49 (20), 11900–11919. doi:10.1093/nar/gkab927
Zhang, J., and Zheng, Y. G. (2016). SAM/SAH Analogs as Versatile Tools for SAM-dependent Methyltransferases. ACS Chem. Biol. 11 (3), 583–597. doi:10.1021/acschembio.5b00812
Keywords: Williams-Beuren syndrome (WBS), methyltransferase (MTase), NMR, protein structure in solution, protein dynamics, S-adenosyl-L-homocysteine (SAH)
Citation: Mariasina SS, Chang C-F, Navalayeu TL, Chugunova AA, Efimov SV, Zgoda VG, Ivlev VA, Dontsova OA, Sergiev PV and Polshakov VI (2022) Williams-Beuren Syndrome Related Methyltransferase WBSCR27: From Structure to Possible Function. Front. Mol. Biosci. 9:865743. doi: 10.3389/fmolb.2022.865743
Received: 30 January 2022; Accepted: 16 May 2022;
Published: 15 June 2022.
Edited by:
Annalisa Pastore, King’s College London, United KingdomReviewed by:
Anna Maria D’Ursi, University of Salerno, ItalyCopyright © 2022 Mariasina, Chang, Navalayeu, Chugunova, Efimov, Zgoda, Ivlev, Dontsova, Sergiev and Polshakov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir I. Polshakov, dnBvbHNoYUBmYm0ubXN1LnJ1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.